pairwise sequence alignment. the most important class of bioinformatics tools – pairwise alignment...
TRANSCRIPT

Pairwise SequenceAlignment

• The most important class of bioinformatics tools – pairwise alignment of DNA and protein seqs. alignment 1 alignment 2
Seq. 1 ACGCTGA ACGCTGASeq. 2 A - - CTGT ACTGT - -
Seeks alignments high seq. identity, few mismatchs and gapsAssumption – the observed identity in seqs. to be aligned is the result
of either random or of a shared evolutionary originIdentity ≠ similaritySequence identity = Homology (a risky assumption)Sequence identity ≠ Homology
Pairwise Sequence Alignment

Pairwise Sequence Alignment
Figure A Common evolutionary events and their effects on alignment
indel
Same true alignment arise through different evolutionary events
Scoring scheme: substitution -1, indel -5, match 3
Score 9 5 4 4

Find the optimal score the best guess for the true alignmentFind the optimal pairwise alignment of two seqs. inserted
gaps into one or both of them maximize the total alignment score
Dynamic programming (DP) – Needleman and Wunsch (1970), Smith and Waterman (1980), this algorithm guarantees that we find all optimal alignments of two seqs. of lengths m and n
BLAST is based on DP with improvement on speed
Prof. Waterman http://www.usc.edu/dept/LAS/biosci/faculty/waterman.html
Pairwise Sequence Alignment

Pairwise Sequence Alignment
),()1,(
),(),1(
),()1,1(
max),(
jcjiS
icjiS
jicjiS
jiS
The score for alignment of i residues of sequence 1 against j residues of sequence 2 is given by
where c(i,j) = the score for alignment of residues i and j and takes the value 3 for a match or -1 for a mismatch,c(-,j) = the penalty for aligning a residue with a gap, which takes the value of -5

• The entry for S(1,1) is the maximum of the following three events:
• S(0,0) + c(A,A) = 0 + 3 = 3 [c(A,A) = c(1,1)]• S(0,1) + c(A, -) = -5 + -5 = -10 [c(A, -) = c(1, -)]• S(1,0) + c(-, A) = -5 + -5 = -10 [c(- ,A) = c(-, 1)]• Similarly, one finds S(2,1) as the maximum of
three values: (-5)-1=-6; 3-5=-2; and (-10)-5=-15 the best is entry is the addition of the C indel to the A-A match, for a score of -2 (see next page).
Pairwise Sequence Alignment

Pairwise Sequence Alignment
The alignment matrix of sequences 1 and 2
TGTCA
A
C
G
C
T
G
A
2520151050
17127235
9416210
1451715
34041220
71191725
224142230
139192735 S(2,1) = max {S(1,0) + c(2,1),S(1,1) + c(2,-), S(2,0) + c(-,1)}
= max { S(1,0) + c(C,A),S(1,1) + c(C,-), S(2,0) + c(-,A) } = max { -5-1, 3-5, -10-5 }= -2

Pairwise Sequence Alignment
Traceback determine the actual alignmentFrom the top right hand corner the (7,5) cell
TGTCA
A
C
G
C
T
G
A
2520151050
17127235
9416210
1451715
34041220
71191725
224142230
139192735
For example the 1 in the (7,5) cell could only be reached by the addition of the mismatch A-T
ACGCTGAA - - CTGTorACGCTGAAC - - TGT4 matches1 mismatch2 indels
Ambiguity – has to do with which C in seq. 1 aligns with the C in seq. 2

Parameters settings - Gap penalties• Default settings are the easiest to use but they are not
necessarily yield the correct alignment• constant penalty independent of the length of gap, A• proportional penalty penalty is proportional to the length L of
the gap, BL (that is what we used in the this lecture)• affine gap penalty gap-opening penalty + gap-extension
penalty = A+BL• There is no rule for predicting the penalty that best suits the
alignment• Optimal penalties vary from seq. to seq. it is a matter of trial
and error• Usually A > B, because of opening a gap (usually A/B ~ 10)• Hint: (1) compare distantly related seqs. high A and very low B
often give the best results penalized more on their existence than on their length, (2) compare closely related seqs., penalize both of extension and extension
Pairwise Sequence Alignment

Exercise - Computing an optimal sequence alignment
Two score schemes(1) Gap penalty = -5, mismatch = -1, match =3(2) Gap penalty = -1, mismatch = -1, match =3
(1) First alignment score = 5*3 + 2*(-1) =13 Second/Third alignment score = 6*3 + 2*(-5) = 8(2) First alignment score = 5*3 + 2*(-1) =13 Second/Third alignment score = 6*3 + 2*(-1) = 16
A more serious problem – identify the wrong alignment

TATGGCA
A
G
C
G
T
A
T
3530
13
2520151050
35
10
15
20
25
30
35
Exercise Computing an optimal sequence alignment
Gap penalty = -5
TATGGCA
A
G
C
G
T
A
T
76
16
543210
31
2
3
4
5
6
7
Gap penalty = -1

