novel thioredoxin inhibitors paradoxically increase hypoxia
TRANSCRIPT

NovelThioredoxin Inhibitors Paradoxically Increase Hypoxia-Inducible Factor-A Expression but Decrease FunctionalTranscriptional Activity, DNA Binding, and DegradationDylan T. Jones,1Christopher W. Pugh,2 Simon Wigfield,1Malcolm F.G. Stevens,3 and Adrian L. Harris1
Abstract Purpose:Hypoxia-inducible factor-a (HIF-a) is a transcription factor that regulates the responseto hypoxia. HIF-a protein is found at high levels in many cancers, and the redox protein thiore-doxin-1 (Trx-1) increases both aerobic and hypoxia-induced HIF-a. Therefore, Trx-1and HIF-aare attractive molecular targets for novel cancer therapeutics.Experimental Design:We investigated whether two novel anticancer drugs AJM290 andAW464 (quinols), which inhibitTrx-1function, can inhibit the HIF pathway.Results:Treatment of several cancer cell lines with AJM290 orAW464 prevented the hypoxia-induced increase of vascular endothelial growth factor (VEGF) at subtoxic concentrations.AJM290 and AW464 also decreasedVEGF in pVHLmutant renal cell carcinoma cells that consti-tutively overexpress HIF-a protein. They surprisingly up-regulated HIF-a expression in breastcancer cell lines in normoxia and hypoxia as well as in pVHL mutant cells. In the MDA-MB-468breast cancer cell line, the compounds inhibited RNA and protein expression of the HIF-a targetgenes, carbonic anhydrase IX,VEGF, and BNIP3, concordantly with HIF-a up-regulation. Bothcompounds specifically inhibited HIF-a-dependent induction of hypoxia regulatory element-luciferase and HIF-1ahypoxia regulatory element-DNA binding. To analyze the HIF-1adomaininhibited byAJM290, we transfected cells with plasmids expressing a fusion protein of Gal linkedto HIF-1aor HIF-1aCOOH-terminal transactivation domain (CAD) with a Gal4-responsive lucif-erase reporter gene. AJM290 inhibitedboth the full-lengthHIF-1aandHIF-1aCAD transcriptionalactivity.Conclusions:AJM290 and AW464 are inhibitors of HIF-1aCAD transcription activity and DNAbinding, but they also inhibit degradation of HIF, in contrast to other Trx inhibitors.
Hypoxia-inducible factor (HIF) is an a,h heterodimerictranscription factor that directs a broad range of responses inhypoxic cells (1). Both proteins are members of the basic helix-loop-helix superfamily of transcription factors in which thebasic helix-loop-helix domains bind to DNA (2). HIF-1h is aconstitutive nuclear localized subunit that binds to availableHIF-a (3). To date, three HIF-a isoforms have been described,with the best characterized being HIF-1a and HIF-2a. In the
presence of oxygen, two prolyl sites within a central degradationdomain of HIF-a are hydroxylated by a set of closely related Fe2+
and 2-OG-dependent dioxygenases (PHD1-3), which leads toHIF-a degradation via the pVHL E3 ubiquitin ligase complexand the 26S proteasome (4). Limiting oxygen levels or the avail-ability of Fe2+ with iron chelators (5) allows HIF-a to escapeproteolysis. In the nucleus, HIF a,h heterodimer interacts withcoactivators, such CBP/p300, and becomes transcriptionallyactive (6). On activation, the HIF-ah complex binds to targetgenes at sites containing the core recognition sequence5¶-RCGTG-3¶, also known as the hypoxia regulatory element(HRE; ref. 7), which finally leads to up-regulation ofgenes involved in angiogenesis, glucose metabolism, and pHregulation (1).The HIF transcription cascade has been shown to contribute
to tumor progression and metastasis and plays an importantpart in the malignant phenotype (8–10). HIF-a is found atincreased levels in a wide variety of human primary tumorscompared with corresponding normal tissue and increasesangiogenesis and other properties that promote increasedvascularity and tumor progression (8–10). Besides physiologichypoxia, genetic abnormalities frequently detected in humancancers, which include key oncogenes (HER-2, FRAP, H-RAS,and SRC) and tumor suppressor genes (pVHL, p53, and PTEN),are also associated with induction of HIF-a activity andexpression of HIF-a-inducible genes (11–15).
Human Cancer Biology
Authors’ Affiliations: 1Cancer Research UK Growth Factor Group,WeatherallInstitute of Molecular Medicine, John Radcliffe Hospital, University of Oxford; 2TheWellcomeTrust Centre for Human Genetics, Oxford, United Kingdom; and 3CancerResearch UK Experimental Cancer Chemotherapy Research Group, University ofNottingham, Nottingham, United KingdomReceived11/1/05; revised 3/15/06; accepted 5/4/06.Grant support: Cancer Research UK and Euroxy EU 6th Framework Grant.The costs of publication of this article were defrayed in part by the payment of pagecharges.This article must therefore be hereby marked advertisement in accordancewith18 U.S.C. Section1734 solely to indicate this fact.Note: AJM290 and AW464 have been licensed to Pharminox and M. Stevens is ashare holder in this company.Requests for reprints: Adrian L. Harris, Cancer Research UK Growth FactorGroup, Weatherall Institute of Molecular Medicine, John Radcliffe Hospital,University of Oxford, Headington, Oxford OX3 9DS, United Kingdom. Phone:44-1865226184; Fax: 44-1865226179; E-mail: [email protected].
F2006 American Association for Cancer Research.doi:10.1158/1078-0432.CCR-05-2380
www.aacrjournals.orgClin Cancer Res 2006;12(18) September15, 2006 5384
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

Due to the involvement of HIF-a in tumor progression andangiogenesis, HIF-a is a promising molecular target fordevelopment of cancer therapeutics (16–18). Thioredoxin-1(Trx-1), which is a ubiquitously expressed small redox proteinwith a conserved catalytic site (19), has been shown to regulatethe activity of enzymes, such as apoptosis signal-regulatingkinase-1 and protein kinases C a, y, q, and ~ in a redox-dependent manner (20, 21). Trx-1 also increases the DNAbinding of redox-sensitive transcription factors, which includesnuclear factor-nB and p53 (22, 23). It has been recentlyreported that increased expression of Trx-1 in cancer cellsincreases HIF-a protein levels and transactivating activity underboth normoxic and hypoxic conditions (24). Trx-1 expression ishighly expressed in several human primary cancers, includingcolon, cervix, lung, pancreatic, liver, colorectal, and squamouscell cancer (25–30).Several inhibitors of Trx pathway have been developed.
PX-12 (Trx-1 inhibitor) and pleurotin and PX478 (Trx-1reductase inhibitors) have already been shown to down-regulate hypoxia-induced increase and constitutive expressionof HIF-1a and HIF-1a transcription factor activity (31, 32).Here, we investigated the effects of novel Trx-1 inhibitorsAJM290 (indole-substituted quinol) and AW464 (benzothia-zole-substituted quinol) on HIF-1a and its downstream targets(Fig. 1A). Both compounds have been shown to have in vitroantitumor activity against colon, renal, and breast cancer celllines and in vivo antitumor activity in mice bearing breast,colon, and renal xenografts (33–36).
Materials and Methods
Cell culture and materials. Human breast cancer cell lines MDA-MB-468 and MDA-MB-231 and melanoma cell line MDA-MB-435 weremaintained in DMEM. The pVHL-deficient RCC4 and 786-0 renalcarcinoma cell lines and their counterpart containing a stably trans-fected pVHL gene were cultured in a-MEM and DMEM, respectively,and maintained in selection with 500 Ag/mL G418. Both a-MEM andDMEM were supplemented with 10% fetal bovine serum, 2 mmol/LL-glutamine, 50 IU/mL penicillin, and 50 Ag/mL streptomycin sulfate.Hypoxic exposures (0.1% O2, 5% CO2, and balance N2) were done in aHeto-Holten CellHouse 170 incubator (RS Biotech, Irvine, Scotland).Cell lines were obtained from the Cancer Research UK. Quinolcompounds AJM290 and AW464 were provided by M.F.G. Stevensand proteasome inhibitor Z-Leu-Leu-Leu-Ala (MG132) was from Sigma(Gillingham, United Kingdom).Viability assay. Cells were seeded at 2.5 � 103 to 10 � 103 per well
100 AL for 16-hour survival assay and 2.5 � 103 per well 100 AL for48-hour survival assay in 96-well plates 24 hours before experimentaltreatments. Cells were treated with compounds at 0.01 to 250 Amol/Lin triplicates and further incubated in hypoxia or normoxia for 16 or48 hours. Cell viability was measured by measuring metabolicconversion (by viable cells) of the dye MTS Cell Titer 96 AQueousOne Solution Cell Proliferation Assay (Promega, Southampton, UnitedKingdom). In each well of a 96-well plate, 20 AL MTS was added, andplates were incubated for 2 to 4 hours in a cell culture incubator. MTSassay results were read in a 96-well format plate reader by measuringabsorbance at 490 nm.Vascular endothelial growth factor ELISA. Vascular endothelial
growth factor (VEGF) secretion into the culture medium was measured
Fig.1. Chemical structures of AJM290 andAW464 (A), effect of AJM290 and AW464on MDA-MB-468 cell viability (B), andVEGF expression (C) following16 hours oftreatment innormoxic or hypoxic conditions.
