nur77 prevents excessive osteoclastogenesis by inducing ubiquitin ligase cbl-b to mediate nfatc1...
DESCRIPTION
Osteoclasts are bone-resorbing cells essential for skeletal remodeling. However, over-active osteoclasts can cause bone degenerative disorders. Therefore, the level of NFATc1, the master transcription factor of osteoclast, must be tightly controlled. Although the activation and amplification of NFATc1 have been extensively studied, how NFATc1 signaling is eventually resolved is unclear. Here, we uncover a novel and critical role of the orphan nuclear receptor Nur77 in mediating an NFATc1 self-limiting regulatory loop to prevent excessive osteoclastogenesis. Nur77 deletion leads to low bone mass owing to augmented osteoclast differentiation and bone resorption. Mechanistically, NFATc1 induces Nur77 expression at late stage of osteoclast differentiation; in turn, Nur77 transcriptionally up-regulates E3 ubiquitin ligase Cbl-b, which triggers NFATc1 protein degradation. These findings not only identify Nur77 as a key player in osteoprotection and a new therapeutic target for bone diseases, but also elucidate a previously unrecognized NFATc1→Nur77→Cblb⎯•NFATc1 feedback mechanism that confers NFATc1 signaling autoresolution.TRANSCRIPT

ACCEPTED MANUSCRIPT
self-limitationNur77 prevents excessive osteoclastogenesis by inducing ubiquitin ligase Cbl-b to mediate NFATc1
Xiaoxiao Li, Wei Wei, HoangDinh Huynh, Hao Zuo, Xueqian Wang, Yihong Wan
http://dx.doi.org/10.7554/eLife.07217DOI:
Cite as: eLife 2015;10.7554/eLife.07217
Published: 14 July 2015Accepted: 13 July 2015Received: 26 February 2015
and proofing.formatted HTML, PDF, and XML versions will be made available after technical processing, editing, This PDF is the version of the article that was accepted for publication after peer review. Fully
elife.elifesciences.org at Sign up for alertsStay current on the latest in life science and biomedical research from eLife.

1
Nur77 Prevents Excessive Osteoclastogenesis by Inducing Ubiquitin Ligase Cbl-b to 1
Mediate NFATc1 Self-Limitation 2
3
Xiaoxiao Li1, Wei Wei1, HoangDinh Huynh1, Hao Zuo1, Xueqian Wang1 and Yihong Wan1 4
5
1Department of Pharmacology, The University of Texas Southwestern Medical Center, Dallas, 6
Texas 75390, USA 7
8
Running Title: Nur77 Induces Cbl-b to Promote NFATc1 Degradation 9
10
Corresponding author: Yihong Wan ([email protected]) 11
12

2
Abstract 13
Osteoclasts are bone-resorbing cells essential for skeletal remodeling. However, over-active 14
osteoclasts can cause bone degenerative disorders. Therefore, the level of NFATc1, the master 15
transcription factor of osteoclast, must be tightly controlled. Although the activation and 16
amplification of NFATc1 have been extensively studied, how NFATc1 signaling is eventually 17
resolved is unclear. Here, we uncover a novel and critical role of the orphan nuclear receptor 18
Nur77 in mediating an NFATc1 self-limiting regulatory loop to prevent excessive 19
osteoclastogenesis. Nur77 deletion leads to low bone mass owing to augmented osteoclast 20
differentiation and bone resorption. Mechanistically, NFATc1 induces Nur77 expression at late 21
stage of osteoclast differentiation; in turn, Nur77 transcriptionally up-regulates E3 ubiquitin 22
ligase Cbl-b, which triggers NFATc1 protein degradation. These findings not only identify Nur77 23
as a key player in osteoprotection and a new therapeutic target for bone diseases, but also 24
elucidate a previously unrecognized NFATc1→Nur77→Cblb⎯•NFATc1 feedback mechanism 25
that confers NFATc1 signaling autoresolution. 26
27

3
Introduction 28
Bone is a dynamic organ that replaces itself every 10 years in humans (Office of the Surgeon 29
General) (2004). It needs to maintain an optimal density to carry out important support, 30
movement and protective functions in organisms. The density of bone is tightly regulated by the 31
bone forming osteoblasts and the bone resorbing osteoclasts. Osteoblasts come from the 32
mesenchymal precursor cells, whereas osteoclast precursors come from monocyte-33
macrophage lineage cells. Upon RANKL signaling, osteoclast precursor cells fuse and become 34
multi-nucleated giant cells that can degrade both the organic and inorganic tissues of the bone. 35
Osteoclast dysregulation has been associated with several human skeletal diseases such as 36
osteoporosis, rheumatoid arthritis and cancer metastasis to the bone (Novack and Teitelbaum, 37
2008). Drugs that can inhibit osteoclast differentiation, activity or survival have been shown to 38
be effective against these diseases (Body et al., 2010; Drake et al., 2008). 39
NFATc1 is a key transcriptional switch that activates osteoclastogenesis. Ectopic NFATc1 40
expression alone in osteoclast precursors is sufficient to produce mature osteoclasts, whereas 41
NFATc1 deletion blocks the ability of the precursors to differentiate into osteoclasts (Takayanagi 42
et al., 2002). NFATc1 binds to its response elements containing a consensus sequence of 43
GGAAA, and its target genes in osteoclasts include cathepsin K, CLC-7 chloride channel, 44
vacuolar proton pump subunit Atp6v0d2, etc. (Kuroda and Matsuo, 2012). NFATc1 has been 45
shown to auto-amplify during osteoclast differentiation, and this auto-amplification process has 46
been suggested to be important for osteoclast lineage commitment (Asagiri et al., 2005). 47
However, so far very few studies have dealt with whether and how NFATc1 signaling is 48
attenuated upon its initial activation. As a result, despite the crucial functions of NFATc1 in 49
osteoclastogenesis, the mechanisms for how NFATc1 signaling is resolved to prevent excessive 50
osteoclast differentiation are still incompletely understood. 51
Nur77 (encoded by Nr4a1), also known as nerve growth factor IB (NGFIB), TR3 or NAK-52
1, is an orphan nuclear receptor in the Nr4a family, which also includes Nurr1 (Nr4a2) and Nor-1 53

4
(Nr4a3). Unlike other nuclear receptors whose functions are mainly modulated by their ligands, 54
Nur77 is mostly regulated at the transcriptional and post-transcriptional levels (Zhao and 55
Bruemmer, 2010). Nur77 can bind to NurRE or NBRE as monomer, homodimer or heterodimer 56
(Philips et al., 1997). Nur77 has been implicated in a variety of physiological processes, 57
including thymocyte negative selection, hypothalamic-pituitary adrenal axis, chronic 58
inflammation, and vascular smooth muscle cell proliferation (Hsu et al., 2004). Nonetheless, it is 59
unknown whether Nur77 can directly regulate skeletal homeostasis or bone cell differentiation. 60
In this study, we uncover a novel and important role of Nur77 in NFATc1 protein degradation 61
during osteoclastogenesis and bone resorption, thus revealing a previously unrecognized 62
mechanism that is essential for the resolution of NFATc1 signaling in which NFATc1 exerts self-63
limitation via an NFATc1→Nur77→Cblb⎯•NFATc1 negative feedback loop. 64
65
Results 66
Nur77 Deletion Enhances Osteoclast Differentiation 67
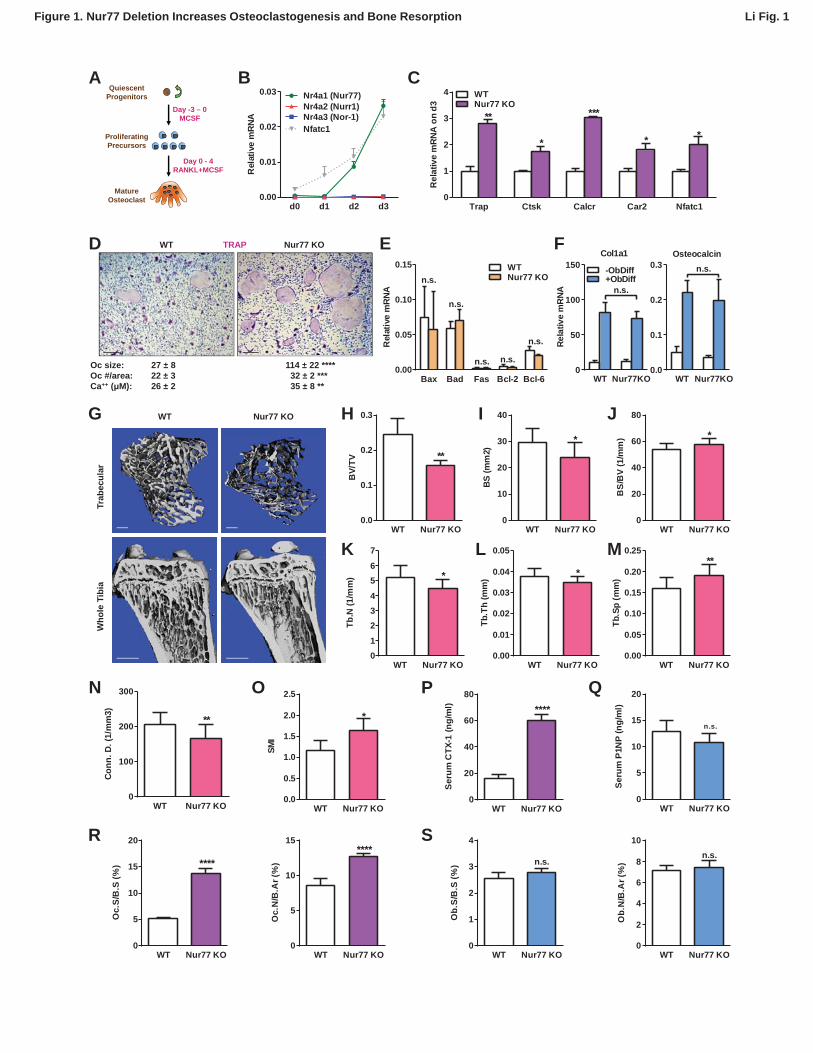
To examine the expression pattern of Nur77 during osteoclast differentiation, we treated bone 68
marrow-derived osteoclast precursor cells with RANKL for 4 days (Figure 1A). A time course 69
analysis showed that Nr4a1 mRNA started to rise on day 2 during osteoclastogenesis (Figure 70
1B). The other two members of the NR4A family, Nr4a2 and Nr4a3, were either not expressed 71
or expressed at much lower levels (Figure 1B). To determine if there is any potential regulatory 72
role of Nur77 in osteoclastogenesis, we compared osteoclast differentiation cultures from bone 73
marrow hematopoietic progenitors of Nur77 knockout (Nur77 KO) mice and WT littermate 74
controls. The results revealed an enhanced osteoclast differentiation in Nur77 KO cultures 75
shown by the higher expression of osteoclast differentiation markers such as tartrate-resistant 76
acid phosphatase (Trap), cathepsin K (Ctsk), calcitonin receptor (Calcr), carbonic anhydrase 2 77
(Car2), as well as the master osteoclastogenic transcription factor Nfatc1 (Figure 1C). 78
Consistent with these results, we have also observed more and larger mature osteoclasts in the 79