• Dynamic Programming do not provide the user with a measure of statistical similarity when regions of local similarity when regions of local similarity are found
• Take into account not just the position-position overlap between two seqs. but the characteristics of the a.a being aligned define scoring matrices
• Protein scoring matrices take three major biological factors into account:
• Conservation – the numbers within the scoring matrix provide a way of representing what a.a. are capable of substituting for other a.a. (characteristics such as charge, size, hydrophobicity)
• Frequency – a.a cannot freely substitute for one another, the matrices need to reflect how often particular a.a occur among the entire proteins.
• Evolution – scoring matrices implicitly represent evolutionary patterns, and matrices can be adjusted to favor the detection of closely related or more distantly related proteins.
BLAST (Scoring matrices)

Scoring matrices and the Log Odds Ratio
where pi[pj] = probability with which a.a i [j] occurs among all proteins
qi,j = how often the two a.a i and j are seen to align with one another in MSA of protein families or in seqs. that are known to have a biological relationship.
BLAST (Scoring matrices)
]log[ ,,
j
jiji pp
qS
i

Amino acid substitution matrix (PAM and BLOSUM)• Leave most adjustable parameters to the default value except the
scoring matrix• Box 2.1 a simple scheme for scoring seq. matches and mismatches
(all mismatches received the same penalty)• Scoring matrix allows some mismatches to be penalized less then
others• Leucine-isoleucine mismatch < leucine-tryptophan mismatch • PAM (Point Accepted Mutations) scoring matrices – derived from
closely related species (evolutionary point of view, avoid the complications of unobserved multiple substitutions at a single position)
• PAM derived from the likelihood of amino acids substitution during the evolutionary process
• PAM matrices with a smaller number represent shorter evolutionary distance
• PAM1 – one a.a change per 100 a.a, or roughly 1% divergence
BLAST (PAM matrices)

PAM
Asp Glu0.95%

BLOSUM (BLOck SUM) – there are evidence it outperform PAM• Block proteins in the same family can be aligned without
introducing a gap (not the individual seqs.)• So any given protein can contain one or more blocks, corresponding
to each of its functional or structural motif • With these protein blocks, it is possible to look for substitution
patterns only in the most conserved regions of a protein block substitution matrices are generated
• BLOSUM scoring matrix – based on data from distantly related seqs. (default BLOSUM62 for general use)
• The most commonly used matrices are PAM120, PAM250, BLOSUM50 and BLOSUM 62
• BLOSUM matrices with a smaller number represent a longer evolutionary distance
BLAST (BLOSUM matrices)
BLAST (BLOSUM matrices)
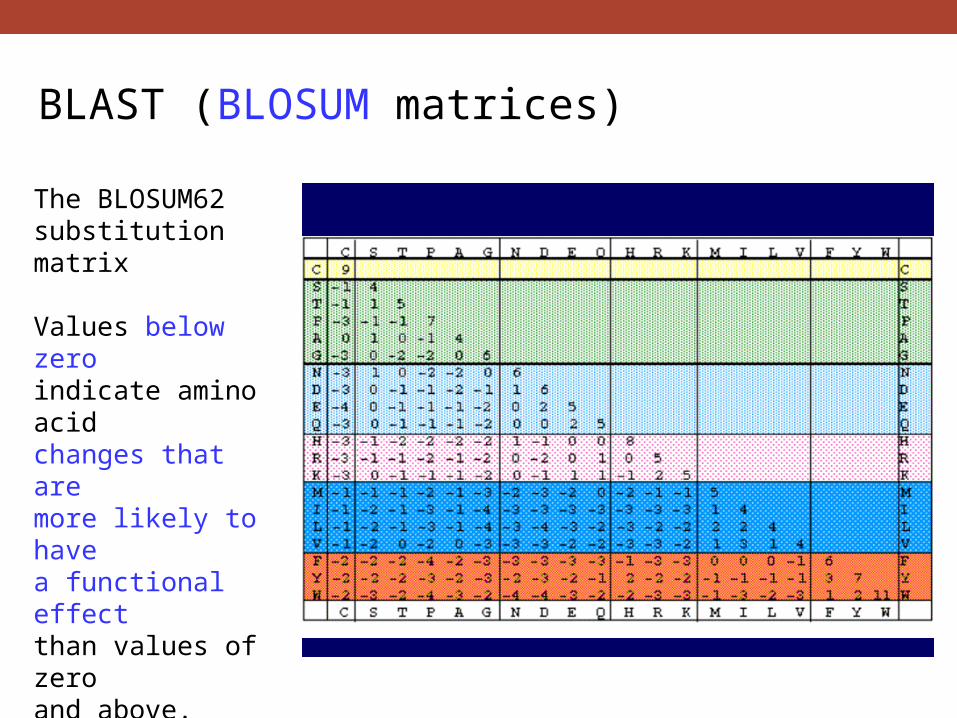
The BLOSUM62 substitution matrix
Values below zero indicate amino acid changes that are more likely to have a functional effect than values of zero and above.

