overview biological motivation methods in gene prediction mapping of large est data sets ...
TRANSCRIPT

Overview
Biological motivationBiological motivation
Methods in gene predictionMethods in gene prediction
Mapping of large EST data setsMapping of large EST data sets
Applications of EST data miningApplications of EST data mining

ESTomicsESTomics
Sorin Istrail

Biological motivation
Model of eukaryotic gene transcription and translationModel of eukaryotic gene transcription and translation
Upstream binding sites
RNA polymerase II promoter
Sp1 C/EBP Oct1
TATA box
Initiator
GeneDNA coding
strand

Biological motivation
Model of eukaryotic gene transcription and translationModel of eukaryotic gene transcription and translation
Upstream binding sites
RNA polymerase II promoter
Sp1 C/EBP Oct1
TATA box
Initiator
GeneDNA coding
strand
Exon 1 Exon 2Intron primary transcript5’ UTR5’ UTR 3’ UTR3’ UTR
AAUAAAAAUAAATranscription(A)n(A)n
capcap
GTGT AGAG

Biological motivation
Model of eukaryotic gene transcription and translationModel of eukaryotic gene transcription and translation
Upstream binding sites
RNA polymerase II promoter
Sp1 C/EBP Oct1
TATA box
Initiator
GeneDNA coding
strand
Exon 1 Exon 2Intron
Splicing
primary transcript
mRNA
5’ UTR5’ UTR 3’ UTR3’ UTR
AAUAAAAAUAAATranscription(A)n(A)n
capcap
GTGT AGAG
5’ UTR5’ UTR 3’ UTR3’ UTR

Biological motivation
Model of eukaryotic gene transcription and translationModel of eukaryotic gene transcription and translation
Upstream binding sites
RNA polymerase II promoter
Sp1 C/EBP Oct1
TATA box
Initiator
GeneDNA coding
strand
Exon 1 Exon 2Intron
Splicing
primary transcript
mRNA
protein(peptide)
5’ UTR5’ UTR 3’ UTR3’ UTR
AAUAAAAAUAAA
Translation
Transcription(A)n(A)n
capcap
GTGT AGAG
5’ UTR5’ UTR 3’ UTR3’ UTR

Biological motivation
GeneDNA coding
strand
Exon 2 Exon 3Intron primary transcript
mRNA
5’ UTR5’ UTR 3’ UTR3’ UTR
ESTs
Expressed Sequence Tags (ESTs) are cDNA fragmentsExpressed Sequence Tags (ESTs) are cDNA fragments
500 bp long on average
may span one or more exons
cDNA: single-stranded DNA complementary to an RNA, synthesized cDNA: single-stranded DNA complementary to an RNA, synthesized from it by reverse transcriptionfrom it by reverse transcription
Exon 1 Intron
Exon 4(non-coding)
Intron

Overview
Biological motivationBiological motivation
Methods in gene predictionMethods in gene prediction
Mapping of large EST data setsMapping of large EST data sets
Applications of EST data miningApplications of EST data mining

Methods in gene finding
Ab initioAb initio analysis of genomic sequences analysis of genomic sequences ((GenScanGenScan, Burge and Karlin 1997; , Burge and Karlin 1997; HMMerHMMer, Haussler et al. 1993, Krogh et al. , Haussler et al. 1993, Krogh et al.
1994; 1994; FGenesHFGenesH, Solovyev and Salamov 1994), Solovyev and Salamov 1994)
Comparison of protein and genomic sequences Comparison of protein and genomic sequences ((ProcrustesProcrustes, Gelfand et al. 1996; , Gelfand et al. 1996; GenewiseGenewise, Birney and Durbin), Birney and Durbin)
Comparison of expressed DNA (ESTs, cDNA, mRNA) and genomic Comparison of expressed DNA (ESTs, cDNA, mRNA) and genomic
sequences sequences ((EST_GENOMEEST_GENOME, Mott 1997; , Mott 1997; SIM4SIM4, Florea et al. 1998), Florea et al. 1998)
Cross-species genomic sequence comparisons Cross-species genomic sequence comparisons ((ROSETTAROSETTA, Batzoglou et al. 2000; , Batzoglou et al. 2000; CEMCEM, Bafna and Huson 2000), Bafna and Huson 2000)

Ab initio gene finders
Use information embedded in the genomic sequence to predict the exon Use information embedded in the genomic sequence to predict the exon modelmodel polyadenylation signal (AATAAA) differential codon usage in coding versus non-coding sections of the gene upstream regulatory signals (TATA boxes) and local characteristics of the
sequence (CpG islands) splice recognition signals (e.g., GT-AG)
Markov models are the predominant predictive methodMarkov models are the predominant predictive method
CaveatsCaveats not effective in detecting alternatively spliced forms, interleaved or overlapping
genes

The GenScan method
High-level organizationHigh-level organization each of the basic functional units of a gene is associated with a state in the
HMM
Lower-level organization Lower-level organization separate sequence prediction module for each of the higher-level elements
exons (marginal, internal, phase-specific) - inhomogeneous 3-periodic fifth order Markov model
introns and intergenic regions - homogeneous 5th order Markov model 5’ and 3’UTRs - homogeneous 5th order Markov model polyadenylation signal donor and acceptor splice sites - WAM and the Maximal Dependence
Decomposition (MDD), i.e., a decision tree-based weighted position matrix