5
differentiation cultures (Figure 1D), as well as higher resorptive activity (Figure 1D). The 80
expression of pro- and anti-apoptotic genes was comparable, indicating an unaffected 81
osteoclast apoptosis (Figure 1E). In contrast, osteoblast differentiation from Nur77 KO bone 82
marrow mesenchymal progenitors was unaltered, shown by the similar induction of osteoblast 83
markers such as collagen, type I, alpha 1 (Col1a1) and Osteocalcin (Figure 1F). These results 84
suggest that Nur77 may specifically suppress osteoclastogenesis during bone remodeling. 85
86
Nur77 Deletion Leads to Bone Loss Due to Excessive Bone Resorption 87
To determine if Nur77 is a physiologically significant regulator of bone, we next examined the in 88
vivo skeletal phenotype of Nur77 KO mice. MicroCT analysis revealed that male Nur77 KO mice 89
had a low-bone-mass compared to WT male littermate controls (Figure 1G), illustrated by a 90
36% lower bone volume/tissue volume ratio (BV/TV) (Figure 1H), 19% less bone surface (BS) 91
(Figure 1I), 8% greater bone volume/bone surface ratio (BV/BS) (Figure 1J), 14% less 92
trabecular number (Tb.N) (Figure 1K), 8% less trabecular thickness (Tb.Th) (Figure 1L) and 93
19% more trabecular separation (Tb.Sp) (Figure 1M). This resulted in a 19% decrease in 94
connectivity density (Conn. D.) (Figure 1N) and a 40% increase in the Structure Model Index 95
(Wei et al., 2014), which quantifies the 3D structure for the relative amount of plates (SMI=0, 96
strong bone) and rods (SMI=3, fragile bone) (Figure 1O). Cortical thickness was reduced by 7% 97
(WT=0.1125±0.003mm; KO=0.1048±0.004mm; p=0.02; n=6) whereas tibia length was 98
unaltered. Female Nur77 KO mice showed a similar phenotype with a 19% lower trabecular 99
BV/TV (WT=0.2025±0.03; KO=0.1632±0.02; p=0.03) and a 5% decreased cortical thickness 100
(WT=0.108±0.003mm; KO=0.102±0.001mm; p=0.04) (3 month old, n=6). 101
ELISA analyses showed that the serum bone resorption marker C-terminal telopeptide 102
fragments of the type I collagen (CTX-1) was 3.7-fold higher in Nur77 KO mice (Figure 1P), 103
whereas the serum bone formation marker N-terminal propeptide of type I procollagen (P1NP) 104
was unchanged (Figure 1Q). Consistent with these observations, histomorphometry of the 105

6
femur showed that osteoclast surface and osteoclast number were significantly increased in 106
Nur77 KO mice (Figure 1R), whereas osteoblast surface and osteoblast number (Figure 1S) 107
were unaltered. Together, these results suggest that Nur77 deletion causes low bone mass 108
primarily through increasing osteoclastogenesis and bone resorption. 109
110
Nur77 Regulation of Bone Resorption is Intrinsic to the Osteoclast Lineage 111
Because Nur77 has been implicated to regulate many other cell types (Hsu et al., 2004), we 112
next performed bone marrow transplantation experiments to examine whether Nur77 regulation 113
of bone resorption stems from the intrinsic effects in the hematopoietic/osteoclast lineage or 114
non-autonomous effects from other tissues or cell types such as osteoblasts, osteocytes or the 115
neuroendocrine system. Two complimentary sets of bone marrow transplantation were 116
performed and serum bone markers were assessed two months later. In the first set, we 117
harvested donor bone marrow cells from both WT and Nur77 KO mice, and transplanted them 118
to irradiated WT recipient mice (Figure 2A). The results showed that WT mice receiving Nur77 119
KO bone marrow cells exhibited significantly higher CTX-1 levels than the control group (Figure 120
2B), but unaltered P1NP levels (Figure 2C), suggesting that Nur77 KO hematopoietic lineage 121
was sufficient to elevate bone resorption. In the second set, we transplanted WT donor bone 122
marrow cells into either Nur77 KO or WT control recipient mice (Figure 2D). Nur77 KO mice 123
receiving WT bone marrow cells showed normalized CTX-1 levels similar to the control group 124
(Figure 2E), with also similar P1NP levels (Figure 2F), suggesting that WT bone marrow can 125
completely rescue the osteoclast defects in the Nur77 KO mice and thus other tissues/cell types 126
play only a minor role if any. The results from these two experiments indicate that Nur77 127
regulation of bone resorption is intrinsic to the hematopoetic lineage. 128
In the bone milieu, osteoblasts and osteocytes provide RANKL and the RANKL decoy 129
receptor OPG to stimulate and inhibit osteoclast differentiation, respectively (Boyce and Xing, 130
2007). Thus, we assessed whether RANKL and OPG levels were different between Nur77 KO 131

7
and WT control mice. We first compared osteoblast differentiation cultures derived from Nur77 132
KO or WT control mice, and found that there was no difference in the expression of Rankl or 133
Opg, resulting in a comparable Rankl/Opg ratio (Figure 2G). Recently osteocytes, the mature 134
long living osteoblasts embedded in the bone matrix, have been shown to provide the majority 135
of RANKL and OPG to osteoclasts (Nakashima et al., 2011; Xiong et al., 2011). Hence, we 136
compared Rankl and Opg expression in femur shafts that contain mainly osteocytes. Our results 137
showed that there was no difference in Rankl expression (Figure 2H); however, Nur77 KO 138
osteocytes express a markedly higher level of Opg (Figure 2H), leading to a significantly lower 139
Rankl/Opg ratio (Figure 2H). The lower Rankl/Opg ratio in the Nur77 KO mice is unlikely to be 140
the cause of the augmented osteoclastogenesis, but presumably an attempt of osteocytes to 141
suppress the osteoclast over-activation. These results, coupled with unaltered in vitro osteoblast 142
differentiation (Figure 1F) and in vivo bone formation (Figure 1Q, S), suggest that 143
osteoblast/osteocyte are not major contributors to the excessive osteoclastogenesis and bone 144
resorption in Nur77 KO mice. 145
Inflammatory cytokines in the bone microenvironment can also promote osteoclast 146
differentiation (Zupan et al., 2013). Thus, we collected bone marrow cells from mouse femurs for 147
gene expression assessment. No significant difference was found in the expression of Tnfα, 148
IL1β or IL6 (Figure 2I). Moreover, it has been shown that serum levels of TNFa, IL1 and IL6 149
were unchanged in mice injected with either Nur77-expressing lentivirus or Nur77 siRNA (Hu et 150
al., 2014), further suggesting that the augmented osteoclastogenesis in Nur77 KO mice is not 151
due to differential levels of these inflammatory cytokines. 152
The above findings, together with the fact that Nur77 KO osteoclast precursors exhibited 153
enhanced osteoclast differentiation independent of the bone and neuroendocrine environment 154
(Figures 1C-D), indicate that Nur77 regulation of osteoclastogenesis is mainly intrinsic and cell-155
autonomous to the osteoclast lineage. 156
157

