pediatrics neuromuscular disorders...
TRANSCRIPT

PEDIATRICS NEUROMUSCULAR DISORDERS (NMD)
1
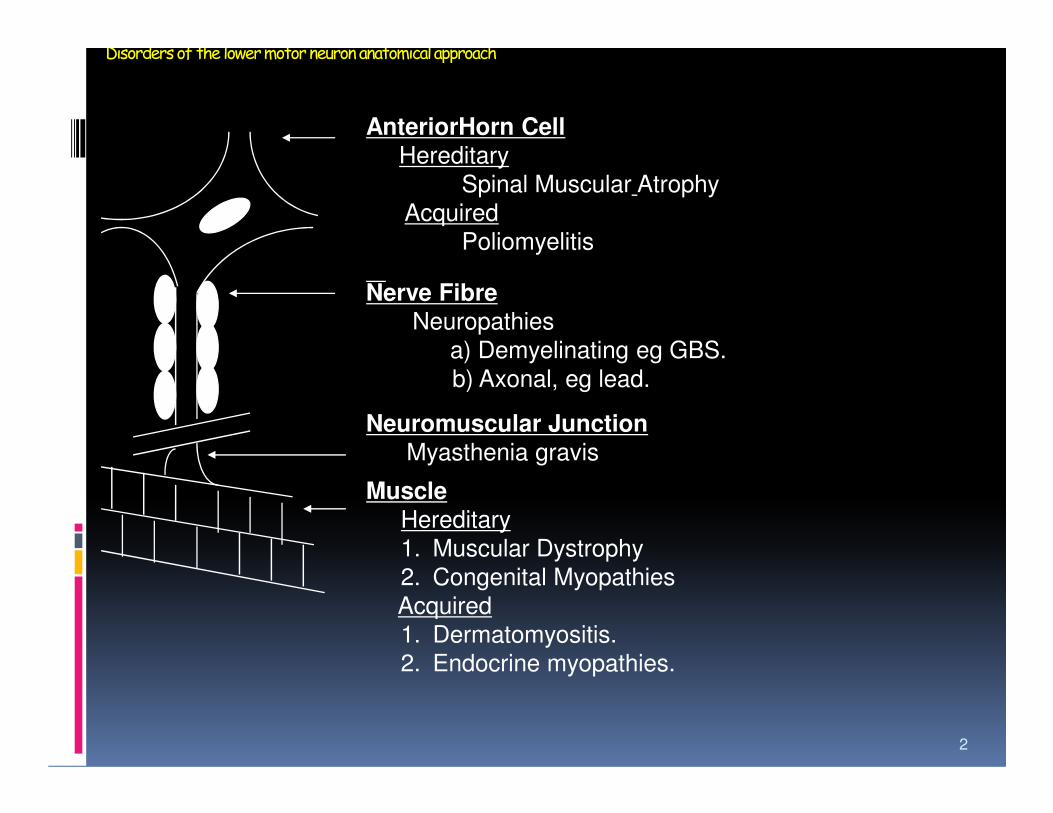
Disorders of the lower motor neuron anatomical approach
AnteriorHorn CellHereditary
Spinal Muscular Atrophy
Acquired
Poliomyelitis
Nerve FibreNeuropathies
a) Demyelinating eg GBS.
b) Axonal, eg lead.
2
Neuromuscular JunctionMyasthenia gravis
Muscle
Hereditary
1. Muscular Dystrophy
2. Congenital Myopathies
Acquired
1. Dermatomyositis.
2. Endocrine myopathies.

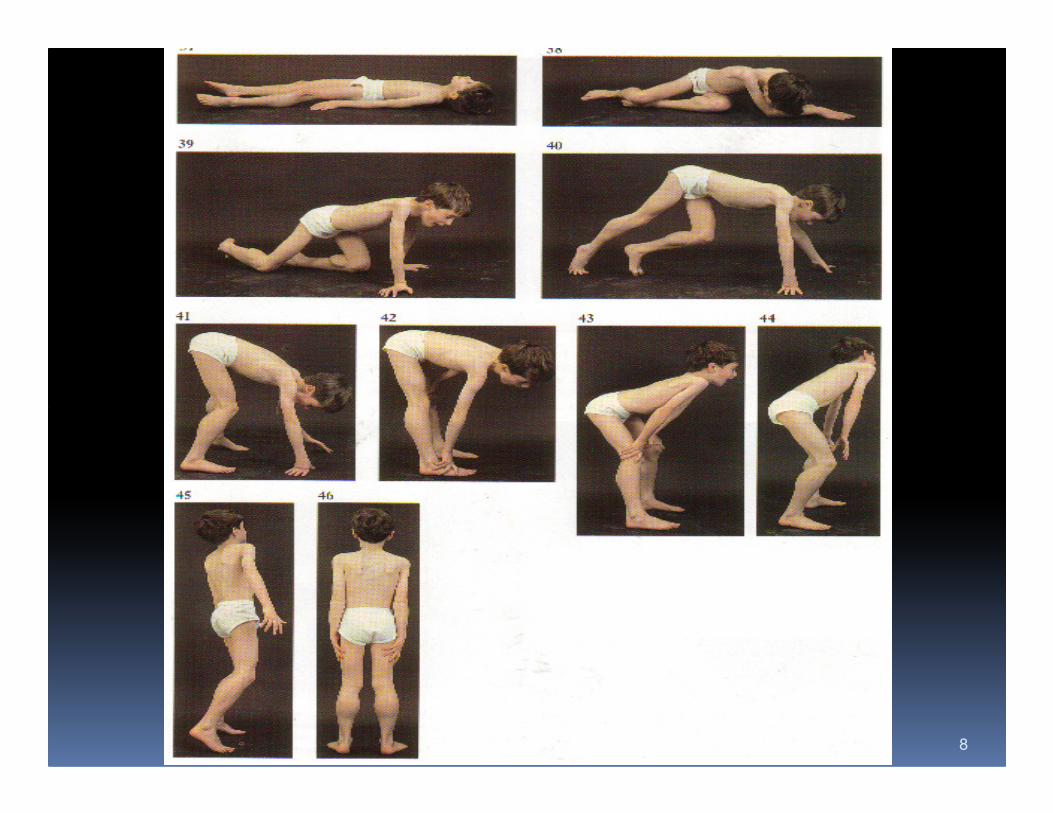
Symptoms of Neuromuscular Disease
1. Abnormal gait
a. Steppage
b. Toe-walking
c. Waddle
2. Easy fatigability
3
2. Easy fatigability
3. Frequent falls
4. Slow motor development
5. Specific disability
a. Arm elevation
b. Climbing stairs
c. Hand grip
d. Rising from floor

Signs of Neuromuscular Disease
Observation1. Atrophy and hypertrophy
2. Fasciculations
3. Functional ability
Palpation
4
Palpation1. Muscle texture
2. Tenderness
Examination1. Joint contractures
2. Myotonia
3. Strength
4. Tendon reflexes

Parental Concern not Weakness
� Trouble with walking and running
� Poor at sports
� Cannot keep up with peers
� Poor coordination
� Tires easily� Tires easily
� Falls frequently
� Delay milestones
� Difficulty climbing stairs
5