GenScan’s HMM for sequence generation
N(intergenic
region)
Reverse (-) stran
d
Forw
ard (+
) strand
E0 +
E1 +
E2 +
I0 +
I1 +
I2 +
Einit+
Eterm+
F +(5’UTR)
T +(3’UTR)
P +(prom)
A +(polyA signal)
Esngl +(single-exon
gene)
A -(polyA signal)
P -(prom)
F -(5’UTR)
T -(3’UTR)
Einit-
Eterm-
I0 -
I1 -
I2 -
E0 -
E1 -
E2 -
Esngl -(single-exon
gene)
(“Prediction of complete gene structures in human genomic DNAPrediction of complete gene structures in human genomic DNA”(1997) Burge and Karlin, JMB 268, p. 86)

Protein-genomic sequence comparisons
Use sequence similaritUse sequence similarityy between the protein and the protein-coding between the protein and the protein-coding regions of the genomic sequence for gene model predictionregions of the genomic sequence for gene model prediction
Algorithmic techniquesAlgorithmic techniques dynamic programming-based sequence alignment algorithms specialized recognition modules for splice junction prediction profile HMMs
ExamplesExamples Procrustes (Gelfand et al. 1996)
combinatorial pairing of putative splice junctions to form introns uses protein-genomic sequence similarity to validate the correct pairings
Genewise (Durbin and Birney) HMM-based sequence profiles uses similarity between the query protein and a database of protein families
organized in profiles (Pfam)
CaveatsCaveats prediction limited to coding regions (excluding 5’ and 3’ UTRs)

cDNA-genomic sequence comparisons
Use similarities between the cDNA (ESTs, mRNAs) and the genomic Use similarities between the cDNA (ESTs, mRNAs) and the genomic sequences to predict the gene model.sequences to predict the gene model.
Algorithmic techniquesAlgorithmic techniques dynamic-programming based sequence alignment algorithms specialized module for splice junction detection (pattern matching techniques, or
statistical modeling)
ExamplesExamples EST_GENOME (Mott 1997)
dynamic programming alignment with an affine scoring scheme uniform scoring for large indels (introns)
SIM4 (Florea et al. 1998) incremental exon detection and refinement with ‘blast’-like and greedy
sequence comparison techniques pattern matching prediction of splice junctions
CaveatsCaveats accuracy depends on the quality of the data source (e.g., cannot detect genomic
contamination by unspliced introns, or spurious priming)

Cross-species genomic sequence comparison
Use the sequence similarity and the ordering of homologous regions Use the sequence similarity and the ordering of homologous regions between genomic sequences from related organisms to infer their between genomic sequences from related organisms to infer their common gene model. common gene model.
Algorithmic techniquesAlgorithmic techniques dynamic programming-based sequence comparison algorithms statistical modeling of the splice junctions and other common transcriptional
elements
ExamplesExamples ROSETTA (Batzoglou et al. 2000), CEM (Conserved Exon Model; Bafna and
Huson 2000) progressive sequence alignment between the various categories of
orthologus regions (based on the expected sequence similarity) statistical methods for splice signal recognition (?)
CaveatsCaveats accuracy depends on the specificity of sequence similarity and the presence of
delimiting transcriptional signals at that locus (similarity may extend past the gene boundaries)
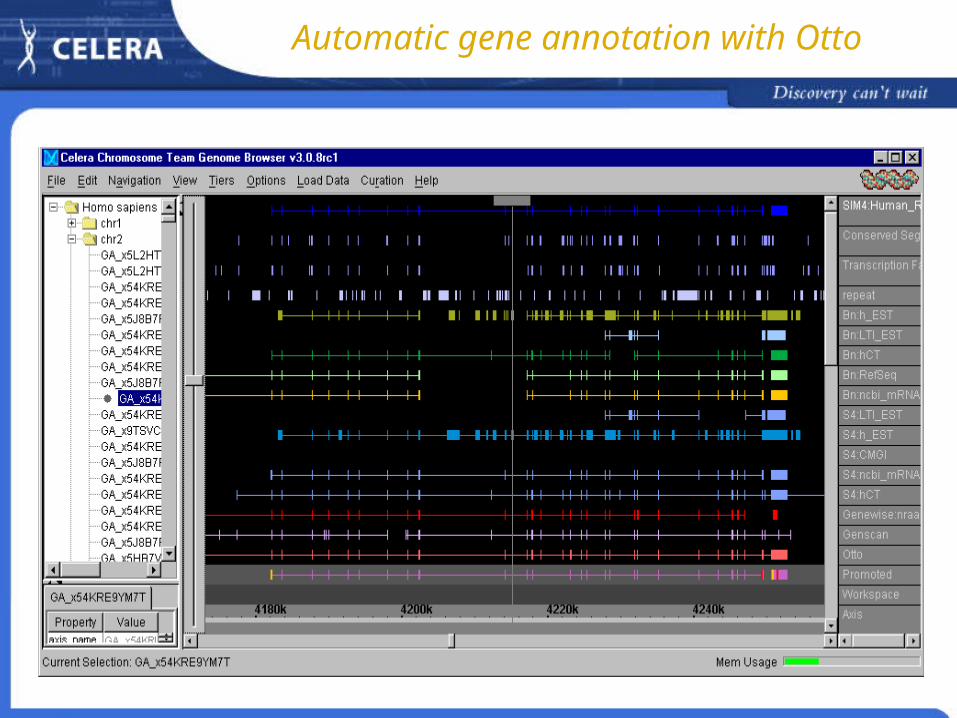
Automatic gene annotation with Otto

Components of the automatic gene annotation
Bn - blastn (dbEST, CHGI, CMGI, RefSeq) S4 - SIM4 (dbEST, CHGI, CMGI, RefSeq) Genewise (nr) GenScan FGenesH repeat - RepeatMasker etc.
Otto automatic gene predictions by Otto Promoted curated transcripts