8
Nur77 Promotes NFATc1 Degradation 158
Given that Nur77 exerts functions within the osteoclast itself, we decided to investigate whether 159
Nur77 affects RANKL signaling pathways. RANKL binding to RANK receptor on osteoclast 160
precursor cells activates AP1 transcription factor via c-Jun phosphorylation, as well as NFκB 161
transcription factor via IκBα degradation, which in turn induces and initiates the 162
autoamplification of NFATc1, the master transcriptional switch of osteoclastogenesis (Kuroda 163
and Matsuo, 2012). Although Nfatc1 mRNA level was 2-fold higher in Nur77 KO osteoclast 164
differentiation cultures on day 3 (Figure 1C), we found that NFATc1 protein level was 7-fold 165
higher in Nur77 KO cultures compared to WT control cultures on day 3 (Figure 3A). It has been 166
reported that NFATc1 protein is degraded on day 3-4 during osteoclast differentiation, despite 167
the continuously rising Nfatc1 mRNA levels (Kim et al., 2010). Indeed, our time course analysis 168
showed that in WT cultures, NFATc1 protein was elevated on day 2 but then rapidly down-169
regulated on day 3 and day 4 (Figure 3A). In contrast, in Nur77 KO cultures, the initial increase 170
of NFATc1 protein was sustained and NFATc1 protein remained high on day 3 and day 4 (Figure 171
3A). We then compared c-Jun phosphorylation and IκBα degradation in the osteoclast 172
differentiation cultures upon RANKL stimulation, but did not observe any significant difference 173
between Nur77 KO and WT control cultures (Figure 3B), which is in agreement to the similar 174
NFATc1 protein induction on day 1 and day 2 (Figure 3A). Moreover, there is no Nur77 binding 175
sequence in a 4Kb region of NFATc1 promoter. These results indicate that Nur77 regulation of 176
NFATc1 protein level resides downstream of transcription. 177
It has been shown that the decrease in NFATc1 protein levels at later stage of osteoclast 178
differentiation is due to ubiquitin-mediated protein degradation; and that MG132, a proteasome 179
inhibitor, can restore NFATc1 protein to a similar level on day 2 (Kim et al., 2010). To examine 180
whether the differences in NFATc1 levels between Nur77 KO and WT mice were due to protein 181
degradation, we treated osteoclast differentiation cultures with MG132 on day 3. As our result 182
shows, MG132 treatment increased NFATc1 protein level in WT cultures to a level similar to 183

9
Nur77 KO cultures; and MG132 treatment could no longer further increase NFATc1 protein level 184
in the Nur77 KO cultures (Figure 3C). In line with these observations, our co-IP experiments 185
reveal that Nur77 (either endogenous or flag-tagged) does not directly interact with NFATc1 (not 186
shown), suggesting that Nur77 does not directly modulate NFATc1’s localization or activity. 187
These results indicate that Nur77 deletion elevates NFATc1 protein levels by suppressing 188
ubiquitin-degradation pathway. 189
As a complementary gain-of-function approach, we tested whether Nur77 over-190
expression could promote NFATc1 protein degradation. We transfected HEK293 cells with 191
NFATc1 together with Nur77 or a GFP control, and then quantified Nfatc1 mRNA and protein 192
levels. The result shows that Nur77 over-expression significantly decreased NFATc1 protein 193
levels (Figure 3D, left) without altering Nfatc1 mRNA levels (Figure 3D, right). Consistent with 194
the lower Nur77 protein abundance, Nur77 over-expression also dosage-dependently reduced 195
the NFATc1 transcriptional output from a luciferase reporter driven by NFATc1 response 196
elements (Figure 3E). This Nur77 reduction of NFATc1 activity was completely abolished by 197
MG132 (Figure 3F), indicating that it was mediated by ubiquitin-degradation pathway. The 198
mRNA expression of Nr4a1 and Nfatc1 in osteoclasts is comparable on day 2-3 (Figure 1B), 199
supporting that Nur77 regulation of NFATc1 is relevant in the osteoclast. These findings further 200
support the notion that Nur77 promotes NFATc1 protein degradation. 201
202
Nur77 transcriptionally up-regulates E3 ligase Cbl-b 203
In the ubiquitin degradation pathway, E3 ligases are responsible for substrate specificity and 204
ubiquitination regulation. We next searched for E3 ligases that could be responsible for NFATc1 205
degradation in osteoclasts. It has been reported that Cbl-b, an E3 ligase in the Cbl family, is a 206
major contributor to the ubiquitin-mediated down-regulation of NFATc1 at late stage of 207
osteoclast differentiation (Kim et al., 2010). Therefore, we tested the hypothesis that Nur77 may 208
promote NFATc1 degradation by inducing Cbl-b. We found that Cblb expression was 209

10
significantly lower in Nur77 KO osteoclast differentiation cultures compared to WT control 210
cultures on day 2 and day 4 (Figure 4A). Conversely, Nur77 over-expression in HEK293 cells 211
significantly increased Cblb expression (Figure 4B). Importantly, a truncated Nur77 mutant in 212
which the DNA binding domain (DBD) was deleted could no longer up-regulate Cblb, suggesting 213
that Nur77 induction of Cblb transcription depends on its DNA binding ability (Figure 4B). The 214
functional connection between Nur77 and Cbl-b is further supported by the similar bone 215
phenotype in Nur77 KO mice (Figure 1) and Cblb KO mice (Nakajima et al., 2009), including 216
increased ex vivo osteoclast differentiation and in vivo bone resorption, but unaltered bone 217
formation, leading to lower bone mass. 218
To investigate whether Cblb is a direct Nur77 target gene, we tested whether Nur77 219
could transcriptionally activate the Cbl-b promoter. We cloned a 1kb segment of Cbl-b promoter 220
upstream of a luciferase reporter and tested its expression in a transient transfection assay in 221
HEK293 cells. The result showed that co-transfection with Nur77 significantly up-regulated the 222
Cbl-b promoter activity by 2.2-fold (Figure 4C), suggesting that Nur77 is able to directly activate 223
Cblb transcription. Bioinformatic analyses revealed a pair of motifs that may comprise a Nur77 224
response element (NurRE) in the Cbl-b promoter at ~600bp upstream of the transcription start 225
site (Figure 4C, inset). To examine whether these putative NurRE motifs are important for 226
Nur77 induction of Cbl-b promoter, we mutated each NurRE motif to derive mutant-1 and 227
mutant-2 luciferase reporters (Figure 4C, inset). Both mutant reporters exhibited a significantly 228
compromised ability to be activated by Nur77 compared to the WT reporter (Figure 4C), 229
indicating that both NurRE motifs are functionally required. To determine whether Nur77 can 230
bind to the endogenous Cbl-b NurRE, we performed Chromatin Immunoprecipitation (ChIP) 231
assay. Nur77 was found to be enriched at the NurRE region, leading to transcription activation 232
shown by the presence of H3K4Me3 histone mark at the transcription start site (Figure 4D). 233
These results suggest that Nur77 can directly induce Cbl-b transcription by binding to a NurRE 234
in the Cbl-b promoter. 235

11
We next sought to elucidate whether Nur77 induction of Cbl-b is functionally required for 236
Nur77 down-regulation of NFATc1 protein. Instead of deleting Cblb, we designed a more 237
prudent strategy to specifically disrupt the NurRE region in the endogenous Cbl-b promoter 238
using CRISPR/Cas9 genome editing tool, thus more precisely dissecting the functional 239
interaction among Nur77, Cbl-b and NFATc1 (Fig. 4E). Compared with WT control cells, the 240
ability of Nur77 to increase Cblb mRNA (Fig. 4F), as well as to decrease NFATc1 protein level 241
(Fig. 4G) and transcriptional output (Fig. 4H), was significantly attenuated in two independent 242
CRISPR mutant clones. These results provide strong evidence that Nur77 promotes NFATc1 243
protein degradation by directly inducing the transcription of Cbl-b E3 ligase. 244
245
NFATc1 Up-regulates Nur77 to Form a Self-limiting Loop 246
Since Nur77 expression consistently rises during osteoclastogenesis (Figure 1B), we 247
hypothesize that there is an upstream regulator that induces Nur77 transcription upon RANKL 248
signaling activation. Interestingly, we found several NFATc1 response elements in the Nur77 249
promoter region, suggesting that NFATc1 up-regulates Nur77 to initiate a negative feedback 250
loop. To test whether NFATc1 itself is sufficient to increase Nur77 expression independent of 251
other RANKL signaling pathways, we performed transfection assays to over-express NFATc1. 252
Compared to a GFP negative control, NFATc1 over-expression significantly increased Nur77 253
expression in both HEK293 cells and mouse myoblast C2C12 cells (Figure 5A). Conversely, 254
treatment of osteoclast differentiation cultures with cyclosporin A, a calcineurin inhibitor that 255
suppresses NFATc1 activity, dosage-dependently decreased Nur77 expression (Figure 5B). 256
We next examined whether NFATc1 could directly activate Nur77 promoter. We cloned a 257
0.8Kb Nur77 promoter region upstream of a luciferase reporter and tested its inducibility by 258
NFATc1 by transient transfection. Compared to a GFP negative control, NFATc1 over-expression 259
significantly elevated the luciferase output (Figure 5C). Moreover, ChIP assay showed that 260
RANKL treatment of osteoclast differentiation cultures markedly increased NFATc1 binding to 261

12
the endogenous Nur77 promoter (Figure 5D), leading to activated Nur77 transcription as shown 262
by the higher level of H3K4Me3 histone mark at the transcription start site (Figure 5D). 263
Together, these results indicate that Nur77 is a direct transcriptional target of NFATc1, and thus 264
revealing a key mechanism for how NFATc1 resolves its own signaling to prevent excessive 265
osteoclastogenesis via an NFATc1→Nur77→Cblb⎯•NFATc1 self-limiting loop (Figure 5E). 266
267
Cbl-b Deletion Abolishes Nur77 Regulation of Osteoclastogenesis and Bone Resorption 268
To further determine whether Cbl-b is required for the anti-osteoclastogenic function of Nur77 in 269
vivo, we conducted genetic experiments by comparing Nur77 Cblb double knockout mice (DKO) 270
with Nur77 KO mice, Cblb KO mice and WT littermate controls. Ex vivo bone marrow osteoclast 271
differentiation assay showed that Nur77 deletion could no longer further enhance osteoclast 272
differentiation in the absence of Cbl-b; furthermore, Cblb KO cultures and Nur77 Cblb DKO 273
cultures showed a similar enhanced osteoclastogenesis as Nur77 KO cultures compared with 274
WT control cultures (Figure 6A-E). In accordance to this ex vivo finding, in vivo analyses 275
showed that when Cbl-b is absent, Nur77 deletion could no long further elevate serum bone 276
resorption (Figure 6F) or reduce bone mass (Figure 6G-K); Cblb KO mice and Nur77 Cblb 277
DKO mice showed a similar high bone resorption and low bone mass phenotype as Nur77 KO 278
mice compared with WT controls (Figure 6F-K). These findings demonstrate that Cbl-b deletion 279
fully recapitulates Nur77 deletion, and Cbl-b deletion completely abolishes Nur77 regulation of 280
osteoclastogenesis and bone resorption. Therefore, this in vivo genetic evidence strongly 281
supports Cbl-b as a major and essential mediator of Nur77 function in the osteoclast lineage. 282
283
Discussion 284
In this study, we have identified the nuclear receptor Nur77 as a critical negative regulator of 285
osteoclastogenesis and bone resorption, revealing its novel bone protective role. Nur77 deletion 286
causes elevated bone resorption and bone loss in mice. Moreover, we have also unraveled a 287

