p&d e produção de insumos
TRANSCRIPT


P&D e Produção de Insumos
Farmacêuticos Ativos
Conteúdo Programático
Módulo 1
IFA: Conceito e
Tecnologias de
Produção
Módulo 2
Desenvolvimento
de IFA sintéticos
Módulo 3
Desenvolvimento
de Métodos
Analíticos de IFA
Módulo 4
DMF (Drug
Master File) ou
DIFA (Dossiê
do IFA)
Módulo 5
Boas Práticas
de Fabricação
de IFA

DMF (Drug Master File) ou DIFA (Dossiê do IFA)
1. Estrutura do DIFA (Carolina)
2. CADIFA (Paulo)
Foco: Discussão sobre aspectos técnicos necessários para composição do
DMF partes aberta e restrita sob a ótica do produtor do IFA (ênfase na
fabricação e caracterização)

DossiêD
InsumoI
FarmacêuticoF
AtivoA
Fonte: https://www.in.gov.br/en/web/dou/-/resolucao-de-diretoria-colegiada-rdc-n-359-de-27-de-marco-de-
2020-250639483. Acesso em 27/03/2021.
Contextualização
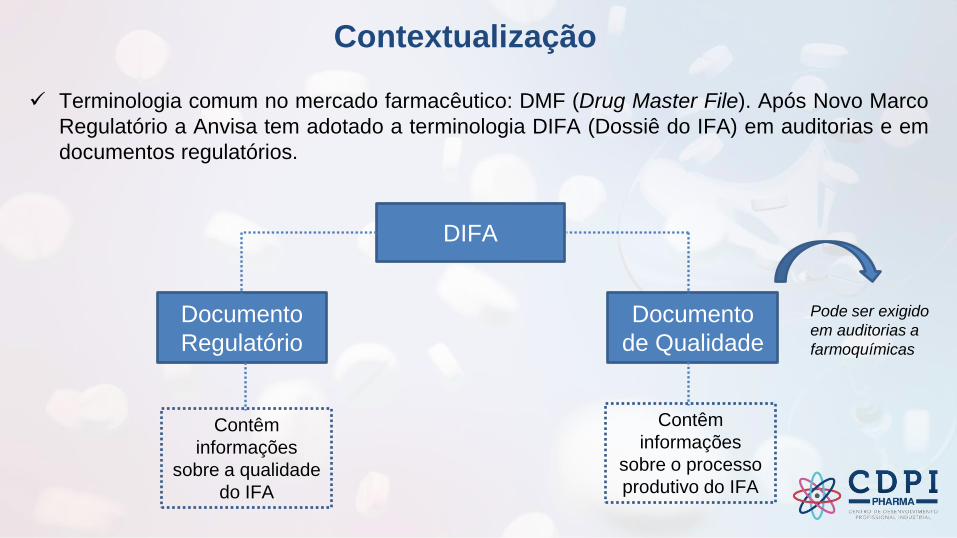
Contextualização
✓ Terminologia comum no mercado farmacêutico: DMF (Drug Master File). Após Novo Marco
Regulatório a Anvisa tem adotado a terminologia DIFA (Dossiê do IFA) em auditorias e em
documentos regulatórios.
DIFA
Documento
Regulatório
Documento
de Qualidade
Contêm
informações
sobre o processo
produtivo do IFA
Contêm
informações
sobre a qualidade
do IFA
Pode ser exigido
em auditorias a
farmoquímicas

Organograma da Anvisa

Auditorias em farmoquímicas

Fonte: https://www.gov.br/anvisa/pt-br/composicao/estrutura. Acesso em 27/03/2021.
Coordenação de Registro de Insumos Farmacêuticos
Ativos - COIFA

Dossiê do
Insumo
Farmacêutico
Ativo XXX
Parte Aberta
✓ Contêm informações sobre atributos
críticos de qualidade do IFA que são
relevantes para o fabricante do
medicamento.
✓ Descrição simplificada sobre o processo
de fabricação.
✓ Compartilhada com clientes via acordo de
confidencialidade
Estrutura do DIFA
Dossiê do
Insumo
Farmacêutico
Ativo XXX
Parte Restrita
✓ Contêm informações detalhadas sobre o
processo de fabricação contendo a lista
técnica, controle de intermediários e
etapas críticas, controle de materiais,
validação de processo.
✓ Compartilhada diretamente com a agência
regulatória.
Versão 1 Versão 1

Estrutura do DIFA
Fonte: https://coifa.anvisa.gov.br/guia.html
Acesso em 27/03/2021.
(a) A parte aberta deve conter, no mínimo, diagrama da rota de síntese e
descrição simplificada do processo de fabricação, desde a introdução do
material de partida.
(b) A parte restrita deve conter todas as informações pertinentes ao processo de
fabricação.
(c) Informações que também sejam relevantes para o solicitante do registro do
medicamento.
(d) Informações relacionadas à descrição detalhada do processo de fabricação e
não relevantes para o solicitante do registro do medicamento.
(e) Para IFA estéreis, quando não houver etapa de esterilização adicional no
processo de fabricação do medicamento.
(f) Informações sobre impurezas potenciais que remetam à narrativa sequencial
do processo de fabricação podem constar na parte restrita, desde que haja
comprovação inequívoca de que não há necessidade de serem controladas no
IFA.
(g) Informações referentes à narrativa sequencial do processo de fabricação,
controle de matérias-primas e validação de processo podem constar na parte
restrita.

Estrutura do DIFA
✓ Detentores do DIFA e responsáveis pela avaliação de DIFA
e/ou intermediação de respostas a exigências entre parceiros
externos e órgão regulatório: sejam redundantes e
justifiquem com clareza a parte técnica.
✓ Não há problema em usar textos para descrever informações
técnicas. Porém, usem tabelas quando possível para
agrupamento de informações – facilitará a análise por parte
do órgão regulatório.
✓ O alinhamento das nomenclaturas de substâncias (ex:
impurezas/MP/intermediários) ao longo do documento é
importante para facilitar o entendimento do avaliador.
O óbvio
precisa
ser dito

Estrutura do DIFA
3.2.S.1.1
3.2.S.1.2
...
Parte Aberta:
Destacar o que será
encontrado apenas
na parte restrita do
documento.
Versão 1.0
Versão 1.1
...
Partes Aberta e
Restrita: data da
modificação e
mudanças realizadas
na nova versão.
Partes Aberta e
Fechada:
siglas utilizadas ao
longo do documento.
SumárioVersionamento e
Histórico de Mudanças Lista de Abreviaturas