Overview
Biological motivationBiological motivation
Methods in gene predictionMethods in gene prediction
Mapping of large EST data setsMapping of large EST data sets
Applications of EST data miningApplications of EST data mining
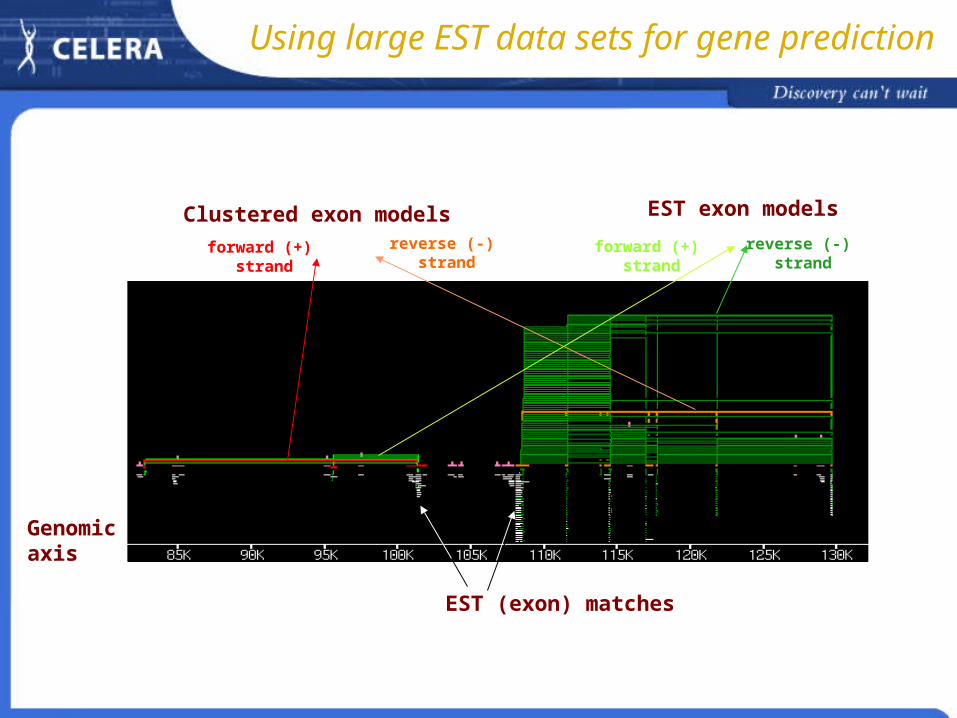
Using large EST data sets for gene prediction
EST exon modelsClustered exon models
EST (exon) matches
Genomicaxis
forward (+) strand
reverse (-) strand
reverse (-) strand
forward (+) strand

Using large EST data sets for gene prediction
Each EST may span one or more of a gene’s exonsEach EST may span one or more of a gene’s exons Overlapping ESTs and mRNAs on the genome can be used to infer gene Overlapping ESTs and mRNAs on the genome can be used to infer gene
modelsmodels Large data sets must be used for completeness Large data sets must be used for completeness
dbEST ( ~3.7 million ESTs) UniGene (~90,000 ESTs and mRNA transcripts, grouped by similarity) proprietary data sets (LifeSeq, CHGI)
Analyzing such large data sets is time and resource-consumingAnalyzing such large data sets is time and resource-consuming
Strategy for EST data miningStrategy for EST data mining determine the occurrences of a large set of cDNA sequences in a target
genome (mapping) group the overlapping EST matches on the genome to infer the underlying gene
model (clustering)

Mapping ESTs to a target genome
MappingMapping
Determine, for a given EST, the exact genomic location(s) and exon Determine, for a given EST, the exact genomic location(s) and exon model(s), i.e.model(s), i.e. exon coordinates in the genomic sequence genomic match strand (forward, or reverse complement) percent sequence identity values (at the exon and EST levels) spliced EST-genomic sequence alignment
ValidationValidation
Criteria for validating putative EST occurrences on the genomeCriteria for validating putative EST occurrences on the genome EST coverage similarity between the EST and genomic sequences e.g., >80% of the EST must match the genome, at >90% sequence identity