NMD in Dept of Childhealth Cipto-Mangunkusumo tHospial
� Duchenne muscular dystrophy (DMD)
� Spinal muscualar atrophy (SMA)
� Guillain-Barre syndrome (GBS)
� Myastenia gravis (MG)
� Chronic inflammatory demyelinating polyneuropathy (CIDP)
� Periodic paralysis� Periodic paralysis
� Myotonia congenita
� Congenital myopathies
� Peroneal muscular atrophy
� Hereditary motor sensory neuropathy
6
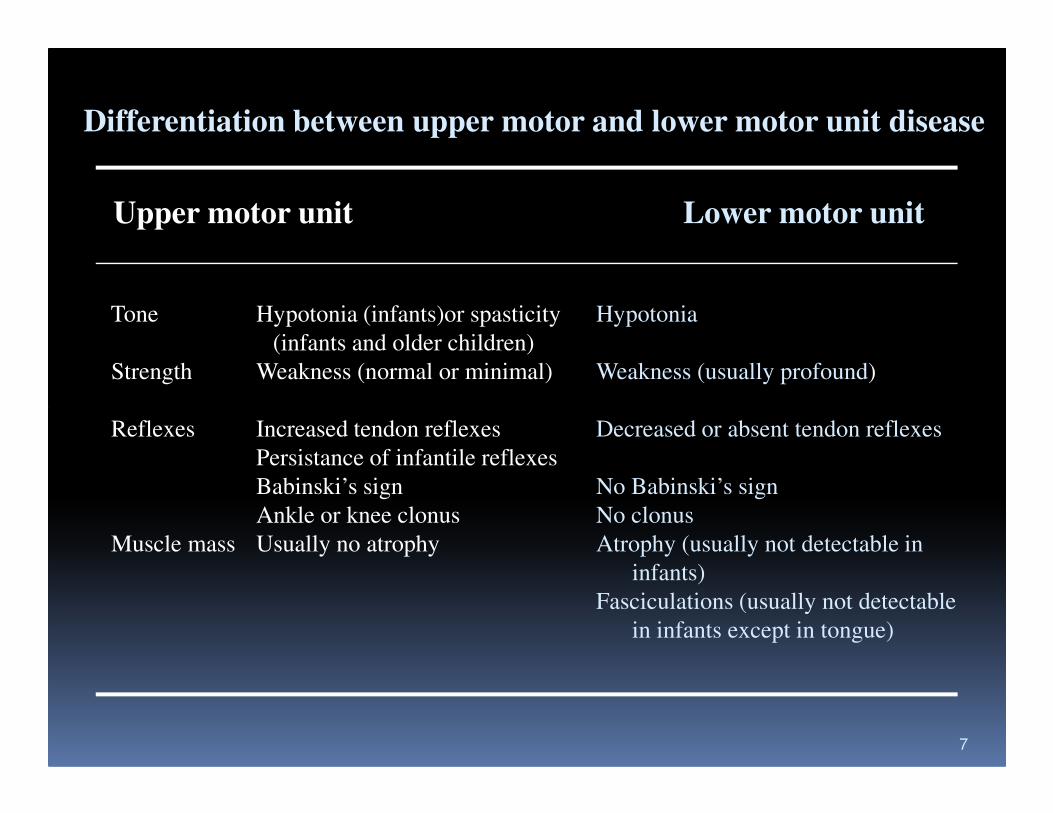
Differentiation between upper motor and lower motor unit disease
Upper motor unit Lower motor unit
Tone Hypotonia (infants)or spasticity Hypotonia
(infants and older children)
Strength Weakness (normal or minimal) Weakness (usually profound)
7
Reflexes Increased tendon reflexes Decreased or absent tendon reflexes
Persistance of infantile reflexes
Babinski’s sign No Babinski’s sign
Ankle or knee clonus No clonus
Muscle mass Usually no atrophy Atrophy (usually not detectable in
infants)
Fasciculations (usually not detectable
in infants except in tongue)
8
9
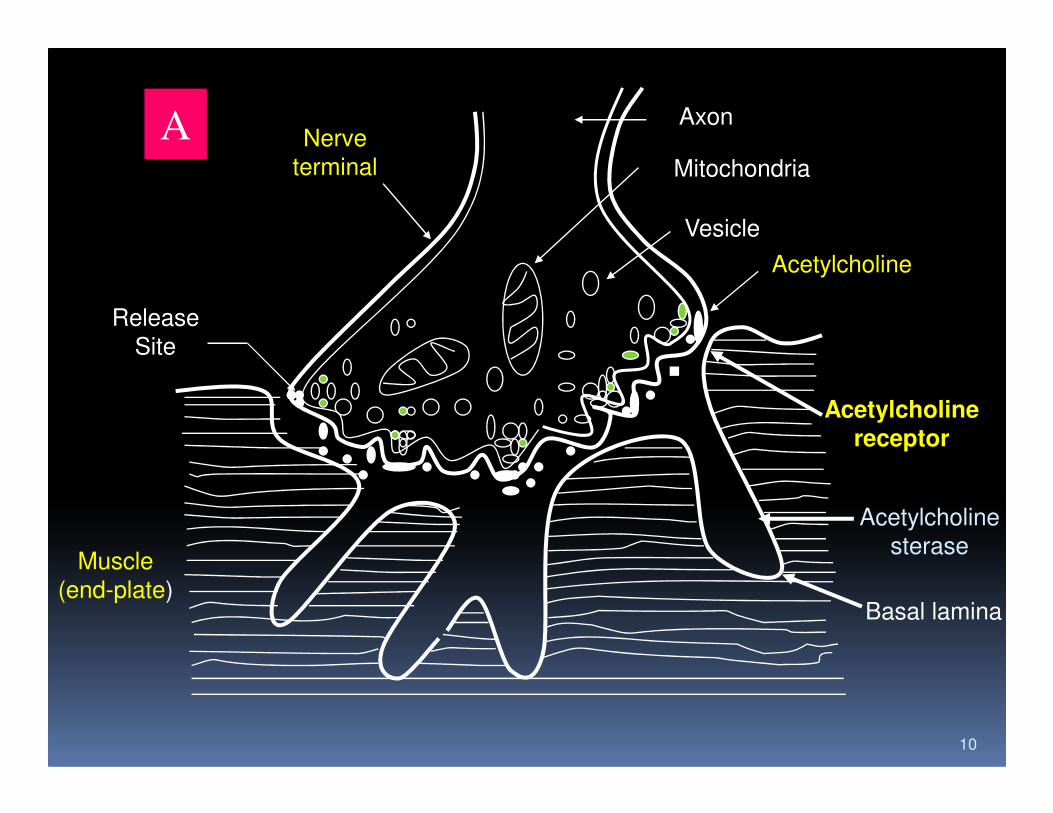
A Nerve
terminal
Axon
Mitochondria
Vesicle
Acetylcholine
Acetylcholine
Release
Site
10
Acetylcholinereceptor
Acetylcholine
sterase
Basal lamina
Muscle
(end-plate)

B
11

Postsynaptic Alterations in MG
1. Reduction of AChR number
2. Destruction and simplification of junctional fold
3. Attachment of antibody to AChR and blocking of
its function
12
its function
4. Increase the gap between nerve terminal and
postsynaptic
FIG.3. Neuromuscular Junction. A, Normal human neuromuscular
Junction. B, Neuromuscular junction of a patient with myasthenia
gravis
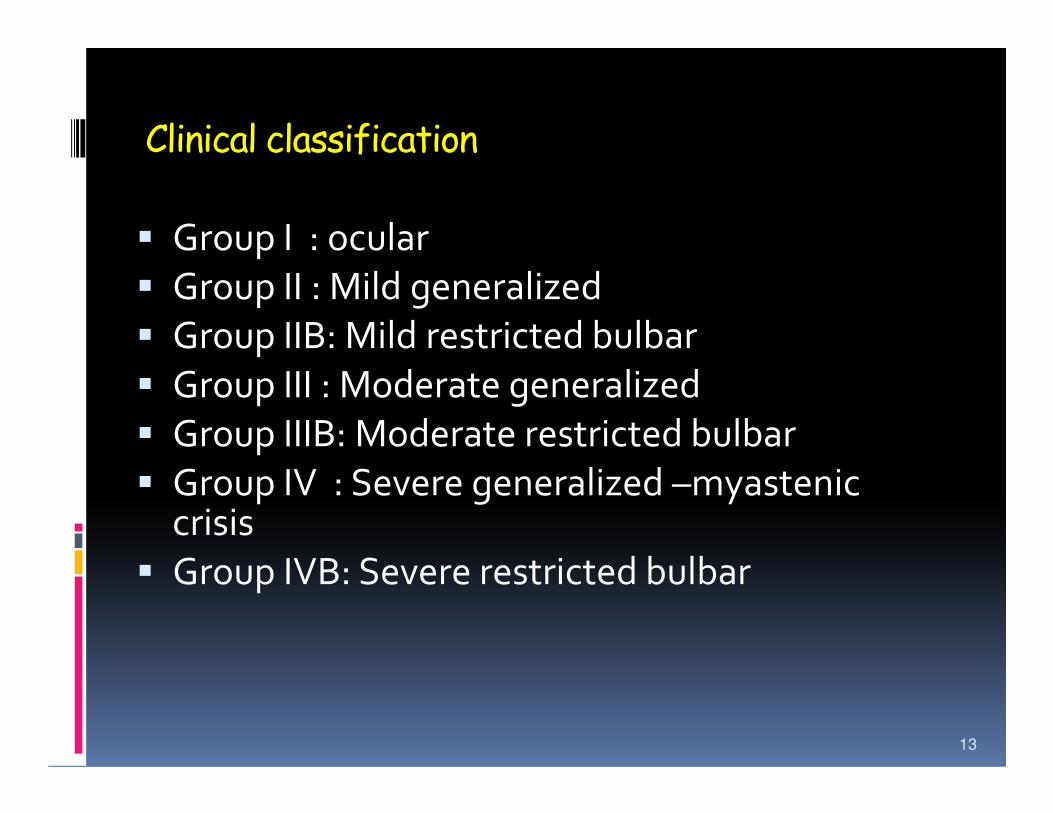
Clinical classification
� Group I : ocular
� Group II : Mild generalized
� Group IIB: Mild restricted bulbar
� Group III : Moderate generalized
� Group IIIB: Moderate restricted bulbar
� Group IV : Severe generalized –myastenic crisis
� Group IVB: Severe restricted bulbar
13