New Thioredoxin Inhibitors Inhibit Hypoxia Pathway
www.aacrjournals.org Clin Cancer Res 2006;12(18) September15, 20065385
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

using DuoSet ELISA Development Human VEGF Immunoassay (R&DSystems, Minneapolis, MN) and 3,3¶,5,5¶-Tetramethylbenzidine LiquidSubstrate System for ELISA (Sigma) by following the manufacturers’protocol. VEGF ELISA assay results were read in a 96-well format platereader by measuring absorbance at 450 nm with correction at 540 nm.HRE reporter assay. Cells were transfected with 2 Ag/mL HIF-1
reporter plasmid or pGL3 promoter control plasmid and 0.02 Ag/mLphRL-cytomegalovirus (CMV) Renilla luciferase plasmid using Fugene 6eukaryote transfection reagent kit (Roche, Welwyn Garden City, UnitedKingdom). The pGL3 firefly luciferase HIF-1 reporter plasmidscontained the HRE from phosphoglycerate kinase or carbonicanhydrase IX (CA-IX). The pGL3 SV40 promoter vector was used forcontrol and phRL-CMV Renilla luciferase plasmid was used as controlfor transfection efficiency (Promega). Twenty-four hours later, cellswere exposed to hypoxia for 16 hours as described previously withAJM290 or AW464. Firefly and Renilla luciferase activity was measuredusing the Dual-Luciferase Reporter Assay System (Promega) accordingto the manufacturer’s instructions.
Western blotting. Whole-cell extracts were made by homogenizingcells in lysis buffer (6.2 mol/L urea, 10% glycerol, 5 mmol/L DTT, 1%SDS + protease inhibitors). Whole-cell extract was separated by 8%or 10% SDS-PAGE and transferred to polyvinylidene difluoridemembrane. Primary antibodies used were mouse anti-HIF-1a, rabbitanti-HIF-2a (BD Transduction Laboratories, Lexington, KY), rabbitanti-hydroxylated-HIF-1a (provided by The Wellcome Trust Centre forHuman Genetics, Oxford, United Kingdom), mouse anti-CA-IX M75monoclonal antibody, mouse anti-BNIP3 (Sigma), mouse anti-Hsp70(Abcam, Cambridge, United Kingdom), mouse anti-Hsp90 (Stressgen,San Diego, CA), and mouse anti-h-tubulin monoclonal antibody(Sigma). Immunoreactivity was visualized with horseradish peroxi-dase– linked goat anti-mouse or anti-rabbit serum and chemilu-minescence.RNA extraction and reverse transcription. Cells were rinsed with PBS
and drained thoroughly. RNA was extracted from the cells using thesolution D method described by Chomczynski and Sacchi (37). Thequantity and quality of RNA extracted were assessed using NanoDrop
Fig. 2. Effect of AJM290 and AW464 oncell viability andVEGF IC50 of hypoxicand pVHL mutant cell lines (A) and viabilityIC50 of hypoxic and normoxic cell linesfollowing16 (B) and 48 (C) hours oftreatment. Results were plotted in triplicate.Viability IC50,VEGF IC50, and 50% VEGFhypoxic to normoxic values were calculatedusing Graph Prism. Bars, SE.
Human Cancer Biology
www.aacrjournals.orgClin Cancer Res 2006;12(18) September15, 2006 5386
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

ND 1000 Spectrophotometer (NanoDrop Technologies, Wilmington,DE) and the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto,CA), respectively. RNA samples were stored at �80jC. cDNA wassynthesized by reverse transcribing RNA using the High-Capacity cDNAArchive kit (Applied Biosystems, Foster City, CA) following themanufacturer’s instruction.Real-time quantitative PCR. Real-time quantitative PCRs were done
in triplicate using the Corbett Research Rotor Gene RG-3000 (Sydney,
New South Wales, Australia). Each reaction was done in an individualtube and made up to 25 AL containing 10 AL cDNA, 12.5 AL TaqManPCR Master Mix (Abgene, Epsom, United Kingdom), 0.25 AL probe,1 AL forward and reverse primer, and 0.2 AL H2O. Conditions for thePCR were 2 minutes at 50jC, 10 minutes at 95jC, and 40 cycles, eachconsisting of 15 seconds at 95jC and 1 minute at 60jC. b-Actin was
used as reference gene using primers (Invitrogen, Paisley, United
Kingdom) forward 5¶-CCCAGCACAATGAAGATCAA-3¶ and reverse
5¶-CGATCCACACGGAGTACTTG-3¶ with probe 63 (Exiqon, Vedbaek,Denmark). Primers against CA-IX were forward 5¶-CTTGGAA-GAAATCGCTGAGG-3¶ and reverse 5¶-TGGAAGTAGCGGCTGAAGTC-3¶with probe 73, BNIP3 forward 5¶-TGCTGCTCTCTCATTTGCTG-3¶ andreverse 5¶-GACTCCAGTTCTTCATCAAAAGGT-3¶ with probe 22, VEGF
forward 5¶-CTACCTCCACCATGCCAAGT-3¶ and reverse 5¶-CCAC-TTCGTGATGATTCTGC-3¶ with probe 29, and HIF-1a forward 5¶-CAGC-TATTTGCGTGTGAGGA-3¶ and reverse 5¶-TGTGCTTTCATGTCATCTT-CAA-3¶ with probe 89 were used for quantitative PCR. Relative
quantitation of gene expression was done using the method described
by Pfaffl (38). In brief, comparisons were made between the number of
cycles required for the fluorescence of a sample to reach a predeter-
mined threshold that lay within the exponential phase and above
nonspecific background. The relative ratio of gene expression was
calculated as follows:
Relative ratio ¼ Etarget DCttarget ðmean comparator �mean sampleÞEref DCtref ðmean comparator �mean sampleÞ
where E target is reaction efficiency of the gene of interest, E ref is reaction
efficiency of the reference gene, and DCt is the cycle difference between
the comparator and the sample. All calculations are based on the mean
of PCRs done in triplicate.Nuclear cell extracts and HRE-binding assay. MDA-MB-468 cells
were grown to 80% confluence in T-75 flasks and treated with
compounds for 16 hours in hypoxia. Nuclear extracts were prepared
using a Transfactor Extraction Kit (BD Biosciences Clontech, Palo Alto,
CA) according to the manufacturer’s instructions. Cells were collected
and lysed, and the cytosolic fraction was removed. The nuclear pellets
were resuspended and homogenized, and the nuclear extracts were
collected. Cytoplasmic and nuclear cell extracts from MDA-MB-468
cells were run on 8% SDS-PAGE and transferred to polyvinylidene
difluoride membrane and staining with anti-HIF-1a (BD TransductionLaboratories), anti-h-tubulin (Sigma), and anti–histone deacetylase-1(Santa Cruz Biotechnology, Santa Cruz, CA) monoclonal antibodies.HIF-1a DNA-binding activity was determined using a BD Mercury
TransFactor kit specific for HIF-1a (BD Biosciences Clontech) accordingto the manufacturer’s instructions. Three aliquots (30 Ag) of nuclearextracts from each treatment group were added to individual wells of a96-well plate coated with oligonucleotides containing the consensus-binding sequence of HIF-1a. Bound HIF-1a was detected by theappropriate primary and horseradish peroxidase– labeled secondaryantibodies. 3,3¶,5,5¶-Tetramethylbenzidine substrate was added andconverted by horseradish peroxidase to a blue product. After 30minutes, the absorbance was measured at 650 nm. Nuclear extractsfrom Cos-7 cells treated with CoCl2 (Active Motif, Carlsbad, CA) andnuclear extracts incubated with a HIF-1-specific competitor oligonucle-otide (500 ng) served as positive and negative controls, respectively.Recombinant HIF-1a plasmids and transactivation assays. The
chimeric activator/reporter system used in transactivation assays wasbased on pGal (a plasmid based on pcDNA3, which contains a SV40
origin of replication and a CMV-promoted, truncated Gal4 geneencoding amino acids 1-147 followed by a polylinker bearingrestriction endonuclease sites, SacII, AscI, and NotI), and the Gal4-responsive luciferase reporter pUAS-tk-Luc (consisting of two copies ofa 17-bp Gal4 DNA-binding site and the thymidine kinase promoter,105 to +50, inserted into the HindIII site of pA3LUC) as describedpreviously by O’Rourke et al. (39). To analyze the effect of AJM290 ontransactivation function of HIF-1a, HIF-1a was amplified by PCR usingPfu polymerase (Stratagene, La Jolla, CA) and forward oligonucleotidescontaining a SacII recognition sequence in the appropriate readingframe and reverse oligonucleotides containing an AscI recognition siteand cloned into pGal. pBluescript/HIF-1a 3.2-3T7 (2) was used astemplate for HIF-1a sequences. The pGal plasmid containing sequencescoding for the herpes simplex virus protein 16 amino acids 410 to 490(VP16) as positive control was produced by insertion of this PCRproduct directly into pGal, preserving the reading frame, whereas pGalplasmid alone was used as background control. The phRL-CMV Renillaluciferase plasmid was used for transfection efficiency control (Prom-ega). For transactivation assays, in 24-well plates, cells were transfectedusing Fugene 6 eukaryote transfection reagent kit (Roche) with activatorplasmid (ranging between 0.5 and 5 ng), reporter plasmid (100-200ng), and the transfection control plasmid phRL-CMV (0.5 ng). Twenty-four hours later, cells were exposed to 1 Amol/L AJM290 in normoxiaor hypoxia for 16 hours. Firefly and Renilla luciferase activity wasmeasured using the Dual-Luciferase Reporter Assay System accordingto the manufacturer’s instructions.
