oedema formation in acute inflammation in the rabbit …
TRANSCRIPT

OEDEMA FORMATION IN ACUTE INFLAMMATION IN THE RABBIT AND THE EFFECTS OF GLUCOCORTICOSTEROIDS
Helen Yarwood
A thesis submitted for the degree of Doctor of Philosophy (PhD)
University of London 1994
Dept. Applied Pharmacology National Heart & Lung Institute
Dovehouse Street London SW3 6 LY

ProQuest Number: 10017330
All rights reserved
INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10017330
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.This work is protected against unauthorized copying under Title 17, United States Code.
Microform Edition © ProQuest LLC.
ProQuest LLC 789 East Eisenhower Parkway
P.O. Box 1346 Ann Arbor, Ml 48106-1346

ABSTRACT
A characteristic feature of acute inflammation is increased microvascular permeability. Mediators that increase microvascular permeability can act in 2 ways; either directly on the endothelial cell eg. bradykinin, or by a mechanism dependent on circulating neutrophils eg. the chemoattractant FMLP. Oedema can be suppressed by both steroid and non-steroid anti-inflammatory drugs. These compounds may be able to act at several levels. This study was designed to investigate mechanisms of oedema formation and the possible sites of action of anti-inflammatory compounds.
FMLP-induced oedema formation was not dependent on endogenous histamine release or pro-inflammatory products of the cyclo-oxygenase pathway. Intravenous infusion of zymosan-activated plasma produced transient neutropenia in rabbits which resulted in inhibition of oedema formation induced by FMLP, but not that induced by bradykinin.
Ibuprofen, selectively inhibited FMLP-induced oedema formation when administered intravenously. This drug did not induce neutropenia and the effect was independent of cyclo-oxygenase inhibition. Ibuprofen may interfere with the interaction between circulating neutrophils and venular endothelial cells.
The microtubule blocking agent colchicine also selectively inhibited FMLP- induced oedema formation even when it was administered at intervals after intradermal FMLP. This suggests that continuing interactions between functionally active neutrophils and endothelial cells are necessary for the protracted plasma protein leakage induced by this chemoattractant.
There is evidence that anti-inflammatory steroids may owe some of their antiinflammatory actions to effects on the target cells of inflammatory mediators eg. microvascular endothelial cells and neutrophils. In an attempt to investigate this possibility in vivo the ability of dexamethasone to modulate oedema formation and * "In-neutrophil accumulation in rabbit skin was investigated. Dexamethasone was effective when administered in three ways. Firstly, local pretreatment of skin with dexamethasone inhibited oedema responses but not * "In-neutrophil accumulation. Secondly, treatment of neutrophil recipient rabbits intravenously with dexamethasone inhibited both oedema responses and "^In-neutrophil accumulation in response to exogenous chemoattractants. Thirdly, systemic treatment of neutrophil donor animals with dexamethasone resulted in suppression of * "In-neutrophil accumulation in response to intradermal chemoattractants in recipients.
Systemic treatment with dexamethasone suppressed neutrophil accumulation oedema formation and the generation of LTB4 induced by intraperitoneal-injection of zymosan in the rabbit. The generation of TXBj and prostacyclin were not inhibited by dexamethasone. Depleting animals of circulating neutrophils inhibited the generation of LTB4 suggesting that accumulating neutrophils are responsible for the generation of this chemoattractant.
This study shows that anti-inflammatory compounds can inhibit oedema formation and neutrophil-accumulation by acting at several different sites. There is evidence for inhibition by effects on the microvascular endothelial cell and the neutrophil.

CONTENTS
Title page 1
Abstract 2
Abbreviations 10
List of figures 13
List of tables 16
Acknowledgements 17
Chapter 1: Introduction 18
1.1 General Introduction 18
1.2 Increased microvascular permeability 19
1.2.1 Introduction 19
1.2.2 Mediators increasing permeability by a direct action on thevessel wall 2 0
1.2.3 Neutrophil-dependent mediators of increased microvascular permeability 2 2
1.2.4 Mechanisms of neutrophil-dependent oedema formation.How do neutrophils increase vascular permeability? 24
1.2.4.1 Introduction 24
1.2.4.2 Lipid mediators 26
1.2.4.3 Oxygen metabolites 27
1.2.4.4 Granule enzymes and proteins 28
1.3 Vasodilation and mediator synergism 31
1.4 Neutrophil accumulation 32
1.4.1 Introduction 32
1.4.2 Neutrophil-endothelial cell interactions 33
1.4.3 Role of cell surface adhesive glycoproteins in neutrophil-endothelial interactions 37

1.4.4 Neutrophil emigration through the endothelial cell layer 44
1.4.5 Passage of neutrophils through the basement membrane 45
1.4.6 Neutrophil locomotion towards the removal of theinflammatory stimulus 45
1.5 The anti-inflammatory glucocorticoids 46
1.5.1 Introduction 46
1.5.2 Inhibition of inflammatory mediator generation 46
1.5.3 Inhibition of inflammatory mediator action 54
1.6 Aims of the study 55
Chapter 2: Materials and Methods 56
2.1 In vivo measurement of inflammatory activity in rabbit skin: therabbit skin bioassay 56
2.1.1 Measurement of vascular permeability in rabbit skin 56
2.1.2 Measurement of neutrophil accumulation in rabbit skin 57
2.1.3 Measurement of local blood flow in rabbit skin 57
2.2 Preparation of ‘"In-neutrophils 58
2.3 Experimental protocols measuring neutrophil accumulation and/oroedema formation or blood flow using the rabbit skin bioassay 60
2.3.1 Investigation of the mechanisms of oedema formation 61
2.3.2 The effect of intravenously administered agents on oedemaformation induced by inflammatory mediators 61
2.3.3 Measurement of the duration of action of inflammatorymediators 62
2.3.4 The effect of local dexamethasone on inflammatoryresponses 62
2.3.5 The effect of systemic dexamethasone on inflammatoryresponses 62
2.3.6 The effect of dexamethasone on the neutrophil in vivo 62
2.3.7 The effect of dexamethasone on blood flow 63

2.4 Preparation of zymosan-activated plasma 63
2.5 Staining procedure for leukocytes 63
2.6 The generation of inflammatory exudate: the rabbit peritonealcavity model 64
2.6.1 Generation and recovery of peritoneal exudate fluid 64
2.6.2 Preparation of zymosan for intraperitoneal injection 65
2.6.3 Measurement of the inflammatory reaction in the peritonealcavity 65
2.6.4 Determination of inflammatory mediator concentration in peritoneal exudate samples 65
2.6.5 Confirmation of immunoreactive LTB4 identity 6 6
2.6.5.1 Separation of LTB4 6 6
2.6.5.2 Rp HPLC 67
2.7 Experimental protocols using the peritoneal cavity model 67
2.7.1 The effect of depletion of circulating neutrophils on the inflammatory response to i.p. zymosan 67
2.7.2 The effect of systemic dexamethasone on the inflammatory response to i.p. zymosan 67
2.8 Treatment of rabbits with mustine hydrochloride 6 8
2.9 Stimulation of blood with A23187 6 8
2.10 Detection of endotoxin levels 69
2.11 Materials 69
2.12 Statistical Analysis 70
Chapter 3: Characteristics of oedema formation induced by FMLP 71
3.1 Introduction 71
3.2 Results 73
3.2.1 The effect of vasodilator prostaglandin E2 on oedemaformation induced by histamine and FMLP in rabbit skin 73

3.2.2 The effect of formy 1-peptides on the skin microvasculature 73
3.2.3 The effect of local treatment with indomethacin on oedemaresponses induced by FMLP 76
3.2.4 The effect of mepyramine on FMLP-induced oedemaformation 76
3.2.5 The effect of intravenous infusion of zymosan-activatedplasma on FMLP induced oedema formation 78
3.2.6 The effect of intravenous ibuprofen on oedema formationin rabbit skin 81
3.2.7 The effect of intravenous colchicine on FMLP-inducedoedema formation 84
3.2.8 The effect of intravenous colchicine on the duration ofaction of permeability-increasing mediators 87
3.2.9 Summary 89
Chapter 4: The effect of glucocorticosterolds on vascular responsesin the rabbit microcirculation induced by inflammatory mediators 90
4.1 Introduction 90
4.2 Results 92
4.2.1 The local treatment of rabbits with dexamethasone 92
4.2.1.1 Effect of local dexamethasone on oedemaformation in rabbit skin 92
4.2.1.2 Effect of local dexamethasone on ‘"In-neutrophil accumulation in rabbit skin 92
4.2.1.3 Effect of local dexamethasone on blood flow inrabbit skin 94
4.2.2 The systemic treatment of recipient rabbits withdexamethasone 97
4.2.2.1 The effect of intravenous dexamethasone on ‘‘‘Inneutrophil accumulation in rabbit skin 97

4.2.2.2 The effect of intravenous dexamethasone on oedema formation in rabbit skin 97
4.2.2.3 The effect of intravenous dexamethasone on oedema formation potentiated with calcitonin gene-related peptide 1 0 0
4.2.3 The systemic treatment of donor rabbits withdexamethasone 1 0 2
4.2.3.1 The effect of pretreating neutrophil donor rabbitswith systemic dexamethasone on * “ In-neutrophil accumulation in recipient animals 1 0 2
4.2.3.2 The effect of pretreating neutrophil donor rabbitswith systemic dexamethasone on oedema formation in recipient animals 1 0 2
4.2.4 Summary 105
Chapter 5: The generation, characertisation and modulation ofinflammatory activity in rabbit peritoneal exudate 107
5.1 Introduction 107
5.2 Results 108
5.2.1 The inflammatory response to intraperitoneal zymosan 108
5.2.2 Time course of the generation of inflammatory mediatorsin zymosan-induced peritoneal cavity 1 1 1
5.2.2.1 LTB4 111
5.2.2.2 TXBj 111
5.2.2.3 PGE2 and 6 -oxo-PGFi„ 111
5.2.3 The role of circulating neutrophils in the response tointraperitoneal zymosan 115
5.2.3.1 The effect of depletion of circulating neutrophilson plasma extravasation and neutrophil accumulation in response to intraperitoneal zymosan 115
5.2.3.2 The generation of LTB4 in response to zymosanin neutropenic animals 117

5.2.3.3 The generation of 6 -oxo-prostaglandin F,„ inneutropenic animals 117
5.2.4 The effect of intravenous dexamethasone on theinflammatory response to intraperitoneal zymsan 117
5.2.4.1 The effect of dexamethasone on plasma extravasation and neutrophil accumulation into zymosan injected cavities 1 2 1
5.2.4.2 The effect of systemic dexamethasone on LTB4
generation in response to zymosan 1 2 1
5.2.4.3 The effect of systemic dexamethasone on TXBg generation following intraperitoneal zymosan 125
5.2.4.4 The effect of dexamethasone on 6 -oxo-PGFi„ generation in response to intraperitoneal zymosan 125
5.2.4.5 Effect of dexamethasone on A23187-inducedLTB4 production in whole blood 125
5.2.5 Authentication of immunoreactive LTB4 128
5.2.6 Summary 133
Chapter 6 : Discussion 134
6.1 Characteristics of FMLP-induced oedema formation in rabbit skin 134
6.2 The effect of intravenous zymosan activated plasma onFMLP-induced oedema formation 135
6.3 The effect of intravenous ibuprofen on FMLP-induced oedemaformation 136
6.4 The effect of intravenous colchicine on FMLP-induced oedemaformation 141
6.5 The effect of colchicine on the duration of the permeabilityresponse to FMLP 143
6 . 6 Systemic treatment of rabbits with dexamethasone 143
6.7 Local treatment of skin sites with dexamethasone 144

6 . 8 Effect of dexamethasone on neutrophils in vivo. Modulation of neutrophil accumulation in vivo by an action of dexamethasone onthe neutrophil 148
6.9 The inflammatory response induced by intraperitoneal zymosan 154
6.9.1 The generation of inflammatory mediators in zymosan-induced peritoneal exudate fluid 155
6.9.1.1 C5aandPGl2 155
6.9.1.2 PGEg 158
6.9.1.3 TXB2 158
6.9.1.4 LTB4 159
6 .10 Effect of systemic dexamethasone on the inflammatory response to intraperitoneal zymosan 162
References 168
Publications 208

ABBREVIATIONS
AA arachidonic acid
BK bradykinin
CGRP Calcitonin gene-related peptide
C5a complement component C5a
DNA deoxy ribonucleic acid
Dex dexamethasone
DMSG dimethyl suphoxide
EDTA ethylene diamine tetraacetic acid
EGTA ethylene glycol-bis (fi amino-ethyl ether)N,N,N’ ,N’-tetraacetic acidFITC fluorescein isothiocyanateFMLP N-formyl-L-methionyl-L-leucyl-L-phenylalanine
FMLP? N-formyl-L-methionyl-leucyl-phenylalanine-phenylalanine
FMMP N-formyl-methionyl-methionyl-phenylalanine
FMP N-formyl-methionyl-phenylalanine
FM N-formyl-methionine
FNLP N-formyl-norleucyl-phenylalanine
GMCSF granulocyte-macrophage colony-stimulating factor
GMP-140 granule membrane protein-140
Hist histamine
Hrs hours
HSA human serum albumin
H2 O2 hydrogen peroxide
12-HETE 1 2 -hydroxyeicosatetraenoic acid
5-HT 5-Hydroxy tryptamine
i.r. immunoreactive
"*In indium 1 1 1
indo indomethacin
ICAM intercellular adhesion molecule
IL-1 interleukin- 1
IL- 8 interleukin- 8
i.d. intra-dermal
i.p intra-peritoneal
i.v. intra-venous
10

1251 Iodine 125
IFNt gamma interferon
LECCAM lectin-cellular adhesion molecule
LAD leukocyte adhesion syndrome
LTB4 leukotriene B4
LTC4 leukotriene C4
LTD4 leukotriene D4
LTE4 leukotriene E4
LPS bacterial lipopolysaccharide
LFA lymphocyte function assoicated antigen- 1
Mac-1 macrophage antigen- 1
mRNA messenger ribonucleic acid
Min minute
mAb monoclonal antibody
NSAID non-steroidal-antiinflammatory drug
6 -oxo-PGFi„ 6 -oxo-prostaglandin Fj«
PMSF phenyl methyl-sulphonyl fluoride
PBS phosphate buffered saline
PLA2 phospholipase A2
PAF platelet-activating factor
PMN leukocytes polymorphonuclear leukocyte
PGE2 prostaglandin E2
PGD2 prostaglandin D2
PGG2 prostaglandin G2
PGI2 prostaglandin I;
PGF,« prostaglandin Fi„
RIA radioimmunoassay
Rp HPLC reversed-phase high performance liquid <
RNA ribonucleic acid
Sal saline
SRS-A slow-reacting substance of anaphylaxis
S.E.M. standard error of the mean
SOD superoxide dismutase
TXB2 thromboxane Bj
11

TNF tumour necrosis factor
VIP vasoactive intestinal peptide
^ ^Xe xenon 133
ZAP xymosan-activated plasma
12

LIST OF FIGURES
Page
3.1 Oedema responses in rabbit skin induced by FMLP and histamine
in the absence and presence of PGE2 74
3.2 Oedema responses in rabbit skin induced by N-formyl peptides 75
3.3 The effect of intravenous zymosan-activated plasma (ZAP) on
oedema responses in rabbit skin 79
3.4 The effect of intravenous Ibuprofen on oedema formation in
response to inflammatory mediators in rabbit skin 82
3.5 The effect of intravenous colchicine on oedema formation induced
by FMLP 4- PGEj and Bk 4- PGE2 in rabbit skin 85
3.6 The effect of intravenous colchicine on the duration of action of
permeability-increasing mediators in rabbit skin 8 8
4.1 The kinetics of the effect of local dexamethasone on oedema
formation in rabbit skin 93
4.2 The effect of local treatment with dexamethasone on PGEj-
stimulated increased blood flow in rabbit skin 96
4.3 The effect of systemic treatment with dexamethasone on ‘"In
neutrophil accumulation in rabbit skin 98
4.4 The effect of systemic treatment with dexamethasone on oedema
formation in rabbit skin 99
4.5 The effect of systemic treatment with dexamethasone on oedema
formation induced by Bk 4- PGEj and Bk 4- CGRP in rabbit skin 101
13

4.6 The effect of systemic treatment of ‘"In-neutrophil donor rabbits
with dexamethasone on " ‘In-neutrophil accumulation in the skin
of untreated recipient animals 103
4.7 The effect of systemic treatment of neutrophil donor rabbits with
dexamethasone on oedema formation in the skin of untreated
recipient animals 104
5.1 Neutrophil accumulation into the rabbit peritoneal cavity following
the intraperitoneal injection of zymosan 1 1 0
5.2 The kinetics of the level of LTB4 in zymosan-induced peritoneal
exudate fluid as determined using radioimmunoassay 1 1 2
5.3 The kinetics of the level of TXBj in zymosan-induced peritoneal
exudate fluid as determined using radioimmunassay 113
5.4 The kinetics of the level of PGE2 in zymosan-induced peritoneal
exudate fluid as measured by radioimmunoassay 114
5.5 The kinetics of the generation of 6 -oxo-PGFi„ in zymosan-induced
peritoneal exudate fluid as measured by radioimmunoassay 114
5.6 The effect of depletion of circulating neutrophils on the generation
of LTB4 in the rabbit peritoneal cavity following intraperitoneal
injection of zymosan 119
5.7 The effect of depletion of circulating neutrophils on the generation
of 6 -oxo-PGFi„ in the rabbit peritoneal cavity following
intraperitoneal injection of zymosan 1 2 0
5.8 The effect of systemic treatment with dexamethasone on the
generation of LTB4 in the peritoneal cavity in response to
intraperitoneal injection of zymosan 124
14

5.9 The effect of systemic treatment with dexamethasone on the
generation of TXB2 in the peritoneal cavity in response to
intraperitoneal injection of zymosan 126
5.10 The effect of systemic treatment with dexamethasone on the
generation of 6 -oxo-PGFi„ in the peritoneal cavity in response to
intraperitoneal injection of zymosan 126
5.11 Rp-HPLC of radiolabelled LTB4, 12-HETE and arachidonic acid
(Arach) 129
5.12 Immunoreactive LTB4 profile following Rp-HPLC fractionation of
zymosan-induced inflammatory exudate 130
5.13 Immunoreactive LTB4 profile following Rp-HPLC fractionation of
zymosan-induced peritoneal exudate fluid generated in animals
treated systemically with dexamethasone 131
5.14 Immunoreactive LTB4 profile following Rp-HPLC fractionation of
zymosan-induced peritoneal exudate fluid generated in animals
depleted of circulating neutrophils 132
15

LIST OF TABLES
3.1 The effect of local treatment with indomethacin on oedemaformation induced by FMLP in rabbit skin 77
3.2 The effect of treatment with intravenous zymosan-activated plasmaon the number of circulating leukocytes and the number of circulating neutrophils in rabbits 80
3.3 The effect of treatment with intravenous ibuprofen on the number of circulating leukocytes and the number of circulating neutrophilsin rabbits 83
3.4 The effect of treatment with intravenous colchicine on the number of circulating leukocytes and the number of circulating neutrophilsin rabbits 8 6
4.1 Kinetics of the effect of local treatment with dexamethasone and indomethacin on * "In-neutrophil accumulation in response to ZAP4- PGEj in rabbit skin 95
5.1 The local accumulation of intravenously-injected Evans blue dye in response to the intraperitoneal injection of zymosan in therabbit 109
5.2 The effect of depletion of circulating neutrophils on theintraperitoneal accumulation of Evans blue dye following intraperitoneal injection of zymosan 116
5.3 The effect of depletion of circulating neutrophils on zymosan-induced neutrophil accumulation in the rabbit peritoneal cavity 118
5.4 The effect of systemic treatment with dexamethasone on the local accumulation of Evans blue dye following intraperitoneal injectionof zymosan 1 2 2
5.5 The effect of systemic treatment with dexamethasone on zymosan-induced neutrophil accumulation in the rabbit peritoneal cavity 123
5.6 The effect of systemic treatment with dexamethasone on the exvivo generation of LTB4 from A23187-stimulated rabbit blood 127
16

ACKNOWLEDGEMENTS
I would like to thank Professor Timothy Williams for his guidance and kindness
throughout this work and for creating, together with his team a happy working
environment. My special thanks go to 3 members of this team: Dr Susan Brain,
Dr Sussan Nourshargh and Dr Lindsay Needham.
I am also grateful to Dr Steve Foster and his group at Imperial Chemical Industries,
Macclesfield for their invaluable support and friendship during my return to the north.
I thank the Medical Research Council and Imperial Chemical Industries for financial
support and Carol for the typing of this thesis.
Finally, thanks to Mum, Dad and James.
17

CHAPTER 1; INTRODUCTION
1.1 General Introduction
Inflammation is the local response of a tissue and its vasculature to infection and
injury; it is intended to eliminate noxious stimuli and facilitate the repair process. The
inflammatory response is triggered by an array of chemical mediators released in the
affected tissue. These mediators act and interact to induce changes in blood
microvessels, tissue cells and blood cells. This brings about the three characteristic and
closely inter-linked features of acute inflammation: vasodilatation of local arterioles
leading to an increased blood supply to the tissue, an increase in the permeability of
microvessels to plasma proteins and the adherence of leukocytes to endothelial cells
followed by their movement into the tissue.
The cardinal signs of inflammation described by Celsus almost 2000 years ago as
redness, heat, swelling and pain reflect the changes in the microcirculation: vasodilatation
underlying heat and redness, and plasma extravasation underlying tissue swelling. It is
likely that these vascular adaptations occur continually in response to small local stimuli
which, because they are rapidly neutralised, do not evoke the clinical signs of
inflammation.
As research has advanced it has become apparent that a plethora of chemical
signals mediate the inflammatory process. Several of these mediators are simultaneously
involved in an inflammatory reaction, they can act in concert with one another and
sometimes exhibit synergistic actions (see section 1.3). Each mediator can have a single
or a combination of actions, the exact effect is determined by species and tissue. The
complexity of the inflammatory process gives the organism a great reserve capacity for
maintaining an adequate response and reflects the vital role that inflammation plays in
host defence. It is when these complex mechanisms become excessive and
inappropriately deployed that the physiological function of the affected organ can become
compromised as in inflammatory diseases such as rheumatoid arthritis and asthma.
This thesis concerns an investigation into mechanisms of oedema formation, the
accumulation of neutrophils, the inter-relationship between these two events and the
modulatory effects of anti-inflammatory compounds concentrating on the
glucocorticosteroids. Oedema formation was measured in rabbit skin in response to
intradermal injections of putative inflammatory mediators. The simultaneous estimation
of neutrophil accumulation in skin sites allowed the inter-relationship between these two
characteristic components of the inflammatory response to be investigated and the
18

potential site(s) of action of glucocorticosteroids in modulating responses in vivo to be
established precisely. The effect of glucocorticosteroids on the generation of
inflammatory mediators in response to zymosan was investigated in the rabbit peritoneal
cavity.
An understanding of the mechanisms underlying inflammation and the
determination of the precise modes of action of anti-inflammatory compounds will aid the
more efficient use of existing drugs and the design of more specific anti-inflammatory
therapy for the future.
It is the purpose of this chapter to discuss the changes that occur in the
microvascular bed in inflammation, the factors that bring about these changes and the
actions of anti-inflammatory agents, specifically the glucocorticosteroids.
1.2 Increased Microvascular Permeability
1.2.1. Introduction
Under normal physiological conditions a state of dynamic fluid equilibrium exists
between a tissue and its blood supply. The hydrostatic pressure gradient tending to move
water and small solutes from blood microvessels to the interstitium is largely counteracted
by an osmotic pressure exerted in the opposite direction by virtue of a high intravascular
protein concentration. Any small net outward movement of fluid resulting from a slight
imbalance in these two forces is cleared by the lymphatic system which, in turn, returns
fluid to the blood. This equilibrium is dependent on the integrity of the vascular
endothelium and its low permeability to plasma proteins. Amongst the plasma-derived
proteins present in extravascular tissue fluid in normal uninflamed tissue are antibodies
and complement components, which play an important role in the recognition of, for
example, invading micro-organisms. In an inflammatory situation the permeability of the
microvascular endothelium rises dramatically which eliminates the pre-existing fluid
equilibrium and protein-rich fluid leaks into the affected tissue. When microvascular
leakage exceeds lymphatic clearance the tissue becomes swollen. In an inflammatory
reaction vascular leakage can result from direct damage to endothelial cells of the
microvascular bed and this is most striking in thermal or radiation injury. When such
direct damage occurs this manifests in a generalised leakiness of microvessels - that is
all vessel types of the microvasculature (ie. small arterioles, capillaries and post capillary
venules) may exhibit an increased permeability (Cotran & Majno, 1964; Cotran, 1965;
19

Cotran, 1967). In contrast, in inflammatory situations initiated by, for example bacterial
infection, other foreign organisms or particles specific endogenous mechanisms have
evolved actively to cause plasma protein leakage. Vascular leakage is brought about by
the stimulus-induced release of endogenous chemicals in the tissue and occurs only in
specialised regions of the microvascular bed - the post capillary venules. These
mechanisms imply that oedema formation induced by chemical mediators, unlike direct
endothelial damage, has a functional role. This is to supply vital plasma proteins such
as antibodies and complement factors to an infected tissue in order to facilitate local
defence mechanisms, for example lysis and opsonisation.
Mediators are also released that may change arteriolar tone, thus altering blood
flow to the affected area and this can modulate the inflammatory response. For example,
increased blood flow has been shown to play an important role in determining the amount
of plasma leakage to the extravascular space. This will be elaborated later in the text
(section 1.3) as will the accumulation of blood neutrophils at the inflammatory site
(section 1.4).
Mediators of increased microvascular permeability can be divided into two groups
based on their mode of action (Wedmore & Williams, 1981a), as discussed below.
1.2.2. Mediators increasing permeabilitv bv a direct action on the vessel wall
Thomas Lewis and his associates were the first to propose that histamine, the
oldest known mediator, was an endogenous mediator of increased vascular permeability
(Lewis, 1927). However, it was not until the classical studies of Majno and colleagues
using carbon labelling in the rat cremaster muscle that histamine, along with serotonin
(5- hydroxy tryptamine) were found to increase vascular leakage exclusively in a specific
region of the microvasculature, the post-capillary venules (Majno et al., 1961). Electron
microscopic studies revealed that these mediators increase permeability by inducing the
formation of gaps between adjacent endothelial cells (Majno & Palade, 1961). In the
hamster cheek pouch prepared for intravital microscopy topical application of histamine
or bradykinin was also observed to cause leakage of intravenously administered
fluorescein-labelled dextran exclusively at post-capillary venules (Svensjo et al., 1973;
Svensjo & Arfors, 1979; Svensjo et al., 1979). By combining the identification of
leakage of FITC-dextran (by intravital microscopy) in the living pouch with the
identification of dextran by electron microscopy in the same pouch after death, Hultstrom
& Svensjo (1979) also observed gaps between endothelial cells in response to bradykinin.
20

Precipitates of dextran were seen in the vascular lumen but also within the gap and the
interstitial tissue outside the gap. Venular endothelial cells are characterised by the
loosest junctional organisation of the entire vascular system, postcapillary venules in
muscle have approximately 25% of junctions open to a gap of 20-60Â. These are the
junctions which are thought to open up when exposed to inflammatory agents (Simionescu
et al.» 1978). Furthermore, it has been demonstrated, using histamine-ferritin conjugates
that histamine receptors on endothelial cells are most prominent in post capillary venules
close to endothelial junctions (Heltianu et a l, 1982). Histamine and bradykinin have
been reported to cause swelling of individual endothelial cells leading to the suggestion
that endothelial cell contraction is involved in opening inter-endothelial cell junctions
(Majno et at., 1969; Joris et al, 1972; Joris et a l, 1987). Furthermore, contracted
endothelial cells were demonstrated by electron microscopy in post capillary venules from
the lungs of a guinea-pig subjected to ovalbumin anaphylaxis (Ryan & Ryan, 1984).
Filamentous structures have been described in endothelial cytoplasm (Lauweryns &
Bousauw, 1973; DeBruyn & Cho, 1974; Gabbiani et a l, 1975) and other investigators
have demonstrated the presence of contractile proteins (Becker & Nachman, 1973; Moore
eta l, 1977; Chamley-Campbell etal., 1977). However, Hammersen (1980) was unable
to produce evidence of mediator-induced endothelial cell contraction and has suggested
that the abundant contractile machinery that endothelial cells undoubtedly contain may
exist to provide tensile strength and attachment only. Endothelial cell contraction induced
by mediators is an attractive way of explaining gap formation and increase in venular
permeability to macromolecules. However, the evidence supporting endothelial
contractility is largely indirect and the detailed mechanics of how contraction may lead
to opening of junctions remains to be determined. Furthermore, it cannot be excluded
that a conformational change in molecules at endothelial cell junctions is induced by some
mechanism unrelated to contractile activity. Whatever the mechanism for the gap
formation may be, it requires active metabolism of the endothelial cell, since cooling
inhibits protein efflux induced by histamine (Rippe & Grega, 1978).
Leukotriene C4 and D4 have been shown to increase microvascular permeability
in guinea-pig skin (Williams & Piper, 1980; Peck et a l, 1981; Drazen et a l, 1980) and
also, together with leukotriene E4 in the hamster cheek pouch (Dahlen et a l , 1981; Bjork
et a l, 1981). LTC4 and LTD4 also have vasoconstrictor activity (Williams & Piper,
1980) which tends to mask the permeability-increasing activity (Peck et a l, 1981 see
section 1.3 ). In human skin LTC4, LTD4 and LTE4 appear to causewheal and flare
21

responses in an equipotent manner at low concentrations (Soter et al., 1983; Camp et al., 1983).
The phospholipid platelet-activating factor (PAF) was first shown in 1981 to
increase microvascular permeability in the rat paw (Vargaftig & Ferreira, 1981). Injected
in skin, PAF has since been shown to induce vascular leakage in rabbit (Wedmore &
Williams, 1981b), guinea- pig (Hwang et al., 1985), rat (Hwang et a l, 1985) and man
(McGivem & Basran, 1984; Page et a l, 1985). Despite the observation that PAF can
also cause the accumulation of PMN-leukocytes in rabbit skin (Humphrey et a l, 1982a)
cutaneous oedema responses are not dependent on PMN-leukocytes in rabbit (Wedmore
& Williams, 1981b) and rat (Gerdin et a l, 1985). Thus it appears that PAF-induced
vascular leakage occurs via a direct action on the endothelium, except in certain
circumstances as discussed below.
1.2.3. Neutrophil-dependent mediators of increased microvascular permeability
The arachidonate lipoxygenase product leukotriene B4 (LTB4 ), FMLP (N-formyl-
methionyl-leucyl-phenylalanine), a synthetic peptide based on substances derived from
bacterial culture filtrates, and the polypeptide fragment of the fifth component of
complement C5a and its stable metabolite C5a des Arg are all highly chemotactic for
neutrophils (Ford-Hutchinson et al, 1980; Schiffmann et a l, 1975b; Schiffmann et al, 1975a; Snyderman et a l, 1969). These chemotactic agents also increase vascular
permeability in vivo in rabbit skin by a mechanism entirely dependent on the presence of
circulating neutrophils. The dependence on the presence of neutrophils was
demonstrated by the fact that oedema formation in response to LTB4 , C5a or FMLP was
absent in the skin of rabbits depleted of circulating neutrophils, although oedema
responses to intradermally-injected histamine, bradykinin and PAF were unaffected
(Wedmore & Williams, 1981b; Wedmore & Williams, 1981a; Issekutz, 1981a).
Neutrophils have since been shown to mediate increased vascular permeability in
numerous models (Staub et a l, 1985; Granger et a l, 1988; Bjork et a l, 1982; Bjork et a l, 1983), including man where human C5a has been shown to induce wheal and flare
reactions and neutrophil infiltration in human skin (Yancey et a l, 1985) and a
requirement for neutrophils has been determined (Williamson et a l, 1986). The
cytokines interleukin- 8 (IL-8 ) and tumour necrosis factor have also been shown to induce
neutrophil accumulation associated with oedema formation (Rampart et al., 1989a; Foster
e ta l, 1989).
22

In the hamster cheek pouch part of the vascular leakage induced with high doses
of PAF was attenuated in neutropenic animals (Bjork & Smedegard, 1983). This
suggests that in addition to a direct action, in some situations PAF-induced vascular
leakage could be augmented by a PMN-leukocyte dependent mechanism. Using the doses
of PAF utilized in this thesis no difference between oedema responses in normal and
neutropenic rabbits were detected (Hellewell & Williams unpublished observation,
(Wedmore & Williams, 1981b). Oedema responses to PAF also seem to be independent
of circulating platelets (Page et a l , 1985).
Direct observation of the microvasculature using intravital microscopy of the
rabbit mesentery and hamster cheek pouch revealed that, in common with other
permeability increasing mediators, (see section 1 .2 .2 .), leakage induced by
chemoattractants occurred in the post-capillary venules (Williams et al., 1984a; Bjork et
al., 1982,1983). In addition, somewhat larger (collecting type) venules were also
observed to be leaking. With direct-action mediators leakage was confined solely to post
capillary venules (Dahlen et al., 1981), see section 1.2.2.). Oedema was accompanied
by intravascular neutrophil adhesion and subsequent extravascular migration observed in
post-capillary and larger venules (Bjork et al., 1982, 1983).
In addition to the spatial differences observed between neutrophil-dependent and
independent leakage, temporal differences have also been shown. Wedmore & Williams
(1981a) found oedema responses in rabbit skin to bradykinin and histamine were fast in
onset, significant microvascular leakage being detectable 1.5 minutes after injection,
whereas there was a latent period of approximately 6 minutes before responses to C5a
were apparent (surprisingly early considering the necessity for neutrophils).
Furthermore, the permeability increasing activity of bradykinin and histamine was of very
short duration (tVi = 4-6 minutes), whereas that of chemotactic substances was
remarkably protracted, (for example, C5a = 90-100 minutes). These times refer to
results obtained with permeability-increasing mediators in the presence of vasodilator
prostaglandins (see section 1.3). From these results the following hypothesis was
formulated; that chemoattractants injected, or generated, extravascularly trigger a very
rapid interaction between circulating neutrophils and venular endothelial cells and that this
interaction results (by some unknown mechanism) in an increase in permeability of the
venule wall to macromolecules. When the accumulation of radiolabelled neutrophils was
measured, together with oedema formation in rabbit skin the kinetics of plasma proteinaccumulation
leakage induced by chemoattractants were found to closely parallel neutrophil^responses
23

were measured in the presence of prostaglandin 5% (Rampart & Williams, unpublished
observations). This is contrary to the old view that the two are distinct processes.
Issekutz et al (1981a) found similar results in response to C5a in rabbit skin although
responses developed more slowly as they were measured in the absence of a vasodilator
(see section 1.3). At low levels of cell accumulation there was no significant leakage of
plasma, whilst at higher levels of cell accumulation the close parallelism with plasma
leakage was observed. This observation may be a possible explanation for the previous
reports by Hurley that events of cell accumulation and plasma leakage are separable
(Hurley, 1963; Hurley, 1964). The low levels of cell accumulation referred to by
Issekutz were similar to those reported by Hurley and may thus explain the apparent
absence of vascular leakage in Hurleys experiments. Direct visualisation of the hamster
cheek pouch has also yielded information regarding the relationship between neutrophil
accumulation and vascular permeability. After application of LTB4 (Bjork et a l, 1982;
Bjork et al., 1982; Dahlen et al., 1981) leukocytes were visibly adhering within
approximately 1 minute, while leakage developed more slowly becoming apparent
approximately 5 minutes after application which is closer to the mean time for neutrophil
extravasation (Katori et a l, 1990). However, the LTB^-induced leakage was prolonged
in nature compared to the quick and short lived response to histamine even despite
continuous application of the latter.
1.2.4. Mechanisms of neutrophil-dependent oedema formation. How do neutrophils
increase vascular permeability?
1.2.4.1. Introduction
The manner by which accumulated neutrophils increase vessel wall permeability
remains undetermined despite many attempts to establish the mechanism or mechanisms
involved. This is inherently a complex process, involving multiple interactions between
two complicated and variable interfaces. Furthermore, a variety of test systems and
models are in use to study the process which may not relate exactly to one another. In
this section some of the possible mechanisms behind neutrophil-mediated vascular leakage
are discussed.
It is perceivable that neutrophils cause vascular leakage simply by the process of
transendothelial passage (see section 1.5.4), particularly if there is not a tight seal
between the neutrophil and the endothelial cells. Electronmicrographs of skin biopsies
24

taken 6 minutes after injection of C5a + PGE2 showed PMN leukocytes already on the
ablumenal surface of endothelial cells, many underneath endothelial cell junctions. If
PMN-leukocytes were to swell (as has been observed in response to C5a in vitro
(O’Flaherty et al., 1978b) junctions could open (Wedmore & Williams, 1981a). The
endothelial cell junctions might fail to close immediately behind the escaping neutrophil,
thus allowing leakage of plasma (Lewis & Granger, 1986). In the rabbit skin model
however, this does not appear to be the case as intradermal IL-1 induced comparable
neutrophil accumulation to C5a with little associated plasma protein leakage (Rampart &
Williams, 1988). In the Rampart & Williams study intravenously administered
radiolabelled cells were used to monitor neutrophil accumulation, thus it is possible that
extravascular cell migration did not occur in response to IL-1 since cells at all stages of
adhesion and transmigration would be measured in such studies. However, in a similar
study where PMN accumulation was assessed histologically Watson et al (1989) were
able to show pronounced extravascular PMNs in response to intradermal IL-1 with only
a minimal increase in permeability, even when IL-1 was coinjected with PGEj. Pettipher
et al (1986) also demonstrated neutrophil infiltration with no concomitant increase in
vascular permeability in response to IL-1 in the rabbit knee. Furthermore, in the hamster
cheek pouch model, intravenous dextran sulphate was found to prevent vascular leakage
but not neutrophil emigration in response to topical LTB4 (Rosengren et al., 1989).
Electron microscopy of LTB^-exposed hamster cheek pouches did not detect endothelial
gaps left in the wake of diapedesing neutrophils but, instead, efficient closing of the
vascular barrier as soon as the neutrophils had passed through was observed
(Thureson-Klein et al., 1986; Lewis & Granger, 1988). Intimate contact between the
migrating neutrophil and endothelial cells has been described in other models (Meyrick
et al., 1984; Huang et al., 1988). Separation of the emigration of the neutrophil from
its ability to induce vascular permeability has been shown in in vitro models of
neutrophil emigration through cultured endothelium (Huang et al., 1988) and through
artery intimai expiants (Meyrick et al., 1984). These data collectively suggest that the
phenomena of neutrophil-induced macromolecular leakage and neutrophil emigration can
be dissociable events.
A possible alternative explanation is that emigrating neutrophils are stimulated by
chemoattractants to induce the release of a secondary, permeability increasing factor(s).
The neutrophil has the ability to release a complex assortment of granular proteins and
proteolytic enzymes and to generate a family of reactive oxygen metabolites (Weiss,
25

1989; Henson & Johnston, 1987). Furthermore, neutrophil-derived lipid-mediators, for
example PAP may also be possible candidates for a chemoattractant-induced secondary
mediator. Potential candidates for this neutrophil-derived permeability increasing factor
are discussed below.
1.2.4.2 Lipid mediators
Chemoattractants could induce the neutrophil, whilst attached either to the lumenal
or ablumenal surface of the endothelium, to release a secondary mediator capable of
inducing inter-endothelial gap formation through, for example, endothelial cell contraction
in an analogous manner to that observed for histamine or bradykinin. The phospholipid
PAP was an early candidate as it is known to be released from neutrophils following
stimulation with C5a in vitro (Lynch et a l, 1979; Camussi et al., 1980) and induced
oedema in rabbit skin independently of the presence of neutrophils (Wedmore &
Williams, 1981b). However, a PAP antagonist (L-652,731) was found to have no effect
on leakage induced by neutrophil chemoattractants in rabbit skin, although leakage
induced by exogenous PAP was inhibited (Hellewell & Williams, 1986). Interestingly,
PAP antagonists do suppress the neutrophil-dependent oedema in an Arthus reaction,
suggesting a role of endogenously formed PAP in this situation (Hellewell & Williams,
1986). Stimulated neutrophils also release the lipoxygenase product LTB4 . In vivo, LTB4
induces neutrophil accumulation and neutrophil-dependent oedema (Wedmore &
Williams, 1981a; Bjork et a l , 1982) and this lipid mediator may contribute to
chemoattractant-induced changes in microvascular permeability. Indeed, Nagai and Katori
(1988) showed that PMLP-induced neutrophil adhesion in the microvasculature of the
hamster cheek pouch was completely inhibited by a selective 5-lipoxygenase inhibitor,
although they were unable to identify the product generated by PMLP as LTB4 .
Furthermore, desensitisation experiments in rabbit skin have indicated that LTB4 partly
mediates the inflammatory response induced by zymosan-activated plasma and platelet-
activating factor (PAP) (Colditz & Movat, 1984b). However, more recently using the
same experimental model an LTB4 antagonist LY-255,283, whilst suppressing neutrophil
accumulation and oedema formation induced by LTB4 , had no significant effect on
inflammatory responses induced by other chemoattractants (Von Uexkull et a l , 1991).
In contrast, evidence has been presented suggesting that lipoxygenase products appear to
be involved in thrombin-induced, neutrophil-mediated vascular permeability in the lung
(Perlman et a l , 1989).
26

Further research is needed to establish the identity of such a secondary mediator,
if one exists. If neutrophils do secrete such as substance, phagocytosis does not seem to
be essential because soluble factors (C5a, FMLP and LTB4) or particulate matter
(zymosan) can induce increased vascular permeability equally well.
I.2.4.3. Oxygen metabolites
In addition to synthesising and releasing mediators, neutrophils also produce
oxygen free radicals (such as superoxide anion, hydroxyl radical and HgOj upon
activation (Babior et a l, 1973) and it is possible these may play a role in neutrophil-
mediated microvascular permeability. Numerous in vitro studies have demonstrated that
oxygen radicals generated from neutrophils stimulated with chemotactic agents or phorbol
esters are capable of inducing endothelial cell damage (Sacks et a l, 1978; Shasby et al, 1983; Weiss et a l, 1981). Interestingly, close approximation of the neutrophil and
endothelial cell appears to be required for this cytotoxicity (Shasby et a l, 1983; Sacks
et a l, 1978). This perhaps reflects the involvement of short-lived mediators or the
requirement for a microenvironment at the cell-cell interface which is protected from
exogenous scavengers. On the other hand it may reflect the finding that neutrophils
adherent to endothelium (and thus neutrophils closer to the endothelium), and,
interestingly, to extravascular matrix proteins produce toxic oxygen radicals more readily
upon activation with chemotactic stimuli (Dahinden eta l, 1983; Nathan, 1987a; Shappell
et a l, 1990). It should be noted, however, that most in vitro assays employ low serum
concentrations whereas the ability of oxygen radicals to damage endothelium is decreased
in the presence of serum components (Holt et a l, 1984; Bishop et a l, 1985).
Erythrocytes also contain large amounts of scavenging substances which may protect the
endothelium from radical damage at sites of inflammation (Toth et a l, 1984).
Furthermore, the endothelium itself contains radical scavenging enzymes (Hoover et a l , 1987; Harlan e ta l, 1984).
Neutrophil-generated toxic oxygen metabolites have also been shown to induce
vascular injury in vivo based on the effect of radical scavenging or inhibitory compounds
such as catalase and superoxide dismutase (SOD) (Till et a l, 1982; Ward et a l, 1985;
Kuroda et a l, 1987). These studies entail systemic activation of neutrophils and their
damage to lung endothelium, a situation which the in vitro studies may model. However,
these studies may not accurately represent the situation in which the extravascular
generation of chemotactic factors and their interaction with intravascular neutrophils
27

brings about a reversible increase in permeability in a specialised area of the vasculature
in order to facilitate host defence. More rarely, complement may be activated
intravascularly eg. in Adult Respiratory Distress Syndrome (for review see Renaldo &
Rogers 1982) or possibly during haemodialysis (Craddock et al., 1977a) and under these
circumstances protein leakage, especially from the lung may be by an entirely different
mechanism from that involved in extravascular generation of mediators, possibly
involving endothelial cell damage as a result of interaction between activated neutrophils
and endothelial cells.
In post-capillary venules there was no electron-microscopical evidence of
endothelial damage after neutrophil migration induced by topical application of LTB4 in
the hamster cheek pouch (Thureson-Klein et al., 1984). Furthermore, the oxygen radical
scavenging enzymes superoxide dismutase and catalase, when administered as an
intravenous infusion or locally, failed to inhibit neutrophil-dependent oedema formation
in response to topical LTB4 or FMLP in the hamster cheek pouch (Rosengren et al.,
1988). Moreover Issekutz (1981a) reported that superoxide dismutase failed to inhibit
neutrophil-mediated macromolecular extravasation in response to intradermal
chemoattractants in the rabbit. However, Rampart et al (1989b) found catalase, but not
SOD, inhibited neutrophil accumulation and neutrophil-dependent oedema in rabbit skin
by a mechanism independent of its enzymic activity. It is possible, as suggested by the
experiments of Vissers Day and Winterboum (1985) that the scavenging enzymes were
unable to gain access to the microenvironment at the interface between the two cells when
given by systemic administration, although this seems to contradict the in vitro studies
of Sacks et al (1978) and Weiss et al (1981) where SOD and/or catalase were effective
when given i.v. despite the need for close apposition between neutrophil and
endothelium. However, it remains possible that in the situation where extravascular
chemoattractant triggers neutrophil accumulation a low level production of oxygen
radicals by the neutrophil in the microenvironment between the two cells may contribute
to the oedema formation. It should be noted that reversible oxidant- induced albumin
leakage over endothelial monolayers has been reported (Shasby et al., 1985).
1.2.4.4. Granule enzymes and proteins
It is well established that neutrophil granules contain a myriad of potentially
destructive enzymes which constitute the oxygen-independent arm of the cells lethal
arsenal used against invasive organisms. These enzymes are able to degrade connective
28

tissue proteins, glycoproteins and proteoglycans (Weiss, 1989). They are usually
implicated in pathological tissue destruction, however, it has been suggested that they
may also play a role in facilitating neutrophil emigration across the blood vessel wall and
promoting neutrophil-dependent increased vascular permeability. Instillation of elastase
into hamster lungs has been shown to induced oedema formation (Senior et a l , 1977) and
neutrophil-dependent vascular leakage in the Arthus reaction has been attributed to
neutrophil-derived elastase (Hamanaka et a l , 1984). Furthermore, Von Ritter et al
(1989) found elastase inhibitors prevented FMLP-induced, neutrophil-dependent changes
in mucosal permeability in the terminal ileum. Chemoattractants are capable of causing
the release of neutrophil granular constituents (Hafstrom et a l , 1981; Rae & Smith,
1981; Chenoweth & Hugli, 1978) as is the mere adherence of neutrophils to surfaces
(Wright & Gallin, 1979; Wright et a l, 1978) and these constituents, having the ability
to decrease vascular integrity, clearly may be a factor involved in neutrophil-induced
oedema. However, studies using the hamster cheek pouch model found elastase
inhibitors, when administered i.v. or locally had no effect on neutrophil extravasation or
neutrophil-dependent vascular leakage induced by topical LTB4 (Rosengren & Arfors,
1990). If the process of neutrophil extravasation and neutrophil-mediated oedema was
dependent on an active enzymatic degradation of extracellular matrix constituents, visible
structural alterations in the transmigrated vessel might be expected, but neutrophil
diapedesis has not been associated with visible changes in vessel wall structure in vivo
(Marchesi & Florey, 1960; Hurley, 1963; Thureson-Klein et a l , 1986) or in vitro (Huber
& Weiss, 1989). Also, the nature of the increased macromolecular leakage induced by
LTB4 in the hamster cheek pouch (ie. temporary and reversable upon withdrawal of the
chemoattractant) suggests the absence of extended trauma to the endothelial barrier. It
remains possible that the vessel wall has the capacity for self repair following, for
example, limited damage. Huber & Weiss 1989 showed that following neutrophil
diapedesis the basement membrane exhibited increased permeability to proteins which was
not dependent on neutrophil elastase or cathepsin G and was resistant to inhibitors of
neutrophil collagenase, gelatinase and heparanase. It is possible that neutrophil-derived
proteolytic enzymes remain neutrophil associated during their emigration across the vessel
wall. Such a localised delivery of enzymes may achieve maximal degradation of
extracellular matrix proteins and yet be highly resistant to soluble inhibitors. In addition
it remains possible that neutrophil-derived enzymes such as elastase may affect
endothelial cells by a mechanism independent of their enzymatic activity but dependent
29

on their cationic nature (Henson & Johnston, 1987). In support of this theory it has been
reported that the highly cationic neutrophil elastase and cathepsin G, even when
inactivated, can induce albumin leakage across endothelial cell monolayers and that this
activity is suppressed if the elastase is complexed with heparin, an anionic compound
(Peterson et al., 1987; Peterson, 1989). Also, in the hamster cheek pouch model
vascular leakage induced by elastase applied with a micropipette near the venule was not
dependent on the enzymatic activity of elastase (Rosengren & Arfors, 1991).
Preincubation of elastase with the anionic molecule dextran sulphate partly inhibited the
vascular leakage (Rosengren & Arfors, 1991). Further, synthetic polycations such as
poly-L-lysine have been shown to increase vascular permeability in rabbit skin (Needham
et at., 1988) and after topical application in the hamster cheek pouch (Rosengren &
Arfors, 1991) by mechanisms not associated with endothelial injury. Polycations either
released from neutrophils undergoing limited local degranulation or applied exogenously
bind to and neutralise anionic sites on the endothelium leading to an increase in
permeability to anionic proteins such as albumin (Rosengren et a l , 1989), (Vehaskari et
a i, 1984; Sunnergren & Rovetto, 1987). In the hamster cheek pouch model (Rosengren
et a l , 1989) found i.v. dextran sulphate, an anionic molecule inhibited LTB^-induced
neutrophil-dependent macromolecular permeability without affecting adhesion or
emigration of neutrophils. Uncharged dextran was without effect. It was suggested that
the dextran sulphate might complex with neutrophil-derived cationic proteins thus
preventing their permeability-increasing effects on endothelial cells. Released polycations
may act directly on the endothelium leading to retraction of endothelial cells. In support
of this, gap formation in endothelial culture without lysis of endothelial cells has been
seen after application of leukocyte elastase (Peterson et al., 1987) Cathepsin G (Toth et
al., 1984) or histone (Ginsburg et al., 1989). No endothelial receptors for cationic
proteins have yet been described, however, increased calcium flux and second messenger
production in cathepsin G exposed endothelial cells has been observed (Peterson et al.,
1989). Furthermore, cathepsin G induced leakage in vitro (Peterson et al., 1989) and
polylysine-induced permeability in the lung (Toyofuku et a l , 1989) have been prevented
by calcium antagonists or calcium channel blocking agents. The site specificity and
tachyphylaxis of leakage induced by polylysine in the hamster cheek pouch also suggests
a receptor-mediator mechanism (Rosengren & Arfors, 1991).
It must be noted, however, that polycations may cause the release of inflammatory
mediators (Fairman et al., 1987; Toyofuku et al., 1989; Lee et al., 1985) which may
30

then possibly play a role in the induced inflammatory response in some studies.
IL-1 does not directly activate neutrophils (Georgilis et al., 1987; Yoshimura et
al., 1987). Moser et al (1989) have found that, following IL-1 induced migration across
endothelial cell monolayers, neutrophils were in a metabolically calm state (ie. no
oxidative burst activation, no granule content release). This may explain the results of
Watson et al (1989) and Rampart & Williams (1988) showing the lack of microvascular
permeability induced by IL-1 despite transendothelial neutrophil passage.
1.3. Vasodilatation and mediator svnergism
Williams and Morley (1973) showed that the ‘E-series’ prostaglandins although
potent vasodilators, were poor mediators of oedema formation when injected into
guinea-pig skin. However, they found that these prostaglandins greatly potentiated the
action of permeability-increasing mediators such as histamine and bradykinin. This
phenomenon was also demonstrated in a number of other species (Moncada & Ferreira,
1973; Williams, 1976; Williams & Peck, 1977; Basran et al., 1982). The mechanism
involved in synergism between prostaglandins and permeability- increasing mediators is
believed to be as follows: prostaglandins dilate arterioles resulting in an increase in
blood flow to the tissue; as a consequence there is an increased intravenular hydrostatic
pressure, aiding the outward passage of plasma, together with the passive venular
distension, thus increasing the vessel wall surface area. The observation that a
correlation exists between the oedema-potentiating ability of different prostaglandins and
their vasodilator activity supports this mechanism of action (Williams, 1976; Williams
& Peck, 1977). Evidence was obtained for oedema induced by synergism between
endogenous vasodilator and permeability-increasing mediators generated in response to
intradermal injection of Bordetella pertussis organisms or yeast cell walls (zymosan) in
the rabbit (Williams & Peck, 1977; Williams, 1979; Williams & Jose, 1981). These
observations formed the basis of the "two-mediator hypothesis" ie. that in response to an
inflammatory stimulus a vasodilator mediator and a permeability-increasing mediator are
released which act synergistically to cause oedema (Williams & Peck, 1977; Williams,
1977).
Vasodilator substances, other than prostaglandins have also been found to
potentiate oedema; agents such as adenosine (Williams & Peck, 1977), vasoactive
intestinal peptide (VIP) (Williams, 1982) and calcitonin gene-related peptide (CGRP)
(Brain & Williams, 1985) can synergise with mediators of increased-vascular
31

permeability. Conversely, vasoconstrictor substances have been found to reduce oedema
formation and blood flow in parallel (Williams & Peck, 1977). Furthermore, synergism
appears to be important in areas with low basal blood flow such as rabbit skin as
compared to tissues with a higher basal blood flow such as the rabbit peritoneum.
Indomethacin was found to inhibit oedema formation in response to intradermal zymosan
in rabbit skin by >80% by preventing the generation of potentiating prostaglandins
(Williams & Jose, 1981). However, in the rabbit peritoneal cavity the oedema produced
in response to intraperitoneal zymosan was suppressed by only 25 % despite the virtual
abolition of the active vasodilator prostacyclin (measured as 6 -oxo PGF^J (Forrest et al.,
1985; Forrest et a l, 1986).
Local vasodilators have also been found to potentiate neutrophil-dependent oedema
formation (Williams & Jose, 1981; Williams, 1982; Wedmore & Williams, 1981a; Brain
& Williams, 1985) and neutrophil accumulation in response to chemotactic stimuli
(Issekutz & Movat, 1979; Issekutz, 1981b). Prostaglandins alone were found to be
extremely weak at inducing neutrophil accumulation (Higgs et al., 1981). It is possible
that vasodilatation enhanced neutrophil-infiltration by increasing the rate of delivery of
blood leukocytes to the area and by providing a larger vascular bed for leukocyte
emigration (Issekutz, 1981b).
In an inflamed tissue it is desirable to increase local blood flow to accommodate
increased metabolic activity. This is especially the case following the infiltration of high
numbers of metabolically active leukocytes (see section 1.4). Increased blood flow acts
to assist the recruitment of leukocytes and the efflux of macromolecules from the blood
necessary for host defense. It also serves to dilute/remove any toxins produced at the
inflammatory site. Thus, increased blood flow in inflammation may be considered to be
a form of functional hyperaemia.
1.4. Neutrophil Accumulation
1.4.1 Introduction
Invading microbes, other foreign organisms or particles and damaged tissue cells
stimulate the generation of chemotactic mediators that induce the local accumulation of
neutrophilic leukocytes - one of the characteristic features of the acute inflammatory
response. Upon exposure to chemotactic mediators neutrophils adhere to the
microvascular endothelium selectivity in small venules by the aid of adhesion molecules
32