13
previously unrecognized mechanism for how Nur77 attenuates NFATc1 signaling at late stage of 288
osteoclast differentiation. Nur77 transcriptionally up-regulates Cbl-b E3 ligase to trigger NFATc1 289
protein degradation, so that NFATc1 signaling can be resolved in a timely fashion to prevent 290
excessive osteoclastogenesis and bone resorption (Figure 5E). 291
When osteoclast differentiation is enhanced in a culture, typically all RANKL-induced 292
osteoclastogenic genes will be elevated, including Nfatc1, because the gene expression 293
analysis is a population-based assay (Figure 1C). In addition to induction by RANKL-activated 294
upstream signaling, NFATc1 also auto-amplifies itself at mRNA level (Asagiri et al., 2005), which 295
makes it harder to discern the origin of NFATc1 regulation in osteoclast cultures. Thus, we 296
subsequently examined the intrinsic ability of Nur77 to regulate NFATc1 via transfection in a 297
heterologous cell type such as HEK293 cells, in the absence of the other RANKL signaling in 298
osteoclast cultures. We show that Nur77 can decrease NFATc1 protein levels without affecting 299
Nfatc1 mRNA levels (Figure 3D). Also, there is no Nur77 binding sequence in NFATc1 300
promoter, further support that Nur77 does not directly regulate NFATc1 transcription. Moreover, 301
our co-IP experiment show that Nur77 (either endogenous or a flag-tagged) does not directly 302
interact with NFATc1, suggesting that Nur77 does not directly modulate NFATc1 localization or 303
activity. As a result, we conclude that Nur77 mainly regulates NFATc1 protein degradation. 304
In searching for the Nur77-induced NFATc1-targeting E3 ubiquitin ligase, we have 305
considered both of the two members in the Cbl family - Cbl and Cbl-b. Findings from our study 306
and previous studies indicate that Cbl-b, but not Cbl, is the major regulator in osteoclast due to 307
the following reasons. 1) Previous studies and our data show that Cbl-b KO mice exhibit 308
enhanced osteoclast differentiation, higher bone resorption and lower bone mass that is similar 309
to the phenotype we observed in Nur77 KO mice (Nakajima et al., 2009) (Figure 6). In contrast, 310
adult Cbl KO mice have no obvious bone phenotype (Chiusaroli et al., 2003). This supports that 311
Cbl-b, but not Cbl, is a physiologically significant regulator of osteoclast in vivo. 2) Nur77 over-312
expression induces the expression of Cbl-b, but not Cbl. 3) A complete NurRE exists only in 313

14
Cbl-b promoter, but not in Cbl promoter. 4) Nur77 KO osteoclast cultures show a significant 314
lower level of Cbl-b, but not Cbl. 5) Finally, our in vivo genetic data comparing Nur77 Cblb DKO 315
mice with Cblb KO mice show that Cbl-b deletion completely abolishes the enhanced osteoclast 316
differentiation, elevated bone resorption and reduced bone mass in Nur77 KO mice, 317
demonstrating that Cbl-b is the major and essential mediator of Nur77 regulation of 318
osteoclastogenesis, which is not compensated by Cbl. 319
In the process of studying the role of Nur77 in NFATc1 regulation and osteoclast 320
differentiation, we inadvertently discovered that Nur77 is not only a regulator of NFATc1 but also 321
a transcriptional target of NFATc1, thus revealing a negative feedback loop where NFATc1 322
induces its own degradation by up-regulating Nur77 and Cbl-b. This mechanism is crucial for 323
proper cellular differentiation and function, since the resolution of signaling is just as important 324
as its initiation and amplification. A breach in the NFATc1→Nur77→Cblb⎯•NFATc1 regulatory 325
loop, exemplified by the Nur77 KO mice, will cause pathologically elevated NFATc1 levels during 326
late stage of osteoclastogenesis and send osteoclasts into overdrive. To the best of our 327
knowledge, we are the first group to propose an NFATc1 self-limiting regulatory mechanism. 328
Therefore, NFATc1 exerts three functions to control osteoclastogenesis. In addition to the 329
previously recognized roles in its auto-amplification and activating osteoclast genes to initiate 330
the differentiation, NFATc1 also plays a role in its auto-resolution to cease the differentiation 331
(Figure 5E). Our findings will pave the road for future investigations to examine whether this 332
NFATc1→Nur77→Cblb⎯•NFATc1 negative feedback loop may be widely applicable to NFATc1 333
regulation of other cellular processes such as T cell activation and cancer development. 334
Identification of novel osteoclast signaling pathways provides insights into potential new 335
therapeutic options to treat bone degenerative diseases by inhibiting osteoclast activity. Current 336
clinically approved osteoclast inhibitors, such as bisphosphonates and denosumab (anti-RANKL 337
antibody), may cause severe side effects such as osteonecrosis of the jaw (ONJ) (Boquete-338

15
Castro et al., 2015; Khan et al., 2009). These side effects could stem from a variety of factors 339
including target specificity. The whole body Nur77 KO mice, however, provides an example 340
where despite the wide spread expression of a gene, it still can be targeted due to the 341
differential sensitivity in different tissues. Although Nur77 has been implicated in numerous 342
physiological functions in vitro, most of these functions did not hold true in vivo based on a 343
general lack of phenotype in Nur77 KO mice (Chao et al., 2013; Lee et al., 1995). Until recently, 344
Nur77 KO mice largely appear healthy and normal, and only exhibit clinical deficiencies under 345
severe stress (Chao et al., 2009; Palumbo-Zerr et al., 2015) or with the deletion of an additional 346
NR4A gene (Mullican et al., 2007). This may be partially due to functional redundancy among 347
NR4A family members so that the compensation by Nurr1 and Nor1 masks the effects of Nur77 348
loss. Interestingly, we found that Nur77 is the predominant NR4A member in the osteoclast 349
lineage with little or no Nurr1 or Nor1 expression (Figure 1B), explaining the critical role of 350
Nur77 in osteoclastogenesis so that the effects on bone is evident by Nur77 deletion alone. This 351
creates an exciting opportunity for selective drug targeting and precision medicine with minimal 352
side effects. Most recently, Tontonoz et al have uncovered a muscle protective role of Nur77 as 353
mice deficient in Nur77 alone exhibit reduced muscle mass and myofiber size (Tontonoz et al., 354
2015). Therefore, Nur77 activation may represent a promising therapeutic strategy for 355
musculoskeletal degenerative diseases with dual benefits on muscle and bone. 356
The crucial regulation of NFATc1 protein degradation by Nur77 and Cbl-b suggests that it 357
may be therapeutically beneficial to accelerate RANKL signaling resolution during 358
osteoclastogenesis. Indeed, defects in the components of ubiquitin and proteasome system 359
have been implicated in diseases including cancer and neurodegenerative disorders (Popovic et 360
al., 2014). Bortezomib, a peptide inhibitor of proteasome, has been approved for clinical usage 361
in pathological settings such as refractory multiple myeloma (Richardson et al., 2005). The 362
discovery of pathway-specific ubiquitin-proteasome activators, however, is somewhat lagging 363
behind. Nonetheless, oleuropein, a small molecule proteasome activator, has been shown to 364

16
delay replicative senescence of human embryonic fibroblast (Katsiki et al., 2007). In addition, 365
oleuropein treatment has been shown to inhibit osteoclast formation and suppress the loss of 366
trabecular bone in ovariectomized mice (Hagiwara et al., 2011), giving hope that small 367
molecules that selectively activate the protein degradation pathway may be a promising future 368
therapeutic strategy for skeletal and other diseases. 369
370

