nos inhibition modulates immune polarization and …...nos inhibition modulates immune polarization...
TRANSCRIPT

Microenvironment and Immunology
NOS Inhibition Modulates Immune Polarizationand Improves Radiation-Induced Tumor GrowthDelayLisa A. Ridnour1, Robert Y.S. Cheng1, Jonathan M.Weiss2, Sukhbir Kaur3,David R. Soto-Pantoja3, Debashree Basudhar1, Julie L. Heinecke1, C. Andrew Stewart2,William DeGraff1, Anastasia L. Sowers1, Angela Thetford1, Aparna H. Kesarwala4,David D. Roberts3, Howard A.Young2, James B. Mitchell1, Giorgio Trinchieri2,Robert H.Wiltrout2, and David A.Wink1
Abstract
Nitric oxide synthases (NOS) are important mediators ofprogrowth signaling in tumor cells, as they regulate angio-genesis, immune response, and immune-mediated woundhealing. Ionizing radiation (IR) is also an immune modulatorand inducer of wound response. We hypothesized that radi-ation therapeutic efficacy could be improved by targetingNOS following tumor irradiation. Herein, we show enhancedradiation-induced (10 Gy) tumor growth delay in a syngeneicmodel (C3H) but not immunosuppressed (Nu/Nu) squa-mous cell carcinoma tumor-bearing mice treated post-IRwith the constitutive NOS inhibitor NG-nitro-L-arginine meth-yl ester (L-NAME). These results suggest a requirement ofT cells for improved radiation tumor response. In supportof this observation, tumor irradiation induced a rapid increasein the immunosuppressive Th2 cytokine IL10, which wasabated by post-IR administration of L-NAME. In vivo suppres-
sion of IL10 using an antisense IL10 morpholino also extend-ed the tumor growth delay induced by radiation in a mannersimilar to L-NAME. Further examination of this mechanismin cultured Jurkat T cells revealed L-NAME suppression ofIR-induced IL10 expression, which reaccumulated in the pres-ence of exogenous NO donor. In addition to L-NAME, theguanylyl cyclase inhibitors ODQ and thrombospondin-1 alsoabated IR-induced IL10 expression in Jurkat T cells andANA-1 macrophages, which further suggests that the immu-nosuppressive effects involve eNOS. Moreover, cytotoxic Th1cytokines, including IL2, IL12p40, and IFNg , as well as acti-vated CD8þ T cells were elevated in tumors receiving post-IRL-NAME. Together, these results suggest that post-IR NOSinhibition improves radiation tumor response via Th1immune polarization within the tumor microenvironment.Cancer Res; 75(14); 2788–99. �2015 AACR.
IntroductionRadiotherapy remains a primary mode of treatment for more
than 50% of cancer patients in North America (1). At the molec-ular level, ionizing radiation (IR) exerts its antitumor effects byinducing direct DNA damage in the form of DNA double-strandbreaks as well as indirect damage by the generation of reactiveoxygen species (2). Although DNA damage has a central role inradiation-induced tumor cell death, it does not fully account fortumor response to local radiation. In addition to stimulation ofDNA repair, IR induces multiple cellular signaling pathways.
Importantly, cell survival depends upon the ratio of activatedpro- and antiproliferative pathways, suggesting that irradiatedcells, which evade death, survive and progress to more aggressiveand therapeutically resistant tumors (3). Radiation-inducedsignaling pathways associated with cancer progression includeelevated epidermal growth factor receptor, hypoxia induciblefactor-1 (HIF1), upregulation, and/or activation of matrix metal-loproteinases (MMP), and overexpression of cytokines, includingvascular endothelial growth factor (VEGF) and other immuno-suppressive mediators that promote cancer survival, invasion,and metastasis (4). Thus, the biology of sublethally irradiatedtumor cells favor survival, invasion, and angiogenesis, suggestingthat therapeutic efficacy could be improved by combining radi-ation treatment with agents that target these or other progrowthpathways induced by radiation (5).
Nitric oxide (NO) is an important mediator of many pro-growth signaling cascades in cancer (6–9). Nitric oxidesynthases (NOS) catalyze the production of NO by the five-electron oxidation of a guanidino nitrogen atom of the sub-strate L-arginine, which requires NADPH, FAD, FMN, heme,and O2 as cofactors (10). Three NOS isoforms are known toexist; neuronal NOS (nNOS or NOS1), inducible NOS (iNOSor NOS2), and endothelial NOS (eNOS or NOS3). NO hasmany diverse roles in normal physiology and tumor biology,which are spatially, temporally, and concentration dependent.
1Radiation Biology Branch, Center for Cancer Research, National Can-cer Institute, Bethesda, Maryland. 2Cancer Inflammation Program,Center for Cancer Research, National Cancer Institute, Frederick,Maryland. 3Laboratory of Pathology, Center for Cancer Research,National Cancer Institute, Bethesda, Maryland. 4Radiation OncologyBranch, Center for Cancer Research, National Cancer Institute,Bethesda, Maryland.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author: Lisa A. Ridnour, National Cancer Institute, NIH, Building10, B3B35, 10 Center Drive, Bethesda, MD 20892. Phone: 301-846-5350; Fax:301-846-6641; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-14-3011
�2015 American Association for Cancer Research.
CancerResearch
Cancer Res; 75(14) July 15, 20152788
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

The constitutive isoforms eNOS and nNOS are tightly regulatedby Ca2þ/calmodulin, and produce low flux (pmol/L) NO overshort periods of time. In contrast, the inducible isoform iNOS isCa2þ independent and generates higher flux NO over a longerperiod of time that can range from nmol/L to mmol/L inconcentration, depending upon the stimulant (11). NOS hasbeen studied extensively in carcinogenesis. Although elevatedNOS3 expression has a role in tumor angiogenesis, increasedNOS2 expression predicts poor therapeutic response, tumorprogression, and decreased patient survival (9, 12–15). To date,our molecular signatures suggest that NO-mediated prosurvi-val, cell migration, angiogenesis, and stem cell marker (i.e.,ERK, Akt, IL8, IL6, S100A8, CD44) signaling in tumors andtumor cells occurs at �400 nmol/L steady-state NO (6, 9).Together, these observations suggest that the NOS enzymes areexploitable therapeutic targets.
NO produced by the constitutive eNOS isoform controlsblood flow and is a key mediator of the pro-angiogenic effectsof vascular endothelial growth factor (VEGF; ref. 16). A clinicalstudy demonstrated reduced tumor blood volume within 1 hourof administration of the competitive NOS inhibitor nitro-L-arginine (L-NNA), which lasted for 24 hours in all patientsstudied (17). Side effects of NOS inhibition included bradycar-dia and hypertension, which were not study limiting and suggestthat NOS inhibition may be a beneficial therapeutic option forcombined modalities (17). Advances in radiotherapy haveincluded the co-administration of anti-angiogenic (AA) drugs,which radiosensitize endothelial cells (18). IR also activatesconstitutive NOS, as well as ERK1/2 kinase prosurvival signal-ing, both of which were blocked by L-NNA (19). In addition,the administration of L-NNA 24 hours prior to 10 Gy irradi-ation of tumor-bearing mice demonstrated reduced tumorblood flow and increased tumor cell apoptosis when comparedto mice receiving radiation or L-NNA alone, further supportingNOS inhibition as a target to improve radiation therapeuticresponse (20).