Results
Effects of AJM290 and AW464 on viability and VEGFexpression. Sixteen-hour exposure of MDA-MB-468 breastcancer cells seeded at 10 � 103 to AJM290 or AW464 induceda dose-dependent decrease in viability and hypoxia-inducedVEGF protein levels in cell medium (Fig. 1B and C). Expressionof hypoxic VEGF was down-regulated at subtoxic concentra-tions by both compounds. In hypoxic cells treated withAJM290, VEGF IC50 was 1.26 Amol/L and cell viability IC50was 4.1 Amol/L. Fifty percent inhibition of hypoxic VEGF levelsto normoxic VEGF levels in cells treated with AJM290 wasachieved at 0.6 Amol/L. AW464 was less toxic and a less potentinhibitor of VEGF than AJM290, with a VEGF IC50 of 2.3 Amol/Land cell viability IC50 of 8.2 Amol/L in hypoxic cells. Fiftypercent inhibition of hypoxic VEGF levels to normoxic VEGFlevel in cells treated with AW464 was achieved at 1.8 Amol/L.Cell viability and VEGF ELISA assays showed that both AJM290and AW464 also inhibited hypoxic VEGF expression in thebreast cancer cell line MDA-MB-231 and melanoma MDA-MB-435 cell line at subtoxic concentrations after 16 hours ofexposure (Fig. 2A). AJM290 and AW464 also decreased VEGF atsubtoxic doses in pVHL mutant RCC4 and 786-0 renal cellcarcinoma cells that constitutively overexpress HIF-1a and/orHIF-2a protein, indicating that HIF-a is inhibited by AJM290and AW464 independently of the pVHL pathway. Because786-0 cell line expresses HIF-2a only and has been shownpreviously by us to regulate VEGF via HIF-2a (40), AJM290 andAW464 may also inhibit HIF-2a transcription activity.As Trx-1 and Trx-1 reductase have been shown to be up-
regulated under hypoxic conditions (28), we investigated ifhypoxic or pVHL mutant cells seeded at 2.5 � 103 per well weremore sensitive to the antiproliferative effect of AJM290 andAW464 following 16 or 48 hours of exposure to hypoxia ornormoxia with compounds (Fig. 2B and C). The pVHL mutantrenal cancer cell lines were only slightly more sensitive than the
New Thioredoxin Inhibitors Inhibit Hypoxia Pathway
www.aacrjournals.org Clin Cancer Res 2006;12(18) September15, 20065387
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

pVHL-transfected cells to antiproliferative effects of AJM290and AW464 following 16 hours of exposure. However, therewere no significant difference between normoxic and hypoxicor pVHL-expressing and pVHL mutant cells following 16 or 48hours of exposure.Effects of AJM290 and AW464 on HIF-1a, HIF-2a, and
HIF-a downstream targets. We incubated MDA-MB-468 cellsunder normoxic or hypoxic conditions for 16 hours in thepresence or absence of 1 to 10 Amol/L AJM290 or AW464 andinvestigated the effects of these compounds on HIF-1a andHIF-2a protein levels and downstream transcription targets ofHIF-a (Fig. 3A). Both HIF-1a and HIF-2a were undetectableunder normoxia but were stabilized under hypoxia. Following16 hours of treatment, surprisingly, both AJM290 and AW464 up-regulated the expression of HIF-1a and HIF-2a in both normoxicand hypoxic cells at a concentration of 2.5 to 10 and 10 Amol/L,respectively. However, the concentration at which this occurredvaried between cell lines. No change in HIF-1h expression wasobserved following treatment with AJM290 (data not shown).To investigate if both AJM290 and AW464 can inhibit HIF-a
transcriptional activity, expression of downstream transcriptiontargets of HIF-1a, CA-IX and BNIP3, was measured (Fig. 3A).Both AJM290 and AW464 inhibited the hypoxic up-regulationof CA-IX and BNIP3 in a dose-dependent manner. CompoundAJM290 was shown to be a more potent inhibitor of HIF-atranscription activity than AW464 in MDA-MB-468 cell line, asboth CA-IX and BNIP3 hypoxic up-regulation was inhibited by1 Amol/L AJM290, whereas 10 Amol/L AW464 was required to
inhibit the expression of CA-IX and BNIP3 in hypoxic cells. It isnotable that these effects occurred at concentrations similar tothose associated with an increase in HIF-a protein.Effects of AJM290 and AW464 compared with the proteasome
inhibitor MG132. To further understand the mechanism ofHIF-a up-regulation in normoxic and hypoxic MDA-MB-468cells following treatment with AJM290, we investigated theeffects of AJM290 on heat shock proteins and hydroxylationof HIF-1a by PHDs and used proteasome inhibitor MG132as a control (Fig. 3B). Both AJM290 and MG132 increased theexpression of HIF-1a and hydroxylated-HIF-1a in normoxiccells. Up-regulation of HIF-1a by MG132 and AJM290 coin-cided with the up-regulation of Hsp70, whereas no change inexpression was observed with Hsp90. Maximum expression ofHsp70 in normoxic cells was observed following treatment with2.5 Amol/L, whereas maximum expression of HIF-1a occurredfollowing treatment with 10 Amol/L, although both proteinswere induced at both drug concentrations. In hypoxic cells,expression of HIF-1a and Hsp70 was up-regulated by treatmentwith 2.5 Amol/L with no further enhancement by 10 Amol/L.Hsp70 was also up-regulated in MDA-MB-468 cells followingtreatment with AW464 (data not shown).Effects of AJM290 and AW464 on pVHL mutant cell lines. To
investigate how these compounds up-regulated HIF-a in cellsthat have defects in HIF degradation, we treated mutantpVHL RCC4+EV cells with 1 to 10 Amol/L AJM290 or AW464for 16 hours (Fig. 3C). HIF-1a and HIF-2a expression isconstitutively expressed in RCC4+EV and, like MDA-MB-468,
Fig. 3. Effect ofTrx-1inhibitors on HIF-aand HIF-a target genes (A), proteasomeinhibitor MG132 and AJM290 onhydroxylated HIF-1aand Hsp70/Hsp90in MDA-MB-468 cell line (B), andTrx-1inhibitors onpVHLmutant RCC4+EVcell line(C) and wild-type pVHL-transfectedRCC4+pVHL cells (D). Cells were exposedto normoxia or hypoxia for16 hours in thepresence of increasing concentrations ofAJM290, AW464, or MG132. Protein levelsin cell extracts were detected usingWesternblotting. h-Tubulin was used as loadingcontrol.