17
Materials and methods 371
Mice 372
Nur77 KO mice (Lee et al., 1995) in a C57BL/6 and 129SvJ hybrid background was originally 373
generated by Jeffrey Milbrandt at Washington University School of Medicine and kindly provided 374
by Orla Conneely at Baylor College of Medicine. Cblb KO mice in a mixed background were 375
originally generated by Josef Penninger (Bachmaier et al., 2000). Mice were fed with standard 376
chow ad libitum and kept on a 12-h light, 12-h dark cycle. Nur77 KO mice were bred with Cblb 377
KO mice to generate Nur77 Cblb double heterozygous mice, which were then bred to generate 378
littermates for Nur77 KO, Cblb KO, Nur77 Cblb DKO and WT control. All experiments were 379
conducted using littermates. Bone marrow transplantation was performed as described 380
(Krzeszinski et al., 2014; Wan et al., 2007). Briefly, bone marrow cells from 2-month-old male 381
donor (WT or Nur77 KO) were intravenously transplanted via retro orbital injection into 2-month-382
old male recipients (WT or Nur77 KO) that were irradiated at lethal dose (1000 roentgen); the 383
mice were analyzed 3 month post transplantation. Our established protocol for lethal irradiation 384
and bone marrow transplantation achieves >95% repopulation of donor cells in the recipient 385
mice, as measured by the percentage of CD45.1 vs. CD45.2. Sample size estimate was based 386
on power analyses performed using SAS 9.3 TS X64_7PRO platform at the UTSW Biostatistics 387
Core. With the observed group differences and the relatively small variation of the in vivo 388
measurements, n=4 and n=3 will provide >90% and >80% power at type I error rate of 0.05 389
(two-sided test), respectively. All protocols for mouse experiments were approved under number 390
2008-0324 by the Institutional Animal Care and Use Committee of UTSW. 391
392
Bone Analyses 393
μCT was performed using a Scanco μCT-35 instrument (Scanco Medical) as described (Wei et 394
al., 2010). Histomorphometry were performed as described (Wan et al., 2007; Wei et al., 2011). 395
Serum CTX-1 bone resorption marker and P1NP bone formation marker were measured with 396

18
RatLapsTM EIA kit and Rat/Mouse PINP EIA kit (Immunodiagnostic Systems), respectively. To 397
analyze osteocyte gene expression, mouse femur was cut off at both ends to allow marrow cells 398
to be flushed out with media. It was then soaked in PBS and spun down to remove residual 399
marrow cells, and snap frozen in liquid nitrogen, stored at -80°C until RNA extraction. 400
401
Ex Vivo Osteoclast and Osteoblast Differentiation 402
Osteoclasts were differentiated from bone marrow cells as described (Wan et al., 2007). Briefly, 403
hematopoietic bone marrow cells were purified with a 40 μm cell strainer, cultured for 16 hours 404
with 5 ng/ml MCSF (R&DSystems) in α-MEM containing 10% FBS. Floating cells were then 405
collected and differentiated with 40 ng/ml of M-CSF in α-MEM containing 10% FBS for 3 days 406
(day -3 to day 0), then with 40 ng/ml of MCSF and 100 ng/ml of RANKL (R&D Systems) for 3-9 407
days (day 0 to day 9). Mature osteoclasts were identified as multinucleated (>3 nuclei) TRAP+ 408
cells on day 9. Osteoclast differentiation and apoptosis were quantified by the RNA expression 409
of osteoclast markers and apoptosis genes on day 3 and 6, respectively, using RT-QPCR 410
analysis. For osteoclast resorptive function analyses, osteoclast differentiation was conducted in 411
OsteoAssay bone plates (Lonza), and osteoclast activity was quantified as calcium release from 412
bone into culture medium using a colorimetric calcium detection kit (Abcam). Osteoblast 413
differentiation from bone marrow cells was performed as previously described (Wei et al., 2012; 414
Wei et al., 2014). Briefly, bone marrow cells were cultured for 4 days in MSC media (Mouse 415
MesenCult® Proliferation Kit, StemCell Technologies), then differentiated into osteoblast with α-416
MEM containing 10% FBS, 5 mM β-glycerophosphate and 100 μg/ml ascorbic acid for 9 days. 417
418
Reagents 419
Antibodies for NFATc1, total-c-Jun and IκBα, as well as cyclosporine A were purchased from 420
Santa Cruz Biotechnology. Phospho (ser73)-c-Jun antibody was from Cell Signaling. Anti-421
Histone H3 (tri methyl K4) antibody was from Abcam. Antibodies for Flag and β-actin were from 422

19
Sigma. MG132 was from Fisher. Western blot and ChIP assays were performed as previously 423
described (Krzeszinski et al., 2014; Wan et al., 2007). NFATc1 expression plasmid was 424
purchased from Open Biosystems. Human Flag-Nur77 expression plasmid was kindly provided 425
by Orla Conneely lab. Nur77-ΔDBD expression plasmid was constructed by deleting the amino 426
acid residues 270-335 from the WT Nur77 expression plasmid. RNA was reverse transcribed 427
into cDNA using an ABI High Capacity cDNA RT Kit, and analyzed using real-time quantitative 428
PCR (SYBR Green) in triplicate. All RNA expression was normalized by the ribosomal gene L19. 429
430
Promoter Analyses 431
Cbl-b-promoter-luc-WT and Nur77-promoter-luc were constructed by cloning 1Kb and 0.8Kb 432
segment upstream of transcription start site into pGL4 luciferase vector. Cbl-b-luc-Mut1 and Cbl-433
b-luc Mut2 were created by introducing mutations to three residues in each NurRE region of 434
Cbl-b promoter, using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent 435
Technologies). NFATc1 transcriptional activity was quantified using pNFAT-Luc reporter (Agilent 436
Technologies). For transient transfection, a luciferase reporter was co-transfected into HEK293 437
cells with expression plasmids for β-gal and factors to be tested using FuGENE HD reagent 438
(Roche). Vector alone or a GFP expression plasmid served as a negative control. Luciferase 439
activity was measured 48 hours later and normalized by β-gal activity. All transfection 440
experiments were performed in triplicates and repeated for at least three times. 441
442
CRISPR Constructs and Clone Screening 443
Plasmids for gRNA cloning and hCas9 expression were from Addgene. Oligos for gRNA were 444
designed to target upstream and downstream of the NurRE in the Cbl-b promoter, and cloned 445
into gRNA vector according to the instruction form George Church Laboratory. Both vectors for 446
gRNAs, and the expression plasmids for hCas9 and GFP marker were co-transfected into 447
HEK293 cells. GFP+ cells were sorted into 96-well plates at 1 cell/well 48 hours later. Each 448

20
clone was expanded, genomic DNA was amplified by PCR and genotyped by sequencing. Two 449
independent clones with NurRE deletion were compared to WT control. 450
451
Statistical Analyses 452
All statistical analyses were performed with Student's t-Test and represented as mean ± 453
standard deviation unless noted otherwise. For in vivo experiments with >=3 groups, statistical 454
analyses were performed with ANOVA followed by the post hoc Tukey pairwise comparisons. 455
The p values were designated as: *, p<0.05; **, p<0.01; ***, p<0.005; ****, p<0.001; n.s. non-456
significant (p>0.05). 457
458 459

21
Acknowledgments 460
We thank Dr. Orla Conneely (Baylor College of Medicine) for Nur77 KO mice and Flag-Nur77 461
plasmid; UT Southwestern Biostatistics Core for their assistance in our studies; Drs. Paul 462
Dechow and Jerry Feng (Baylor College of Dentistry) for assistance with μCT and 463
histomorphometry. Y. Wan is a Virginia Murchison Linthicum Scholar in Medical Research. This 464
work was in part supported by NIH (R01 DK089113, YW), CPRIT (RP130145, YW), DOD BCRP 465
Idea Award (W81XWH-13-1-0318, YW), March of Dimes (#6-FY13-137, YW), The Welch 466
Foundation (I-1751, YW) and UTSW Endowed Scholar Startup Fund (YW). The authors declare 467
that they have no financial conflict of interest. 468
469
470
471

22
References 472
Office of the Surgeon General (US) (2004). In Bone Health and Osteoporosis: A Report of the 473 Surgeon General (Rockville, MD). 474
Asagiri, M., Sato, K., Usami, T., Ochi, S., Nishina, H., Yoshida, H., Morita, I., Wagner, E.F., Mak, 475 T.W., Serfling, E., et al. (2005). Autoamplification of NFATc1 expression determines its 476 essential role in bone homeostasis. The Journal of experimental medicine 202, 1261-1269. 477
Bachmaier, K., Krawczyk, C., Kozieradzki, I., Kong, Y.Y., Sasaki, T., Oliveira-dos-Santos, A., 478 Mariathasan, S., Bouchard, D., Wakeham, A., Itie, A., et al. (2000). Negative regulation of 479 lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 403, 211-480 216. 481
Body, J.J., Lipton, A., Gralow, J., Steger, G.G., Gao, G., Yeh, H., and Fizazi, K. (2010). Effects of 482 denosumab in patients with bone metastases with and without previous bisphosphonate 483 exposure. Journal of bone and mineral research : the official journal of the American Society 484 for Bone and Mineral Research 25, 440-446. 485
Boquete-Castro, A., Gomez-Moreno, G., Calvo-Guirado, J.L., Aguilar-Salvatierra, A., and 486 Delgado-Ruiz, R.A. (2015). Denosumab and osteonecrosis of the jaw. A systematic analysis 487 of events reported in clinical trials. Clinical oral implants research. 488
Boyce, B.F., and Xing, L. (2007). The RANKL/RANK/OPG pathway. Current osteoporosis 489 reports 5, 98-104. 490
Chao, L.C., Soto, E., Hong, C., Ito, A., Pei, L., Chawla, A., Conneely, O.M., Tangirala, R.K., 491 Evans, R.M., and Tontonoz, P. (2013). Bone marrow NR4A expression is not a dominant 492 factor in the development of atherosclerosis or macrophage polarization in mice. Journal of 493 lipid research 54, 806-815. 494
Chao, L.C., Wroblewski, K., Zhang, Z., Pei, L., Vergnes, L., Ilkayeva, O.R., Ding, S.Y., Reue, K., 495 Watt, M.J., Newgard, C.B., et al. (2009). Insulin resistance and altered systemic glucose 496 metabolism in mice lacking Nur77. Diabetes 58, 2788-2796. 497
Chiusaroli, R., Sanjay, A., Henriksen, K., Engsig, M.T., Horne, W.C., Gu, H., and Baron, R. 498 (2003). Deletion of the gene encoding c-Cbl alters the ability of osteoclasts to migrate, 499 delaying resorption and ossification of cartilage during the development of long bones. 500 Developmental biology 261, 537-547. 501
Drake, M.T., Clarke, B.L., and Khosla, S. (2008). Bisphosphonates: mechanism of action and 502 role in clinical practice. Mayo Clinic proceedings 83, 1032-1045. 503
Hagiwara, K., Goto, T., Araki, M., Miyazaki, H., and Hagiwara, H. (2011). Olive polyphenol 504 hydroxytyrosol prevents bone loss. European journal of pharmacology 662, 78-84. 505
Hsu, H.C., Zhou, T., and Mountz, J.D. (2004). Nur77 family of nuclear hormone receptors. 506 Current drug targets. Inflammation and allergy 3, 413-423. 507
Hu, Y.W., Zhang, P., Yang, J.Y., Huang, J.L., Ma, X., Li, S.F., Zhao, J.Y., Hu, Y.R., Wang, Y.C., 508 Gao, J.J., et al. (2014). Nur77 decreases atherosclerosis progression in apoE(-/-) mice fed a 509 high-fat/high-cholesterol diet. PloS one 9, e87313. 510
Katsiki, M., Chondrogianni, N., Chinou, I., Rivett, A.J., and Gonos, E.S. (2007). The olive 511 constituent oleuropein exhibits proteasome stimulatory properties in vitro and confers life 512 span extension of human embryonic fibroblasts. Rejuvenation research 10, 157-172. 513
Khan, A.A., Sandor, G.K., Dore, E., Morrison, A.D., Alsahli, M., Amin, F., Peters, E., Hanley, 514 D.A., Chaudry, S.R., Lentle, B., et al. (2009). Bisphosphonate associated osteonecrosis of 515 the jaw. The Journal of rheumatology 36, 478-490. 516
Kim, J.H., Kim, K., Jin, H.M., Song, I., Youn, B.U., Lee, S.H., Choi, Y., and Kim, N. (2010). 517 Negative feedback control of osteoclast formation through ubiquitin-mediated down-518 regulation of NFATc1. The Journal of biological chemistry 285, 5224-5231. 519
Krzeszinski, J.Y., Wei, W., Huynh, H., Jin, Z., Wang, X., Chang, T.C., Xie, X.J., He, L., Mangala, 520 L.S., Lopez-Berestein, G., et al. (2014). miR-34a blocks osteoporosis and bone metastasis 521