In addition to targeting tumor vasculature, IR modulates hostimmunity and mimics vaccine response by enhancing therelease of damage-associated molecular patterns (DAMP) fromdying cells, which then activate cytotoxic lymphocytes (CTL)through toll-like receptor activation (21). IR facilitates antigen-presenting cell and T-cell penetration into the tumor (22), andalso impacts host immunity through modulation of both pro-and antitumor responses depending upon the Th1 (cytotoxic)versus Th2 (immunosuppressive) cytokine milieu and associ-ated immune cell mediators (21). Macrophages exposed toTh1 cytokines exhibit increased levels of pro-inflammatorycytokine production, antigen presentation, and cytotoxic activ-ity. In contrast, macrophages exposed to Th2 cytokines exhibitan immunosuppressive phenotype associated with blockedCTL activity, increased angiogenesis, tissue restoration, andwound healing response (23). T-regulatory cells (Tregs) arepivotal mediators of immune suppression and the develop-ment of immunologic tolerance through their ability to limitantitumor immune responses (24). Tregs mediate tumorimmune tolerance in part through the secretion of IL10, TGFb,or IL35 immunosuppressive molecules (24). Indeed, IL10 andTGFb derived from Tregs promote tumor progression throughantitumor immune suppression (25). NO also mediates bothcGMP-dependent and -independent Th1–Th2 immune transi-tion and may be important in the tumor response to radiation-
induced injury (26, 27). To explore this hypothesis, we exam-ined cytokine expression profiles and T-cell activation duringradiation-induced tumor growth delay in a syngeneic modeltreated post-IR with the NOS inhibitor NG-nitro-L-argininemethyl ester (L-NAME).
Materials and MethodsCell culture
Jurkat cells (Jurkat clone E6-1) were obtained from the ATCCand maintained in 5% CO2, RPMI-1640 culture medium with10% FBS and 100 IU/mL Pen Strep antibiotics (Life Techno-logies). Jurkat cells were treated with IR (0, 1, or 5 Gy) in thepresence or absence of various concentrations of DETA/NO(0, 30, 60, 100, 300, 500, and 100 mmol/L), L-NAME (0, 500,or 1,000 mmol/L), or the guanylyl cyclase inhibitor ODQ (10mmol/L) or thrombospondin-1 (TSP-1: 1 mg/mL). The NONOatedonors, includingDETA/NO, are stable whenmaintained in basicconditions (10 mmol/L NaOH) but release NO at defined ratesat physiologic pH (28); 10 mmol/L NaOH served as vehiclecontrol in experiments utilizing the NO donor DETA/NO.The ANA-1 macrophage cell line used in this study was establish-ed by immortalization of bone marrow macrophages fromC57BL/6 mice with J2 recombinant retrovirus-expressingv-myc/v-raf oncogenes (29). ANA-1 cells were grown in DMEMsupplemented with 10% FBS and 1% penicillin–streptomycin,plated at a density of 5� 105 perwell in a 12-well plate and grownovernight.
Suppression of IL10Silencing of IL10 protein translation was accomplished by
using an antisense 25-mer oligo (Gene Tools) designed specifi-cally to block the AUG translational start site of mouse IL10(GenBank accession no. NM_010548: oligo sequence, 5-AGCTCTCTTTTCTGCAAGGCTGCTT). This oligo complementsthe sequence from �31 to �6 relative to the initiation codon.Suppression of secreted IL10 protein levels were verified in lipo-polysaccharide (LPS)-stimulated Raw 267.4 (30) cells pretreatedwith control or IL10morpholino.Cell culturemediawas collectedat 24 and 48 hours and IL10 protein levels were measured byELISA assay (R&D Systems) according to the manufacturer'srecommendations.
In vivo mouse tumor modelThe Animal Care and Use Committee (National Cancer
Institute, NIH, Bethesda, MD) approved mouse protocols.Female C3H/Hen or athymic nude mice were supplied by theFrederick Cancer Research and Development Center AnimalProduction Area (Frederick, MD). The animals were receivedat 6 weeks of age, housed five per cage, and given autoclavedfood and water ad libitum. Experiments were performed at 9 to10 weeks of age and in accordance with principles outlined inthe Guide for the Care and Use of Laboratory Animals (Institute ofLaboratory Animal Resources, National Research Council,Washington, DC). Squamous cell carcinoma VII/SF tumor cells(SCC) were derived from spontaneous abdominal wall squa-mous cell cancer (obtained from Dr. T. Phillips, UCSF, SanFrancisco, CA) and propagated in C3H/Hen mice (31). Forgrowth delay studies, 2 � 105 viable SCC cells were injected intothe subcutaneous space of the right hind leg of 8-week-old
NOS Inhibition Alters Post-IR Tumor Immune Response
www.aacrjournals.org Cancer Res; 75(14) July 15, 2015 2789
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

C3H/Hen or nude mice and grown for 1 week when tumor sizereached approximately 200 mm3 in size. Similarly, humancolon carcinoma HT29 tumor xenografts were grown in nudemice injected with 1 � 106 cells. C57BL/6 WT or eNOS�/� (TheJackson Laboratory Stock No. 002684) mice on the samebackground were injected with 1 � 106 B16 melanoma cells.SNP analysis (DartMouse, The Geisel School of Medicine atDartmouth, Dartmouth, NH) demonstrated background puri-ties of C57BL/6 WT and eNOS�/� mice to be 99.8% and 98.9%,respectively, when compared to the in-house control. Tumorvolume was measured by caliper and calculated as mm3 ¼[width2 � length]/2 where width was the smaller dimension.Tumor irradiation was accomplished by securing each animal ina specially designed Lucite jig fitted with lead shielding thatprotected the body from radiation while allowing exposure ofthe tumor-bearing leg. A Therapax DXT300 X-ray irradiator(Pantak, Inc.) using 2.0 mm A1 filtration (300 KVp) at a doserate of 2.53 Gy/min was used as the X-ray source. Irradiatedtumors received one 10 Gy dose. Designated groups of animalswere treated with NOS inhibitor L-NAME or IL10 suppressingagents. NOS inhibition was achieved by administering L-NAMEpost-IR in the drinking water at a concentration of 0.5 g/L forthe duration of the experiment (20). IL10 protein levels weresuppressed using an IL10 morpholino; mice were injected witha 750 mL volume of 10 mmol/L IL10 morpholino (Gene Tools)or a four base-mismatched control morpholino in saline 48hours prior to irradiation. After irradiation, the mice werereturned to their cages, and tumors were measured three timeseach week thereafter to assess tumor growth. Animals wereeuthanized when tumor growth approached the maximumallowable limit.
Cytokine screenControl and irradiated tumors (�L-NAME) were collected at 0,
0.25, 1, 2, 3, 4, and 7 days post-irradiation. Cytokine proteinexpression was evaluated by Q-Plex multiplex ELISA arrays(QUANSYS Biosciences).
Isolation of leukocytes from spleenSpleens were harvested from tumor-bearing animals, placed in
sterile saline, and filtered through a two-chamber sterile Filtra-Bag(Fisher Scientific). Splenocytes were counted by Sysmex KX-21(Roche Diagnostics).