Informações Gerais
✓ Nome compendial e
outras
nomenclaturas
✓ Nome químico
✓ Número CAS
✓ DCB e INN
✓ Abreviações
internas
3.2.S.1.1
✓ Estrutura química
✓ Estereoquímica
✓ Fórmula Molecular
✓ Peso molecular
✓ Composição
Elementar
3.2.S.1.2
✓ Solubilidade;
✓ pKa;
✓ Polimorfismo;
✓ Isomerismo;
✓ Coeficiente de
partição (logP);
✓ Permeabilidade;
✓ Higroscopicidade.
✓ Tamanho de
partícula
3.2.S.1.3

Fabricação
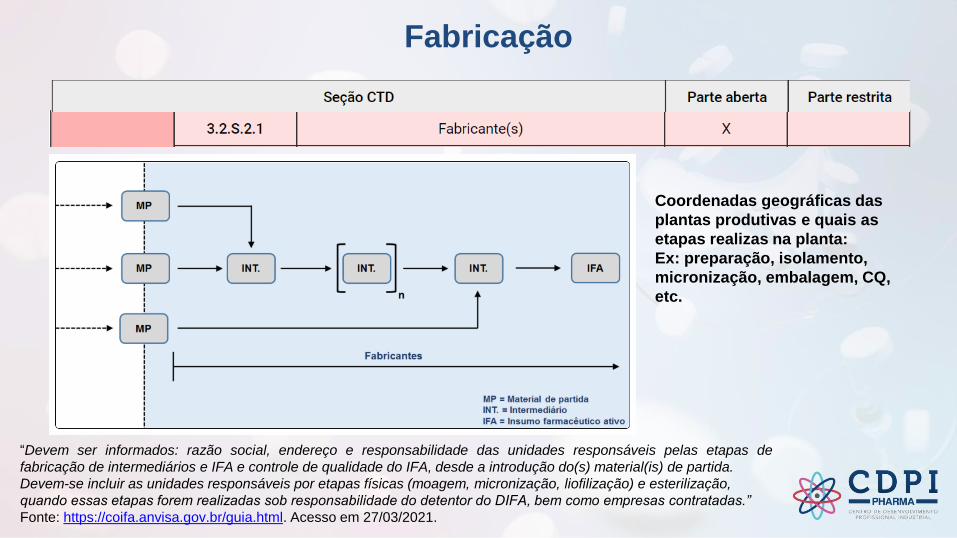
Fabricação
“Devem ser informados: razão social, endereço e responsabilidade das unidades responsáveis pelas etapas de
fabricação de intermediários e IFA e controle de qualidade do IFA, desde a introdução do(s) material(is) de partida.
Devem-se incluir as unidades responsáveis por etapas físicas (moagem, micronização, liofilização) e esterilização,
quando essas etapas forem realizadas sob responsabilidade do detentor do DIFA, bem como empresas contratadas.”
Fonte: https://coifa.anvisa.gov.br/guia.html. Acesso em 27/03/2021.
Coordenadas geográficas das
plantas produtivas e quais as
etapas realizas na planta:
Ex: preparação, isolamento,
micronização, embalagem, CQ,
etc.
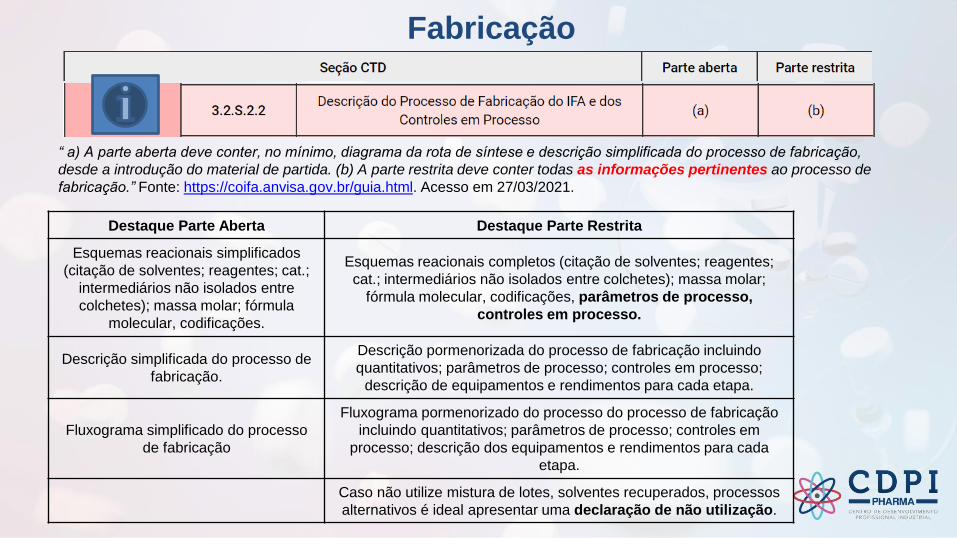
Fabricação
“ a) A parte aberta deve conter, no mínimo, diagrama da rota de síntese e descrição simplificada do processo de fabricação,
desde a introdução do material de partida. (b) A parte restrita deve conter todas as informações pertinentes ao processo de
fabricação.” Fonte: https://coifa.anvisa.gov.br/guia.html. Acesso em 27/03/2021.
Destaque Parte Aberta Destaque Parte Restrita
Esquemas reacionais simplificados
(citação de solventes; reagentes; cat.;
intermediários não isolados entre
colchetes); massa molar; fórmula
molecular, codificações.
Esquemas reacionais completos (citação de solventes; reagentes;
cat.; intermediários não isolados entre colchetes); massa molar;
fórmula molecular, codificações, parâmetros de processo,
controles em processo.
Descrição simplificada do processo de
fabricação.
Descrição pormenorizada do processo de fabricação incluindo
quantitativos; parâmetros de processo; controles em processo;
descrição de equipamentos e rendimentos para cada etapa.
Fluxograma simplificado do processo
de fabricação
Fluxograma pormenorizado do processo do processo de fabricação
incluindo quantitativos; parâmetros de processo; controles em
processo; descrição dos equipamentos e rendimentos para cada
etapa.
Caso não utilize mistura de lotes, solventes recuperados, processos
alternativos é ideal apresentar uma declaração de não utilização.