Human Cancer Biology
www.aacrjournals.orgClin Cancer Res 2006;12(18) September15, 2006 5388
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
was up-regulated by AJM290 and AW464 but at a lowerconcentration of these drugs. In RCC4+EV cells, HIF-2a wasstrongly up-regulated following treatment with 1 Amol/LAJM290 and AW464 and weakly up-regulated with 2.5 Amol/L,whereas HIF-1a was up-regulated to maximum levels by 1 and2.5 Amol/L AJM290 and 1 Amol/L AW464. The highestconcentration of 10 Amol/L of either drug down-regulated theexpression of HIF-1a and HIF-2a.Unlike MDA-MB-468 cells, with 1 Amol/L AJM290, the
increased HIF expression following treatment did not correlatewith down-regulation of CA-IX. However, CA-IX expressionwas down-regulated at higher concentrations. AJM290 at2.5 Amol/L did weakly down-regulate CA-IX, where HIF-1a isat its maximum level, whereas 10 Amol/L AJM290 and AW464significantly down-regulated the expression of CA-IX, whichcoincided with the loss of HIF-1a and HIF-2a expression.To investigate if both AJM290 and AW464 can also inhibit
HIF-a transcription in hypoxic RCC4 that have been stablytransfected with wild-type pVHL, cells were treated for 16 hourswith 1 to 10 Amol/L AJM290 or AW464 in normoxic or hypoxicconditions (Fig. 3D). As in the RCC4+EV cells, both AJM290and AW464 up-regulated HIF-1a and HIF-2a expression butat higher concentrations in normoxic cells, with maximumexpression of HIF-a being observed following treatmentwith 2.5 Amol/L AJM290 or AW464. However, in hypoxicRCC4+pVHL cells, maximum HIF-a expression was observedfollowing treatment with 1 Amol/L AJM290 or AW464. Similarto what was observed in MDA-MB-468 cells, lower concen-trations of AJM290 and AW464 up-regulated HIF-a in hypoxicconditions. As with RCC4+EV cells, 10 Amol/L AJM290 andAW464 resulted in the loss of HIF-1a expression. However,in this pVHL-transfected cell line, the further up-regulation ofHIF-a in hypoxia was associated with decreased transcriptionaleffect on downstream gene BNIP3 at 1 to 2.5 Amol/L. Expres-sion of CA-IX could not be detected in RCC4 cells expressingpVHL. Hsp70 was also up-regulated in RCC4 cells followingtreatment with AJM290 or AW464 (data not shown).Thus, in MDA-MB-468 cells and in the renal cancer cell line
RCC4, reconstituted with wild-type pVHL, similar HIF-aresponses occurred. However, in the RCC4+EV cell lines afterHIF-a protein increased at the lower drug concentrations, at10 Amol/L drug concentrations, HIF-a protein decreased, whichwas not observed in MDA-MB-468 cells. These results show thatthere are cell line differences following treatment with highconcentrations of Trx-1 inhibitors and this may reflect differ-ences in uptake and additional mechanisms involved in toxicityat higher doses.Effect of AJM290 and AW464 on mRNA of HIF-1a and
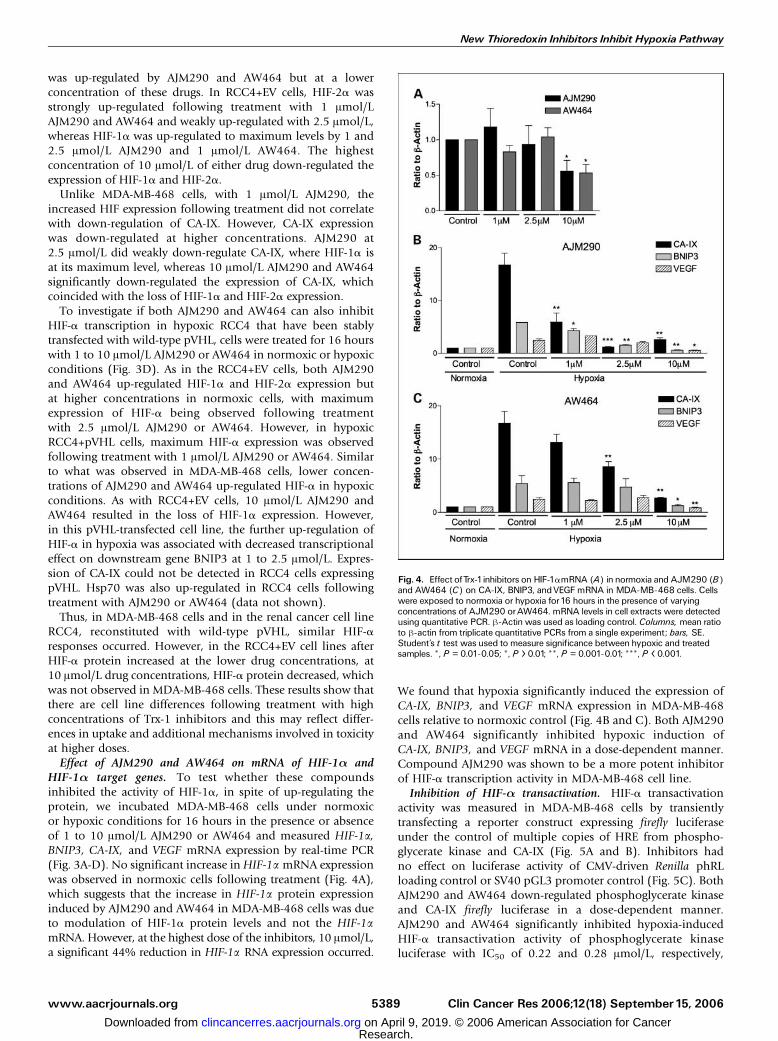
HIF-1a target genes. To test whether these compoundsinhibited the activity of HIF-1a, in spite of up-regulating theprotein, we incubated MDA-MB-468 cells under normoxicor hypoxic conditions for 16 hours in the presence or absenceof 1 to 10 Amol/L AJM290 or AW464 and measured HIF-1a,BNIP3, CA-IX, and VEGF mRNA expression by real-time PCR(Fig. 3A-D). No significant increase inHIF-1a mRNA expressionwas observed in normoxic cells following treatment (Fig. 4A),which suggests that the increase in HIF-1a protein expressioninduced by AJM290 and AW464 in MDA-MB-468 cells was dueto modulation of HIF-1a protein levels and not the HIF-1amRNA. However, at the highest dose of the inhibitors, 10 Amol/L,a significant 44% reduction in HIF-1a RNA expression occurred.
We found that hypoxia significantly induced the expression ofCA-IX, BNIP3, and VEGF mRNA expression in MDA-MB-468cells relative to normoxic control (Fig. 4B and C). Both AJM290and AW464 significantly inhibited hypoxic induction ofCA-IX, BNIP3, and VEGF mRNA in a dose-dependent manner.Compound AJM290 was shown to be a more potent inhibitorof HIF-a transcription activity in MDA-MB-468 cell line.Inhibition of HIF-a transactivation. HIF-a transactivation
activity was measured in MDA-MB-468 cells by transientlytransfecting a reporter construct expressing firefly luciferaseunder the control of multiple copies of HRE from phospho-glycerate kinase and CA-IX (Fig. 5A and B). Inhibitors hadno effect on luciferase activity of CMV-driven Renilla phRLloading control or SV40 pGL3 promoter control (Fig. 5C). BothAJM290 and AW464 down-regulated phosphoglycerate kinaseand CA-IX firefly luciferase in a dose-dependent manner.AJM290 and AW464 significantly inhibited hypoxia-inducedHIF-a transactivation activity of phosphoglycerate kinaseluciferase with IC50 of 0.22 and 0.28 Amol/L, respectively,
Fig. 4. Effect ofTrx-1inhibitors on HIF-1amRNA (A) in normoxia and AJM290 (B)and AW464 (C) on CA-IX, BNIP3, andVEGF mRNA in MDA-MB-468 cells. Cellswere exposed to normoxia or hypoxia for16 hours in the presence of varyingconcentrations of AJM290 orAW464. mRNA levels in cell extracts were detectedusing quantitative PCR. h-Actin was used as loading control. Columns, mean ratioto h-actin from triplicate quantitative PCRs from a single experiment; bars, SE.Student’s t test was used to measure significance between hypoxic and treatedsamples. *, P = 0.01-0.05; *, P > 0.01; **, P = 0.001-0.01; ***, P < 0.001.