(for review, (Bevilacqua, 1993; Springer, 1990) and see section 1.4.3.)- They
subsequently traverse the endothelial cell layer, penetrate the basement membrane and
accumulate in the extravascular tissue affected by the inflammatory agent. Once at the
site of tissue injury the neutrophil has the essential function of killing foreign organisms
and removing these and other unwanted solid material from tissues. Neutrophils are
regarded as the first line of defence against invading organisms (Dale, 1984). The
mechanisms underlying neutrophil accumulation in a tissue are complex and despite
intensive investigation remain unclear and to some extent controversial. The
interpretations of many of the studies are conflicting. In this section the steps involved
in this complex process, which have been investigated using both in vivo and in vitro
studies, will be discussed in sequence.
1.4.2. Neutrophil-endothelial cell interactions
Neutrophil accumulation is fundamentally dependent on the adhesive interaction
between the neutrophil and the vascular endothelial cell, triggered by the chemotactic
signal. Leukocyte emigration is responsible for the successful host response to tissue
injury and infection, but is also potentially harmful and contributes to the pathology of
many diseases and inflammatory disorders. The factors involved in this interaction are
of considerable experimental interest and potential clinical relevance.
The first step in this process is margination, when leukocytes leave the central
stream of flowing blood cells in a postcapillary venule and roll along the endothelial
lining of a vessel, as observed more than 1(X) years ago using intravital microscopy
(Cohnheim, 1889). Leukocyte rolling in post capillary venules does not appear to
account for the "marginating pool" of about 50% of leukocytes that are intravascular
mainly in capillary beds in the lung and enter the circulation in response to exercise or
epinephrine (Athens et a l, 1961; Worthen et a l , 1987). Postcapillary venules are the
major sites of leukocyte emigration in inflammation and in the healthy state these venules
have few or no rolling leukocytes (Fiebig et a l, 1991). The number of rolling cells
increases dramatically during the course of an inflammatory reaction (Atherton & Bom,
1972) and is believed to be important in the accumulation of cells at the site (Fiebig et
a l, 1991). Both the rheology of blood and specific adhesive interactions (see Section
1.4.3.) may regulate the rolling response. In inflammation arterioles dilate increasing the
blood supply to the affected tissue. Vascular permeability is increased, leading to plasma
leakage and an increased haematocrit which leads to erythrocyte rouleaux formation. A
33

combination of these factors causes leukocytes to be displaced to the marginal region of
flow near the vessel wall (Chien, 1982). Because fluid velocity increases with distance
from the wall, cells near the wall have torque exerted on them and will tumble even if
not in contact with the wall. However, the velocity at which cells tumble in a shear flow
near to the vessel wall is much faster than observed for rolling cells in inflammatory
reactions, suggesting that adhesive interactions occur between the leukocyte and vessel
endothelium (Atherton & Bom, 1973). Rolling neutrophils are probably most able to
detect and respond to chemotactic signals generated by the presence of extravascular
inflammatory stimuli. Neutrophils then become firmly attached to the vessel wall. A
fundamental question which remains open is whether chemoattractants generated locally,
or applied extravascularly act on the endothelial cell or the neutrophil, or both, to initiate
this process.
In most tissues neutrophil-endothelial interaction takes place in the venules. The
exception is the pulmonary circulation where the capillary is the major site. This
selectivity of adherence site implies either an active change in the surface of specialised
endothelial cells induced by the chemoattractant, or a favoured site for the adherence of
activated neutrophils. Following direct damage to the endothelium, neutrophils adhere
only to that side of the vessel that has received such an injury and do not adhere to
undamaged endothelium in vessels downstream when they are occasionally dislodged
(Allison et al., 1955; Clark & Clark, 1935), suggesting that the endothelium adjacent to
the site of damage becomes more adhesive. However, it is not clear whether such a
rapid change in the endothelial cell surface can occur in response to chemoattractants.
Several attempts have been made to shed light on this question using in vitro cultures of
endothelial cells, but the results have not been conclusive. Hoover et al, (1980; 1984)
observed that pretreatment of endothelium with C5a, FMLP or LTB4 followed by
washing and incubation with untreated neutrophils resulted in enhanced neutrophil
adherence. This result was shared by (Zimmerman & Hill, 1984) using zymosan-
activated plasma (as a source of C5a) or FMLP. Furthermore, Palmblad et al (1990) and
Lindstrom et al (1990) have reported that LTB4 can enhance the adhesiveness of cultured
EC for neutrophils. These studies indicate that chemotactic agents may bind to or alter
the endothelium, resulting in enhanced adherence. Furthermore, it has been suggested
that endothelial cells have specific binding sites for FMLP (Hoover et al., 1980) and that
FMLP can cause marked restructuring of the endothelial plasma membrane (Kirkpatrick
& Melzner, 1984). It is not clear whether these receptors exist on the lumenal or
34

ablumenal surface of the endothelial cell. Specific endothelial cell membrane receptors
for other chemotactic factors eg C5a or leukotriene B4 have not been described, however.
Furthermore, Tonnesen et al (1984) could not repeat the experiments of Hoover et al
(1980; 1984) and (Zimmerman & Hill, 1984), finding that pretreatment of the neutrophil
but not the endothelial cell with chemotactic agents produced increased adhesion. Oseas
et al (1982) and (Huang et al., 1988) reached a similar conclusion. Tonnesen et al
suggested that the results of Hoover et al could be explained by inadequate washing of
the endothelial monolayer. Furthermore, the length of preincubation of the endothelial
cells with the chemotactic factors differs between the various studies. Hoover et al
(1980; 1984) pretreated endothelial cultures for 1-30 minutes with chemoattractants
whereas, in contrast Huang et al (1988) pretreated for 2 hours. Therefore, it remains
possible that chemotactic agents may induce a transient change in the endothelium. In
this regard the endothelium hyperadherence found by Palmblad et al (1990) and Lindbom
et al (1990) occurred following only l-5min stimulation with LTB4 and was immediate
and transient in nature. A major concern in these studies is that cultured endothelium
may not react like endothelium in vivo. Also, large vessel endothelium, most often used
in the studies, may not be the appropriate substrate since neutrophil adherence in vivo
occurs primarily in the microcirculation.
In vitro the presence of specific high affinity chemoattractant receptors on
neutrophils has been well characterised (Chenoweth & Hugli, 1978; Goldman et al.,
1987). These receptors would allow neutrophils to recognise and respond to
chemoattractants present in the local environment. Neutrophils exposed to
chemoattractants in vitro have been shown to exhibit increased adhesion to each other,
to artificial substrates and to endothelium (Craddock et al., 1977b; Gimbrone et al.,
1984; Tonnesen et al., 1989). Thus, it is possible that chemoattractants generated
extravascularly diffuse into the lumen of microvessels and act on the receptors of passing
neutrophils to increase adherence. Any chemoattractant which diffused into the lumen
would be rapidly diluted by flowing blood and it is not known whether neutrophils are
exposed to a sufficiently high concentration of chemoattractant within the lumen to cause
an increase in adhesive properties. Chemoattractants may act on neutrophils during their
passage along capillaries (see below) when the area of contact between neutrophil
membrane and vessel wall is maximal, thus allowing leukocytes to receive information
about gradients of chemotactic factors diffusing between the endothelial cells or through
pores in the endothelial cells. It is not clear what change takes place in the neutrophil
35

membrane which results in increased adherence, although chemoattractants are known to
reduce cell surface charge thus decreasing repulsive electrostatic forces between Gallin a/., 1975 Hoover aA, 1980
neutrophils and endothelium ( ). Also, chemoattractants stimulate the release
of the specific granule protein lactoferrin which has been proposed to promote neutrophil
adherence (Boxer et a l , 1982; Oseas et a l , 1981). It is noteworthy, however, that other
investigations have found conflicting results with regard to the requirement for both
changes in net surface charge and specific granule contents for neutrophil adhesion
(Hoover et a/., 1980; Gallin et a l, 1982). There is now a wealth of evidence which
suggests that chemoattractants stimulate the presentation of certain glycoproteins on the
neutrophil surface that play a key role in neutrophil adherence to endothelium (Amout
e ta l , 1983; Springer gf aZ., 1985) & see section 1.4.3). The stimulated neutrophils may
then adhere downstream in the venules in most tissues; the apparent predilection of
neutrophil adhesion for postcapillary venules is governed either by differences in
endothelial cell surfaces or rheological factors since the postcapillary venule is the site
of the first major decrease in vessel wall shear stress.
The in vivo study of Katori and colleagues provides evidence in support of the
concept that the chemoattractant signal may be picked up by the neutrophil as it passes
through the capillary. Nagai and Katori (1988) reported that in the hamster cheek pouch,
injection of LTB4 or FMLP by a glass capillary pipette into the interstitial space close to
capillaries resulted in neutrophil adhesion in venules downstream. Furthermore, a similar
injection of these stimuli close to the venule did not cause adhesion of leukocytes,
suggesting that in vivo chemoattractants act on the neutrophil and not on the endothelium
to induce neutrophil-endothelial cell interaction. These findings are supported by the
experiments of Nourshargh et al (1990) in which pretreatment of radiolabelled neutrophils
with pertussis toxin, which inhibits receptor-mediated responses induced by
chemoattractants in vitro, was found to inhibit neutrophil accumulation in vivo in rabbit
skin. These results indicate that a receptor-mediated event on the neutrophil is crucial
in neutrophil accumulation in response to chemoattractants such as C5a, FMLP and
LTB4. However, the above reports are apparently in conflict with the observations of
Colditz and Movat which suggest an active involvement of the endothelial cell in this
process. Colditz and Movat (1984b; 1984a) reported that skin sites in the rabbit could
be specifically desensitised to particular chemoattractants by a previous intradermal
injection of that substance. They also found that the kinetics of neutrophil influx were
independent of the concentration of chemotaxin used to induce the lesion. Together these
36

experiments, using radiolabelled leukocytes offer the intriguing possibility that receptors
for chemoattractants may be present upon endothelial cells or adjacent tissue cells. A
possible explanation for these results is that chemoattractants stimulate the ablumenal
surface of the endothelial cell which induces an increased adhesiveness of its lumenal
surface or, as discussed by Tonnesen et al (1982) mediators diffuse through endothelial
cell junctions and fix on the lumenal surface of the cell. The latter theory has recently
been revived in describing the actions of IL- 8 in vivo. Autoradiographic study of
^^I-labelled IL- 8 injected intradermally in rat and rabbit revealed binding sites on the
endothelial cells of post capillary and collective venules (Rot, 1992).
Alternatively, chemoattractant receptors on the ablumenal surface of endothelial
cells may mediate the translocation of chemoattractant molecules through the cell, perhaps
bound to cell membrane using a membrane cycling phenomenon resulting in receptor-
bound chemoattractants being presented to lumenal neutrophils (Williams et at., 1984b).
Such a mechanism, for which there is now in vitro evidence (Rotrosen et al., 1987),
supports the possibility for an active role of vascular endothelial cells in chemoattractant-
induced neutrophil accumulation in vivo.
1.4.3. Role of cell surface adhesive glycoproteins in neutrophil-endothelial
interactions
Some of the adhesive mechanisms utilized by neutrophils to determine their
localization at sites of inflammation have been defined at a molecular level. A number
of adhesive molecules on both migratory cells and endothelium seem critical for the
interaction of leukocyte and vessel wall endothelium. These pro-adhesive molecules have
diverse structures and mechanisms of expression and vary with the nature of the
inflammatory stimulus.
Much information has been obtained in vitro from studies using cultured
endothelial cells and isolated blood neutrophils. Several lines of evidence have implicated
the &2 integrins, leukocyte cell surface glycoproteins known collectively as the
CD 11/CD 18 antigen complex (Kishimoto et al., 1989b; Bernstein & Self, 1985; Amaout,
1990). This complex is composed of three structurally and functionally related
glycoprotein heterodimers, CD 1 la/CD 18 (LFA-1), CDllb/CD18 (Mac-1, also known
as MOl or complement receptor type 3), CDllc/CD18 (P150,95 or CR4). Each
glycoprotein consists of an immunologically distinct a-subunit (CD ll) that is
non-covalently associated with a common subunit (CD 18). These glycoproteins are
37

members of the supergene family of cell surface receptors termed integrins. Three
subfamilies of integrins can be distinguished by their B subunits: these are known as the
Bi (CD29), B2 (CD 18) and 63 (CD61) integrins. The Bj integrin subfamily includes
receptors for extracellular matrix glycoproteins such as fibronectin, vitronectin, collagen
and laminin (Hynes, 1987). Valuable evidence of the importance of the CD11/CD18
glycoprotein complex was obtained following the recognition of a rare and inheritable
disorder now known as the leukocyte adherence syndrome (LAD). The striking clinical
manifestations of LAD include persistent and recurrent bacterial infections, impaired pus
formation and wound healing. These clinical features appear to reflect a severe
impairment of neutrophil mobilization into extravascular inflammatory sites, although
these patients are generally neutrophilic (Kishimoto et al., 1989b). Furthermore, in vitro,
neutrophils from these patients have apparently normal adhesion-independent functions
in suspension (shape change, respiratory burst and granule release) but have severely
depressed adhesion-dependent responses such as chemotaxis, aggregation, adherence and
phagocytosis of iC3b-opsonized particles and adherence to and migration through
endothelial monolayers in response to stimulation with chemotactic factors (Harlan etal.,
1985; Anderson et al., 1985; Kishimoto et al., 1989b). These in vivo and in vitro
features of LAD syndrome are attributable to a severe or total deficiency of the common
subunit of the CD ll/CD 18 complex (Springer et a l , 1984; Anderson & Springer,
1987). This concept has been further substantiated by the demonstration that treatment
of normal neutrophils with monoclonal antibodies directed against the common subunit
(CD 18) completely inhibits chemotactic factor-induced adherence to endothelial
monolayers (Harlan et al., 1985; Zimmerman & McIntyre, 1988; Schwartz et al., 1985;
Wallis et a l , 1986). Further comparative studies with panels of monoclonal antibodies
directed at individual subunits or combinations of subunits within the complex have
suggested that a functionally active site on CD 11b (Mac-1) is primarily involved in
neutrophil adherence stimulated by chemotactic factors (Zimmerman & McIntyre, 1988;
Anderson et al., 1986; Tonnesen et al., 1989; Wallis et al., 1986). In vivo studies have
also shown that systemic administration of monoclonal antibodies recognising CD 18, for
example, 60.3 and IB4 inhibit neutrophil accumulation into skin sites injected with
chemoattractants (Lundberg & Wright, 1990; Arfors et al., 1987) or into subcutaneousI
sponges soaked with chemoattractants (Price et al., 1987). Intravenous anti CD 18 mAb
also inhibited neutrophil-dependent plasma extravasation into rabbit skin (Lundberg &
Wright, 1990). Furthermore, pretreatment of purified rabbit neutrophils with 60.3 prior
38

to infusion into untreated recipient animals prevented their accumulation into
inflammatory skin sites (Rampart & Williams, 1988; Nourshargh et a l, 1989).
Interestingly, using intravital microscopy direct visual evidence was obtained that
intravenous 60.3 prevented stimulated, but not unstimulated neutrophil adherence to
endothelium (Arfors et a l , 1987). In this study neutrophil adherence and migration into
the tissues in response to extravascular chemoattractant was inhibited, while rolling of
neutrophils was unaffected suggesting that the mechanisms governing the interaction of
‘unstimulated’ neutrophils with endothelium are CD 18-independent (see this section).
Rapid increases in neutrophil adhesion, stimulated by chemoattractants such as
FMLP, C5a and LTB4 , have been shown to be mediated via effects on Mac-1 (Tonnesen
et a l , 1989). Unlike LFA-1, Mac-1 and gp 150/95 are found in intracellular storage
granules (Miller et a l, 1987). On exposure to chemoattractants neutrophils exhibit an
increase (2 to 14 fold) surface expression of GDI lb/CD 18, mobilised from the
intracellular stores (1058,1354). However, several reports have suggested that stimulus-
induced upregulation of cell surface glycoprotein expression and stimulus-induced
adherence to endothelium can be dissociated (Vedder & Harlan, 1988; Philips et a l,
1988; Buyon et a l , 1988). Furthermore, there is in vivo evidence suggesting that
neutrophil accumulation in response to chemoattractants does not require an increase in
the surface expression of CD 18 (Nourshargh et a l , 1989). Therefore, it seems that
upregulation may not be the only regulatory event; modification(s) of the adhesive
glycoprotein molecules on the cell surface such as altered density distribution within the
membrane, conformational or biochemical changes or interaction with other membrane
components may be necessary for adherence to occur. It is perhaps possible that the
increased surface expression of Mac-1 may instead play a role in the alternative effector
function of phagocytic cells such as the Mac-1-dependent secretion of hydrogen peroxide
by adherent neutrophils (Nathan et a l , 1989; Entman et a l , 1990).
The integrins are novel with respect to the inside out activation phenomena, in
which signals from the cytosol are transduced across the membrane to generate changes
in extracellular functions such as adhesion (for review see Hynes (1992). Activation of
leukocytes, for example with phorbol esters or various inflammatory mediators such as
TNF, C5a, PAF, FMLP is required for expression of the various ligand-binding activities
of the B2 integrins. It is thought that activation of leukocytes is accompanied by a
conformational change(s) in the 8 2 integrin ligand binding site resulting in a transient
change in avidity (Keizer et a l , 1988; Buyon et a l , 1988; Lo et a l , 1989a). The
39

cytoplasmic domains of integrins, perhaps by interaction with other cellular proteins, may
signal the changes in the extracellular domains that regulate avidity (Hibbs et al., 1990).
A counter-receptor for CD 11 a/CD 18 (LFA-1) on the endothelial cell surface has
been identified as a glycoprotein designated intercellular adhesion molecule 1 (ICAM-1
or CD54) (Marlin & Springer, 1987; Kishimoto et al., 1989b). ICAM-1 is a member
of the immunoglobulin superfamily (Simmons et al., 1988; Staunton et a l , 1988). In the
absence of an inflammatory response ICAM-1 is constitutively present on the surface of
vascular endothelial cells, as well as fibroblasts, epithelial cells and some leukocytes
(Dustin et a l , 1986). Its cell surface expression is induced during inflammation in vivo
and by treatment of endothelial monolayers in vitro with lipopolysaccharide, and the
cytokines gamma-interferon (gamma IFN), interleukin-1 (IL-1) and tumour necrosis
factor (TNF) (Pober et a l , 1986), thus causing greatly increased binding of leukocytes
through their cell surface LFA-1. ICAM-1 can be expressed on a wide variety of cells
during inflammation and its induction is an important means of regulating LFA-l/ICAM-
1 interactions and thus the localisation of circulating cells at an inflammatory site.
ICAM-1 induction is largely regulated at the mRNA level (Simmons et a l, 1988;
Staunton et a l , 1988). Increased surface expression after cytokine induction is detectable
in vitro or in vivo after 4-6h, and is maximal by 9-24h (Kishimoto et a l , 1989b; Munro
et a l , 1989). A more recently described member of the immunoglobulin superfamily
ICAM-2 is also found constitutively on a variety of cell types including endothelium and
also binds leukocyte LFA-1, however, expression of ICAM-2 is not increased by
cytokines (Staunton et a l, 1989).
Mac-1 was originally identified as the receptor for the surface-bound complement
protein C3bi (Wright et a l, 1983). Further investigations from several laboratories have
provided evidence that Mac-1 can also recognise other ligands, for example fibrinogen
(Wright et a l , 1988), bacterial lipopolysaccharide (Wright et a l , 1989), filamentous
hemagglutinin of Bordetella pertussis (Reiman et a l , 1990) and possibly thrombospondin
and laminin (Nathan et a l , 1989). With regard to the ligand for Mac-1 on the
endothelium; in addition to the ICAM-1/LFA-1 interaction being involved in neutrophil
adhesion Smith & co-workers, using specific monoclonal antibodies, have provided
evidence that chemotactic stimulation of neutrophils may enable Mac-1 on the neutrophil
to interact with ICAM-1 on the surface of the endothelial cell (Smith et a l , 1988; Smith
et a l , 1989). Diamond et al (1991) have shown that purified Mac-1 binds to domain 3
40

of ICAM-1 whereas LFA-1 binds domain 1. A report from Lo et al (1989b) has also
suggested the existence of a novel alternative endothelial ligand for Mac-1 which is
consistent with its multivalent nature.
A recent study by Argenbright et al (1991) using the technique of intravital
microscopy to visualise the microcirculation of the rabbit mesentery showed that
GDI lb/CD 18 contributes to the adhesion of C5a-stimulated leukocytes to the
microvasculature in vivo. They were also able to demonstrate that CDlla/CD18
participates in this binding in vivo. That these leukocyte receptors were binding to
ICAM-1 was supported by the strong inhibition of adherence by an antibody against
ICAM-1. This data provides evidence in vivo that there is sufficient basal ICAM-1
expression to support CD 18-dependent adhesion. Incubation of C5a with human
umbilical vein endothelial cells for 24 hours was not able to induce ICAM-1 expression
(Argenbright et al., 1991).
In addition to the CDll/CD 18 antigen complex, the leukocyte cell surface
glycoprotein originally defined in the mouse by the MEL-14 monoclonal antibody
(Gallatin et al., 1983) has also been implicated in neutrophil-endothelial cell interactions.
The human homologue of the murine MEL-14 antigen (known as leu-8 , TQ-1 or LAM-1)
has been identified and cloned (Tedder et al., 1989; Camerini et a l , 1989; Siegelman
& Weissman, 1989; Bowen et al., 1989) and is now known to be a member of the most
recently recognised family of adhesion molecules, the selectins or LECCAM (lectin-
cellular adhesion molecule) family and has been renamed L-selectin or LEG AM-1 (for
review see Zimmerman et al 1992). Evidence that the MEL-14 antigen or L-selectin is
a neutrophil adhesion molecule came from studies in murine models of inflammation.
Systemic administration of the MEL-14 monoclonal antibody was found to inhibit murine
neutrophil localisation into subcutaneous inflammatory sites (Lewinsohn et al., 1987) and
to the inflamed peritoneum in vivo (Lewinsohn et a l , 1987; Jutila et a l , 1989) and
blocked binding of neutrophils to vascular endothelium in vitro (Lewinsohn et a l , 1987;
Jutila et a l , 1989). Human L-selectin is present on neutrophils, monocytes and a sub
population of lymphocytes that migrate to peripheral lymph nodes (Tedder et al
1990, Gallatin et a l , 1983). Using the DREG series of monoclonal antibodies which
recognise human neutrophil L-selectin (Jutila et a l , 1990) a role for this molecule in
CD 18-independent adhesion of neutrophils to cytokine- stimulated endothelium in vitro
has been indicated (Kallmann et a l , 1991; Smith et a l , 1991; Spertini et a l , 1991). In
contrast to Mac-1 activity, L-selectin-mediated binding does not appear to require
41

neutrophil activation. Indeed activation of neutrophils (eg. with chemotactic factors)
results in rapid shedding of L-selectin from the surface of murine neutrophils (Kishimoto
et al,, 1989a) and human neutrophils (Jutila et al., 1990). However, prior to its shedding
following neutrophil activation, L-selectin shows a rapid and transient enhancement of
avidity for its endothelial ligand (Spertini et a l , 1991). The rapid down-regulation of
L-selectin is coincident with rapid upregulation of Mac-1 expression and function
(Kishimoto et a l , 1989a) and was also found to occur early during the process of
neutrophil extravasation in vivo (Kishimoto et a l , 1989a; Jutila et a l , 1989). These
results suggest that Mac-1 and L-selectin on the neutrophil surface mediate fundamentally
distinct adhesion events (see this section later).
In addition to L-selectin two other members of the selectin family have been
described, which are expressed on endothelial cells. Neither is constitutively present on
the surface of resting endothelium but each is expressed, by different mechanisms, when
the endothelium is activated. Endothelial leukocyte adhesion molecule, ELAM-1 or
E-selectin, is transiently expressed on endothelial cells 2-8hr after stimulation in vitro
with IL-1 and other inflammatory cytokines, and mediates adhesion of unactivated
neutrophils by a pathway distinct from that mediated by ICAMs and leukocyte integrins
(Bevilacqua et a l, 1989; Luscinskas et a l , 1989). Monocytes and a subpopulation of
lymphocytes have also been shown to bind E-selectin (Picker et a l , 1991a).
The counter receptor(s) for L-selectin on cultured endothelial cells has not been
characterised. L-selectin may interact with E-selectin (Kishimoto et a l , 1991) and also
the third selectin, P selectin (Picker et a l, 1991b). However, neither of these
interactions are thought to be unique (Kishimoto et a l, 1991; Geng et a l, 1990;
Bevilacqua et a l , 1989). There is evidence that both E-and P-selectins recognize
sialyl-Lewis x (sLe*) found in glycoproteins and glycolipids (Larsen et a l , 1990; Moore
et a l , 1991; Polley et a l , 1991; Phillips et a l, 1990; Lowe et a l , 1990; Walz et a l,
1990). However, this is not thought to be the complete story. Neutrophil L-selectin
bears sLe* that is recognized by the other two selectins (Picker et a l , 1991b).
P-selectin, otherwise known as PADGEM or GMP (granule membrane protein)
140 or CD62 is stored in «granules of platelets and weibel-palade bodies of endothelial
cells. It is rapidly mobilized to the surface of endothelial cells within seconds after
stimulation with thrombin or histamine where it mediates adhesion of neutrophils and
monocytes (Johnston et a l , 1989; Larsen et a l , 1989; Hattori et a l , 1989). It is rapidly
reintemalized, resulting in transient neutrophil adhesion. In common with the other
42

selectins P-selectin tethering of neutrophils does not require neutrophil activation or the
&2 integrin system (Geng et a l , 1990; Lorant et al., 1991). In addition, most studies
have indicated that the selectins are far more efficient than CD 18 molecules in operating
at physiologic shear stress (Lawrence et at., 1990; Lawrence & Springer, 1991; Smith
e ta l , 1991).
Based on the accumulated data on the selectins and integrin-mediated adhesion a
general model of leukocyte-endothelial recognition has been proposed involving at least
three sequential events (for review see Butcher, 1991). Firstly, the selectins and their
counter receptors are believed to be important in the initial rolling interaction between
unstimulated neutrophils in the circulation and the inflamed endothelium under conditions
of flow. Rolling is thought to slow the transit of leukocytes through the inflamed vessel
and so allow for close proximity and sufficient time of exposure to neutrophil activating
factors in the local environment or on the endothelial surface. Neutrophil activation leads
to shedding of L-selectin and the third step of the process, functional upregulation of
Mac-1 and integrin-mediated adhesion strengthening and transendothelial migration. The
specific factor(s) responsible for activation of rolling leukocytes in situ are likely to vary
with the physiological setting. They may include chemoattractants that are diffusing from
tissue through endothelial cell junctions (Lo et a l , 1991) or signalling molecules
synthesized by activated endothelial cells, for example, PAF (Zimmerman et a l , 1990;
Lorant et a l , 1991), IL- 8 (Carveth et a l , 1989) and possibly granulocyte-macrophage
colony-stimulating factor (GMCSF), nitric oxide or adenosine. Interestingly E-selectin
itself has been reported to activate integrin-mediated adhesion of neutrophils (Lo et a l ,
1991) and P-selectin has been reported to potentiate responses to activators (Lorant et a l ,
1991). There is evidence that this series of events may mediate adhesion and emigration
of neutrophils in vivo (von Andrian et a l , 1991; Kubes et a l , 1990; Gasic et a l , 1991;
Arfors et a l , 1987; Argenbright et a l , 1991). However most of the information to date
has been derived from in vitro studies and much has concentrated on cytokine stimulation
of endothelial cells. It is well established in vivo that extravascular chemoattractants are
able to induce the rapid ‘capture’ of neutrophils from the circulation followed by
transmigration into the tissue, but the specific molecules involved and their roles remain
to be clearly defined. For example, do chemoattractants stimulate the expression of P-
selectin on the endothelium allowing the capture of the neutrophil, and if so why does
histamine, an effective upregulator of P-selectin in vitro not induce leukocyte
accumulation in vivo. The role, if any of L-selectin in the response to chemoattractants
43

is also unknown. Although three-step adhesion may be necessary for circulating
leukocytes under physiological conditions the possibility remains that one step, immediate
adhesion may occur in pathological conditions. Altieri (1991) and Elices et al (1991)
have shown that cations such as Mn " and Mg ' can increase the avidity of integrins for
their ligands and it has been proposed that release of intracellular stores of these cations
at the sites of tissue or vascular injury may provide a mechanism for rapid upregulation
of integrin-mediated adhesion.
1.4,4. Neutrophil emigration through the endothelial cell laver
The passage of neutrophils through the endothelial cell barrier was investigated
in detail in early electron microscopic studies. Marchesi & Florey (1960) and Florey and
Grant (1961) observed a clear pseudopodium emerging from the flattened surface of
neutrophils that penetrated into the junctions between adjacent endothelial cells, rapidly
followed by the rest of the neutrophil. The junctions appear to ‘reseal’ after the
neutrophil (Marchesi & Florey, 1960). How chemoattractants induce neutrophil
extravasation is not known. Neutrophils migrate up concentrations gradients of
chemotactic agents in vitro and such a gradient, at inter-endothelial junctions may be
necessary in vivo to stimulate neutrophil emigration. With regard to this Hopkins et al
(1984) have reported gradient-dependent chemotaxis of neutrophils across endothelial
monolayers. Interestingly, a reverse gradient, with chemoattractant present
intravascularly has been reported to cause neutrophil/endothelial cell adherence with no
emigration, at least in the lung (Henson et al., 1982b). It is still conjectural whether a
cement substance is present at endothelial junctions and whether this substance is digested
by neutrophils during their passage (Marchesi & Florey, 1960).
Much evidence concerning the molecular mechanisms governing trans-endothelial
migration has come from in vitro studies using confluent endothelial cell monolayers
cultured on synthetic filters, on collagen gels, or on amniotic membranes. Two lines of
evidence point to the importance of CD 18-dependent mechanisms in neutrophil
transmigration. First, transmigration is inhibited in the presence of anti-CD 18
monoclonal antibodies (Smith et a l , 1988; Smith et a l , 1989; Furie et a l , 1991), and
secondly, CD 18 deficient neutrophils from patients with LAD fail to transmigrate in vitro
or in vivo (Davies et a l, 1991). During transmigration the leukocyte 8 2 integrins are
believed to interact with ICAM-1 molecules located on the luminal, lateral and basal
surfaces of endothelial cells (Oppenheimer-Marks et a l , 1991; Smith et a l , 1988; Smith
44

et a l , 1989; Furie et a l, 1991).
1.4.5. Passage of neutrophils through the basement membrane
After penetrating the intercellular junction neutrophils appear to hesitate,
sandwiched between endothelial cells and the basement membrane (Marchesi & Florey,
1960). Florey & Grant (1961) observed that neutrophils may migrate parallel to the
basement membrane possibly "looking for" a suitable site to penetrate the tough matrix
barrier. Interestingly, it has been suggested that specific neutrophil receptors for laminin,
a glycoprotein ubiquitous to all basement membranes, may possibly play a role in the
neutrophils negotiation of the basement membrane (Thorgeirsson et a l , 1985). Laminin
has been reported to be chemotactic for neutrophils (Terranova et al., 1986). Thus,
neutrophils may penetrate the basement membrane by using laminin as an attractant and
an attachment factor. Penetration of the vessel wall and basement membrane may require
that the neutrophils "digest" their way through interendothelial cell junctions and
subendothelial matrix proteins by limited release of extracellular enzymes. In an
interesting study Huber & Weiss (1989) have provided evidence suggesting that the
neutrophil-induced loss of basement membrane integrity associated with neutrophil
diapedesis in vitro is transient and that basement membrane defects can be repaired by
the overlying endothelium via a process dependent on protein synthesis. This is in
agreement with reports showing that microscopic examinations of vessel walls exposed
to neutrophil traffic in vivo fail to identify defects or alterations in basement membrane
structure (Hurley, 1963).
1.4.6. Neutrophil locomotion towards and removal of the inflanmiatorv stimulus
Once outside the vessel wall the neutrophil can move under the influence of the
concentration gradient towards the site of generation of the chemoattractant. Neutrophils
can detect very small changes in stimulus concentration across their surface, which allows
them to move in the direction of higher concentration. On exposure to a gradient of
chemoattractant the neutrophil becomes elongated and polarized towards the direction of
the stimulus, with a broad anterior lamellipodium and a thin posterior uropod (Snyderman
& Goetzl, 1981).
The chemotactic and chemokinetic activity of chemoattractants exhibits a bell
shaped dose response curve in vitro (Webster et at., 1980). The higher concentration of
chemoattractant mediator at the centre of inflammatory lesions may reduce the mobility
45

of neutrophils, thus holding cells at the inflammatory focus. At the focus of the
inflammatory response the neutrophil is capable of phagocytosing encountered microbes
into phagocytic vesicles. To assist in the destruction of internalised organisms the
neutrophil has several potent devices. Antimicrobial and proteolytic enzymes are released
into the phagosome from neutrophil granules. Another class of antimicrobial substances
are generated by the action of NADPH-oxidase during a response known as the
respiratory burst (reviewed by Baboir, 1984), these agents are collectively known as
oxygen free radicals.
1.5. The anti-inflammatory glucocorticosteroids
1.5.1. Introduction
The glucocorticosteroids are potent anti-inflammatory drugs, widely used clinically
against many inflammatory disease states and able to suppress almost all phases of
inflammatory lesions in many animal models. Clearly no single mechanistic explanation
could suffice to account for all the biological effects of glucocorticosteroids. In light of
this it is not surprising that numerous anti- inflammatory actions have been attributed to
these drugs. This discussion of mechanisms of anti-inflammatory steroid action focuses
specifically on mechanisms underlying inhibition of the exudation of plasma proteins, as
well as leukocyte accumulation at sites of inflammation.
From the discussion of inflammatory mechanisms presented in the previous
sections there are clearly several potential target sites through which glucocorticosteroids
can attenuate oedema formation. The most likely sites of action are discussed below.
1.5.2. Inhibition of inflammatory mediator generation
There is an extensive literature on inhibition of mediator production or release by
glucocorticosteroids. In the last 10 years the inhibitory effect of steroids on the
generation of the pro-inflammatory arachidonic acid metabolites, prostaglandins and
leukotrienes, as well as on the generation of platelet-activating factor has been most
extensively investigated. During these investigations it became apparent that "anti
inflammatory" actions of steroids occur by means of the "classical pathway" of steroid
hormone action responsible for their biological effects, that is they are mediated by
cytosolic steroid receptors which interact with nuclear DNA resulting in the stimulation
of transcription of specific mRNA necessary for the synthesis of effector proteins (Baxter
& Tomlins, 1977; Buller & O’Malley, 1976; Chan & O’Malley, 1976; Wira & Munck,
46

1974; Munck & Leung, 1977).
Several early studies showed inhibition of prostaglandin production in the presence
of glucocorticoids (Lewis & Piper, 1975; Gryglewski et a l , 1975; Chang et a l, 1977).
Experiments reported by several groups found that this was not a consequence of cyclo-
oxygenase inhibition unlike the non-steroidal anti-inflammatory drugs, but suggested the
glucocorticoids were inhibiting arachidonic acid liberation from membrane phospholipids
(Gryglewski et a l , 1975; Hong & Levine, 1976; Nijkamp et a l , 1976). Further
experiments to pinpoint the site of glucocorticoid action revealed that steroids inhibited
the enzyme responsible for release of arachidonic acid, phospholipase A% (Blackwell et
a l , 1978). In many of these studies it was observed that the onset of the steroid effect
occurred after a time delay. Subsequently the inhibitory action of glucocorticoids on
prostaglandin production was shown to be mediated through receptor occupancy and was
prevented by actinomycin D and cycloheximide, indicating a dependence on RNA and
protein synthesis (Danon & Assouline, 1978; Flower & Blackwell, 1979; Russo-Marie
et a l , 1979; Di Rosa & Persico, 1979). At about the same time Tsurufuji and his
colleagues also demonstrated that the anti- inflammatory action of dexamethasone in rats
depended on receptor occupancy and unimpaired RNA and protein synthesis (Tsurufuji
et a l , 1979). More recently glucocorticoid receptor antagonists have been shown to
block the anti-inflammatory actions of these drugs providing supporting evidence for the
"classical pathway" of steroid action in acute inflammation (Peers et a l , 1988).
Three groups working independently produced evidence at about the same time
that glucocorticoids inhibit prostaglandin production as a result of the de novo synthesis
of a protein inhibitor of the enzyme phospholipase Ag which they christened
"macrocortin" derived from macrophages and perfused lungs
^Blackwell et a l , 1980; Flower & Blackwell, 1979), "lipomodulin" derived from
neutrophils (Hirata et a l, 1980) and ‘renocortin’ derived from renal cells (Cloix et a l,
1983; Russo-Marie & Duval, 1982). Subsequent rigorous examination of the conditions
of generation, molecular weights, antiphospholipase A2 and immunological properties of
these proteins led to the conclusion that they were all functionally identical and may be
active fragments of the same precursor, thus the family of proteins were renamed the
"lipocortins" (Di Rosa et a l , 1984). The lipocortins were defined as phospholipase Ag
(PLA2 ) inhibitory proteins the synthesis or secretion of which is stimulated by
glucocorticosteroids (Di Rosa et a l , 1984). The PLA2 enzyme can control the liberation
of arachidonic acid and thus its availability for conversion to prostaglandins and
47

thromboxanes via the cyclo-oxygenase enzyme, and leukotrienes via the lipoxygenase
pathway. Phospholipase Aj activation also controls the liberation of lyso-PAF, the
precursor of the potent lipid mediator PAF (Albert & Snyder, 1983). The significance
of these phospholipase A2 inhibitory proteins in vivo will depend upon the mediators
involved in particular types of inflammatory reactions, although it is also possible that
lipocortins may have other anti- inflammatory actions.
In several in vitro assays the lipocortins inhibited phospholipase Ag from pig
pancreas, snake and bee venoms (Hirata, 1981). Hirata (1983) suggested that a
stoichiometric complex formed between the enzyme and the inhibitor. He further
suggested that lipocortin may bind Ca " at the active site and that this action may underlie
its inhibitory ability (Hirata, 1981). Despite confirmation of the calcium binding
properties of lipocortin (Schlaepfer & Haigler, 1987) this cannot entirely explain the
inhibitory action since lipocortin also inhibits some calcium-independent phospholipases.
Human recombinant lipocortin (see later) has also been shown to inhibit PLAg activity
from a variety of sources in vitro (Wallner et a l , 1986) and it appears that a region
contained within residues 83-212 is important for this activity (Huang et a l, 1986).
Using the recombinant material. Peers et al (1987) have produced in vitro data
confirming the work of Hirata (1983) cited above. Furthermore Cirino et al (1989)
showed that recombinant lipocortin 1 inhibited phospholipase-induced oedema in vivo and
that the maximum inhibition occurred with stoichiometric quantities of lipocortin and
enzyme. Recently, however, it has been suggested that this ‘phospolipase inhibition’
could be due to lipocortin binding to substrate in a Ca^"^-dependent manner, thereby
blocking access to PLA2 , since the inhibition seen in some assays can be overcome by
increasing the substrate concentration (Aarsman et a l, 1987; Davidson et a l , 1987;
Schlaepfer & Haigler, 1987). Lipocortin does not, however, seem to bind to
phosphatidylcholine vesicles (Schlaepfer & Haigler, 1987) therefore this theory does not
explain the antiphospholipase action of lipocortin on this substrate (see Hirata 1981).
Furthermore, it has been reported that PLA2 activity can be inhibited by lipocortin under
conditions of substrate excess (Pepinsky et a l , 1988). Perhaps the simplest explanation,
accommodating all sets of data, is that lipocortin can have effects on both the enzyme and
its substrate. Although the exact mechanism whereby lipocortin inhibits phospholipase
A2 awaits resolution there is growing evidence supporting lipocortin as the candidate
mediating steroid inhibition of lipid mediator production. The anti-phospholipase proteins
mimic closely the action of glucocorticosteroids in the inhibition of eicosanoid synthesis,
48

although there is no lag phase and their effect is resistant to the action of protein/RNA
synthesis inhibitors. Purified lipocortins have been shown to inhibit the production by
leukocytes of cyclo-oxygenase (PGEj) and lipoxygenase (LTB4 ) products in vitro (Parente
et a l , 1984). Unlike the NSAIDs, but like the steroids themselves, the inhibitory effect
of these proteins was readily reversed by the addition of exogenous arachidonic acid,
highlighting the different sites of action of these two types of drugs. Lipocortin has also
been shown to inhibit thromboxane A% release from perfused guinea pig lung elicited in
response to histamine, leukotrienes and FMLP but not to Bk or AA, as with the steroids
(Blackwell et a l , 1980). The release of lyso-PAF from resident leukocytes in response
to opsonized zymosan was also inhibited by lipocortin (Parente & Flower, 1985). Fradin
et al (1988) demonstrated impaired production of LTB4 and PAF in rat neutrophils treated
with lipocortin. Purified lipocortins have also been shown to mimic the suppressive
action of glucocorticosteroids in vivo in models driven by the release of these lipid
mediators. Lipocortin inhibited oedema and leukocyte infiltration in the rat carrageenin-
induced pleurisy model (Blackwell et al., 1982). Earrasfa and Russo-Marie (1989)
demonstrated inhibition of eicosanoid generation and neutrophil accumulation by purified
lipocortin in a mouse model of acute inflammation. Parente et al (1984) and Hirata
(1983) demonstrated inhibition of carrageenin-induced rat paw oedema by lipocortin.
Again the effect was reversed by supra-injection of arachidonic acid. Interestingly the
lipocortin did not mimic the suppressive action of steroids against dextran-induced
oedema (Calignano et al., 1985).
Recently the protein now known as human lipocortin I has been cloned and
expressed in E.coli (Wallner et al., 1986). Recombinant material has been shown to
inhibit the synthesis of arachidonate metabolites in vitro from human umbilical vein
fragments (Cirino & Flower, 1987a) from rat peritoneal macrophages (Cirino & Flower,
1987b) and from isolated perfused guinea-pig lung with the same profile of action that
naturally occurring lipocortins and steroids show (Cirino et al., 1987).
Lipocortin I given locally has been shown to inhibit the eicosanoid but not the
amine-dependent component of carrageenin oedema in the rat paw (Cirino et al., 1988).
Lipocortin 1 also appeared to inhibit an eicosanoid-dependent component of compound
48/80-induced oedema. Lipocortin 1 was inactive as an inhibitor of oedema induced by
dextran (driven by the release of mast cell amines), PAF, Bk or serotonin, and thus tends
to show a similar profile of action to indomethacin (Cirino et a l , 1988). Only when
mast cell degranulation was induced by phospholipase Aj was lipocortin effective.
49

Because glucocorticoids are effective against all forms of oedema obviously an additional
mechanism must be invoked to explain how glucocorticoids bring about other types of
anti-inflammatory action.
Administration of a fragment of human recombinant lipocortin 1 has been shown
to mimick dexamethasone and prevent the pyrogenic actions of cytokines in rats (Carey
et al., 1990). Davidson et al (1991) were also able to demonstrate anti-pyretic effects
of recombinant human lipocortin 1 in rabbits. In addition, Carey et al (1990) were able
to reverse the anti-pyretic effects of dexamethasone following administration of a
polyclonal antisera raised to the lipocortin fragment. Likewise Duncan et al (1993) using
the same anti serum as Carey et al (1990) were able to show reversal of the local
antiinflammatory action of dexamethasone in the rat carrageenin oedema model. These
studies provide evidence suggesting that the antiinflammatory action of dexamethasone
is at least partially dependent upon the generation of lipocortin 1. In addition, the fact
that externally applied antisera can reverse the action of the glucocorticoids implies
strongly that the lipocortin target site can be extracellular. This is supported by the
numerous studies showing immediate antiinflammatory effects of externally applied
lipocortin on cells and in animals (Cirino et al., 1989; Carey et al., 1990; Davidson et
al., 1991; Errasfa & Russo-Marie, 1989). Interestingly, Goulding et al (1990b) have
identified specific lipocortin 1 binding molecules on the surface of peripheral blood
monocyte and polymorphonuclear leukocytes. Binding of lipocortin was found to be
through a calcium-dependent mechanism (Goulding et a l , 1990b).
Wallner et al (1986) showed that lipocortin 1 is a very polar molecule,
approximately a third of its amino acids are charged. The protein contains a single
potential glycosylation site, and two potential phosphorylation sites. This is significant
since it had already been demonstrated that the naturally occurring molecule could be
phosphorylated and that the phosphorylated form was inactive as a phospholipase inhibitor
(Hirata, 1981). However, a correlation between levels of phosphorylation of a
"lipocortin" and eicosanoid biosynthesis in vivo has yet to be demonstrated.
Huang et al (1986) observed two types of lipocortin in placental extracts. One
identical to the recombinant protein previously cloned (subsequently christened lipocortin
I and to which the work cited above using recombinant material refers) and a second
protein (lipocortin II) shows 50% sequence homology with lipocortin 1. Both proteins
inhibited phospholipase activity. More recently it has been found that lipocortins I and
II are members of a family of structurally related proteins and that these are identical to
50

other calcium and phospholipid binding proteins, including the calpactins and calelectrins
(Crompton et al., 1988; Sudhof et a l , 1988; Saris et al., 1986). It has been suggested
that lipocortin I is, in fact, the same protein as the substrate for the EGF receptor kinase,
p35 also termed calpactin II (Huang et al., 1986) and lipocortin II is the same protein as
the pp60'"= kinase substrate, p36, termed calpactin 1 (Kristensen et al., 1986; Huang et
al., 1986). The use of different names for the same members of this superfamily caused
considerable confusion. For this reason a common nomenclature was proposed
(Crumpton & Dedman, 1990) using the term annexin. Lipocortin 1 or calpactin II was
named annexin 1 and lipocortin II or calpactin 1 was named annexin II (Crumpton &
Dedman, 1990). At least 10 members of this family have been characterised to date, all
sharing approximately 50% sequence identity (Pepinsky et al., 1988). Most conserved
is a 70 amino acid repeat unit of which the proteins contain multiple copies and which
is thought to be responsible for calcium and phospholipid binding (Kretsinger & Creutz,
1986; Saris et al., 1986). Distinct form the core structure of repeat units is a short N-
terminal segment that is unique to each protein and may confer distinct biological
activities (Glenney & Zokas, 1988). Lipocortin lacks a leader or signal sequence usually
required for proteins to be secreted by conventional means. However, some secretory
fluids have been found to contain high concentrations of lipocortin (Christmas et al.,
1991).
Although, as outlined above, biological evidence has suggested that lipocortin 1
is anti-inflammatory and that members of the lipocortin family are produced by
glucocorticoids, whether lipocortins are actually induced or regulated by
glucocorticosteroids, especially in a way that correlates with antiinflammatory activity,
has proved to be a more complex question than expected and is surrounded by much
controversy. Glucocorticoids have been shown to induce lipocortin 1 mRNA in
leukocytes (Wallner et al., 1986). However, no accompanying increase in protein was
seen. Adrenalectomy lead to a fall in lipocortin mRNA and protein in animals
(Vishwanath et al., 1992) and conversely administration of hydrocortisone in vivo in man
lead to induction of intracellular and cell surface lipocortin 1 in monocytes, but not
neutrophils (Goulding et a l , 1990a). Furthermore, Peers et al (1993) found that both
lipocortin 1 and II, but not the closely related species lipocortin 5, were elevated in rat
mixed peritoneal cells by acute glucocorticoid treatment. Ambrose & Hunninghake
(1990) found corticosteroids increased lipocortin 1 in alveolar epithelial cells. However,
in contrast Northup et al (1988) were unable to detect an increase in the synthesis/release
51

of lipocortin 1 protein in peritoneal macrophages following glucocorticoid treatment of
the cells in vitro. Also Bienkowski et al (1989) and Isacke et al (1989) failed to detect
a steroid-induced expression of lipocortin at both the protein and mRNA level in the
U937 cell line even though glucocorticoids inhibited eicosanoid formation in these cells
(Bienkowski et a l , 1989). Likewise Bronnegard et al (1988) reported that lipocortin 1
is not inducible at the mRNA level by dexamethasone in several different cell types,
including human macrophages. Furthermore, work by Hullin et al (1989) on human
endothelial cells showed that, under conditions in which dexamethasone treatment led to
a decrease in PGI2 production, no change in lipocortin level was observed. Such
discrepancies might reflect differences in cell responses to glucocorticoids as well as
variations in the experimental procedure. Goulding et al (1990a) found that if cells were
first separated from blood before exposure to steroids they were unable to detect
induction of lipocortin 1. The capacity of cells to respond well to glucocorticoids may
be lost or in some way impaired following long term culture. Solito et al (1991)
suggested that the state of differentiation of the cell may have a bearing on steroid
induction of lipocortin and Phillips et al (1989) found the presence of growth factors is
required for steroid induction. Further clarification is needed.
Another line of evidence pointing indirectly to the importance of lipocortin as a
mediator of steroid action is the detection of autoantibodies to lipocortin in the plasma Hirata et al., 1981; Podgorsky et al., 1987; Goulding et a i, 19W; 1992
of patients with chronic inflammatory diseases ( ■ ). In certain
groups of patients oral steroids raised the antibody titre, a high titre correlated strongly
with the clinical phenomenon of ‘steroid-resistance’ (Podgorski et al., 1987).
Interestingly a deficiency in the expression of lipocortin receptors has recently been
demonstrated in active rheumatoid arthritis (Goulding et al., 1992).
Thus, in summary, there is a body of experimental evidence suggesting that
glucocorticoids are able to inhibit the generation of pro-inflammatory arachidonic acid
metabolites by inducing the synthesis of annexins (lipocortins) which can act to inhibit
phospholipase A;. As outlined in this section this mechanism of inhibition has not been
found to be consistent with all experimental evidence obtained in different models. The
subject is still controversial and awaits some clarification. Furthermore, there is evidence
indicating an inhibitory action of glucocorticoids at a later stage of arachidonic acid
metabolism than phospholipase A%. It is becoming apparent that glucocorticoids may
modulate transcription of the cyclooxygenase gene. Dexamethasone has been shown to
block the stimulus-induced synthesis of cyclo-oxygenase in vitro in human dermal
52

fibroblasts (Raz et al., 1989) and vascular smooth muscle cells (Bailey et al., 1988),
where corticosteroids appeared to suppress cyclo-oxygenase mRNA (Bailey et al., 1988).
Furthermore, Masferrer et al (1990) demonstrated dexamethasone inhibition of LPS-
induced cyclooxygenase synthesis in vivo in mouse macrophages. Thus it seems
glucocorticoids, in some situations in a variety of cells are able to exert a more complex
effect on arachidonic acid metabolism than a generalised inhibition of PLAg which seems
to involve effects on induction of cyclo-oxygenase. This inducible cylcooxygenase is now
known to be a distinct isoform "CO-2".
In addition to their effects on the generation of arachidonic acid metabolites anti
inflammatory steroids have also been reported to inhibit histamine release, the major
sources of which are the mast cells and basophils. Early studies demonstrated that mast
cell histamine release was inhibited by steroids, however, high concentrations of drug
were required (Lewis & Whittle, 1977; Dembinska-Kiec et al., 1978). More recent
studies, using lower concentrations of dexamethasone demonstrated no inhibition of
mediator release from purified human lung mast cells (Schleimer et al., 1983).
However, in the guinea-pig dexamethasone treatment did inhibit antigen-induced release
of lung mast cell mediators (Schleimer et al., 1987). Furthermore, histamine release
from human basophils was readily inhibited with low concentrations of glucocorticoids Schleimer a/.,
(1981; 1982). A protein, distinct from lipocortins and with an apparent molecular weight
of lOOkDa has been found in the peritoneal lavage fluid of dexamethasone pretreated rats
(Camuccio et al., 1987). This protein, like corticosteroids, but unlike lipocortins
inhibited dextran oedema in the rat when injected either locally or systemically
(Camuccio et al., 1987; lalenti et al., 1990) and has been named vasocortin since it
modulates vascular permeability and is induced by corticosteroids. Vasocortin is distinct
from lipocortins as its biological activity is not correlated with PLA2 inhibition and is lost
after heating at 70°C for 5min, unlike that of lipocortins. Vasocortin-like proteins with
an apparent molecular weight less than 40kDa have been found in vascular tissues
following glucocorticoid treatment (Camuccio et al., 1989b). Further studies revealed
that vasocortin selectively inhibited histamine release from rat peritoneal cells induced by
dextran or concanavalin A but not that induced by calcium ionophore A23187 or
compound 48/80 (Camuccio et al., 1989a) suggesting that vasocortin may be the protein
responsible for the glucocorticoid inhibition of histamine release. Such an action is also
consistent with the ability of vasocortin to inhibit dextran oedema since this inflammatory
reaction has been shown to be mainly, if not exclusively, brought about by the release
53

of histamine and 5-hydroxy tryptamine (Di Rosa & Willoughby, 1971). Preliminary data
has shown that the inhibition of histamine release may depend on the ability of vasocortin
to prevent the Ca^ influx into the cells (lalenti & Di Rosa, 1990).
Glucocorticoids have also been shown to inhibit the generation of cytokines IL-1
and IL- 8 which cause neutrophil recruitment (Knusden et al., 1987; Watson et al., 1987;
Bochner et al., 1987a).
1.5.3 Inhibition of inflammatory mediator action
In addition to suppressing the generation of permeability-increasing or vasodilator
mediators as described above, there is also evidence that glucocorticoids can inhibit the
action of permeability increasing mediators in vivo in models unlikely to be mediated by
eicosanoid synthesis. Dexamethasone inhibited oedema formation induced by bradykinin
and 5-hydroxytryptamine (5HT) in a model of mouse foot pad oedema (Tsurufuji et al.,
1979). In contrast, these oedema responses were unaffected by indomethacin indicating
their independence of local prostaglandin production and thus suggesting the inhibitory
effect of dexamethasone was unlikely to be due to phospholipase Aj inhibition. Further,
the inhibitory effect was protein synthesis dependent indicating a requirement for the
synthesis of an endogenous anti-inflammatory protein (Tsurufuji et al., 1979). These
observations have been supported by several studies using different animal species and
an array of inflammatory mediators. Glucocorticoids inhibited histamine-induced oedema
in rat skin and mouse ear (Church & Miller, 1978) and plasma protein leakage induced
by exogenous histamine, Bk, PAP and leukotriene C4 (LTC4 ) in the hamster cheek pouch
(Bjork et a l , 1985; Svensjo & Roempke, 1985). It has been suggested that these
observations are unlikely to be a result of inhibition of mediator generation and that
dexamethasone or more likely a steroid induced protein(s) may inhibit oedema formation
by a direct effect on the endothelium rendering it less responsive to the action of
mediators of increased microvascular permeability (Tsurufuji et al., 1979). The
involvement of lipocortin in mediating these actions of dexamethasone seems unlikely as
recombinant lipocortin 1 was ineffective against oedema induced by 5HT, Bk and PAP
in the rat paw (Cirino et a l , 1989). Glucocorticoids have also been shown to inhibit
neutrophil-dependent oedema induced by exogenous chemotactic mediators (Poster &
McCormick, 1985; Griffiths & Blackham, 1988; Peers & Flower, 1991). Furthermore,
Poster & McCormick 1985 demonstrated dexamethasone-induced inhibition of neutrophil
accumulation in response to LTB4 . It remains unclear whether the primary target of
54

steroids in these studies is the endothelial cell, the neutrophil, or both (see chapter 4).
1.6 Aims of study
1. To characterise neutrophil-dependent oedema formation induced by FMLP and
structurally-related formyl peptides in rabbit skin.
2. To further investigate FMLP-induced oedema formation in rabbit skin using a
variety of modulatory agents, with a view to better understanding mechanisms of
neutrophil-dependent oedema formation.
3 To investigate the effect of glucocorticosteroids on vascular responses in rabbit
skin induced by neutrophil-dependent and direct-acting mediators of increased
microvascular permeability. The effect of steroids on three aspects of the
inflammatory response will be investigated; blood flow, vascular permeabilityand
neutrophil accumulation. By investigating action of steroids administered by
different routes the possible sites of action of steroids in inhibiting plasma protein
leakage and neutrophil accumulation in vivo will be investigated.
4. To further characterise the inflammatory response to zymosan in the rabbit
peritoneal cavity specifically the involvement of LTB4. To investigate the role of
circulating neutrophils in the development of the inflammatory response to
intraperitoneal zymosan.
5. To investigate the effect of glucocorticosteroids on the inflammatory response
induced by zymosan in the peritoneal cavity. Using a more complex model of
inflammation the possible contribution of steroid inhibition of both inflammatory
mediator generation and mediator action in vivo will be investigated.
55

CHAPTER 2 MATERIALS AND METHODS
2.1 In vivo measurement of inflammatory activity in rabbit skin: the rabbit skin
bioassay
Rabbit dorsal skin allows the assessment and modulation of the inflammatory
reaction induced by a variety of stimuli in a single experiment. Local oedema formation
(plasma protein exudation) and neutrophil accumulation can be monitored simultaneously
and changes in local blood flow can also be investigated, all in a quantitative manner in
the same versatile model. Modulating agents can be injected intradermally or
intravenously allowing thorough investigation of both the underlying mechanism of the
inflammatory reaction and the mode of action of the drug under test.
2.1.1 Measurement of vascular permeability in rabbit skin
Increased vascular permeability was assessed in rabbit skin by the measurement
of the dermal accumulation of intravenously administered ‘ ^I-human serum albumin
(HSA) (Wedmore & Williams, 1981a). Male, New Zealand White rabbits (2.0-3.5kg)
were anaesthetised with sodium pentobarbitone (Sagatal, 30mg/kg i.v., with maintenance
doses as required). The dorsal skin was then shaved and * I-HSA (5^Ci in 1ml sterile
saline) was injected intravenously. To allow a visual record of the inflammatory reaction
and aid accurate excision of injection sites Evans blue dye (2.5% w/v in sterile saline)
was also administered intravenously. After 15min test agents were injected intradermally
in 100/d volumes via a 27 gauge needle. Each agent was injected into 6 skin sites
distributed over the dorsal skin according to a balanced site plan. At the end of the
accumulation period animals were sacrificed by i.v. administration of an overdose of
pentobarbitone. The dorsal skin was removed and blood expressed from major vessels
before individual treatment sites were excised with a 17mm diameter metal punch. The
radioactivity associated with each skin site, together with 3x1 ml plasma samples obtained
by cardiac puncture at the time of death was measured in a manual multiwell gamma
counter (1260 multigamma 2; IKB-Wallac Ltd). Samples were counted for 30 seconds
over the energy range 10 kev-500 kev. All counts were automatically corrected for
background andcross over and detectors normalised for each radioisotope.
Oedema formation was quantitated as /il plasma per skin site by comparing the
^^^I-counts in each site with the *^ I-counts in 1/xl plasma. For each test agent results are
expressed as mean ± s.e.mean /il plasma/site for n animals, each animal respresenting
56

the mean value from 6 replicate skin site measurements.
2.1.2 Measurement of neutrophil accumulation in rabbit skin
In some experiments neutrophil accumulation, measured as the local accumulation
of i.v. injected "^In-labelled neutrophils was simultaneously monitored together with
oedema formation in response to i.d. test agents. In such experiments "^In-rabbit
neutrophils (50-200/iCi per 2-5x10^ cells) in 3ml autologous plasma (see section 2.2)
were injected intravenously at the same time as 15^Ci *^I-HSA. To allow detection of
both *“ I- and " ‘In radioisotopes in the same samples each isotope was counted separately
in limited energy ranges ("^I lO-lOOkeV; ‘"In 100-5(X) keV) for 60 seconds. The counts
were automatically corrected for spill up (0.03%) and spill down (25.03%) in a manual
multiwell gamma counter (1260 multigamma 2; LKB-Wallac, Ltd).
Neutrophil accumulation was expressed as the number of " ‘In-neutrophils per skin
site by comparing the " ‘In-counts in each skin site with the " ‘In-counts per neutrophil
(see section 2.2). For each test agent results are expressed as mean ± s.e.mean " ‘In
neutrophils per skin site for n animals (each animal representing the mean value of 6
replicates).
2.1.3 Measurement of local blood flow in rabbit skin
Changes in rabbit skin blood flow were monitored using a multiple site ‘% e
clearance technique (Williams, 1979). The vasodilator prostaglandin Eg (PGEg) was
mixed with a solution of ‘” Xe in saline (5-10/xCi/injection) and rapidly injected i.d.
(0.1ml volumes) into the shaved dorsal skin of New Zealand White rabbits (anaesthetised
and prepared as described in section 2.1.1). Intradermal injections were given in
random-block order according to a predetermined balanced site plan with 6 replicates for
each treatment per animal. ‘” Xe in saline was injected as control. After a 15 minute
clearance period animals were killed with an overdose of i.v. sodium pentobarbitone, the
dorsal skin excised and injection sites removed with a 16mm diameter punch. Skin
samples and 0 . 1 ml aliquots of injection fluids, under paraffin oil ( 1 ml) in sealed tubes
(to prevent xenon loss), were counted limmediàtely in an automatic gamma-counter (LKB
Wallac 1260 Multigamma II). Samples were counted for 30 seconds over the energy
range 10keV-115keV. The change in local blood flow produced by i.d. injection of test
agents was calculated as a percentage increase of ‘% e washout over that in saline-
injected control sites using the following equation:
57