Isolation of tumor-infiltrating leukocytesTumors were dissected and filtered through a two-chamber
sterile Filtra-Bag (Fisher Scientific), then digested in RPMIcontaining 5% fetal calf serum, 700 units/mL collagenase (Invi-trogen), 100 mg/mL DNAse I (Boehringer Mannheim), and 1mmol/L EDTA (pH8.0), at 37�C for 45minutes. The homogenatewas then processed in a tissue stomacher-80 (Seward) for 30seconds, washed with HBSS (BioWhittaker), and resuspended in40% Percoll (Amersham Pharmacia) in DMEM medium (Bio-Whittaker). The suspension was underlaid with 80% Percoll andcentrifuged for 25minutes at 1,000� g. Leukocytes were collectedfrom the interphase, washed, and counted.
Flow cytometryCells (1 � 106) were incubated in cell staining buffer (0.1%
BSA, 0.1% sodium azide) containing 250 mg/mL 2.4G2 ascites,which blocks nonspecific Fc receptor antibody binding for 15
minutes. Cells were stained with fluorescently conjugated anti-bodies (BD Pharmingen) for 20 minutes. Labeled cells werewashed twice in cell staining buffer and analyzed on a BD FacsCanto II flow cytometer (Becton Dickinson).
RNA extraction, reverse transcription, and quantitative real-time PCR
Total RNA was extracted with TRizol (Invitrogen) accordingto the manufacturer's protocol. RNA samples were reversetranscribed into cDNA using Sprint RT Complete 8-well strips(Clontech) according to the manufacturer's recommendation.Primer pairs were designed for IL10 that recognized F:TTAAGGGTTACCTGGGTTGC and R: GCCTGAGGGTCTT-CAGGTTC sequences using the IL10 gene Ref Seq sequences byPrimer3 (32). All quantitative real-time PCR reactions weredesigned to follow a universal real time PCR condition: 94�C 2minutes, 45 cycles of 94�C 30 seconds, and 60�C 30 seconds inPowerSYBR Master Mix (Applied Biosystems). Amplicon speci-ficity was checked by BLAST search and on a 2% FlashGel (Lonza)to ensure that neither nonspecific amplicons nor primer–dimercomplexes were formed. Relative expression was calculated usingthe ddCt formula. Amplification efficiencies for primers werechecked against the 18S housekeeping gene to ensure signalswere detected in PCR exponential phase and that primers hadsimilar amplification efficiencies.
Jurkat and CD47-deficient JinB8 Jurkat cells (�1� 106; ref. 33)were plated on 12-well plates (Corning) using RPM1 medium þ2% FBS at 37�C in 5% CO2 and treated with 1 mg/mL TSP-1 for6 hours. Untreated cells were used as controls. Total RNA wasextracted using TriPure Isolation Reagent (Roche). The firststrand cDNA was made using Maxima First Strand cDNASynthesis Kit for RT-qPCR (Thermo Scientific). Primersequences for hypoxanthine phosphoribosyltransferase 1(HPRT1; 50-ATT GTA ATG ACC AGT CAA CAG GG-30/50-GCATTG TTT TGC CAG TGT CAA-30) and IL10 (50-AAA TTA GCCGGG CAT GGT GG-30/50-CTG CAA CTT CCA TCT CCT GGG T-30) were used for real-time PCR performed using SYBR Green(Roche) on an MJ Research Opticon I instrument (Bio-Rad)with the following amplification program: 95�C for 15 min-utes, followed by 40 cycles of 95�C for 15 seconds, 58�C for 20seconds, 72�C for 25 seconds, and 72�C for 1 minute. Meltingcurves were performed for each product from 30�C to 95�C,reading every 0.5�C with a 6-second dwell time. Fold change inmRNA expression was calculated by normalizing to HPRT1mRNA level. Two-factor ANOVA with replication was used forstatistics analysis.
Automated capillary Western blotWestern blots were performed using WES, an automated
capillary-based size sorting system (ProteinSimple; ref. 34). Allprocedures were performed with manufacturer's reagentsaccording to their user manual. Briefly, 8 mL of diluted proteinlysate was mixed with 2 mL of 5� fluorescent master mix andheated at 95�C for 5 minutes. The samples (1 mg), blockingreagent, wash buffer, primary antibodies, secondary antibodies,and chemiluminescent substrate were dispensed into designat-ed wells in a manufacturer provided microplate. The platewas loaded into the instrument and protein was drawn intoindividual capillaries on a 25 capillary cassette provided bythe manufacturer. Protein separation and immunodetectionwas performed automatically on the individual capillaries
Ridnour et al.
Cancer Res; 75(14) July 15, 2015 Cancer Research2790
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

using default settings. The data were analyzed using Compasssoftware (ProteinSimple; ref. 34). Primary antibodies usedwere nNOS, eNOS (Cell Signaling), and iNOS (Santa CruzBiotech) HPRT was used as loading control (Santa CruzBiotech).
Statistical analysisAll results are expressed as the mean � SEM. The differences in
means of groups were determined by the Student t test with theminimum level of significance set at P � 0.05.
ResultsNOS inhibition enhances radiation-induced tumor growthdelay in syngeneic mice
Tumor growth delay is described by the substance enhance-ment ratio (SER), which is the ratio of time required for treatedversus control tumors to reach a defined size (1,000 mm3). Theeffect of NOS inhibition by L-NAME, a NOS inhibitor that is
more selective for the constitutive isoforms (eNOS and nNOS;ref. 35), on radiation-induced tumor growth delay was examinedin a syngeneic murine model of SCC tumor-bearing C3H mice.L-NAMEwas administered in the animals' drinkingwater (0.5 g/L)following tumor irradiation (post-IR, 10 Gy). Because NO reg-ulates vascular tonicity, we chose post-IR administrationof L-NAME to minimize vascular constriction and maintaintumor pO2 prior to and during tumor irradiation while targetingvascular constriction post-IR. Figure 1A demonstrates enhancedradiation-induced tumor growth delay in mice that receivedpost-IR L-NAME (SER �3.3) when compared to tumors treatedwith 10 Gy IR alone (SER �1.8). The iNOS-specific inhibitoraminoguanidine was also tested, and yielded an SER of 2.1.Interestingly, L-NAME–mediated NOS inhibition had no effecton the radiation-induced tumor growth delay of HT29 humanadenocarcinoma cells or SCC xenografts in immunosuppressednude mice lacking T cells (Fig. 1B and C, respectively). Theseresults indicate a requirement of T cells for L-NAME potentiationof radiation-induced tumor growth delay.
A
B
C
151050
0
1,000
2,000
3,000
4,000
Day
Control
15 Gy
L-NAME
15 Gy + L-NAME
403020100
0
500
1,000
1,500
2,000
Day
Control
10 Gy
L-NAME
10 Gy + L-NAME
151050
0
1,000
2,000
3,000
Day
Tum
or v
olum
e (m
m3 )
Tum
or v
olum
e (m
m3 )
Tum
or v
olum
e (m
m3 )
Control
10 Gy
10 Gy + L-NAME
10 Gy + AG
* P = 0.048 day 14
** P = 0.01 day 16**
*
10 Gy + L-NAME vs. 10 Gy + AG P value
Figure 1.Radiation-induced tumor growth delay isenhanced by post-IR administration of L-NAMEand requires cytolytic T cells in SCC tumorxenografts grown in female syngeneic C3H/Henmice. Post-IR aminoguanidine, a selective iNOSinhibitor, was less effective than L-NAME atextending radiation-induced tumor growthdelay (A). Human HT29 colon carcinoma (B) andSCC (C) xenografts grown in female athymicnude mice showed no effect of post-IR L-NAMEon radiation-induced tumor growth delay. Micewere injected with 2 � 105 SCC (A and C) or1 � 106 HT29 (B) tumor cells in the right hindleg and grown for 1 week to allow formation ofpalpable tumors of uniform size (�200 mm3).On day 7, animals received tumorirradiation � post-IR L-NAME (oraminoguanidine) in the animals drinkingwater (0.5 g/L). Data, mean � SEM; n � 5animals per group.