Exemplos
Descrição simplificada do processo de fabricação
Etapa 1: Reação
A um determinado volume de (nome do solvente) é adicionado (nome dos reagentes) a um vaso de
reação. A mistura é mantida sob agitação e temperatura controladas por um tempo determinado. (...)
Descrição pormenorizada do processo de fabricação
Etapa 1: Reação
A XX Kg (nome do solvente) é adicionado XX Kg (nome do reagente A) e XX Kg (nome do reagente B)
a um reator de (material do reator; Inox; Vitrificado) de volume XX e TAG XX. A mistura é mantida sob
agitação a XX RPM por XX h a temperatura de XX °C. (...)
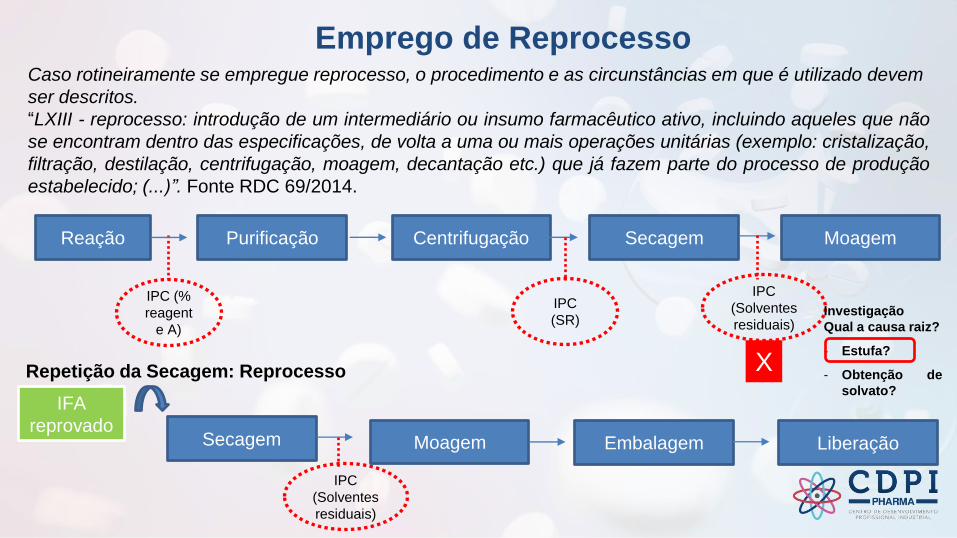
Emprego de Reprocesso Caso rotineiramente se empregue reprocesso, o procedimento e as circunstâncias em que é utilizado devem
ser descritos.
“LXIII - reprocesso: introdução de um intermediário ou insumo farmacêutico ativo, incluindo aqueles que não
se encontram dentro das especificações, de volta a uma ou mais operações unitárias (exemplo: cristalização,
filtração, destilação, centrifugação, moagem, decantação etc.) que já fazem parte do processo de produção
estabelecido; (...)”. Fonte RDC 69/2014.
Purificação Centrifugação Secagem Moagem
IPC
(SR)
IPC
(Solventes
residuais)
IPC (%
reagent
e A)
Reação
IPC
(Solventes
residuais)
Repetição da Secagem: Reprocesso
Secagem Moagem Embalagem Liberação
IFA
reprovado
X
Investigação
Qual a causa raiz?
- Estufa?
- Obtenção de
solvato?

Estudos de Caso
O fornecedor do IFA recebeu os seguintes itens de exigência, descritos abaixo, da COIFA. Ele pediu a sua
ajuda para responder aos questionamentos da agência. Dessa forma, construa um plano para responder a
exigência.
Item 1. Fabricação. 3.2.S.2.2. Processos alternativos
A empresa apresentou dois 4 processos alternativos para a preparação do intermediário avançado de
síntese do IFA em seu DIFA. Em todos os processos alternativos os mesmos controles em processo são
utilizados e a especificação do IFA é a mesma. O fabricante do IFA não comprovou que não há impactos na
qualidade do IFA obtidos pelos diferente processos. Cabe destacar que de acordo com a RDC 359/2020 “Art.
23. Processos alternativos com rotas de síntese substancialmente diferentes devem constituir DIFA distintos,
ainda que se mantenha a especificação e perfil de impurezas de intermediários terminais e do IFA.”
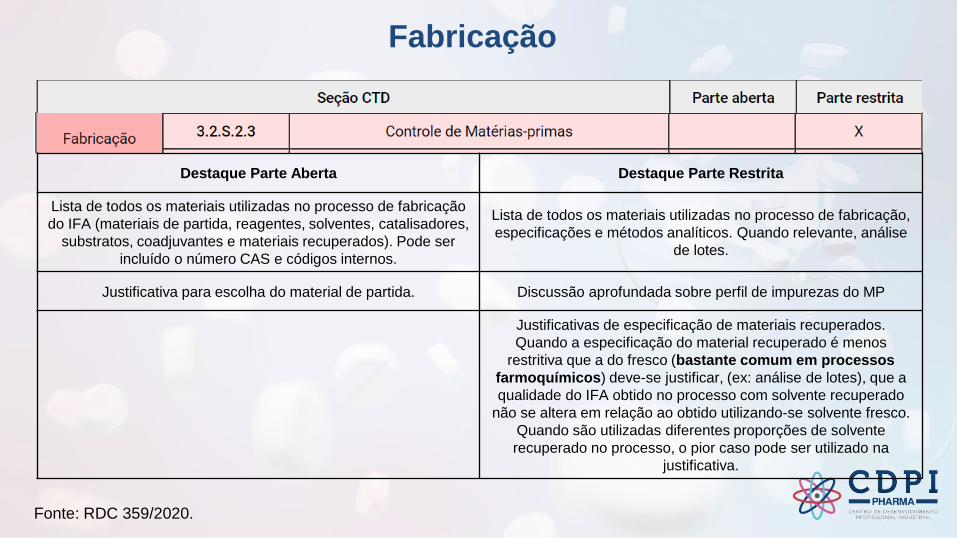
Fabricação
Destaque Parte Aberta Destaque Parte Restrita
Lista de todos os materiais utilizadas no processo de fabricação
do IFA (materiais de partida, reagentes, solventes, catalisadores,
substratos, coadjuvantes e materiais recuperados). Pode ser
incluído o número CAS e códigos internos.
Lista de todos os materiais utilizadas no processo de fabricação,
especificações e métodos analíticos. Quando relevante, análise
de lotes.
Justificativa para escolha do material de partida. Discussão aprofundada sobre perfil de impurezas do MP
Justificativas de especificação de materiais recuperados.
Quando a especificação do material recuperado é menos
restritiva que a do fresco (bastante comum em processos
farmoquímicos) deve-se justificar, (ex: análise de lotes), que a
qualidade do IFA obtido no processo com solvente recuperado
não se altera em relação ao obtido utilizando-se solvente fresco.
Quando são utilizadas diferentes proporções de solvente
recuperado no processo, o pior caso pode ser utilizado na
justificativa.
Fonte: RDC 359/2020.

Fabricação
Destaque Parte Restrita: Material de Partida (RDC 359/2020).
“Apresentar nome e estrutura química; especificação; métodos analíticos; razão social e endereço dos fabricantes; rota de
síntese de cada fornecedor de material de partida, incluindo reagentes, solventes e catalisadores; análise de lotes, justificativa de
seleção do MP.(...)”
“Justificar as especificações dos materiais de partida incluindo, se aplicável, testes para impurezas especificadas e não
especificadas, impurezas totais, solventes, catalisadores, impurezas elementares e impurezas mutagênicas (...)”. Pode-se anexar
documentos técnicos dos fabricantes do MP como o TDP (Technical Data Package).
“Quando houver mais de um fornecedor para o mesmo material de partida, a especificação do fabricante do intermediário ou do
IFA para o material de partida deve ser discutida compreendendo as possíveis diferenças entre as formas de obtenção
propostas.(...)”
Fonte: RDC 359/2020.
Pode-se utilizar uma avaliação do perfil de impurezas comparativo entre os materiais de partida, avaliações
teóricas do perfil toxicológico das impurezas presentes no MP, cálculos de fator de purga, análises de lotes para
justificar que não há necessidade de controle de uma determinada impureza na rotina, etc.