23
by inhibiting osteoclastogenesis and Tgif2. Nature 512, 431-435. 522 Kuroda, Y., and Matsuo, K. (2012). Molecular mechanisms of triggering, amplifying and targeting 523
RANK signaling in osteoclasts. World journal of orthopedics 3, 167-174. 524 Lee, S.L., Wesselschmidt, R.L., Linette, G.P., Kanagawa, O., Russell, J.H., and Milbrandt, J. 525
(1995). Unimpaired thymic and peripheral T cell death in mice lacking the nuclear receptor 526 NGFI-B (Nur77). Science 269, 532-535. 527
Mullican, S.E., Zhang, S., Konopleva, M., Ruvolo, V., Andreeff, M., Milbrandt, J., and Conneely, 528 O.M. (2007). Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of 529 acute myeloid leukemia. Nature medicine 13, 730-735. 530
Nakajima, A., Sanjay, A., Chiusaroli, R., Adapala, N.S., Neff, L., Itzsteink, C., Horne, W.C., and 531 Baron, R. (2009). Loss of Cbl-b increases osteoclast bone-resorbing activity and induces 532 osteopenia. Journal of bone and mineral research : the official journal of the American 533 Society for Bone and Mineral Research 24, 1162-1172. 534
Nakashima, T., Hayashi, M., Fukunaga, T., Kurata, K., Oh-Hora, M., Feng, J.Q., Bonewald, L.F., 535 Kodama, T., Wutz, A., Wagner, E.F., et al. (2011). Evidence for osteocyte regulation of bone 536 homeostasis through RANKL expression. Nature medicine 17, 1231-1234. 537
Novack, D.V., and Teitelbaum, S.L. (2008). The osteoclast: friend or foe? Annual review of 538 pathology 3, 457-484. 539
Palumbo-Zerr, K., Zerr, P., Distler, A., Fliehr, J., Mancuso, R., Huang, J., Mielenz, D., Tomcik, M., 540 Furnrohr, B.G., Scholtysek, C., et al. (2015). Orphan nuclear receptor NR4A1 regulates 541 transforming growth factor-beta signaling and fibrosis. Nature medicine 21, 150-158. 542
Philips, A., Lesage, S., Gingras, R., Maira, M.H., Gauthier, Y., Hugo, P., and Drouin, J. (1997). 543 Novel dimeric Nur77 signaling mechanism in endocrine and lymphoid cells. Molecular and 544 cellular biology 17, 5946-5951. 545
Popovic, D., Vucic, D., and Dikic, I. (2014). Ubiquitination in disease pathogenesis and 546 treatment. Nature medicine 20, 1242-1253. 547
Richardson, P.G., Sonneveld, P., Schuster, M.W., Irwin, D., Stadtmauer, E.A., Facon, T., 548 Harousseau, J.L., Ben-Yehuda, D., Lonial, S., Goldschmidt, H., et al. (2005). Bortezomib or 549 high-dose dexamethasone for relapsed multiple myeloma. The New England journal of 550 medicine 352, 2487-2498. 551
Takayanagi, H., Kim, S., Koga, T., Nishina, H., Isshiki, M., Yoshida, H., Saiura, A., Isobe, M., 552 Yokochi, T., Inoue, J., et al. (2002). Induction and activation of the transcription factor 553 NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. 554 Developmental cell 3, 889-901. 555
Tontonoz, P., Cortez-Toledo, O., Wroblewski, K., Hong, C., Lim, L., Carranza, R., Conneely, O., 556 Metzger, D., and Chao, L.C. (2015). The orphan nuclear receptor Nur77 is a determinant of 557 myofiber size and muscle mass in mice. Molecular and cellular biology. 558
Wan, Y., Chong, L.W., and Evans, R.M. (2007). PPAR-gamma regulates osteoclastogenesis in 559 mice. Nat Med 13, 1496-1503. 560
Wei, W., Dutchak, P.A., Wang, X., Ding, X., Bookout, A.L., Goetz, R., Mohammadi, M., Gerard, 561 R.D., Dechow, P.C., Mangelsdorf, D.J., et al. (2012). Fibroblast growth factor 21 promotes 562 bone loss by potentiating the effects of peroxisome proliferator-activated receptor gamma. 563 Proc Natl Acad Sci U S A 109, 3143-3148. 564
Wei, W., Motoike, T., Krzeszinski, J.Y., Jin, Z., Xie, X.J., Dechow, P.C., Yanagisawa, M., and 565 Wan, Y. (2014). Orexin regulates bone remodeling via a dominant positive central action and 566 a subordinate negative peripheral action. Cell Metab 19, 927-940. 567
Wei, W., Wang, X., Yang, M., Smith, L.C., Dechow, P.C., Sonoda, J., Evans, R.M., and Wan, Y. 568 (2010). PGC1beta mediates PPARgamma activation of osteoclastogenesis and 569 rosiglitazone-induced bone loss. Cell Metab 11, 503-516. 570
Wei, W., Zeve, D., Suh, J.M., Wang, X., Du, Y., Zerwekh, J.E., Dechow, P.C., Graff, J.M., and 571 Wan, Y. (2011). Biphasic and dosage-dependent regulation of osteoclastogenesis by beta-572

24
catenin. Mol Cell Biol 31, 4706-4719. 573 Xiong, J., Onal, M., Jilka, R.L., Weinstein, R.S., Manolagas, S.C., and O'Brien, C.A. (2011). 574
Matrix-embedded cells control osteoclast formation. Nature medicine 17, 1235-1241. 575 Zhao, Y., and Bruemmer, D. (2010). NR4A orphan nuclear receptors: transcriptional regulators 576
of gene expression in metabolism and vascular biology. Arteriosclerosis, thrombosis, and 577 vascular biology 30, 1535-1541. 578
Zupan, J., Jeras, M., and Marc, J. (2013). Osteoimmunology and the influence of pro-579 inflammatory cytokines on osteoclasts. Biochemia medica 23, 43-63. 580
581 582

25
FIGURE LEGEND 583
Figure 1. Nur77 Deletion Increases Osteoclastogenesis and Bone Resorption. 584
(A) A schematic diagram of ex vivo bone marrow osteoclast differentiation. 585
(B) Expression of Nur77, Nurr1 and Nor-1 encoding genes (Nr4a1, Nr4a1, Nr4a3) during a time 586
course of RANKL-induced osteoclast differentiation (n=3 mice). Nfatc1 mRNA level was similar 587
to Nr4a1 mRNA level on day 2-3. 588
(C-D) Osteoclast differentiation was enhanced in Nur77 KO cultures compared to WT control 589
cultures. (C) Expression of osteoclast differentiation markers on day 3 (n=4 mice). (D) 590
Representative images of the TRAP-stained osteoclast differentiation cultures. Mature 591
osteoclasts were identified as multinucleated TRAP+ (purple) cells on day 9. Scale bar, 25μm. 592
Quantification of osteoclast size, osteoclast number per area, and osteoclast resorptive activity 593
by calcium release from bone plate to culture medium are shown (n=4 mice in triplicate 594
cultures). Oc, osteoclast. 595
(E) Osteoclast apoptosis was unaltered, quantified by the expression of apoptosis genes on day 596
9 of osteoclast differentiation cultures (n=4). 597
(F) Osteoblast differentiation was unaltered in Nur77 KO cultures, measured by the expression 598
of osteoblast markers (n=4). 599
(G-O) Nur77 KO mice exhibited bone loss. Tibiae from Nur77 KO mice or WT littermate controls 600
(3 month old, male, n=6) were analyzed by μCT. (G) Representative images of the trabecular 601
bone of the tibial metaphysis (top) (scale bar, 10μm) and the entire proximal tibia (bottom) (scale 602
bar, 1mm). (H-O) Quantification of trabecular bone volume and architecture. (H) BV/TV, bone 603
volume/tissue volume ratio. (I) BS, bone surface. (J) BS/BV, bone surface/bone volume ratio. 604
(K) Tb.N, trabecular number. (L) Tb.Th, trabecular thickness. (M) Tb.Sp, trabecular separation. 605
(N) Conn.D., connectivity density. (O) SMI, structure model index. 606
(P) Serum CTX-1 bone resorption marker was increased (3 month old, male, n=6). 607
(Q) Serum P1NP bone formation marker was unaltered (3 month old, male, n=6). 608