Technical challenges
cDNAcDNA Sequencing errors and polymorphisms Interspecies contamination Low quality EST data
Gene modelGene model Multiple gene homologues Alternative splicing Interleaving and overlapping of genes
Genomic sequenceGenomic sequence Repetitive elements Genomic contamination Genomic sequence representation
Large data sizeLarge data size ~3 billion bp in the human genome ~2.8 billion bp in dbEST
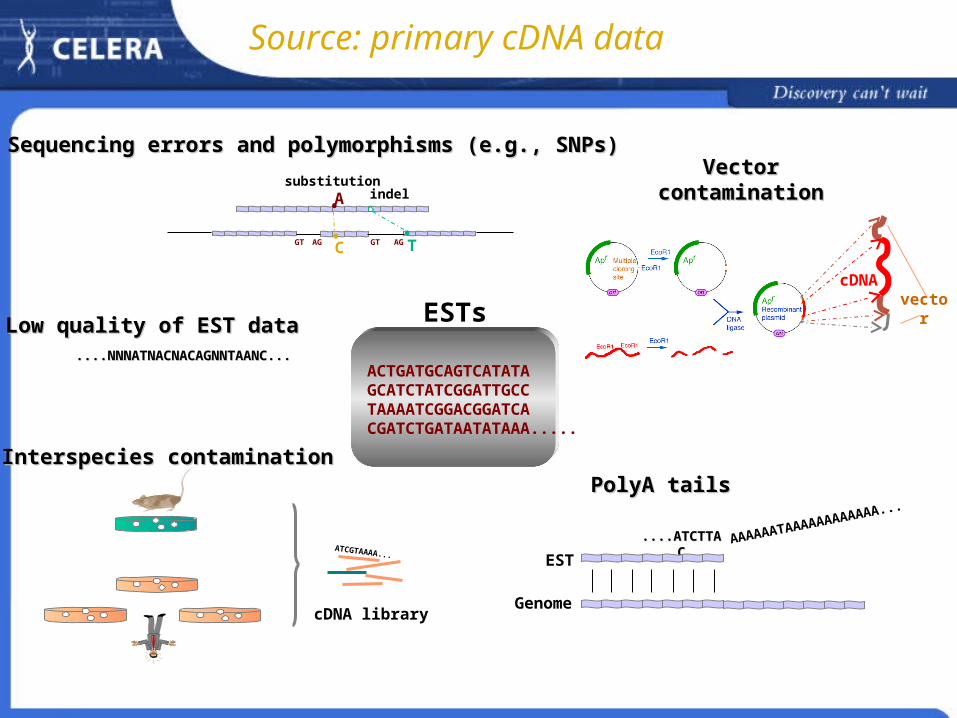
Source: primary cDNA data
ACTGATGCAGTCATATAGCATCTATCGGATTGCCTAAAATCGGACGGATCACGATCTGATAATATAAA.....
ESTs
cDNA library
ATCGTAAAA...
Interspecies contaminationInterspecies contaminationPolyA tailsPolyA tails
....ATCTTAC....ATCTTAC AAAAAATAAAAAAAAAAAA...
EST
Genome
Low quality of EST dataLow quality of EST data ....NNNATNACNACAGNNTAANC.......NNNATNACNACAGNNTAANC...
TGT AG
A
C AGGT
substitutionindel
Sequencing errors and polymorphisms (e.g., SNPs)Sequencing errors and polymorphisms (e.g., SNPs)
Vector contaminationVector contamination
vector
cDNA
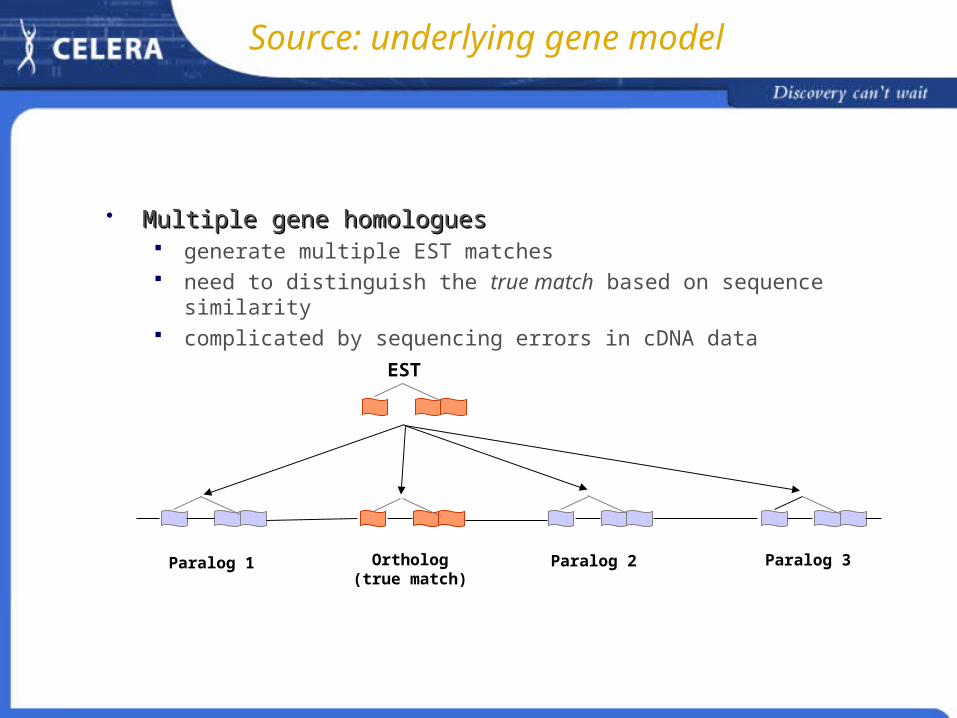
Source: underlying gene model
Multiple gene homologuesMultiple gene homologues generate multiple EST matches need to distinguish the true match based on sequence similarity complicated by sequencing errors in cDNA data
EST
Ortholog(true match)
Paralog 1 Paralog 2 Paralog 3
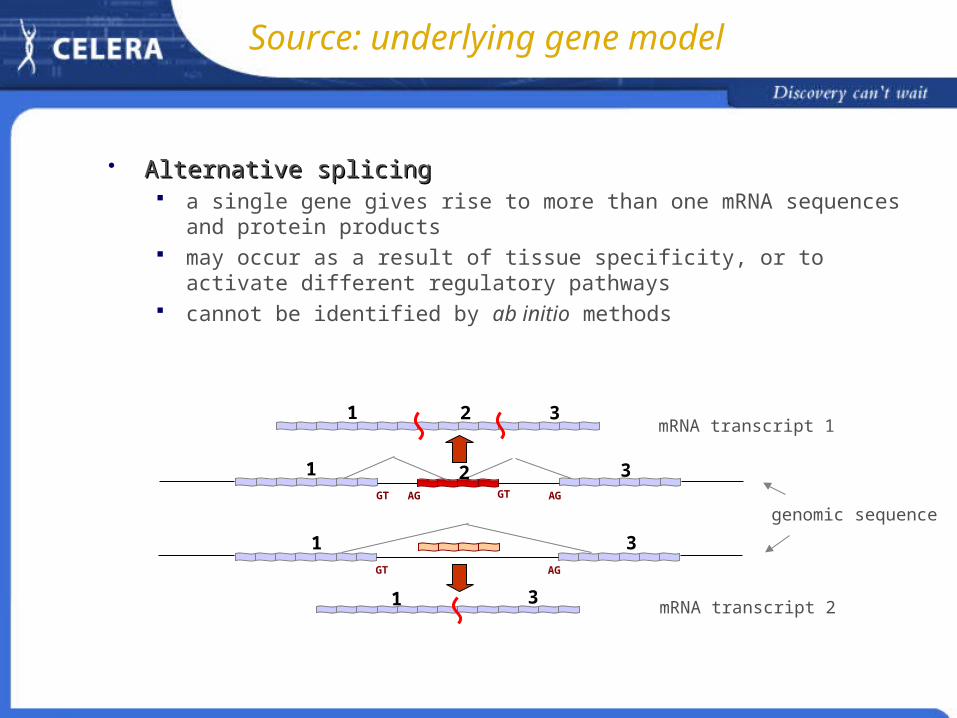
Source: underlying gene model
Alternative splicingAlternative splicing a single gene gives rise to more than one mRNA sequences and protein
products may occur as a result of tissue specificity, or to activate different regulatory
pathways cannot be identified by ab initio methods
GT AG
1 3
GT AG GT AG
1 32
1 2 3
1 3
mRNA transcript 1
mRNA transcript 2
genomic sequence