Estudos de Caso
O fornecedor do IFA recebeu os seguintes itens de exigência, descritos abaixo, da COIFA. Ele pediu a sua
ajuda para responder aos questionamentos da agência. Dessa forma, construa um plano para responder a
exigência.
Item 2. Fabricação. 3.2.S.2.3. Controle de Matérias-primas.
Verificou-se a utilização de tolueno recuperado na etapa de reação e extração durante o processo de
fabricação do IFA. São utilizadas proporções que variam de 50% a 100% de solventes recuperado versus
fresco. Verificou-se também que a especificação do solvente recuperado é menos restritiva que o do solvente
fresco e que não há controle de benzeno no solvente recuperado. Uma vez que o procedimento se enquadra
como uma variável significativa do processo de fabricação, justifique a qualidade do IFA.
Item 3. Fabricação. 3.2.S.2.3. Controle de Matérias-primas.
Para a fabricação do IFA são utilizados os solventes tolueno (etapa de reação) e acetato de etila (etapa de
purificação). Na especificação do tolueno há controle do benzeno residual com limite de 5 ppm. Já para o
solvente acetato de etila, não há controle desse solvente na especificação da matéria-prima. Adicionalmente,
na especificação do IFA, não há controle de benzeno. Justifique a estratégia de controle desse solvente
residual a luz do ICH Q3C.

Fabricação
“(c) Informações que também sejam relevantes para o solicitante do registro do medicamento.
(d) Informações relacionadas à descrição detalhada do processo de fabricação e não relevantes para o solicitante do
registro do medicamento.” Fonte: https://coifa.anvisa.gov.br/guia.html. Acesso em 27/03/2021.
Em geral esse módulo compõe apenas a parte restrita:
✓Etapas críticas de processo:
“Devem ser apresentados testes e critérios de aceitação, com justificativa baseada em dados
experimentais, para as etapas críticas identificadas na narrativa sequencial do processo de fabricação.”
Apresentar as etapas críticas, controles em processo, parâmetros críticos e justificativas.
✓ Intermediários isolados: especificações e métodos analíticos.
✓ Intermediários não isolados: testes e parâmetros utilizados para se determinar o fim da reação
química devem ser apresentados ou sua ausência justificada.

Fabricação
3.2.S.2.6. IFA que se enquadre como nova entidade química deve apresentar ensaios pré-clínicos,
clínicos, aumento de escala, etc. Veja Subseção VI, Art. 37. da RDC 359/2020.
3.2.S.2.5. IFAs estéreis – foco principal. Veja Subseção V da RDC 359/2020.
Para IFAs não estéreis: pode-se apresentar na parte restrita uma declaração de validação do
processo citando o relatório de validação e os lotes que foram utilizados para a validação do
processo.
“(e) Para IFA estéreis, quando não houver etapa de esterilização adicional no processo de fabricação do medicamento.”
Fonte: https://coifa.anvisa.gov.br/guia.html. Acesso em 27/03/2021.

Caracterização
A) Elucidação estrutural do IFA:
Exemplos de análises:
- Análise Elementar
- Rotação ótica específica (quando aplicável)
- Espectroscopia de Infravermelho
- Ressonância Magnética Nuclear
- Espectroscopia no Ultravioleta
- Espectrometria de massas
B) Estrutura Cristalina
Exemplos de análises:
- Calorimetria exploratória diferencial
- Análise Termogravimétrica
- Difração de Raios-X.
C) Distribuição do tamanho de partículas
- Apresentar resultados de PSD de 3 ou mais
lotes consecutivos.
Importante citar referencias, modelos de equipamentos e endereços dos locais onde as análises
foram realizadas.
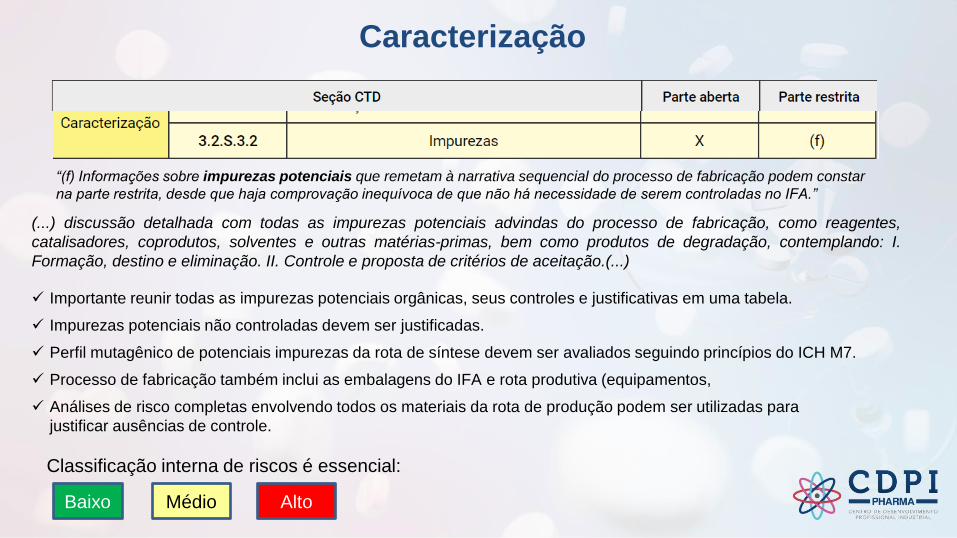
Caracterização
(...) discussão detalhada com todas as impurezas potenciais advindas do processo de fabricação, como reagentes,
catalisadores, coprodutos, solventes e outras matérias-primas, bem como produtos de degradação, contemplando: I.
Formação, destino e eliminação. II. Controle e proposta de critérios de aceitação.(...)
“(f) Informações sobre impurezas potenciais que remetam à narrativa sequencial do processo de fabricação podem constar
na parte restrita, desde que haja comprovação inequívoca de que não há necessidade de serem controladas no IFA.”
✓ Importante reunir todas as impurezas potenciais orgânicas, seus controles e justificativas em uma tabela.
✓ Impurezas potenciais não controladas devem ser justificadas.
✓ Perfil mutagênico de potenciais impurezas da rota de síntese devem ser avaliados seguindo princípios do ICH M7.
✓ Processo de fabricação também inclui as embalagens do IFA e rota produtiva (equipamentos,
✓ Análises de risco completas envolvendo todos os materiais da rota de produção podem ser utilizadas para
justificar ausências de controle.
Baixo Médio Alto
Classificação interna de riscos é essencial:

Estudos de Caso
O fornecedor do IFA recebeu os seguintes itens de exigência, descritos abaixo, da COIFA. Ele pediu a sua
ajuda para responder aos questionamentos da agência. Dessa forma, construa um plano para responder a
exigência.
Item 4. Caracterização. 3.2.S.3.2. Impurezas
Na síntese do intermediário avançado do processo de fabricação do IFA há formação de uma impureza
orgânica potencial que não é controlada na especificação desse material e também não é controlada na
especificação do IFA final. Justifique a qualidade do IFA.
Item 5. Caracterização. 3.2.S.3.2. Impurezas
Na etapa de reação do IFA há possibilidade de formação do isômero minoritário. Esse isômero não é
controlado na especificação do IFA. O fabricante do IFA justificou que a impureza potencial é controlada
como impureza desconhecida no limite de 0,1%. Entretanto, não foi encontrado no DIFA nenhum estudo que
que justifique ausência de controle dessa impureza.

DMF (Drug Master File) ou DIFA (Dossiê do IFA)
1. Estrutura do DIFA
2.CADIFA

RDC 57/09: Registro de IFA
IN 15/09: Lista de IFAs
prioritários
1999 a
2005
Regulação em normas
relacionadas ao registro de
medicamento
2005
RDC 250/05: Cria o programa
de IFA
2005
RDC 249/05: PF de Produtos
Intermediários e IFAs
RDC 69/14
2008
RDC 30/08: Cadastro de IFA
2009
Contexto do IFA na Anvisa
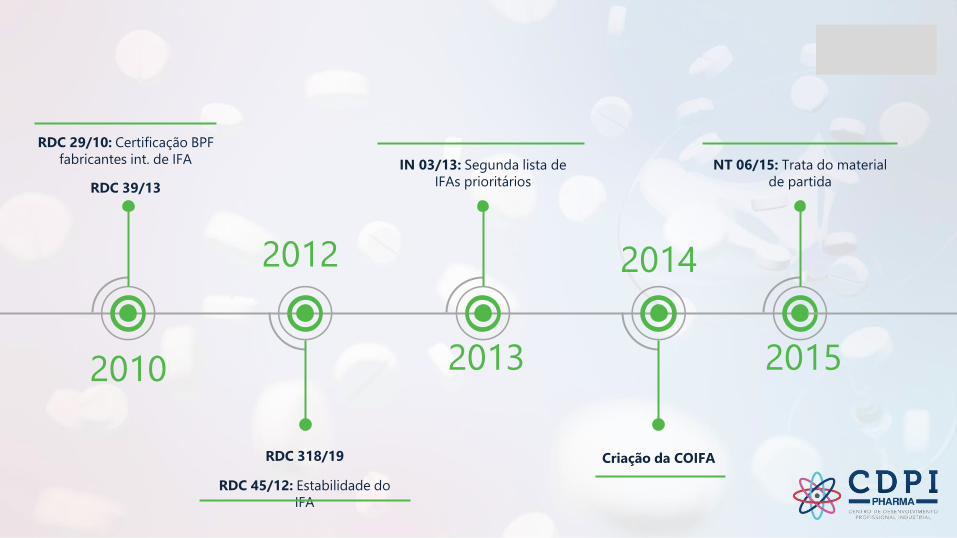
2010
RDC 29/10: Certificação BPF
fabricantes int. de IFA
RDC 39/13
2012
RDC 318/19
RDC 45/12: Estabilidade do
IFA
2013
IN 03/13: Segunda lista de
IFAs prioritários
2014
Criação da COIFA
2015
NT 06/15: Trata do material
de partida
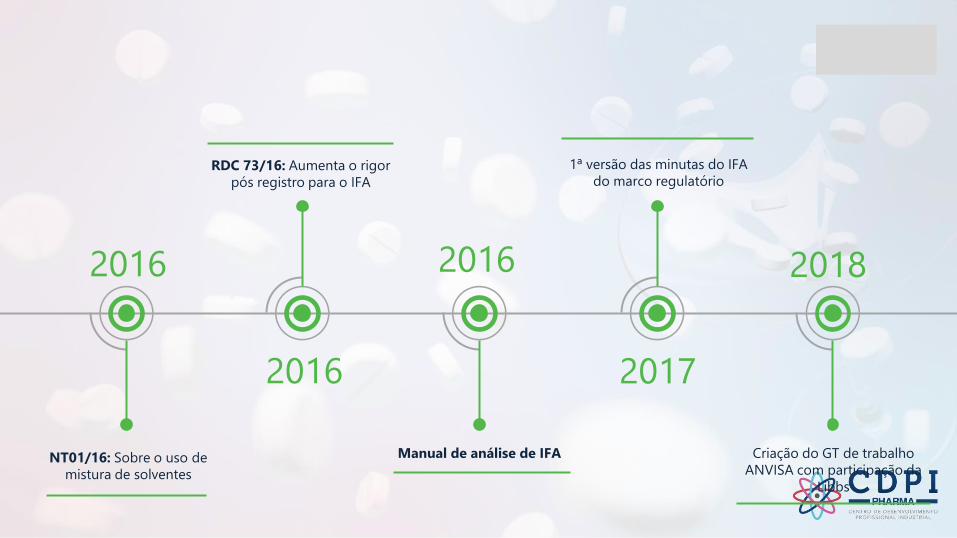
2016
Manual de análise de IFA
2017
1ª versão das minutas do IFA
do marco regulatório
2018
Criação do GT de trabalho
ANVISA com participação da
Libbs
2016
RDC 73/16: Aumenta o rigor
pós registro para o IFA
NT01/16: Sobre o uso de
mistura de solventes
2016
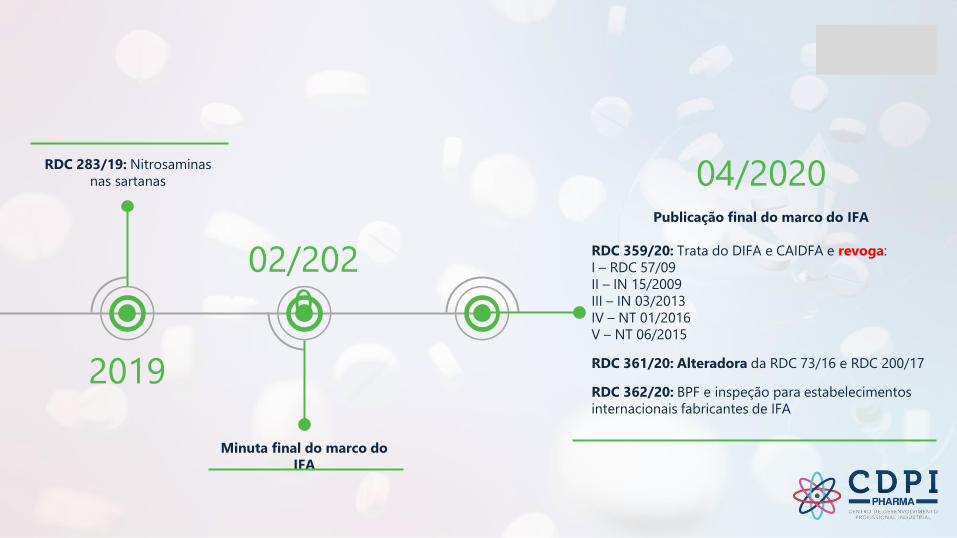
2019
RDC 283/19: Nitrosaminas
nas sartanas
02/202
0
Minuta final do marco do
IFA
04/2020Publicação final do marco do IFA
RDC 359/20: Trata do DIFA e CAIDFA e revoga:
I – RDC 57/09
II – IN 15/2009
III – IN 03/2013
IV – NT 01/2016
V – NT 06/2015
RDC 361/20: Alteradora da RDC 73/16 e RDC 200/17
RDC 362/20: BPF e inspeção para estabelecimentos
internacionais fabricantes de IFA