% change in blood flow = (In ‘ Xe.-In *” Xe test)
----------------------------------------------------- xlOO
(In ‘” Xej-In *” Xe.)
where: In‘” Xe, = natural log (In) cpm of saline injected sites
In*” Xe test = In cpm of sites injected with test agents
In*” Xej = In cpm of 0.1ml aliquots of injected xenon solution
Results are expressed as mean % change in blood flow compared with
saline-treated skin ± SEM for n separate experiments.
2.2 Preparation of “ ^In-neutrophils
Neutrophils isolated from the blood of a donor rabbit were labelled with " ‘In and
injected intravenously into recipient animals where their accumulation in the skin was
monitored. The swift preparation of ‘"In-neutrophils was carried out under sterile
conditions (Nourshargh et al., 1989; Rampart & Williams, 1988).
Male, specific pathogen free. New Zealand White rabbits were anaesthetised with
sodium pentobarbitone (30mg/kg i.v.). A carotid artery was exposed and cannulated with
a 16 gauge abocath catheter (venisystems). A total of 72ml blood was collected equally
into eight 50ml falcon tubes, each containing 1ml of acid citrate dextrose (160mM
disodium hydrogen citrate + 0.28M glucose in sterile water). A further 13ml blood was
collected into tri-sodium citrate (3.5% in sterile water), and used to produce autologous
citrated plasma (centrifuged twice at 3000g; 7min 20°C). To each falcon tube of blood
15ml hydroxyethyl starch (Hespan, 3% final concentration) was added to facilitate
sedimentation of the majority of red blood cells. Tubes were centrifuged (25g; 15 min
20°C) to further aid red blood cell sedimentation. The leukocyte-rich supernatant was
removed and centrifuged (300g; 7min at 20®C) to form a leukocyte pellet and platelet-
rich plasma supernatant. The leukocyte pellets were resuspended gently in a total of 5ml
autologous citrated plasma. Meanwhile autologous platelet poor plasma (PPP) was
prepared from the platelet rich plasma (by centifugation twice; 2 0 0 0 g for 1 0 min,
followed by 2500g for lOmin).
Neutrophils were separated from mononuclear cells and remaining erythrocytes
using a two-layer discontinuous percoll-plasma gradient. Gradients were prepared by
carefully layering 3ml of 50% percoll in PPP over 3ml of 70% percoll in plasma. The
58

cell suspension was layered carefully onto two such gradients and centrifuged (400g;
17min 20°C). Neutrophils were harvested from the 70%/50% percoll interface in
1.0-1.4ml. A 10/xl sample of cell suspension recovered from the percoll/plasma gradient
was diluted 2000-fold in azide-free balanced electrolyte solution (Isoton 2). The total
number of red and white blood cells was measured in a Coulter Counter (Model M2,
Coulter Electronics Ltd). Erythrocytes were then lysed using approximately 20^1
Zaponin (potassium cyanide in Isoton 2) and the number of white blood cells measured.
From this value the total number of neutrophils recovered from the gradient was
determined.
Neutrophils (approximately 3.5-5.0xl0^cells in l-2ml volume) were incubated with
"^InClg (50-200/xCi in 10-15/xl of 0.04m hydrochloric acid) chelated to
2-mercaptopyridine-N-oxide (4/xg in 100/xl phosphate buffered saline; pH7.5) for 15min
at room temperature. Labelled leukocytes were then washed three times in 10ml of PPP
(400g; 7 minutes at 20®C) and finally resuspended in 3ml of PPP and injected, via a 19
gauge butterfly into the ear vein of recipient animals.
The purity of the resulting neutrophil suspension was greater than 95% as
determined by light microscopy of Grunwald and Giemsa stained films (see section 2.5)
and viability was greater than 95%, as judged by trypan blue exclusion.
A 10 1 sample was taken from the 3ml cell suspension just prior to i.v. injection.
This sample was diluted 100-fold and the radioactivity in counts per minute (CPM)
determined. From this the total number of neutrophil-bound " ‘In counts was calculated
as:
cpm per neutrophil = cpm injected (ie. cpm in 10/xlx300)
No of neutrophils injected
A 10/xl sample was removed from the supernatant following each " ‘In-neutrophil
wash step. Each sample was diluted l(X)-fold and counted in a gamma-counter. From
these, and the total cpm injected, the total cpm used during the labelling procedure was
calculated as:
total cpm = cpm injected 4- cpm in washes 14-2-H3
59

The percentage binding of " ‘In of the neutrophils in vitro was then calculated as;
binding = cell associated after wash 3 x 100
total cpm
From the blood sample taken by cardiac puncture just prior to death (see section
2.1.1) three 1ml samples were centrifuged (78(X)g; 5 minutes) and the resulting plasma
was separated from the corresponding cell pellets. The volume of all tubes was made up
to 1 ml and the cpm measured in a gamma counter.
The cell-bound and the free (plasma) cpm were used to calculate the percentage
free " ‘In in the blood in the following way;
% free " ‘In = cpm in plasma xlOO
cpm in plasma + cell bound cpm
Data from experiments where a high (>10%) level of free circulating " ‘In was
found were discarded.
The percentage circulating " ‘In-neutrophils at the end of experiments was
estimated as follows:
% circ. cells = cpm in 1ml blood x total blood volume xl(X3
cpm injected
where the toal blood volume was calculated as 8 .0 % of the body weight.
2.3 Experimental protocols measuring oedema formation or oedema formation
together with neutrophil accumulation using the rabbit skin bioassav
In the standard protocol used for the investigation of local plasma protein leakage
and, where appropriate, the local accumulation of " ‘In-neutrophils, ‘ 1 -HSA and
‘"In-neutrophils were injected intravenously 15 minutes prior to the intradermal injection
of agents under test, coinjected with a vasodilator prostaglandin (PGEj). Control skin
60

sites were injected with sterile saline or PBS. The accumulation of labels in skin sites
was then measured over a 30 minute in vivo test period. Oedema formation and
"Un-neutrophil accumulation in response to test agents was compared to the baseline
response to saline and prostaglandin Eg. This protocol was adapted according to
experimental requirements as follows:
2.3.1 Investigation of the mechanisms of oedema formation
In order to investigate mechanisms of oedema formation induced by inflammatory
mediators in the absence of a vasodilator the accumulation of i.v. *^I-HSA into skin sites
was measured over 4 hours in addition to 30 minutes in the same animals. Skin sites
were injected i.d. 15 minutes after i.v. "U-HSA and separate sites were then injected 3'A
hours later, 30 minutes prior to termination of the experiment (see section 3.2.3).
The involvement of the generation of various inflammatory mediators to oedema
responses was investigated by pre-treating skin sites i.d. with various blocking agents,
for example, indomethacin and mepyramine maleate for 1 0 minutes prior to the injection
of test agents into the pretreated sites (see sections 3.2.3 and 3.2.4). Control sites were
pretreated intradermally with saline. The activity of the various blocking agents was
confirmed by i.d. injection of the appropriate inflammatory mediator into pretreated. sites.
2.3.2 The effect of intravenously administered agents on oedema formation induced
bv inflammatorv mediators
A "two-stage" protocol was adopted to test the action of various intravenously
administered agents on oedema formation induced by i.d. inflammatory agents (see
sections 3.2.5, 3.2.6 and 3.2.7). These experiments were designed such that each animal
served as its own control. Animals were injected i.v. with "U-HSA, followed by i.d.
injection of inflammatory mediators in the presence of PGBg. The accumulation of label
was allowed to proceed for 30min before i.v. injection of agent under investigation,
followed by further i.d. injections of the same inflammatory mediators into a second set
of injection sites. Responses were allowed to proceed for a further 30 minutes. Oedema
responses over the first 30 minutes in the absence of i.v. test agent were then compared
with those obtained to the same inflammatory mediators over the second period of 30
minutes in the presence of the i.v. test agent. Control animals received an i.v. injection
of sterile saline prior to the second series of i.d. injections.
61

2.3.3 Measurement of the duration of action of inflammatory mediators
In order to measure the duration of the permeability-increasing activity of
inflammatory mediators animals were injected i.d. with mediators of increased
microvascular permeability at various times before an i.v. injection of *^I-HSA and
Evans blue dye at t=0. Prostaglandin E% (3xlO'*°mol per site) was then injected into all
skin sites previously injected with permeability-increasing mediators. Any oedema
increasing activity remaining in skin sites would then be potentiated by vasodilator and
was assessed after 30 minutes. To investigate a possible effect of colchicine on the
duration of permeability increasing action of inflammatory mediators colchicine was
injected i.v. at the time of administration of ^^ I-HSA, control animals received i.v.
saline. The duration of the permeability increasing action of colchicine treated animals
was compared with that in saline treated animals (see section 3.2.8).
2.3.4 The effect of local dexamethasone on inflammatorv responses
In experiments where the effect of local dexamethasone was investigated (see
section 4.2.1 4- 4.2.2) skin sites were pretreated i.d. with dexamethasone (2xl0‘*°mol per
site) or indomethacin (10'*mol per site) for various times (15, 90 or 240 minutes) before
the i.v. administration of " ‘In-neutrophils, ‘ ^I-HSA and Evans blue dye. Test agents
were then injected i.d. together with PGE; into the pretreated sites. Control sites were
pretreated i.d. with saline. The accumulation of labels in skin sites was then measured
over a 30 minute in vivo test period.
2.3.5 The effect of systemic dexamethasone on inflammatorv responses
The experiments described in section 4.2.4 were designed to evaluate the effect
of systemic treatment of recipient rabbits with dexamethasone. Washed labelled
neutrophils separated from the blood of a donor animal were divided equally for i.v.
injection, together with ‘ I-HSA into two recipient animals, one test and one control
rabbit. Test recipient rabbits were treated systemically with dexamethasone (3mg/kg) 4h
before i.v. injection of labelled cells and ‘ ^I-albumin. Control animals received an equal
volume of saline. All recipient animals received i.d. inflammatory mediators and the
accumulation of labels into skin sites was allowed to proceed for 30 minutes.
2.3.6 The effect of dexamethasone on the neutrophil in vivo
In order to investigate the effect of dexamethasone specifically on the neutrophil
62

in vivo the experiments described in sections 4.2.6 and 4.2.7 were designed. Pairs of
neutrophil donor rabbits were treated i.v. with dexamethasone (3 mg/kg) or an equal
volume of saline. 4h later blood from each pair was collected, neutrophils isolated and
"^In-labelled in parallel as described in section 2.2. "^In-cells from treated and control
donor animals were then injected i.v. into two untreated recipient animals, together with
*^I-albumin. Cell numbers were adjusted if necessary so that each pair of recipient
animals received the same number of labelled neutrophils. Neutrophil accumulation and
oedema formation were measured in skin sites over 30 minutes.
All procedures for each pair of saline and dexamethasone treated rabbits were
carried out in parallel.
2,3.7 The effect of dexamethasone on blood flow
The effect of dexamethasone on blood flow was assessed by pretreating skin sites
locally with dexamethasone (80ng/site, equivalent to 2 x 1 0 "*° moles per site) or saline as
a control for 4h prior to re-injection of the sites with doses of PGEg or saline mixed with
"^Xe as described in section 2.1.3.
2.4 Preparation of zymosan-activated plasma
Zymosan activated plasma was used as a source of C5a des Arg. New Zealand
White rabbits were anaesthetised with sodium pentobarbitone, cannulated via the carotid
artery and bled into heparinised tubes (lOU/ml). Rabbit plasma was prepared and
incubated for 30 minutes at 37°C with zymosan 1 mg/ml under shaking conditions.
Zymosan was removed by centrifugation (2500g for 2x10 minutes). Before use the
zymosan activated plasma was desalted by passing over a Sephadex G-25M (PD-10)
column. Activated plasma was then stored in aliquots of 1ml at -20®C. The C5a des
Arg content of activated plasma was measured by radioimmunoassay by Dr P Jose (Jose
et al 1983) and was approximately 3xl0‘"moles/0.1ml.
2.5 Staining procedure for leukocytes
Blood smears were air dried for at least 1 hour and then fixed in methanol (5
minutes). The slides were directly transferred to 50% May Grunwald stain and then 10%
Giemsa, both for 5 minutes. After washing in tap water the slide was ready for
differential cell counting. From the Coulter counter analysis (white leukocyte count see
section 2 .2 ) and the differential count, the number of neutrophils per ml of whole blood
63

was calculated.
2.6 The generation of inflammatory exudate; the rabbit peritoneal cavity model
2.6.1 Generation and recovery of peritoneal exudate fluid
To investigate the generation of inflammatory mediators in vivo large quantities
of easily recoverable inflammatory material must be obtained. An acute inflammatory
response was induced in the peritoneal cavity of rabbits, the accumulated inflammatory
material was then drawn off from the cavity for identification of inflammatory mediators
(Forrest et a l , 1986).
Male, specific pathogen free, New Zealand white rabbits (2.5-4.0kg) were
anaesthetised with an intravenous injection of Sagatal, 30mg/kg initially followed by
maintenance doses as required. Evans blue dye (2.5% w/v in sterile saline, 0.22/xM
Millex sterile filtered, 0.5ml/kg) was injected intraveously to monitor plasma protein
extravasation into the peritoneal cavity. The abdomen was shaved and a sterile 14-gauge
concentric polythene cannula and steel needle (Arterioveine 11720) was inserted into the
peritoneum. The needle was withdrawn and the cannula was held in place with surgical
adhesive tape. Approximately 1ml of sterile pyrogen free saline was flushed through the
cannula and drawn back to ensure the needle had not penetrated the intestines. Any
animals where a coloured discharge was visible were discarded. A suspension of
zymosan A (lOmg/ml) in 50ml sterile pyrogen free saline (prepared as described in
section 2.6.2) prewarmed to 37®C was injected into the peritoneum via the cannula.
Control rabbits were injected with 50ml sterile saline at 37°C. The end of the cannula
was stoppered and the peritoneum gently massaged to disperse the injected liquid.
At various times thereafter (5 mintues, up to 8 h) the stopper was removed and
1 .8 ml of exudate was withdrawn from the cavity, via the catheter, into 2 0 0 /d of freshly
prepared "cocktail" containing lOmM sodium EDTA pH7.2, 1% azide, 1 mg/ml
indomethacin, 1 mg/ml phenidone and 1 mg/ml phenylmethyl-sulphonyl-fluoride (PMSF),
final concentrations respectively to prevent in vitro generation of inflammatory mediators.
Following immediate mixing an aliquot of the exudate was removed for cell counting (see
section 2.6.3) and the remainder of the sample was immediately centrifuged (1950g;
lOmin at 4°C) to remove accumulated leukocytes and particulate matter. A 100/d aliquot
of the cell-free supernatant was removed for spectrophotometric analysis of blue dye
concentration (see section 2.6.3) and the remainder of the supernatant was immediately
64

stored at -20°C for subsequent determination of inflammatory mediator concentration by
radioimmunoassay (see section 2.6.4).
2.6.2 Preparation of zvmosan for intraperitoneal injection
Zymosan A (from saccharomyces cerevisiae) which had previously been stored
as a solid at 0-5 °C was used in all experiments. Suspensions of zymosan^Omg/ml) were
prepared in 50ml aliquots in sterile pyrogen free saline containing 40^M polymixin B
sulphate (in order to negate the effects of any endotoxin present, (Collins et al., 1991a).
Zymosan suspensions were mixed for 1 hour at 37®C before centrifugation (1200g;
lOmin). Zymosan pellets were then resuspended in sterile pyrogen free saline, pooled
under sterile conditions and aliquotted into sterile falcon tubes (50ml/tube). After
centrifugation (1200g; lOmin) zymosan pellets were stored at -20®C until used. Pellets
were thawed and resuspended in 50ml of sterile pyrogen free saline (37°C) before i.p.
injection.
2.6.3 Measurement of the inflammatorv reaction in the peritoneal cavity
Plasma protein extravasation into the peritoneal cavity was monitored as the
intraperitoneal accumulation of intravenously-injected Evans blue dye (which binds to
plasma albumin) using spectrophotometric analysis at 620nm. Using the absorbance of
plasma, obtained from blood removed at the end of the experiment, the exudate: plasma
blue dye ratio was determined.
The number of polymorphonuclear leukocytes in peritoneal exudate samples was
determined by diluting the exudate in an equal volume of gentian violet ( 1 ml 1 % crystal
violet; 1.5ml glacial acetic acid; 97.5ml distilled water) and counting the PMNLs in an
improved Neubauer chamber microscopically.
2.6.4 Determmation of inflammatorv mediator concentration in peritoneal exudate
samples
Levels of immunoreactive leukotriene B4 (i.r. LTB4 ), immunoreactive
thromboxane Bj (i.r.TxBz), immunoreactive prostaglandin Ej (i.r.PGE2 ) and
immunoreactive 6 -oxo-prostaglandin F,„ (i.r. 6 -0 x0 PGP,J were measured in cell-free
exudate samples by specific radioimmunoassays (RIA) developed and fully characterised
by Imperial Chemical Industries (Carey & Border, 1986; Border & Carey, 1984; Carey
era/., 1989).
65

Samples and standards were diluted when appropriate in the relevant RIA buffer
(0.04M sodium dihydrogen phosphate in 0.9% sodium chloride containing 0.1% bovine
gamma globulin, pH7.4). They were prepared in duplicate in glass tubes in a final
volume of 0.1ml and incubated for 16-24h at 4°C with 0.1ml pH] ligand (6x10 dpM)
and 0.2ml of appropriately diluted specific antibody in a competitive binding assay. The
dilution of each specific antibody used was such that in the absence of authentic material,
40-50% of the pH] ligand was bound to antibody, with non-specific binding less than 5%
(i.e. binding in the absence of antibody). Control samples of total radioactivity added,
total binding of label in the absence of cold material, and non-specific binding in the
absence of antibody were also produced. Free and antibody bound ligand were separated
by addition of 0.2ml of dextran coated charcoal (0.5% (w/v) dextran T70 and 1% (w/v)
charcoal) for 15min at 4°C, followed by centrifugation (1500g; 15min 4®C). The
supernatant (0.45ml) was removed and radioactivity determined by scintillation counting
using an LKB Rack Beta 1216 counter. The concentration of material in unknown
samples was read from the standard curve, dilutions of unknown samples were used such
that the level of radiolabelled ligand displaced was a value on the linear portion of the
standard curve. Results are expressed as ng material/ml exudate. The minimum
detectable levels of material are approximately 5pg/ml.
2.6.5 Confirmation of immunoreactive LTB identity
Authentication of immunoreactive LTB4 detected in inflammatory exudates was
kindly carried out by Dr D Masters at Imperial Chemical Industries using reversed-phase
HPLC (Rp HPLC) fractionation. All solvents used were of the highest purity available.
2.6.5.1 Separation of LTB^
Prior to Rp HPLC LTB4 present in inflammatory exudate was isolated on Bond-
Elut cyanopropyl mini-columns (C-18 columns). Mini-columns were preconditioned with
two 0.5ml aliquots of methanol followed by 2x0.5ml of 1% methanol. Samples of
inflammatory exudate were adjusted to pH5.6 with IM citric acid (25% v/v) and loaded
onto the C-18 columns under nitrogen pressure. Samples were eluted using 3x0.5ml of
methanol, and lOO/il of each was immediately analysed by Rp HPLC. Using this mini
column separation procedure overall recoveries of LTB4 of approximately 80-90% have
been obtained.
66

2.6.S.2 Rp HPLC
Rp HPLC was carried out using a Schimadzu LC320 pump and UV detector with
a 5(1 spherisorb OD52 column (24x0.46cm) and linear gradient of 70-95% methanol in
0.1% (v/v) glacial acetic acid and water, pH5.6 as the mobile phase at a flow rate of
2ml/min. The column was calibrated using a "cocktail" of radiolabelled pH] LTB4 ,
pH]12-HETE and pH] arachidonic acid in 70% methanol, of which 10^1 was loaded onto
the column and 70, 0.2min fractions were collected. Samples (0.1ml) were loaded at a
flow rate of 0.2ml/min for 4 minutes and were similarly eluted as 40, 0.35min fractions
(700^1) at a flow rate of 2ml/min (ie. over 14 minutes, as for standards). Fractions
(standards and samples) were evaporated to dryness and immediately reconstituted in
sample buffer prior to scintillation counting using an LKB Rack Beta 1216 counter and
RIA (see Section 2.12). The retention time of immunoreactivity in exudate samples was
then compared with that of the authentic pH] LTB4 .
2.7 Experimental protocols using the peritoneal cavitv model
The inflammatory reaction in response to intraperitoneal zymosan was investigated
further in two separate series of experiments using adapted versions of the protocol
described in section 2 .6 . 1 ,
2.7.1 The effect of depletion of circulating neutrophils on the inflammatorv
response to i.p. zvmosan
To determine the possible contribution of accumulating neutrophils to the
inflammatory response to zymosan, animals were depleted of circulating neutrophils using
mustine hydrochloride (see section 2 .8 ) prior to challenge with i.p. zymosan and
monitoring of the subsequent inflammatory response (see sections 2.6.3 and 2.6.4). The
inflammatory response to i.p. zymosan was monitored in paired control animals that had
not been depleted of neutrophils.
2.7.2 The effect of dexamethasone on the inflammatorv response to i.p. zvmosan
Pairs of animals were treated i.v. with either dexamethasone (3mg/kg) or saline
as a control. 4h later test animals were treated with a second i.v. injection of
dexamethasone ( 1 mg/kg), whilst control animals again received an equal volume of
saline. Test and control animals were then anaesthetised with i.v. Sagatal and given i.v.
Evans blue dye and i.p. zymosan (500mg in 50ml sterile saline). Inflammatory exudate
67

samples were withdrawn for up to 6 hours and the inflammatory response generated in
control animals was compared to that obtained in test, dexamethasone treated animals (see
section 5.2.4).
To extend this series of experiments investigating the effect of i.v. dexamethasone
treatment on the generation of inflammatory mediators in response to i.p. zymosan these
animals were also used to look at the generation of LTB4 ex vivo from calcium ionophore
challenged peripheral blood leukocytes.
2.8 Treatment of rabbits with mustine hydrochloride
On day one mustine hydrochloride (1.75mg/ml in sterile saline, 1 ml/kg) was
injected intravenously into rabbits following the removal of a 1 ml peripheral blood
sample into heparin. On the fourth day a further blood sample was taken and then
animals were used to investigate the inflammatory response to i.p. zymosan in the
absence of neutrophils (see section 5.2.3). Animals entering the control group received
saline 1 ml/kg i.v. on day one followed by i.p. zymosan on day 4. Blood samples were
also obtained from control rabbits.
From each blood sample a 10/xl aliquot was taken to determine the whole
leukocyte count using a Coulter counter (as described in section 2:2). Blood smears were
also taken and stained enabling the number of polymorphonuclear leukocytes as a
percentage of the total white blood cell count to be obtained (see section 2.5).
2.9 Stimulation of blood with A23187
Blood "samples” were taken into lOOU/ml heparin, before and 4 hours following
intravenous dexamethasone treatment (3mg/kg) and challenged with the calcium
ionophore A23187. Parallel blood samples from control saline treated animals were
similarly challenged. At each time point, for each animal 250/xl aliquots of blood were
preincubated for lOmin at 37°C prior to the addition of 5/xl A23187 (0.5mg/ml in
DMSO), control incubations received an equal volume of DMSO vehicle (triplicate
samples for each). Reactions were allowed to proceed for 15min at 37°C before being
terminated by the addition of 50/xl of "cocktail" (ImM BW755C, ImM indomethacin,
l(X)mM EGTA, 1 % sodium azide, pH7.2). Plasma was then harvested by centrifugation
in an Eppendorf microfuge (15000g; 2min) and stored at -20°C for subsequent
determination of LTB4 content by RIA (see Section 2.6.4).
68

2.10 Detection of endotoxin levels present in solutions
The amount of bacterial endotoxin in solutions for i.v. administration was kindly
determined by Mr Gary Douglas using a modified Limulus Amebocyte Lysate (LAL) test
and a synthetic colour producing substrate to detect endotoxin chromogenically. Samples
were mixed with the Limulus Amebocyte Lysate supplied in the test kit and incubated at
37°C for 10 minutes. Any gram-negative bacterial endotoxin present in the sample
catalyzes the activation of a proenzyme in the Limulus Amebocyte Lysate. A colourless
substrate solution was then mixed with the LAL-sample and incubated at 37°C for an
additional 3 minutes. Any activated enzyme present catalyzes the splitting of P-
nitroaniline from the colourless substrate to form a yellow colour. The absorbance of the
sample was then determined spectrophotometrically at 405nm, having stopped the reaction
by addition of 25% acetic acid. The correlation between the absorbance and the
endotoxin concentration is linear, and the concentration of endotoxin in samples was
calculated from a standard curve.
2.11 Materials
Rabbits were from Froxfield Farm, Froxfield, or Hacking and Churchill,
Huntingdon. Sagatal and Expirai (Sodium pentobarbitone) were from May and Baker
Ltd, Dagenham. "'InClg (2mCi in 0.2ml sterile, pyrogen free 0.04N hydrochloric acid),
‘ ^I-human serum albumin (20mg albumin per ml of sterile isotonic saline, 50/xCi/ml) and
'% enon (lOmCi in 3ml sterile pyrogen free saline) were from Amersham International
Pic, Little Chalfont. Hespan (6 % hydroxy-ethyl starch in 0.9% NaCl) was from Dupont
Ltd, Stevenage. Percoll and Sephadex G-25M (PD-10) columns were from Pharmacia
Ltd, Milton Keynes. Steriflex (sterile, pyrogen free isotonic saline solution) was from
the Boots Company, Nottingham. Bradykinin, N-formyl-methionyl-leucy 1-phenylalanine
(FMLP), N-formyl-norleucyl-phenylalanine (FNLP), N-formyl-methionyl-phenylalanine
(FMP), N-formyl-methionine (FM), indomethacin, 2-mercaptopyridine-N-oxide,
prostaglandin Eg (PGEg), arachidonic acid (AA), zymosan A were purchased from Sigma
Chemical Company, Poole. Heparin sodium BP (0.3% cresol as preservative) was from
Paines and Bryne Ltd, Greenford. Platelet activating factor (PAF) was from Bachem,
Saffron Walden, Essex. Evans blue dye, histamine acid phosphate, di-sodium
ethylenediamine tetraacetic acid (EDTA) were from British Drug Houses Ltd, Poole.
Arterioveine 11720 14 gauge catheters were from Vygon, Ecoven, France. Mepyramine
Maleate was from May and Baker Ltd, Dagenham. Dexamethasone phosphate (8 mg,
69

dexamethasone phosphate in 2ml ampoules) was from David Bull Laboratories, Warwick.
N-formyl-methionyl-methionyl-phenylalanine (FMMP) was from Miles Laboratories,
Buckinhamshire. The following were generous gifts: Human CGRP from Dr U Ney,
Celltech Ltd, Slough. LTB4 from Dr SJ Foster, Imperial Chemical Industries, Cheshire.
N-formyl-methionyl-leucyl-phenylalanine-phenylalanine(FMLPP) was from Dr RJ Freer,
Medical College of Virginia, Richmond, Virginia USA. Sodium ibuprofen dihydrate was
from Dr IM Hunneyball, The Boots Company.
2.12 Statistical Analysis
Results are expressed as the means ± S.E.M. for n animals or n pairs of animals
where each datum unit is the average of 6 replicate sites. Where appropriate data were
analysed by two-way analysis of variance (ANOVA) of log transformed data and
statistical significance determined with the Neuman-Keuls procedure for multiple
comparisons (Snedecor & Cochran, 1967). A p value of less than 0.05 was considered
statistically significant.
70

CHAPTER 3 CHARACTERISTICS OF OEDEMA FORMATION INDUCED
BY FMLP
3.1 Introduction
In 1975 Schiffmann and his colleagues showed that synthetic formylmethionyl
peptides were potent chemoattractants for leukocytes and that they resembled chemotactic
substances in bacterial cultures (Schiffmann et aL, 1975b; Schiffmann et al. y 1975a).
Bacterial proteins are synthesised with a formyl-blocked N terminal methionine which is
subsequently cleaved. Schiffmann et al (1975a) thus suggested that eukaryotic cells
accumulating at foci of bacterial infections may be responding, at least in part, to these
bacterial products. In a structure-activity relationship study of 24 synthetic peptides N-
formyl-methionyl-leucyl-phenylalanine (FMLP) was found to be the most potent
chemotactic and enzyme-releasing stimulus (Showell et al., 1976). Subsequently this
same peptide was identified as the major chemotactic factor in E.coli culture broths
(Marasco et a l , 1984). Furthermore Carp (1982) demonstrated that, in addition to
prokaryotes being a source for n-formyl peptides, eukaryotic mitochondrial proteins also
had N-terminal formyl methionine which was chemotactic for neutrophils. Receptor
antagonists to these formyl peptides have been described (Freer et al., 1980; Becker,
1987). While the role of these peptides as endogenous chemotactic substances remains
unclear, the above findings demonstrate that, either through infection or tissue damage,
such substances are likely to be present and could contribute to the recruitment of
neutrophils into tissues in vivo. In addition to being a potent chemotaxin, FMLP shares
with leukotriene B4 and the complement fragment C5a the ability to promote neutrophil
adherence to vascular endothelium, degranulation and production of toxic oxygen species
(Boxer et al., 1989; Hafstrom et al., 1981; Hoover et a l , 1984; Palmblad et al., 1984;
Showell et al., 1976; Tonnesen et al., 1984; Webster et a l , 1980). In vivo FMLP has
been shown to increase neutrophil accumulation in the lung (Desai et a l , 1979), hamster
cheek pouch (Nagai & Katori, 1988) and rabbit skin (Colditz & Movat, 1984b; Rampart
& Williams, 1988). Wedmore & Williams (1981a) showed that FMLP together with C5a
and LTB4 shared the distinction of being neutrophil dependent in their ability to enhance
vascular permeability (see also section 1.2.3),
The goals of the present study were to compare the activity of formyl peptides in
the skin microvasculature and to investigate the characteristics and underlying
mechanisms of FMLP-induced oedema formation.
71

Measurement of oedema formation can be achieved using a variety of techniques
covering a wide range of sophistication. Determination of tissue thickness or volume are
simple but adequate methods. Evans blue dye binds avidly to albumin when injected
intravenously. Thus, the amount of dye accumulating in a tissue can be used to estimate
protein leakage, but quantification is difficult. Measurement of the extravasation of
intravenously administered radiolabelled albumin provides a rapid and accurate
assessment of tissue oedema which can easily be quantified, with no subjective estimation
involved, using a gamma counter.
The rabbit dorsal skin provides a convenient model for the study of inflammation.
It is a large accessible organ, which allows multiple treatment sites in the same animal.
By use of a balanced site plan the effect of several different treatments can be assessed
simultaneously or time-courses followed within an experiment.
72

3.2 Results
3.2.1 The effect of vasodilator prostaglandin on oedema formation induced bv
histamine and FMLP in rabbit skin
Figure 3.1 shows that over a 30 minute period histamine (2.5xlO ’moles/site) and
FMLP (at doses of 5x10" and 5xlO'"moles/site) injected alone induced a significant
(p<0.05) but small increase in plasma leakage when compared with control sites injected
with phosphate buffered saline (PBS). As previously documented, coinjection of the
vasodilator substance PGE2 (3xlO*‘°moles/site) resulted in large potentiated oedema
responses by all doses of histamine and FMLP (figure 3.1). This potentiation occurs as
a result of synergism between increased microvascular permeability induced by mediators
such as histamine and FMLP, and the increased microvascular blood flow induced by
PGEj (Williams & Morley, 1973; Williams & Peck, 1977; Wedmore & Williams,
1981a). PGE2 (3xlO *°moles/site) injected alone induced little plasma leakage as shown.
On a molar basis FMLP was 100-1000 times more active than histamine in increasing
microvascular permeability (figure 3.1).
3.2.2 The effect of formvl-peptides on the skin microvasculature
To investigate the relative ability of six formyl peptides to increase microvascular
permeability the peptides were co-administered intradermally with prostaglandin Eg (3x10
*°moles/site) in rabbit skin. The accumulation of intravenously administered ^^ I-albumin
was measured over 30 minutes. Figure 3.2 shows the oedema responses induced by the
various doses of formyl-peptides. The peptide most potent in increasing permeability was
N-formyl-methionyl-leucyl-phenylalanyl-phenylalanine (FMLPP); significant oedema
responses (p<0.05) being obtained with 5xlO'*'*moles/site when compared with responses
to PGEg alone. At higher doses (5x10"* and 5xl0‘"moles/site) FMLPP showed similar
activity to N-formyl-methionyl-leucyl-phenylalanine (FMLP), giving responses of 60±5
and 65±9/xl/site respectively at the highest dose. The analogues N-formyl-norleucyl-
phenylalanine (FNLP) and N-formyl-methionyl-methionyl-phenylalanine (FMMP) were
equi-active and approximately ten times less active than FMLP, giving responses of
42±4 and 40±5/xl/site respectively at a dose of 5xlO "moles/site. N-formyl-methionyl-
phenylalanine (FMP) was considerably weaker giving only 20±2/xl of oedema/site at
5x10 "moles/site. No oedema formation in response to 5x10 " moles/site of N-formyl-
methionine (FM) was detected, ie the response to intradermal injection of FM + PGEg
was no different to that obtained with PGEg alone.
73

9 0 -oTr 8 0 - (0
& 7 0 -
3 6 0 -
I 5 0 - Ô> 4 0 -
I 3 0 - ro
20 -
W 1 0 - PBS0 -^
10-* 3x10 ’°mol per s ite PGE,
Figure 3.1
Oedema responses in rabbit skin induced by i.d. injection of N-formyl-methionyl-leucyl-
phenylalanine (FMLP) and histamine (HA) at the concentrations shown (mol per site).
Mediators were injected either alone (open circles FMLP, open triangles histamine), or
in the presence of a fixed potentiating dose of prostaglandin Ej (PGEg, 3xl0'^°mol per
site) represented by closed circles for FMLP+PGEj; closed triangles for HA+PGE 2 .
Oedema responses were measured over 30 min. The dashed line shows the control value
obtained after i.d. injection of PBS. Each point represents the mean ± s.e.mean for n = 6
rabbits, * p <0.05 with respect to PBS.
74

»CO
I3.I3I I8
Q.C
$
80-1
7 0 -
FMLPFMLPP6 0 -
5 0 -
FNLPFMMP4 0 —
3 0 -
FMP20 -
FM PGE1 0 -
PBS
0 ->5x10 5x10'" 5x10 '
mol per s ite5x 1 0 " 3x10 '°
Figure 3.2
Oedema responses in rabbit skin induced by i.d. injection of a range of N-formyl
peptides; N-formyl-methionyl-leucyl-phenylalanine (FMLP), N-formyl-methionyl-leucyl-
phenylalanyl-phenylalanine (FMLPP), N-formyl-norleucyl-phenylalanine (FNLP),
N-formyl-methionyl-methionyl-phenylalanine (FMMP), N-formyl-methionyl-phenylalanine
(FMP), N-formyl-methionine (FM). Concentrations are given as mol per site. All
peptides were coinjected with a fixed potentiating dose of prostaglandin Eg (PGEg, SxlO" ®
mol per site). Responses were measured over 30min. The dashed line shows the value
obtained after i.d. injection of PBS. Each point represents the mean ± s.e.mean for n = 6
replicate experiments.
75

Further experiments to investigate the characteristics and mechanisms of oedema
formation were performed using FMLP.
3.2.3 Effect of local treatment with indomethacin on oedema responses
induced bv FMLP
The accumulation of intravenous ^^I-albumin in response to intradermal FMLP
(5xl0*“ moles/site) was measured over 4 hour and 30 minute periods in the same animals.
In six rabbits significantly elevated plasma accumulation was observed after 4 hours
FMLP administration when compared with 30 minutes (17±3 and 6 ±2/xl/site respectively
results subtracted for PBS values, p<0.05). To investigate any possible contribution of
pro-inflammatory products of the cyclo-oxygenase pathway to the FMLP-induced oedema
response skin sites were treated locally with the cyclooxygenase inhibitor indomethacin
(10'*moles/site) for 10 minutes prior to injection of the same dose of FMLP (5xlB
"moles/site). Neither 4 hour or 30 minute oedema responses were affected by
indomethacin (responses were 18+3 and 5+2/xl oedema/skin site respectively, values
subtracted for PBS sites, as shown in table 3.1). These results suggest that inflammatory
products of the cyclooxygenase pathway did not contribute to the FMLP-induced
responses observed.
3.2.4. The effect of mepvramine on FMLP-induced oedema formation
It has been reported by Desai et al (1979) that FMLP-induced neutrophil
accumulation in hamster lung was accompanied by a small permeability change which it
was suggested might be due to histamine release. Furthermore, N-formyl-peptides have
been shown to stimulate histamine release from human basophil-rich leukocytes (Hook
et a l , 1976). To investigate the possibility that endogenous histamine is released in
rabbit skin in response to FMLP skin sites were treated locally with the anti-histamine
mepyramine maleate (3xlO'^moles/site) just prior to injection of FMLP (2.5x10’
"moles/site) + PGE; (3x10'^°moles/site). Control skin sites were injected with an equal
volume (100/xl) of PBS prior to injection of the same doses of FMLP + PGE . The
susceptibility of oedema responses induced by FMLP + PGEg to inhibition by
mepyramine showed some inter-animal variation, however in six rabbits the mean effect
was minimal; 18+6% suppression (p>0.05). In the same group of animals oedema
responses to injection of a mixture of histamine (2.5xlO’*moles/site) + PGEj (3x10
^°moles/site) were suppressed by a mean 84+3% by the local administration of the same
76

Time after i.d. FMLP
(mins)
Microlitres plasma per site induced by i.d. FMLP
(5x1 O'" moles/site)
- Indomethacin + Indomethacin
30 6 ± 2 5±2
240 17±3* 18±3*
Table 3.1
Increased vascular permeability in rabbit skin in response to N-formyl-methionyl-leucyl-
phenylalanine (FMLP). The indicated dose of FMLP was injected i.d. and plasma
accumulation measured over 4h and 30min, in the absence and presence of local
indomethacin (10'®mol per site for 10 minutes). Values are the mean ± s.e.mean for
n = 6 rabbits and are corrected for the low levels in PBS injected sites. * Significantly
increased compared with sites injected for 30 minutes, p<0.05.
77

dose of mepyramine (p<0.05) which was thus evidently blocking histamine action. The
background value for intradermal injection of PBS alone was subtracted from the data
before calculating percentage inhibitions.
Overall these results demonstrate that, whilst mepyramine was able to inhibit the
local inflammatory actions of histamine 4- PGEj effectively it was unable to influence
oedema formation in response to FMLP + PGEj significantly in this model.
3.2.5. The effect of intravenous infusion of zvmosan-activated plasma on FMLP-
induced oedema
In the initial experiments of Wedmore and Williams (1981a) investigating the
neutrophil dependency of oedema formation, neutropenia was induced using nitrogen
mustard 3/4 days prior to experimentation. In this investigation experiments were carried
out to determine if infusion of zymosan-activated plasma (as a source of C5a des Arg)
was able to cause transient neutropenia and selectively effect neutrophil-dependent
oedema responses.
Animals were injected intradermally with FMLP, C5a des Arg (each at 5x10"
"moles/site) and bradykinin (10 *°moles/site), together with prostaglandin Eg (3x10
*°moles/site) and the accumulation of ‘ ^I-albumin was allowed to proceed for 30 minutes.
ZAP (previously filtered, 0.22/xm millex) was then infused intravenously at 6 ml/min for
30 seconds, then at 0.5ml/min for the remainder of the experiment. Five minutes after
commencing ZAP infusion the remaining skin sites were injected with the same
combination of test agents and ‘ ^I-albumin accumulation measured for a final 30
minutes. This experimental design allowed each animal to serve as its own control.
As indicated in figure 3.3 this technique for rapid systemic neutrophil depletion
was found to be effective at selectively inhibiting neutrophil-dependent oedema responses.
Responses to FMLP and C5a des Arg were significantly inhibited to 67±7% and 74±4%
respectively (p<0.05). In contrast the response to bradykinin was not significantly
inhibited (mean percentage inhibition of 9 ± 2 %).
Total circulating white cell numbers (xlOVml, n=3) were measured using a
coulter counter before ZAP, 5 minutes after ZAP infusion and 35 minutes after ZAP (ie.
the end of the experiment) and are shown in table 3.2, together with the corresponding
neutrophil numbers (xlOVml) assessed using May-Grunwald-Giemsa stain. This table
clearly shows that ZAP infusion caused an immediate (after 5 mins) significant (p< 0.05)
decline in both the total number of circulating white cells and the number of circulating
78

160n
o'r 140-CO
& 1 2 0 - 3.13Ô>
ECOCDQ.C
2
(/)
100 -
8 0 -
6 0 —
4 0 -
20 -
0 ^
I
I
I
FMLP C 5a d e s Arg Bradykinin PGE^ PBS
Figure 3.3
Effect of i.v. infusion of zymosan-activated plasma (ZAP) on oedema responses in rabbit
skin induced by N-formyl-methyl-leucyl-phenylalanine (FMLP) + prostaglandin Eg
(PGEg), C5a des Arg + PGEg and bradykinin (Bk) + PGEg. The open columns
represent responses obtained before, and black columns represent responses after infusion
of ZAP. FMLP and C5a des Arg were injected at SxlO’ mol per site, Bk at 10^°mol per
site and PGEg at 3xlO*‘°mol per site. The dashed line shows oedema after i.d. injection
of PBS. Each point represents the mean ± s.e.mean from n = 6 animals, *P<0.05,
compared with control pre-ZAP values.
79

Time of Sample Total No of Leukocytes
(xlO^/ml)
No of neutrophils
Before ZAP 6.6±0.9 2.3±0.4
5 mins after ZAP infusion 3.2±0.6’ 0.1 ±0.04*
35mins after ZAP infusion 4.6±0.7* 0.8+0.3*
Table 3.2
The total number of leukocytes and the number of neutorphils circulating in animals
before and at the indicated times after i.v. zymosan activated plasma (ZAP). Total
leukocyte numbers were determined in a coulter counter and May-Grunwald-Giemsa stain
was used for assessing neutrophil numbers. The values represent the number of cells
xlOVml and are the mean ± s.e.mean for n=3 rabbits. * p<0.05 compared to control,
pre ZAP numbers.
80

neutrophils (from 6.6±0.9 before ZAP to 3.2±0.6, 5 minutes after ZAP, and from
2.3±0.4 before ZAP to 0.1 ±0.04, 5 minutes after ZAP for total white cells and
neutrophils respectively). The total white cell and neutrophil numbers rose slightly by
the 35 minute time point but were both still significantly (p<0.05) reduced compared to
pre-ZAP values (table 3.2).
3.2.6 The effect of intravenous Ibuprofen on oedema formation in rabbit skin
Several reports have indicated that the non-steroidal anti-inflammatory compound
Ibuprofen can inhibit certain PMN leukocyte responses (including aggregation,
locomotion, enzyme release and oxygen burst) in vitro (Maderazo et a l , 1984; Flynn et
a l, 1984; Kaplan et a l , 1984) and ex vivo (Kaplan et a l , 1984) by a mechanism or
mechanisms independent of cyclooxygenase inhibition. Thus the present investigation
was designed to establish possible cyclooxygenase-independent effects of Ibuprofen on
inflammatory oedema in vivo.
Pairs of animals were injected with doses of FMLP and histamine mixed with
prostaglandin Eg. The top dose of each permeability increasing mediator was also
coinjected with the prostaglandin precursor arachidonic acid (AA, 3x10 moles/site) to
distinguish cyclo-oxygenase-dependent and independent actions of Ibuprofen.
Accumulation of *^I-albumin was allowed to proceed for 30 minutes before intravenous
administration of either ibuprofen (2 0 mg/kg) or in the case of control animals an equal
volume of saline. Five minutes after the intravenous injections further intradermal
injections of the same test agents were made in fresh skin sites and *^I-albumin
accumulation measured for a further 30 minutes. The intravenous administration of
saline had no significant effect on oedema responses to all combinations of agents tested
(ie. responses over 35-65 minutes were the same as those measured 0-65 minutes, figure
3.4a). In contrast, figure 3.4b clearly shows that systemic treatment with ibuprofen
resulted in significant suppression of leakage responses to both FMLP and histamine
when they were potentiated by A A. This is due to inhibition of cyclo-oxygenase and
reduction of potentiating vasodilator prostaglandin production. However, when leakage
responses to FMLP and histamine were potentiated by the simultaneous injection of PGEg
to eliminate effects of cyclo-oxygenase inhibition, only the oedema induced by all doses
of FMLP + PGEg was significantly suppressed by the systemic administration of
ibuprofen. Responses to histamine + PGBg were unaffected by ibuprofen treatment, at
all doses of histamine tested (figure 3.4b).
81
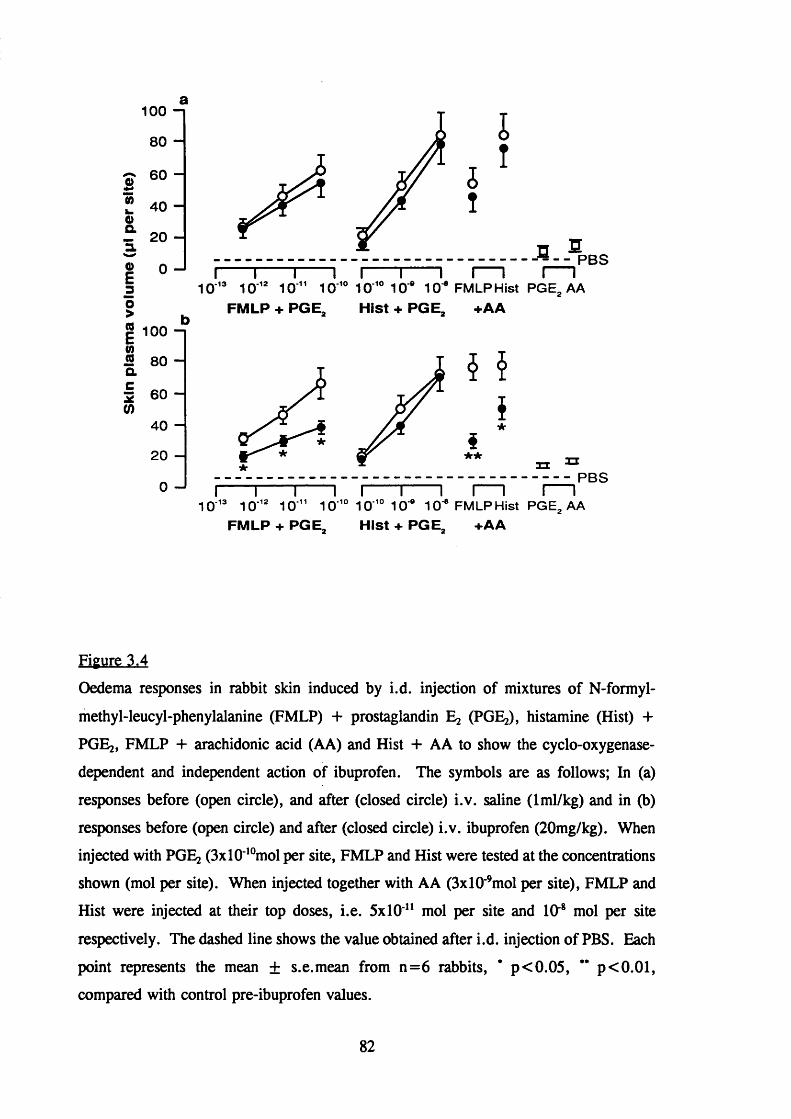
£0 )
I
I30>(0
12 Q.C
2
(0
100
80
60
40
20
0
100
80
60
40
200
PBSI 1- - - - - - 1- - - - - - 1 r
10 '® 10 '® 1 0 " 10 '° 10 '° 10 * 10'® FMLP Hist PGE, AAFMLP + PGE, Hist + PGE, +AA
*
XXPBS
10 '® 1 0 '® 1 0 " 1 0 '° 10 '° 1 0 ° 10 ® FMLP Hist PGE^ AAFMLP + PGE, Hist + PGE, +AA
Figure 3.4
Oedema responses in rabbit skin induced by i.d. injection of mixtures of N-formyl-
methyl-leucyl-phenylalanine (FMLP) + prostaglandin Eg (PGEg), histamine (Hist) +
PGEg, FMLP 4- arachidonic acid (AA) and Hist + AA to show the cyclo-oxygenase-
dependent and independent action of ibuprofen. The symbols are as follows; In (a)
responses before (open circle), and after (closed circle) i.v. saline ( 1 ml/kg) and in (b)
responses before (open circle) and after (closed circle) i.v. ibuprofen (20mg/kg). When
injected with PGEg (3xlO'*°mol per site, FMLP and Hist were tested at the concentrations
shown (mol per site). When injected together with AA (3xlO ’mol per site), FMLP and
Hist were injected at their top doses, i.e. 5x10 " mol per site and lO"* mol per site
respectively. The dashed line shows the value obtained after i.d. injection of PBS. Each
point represents the mean ± s.e.mean from n = 6 rabbits, * p<0.05, ** p<0.01,
compared with control pre-ibuprofen values.
82

Time of sample Total no. of leukocytes
(xlO^cells/ml)
No of neutrophils
Before Ibuprofen 5.6±1.4 2.0±0.5
5 mins after Ibuprofen 4.9±1.3 1.9+0.4
35 mins after Ibuprofen 5.5±1.3 2.1±0.5
Table 3.3
The total number of leukocytes and the number of neutrophils circulating in rabbits
before and at the indicated times after i.v. adminsitration of ibuprofen (20mg/kg). Cell
numbers were determined as for table 3.2. The values represent the number of cells xlO
circulating/ml and are the mean± s.e.mean for n=6 rabbits.
83

To eliminate the possibility that endotoxin contamination of the ibuprofen may
have accounted for the observed effects the amount of endotoxin in the ibuprofen solution
(20mg/ml) was measured by use of Limulus amoebocyte lysate and a chromogenic
substrate. The level of endotoxin in the ibuprofen solution was 0.8ng/ml which was
considered to be an insufficient amount to account for the observed effects of the drug
on oedema responses to FMLP + PGE2 (Haslett et al,, 1987).
Total leukocyte numbers (xlOVml) and neutrophil numbers (xlO^/ml) were
measured before, 5 and 35 minutes after intravenous ibuprofen and are shown in table
3.3. Ibuprofen did not affect either the total number of leukocytes or the number of
neutrophils over the 35 minutes of the experiment (table 3.3).
These results demonstrate that intravenous Ibuprofen was able to reduce
selectively oedema responses induced by FMLP + PGE2 (neutrophil dependent) by a
cyclo-oxygenase-independent method.
3.2.7 The effect of colchicine on FMLP-induced oedema formation
The microtubule blocking agent colchicine has been reported to interfere with
several aspects of neutrophil function in vitro and in vivo (Malech et al., 1977a; Valerius,
1978; Cunningham & Smith, 1982; Higgs et al., 1983; Chang, 1975). In an attempt to
investigate the underlying mechanisms involved in neutrophil-dependent oedema
formation, colchicine was administered intravenously and the effect on oedema formation
was monitored. The same protocol as that described in sections 3.2.5 and 3.2.6 was
adopted ie. internally controlled experiments were performed.
Oedema responses to both FMLP + PGE2 and Bk 4- PGE2 at all doses tested
were unchanged in control animals which received 1 ml/kg saline intravenously (figure
3.5a). In contrast, as shown in figure 3.5b oedema responses to FMLP 4- PGE; were
virtually abolished after intravenous colchicine (1 mg/kg), while bradykinin 4- PGE2
responses were unaffected. The small amount of endotoxin (0.05ng/ml) in the colchicine
solution (1 mg/ml) was not thought to have been sufficient to affect oedema responses
(Haslett et a l , 1987).
Circulating leukocyte counts and neutrophil counts were measured in blood
samples taken before, 10 and 40 minutes after colchicine treatment (table 3.4). Over the
40 minute period colchicine caused a fall in circulating neutrophil numbers from 2.8±0.8
to 0.7+0.2 X lO^cells/ml (p<0.05), however, this was a slow decline since 10 minutes
after colchicine a significant (p<0.05) but only relatively small fall to
84

9 0 —1
8 0 -M& 70-3.
I3
1
ISO
SO —
20-c(/) 1 0 -
P B SO—'
5 x 1 0 ’* 5 x 1 0 ’* 5 x 1 0 ” 1 0 ’* 1 0 3 x 1 0 ’°
FM LP B ra d y k in in
m o i p e r s i t e
P G E .
9 0 —1s
8 0 —(0
7 0 —3.
GO—O)E3 5 0 -
SiI 3 0 —
20—c
tn io —P B S
o—'5 x 1 0 ’* 5 x 1 0 * 5 x 1 0 ” 1 0 ’* 1 0 ” 1 0 ’° 3 x 1 0 ’°
FM LP B r a d y k in in
m o i p e r s i t e
P G E .
Figure 3.5
Oedema responses in rabbit skin induced by i.d. injection of mixtures of N-formyl- methyl-leucyl-phenylalanine (FMLP) + prostaglandin Ej (PGEg) and bradykinin (Bk) + PGE2 to show the selective effect of i.v. colchicine on neutrophil-dependent oedema formation. The symbols represent; In (a) responses before (open circle) and after (closed circle) i.v. saline (1 ml/kg), and in (b) responses before (open circles) and after (closed circles) i.v. colchicine (1 ml/kg). FMLP and Bk were injected at the concentrations shown (mol per site) mixed with PGE2 (3xlO‘*° mol per site). The dashed line represents oedema in sites injected i.d. with PBS. Each point represents the mean ± s.e.mean from n=4 rabbits. * P<0.05, ** P<0.01 compared with control pre-colchicine oedema responses.
85

Time of sample Total no of leukocytes No of neutrophils
(xlO^cells/ml)
Before colchicine 5.9±0.9 2.8±0.8
lOmin after colchicine 4.8+0.6 1.2±0.3*
40min after colchicine 3.8±0.6 0.7+0.2*
Table 3.4
The total number of leukocytes and the number of neutrophils circulating in rabbits
before and at the indicated times after i.v. colchicine (Img/kg). The values represent the
number of cells xlO circulating/ml and are the mean ± s.e.mean for n=4 rabbits.
* P<0.05 compared to control, pre colchicine numbers.
86