Bedside clinical clues in the diagnosis of MG *
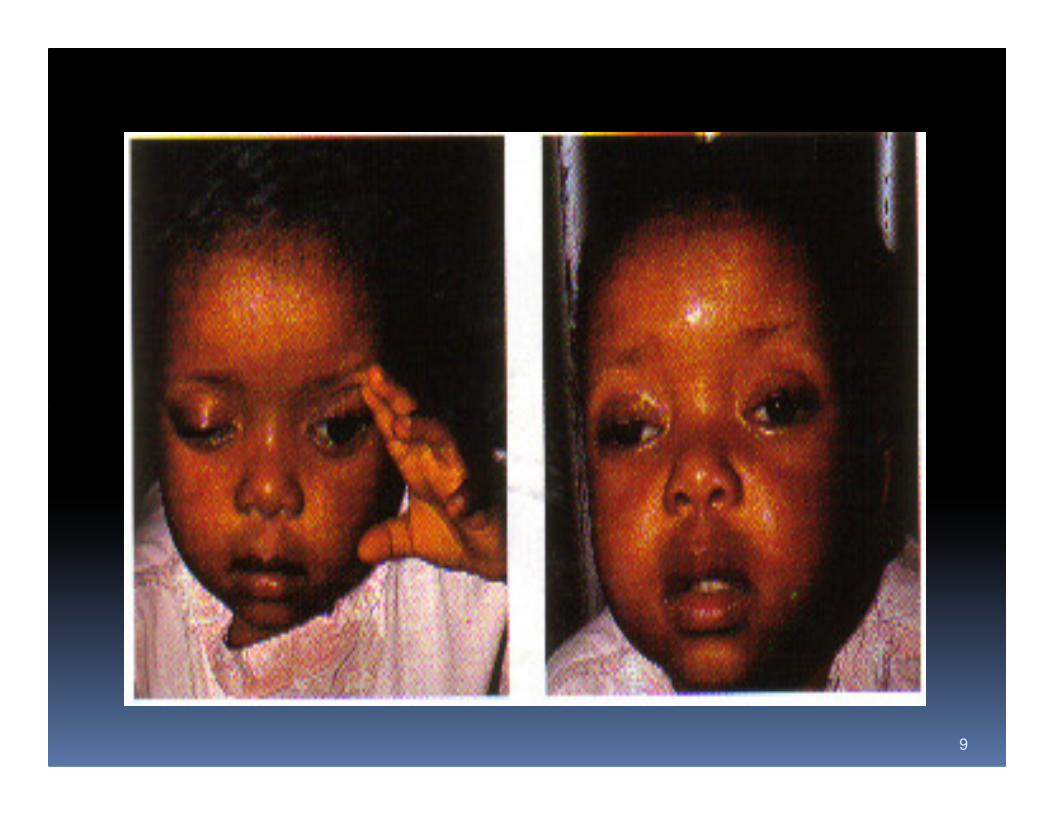
History
Onset of fluctuating ptosis or diplopia that worsens with repeated use
and improves with rest
Onset of fluctuating dysarthria, dysphagia, dysphonia with or without
ocular symptoms or generalized weakness that worsens with repeated
use and improves with rest
14
Physical examination
Weakness referable to ocular, bulbar, or limb muscle
Limb Weakness prominent in proximal flexor groups
Normal muscle tone and bulk
Normal reflexes and sensation
Induction of muscle weakness with exercise when weakness is subtle
* For otherwise healthy persons

Clinical feature
� Ocular symptoms (53%)
� 25% ptosis, 25% diplopia, 3 % blurred visio
� Bulbar symptoms 16%
� 6% difficulty swallowing, 5% slurred, 4% difficulty chewing,
1% dyspnea
� Most patients exhibit progession of disease 86%� Most patients exhibit progession of disease 86%
� 40% purely ocular, 35% generalized, 15% bulbar or ocular-bulbar
� If an ocular MG is going to develop general symptoms
� 56% by 6 months
� 78% by the first years
� 85% by the second years
� 92% by the third years
15

Symtomps and Signs in 35 Patients WithJuvenile Myasthenia Gravis
Symptom or Sign Number of patients
Ptosis 32
Diplopia 30
Facial weakness 29
Dysphonia 29
16
Dysphonia 29
Weakness of arms 29
Weakness of legs 29
Chewing weakness 22
External opthalmoplegia 18
Respiratory difficulties 12
After Millichap and Dodge (69)

TABLE 3. Diagnostic tests *
Studies to demonstrate neuromuscular transmission defectsPharmacologic
Edrophonium (Tensilon)
Neostigmine (Prostigmin)
Electrophysiologic
Repetitive nerve stimulation
Needle electromyography
17
Needle electromyography
Single-fiber electromyography
Studies to demonstrate an abnormal immune respons against the endplate
and muscleAcetylcholine receptor antibodies
Anti-strational muscle antibodies
* Diagnostic test that are currently used in practice

Clinical test for MG
� Edrophonium chloride (Tensilon) (IV)
� Full dose 0,2 mg /kg (maximum dose 10 mg). A small
test 1/10 dose initially is needed
� Should not be given to young infant
� Neostigmine (IM)� Neostigmine (IM)
� Initial test 0,02-0,04 mg/kg is negative, retested 4 hr
later with 0,08 mg/kg
� 0,5 – 1,5 mg IM
� 0,01 mg/kg of atropine before neostigmine
18

TABLE . Therapeutic steps in ocular myasthenia gravis
1. Begin by optimizing the response to pyridostigmine2. If a satisfactory response is obtained, continue pyridostigmine
with lowest effective dose
19
with lowest effective dose3. If the response to pyridostigmine is unsatisfactory, consider
alternate-day low-dose prednisone or immunosuppressive drug

Table 2. Commonly Used Drugs in the Treatment of Myasthenic Disorders
Drug Available Dose Dosage Frequency
Endrophonium chloride (Tensilon) 10 mg/ml 0.2 mg/kg iv
20
Endrophonium chloride (Tensilon) 10 mg/ml 0.2 mg/kg iv
Neostigmine bromide (Prostigmin) 15-mg tablet 7.5-15 mg/dose po 3-4 hours
Neostigmine methylsulfate (Prostigmin) 0.25, 0.5, 1.0 mg/ml 0.04 mg/kg im 3-4 hours
0.02 mg/kg iv
Pyridostigmin bromide (Mestinon) 60-mg tablet 30-60 mg/dose po 4-6 hours
Prednisone 1-, 2.5-, 5-, 20-, 2-3 mg/kg/day alternate day
20-, 50-mg tablets

TABLE 7. Therapeutic options in myasthenia gravis
Symptomatic therapy
Short-acting cholinesterase inhibitors
Pyridostigmine bromide (Mestinon)
Longer-acting cholinesterase inhibitors
Neostigmine (Prostigmin)
Mestinon Timespan
Long-term immunosuppressive drug therapy
21
Long-term immunosuppressive drug therapy
Corticosteroids
Immunosuppressive drugs
Azathioprine (Imuran)
Long-term surgical Immunosuppression
Thymectomy
Short-term Immunosuppression
Plasma exchange
Intravenous administration of immune globulin
22

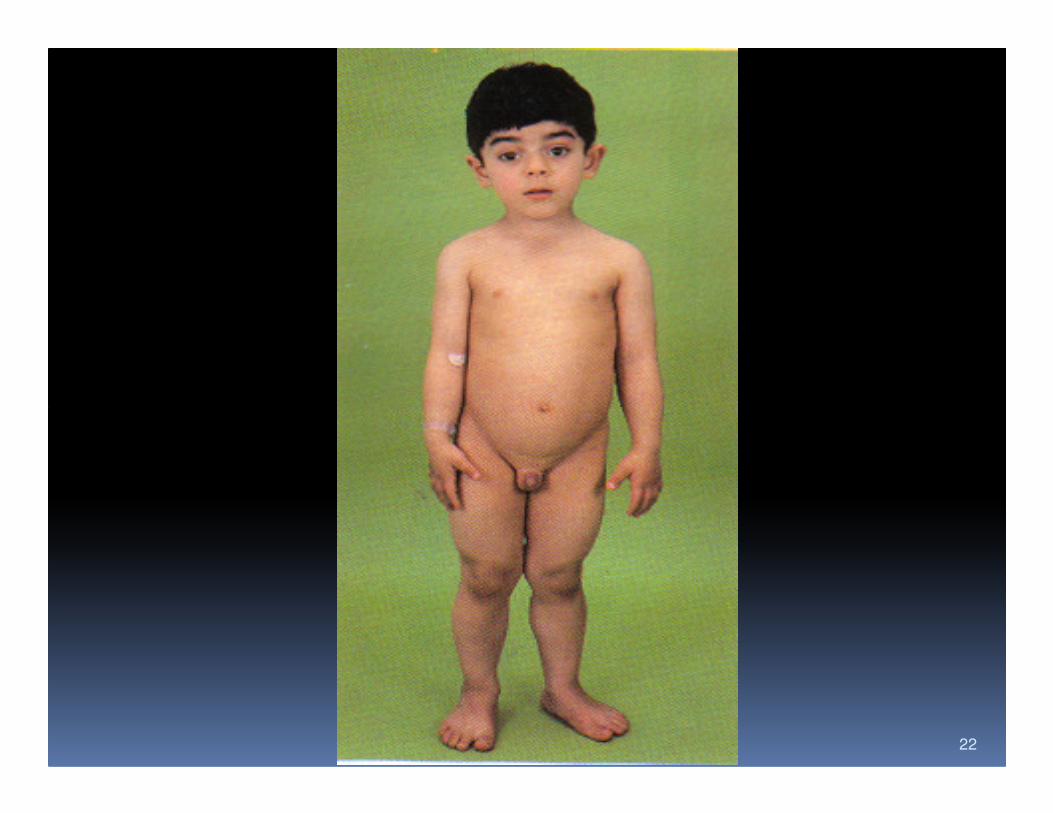
Duchenne muscular dystrophy
� Commonest and most serious type of muscular dystrophy
� 30 per 100,000 live born male affected
� Inheritance is as a sex-linked recessive but mutation are frequent (30%)mutation are frequent (30%)
� Symptoms appear in the first five years
� Prominence of the calf muscle is an early feature (pseudo-hypertrophy).
� Unable to walk by the age of 8 to 11 years
23