26
(R-S) Bone histomorphometry (3 month old, male, n=6). 609
(R) Quantification of osteoclast surface (Oc.S/B.S) and osteoclast number (Oc.N/B.Ar). 610
(S) Quantification of osteoblast surface (Ob.S/B.S) and osteoblast number (Ob.N/B.Ar). B.S, 611
bone surface; B.Ar, bone area. Error bars, SD. 612
613
Figure 2. Nur77 Regulation of Bone Resorption is Intrinsic to the Hematopoietic Lineage. 614
(A-F) Bone marrow transplantation. Bone marrow cells from 2-month-old male donor mice were 615
transplanted into 2-month-old irradiated male recipient male mice (n=5), and analyzed 3 months 616
later at 5 month old. 617
(A-C) Transplantation of Nur77 KO donor bone marrow cells into WT recipients conferred 618
elevated bone resorption compared to WT control donor bone marrow cells. (A) A schematic 619
diagram. (B) Serum CTX-1. (C) Serum P1NP. 620
(D-F) Transplantation of WT donor bone marrow cells into Nur77 KO recipients rescued the 621
bone resorption to a level similar to the WT control recipients. (D) A schematic diagram. (E) 622
Serum CTX-1. (F) Serum P1NP. 623
(G) Expression of RANKL and OPG, as well as RANKL/OPG ratio, in Nur77 KO ex vivo 624
osteoblast differentiation cultures compared to WT control cultures (n=3). 625
(H) Expression of Rankl and Opg, as well as Rankl/Opg ratio, in osteocytes from femur shaft in 626
Nur77 KO mice compared to WT control mice (n=3). 627
(I) Expression of pro-osteoclastogenic cytokines in bone marrow cells from Nur77 KO mice 628
compared to WT control mice (n=3). Error bars, SD. 629
630
Figure 3. Nur77 Inhibits Osteoclast Differentiation by Promoting NFATc1 Degradation. 631
(A) NFATc1 protein levels during a time course of osteoclast differentiation from the bone 632
marrow cells of Nur77 KO mice or WT control mice. Left, representative western blot image. 633
Right, quantification of NFATc1/β-actin ratio. 634

27
(B) c-Jun phosphorylation and IκBα degradation post RANKL treatment in osteoclast 635
differentiation cultures from the bone marrow cells of Nur77 KO mice or WT control mice. P-c-636
Jun, phosphorylated c-Jun; t-c-Jun, total c-Jun. 637
(C) Effects of MG132 on NFATc1 protein levels in Nur77 KO or WT bone marrow osteoclast 638
differentiation cultures. Cells were treated with 25μM MG132 for 6 hours 3 days after RANKL 639
stimulation. Left, representative western blot image. Right, quantification of NFATc1/β-actin ratio. 640
(D) Effects of Nur77 over-expression on NFATc1 protein and mRNA levels. HEK293 cells were 641
transfected with NFATc1, together with either Flag-Nur77 or GFP control. Left, representative 642
western blot image with quantification of NFATc1/β-actin ratio. Right, relative Nfatc1 mRNA. 643
(E) Effects of Nur77 over-expression on NFATc1 transcriptional output (n=3). HEK293 cells were 644
transfected with NFATc1 and its luciferase reporter, together with increasing amount of Nur77. 645
(F) MG132 abolished the effects of Nur77 over-expression on NFATc1 transcriptional output 646
(n=3). HEK293 cells were treated with MG132 (25μM) 1 day after transfection for 6 hours before 647
harvesting. All data are representative of at least three experiments. Error bars, SD. 648
649
Figure 4. Nur77 Transcriptionally Up-regulates E3 Ligase Cbl-b. 650
(A) Cblb expression during a time course of osteoclast differentiation from the bone marrow 651
cells of Nur77 KO mice or WT control mice (n=3). 652
(B) Nur77 over-expression increased Cblb mRNA in a DNA-binding-dependent manner. 653
HEK293 cells were transfected with vector control, WT Nur77 or a mutant Nur77 with a deletion 654
of the DNA binding domain (DBD) (n=3). 655
(C) Nur77 activated Cbl-b promoter via NurRE. HEK293 cells were transfected with Nur77, 656
together with a luciferase vector control or a luciferase reporter driven by 1Kb Cbl-b promoter 657
containing either a WT NurRE or a mutant NurRE (n=3). Inset shows the mutations in the two 658
mutant reporters. 659
(D) ChIP assay of Nur77 binding and H3K4me3 levels at the endogenous Cbl-b promoter. 660

28
HEK293 cells were transfected with Flag-Nur77, Nur77 binding were detected with anti-Flag 661
antibody and compared with IgG control antibody (n=3). 662
(E-H) CRISPR/Cas9 deletion of NurRE in the endogenous Cbl-b promoter abolished Nur77 663
regulation of Cbl-b and NFATc1. (E) A schematic representation of CRISPR/Cas9 gRNAs and 664
their target locus in the Cbl-b promoter. (F-H) Effects of Nur77 over-expression on Cbl-b mRNA 665
(F) (n=4), NFATc1 protein (G) and NFATc1 transcriptional output (H) (n=3) in two independent 666
HEK293 CRISPR mutant clones and WT controls. Error bars, SD. 667
668
Figure 5. NFATc1 Induces Nur77 Transcription to Elicit a Self-Limiting Loop. 669
(A) NFATc1 over-expression increased Nur77 mRNA. Mouse myoblast cell line C2C12 or 670
human embryonic kidney cell line HEK293 were transfected with NFATc1 or GFP control. 671
(B) NFATc1 inhibition by Cyclosporin A dosage-dependently decreased Nur77 mRNA. 672
Osteoclast differentiation cultures were treated with Cyclosporin A on day 2 for 24 hours (n=4). 673
(C) NFATc1 over-expression enhances Nur77 promoter activity. HEK293 cells were transfected 674
with a luciferase vector control or a luciferase reporter driven by a 0.8Kb Nur77 promoter, 675
together with NFATc1 or GFP control (n=3). 676
(D) ChIP assay of NFATc1 binding and H3K4me3 level at the endogenous Nur77 promoter in 677
RAW264.7 mouse macrophage cell line with or without 2 day RANKL stimulation. 678
(E) A working model of an NFATc1 self-limiting loop in which NFATc1 elicits its own degradation 679
by inducing Nur77 and consequently Cbl-b to resolve NFATc1 signaling. Error bars, SD. 680
681
Figure 6. Cbl-b Deletion Abolishes Nur77 Regulation of Osteoclastogenesis and Bone 682
Resorption. 683
Nur77 Cblb DKO mice were compared with Nur77 KO mice, Cblb KO mice and WT littermate 684
control mice (3 month old, male, n=4). (A-E) Ex vivo bone marrow osteoclast differentiation (n=4 685
mice in triplicate cultures). (A) Representative images of the TRAP-stained osteoclast 686

29
differentiation cultures. Mature osteoclasts were identified as multinucleated TRAP+ (purple) 687
cells on day 9. Scale bar, 25μm. (B) Osteoclast size. (C) Osteoclast number per area. (D) 688
Osteoclast resorptive activity by calcium release from bone plate to culture medium. (E) 689
Expression of osteoclast differentiation markers on day 3. (F) ELISA analysis of serum CTX-1 690
bone resorption marker (n=4). (G-K) μCT analysis of trabecular bone parameters (n=4). (G) 691
BV/TV, bone volume/tissue volume ratio. (H) BS, bone surface. (I) Tb.N, trabecular number. (J) 692
Tb.Sp, trabecular separation. (K) Conn.D., connectivity density. Statistical analyses were 693
performed with ANOVA followed by the post hoc Tukey pairwise comparisons. Error bars, SD. 694
Conn
. D. (
1/m
m3)
WT Nur77 KO0
100
200
300
**
BA
Rel
ativ
e m
RNA
d0 d1 d2 d30.00
0.01
0.02
0.03 Nr4a1 (Nur77)
Nr4a3 (Nor-1)Nr4a2 (Nurr1)
Nfatc1
Rel
ativ
e m
RNA
on
d3
Trap Ctsk Calcr Car2 Nfatc10
1
2
3
4Nur77 KOWT
*****
* * *
C
G
BV/
TV
WT Nur77 KO0.0
0.1
0.2
0.3
**
BS (m
m2)
WT Nur77 KO0
10
20
30
40
*
BS/
BV
(1/m
m)
WT Nur77 KO0
20
40
60
80
*
Tb.N
(1/m
m)
WT Nur77 KO01
234
56
7
*
Tb.T
h (m
m)
WT Nur77 KO0.00
0.01
0.02
0.03
0.04
0.05
*
Tb.S
p (m
m)
WT Nur77 KO0.00
0.05
0.10
0.15
0.20
0.25**
SMI
WT Nur77 KO0.0
0.5
1.0
1.5
2.0
2.5
*
Seru
m C
TX-1
(ng/
ml)
WT Nur77 KO0
20
40
60
80
****
Seru
m P
1NP
(ng/
ml)
WT Nur77 KO0
5
10
15
20
n.s.
WT Nur77 KO
Trab
ecul
arW
hole
Tib
ia
K L M
H I J
N O P Q
Figure 1. Nur77 Deletion Increases Osteoclastogenesis and Bone Resorption
QuiescentProgenitors
ProliferatingPrecursors
MatureOsteoclast
Day -3 – 0MCSF
Day 0 - 4RANKL+MCSF
D
Rela
tive
mRN
A
WT Nur77KO0
50
100
150
n.s.
Col1a1
-ObDiff+ObDiff
WT Nur77KO0.0
0.1
0.2
0.3 n.s.Osteocalcin
E
R S
Oc.
S/B
.S (%
)
WT Nur77 KO0
5
10
15
20
****
Oc.
N/B
.Ar (
%)
WT Nur77 KO0
5
10
15****
Ob.
S/B
.S (%
)
WT Nur77 KO0
1
2
3
4
n.s.
Ob.
N/B
.Ar (
%)
WT Nur77 KO0
2
4
6
8
10n.s.
Li Fig. 1
WT Nur77 KOTRAP
Oc size: 27 ± 8 114 ± 22 ****Oc #/area: 22 ± 3 32 ± 2 ***Ca++ ( M): 26 ± 2 35 ± 8 **
Bax Bad Fas Bcl-2 Bcl-60.00
0.05
0.10
0.15
Rel
ativ
e m
RNA
WTNur77 KOn.s.
n.s.
n.s. n.s.
n.s.
F