NOS Inhibition Alters Post-IR Tumor Immune Response
www.aacrjournals.org Cancer Res; 75(14) July 15, 2015 2791
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

Effect of L-NAME on radiation-induced cytokine expressionin syngeneic mice
T cells are lymphocytes that direct cell-mediated immunityand are distinguished from other lymphocytes by the presenceof cell surface T-cell receptors. There are several subsets ofT cells, each with a distinct function; proliferating helper Tcells differentiate into two major types of effector T cells knownas Th1 and Th2 cells, which secrete specific cytokines thatmediate different immune responses. Th1 cells are proinflam-matory, mediate host immunity to foreign pathogens, and areinduced by IL2, IL12, and their effector cytokine IFNg (36).In contrast, Th2 cells are immunosuppressive, secrete IL4, IL5,IL10 as well as TGFb, and mediate wound resolution followingpro-inflammatory assault (37). To explore a potential role forNOS-derived NO during radiation-induced T-cell response,QPlex was used to examine alterations in tumor cytokineexpression. Supplementary Table S1 summarizes the impactof NOS inhibition by L-NAME, on the trend of Th1 versus Th2cytokine protein expression induced by 10 Gy tumor irradia-tion, whereas Supplementary Table S2 summarizes pg/mg cyto-kine levels as well as P-values and fold-change, as a function oftime after irradiation. The trend of Th1 versus Th2 cytokineprotein expression summarized in Supplementary Table S1suggests that tumors receiving radiation alone rapidly acquire(within 24 hours) an overall Th2 signaling profile as defined byearly elevation of IL10 (Day 1) followed by increased IL5, IL3,and IL4 tumor expression (Day 2–4). In contrast, tumors fromanimals that received post-IR L-NAME exhibited a Th1 profile asdefined by elevated IL2 (6 hours, Day 1), IL12, and IFNg (Day 3,4, 7) tumor expression. The most profound observation per-tained to the dramatic early induction of IL10 24-hour post-IR,which was abated by L-NAME (Fig. 2A). These results suggestthe rapid induction of an IL10-mediated immunosuppressive
phenotype in response to tumor irradiation, which was abol-ished by post-IR NOS inhibition. We also examined tumorNOS isoform protein expression 24-hour post-IR. When com-pared to control, Fig. 2B shows increased iNOS protein expres-sion in the 10 and 10 Gy þ L-NAME tumors. This is aninteresting observation considering that constitutive NOSinhibition by L-NAME was more effective in extending theradiation-induced tumor growth delay. This may be explainedby the findings of Connelly and colleagues (38) who demon-strated that eNOS is required for the full activation of iNOS.
NO–induced IL10 expression in Jurkat T cells and ANA-1macrophages
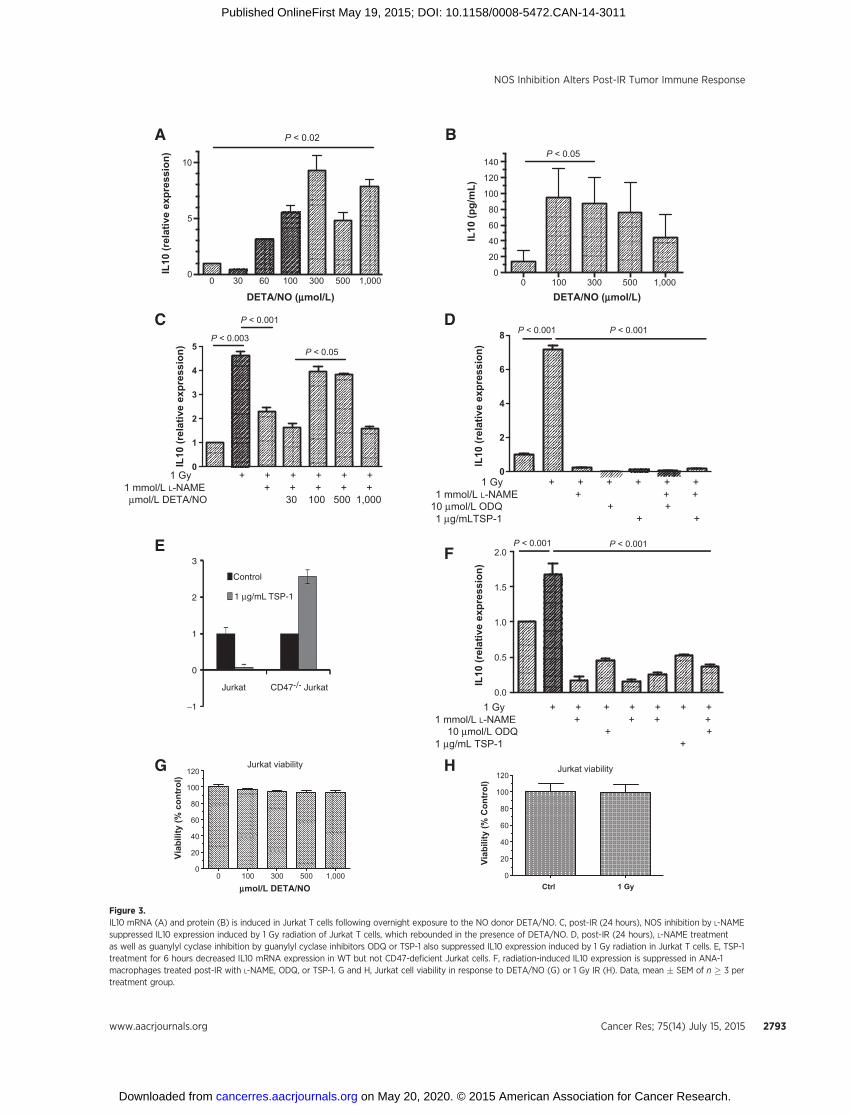
IL10 is generally produced by differentiated monocytes andlymphocytes (i.e., macrophages and T cells, respectively). Tofurther examine the involvement of NO during radiation-induced IL10 expression, we used Jurkat cells, which are Tlymphocytes that express IL10 and are commonly employedto study T-cell signaling. Cytokine expression profiles wereexamined in cells exposed to the slow releasing NO donorDETA/NO, which mimics NO flux under inflammatory condi-tions. Figure 3 demonstrates NO concentration-dependentinduction of IL10 mRNA (A) and protein (B) in Jurkat cells,which peaked at 300 mmol/L DETA/NO. Next, Jurkat cells wereexposed to 1 Gy irradiation, then treated with or withoutL-NAME, and incubated overnight to mimic the tumor xeno-graft irradiation protocol. Figure 3C shows a greater than four-fold increase in Jurkat IL10 expression 24 hours after 1 Gyirradiation, which was abated by L-NAME and is similar to theL-NAME effect on radiation-induced tumor IL10 expressionshown in Fig. 2. Interestingly, the L-NAME suppressed IL10levels re-accumulated to that induced by 1 Gy irradiation in thepresence of the exogenous NO donor DETA/NO at concentra-tions of 100 to 500 mmol/L or �400 nmol/L steady-state NO(Fig. 3C; refs. 6, 7, and 12). To date, our breast cancer bio-marker signatures suggest that NO-mediated prosurvival, cellmigration, angiogenesis, and stem cell marker (i.e., ERK, Akt,IL8, IL6, S100A8, CD44) signaling in tumors and tumor cellsoccurs at �400 nmol/L steady-state NO (6, 7, 9, 12, 39). Whenconsidering this molecular signature, the results shown in Fig.3A–C are consistent with our earlier reports and suggest that�400 nmol/L steady-state NO modulates radiation-inducedIL10 expression in Jurkat cells. Also, the NO flux-dependentregulatory trend of IL10 shown in Fig. 3C resembles a bell-shaped curve, which is consistent with low flux NO regulationof wound response vs. high flux NO-mediated toxicity (11,40–42).