New Thioredoxin Inhibitors Inhibit Hypoxia Pathway
www.aacrjournals.org Clin Cancer Res 2006;12(18) September15, 20065389
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
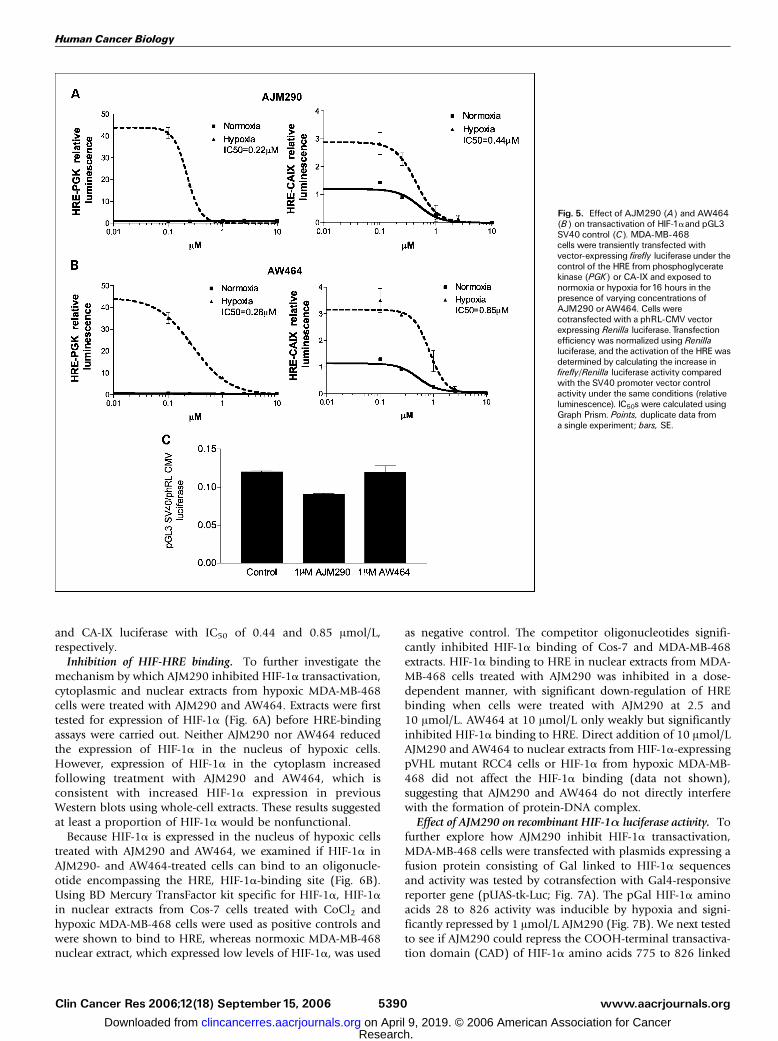
and CA-IX luciferase with IC50 of 0.44 and 0.85 Amol/L,respectively.Inhibition of HIF-HRE binding. To further investigate the
mechanism by which AJM290 inhibited HIF-1a transactivation,cytoplasmic and nuclear extracts from hypoxic MDA-MB-468cells were treated with AJM290 and AW464. Extracts were firsttested for expression of HIF-1a (Fig. 6A) before HRE-bindingassays were carried out. Neither AJM290 nor AW464 reducedthe expression of HIF-1a in the nucleus of hypoxic cells.However, expression of HIF-1a in the cytoplasm increasedfollowing treatment with AJM290 and AW464, which isconsistent with increased HIF-1a expression in previousWestern blots using whole-cell extracts. These results suggestedat least a proportion of HIF-1a would be nonfunctional.Because HIF-1a is expressed in the nucleus of hypoxic cells
treated with AJM290 and AW464, we examined if HIF-1a inAJM290- and AW464-treated cells can bind to an oligonucle-otide encompassing the HRE, HIF-1a-binding site (Fig. 6B).Using BD Mercury TransFactor kit specific for HIF-1a, HIF-1ain nuclear extracts from Cos-7 cells treated with CoCl2 andhypoxic MDA-MB-468 cells were used as positive controls andwere shown to bind to HRE, whereas normoxic MDA-MB-468nuclear extract, which expressed low levels of HIF-1a, was used
as negative control. The competitor oligonucleotides signifi-cantly inhibited HIF-1a binding of Cos-7 and MDA-MB-468extracts. HIF-1a binding to HRE in nuclear extracts from MDA-MB-468 cells treated with AJM290 was inhibited in a dose-dependent manner, with significant down-regulation of HREbinding when cells were treated with AJM290 at 2.5 and10 Amol/L. AW464 at 10 Amol/L only weakly but significantlyinhibited HIF-1a binding to HRE. Direct addition of 10 Amol/LAJM290 and AW464 to nuclear extracts from HIF-1a-expressingpVHL mutant RCC4 cells or HIF-1a from hypoxic MDA-MB-468 did not affect the HIF-1a binding (data not shown),suggesting that AJM290 and AW464 do not directly interferewith the formation of protein-DNA complex.Effect of AJM290 on recombinant HIF-1a luciferase activity. To
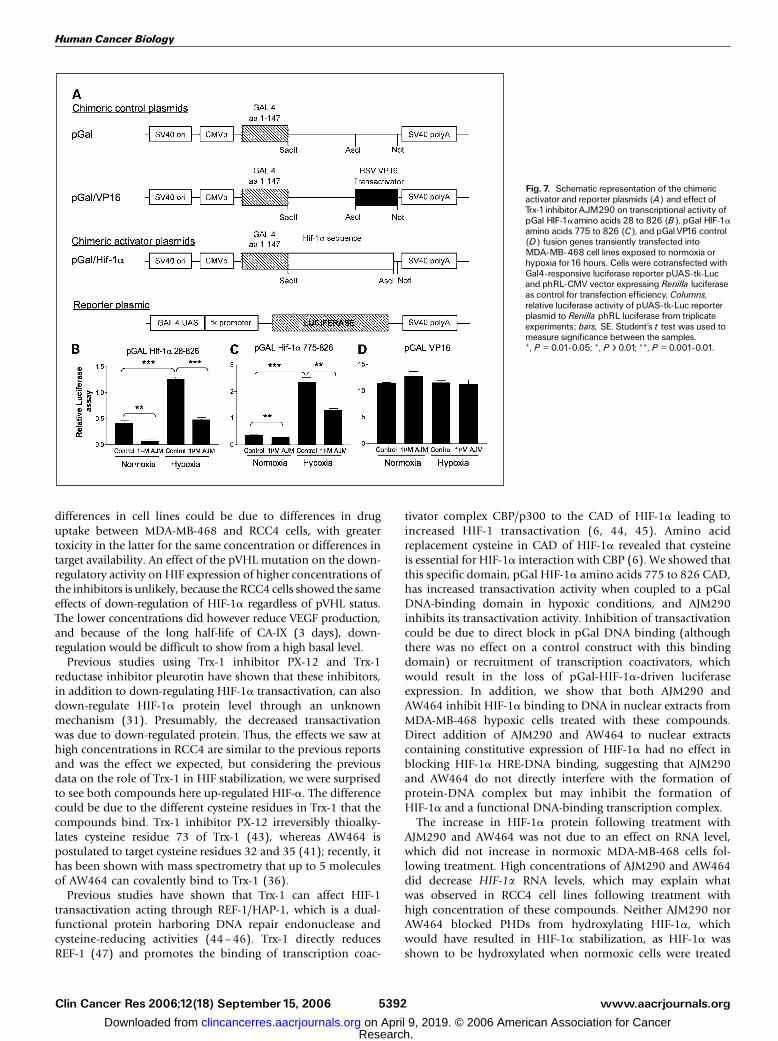
further explore how AJM290 inhibit HIF-1a transactivation,MDA-MB-468 cells were transfected with plasmids expressing afusion protein consisting of Gal linked to HIF-1a sequencesand activity was tested by cotransfection with Gal4-responsivereporter gene (pUAS-tk-Luc; Fig. 7A). The pGal HIF-1a aminoacids 28 to 826 activity was inducible by hypoxia and signi-ficantly repressed by 1 Amol/L AJM290 (Fig. 7B). We next testedto see if AJM290 could repress the COOH-terminal transactiva-tion domain (CAD) of HIF-1a amino acids 775 to 826 linked
Fig. 5. Effect of AJM290 (A) and AW464(B) on transactivation of HIF-1aand pGL3SV40 control (C). MDA-MB-468cells were transiently transfected withvector-expressing firefly luciferase under thecontrol of the HRE from phosphoglyceratekinase (PGK) or CA-IX and exposed tonormoxia or hypoxia for16 hours in thepresence of varying concentrations ofAJM290 orAW464. Cells werecotransfected with a phRL-CMV vectorexpressing Renilla luciferase.Transfectionefficiency was normalized using Renillaluciferase, and the activation of the HREwasdetermined by calculating the increase infirefly/Renilla luciferase activity comparedwith the SV40 promoter vector controlactivity under the same conditions (relativeluminescence). IC50s were calculated usingGraph Prism. Points, duplicate data froma single experiment; bars, SE.