1.2±0.3xl0^cell/ml was observed. A similar profile of decline was also obtained for the
total number of leukocytes (table 3.4). Thus, intravenous injection of colchicine resulted
in a slow decline in both the total number of circulating leukocytes and the number of
neutrophils.
3.2.8 The effect of intravenous colchicine on the duration of action of permeabilîtv-
increasing mediators
When compared with direct-acting mediators such as bradykinin and histamine
plasma leakage responses induced by the neutrophil chemoattractants are of a long
duration of action ((Wedmore & Williams, 1981a), see section 1.2.3). The t'A for the
duration of the permeability-increasing activity of bradykinin and histamine is
approximately 4 minutes and that of FMLP is approximately 50 minutes. The
mechanism of this protracted response is unknown. In an attempt to investigate the
protracted nature of the response, colchicine was administered intravenously after
intradermal FMLP injections and the effect on the development of oedema was measured.
Figure 3.6 shows the effect of colchicine administration (1 mg/kg intravenously)
on the duration of action of bradykinin and FMLP in rabbit skin. In these experiments
animals were pretreated with mepyramine (3mg/kg i.v.) to block the action of any
endogenous histamine released by FMLP. They were then injected intradermally with
FMLP or Bk at various time intervals before intravenous injection of ‘“ I-albumin and
Evans blue dye and either saline in the case of control animals or colchicine (1 mg/kg).
A potentiating dose of PGEj (3xl0 ‘°moles/site) was immediately injected into all sites
and oedema responses were then measured over 30 minutes. As shown in figure 3.6
colchicine had no effect on the duration of action of bradykinin, the response half-life in
controls being 5± Imin, compared with 4± Imin in the colchicine group. In contrast, the
long duration of action of FMLP (response half-life of 36±3min open circles figure 3.6)
was significantly reduced in the presence of colchicine (closed circles figure 3.6). These
results suggest that continuing interactions between functionally active neutrophils and
endothelial cells are necessary for a protracted protein leakage response.
87

a
B0 )
I
I3I I1ac
2
</)
90-1
8 0 -
7 0 -
6 0 -
5 0 -
4 0 —
3 0 -
20-
1 0 -
0 I— I— I— I I— I— I— I— I— I-----1— I----1— IPBS
0 10 20 30 0 20 40 60 80 PGE,
Interval before album in Injection (mln)
Figure 3.6
Effect of colchicine on the duration of action by (a) bradykinin (Bk) and (b) N-formyl-
methyl-leucyl-phenylalanine (FMLP) in rabbit skin. FMLP (5x10" ° mol per site) and Bk
(10 *° mol per site) were injected i.d. at the time intervals shown before an i.v. injection
of *^I-albumin and Evans blue dye and either saline (1 ml/kg) or colchicine (1 mg/kg) at
t=0. PGEj (3x10**° mol per site) was injected i.d. into all skin sites and oedema
formation allowed to proceed for 30min. In (a) responses to Bk and (b) responses to
FMLP in saline (open circles) and colchicine-treated rabbits (closed circles). The dashed
line shows the response obtained after i.d. injection of PBS. Each point represents the
mean ± s.e.mean for n=6 rabbits, * P<0.05 with respect to saline treated animals.
88

3.2.9 Summary
1) Oedema responses in rabbit skin to mediators of increased microvascular
permeability are potentiated by coinjection of vasodilator substances such as PGEj.
2) The rank order of potency of 6 structurally-related formyl peptides in inducing
oedema formation in rabbit skin when coinjected with PGE2 is
FMLPP > FMLP> FNLP/FMMP> FMP> FM.
3) In this model the inflammatory oedema induced by FMLP increases with time of
FMLP treatment (at least up to 4 hours) and is not a consequence of the generation of
endogenous histamine or pro-inflammatory products of the cyclo-oxygenase pathway.
4) The intravenous infusion of ZAP is able to cause an immediate depletion of
cirulating neutrophils and the selective inhibition of neutrophil-dependent oedema
formation in the skin induced by mediators such as FMLP, but leaves oedema formation
in response to direct-acting mediators unaffected.
5) The non-steroidal anti-inflammatory compound Ibuprofen administered
intravenously is able to inhibit selectively oedema responses induced by FMLP + PGEj
(neutrophil-dependent) by a cyclooxygenase-independent mechanism(s) which is not due
to depletion of circulating neutrophils.
6) Intravenous administration of the microtubule blocking agent colchicine abolishes
oedema responses to FMLP 4- PGEj but not histamine + PGEg. Over 30 minutes
colchicine administration causes a slow decline in the total number of circulating
leukocytes and in the number of neutrophils.
7) The plasma leakage response induced by FMLP is of a long duration of action
when compared to that of the direct acting mediator bradykinin. Intravenous colchicine
is able to reduce significantly the long duration of FMLP-induced permeability increasing
activity but has no effect on the duration of permeability increasing activity of bradykinin.
89

CHAPTER 4 THE EFFECT OF GLUCQ CORTICOSTEROIDS ON
V A S C U L A R R E S P O N S E S IN T HE R A B B I T
MICROCIRCULATION INDUCED BY INFLAMMATORY
MEDIATORS
4.1 Introduction
As discussed in section 1.5 glucocorticoids are highly effective at reducing almost
every aspect of the inflammatory response. This very complexity can hinder analysis of
their mode of action. In many situations their target and mechanism of action remain
unclear. Such is the case with increased endothelial cell permeability a phenomenon
essential for the development of inflammatory oedema. It is clear that glucocorticoids
can act by suppressing the generation of inflammatory mediators to inhibit permeability
changes. However, since glucocorticoids also inhibit oedema induced by an array of
inflammatory mediators including Bk, 5-HT, histamine and FAF in a variety of animal
species (Tsurufuji et a l, 1979; Church & Miller, 1978; Bjork et a l, 1985; Svensjo &
Roempke, 1985) it has been suggested this is unlikely to be a result of inhibition of
mediator generation and more likely to be a direct anti-exudate effect of glucocorticoids
on the target cell of inflammatory mediators, for example the microvascular endothelial
cell.
In addition, glucocorticoids have also been shown to inhibit oedema formation
induced by chemotactic mediators (Foster & McCormick, 1985; Griffiths & Blackham,
1988; Peers & Flower, 1991) and it remains unclear whether the primary target of
steroids is the endothelial cell, the neutrophil or both. To further complicate matters
glucocorticosteroids have been reported to have vasocontrictor activity. Microvascular
protein leakage and leukocyte accumulation have been shown to be dependent on blood
flow, thus vasoconstriction in vivo can have anti-permeability consequences.
The aim of the present study was to investigate the effects of glucocorticoids on
vascular responses in the rabbit microcirculation induced by a variety of inflammatory
mediators. To achieve this the rabbit skin assay already utilized in Chapter 3 to measure
oedema formation was extended to incorporate the simultaneous measurement of
neutrophil accumulation and the assessment of blood flow. This allowed the effect of
steroids on three aspects of the inflammatory response to be investigated; blood flow,
vascular permeability and neutrophil accumulation.
Skin blood flow was measured using a ‘ ^Xenon clearance technique (Williams,
90

1979) which allows multiple treatments and|replicates to be performed simultaneously on
a single animal. Neutrophil accumulation at inflammatory sites was monitored using a
radioisotopic method employing ‘"In-labelling of neutrophils isolated from the blood of
a donor animal. This method allowed protocols to be adopted where either the neutrophil
donor rabbit or the neutrophil recipient rabbit were treated with dexamethasone. This
approach coupled with a comparison of the effects of local and systemic steroid treatment
facilitated a detailed investigation of the possible sites of action of steroids in inhibiting
vascular permeability and neutrophil accumulation induced by a variety of stimuli in vivo.
A detailed knowledge of the way these powerful drugs work may provide the basis
for rational therapies designed to control inflammatory conditions with minimal side
effects. Conversely, pharmacological manipulation may shed light on mechanisms of
action and interaction of chemical mediators in inflammation.
91

4.2 Results
4.2.1 The local treatment of rabbits with dexamethasone
4.2.1.1 Effect of local dexamethasone on oedema formation in rabbit skin
Skin sites were treated intradermally with dexamethasone (80ng/site) or saline at
various time intervals before intravenous *^I-albumin and Evans blue dye. Skin sites
were then re-injected with combinations of mediators under test and oedema formation
measured over 30 minutes.
As shown in figure 4.1a local pretreatment of skin sites with dexamethasone
caused a time-dependent inhibition of oedema formation induced by both the direct-acting
mediator bradykinin (shown as open bars) and the neutrophil-dependent mediator ZAP,
used as a source of C5a des Arg (shown as solid bars) when coinjected with PGE;: ie.
after 240 minutes dexamethasone pretreatment the percentage inhibition was 62.0+5.4
(p<0.05) and 62.4+3.8 (p<0.05) for Bk + PGE; and ZAP + PGE; respectively. This
inhibition was considerably greater than that observed with 15 minutes pretreatment
where no significant inhibition was obtained or 90 minutes where a significant, but only
minimal, suppression of the Bk + PGE; response was observed (27.8±5.6% inhibition,
p<0.05). In contrast, pretreatment of skin sites with the cyclooxygenase inhibitor
indomethacin (3^g/site) had no effect on oedema responses to bradykinin or ZAP when
coinjected with PGE; (figure 4.1b). However, indomethacin pretreatment did inhibit the
exudation potentiation produced by coinjection of mediators with the prostaglandin
precursor arachidonic acid and was thus evidently blocking cyclooxygenase at all
pretreatment times tested (cross hatched bars, p < 0.05, figure 4. lb). The effect of local
indomethacin was remarkably persistent; inhibition was not different for the 15min and
240min pretreatment times. Like its metabolite, arachidonic acid was a poor inducer of
plasma protein leakage when injected alone (mean 8.3±3.0/xl oedema/skin site, data not
shown).
These results demonstrate that the inhibitory effect of dexamethasone on oedema
formation was only evident after a lag period of approximately 240 minutes and that this
inhibition was not a consequence of inhibition of vasodilator prostaglandin generation.
4.2.1.2 Effect of local dexamethasone on “ In-neutrophil accumulation in
rabbit skin
In some of the animals discussed in section 4.2.1.1 the accumulation
o f “ In-neutrophils in skin sites was simultaneously measured over the 30 minute period.
92

a. Local dexamethasone 100-1
Eo>T30 >Ooco
it it
pretreatm ent time (mln)
b. Local indomethacin
1 0 0 - 1
90 -(0E 80 -o>■00> 70 -0 600c 50 -0 40 -5!E 30 -c
2 0 -1 0 -
0 -15 90 240
pretreatm ent time (min)
Figure 4.1Effect of local i.d. pretreatment with dexamethasone (a) and indomethacin (b) on
oedema formation induced by i.d. bradykinin (Bk) 4- prost^landin Eg (PGEg) (open columns), zymosan activated plasma (ZAP) 4- PGE2 (solid columns) and Bk 4- arachidonic acid (AA, hatched columns in b). Skin sites were pretreated locally with either dexamethasone (2xl0‘*®mol per site), indomethacin (lO^mol per site) or saline for the times indicated prior to i.v. ‘ -albumin and "'In-neutrophils. Pretreated sites were then reinjected with mixtures of mediators under test and the response allowed to proceed for 30min as described in methods. Concentrations of mediators used were as fbllo\ys;Bk (10^°mol per site), ZAP (1(X)%), PGE2 (3xl(X °mol per site), AA (3xl(f’mol per site). For each time point, responses in drug pretreated sites were cAculated as a percentage of those obtained in saline pretreated sites in the same rabbit. Calculations were performed using data after subtraction of the relevant background response for each animal (saline injected skin). Responses are mean ± s.e.mean for n=4-7 rabbits. Significant inhibition, *P<0.05.
93

"^In-neutrophils separated from the blood of donor rabbits were injected intravenously,
together with *^I-albumin and Evans blue dye. In contrast to the effect on neutrophil-
dependent oedema formation, local dexamethasone pretreatment did not significantly
inhibit "^In-neutrophil accumulation induced by ZAP + PGE2 , even after 240 minutes
pretreatment (table 4.1). Responses to ZAP + PGEj expressed as number of * "In
neutrophils/site were as follows: in sites pretreated for 15 minutes with saline
1799.0±359, with dexamethasone 1723.8±335.9, in skin sites pretreated for 240 minutes
with saline 1803.9±383.8, dexamethasone 1551.0±362.6. Indomethacin had no
significant effect on cell accumulation at any pretreatment time (table 4.1).
4.2.1.3 Effect of local dexamethasone on blood flow in rabbit skin
Since anti-inflammatory steroids have been shown to constrict local blood vessels
when applied to human skin it has been suggested that a 'vasoconstrictor action may be
responsible for the anti-exudative effect of these steroids (Marks & Sawyer, 1986; Reid
& Brookes, 1968; Kaidbey & Kligman, 1974). Figure 4.2 shows the results of
experiments designed to determine whether the anti-permeability effects of dexamethasone
(figure 4.1a) were accompanied by local vasoconstriction. These experiments were
performed by measuring prostaglandin Ej stimulated increased blood flow in skin sites
which had been preinjected 4 hours previously with either dexamethasone (80ng/site,
closed circles figure 4.2) or saline (open circles, figure 4.2). Blood flow was measured
using "^Xe clearance. The effect of dexamethasone pretreatment on blood flow at
saline-injected sites was also investigated as a means of monitoring basal blood flow.
Figure 4.2 clearly shows that whilst dexamethasone pretreatment caused a slight mean
percentage decrease in basal blood flow (13.2+3.8%, open square) there was no
significant effect of dexamethasone on the dose related increase in blood flow observed
with prostaglandin Ej. At the dose of PGEj used to potentiate oedema responses (3x10
^°moles/site) a 48.6+1.8% increase in blood flow was observed in saline pretreated sites
compared to a 46.2±4.6% increase in dexamethasone pretreated sites (n=5 animals).
These results indicate that the inhibitory effect of dexamethasone on oedema
formation (figure 4.1a) was not a consequence of local vasoconstrictor action.
94

pretreatment time Sal Dex Indo
(mins)
15 1799.0±359.0 1723.8±335.9 2116.0+488.0
240 1803.9±383.8 1551.0±362.6 2258.0±372.0
Table 4.1
Effect of local i.d. pretreatment with dexamethasone (Dex, 80ng/site),
indomethacin (Indo, 3/xg/site), or saline (Sal) on "'In-neutrophil accumulation induced
by i.d. ZAP + PGEj. Skin sites were treated as described in legend for figure 4.1.
Responses are expressed as the number of "'In-neutrophils per skin site and are the mean
± s.e.m for n=4 rabbits; all data subtracted for saline background levels.
95
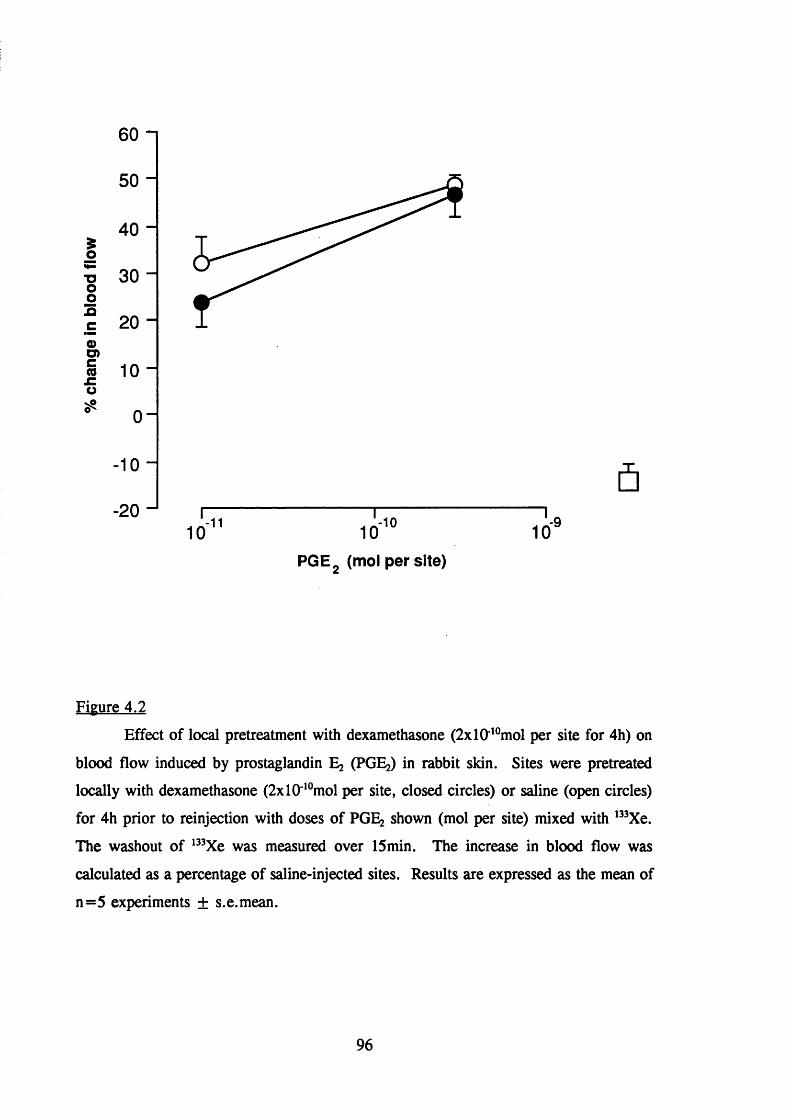
IH—
%Onc0 )O)g€nP
60
50
40
30
20
10
0
- 1 0
-201 0
1 1
1 0
10 10-9
PGE- (mol per site)
Figure 4.2
Effect of local pretreatment with dexamethasone (2xlO^°mol per site for 4h) on
blood flow induced by prostaglandin 5% (PGEg) in rabbit skin. Sites were pretreated
locally with dexamethasone (2 x l 0 *°mol per site, closed circles) or saline (open circles)
for 4h prior to reinjection with doses of PGE2 shown (mol per site) mixed with ^%e.
The washout of '% e was measured over 15min. The increase in blood flow was
calculated as a percentage of saline-injected sites. Results are expressed as the mean of
n=5 experiments ± s.e.mean.
96

4.2.2 The systemic treatment of rabbits with dexamethasone
4.2.2.1 The effect of intravenous dexamethasone on * "In-neutrophil
accumulation in rabbit skin
To investigate the effect of systemic treatment with dexamethasone on "^In-
neutrophil accumulation and oedema formation, pairs of animals were treated
intravenously with either dexamethasone (3mg/kg) or saline (1 ml/kg) 4 hours prior to the
intravenous administration of " 'In-neutrophils, "^I-albumin and intradermal inflammatory
mediators, mixed with PGE2 . The accumulation of labels was measured over 30 minutes.
Figure 4.3 shows that in control animals there was a dose-related increase in "^In-
neutrophil accumulation in response to the three chemoattractants FMLP 4- PGEg, LTB4
+ PGE2 and ZAP 4- PGE2 (open circles). Systemic pretreatment of animals with
dexamethasone significantly inhibited the accumulation of "'In-neutrophils in response
to all doses of chemoattractants tested (closed circles) except at 5xl(y'°moles/site LTB4
where the apparent inhibition was not significant (figure 4.3). This inhibitory effect of
dexamethasone was not due to a difference in the number of '"In-neutrophils circulating
in the two groups of animals ie. the percentage of labelled neutrophils circulating in
control and dexamethasone treated rabbits at the end of the 30 minute accumulation
period were 43.7± 10.7% and 39.3±11.8 (mean ± SEM, n=9 pairs of rabbits)
respectively. The neutrophil-independent mediators platelet-activating factor and
bradykinin caused minimal cell accumulation in either group of animals (figure 4.3).
Intradermal injection of PGEj alone caused little cell accumulation (figure 4.3).
4.2.2.2. The effect of intravenous dexamethasone on oedema formation in
rabbit skin
The simultaneous measurement of plasma protein leakage in these animals which
is illustrated in figure 4.4 shows that oedema responses to both neutrophil-dependent
(except SxlO^'^moles/site LTB4) and neutrophil-independent mediators of increased
microvascular permeability were significantly (p < 0 .0 1 ) suppressed in dexamethasone
pretreated animals. PGEj injected alone cause little oedema formation (figure 4.4).
Together the results of sections 4.2.2.1. and 4.2.2.2. show that systemic treatment
with dexamethasone inhibited cell accumulation and oedema formation in response to all
combinations of mediators tested.
97

5000
4000
3000
2000B(0o 1000Q.
O 0
*** **
jg 3E3 Ou (0= 3000-1 x:Q.2 4-»3Ç) 2 0 0 0 - 1
1000 -
-1210% 1 0 0 % 5 x 1 0
ZAP FMLP
-11 -11 5 x 1 0 5 x 1 0 5 x 1 0
10
LTB>
1 0 ‘® 1 0 '^PAR BK
1 0 '^° 3x10"^°PGEo
Figure 4.3Effect of i.v. dexamethasone (3mg/kg) on "Un-labelled neutrophil accumulation
in response to i.d. injections of inflammatory mediators. Concentrations are given as mol per site for N-formyl-methionyl-leucyl-phenylalanine (FMLP), leukotriene B4 (LTB4), platelet activating factor (FAF) and bradykinin (Bk). Zymosan activated plasma (ZAP) was used undiluted (1(X)%) or as a 1:10 dilution in heparinized rabbit plasma (10%). All stimuli were co-injected with a fixed potentiating dose of prostaglandin Ej (PGE2 ), 3xlO'*°mol per site. "^In-neutrophil accumulation was measured in recipient animals given i.v. dexamethasone (closed circles) or saline (open circles) 4h prior to i.v. * "In-neutrophils and i.d. mediators. Responses are corrected for the low levels in saline- injected sites. Each point represents the mean±s.e.mean for n=9 pairs of rabbits. Significant difference from control: *P<0.05, and **P<0.01.
98

50
40
2 30w 20
I 10
â 0
**
**
Q)E3O>(0ESQ.C!S(0
1 0 0
90"80-7060150403012010
0 "
10ZAP
1 0 0 % 5 x 1 0 " 5 x 1 0FMLP
10 •101 0 ' 1 0 '^ 10 10
**
- 1 1
5 x 1 0• 1 1
5 x 1 0-10
LTB.
PAR BK
3 x 10 PGE
-10
Figure 4.4Effect of i.v. dexamethasone (3mg/kg) on plasma protein extravasation in response
to inflammatory mediators: oedema responses measured in the same recipient animals as in Figure 4.3. Concentrations are given as mol per site for N-formyl-methionyl-leucyl- phenylalanine (FMLP), leukotriene B4 (LTBJ, platelet-activating factor (FAF) and bradykinin (Bk). Zymosan activated plasma (ZAP) was used undiluted (100%) or as a 1:10 dilution in heparinized rabbit plasma (10%). Oedema formation was measured in dexamethasone pretreated recipients (closed circles) and recipient animals pretreated with an equal volume of saline (open circles). Responses are corrected for the low levels in saline-injected sites. Each point represents the mean ± s.e.mean for n=9 pairs of rabbits. Significant difference from control **P<0.01.
99

4.1.2.3 Effect of intravenous dexamethasone (3mg/kg) on oedema formation
potentiated with calcitonin gene-related peptide
The neuropeptide calcitonin gene-related peptide (CGRP) is an extremely potent
microvascular dilator with a long duration of action in human and animal skin (Brain et
al., 1985; Brain et al., 1986b). Studies in the cutaneous microvasculature have shown
that CGRP, as a consequence of its vasodilator activity, acts synergistically with
mediators of increased microvascular permeability to potentiate inflammatory oedema
formation (Brain & Williams, 1985; Brain et a l , 1986a; Buckley et a l , 1988). An
effect of systemic dexamethasone on the action of prostaglandin Eg, removing its
potentiating action, might possibly have explained the inhibition of "^In-neutrophil
accumulation and oedema formation observed in animals that underwent dexamethasone
treatment. To investigate this possibility further, microvascular permeability in response
to a fixed dose of bradykinin ( 1 0 '^°moles/site) was potentiated with prostaglandin E; or
CGRP in the same animals and the effect of systemic dexamethasone (3mg/kg, 4h) was
investigated. Control animals received intravenous saline. Oedema formation was
measured over 30 minutes. Figure 4.5 shows that in animals receiving i.v. saline the
plasma protein leakage induced by Bk injected alone (28±6/xl oedema/site, open squares)
was clearly potentiated by coinjection with prostaglandin E2 or CGRP. The extent of this
potentiation was dependent on the dose of vasodilator. CGRP was clearly more potent
than PGE2 at potentiating the oedema response, 53±6/xl oedema/site was obtained with
a potentiating dose of 3x10" moles/si te PGEj as compared with 77±8/il oedema/site with
the same dose of CGRP. Such potentiation of oedema was only matched with a dose of
3xlO '°moles/site PGE2 (80±10/xl oedema/site). CGRP (top dose 3xlO"moles/site)
injected alone, like PGE2 (top dose 3xlO ‘°moles/site) induced little plasma leakage (figure
4.5). In animals treated with intravenous dexamethasone, oedema responses to Bk
potentiated by all doses of both PGEg and CGRP were significantly (p<0.01) suppressed
(figure 4.5, closed symbols). The oedema response to i.d. injection of Bk alone was also
significantly (p < 0 .0 1 ) inhibited in dexamethasone treated animals when compared to
animals which underwent saline treatment (8 ± 1.5^1 oedema/site and 28±6/xl oedema/site
respectively).
These results suggest that the inhibitory effect of systemic dexamethasone on
oedema formation was unlikely to be a result of inhibition of vasodilator activity.
100

80
B(0
II30 >
1 (/) OTQ.C
%
60
40
20
0 ■13 -12 -113 x 10 3 x 10 3 x 10 3 x 10
concentration of dilator (mol per site)
■10
Figure 4.5
Effect of i.v. dexamethasone (3mg/kg) on plasma protein extravasation induced
by i.d. bradykinin (Bk, square symbols) potentiated by co-injection with prostaglandin
Eg (PGE2 , circular symbols) or calcitonin gene-related peptide (CGRP, triangular
symbols). Concentrations of vasodilators are given as mol per site as shown, Bk was
used at a concentration of lO^^mol per site. Oedema formation was measured over 30
minutes in animals pretreated for 4 hours with dexamethasone (closed symbols) and
animals pretreated with an equal volume of saline (open symbols). Responses are
corrected for the low levels in saline-injected sites. Each point represents the mean ±
s.e.mean for n=4 rabbits.
101

4.2.3 The systemic treatment of donor rabbits with dexamethasone
4.2.3.1 Effect of pretreating neutrophil donor rabbits with systemic
dexamethasone on "Tn-neutrophil accumulation in recipient animals
The experiments shown in figure 4.6 were designed to investigate a possible effect
of steroid, or a steroid-induced product on the neutrophil in vivo. For these experiments
neutrophil donor animals were treated intravenously with either dexamethasone (3mg/kg)
or an equal volume of saline 4 hours prior to collection of blood, neutrophil isolation and
* "In-labelling. * "In-neutrophils from treated and control donor animals were injected
intravenously into two untreated recipient animals respectively, together with *“ l-albumin.
Cell accumulation and oedema formation were measured in recipient rabbits in response
to intradermal inflammatory mediators mixed with PGEj. Figure 4.6 shows that " ‘In
neutrophils from control saline-treated donor animals accumulated in recipients to the
three chemoattractants ZAP, FMLP and LTB4 in a dose related manner (open circles).
Treatment of neutrophil donor animals with dexamethasone significantly (P<0.05)
inhibited the accumulation of treated ‘"In-neutrophils induced by all (except 5x10*
‘ moles/site FMLP) doses of chemoattractants in recipients. At the end of the 30 minute
accumulation period the percentage of circulating " ‘In-neutrophils did not differ
significantly between the two groups and, therefore, this did not account for the
differences in cell accumulation observed. The percentage of treated and control
" ‘In-neutrophils circulating after 30 minutes were 37± 10 and 35+8 (mean ± SEM, n=9
pairs) respectively. The neutrophil-independent mediators platelet-activating factor and
bradykinin induced very little cell accumulation in either group of recipients (data not
shown). Little cell accumulation was observed in response to PGEj injected alone (data
not shown).
These results suggest that dexamethasone treatment of neutrophils in vivo is able
to suppress their ability to accumulate in vivo in response to chemoattractants.
4.2.3.2. Effect of pretreating neutrophil donor rabbits i.v. with dexamethasone
on oedema formation in recipient animals.
The data illustrated in figure 4.7. shows oedema responses measured in the same
recipient animals as those shown in figure 4.6. Figure 4.7 clearly demonstrates that there
was no significant difference in oedema responses to either direct-acting or neutrophil-
dependent mediators between the two groups of recipient rabbits (i.e. those receiving
" ‘In-neutrophils from dexamethasone treated donor animals and those receiving
102

s1 3000-OQ.CO% 2000- 3E3§ 1 0 0 0 -
Q.sg 0 0 )
-1210% 1 0 0 % 5 x 1 0
ZAP FMLP
. - 1 1
r -101 1
8 I
LTB3 x 10
PGEg
-10
Figure 4.6
Accumulation of neutrophils isolated from saline (open circles) or dexamethasone
(3mg/kg, closed circles) - pretreated donor rabbits in untreated recipient animals.
‘"In-neutrophil accumulation in recipient rabbits was measured over 30 min in response
to i.d. injections of doses of stimuli shown as mol per site for N-formyl-methionyl-
phylalanine (FMLP) and leukotriene B4 (LTB4) and 100% and 10% zymosan activated
plasma (ZAP), all mixed with a fixed potentiating dose of prostaglandin Eg (PGEg,
3x10 ‘°mol per site). Responses are corrected for the low levels in saline injected sites.
Each point represents the mean ± s.e.mean for n=9 pairs of rabbits. Significant
difference from control, *P<0.05.
103

801
50
40
g 30 w 20a 10
0 >E3O>(0E( 0
Q.cZco
908070605040302010
0
10 100% 5 x 10-12 -11 -11
5 x 1 0 5 x 1 0 5 x 1 0-10
ZAP FMLP LTBy
10 -101 0 '^ lO'"* 10 -10
PAF BK
6
I3 x 1 0
PGE
-10
Figure 4.7
Oedema formation in untreated rabbits receiving labelled-neutrophils isolated from
saline (open circles) or dexamethasone (3mg/kg, closed circles) pretreated donor rabbits.
Oedema responses were measured in the same recipient animals as in Figure 4.6.
Responses are corrected for the low levels in saline-injected sites. Each point represents
the mean±s.e.mean for n=9 pairs of rabbits.
104

“ ^In-neutrophils from saline treated donor animals). In both groups of animals a dose
related increase in oedema formation was observed in response to all mediators under
test, coinjected with PGE;. ‘"In-labelled cells account for approximately 3% of the total
circulating neutrophils in recipients and therefore do not make a major functional
contribution to oedema formation.
4.2.4. Summary
1 Local treatment of skin sites with dexamethasone inhibits oedema responses
induced by direct-acting and neutrophil-dependent mediators of increased
microvascular permeability coinjected with prostaglandin Eg. The inhibitory
action is time-dependent, significant suppression of oedema formation is only
evident after treatment of skin sites for 4 hours prior to injection of inflammatory
mediators. Inhibition is not a consequence of inhibition of the generation of pro-
inflammatory products of the cyclooxygenase pathway.
2 Local treatment of skin sites with dexamethasone does not result in inhibition of
neutrophil accumulation in response to intradermal chemoattractants + PGEj even
after 4 hours pretreatment with dexamethasone. Local indomethacin similarly has
no effect on neutrophil accumulation.
3 Treatment of skin sites for 4 hours with dexamethasone has no significant effect
on blood flow induced by prostaglandin Ej although a slight decrease in basal
blood flow is observed.
4 Treatment of animals with intravenous dexamethasone for 4 hours inhibits oedema
formation induced by direct acting and neutrophil dependent mediators. Oedema
formation potentiated by either PGE; or CGRP is inhibited by intravenous
dexamethasone.
5 Treatment of ‘ "In-neutrophil recipient rabbits with intravenous dexamethasone for
4 hours inhibits ‘"In-neutrophil accumulation in skin in response to FMLP 4-
PGE;, ZAP + PGE; and LTB4 + PGE;.
105

Intravenous administration of dexamethasone to neutrophil donor animals 4 hours
prior to bleeding and neutrophil isolation inhibits the accumulation of treated
neutrophils into the skin of untreated recipient animals injected intradermally with
chemoattractants + PGE;.
106

CHAPTER 5 GENERATION. CHARACTERISATION AND MODULATION
OF INFLAMMATORY ACTIVITY IN RABBIT
PERITONEAL EXUDATE
5.1 Introduction
The inflammatory response induced by intraperitoneal zymosan in the rabbit has
been investigated (Forrest et a l , 1986; Jose et al., 1985; Collins et a l , 1991a).
However, the involvement of the lipoxygenase metabolite of arachidonic acid LTB4 was
not studied. LTB4 is known to increase vascular permeability (Wedmore & Williams,
1981a), and to promote neutrophil adhesion and transmigration of endothelial cells (Bjork
et a l , 1982; Dahlen et a l , 1981). In the present study the involvement of LTB4, and
also TXB2 in the inflammatory response to zymosan was investigated by measuring levels
of these mediators in inflammatory exudate recovered from the peritoneal cavity. In
addition, in order to further understand the underlying mechanisms involved in this model
the role of circulating neutrophils in the development of the inflammatory response was
investigated.
From the results of chapter 4 it is clear that dexamethasone can act in vivo on both
the endothelial cell and the neutrophil to inhibit the action of inflammatory mediators.
In the present investigation the effect of steroids in a more complex model involving both
mediator generation and action was studied. This study was undertaken in an attempt to
gain insight into the relative contribution of steroid inhibition of inflammatory mediator
action and mediator generation. The effect of systemic dexamethasone on the
inflammatory response induced by zymosan in the peritoneal cavity was assessed.
107

5.2 Results
5.2.1 The inflammatory response to intraperitoneal zvmosan
To investigate the response to zymosan, pairs of anaesthetised animals were
injected intraperitoneally with either sterile saline (50ml) or a suspension of zymosan
(5(X)mg in 50ml) following intravenous injection with Evans blue dye (2.5%, 0.5ml/kg).
Sequential samples of exudate were withdrawn from the cavity at the times indicated for
subsequent analysis.
Table 5.1 clearly shows that the intraperitoneal injection of zymosan induced
plasma protein extravasation into the cavity as measured by the accumulation of albumin-
bound Evans blue dye. The exudate: plasma blue dye ratio rose with the time of exudate
collection (0.013 at 5 minutes, 0.39 at 8 hours, n=7) and was indicative of an
inflammatory reaction in the cavity. Leakage was first detectable at 15-30 minutes; the
dye concentration increased most rapidly during the first hour, thereafter the
concentration rose progressively but at a slower rate, and little change occurred between
6 and 8 hours although the concentration of dye remained high. In contrast, little dye
was apparent in control fluids obtained after injection of saline (exudate: plasma dye ratio
of 0.032 at 1 hour compared to 0.19 in animals receiving zymosan for one hour, n=7,
table 5.1). The exudate: plasma dye ratio increased slightly with time in control animals
reaching a value of 0.08 at 8 hours (approximately 2 0 % of test levels at 8 hours).
Figure 5.1 shows the appearance of neutrophils in exudates. Accurate estimates
of the number of neutrophils present in early exudates from zymosan-injected animals
were difficult to obtain because of the preponderance of zymosan particles and small
numbers of cells. Two hours after injection of zymosan, significant numbers of
neutrophils were found in exudate fluid; 4.0±0.68xl0* neutrophils/ml, compared with
2.0±0.5xl0^cells/ml in paired exudates from saline treated animals. The number of
neutrophils accumulating in zymosan injected cavities increased slowly between 2 and 4
hours followed by a progressive increase in the rate of accumulation with time. A few
neutrophils were detected in some control exudates, reaching a maximum number of
2.09±0.7xl0‘* neutrophils/ml in 8 hour exudates, compared to 3± 0.62x 1 O^neutrophils/ml
in paired exudates from test animals.
108

Time of sample recovery
from peritoneal cavity
(mins)
exudate O.D.
plasma O.D.
saline
n=7
zymosan
n=7
5 0.02±0.004 0.01 ±0.004
15 0 .0 1 + 0 . 0 0 2 0.08±0.009
30 0 .0 2 + 0 . 0 0 1 0 . 1 0 + 0 . 0 1 0
60 0.03+0.002 0.19±0.030
1 2 0 0.05+0.020 0.23±0.040
240 0.08 +0.040 0.33+0.040
360 0.09+0.010 0.38+0.120
480 0.08+0.050 0.39±0.080
Table 5.1
Measurement of microvascular plasma protein leakage after intraperitoneal
injection of saline (50mls) or zymosan (500mg in 50mls) in rabbits. Values are the
exudate: plasma ratio of i.v. injected Evans blue dye concentration in samples taken at
the various times indicated following i.p. injection. Results are expressed as the mean
± s.e.mean for n==7 rabbits in each group.
109
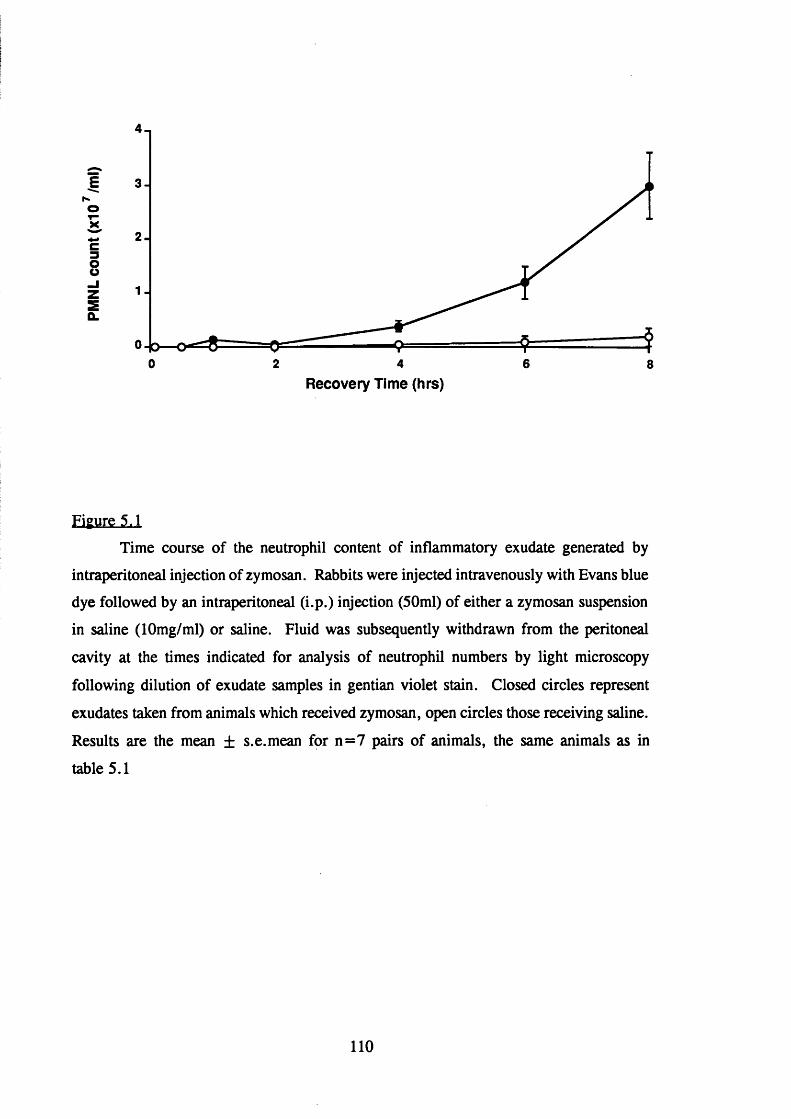
38
sû.
0 2 64 8
Recovery Time (hrs)
Figure 5.1
Time course of the neutrophil content of inflammatory exudate generated by
intraperitoneal injection of zymosan. Rabbits were injected intravenously with Evans blue
dye followed by an intraperitoneal (i.p.) injection (50ml) of either a zymosan suspension
in saline (lOmg/ml) or saline. Fluid was subsequently withdrawn from the peritoneal
cavity at the times indicated for analysis of neutrophil numbers by light microscopy
following dilution of exudate samples in gentian violet stain. Closed circles represent
exudates taken from animals which received zymosan, open circles those receiving saline.
Results are the mean ± s.e.mean for n=7 pairs of animals, the same animals as in
table 5.1
110

5.2.2 Time course of the generation of inflammatory mediators in zvmosan-induced
peritoneal exudate
Samples of exudate fluids recovered at the times indicated from the pairs of
animals were also analysed for the levels of various inflammatory mediators using
radioimmunoassay.
5.2.2.1. LTB^
Figure 5.2 shows the concentration of immunoreactive LTB4 (i.r. LTB4) with time
in exudate fluids obtained from zymosan (closed circles) and saline (open circles) injected
animals. Low levels of LTB4 were detected in exudate fluids at all time points tested
following intraperitoneal injection of saline. Levels in these fluids were not significantly
different from that detected in zymosan suspensions prior to injection into the cavity
(0.9±0.2ng LTB4 /ml). Exudate fluids from test animals were found to contain i.r. LTB4.
Significantly elevated levels of LTB4 were detected 30 minutes after zymosan injection
(2.7±0.36ng/ml). Levels continued to rise reaching a maximum of 6.04±0.79ng/ml at
4 hours. The LTB4 content of 6 and 8 hour exudates fell progressively although it
remained elevated above levels in saline treated animals even after 8 hours.
5.2.2.2. Thromboxane (TXB )
Figure 5.3 shows the time course of generation of immunoreactive TXB2 .
Zymosan caused a rapid and pronounced rise in TXB2 ; maximal levels were detected 5
minutes after zymosan injection (4.74±0.9ng/ml, figure 5.3 closed circles). The level
was maintained at the 15 minute time point but was followed by a relatively rapid decline
over the next 1 0 0 minutes to levels similar to those detected in saline injected animals.
Levels of TXBj then remained low (under l.Ong/ml) throughout the course of the
experiment as did the TXB2 content in saline exudates (approximately 0.5ng/ml or less
TXB2 was consistently found throughout the time period investigated, figure 5.3, open
circles).
5.2.2.3. Prostaglandin and 6-oxo-prostaglandin
Figures 5.4 and 5.5. show the concentrations of the vasodilator substances PGB2
and PGI2 (detected as the stable break-down product 6 -oxo-PGFi J respectively in exudate
fluids. Low concentrations of immunoreactive PGE2 were found in saline exudates
111

7-,
6 -
2 4 6 80
Recovery Time (hrs)
Figure 5.2
Kinetics of the generation of leukotriene B4 (LTB4) induced by zymosan.
Anaesthetised rabbits were injected intravenously with Evans blue dye followed by an
intraperitoneal injection (50ml) of either a zymosan suspension in sterile saline (lOmg/ml)
or sterile saline. Peritoneal fluid (closed circles zymosan, open circles saline) was then
withdrawn through an indwelling cannular at timed intervals for analysis.
Radioimmunoassay was used to measure immunoreactive LTB4 in exudate fluids. Each
point represents the mean result of exudates obtained from 7 rabbits in each group,
vertical lines show s.e.mean. *P<0.05 compared with control exudates.
112

5 .( H i’
O)3 .
2 4 60 8
Recovery Time (hrs)
Figure 3,3
Kinetics of the generation of Thromboxane (TXB2 ) induced by zymosan.
Levels of immunoreactive TXBj in exudates generated from the same animals as in figure
5.2; closed circles zymosan and open circles saline treated animals. Results are the mean
± s.e.mean from 7 pairs of rabbits, *P<0.05 compared with control exudates.
113

2JS .
2.0 .
I 1.5 -
UJ
2
1.0 -
0.064 820
Recovery Time (hrs)
200 n
Si100 -
CO
86420Recovery Time (hrs)
Figure 5.4 and 5.5
Kinetics of the generation of prostaglandin Eg (PGEg, figure 5.4) and 6 -oxo-PGFj^
(figure 5.5) induced by zymosan. Levels of inflammatory mediators were measured by
radioimmunoassay in the same exudate samples as in figure 5.2. Closed circles represent
levels in exudates from zymosan treated animals, open circles levels in exudates from
saline treated animals. Values are the mean ± s.e.mean of exudates obtained from 7
pairs of rabbits. Figure 5.5., *P < 0.05 compared with the paired saline control exudates.
114

(consistently below l.Ong/ml). Levels in zymosan exudates were not significantly
different from control animals at any time points (Figure 5.4). In comparison a large
increase in immunoreactive 6 -oxo-PGFi„ was apparent following zymosan injection
(figure 5.5). A significant increase was detected 5 minutes after zymosan injection
(135.2±30.4ng/ml, closed circles figure 5.5). Levels remained similar up to 30 minutes
post-zymosan. Although levels fell slightly by one hour (120±22.2ng/ml) this
concentration was maintained up to 2 hours before falling to 60±15ng/ml by 4 hours.
At 6 and 8 hours, levels in zymosan exudates were not significantly different from the
low levels found in exudates from saline injected animals (below 1 2 ng/ml throughout the
time course).
5.2.3 The role of circulating neutrophils in the response to intraperitoneal zvmosan
To investigate the role of circulating neutrophils in the inflammatory response to
intraperitoneal zymosan, animals were rendered neutropenic by a single i.v. injection of
nitrogen mustard at a dose of 1.75mg/kg three days prior to the experiment (Wedmore
& Williams, 1981a). Control animals received an equal volume of saline. The
circulating neutrophil count three days after nitrogen mustard was approximately 1 % of
the pretreatment value. Neutropenic and control animals were then injected intravenously
with Evans blue dye (2.5%, 0.5ml/kg) and intraperitoneally with 500mg of zymosan
suspension in 50mls of sterile saline. Samples of exudate fluid were taken at timed
intervals after zymosan administration for subsequent analysis of the inflammatory
response.
5.2.3.1 The effect of depletion of circulating neutrophils on plasma
extravasation and neutrophil accumulation in response to
intraperitoneal zvmosan
Table 5.2 shows the exudate: plasma dye ratio of exudates generated in control
and neutropenic animals. In control animals i.p. zymosan caused the accumulation of
albumin bound Evans blue dye into the cavity. In contrast the exudate: plasma dye ratio
in mustine treated animals was significantly (p<0.05) reduced compared to controls at
one hour post zymosan and all subsequent time points. The exudate: plasma dye ratio
in neutropenic animals was reduced by approximately 60-70% compared with control
animals, even after 6 hours of zymosan treatment.
The accumulation of neutrophils into the cavity which occurred in control animals
115

Time of sample recovery
from peritoneal cavity
(mins)
exudate O.D.
plasma O.D.
control
animals
n=9
neutropenic
animals
n=9
5 0.003 +0.002 0 . 0 0 2 ± 0 . 0 0 1
30 0.080+0.014 0.028 ±0.007
60 0.170±0.040 0.068+0.013*
1 2 0 0.270+0.048 0.086+0.029*
240 0.30+0.060 0.140+0.044*
360 0.370+0.10 0.152+0.090*
Table 5.2
Measurement of microvascular plasma protein leakage following intraperitoneal
injection of zymosan (500mg) in rabbits rendered neutropenic by prior treatment with
mustine hydrochloride and in control untreated rabbits. Values are the exudate: plasma
blue dye ratio in exudate fluid collected at the times indicated following i.p. injection.
Results are expressed as the mean ± s.e.mean of values obtained from 9 rabbits in each
group. *, significantly decreased compared with numbers in control exudates recovered
at the same time, P < 0.05.
116

(table 5.3) did not occur in the neutropenic group of animals. 2 hour post zymosan
2.63±0.44 xlO^cells/ml accumulated in control animals rising to 8.36±0.82xl0^ cells/ml
6 hours post zymosan. The number of cells found in exudate from neutropenic animals
was 3.3+0.8x10^ cells/ml and 3.2±0.4xl0^cells/ml at the same time points (table 5.3).
5.2.3.2. The generation of LTB^ in response to zvmosan in neutropenic animals
There was no significant difference between neutropenic and control rabbits in the
low levels of i.r. LTB4 found in early (up to 1 hour post zymosan) exudate fluids (figure
5.6). Levels of i.r. LTB4 at 1 hour were 2.5±0.4ng/ml in mustine treated animals and
2.8±0.5 ng/ml in control animals. At 2 hours post zymosan levels of i.r. LTB4 in
control animals began to rise and continued to rise reaching levels of 5.9±l.Ong/ml and
5.7±1.2ng/ml at 4 and 6 hours respectively. However, neutropenic rabbits failed to
mount an LTB4 response during this time period and levels at 4 and 6 hours were
significantly (p<0.05) reduced compared to controls being 2.5 ±0.19 and 2.0±0.2ng/ml
respectively.
5.2.3.3. The generation of 6 -oxo-prostaglandin F,, in neutropenic animals
As shown in figure 5.7 there was no significant difference in the zymosan induced
generation of i.r. 6 -oxo-PGFi„ between neutropenic and control animals at any time
point. The response obtained in both groups of animals showed a similar pattern to that
obtained in figure 5.5.
5.2.4. The effects of intravenous dexamethasone on the inflammatorv
response to intraperitoneal zvmosan
To investigate the effect of systemic dexamethasone treatment on the inflammatory
response to zymosan, pairs of animals were treated intravenously with either
dexamethasone (3 mg/kg) or an equal volume of saline four hours prior to the
intraperitoneal injection of a suspension of zymosan (lOmg/ml, 50mls/animal). Just
before the administration of zymosan, dexamethasone-treated animals were given a
second intravenous injection of dexamethasone ( 1 mg/kg) while control animals received
an equal volume of saline. Both groups of animals received i.v. Evans blue due (2.5%,
0.5mg/kg). Samples of exudate fluid were then removed at timed intervals for
subsequent analysis of the inflammatory response.
117

Time of sample recovery
from peritoneal cavity
(mins)
no of neutrophils (xlO^/ml)
control animals
n=9
neutropenic animals
n=9
60 0.07+0.06 0.015+0.010
1 2 0 0.26+0.04 0.003 +0.001
240 2.32+1.20 0.006+0.001
360 8.36+0.82 0.003 +0.008
Table 5.3
The appearance of neutrophils in the peritoneal cavity of rabbits following
intraperitoneal injection of zymosan (500mg in 50mls). Animals were treated 3 days
previously with mustine hydrochloride to induce neutropenia, control animals received
pretreatment with saline. Values are the number of neutrophils xlO^/ml of exudate in
fluid recovered at the times indicated after zymosan. Results are the mean ± s.e.mean
for n=9 pairs of animals.
118

7-1
5 -Ic 4 -
S 3 -
4 5 631 20
Recovery Time (hrs)
Figure 5.6
The effect of neutropenia on the generation of leukotriene B4 (LTB4) induced by
intraperitoneal zymosan. Neutropenic or control animals were injected i.p. with zymosan
(500mg in 50ml of sterile saline) and exudate fluid was removed at timed intervals for
analysis of the inflammatory response (tables 5.2 and 5.3) and levels of inflammatory
mediators by radioimmunoassay. The symbols represent; open triangles, levels of
immunoreactive LTB4 in exudates from neutropenic animals, closed triangles levels in
exudates from control animals. Values are the mean ± s.e.mean for 9 pairs of animals.
*P<0.05 compared to levels in control exudates.
119

Iu_2I
300 H
200 -
100 -
2 3 4 50 1 6
Recovery Time (hrs)
Figure 5.7
The effect of neutropenia on the generation of 6 -oxo-PGFj^ induced by
intraperitoneal zymosan. Levels of immunoreactive 6 -oxo-PGFi„ in the same exudates
as in figure 5.6. Values are the mean ± s.e.mean of exudates obtained from 7
neutropenic animals (open traingles) and 7 control animals (closed triangles).
120

5.2.4.1. The effect of dexamethasone on plasma extravasation and neutrophil
accumulation into zvmosan injected cavities
Table 5.4 shows the exudate:plasma dye ratio of exudates generated from control
and dexamethasone treated animals. Dexamethasone significantly (p < 0.05) inhibited the
extravasation of Evans blue dye into the cavity which occurred in control animals.
The zymosan-induced accumulation of neutrophils in exudate fluids recovered at
various times from the cavity of dexamethasone-treated and control animals is shown in
table 5.5. In control animals, zymosan induced neutrophil accumulation; 2 hours post
zymosan 3.26± 1.0x10^ cells/ml had accumulated and the number continued to rise at 4
and 6 hours. This neutrophil accumulation did not occur in animals pretreated
systemically with dexamethasone prior to intraperitoneal zymosan. Significantly
(P<0.05) fewer neutrophils accumulated in response to intraperitoneal zymosan in
animals treated with i.v. dexamethasone at all time points.
Exudate fluids taken at 6 hours from dexamethasone-treated animals were, in the
main found to still contain large numbers of zymosan particles whereas control exudate
fluids were found to be mostly clear of zymosan particles by 4 hours and definitely by
6 hours.
5.2.4.2. The effect of systemic dexamethasone on LTB generation in response
to zvmosan
Figure 5.8 shows the generation of LTB4 in zymosan-induced exudate fluids from
animals treated i.v. with dexamethasone (open squares) or saline (closed squares). Up
to one hour post-zymosan there was no significant difference between the two groups of
animals - levels of LTB4 were between 1.9 and 2.7ng/ml during this time period which
is of the same order of magnitude as that found previously (Figure 5.2 closed circles).
At 2, 4 and 6 hours post-zymosan injection there was a significant (p<0.05) difference
in the concentration of LTB4 found in animals treated intravenously with dexamethasone
compared to paired saline treated animals. In dexamethasone-treated animals the level
of LTB4 remained around 2.0ng/ml during this time period. In contrast, in animals
receiving saline prior to intraperitoneal zymosan the levels of LTB4 were significantly
elevated reaching a maximum of 6.9± 1.4 (figure 5.8) at 4 hours falling to 5.3±0.9ng/ml
by 6 hours. This pattern was very similar to that observed previously (see Section
5.2.2.1., figure 5.2 closed circles).
121

Time of sample recovery
from peritoneal cavity
(mins)
exudate O.D.
plasma O.D.
Saline i.v.
n=9
Dexamethasone i.v.
n=9
5 0 . 0 1 ± 0 . 0 1 0 .0 0 2 + 0 . 0 0 1
15 0.04±0.01 0.015+0.010*
30 0 . 1 0 ± 0 . 0 1 0.017+0.006*
60 0.23+0.09 0.029+0.013*
1 2 0 0.25+0.08 0.090+0.040*
240 0.29+0.07 0.090+0.060*
360 0.28+0.10 0.100+0.090*
Table 5.4
The effect of intravenous dexamethasone treatment on the increase in
microvascular plasma protein leakage induced by intraperitoneal zymosan (500mg in
50mls). Animals treated i.v. with either saline or dexamethasone (3mg/kg) for 4 hours
were then injected i.v. with Evans blue dye (2.5%, 0.5ml/kg) and, in the case of treated
animals a second dose of dexamethasone ( 1 mg/kg), for control animals on equal volume
of saline, followed for all animals by i.p. zymosan. The exudate:plasma blue dye ratio
for exudate fluids recovered at the times indicated were calculated and are expressed as
the mean ± s.e.mean for n=9 pairs of animals. *, significantly decreased compared with
paired exudates generated in animals receiving i.v. saline, P<0.05.
122

Time of sample recovery
from peritoneal cavity
(mins)
Number of neutrophils (xlO^/ml)
i.v. Saline
n=9
i.v. Dexamethasone
n=9
60 0.16±0.013 0 . 0 1 ± 0 .0 0 1 *
1 2 0 0.33+0.10 0 .0 2 ± 0 .0 0 1 *
240 2.40+1.00 0.30+0.20*
360 8.00+1.06 0.50+0.45*
Table 5.5
The effect of intravenous dexamethasone on the accumulation of neutrophils in
response to intraperitoneal zymosan (500mg). Animals were treated with saline or
dexamethasone (3mg/kg 4 hours, and 1 mg/kg 10 min) prior to i.p. zymosan. Exudate
fluid was collected at the times indicated and the number of neutrophils in samples was
assessed using light microscopy. The values represent the number of neutrophils
xlOVml of exudate and are expressed as the mean ± s.e.mean for n=9 rabbits in each
treatment group. *P<0.05 compared with control exudates.
123

9 - ,
8 .
O)
5 .
4 .
3 .
2 -
3 52 4 60 1
Recovery Time (hrs)
Figure 5.8
The effect of dexamethasone on the generation of leukotriene B4 (LTBJ in
zymosan-induced inflammatory exudate. Animals were treated with either i.v. saline or
i.v. dexamethasone (3mg/kg 4h and 1 mg/kg 10 min) prior to i.p. zymosan (500mg in
50ml sterile saline). Exudate fluid was removed at timed intervals for analysis of the
inflammatory response (tables 5.4 and 5.5) and for the levels of inflammatory mediators
by radioimmunoassay. Levels of LTB4 in exudates from dexamethasone (open squares)
and control (closed squares) animals are shown, n=7 animals in each group. *P<0.05
significantly reduced levels compared with untreated animals.
124

S.2.4.3. The effect of systemic dexamethasone on TXBj generation following
intraperitoneal zvmosan
As shown in figure 5.9 intraperitoneal zymosan caused a rapid (within 5 minutes)
increase in the concentration of TXBj in exudate fluid. Levels remained elevated for 15
minutes post-zymosan and then fell dramatically by one hour. The same pattern of
generation with time was found in both dexamethasone- and saline-treated animals and
there was no significant difference between the two groups at all time points except 1
hour where a significantly (p<0.05) lower concentration of TXBj was found in the
dexamethasone treated group of animals compared to control (2.7±0.6ng/ml in saline
treated animals, falling to 1.2±0.3ng/ml in dexamethasone treated animals).
5.2.4.4 The effect of dexamethasone on 6-oxo-PGF|^ generation in response to
zvmosan
In both the animals treated with dexamethasone and those treated with saline there
was considerable inter-animal variation in the concentration of 6 -oxo-PGFi„ detected in
exudate fluids (figure 5.10), also the magnitude of the levels detected were somewhat
greater than those found previously (see Section 5.2, figure 5.5. closed circles).
However, the time course of generation of 6 -oxo-PGFi„ followed an identical pattern to
that found in figure 5.5; that is a rapid and pronounced rise 5 minutes post-zymosan
followed by a slow decline over one and two hours reaching low levels by 6 hours
post-zymosan. The concentration of b-oxo-PGF,^ and the pattern of generation with
time did not differ in animals treated systemically with dexamethasone from those treated
systemically with saline prior to the zymosan challenge (figure 5.10).
5.2.4.5 Effect of dexamethasone on A23187-induced LTB production in whole
blood
Table 5.6 shows the generation of i.r. LTB4 by A23187-challenged blood cells.
Heparinized blood samples, taken from animals before and 4 hours after i.v. saline and
dexamethasone were incubated for 15min at 37 ®C with A23187 (10|tg/ml). Control
incubations with a similar volume of DMSO vehicle were carried out in parallel at all
time points. A23187-challenged blood gave i.r. LTB4 concentrations of around lOOng/ml
(table 5.6), compared with 17.9+1.Ong/ml in control incubations with DMSO vehicle.
The levels of i.r. LTB4 generated from ionophore-challenged blood cells did not differ
in animals treated i.v. with saline or dexamethasone, either before or after i.v. treatment
125