L-NAME is more selective for the constitutive NOS isoforms,which implicates possible eNOS/cGMP-dependent signaling(35, 43). To explore the potential of cGMP-dependent signalingduring radiation-induced IL10 expression, Jurkat cells wereexposed to 1 Gy IR � the guanylyl cyclase inhibitor ODQ, whichcompletely abolished IL10 expression induced by 1 Gy IR (Fig.3D). TSP-1 inhibits NO signaling through its receptor CD47 byinhibiting eNOS activation and abating NO-dependent cGMPsynthesis and cGMP-dependent protein kinase signaling in vas-cular cells and Jurkat T cells (41, 42). Exogenous TSP-1 alsoblocked radiation-induced Jurkat IL10 expression, suggestingeNOS/cGMP-dependence for this process (Fig. 3D). Inhibitionof IL10 expression by TSP-1 is CD47-dependent because
7.004.003.002.001.000.250
1
2
3
4
5
Day
IL10
(p
g/m
g)
Control10 GyL-NAME10 Gy + L-NAME
A
B
< eNOS
< nNOS
< iNOS
< HPRT
Control 10 Gy 10 Gy + L-NAME
Figure 2.Radiation-induced tumor IL10 protein expression is abolished by post-IRL-NAME. Data plotted as mean � SEM of two tumor lysate samples, eachmeasured in triplicate (A). B, NOS isoformprotein expression in control, 10Gy,and 10 Gy þ L-NAME tumors 24 hours following tumor irradiation.
Ridnour et al.
Cancer Res; 75(14) July 15, 2015 Cancer Research2792
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011
A
C
E
D
F
G H
B
1 Gy + + + + + +1 mmol/L L-NAME + + +
10 µmol/L ODQ + +1 µg/mLTSP-1 + +
0
2
4
6
8
IL10
(rel
ativ
e ex
pres
sion
)
P < 0.001 P < 0.001
1 Gy + + + + + +1 mmol/L L-NAME + + + + +µmol/L DETA/NO 30 100 500 1,000
0
1
2
3
4
5
IL10
(rel
ativ
e ex
pres
sion
)
P < 0.003
P < 0.001
P < 0.05
1 Gy + + + + + + +1 mmol/L L-NAME + + + +
10 µmol/L ODQ + +1 µg/mL TSP-1 +
0.0
0.5
1.0
1.5
2.0
IL10
(rel
ativ
e ex
pres
sion
)
P < 0.001P < 0.001
1,000500300100603000
5
10IL
10 (r
elat
ive
expr
essi
on)
DETA/NO (mmol/L) DETA/NO (mmol/L)
P < 0.02
1,00050030010000
20
40
60
80
100
120
140
IL10
(pg/
mL)
P < 0.05
1 GyCtrl0
20
40
60
80
100
120
Viab
ility
(% C
ontr
ol)
Jurkat viability
1,00050030010000
20
40
60
80
100
120
mmol/L DETA/NO
Viab
ility
(% c
ontr
ol)
Jurkat viability
3
2
1
0
−1
Jurkat CD47-/- Jurkat
Control
1 µg/mL TSP-1
Figure 3.IL10 mRNA (A) and protein (B) is induced in Jurkat T cells following overnight exposure to the NO donor DETA/NO. C, post-IR (24 hours), NOS inhibition by L-NAMEsuppressed IL10 expression induced by 1 Gy radiation of Jurkat T cells, which rebounded in the presence of DETA/NO. D, post-IR (24 hours), L-NAME treatmentas well as guanylyl cyclase inhibition by guanylyl cyclase inhibitors ODQ or TSP-1 also suppressed IL10 expression induced by 1 Gy radiation in Jurkat T cells. E, TSP-1treatment for 6 hours decreased IL10 mRNA expression in WT but not CD47-deficient Jurkat cells. F, radiation-induced IL10 expression is suppressed in ANA-1macrophages treated post-IR with L-NAME, ODQ, or TSP-1. G and H, Jurkat cell viability in response to DETA/NO (G) or 1 Gy IR (H). Data, mean � SEM of n � 3 pertreatment group.
NOS Inhibition Alters Post-IR Tumor Immune Response
www.aacrjournals.org Cancer Res; 75(14) July 15, 2015 2793
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

inhibition of basal IL10 mRNA expression was lost in the CD47-deficient Jurkat mutant JinB8 (Fig. 3E). The 2-fold stimulationof IL10 mRNA by TSP-1 in the CD47 mutant is consistent withreported positive effects of TSP-1 on IL10 expression mediatedby the TSP-1 receptor CD36 (44). Radiation also induced IL10in murine ANA-1 macrophages, which was abated by L-NAME,ODQ and TSP-1, indicating that cGMP-dependent regulationof IL10 is not restricted to T cells (Fig. 3F). Collectively, theseresults indicate that radiation-induced IL10 expression in T cells istightly controlled by low constitutive NOS-derived NO flux.
IL10 suppression enhances radiation-induced tumor growthdelay
Post-IR NOS inhibition by L-NAME enhanced radiation-induced tumor growth delay and abated radiation-induced IL10expression (Figs. 1A and 2, respectively). To examine a role of IL10in the recovery from post-IR tumor growth delay, IL10 proteintranslation was suppressed by treatment with an IL10 morpho-lino (45, 46). Confirmation of the morpholino efficacy for IL10protein suppression was verified using LPS-stimulated Raw 267.4cells because LPS is a strong inducer of IL10 in these cells (30) asshown in Fig. 4A. Next, tumor-bearing animals were treated withIL10 or control morpholino 48 hours prior to tumor irradiation.IL10 morpholino treatment enhanced the radiation-inducedtumor growth delay (SER �2.7) as shown in Fig. 4B in a mannersimilar to that observed by L-NAME (Fig. 1A) but had no effect ontumor growth in the absence of radiation. These results suggestthat radiation-induced tumor growth delay can be improved by
inhibiting IL10-mediated immunosuppressive signaling in theC3H/SCC syngeneic model.