Human Cancer Biology
www.aacrjournals.orgClin Cancer Res 2006;12(18) September15, 2006 5390
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

to pGal (Fig. 7C). Hypoxia up-regulated pGal HIF-1a CADactivity and was repressed significantly by 1 Amol/L AJM290.Hypoxia and 1 Amol/L AJM290 had no significant effect oncontrol pGal VP16 (Fig. 7D). This showed that the compoundscan reduce pGal-HIF-1a CAD transactivation of Gal responsivesequences.
Discussion
Here, we investigated the effects of novel Trx-1 inhibitorsAJM290 and AW464 on HIF-a expression and HIF-a trans-activation in vitro . AW464 is a benzothiazole-substituted quinolcompound that has been shown by mass spectrometry to bindto Trx-1 and has been proposed to cross-link irreversibly tocysteine residues 32 and 35 of Trx-1 active site via its twoh-carbon atoms; the first link is reversible, whereas the secondcross-link is thought to be irreversible (36, 41). AW464 caninhibit VEGF expression in hypoxic colorectal cells, andhypoxia has been shown to sensitize colorectal cancer cells toantiproliferative effect of AW464 (35). Further chemicalsyntheses and structure activity screening uncovered AJM290,an indole-substituted quinol that possess enhanced potencyin vitro against human derived colon, renal, and mammarycarcinoma cell lines and in vivo antitumor activity againstbreast, colon, and renal mouse xenografts (36, 42).In this study, we showed that AJM290 and AW464 are potent
inhibitors of the hypoxia pathway. Both compounds inhibitedVEGF expression at subtoxic concentrations in hypoxic breast
and melanoma cell lines and pVHL mutant renal cell lines, withAJM290 being the more potent compound. Studies carried outby Mukherjee et al. showed that hypoxia reduced the IC50 ofAW464 byf5-fold in all colorectal cancer cell lines tested (35).However, we saw no changes in IC50 between hypoxic andnormoxic cells and pVHL mutant and stable pVHL-transfectedcells following 16 or 48 hours of treatment with AJM290 orAW464. The difference in results could be due to the cell linesused (35).AJM290 and AW464 in the MDA-MB-468 cell line inhibited
expression of downstream HIF-a target genes, including theangiogenesis factor VEGF, pH regulatory protein CA-IX, andproapoptotic protein BNIP3, which coincided with the increasein HIF-a expression. Differences were observed betweenRCC4+EV and MDA-MB-468 cell lines. In RCC4+EV cells,lower concentration of AJM290 and AW464 increased HIF-1a,whereas CA-IX did not decrease. Higher concentrations ofAJM290 did reduce CA-IX expression, whereas HIF-1a was at itshighest level, but the highest concentrations of AJM290 andAW464 markedly reduced HIF-a expression, which coincidedwith the loss of CA-IX.Thus, in MDA-MB-468 cells, the increase in HIF-1a expres-
sion resulted in the decrease of HIF-a transcription, whereas inRCC4+EV cell line it seems that it is the loss of HIF-a that leadsto the decrease in HIF-a transcription targets. However, pVHL-transfected RCC4 cells behaved the same as MDA-MB-468 whentreated with AJM290 and AW464 at lower concentrations.In hypoxic RCC4+pVHL cells, AJM290 and AW464 down-regulated BNIP3, whereas levels of HIF-awere up-regulated. The
Fig. 6. Effect of Trx-1inhibitors on HIF-1anuclear localization (A) and HIF-1aDNA-binding assay (B). MDA-MB-468cells were exposed to normoxia or hypoxiafor16 hours in the presence of1to10 Amol/LAJM290 or10 Amol/L AW464. Proteinlevels in cytoplasmic and nuclear cellextracts were detected usingWesternblotting with antibody against HIF-1a.h-Tubulin was used as loading control forcytoplasmic cell extracts, and histonedeacetylase-1was used as loading controlfor nuclear cell extracts. HIF-1aDNA-bindingassay was carried out on the sameMDA-MB-468 nuclear extracts treated withTrx-1inhibitors using BDMercuryTransFactor kit specific for HIF-1a. HIF-1a innuclear extracts from Cos-7 cells treatedwith CoCl2 andhypoxicMDA-MB-468 cellswere used as positive controls. Nuclearextracts from normoxic MDA-MB-468 andnuclear extracts incubated with aHIF-1a-specific competitor oligonucleotideserved as negative controls.Columns, meanof triplicate set of data from a singleexperiment; bars, SE. Student’s t test wasused to measure significance betweenhypoxic or CoCl2 and treated samples.*, P = 0.01-0.05; *, P > 0.01;**, P = 0.001-0.01.
New Thioredoxin Inhibitors Inhibit Hypoxia Pathway
www.aacrjournals.org Clin Cancer Res 2006;12(18) September15, 20065391
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
differences in cell lines could be due to differences in druguptake between MDA-MB-468 and RCC4 cells, with greatertoxicity in the latter for the same concentration or differences intarget availability. An effect of the pVHL mutation on the down-regulatory activity on HIF expression of higher concentrations ofthe inhibitors is unlikely, because the RCC4 cells showed the sameeffects of down-regulation of HIF-1a regardless of pVHL status.The lower concentrations did however reduce VEGF production,and because of the long half-life of CA-IX (3 days), down-regulation would be difficult to show from a high basal level.Previous studies using Trx-1 inhibitor PX-12 and Trx-1
reductase inhibitor pleurotin have shown that these inhibitors,in addition to down-regulating HIF-1a transactivation, can alsodown-regulate HIF-1a protein level through an unknownmechanism (31). Presumably, the decreased transactivationwas due to down-regulated protein. Thus, the effects we saw athigh concentrations in RCC4 are similar to the previous reportsand was the effect we expected, but considering the previousdata on the role of Trx-1 in HIF stabilization, we were surprisedto see both compounds here up-regulated HIF-a. The differencecould be due to the different cysteine residues in Trx-1 that thecompounds bind. Trx-1 inhibitor PX-12 irreversibly thioalky-lates cysteine residue 73 of Trx-1 (43), whereas AW464 ispostulated to target cysteine residues 32 and 35 (41); recently, ithas been shown with mass spectrometry that up to 5 moleculesof AW464 can covalently bind to Trx-1 (36).Previous studies have shown that Trx-1 can affect HIF-1
transactivation acting through REF-1/HAP-1, which is a dual-functional protein harboring DNA repair endonuclease andcysteine-reducing activities (44–46). Trx-1 directly reducesREF-1 (47) and promotes the binding of transcription coac-
tivator complex CBP/p300 to the CAD of HIF-1a leading toincreased HIF-1 transactivation (6, 44, 45). Amino acidreplacement cysteine in CAD of HIF-1a revealed that cysteineis essential for HIF-1a interaction with CBP (6). We showed thatthis specific domain, pGal HIF-1a amino acids 775 to 826 CAD,has increased transactivation activity when coupled to a pGalDNA-binding domain in hypoxic conditions, and AJM290inhibits its transactivation activity. Inhibition of transactivationcould be due to direct block in pGal DNA binding (althoughthere was no effect on a control construct with this bindingdomain) or recruitment of transcription coactivators, whichwould result in the loss of pGal-HIF-1a-driven luciferaseexpression. In addition, we show that both AJM290 andAW464 inhibit HIF-1a binding to DNA in nuclear extracts fromMDA-MB-468 hypoxic cells treated with these compounds.Direct addition of AJM290 and AW464 to nuclear extractscontaining constitutive expression of HIF-1a had no effect inblocking HIF-1a HRE-DNA binding, suggesting that AJM290and AW464 do not directly interfere with the formation ofprotein-DNA complex but may inhibit the formation ofHIF-1a and a functional DNA-binding transcription complex.The increase in HIF-1a protein following treatment with
AJM290 and AW464 was not due to an effect on RNA level,which did not increase in normoxic MDA-MB-468 cells fol-lowing treatment. High concentrations of AJM290 and AW464did decrease HIF-1a RNA levels, which may explain whatwas observed in RCC4 cell lines following treatment withhigh concentration of these compounds. Neither AJM290 norAW464 blocked PHDs from hydroxylating HIF-1a, whichwould have resulted in HIF-1a stabilization, as HIF-1a wasshown to be hydroxylated when normoxic cells were treated
Fig. 7. Schematic representation of the chimericactivator and reporter plasmids (A) and effect ofTrx-1inhibitorAJM290 on transcriptional activity ofpGal HIF-1aamino acids 28 to 826 (B), pGal HIF-1aamino acids 775 to 826 (C), and pGalVP16 control(D) fusion genes transiently transfected intoMDA-MB-468 cell lines exposed to normoxia orhypoxia for16 hours. Cells were cotransfected withGal4-responsive luciferase reporter pUAS-tk-Lucand phRL-CMV vector expressing Renilla luciferaseas control for transfection efficiency. Columns,relative luciferase activity of pUAS-tk-Luc reporterplasmid to Renilla phRL luciferase from triplicateexperiments; bars, SE. Student’s t test was used tomeasure significance between the samples.*, P = 0.01-0.05; *, P > 0.01; **, P = 0.001-0.01.