Figure 5.9
7 -
O)
4 .
2 .
63 4 520 1
I5
U_
268(6
Figure 5.10
Recovery Time (hrs)
400
300
200
100
653 420 1
Recovery Time (hrs)
Figure 5.9 and 5.10
The effect of dexamethasone on the generation of inflammatory mediators,
thromboxane B2 (TXB2 ) and 6 -oxo-PGFi„ induced by zymosan. Levels of TXBj (figure
5.9) and 6 -oxo-PGF,„ (figure 5.10) were measured in the same exudate samples as figure
5.8 and are represented as the mean ± s.e.mean for n=9 rabbits in each group (open
squares dexamethasone treated, closed squares control animals).
126

Time measurement taken i.r. LTB4 (ng/ml)
Before i.v. Saline 95.9+9.6
4h after i.v. Saline 107.2+12.4
Before i.v. Dexamethasone 100.6+23.3
4h after i.v. Dexamethasone 119.6+24.6
Table 5.6
The effect of intravenous dexamethasone (3mg/kg) on the generation of
immunoreactive leukotriene B4 (i.r. LTB4 ) by A23187-stimulated blood cells. Levels of
i.r. LTB4 were measured in samples before and 4h after i.v. treatment. Values are the
mean ± s.e.mean for n=9 rabbits in each group. Blood samples taken in parallel and
stimulated with a similar volume of DMSO generated 17.9±0.8ng i.r. LTB4 /ml, n=36).
127

(table 5.6). Furthermore, in some animals an additional challenge was performed on
blood samples taken 6 h after i.p. zymosan injection. There was also no difference in the
levels of i.r. LTB4 in samples taken from saline or dexamethasone treated animals
(84.6±8.1ng/ml, n=3 and 102.3±20.6 ng/ml, n = 6 respectively).
5.2.5. Authentication of immunoreactive LTB^
Authentication of the immunoreactive LTB4 in inflammatory exudates was kindly
carried out by Dr D. Masters (ICI) using reversed-phase HPLC (Rp-HPLC) fractionation.
A 5 micron spherisorb 0DS2 column was used and a linear gradient of 70-95% methanol
in 0.1 % (v/v) glacial acetic acid and water. The Rp-HPLC column was calibrated using
a cocktail of radiolabelled pH] LTB4 , pH] 12-HETE and pH] arachidonic acid (AA) in
70% methanol. The authentic standards were eluted as 70, 0.2 minute fractions and the
elution profile is shown in figure 5.11. The authentic LTB4 , 12-HETE and AA standards
appeared as three distinct peaks (figure 5.11). Samples of inflammatory exudate fluid in
70% methanol (100^1) were loaded onto the column and similarly eluted over 14 minutes
at 2ml/min. Fractions were evaporated to dryness and resuspended in RIA buffer and
i.r. LTB4 measured in 100/xl aliquots of each fraction. Figure 5.12 shows the i.r. LTB4
profile of an inflammatory exudate sample taken from an animal 4 hours after i.p.
zymosan. A major peak of immunoreactivity from the exudate occurred at a retention
time identical to that of authentic LTB4 (figure 5.12) thus confirming the identity of the
i.r. LTB4 in exudate fluid. Immunoreactive LTB4 was not detected in similarly processed
4 hour post zymosan-exudates derived from either animals receiving intravenous
dexamethasone (figure 5.13) or nitrogen mustard treated animals (figure 5.14).
128

10-,ARACH
9 -
12-HETE8 —
S
i = -O LTB4
5 -sÛ.
t -
i 3 -ORIGIN
2 —
1 -
10 148 1260 42
Elution Time (mlns)
Figure 5.11
Elution profile of authentic standards. Radiolabelled LTB4, 12-HETE and
arachidonic acid (arach) were used to calibrate the column and 70, 0.2min fractions
(400/zl) were collected over 14 minutes.
129

c01&
1 8
itoc3EE2
1.5 -
1.4 -
1.3 -
1.2 -
1.1 -
1 -
0.9 —
0 8 -
0.7 -
0 .6 _
0.5 _
0.4 _
0.3 _
0.2 _
0.1
0
0I I I I I I I I I I I I I I I I I I I I I I I I T ! I I I I I I I I I I I I I I I
2 4 6 8 10 12 14
Elution Time (mins)
Figure 5.12
Immunoreactive LTB4 profile following fractionation of inflammatory exudate in 70%
methanol by reversed-phase HPLC. 40, 0.35min fractions were collected and i.r. LTB4
was measured in 100/xl samples of each fraction. Figure 5.12 represents the i.r. LTB4
profile of inflammatory exudate collected 4 hours following i.p. zymosan, figures 5.13
and 5.14 that of similar exudate samples in animals treated with i.v. dexamethasone or
rendered neutropenic respectively.
130

1.5 —1 .4 -
1.3
1.2 -
5 c 0 8 -
9> ? 0 .7 _I 0.6 _
0 .5 _
0 .4 _
0.3 _
0 2 _
0.1
0 8 102 4 6 12 14
Elution Time (mins)
1131

pg immunoreactive LTB* per fraction (Thousands)
O O O O O O O O O —* —m(a>
Wto
EDco3
d3(D
33(0
O'

5.2.6. Summary
The injection of zymosan into the peritoneal cavity of rabbits induces an
inflammatory reaction that is characterised by an increased extravasation of
plasma proteins into the peritoneum and the accumulation of neutrophils.
There is an increased generation of pro-inflammatory mediators including LTB4,
TXB2 and 6 -oxo-PGFi„, but not PGEg in peritoneal exudate fluid taken from
zymosan injected rabbits.
Depletion of circulating neutrophils using mustine hydrochloride inhibits plasma
extravasation, neutrophil accumulation and the generation of the pro-inflammatory
mediator LTB4 induced by intraperitoneal zymosan. Neutropenia does not affect
the generation of 6 -oxo-PGFi„.
The anti-inflammatory steroid dexamethasone administered intravenously 4 hours
and immediately prior to intraperitoneal zymosan inhibits the plasma extravasation
and neutrophil accumulation which zymosan induces.
Treatment of animals with intravenous dexamethasone for 4 hours and
immediately inhibits the generation of LTB4 induced following zymosan
administration. Intravenous dexamethasone does not significantly inhibit the
generation of TXBj, or 6 -oxo-PGFi„ induced by intraperitoneal zymosan.
Intravenous dexamethasone does not inhibit the ex vivo generation of i.r. LTB4
by A23187-stimulated blood cells.as expected
133

CHAPTER 6 DISCUSSION
6.1 Characteristics of FMLP-lnduced oedema formation in rabbit skin
It has been established using techniques of systemic neutrophil depletion that
FMLP, together with other chemoattractants LTB4 and C5a increase microvascular
permeability by a mechanism entirely dependent on the presence of circulating neutrophils
in the rabbit (Wedmore & Williams, 1981a). In the present study the characteristics of
neutrophil-dependent oedema formation induced by FMLP were investigated in rabbit
skin and its activity was compared with structurally-related formyl peptides and with the
direct-acting mediators histamine or bradykinin in the same animals.
Consistent with observations for other mediators of increased microvascular
permeability and published data, oedema responses to both FMLP and histamine were
greatly potentiated by the vasodilator prostaglandin Ej (figure 3.1). Many studies have
provided evidence that this potentiation occurs as a result of synergism between increased
microvascular permeability and increased microvascular blood flow causing increased
hydrostatic pressure within the venule lumen (Williams & Morley, 1973; Williams &
Peck, 1977; Wedmore & Williams, 1981a).
The activity of a range of N-formyl peptides in increasing vascular permeability
in vivo in the presence of PGEj (figure 3.2) correlated well with their activity in vitro as
neutrophil chemoattractants and inducers of lysosomal enzyme secretion (Showell et al. ,
1976; Freer et al., 1982). Further, one of the most active peptides, FMLP, which has
been shown to be the major chemotactic component of bacterial culture filtrates (Marasco
et al., 1984) was approximately 100-l(X)0x more active on a molar basis than histamine
(figure 3.2) and equipotent with C5a. This suggests that, although a role for these
peptides as endogenous chemotactic substances remains unclear, at least their presence
at the inflammatory site through infection or tissue damage could make a significant
contribution to oedema formation and the recruitment of neutrophils into tissue and
prompts investigation into the consequences and mechanisms of action of FMLP in vivo.
Pro-inflammatory products of the cyclo-oxygenase pathway were not found to
contribute to the oedema response to FMLP as evidenced by the fact that local
indomethacin was not able to modulate the response to FMLP when oedema responses
were measured over 4 hours (table 3.1). Indomethacin remained active in skin sites for
at least 4 hours (see figure 4.1b) indicating that the lack of effect of indomethacin was
not due to clearance of the drug from skin sites. Further, oedema responses to FMLP
134

4- PGE2 were also found to be independent of endogenous histamine release (section
3.2.4).
To investigate further the oedema response to FMLP the modulating effect of
various agents was studied.
6.2 The effect of intravenous zvmosan activated plasma (ZAP) on FMLP-induced
oedema formation
The infusion of activated plasma induced a rapid (within 5 minutes) and profound
fall in circulating neutrophil numbers. Although this neutropenia was maintained for 35
minutes, at the end of this time period circulating cell numbers were beginning to rise
despite the continuous infusion of ZAP indicating that the mechanism involved was
sufficient to maintain the neutropenia for only a brief period of time. Oedema responses
to neutrophil dependent but not independent mediators were suppressed following ZAP
infusion (figure 3.3) which is consistent with earlier observations using animals rendered
neutropenic by treating 3 days previously with nitrogen mustard (Wedmore & Williams,
1981a). The infusion of ZAP therefore provides a rapid method of assessing the
neutrophil dependency of inflammatory mediators in this model.
Several other studies have shown that intravascular complement activation or
infusion of chemoattractants leads to an immediate and profound, but transient,
neutropenia (O’Flaherty et ah, 1977; O’Flaherty et ah, 1978a; McCall et ah, 1974;
Doerschuk et ah, 1989). This fall in circulating neutrophils has been shown to be
associated with a marked, rapid and reversible sequestration of neutrophils in the
microvasculature of mainly the pulmonary but also the systemic circulation (Doerschuk
et ah, 1989; Hammerschmidt et ah, 1981; Issekutz & Ripley, 1986; Henson et ah,
1982a; Doerschuk, 1992; Lundberg & Wright, 1990). It has been suggested that this
margination may result from both stimulus-induced decreased neutrophil deformability,
increased neutrophil-endothelial cell adherence or intravascular neutrophil aggregation
(Inano et ah, 1992; Hammerschmidt et ah, 1981 ; Henson et ah, 1982a). Under normal
conditions the microvascular bed in the lung is a major site of neutrophil margination.
It has been shown that many inflammatory stimuli that activate neutrophils, including
ZAP, can cause a rapid decrease in neutrophil derformability which is associated with an
increase in the amount of f-actin present within the cytoskeleton (Worthen et ah, 1989;
Inano et ah, 1992; Nash et ah, 1988; Frank, 1990; Downey et ah, 1991). It has been
suggested that less deformable activated neutrophils may thus have greater difficulty and
135

require more time to negotiate narrow capillaries resulting in further neutrophil
sequestration and sudden neutropenia.
It has been well documented that the rapid recruitment of neutrophils into the
tissues in response to locally injected chemoattractants can be blocked by mAbs directed
against CD 18 (Arfors et al., 1987; Price et al., 1987; Rampart & Williams, 1988;
Nourshargh et a l , 1989; Lundberg & Wright, 1990). However, in contrast, the rapid
and transient fall in circulating neutrophils caused by intravascular injection of
chemoattractants has been shown to be independent of the GDI 1/CD 18 complex
(Lundberg & Wright, 1990; Doerschuk, 1992). Doerschuk 1992 presented evidence that,
whilst the initial (within 1 minute) neutropenia and intravascular sequestration occurred
by mechansims independent of CD 11/CD 18-mediated adhesion, the continued
sequestration and massive neutrophil accumulation in the lung resulting in prolonged
neutropenia required CD 11 /CD 18-mediated adhesion. The authors suggested that the
CD 18-independent step may be required to slow neutrophil transit time and allow CD 18-
dependent adhesion to the endothelium. They further suggested that the initial step may
occur through either a stimulus-induced decrease in neutrophil deformability or a CD 18-
independent adhesion system, although L-selectin has been shown not to be a candidate
molecule (Doyle et a l, 1992).
These mechanisms of neutrophil sequestration may be relevant to such conditions
as the adult respiratory distress syndrome since neutrophils as well as complement
activation have been implicated in the pathogenesis of such acute lung injuries. By
sequestering in the lung and producing a local injury neutrophils can cause an increase
in vascular permeability resulting in pulmonary oedema and decreased oxygen delivery
(Tate & Repine, 1983; Sibille & Reynolds, 1990; Boxer et a l , 1990). By diverting
neutrophils to the lung neutrophil-dependent responses in other tissues are suppressed as
demonstrated in figure 3.3.
6.3 The effect of intravenous Ibuprofen on FMLP-induced oedema formation
Systemically administered Ibuprofen suppressed oedema formation induced by
both FMLP and histamine when the responses to these mediators were potentiated by
arachidonic acid (figure 3.4b). Similar observations have been obtained by others using
a variety of permeability-increasing mediators and both local and systemically
administered Ibuprofen (Rampart & Williams, 1986b). These observations are consistent
with those found for indomethacin (Williams & Peck, 1977; Williams, 1979) and can be
136

explained in terms of the known cyclooxygenase inhibitory activity of both of these
drugs; by inhibiting the conversion of endogenous arachidonic acid to vasodilator
prostaglandins, PGE2 /PGI2 in the skin they are able to prevent the potentiation of the
oedema response. It has been shown that local cyclooxygenase inhibitors including
Ibuprofen have no effect on oedema responses potentiated by exogenous PGEg indicating
the cyclooxygenase independent nature of these responses (Rampart & Williams, 1986b
and also see Chapter 4).
In contrast to the known local effect of cyclooxygenase inhibitors systemic
Ibuprofen was found in the present study to suppress local oedema induced by FMLP
coinjected with PGE2 but was without effect on responses to histamine 4- PGEg (Figure
3.4b). Since the response to FMLP is known to be neutrophil-dependent and local
Ibuprofen was ineffective it seems likely that systemic Ibuprofen was targetting the
interaction between circulating neutrophils and venular endothelium by an effect on the
neutrophil, and this interaction is a prerequisite for neutrophil-dependent oedema
formation. This is supported by the study of Rampart & Williams (1986b) who found
that systemic Ibuprofen inhibited oedema formation to C5a + PGEj but was without
effect on the response to BK + PGEj.
The simplest explanation for the results obtained is that a cyclooxygenase product
of arachidonic acid may be involved in mediating neutrophil-dependent oedema
formation. In this regard Palder et al (1986) found some evidence for the involvement
of TXB2 in the accumulation of neutrophils in rabbit skin induced by chemoattractants.
They found that i.v. Ibuprofen or thromboxane syntase inhibitors prevented both the
neutrophil accumulation and TXB2 production in blister chambers filled with chemotactic
agents. These authors suggested that thromboxane may be produced by the neutrophil,
or alternatively by the endothelium in response to chemoattractants (Ingerman et a l,
1980; Dunham et al., 1984) and act on the endothelium causing contraction so facilitating
diapedesis and increased microvascular permeability. Alternatively, they theorized that
thromboxane may be acting indirectly via platelet activation, and that products generated
from activated platelets may modulate neutrophil-endothelial interactions. This is an
interesting idea especially in light of the recent study by Pons et al 1993 who showed that
local C5a induces platelet accumulation in rabbit skin. Pons et al 1993 in fact suggested
that platelet accumulation may be secondary to the release of platelet stimulatory activity
from neutrophils. It also remains possible that platelets accumulating together with
neutrophils are the source of thromboxane which is then able to mediate neutrophil-
137

dependent oedema formation. In fact it has been shown that platelet-derived thromboxane
A; can mediate increased neutrophil adhesiveness (Spagnuolo et al., 1980).
In the present investigation several lines of evidence indicate that the effect of
systemic Ibuprofen was independent of cyclooxygenase activity. Firstly, oedema
responses to chemoattractants potentiated by PGE^ have been shown to be unaffected by
systemic administration of indomethacin at a dose evidently blocking cyclooxygenase
(Rampart & Williams, 1986b; Peers & Flower, 1991). Secondly, a thromboxane
synthesis inhibitor, dazmegrel was also found to be ineffective when given i.v. (Rampart
& Williams, 1986b). However, the possibility remains that platelets accumulating
together with neutrophils by an as yet undefined, neutrophil-dependent (but apparently
cyclooxygenase-independent) mechanism hold the key to the mechanism of
neutrophil-dependent oedema formation and that a platelet-derived product acts on the
endothelium to increase microvascular permeability. Such a platelet-derived product
would have to be independent of cyclooxygenase or PAF. This theory could be tested
by monitoring neutrophil-dependent oedema formation in platelet-depleted animals.
Thus the results of the present study indicate that ibuprofen can inhibit
inflammatory oedema by an action on the circulating neutrophil independent of
cyclooxygenase inhibition. These observations in vivo may be related to the numerous
studies on the effects of ibuprofen on multiple neutrophil functions stimulated by
chemotactic factors in vitro and ex vivo. Ibuprofen has been shown to inhibit neutrophil
aggregation (Kaplan et al., 1984; Maderazo et al., 1984), degranulation (Nielson &
Webster, 1987; Kaplan et al., 1984; Flynn et al., 1984; Maderazo et al., 1984),
generation of superoxide anion radicals (Nielson & Webster, 1987; Flynn et al., 1984)
and chemotaxis (Maderazo et a l , 1984; Brown & Collins, 1977; Partsch et a l , 1990;
Nielson & Webster, 1987). Since a role for oxygen-derived free radicals and neutrophil
granule enzymes has been suggested in neutrophil-dependent oedema formation, such
actions of ibuprofen on the circulating neutrophil may lead to the inability of
accumulating neutrophils to increase microvascular permeability. The mechanisms
underlying the inhibitory effects of ibuprofen on neutrophils are not known. It appears
the mechanisms are independent of cyclooxygenase inhibition since the various NSAID
show differential effects and the addition of prostaglandin does not abrogate the inhibition
observed (Abramson & Weissmann, 1989; Nielson & Webster, 1987). Furthermore,
higher concentrations are usually required to inhibit neutrophils than to inhibit
prostaglandin synthesis. There are considerable differences in the literature concerning
138

the effects of ibuprofen on neutrophils. Kaplan et al (1984) and Abramson et al (1984)
found no evidence of inhibition of superoxide anion generation by ibuprofen and Neal et
al (1987) found inhibition only at high concentrations. Furthermore Neal et al (1987)
found no effect of ibuprofen on neutrophil degranulation and several studies found
inhibition only at high concentrations (Nielson & Webster, 1987; Maderazo et a l , 1984)
which also seems to be the case for effects on chemotaxis (Maderazo et al., 1984; Brown
& Collins, 1977). The reasons for the discrepancies in the data are not apparent, but
may reflect differences in experimental procedure. The effects of antiinflammatory
agents on neutrophil activation in vitro does not necessarily predict their in vivo actions
in acute inflammation and such inconsistencies make extrapolation of in vitro findings to
in vivo even more contentious. Furthermore, indomethacin has also been found to be an
effective inhibitor of neutrophil degranulation (Smolen & Weissmann, 1980; Neal et al.,
1987), superoxide anion production (Neal et al., 1987; Smolen & Weissmann, 1980) and
aggregation (Kaplan et a l, 1984) in vitro whilst only ibuprofen was effective in vivo in
the present study.
It has been suggested that some NSAID, including indomethacin can interfere with
the binding of FMLP to its receptor on the neutrophil surface (Cost et a l , 1981; Abita,
1981; Minta & Williams, 1985). However, it is unlikely such an action could explain
the present in vivo findings since ibuprofen inhibited neutrophil-dependent oedema in
response to both FMLP (figure 3.4b) and C5a (Rampart & Williams, 1986b) equally
well. Furthermore, Kaplan et al (1984) found ibuprofen, together with indomethacin had
no effect on FMLP binding to neutrophils.
Ibuprofen may be acting in the present study on the circulating neutrophil to
prevent adhesion to the endothelial lining, a necessary antecedent to neutrophil-dependent
oedema formation. Nielson & Webster (1987) found ibuprofen inhibited C5a or FMLP-
stimulated adhesion of human neutrophils to plastic or bovine endothelial cells. Venezio
et al (1985) showed that ibuprofen administered in vivo to volunteers suppressed in vitro
attachment of neutrophils to nylon-wool columns 4 and 24 hours following ibuprofen.
Furthermore, they were able to demonstrate inhibition of neutrophil adherence after
incubation of normal neutrophils with plasma or serum specimens obtained from
volunteers treated with ibuprofen, even 24 hours after treatment when the drug levels
were undetectable in plasma, suggesting ibuprofen may be converted to as yet
unrecognised neutrophil inhibitory factor(s) with prolonged action, or alternatively
ibuprofen may stimulate the release of an endogenous factor that inhibits leukocyte
139

adherence. This may explain differences in in vitro and in vivo actions of NSAID.
It is possible that ibuprofen may affect the cell surface expression of adhesive
glycoproteins on neutrophils which are crucial for chemoattractant-induced adhesion and
extravascular recruitment of neutrophils, and are important for other neutrophil functions
for example chemotaxis (see section 1.4.3). In this regard Crowell and Van-Epps (1990)
found that piroxicam and indomethacin inhibited FMLP but not C5a or ionomycin-
induced upregulation of GDI lb and CD 11c on neutrophils, raising the interesting
possibility that these two drugs can interfere with postreceptor signalling events specific
to neutrophil stimulation by FMLP. However, Ottonello et al (1992) found ibuprofen
was without effect on the cell surface expression of GDI lb or CD 18. NSAID are planar,
lipophilic molecules that are able to partition into lipid environments such as lipid
bilayers of plasma membranes and disrupt plasma membrane fluidity (Abramson &
Weissmann, 1989; Abramson et al., 1990). Ibuprofen may, therefore, prevent the
clustering or conformational change of cell surface integrins thought to be vital for their
interaction with endothelial cell surface ICAM (Nourshargh et a l , 1989).
It has been suggested that the capacity of NSAID to insert into the lipid bilayer
results in the disruption of signal transduction through the plasma membrane following
receptor-ligand interactions (Abramson & Weissmann, 1989). In fact, Abramson et al
(1988) presented indirect evidence that piroxicam was able to block the pertussis toxin
dependent ADP-ribosylation of the G-protein in the neutrophil. These authors have
suggested, therefore, that NSAID, like pertussis toxin are able to inhibit neutrophil
function by interfering with the guanine nucleotide binding protein which couples
neutrophil chemoattractant receptor occupancy to the production of second messenger
molecules such as inositol 1,4,5-triphosphate and 1,2-diacylglycerol. In keeping with this
argument Nourshargh & Williams (1990) found that a pertussis toxin-sensitive GTP-
binding protein on the neutrophil was essential for chemoattractant-induced neutrophil
accumulation in vivo in rabbit skin. It is possible, therefore, that ibuprofen was acting
on the neutrophil in a similar manner in vivo in the present study. It would be interesting
to ascertain, using the accumulation of * “ In-labelled neutrophils if ibuprofen was limiting
the accumulation of neutrophils at the site or preventing the accumulated neutrophil from
increasing microvascular permeability.
The ability of ibuprofen, but not indomethacin, to inhibit neutrophil-dependent
oedema formation observed in the present study may be related to several other reports
on the action of this drug in vivo. Earlier studies suggested that ibuprofen differed from
140

other NSAID in that it was effective in reducing the myocardial infarct size in
experimentally-induced myocardial infarction. This effect was associated with the
absence of infiltrating neutrophils in areas bordering the infarct and was shown to parallel
inhibition of neutrophil function (Flynn et al., 1984; Romson et a l , 1982; Lefer &
Polansky, 1979; Ogletree & Lefer, 1976). Neutrophil-mediated local tissue damage has
been implicated by several, but not all studies in the extension of the zone of ischaemia
following myocardial infarction (Romson et a l , 1983; Mullane et a l , 1984; Litt et a l,
1989; Châtelain et a l , 1987). Ibuprofen has also been found to be an effective agent in
vivo in other animal models involving neutrophil-mediated tissue injury. Ibuprofen was
found to have a protective effect in a model of thrombin-induced lung vascular injury in
sheep (Perlman et a l , 1987) and this effect was thought to be related to an ibuprofen-
induced alteration in neutrophil function resulting in a reduction in the migration of
neutrophils and in the adherence of neutrophils to the endothelium. These results
encourage speculation that ibuprofen may provide a lead for the development of
compounds which have a useful role in the treatment of human diseases where stimulated
neutrophils are thought to play a role, such as the adult respiratory distress syndrome and
myocardial infarction, and highlight the importance of further characterising the
mechanisms of action of ibuprofen.
6.4 The effect of intravenous colchicine on FMLP-induced oedema formation
The microtubule blocking agent colchicine caused a profound suppression of
oedema formation induced by FMLP + PGE2 following intravenous administration
(figure 3.5b). This inhibition was unlikely to be mediated by a non-specific action of
colchicine on the local blood vessels since responses to the direct-acting mediator Bk -H
PGE2 were unaffected (figure 3.5b). It seems likely, therefore, that colchicine, by itself,
or by a microtubule mediated process interferes with neutrophil-endothelial cell
interactions by acting on the circulating neutrophil. In contrast to the decline in
circulating neutrophils observed following i.v. ZAP, Img colchicine/kg i.v. caused a very
slow, less profound but sustained decline in the numbers of circulating neutrophils over
the duration of the experiment (table 3.4). A similar result was found with the same dose
of colchicine in rabbits by Inano et al (1992), although other groups have found this dose
of colchicine to be non-leukopenic in rats (Higgs et al., 1983). It is difficult to determine
the extent to which such as small decline in circulating neutrophil numbers would affect
neutrophil-dependent permeability responses. It seems unlikely that such a decline would
141

result in the profound inhibition of oedema responses observed. Alternatively, and more
likely, colchicine may be acting on the normal function of circulating neutrophils to
prevent their accumulation at the inflammatory site or possibly to prevent accumulating
neutrophils from acting to increase microvascular permeability.
Colchicine has also been shown to have potent inhibitory action on oedema
formation and neutrophil accumulation in a variety of other animal models (Griffiths et
a l , 1988; Griffiths er a/., 1991; Brooks efû/., 1987; Griswold e ta l , 1991; Higgs era/.,
1983; Simmons et a l , 1983). It is commonly used to treat a number of illnesses which
are characterised by the pathological accumulation of neutrophils, including gout, Bechets
disease and vasculitis. However, it remains unclear how colchicine exerts its
antiinflammatory actions. From in vitro studies possible mechanisms include inhibition
of the ability of neutrophils to undergo chemotaxis (Malech et a l , 1977b; Caner, 1965),
to degranulate (Hoffstein et a l, 1977; Zurier et a l, 1974) or generate LTB4 (Reibman
et a l , 1986; Ouyang et a l , 1989; Langlois & Gawryl, 1988; Serhan et a l, 1984).
However, conflicting statements exist in the literature both regarding the effect of
colchicine on neutrophil function in vitro and whether any observed effects are mediated
via microtubule disruption. For example, colchicine was found to be ineffective in
inhibiting secretion induced by the calcium ionophore A23187 (Hoffstein & Weissmann,
1978) and in fact augmented degranulation induced by FMLP (Reibman et a l , 1991).
It has been suggested that microtubules are involved in the cycling of specific receptors
or the generation of specific intracellular signals required for the signal transduction
process and can specifically participate in a pertussis toxin-sensitive pathway of activation
(Reibman et a l , 1991). Prevention of such action by microtubule disrupting agents may
therefore prevent neutrophil accumulation in vivo since this has been shown to be a
pertussis toxin sensitive process (Nourshargh & Williams, 1990).
Recently, Asako et al (1992) found that colchicine inhibited LTB^-induced
neutrophil adherence and emigration in the microvasculature of the rat, but did not
influence the ability of LTB4 to upregulate CD ll/CD 18 on the neutrophil surface. Molad
et al (1992) also failed to demonstrate an effect of colchicine on the expression of
GDI lb/CD 18 on neutrophils. However, these authors were able to show that colchicine,
by disrupting microtubules, diminished the cell surface expression of L-selectin on
neutrophils. Thus, the antiinflammatory action of colchicine may be due, at least in part,
to its capacity to diminish the expression of adhesive molecules on the neutrophil. It also
remains possible that colchicine affects the clustering or conformational change of cell
142

surface integrins thought to be important for cell accumulation (Nourshargh et al., 1989).
Of possible relevance to this, data has been presented suggesting a dynamic interaction
between the composition and arrangement of membrane lipids and the cytoskeletal system
(Berlin & Fera, 1977; Pike et al., 1980; Hoover et al., 1981). Microtubule-dependent
anchoring of membrane lipids may in turn affect clustering or changes in conformation
of cell surface adhesion molecules.
6.5 The effect of colchicine on the duration of the permeability response to FMLP
The increased microvascular permeability induced by a single intradermal injection
of FMLP is of long duration compared to that induced by direct-acting mediators of
increased microvascular permeability ( Wedmore & Williams, 1981a , figure 3.6). The
mechanism of this protracted response is unknown, for example it is not clear if this long
duration is because of a protracted change in the endothelium or a protracted efflux of
neutrophils. The intravenous administration of colchicine at different times after
intradermal FMLP injection almost abolished the protracted nature of the FMLP response
whilst having no effect on responses to bradykinin (figure 3.6). This suggests that
continuing interactions between functionally active neutrophils and endothelial cells are
necessary for the protracted plasma protein leakage induced by this chemoattractant,
rather than an initial neutrophil-endothelial interaction resulting in a protracted change
in the endothelium. Increased permeability during neutrophil migration has also been
shown using epithelial cell cultures and this increase appeared dependent upon the number
of neutrophils present and prolonged contact between the neutrophils and the epithelium
(Milks et al., 1986; Parsons et al., 1987). The nature of the neutrophil-endothelial
interaction resulting in increased permeability remains elusive and represents a very
interesting area for future study.
6.6 Systemic treatment of rabbits with dexamethasone
Systemic treatment of rabbits with dexamethasone for 4 hours inhibited neutrophil-
dependent and independent oedema formation induced by exogenously administered
inflammatory mediators mixed with PGEj (figure 4.4). These results are in agreement
with other studies (Tsurufuji et al., 1979; Tsurufuji et a l , 1980; Church & Miller, 1978;
Bjork et a l , 1985; Griffiths & Blackham, 1988; Foster & McCormick, 1985; Peers &
Flower, 1991; Ahluwalia et al., 1992). In addition, dexamethasone when administered
intravenously inhibited neutrophil accumulation induced by LTB4 (as shown previously
143

by Foster & McCormick, 1985), FMLP and C5a des Arg (figure 4.3). These results
clearly demonstrate the anti-inflammatory properties of dexamethasone, but alone tell us
little of the mechanisms involved. These results are therefore best discussed together
with, and in relation to results obtained using local dexamethasone treatment and the
treatment of neutrophil donor rabbits.
6.7 Local treatment of skin sites with dexamethasone
In this model of acute inflammation, locally administered dexamethasone, at a
concentration as low as SOng/site (2xl0'^°moles/site) inhibited oedema formation induced
by both neutrophil-dependent and direct-acting inflammatory mediators, co-administered
with prostaglandin Ej (figure 4. la). Similar results have been described in other studies
using different species, for example (Ahluwalia et a l , 1992; Svensjo & Roempke, 1985;
Erlansson et al. , 1989) although most studies addressing the action of glucocorticosteroids
against oedema induced by inflammatory mediators have involved systemic treatment
regimes (see section 6 .6 ). Local dexamethasone failed to inhibit "'In-neutrophil
accumulation in response to C5a des Arg. It is likely that definite local mechanisms were
responsible for the action of dexamethasone observed since responses in saline treated
skin sites were of an expected magnitude.
Many of the reported effects of steroids on oedema formation and neutrophil
accumulation (Sato et a l , 1980; Kurihara et a l, 1984a; Tsurufuji et at., 1984a; Cirino
et a l , 1989; Errasfa & Russo-Marie, 1989) can most probably be largely attributed to
their well-documented action to inhibit the generation of pro-inflammatory prostaglandins,
leukotrienes and platelet-activating factor by inducing the formation of lipocortins which
have been reported to inhibit phospholipase Aj (see section 1.5.2). The use of preformed
inflammatory mediators and chemoattractants in this study eliminates such an explanation
being likely for the steroid effects observed. The following points further substantiate
this argument;
1. Experiments were designed so that all mediators were coinjected intradermally
with a dose of the vasodilator PGEj that caused maximum potentiation of oedema
formation and/or neutrophil accumulation. This ensured that the effect of any
endogenous vasodilator prostaglandin generation in skin sites was overriden; thus
eliminating the possibility of a glucocorticoid effect being due to inhibition of
prostaglandin synthesis. This was further confirmed by the failure of local indomethacin
144

treatment to mimick the effect of dexamethasone at a dose that was evidently blocking
cyclooxygenase (figure 4. lb). Other studies have shown similar effects of indomethacin
(Wedmore & Williams, 1981a; Peers & Flower, 1991) which also serves to eliminate any
possible effect of dexamethasone on cyclooxygenase which has been suggested in some
studies (Raz et al,, 1989; Masferrer et ah, 1990).
2. PAF generation was found not to be a component of the responses to these
inflammatory agents in the rabbit skin. This was demonstrated using a variety of specific
PAF antagonists which inhibited oedema formation in response to intradermal PAF but
had no effect on leakage induced by other mediators (Hellewell & Williams, 1986;
Hellewell & Williams, 1989).
3. Responses of the hamster cheek pouch microvasculature to FMLP have been
reported to be mediated by products of the lipoxygenase pathway of arachidonic acid
metabolism (Nagai & Katori, 1988). C5a has been shown in a number of systems to be
able to act via the release of lipoxygenase products (Regal & Pickering, 1981; Stimler
et ah, 1982). However, in the rabbit skin responses to chemoattractants were not found
to be mediated by lipoxygenase products (Von Uexkull et ah, 1991; Forrest et ah,
1981b).
4. In addition to inhibiting the generation of pro-inflammatory arachidonic acid
metabolites there have also been reports suggesting glucocorticoids can inhibit the
generation of histamine (Schleimer et ah, 1981). However, no evidence for the
involvement of histamine release in responses to inflammatory mediators in rabbit skin
was found (Williams & Jose, 1981, see section 1.5.2).
Glucocorticosteroids have been shown to constrict local blood vessels when
applied topically (Reid & Brookes, 1968; Kaidbey & Kligman, 1974; Marks & Sawyer,
1986). The mechanism of this vasoconstrictor action is unknown, it may be related to
the induction of vasoconstrictor substances or the inhibition of formation/enhanced
breakdown of a vasodilator. It has been suggested that glucocorticoids may enhance
vascular responsiveness to catecholamines and other vasoconstrictors (Besse & Bass,
1966; Rascher et ah, 1980; Kalsner, 1969). Glucocorticoids have been reported to
induce the synthesis of angiotensin converting enzyme (ACE) thus increasing the
formation of the constrictor angiotensin II and the inactivation of the vasodilator
145

bradykinin. In addition Radomski et al (1990) have shown that induction of nitric oxide
synthase in endothelial cells in response to cytokines and endotoxin was inhibited by
glucocorticoids. Furthermore, there have been suggestions that steroids can directly
affect prostanoid receptor numbers resulting in down-regulation of the receptor (Sessa et
al., 1990; Sessa & Nasjletti, 1990). Whatever the mechanism or mechanisms the
vasoconstrictor property of steroids may contribute to the anti-oedema actions of
dexamethasone observed, given the active role vasodilatation has been shown to play in
oedema formation (see section 1.3). Two lines of evidence suggested that modulation of
blood flow was not a possible site of action of dexamethasone in this model. Firstly,
oedema formation potentiated with either the non-prostanoid, potent peptide vasodilator
CGRP or PGEj was inhibited equally well by systemic dexamethasone (figure 4.5)
indicating the steroid effect was not mediated by an action on prostanoid receptors.
Secondly, although local dexamethasone had a slight inhibitory effect on basal blood flow
it had no effect on increased blood flow induced by PGEg (figure 4.2). Furthermore,
the induction of nitric oxide synthase was found to be a slow, protein synthesis dependent
process with a lag period of 2 hours (Radomski et al., 1990) whereas oedema responses
were measured over only 30 minutes indicating that, although interference with nitric
oxide production may partly mediate the effects of steroids in some models such a
mechanism was unlikely here.
Thus, the inhibition of oedema formation observed with both local and
systemically-administered steroid against all inflammatory agents was not mediated by a
steroid effect on blood flow or inhibition of mediator generation. Together with the lack
of effect of local dexamethasone on neutrophil accumulation and the fact that mediators
such as Bk act directly on endothelial cells to allow leakage, these results indicate that
dexamethasone can act at the level of the microvascular endothelial cell to inhibit oedema
formation to both direct acting and neutrophil-dependent mediators.
The inhibitory effect of dexamethasone on the endothelium was time-dependent;
a 4 hour pretreatment of skin sites locally with dexamethasone was required before a
substantial significant inhibition of oedema formation was obtained. The magnitude of
inhibition obtained for both direct acting and neutrophil-dependent stimuli after 4 hours
pretreatment was comparable. This, together with the comparable lag period required,
possibly suggests the mechanism by which glucocorticoids inhibit endothelial cell
responses to direct acting stimuli is the same as that by which responses to neutrophil
dependent stimuli are inhibited. The inhibitory action of systemic dexamethasone was
146

also found to be time dependent (Peers & Flower, 1991). The requirement for a lag
period suggests the effect of dexamethasone was protein synthesis dependent. Previous
studies have shown that RNA and protein synthesis are essential for manifestation of the
anti-exudative effect of dexamethasone against direct-acting mediators (Tsurufuji et al.,
1979; Tsurufuji et al., 1980; Peers & Flower, 1991) together with binding to the
glucocorticoid receptor (Tsurufuji et al., 1979; Tsurufuji et al., 1980). Thus, it seems
dexamethasone can induce the synsthesis of a protein that can inhibit microvascular
responses by an action on the endothelium. However, the situation with neutrophil-
dependent mediators is less clear. Tsurufuji demonstrated a role for the glucocorticoid
receptor and RNA synthesis in the inhibiton of leukocyte accumulation by local
glucocorticoids (Tsurufuji et al., 1984b). However, in this study the action of
dexamethasone reflected, at least in part, an inhibition of mediator generation and the
model used was unsuitable for measuring changes in plasma extravasation. Peers et al
(1991) were able to show a requirement for RNA synthesis in the ability of systemic
dexamethasone to inhibit local oedema formation to neutrophil dependent mediators.
However, the role of the glucocorticoid receptor and protein synthesis were not addressed
and neutrophil accumulation was not monitored. To clearly establish the molecular
mechanism involved in the glucocorticoid action on permeability induced by both
direct-acting and neutrophil-dependent mediators and on neutrophil accumulation in the
rabbit skin model the glucocorticoid receptor antagonist RU 38486 and inhibitors of
mRNA and protein synthesis could be used.
The identity of the anti-permeability protein is presently unknown. Lipocortin has
been found in endothelial cells (Hullin et al., 1989). However, it is not thought to be
involved as Cirino et al (1989) found that recombinant lipocortin 1 injected locally was
not effective against oedema induced by a host of preformed inflammatory mediators
including 5HT, Bk and PAF in the rat paw. The involvement of lipocortin in modulation
of oedema induced by chemoattractants has not been established. It would be interesting
to ascertain this using the rabbit skin model and recombinant lipocortin and/or
neutralising antibodies. A second glucocorticoid-induced anti-inflammatory protein
named vasocortin has been identified in the peritoneal lavage fluid from dexamethasone
treated rats (Camuccio et al., 1987; Camuccio et al., 1989a; Di Rosa et al., 1985). This
protein has distinct physicochemical properties from lipocortin and was shown to be
inhibitory against rat dextran oedema (Camuccio et al., 1987) like the
glucocorticosteroids (Calignano et al., 1985) but in contrast to lipocortin (Cirino et al.,
147

1989). In a further study Camuccio et al (1989a) showed that vasocortin prevented
histamine release. It remains possible that vasocortin may act on the endothelial cell to
inhibit oedema formation, no studies to date have addressed this question.
The mechanism(s) underlying the effect of glucocorticoids or glucocorticoid-
induced proteins on the endothelium remains to be determined. This represents a very
interesting area for future research. Such studies may not only help the development of
more specific anti-inflammatory therapy but could also throw new light on mechanisms
of increased microvascular permeability. The fact that steroids blocked the formation of
inflammatory oedema to all stimuli tested suggests that a very fundamental change was
occurring in the endothelial cell after exposure to steroids. A block in the signal
transduction pathway operating for these agents may be the locus of action of steroids,
or possibly interference with the contractile machinery of the endothelial cell. Studies
investigating the interaction of glucocorticoids with intracellular second messengers and
comparing steroid effects with those of compounds and drugs of known action may help
to unravel the mechanism of steroid action.
With regard to the mechanism of neutrophil-dependent oedema formation, the
observation that plasma leakage but not neutrophil accumulation was affected by local
dexamethasone provides further evidence that neutrophil-dependent oedema formation
cannot entirely be a passive process resulting from neutrophil emigration (see section
1.2.4.1). However, since radiolabelled cells were used to monitor cell accumulation it
is not possible to make such a conclusion unequivocally, which would need microscopic
examination of histologic skin samples. Whilst time-consuming this technique would
provide not only useful information about the tissue location of cells but would also allow
different subtypes of leukocytes to be investigated.
Since systemic, but not local dexamethasone inhibited chemoattractant-induced
neutrophil accumulation it was conceivable that the target site for steroid action was the
neutrophil itself. A series of experiments were designed to examine such a possibility
in vivo.
6 . 8 Effect of dexamethasone on neutrophils in vivo. Modulation of neutrophil
accumulation in vivo bv an action of dexamethasone on the neutrophil
For these studies neutrophils were isolated from the blood of steroid-treated donor
animals and their accumulation was monitored in vivo in untreated recipients. Control
recipient animals received an equal number of '"In-neutrophils prepared at the same time
148

from a saline treated donor animal. Treatment of neutrophil donor rabbits with
dexamethasone resulted in inhibition of the accumulation of treated " ‘In-neutrophils in
untreated recipients (figure 4.6). The results were not explicable by transfer of some
dexamethasone along with the treated donor cells because there was no difference in the
ability of recipient animals to mount an oedema response to both direct-acting and
neutrophil-dependent mediators (figure 4.7). These observations are consistent with the
concept that steroids, or more likely steroid-induced products, can suppress neutrophil
accumulation in vivo by an effect on the neutrophil itself, in addition to a possible effect
on the endothelium.
The process of neutrophil accumulation at an inflammatory site is complex with
multiple components. These include rolling of the neutrophil along the vessel wall, firm
adhesion followed by passage between the endothelium and finally penetration of the
basement membrane to the interstitial space (see sections 1.4.3, 1.4.4 and 1.4.5). It is
possible that dexamethasone suppresses chemoattractant-induced neutrophil accumulation
by inhibiting the neutrophil response at any or several of these steps in addition to a
possible effect on the endothelium. The evidence for steroid effects at these different
stages will be discussed.
Following glucocorticoid administration in vivo in man a significant increase in
the number of circulating neutrophils has been found (Athens et al., 1961) and it has been
suggested that this neutrophilia results in part from reduced neutrophil adherence (Bishop
et al., 1968; Mischler, 1977). Similarly, in vivo treatment of man and animals with
glucocorticoids followed by phlebotomy was found to yield neutrophils with reduced
adherence to nylon fibres (Clark et al., 1979; Stecher & Chinea, 1978; Ackerman et al.,
1982). The mechanism by which glucocorticoids prevented attachment of leukocytes was
unknown and remains contentious. Whether glucocorticoids can modulate neutrophil
functions in vitro has been addressed in many studies. However, many of these studies
used very high concentrations of steroids and it is therefore doubtful whether the results
obtained bear any relevance to glucocorticoid receptor mediated anti-inflammatory effects
in vivo. Furthermore, few studies have been performed analysing the effects of culture
with glucocorticoids for appropriate lengths of time on the function of neutrophils. It is
now clear glucocorticoid action both in vivo and in vitro requires a considerable amount
of time to become evident, although too long a treatment time may also cloud the
interpretation of results (Ackerman et al., 1982; Cronstein et al., 1992). The suggestion
that glucocorticoid effects on target cells may differ depending on whether the steroid was
149

administered in vivo, followed by subsequent separation of cells or whether isolated
"virgin" cells were treated in vitro with steroid (Goulding et al., 1990a) further
complicates interpretation of results. Furthermore, in trying to reconcile findings from
different studies it must also be recognised that the behaviour of inflammatory neutrophils
and their response to pharmacological agents has been found to differ from that of
peripheral blood cells (Cunningham et al., 1979; Thieme et a l, 1982).
Culture of human neutrophils with glucocorticoids for 24 hours led to inhibition
of basal (unstimulated) adherence to cultured vascular endothelial cells (Schleimer, 1985;
Schleimer et a l, 1989). A similar result was found by Rosenbaum et al (1986) using 4
hour pretreatment of rabbit peripheral blood neutrophils and rabbit microvascular
endothelium. However, Schleimer and co-workers found that the treated neutrophils
showed an undiminished ability to increase adherence to endothelial cells following
exposure to neutrophil activators such as FMLP, as well as endothelial activators such
as IL-1 and LPS. Furthermore, 24 hour treatment of endothelial cells with steroid did
not inhibit their acquisition of adhesive properties stimulated by IL-1 (Schleimer et a l,
1989; Schleimer, 1985; Cybulsky et a l , 1987). In a more recent study Forsyth & Talbot
found similar results in that prior co-culture of endothelial cells for 6 hours with
dexamethasone failed to inhibit neutrophil adhesion to endotoxin-stimulated endothelial
cells in the presence or absence of FMLP (Forsyth & Talbot, 1992). Furthermore,
dexamethasone was without effect on increased expression of the adhesion molecules E-
selectin and ICAM-1 on the endothelium. In contrast, Cronstein et al (1992) found
dexamethasone treatment of endothelium for 4 hours dramatically inhibited neutrophil
adhesion to LPS-stimulated endothelium, an effect which was found to be mediated by
glucocorticoid receptors. They were also able to demonstrate that dexamethasone
inhibited LPS-stimulated expression of ICAM-1 and E-selectin (Cronstein et a l, 1992).
It is difficult to reconcile these studies, obvious differences were the time of treatment
of endothelial cells with dexamethasone and whether the endothelial stimulus was added
together with dexamethasone or later. Cronstein et al (1992) have suggested that the
prolonged treatment with dexamethasone undertaken in the studies by Schleimer and co
workers may lead to "desensitization" of glucocorticoid receptors or that glucocorticoids
effects to regulate E-selectin/ICAM are transient permitting subsequent activation in some
studies. In both the study by Cronstein et al (1992) and Forsyth and Talbot (1992)
dexamethasone did not alter basal expression of endothelial adhesion molecules. This is
particularly interesting in relation to the findings in this thesis since chemoattractant-
150

induced neutrophil accumulation is believed to involve binding of neutrophil
CDlla/CD18 and CD 1 lb/CD 18 with basally expressed ICAM-1 (Argenbright et a l,
1991). With regard to steroid modulation of neutrophil expression of adhesion
molecules; few studies have addressed this question. Forsyth & Talbot (1992) found
dexamethasone pretreatment of neutrophils failed to affect upregulation of integrins by
FMLP, however, the authors chose to study only a fixed time (2 hours) of dexamethasone
treatment. A more thorough study possibly involving a time course of dexamethasone
treatment is required. Petroni et al (1988) demonstrated that overnight culture with
glucocorticoids caused a slight reduction in CDllb/CD18 expression on neutrophils. It
has been suggested that such a reduction may explain the decreased adhesion of
unstimulated neutrophils to endothelium or nylon fibres (Schleimer et a l, 1989; Clark
et a l, 1979) and the "demargination" of neutrophils seen with glucocorticoid treatment
in vivo (Athens et a l, 1961; Mischler, 1977). Petroni et al (1988) also observed that
IFN^ was a strong inducer of CDllb/CD18 on neutrophils and that this induction was
inhibited substantially by treatment of the neutrophils with dexamethasone. This area is
worthy of further research, it would be interesting to monitor the integrin expression on
cells taken from the animals used in the donor treatment studies. Since some kind of
conformational or biochemical change, possibly involving interactions with other
membrane components is thought to be important in integrin mediated adhesion in vitro
(Philips et a l, 1988) and in vivo (Nourshargh et a l, 1989) rather than surface
upregulation it may also be important to establish if steroids can interfere at this level
(this is discussed further later).
Watanabe and colleagues recently showed that dexamethasone inhibited neutrophil
adhesion to thrombin- and histamine-activated endothelial cells by an effect on the
neutrophil (Watanabe et a l, 1991). Since thrombin and histamine have been shown to
upregulate the expression of the adhesion molecule P selectin on cultured endothelial cells
(McEver et a l, 1989; Lorant et a l, 1991), the study of Watanabe and colleagues 1991
suggests dexamethasone may modulate the expression of the ligand for endothelial P
selectin on the neutrophil. Such an effect has yet to be investigated. Neutrophil L
selectin is thought to bind P-selectin. It is as yet unknown whether chemoattractants are
able to affect expression of P-selectin on the endothelium and therefore whether steroid
modulation of the neutrophil ligand would be important in chemoattractant-induced
neutrophil accumulation. Other as yet unidentified ligands for both P selectin and L
selectin may also be modulated by steroids. Interestingly, upregulation of P selectin by
151

H2 O2 has been demonstrated and it has been suggested that neutrophils may release H2 O2
during neutrophil:endothelial cell interactions stimulated by chemoattractants (see section
1.2.4.3). This may act as a positive feedback mechanism allowing further neutrophil
accumulation which steroids may be able to modulate.
Evaluation of the effect of dexamethasone on neutrophil chemotaxis, using either
neutrophils from steroid-treated donors or co-culture of neutrophils with dexamethasone
have indicated that chemotaxis can be either enhanced, inhibited or unaffected by
glucocorticoids (Clark et a l, 1979; Roth & Kaeberle, 1981; Stevenson, 1986; Ackerman
et a l, 1982; Hirata et a l, 1980; Kurihara et a l, 1984b; Schleimer et a l, 1989). There
may be some species differences in this response; rabbit neutrophil chemotaxis, has been
found to be inhibited by dexamethasone (Hirata et a l, 1980).
Dexamethasone may suppress an alternative step during the process of diapedesis.
In fact Katori et al (1990) using the hamster cheek pouch preparation stimulated with
topical L T B 4 found that leukocyte adhesion to the endothelium was not affected by
systemic dexamethasone treatment, neither was the time required for the leukocytes to
migrate out of the vascular lumen. However leukocyte migration into the interstitial
space was substantially depressed by dexamethasone indicating that, in this model, the
steroid was inhibiting passage of the neutrophil across the perivascular basement
membrane. Rosengren et al 1987 using the same model and chemotactic stimulus failed
to demonstrate such a steroid effect. In the rabbit skin model the use of " ‘In-cells to
monitor neutrophil accumulation makes it unlikely that the sole site of action of
dexamethasone was inhibiting neutrophil penetration of the perivascular basement
membrane. In such a situation trapped radiolabelled cells would still have been counted
in the skin site whereas there was a significant reduction in the number of radiolabelled
cells in skin samples. It remains possible, although unlikely, that the trapping of some
cells in such a position would prevent other cells from being able to traverse the
endothelial layer. To confirm the precise location of emigrating cells a rabbit model of
intravital microscopy, for example the mesentery or ear chamber preparation could be
used and donor cells labelled with a fluorescent tag, for example calcein.
The results of this study clearly demonstrate that dexamethasone, or more likely
a dexamethasone-induced factor, exerts an inhibitory effect on chemoattractant-stimulated
neutrophil accumulation in vivo by an action on the circulating neutrophil. The
mechanisms involved remain unknown. The well characterised model used in these
studies, together with models of intravital microscopy which afford a precise analysis of
152

the steps involved in neutrophil-extravasation may be useful in further investigating this
steroid action. It would be interesting to obtain a profile of steroid action against
different inflammatory stimuli which activate by different mechanisms (the neutrophil,
endothelial cell or both) and involve different pairs of adhesion molecules. The
molecular mechanisms of glucocorticoid action could be investigated using the steroid
receptor antagonist RU486 and inhibitors of protein and mRNA synthesis. It has become
increasingly evident that anti-inflammatory actions of steroids are invariably mediated by
the generation of proteins, it is likely, therefore that this mechanism is responsible for
the observed effect of dexamethasone on the circulating neutrophil. The availability of
purified and more recently recombinant lipocortin has facilitated investigations into its
mode of action. Clearly, the ability of lipocortin 1 to inhibit eicosanoid generation (see
section 1.5.2) cannot account for all the anti-inflammatory actions observed. Russo-
Marie et al (1989) found purified mouse lipocortin inhibited chemotaxis of mouse
neutrophils in vitro and suggested that this activity may contribute to the inhibition of cell
migration elicited by polyacrylamide gel injection following i.v. administration of
lipocortin in vivo. Cirino et al (1989) also presented data suggesting recombinant human
lipocortin may be exerting some of its actions by decreasing the migration of neutrophils
to the inflammatory site in vivo. Recently Perretti & Flower (1993) showed that
intravenous, but not local human recombinant lipocortin 1, inhibited IL-1 induced
neutrophil migration which was not due to inhibition of eicosanoid or PAP generation.
The authors suggested that lipocortin was interacting with circulating neutrophils to
inhibit their accumulation at the inflammatory site. Lipocortin 1 has also been shown to
diminish other neutrophil functions such as hydrogen peroxide generation (Stevens et al
1988). Furthermore, Goulding et al (1990b) were able to demonstrate the existence of
specific binding sites for lipocortin on the surface of human peripheral blood neutrophils,
although they failed to detect lipocortin induction in human neutrophils following
glucocorticoid treatment (Goulding et al., 1990a).
These data suggest it is possible that the inhibitory effect of dexamethasone on the
neutrophil observed in the experiments presented in this thesis is mediated by
dexamethasone-induced lipocortin. It would be very interesting to investigate the effect
of systemic and local treatment with recombinant lipocortin on responses to
chemoattractants in rabbit skin, and the treatment of neutrophils with lipocortin prior to
their i.v. injection into a recipient animal.
Further considerations can be drawn from the local treatment experiments.
153