Duchenne muscular dystrophy� Often develop scoliosis and an equinus deformity.
� Genetic counselling is important, and carriers may be detected by finding a moderately raised serum creatine kinase.
� The serum creatine kinase is always very elevated before clinically evidence.before clinically evidence.
� The diagnosis: serum creatine kinase 30 to 200 times higher, myopathic changes on EMG, characteristic muscle biopsy feature.
24

Blood creatine kinase
� The Serum CPK is always elevated to 50 to 300 times normal. Characteristically is 15.000 –45.000 IU/L (normal, < 150 IU/L).
� Elevations have been demonstrated in placental blood of affected male fetuses of 16-20 weeks gestation.
� The CK level is decreased in pregnancy by as much as 30%.
� About 70% of genetically definite carriers will show a raised CPK
25

Dystrophin
� Is a protein which is found on the inner side of
the membrane surrounding each muscle
fibres.
� If dystrophin is missing the muscle fibres � If dystrophin is missing the muscle fibres
break down (degenerated).
26

Duchenne Normal
27
40 50 60 70 80 90 100 110 120 130 140
IQ
Figure 2-17. Stylized distribution curve of IQ in Duchenne dystrophy showing normal bell-shape but a shift to the left.
28

Guillain-Barre syndrome
� Commonest cause of acute generalised paralysis
� 0.6 – 1.1 per 100,000 (<15 yrs)
� Any time during childhood (4 and 9 yrs)
� Core symptoms
� Progresive symmetric weakness
� Cease by 4 weeks
� areflexia
� Considerable clinical variability
� Untreated mortality 15% (at least)
29

Guillain-Barre Syndrome
� Postinfectious polyneuropathy that causes
demyelination in mainly motor but sometime
also sensory nerves.
� This syndrome affects people of all ages and � This syndrome affects people of all ages and
is not hereditary.
30

Guillian-Bare Syndromes
AcuteInflammatoryDemyelinatingpolyneuropathy
AcuteMotor axonalNeuropathy(AMAN)
Fishersyndrome
Acute motor-
IncreasingSeverity
Of the immune
31
AIDP withSecondaryAxonal
degeneration
Acute motor-Sensory axonalNeuropathy (AMSAN)
Of the immuneattack
Fig. 22-3. Proposed interrelationships of the forms of GBS. (Reprinted with permission
From Griffin et al., Pathology of the motor-sensory axonal Guillian-Barre syndrome,
Ann Neurol 39:17 – 28, 1996 [41].)

Clinical Characteristics of 56 children with GBS
� Antecedent infection 70%
� Distal weakness predominantly 44%
� Cranial nerve weakness 43%
� Paresthesia and pain 43%� Paresthesia and pain 43%
� Meningeal irritation 17%
� CSF protein > 45 mg/dl 88%
� Asymmetry of involvement 9%
� Full recovery or mild impairment 77%
� Relapses 7%
� Mortality 4%
32

Diagnostic Features of Guillain-Barre Syndrome
Features strongly supportive of the diagnosis :� Progression over days to a few weeks
� Relative symmetry
� Mild sensory loss
Features required for diagnosis :• Progressive motor weakness of more than one limb
• Areflexia or marked hyporeflexia
� Mild sensory loss
� Onset with extremity pain or discomfort
� Cranial nerve involvement
� Onset of recovery 2 to 4 weeks after halt of progression
� Autonomic dysfunction
� Initial absence of fever
� Elevated CSF protein level after 1 week of symptoms
� Abnormal electrodiagnosis with slowed conduction or prolonged F Waves
33
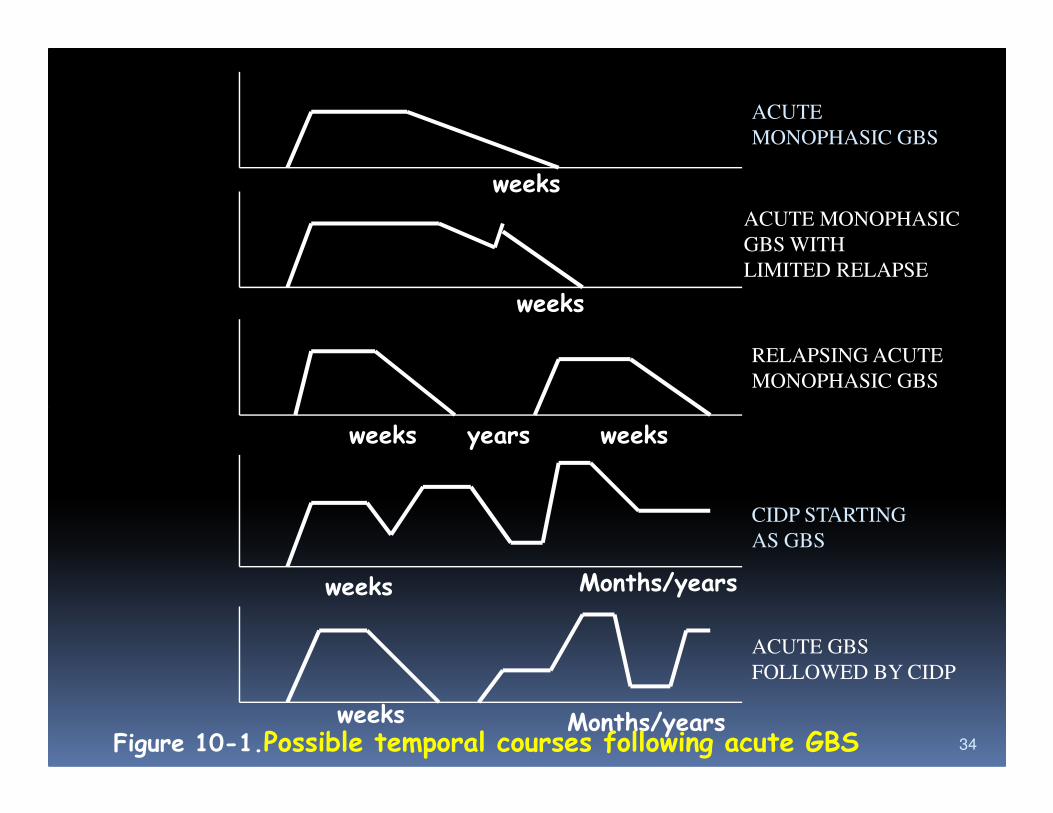
weeks
weeks
ACUTE
MONOPHASIC GBS
ACUTE MONOPHASIC
GBS WITH
LIMITED RELAPSE
RELAPSING ACUTE
MONOPHASIC GBS
34
weeks years weeks
weeks Months/years
weeks Months/yearsFigure 10-1.Possible temporal courses following acute GBS
CIDP STARTING
AS GBS
ACUTE GBS
FOLLOWED BY CIDP
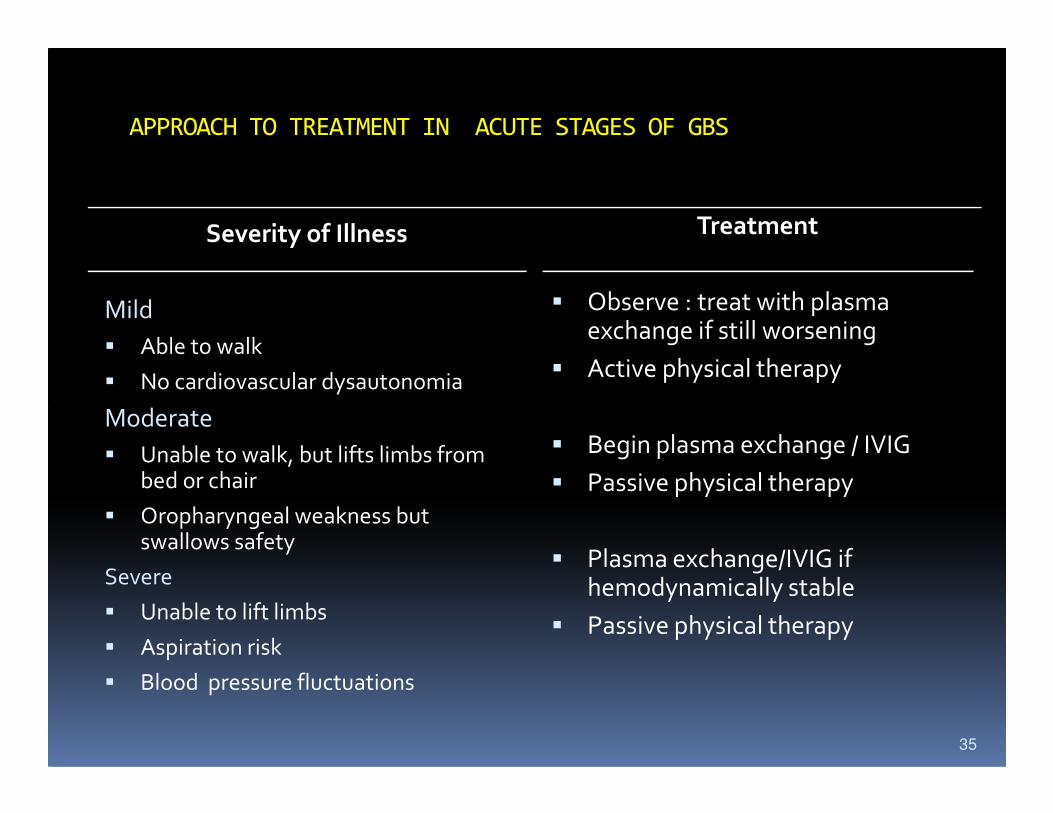
APPROACH TO TREATMENT IN ACUTE STAGES OF GBS
Severity of Illness
Mild
� Able to walk
� No cardiovascular dysautonomia
Moderate
Treatment
� Observe : treat with plasma exchange if still worsening
� Active physical therapy
Moderate
� Unable to walk, but lifts limbs from bed or chair
� Oropharyngeal weakness but swallows safety
Severe
� Unable to lift limbs
� Aspiration risk
� Blood pressure fluctuations
� Begin plasma exchange / IVIG
� Passive physical therapy
� Plasma exchange/IVIG if hemodynamically stable
� Passive physical therapy
35
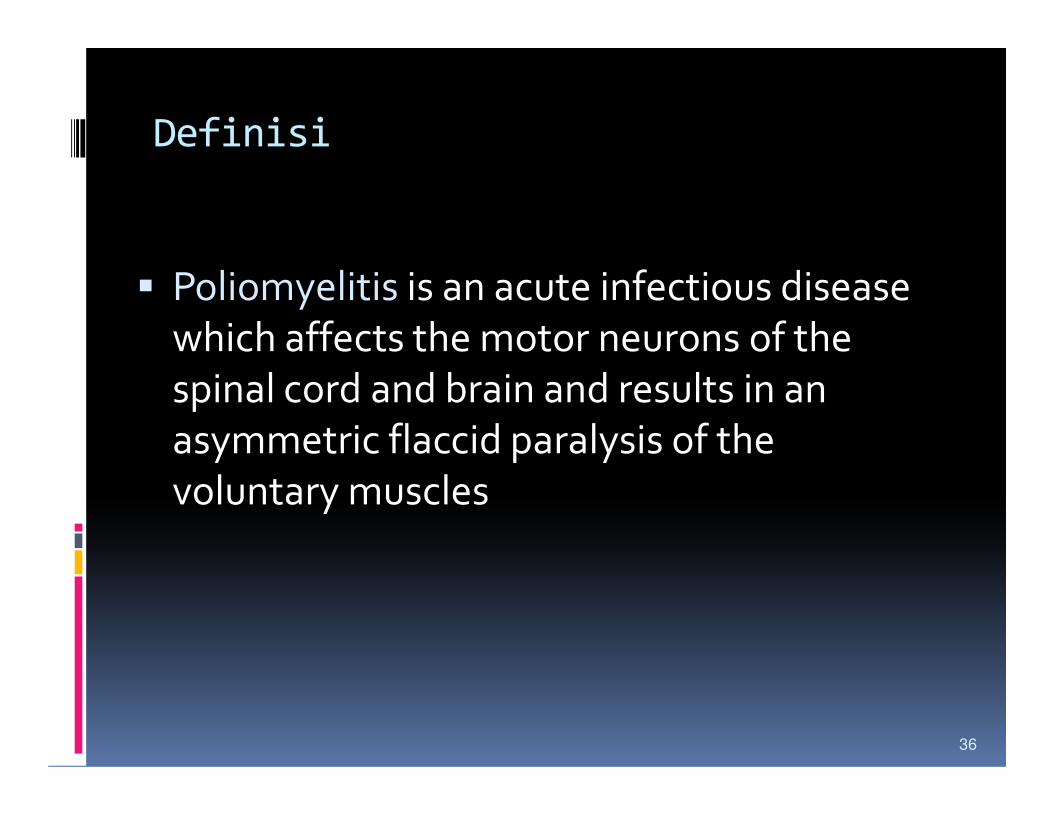
Definisi
� Poliomyelitis is an acute infectious disease
which affects the motor neurons of the
spinal cord and brain and results in an spinal cord and brain and results in an
asymmetric flaccid paralysis of the
voluntary muscles
36