Human Cancer Biology
www.aacrjournals.orgClin Cancer Res 2006;12(18) September15, 2006 5392
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

with AJM290 and AW464. The increase in HIF-a protein expres-sion could be due to a block in HIF-a protein degradation.However, it is unlikely to involve pVHL directly becauseAJM290 and AW464 increased HIF-a in RCC4 cell lines, whichlack pVHL. Previous studies have shown that Hsp90/Hsp70can stabilize HIF-1a by preventing its degradation via a pVHL-independent pathway (48). It is possible that Hsp70, whichwas shown to be up-regulated in cells treated with AJM290 orAW464, may prevent the degradation of HIF-a. Microarrayanalysis have shown that Hsp70 is induced by high doses ofAW464 (35). Previous studies have also shown that HIF-1a cancoimmunoprecipitate with Hsp90/Hsp70 and that Hsp70directly binds to the oxygen-dependent degradation domainof HIF-1a (48), so it is possible that Hsp70 directly stabilizesHIF-a and inhibit degradation of HIF-a through a pVHL-independent pathway (49), but this would not account for theblock in transactivation.The phenotype induced by AJM290 and AW464 was similar
to that produced by the proteasome inhibitor MG132. LikeAJM290, MG132 up-regulated HIF-1a and Hsp70 in normoxic(Fig. 2B) and hypoxic cells and these changes were concordantwith down-regulation of the target genes (data not shown). Pre-vious studies have shown that MG132 blocks the proteasomedegradation of hydroxylated HIF-a protein, resulting in increasedlevels of stabilized polyubiquitinated forms of HIF-1a, whichhave reduced transcriptional activity (50). HIF-1a up-regulatedin hypoxic cells by AJM290 or MG132 (data not shown) wasmuch less hydroxylated and therefore would be much lessubiquitinated by the ubiquitinase pVHL complex. Nevertheless,prolyl hydroxylases have some function even at low oxygentension, which would then contribute to accumulation ofmodified ubiquitinated HIF. This residual function may explainthe differences between pVHL mutant cells and the MDA-MB-468 cells at lower drug concentrations. In the former, there isminimal ubiquitination, so proteasome inhibition, although up-
regulating HIF, does not result in loss of function, whereas inMDA-MB-468 cells the accumulated HIF will be ubiquitinated.At higher drug concentrations, HIF was down-regulated asreported for other proteasome inhibitors.Additionally, HIF-1a may not be in a reduced state following
treatment with anti-Trx compounds, which is essential for itsactivity. Trx has also been shown to have a role in the pro-teasome function (51), and it is possible that inhibitors ofTrx that bind to different cysteines will differentially modifyproteasome interaction with key targets. This is the most likelyexplanation of an increase in hydroxylated HIF-a in normoxiccells and nonhydroxylated HIF-1a in hypoxic cells. Thecombined effects would then be a protein that is nonfunctionalin DNA binding or transactivation but increased in amount. Athigher concentration, additional effects occur with down-regulation of HIF-1a in RCC4 versus breast cancer cell lines.We conclude that AJM290 and AW464 are effective drugs
against the HIF pathway in breast cancer cells, and this isprobably because of their unique activity compared with otherTrx inhibitors. They decreased HIF-1a transactivation via theCAD, inhibited binding to HREs in DNA, increased cytoplasmicaccumulation of HIF-a, and blocked the induction of HIF-atarget genes. Both compounds have also been shown previouslyto have antitumor activity in vivo , and AW464 has been shownto exert antiproliferative effects on endothelial cells, whereasnormal fibroblasts are resistant (33–36). Inhibition of Trx-1redox system may therefore represent an effective way oftreating cancer cells resulting in decreased hypoxia response,angiogenesis, and resistance to apoptosis.
Acknowledgments
We thankTracey Bradshaw (University of Nottingham, Nottingham, UnitedKingdom) for collaboration and all members of A.L. Harris laboratory for technicalinput and support.
References1. Maxwell PH, Dachs GU, Gleadle JM, et al. Hypoxia-inducible factor-1 modulates gene expression insolid tumors and influences both angiogenesis andtumor growth. Proc Natl Acad Sci U S A 1997;94:8104^9.
2.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor1is a basic-helix-loop-helix-PAS hete-rodimer regulated by cellular O2 tension. Proc NatlAcad Sci US A1995;92:5510^4.
3.Wood SM, Gleadle JM, Pugh CW, Hankinson O,Ratcliffe PJ. The role of the aryl hydrocarbon receptornuclear translocator (ARNT) in hypoxic induction ofgene expression. Studies in ARNT-deficient cells.JBiol Chem1996;271:15117^23.
4. Maxwell PH, Pugh CW, Ratcliffe PJ. The pVHL-hIF-1system. A key mediator of oxygen homeostasis. AdvExp Med Biol 2001;502:365^76.
5. Jaakkola P,Mole DR,TianYM, et al.Targeting of HIF-ato the von Hippel-Lindau ubiquitylation complex byO2-regulated prolyl hydroxylation. Science 2001;292:468^72.
6. Ema M, Hirota K, Mimura J, et al. Molecular mecha-nisms of transcription activation by HLF and HIF1a inresponse to hypoxia: their stabilization and redoxsignal-induced interaction with CBP/p300. EMBO J1999;18:1905^14.
7. Minchenko A, Salceda S, Bauer T, Caro J. Hypoxiaregulatory elements of the human vascular endothe-lial growth factor gene. Cell Mol Biol Res 1994;40:35^9.
8. Blancher C, MooreJW,Talks KL, Houlbrook S, HarrisAL. Relationship of hypoxia-inducible factor (HIF)-1aand HIF-2a expression to vascular endothelial growthfactor induction and hypoxia survival in human breastcancer cell lines. Cancer Res 2000;60:7106^13.
9. Semenza GL. Hypoxia, clonal selection, and the roleof HIF-1 in tumor progression. Crit Rev Biochem MolBiol 2000;35:71^103.
10. Bos R, Zhong H, Hanrahan CF, et al. Levels ofhypoxia-inducible factor-1aduring breast carcinoge-nesis. JNatl Cancer Inst 2001;93:309^14.
11. Laughner E, Taghavi P, Chiles K, Mahon PC,Semenza GL. HER2 (neu) signaling increases the rateof hypoxia-inducible factor 1a (HIF-1a) synthesis:novel mechanism for HIF-1-mediated vascular endo-thelial growth factor expression. Mol Cell Biol 2001;21:3995^4004.
12. Zhong H, Chiles K, Feldser D, et al. Modulation ofhypoxia-inducible factor 1aexpression by the epider-mal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancercells: implications for tumor angiogenesis and thera-peutics. Cancer Res 2000;60:1541^5.
13. Chen C, Pore N, Behrooz A, Ismail-Beigi F, MaityA.Regulation of glut1 mRNA by hypoxia-induciblefactor-1. InteractionbetweenH-ras andhypoxia. JBiolChem 2001;276:9519^25.
14. Jiang BH, Agani F, Passaniti A, Semenza GL.V-SRCinduces expressionof hypoxia-inducible factor1 (HIF-1)and transcription of genes encoding vascular endo-
thelial growth factor and enolase 1: involvement ofHIF-1 in tumor progression. Cancer Res 1997;57:5328^35.
15. Ravi R, Mookerjee B, Bhujwalla ZM, et al. Regu-lation of tumor angiogenesis by p53-induced degra-dation of hypoxia-inducible factor 1a. Genes Dev2000;14:34^44.
16. Powis G, Kirkpatrick L. Hypoxia inducible factor-1aas a cancer drug target. Mol Cancer Ther 2004;3:647^54.
17. Semenza GL.Targeting HIF-1for cancer therapy. NatRev Cancer 2003;3:721^32.
18. Yeo EJ, Chun YS, Cho YS, et al. YC-1: a potentialanticancer drug targeting hypoxia-inducible factor 1.J Natl Cancer Inst 2003;95:516^25.
19. Powis G, Mustacich D, Coon A. The role of theredox protein thioredoxin in cell growth and cancer.Free Radic Biol Med 2000;29:312^22.