L-NAME increases tumor-associated CD8þ cytolytic T cellspost-IR
Cytotoxic T lymphocytes are a subgroup of T cells that whenactivated kill invading pathogens and tumor cells. These cells arecommonly referred to as CD8þ T cells because they express cellsurface CD8 glycoprotein. Importantly, immunosuppressivemolecules, including IL10, can inactivate CD8þ T cells. To identifythe presence of CD8þ T cells, markers of tumor lymphocyteinfiltration were examined in control and 10 Gy � L-NAMEtumors, as well as spleen from tumor-bearing mice. Figure 5Ashows increased tumor-associated CD8þ T cells in irradiatedtumors treated with L-NAME but not spleen taken from the sameanimals (Fig. 5B). CD69 is amarker of T-cell activation. Figure 5Cdemonstrates increased CD8þ CD69þ mean fluorescence inten-sity in infiltrating lymphocytes from irradiated tumors treatedwith L-NAME but not in spleen taken from the same animals (Fig.5D). Importantly, these results implicate a localized tumorresponse culminating in the elevation of activated cytotoxic Tcells in post-IR NOS inhibited tumors. Neutrophils, dendriticcells, and immature myeloid cells from post-IR treated L-NAMEtumors were elevated on day 3, when compared to irradiatedtumors (Fig. 5E–G). In contrast, Tregs and natural killer cells didnot change (Fig. 5H and I). Collectively, these results demonstratethat altered NO flux via L-NAME–mediated NOS inhibition canimprove the efficacy of therapeutic radiation by immune polar-ization favoring a proinflammatory phenotype within the tumormicroenvironment in an SCC/C3H syngeneic model.
L-NAME is more selective for inhibiting the constitutiveNOS isoforms (eNOS and nNOS) and our cell culture resultsimplicate eNOS/cGMP-dependent signaling (Fig. 3) in theimmune polarization shown in Fig. 5. To further explore a roleof eNOS in the potentiation of radiation therapeutic efficacy, weexamined the radiation-induced tumor growth delay of murineB16 melanoma xenografts in wild-type (WT) and eNOS knock-out (eNOS�/�) mice on the C57BL/6 background (Fig. 6A).When compared to control, the irradiated tumor in WT miceyielded an SER of �2 (Fig. 6A), which is consistent with theSCC/C3H syngeneic model shown in Fig. 1A. Remarkably, theirradiated tumor in eNOS�/� animals exhibited an SER of �5.5when compared to the WT-irradiated tumor (Fig. 6A). Interest-ingly, IL10 protein was not detected in B16 xenografts grown inWT C57BL/6 or eNOS�/� on the same background under theseconditions; however, the cytokine protein expression profile ofirradiated tumors in eNOS�/� mice exhibited significantly ele-vated Th1 cytokines, including IL2, TNFa, and IFNg , as shownin Fig. 6B–D. Importantly, radiation has been shown to induceeNOS expression, which promotes tumor recovery from radiationinjury (47). Together, these results suggest that eNOS has a vitalrole in acute tumor radiation response and that improved phar-macology of NOS inhibitors in combination with radiation maybe therapeutically beneficial.
DiscussionThe role of NO flux within the tumor microenvironment
as it relates to therapeutic efficacy is complex. Studies haveshown that steady-state NO modulation within the tumormicroenvironment leads to improved radiation therapeutic
A
B
20151050
0
500
1,000
1,500
2,000
2,500
3,000
3,500
Day
Tu
mo
r vo
lum
e (m
m3 ) Control
IL10 M + 10 Gy * P < 0.05
Cont M + 10 Gy
* * * *
Co
ntr
ol
LP
S
LP
S +
IL10
M
LP
S +
Co
nt
M
0
500
1,000
1,500
2,00024 h48 h
IL10
(p
g/m
L)
Figure 4.A, LPS is a strong inducer of IL10 protein in Raw 267.4 cells, which is abatedby IL10 morpholino (IL10 M). B, IL10 morpholino was administeredintraperitoneally to SCC tumor-bearing C3H mice 48 hours prior to tumorirradiation. IL10 suppression by IL10 morpholino enhanced radiation-inducedtumor growth delay of SCC xenografts. Data, mean � SEM of 5 animals pergroup.
Ridnour et al.
Cancer Res; 75(14) July 15, 2015 Cancer Research2794
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

4320
500
1,000
1,500
Day
% C
D69
/CD
8
Tumor CD8 CD69 MFI
Control
10 Gy
10 Gy + L-NAME
P = 0.003 P = 0.003
4320
1
2
3
4
Day
% C
D8+
cel
ls% CD8+ cells tumor
Control
10 Gy
10 Gy + L-NAME
P < 0.008
10Gy+LN10 GyL-NAMEControl0
10
20
30
% N
K C
ells
% NK cells tumor
4320
100
200
300
400
500
Day
CD
8 C
D69
MF
I (S
ple
en) Control
10 Gy
10 Gy + L-NAME
Spleen CD8 CD69 MFI
% CD8+ cells spleen
4320
5
10
15
Day
% C
D8+
cel
ls (
sple
en) Control
10 Gy
10 Gy + L-NAME
10 Gy+LN10 GyL-NAMEControl0.0
0.1
0.2
0.3
0.4
0.5
% T
reg
s/to
tal %
CD
4s
% Tregs/total % CD4s
% Neutrophils tumor
Day
% N
eutr
op
hils
4320
10
20
30
40
50Control
10 Gy
10 Gy + L-NAMEP < 0.05
% Dendritic cells tumor
4320
5
10
15
Day
% D
end
riti
c ce
lls
Control
10 Gy
10 Gy + L-NAMEP < 0.04
4320
20
40
60
80
% C
D11
b G
r1 L
o
Day
Control
10 Gy
10 Gy + L-NAME
CD11b Gr1 Lo tumor
P < 0.001
A
C
E
G H I
F
D
B
Figure 5.Post-IR NOS inhibition by L-NAME increased percentage of CD8þ T cells and activation measured by CD8 CD69 MFI in tumors (A and C) but not spleen(B and D) in SCC xenografts. Elevations in the percentage of tumor neutrophils, dendritic cells, and immature myeloid cells were also observed on day3 in post-IRþL-NAME tumors, when compared to tumors receiving 10 Gy radiation alone (E–G). Cell surface marker expression was measured byflow cytometry 2 to 4 days (A–G) or 24 hours (H and I) post-IR in SCC xenografts.
NOS Inhibition Alters Post-IR Tumor Immune Response
www.aacrjournals.org Cancer Res; 75(14) July 15, 2015 2795
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

efficacy (20, 47, 48). NO mediates numerous effects at thecellular and physiological levels. Similar to molecular O2, theouter p orbital of NO has an unpaired electron, which imparts ahigh affinity for other radicals, including radiation-inducedcarbon radicals on DNA, which leads to fixed DNA damageand enhanced NO-mediated radiosensitivity (49). In supportof this early observation, a recent study demonstrated thatthe presence of low steady-state NO dramatically enhanced
radiosensitization as well as increased the time of DNA repair,when compared to anoxic and aerated control tumors (50).Similarly, site-specific iNOS transgene expression driven by theradiation-inducible pE9 promoter demonstrated enhancedtumor radiation response under hypoxic conditions (51).In addition to these direct effects of NO-mediated radiosensi-tization, altered NO gradients prior to tumor irradiation havebeen shown to normalize tumor vasculature, which increasedtumor oxygen tension and tumor response to radiation (52).In contrast, administration of the NOS inhibitor L-NAMEbefore and after radiation minimized the cytotoxic effect ofNO under conditions of hypoxia (48). In this context, NOimproved radiation therapeutic efficacy by enhanced tumorperfusion and oxygen effect (53). Thus, NO modulation priorto and at the time of radiation is therapeutically beneficial.Together, these studies demonstrate the contextual depend-ence of timing and distinct mechanisms directed by NO fluxfor improved tumor response to radiotherapy.