Marrow transplant
Marrow transplant
WT
Nur77 KO
A
WT
Marrow transplant
Marrow transplant
Seru
m C
TX-1
(ng/
ml)
0
10
20
30
40
50 **
Donor WT Nur77KO
Recipient WT WT
Seru
m P
1NP
(ng/
ml)
0
5
10
15
20
25n.s.
Donor WT Nur77KO
Recipient WT WT
Seru
m C
TX-1
(ng/
ml)
0
10
20
30
40
50
n.s.
Donor WT WT
Recipient WT Nur77KO
Seru
m P
1NP
(ng/
ml)
0
2
4
6
8
10
n.s.
Donor WT WT
Recipient WT Nur77KO
CB
D FE
Li Fig. 2
Rel
ativ
e m
RNA
Rankl Opg Rankl/Opg0
1
2Nur77 KOWT
Osteoblast Culture
n.s. n.s.n.s.
Rel
ativ
e m
RNA
Rankl Opg Rankl/Opg0
2
4
6
8Nur77 KOWT
Osteocyte (Femur Shaft)
n.s.
*
n.s.
Rel
ativ
e m
RNA
Tnfa IL1 IL60
1
2Nur77 KOWT
Marrow Cells
n.s.
n.s.n.s.
G IH
Figure 2. Nur77 Regulation of Bone Resorption is Intrinsic to the Hematopoietic Lineage

Li Fig. 3
NFAT
c1/
-Act
in P
rote
in
d1 d2 d3 d40
1
2Nur77 KOWT
Rela
tive
Nfat
c1 m
RNA
GFP Nur770
2
4
6
8
10 n.s.lu
c/-g
al
0
1000
2000
3000
***
***
*
n.s.
NFATc1Nur77
--
+-
+0.5
+1.5
+4.5
NFATc1 Transcriptional Activity
NFAT
c1/
-Act
in P
rote
in
Veh MG1320
1
2
3 WTNur77 KO
C
B
D
E
WT Nur77KO WT Nur77KO
Veh MG132
NFATc1
-actin
1 2 3 4 1 2 3 4
NFATc1
-actin
Day
WT Nur77 KOA
p-c-Jun
0 15 30 0 15 30 min
t-c-Jun
I B
-actin
WT Nur77 KO
NFATc1+ Nur77
NFATc1
-actin
NFATc1+ GFP
3.73±0.17 2.95±0.01 *NFATc1/ -actin
Figure 3. Nur77 Inhibits Osteoclast Differentiation by Promoting NFATc1 Degradation
F
Veh MG1320
500
1000
1500
2000
2500
NFATc1 Transcriptional Activity
luc/
-gal
VectorNFATc1NFATc1 + Nur77
*** ***n.s.
*****
n.s.

0
1
2
luc/
-gal
Cblb-luc-WTCblb-luc-WT**** ****
***Cblb-luc-Mut1Cblb-luc-Mut2
Nur77 - + + +
Rel
ativ
e Cb
lb m
RNA
d0 d2 d40.0
0.5
1.0
1.5
2.0
2.5Nur77 KOWT
***
353
270
265
335
AF1 DBD LBD
Nur77-WT
Nur77- DBD
Rel
ativ
e Cb
lb m
RNA
Vector Nur77-WT Nur77- DBD0.000
0.002
0.004
0.006
0.008
n.s.
****
Li Fig. 4
A
C
B
WT AAATATCAgaacatcTGAGATGAMut1 AACCAGCAgaacatcTGAGATGAMut2 AAATATCAgaacatcTGCGACGC
IgG Flag-Nur770.000
0.005
0.010
0.015
0.020
0.025
ChIP
/10%
Inpu
t ****
IgG H3K4Me30.0
0.2
0.4
0.6
0.8
****
D
E F
5’……aacCCAatgtaAAATATCAgaacatcTGAGATGACCAagct……3’
3’……ttgGGTtacatTTTATAGTcttgtagACTCTACTGGCtcga……5’
PAM PAMNurRE NurRE
gRNA-1 + gRNA-2
Rel
ativ
e Cb
lb m
RNA
Nur77 - + - + - + - +
WT1 WT2 Mut1 Mut2
NFATc1
-actin
NFATc1/actin 1.6 0.8 1.9 1.1 3.3 2.9 2.1 1.8
51% 43% 12% 13%Reduction by Nur77
Fold
(Luc
/-g
al)
G H
Figure 4. Nur77 Transcriptionally Up-regulates E3 Ligase Cbl-b

1. Autoamplify2. Turn on genes for osteoclastogeneiss
3. Autoresolve by turning on Nur77-Cbl-b loop
NFATc1 Nur77
Cbl-b
Ca2+
cFoscJun
RANKL
Activation Resolution
Li Fig. 5
GFP NFATc10.00
0.05
0.10
0.15
0.20
0.25Re
lativ
e Nu
r77
mRN
A ****
C2C12
GFP NFATc10.000
0.001
0.002
0.003**
HEK293A CB
Veh 0.1 M 1 M0.000
0.001
0.002
0.003
0.004
0.005
Rela
tive
Nur7
7 m
RNA
****
Osteoclast
CsA
*
GFP NFATc10.0
0.1
0.2
0.3
0.4
luc/
-gal
**
Nur77-LucVector
NFATc1 H3K4Me30
5
10
15
20
25
Fold
(ChI
P/10
% In
put) -RANKL
+RANKL**
****
D E
Figure 5. NFATc1 Induces Nur77 Transcription to Elicit a Self-Limiting Loop

Li Fig. 6
A B
E
Figure 6. Cblb Deletion Abolishes Nur77 Regulation of Osteoclastogenesis and Bone Resorption
WT Nur77 KOTRAPW
TC
blb
KO
Rela
tive
Trap
mR
NA
WT
Nur77 KO
Cblb KO
Nur77 Cblb DKO
0
2
4
6
*** n.s.
*n.s.
ANOVA p<0.05
Rela
tive
Ctsk
mR
NA
WT
Nur77 K
O
Cblb KO
Nur77 Cblb DKO
0.00
0.05
0.10
0.15
**n.s.
*n.s.
ANOVA p<0.05
BV/
TV
WT
Nur77 KO
Cblb KO
Nur77 Cblb DKO
0.0
0.1
0.2
0.3*
n.s.
*
n.s.
ANOVA p<0.05
DBS
(mm
2)
WT
Nur77 KO
Cblb KO
Nur77 Cblb DKO
0
10
20
30
40*
n.s.
*
n.s.
ANOVA p<0.05
Tb.N
(1/m
m)
WT
Nur77 K
O
Cblb KO
Nur77 C
blb DKO0
2
4
6
8
*
n.s.
*
n.s.
ANOVA p<0.05
Tb.S
p (m
m)
WT
Nur77 KO
Cblb KO
Nur77 Cblb DKO
0.0
0.1
0.2
0.3
0.4
0.5
*n.s.
*n.s.
ANOVA p<0.05
Con
n. D
. (1/
mm
3)
WT
Nur77 K
O
Cblb KO
Nur77 C
blb DKO0
100
200
300 *
n.s.
*
n.s.
ANOVA p<0.05
C
Calc
ium
(M
)
WT
Nur77 KO
Cblb KO
Nur77 Cblb DKO
0
20
40
60
80
***n.s.
***n.s.
ANOVA p<0.05
Oc
size
WT
Nur77 KO
Cblb KO
Nur77 Cblb DKO
0
100
200
300
**** n.s.
****n.s.
ANOVA p<0.05
Oc
# / a
rea
WT
Nur77 KO
Cblb KO
Nur77 Cblb DKO
0
20
40
60
80
100
**** n.s.
****n.s.
ANOVA p<0.05
F G
H I J K
Seru
m C
TX-1
(ng/
ml)
WT
Nur77 KO
Cblb KO
Nur77 Cblb DKO
0
20
40
60
80
* n.s.
*n.s.
ANOVA p<0.05