Poliomyelitis
� Is a generalized viral infection with an affinity
for lower motor neurons
� Polios, meaning gray, reflecting the
involvement of the anterior horn gray matter involvement of the anterior horn gray matter
in the spinal cord.
� The paralytic rate also varies with the
virulence of the strain of poliovirus
37
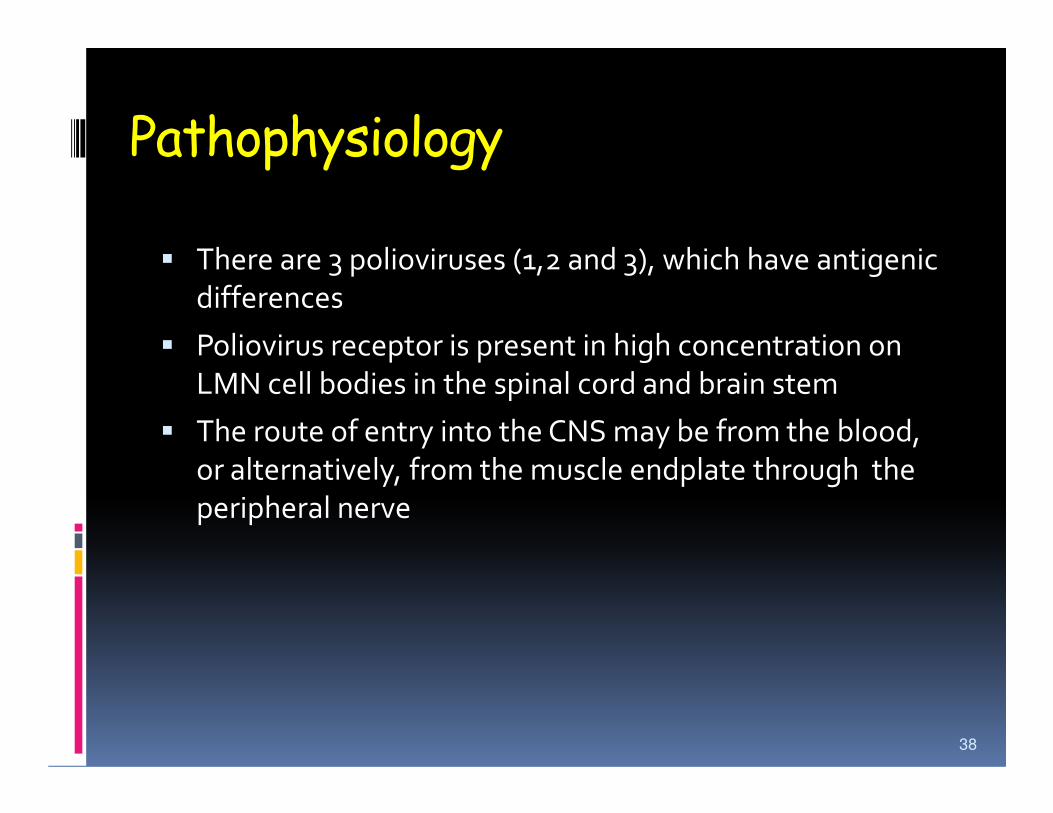
Pathophysiology
� There are 3 polioviruses (1,2 and 3), which have antigenic
differences
� Poliovirus receptor is present in high concentration on
LMN cell bodies in the spinal cord and brain stem
� The route of entry into the CNS may be from the blood,
or alternatively, from the muscle endplate through the
peripheral nerve
38
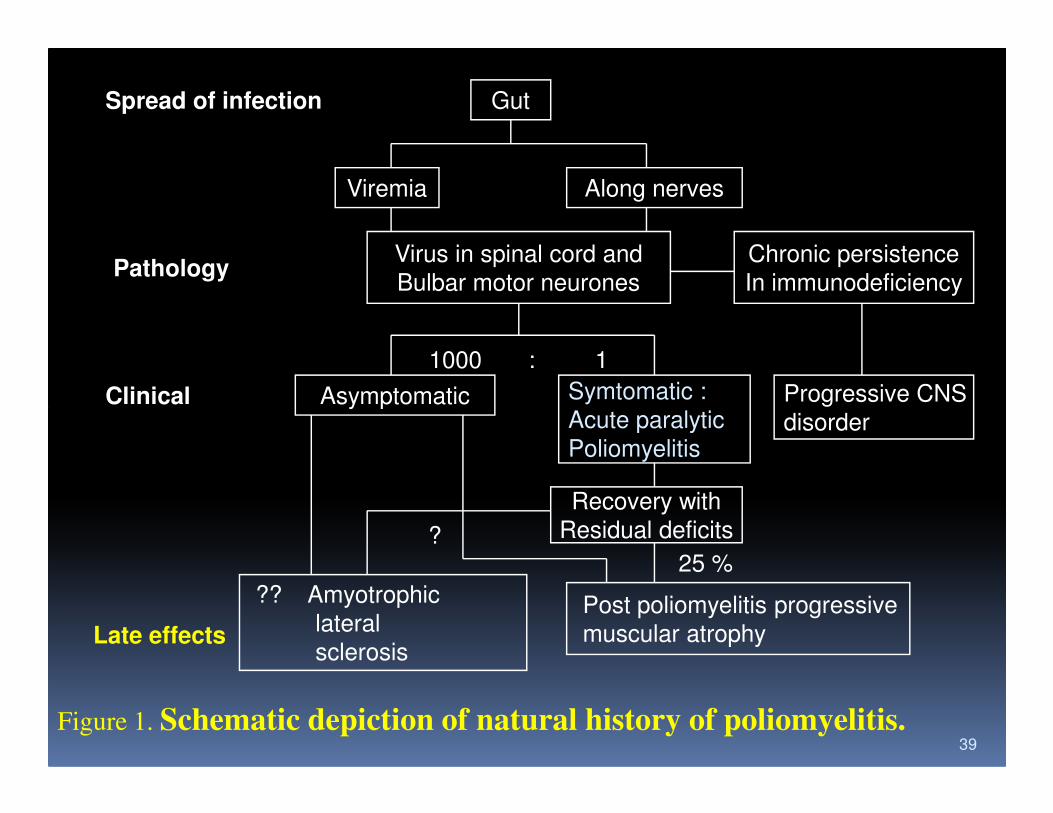
Spread of infection Gut
Viremia Along nerves
Virus in spinal cord and
Bulbar motor neurones
Chronic persistence
In immunodeficiency
1000 : 1
Asymptomatic Symtomatic :
Acute paralyticProgressive CNS
Pathology
Clinical
39
AsymptomaticAcute paralytic
Poliomyelitisdisorder
Recovery with
Residual deficits
Post poliomyelitis progressive
muscular atrophy
?? Amyotrophic
lateral
sclerosis
25 %?
Clinical
Late effects
Figure 1. Schematic depiction of natural history of poliomyelitis.