Lipocortin 1 seems to be produced at discrete sites throughout the body (Fava et al. ,
1989) where its concentration can be as high as 100/zg/ml (compared to 50ng/ml in blood
plasma). Furthermore, putative receptors for this protein have been demonstrated on
neutrophils. Based on these findings Goulding and Guyre (1992) have suggested that
neutrophils migrating into tissues would have to pass through regions of elevated
lipocortin 1 concentration which would be expected to progressively saturate their
putative lipocortin receptors resulting in suppression of migratory and/or pro-
inflammatory activities. One such area where lipocortin has been found is the basal
kératinocytes of the epidermis (Fava et a l, 1989). Thus, Ut remains possible that
neutrophils accumulating at the inflammatory site treated locally with dexamethasone
encountered increasing levels of lipocortin which may have prevented the neutrophil from
acting to increase microvascular permeability. Pertinent to this argument is the finding
that lipocortin has been shown to have inhibitory effects on the release of reactive oxygen
species (Stevens et al., 1988).
By using a relatively simple model of acute inflammation in which the actions and
interactions of a range of inflammatory mediators have been well characterised the
possible sites of anti-inflammatory action of dexamethasone could be accurately pin
pointed. Clearly dexamethasone can have actions on the endothelial cell and the
neutrophil. To extend these observations further and investigate the relative contribution
of the actions of dexamethasone a more complex model of inflammation involving
mediator generation and action was studied. The inflammatory response induced by
zymosan in the peritoneal cavity was investigated and the effect of systemic
dexamethasone assessed.
6.9 The inflammatorv response induced bv intraperitoneal zvmosan
The intraperitoneal administration of zymosan caused an inflammatory response
in the cavity, characterised by an increase in plasma exudation (table 5.1) and neutrophil
accumulation (figure 5.1). These results confirmed the observations of Forrest et al
(1986). The extravasation of plasma proteins was assessed by measuring the
exudateiplasma blue dye ratio. As discussed by Forrest et al (1986) the blue dye
concentration in the cavity at any time will depend on several opposing factors, in
addition to dye-albumin extravasation, these include dilution by the injection fluid,
lymphatic clearance of dye-albumin from the inflammatory site and systemic clearance
of intravascular dye-albumin. Clearly an increased extravasation of plasma occurred in
154

zymosan treated animals compared to controls, and this increase occurred early in the
response to zymosan (the maximum rate of extravasation of dye occurred in the first hour
post-zymosan). The small accumulation of dye in the cavities of control animals was
probably a result of some low level inflammation caused by saline or the presence of the
indwelling cannula together with any normal basal microvascular permeability to plasma
protein (Forrest et al., 1986). In contrast to plasma extravasation, the accumulation of
neutrophils in exudate fluid was slower, significant numbers appeared at 2 hours
post-zymosan and numbers increased progressively with time. Griffiths et al (1991) also
found an increase in vascular permeability in response to intraperitoneal injection of
zymosan in the rat. In this study the authors found two phases of increased permeability,
an initial rapid phase in the first half hour and a later larger phase which commenced
2V6-3 hours post-zymosan and which coincided with the onset of leukocyte infiltration.
These changes in the local microenvironment occurred as a result of the
generation, action and interaction of a range of inflammatory mediators triggered by, and
designed to facilitate the removal of the inflammatory stimulus.
6.9.1 The generation of inflammatorv mediators in zvmosan-induced
peritoneal exudate fluid
6.9.1.1 C5a and PGIj
It has been shown previously that local extravascular complement activation
is an important part of the host defence reaction to zymosan (Forrest et al., 1986). These
authors detected the early generation (within 10 minutes) of C5a in inflammatory exudate
fluid which rose to high levels at 1, 2 and 4 hours post zymosan, the peak occurred at
2 hours (Forrest et al., 1981a; Jose et a l, 1983; Forrest et al., 1986). Based on their
findings in the peritoneal cavity and rabbit skin these authors have proposed that C5a
generation, plasma protein extravasation and neutrophil accumulation in response to
intraperitoneal zymosan are closely linked. The zymosan-induced activation of
complement in the local extravascular tissue fluid via the alternative pathway leads to the
opsonisation of particulate material so facilitating phagocytosis by neutrophils and resident
macrophages, and in addition also generates the by-product C5a, which is rapidly
converted to its physiologically more stable metabolite C5a des Arg by the action of
tissue fluid carboxypeptidase N (Hugli & Muller-Eberhard, 1978). C5a and C5a des Arg
are able to cause the neutrophil accumulation and rapid increase of venular permeability
155

observed. This sequence of events functions to supply neutrophils and more complement
components to the site in order to effect extravascular microbial lysis, opsonisation,
phagocytosis and so removal of foreign material from host tissue. Similar findings have
been reported in different types of inflammatory reactions in other species (Snyderman
et al., 1971; Damerau & Vogt, 1976; Jose & Williams, 1987) including the rheumatoid
joint (Ward & Zvaifler, 1971; Haslett et al., 1989).
In addition to C5a, Forrest et al (1986), Jose et al (1985) also demonstrated the
presence of 6 -oxo-PGFi„, the stable breakdown product of the vasodilator prostacyclin
(PGI2 ), but not PGE2 in zymosan induced inflammatory exudate fluid. These results have
been confirmed in the present study (figures 5.4 & 5.5). A more detailed time course
was followed in the present investigation which revealed the early (in the first 5 minutes)
generation of large amounts of prostacyclin (figure 5.5).
The increase in prostacyclin occurred in parallel with the increase in C5a (Forrest
et al., 1986) suggesting these events may be causally related. A correlation between
intravascular complement activation and PGI2 biosynthesis has been obtained in rabbits
following i.v. administration of endotoxin (Rampart et al., 1982). Furthermore, C5a and
C5a des Arg have been shown to trigger PGI2 formation from rabbit aorta endothelium
in vitro (Rampart et al., 1983b). In a further study Rampart et al (1989c) showed that
this effect was greatly enhanced when C5a des Arg and leukocytes were incubated
together on the endothelium, an effect which was mimicked by other chemoattractants and
was found to be, at least partly mediated by release of H2 O2 from activated leukocytes.
It is possible that these in vitro findings are relevant to the in vivo model described in this
thesis and that C5a induced interaction of neutrophils with vascular endothelial cells
might lead to PGI2 formation from the endothelium.
The results obtained with animals depleted of circulating neutrophils suggested that
the sequence of events described above was unlikely to explain the presence of PGI2 .
Neutrophil depletion had no effect on the level of 6 -oxo-PGFi„ detected in exudate fluid
at any time (figure 5.5). This result also eliminated the possibility that phagocytosis of
zymosan particles by accumulating neutrophils could account for the later generation of
prostacyclin observed. Furthermore, C5a injected intradermally in rabbit skin without
addition of exogenous prostaglandin caused only a small amount of leakage even over 4
hours which was not inhibited by indomethacin (Jose et al., 1981; Williams & Jose,
1981). It is possible, however, that the sensitivity of different blood vessels to C5a,
differences in their capacity to form various prostanoids and the sensitivity of the blood
156

vessel to the individual prostanoids released contribute to different outcomes in different
vascular beds.
It has been demonstrated in vitro that the by-products of complement activation
C3a and C5a are able to cause the biosynthesis of PGI2 by isolated rabbit peritoneum
(Rampart et a l, 1983a) an effect mediated via enhanced availability of arachidonic acid.
It is possible, therefore, that such a mechanism may be of importance in vivo. It would
be interesting to ascertain the levels of 6 -oxo-PGFi„ obtained in exudate fluid generated
by the intraperitoneal injection of pure C5a des Arg. Such an involvement of
complement in the initiation of prostacyclin generation at the inflammatory site would
serve to enhance neutrophil accumulation and oedema formation induced by C5a
(Rampart & Williams, 1986a).
It would be interesting to ascertain the effect of neutrophil depletion on the
presence of C5a in inflammatory exudate fluid. Such data would help to confirm the
sequence of events put forward by Forrest et al (1986) linking complement activation,
neutrophil accumulation and oedema formation, outlined above. It would also help to
ascertain if complement generation and prostacyclin production are linked. Neutropenia
would lead to the absence of neutrophil dependent oedema formation and hence the
further supply of complement to the area and therefore the generation of C5a would be
expected to be limited which in turn may affect prostacyclin production. Wedmore &
Williams (1981a) abolished the inflammatory response to zymosan in rabbit skin by
rendering the animals neutropenic. Neutropenia did not affect the generation of
prostacyclin in the peritoneal cavity (figure 5.7 ). Although neutropenia reduced the
accumulation of albumin-bound dye (by approximately 60-70%, table 5.2) compared with
normal animals there was still some residual oedema formation presumably mediated by
PMN-independent mediators. Thus, it is possible that sufficient complement was
supplied and hence C5a was present at the inflammatory site to maintain unchanged
prostacyclin production. Alternatively, it remains possible that prostacyclin production
in not dependent on C5a generation. Other mediators generated in response to zymosan
may lead to prostacyclin biosynthesis. Resident macrophages may generate
prostacyclin during phagocytosis of opsonized or possibly unopsonized zymosan as has
been demonstrated in vitro (Warr, 1980; Sung et a l, 1983; Czop, 1986; Nathan, 1987b;
Humes et a l, 1977). The mere presence of zymosan in the cavity may trigger
prostacyclin generation by mechanical stimulation.
157

6.9.1.2 PGE^
PGE2 has been found in inflammatory exudate generated in other models
of inflammation (Mikami & Miyasaka, 1983; Sedgwick & Lees, 1986b) and it has been
suggested that incoming neutrophils may be the source (Higgs & Salmon, 1979). Only
low levels of PGEj were detected in exudates in the present study (figure 5.4) despite the
presence of neutrophils in exudate fluid (figure 5.1). A similar result was found
(Sedgwick & Lees, 1986b) using a pleurisy model of inflammation. The disparity
between studies most likely reflects different species, stimulus, dose of stimulus, models
and methods of detection used.
6.9.1.3 Thromboxane
Thromboxane was only detected in early inflammatory exudate, the peak
level occurred 5-15 minutes post zymosan and levels fell dramatically by 1-2 hours. It
is possible that the initial presence of zymosan triggered the production of thromboxane
by mechanical stimulation. Resident macrophages are also a possible source of
thromboxane (Humes et a l, 1977).
Neutrophils are known to be a rich source of thromboxane A2 (Morley et al.,
1979; Higgs et ah, 1976) it is possible, therefore, that migrating neutrophils contributed
to the thromboxane observed as was shown by Higgs et al (1983), using animals depleted
of circulating neutrophils. Although the rise in thromboxane occurred before the
accumulation of neutrophils in the cavity it is possible thromboxane was generated by the
neutrophil during transmigration of the endothelium or perivascular basement membrane
or during passage through connective tissue and mésothélium. During their passage the
increase in neutrophil-dependent oedema formation would serve to carry any thromboxane
generated into the cavity before the arrival of the neutrophils. Such an explanation has
been proposed by Forrest et al (1986) for the different kinetics of neutrophil accumulation
and oedema formation in the cavity. Unfortunately thromboxane levels were not
measured in exudate fluid from neutropenic animals in the present study.
Since thromboxane A; is known to be the major cyclooxygenase product produced
by platelets (Hamberg et al., 1975) its detection in inflammatory exudate may indicate
the involvement of platelets in the response to zymosan. Platelet accumulation has been
observed in different animal models of acute inflammation including carrageenin-induced
paw oedema (Vincent et al., 1978), the dermal reversed passive Arthus reaction (Henson
& Cochrane, 1969) and the local Shwartzman reaction (Movat & Burrows, 1985).
158

However, the sequence of events leading to platelet accumulation, the role of platelets
and their contribution to the development of inflammation remains unclear. Higgs et al
(1983) found the concentration of thromboxane B2 in inflammatory exudates induced by
carrageenin was not altered in platelet-depleted animals neither was oedema formation
(Vargaftig, 1978). Furthermore, Cochrane and Janoff (1974) found no modification of
the Arthus reaction in the absence of platelets, although this was not confirmed by other
studies (Henson & Cochrane, 1969). The detection of thromboxane 8 % in
zymosan-induced exudate in the present study is particularly interesting in light of the
recent study by Pons et al (1993) showing “ ^In-platelet accumulation in rabbit skin in
response to intradermal zymosan. Pons et al 1993 found C5a also induced platelet
accumulation in rabbit skin and the authors have suggested the possibility that neutrophil
accumulation and platelet accumulation may be linked in this model, for example via
release of platelet stimulatory activity from neutrophils for which Chignard et al (1986)
have presented some evidence. The early generation of C5a observed in the peritoneal
cavity in response to zymosan (Forrest et al., 1986) may trigger platelet accumulation,
possibly secondary to neutrophil accumulation, the platelets may then produce the
thromboxane Bg observed possibly during adherence to or transmigration of the
endothelium. In this regard it would be interesting to compare thromboxane 8 %
production in both neutrophil-depleted and platelet-depleted animals; the accumulation of
platelets in neutrophil-depleted animals would also be interesting to ascertain.
6 . 9 . 1.4 L T B ,
Immunoreactive L T 8 4 was detected in exudates from zymosan-treated
animals (figure 5.2) and the immunoreactive material was confirmed to be L T 8 4 by
reversed phase HPLC (figure 5.12). L T 8 4 has been shown to be metabolised through w-
oxidation by neutrophils (Hansson et al., 1981; Salmon et al., 1982). Therefore the level
of L T 8 4 detected was likely to be a balance between the rate of synthesis and rate of
metabolism of L T 8 4 .
L T 8 4 has also been found in carrageenin-induced (Simmons et al., 1983;
Sedgwick & Lees, 1986a) and zymosan-induced (Ford-Hutchinson et al., 1984)
inflammatory exudate fluid in rats, and infact showed a similar time course of generation,
maximum levels being detected 4-6 hours post stimulus. Leukotriene 8 4 levels were,
therefore, highest during the later phase of the zymosan response when C5a levels had
decreased. Leukotriene 8 4 has been shown to cause neutrophil accumulation and
159

neutrophil dependent oedema formation in vivo (Wedmore & Williams, 1981a; Bray et
a l, 1981; Dahlen et a/., 1982). Thus, the LTB4 detected in this study may contribute
to the inflammatory response observed post-C5a. In this regard Ford-Hutchinson et al
(1980) found LTB4 caused neutrophil chemotaxis and chemokinesis in vitro over the
concentration range 10pg-5ng/ml and Smith et al (1980) found tbat lOOng LTB4 injected
intraperitoneally into guinea-pigs resulted in a significant increase in neutrophil
accumulation 5 hours later. However, Simmons et al (1983) and Ford-Hutchinson et al
(1984) were unable to demonstrate a correlation between the presence of this mediator
in inflammatory exudate and the accumulation of leukocytes. The present study did not
address whether the LTB4 found was an important mediator of leukocyte accumulation
in this model. It may be that sufficient LTB4 was not present to make a direct
contribution to the inflammatory response. It is possible that the time or site of
generation of LTB4 is important and that LTB4 could act indirectly, for example to induce
the synthesis of further mediators or to modulate the actions of other mediators present
during the inflammatory response. The significance of LTB4 to the inflammatory
response could be ascertained with specific leukotriene biosynthesis inhibitors or LTB4
antagonists. Recently, Rossi et al (1992) found that a specific LTB4 antagonist was
without effect on the oedema response to zymosan in rabbit skin suggesting that LTB4
does not play a major role in this response, perhaps because its action in this model is
masked by the generation of more powerful oedema-inducing mediators.
In neutrophil-depleted animals the rise in LTB4 concentration in exudate fluid was
prevented (figure 5.6) indicating that the infiltrating neutrophil was the source of LTB4
biosynthesis in this model. Similar results have been found by other groups using the
carrageenin-soaked sponge model and treating animals with colchicine to inhibit leukocyte
migration (Simmons et al., 1983). Furthermore, neutrophils have been shown to produce
LTB4 upon appropriate stimulation in vitro (Borgeat et a l, 1976; Borgeat & Samuelsson,
1979; O’Flaherty et a l, 1981). However, Sedgwick & Lees, (1986a) found LTB4 levels
and neutrophil accumulation were not related in a carrageenin-induced 6 day air pouch
model. This may reflect differences between the experimental models. A six day air
pouch was used by (Sedgwick & Lees, 1986b) and it is possible, as suggested by the
authors, that the lining of, for example macrophages and fibroblasts by six days
influenced the profile of inflammatory mediators found.
Opsonised zymosan and C5a have both been shown to cause the release of L T B 4
from neutrophils in vitro as has arachidonic acid (Palmer & Salmon, 1983; Clancy et a l,
160

1983). The release caused by opsonised zymosan was potentiated by exogenous
arachidonic acid (Palmer & Salmon, 1983). It is possible that in vivo arachidonic acid
released from membrane phospholipids of cells at the inflammatory site may synergise
with opsonised zymosan to release LTB4 from neutrophils. The stimulus for the release
of LTB4 from neutrophils was not investigated in the present study; it would be
interesting to ascertain if LTB4 was found in exudate generated following intraperitoneal
C5a.
Other inflammatory mediators both known and possibly novel are likely to be
generated either directly or indirectly in response to intraperitoneal zymosan. In the in
vivo environment populations of resident macrophages, mast cells and other tissue cells
and infiltrating neutrophils and possibly monocytes might contribute to the production of
such mediators. Bradykinin, histamine and FAF are possible candidates. However,
when the oedema-inducing activity of inflammatory exudate fluid was assessed in the skin
of rabbits in the presence of specific antagonists and inactivators of these mediators no
suppression was obtained (Collins et a l, 1991b; Forrest et a l, 1986). These results
indicate that none of these mediators have an important role in the response to zymosan
and are in agreement with the findings of Pons et al 1993, and Williams & Jose (1981)
and Rossi et al (1992) using zymosan injected intradermally in rabbits. However, the
presence of inactive metabolites in exudate was not excluded. There remains a possible
contribution therefore of these mediators to the zymosan-induced peritoneal response.
Generation of mediators might be induced by C5a (which is known to be a powerful
secretagogue (Clancy et a l , 1983) acting upon mast cells, macrophages and neutrophils,
with mediators being rapidly inactivated by extravascular enzymes - histaminases,
kininases and acetylhydrolases.
The endogenous cytokine IL-1 which can be generated from a variety of cells in
response to a wide range of stimuli may also be generated in the peritoneal cavity. IL-1
is known to be a potent mediator of neutrophil accumulation in vivo in rabbit skin
(Rampart & Williams, 1988), mouse peritoneal cavity (Sayers et a l, 1988) and mouse
air pouch (Perretti & Flower, 1993). The endogenous production of IL-1 has been
demonstrated after challenge with casein, turpentine and zymosan (Goto et a l, 1984;
Gershenwald et a l, 1990; Perretti et a l, 1992). Rampart and Williams (1988) found
that neutrophil accumulation in response to i.d. zymosan in the rabbit was partially
mediated by a cycloheximide sensitive process, which has been shown to be the case for
lL-1-induced neutrophil accumulation (Rampart & Williams, 1988).
161

TNF, also known to induce neutrophil accumulation and oedema formation in vivo
(Rampart et al. , 1989a; Sayers et al., 1988) may contribute to the inflammatory response
to zymosan. Collins et al (1991b) in a study similar to the one presented here
demonstrated the presence of IL- 8 in inflammatory exudate fluid. IL- 8 appeared later
than C5a, increasing in 4 and 6 hour exudate samples. IL- 8 is known to cause both
oedema formation and neutrophil accumulation in vivo (Foster et al. , 1989; Colditz et al.,
1989; Rampart et al., 1989d) and is likely to contribute to the later (post-C5a) neutrophil
accumulation and oedema formation in the peritoneal cavity. The stimulus for IL- 8
production was not identified although it was found not to be C5a des Arg (Collins et al.,
1991b), its source also remains unknown.
6.10 Effect of systemic dexamethasone on the inflammatorv response to
intraperitoneal zvmosan
Systemic administration of dexamethasone resulted in inhibition of both the
accumulation of inflammatory exudate protein (table 5.4) and the migration of neutrophils
into the exudate (table 5.5). Dexamethasone inhibited oedema formation and neutrophil
accumulation to levels comparable to those obtained in the group of animals that received
ip saline (table 5.1, figure 5.1). As already discussed, the glucocorticoids have been
shown to prevent oedema formation and cell migration in many models of inflammation.
The inhibition observed could be mediated by steroid effects on; 1) the
generation of chemotactic factors, and/or direct-acting mediators of increased
microvascular permeability, and/or mediators which may modulate neutrophil
accumulation and oedema formation, for example vasodilators, and/or 2 ) the action of
inflammatory mediators i.e. effects on the target cells of inflammatory mediators as
observed in Chapter 4.
Glucocorticoids have been shown by many studies both in vitro and in vivo to
inhibit the generation of pro-inflammatory arachidonic acid metabolites. This is felt to
be an important anti-inflammatory action of steroids and is believed to be mediated by
the anti-phospholipase protein, lipocortin-1. Lipocortin has been found in many studies
to inhibit eicosanoid formation and mimic closely this action of glucocorticosteroids
(discussed in detail in section 1.5.2). Inhibition by steroids and lipocortin has been noted
in vitro in a variety of cells and species including; inhibition of thromboxane (TXA2 )
162

release from isolated perfused guinea-pig lung and human lung (together with other
eicosanoids) in response to a variety of stimuli (Ervine & Shaw, 1986; Blackwell et a l,
1980; Cirino ere/., 1987; Schleimer er û/. , 1983; Schleimer er û/. , 1986). Furthermore,
inhibition by steroids and lipocortin of PGI2 release from rabbit coronary microvessel
(Rosenbaum et a l, 1986) and human umbilical artery (Cirino & Flower, 1987a)
endothelium and from rat peritoneal macrophages stimulated with opsonized zymosan
(Cirino & Flower, 1987b) has been demonstrated, as has inhibition of prostaglandin E2
from phagocytosing leukocytes and renomedullary cells (Camuccio et al., 1980; Parente
et al., 1984; Russo-Marie & Duval, 1982; Di Rosa & Persico, 1979). Finally, LTB4
release from leukocytes in response to heat-killed Bordatella pertussis (Parente et al.,
1984) and opsonized zymosan (Kurihara et al., 1984c) has been inhibited by lipocortin
and steroids.
In vivo studies have also demonstrated inhibition of eicosanoid formation by
glucocorticosteroids. However, these studies are less numerous and in some cases may
be open to alternative interpretations other than a direct effect of steroids on mediator
generation (discussed later). In a model of allergic air pouch inflammation Ohuchi et al
(1982; 1984) found that local dexamethasone reduced levels of PGEj, PGI2 , 12-HETE
and the lipoxygenase products LTC4 /LTD4 in inflammatory exudate. In a carrageenin air
pouch model in rats, systemic dexamethasone inhibited PGE2 , production (Sato et al.,
1980) and in a sponge-implant model PGE2 , TXB2 and LTB4 were reduced (Salmon et
al., 1983). Steroid treatment reduced oedema formation and leukocyte accumulation in
all studies. Tsurufuji et al (1984a) found local dexamethasone inhibited both LTB4
formation and leukocyte accumulation in an allergic air pouch model. Based on the
finding that LTB4 appeared in exudate fluid before leukocytes, these authors concluded
the inhibition of leukocyte accumulation was an indirect effect resulting from reduced
generation of chemotactic activity (LTB4 ) at the inflammatory site. Interestingly, Errasfa
& Russo-Marie (1989) found both dexamethasone and lipocortin administered i.v.
inhibited production of PGEg, LTB4 and neutrophil accumulation at the inflammatory site
induced by subcutaneous polyacrylamide gel in mice. Furthermore, Flower et al (1986)
and Vincent et al (1986) found that fluid exudation, cell migration and eicosanoid
production in a carrageenin-induced pleurisy model were greatly increased in
adrenalectomised animals compared to sham-operated controls. Glucocorticosteroids have
been shown to inhibit rat paw oedema induced by carrageenin which is thought to act
principally by releasing eicosanoids, and local lipocortin inhibited the eicosanoid
163

component of this oedema (Cirino et ah, 1989).
Surprisingly, in light of the wealth of studies to the contrary outlined above, in
the present study systemic dexamethasone failed to inhibit the level of both 6 -oxo-PGF,„
(figure 5.10) and TXB2 (figure 5.9) in zymosan-induced inflammatory exudate.
Furthermore, dexamethasone was not found to inhibit LTB4 or PGE2 production in
A23187 stimulated blood ex vivo (table 5.6). Dexamethasone was effective in inhibiting
the appearance of LTB4 in inflammatory exudate (figure 5.8) and showed the same profile
of activity as neutropenia (figure 5.6). It seems likely, therefore, that the inhibition
observed with dexamethasone was a result of inhibition of the accumulation of neutrophils
rather than a direct effect on LTB4 synthesis by these cells. However, it remains possible
that a contribution of the LTB4 inhibition was mediated by a direct effect on LTB4
production by any residual accumulation of neutrophils to the inflammatory site (table
5.5). Given the substantial inhibition of neutrophil accumulation and oedema formation
observed with dexamethasone it seems unlikely that inappropriate treatment time or
insufficient dexamethasone could explain the failure to inhibit eicosanoid production.
Specifically at odds with the results obtained are the following two findings, firstly Van
de Velde et al (1986a; 1986b) showed that mésothélial PGI2 production in vitro using
both cultured cells and isolated rabbit peritoneum was inhibited by dexamethasone, as was
the C5a-induced release of PGI; from isolated rabbit peritoneum (Rampart et al., 1983c).
However, as discussed previously, it is possible that such a mechanism was not
responsible for PGI2 production in vivo. Secondly, this site, the peritoneal cavity was
used as a souce of steroid-induced lipocortin (Blackwell et al., 1982). However, this
study was in rat.
The results obtained in this thesis are in agreement with those presented by
Griffiths and Blackham (1991) using the same model of inflammation but in rats. These
authors found that oral or subcutaneous dexamethasone failed to inhibit eicosanoid
production in peritoneal exudate fluid and in A23187 stimulated blood ex vivo.
Furthermore, only partial inhibition of eicosanoid production in zymosan-induced arthritis
was observed, although the later of two phases of increased vascular permeability and
neutrophil accumulation were inhibited. The same authors have also obtained similar
data in rabbit skin. In these experiments i.v. dexamethasone inhibited plasma exudation
and neutrophil accumulation but failed to inhibit LTB4 and PGE2 production in response
to intradermal phospholipase A2 (Griffiths & Blackham, 1988). A further study by Ford-
Hutchinson et al (1984) using zymosan impregnated sponges in rats found dexamethasone
164

had no significant effect on the generation of LTB ; in this study neutrophil accumulation
was also unaffected. These results are difficult to explain and raise the question of
whether the inhibition of phospholipase Aj by lipocortins is important in vivo. The nature
of the interaction and the physiological relevance of the inhibitory effect of lipocortin on
PLA2 activity has been questioned and the topic remains controversial (Davidson &
Dennis, 1989).
In a study investigating the effects of steroids on arachidonic acid metabolite
release from human lung fragments Schleimer et al (1986) found that the inhibitory effect
of dexamethasone varied depending on the stimulus used and the metabolite in question.
Carrageenin rather than zymosan has often been used as the inflammatory stimulus where
dexamethasone-induced inhibition of eicosanoid production has been demonstrated in vivo
(Flower et al., 1986; Salmon et al., 1983). It is possible, therefore, that differences in
stimuli used may explain, at least in part the conflicting results obtained in different
studies, as might the variety of inflammatory models. Sedgwick and Lees (1986b) have
presented some evidence in support of the latter point. These authors found that
carrageenin-induced air pouch, sponge and pleurisy models of inflammation in rats
showed different susceptibility to dexamethasone with regard to inhibition of plasma
exudation, cell accumulation and eicosanoid formation.
The reason dexamethasone failed to inhibit the generation of pro-inflammatory
arachidonic acid metabolites in this model remains unanswered, but this finding, together
with the fact that dexamethasone was effective at inhibiting oedema formation and
neutrophil accumulation suggest several points. Firstly, these results support those found
with neutropenic animals with regard to the conclusion that prostacyclin generation in the
cavity is independent of neutrophil infiltration and indicate that TXBj generation is
unlikely to be mediated by the infiltrating neutrophil. The slight reduction in TXBj in
dexamethasone-treated animals which did not reach significance may reflect a component
of neutrophil-derived thromboxane. Secondly, the results indicate that the formation of
eicosanoids was not obligatory for the inflammatory response to occur in this model and,
thirdly, that dexamethasone exerts alternative antiinflammatory actions in this model.
The latter two points complement the findings of Jose et al (1985). These authors, using
the same model found that i.v. indomethacin in contrast to dexamethasone almost
abolished the production of 6 -oxo-PGF,„ (greater than 90% inhibition compared to
controls) but only minimally affected plasma albumin extravasation (30% inhibition).
Alternative anti-inflammatory actions for dexamethasone in this model could
165

include a direct effect of steroid on the neutrophil and/or the endothelium as observed in
Chapter 4 and inhibition of the generation of non-arachidonic acid derived mediators.
These two events could be related in this model. C5a has been shown to be an important
chemotactic factor generated in response to i.p. zymosan. Since neutrophil accumulation,
oedema formation and C5a generation have been postulated to be intimately related as
discussed previously, any direct effect of steroids on neutrophils (and/or endothelium) to
prevent their accumulation would be expected to reduce C5a generation indirectly by
reducing neutrophil dependent oedema formation. An effect of dexamethasone on the
generation or action of histamine (Schleimer et al., 1981) PAF (Parente et al., 1985) IL-
1 (Knusden et a l, 1987; Snyder & Unanue, 1982; Bochner et a l, 1987b) TNF (Watson
et a l, 1987) or IL- 8 (Watson et a l, 1987) may also contribute.
It is difficult to decifer the relative contribution or importance of direct actions of
dexamethasone on cells and inhibition of mediator generation in this model. It is clear
that dexamethasone action may be multifactorial and the final outcome in any given
model may depend on the sensitivity of the reaction to inhibition of mediator generation
and of the target cells involved and the nature of their interactions with each other. The
route of administration of the steroid may also be important in determining its actions.
Systemically administered steroid would allow circulating neutrophils (and possibly other
cell types) to be exposed and so possibly affected directly. In contrast, neutrophils would
not be affected directly by local steroids and thus the balance of steroid action may tip
towards the contribution and importance of the inhibition of mediator generation. Several
studies have provided evidence which may support such a theory. Perretti & Flower
(1993) using a model of IL-1-induced inflammation in mouse air pouch found that
systemic but not local lipocortin inhibited neutrophil accumulation whereas both systemic
(via lipocortin induction) and local dexamethasone were inhibitory. These authors
concluded that systemic inhibition was likely to be mediated by an effect on the
circulating neutrophil whilst local dexamethasone could possibly be interfering with the
generation of chemotactic factors induced by IL-1 (Watson et a l, 1988). Furthermore,
Errasfa & Russo-Marie (1989) using an i.v. route of administration concluded that both
inhibition of PLAj and a direct effect on the circulating neutrophil was contributing to
the inhibition of neutrophil accumulation by dexamethasone and lipocortin. Tsurufuji
et al (1984a) using local dexamethasone concluded that the inhibition of neutrophil
accumulation was due solely to inhibition of LTB4 generation at the site. However, an
alternative explanation for the results obtained by Tsurufuji et al (1984a), which does not
166

involve a steroid effect on mediator generation remains possible. The L T B 4 detected may
be generated by neutrophils during diapedesis so that a steroid effect on the neutrophil
to prevent its adhesion and transmigration would therefore also lead to reduced LTB4
levels, as was found in the present study. Such an explanation casts doubt on steroid
action being to inhibit mediator generation in other models in vivo.
As we find out more about the actions of antiinflammatory steroids and the
importance of these actions it may become possible to identify key effector mechanisms
which mediate particular aspects of steroid action. This in turn may lead to more
targetted antiinflammatory therapy.6.11 Further work
1. In view of the small but significant neutropenia induced by i.v. colchicine at a dose of1 mg/kg it would be interesting to establish if lower doses could induce the profound effects observed on neutrophil-dependent oedema formation without inducing a neutropenia.These experiments would help to establish to what extent the decline in circulating neutrophils observed with 1 mg/kg colchicine i.v. contributed to the profound suppression of neutrophil-dependent oedema formation.
2. To clearly establish the molecular mechanism involved in the glucocorticoid action on permeability induced by both direct-acting and neutrophil-dependent mediators and on neutrophil accumulation in the rabbit skin model the receptor antagonist RU 38486 and inhibitors of mRNA and protein synthesis could be used.
3. The involvement of lipocortin in the glucocorticoid-induced modulation of oedema and neutrophil accumulation induced by chemoattractants could be investigated using recombinant lipocortin and/or neutralising antibodies. Lipocortin could be administersd i.d., i.v. or used to pretreat 111 In-neutrophils in vitro prior to their i.v, administration to a recipient animal. Such studies would establish the site of action of any anti-inflammatory action of lipocortin. It would also be interesting to investigate the possible role of vasocortin in mediating the anti-exudative actions of dexamethasone. Such studies could be performed using purified vasocortin if available.
4. The effect of glucocorticoids or glucocorticoid-induced proteins on the endothelium could be investigated in vitro. A block in the signal transduction pathway in EC may be the locus of action of steroids, or possible interferance with the contractile machinery of the endothelial cell. Studies investigating the interaction of glucocorticoids with intracellular second messengers and comparing steroid effects with those of compounds and drugs of known action may help to unravel the mechanism of steroid action.
5. To complement and extend the studies where neutrophil donor rabbits were treated i.v. with dexamethasone prior to the isolation and 1 1 IJn-labelling of neutrophils it would be interesting to treat isolated neutrophils in vitro with dexamethasone prior to their i.v. administration to animals. Such studies would help clarify whether the steroid was able to act directly on the neutrophil or whether steroids were acting initially on other cell types with a resultant secondary effect on the neutrophil. Such experiments may be technically difficult as it is not wise to keep isolated neutrophils in culture for periods longer than one hour prior to their introduction in vivo. The success of the experiment would depend on the time necessary for neutrophil treatment with dexamethasone in vitro.
6 . It would be interesting to ascertain the effect of neutrophil depletion on the presence of C5a in zymosan induced inflammatory exudate fluid. Such data would help to establish the relationship between complement activation, neutrophil accumulation and oedema formation. Furthermore, it would also help to ascertain if complement generation and prostacyclin generation are linked.

References
AARSMAN, A.J., MYNBEEK, G., VAN DEN BOSCH, H., ROTHHUT, B., PRIEUR, B., COMERA, C., JORDAN, L., & RUSSO-MARIE, F. (1987). Lipocortin inhibition of extracellular and intracellular phospholipases A2 is substrate concentration dependent. FEBS Letts. 219: 176-180.
ABITA, J.P. (1981). Indomethacin is a competitive inhibitor of the binding of the chemotactic peptide formyl-Met-Leu-Phe to human polymorphonuclear leukocytes. Agents Actions 11: 610-612.
ABRAMSON, S., EDELSON, H., KAPLAN, H., LUDEWIG, R., & WEISSMANN, G.(1984). Inhibition of neutrophil activation by nonsteroidal anti-inflammatory drugs. Am. J. Med. 77: 3-6.
ABRAMSON, S., HAINES, K., LESZCZYNSKA, J., REIBMAN, J., & WEISSMANN, G. (1988). Nonsteroidal anti-inflammatory drugs (NSADDs) inhibit neutrophil (PMN) functions via effects at the G protein of the plasma membrane. Clin. Res. 36: 548A.
ABRAMSON, S B., CHERKSEY, B., GUDE, D., LESZCZYNSKA PIZIAK, J., PHILIPS, M R., BLAU, L., & WEISSMANN, G. (19%). Nonsteroidal antiinflammatory drugs exert differential effects on neutrophil function and plasma membrane viscosity. Studies in human neutrophils and liposomes. Inflammation 14: 11-30.
ABRAMSON, S B. & WEISSMANN, G. (1989). The mechanisms of action of nonsteroidal antiinflammatory drugs. Arthritis Rheum. 32: 1-9.
ACKERMAN, N., MARTINEZ, S., THIEME, T., & MIRKOVICH, A. (1982). Relationship between adherence, chemotaxis and the accumulation of rat polymorphonuclear leukocytes at an inflammatory site. J. Pharmacol. Exp. Ther. 221: 701-707.
AHLUWALIA, A., PEERS, S.H., & FLOWER, R.J. (1992). Steroid inhibition of oedema formation in the rat skin. Br. J. Pharmacol. 106: 628-631.
A L B E R T , D . H . & S N Y D E R , F. ( 1 9 8 3 ) . B i o s y n t h e s i s of l-Alkyl-2-acetyl-Sn-glycero-3-phosphocholine (Platelet-Activating factor) from l-alkyl-2-acyl-3n-glycerophosphocholine by rat alveolar macrophages. Phospholipase A% and acetyltransferase activities during phagocyosis and ionophore stimulation. J. Biol. Chem. 258: 97-102.
ALLISON, F., SMITH, M R., & WOOD, W.B. (1955). Studies on the pathogenesis of acute inflammation. The action of cortisone on the inflammatory response to thermal injury. J. Exp. Med. 102: 669.
ALTIERI, D C. (1991). Occupancy of CDllb/CD18 (Mac-1) divalent ion binding site(s) induces leukocyte adhesion. J. Immunol. 147: 1891-1898.
AMBROSE, M.P. & HUNNINGHAKE, G.W. (1990). Corticosteroids increase lipocortin 1 in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol 3: 349-353.
ANDERSON, D.C., SCHMALSTEIG, F.C., FINEGOLD, M.J., HUGHES, B.J., ROTHLEIN, R., MILLER, L.J., KOHL, S., TOSI, M.F., JACOBS, R.L., WALDROP, T.C., GOLDMAN,A.S., SHEARER, W.T., & SPRINGER, T.A. (1985). The severe and moderate phenotypes of heritable Mac-1, LFA-1 deficiency: their quantiative definition and relation to leukocyte dysfunction and clinical features. J. Irfect. Dis. 152: 668-689.
168

ANDERSON, D.C., MILLER, L.J., SCHMALSTIEG, F.C., ROTHLEIN, R., & SPRINGER, T.A. (1986). Contributions of the Mac-1 glycoprotein family to adherence-dependent granulocyte functions: structure-function assessments employing subunit-specific monoclonal antibodies. J. Immunol 137: 15-27.
ANDERSON, D C. & SPRINGER, T.A. (1987). Leukocyte adhesion deficiency: An inherited defect in the mac-1, LFA-1 and pl50,95 glycoproteins. Ann. Rev. Med. 38: 175-194.
ARFORS, K. E., LUNDBERG, C., LINDBOM, L., LUNDBERG, K., BEATTY, P.G., & HARLAN, J.M. (1987). A monoclonal antibody to the membrane glycoprotein complex CD18 inhibits polymorphonuclear leukocyte accumulation and plasma leakage in vivo. Blood 69: 338-340.
ARGENBRIGHT, L.W., LETTS, L.G., & ROTHLEIN, R. (1991). Monoclonal antibodies to the leukocyte membrane CD18 glycoprotein complex and to intercellular adhesion molecule-1 inhibit leukocyte-endothelial adhesion in rabbits. J. Leuk. Biol 49: 253-257.
ARNAOUT, M.A. (1990). Leucocyte adhesion molecules deficiency: Its structural basis, pathophysiology and implications for modulating the inflammatory response. Immunol Rev. 114:145-180.
ARNAOUTM.A., TODCHI, R.F., DANA, N., MELAMED, J., SCHLOSSMAN, S.F., & COLTEN, H R. (1983). Inhibition of phagocytosis of complement C3 or immunoglobulin G-coated particles and of C3bi binding by monoclonal antibodies to a monocyte-granulocyte membrane glycoprotein (Mo-1). J. Clin. Invest. 72: 171-179.
ASAKO, H., KUBES, P., BAETHGE, B.A., WOLF, R.E., & GRANGER, D.N. (1992). Colchicine and methotrexate reduce leukocyte adherence and emigration in rat mesenteric venules. Irtflammation 16: 45-56.
ATHENS, J.W., HAAB, O.P., RAAB, S O., MAUER, A.M., ASHENBRUCKER, H., CARTWRIGHT, G.E., & WINTROBE, M.M. (1961). Leukokinetic studies. IV. The total blood, circulating and marginal granulocyte pools and the granulocyte turnover rate in normal subjects. J. Clin. Invest. 40: 989-995.
ATHERTON, A. & BORN, G.V.R. (1972). Quantitative investigations of the adhesiveness of circulating polymorphonuclear leucocytes to blood vessel walls. J. Physiol 222: 447-474.
ATHERTON, A. & BORN, G.V.R. (1973). Relationship between the velocity of rolling granulocytes and that of blood flow in venules. J. Physiol 233: 157-165.
BABIOR, B.M., KIPNES, R.S., & CURNUTTE, J.T. (1973). The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Invest. 52: 741-744.
BABIOR B.M. (1984). Oxidants from phagocytes: agents of defense and destruction. Blood 64: 959-966.
BAILEY, J.M., MAKHEJA, A.N., PASH, J., & VERM A, M. (1988). Corticosteroids suppress cyclooxygenase messenger RM A levels and prostanoid synthesis in cultured vascular cells. Biochem. Biophys. Res. Commun. 157: 1159-1163.
BASRAN, G.S., MORLEY, J., PAUL, W., & TURNER-WARWICK, M. (1982). Evidence in man of synergistic interaction between putative mediators of acute inflammation and asthma. Lancet 1: 935-937.
169

BAXTER, J.D. & TOMLINS, G.M. (1977). Specific cytoplasmic glucocorticoid hormone receptors in hepatoma tissue culture cells. Proc. Neal Acad. Sal USA 6 8 : 932-937.
BECKER, C.G. & NACHMAN, R.L. (1973). Contractile proteins of endothelial cells, platelets and smootii muscle. Am. J. Pathol 71: 1-22.
BECKER, E.L. (1987). The formyl peptide receptor of the neutrophil. A search and conserve operation. Am. J. Pathol 129: 16-24.
BERLIN, R. & FERA, J. (1977). Changes in membrane viscosity associated with phagocytosis: effects of colchicine. /Voc. Natl Acad. Sci. USA 74: 1072.
BERNSTEIN, I.D. & SELF, S. (1985). The joint report of the myeloid section of the Second International Workshop on human leukocyte differentiation antigens. In Leukocyte Typing IIReport of the Second International Workshop on human leukocyte differentiation antigens, eds. ReiiAerz E. L., Haynes B. S., Nadler L. M., & Bernstein I. D. pp. 1. New York: Springer-Verlag.
BESSE, J.C. & BASS, A.D. (1966). Potentiation by hydrocortisone of responses to catecholamines in vascular smooth muscle. J. Pharmacol Exp. Ther. 154: 224-238.
BEVILACQUA, M.P., STENGELIN, S., GIMBRONE, M.A., & SEED, B. (1989). Endothelial leukocyte adhesion molecule 1 : an inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science 243: 1160-1164.
BEVILACQUA, M.P. (1993). Endothelial-leukocyte adhesion molecules. Ann. Rev. Immunol 11: 767-804.
BIENKOWSKI, M.J., PETRO, M.A., & ROBINSON, L.J. (1989). Inhibition of thromboxane A synthesis in U937 cells by glucocorticoids. Lack of evidence for lipocortin 1 as the second messenger. J. Biol Chem. 264: 6536-6544.
BISHOP, C.R., ATHENS, J.W., BOGGS, D R., WARNER, H R., CARTWRIGHT, G.E., & WINTROBE, M.M. (1968). Leukokinetic studies. 13. A non-steady-state kinetic evaluation of the mechanism of cortisone-induced granulocytosis. J. Clin. Invest. 47: 249-260.
BISHOP, C.T., MIRZA, Z., CRAPO, J.D., & FREEMAN, B.A. (1985). Free radical damage to cultured procine aortic endothelial cells and lung fibroblasts: modulation by culture conditions. In Vitro Cell Dev. Biol 21: 229-236.
BJORK, J , ARFORS, K-E., DAHLEN, S.E., & HEDQVIST, P. (1981). Effects of leukotrienes on vascular permeability and leukocyte adhesion. In The inflammatory process, eds. Venge P. & Lindbom A. pp. 103. Stockholm: Almqvist & Wiksell International.
BJORK, J., ARFORS, K-E., HEDQUIST, P., DAHLEN, S.E., & LINDGREN, J.A. (1982). Leukotriene B4 causes leukocyte emigration from postcapillary venules in the hamster cheek pouch. Microcirculation 2: 271-281.
BJORK, J., HUGLI, T.E., & SMEDEGARD, G. (1983). Microvascular effects of anaphylatoxins C3a and C5a. J. Immunol 134: 1115-1119.
BJORK, J., GOLDSCHMIDT, T., SMEDEGARD, G., & ARFORS, K-E. (1985). Methylprednisolone acts at the endothelial cell level reducing inflammatory responses. Aaa Physiol Scand. 123: 221-224.
BJORK, J. & SMEDEGARD, G. (1983). Acute microvascular effects of PAF-acether as studied by intravital microscopy. Eur. J. Pharmacol 96: 87-94.
170

BJORK, J., HEDQUIST, P., & ARFORS, K-E. (1982). Increase in vascular permeability induced by Leukotriene B4 and the role of polymorphonuclear leukocytes. Irtflammarion 6 : 189-2(X).
BLACKWELL, G.J., FLOWER, R.J., NUKAMP, F.P., & VANE, J R. (1978). Phospholipase A2 activity of guinea-pig isolated perfused lungs: stimulation and inhibition by anti-inflammatory steroids. Br. J. Pharmacol 62: 79-89.
BLACKWELL, G.J., CARNUCCIO, R., DI ROSA, M., FLOWER, R.J., PARANTE, L., & PERSICO, P. (1980). Macrocortin: a polypeptide causing the anti-phospholipase effect of glucocorticoids. Nature 287: 147-149.
BLACKWELL, G.J., CARNUCCIO, R., ROSA, M.D., FLOWER, R.J., LANGHAM, C.S.J., PARENTE, L., PERSICO, P., RUSSEU^SMITH, N.C., & STONE, D. (1982). Glucocorticoids induce the formation and release of anti-inflammatory and anti-phospholipase proteins into the peritoneal cavity of the rat. Br. J. Pharmacol 76: 185-194.
BOCHNER, B.S., LANDY, S.D., PLAUT, M., DINARELLO, C.A., & SCHLEIMER, R.P. (1987a). Interleukin 1 production by human lung tissue: I. Identification and characterization. J. Immunol 139: 2297-2302.
BOCHNER, B.S., RUTLEDE, B.K., & SCHLEIMER, R.P. (1987b). Interleukin 1 production by human lung tissue. II. Inhibition by anti-inflammatory steroids. J. Immunol 139: 2303-2307.
BORGEAT, P., HAMBERG, M., & SAMUELSSON, B. (1976). Transformation of arachidonic acid and homo-gamma-linolenic acid by rabbit polymorphonuclear leukocytes. Monohydroxy acids from novel lipoxygenases. J. Biol Chem. 251: 7816-7820.
BORGEAT, P. & SAMUELSSON, B. (1979). Arachidonic acid metabolism in polymorphonuclear leukocytes: effects of ionophore A23187. Proc. Natl Acad. Scl U. S. A. 76: 2148-2152.
BOWEN, B.R., NGUYEN, T., & LASKY, L A. (1989). Characterization of a human homologue of the murine peripheral lymph node homing receptor. J. Cell Biol 109: 421.
BOXER, L.A., HAAK, R.A., YANG, H.H., WOLACH, J.B., WHITCOMB, J.A., BUTTERICK, C.J., & BAEHNER, R.L. (1982). Membrane-bound lactoferrin alters the surface properties of polymorphonuclear leukocytes. J. Qin. Invest. 70: 1049-1057.
BOXER, L.A., YODER, M., BONSIB, S., SCHMIDT, M., HO, P., JERSILD, R., & BAEHNER, R.L. (1989). Effects of a chemotactic factor N formyl-methionyl peptide on adherence, superoxide anion generation, phagocytosis, and microtubule assembly of human polymorphonuclear leukocytes. J. Lab. Clin. Med. 93: 506-574.
BOXER, L.A., AXTELL, R., & SUCHARD, S. (1990). The role of the neutrophil in inflammatory diseases of the lung. Blood Cells 16: 25-40.
BRAIN, S.D., WILLIAMS, T.J., TIPPINS, J R., MORRIS, H R., & MACINTYRE, I. (1985). Calcitonin gene-related peptide (CGRP) is a potent vasodilator. Nature 313: 54-56.
BRAIN, S.D., MACINTYRE, I., & WILLIAMS, T.J. (1986a). A second form of human calcitonin gene-related peptide which is a potent vasodilator. Eur. J. Pharmacol 124: 349-352.
BRAIN, S.D., TIPPINS, JR. , MORRIS, H R , MACINTYRE, I., & WILLIAMS, T.J. (1986b). The potent vasodilator activity of calcitonin gene-related peptide in human skin. J. Invest. Dermatol 87: 533-536.
171

BRAIN, S.D. & WILLIAMS, T.J. (1985). Inflammatory oedema induced by synergism between calcitonin gene-related peptide (CGRP) and mediators of increased vascular permeability. Br. J. Pharmacol 8 6 : 855-860.
BRAY, M.A., CUNNINGHAM, P.M., FORD-HUTCHINSON, A.W., & SMITH, M.J.H. (1981). Leukotriene B : a mediator of vascular permeability. Br. J. Pharmacol 72: 483-486.
BRONNEGARD, M., ANDERSSON, O., EDWALL, D., LUND, J., NORSTEDT, G., & CARSTEDT-DUKE, J. (1988). Human calpactin II (lipocortin 1) messenger ribonucleic acid is not induced by glucocorticoids. Mol Endocrinol 2: 732-739.
BROOKS, P.M., BURTON, D., & FORREST, M.J. (1987). Crystal-induced inflammation in the rat subcutaneous air-pouch. Br. J. Pharmacol 90: 413-419.
BROWN, K.A. & COLLINS, A.J. (197* . Action of nonsteroidal, antiinflammatory drugs on human and rat peripheral leukocyte migration. Ann. Rheum. Dis. 36: 239-243.
BUCKLEY, T.L., BRAIN, S.D., RAMPART, M., & WILLIAMS, T.J. (1988). The potential of calcitonin gene-related peptide as an inflammatory mediator. Regulatory Peptides 22 No.4: 393 (Abst).
BULLER, R E. & O’MALLEY, B.W. (1976). The biology and mechanism of steroid hormone receptor interaction with the eukaryotic nucleus. Biochem. Pharmacol 25: 1-12.
BUTCHER, E C. (1991). Leukocyte-endothelial cell recognition - three (or more) steps to specificity and diversity. Cell 67: 1033-1036.
BUYON, J.P., ABRAMSON, SB., PHILIPS, MR. , SLADE, S.G., ROSS, G.D., WEISSMANN, G., & WINCHESTER, R.J. (1988). Dissociation between increased surface expression of Gp165/95 and homotypic neutrophil aggregation. J. Immunol 140: 3156-3160.
CALIGNANO, A., CARNUCCIO, R., DI ROSA, M., lALENTI, A., & MONCADA, S.(1985). The anti-inflammatory effect of glucocorticoid-induced phospholipase inhibitory proteins. Agents Actions 16: 60-62.
CAMERINI, D., JAMES, S.P., STAMENKOVIC, I., & SEED, B. (1989). Leu-8 /TQl is the human equivalent of the Mel-14 lymph node homing receptor. Nature 342: 78-82.
CAMP, R.D.R., COUTTS, A.A., GREAVES,M.W., KAY, A.B., & WALPORT, M.J. (1983). Responses of human skin to intradermal injection of leukotrienes C4 , D4 and B4 . Br. J. Pharmacol 80: 497-502.
CAMUSSI, G., TETTA, C., BUSSOLINO, F., CAPPIO, F.C., CODA, R., MASERA, C., & SEGOLONI, G. (1980). Mediators of immune complex-induced aggregation of polymorphonuclear neutrophils. Int. Archs. Allergy appl Immunol 62: 1-15.
CANER, J.E.Z. (1965). Colchicine inhibition of chemotaxis. Arthritis Rheum. 8 : 757.
CAREY, F., FORDER, R.A., GIBSON, K.H., & HAWORTH, D. (1989). Radioimmunoassay of LTB4 in plasma from different species: A cautionary note. Prostaglandins Leukotrienes and Essential Fatty Acids 36: 57-61.
CAREY, F., FORDER, R., EDGE, M.D., GREENE, A.R., HORAN, M.A., STRUBOS, P.J., & ROTHWELL, N.J. (1990). Lipocortin 1 fragment modifies pyrogenic actions of cytokines in rats. Am. J. Physiol 259: R266-^69.
172

CAREY, F. & FORDER, R.A. (1986). Radioimmunoassay of LTB» and 6 -trans LTB : analytical and pharmacological characterisation of immunoreactive LTB4 in ionophore stimulated human blood. Prostaglandins Leukotrienes Medicine 22: 57-70.
CARNUCCIO, R., DI ROSA, M., & PERSICO, P. (1980). Hydrocortisone-induced inhibitor of prostaglandin biosynthesis in rat leucocytes. Br. J. PharmacoL 6 8 : 14-16.
CARNUCCIO, R., ROSA, M.D., GUERRASIO, B., lUVONE, T., & SAUTEBIN, L. (1987). Vasocortin: a novel glucocorticoid-induced anti-inflammatory protein. Br. J. Pharmacol 90: 443-445.
CARNUCCIO, R., DI ROSA, M., lALENTI, A., lUVONE, T., & SAUTEBIN, L. (1989a). Selective inhibition by vasocortin of histamine release induced by dextran and concanavalin-A from rat peritoneal cells. Br. J. Pharmacol 98: 32-34.
CARNUCCIO, R., DI ROSA, M., lUVONE, T., MAROTTA, P., & SAUTEBIN, L. (1989b). Vasocortin-like proteins induced by glucocorticoids in vascular tissue. Eur. J. Pharmacol 166: 535-539.
CARP, H. (1982). Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J. Exp. Med. 155: 264-275.
CARVETH, H.J., BOHNSACK, J.F., MCINTYRE, T.M., BAGGIOLINI, M., PRESCOTT,S.M., & ZIMMERMAN, G.A. (1989). Neutrophil activating factor (NAl^ induces polymorphonuclear leukocyte adherence to endothelial cells and to subendothelial matrix proteins. Biochem. Biophys. Res. Commun. 162: 387-393.
CHAMLEY-CAMPBELL, J., CAMPBELL, G.R., GROSCHEL-STEWART, U., & BURNSTOCK, G. (1977). FITC-labelled antibody staining of tropomyosin-containing fibrils in smooth, cardiac and skeletal muscle cells, perfusion myoblasts, fibroblasts, endothelial cells and 3T3 cells in culture. Cell Tissue Res. 183: 153-166.
CHAN, L. & O’MALLEY, B.W. (1976). Mechanism of action of the sex steroid hormones. N. Engl J. Med. 294: 1372-1379.
CHANG, J., LEWIS, G.P., & PIPER, P.J. (1977). Inhibition by glucocorticosteroids of prostaglandin release from adipose tissue in vitro. Br. J. Pharmacol 59: 425-432.
CHANG, Y-H. (1975). Mechanisms of action of colchicine 1. Effect of colchicine and its analogs on the reversed passive Arthus reaction and the carrageenan-induced hind paw oedema in the rat. J. Pharmacol Exp. Ther. 194: 154-158.
CHATELAIN, P., LATOUR, J.G., TRAN, D., DE LORGERIL, M., DUPRAS, G., & BOURASSA, M. (1987). Neutrophil accumulation in experimental myocardial infarcts: relation with extent of injury and effect of reperfusion. Grculation 75: 1083-1090.
CHENOWETH, D.E. & HUGLI, T.E. (1978). Demonstration of specific C5a receptor on intact human polymorphonuclear leukocytes. Proc. Natl Acad. Scl USA 75: 3943-3947.
CHIEN, S. (1982). Rheology in the microcirculation in normal and low flow states. Adv. Shock res. 8 : 71-80.
CHIGNARD, M., SELAK, M.A., & SMITH, J.B. (1986). Direct evidence for the existence of a neutrophil-derived platelet activator (neutrophilin). Proc. Natl Acad. Scl USA 83: 8609-8613.
173

CHRISTMAS, P., CALLAWAY, J., FALLON, J., JONES, J., & HAIGLER, H.T. (1991). Selective secretion of annexin 1, a protein without a signal sequence, by the human prostate gland. J. Biol Oienu 266: 2499-2507.
CHURCH, M.K. & MILLER, P. (1978). Time courses of the anti-anaphylactic and anti-inflammatory effects of dexamethasone in the rat and mouse. Br. J. Pharmacol 62:481-486.
CIRINO, G, FLOWER, R.J., BROWNING, J.L., SINCLAIR, L.K., & PEPINSKY, R.B.(1987). Recombinant human lipocortin 1 inhibits thromboxane release from guinea-pig isolated perfused lung. Nature 328: 270-272.
CIRINO, G., PEERS, S.H., FLOWER, R.J., BROWNING, J.L., & PEPINSKY, R.B. (1988). Human recombinant lipocortin 1 has acute local anti-inflammatory properties in the rat paw oedema test. Proc. Natl Acad. Scl USA 8 6 : 3428-3432.
CIRINO, G., PEERS, S.H., FLOWER, R.J., BROWNING, J.L., & PEPINSKY, R.B. (1989). Human recombinant lipocortin 1 has acute local anti-inflammatory properties in the rat paw edema test. Proc. Natl Acad. Scl USA 8 6 : 3428-3432.
CIRINO, G. & FLOWER, R.J. (1987a). Human recombinant lipocortin 1 inhibits prostacyclin production by human umbilical artery in vitro. Prostaglandins 34: 59-62.
CIRINO, G. & FLOWER, R.J. (1987b). The inhibitory effect of lipocortin on eicosanoid synthesis is dependent upon divalent cation. Br. J. Pharmacol 92: 521.
CLANCY, R.M., DAHINDEN, C.A., & HUGLI, T.E. (1983). Arachidonate metabolism by human polymorphonuclear leukocytes stimulated by N-formyl-Met-Leu-Phe or complement component C5a is independent of phospholipase activation. Proc. Natl Acad. Scl USA 80: 7200-7204.
CLARK, E.R. & CLARK, E.L. (1935). Observations on changes in blood vascular endothelium in the living animal. Am. J. Anat. 57: 385-438.
CLARK, R.A.F., GALLIN, J.I., & FAUCI, A S. (1979). Effects of in vivo perdnisone on in vitro eosinophil and neutrophil adherence and chemotaxis. Blood 53: 633-641.
CLOK, J.F., COLARD, O., ROTHHUT, B., & RUSSO-MARIE, F. (1983). Characterization and partial purification of ’renocortins’: two polypeptides formed in renal cells causing the anti-phospholipase-like action of glucocorticoids. Br. J. Pharmacol 79: 313-321.
COCHRANE, G.C. & JANOFF, A. (1974). The Arthus reaction: A model of neutrophil and complement-mediated injury. In The inflammatory process, eds. Zweifach B. W., Grant L., & McCluskey R. T. pp. 85-162. New York: Academic Press.
COHNHEIM, J. (1889). Inflammation. In Lectures on General Pathology. Section 1. The pathology of the circulation, pp. 242-382. London: New Sydenham Society.
COLDITZ, 1., ZWAHLEN, R., DEWALD, B., & BAGGIOLINI, M. (1989). In vivo inflammatory activity of neutrophil-activating factor, a novel chemotactic peptide derived from human monocytes. Am. J. Pathol 134: 755-760.
COLDITZ, l.G. & MOVAT, H Z. (1984a). Kinetics of neutrophil accumulation in acute inflammatory lesions induced by chemotaxins and chemotaxinogens. J. Immunol 133: 2169-2173.
COLDITZ, l.G. & MOVAT, H Z. (1984b). Desensitization of acute inflammatory lesions to chemotaxins and endotoxin. J. Immunol 133: 2163-2168.
174