A RDC 57/09
não é
isonômicaA RDC 57/09
possui pouca
capilaridade
A RDC 57/09 não
contempla
moléculas novas
Existência de três
normativas não
equivalentes em requisitos
técnicos para o controle
sanitário de IFA
CBPF Anvisa
somente para
IFAs da RDC
57/09
Ausência de norma
que trata do pós
registro de IFA
Fluxo de
comunicação via
indústria
farmacêutica
Análise de
documentos
repetidos pela
Anvisa
O que motivou a
Anvisa implementar
o marco
regulatório de IFA?
Harmonização com
agências
internacionais

Vou aplicar as
normas do marco
do IFA para
qualquer tipo de
registro?
Registro e pós registro de
medicamentos novos, inovadores,
genéricos e similares.
Produto farmacêutico notificado,
produto biológico, fitoterápico,
produto tradicional fitoterápico,
medicamento específico, medicamento
dinamizado e IFA atípico. Incluindo
suas associações com IFA sintético ou
semissintético medicamento novo,
inovador, genérico ou similar.

Lote do medicamento: 01/02/2022
E
Petição: 01/08/2023
01 de março
de 2021
Fabricação lote medicamento: 01/02/22
E
Peticionamento: 01/08/2023
Outros
IFAs
IFAs da
RDC
57/09
Como será a
transitoriedade
da norma?

Linha do tempo – IFAs RDC 57/09
Publicação do Marco
regulatório de IFA
01/04/20Início do vigoramento do
marco regulatório de IFA
03/08/20Limite para protocolar a
petição
01/03/21
Não aplicável
Período de transição p/ petição
Nova norma
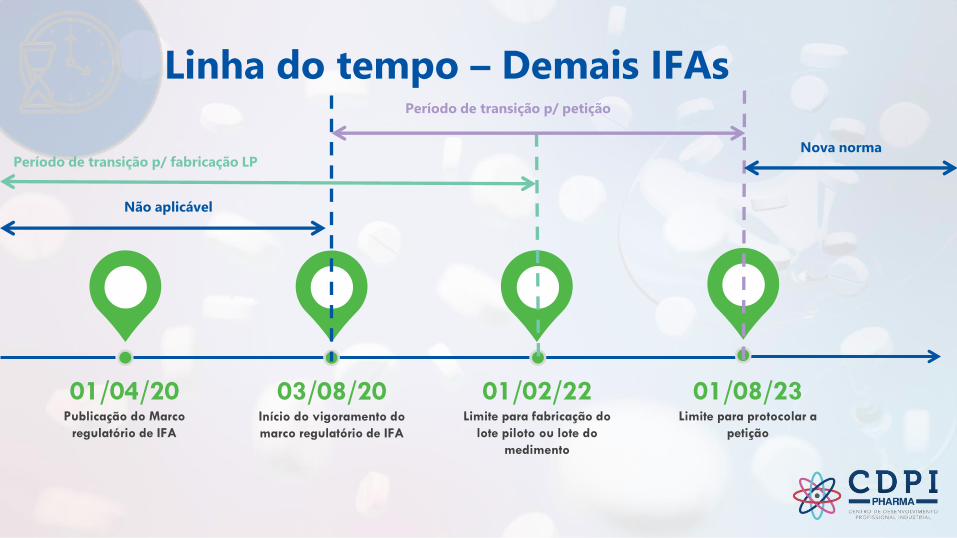
Linha do tempo – Demais IFAs
Publicação do Marco
regulatório de IFA
01/04/20Início do vigoramento do
marco regulatório de IFA
03/08/20Limite para fabricação do
lote piloto ou lote do
medimento
01/02/22Limite para protocolar a
petição
01/08/23
Não aplicável
Período de transição p/ fabricação LPNova norma
Período de transição p/ petição

Detentor
do DIFA

Detentor
do DIFA
Empresa que detém o
conhecimento de
todo o processo de
fabricação do
Insumo Farmacêutico
Ativo (IFA)
É a responsável pela
fabricação do IFA,
desde a introdução
do material de
partida na rota de
síntese.
Pode não ser a
unidade
responsável por
fabricar o IFA

CADIFACADIFA

CADIFACADIFA
arta de
dequação do
Instrumento
administrativo
que atesta que o
DIFA foi
considerado
adequado pela
Anvisa
A solicitação
pode ocorrer
de forma
associada à
petição de
medicamento
Não possui
prazo de
validade. Só
precisa manter o
ciclo de vida
A CADIFA é
emitida pela
Anvisa com
base nas
informações do
DIFA
submetido.

CADIFA
CADIFAI - número e data de emissão da CADIFA;
II - nome e número de DCB e CAS do IFA;
III - razão social e endereço do detentor do DIFA;
IV - razão social e endereço dos locais de
fabricação;
V - especificação do IFA e, se aplicável, referência
compendial;
VI - descrição da embalagem;
VII - condições de armazenamento do IFA;
VIII - prazo de reteste ou validade do IFA;
IX - campo para declaração de acesso.
As informações do inciso IV
contemplarão:
I - locais de fabricação do IFA
e intermediários; e
II - locais de esterilização ou
de etapas físicas
A CADIFA poderá conter
outras informações
consideradas relevantes
pela Anvisa