Epidemiology
� In infancy the illness was usually benign and
only rarely produced paralysis
� The potensial for paralysis increases with age
� 90-95% of cases asymptomatic; in 4-8% is � 90-95% of cases asymptomatic; in 4-8% is
nonspesific viral syndrome; only 1-2% of
cases are associated with paralysis
40
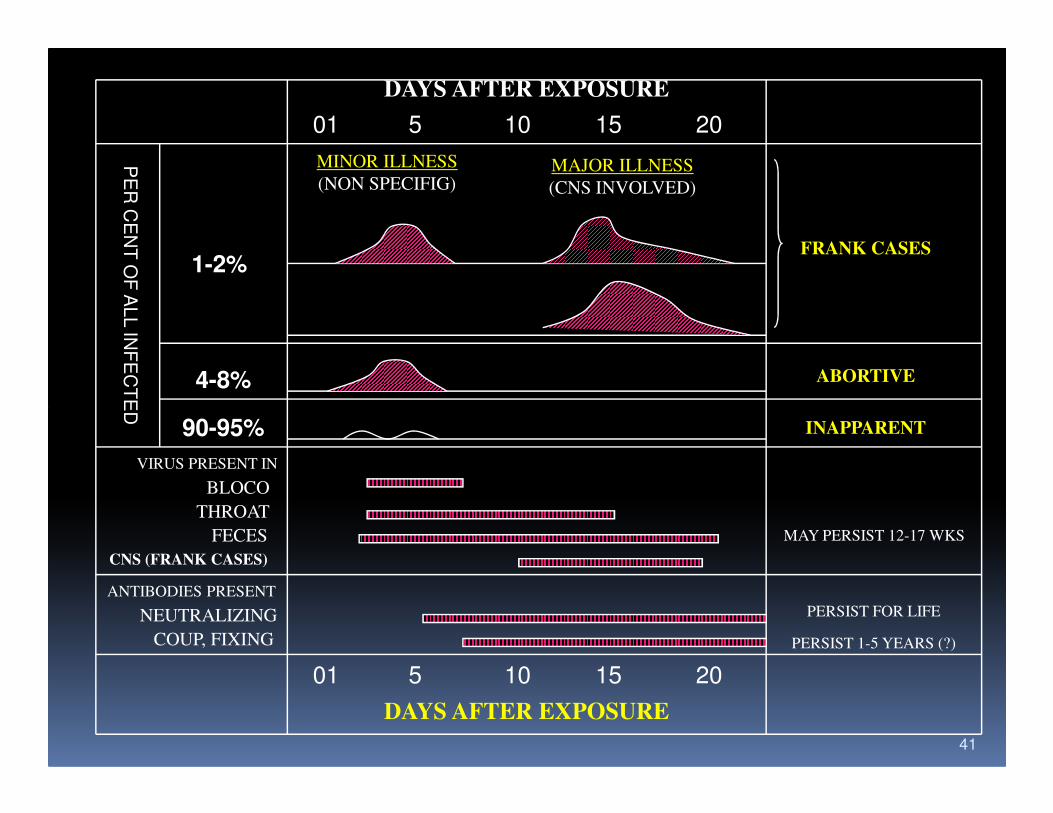
DAYS AFTER EXPOSURE
01 5 10 15 20
MINOR ILLNESS
(NON SPECIFIG)MAJOR ILLNESS
(CNS INVOLVED)
1-2%
4-8%
FRANK CASES
ABORTIVE
PE
R C
EN
T O
F A
LL IN
FE
CT
ED
41
90-95% INAPPARENT
VIRUS PRESENT IN
BLOCO
THROAT
FECES
CNS (FRANK CASES)
MAY PERSIST 12-17 WKS
ANTIBODIES PRESENT
NEUTRALIZING
COUP, FIXING
PERSIST FOR LIFE
PERSIST 1-5 YEARS (?)
01 5 10 15 20
DAYS AFTER EXPOSURE
PE
R C
EN
T O
F A
LL IN
FE
CT
ED
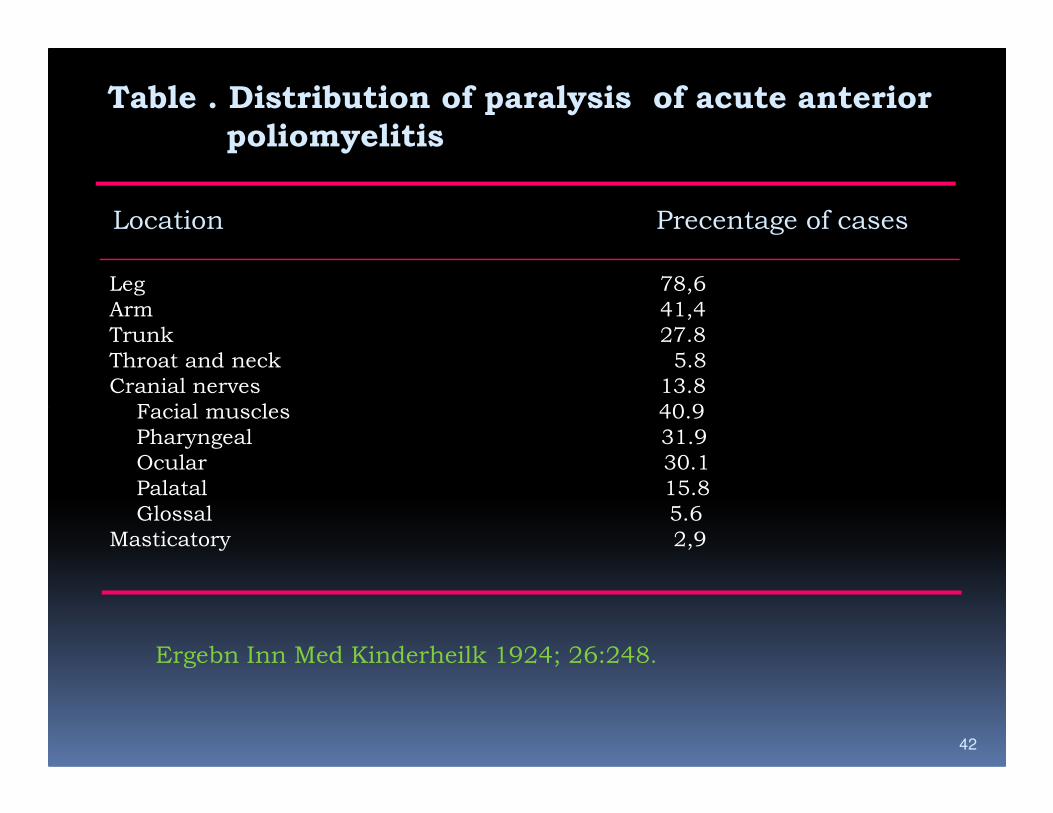
Table . Distribution of paralysis of acute anterior
poliomyelitis
Location Precentage of cases
Leg 78,6
Arm 41,4
Trunk 27.8
Throat and neck 5.8
Cranial nerves 13.8
Facial muscles 40.9
42
Facial muscles 40.9
Pharyngeal 31.9
Ocular 30.1
Palatal 15.8
Glossal 5.6
Masticatory 2,9
Ergebn Inn Med Kinderheilk 1924; 26:248.