Source: underlying gene model
Interleaving and overlapping of genesInterleaving and overlapping of genes
genes located in the introns of another gene overlapping exons from different genes difficult to detect with ab initio methods
Gene 1
Gene2

Source: genomic sequence
Repetitive elementsRepetitive elements classes:
LINEs (Long Interspersed Nuclear Elements) -- 7,000bp SINEs (Short Interspersed Nuclear Elements) -- 300bp -- e.g., Alu low complexity regions -- e.g., ACACACACACACACAC tandem repeats -- e.g., CAGCAGCAGCAG
occur in large numbers in the genome considerably increase the size of the computation
CGGATAGACATAACCGGATAGACATAAC
CGGATAGACATAAC
CAGCAGCAGCAGCACAGCAGCAGCAGCA
CAGCAGCAGCAGCA

Source: genomic sequence
Genomic contaminationGenomic contamination unspliced introns (A) internal priming (B) these artifacts can only be resolved by clustering the ESTs on the genomic
axis, or in conjunction with other prediction methods
genome
EST
unspliced intron
(A)
EST
genomeAATATAAA
(B)
false (non-genic) primer
Source: genomic sequence
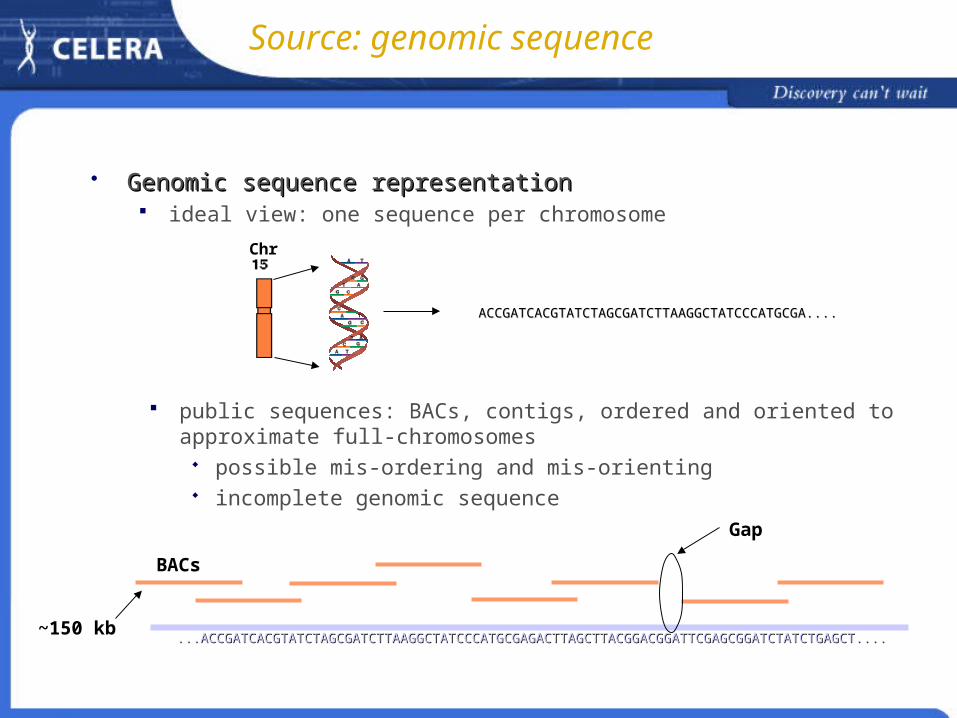
Genomic sequence representationGenomic sequence representation ideal view: one sequence per chromosome