CADIFA
Será
obrigatória a
certificação
BPF da
AnvisaCaso o fabricante do IFA
reprove na inspeção BPF
Anvisa a CADIFA será
negada e o registro do
medicamento NÃO
SERÁ DEFERIDO
cBPF
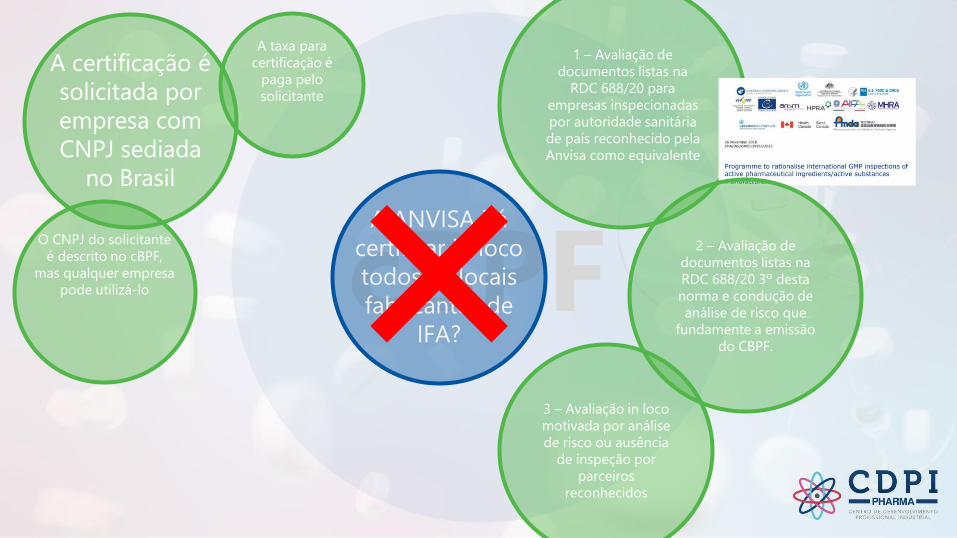
cBPF
A certificação é
solicitada por
empresa com
CNPJ sediada
no Brasil
A taxa para
certificação é
paga pelo
solicitante
O CNPJ do solicitante
é descrito no cBPF,
mas qualquer empresa
pode utilizá-lo
A ANVISA irá
certificar in loco
todos os locais
fabricantes de
IFA?
1 – Avaliação de
documentos listas na
RDC 688/20 para
empresas inspecionadas
por autoridade sanitária
de país reconhecido pela
Anvisa como equivalente
3 – Avaliação in loco
motivada por análise
de risco ou ausência
de inspeção por
parceiros
reconhecidos
2 – Avaliação de
documentos listas na
RDC 688/20 3º desta
norma e condução de
análise de risco que
fundamente a emissão
do CBPF.

Fluxos
ANVISA

Fluxos
ANVISA
Fluxo de trabalho atual – Registro e inclusão de novo fornecedor de IFA
Parte
aberta
Registro produto 01Resp.
exigência
Exigência 01
Exigência 02
Exigência 03
Exigência 01
Resposta
carta
Resposta
carta
Detentor
do DIFA
Farmacêutica
01
Farmacêutica
02
Farmacêutica
03
Parte
aberta
Parte
aberta
Parte
aberta
Parte
aberta
Parte
aberta
Registro produto 02
Registro produto 03Resp.
exigência
Resp.
exigência
Exigência 02
Exigência 03
Parte restrita
01
Resposta
carta
Parte restrita
02
Parte restrita
03

Fluxos
ANVISA
Fluxo de trabalho após marco do IFA - Registro e inclusão de novo fornecedor de IFA
Parte
aberta
Registro produto 01
Detentor
do DIFA
Farmacêutica
01
Farmacêutica
02
Farmacêutica
03
Autorizaçã
o do
detentor
do DIFA.
CADIFA
Registro produto 02
Registro produto 03
Parte restritaParte
aberta
Resposta carta
CADIFA
CADIFA
Exigência 01
Parte
aberta
Parte
aberta
CADIFA
CADIFA

Fluxos
ANVISA
Fluxo de trabalho atual – Pós registros
Notificação
Pós registro produto 01Resp.
exigência
Exigência 01
Exigência 02
Exigência 03
Exigência 01
Resposta
exigência
Resposta
exigência
Detentor
do DIFA
Farmacêutica
01
Farmacêutica
02
Farmacêutica
03
Notificação
Notificação
Pós registro
Pós
registro
Pós
registro
Pós registro produto 02
Pós registro produto 03Resp.
exigência
Resp.
exigência
Exigência 02
Exigência 03
Resposta
exigência
Fluxos
ANVISA
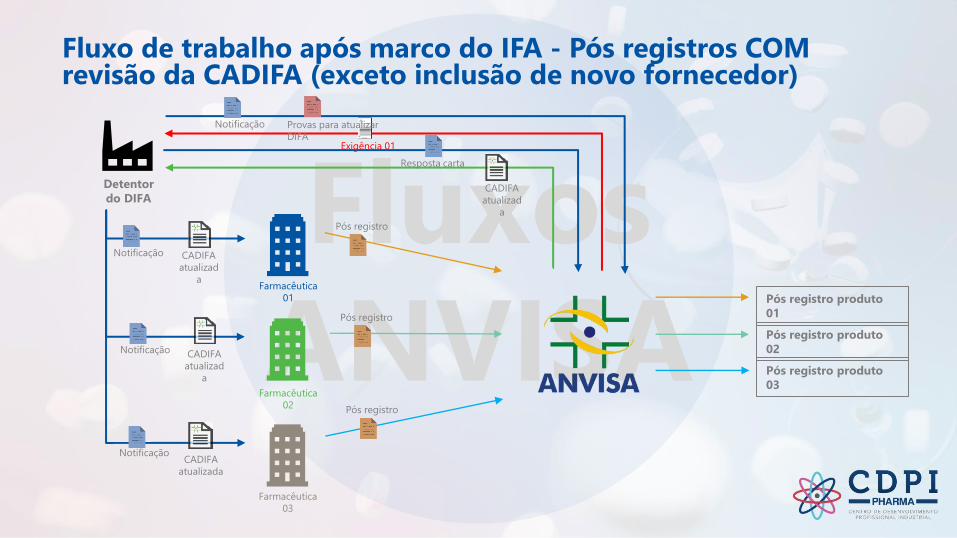
Fluxo de trabalho após marco do IFA - Pós registros COM revisão da CADIFA (exceto inclusão de novo fornecedor)
Notificação
Pós registro produto
01
Detentor
do DIFA
Farmacêutica
01
Farmacêutica
02
Farmacêutica
03
Pós registro produto
02
Pós registro produto
03
Provas para atualizar
DIFA
Notificação
Resposta carta
CADIFA
atualizad
a
Exigência 01
Notificação
Notificação
CADIFA
atualizad
a
CADIFA
atualizad
a
CADIFA
atualizada
Pós registro
Pós registro
Pós registro