Although the modulation of tumor NO flux prior to irradi-ation improves tumor oxygenation and radiation efficacy,NO also promotes angiogenesis in the context of immune-mediated wound response (40–42), which may facilitate post-irradiation recovery of a sublethally irradiated tumor. Indeed,macrophages employ NO generated by both eNOS and iNOSduring wound response (40, 54) and in vivo models haveshown delayed wound closure in iNOS knockout mice (55).Toward this end, IR-induced angiogenesis (56) through NOsignaling (47), which promoted tumor recovery followingradiation injury. These observations suggest that post-irradia-tion inhibition of angiogenesis may be beneficial. Thus, wehypothesized that improved radiation therapeutic efficacy andextended tumor growth delay may be achievable by targetingNO flux through NOS inhibition following tumor irradiation.
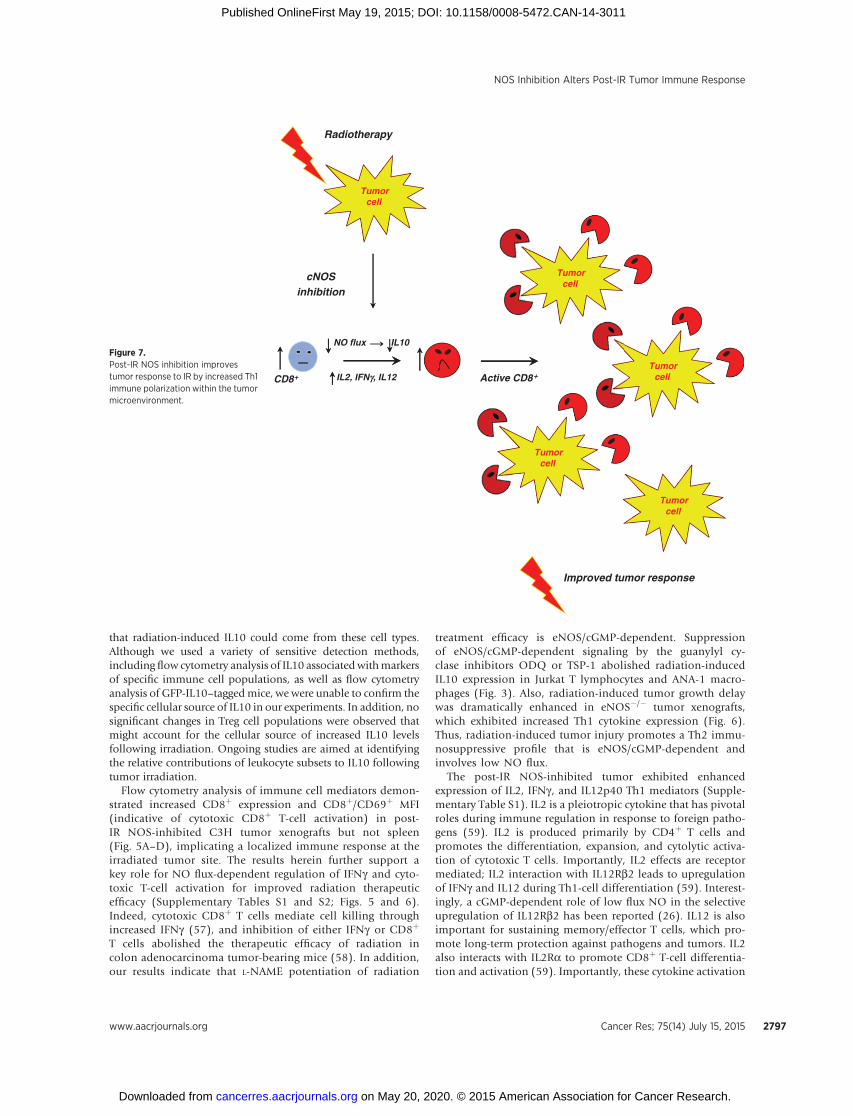
Interestingly, post-IR administration of the constitutive NOSinhibitor L-NAME extended radiation-induced tumor growthdelay and was more effective than the selective iNOS inhibitoraminoguanidine (Fig. 1A). Moreover, L-NAME extended theradiation-induced tumor growth delay only in syngeneic micebut not nude mice. This observation implicates the involve-ment of innate immunity and cytotoxic T cells in enhancedradiosensitivity, which is regulated by NO flux, and furthersupported by the cytokine expression profile of post-IR NOS-inhibited tumors that expressed high levels of cytotoxic Th1cytokines, including IL2, IFNg , and IL12p40, as summarizedin Fig. 7. In contrast, tumors receiving radiation alone exhibitedimmunosuppressive Th2 signaling, as indicated by increasedIL10, IL5, and IL4 cytokine expression (Supplementary TablesS1 and S2). Moreover, tumor cytokine expression analysisrevealed enhanced IL10 protein levels 24 hours followingtumor irradiation in SCC-tumor bearing C3H mice, which wasabolished by L-NAME (Fig. 2) and confirmed in irradiatedJurkat T lymphocytes and ANA-1 macrophages (Fig. 3). Impor-tantly, in vivo IL10 protein suppression extended radiation-induced tumor growth delay in C3H mice in a manner similarto that of L-NAME. These findings implicate a novel role forNO as a stimulator of IL10-mediated tumor immunosuppres-sive signaling, which accelerates tumor recovery and regrowthin response to radiation injury in the C3H model.
Cytokine expression analysis of ANA-1 macrophages, andJurkat T cells demonstrated increased IL10 expression 24 hoursafter 1 Gy irradiation, which was abated by L-NAME, suggesting
0
50
100
150
200
IFN
g (p
g/m
g)
IFNg 72 hWT 72 eNOS–/– 72WT IR 72eNOS–/– IR 72
P = 0.0047
P = 0.0409
0
2
4
6
8
10
IL2
(pg
/mg
)
IL2 72 h WT cont 72
eNOS–/– cont 72
WT IR 72
eNOS–/– IR 72
P = 0.0375
0
20
40
60
80
100
TN
Fa
(pg
/mg
)
TNFa 72 h WT control
eNOS–/– control
WT IR 72
eNOS–/– IR 72
P = 0.049
181614121086420
0
1,000
2,000
3,000
4,000
Day
Tu
mo
r vo
lum
e (m
m3 )
WT
WT+IR SER ~ 2
eNOS-/- SER ~ 2.4
eNOS-/- + IR SER ~ 5.5 * P < 0.01
* * *
A
B
C
D
Figure 6.A, radiation-induced tumor growth delay is enhanced in eNOS�/� mice.C57BL/6 WT or eNOS�/� mice on C57BL/6 background were injected with2 � 105 B16 tumor cells in the right hind leg and grown for 1 week to allowformation of palpable tumors of uniform size (�200mm3). On day 7, animalsreceived tumor irradiation. Data, mean � SEM; n > 5 animals per group. B–D,irradiated tumors fromeNOS�/�mice exhibited elevated protein levels of IL2,TNFa, and IFNg proinflammatory Th1 cytokines.
Ridnour et al.