PAM250 equivalent to BLOSUM45PAM160 equivalent to BLOSUM62PAM120 equivalent to BLOSUM80
BLAST (relating PAM to BLOSUM)
Matrix Best use Similarity(%)
BLOSUM90Short alignments that are highly similar
70-90
BLOSUM80Detecting members of a protein family
50-60
BLOSUM62Most effective in finding all potential similarities
30-40
BLOSUM30Longer alignment of more divergent seqs.
<30
Selecting an appropriate scoring matrix

BLAST (Sensitivity and Specificity)

BLAST (Sensitivity and Specificity)
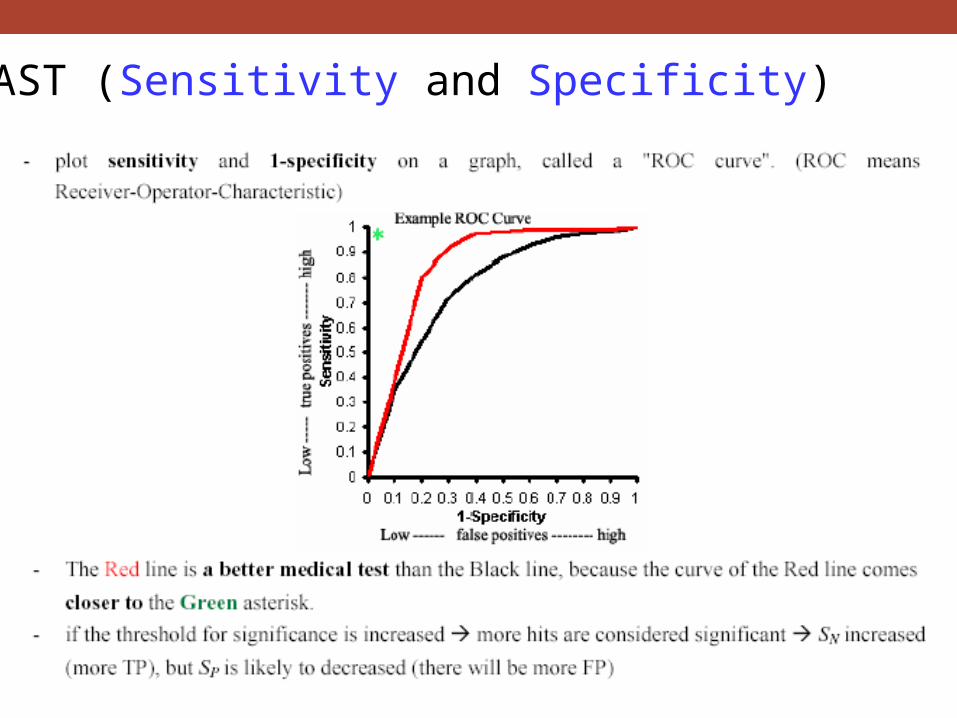
BLAST (Sensitivity and Specificity)
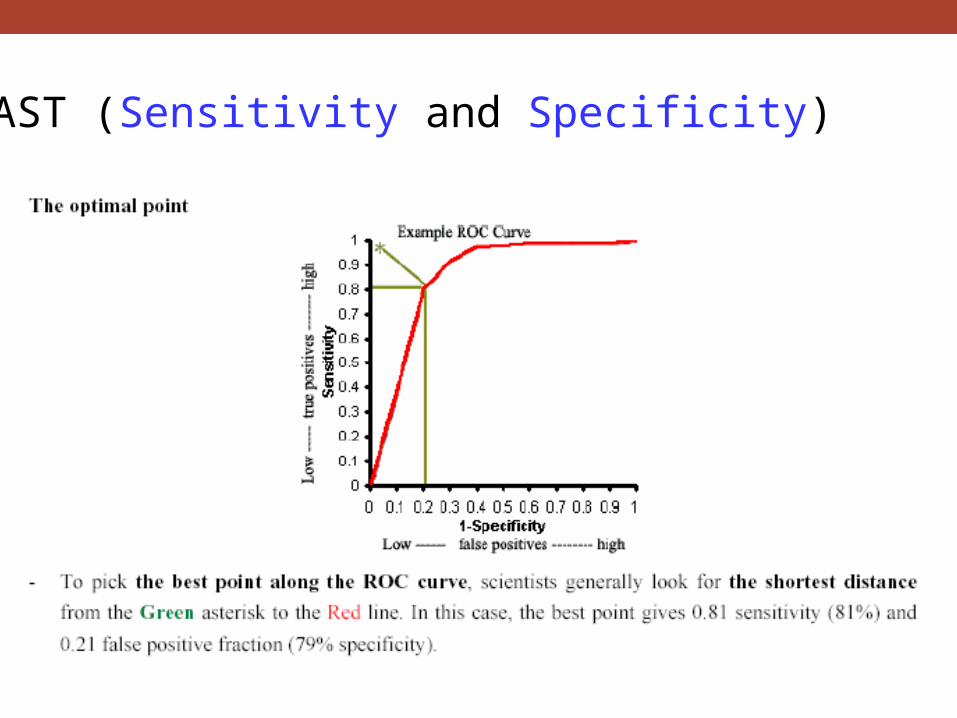
BLAST (Sensitivity and Specificity)