Paralytic Poliomyelitis
� Spinal form
� Bulbar form ( paralysis of motor cranial nerve
with or without involvement vital centre)
� Bulbospinal form� Bulbospinal form
� Encephalitic form
43
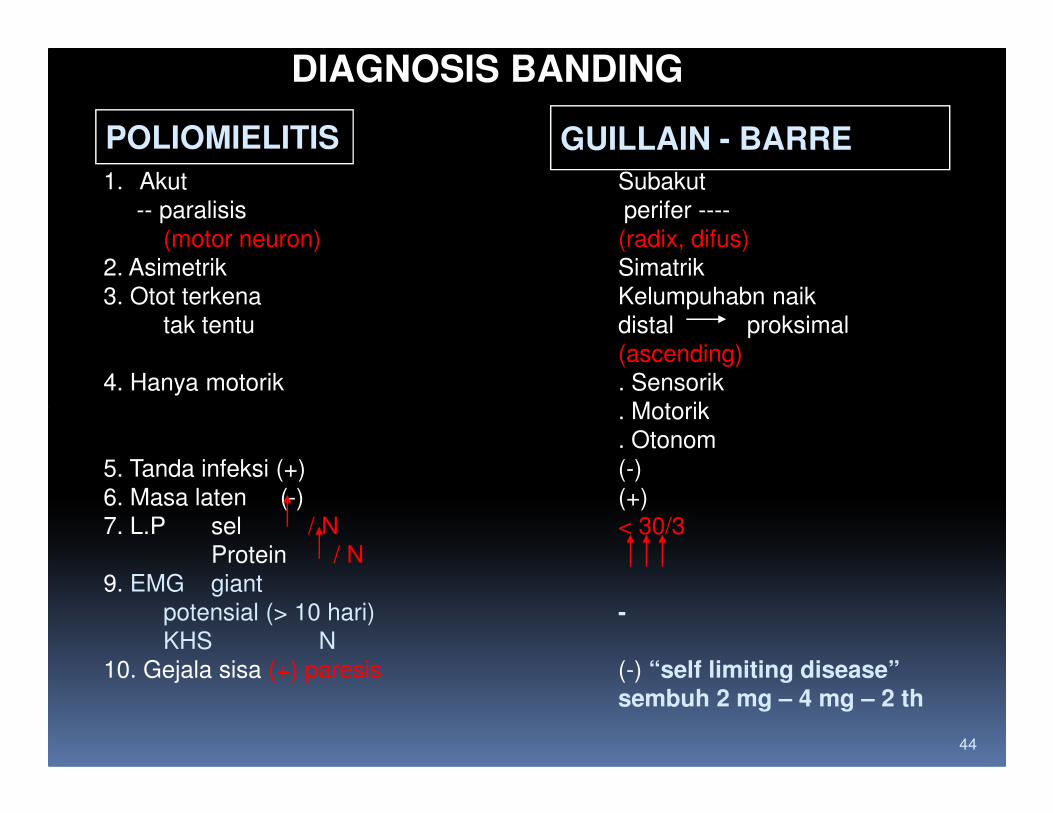
DIAGNOSIS BANDING
POLIOMIELITIS GUILLAIN - BARRE
1. Akut Subakut
-- paralisis perifer ----
(motor neuron) (radix, difus)
2. Asimetrik Simatrik
3. Otot terkena Kelumpuhabn naik
tak tentu distal proksimal
(ascending)
4. Hanya motorik . Sensorik
. Motorik
44
. Motorik
. Otonom
5. Tanda infeksi (+) (-)
6. Masa laten (-) (+)
7. L.P sel / N < 30/3
Protein / N
9. EMG giant
potensial (> 10 hari) -KHS N
10. Gejala sisa (+) paresis (-) “self limiting disease”sembuh 2 mg – 4 mg – 2 th

Spinal Muscular Atrophies
� SMA are degenerative disease of motor
neurons that begin in fetal life and continue
to be progressive in infancy and childhood
45

Classification SMA
� SMA type I, severe infantile form (Werdnig-
Hoffmann disease)
� SMA type 2, a late infantile form (more slowly
progressive form)progressive form)
� SMA type 3, juvenile form
(Kugelberg-Welander disease)
46

SMA type I (in utero or 2-3 months)� Severe hypotonia, general weakness
� The lower limbs are more severely affected
than the upper
� Fasciculation and atrophy of the tongue� Fasciculation and atrophy of the tongue
� Intercostal muscles are severely affected ‘a
bell-shaped appearance’
� Contracture occurs in about 10%
� More than 2/3 die by 2 yr of age.
47

SMA type 2 ( 3 to 15 months)
� Never achieves the ability to stand or walk
� Symmetrical, proximal muscles are more affected than the distal
� Tendon reflexes are usually depressed or � Tendon reflexes are usually depressed or absent
� Fasciculation and atrophy of the tongue and tremor of the hand are common
� The disastrous complication is scoliosis
48

SMA type 3 ( > 24 months)
� Normal milestones in the first years and
achieves the ability to walk
� Muscular dystrophy fashion: waddling gait,
Gowers’ manouver, walk flat-footedGowers’ manouver, walk flat-footed
with eversion of the feet. In contrast to
tendency to inversion in Duchenne
� Reflexes may be normal or depressed
49
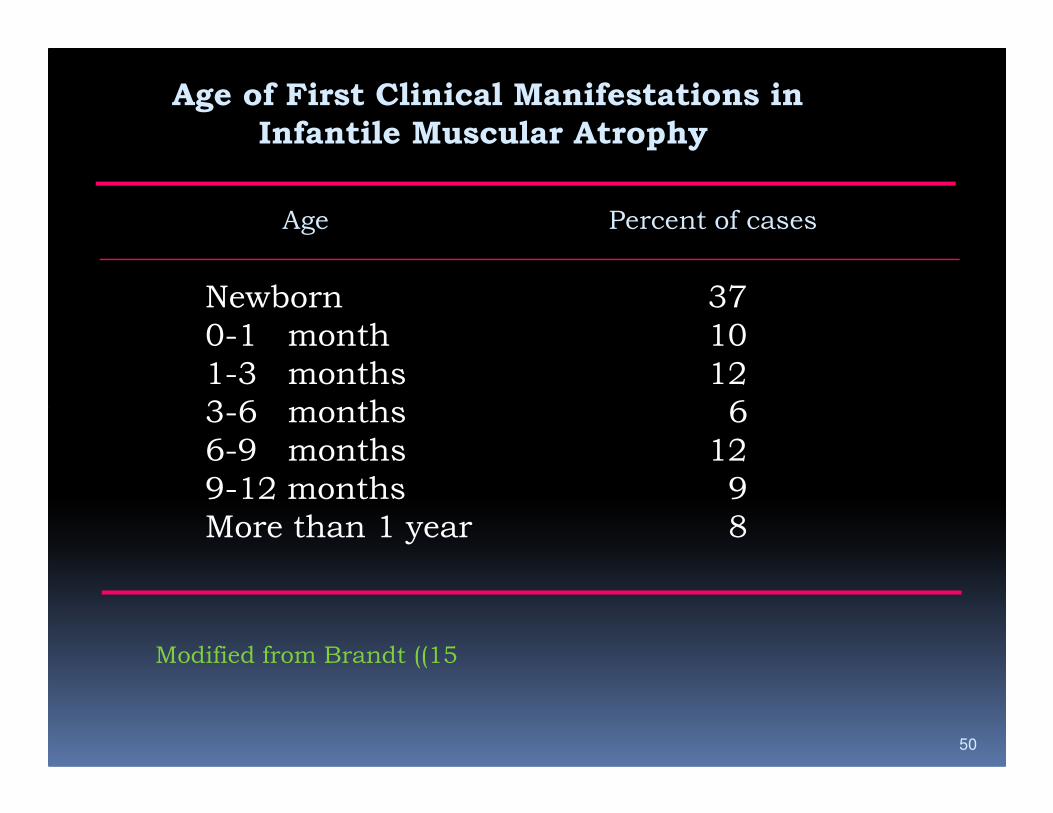
Age of First Clinical Manifestations in
Infantile Muscular Atrophy
Age Percent of cases
Newborn 37
0-1 month 10
1-3 months 12
3-6 months 6
50
3-6 months 6
6-9 months 12
9-12 months 9
More than 1 year 8
Modified from Brandt ((15
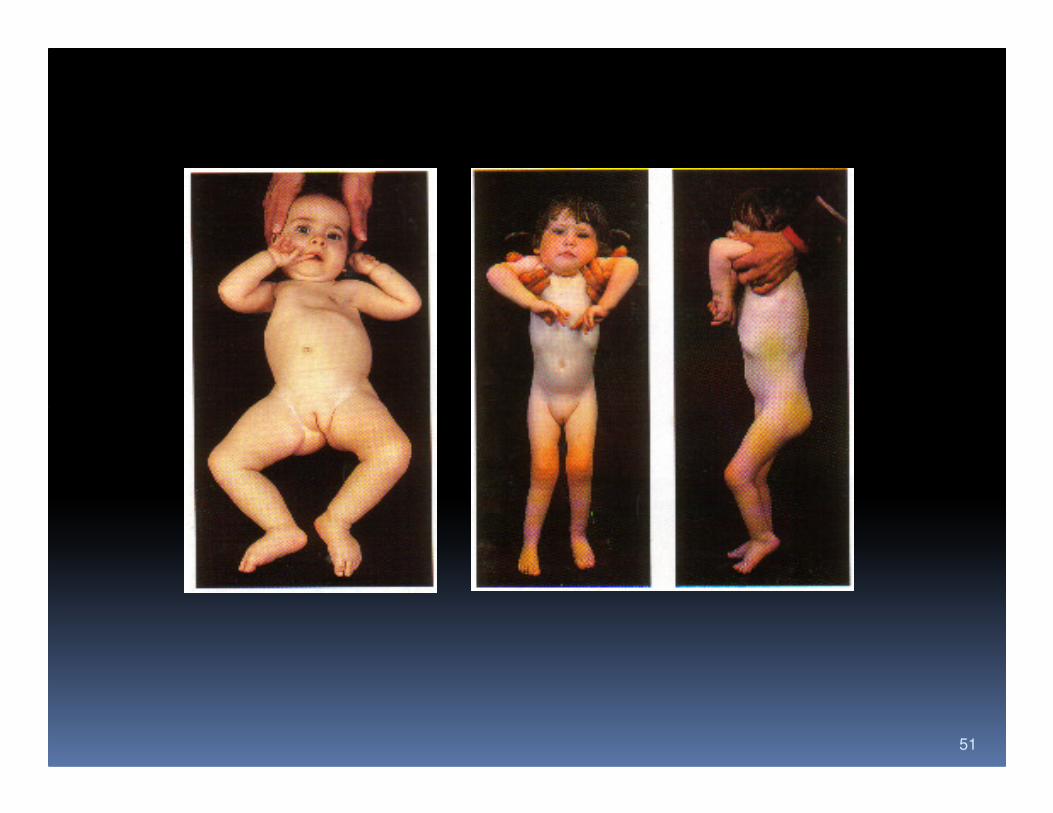
51