COLLINS, P.D., JOSE, P.J., & WILLIAMS, T.J. (1991b). The sequential generation of neutrophil chemoattractant proteins in acute inflammation in the rabbit in vivo: relationship between C5a and a protein with the characteristics of IL-8 . J. Immunol 146: 677-684.
COST, H., GESPACH, C., & ABITA, J.P. (1981). Effect of indomethacin on the binding of the chemotactic peptide formyl-Met-Leu-Phe on human polymorphonuclear leukocytes. FEBS Lett. 132: 85-88.
COTRAN, R.S. (1965). The delayed and prolonged vascular leakage in inflammation. II. An electron microscopic study of the vascular response after thermal injury. Am. J. Pathol 46: 589-620.
COTRAN, R.S. (1967). The delayed and prolonged vascular leakage in inflammation. III. Immediate and delayed reactions in skeletal muscle. Exp. Mol Pathol 6 : 143-155.
COTRAN, R.S. & MAJNO, G. (1964). The delayed and prolonged vascular leakage in inflammation. I. Topography of the leaking vessels after thermal injury. Am. J. Pathol 45: 261-281.
CRADDOCK, P R., FEHR, J., BRIGHAM, K.L., KRONENBERG, R.S., & JACOB, H.S. (1977a). Complement and leukocyte mediated pulmonary dysfunction in hemodialysis. N. Engl J. Med. 296: 769-773.
CRADDOCK, P.R., HAMMERSCHMIDT, D , WHITE, J.G., DALMASSO, A.P., & JACOB, H.S. (1977b). Complement (C5a)-induced granulocyte aggregation in vitro. J. Clin. Invest. 60: 260-264.
CROMPTON, M R., MOSS, S.E., & CRUMPTON, M.J. (1988). Diversity in the lipocortin/calpactin family. Cell 55: 1-3.
CRONSTEIN, B.N., KIMMEL, S.C., LEVIN, R.I., MARTINIUK, F., & WEISSMANN, G. (1992). A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc. Natl Acad. Scl U. S. A. 89: 9991-9995.
CROWELL, R E. & VAN EPPS, D.E. (1990). Nonsteroidal antiinflammatory agents inhibit upregulation of CDllb, CDllc, and CD35 in neutrophils stimulated by formyl-methionine-leucine-phenylalanine. Irtflammation 14: 163-171.
CRUMPTON, M.J. & DEDMAN, J R. (1990). Protein terminology tangle. Nature 345: 212.
CUNNINGHAM, P.M., SMITH, M.J., FORD HUTCHINSON, A.W., & WALKER, J R. (1979). Migration of peritoneal polymorphonuclear leucocytes in the rat. J. Pathol 128: 15-20.
CUNNINGHAM, P.M. & SMITH, M.J.H. (1982). Leukotriene B : biological activities and the cytoskeleton. Br. J. Pharmacol 75: 383-387.
175

CYBULSKY, M.I., MCCOMB, D.J., DINARELLO, C.A., & MOVAT, H.Z. (1987). Mediation by interleukin-1 of neutrophil leukocyte emigration induced by endotoxin. In Leukocyte emigration and its sequelae, ed. Movat H. Z. pp. 38-50. Basel: Karger.
CZOP, J.K. (1986). Phagocytosis of particulate activators of the alternative complement pathway: effects of fibronectin. Adv, Immunol 38: 361-398.
DAHINDEN, C.A., FEHR, J., & HUGLJ, T.E. (1983). Role of cell surface contact in the kinetics of superoxide production by granulocytes. J. Clin. Invest. 72: 113-121.
DAHLEN, S-E., BJORK, J., HEDQUIST, P., ARFORS, K-E., HAMMARSTROM, S., LINDGREN, J-A., & SAMUELSSON, B. (1981). Leukotrienes promote plasma leakage and leukocyte adhesion in post capillary venules. In vivo effects with relevance to the acute inflammatory response. Proc. Natl Acad. Sci. USA 78: 3887-3891.
DAHLEN, S.E., HEDQVIST, P., & ARFORS, K.E. (1982). Increase in vascular permeability induced by leukotriene B4 and the role of polymorphonuclear leukocytes. Inflammation 6 :189-2CX).
DALE, M.M. (1984). The neutrophil leukocyte. In Textbook oflmmunopharmacology. eds. Dale M. M. & Forman J. C. pp. 36-52. Oxford: Blackwell.
DAMERAU, B. & VOGT, W. (1976). Effect of hog anaphylatoxin (C5a) on vascular permeability and leakocyte emigration in vivo. Naunyn-Schmiedeberg’s Arch. Pharmacol 255: 237-241.
DANON, A. & ASSOULINE, G. (1978). Inhibition of prostaglandin biosynthesis bycorticosteroids requires RNA and protein synthesis. Nature 273: 552-554.
DAVIDSON, F.F., DENNIS, E.A., POWELL, M., & GLENNEY JR, J R. (1987). Inhibition of phospholipase A2 by "lipocortins" and calpactins: An effect of binding to substrate phospholipids. J. Biol Oiem. 262: 1698-1706.
DAVIDSON, F.F. & DENNIS, E.A. (1989). Biological relevance of lipocortins and relatedproteins as inhibitors of phospholipase Ag. Biochem. Pharmacol 38: 3645-3651.
DAVIDSON, J., FLOWER, R.J., MILTON, A S., PEERS, S.H., & ROTONDO, D. (1991). Antipyretic actions of human recombinant lipocortin-1. Br. J. Pharmacol 102: 7-9.
DAVIES, K.A., TOOTHILL, V.J., SAVILL, J., HOTCHIN, N., PETERS, A.M., PEARSON, J.D., HASLETT, C., BURKE, M., LAW, S.K., & MERCER, N.F. (1991). A 19-year-old man with leucocyte adhesion deficiency. In vitro and in vivo studies of leucocyte function. Clin. Exp. Immunol 84: 223-231.
DEBRUYN, P.P.H. & CHO, Y. (1974). contractile structures in endothelial cells of splenic sinusoids. Journal o f Ultrastruct Research 49: 24-33.
DEMBINSKA-KIEC, A., GRODZINSKA, L., PANCZENKO, B., & GRYGLEWSKI, R.J. (1978). Effect of corticosteroids on the release of histamine and prostaglandins from fragments of mesentery of immunized guinea pigs. Pharmacol Res. Commun. 10: 153-160.
DESAI, U., KREUTZER, D.L., SHOWELL, H., ARROYAVE, C.V., & WARD, P. A. (1979). Acute inflammatory pulmonary reactions induced by chemotactic factors. Am. J. Pathol 96: 71-84.
DI ROSA, M., FLOWER, R.J., HIRATA, F., PARENTE, L., & RUSSO-MARIE, F. (1984). Nomenclature announcement: Antiphospholipase proteins. Prostaglandins 28: 441-442.
176

DI ROSA, M., CALIGNANO, A., CARNUCCIO, R., lALENTI, A., & SAUTEBIN, L.(1985). Multiple control of inflammation by glucocorticoids. Agents Actions 17: 284-289.
DI ROSA, M. & PERSICO, P. (1979). Medianism of inhibition of prostaglandin biosynthesis by hydrocortisone in rat leucocytes. Br. J. Pharmacol 66: 161-163.
DI ROSA, M. & WILLOUGHBY, D A. (1971). Screens for anti-inflammatory drugs. J. Pharm. Pharmacol 23: 297-298.
DIAMOND, M.S., STAUNTON, D.E., MARUN, S.D., & SPRINGER, T.A. (1991). Binding of the integrin Mac-1 (GDI lb/CD 18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Ceü 65: 961-971.
DOERSCHUK, C M., ALLARD, M.F., & HOGG, J.C. (1989). Neutrophil kinetics in rabbits during infusion of zymosan-activated plasma. J. Appl Physiol 67: 88-95.
DOERSCHUK, C M. (1992). The role of CD18-mediated adhesion in neutrophil sequestration induced by infusion of activated plasma in rabbits. Am. J. Respir. Cell Mol Biol 7: 140-148.
DOWNEY, G.P., ELSON, E.L., SCHWAB, B., ERZURUM, S.C., YOUNG, S.K., & WORTHEN, G.S. (1991). Biophysical properties and microfilament assembly in neutrophils: modulation by cyclic AMP. J. Cell Biol 114: 1179-1190.
DOYLE, N.A., QUINLAN, W.M., GRAHAM, L., & DOERSCHUK, C M. (1992). The role of LECAM-1 in neutrophil sequestration induced by infusion of complement fragments. Am. Rev. Respir. Dis. 145: A187.
DUNCAN, G.S., PEERS, S.H., CAREY, P., FORDER, R., & FLOWER, R.J. (1993). The local anti-inflammatory action of dexamethasone in the rat carrageenin oedema model is reversed by an antiserum to lipocortin 1. Br. J. Pharmacol 108: 62-65.
DUNHAM, B., SHEPRO, D., & HECHTMAN, H.B. (1984). Leukotriene induction of TxB2 in cultured bovine aortic endothelial cells. Jrtflammatlon 8 : 313-321.
DUSTIN, M.L., ROTHLEIN, R., BAHN, A.K., DINARELLO, C.A., & SPRINGER, T.A.(1986). Induction by IL-1 and interferon-gamma: tissue distribution, biochemistry and function of a natural adherence molecule (ICAM-1). J. Immunol 137: 245-254.
ELICES, M.J., URRY, L.A., & HEMLER, M E. (1991). Receptor functions for the integrin VLA-3: fibronectin, collagen, and laminin binding are differentially influenced by Arg-Gly-Asp peptide and by divalent cations. J. Cell Biol 112: 169-181.
ENTMAN, M L., YOUKER, K., SHAPPELL, S B., SIEGEL, C., ROTHLEIN, R.,DREYER, W.J., SCHMALSTIEG, F.C., & SMITH, C.W. (1990). Neutrophil adherence to isolated adult canine myocytes. Evidence for a CD 18-dependent mechanism. J. CUn. Invest. 85: 1497-1506.
ERLANSSON, M., SVENSJO, E., & BERGQVIST, D. (1989). Leukotriene ^.-induced permeability increase in postcapillary venules and its inhibition by three different anti-inflammatory drugs. Inflammation 13: 693-705.
ERRASFA, M. & RUSSO-MARIE, F. (1989). A purified lipocortin shares the anti-inflammatory effect of glucocorticosteroids in vivo in mice. Br. J. Pharmacol 97: 1051-1058.
ERVINE, G.B. & SHAW, C. (1986). High-performance gel permeation chromatography af proteins and peptides on columns of TSK-G2(XX)-SW and TSK-G3(XX)-SW: A volatile solvent
177

giving separation based on charge and size of polypeptides. AnaL Biochem, 155: 141-148.
FAIRMAN, R.P., SESSLER, C.N., BIERMAN, M., & GLAUSER, F.L. (1987). Protamine sulphate causes pulmonary hypertension and oedema in isolated rat lungs. J. Appl Physiol 62: 1363-1367.
FAVA, R.A., MCKANNA, J., & COHEN, S. (1989). Lipocortin I (p35) is abundant in a restricted number of differentiate cell types in adult organs. J, Cell Physiol 141: 284-293.
FIEBIG, E., LEY, K., & ARFORS, K-E. (1991). Rapid leukocyte accumulation by "spontaneous" rolling and adhesion in the exteriorized rabbit mesentery. Int. J. Microcirc. Œn. Exp. 10: 127-144.
FLOREY, H.W. & GRANT, L.H. (1961). Leukocyte migration from small blood vessels stimulated with ultraviolet light: An electron-microscope study. J. Pathol 82: 13-17.
FLOWER, R.J., PARENTE, L., PERSICO, P., & SALMON, J.A. (1986). A comparison of the acute inflammatory response in adrenalectomised and sham-operated rats. Br. J. Pharmacol 87: 57-62.
FLOWER, R.J. & BLACKWELL, G.J. (1979). Anti-inflammatory steroids induce biosynthesis of a phospholipase A2 inhibitor which prevents prostaglandin generation. Nature 278: 456-459.
FLYNN, P.J., BECKER, W.K., VERCELLOTTI, G.M., WEISDORF, D.J., CRADDOCK, PR., HAMMERSCHMIDT, D.E., LILLEHEI, R.C., & JACOB, H.S. (1984). Ibuprofen inhibits granulocyte responses to inflammatory mediators. A proposed mechanism for reduction of experimental myocardial infarct size. Inflammation 8 : 33-44.
FORD-HUTCHINSON, A.W., BRAY, M.A., DOIG, M.V., SHIPLEY, M.E., & SMITH, M.J.H. (1980). Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 286: 264-265.
FORD-HUTCHINSON, A.W., BRUNET, G., SAVARD, P., & CHARLESON, S. (1984). Leukotriene B4 , polymorphonuclear leukocytes and inflammatory exudates in the rat. Prostaglandins 2S: 13-18.
FORDER, R.A. & CAREY, F. (1984). Human 5-lipoxygenase: measurement byradioimmunoassay of LTB4 in calcium ionophore stimulated blood. Prostaglandins 28: 6 6 6 .
FORREST, M.J., JOSE, P.J., & WILLIAMS, T.J. (1981a). Evidence that the complement fragment C5a mediates increased vascular permeability in response to zymosan in the peritoneal cavity of the rabbit. Br. J. Pharmacol 74: 787P.
FORREST, M.J., PECK, M.J., & WILLIAMS, T.J. (1981b). Is increased vascular permeability induced by C5a dependent on the generation of lipoxygenase products. Br. J. Pharmacol 74: 921-922P.
FORREST, M.J., JOSE, P.J., & WILLIAMS, T.J. (1985). The role of the complement-derived polypeptide C5a in inflammatory reactions. In Ir0ammatory Mediators, eds. Higgs G. A. & Williams T. J. pp. 99-115. Basingstoke: MacMillan Press.
178

FORREST, M.J., JOSE, P.J., & WILLIAMS, T.J. (1986). Kinetics of the generation and action of chemical mediators in zymosan-induced inflammation of the rabbit peritoneal cavity. Br. J. Pharmacol 89: 719-730.
FORSYTH, K.D. & TALBOT, V. (1992). Role of glucocorticoids in neutrophil and endothelial adhesion molecule expression and function. Mediators of Inflammation 1: 101-106.
FOSTER, S.J., AKED, D M., SCHRODER, J.M., & CHRISTOPHERS, E. (1989). Acute inflammatory effects of a monocyte-derived neutrophil-activating peptide in rabbit skin. Immunology 61 \ 181-183.
FOSTER, S.J. & MCCORMICK, M.E. (1985). The mechanism of the anti-inflammatory activity of glucocorticosteroids. Agents Actions 16: 58-59.
FRADIN, A., ROTHHUT, B., POINCELOT CANTON, B., ERRASFA, M., & RUSSO MARIE, F. (1988). Inhibition of eicosanoid and PAF formation by dexamethasone in rat inflammatory polymorphonuclear neutrophils may implicate lipocortin ’s’. Biochim. Biophys. Acta 963: 248-257.
FRANK, R.S. (1990). Time-dependent alterations in the deformability of human neutrophils in response to chemotactic activation. Blood 76: 2606-2612.
FREER, R.J., DAY, A.R., RADDING, J.A., SCHIFFMANN, E., ASWANIKUMAR, S., SHOWELL, H.J., & BECKER, E.L. (1980). Further studies on die structural requirements for synthetic peptide chemoattractants. Biochemistry 19: 2404-2410.
FREER, R.J., DAY, A.R., MUTHUKUMARASWARMY, N., PINON, D., WU, A., SHOWELL, H.J., & BECKER, E.L. (1982). Formyl peptide chemoattractants: a model of the receptor on rabbit neutrophils. Biochemistry 21: 257-263.
FURIE, M B., TANCINCO, M.C.A., & SMITH, C.W. (1991). Monoclonal antbodies to leukocyte integrins CDlla/CD18 and CD 1 lb/CD 18 or intercellular adhesion molecule-1 inhibit chemoattractant-stimulated neutrophil transendothelial migration in vitro. Blood 78: 2089-2097.
GABBIANI, G., BADONNEL, M-C., & RONA, G. (1975). Cytoplasmic contractile apparatus in aortic endothelial cells of hypertensive rats. Lab. Invest. 32: 227-234.
GALLATIN, W.M., WEISSMAN, I.L., & BUTCHER, E C. (1983). A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature 304: 30-39.
GALLIN, J., DUROCHER, J., & KAPLAN, A. (1975). Interaction of leukocyte chemotactic factors with the cell surface 1. Chemotactic factor-induced changes in human granulocyte surface charge. J. Oin. Invest. 55: 967.
GALLIN, J., FLETCHER, M., SELIGMANN, B., HOFFSTEIN, S., CEHN, K., & MOUNESSA, N. (1982). Human neutrophil-specific granule deficiency: a model to assess the role of neutrophil-specific granules in the evolution of the inflammatory response. Blood 59: 1317-1329.
GALLIN, J.I. (1980). Degranulating stimuli decrease the negative surface charge and increase the adhesiveness of human neutrophils. J. Clin. Invest. 65: 298-306.
GASIC, A C., MCGUIRE, G., KRATER, S., FARHOOD, A.I., GOLDSTEIN, M.A., SMITH, C.W., ENTMAN, M L , & TAYLOR, A. A. (1991). Hydrogen peroxide pretreatment of perfused canine vessels induces ICAM-1 and CD18^ependent neutrophil adherence. Circulation 84: 2154-2166.
179

GENG, J-G., BEVILACQUA, M.P., MOORE, K.L., MCINTYRE, T.M., PRESCOTT, S.M., KIM, J.M., BLISS, G.A., ZIMMERMAN, G.A., & MCEVER, R.P. (1990). Rapid neutrophil adhesion to activated endothelium mediated by GMP-140. Nature 343: 757-760.
GEORGILIS, K., SHAEFER, C., DINARELLO, C.A., & KLEMPNER, M.S. (1987). Human recombinant interleukin 1/5 has no effect on intracellular calcium or on functional responses of human neutrophils. J. Immunol 138: 3403-3407.
GERDIN, B., LUNDBERG, C., & SMEDEGARD, G. (1985). Permeability-increasing ability of PAF-acether in rat skin. Irtflammation 9 : 107-112.
GERSHENWALD, I.E., FONG, Y.M., FAHEY, T.J., CALVANO, S.E., CHIZZONITE, R., KILIAN, P.L., LOWRY, S.F., & MOLDAWER, L.L. (1990). Interleukin 1 receptor blockade attenuates the host inflammatory response. Proc. Natl Acad. Scl U. S. A. 87: 4966-4970.
GIMBRONE, M.A., BROCK, A.F., & SCHAFER, A.I. (1984). Leukotriene B» stimulates polymorphonuclear leukocyte adhesion to cultured vascular endothelial cells. J. Œn. Invest. 74: 1552-1555.
GINSBURG, I., GIBBS, D.F., SCHUGER, L., JOHNSON, K.J., RYAN, U.S., WARD, P.A., & VARANI, J. (1989). Vascular endothelial cell killing by combinations of membrane-active agents and hydrogen peroxide. Free Radical Biology & Medicine 7: 369-376.
GLENNEY, J. & ZOKAS, L. (1988). Antibodies to the N-terminus of calpactin II (p35) affect Ca '*' binding and phosphorylation by the epidermal growth factor receptor in vitro. Biochemistry 27: 2069-2076.
GOLDMAN, D.W., GIFFORD, L.A., MAROTTI, T., KOO, C.H., & GOETZL, E.L. (1987). Molecular and cellular properties of human polymorphonuclear leukocyte receptors for leukotriene B4 . Fedn. Proc. 46: 200-203.
GOTO, K., NAKAMURA, S., GOTO, F., & YOSHINAGA, M. (1984). Generation of an interleukin-1 -like lymphocyte-stimulating factor at inflammatory sites: correlation with the infiltration of polymorphonuclear leukocytes. Br. J. Exp. Pathol 65: 521-532.
GOULDING, N.J., PODGORSKI, M R., HALL, N.D., FLOWER, R.J., BROWNING, J.L., PEPINSKY, R.B., & MADDISON, P.J. (1989). Autoantibodies to recombinant lipocortin-1 in rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 48: 843-850.
GOULDING, N.J., GODOLPHIN, J.L., SHARLAND, P R., PEERS, S.H., SAMPSON, M., MADDISON, P.J., & FLOWER, R.J. (1990a). Anti-inflammatory lipocortin 1 production by peripheral blood leucocytes in response to hydrocortisone. Lancet 335: 1416-1418.
GOULDING, N.J., LUYING, P., & GUYRE, P.M. (1990b). Characteristics of lipocortin 1 binding to the surface of human peripheral blood leucocytes. Biochem. Soc. Trans. 18:1237-1238.
GOULDING, N.J., JEFFERISS, C M., PAN, L., RIGBY, W.F., & GUYRE, P.M. (1992). Specific binding of lipocortin-1 (annexin I) to monocytes and neutrophils is decreased in rheumatoid arthritis. Arthritis Rheum. 35: 1395-1397.
GOULDING, N.J. & GUYRE, P.M. (1992). Regulation of inflammation by lipocortin 1. Immunol Today 13: 295-297.
180

GRANGER, D.N., ZIMMERMAN, B.J., SEKIZUKA, E., & GRISHAM, M B. (1988). Intestinal microvascular exchange in the rat during luminal perfusion with formyl-methionyl-leucyl-phenylalanine. Gastroenterology 94: 673-681.
GRIFFITHS, R.J., WOOD, B E., & BLACKHAM, A. (1988). Pharmacological modification of 1 2 -0 -tetra decanoyIphorbol-13-acetate induced inflammation and epidermal cell proliferation in mouse skin. Agents Actions 25: 344-351.
GRIFFITHS, R.J., LI, S.W., WOOD, B.E., & BLACKHAM, A. (1991). A comparison of the anti-inflammatory activity of selective 5-lipoxygenase inhibitors with dexamethasone and colchicine in a model of zymosan induced inflammation in the rat knee joint and peritoneal cavity. Agents Actions 32: 312-320.
GRIFFITHS, R.J. & BLACKHAM, A. (1988). The effects of dexamethasone on inflammatory response in rabbit skin. Br. J. Pharmacol 95: 535P.
GRISWOLD, D.E., WEBB, E.F., & HILLEGASS, L.M. (1991). Induction of plasma exudation and inflammatory cell infiltration by leukotriene C4 and leukotriene B4 in mouse peritonitis. hÿUmunation 15: 251-258.
GRYGLEWSKI, R.J., PANCZENKO, B., KORBUT, R., GRODYINSKA, L., & OCETKIEWICZ, A. (1975). Corticosteroids inhibit prostaglandin release from perfused mesentric blood vessels of rabbit and perfused lungs of sensitized guinea pig. Prostaglandins 10: 343-355.
HAFSTROM, J., PALMBLAD, J., MALMSTEN, C.L., RADMARK, O., & SAMUELSSON,B. (1981). Leukotriene B4 - a stereospecific stimulator for release of lysosomal enzymes from neutrophils. FEBS Lett. 130: 146-148.
HALLMANN, R., JUTILA, M.A., SMITH, C.W., ANDERSON, D C., KISHIMOTO, T.K., & BUTCHER, E.C. (1991). The peripheral lymph node homing receptor, LECAM-1, is involved in CD 18-independent adhesion of human neutrophils to endothelium. Biochem. Biophys. Res. Commun. 174: 236-243.
HAMANAKA, K., TOKI, N., TSUSHIMA, H., & TAKASUGI, S. (1984). Studies on the role of polymorphonuclear leukocyte granule elastase in Arthus reaction. Arch. Dermatol Res. 276: 370-374.
HAMBERG, M., SVENSSON, J., & SAMUELSSON, B. (1975). Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc. Natl Acad. Scl U. S. A. 72: 2994-2998.
HAMMERSCHMIDT, D.E., HARRIS, P.D., WAYLAND, J.H., CRADDOCK, PR., & JACOB, H.S. (1981). Complement-induced granulocyte aggregation in vivo. Am. J. Pathol 102:146-150.
HAMMERSEN, F. (1980). Endothelial contractivity - does it exist? In Advances in microcirculation, ed. Altura B. M. pp. 95-134. Basel: S.Karger.
HANSSON, G., LINDGREN, J.A., DAHLEN, S.E., HEDQVIST, P., & SAMUELSSON, B.(1981). Identification and biological activity of novel omega-oxidized metabolites of leukotriene B4 from human leukocytes. FEBS Lett. 130: 107-112.
HARLAN, J.M., LEVINE, J.D., CALLAHAN, K.S., SCHWARTZ, B.R., & HARKER, L A.(1984). Glutathione redox cycle protects cultured endothelial cells against lysis by extracellularly generated hydrogen peroxide. J. Clin. Invest. 73: 706-713.
181

HARLAN, J.M., KILLEN, P.D., SENEGAL, F., SCHWARTZ, B.R., YEE, E.K., TAYLOR, R.F., BEATTY, P.G., PRICE, T., & OCHS, H.D. (1985). The role of neutrophil membrane glycoprotein GP-150 in neutrophil adherence to endothelium in vitro. Blood 6 6 : 167-178.
HASLETT, C., WORTHEN, G.S., GICLAS, P.C., MORRISON, D C., HENSON, J.E., & HENSON, P.M. (1987). The pulmonary vascular sequestration of neutrophils in endotoxemia is initiated by an effect of endotoxin on the neutrophil in tiie rabbit. Am, Rev. Respir. Dis. 136:9-18.
HASLETT, C., JOSE, P.J., GICLAS, P.C., WILLIAMS, T.J., & HENSON, P.M. (1989). Cessation of neutrophil influx in C5a-induced acute experimental arthritis is associated with loss of chemoattractant activity from the joint space. J. Imnumol 142: 3510-3517.
HATTORI, R., HAMILTON, K.K., FUGATE, R.D., MCEVER, R.P., & SIMS, P.J. (1989). Stimulated secretion of endothelial von Willebrand factor is accompanied by rapid redistribution to the cell surface of the intracellular granule membrane protein GMP-140. J. Biol Oiem. 264: 7768-7771.
HELLEWELL, P.G. & WILLIAMS, T.J. (1986). A specific antagonist of platelet-activating factor suppresses oedema formation in an Arthus reaction but not oedema induced by leukocyte chemoattractants in rabbit skin. J. Immunol 137: 302-307.
HELLEWELL, P.G. & WILLIAMS, T.J. (1989). Antagonism of PAF-induced oedema formation in rabbit skin: a comparison of different antagonists. Br. J. Pharmacol 97: 171-180.
HELTIANU, C., SIMIONESCU, M., & SIMIONESCU, N. (1982). Histamine receptors of the microvascular endothelium revealed in situ with a histamine-ferritin conjugate: characterisitic high-affinity binding sites in venules. J. Cell Physiol 93: 357-364.
HENSON, P.M., LARSEN, G.L., WEBSTER, R.O., MITCHELL, B.C., GOINS, A.J., & HENSON, J.E. (1982a). Pulmonary microvascular alterations and injury induced by complement fragments: synergistic effect of complement activation, neutrophil sequestration, and prostaglandins. Ann. N. Y. Acad. Scl 384: 287-300.
HENSON, P.M. & COCHRANE, C.G. (1969). Immunological induction of increased vascular permeability: a rabbit passive cutaneous anaphylactic reaction requiring complement, platelets and neutrophils. J. Exp. Med. 129: 153.
HENSON, P.M. & JOHNSTON, R.B.JR. (1987). Tissue injury in inflammation. Oxidants, proteinases and cationic proteins. J. Clin. Invest. 79: 669-674.
HIBBS, M L., WARDLAW, A.J., STACKER, S.A., ANDERSON, DC., LEE, A., ROBERTS, T.M., & SPRINGER, T.A. (1990). Transfection of cells from patients with leukocyte adhesion deficiency with an integrin beta subunit (CD 18) restores lymphocyte function-associated antigen-1 expression and function. J. Clin. Invest. 85: 674-681.
HIGGS, G.A., BUNTING, S., MONCADA, S., & VANE, J R. (1976). Polymorphonuclear leukocytes produce thromboxane A2-like activity during phagocytosis. Prostaglandins 12: 749-757.
182

HIGGS, G.A., SALMON, J.A., & SPAYNE, M. (1981). The inflammatory effect of hydroperoxy and hydroxy acid products of arachidonate lipoxygenase in rabbit skin. Br. J. Pharmacol 74: 429-433.
HIGGS, G.A., MONCADA, S., SALMON, J.A., & SEAGER, K. (1983). The source of thromboxane and prostaglandins in experimental inflammation. Br. J. Pharmacol 79: 863-868.
HIGGS, G.A. & SALMON, J.A. (1979). Cylco-oxygenase products in carrageenan induced inflammation. Prostaglandins 17: 737-746.
HIRATA, F., SCHIFFMANN, E., VENKATASUBRAMANIAN, K., SALMON, D., & AXELROD, J. (1980). A phospholipase A2 inhibitory protein in rabbit neutrophils induced by glucocorticoids. Proc. Natl Acad. Scl USA 77: 2533-2536.
HIRATA, F. (1981). The regulation of lipomodulin, a phospholipase inhibitory protein, in rabbit neutrophils by phosphorylation. J. Biol CJiem. 256: 7730-7733.
HIRATA, F., DEL CARMINE, R., NELSON, C.A., AXELROD, J., SCHIFFMANN, E., WARABI, A., DE BLAS, A.L., NIRENBERG, M., MANGANIELLO, V., VAUGHAN, M., KUMAGAI, S., GREEN, I., DECKER, J.L., & STEINBERG, A.D. (1981). Presence of autoantibody for phospholipase inhibitory protein, lipomodulin, in patients with rheumatic diseases. Proc. Natl Acad. Scl U. S. A. 78: 3190-3194.
HIRATA, F. (1983). Lipomodulin: a possible mediator of the action of glucocorticoids. In Advances in prostaglandin, thromboxane and leukotriene research, eds. Samuelsson B., Paoletti R., & Ramwell P. pp. 73-78. New York: Raven Press.
HOFFSTEIN, S., GOLDSTEIN, I.M., & WEISSMANN, G. (1977). Role of microtubule assembly in lysosomal enzyme secretion from human polymorphonuclear leukocytes. J. Cell Biol. 73: 242.
HOFFSTEIN, S. & WEISSMANN, G. (1978). Microfilaments and microtubules in calcium ionophore-induced secretion of lysosomal enzymes from human polymorphonuclear leukocytes. J. Cell Biol 78: 769.
HOLT, M.E., RYALL, M.E.T., & CAMPBELL, A.K. (1984). Albumin inhibits human polymorphonuclear leucocyte luminol-dependent chemiluminescence: evidence for oxygen radical scavenging. Br. J. Exp. Pathol 65: 231-241.
HONG, S.L. & LEVINE, L. (1976). Inhibition of arachidonic acid release from cells as the biochemical action of anti-inflammatory corticosteroids. Proc. Natl Acad. Scl USA 73: 1730-1734.
HOOK, W.A., SCHIFFMANN, E., ASWANIKMUR, S., & SIRAGANIAN, R.P. (1976). Histamine release by chemotactic formyl methionine-containing peptides. J. Immunol 117: 594-596.
HOOVER, R.L., FOLGER, R., HAERING, W.A., WARE, B.R., & KARROVS, M.J. (1980). Adhesion of leukocytes to endothelium: Roles of divalent cations, surface charge, chemotactic agents and substrate. J. Cell Scl 45: 73-86.
HOOVER, R.L., FUJUWARA, K., KLAUSNER, R.D., BHALLA, D.K., TUCKER, R., & KARNOVSKY, M.J. (1981). Effects of free fatty acids on the organisation of cytoskeletal elements in lymphocytes. Molecular & Cellular Biology 1: 939.
183

HOOVER, R.L., KARNOFSKY, M.J., AUSTEN, K.F., COREY, E.J., & LEWIS, R.A.(1984). Leukotriene B4 action on endothelium mediates augmented neutrophil/endothelial adhesion. Proc. Naxl Acad. Sci. USA 81: 2191-2193.
HOOVER, R.L., ROBINSON, J.M., & LARNOVSKY, M.J. (1987). Adhesion of polymorphonuclear leukocytes to endothelium enhances the efficiency of detoxification of oxygen free radicals. Am. J. Pathol 126: 258-268.
HOPKINS, N.K., SCHAUB, R.G., & GORMAN, R.R. (1984). Acetyl glyceryl ether phophoryldioline (PAF-acether) and leukotriene B4-mediated neutrophil chemotaxis through an intact endothelial cell monolayer. Biochem. Biophys. Acta. 805: 30-36.
HUANG, A.J., FURIE, M.B., NICHOLSON, S.C., FISCHBORG, J., LIEBOVITCH, L.S., & SILVERSTEIN, S.C. (1988). Effects of human neutrophil chemotaxis across human endothelial cell monolayers on the permeability of these monolayers to ions and macromolecules. J. Cell Physiol 135: 355-366.
HUANG, K-S., WALLNER, B.P., MATTALIANO, R.J., TIZARD, R., BURNE, C., FREY,A., HESSION, C., MCGRAY, P., SINCLAIR, L.K., CHOW, E.P., BROWNING, J.L., RAMACHANDRAN, K.L., TANG, J , SMART, J.E., & PEPINSKY, R.B. (1986). Two human 35KD inhibitors of phospholipase Ag are related to substrates of pp60V-S RC and of the epidermal growth factor receptor/kinases. Cell 46: 191-199.
HUBER, A.R. & WEISS, S.J. (1989). Disruption of the subendothelial basement membrane during neutrophil diapedesis in an in vitro construct of a blood vessel wall. J. Clin. Invest. 83: 1122-1136.
HUGLI, T.E. & MULLER-EBERHARD, H.J. (1978). Anaphylatoxins: C3aand C5a. Advances in Immunology 26: 1-53.
HULLIN, F., RAYNAL, P., RAGAB-THOMAS, J.H.F., FAUVEL, J., & CHAP, H. (1989). Effect of dexamethasone on prostaglandin synthesis and on lipocortin status in human endothelial cells. J. Biol Oiem. 264: 3506-3513.
HULSTROM, D. & SVENSJO, E. (1979). Intravital and electron microscopic study of bradykinin-induced vascular permeability changes using FITC-Dextrans as a tracer. Journal of Pathology 129: 125-133.
HUMES, J.L., BONNEY, R.J., PELUS, L., DAHLGREN, M.E., SADOWSKI, S.J., KUEHL, F.A.J., & DAVIES, P. (1977). Macrophage synthesis and release prostaglandins in response to inflammatory stimuli. Nature 269: 149-151.
HUMPHREY, DM., HANAHAN, D.J., & PINCKARD, R.N. (1982a). Induction of leukocytic infiltrates in ri)bit skin by acetyl glyceryl ether phosphorylcholine. Lxib. Invest. 47: 227-234.
HURLEY, J.V. (1963). An electron microscopic study of leukocyte emigration and vascular permeability in rat skin. Aus. J. Exp. Biol Med. Scl 41: 171-186.
HURLEY, J.V. (1964). Acute inflammation: the effect of concurrent leucocyte emigration and increased permeability on particle retention by the vascular wall. Br. J. Exp. Pathol 45: 627-633.
HWANG, S-B., LI, G.L., LAM, M-H., & SHEN, T-Y. (1985). Characterisation of cutaneous vascular permeability induced by platelet-activating factor in guinea-pigs and rats and its inhibition by a platelet-activating factor receptor antagonist. Lab. Invest. 52: 617-630.
HYNES, R.O. (1987). Integrins: A family of cell surface receptors. Cell 48: 549-554.
184

HYNES, R.O. (1992). Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69: 11-25.
lALENTI, A., DOYLE, P.M., HARDY, G.N., SIMPKIN, D.S., & DI ROSA, M. (1990). Anti-inflammatory effects of vasocortin and nonapeptide fragments of uteroglobin and lipocortin I (antiflammins). Agents Actions 29: 48-49.
lALENTI, A. & DI ROSA, M. (1990). Vasocortin inhibits Ca2+ influx and histamine release induced by concanavalin A in rat mast cells. Aaa Physiol Hung. 75 Suppl: 147-148.
INANO, H., ENGLISH, D., & DOERSCHUK, C M. (1992). Effect of zymosan-activated plasma on the deformability of rabbit polymorphonuclear leukocytes. J. Appl Physiol 73: 1370-1376.
INGERMAN, C M., AHARONY, D., SILVER, M.J., SILVER, J.B., NISSENBAUM, M., SEDAR, A.W., & MACARAK, E. (1980). Prostaglandin 1 and thromboxane Aj can be produced by endotiielial cells in situ and in culture. Fedn. Proc. 39: 391.
ISACKE, C M., LINDBERG, R.A., & HUNTER, T. (1989). Synthesis of p36 and p35 is increased when U937 cells differentiate in culture but expression is not inducible by glucocorticoids. Molecular & Cellular Biology 9: 232-240.
ISSEKUTZ, A C. (1981a). Vascular responses during acute neutrophilic inflammation. Their relationship to in vitro neutrophil emigration. Lab. Invest. 45: 435-441.
ISSEKUTZ, A C. (1981b). Effect of vasoactive agents on polymorphonuclear leukocyte emigration in vivo. Uib. Invest. 45: 234.
ISSEKUTZ, A C. & MOVAT, H.Z. (1979). The effect of vasodilator prostaglandins on polymorphonuclear leukocyte infiltration and vascular injury. Pathology 107: 3(X)-309.
ISSEKUTZ, A C. & RIPLEY, M. (1986). The effect of intravascular neutrophil chemotactic factors on blood neutrophil and platelet kinetics. Am. J. Hematol 21: 157-171.
JOHNSTON, G.I., COOK, R.G., & MCEVER, R.P. (1989). Cloning of GMP-140, a granule membrane protein of platelets and endothelium: sequence similarity to proteins involved in cell adhesion and inflammation. Cell 56: 1033-1044.
JORIS, I., MAJNO, G., & RYAN, G.B. (1972). Endothelial contraction in vivo: a study of the rat mesentery. Virchows Arch 12: 73-83.
JORIS, I., MAJNO, G., COREY, E.J., & LEWIS, R.A. (1987). The mechanism of vascular leakage induced by leukotriene E : endothelial contraction. Am. J. Pathol 126: 19-24.
JOSE, P.J., FORREST, M.J., & WILLIAMS, T.J. (1981). Human C5a des Arg increases vascular permeability. J. Immunol 127: 2376-2380.
JOSE, P.J., FORREST, M.J., & WILLIAMS, T.J. (1983). Detection of the complement fragment C5a in inflammatory exudates from the rabbit peritoneal cavity using radioimmunoassay. J. Exp. Med. 158: 2177-2182.
JOSE, P.J., FORREST, M.J., & WILLIAMS, T.J. (1985). Generation of C5a and prostacyclin (PGI2 ) in an inflammatory response to zymosan and a reversed passive Arthus-type reaction in the peritoneal cavity of the rabbit. Agents Actions 16: 39-40.
185

JOSE, P.J. & WILLIAMS, T.J. (1987). Suppression of acute inflammatory responses by anti-C5a antibodies. Complement 4: 175 (Abstr).
JUTILA, M.A., ROTT, L., BERG, E.L., & BUTCHER, E.C. (1989). Function and regulation of the neutrophil MEL-14 antigen in vivo: comparison with LFA-1 and MAC-1.7. Immunol 143: 3318-3324.
JUTILA, M.A., KISHIMOTO, T.K., & BUTCHER, E.C. (1990). Regulation and lectin activity of the human neutrophil peripheral lymph node homing receptor. Blood 76: 178-183.
KAIDBEY, K.H. & KLIGMAN, A.M. (1974). Assay of topical corticosteroids by suppression of experimental inflammation in humans. J. Invest. Dermatol 63(3): 292-297.
KALSNER, S. (1969). Steroid potentiation of responses to sympathomimetic amines in aorticstrips. Br. J. Pharmacol 36: 582-593.
KAPLAN, B.K., EDELSON, H.S., KORCHAK, H.M., GIVEN, W.P., ABRAHAMSON, S., & WEISSMANN, G. (1984). Effects of non-steroidal anti-inflammatory agents on humanneutrophil functions in vitro and in vivo. Biochem. Pharmacol 33: 371-378.
KAPLAN, H.B., EDELSON, H.S., FRIEDMAN, R., & WEISSMANN, G. (1982). . Biochim. Biophys. Acta 721: 55.
KATORI, M., ODA, T., & NAGAI, K. (1990). A site of action of dexamethasone on leukocyte extravasation in microcirculation. Agents Actions 29: 24-26.
KEIZER, G.D., VISSER, W., VLIEM, M., & HGDOR, C.G. (1988). A monoclonal antibody (NK1-L16) directed against a unique epitope on the alpha-chain of human leukocyte function-associated antigen 1 induces homotypic cell-cell interactions. J. Immunol 140: 1393-1400.
KIRKPATRICK, C.J. & MELZNER, I. (1984). Alterations in the biophysical properties of the human endothelial cell plasma membrane induced by a chemotactic tripeptide: correlation with enhanced adherence to granulocytes. J. Pathol 144: 201-211.
KISHIMOTO, T.K., JUTILA, M.A., BERG, E.L., & BUTCHER, E.C. (1989a). Neutrophil Mac-1 and MEI^14 adhesion proteins inversely regulated by chemotactic factors. Science 245: 1238-1241.
KISHIMOTO, T.K., LARSON, R.S., CORBI, A.L., DUSTIN, M L., STAUNTON, D.E., & SPRINGER, T.A. (1989b). The leukocyte integrins. Advances in Immunology 46: 149-182.
KISHIMOTO, T.K., WARNOCK, R.A., JUTILA, M.A., BUTCHER, E C., LANE, C., ANDERSON, D C., & SMITH, C.W. (1991). Antibcxiies against human neutrophil LECAM-1 (LAM-l/Leu-8/DREG-56 antigen) and endothelial cell ELAM-1 inhibit a common CDl 8 -independent adhesion pathway in vitro. Blood 78: 805-811.
KNUSDEN, P.J., DINARELLO, C.A., & STROM, T.B. (1987). Glucocorticoids inhibit transcriptional and post-transcriptional expression of interleukin 1 in U937 cells. J. Immunol 139: 4129^134.
186

KRETSINGER, R.H. & CREUTZ, C.E. (1986). Cell biology. Consensus in exocytosis [news] [published erratum appears in Nature 1986 May 8-14;321(6066):lll]. Nature 320: 573.
KRISTENSEN, T., SARIS, C.J., HUNTER, T., HICKS, L.J., NOONAN, D.J., GLENNEY, J.R.J., & TACK, B.F. (1986). Primary structure of bovine calpactin I heavy chain (p36), a major cellular substrate for retroviral protein-tyrosine kinases: homology with the human phospholipase A2 inhibitor lipocortin. Biochemistry 25: 4497-4503.
KUBES, P., IBBOTSON, G., RUSSELL, J., WALLACE, J.L., & GRANGER, D.N. (1990). Role of platelet-activating factor in ischemia/reperfusion-induced leukocyte adherence. Am. J. Physiol 259: G300-G305.
KURIHARA, A., OHUCHI, K., & TSURUFUJI, S. (1984a). Reduction by dexamethasone of chemotactic activity in inflammatory exudates. Eur. J. Pharmacol 101: 11-16.
KURIHARA, A., OJIMA, F., & TSURUFUJI, S. (1984b). Analysis of the effect of an anti-inflammatory steroid, dexamethasone, on neutrophil chemotaxis in the Boyden chamber with a modified 51Cr-labeling method. J. Phamuicobiodyn. 7: 747-754.
KURIHARA, A., OJIMA, F., & TSURUFUJI, S. (1984c). Chemotactic factor production by rat polymorphonuclear leukocytes: stimulation with opsonized zymosan particles and inhibition by dexamethasone. Biochem. Biophys. Res. Commun. 119: 720-725.
KURODA, M., MURAKAMI, K., & ISHIKAWA, Y. (1987). Role of hydroxyl radicals derived from granulocytes in lung injury induced by phorbol myristate acetate. Am. Rev. Respir. Dis. 136: 1435-1444.
LANGLOIS, P.F. & GAWRYL, M.S. (1988). Detection of the terminal complement complex in patient plasma following acute myocardial infarction. Atherosclerosis 70: 95-105.
LARSEN, E., CELI, A., GILBERT, G.E., FURIE, B.C., ERBAN, J.K., BONFANTI, R., WAGNER, D.D., & FURIE, B. (1989). PADGEM Protein: A receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 59: 305-312.
LARSEN, E., PALABRICA, T., SAJER, S., GILBERT, G.E., WAGNER, D.D., FURIE, B.C., & FURIE, B. (1990). PADGEM-dependent adhesion of platelets to monocytes and neutrophils is mediated by a lineage-specific carbohydrate, LNF III (CD15). Cell 63: 467-474.
LAUWERYNS, J.M. & BOUSAUW, L. (1973). Striated filamentous bundles associated with centrioles in pulmonary lymphatic endothelial cells. Journal of Ultrastruct Research 42: 25-28.
LAWRENCE, M B., SMITH, C.W., ESKIN, S.G., & MCINTIRE, L.V. (1990). Effect of venous shear stress on CD18-mediated neutrophil adhesion to cultured endothelium. Blood 75: 227-237.
LAWRENCE, M B. & SPRINGER, T.A. (1991). Leukocytes roll on a selectin at phyiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell 65: 859-873.
LEE, T.D., SWIETER, M., BIENENSTOCK, J., & BEFUS, A.D. (1985). Heterogeneity in mast cell populations. Clin. Immunol Rev. 4: 143-199.
LEFER, A.M. & POLANSKY, E.W. (1979). Beneficial effects of ibuprofen in acute myocardial ischemia. Cardiology 64: 265-279.
LEWINSOHN, D.M., BARGATZE, R.F., & BUTCHER, E.C. (1987). Leukocyte-endothelial cell recognition: evidence of a common molecular mechanism shared by neutrophils, lymphocytes,
187

and other leukocytes. J. Immunol 138: 4313-4321.
LEWIS, G.P. & PIPER, P.J. (1975). Inhibition of release of prostaglandins as an explanation of some of the actions of anti-inflammatory corticosteroids. Nature 254: 308-311.
LEWIS, G.P. & WHITTLE, B.J.R. (1977). The inhibition of histamine release from rat peritoneal mast cells by non-steroid anti-inflammatory drugs and its reversal by calcium. Br. J. Pharmacol 61: 229-235.
LEWIS, R E. & GRANGER, H.J. (1986). Neutrophil-dependent mediation of microvascular permeability. Fedn. Proc. 45: 109-113.
LEWIS, R E. & GRANGER, H.J. (1988). Diapedesis and the permeability of venous microvessels to protein macromolecules: the impact of leukotriene B4 (LTBJ. Microvascular Research 35: 27-47.
LEWIS, T. The blood vessels of the human skin and their response. London: Shaw & Sons, 1927,
LINDSTROM, P., LERNER, R., PALMBLAD, J., & PATARROYO, M. (1990). Rapid adhesive responses of endothelial cells and of neutrophils induced by leukotriene B4 are mediated by leucocytic adhesion protein CD 18. Scand. J. Immunol 31: 737-744.
LITT, M R., JEREMY, R.W., WEISMAN, H.F., WINKELSTEIN, J.A., & BECKER, L.C. (1989). Neutrophil depletion limited to reperftision reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil-mediated reperfusion injury. Grculation 80: 1816-1827.
LO, S.K., DETMERS, P.A., LEVIN, S.M., & WRIGHT, S.D. (1989a). Transient adhesion of neutrophils to endothelium. J. Exp. Med. 169: 1779-1793.
LO, S.K., VAN-SEVENTER, G.A., LEVIN, S.M., & WRIGHT, S.D. (1989b). Two leukoc^ receptors (CDl 1 a/CD 18 and CDl lb/CD 18) mediate transient adhesion to endothelium by binding to different ligands. J. Immunol 143: 3325-3329.
LO, S.K., LEE, S., RAMOS, R.A., LOBB, R., ROSA, M., CHI-ROSSO, G., & WRIGHT, S.D. (1991). Endothelial-leukocyte adhesion molecule 1 stimulates the adhesive activity of leukocyte integrin CR3 (CDllb/CD18, Mac-1, on human neutrophils. J. Exp. Med. 173: 1493-1500.
LORANT, D.E., PATEL, K.D., MCINTYRE, T.M., MCEVER, R.P., PRESCOTT, S.M., & ZIMMERMAN, G.A. (1991). Coexpression of GMP-140 and PAF by endothelium stimulated by histamine or thrombin: A juxtacrine system for adhesion and activation of neutrophils. J. Ceü Biol 115: 223-234.
LOWE, J.B., STOOLMAN, L.M., NAIR, R.P., LARSEN, R.D., BERHEND, T.L., & MARKS, R.M. (1990). ELAM-1-dependent cell adhesion to vascular endothelium determined by a transfected human fticosyltransferase cDNA. Cell 63: 475-484.
LUNDBERG, C. & WRIGHT, S.D. (1990). Relation of the CD11/CD18 family of leukocyte antigens to the transient neutropenia caused by chemoattractants. Blood 76: 1240-1245.
LUSCINSKAS, F.W., BROCK, A.F., ARNAOUT, M.A., & GIMBRONE, M.A. (1989). Endothelial-leukocyte adhesion molecule-1-dependent and leukocyte (CD 11 /CD 18)-dependent mechanisms contribute to polymorphonuclear leukocyte adhesion to cytokine-activated human vascular endothelium. J. Immunol 142: 2257-2263.
188

LYNCH, J.M., LOTNER, G.Z., BETZ, S.J., & HENSON, P.M. (1979). The release of a platelet-activating factor by stimulated rabbit neutrophils. J. Immunol 123: 1219-1226.
MADERAZO, E.G., BREAUX, S.P., & WORONICK, C.L. (1984). Inhibition of human polymorphonuclear leukocyte cell responses by ibuprofen. J. Pharmac. Scl 73: 1403-1406.
MAJNO, G., SCHOEFL, G.I., & PALADE, G. (1961). Studies on inflammation. The site of action of histamine and serotonin on the vascular tree; a topographic study. J. Cell Biol II: 607-626.
MAJNO, G., SHEA, S.M., & LEVENTHAL, M. (1969). Endothelial contraction induced by histamine-type mediators. An electron microscopic study. J. Cell Biol 42: 647-672.
MAJNO, G. & PALADE, G.E. (1961). Studies on inflammation. 1. The effect of histamine and serotonin on vascular penneability: An electron microscopic study. 7. Biol Phys. Biochem, Cytol 11: 571-605.
MALECH, H.L., ROOT, R.K., & GALLIN, J.I. (1977b). Structural analysis of human neutrophil migration. Centriole, microtubule, and microfilament orientation and function during chemotaxis. J. Cell Biol 75: 6 ^ -G 9 3 ,
MARASCO, W.A., PHAN, S.H., KRUTZSCH, H., SHOWELL, H.J., FELTNER, D.E., NAIRN, R., BECKER, E.L., & WARD, P.A. (1984). Purification and identification of formy 1-methiony 1-leucyl-pheny lalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J. Biol Chem. 259: 5430-5435.
MARKS, R. & SAWYER, N. (1986). Glucocorticoid-induced vasoconstriction in human skin. An inhibitory role of phospholipase A2 activity. Arch. Dermatol 122: 881-883.
MARLIN, S.D. & SPRINGER, T.A. (1987). Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1). Cell 51: 813-819.
MASFERRER, J.L., ZWEIFEL, B.S., SEIBERT, K., & NEEDLEMAN, P. (1990). Selective regulation of cellular cyclooxygenase by dexamethasone and endotoxin in mice. J. Clin. Invest. 8 6 : 1375-1379.
MCCALL, C.E., DE CHATELET, L.R., BROWN, D., & LACHMANN, P. (1974). New biological activity following intravascular activation of the complement cascade. Nature 249: 841-843.
MCEVER, R.P., BECKSTEAD, J.H., MOORE, K.L., MARSHALL-CARLSON, L., & BAINTON, D.F. (1989). GMP-140, a platelet a-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J. Clin. Invest. 84: 92-99.
MCGIVERN, D.V. & BASRAN, G.S. (1984). Synergism between platelet-activating factor (PAF-acether) and prostaglandin Eg in man. Eur. J. Pharmacol 102: 183-185.
MEYRICK, B., HOFFMAN, L.H., & BRIGHAM, K.L. (1984). Chemotaxis of granulocytes across bovine pulmonary artery intimai expiants without endothelial cell injury. Tissue & Cell 16: 1-16.
189

MIKAMI, T. & MIYASAKA, K. (1983). Effects of several anti-inflammatory drugs on the various parameters involved in the inflammatory response in rat carrageenin-induced pleurisy. Eur. J. Phar^col. 95: 1-12.
MILKS, L.C., CONYERS, G.P., & CRAMER, E.B. (1986). The effect of neutrophil emigration on epithelial permeability. J. Cell Biol 103: 2729-2738.
MILLER, L.J., BAINTON, D.F., BORREGARD, N., & SPRINGER, T.A. (1987). Stimulated mobilization of monocyte Mac-1 and P150,95 adhesion proteins from an intracellular vesicular compartment to the cell surface. J. Clin. Invest. 80: 535-544.
MINTA, J O. & WILLIAMS, M.D. (1985). Some nonsteroidal antiinflammatory drugs inhibit the generation of superoxide anions by activated polymorphs by blocking ligand-receptor interactions. J. Rheumatol 12: 751-757.
MISCHLER, J.M. (1977). The effect of corticosteroids on mobilization and fuction of neutrophils. Exp. Hematol S^upp!.: 15-32.
MONCADA, S. & FERREIRA, S.H. (1973). Prostaglandins, asprin-like drugs and the oedema of inflammation. Nature 246: 217-219.
MOORE, A., JAFFE, E.A., BECKER, C.G., & NACHMANN, R.L. (1977). Myosin in cultured human endothelial cells. Br. J. Haematol 35: 71-79.
MOORE, K.L., VARKI, A., & MCEVER, R.P. (1991). GMP-140 binds to a glycoprotein receptor on human neutrophils: evidence for a lectin-like interaction. J. Cell Biol 112: 491-499.
MORLEY, J., BRAY, M.A., JONES, R.W., NUGTEREN, D.H., & VAN DORP, D A. (1979). Prostaglandin and thromboxane production by human and guinea-pig macrophages and leucocytes. Prostaglandins 17: 729-746.
MOSER, R., SCHIEIFFENBAUM, B., GROSGURTH, P., & FEHR, J. (1989). Interleukin-1 and tumor necrosis factor stimulate human vascular endothelial cells to promote transendothelial neutrophil passage. J. Clin. Invest. 83: 444-455.
MOVAT, H.Z. & BURROWS, C.E. (1985). The local Schwartzman reaction: endotoxin-mediated inflammatory and thrombo-hemorrhagic lesions. In Handbook of endotoxin, volume 3: Cellular biology of endotoxin, ed. Berry L. J. pp. 260-302. Amsterdam: Elsevier Science Publications.
MULLANE, K.M., READ, N., SALMON, J.A., & MONCADA, S. (1984). Role of leukocytes in acute myocardial infarction in anesthetized dogs: relationship to myocardial salvage by anti-inflammatory drugs. J. Pharmacol Exp. Ther. 228: 510-522.
MUNCK, A. & LEUNG, K. (1977). Glucocorticoid receptors and mechanisms of action. In Receptors and mechanism of action of steroid hormones, ed. Pasqualini J. R. pp. 311-319. New York: Marcel Dekker.
MUNRO, J.M., POBER, J.S., & COTRAN, R.S. (1989). Tumor necrosis factor and interferon-gamma induce distinct patterns of endothelial activation and associated leukocyte accumulation in skin of papio anubis. Am. J. Pathol 135: 121-133.
NAGAI, K. & KATORI, M. (1988). Possible changes in the leukocyte membrane as a mechanism of leukocyte adhesion to the venular walls induced by leukotriene B4 and fMLP in the microvasculature of the hamster cheek pouch. Int. J. Microcirc. 7: 305-314.
190