ACCGATCACGTATCTAGCGATCTTAAGGCTATCCCATGCGA....ACCGATCACGTATCTAGCGATCTTAAGGCTATCCCATGCGA....
Chr
public sequences: BACs, contigs, ordered and oriented to approximate full-chromosomes
possible mis-ordering and mis-orienting incomplete genomic sequence
BACs
~150 kb...ACCGATCACGTATCTAGCGATCTTAAGGCTATCCCATGCGAGACTTAGCTTACGGACGGATTCGAGCGGATCTATCTGAGCT.......ACCGATCACGTATCTAGCGATCTTAAGGCTATCCCATGCGAGACTTAGCTTACGGACGGATTCGAGCGGATCTATCTGAGCT....
Gap
Source: genomic sequence
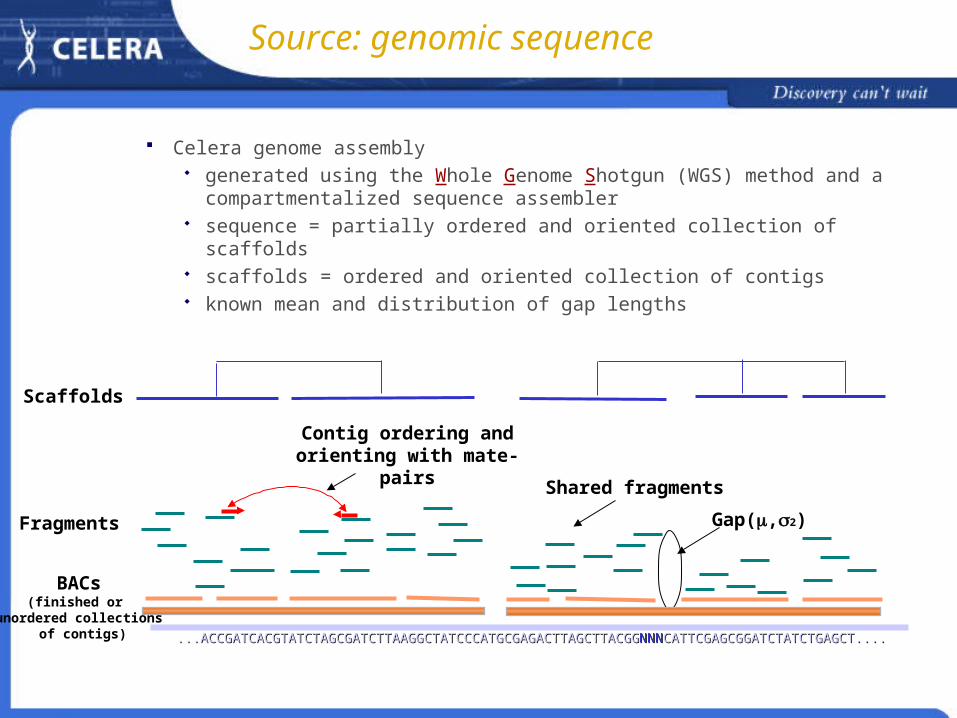
Celera genome assembly generated using the Whole Genome Shotgun (WGS) method and a
compartmentalized sequence assembler sequence = partially ordered and oriented collection of scaffolds scaffolds = ordered and oriented collection of contigs known mean and distribution of gap lengths
BACs(finished or
unordered collections of contigs) ...ACCGATCACGTATCTAGCGATCTTAAGGCTATCCCATGCGAGACTTAGCTTACGG...ACCGATCACGTATCTAGCGATCTTAAGGCTATCCCATGCGAGACTTAGCTTACGGNNNNNNCATTCGAGCGGATCTATCTGAGCT....CATTCGAGCGGATCTATCTGAGCT....
Gap(,2)Fragments
Contig ordering and orienting with mate-pairs
Shared fragments
Scaffolds
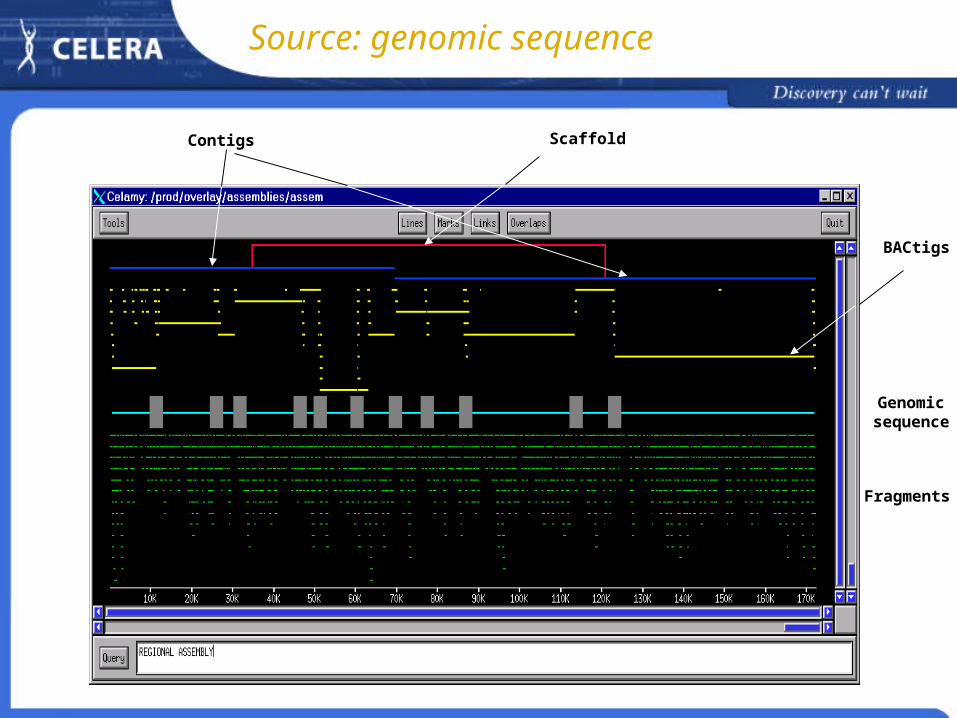
Source: genomic sequence
Fragments
Genomicsequence
BACtigs
Contigs Scaffold