Fluxos
ANVISA
Fluxo de trabalho após marco do IFA - Pós registros SEM revisão da CADIFA (exceto inclusão de novo fornecedor)
Notificação
Detentor
do DIFA
Farmacêutica
01
Farmacêutica
02
Farmacêutica
03
Provas para ciclo de vidaNotificação
Resposta carta
Manifestação
favorável
Exigência 01
Notificação
Notificação
“§ 1º-E Para IFA com CADIFA,
quando a mudança do DIFA não
implicar
revisão da CADIFA, o detentor do
registro do medicamento não deve
protocolar
mudança pós-registro do
medicamento.”
RDC
361/2020 Art.
3º

Registros
novos
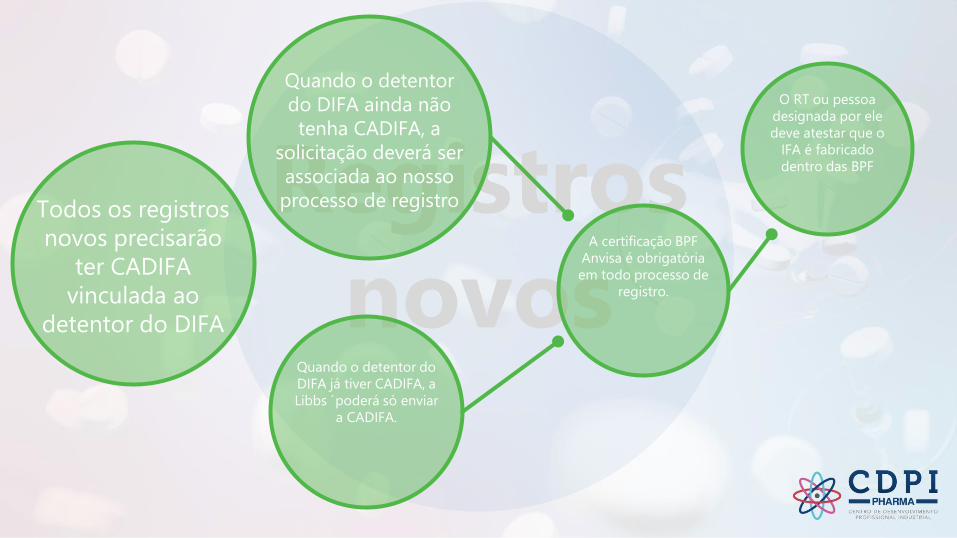
Registros
novosTodos os registros
novos precisarão
ter CADIFA
vinculada ao
detentor do DIFA
Quando o detentor
do DIFA ainda não
tenha CADIFA, a
solicitação deverá ser
associada ao nosso
processo de registro
Quando o detentor do
DIFA já tiver CADIFA, a
Libbs ´poderá só enviar
a CADIFA.
O RT ou pessoa
designada por ele
deve atestar que o
IFA é fabricado
dentro das BPF
A certificação BPF
Anvisa é obrigatória
em todo processo de
registro.

Pós
registro

Pós
registroE os registros já
existentes que não
haverá alteração de
fornecedor ou local
de produção c/
alterações no
processo
produtivo?
Todos os pós registros
relacionados à inclusão
de novo fornecedor ou
alteração de site
produtivo com
alterações no processo
precisarão de CADIFA
Nenhuma ação
é necessária.
Não precisa
pedir CADIFA
para fabricantes
já registrados

Pós
registro
Demais
alterações
pós
registros
Controle das alterações que
ocorrem para registros de
medicamentos que possuem
CADIFA vinculada ficou
harmonizado com os que não
possuem (registros antigos)
Aumento na
abrangência do controle
de questões
relacionadas
exclusivamente ao IFA
Controle de ciclo na própria
CADIFA.
Registros de medicamentos
vinculados à CADIFA, haverá
menos processos de alterações
pós registro
Somente alterações que
mudem o conteúdo da
CADIFA ou alterações
maiores.

Desafios unidade
farmacêutica
Prospecção do
fabricante do
IFA
Falta de controle
sobre exigências
do IFA
ANVISA poderá ter
conhecimento sobre as
alterações do fabricante
do IFA antes dos
clientes
Solicitação e
pagamento da
cBPF Anvisa
Ações nos
cronogramas
Risco de
desabastecimento
da condição
aprovada.
Submissão do
DIFA pelo
detentor do DIFA
Sistema de
petição em
português
Comprometimento para
atender do prazo para
submissão do DIFA
Interesse no
mercado
Brasileiro
Custo para
o projeto
Auditorias
suspensas
devido o cenário
global
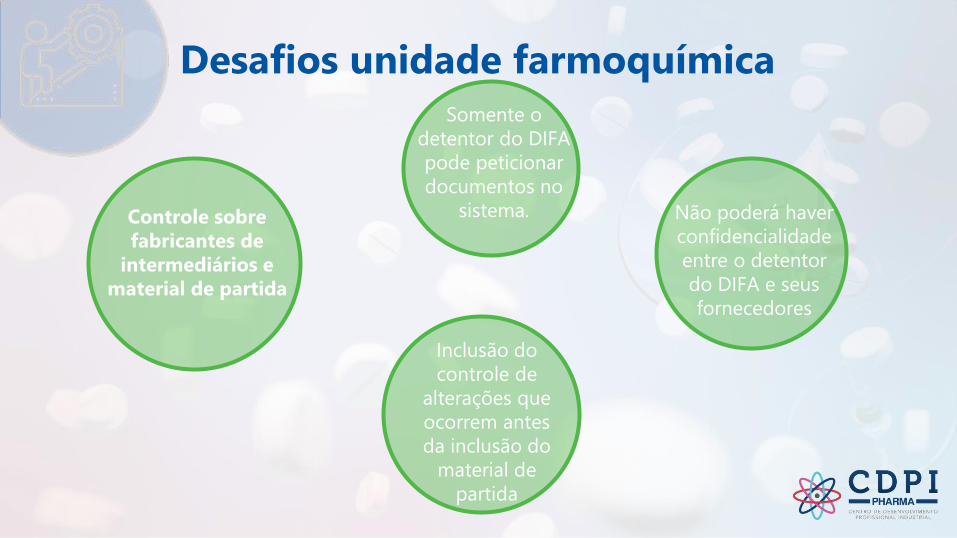
Desafios unidade farmoquímica
Controle sobre
fabricantes de
intermediários e
material de partida
Somente o
detentor do DIFA
pode peticionar
documentos no
sistema. Não poderá haver
confidencialidade
entre o detentor
do DIFA e seus
fornecedores
Inclusão do
controle de
alterações que
ocorrem antes
da inclusão do
material de
partida

Resumo Modulo 4
✓ O que é o DIFA
✓ Estrutura do DIFA com ênfase em fabricação e caracterização:
partes aberta e restrita
✓ Estudos de caso
✓ CADIFA

Dúvidas e Comentários

Carolina Martins Avila de Sant’Ana
Paulo Eliandro da Silva Junior
OBRIGADO!