NASH, G.B., JONES, J.G., MKITA, J., & DORMANDY, J.A. (1988). Methods and theory for analysis of flow of white cell subpopulations through micropore filters. Br. J. Haematol 70: 165-170.
NATHAN, C., SRIMAL, S., FARBER, C., SANCHEZ, E., KABBASH, L., ASCH, A., GAILTT, J., & WRIGHT, S.D. (1989). Cytokine-induced respiratory burst of human neutrophils: dependence on extracellular matrix proteins and CD11/CD18 integrins. J. Cell Biol 109: 1341-1349.
NATHAN, C.F. (1987a). Neutrophil activaton on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J. Clin. Invest. 80: 1550-1560.
NEAL, T.M., VISSERS, M.C., & WINTERBOURN, C.C. (1987). Inhibition by nonsteroidal anti-inflammatory drugs of superoxide production and granule enzyme release by polymorphonuclear leukocytes stimulated with immune complexes or formyl-methionyl-leucyl-phenylalanine. Biochem. Pharmacol 36: 2511-2517.
NEEDHAM, L., HELLEWELL, P.G., WILLIAMS, T.J., & GORDON, J.L. (1988). Endothelial functional responses and increased vascular permeability induced by poly cations. Lab. Invest. 59: 538-548.
NIELSON, V.G. & WEBSTER, R.O. (1987). Inhibition of human polymorphonuclear leukocyte functions by ibuprofen. Immunopharmacology 13: 61-71.
NUKAMP, F.P., FLOWER, R.J., MONCADA, S., & VANE, J R. (1976). Partial purification of RCS-RF (Rabbit Aorta contracting substance - Releasing Factor) and inhibition of its activity by anti-inflammatory steroids. Nature 263: 479-482.
NORTHUP, J.K., VALENTINE-BRAUN, K.A., JOHNSON, L.K., SEVERSON, D.L., & HOLLENBERG, M.D. (1988). Evaluation of the anti-inflammatory and phospholipase-inhibitory activity of calpactin Il/lipocortin 1. J. Clin. Invest. 82: 1347-1352.
NOURSHARGH, S., RAMPART, M., HELLEWELL, P.G., JOSE, P.J., HARLAN, J.M., EDWARDS, A.J., & WILLIAMS, T.J. (1989). Accumulation o f‘"In-neutrophils in rabbit skin in allergic and non-allergic inflammatory reactions in vivo: inhibition by neutrophil pretreatment in vitro with a monoclonal antibody recognising the CDl8 antigen. J. Immunol 142: 3193-3198.
NOURSHARGH, S. & WILLIAMS, T.J. (1990). Evidence that a receptor operated event on the neutrophil mediates neutrophil accumulation in vivo: pretreatment of ‘"In-neutrophils with pertussis toxin in vitro inhibits their accumulation in vivo. J. Immunol 145: 2633-2638.
O’FLAHERTY, J.T., SHOWELL, H.J., & WARD, P.A. (1977). Neutrophils induced by sytemic infusion of chemotactic factors. J. Immunol 118: 1586-1589.
O’FLAHERTY, J.T., CRADDOCK, P R., & JACOB, H.S. (1978a). Effect of intravascular complement activation on granulocyte adhesiveness and distribution. Blood 51: 731-739.
O’FLAHERTY, J.T., KREUTZER, D R., & WARD, P.A. (1978b). Chemotactic factor influences on the aggregation, swelling, and foreign surface adhesiveness of human leukocytes. Am. J. Pathol 90: 537-550.
O’FLAHERTY, J.T., SHOWELL, H.J., WARD, P.A., & BECKER, E.L. (1979). A possible role of arachidonic acid in human neutrophil aggregation and degranulation. Am. J. Pathol 96: 799-810.
191

O’FLAHERTY, J.T., WYKLE, R.L., LEES, C.L, SHEWMAKE, T., MCCALL, C.E., & T H O M A S , M. J . ( 1 981) . N e u t r o p h i l - d e g r a n u l a t i n g ac t i on of 5 , 1 2 - d i h y d r o X y - 6 , 8 , 10 , 1 4 - e i c 0 s a t e t r a e n o i c a c i d a n d l-o-alkyl-2-o-acetyl-sn-glycero-3-phosphocholine. Comparison with other degranulating agents. Am. J. Pathol. 105: 264-269.
OGLETREE, M.L. & LEFER, A.M. (1976). Influence of nonsteroidal anti-inflammatory agents on myocardial isdiemia in the cat. J. Pharmacol Exp. Ther. 197: 582-593.
OHUCHI, K., YOSHINO, S., KANAOKA, K., TSURUFUJI, S., & LEVINE, L. (1982). A possible role of arachidonic metabolism in allergic air pouch inflammation in rats. Anti-inflammatory effect of indomethacin and dexamethasone and the level of prostaglandin Eg in the exudate. Ira. Archs. Allergy appl Immunol 6 8 : 326-331.
OHUCHI, K., WATANABE, M., & LEVINE, L. (1984). Arachidonate metabolites in acute and chronic allergic air pouch inflammation in rats and the anti-inflammatory effects of indomethacin and dexamethasone. Ira. Arch. Allergy Immunol 75: 157-163.
OPPENHEIMER-MARKS, N., DAVIS, L.S., BOGUE, D.T., RAMBERG, J., & LIPSKY, P.E.(1991). Differential utilization of ICAM-1 and VCAM-1 during the adhesion and transendoüielial migration of human T lymphocytes. J. Immunol 147: 2913-2921.
OSEAS, R , YANG, H.H., BAEHNER, R.L., & BOXER, LA. (1981). Lactoferrin: a promoter of polymorphonuclear leukocyte adhesiveness. Blood 57: 939-945.
OSEAS, R.S., ALLEN, L, YANG, H-H., BAEHNER, R.L., & BOXER, L A. (1982). Mechanism of dexamethasone inhibition of chemotactic factor induced granulocyte aggregation. Blood 59. 265-269.
OTTONELLO, L., PASTORINO, G., DAPINO, P., BERETTA, A., &DALLEGRI, F. (1992). Ibuprofen inhibits adhesion-dependent neutrophil response by a glycoprotein-unrelated mechanism. Drugs Exp. Clin. Res. 18: 23-27.
OUYANG, Y., WANG, W., BHUTA, S., & CHANG, Y-H. (1989). Mechanism of action of colchicine VI: Effect of colchicine on generation of leukotriene B4 by human polymorphonuclear leukocytes. Clin. Exp. Rheumatol 7: 397-402.
PAGE, C.P., ARCHER, C.B., & MORLEY, J. (1985). Properties of PAF-acether ^propriate to a mediator of inflammation. In hÿlammatory mediators, eds. Higgs G. A. & Williams T. J. pp. 57-64. Basingstoke: MacMillan.
PAGE, W., PAGE, C.P., CUNNINGHAM, F.M., & MORLEY, J. (1985). The plasma protein extravasation response to PAF-acether is independent of platelet accumulation. Agents Actions 15: 80-82.
PALDER, S B., HUVAL, W., LELCUK, S., ALEXANDER, F., SHEPRO, D., MANNICK, J.A., & HECHTMAN, H.B. (1986). Reduction of polymorphonuclear leukocyte accumulations by inhibition of cyclooxygenase and thromboxane syntase in the rabbit. Surgery 99: 72-81.
PALMBLAD, L, GYLLENHAMMAR, H., LINDGREN, J.A., & MALMSTEN, C.L. (1984). Effects of leukotrienes and f-met-leu-phe on oxidative metalx)lism of neutrophils and eosinophils. J. Immunol 132: 3041-3045.
PALMBLAD, L, LINDSTROM, P., & LERNER, R. (1990). Leukotriene B4 induced hyperadhesiveness of endothelial cells for neutrophils. Biochem. Biophys. Res. Commun. 166: 848-851.
192

PALMER, M.J. & SALMON, J.A. (1983). Release of leukotriene B4 from human neutrophils and its relationship to degranulation induced by N-formyl-methionyl-leucyl-phenylalanine, serum-treated zymosan and the ionophore A23187. Immunology 50: 65-73.
PARENTE, L., DI ROSEA, M., FLOWER, R.J., GHIARA, P., MELI, R., PERSICO, P., SALMON, J.A., & WOOD, J.N. (1984). Relationship between the anti-phospholipase and anti-inflammatory effects of glucocorticoid-induced proteins. Eur. J. Pharmacol 99: 233-239.
PARENTE, L., FITZGERALD, M.F., FLOWER, R.J., & DENUCCI, G. (1985). The effect of glucocorticoids on lyso-PAF formation in vitro and in vivo. Agents Actions 17: 312.
PARENTE, L. & FLOWER, R.J. (1985). Hydrocortisone and macrocortin inhibit the zymosan-induced release of lyso-PAF from rat peritoneal leucocytes. Life Sciences 36:1225-1231.
PARSONS, P.E., SUGAHARA, K., COTT, G.R., MASON, R.J., & HENSON, P.M. (1987). The effect of neutrophil migration and prolonged neutrophil contact on epithelial permeability. Am. J. Pathol 129: 302-312.
PARTSCH, G., SCHWARZER, C., & EBERL, R. (1990). The effects of ibuprofen and diclofenac on the chemotaxis and adenosine triphosphate level of polymorphonuclear cells in vitro. J. Rheumatol 17: 583-588.
PEERS, S.H., TAYLOR, R.D., & FLOWER, R.J. (1987). A novel binding assay for phospholipase Aj. Biochem. Pharmacol 36: 4287^291.
PEERS, S.H., MOON, D., & FLOWER, R.J. (1988). Reversal of the anti-inflammatory effects of dexamethasone by the glucocorticoid antagonist RU 38486. Biochem. Pharmacol 37:556-557.
PEERS, S.H., SMILLIE, F., ELDERFIELD, A.J., & FLOWER, R.J. (1993). Glucocorticoid-and non-glucocorticoid induction of lipocortins (annexins) 1 and 2 in rat peritoneal leucocytes in vivo. Br. J. Pharmacol 108: 66-72.
PEERS, S.H. & FLOWER, R.J. (1991). Site of anti-inflammatory action of dexamethasone in rabbit skin. Eur. J. Pharmacol 196: 37-41.
PEPINSKY, R.B., TIZARD, R., MATTALIANO, R.J., SINCLAIR, L.K., MULLER, G.T., BROWNING, J.L., CHOW, E.P., BURNE, C., HUANG, K-S., PRATT, D., WATCHER, L., HESSION, C., FREY, A.Z., & WALLNER, B.P. (1988). Five distinct calcium and phospholipid binding proteins share homology with lipocortin 1. J. Biol Oiem. 263: 10799-10811.
PERLMAN, MB., JOHNSON, A., & MALIK, A.B. (1987). Ibuprofen prevents thrombin-induced lung vascular injury: mechanism of effect. Am. J. Physiol 252: H605-H614.
PERLMAN, M B., JOHNSON, A., JUBIZ, W., & MALIK, A.B. (1989). Lipoxygenase products induce neutrophil activation and increase endothelial permeability after thrombin-induced pulmonary microembolism. Ore. Res. 64: 62-73.
PERRETTI, M., SOUTO, E., & PARENTE, L. (1992). Evidence that endogenous interleukin-1 is involved in leukocyte migration in acute experimental inflammation in rats and mice. Agents Actions 35: 71-78.
PERRETTI, M. & FLOWER, R.J. (1993). Modulation of IL-1-induced neutrophil migration by dexamethasone and lipocortin 1. J. Immunol 150: 992-999.
193

PETERSON, M.W., STONE, P., & SHASBY, D M. (1987). Cationic neutrophil proteins increase transendothelial albumin movement. J. Appl Physiol 62: 1521-1530.
PETERSON, M.W. (1989). Neutrophil cathepsin G increases transendothelial albumin flux. J. Lab. CUn. Med. 113: 297-308.
PETERSON, M.W., GRUENHAUPT, D., & SHASBY, D.M. (1989). Neutrophil cathepsin G increases calcium flux and inositol polyphosphate production in cultured endothelial cells. J. Imnumol 143: 609-616.
PETRONI, K.C., SHEN, L., & GUYRE, P.M. (1988). Modulation of human polymorphonuclear leukocyte IgG Fc receptors and Fc receptor-mediated functions by IFN-t and glucocorticoids. J. Imnumol 140: 3467-3472.
PETTIPHER, E.R., HIGGS, G.A., & HENDERSON, B. (1986). Interleukin 1 induces leukocyte infiltration and cartilage proteoglycan degradation in the synovial joint. Proc. Natl Acad. Scl USA 83: 8749-8753.
PHILIPS, M.R., BUYON, J.P., WINCHESTER, R., WEISSMANN, G.,&ABRAMSON, S.B.(1988). Up-regulation of the iC3b receptor (CR3) is neither necessary nor sufficient to promote neutrophil aggregation. J. Clin. Invest. 82: 495-501.
PHILLIPS, C., ROSE-JOHN, S., RINCKE, G., FURSTENBERGER, G., & MARKS, F.(1989). cDNA-cloning, sequencing and expression in glucocorticoid-stimulated quiescent swiss 3T3 fibroblasts of mouse lipocortin 1. Biochem. Biophys. Res. Commun. 159: 155-162.
PHILLIPS, M L., NUDELMAN, E., GAETA, F.C.A., PEREZ, M., SINGHAL, A.K., HAKOMORI, S-I., & PAULSON, J.C. (1990). ELAM-1 mediates cell adhesion by recognition of a carbohydrate ligand, sialyl-Le*. Science 2 ^ : 1130-1135.
PICKER, L.J., KISHIMOTO, T.K., SMITH, C.W., WARNOCK, R.A., & BUTCHER, E.C. (1991a). ELAM-1 is an adhesion molecule for skin-homing T cells. Nature 349: 796-738.
PICKER, L.J., WARNOCK, R.A., BURNS, A.R., DOERSCHUK, C M., BERG, E.L., & BUTCHER, E.C. (1991b). The neutrophil selectin LÉCAM-1 presents carbohydrate ligands to the vascular selectins ELAM-1 and GMP-140. Cell 6 6 : 921-933.
PIKE, M.C., KREDICH, N.M., & SNYDERMAN, R. (1980). Influence of cytoskeletal assembly on phosphatidylcholine synthesis in intact phagocytic cells. Cell 20: 373.
POBER, J.S., GIMBRONE, M.A., LAPIERRE, L.A., MENDRICK, D.L., FIERS, W., ROTHlilN, R., & SPRINGER, T.A. (1986). Overlapping patterns of activation of human endothelial cells by interleukin 1, tumor necrosis factor, and immune interferon. J. Imnumol 137: 1893-1896.
PODGORSKI, M., GOULDING, N.J., HALL, N.D., FLOWER, R.J., MADDISON, P.J., & PEPINSKLY, R.B. (1987). . Brit. J. Rheum. 26: 76P.
PODGORSKI, M R., GOULDING, N.J., HALL, N.D., FLOWER, R.J , & MADDISON, P.J. (1992). Autoantibodies to lipocortin-1 are associated with impaired glucocorticoid responsiveness in rheumatoid arthritis. J. Rheumatol 19: 1668-1671.
194

POLLEY, M.J., PHILLIPS, M L., WAYNER, E., NUDELMAN, E., SINGHAL, A.K., HAKOMORI, S-L, & PAULSON, J.C. (1991). CD62 and endothelial cell-leukocyte adhesion molecule 1 (ELAM-1) recognize the same carbohydrate ligand, sialyl-Lewis x. Proc. Natl. Acad. Sci. USA 8 8 : 6224-6228.
PONS, P., ROSSI, A.G., NORMAN, K.E., WILLIAMS, T.J., & NOURSHARGH, S. (1993). Role of platelet-activating factor in platelet accumulation in rabbit skin. Effect of the novel long-acting PAF antagonist, UK-74,505. Br. J. Pharmacol 109: 234-242.
PRICE, T.H., BEATTY, P.G., & CORPUZ, S.R. (1987). In vivo inhibition of neutrophil function in the rabbit using monoclonal antibody to CD18. J. Immunol 139: 4174-4177.
RADOMSKI, M.W., PALMER, R.M.J., & MONCADA, S. (1990). Glucocorticoids inhibit the expression of an inducible, but not the constitutive, nitric oxide synthase in vascular endothelial cells. Proc. Natl Acad. Scl USA 87: 10043-10047.
RAE, S.A. & SMITH, M.J.H. (1981). The stimulation of lysosomal enzyme secretion from human polymorphonuclear leukocytes by leukotriene B4 . J. Pharm. Pharmacol 33: 616-617.
RAMPART, M., BULT, H., & HERMAN, A.G. (1982). Contribution of complement activation to the rise in blood levels of 6 -oxo-prostaglandin FI alpha during endotoxin-induced hypotension in rabbits. Eur. J. Pharmacol 79: 91-99.
RAMPART, M., BULT, H., & HERMAN, A.G. (1983b). Activated complement and anaphylatoxins increase the in vitro production of prostacyclin by rabbit aorta endothelium. Naunyn-Schmiedeberg’s Arch. Pharmacol 322: 158-165.
RAMPART, M., VAN HOVE, C., BULT, H., CLAEYS, M., & HERMAN, A.G. (1983c). Mechanism of complement-induced stimulation of prostacyclin production by isolated rabbit peritoneum. Prostaglandins 2S\ 245-261.
RAMPART, M., DE SMET, W., FIERS, W., & HERMAN, A.G. (1989a). Inflammatory properties of recombinant tumor necrosis factor in rabbit skin in vivo. J. Exp. Med. 169: 2227-2232.
RAMPART, M., HERMAN, A.G., DE SMET, W., NOURSHARGH, S., & WILLIAMS, T.J. (1989b). Anti-inflammatory action of catalase independent of its enzyme activity. EASES J. 3: A3972.
RAMPART, M., JOSE, P.J., & WILLIAMS, T.J. (1989c). Rabbit leukocytes activated by C5a des Arg and N-formy 1-methiony 1-leucyl-pheny lalanine promote endothelial prostacyclin production. Eicosanoids 2: 109-115.
RAMPART, M., VAN DAMME, J., ZONNEKEYN, L., & HERMAN, A.G. (1989d). Granulocyte chemotactic protein/interleukin- 8 induces plasma leakage and neutrophil accumulation in rabbit skin. Am. J. Pathol 135: 21-25.
RAMPART, M. & WILLIAMS, T.J. (1986a). Polymorphonuclear leukocyte-dependent plasma leakage in the rabbit skin is enhanced or inhibited by prostacyclin, depending on the route of administration. Am. J. Pathol 124: 66-73.
195

RAMPART, M. & WILLIAMS, T.J. (1986b). Suppression of inflammatory oedema by ibuprofen involving a mechanism independent of cyclo-oxygenase inhibition. Biochem, Pharmacol 35: 581-586.
RAMPART, M. & WILLIAMS, T.J. (1988). Evidence that neutrophil accumulation induced by interleukin-1 requires both local protein biosynthesis and neutrophil CDl8 antigen expression in vivo. Br. J. Pharmacol 94: 1143-1148.
RASCHER, W., DIETZ, R., SCHOMIG, A., BURKART, G., & GROSS, F. (1980). Reversal of corticosterone-induced supersensitivity of vascular smooüi muscle to noradrenaline by arachidonic acid and prostacyclin. Eur. J. Pharmacol 6 8 : 267-273.
RAZ, A., WYCHE, A., & NEEDLEMAN, P. (1989). Temporal and pharmacological division of fibroblast cyclooxygenase expression into transcriptional and translational phases. Proc. Natl Acad. Sci. USA 86: 1657-1661.
REGAL, J.P. & PICKERING, R.J. (1981). C5a-induced tracheal contraction: effect of an SRS-A antagonist and inhibitors of arachidonate metabolism. J. Immunol 126: 313-316.
REIBMAN, J., HAINES, K.A., RICH, A.M., CRISTELLO, P., GIEDD, K.N., & WEISSMANN, G. (1986). Colchicine inhibits ionophore-induced formation of leukotriene B4 by human neutrophils: The role of microtubules. J. Immunol 136: 1027-1032.
REIBMAN, J., HAINES, K.A., GUDE, P., & WEISSMANN, G. (1991). Differences in signal transduction between F ct receptors (F ct RII, F ct RIII) and FMLP receptors in neutrophils. Effects of colchicine on pertussis toxin sensitivity and diacylglycerol formation. J. Immunol 146: 988-995.
REID, J. & BROOKES, D.B. (1968). Topical corticosteroids: an experimental evaluation of the vasoconstrictor test as an index of anti-inflammatory activity. Br. J. Dermatol 80: 328-336.
RELMAN, D., TUOMANEN, E., FALKOW, S., GOLENBOCK, D.T., SAUKKONEN, K., & WRIGHT, S.D. (1990). Recognition of a bacterial adhesin by an integrin: macrophage CR3 (aM/32, CDl lb/CD18) binds filamentous hemagglutin of bordetella pertusis. Cell61: 1375-1382.
RINALDO, J.E. & ROGERS, R.M. (1982). Adult respiratory distress syndrome. Changing concepts of lung injury and repair. N. Engl J. Med. 306: 900-909.
RIPPE, B. & GREG A, G.J. (1978). Effects of isoprenaline and cooling on histamine induced changes of capillary permeability in the rat hindquarter vascular bed. Aaa Physiol Scand. 103: 252-262.
ROMSON, J.L., BUSH, L.R., JOLLY, S R., & LUCCHESI, B.R. (1982). Cardioprotective effects of ibuprofen in experimental regional and global myocardial ischaemia. J. Cardiovasc. Pharmacol. 4: 187-196.
ROMSON, J.L., HOOK, B.G., KUNKEL, S.L., ABRAMS, G.D., SCHORK, M.A., & LUCCHESI, B.R. (1983). Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Grculation 67: 1016-1023.
ROSENBAUM, R.M., CHELI, C D., & GERRITSEN, M.E. (1986). Dexamethasone inhibits prostaglandin release from rabbit coronary microvessel endothelium. Am. J. Physiol 250: C970-C977.
ROSENGREN, S., BJORK, J., & SMEDEGÂRD, G. (1988). Oxygen radicals are not a prerequisite for neutrophil-medial increased vascular permeability. Lab. Invest. 58: 559-564.
196

ROSENGREN, S., LEY, K., & ARFORS, K.E. (1989). Dextran sulfate prevents LTBj-induced permeability increase, but not neutrophil emigration, in the hamster cheek pouch. Microvascular Research 38: 242-254.
ROSENGREN, S. & ARFORS, K.E. (1990). Neutrophil-mediated vascular leakage is not suppressed by leukocyte elastase inhibitors. Am. J. Physiol 259: H1288-H1294.
ROSENGREN, S. & ARFORS, K.E. (1991). Polycations induce microvascular leakage of macromolecules in hamster cheek pouch. Inflammation 15: 159-172.
ROSSI, A.G., NORMAN, K.E., DONIGI-GALE, D., SHOUPE, T.S., EDWARDS, R., & WILLIAMS, T.J. (1992). The role of complement, platelet-activating factor and leukotriene B4
in the reversed passive Arthus reaction. Br. J. Pharmacol 107: 44-49.
ROT, A. (1992). Endothelial cell binding of NAP-l/IL-8 : role in neutrophil emigration. Immunology Today 13: 291-294.
ROTH, J.A. & KAEBERLE, M.L. (1981). Effects of in vivo dexamethasone administration on in vitro bovine polymorphonuclear leukocyte function. Irfea. Immun. 33: 434-441.
ROTROSEN, D., MALECH, H.L., & GALLIN, J.I. (1987). Formyl peptide leukocyte chemoattractant uptake and release by cultured human umbilical vein endothelial cells. J. Immunol 139: 3034-3040.
RUSSO-MARIE, F., PAING, M., & DUVAL, D. (1979). Involvement of glucocorticoid receptors in steroid-induced inhibition of prostaglandin secretion. The Journal of Biological Chemistry 254: 8468-8504.
RUSSO-MARIE, F. & DUVAL, D. (1982). Dexamethasone-induced inhibition of prostaglandin production does not result from a direct action on phospholipase activities but is mediated through a steroid-inducible factor. Biochemica et Biophysica Aaa 712: 177-185.
RYAN, U.S. & RYAN, J.W. (1984). Inflammatory mediators, contraction and endothelial cells. In Progress in Microcirculation Research IL eds. Courtice F. C., Garlick D. G., & Perry M. A. University of NSW, Australia: Postgraduate Medical Education.
SACKS, T., MOLDOW, C F., CRADDOCK, P R., BOWERS, T.K., & JACOB, H.S. (1978). Oxygen radicals mediate endothelial cell damage by complement-stimulated granulocytes. An in vitro model of immune vascular damage. J. Clin. Invest. 61: 1161-1167.
SALMON, J.A., SIMMONS, P.M., & PALMER, R.M. (1982). Synthesis and metabolism of leukotriene B4 in human neutrophils measured by specific radioimmunoassay. FEBS Lett. 146: 18-22.
SALMON, J.A., SIMMONS, P.M., & MONCADA, S. (1983). The effects of BW755C and other anti-inflammatory drugs on eicosanoid concentrations and leukocyte accumulation in experimentally-induced acute inflammation. J. Pharm. Pharmacol 35: 808-813.
SARIS, C.J., TACK, B.F., KRISTENSEN, T., GLENNEY, J.R.J., & HUNTER, T. (1986). The cDNA sequence for the protein-tyrosine kinase substrate p36 (calpactin I heavy chain) reveals a multidomain protein with internal repeats. Cell 46: 201-212.
197

SATO, H., HASHIMOTO, M., SUGIO, K., OHUCHI, K., & TSURUFUJI, S. (1980). Comparative study between steroidal and non steroidal anti-inflammatory drugs on the mode of their actions on vascular permeability in rat carrageenin-air-pouch inflammation. J. Pharmacobia-Dyn, 3: 345-352.
SAYERS, T.J., WILTROUT, T. A., BULL, C.A., DENN, A.C., PILAEO, A.M., & LOKESH,B. (1988). Effect of cytokines on polymorphonuclear neutrophil infiltration in die mouse. J. Imnumol 141: 1670-1677.
SCHIFFMANN, E., CORCORAN, B.A., & WAHL, S.A. (1975a). N-formylmethionylpeptides as chemoattractants for leukocytes. Proc. Natl Acad. Scl USA 72: 1059-1062.
SCHIFFMANN, E., SHOWELL, H.J., CORCORAN, B.A., WARD, P.A., SMITH, E., & BECKER, E.L. (1975b). The isolation and partial characterization of neutrophil chemotactic factors from Escherichia coli. J. Immunol 114: 1831-1837.
SCHLAEPFER, D.D. & HAIGLER, H.T. (1987). Characterisation of Ca ' -dependent phosphilipid binding and phosphorylation of lipocortin 1. J. Biol Chem. 262: 6931-6937.
SCHLEIMER, R.P., LICHENSTEIN, L.M., & GILLESPIE, E. (1981). Inhibition of basophil histamine release by anti-inflammatory steroids. Nature 292: 454-455.
SCHLEIMER, R.P., MACGLASHAN, D.W.J., GILLESPIE, E., & LICHTENSTEIN, L.M.(1982). Inhibition of basophil histamine release by anti-inflammatory steroids. II. Studies on the mechanism of action. J. Immunol 129: 1632-1636.
SCHLEIMER, R.P., SCHULMAN, E.S., MACGLASHAN, D.W., PETERS, S.P., HAYES, E.C., ADAMS, G.K., LICHENSTEIN, L.M., & ADKINSON, N.F. (1983). Effects of dexamethasone on mediator release from human lung fragments and purified human lung mast cells. J. Clin. Invest. 71: 1830-1835.
SCHLEIMER, R.P. (1985). The mechanism of anti-inflammatory steroid action in allergic diseases. Ann. Rev. Pharmacol Toxicol 25: 381-412.
SCHLEIMER, R.P., DAVIDSON, D.A., LICHTENSTEIN, L.M., & ADKINSON, N.F.J. (1986). Selective inhibition of arachidonic acid metabolite release from human lung tissue by antiinflammatory steroids. J. Immunol 136: 3006-3011.
SCHLEIMER, R.P., UNDEM, B.J., MEEKER, S., BOLLINGER, M E., ADKINSON, N.F., LICHENSTEIN, L.M., & ADAMS, G.K. (1987). Dexamethasone inhibits the antigen-induced contractile activity and release of inflammatory mediators in isolated guinea pig lung tissue. Am. Rev. Respir. Dis. 135: 562-566.
SCHLEIMER, R.P., FREELAND, H.S., PETERS, S.P., BROWN, K.E., & DERSE, C.P.(1989). As assessment of the effects of glucocorticoids on degranulation, chemotaxis, binding to vascular endotelium and format ion of leukotriene B4 by purified human neutrophils. J. Pharmacol Exp. Ther. 250: 598-605.
SCHWARTZ, B.R., OCHS, H.D., BEATTY, P.G., & HARLAN, J.M. (1985). A monoclonal antibody-defined membrane antigen complex is require for neutrophil-neutrophil aggregation. Blood 65: 1553-1556.
SEDGWICK, A.D. & LEES, P. (1986a). Studies of eicosanoid production in the air pouch model of synovial inflammation. Agents Actions 18: 429-438.
198

SEDGWICK, A.D. & LEES, P. (1986b). A comparison of air pouch, sponge and pleurisy models of acute carrageenan inflammation in the rat. Agents Actions 18: 439-446.
SENIOR, R.M., TEGNER, H., KUHN, C., OHLSSON, K., STARCHER, B.C., & PIERCE, J.A. (1977). The induction of pulmonary emphysema wiüi human leukocyte elastase. Am. Rev. Respir. Dis. 116: 469-475.
SERHAN, C.N., LUNDBERG, U., WEISSMANN, G., & SAMUELSSON, B. (1984). Formation of leukotrienes and hydroxy acids from human neutrophils and platelets exposed to monosodium urate. Prostaglandins 27: 563.
SESSA, W.C., LIN, L., & NASJLETTI, A. (1990). Reciprocal effects of dexamethasone on vasodilatory responses to arachidonic acid and prostanoids in the isolated perfused rabbit kidney. Hypertension 15: 193-196.
SESSA, W.C. & NASJLETTI, A. (1990). Dexamethasone selectively attenuates prostanoid-induced vasoconstrictor responses in vitro. Grc. Res. 6 6 : 383-388.
SHAPPELL, S B , TOMAN, C., ANDERSON, D C., TAYLOR, A.A., ENTMAN, M.L., & SMITH, C.W. (19%). Mac-1 (CDl lb/CD 18) mediates adherence-dependent hydrogen peroxide production by human and canine neutrophils. J. Immunol. 144: 2702-2711.
SHASBY, D M., SHASBY, S.S., & PEACH, M.J. (1983). Granulocytes and phorbol myristate acetate increase permeability to ovalbumin of cultured endothelial monolayers and isolated perfused lungs: role of oxygen radicals and granulocyte adherence. Am. Rev. Respir. Dis. 127: 72-76.
SHOWELL, H.J., FREER, R.J., ZIGMOND, S.H., SCHIFFMANN, E., ASWANIKUMAR, S., CORNORAN, B., & BECKER, E.L. (1976). The structure-activity relations of synthetic peptides as chemotactic factors and inducers of lysosomal enzyme secretion for neutrophils. J. Exp. Med. 143: 1154-1169.
SIBILLE, Y. & REYNOLDS, H.Y. (1990). Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am. Rev. Respir. Dis. 141; 471-501.
SIEGELMAN, M.H. & WEISSMAN, I.L. (1989). Human homologue of mouse lymphnode homing receptor: Evolutionary conservation at tandem cell interaction domains. Proc. Natl. Acad. Sci. USA 8 6 : 5562.
SIMIONESCU, N., SIMIONESCU, M., & PALADE, G.E. (1978). Open Junctions in the endothelium of the postcapillary venules of the diaphragm. Journal of Cell Biology 79: 27-44.
SIMMONS, D., MAKGOBA, M.W., & SEED, B. (1988). ICAM, an adhesion ligand of LFA-1, is homologous to the neural cell adhesion molecule NCAM. Nature 331: 624-627.
SIMMONS, P.M., SALMON, J.A., & MONCADA, S. (1983). The release of leukotriene during experiment^ inflammation. Biochem. Pharmacol. 32: 1353-1359.
SMITH, C.W., ROTHLEIN, R., HUGHES, B.J., MARISCALCO, M.M., RUDLOFF, H.E., SCHMALSTIEG, F.C., & ANDERSON, D C. (1988). Recognition of an endothelial determinant for CDl 8 -dependent human neutrophil adherence and transendothelial migration. J. Clin. Invest. 82: 1746-1756.
199

SMITH, C.W., MARLIN, S.D., ROTHLEIN, R., TOMAN, C., & ANDERSON, D C. (1989). Co-operative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J. Clin, Invest. 83: 2008-2017.
SMITH, C.W., KISHIMOTO, T.K., ABBASS, O., HUGHES, B., ROTHLEIN, R., MCINTIRE, L.V., BUTCHER, E., & ANDERSON, D.C. (1991). Chemotactic factors regulate lectin adhesion molecule 1 (LECAM-l)-dependent neutrophil adhesion to cytokine-stimulated endothelial cells in vitro. J. Clin. Invest. 87: 609-618.
SMITH, M.J., FORD HUTCHINSON, A.W., & BRAY, M.A. (1980). Leukotriene B: a potential mediator of inflammation. J. Pharm. Pharmacol 32: 517-518.
SMOLEN, J.E. & WEISSMANN, G. (1980). Effects of indomethacin, 5,8,11,14-eicosatetraynoic acid, and p-bromophenacyl bromide on lysosomal enzyme release and superoxide anion generation by human polymorphonuclear leukocytes. Biochem. Pharmacol 29: 533-538.
SNEDECOR, G. W. AND W. G. COCHRAN. Statistical Methods. Iowa: Iowa State UP, 1967,
SNYDER, D.S. & UNANUE, E.R. (1982). Corticosteroids inhibit murine macrophage la expression and interleukin 1 production. J. Immunol 129: 1803-1805.
SNYDERMAN, R., SHIN, H.S., PHILLIPS, J.K., GEWURZ, H., & MERGENHAGEN, S.E. (1969). A neutrophil chemotactic factor derived from C5 upon interaction of guinea-pig serum with endotoxin. J. Immunol 103: 413-422.
SNYDERMAN, R., PHILLIPS, J.K., & MERGENHAGEN, S.E. (1971). Biological activity of complement in vivo. Role of C5 in the accumulation of polymorphonuclear leukocytes in inflammation. J. Exp. Med. 134: 1131-1143.
SNYDERMAN, R. & GOETZL, E.J. (1981). Molecular and cellular mechanisms of leukocyte chemotaxis. Science 213: 830-837.
SOLITO, E., RAUGEI, G., MELLI, M., & PARENTE, L. (1991). Dexamethasone induces the expression of the mRNA of lipocortin 1 and 2 and the release of lipocortin 1 and 5 in differentiated but not undifferentiated U937 cells. FEES Letts. 291: 238-244.
SOTER, N.A., LEWIS, R.A., COREY, E.J., & AUSTEN, K.F. (1983). Local effects of synthetic leukotrienes (LTC4, LTD4, LTE4 and LTB4) in human skin. J. Invest. Dermatol 80: 115-119.
SPAGNUOLO, P.J., ELLNER, J.J., HASSID, A., & DUNN, M.J. (1980). Thromboxane A2 mediates augmented polymorphonuclear leukocyte adhesiveness. J. Clin. Invest. 6 6 : 406-414.
SPERTINI, O., LUSCINSKAS, F.W., KANSAS, G.S., MUNRO, J.M., GRIFFIN, J.D., GIMBRONE, M.A., & TEDDER, T.F. (1991). Leukocyte adhesion molecule-1 (LAM-1, L-selectin) interacts with an inducible endothelial cell ligand to support leukocyte adhesion. J. Immunol 147: 2565-2573.
SPRINGER, T.A., THOMPSON, W.S., MILLER, L.G., SCHMALSTIEG, F.C., & ANDERSON, D.C. (1984). Inherited deficiency of the Mad, LFA-1, p150/95 glycoprotein family and its molecular basis. J. Exp. Med. 160: 1901-1918.
SPRINGER, T.A., TEPLOW, D.B., & DREYER, W.J. (1985). Sequence homology of the LFA-1 and the Mac-1 leukocyte adhesion glycoproteins and unexpected relation to leukocyte interferon. Nature 314: 540-542.
200

SPRINGER, T A. (1990). Adhesion receptors of the immune system. Nature 346: 425-434.
STAUB, N.C., SCHULTZ, E.L., KOIKE, K., & ALBERTINE, K.H. (1985). Effect of neutrophil migration induced by leukotriene B4 on protein permeability in sheep lung. Fedn. Proc. 44: 30-35.
STAUNTON, D.E., MARLIN, S.D., STRATOWA, C., DUSTIN, M L., & SPRINGER, T.A. (1988). Primary structure of ICAM-1 demonstrates interaction between members of the immunoglobulin and integrin supergene families. Cell 52: 925-933.
STAUNTON, D.E., DUSTIN, M L., & SPRINGER, T.A. (1989). Functional cloning of ICAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1. Nature 339: 61-67.
STECHER, V.J. & CHINEA, G.L. (1978). The neutrophil adherence assay as a method for detecting unique anti-inflammatory agents. Agents Actions 8 : 258-262.
STEVENS, T.R.J., BRASDO, A.L., PEERS, S.H., HALL, N.D., & FLOWER, R.J. (1988). Stimulus-specific inhibition of human neutrophil H2O2 production by human recombinant lipocortin 1. Br. J. Pharmacol 93: 139P.
STEVENSON, R.D. (1986). Effect of steroid therapy on in vitro polymorph migration. Clin. Exp. Immunol 23: 285-289.
STIMLER, N.P., BACH, M.K., BLOOR, CM., & HUGLI, T.E. (1982). Release of leukotrienes from guinea pig lung stimulated by C5a des Arg anaphylatoxin. J. Immunol 128: 2247-2252.
SUDHOF, T.C., SLAUGHTER, C.A., LEZNICKI, I., BARJON, P., & REYNOLDS, G.A. (1988). Human 67-kDa calelectrin contains a duplication of four repeats found in 35-kDa lipocortins. Proc. Natl Acad. Scl U. S. A. 85: 664-668.
SUNG, S.S., NELSON, R.S., & SILVERSTEIN, S.C. (1983). Yeast mannans inhibit binding and phagocytosis of zymosan by mouse peritoneal macrophages. J. Cell Biol 96: 160-166.
SUNNERGREN, K.P. & ROVETTO, M.J. (1987). Myocyte and endothelial injury with ischemia reperfusion in isolated rat hearts. Am. J. Physiol 252: H1211-H1217.
SVENSJO, E., SHARP, D.E., & ARFORS, K-E. (1973). Leakage induced by histamine in the post-capillary venules of the hamster cheek pouch. Microvascular Research 6 : 257.
SVENSJO, E., ARFORS, K-E., RAYMOND, R.M., & GREGA, G.J. (1979). Morphological and physiological correlation of bradykinin-induced macromolecular efflux. Am. J. Physiol 236: H600-H606.
SVENSJO, E. & ARFORS, K-E. (1979). Dimensions of postcapillary venules sensitive to bradykinin- and histamine- induced leakage of macromolecules. Uppsala Journal of Medical Science 84: 47-60.
SVENSJO, E. & ROEMPKE, K. (1985). Time-dependent inhibition of bradykinin- and histamine-induced microvascular permeability increase by local glucocorticoid treatment. Progress in Respiration Research 19: 173-180.
TATE, R.M. & REPINE, J.E. (1983). Neutrophils and the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 128: S52-59.
201

TEDDER, T.F., ISAACS, CM., ERNST, T.J., DEMETRI, G.D., ADLER, D.A., & DISTECHE, C.M. (1989). Isolation and chromosomal localization of cDNAs encoding a novel human lymphocyte cell surface molecule, LAM-1 homology with the mouse lymphocyte homing receptor and other human adhesion proteins. J. Exp. Med. 170: 123-133.
TERRANOVA, V.P., DIFLORIO, R., HUJANEN, E.S., LYALL, R.M., LIOTTA, L.A., THORGEIRSSON, U., SIEGAL, G.P., & SCHIFFMANN, E. (1986). Laminin promotes rabbit neutrophil motility and attachment. J. Clin. Invest. 77: 1180-1186.
THIEME, T.R., MIRKOVICH, A., MALONEY, P., & GOODWIN, D A. (1982). In vivo and in vitro effects of dexamethasone on leukocyte migration in the rat adjuvant ardiritis model. Ir0ammation 6: 371-386.
THORGEIRSSON, U P., TURPEENNIEMI-HUJANEN, T., & LIOTTA, L.A. (1985). Cancer cells, components of basement membranes, and proteolytic enzymes. Int. Rev. Exp. Path. 27: 203-234.
THURESON-KLEIN, A., HEDQVIST, P., & LINDBOM, L. (1984). Ultrastructure of polymorphonuclear leukocytes in post capillary venules after exposure to leukotriene B4 in vivo. Acta Physiol. Scand. 122: 221-224.
THURESON-KLEIN, A., HEDQVIST, P., & LINDBOM, L. (1986). Leukocyte diapedesis and plasma extravasation after leukotriene B : lack of structural injury to the endothelium. Tissue & Cell 18: 1-12.
TILL, G O., JOHNSON, K.J., KUNKEL, R., & WARD, P.A. (1982). Intravascular activation of complement and acute lung injury. Dependency on neutrophils and toxic oxygen metabolites. J. Clin. Invest. 69: 1126-1135.
TONNESEN, M.G., SMEDLEY, L., GOINS, A., & HENSON, P.M. (1982). Interaction between neutrophils and vascular endothelial cells. Agents Actions XI: 25-38.
TONNESEN, M.G., SMEDLEY, L.A., & HENSON, P.M. (1984). Neutrophil-endothelial cell interactions. Modulation of neutrophil adhesiveness induced by complement fragments C5a and C5a des arg and formyl-methionyl-leucyl-phenylalanine in vitro. J. Clin. Invest. 74: 1581-1592.
TONNESEN, M.G., ANDERSON, D C., SPRINGER, T.A., KNEDLER, A., AVDI, N., & HENSON, P.M. (1989). Adherence of neutrophils to cultured human microvascular endothelial cells: Stimulation by chemotactic peptides and lipid mediators and dependence upon the Mac-1, LFA-1, pl50,95 Glycoprotein Family. J. Clin. Invest. 83: 637-646.
TOTH, K.M., CLIFFORD, D.P., BERGER, E.M., WHITE, C.W., & REPINE, J.E. (1984). Intact human erythrocytes prevent hydrogen peroxide-mediated damage to isolated perftised rat lungs and cultured bovine pulmonary artery endothelial cells. J. Clin. Invest. 74: 292-295.
TOYOFUKU, T., TOYAMA, S., KOBAYASHI, T., KUSAMA, S., & VEDA, G. (1989). Effects of polycations on pulmonary vascular permeability on conscious sheep. J. Clin. Invest. 83: 2063-2069.
TSURUFUJI, S., SUGIO, K., & TAKEMASA, F. (1979). The role of glucocorticoid receptor and gene expression in the anti-inflammatory action of dexamethasone. Nature 280: 408-410.
TSURUFUJI, S., SUGIO, K., TAKEMASA, F., & YOSHIZAWA, S. (1980). Blockade by antiglucocorticoids, antinomycin D and cycloheximide of anti-inflammatory action of dexamethasone against bradykinin. J. Pharmacol. Exp. Ther. 212: 225-231.
202

TSURUFUJI, S., KURIHARA, A., KISO, S., SUZUKI, Y., & OHUCHI, K. (1984a). Dexamethasone inhibits generation in inflammatory sites of the chemotactic activity attributable to leukotriene B4 . Biochem, Biophys. Res. Commun. 119: 884-890.
TSURUFUJI, S., KURIHARA, A., & OJIMA, E. (1984b). Mechanisms of anti-inflammatory action of dexamethasone: blockade by hydrocortisone mesylate and actinomycin D of the inhibitory effect of dexamethasone on leukocyte infiltration in inflammatory sites. J. Pharmacol. Exp. Ther. 229: 237-243.
VALERIUS, N.H. (1978). In vitro effect of colchicine on neutrophil granulocyte locomotion. Aaa Pathol Microbiol Immunol Scand. 8 6 : 149-154.
VAN DE VELDE, V.J., BULT, H., & HERMAN, A.G. (1986a). Dexamethasone and prostacyclin biosynthesis by serosal membranes of the rabbit peritoneal cavity. Agents Actions 17: 308-309.
VAN DE VELDE, V.J., HERMAN, A.G., & BULT, H. (1986b). Effects of dexamethasone on prostacyclin biosynthesis in rabbit mésothélial cells. Prostaglandins 32: 169-178.
VARGAFTIG, B.B. (1978). Platelets in inflammation. Agents Actions 3: 75-92.
VARGAFTIG, B.B. & FERREIRA, S.H. (1981). Blockade on the inflammatory effects of PAF by cyclo-oxygenase inhibitors. Braz. J. Med. Res. 14: 187-189.
VEDDER, N.B. & HARLAN, J.M. (1988). Increased surface expression of CDllb/CD18 (Mac-1) is not required for stimulated neutrophil adherence to cultured endothelium. J. Clin. Invest. 81: 676-682.
VEHASKARI, V.M., CHANG, C.T.C., STEVENS, J.K., & ROBSON, A.M. (1984). The effects of poly cations on vascular permeability in the rat: a proposed role for charge sites. J. Gin. Invest. 73: 1053-1061.
VENEZIO, F.R., DIVINCENZO, C., PEARLMAN, F., & PHAIR, J.P. (1985). Effects of the newer nonsteroidal anti-inflammatory agents, ibuprofen, fenoprofen, and sulindac, on neutrophil adherence. J. Infect. Dis. 152: 690-694.
VINCENT, J.E., BONTA, I.L., & ZULSTRA, F.J. (1978). Accumulation of blood platelets in carrageenin rat paw oedema. Possible role in the inflammatory process. Agents Actions 8 : 291-295.
VINCENT, J.E., ZULSTRA, F.J., VAN DEN BVOCK, A.M., & GEZEL, T.E. (1986). Opposite effects of adrenalectomy on eicosanoid release in rat peritoneal macrophages and spleen. Prostaglandins 32: 132-136.
VISHWANATH, B.S., FREY, F.J., BRADBURY, M., DALLMAN, M.F., & FREY, B.M.(1992). Adrenalectomy decreases lipocortin 1 messenger ribonucleic acid and tissue protein content in rats. Endocrinology 130: 585-591.
VISSERS, M.C.M., DAY, W.A., & WINTERBOURN, C.C. (1985). Neutrophils adherent to a non-phagocytosable surface (glomerular basement membrane) produce oxidants only at the site of attachment. Blood 6 6 : 161-166.
VON ANDRIAN, U.H., CHAMBERS, J.D., MCEVOY, L.M., BARGATZE, R.F., ARFORS, K-E., & BUTCHER, E C. (1991). Two-step model of leukocyte-endothelial cell interaction in inflammation: Distinct roles for LECAM-1 and the leukocyte ^ 2 integrins in vivo. Proc. Natl Acad. Scl USA 8 8 : 7538-7542.
203

VON RITTER, C., BE, R., & GRANGER, D.N. (1989). Neutrophilic proteases: mediators of formyl-methionyl-leucyl-phenylalanine-induced ileitis in rats. Gastroenterology 97: 605-609.
VON UEXKULL, C., NOURSHARGH, S., & WILLIAMS, T.J. (1991). Characterisation of inflammatory responses induced by live E.coli in rabbit skin. Br. J. Pharmacol 102: 96P (Abstr).
VON UEXKULL, C., NOURSHARGH, S., & WILLIAMS, T.J. (1992). Vasodilator mediators induced by live E.coli in the skin. Br. J. Pharmacol 105: 209P.(Abstract)
WALLIS, W.J., HICKSTEIN, D.D., SCHWARTZ, B.R., JUNE, C H., OCHS, H.D., BEATTY, P.G., KLEBANOFF, S.J., & HARLAN, J.M. (1986). Monoclonal antibody-defined functional epitopes on the adhesion-promoting glycoprotein complex (CDwl8 ) of human neutrophils. Blood 67: 1007-1013.
WALLNER, B.P., MATTALIANO, R.J., HESSION, C., CATE, R.L., TIZARD, R., SINCLAIR, L.K., FOELLER, C., CHOW, E.P., BROWNING, J.L., RAMACHANDRAN, K.L., & PEPINSKY, R.B. (1986). Cloning and expression of a human lipocortin, a phospholipase A2 inhibitor with potential anti-inflammatory activity. Nature 320: 77-81.
WALZ, G., ARUFFO, A., KOLANUS, W., BEVILACQUA, M., & SEED, B. (1990). Recognition by ELAM-1 of the Sialyl-Le* determinant on myeloid and tumor cells. Science 250: 1132-1135.
WARD, P.A., TILL, G O., HATHERILL, J R., ANNESLEY, T.M., & KUNKEL, R.G.(1985). Systemic complement activation, lung injury, and products of lipid peroxidation. J. Œn. Invest. 76: 517-527.
WARD, P.A. & ZVAIFLER, N.J. (1971). Complement-derived leukotactic factors in inflammatory synovial fluids of humans. J. Clin. Invest. 50: 606-616.
WARR, G.A. (1980). A macrophage receptor for (mannose/glucosamine)-glycoproteins of potential importance in phagocytic activity. Biochem. Biophys. Res. Commun. 93: 737-745.
WATANABE, M., VAGI, M., OMATA, M., HIRASAWA, N., MUE, S., TSURUFUJI, S., & OHUCHI, K. (1991). Stimulation of neutrophil adherence to vascular endothelial cells by histamine and thrombin and its inhibition by PAF antagonists and dexamethasone. Br. J. Pharmacol 102: 239-245.
WATSON, M L., LEWIS, G.P., & WESTWICK, J. (1987). A neutrophil chemokinetic factor produced by interleukin 1-stimulated human synovial fibroblasts. Br. J. Pharmacol 92: 512P.
WATSON, M L., LEWIS, G.P., & WESTWICK, J. (1988). Neutrophil stimulation by recombinant cytokines and a factor from IL-1-treated human synovial cell cultures. Immunology 65: 567-572.
WATSON, M L., LEWIS, G.P., & WESTWICK, J. (1989). Increased vascular permeability and polymorphonuclear leukocyte accumulation in vitro in response to recombinant cytokines and supernatant from interleukin 1-treated human synovial cell cultures. Br. J. Ejq). Pathol 70: 93-101.
WEBSTER, R.O., HONG, S R., JOHNSTON, R.B., & HENSON, P.M. (1980). Biological effects of the human complement fragments C5a and CSa^ oi' neutrophil function. Immunopharmacology 2: 201-219.
204

WEDMORE, C.V. & WILLIAMS, T.J. (1981a). Control of vascular permeability by polymorphonuclear leukocytes in inflammation. Nature 289: 646-650.
WEDMORE, C.V. & WILLIAMS, T.J. (1981b). Platelet-activating factor (PAF), a secretory product of polymorphonuclear leukocytes, increases vascular permeability in rabbit skin. Br. J. Pharmacol 74: 916-917P.
WEISS, S.J., YOUNG, J., LO BUGLIO, A.F., SLIVKA, A., & NIMEN, N.F. (1981). Role of hydrogen peroxide in neutrophil-mediated destruction of cultured endothelial cells. J. Œn. Invest. 6 8 : 714.
WEISS, S.J. (1989). Tissue destruction by neutrophils. N. Engl J. Med. 320: 365-375.
WILLIAMS, T.J. (1976). The pro-inflammatory activity of E-, A-, D- and F-type prostaglandins and analogues 16, 16-dimethyl-PGE2 and (15S)-15-methyl-PGEz in rabbit skin; the relationship between potentiation of plasma exudation and local blood flow changes. Br. J. Pharmacol 56: 341-342P.
WILLIAMS, T.J. (1977). Chemical mediators of vascular responses in inflammation: a two-mediator hypothesis. Br. J. Pharmacol 62: 447-448P.
WILLIAMS, T.J. (1979). Prostaglandin E2 , prostaglandin I2 and the vascular changes of inflammation. Br. J. Pharmacol 65: 517-524.
WILLIAMS, T.J. (1982). Vasoactive intestinal polypeptide is more potent than prostaglandin E2 as a vasodilator and oedema potentiator in rabbit skin. Br. J. Pharmacol 77: 505-509.
WILLIAMS, T.J., JOSE, P.J., FORREST, M.J., SMAJE, L.H., & CLOUGH, G.F. (1984a). Inflammatory oedema induced by synergism between prostaglandins and C5a; the importance of the interaction between neutrophils and venular endothelial cells. In Progress in Microcirculation Research II. eds. Courtice F. C., Garlick D. G., & Perry M. A. pp. 439-448. University of N S W. Australia: Committee in Postgraduate Medical Educaiton.
WILLIAMS, T.J., JOSE, P.J., FORREST, M.J., WEDMORE, C.V., & CLOUGH, G.F. (1984b). Interactions between neutrophils and microvascular endothelial cells leading to cell emigration and plasma protein leakage. In White Blood Cell Mechanics: Basic Science and Clinical Aspects (Kroc Foundation Series), eds. Meiselman H. J., Lichtman M. A., & LaCelle P. L. pp. 195-208. New York: Liss.
WILLIAMS, T.J. & JOSE, P.J. (1981). Mediation of increased vascular permeability after complement activation: histamine-independent action of rabbit C5a. J. Exp. Med. 153: 136-153.
WILLIAMS, T.J. & MORLEY, J. (1973). Prostaglandins as potentiators of increased vascular permeability in inflammation. Nature 246: 215-217.
WILLIAMS, T.J. & PECK, M.J. (1977). Role of prostaglandin-mediated vasodilatation in inflammation. Nature 270: 530-532.
WILLIAMS, T.J. & PIPER, P.J. (1980). The action of chemically pure SRS-A on the microcirculation in vivo. Prostaglandins 19: 779-789.
WILLIAMSON, L.M., SHEPPARD, K., DAVIES, J.M., & FLETCHER, J. (1986). Neutrophils are involved in the increased vascular permeability produced by activated complement in man. Br. J. Haematol 64: 375-384.
205

WIRA, C.R. & MUNCK, A. (1974). Glucocorticoid-receptor complexes in rat thymus cells "cytoplasmic" - nuclear transformations. J. Biol Oiem. 249: 5328-5336.
WORTHEN, G.S., HASLETT, C., REES, A.J., GUMBAY, R.S., HENSON, J.E., & HENSON, P.M. (1987). Neutrophil-mediated pulmonary vascular injury: synergistic effect of trace amounts of lipopolysaccharide and neutrophil stimuli on vascular permeability and neutrophil sequestration in the lung. Am, Rev. Respir. Dis. 136: 19-28.
WORTHEN, G.S., ELSON, E.L., & DOWNEY, G.F. (1989). Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science 245: 183-186.
WRIGHT, D.G., KAUFFMANN, J.C., TERPSTRA, G.K., GRAW, R.G., DEISSEROTH, A.B., & GALLIN, J.I. (1978). Mobilization and exocytosis of specific (secondary) granules by human neutrophils during adherence to nylon wool infiltration leukapheresis (FL). Blood 52: 770.
WRIGHT, D.G. & GALLIN, J.I. (1979). Secretory responses of human neutrophils: exocytosis of specific (secondary) granules by human neutrophils during adherence in vitro and during exudation in vivo. J. Immunol 123: 285-294.
WRIGHT, S.D., RAO, P.E., VAN VOORHIS, W.C., CRAIGMYLE, L.S., IIDA, K., TALLE, M.A., WESTBERG, E.F., GOLDSTEIN, G., & SILVERSTEIN, S.C. (1983). Identification of the C3bi receptor on human monocytes and macrophages by using monoclonal antibodies. Proc. Natl Acad. Sci. USA 80: 5699-5703.
WRIGHT, S.D., WEITZ, J.I., HUANG, A.J., LEVIN, S.M., SILVERSTEIN, S.C., & LOIKE, J.D. (1988). Complement receptor type three (CR3, CD 1 lb/CD 18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc. Natl Acad. Sci. USA 85: 7734.
WRIGHT, S.D., LEVIN, S.M., JONG, M.T.C., CHAD, Z., & KABBASH, L.G. (1989). CR3 (CDllb/CD18) expresses one binding site for Arg-Gly-Asp-containing peptides and a second site for bacterial lipopolysaccharide. J. Exp. Med. 169: 175-183.
YANCEY, K.B., HAMMER, C H., HARVATH, L., RENFER, L., FRANK, M.M., & LAWLEY, T.J. (1985). Studies of human C5a as a mediator of inflammation in normal human skin. J. Clin. Invest. 75: 486-495.
YOSHIMURA, T., MATSUSHIMA, K., OPPENHEIM, J.J., & LEONARD, E.J. (1987). Neutrophil chemotactic factor produced by lipopolysaccharide (LPS)-stimulated human blood mononuclear leukocytes: partial characterization and separation from interleukin 1 (IL 1). J. Immunol 139: 788-793.
ZIMMERMAN, G.A., MCINTYRE, T.M., MEHRA, M., & PRESCOTT, S.M. (1990). Endothelial cell-associated platelet-activating factor: a novel mechanism for signaling intercellular adhesion, y. 110:529-540.
ZIMMERMAN, G.A., PRESCOTT, S.M., & MCINTYRE, T.M. (1992). Endothelial cell interactions with granulocytes: tethering and signaling molecules. Immunol Today 13: 93-100.
ZIMMERMAN, G.A. & HILL, H R. (1984). Inflammatory mediators stimulate granulocyte adherence to cultured human endothelial cells. Thromb. Res. 35: 203.
ZIMMERMAN, G.A. & MCINTYRE, T.M. (1988). Neutrophil adherence to human endothelium in vitro occurs by CDwl8 (Mol,Mac-1/LFA-1/GP150,95) glycoprotein-dependent and -independent mechanisms. J. Clin. Invest. 81: 531-537.
206

ZURIER, R.B., WEISSMAN, G., HOFFSTEIN, S., CAMERON, S., & TAI, H.H. (1974). Mechanisms of lysozomal enzyme release from human leukocytes. II. Effects of cAMP and cGMP autonomic agonists and agents which effect microtubule function. J. Œn. Invest. 53: 217. (Abstract)
207

Publications arising from this thesis
Papers
Hellewell PG, Yarwood H, Williams TJ. (1989) Characteristics of oedema formation induced by N-formyl-methionyl-leucyl-phenylalanine in rabbit skin. BrJ.Pharmacol 97, 181-189.
Yarwood H, Nourshargh S, Brain SD, Williams TJ. (1993) Effect of dexamethasone on neutrophil accumulation and oedema formation in rabbit sl^ : an investigation of site of action. BrJ.Pharmacol 108, 959-966.
ReviewWillliams TJ, Yarwood H. (1990) Effect of glucocorticosteroids on microvascular permeability. Am.Rev.Resp.Dis. 141, S39-S43.
AbstractsYarwood H, Nourshargh S, Brain SD and Williams TJ. (1988) Suppression of neutrophil accumulation and neutrophil-dependent oedema by dexamethasone in rabbit skin. Br.J.Pharmacol 95, 726P.
Yarwood H, Nourshargh S, Brain SD and Williams TJ. (1989) Inhibition of neutrophil accumulation by dexamethasone in vivo: evidence for site of action. Br.J.Pharmacol 96, 284P.
208