Creatine kinase (CK) test
� An enzyme (protein) that is important for
energy production within muscle fibres.
� Normally there is only small amount in the
bloodblood
52
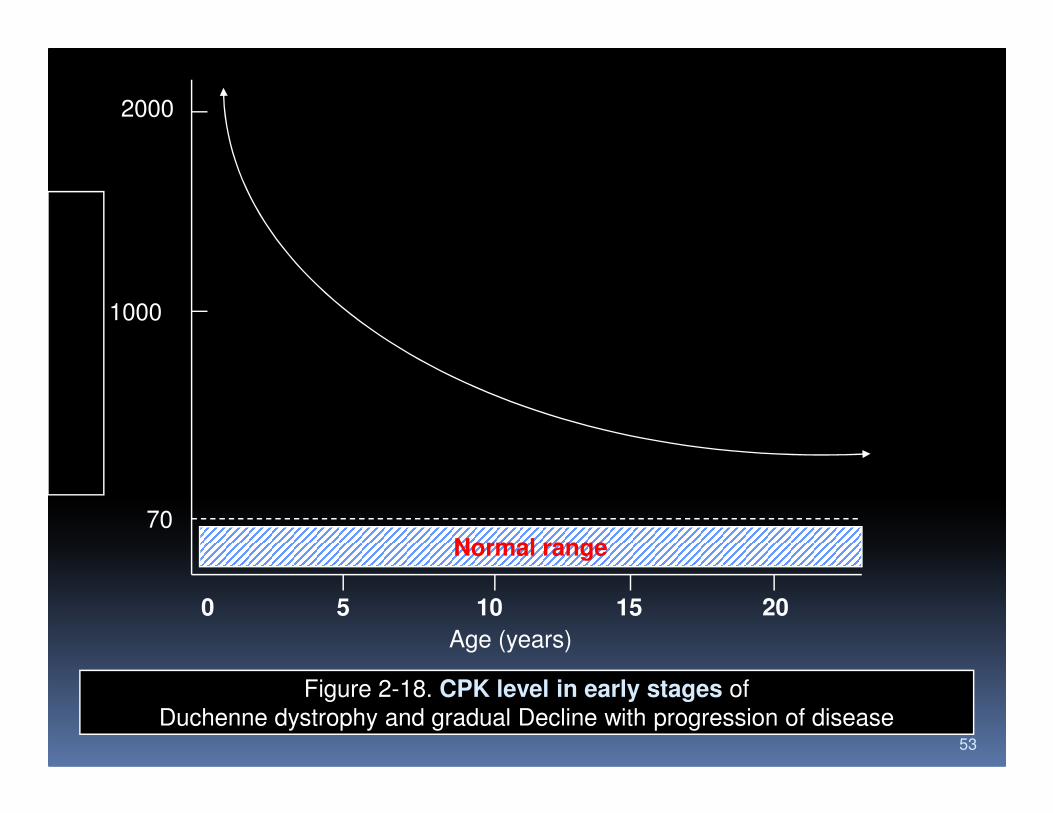
2000
1000
53
Normal range
0 5 10 15 20
70
Age (years)
Figure 2-18. CPK level in early stages of
Duchenne dystrophy and gradual Decline with progression of disease

Electromyo/neurography (EMG/ENG)� Diagnostic aids in detecting NMD
� To determine and to differentiate axonal or myelin disorder
� Not pathognomonic, when present � Not pathognomonic, when present strongly suggest the diagnosis
� Motor conduction, sensory conduction, F wave and H wave.
� Needle electromyography.
54
Muscle and nerve biopsy
Muscle biopsy is essential for any patient with
a suspected neuromuscular disorder in order
to establish a definitive diagnosis
55

Cerebrospinal fluid (CSF)
� Poliomyelitis
� Pleositosis, 20-300 WBC/mm3 lymphocytic,
the cell drops within the first 2-3 weeks
� Total protein increases in the first weeks, < 200 mg/dl. � Total protein increases in the first weeks, < 200 mg/dl.
� Guillain Barre syndrome
� Dissociation between high CSF protein and a lack of
cellular response.
56
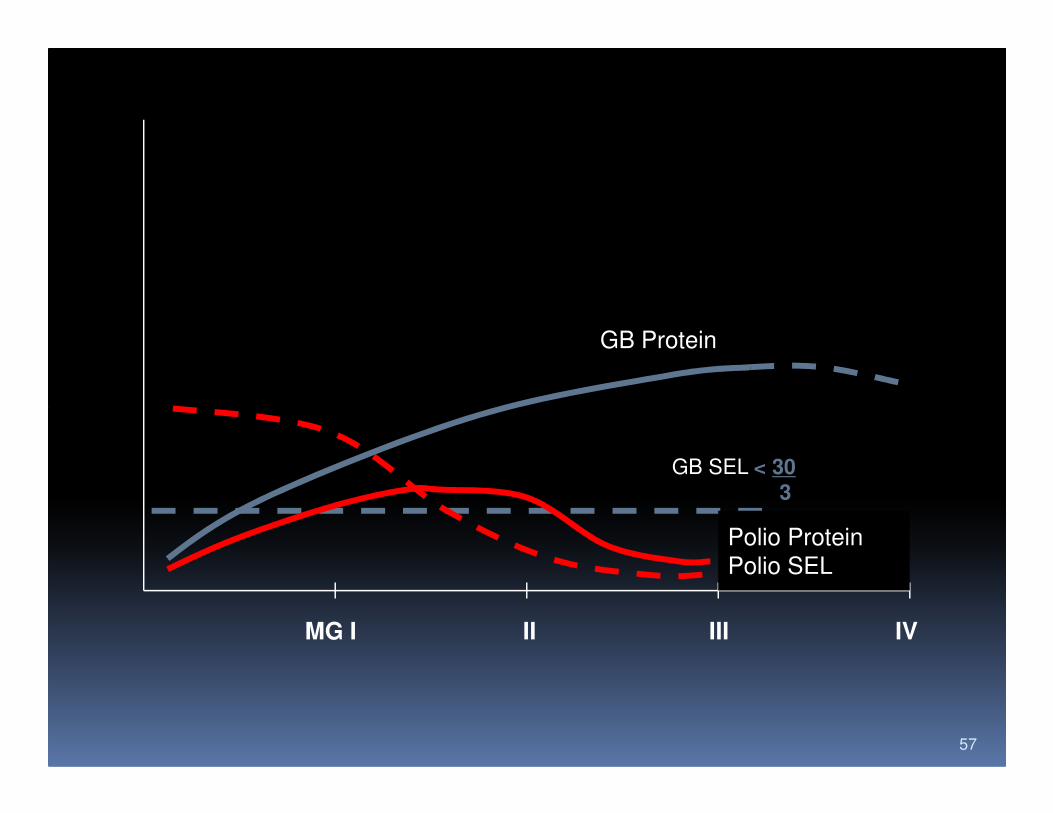
GB Protein
57
MG I II III IV
GB SEL < 30
3
Polio Protein
Polio SEL

58