Strategies for large scale EST mapping
Direct mapping with an exact cDNA-genomic sequence alignment Direct mapping with an exact cDNA-genomic sequence alignment method (method (SIM4, EST_GENOMESIM4, EST_GENOME)) divide the genome in n overlapping fragments align the EST against each of the genomic fragments
Time requiredTime required SIM4 - 0.3s per EST/Mb (1 EST vs. genome in 15 minutes) EST_GENOME - even slower Too expensive!
genome
1 Mb
EST 1 2

Strategies for large scale EST mapping
Mapping of ESTs to the genome via the (predicted) mRNA transcriptsMapping of ESTs to the genome via the (predicted) mRNA transcripts map each of the ESTs on the set of (predicted) mRNA transcripts, or genes
with known genomic locations align the EST against the genomic fragment containing the gene for the
EST with an exact alignment method
3’5’
EST 2EST 3EST 4
EST 1
mRNA transcript
GenomeExon 1
(5’UTR)Exon 2
(coding)Exon 3
(coding)Exon 4
(coding)Exon 5
(3’UTR)
Faster than exact mappingFaster than exact mapping Can be used to improve Can be used to improve existingexisting gene models, but not to discover new gene models, but not to discover new
onesones

Strategies for large scale EST mapping
Two-stage mapping of ESTs to the genome Two-stage mapping of ESTs to the genome
detect potential EST matches on the genome with a fast similarity search program (signal findingsignal finding)
blastn, MUMer, tfastx
align the EST against the bounded genomic region containing the signal with an exact alignment method (polishingpolishing)
SIM4, EST_GENOME
genome
EST 1 2
bounded genomic regions containing the EST signal
EST signal

Repeat detection and resolution
Repeats represent ~40% of the sequence of the human genomeRepeats represent ~40% of the sequence of the human genome
Some repeats can be found in the 3’ UTRs of the genesSome repeats can be found in the 3’ UTRs of the genes
Spurious priming can produce repetitive ESTsSpurious priming can produce repetitive ESTs
In tests using dbESTIn tests using dbEST1% of the ESTs found accounted for 99% of the EST signals1% of the ESTs found accounted for 99% of the EST signals
Resolution StrategiesResolution Strategies repeat mask the genome prior to mapping using, e.g., RepeatMaskerRepeatMasker repeat mask the EST data prior to mapping selectively mask only those ESTs with large numbers of occurrences,
during mapping

Overview
Biological motivationBiological motivation
Methods in gene predictionMethods in gene prediction
Mapping of large EST data setsMapping of large EST data sets
Applications of EST data miningApplications of EST data mining

EST data mining
Gene prediction by genomic EST clustering (previously discussed)Gene prediction by genomic EST clustering (previously discussed)
Generation of gene indices by EST clustering and assemblyGeneration of gene indices by EST clustering and assembly
5’ and 3’ UTR reconstruction5’ and 3’ UTR reconstruction
Detection of alternatively spliced gene variantsDetection of alternatively spliced gene variants

Gene indices
Quality and vector trim the EST sequencesQuality and vector trim the EST sequences
Cluster the ESTs in groups based on sequence similarityCluster the ESTs in groups based on sequence similarity
Assemble the ESTs in each cluster using a multiple alignment programAssemble the ESTs in each cluster using a multiple alignment program
For each cluster, select a consensus sequence = For each cluster, select a consensus sequence = EST assemblyEST assembly
Each EST assembly is a potential mRNA transcriptEach EST assembly is a potential mRNA transcript
Detect potential splice variants by pairwise comparisons between highly Detect potential splice variants by pairwise comparisons between highly
similar EST assembliessimilar EST assemblies

5’ and 3’ UTR reconstruction
Map the ESTs on the genomic axisMap the ESTs on the genomic axis
Cluster the EST matches along the genomic axis in the area Cluster the EST matches along the genomic axis in the area
surrounding the predicted transcripts, in a manner consistent with the surrounding the predicted transcripts, in a manner consistent with the
GenBank annotationGenBank annotation
Determine putative 3’ mRNA transcript ends in the vicinity of the 3’-Determine putative 3’ mRNA transcript ends in the vicinity of the 3’-
most EST-genomic alignmentsmost EST-genomic alignments
Use genomic information (e.g., poly-adenylation signals AATAAA) to Use genomic information (e.g., poly-adenylation signals AATAAA) to
validate the 3’ UTR endsvalidate the 3’ UTR ends

Detection of alternative splices
Using EST consensus informationUsing EST consensus information cluster the ESTs to create gene indices determine the consensus sequence for each cluster compare highly similar consensus sequences to detect putative alternatively
spliced exons (indel blocks)
Using the EST-genomic sequence alignmentsUsing the EST-genomic sequence alignments cluster the EST matches along the genomic axis to infer possible exon
models determine (internal) exons that are present in some, but not all, ESTs in the
cluster (alternatively spliced) collect EST evidence for alternatively spliced variants