20. Saitoh M, Nishitoh H, Fujii M, et al. Mammalianthioredoxin is a direct inhibitor of apoptosis sig-nal-regulating kinase (ASK) 1. EMBO J 1998;17:2596^606.
21.Watson JA, Rumsby MG, Wolowacz RG. Phagedisplay identifies thioredoxin and superoxide dismu-tase as novel protein kinase C-interacting proteins:thioredoxin inhibits protein kinase C-mediated phos-phorylation of histone. Biochem J 1999;343 Pt 2:301^5.
22.HayashiT, UenoY, OkamotoT. Oxidoreductive regu-lation of nuclear factor nB. Involvement of a cellular
New Thioredoxin Inhibitors Inhibit Hypoxia Pathway
www.aacrjournals.org Clin Cancer Res 2006;12(18) September15, 20065393
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

reducing catalyst thioredoxin. J Biol Chem1993;268:11380^8.
23. Ueno M, Masutani H, Arai RJ, et al. Thioredoxin-dependent redox regulation of p53-mediated p21activation. JBiol Chem1999;274:35809^15.
24.Welsh SJ, BellamyWT, Briehl MM, Powis G. Theredox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1aprotein expression: Trx-1 overex-pression results in increased vascular endothelialgrowth factor production and enhanced tumor angio-genesis. Cancer Res 2002;62:5089^95.
25. Baker A, Payne CM, Briehl MM, Powis G. Thiore-doxin, a gene found overexpressed in human cancer,inhibits apoptosis in vitro and in vivo. Cancer Res1997;57:5162^7.
26. Gasdaska PY, Oblong JE, Cotgreave IA, Powis G.The predicted amino acid sequence of human thiore-doxin is identical to that of the autocrine growth factorhuman adult T-cell derived factor (ADF): thioredoxinmRNA is elevated in some human tumors. BiochimBiophys Acta1994;1218:292^6.
27. Raffel J, Bhattacharyya AK, Gallegos A, et al.Increased expression of thioredoxin-1 in human colo-rectal cancer is associated with decreased patientsurvival. JLab Clin Med 2003;142:46^51.
28. Berggren M, Gallegos A, Gasdaska JR, GasdaskaPY,Warneke J, Powis G. Thioredoxin and thioredoxinreductase gene expression in human tumors and celllines, and the effects of serum stimulation andhypoxia.AnticancerRes1996;16:3459^66.
29. GroganTM, Fenoglio-Prieser C, Zeheb R, et al. Thi-oredoxin, a putative oncogene product, is overex-pressed in gastric carcinoma and associated withincreased proliferation and increased cell survival.Hum Pathol 2000;31:475^81.
30. Nakamura H, Bai J, NishinakaY, et al. Expression ofthioredoxin and glutaredoxin, redox-regulating pro-teins, in pancreatic cancer. Cancer Detect Prev 2000;24:53^60.
31.Welsh SJ,Williams RR, Birmingham A, Newman DJ,Kirkpatrick DL, Powis G.The thioredoxin redox inhibi-tors 1-methylpropyl 2-imidazolyl disulfide and pleuro-
tin inhibit hypoxia-induced factor 1a and vascularendothelial growth factor formation. Mol CancerTher2003;2:235^43.
32.Welsh S, Williams R, Kirkpatrick L, Paine-MurrietaG, Powis G. Antitumor activity and pharmacody-namic properties of PX-478, an inhibitor of hypox-ia-inducible factor-1a. Mol Cancer Ther 2004;3:233^44.
33.Wells G, BerryJM, BradshawTD, et al. 4-Substituted4-hydroxycyclohexa-2,5-dien-1-ones with selectiveactivities against colon and renal cancer cell lines.JMed Chem 2003;46:532^41.
34.Wells G, BradshawTD, Diana P, et al. Antitumourbenzothiazoles. Part 10.The synthesis and antitumouractivity of benzothiazole substituted quinol deriva-tives. Bioorg Med Chem Lett 2000;10:513^5.
35. Mukherjee A,Westwell AD, BradshawTD, et al.Cytotoxic and antiangiogenic activity of AW464(NSC 706704), a novel thioredoxin inhibitor : anin vitro study. BrJCancer 2005;92:350^8.
36. Bradshaw TD, Matthews CS, Cookson J, et al.Elucidation of thioredoxin as a molecular target forantitumor quinols. Cancer Res 2005;65:3911^9.
37. Chomczynski P, Sacchi N. Single-step methodof RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987;162:156^9.
38. Pfaffl MW. A new mathematical model for relativequantification in real-time RT-PCR. Nucleic Acids Res2001;29:e45.
39. O’Rourke JF, Tian YM, Ratcliffe PJ, Pugh CW.Oxygen-regulated and transactivating domains inendothelial PAS protein 1: comparison with hypoxia-inducible factor-1a. J Biol Chem 1999;274:2060^71.
40. Raval RR, Lau KW,TranMG, et al. Contrastingprop-erties of hypoxia-inducible factor 1 (HIF-1) and HIF-2in von Hippel-Lindau-associated renal cell carcinoma.Mol Cell Biol 2005;25:5675^86.
41. PallisM, Bradshaw TD,Westwell AD, et al. Inductionofapoptosiswithoutredoxcatastropheby thioredoxin-inhibitory compounds. Biochem Pharmacol 2003;66:1695^705.
42. BerryJM, BradshawTD, Fichtner I, et al. Quinols asnovel therapeutic agents. 2.(1) 4-(1-Arylsulfonylin-dol-2-yl)-4-hydroxycyclohexa-2,5-dien-1-ones andrelated agents as potent and selective antitumoragents. JMed Chem 2005;48:639^44.
43. Kirkpatrick DL, Kuperus M, Dowdeswell M, et al.Mechanisms of inhibition of the thioredoxin growthfactor system by antitumor 2-imidazolyl disulfides.Biochem Pharmacol1998;55:987^94.
44. Carrero P, Okamoto K, Coumailleau P, et al.Redox-regulated recruitment of the transcriptionalcoactivators CREB-binding protein and SRC-1 tohypoxia-inducible factor 1a. Mol Cell Biol 2000;20:402^15.
45. Huang LE, Arany Z, Livingston DM, Bunn HF.Activation of hypoxia-inducible transcription factordepends primarily upon redox-sensitive stabiliza-tion of its a subunit. J Biol Chem 1996;271:32253^9.
46. Barzilay G,Walker LJ, Rothwell DG, Hickson ID.Role of the HAP1protein in repair of oxidative DNAdamage and regulation of transcription factors. Br JCancer Suppl 1996;27:S145^50.
47. Hirota K, Matsui M, Iwata S, et al. AP-1 transcrip-tional activity is regulated by a direct associationbetween thioredoxin and Ref-1. Proc Natl Acad SciUS A1997;94:3633^8.
48. Zhou J, Schmid T, Frank R, Brune B. PI3K/Akt isrequired for heat shock proteins to protect hypoxia-inducible factor 1a from pVHL-independent degrada-tion. JBiol Chem 2004;279:13506^13.
49. Isaacs JS, Jung YJ, Mimnaugh EG, et al. Hsp90regulates a von Hippel Lindau-independent hypoxia-inducible factor-1a-degradative pathway. JBiol Chem2002;277:29936^44.
50. Kallio PJ,WilsonWJ, O’Brien S, MakinoY, PoellingerL. Regulation of the hypoxia-inducible transcriptionfactor 1aby the ubiquitin-proteasome pathway. JBiolChem1999;274:6519^25.
51. Shibata T, YamadaT, Ishii T, et al. Thioredoxin as amolecular target of cyclopentenone prostaglandins.JBiol Chem 2003;278:26046^54.
Human Cancer Biology
www.aacrjournals.orgClin Cancer Res 2006;12(18) September15, 2006 5394
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

2006;12:5384-5394. Clin Cancer Res Dylan T. Jones, Christopher W. Pugh, Simon Wigfield, et al. DegradationFunctional Transcriptional Activity, DNA Binding, and
Expression but DecreaseαHypoxia-Inducible Factor-Novel Thioredoxin Inhibitors Paradoxically Increase
Updated version
http://clincancerres.aacrjournals.org/content/12/18/5384
Access the most recent version of this article at:
Cited articles
http://clincancerres.aacrjournals.org/content/12/18/5384.full#ref-list-1
This article cites 51 articles, 29 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/12/18/5384.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://clincancerres.aacrjournals.org/content/12/18/5384To request permission to re-use all or part of this article, use this link
Research. on April 9, 2019. © 2006 American Association for Cancerclincancerres.aacrjournals.org Downloaded from