References
Lewin B (2000) Genes VII, Lewin B (2000) Genes VII, Oxford University Press Inc.Oxford University Press Inc., New York, ISBN 0-19-, New York, ISBN 0-19-879276-X.879276-X.
Burge C, and Karlin S. (1997) Prediction of complete gene structures in human Burge C, and Karlin S. (1997) Prediction of complete gene structures in human genomic DNA, genomic DNA, J Mol BiolJ Mol Biol. . 268268(1):78-94.(1):78-94.
Kulp D, Haussler D, Reese MG, and Eeckman FH. (1996) A generalized hidden Kulp D, Haussler D, Reese MG, and Eeckman FH. (1996) A generalized hidden Markov model for the recognition of human genes in DNA, Markov model for the recognition of human genes in DNA, Proc Int Conf Intell Proc Int Conf Intell Syst Mol Biol.Syst Mol Biol. 44:134-42.:134-42.
Krogh A, Mian IS, and Haussler D. (1994) A hidden Markov model that finds Krogh A, Mian IS, and Haussler D. (1994) A hidden Markov model that finds genes in E. coli DNA, genes in E. coli DNA, Nucleic Acids Res. Nucleic Acids Res. 2222(22):4768-78.(22):4768-78.
Solovyev VV, Salamov AA, and Lawrence CB. (1994) Predicting internal exons Solovyev VV, Salamov AA, and Lawrence CB. (1994) Predicting internal exons by oligonucleotide composition and discriminant analysis of spliceable open by oligonucleotide composition and discriminant analysis of spliceable open reading frames, reading frames, Nucleic Acids Res.Nucleic Acids Res. 2222(24):5156-63.(24):5156-63.
Salamov AA, and Solovyev VV. (2000) Ab initio gene finding in Drosophila Salamov AA, and Solovyev VV. (2000) Ab initio gene finding in Drosophila genomic DNA, genomic DNA, Genome Res. Genome Res. 1010(4):516-22.(4):516-22.

References
Gelfand MS, Mironov AA, and Pevzner PA (1996) Gene recognition via spliced Gelfand MS, Mironov AA, and Pevzner PA (1996) Gene recognition via spliced sequence alignment, sequence alignment, Proc Natl Acad Sci USAProc Natl Acad Sci USA 9393(17):9061-6.(17):9061-6.
Mott R. (1997) EST_GENOME: a program to align spliced DNA sequences to Mott R. (1997) EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA, unspliced genomic DNA, Comput Appl Biosci.Comput Appl Biosci. 1313(4):477-8.(4):477-8.
Florea L, Hartzell G, Zhang Z, Rubin GM, and Miller W. (1998) A computer Florea L, Hartzell G, Zhang Z, Rubin GM, and Miller W. (1998) A computer program for aligning a cDNA sequence with a genomic DNA sequence, program for aligning a cDNA sequence with a genomic DNA sequence, Genome Res.Genome Res. 88(9):967-74.(9):967-74.
Florea, L. and Walenz, B. (in preparation) ESTMapper: Massive EST Mapping.Florea, L. and Walenz, B. (in preparation) ESTMapper: Massive EST Mapping.
Batzoglou S, Pachter L, Mesirov JP, Berger B, and Lander ES. (2000) Human Batzoglou S, Pachter L, Mesirov JP, Berger B, and Lander ES. (2000) Human and mouse gene structure: comparative analysis and application to exon and mouse gene structure: comparative analysis and application to exon prediction, prediction, Genome Res.Genome Res. 1010(7):950-8.(7):950-8.
Bafna V, and Huson DH. (2000) The conserved exon method for gene finding, Bafna V, and Huson DH. (2000) The conserved exon method for gene finding, Proc Int Conf Intell Syst Mol Biol.Proc Int Conf Intell Syst Mol Biol. 88:3-12.:3-12.
Quackenbush J, Liang F, Holt I, Pertea G, and Upton J. (2000) The TIGR gene Quackenbush J, Liang F, Holt I, Pertea G, and Upton J. (2000) The TIGR gene indices: reconstruction and representation of expressed gene sequences, indices: reconstruction and representation of expressed gene sequences, Nucleic Acids Res.Nucleic Acids Res. 2828(1):141-5.(1):141-5.

References
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Yandell M, Evans CA, Holt RA, et al.et al. (2001) The sequence of the human (2001) The sequence of the human genome, genome, Science Science 291291(5507):1304-51.(5507):1304-51.
Gautheret D, Poirot O, Lopez F, Audic S, and Claverie JM. (1998) Alternate Gautheret D, Poirot O, Lopez F, Audic S, and Claverie JM. (1998) Alternate polyadenylation in human mRNAs: a large-scale analysis by EST clustering, polyadenylation in human mRNAs: a large-scale analysis by EST clustering, Genome Res.Genome Res. 88(5):524-30.(5):524-30.
Kan Z, Rouchka EC, Gish WR, and States DJ. (2001) Gene structure prediction Kan Z, Rouchka EC, Gish WR, and States DJ. (2001) Gene structure prediction and alternative splicing analysis using genomically aligned ESTs, and alternative splicing analysis using genomically aligned ESTs, Genome Res.Genome Res. 1111(5):889-900.(5):889-900.
Kan Z, Gish W, Rouchka E, Glasscock J, and States D. (2000) UTR Kan Z, Gish W, Rouchka E, Glasscock J, and States D. (2000) UTR reconstruction and analysis using genomically aligned EST sequences, reconstruction and analysis using genomically aligned EST sequences, Proc Int Proc Int Conf Intell Syst Mol Biol. Conf Intell Syst Mol Biol. 88:218-27.:218-27.
Ji H, Zhou Q, Wen F, Xia H, Lu X, and Li Y. (2001) AsMamDB: an alternative Ji H, Zhou Q, Wen F, Xia H, Lu X, and Li Y. (2001) AsMamDB: an alternative splice database of mammals, splice database of mammals, Nucleic Acids Res.Nucleic Acids Res. 2929(1):260-3.(1):260-3.