Cancer Res; 75(14) July 15, 2015 Cancer Research2796
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011
that radiation-induced IL10 could come from these cell types.Although we used a variety of sensitive detection methods,including flow cytometry analysis of IL10 associatedwithmarkersof specific immune cell populations, as well as flow cytometryanalysis of GFP-IL10–taggedmice, we were unable to confirm thespecific cellular source of IL10 in our experiments. In addition, nosignificant changes in Treg cell populations were observed thatmight account for the cellular source of increased IL10 levelsfollowing irradiation. Ongoing studies are aimed at identifyingthe relative contributions of leukocyte subsets to IL10 followingtumor irradiation.
Flow cytometry analysis of immune cell mediators demon-strated increased CD8þ expression and CD8þ/CD69þ MFI(indicative of cytotoxic CD8þ T-cell activation) in post-IR NOS-inhibited C3H tumor xenografts but not spleen(Fig. 5A–D), implicating a localized immune response at theirradiated tumor site. The results herein further support akey role for NO flux-dependent regulation of IFNg and cyto-toxic T-cell activation for improved radiation therapeuticefficacy (Supplementary Tables S1 and S2; Figs. 5 and 6).Indeed, cytotoxic CD8þ T cells mediate cell killing throughincreased IFNg (57), and inhibition of either IFNg or CD8þ
T cells abolished the therapeutic efficacy of radiation incolon adenocarcinoma tumor-bearing mice (58). In addition,our results indicate that L-NAME potentiation of radiation
treatment efficacy is eNOS/cGMP-dependent. Suppressionof eNOS/cGMP-dependent signaling by the guanylyl cy-clase inhibitors ODQ or TSP-1 abolished radiation-inducedIL10 expression in Jurkat T lymphocytes and ANA-1 macro-phages (Fig. 3). Also, radiation-induced tumor growth delaywas dramatically enhanced in eNOS�/� tumor xenografts,which exhibited increased Th1 cytokine expression (Fig. 6).Thus, radiation-induced tumor injury promotes a Th2 immu-nosuppressive profile that is eNOS/cGMP-dependent andinvolves low NO flux.
The post-IR NOS-inhibited tumor exhibited enhancedexpression of IL2, IFNg , and IL12p40 Th1 mediators (Supple-mentary Table S1). IL2 is a pleiotropic cytokine that has pivotalroles during immune regulation in response to foreign patho-gens (59). IL2 is produced primarily by CD4þ T cells andpromotes the differentiation, expansion, and cytolytic activa-tion of cytotoxic T cells. Importantly, IL2 effects are receptormediated; IL2 interaction with IL12Rb2 leads to upregulationof IFNg and IL12 during Th1-cell differentiation (59). Interest-ingly, a cGMP-dependent role of low flux NO in the selectiveupregulation of IL12Rb2 has been reported (26). IL12 is alsoimportant for sustaining memory/effector T cells, which pro-mote long-term protection against pathogens and tumors. IL2also interacts with IL2Ra to promote CD8þ T-cell differentia-tion and activation (59). Importantly, these cytokine activation
CD8+ Active CD8+
NO flux IL10
Radiotherapy
Improved tumor response
Tumorcell
Tumorcell
Tumorcell
Tumorcell
IL2, IFNg, IL12
Tumorcell
cNOSinhibition
Figure 7.Post-IR NOS inhibition improvestumor response to IR by increased Th1immune polarization within the tumormicroenvironment.
NOS Inhibition Alters Post-IR Tumor Immune Response
www.aacrjournals.org Cancer Res; 75(14) July 15, 2015 2797
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

profiles are consistent with the time course analysis showingelevated IL2, IFNg , and IL12p40 in the post-IR NOS-inhibitedtumors summarized in Supplementary Table S1, as well asCD8þ T-cell regulation shown in Fig. 5. In contrast, tumorsreceiving irradiation alone demonstrated increased IL10 fol-lowed by elevated Th2 mediators IL5, IL3, and IL4. Interest-ingly, IL2 was also elevated in these irradiated tumors andmay have played a role in the upregulation of IL4 and IL5 Th2cell differentiation, which is IL4Ra dependent (59). The pro-inflammatory cytokine IL1a was also observed in the irra-diated tumor. Despite its proinflammatory status, IL1a releasedby solid tumors acts as a chemoattractant to facilitate malig-nancy-associated inflammatory responses (60).
Collectively, the results herein suggest a novel mechanism oflow flux NO during Th1–Th2 transition, tumor immunosuppres-sive signaling, and accelerated wound recovery in the tumorresponse to ionizing irradiation. Importantly, CD8þ T-cell regu-lation and IFNg expression seem to be determined by NO flux-dependent IL10 versus IL2 signaling cascades, which can bemodulated by pharmacological NOS inhibition andmay providea novel immunotherapeutic approach for improved radiationtherapeutic efficacy.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: L.A. Ridnour, R.Y.S. Cheng, D.D. Roberts, H.A. Young,J.B. Mitchell, G. Trinchieri, D.A. WinkDevelopment of methodology: R.Y.S. Cheng, J.M. Weiss, D.R. Soto-Pantoja,A.H. Kesarwala, D.A. WinkAcquisition of data (provided animals, acquired and managed patients, pro-vided facilities, etc.): L.A. Ridnour, R.Y.S. Cheng, J.M. Weiss, S. Kaur, D.R. Soto-Pantoja, D. Basudhar, J.L. Heinecke, W. DeGraff, A.L. Sowers, J.B. MitchellAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): L.A. Ridnour, R.Y.S. Cheng, J.M. Weiss, S. Kaur,D.R. Soto-Pantoja, D. Basudhar, C.A. Stewart, D.A. WinkWriting, review, and/or revisionof themanuscript: L.A. Ridnour, R.Y.S. Cheng,D. Basudhar, J.L. Heinecke, A.H. Kesarwala, D.D. Roberts, H.A. Young,G. Trinchieri, R.H. Wiltrout, D.A. WinkAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): W. DeGraffStudy supervision: D.D. RobertsOther (performed animal experiments): A. Thetford
Grant SupportThis researchwas supportedby the IntramuralResearchProgramof theNational
InstitutesofHealth,NationalCancer Institute, andCenter forCancerResearch (D.A.Wink, D.D. Roberts, H.A. Young, J.B. Mitchell, G. Trinchieri, and R.H. Wiltrout).
The costs of publication of this article were defrayed in part by the paymentof page charges. This article must therefore be hereby marked advertisementin accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received October 10, 2014; revised April 21, 2015; accepted May 8, 2015;published OnlineFirst May 19, 2015.
References1. El Kaffas A, Tran W, Czarnota GJ. Vascular strategies for enhancing
tumour response to radiation therapy. Technol Cancer Res Treat 2012;11:421–32.
2. Bernier J, Hall EJ, Giaccia A. Radiation oncology: a century of achievements.Nat Rev Cancer 2004;4:737–47.
3. Kargiotis O, Geka A, Rao JS, Kyritsis AP. Effects of irradiation on tumorcell survival, invasion and angiogenesis. J Neurooncol 2010;100:323–38.
4. Bergers G, HanahanD.Modes of resistance to anti-angiogenic therapy. NatRev Cancer 2008;8:592–603.
5. Prise KM, Schettino G, Folkard M, Held KD. New insights on cell deathfrom radiation exposure. Lancet Oncol 2005;6:520–8.
6. Ridnour LA, BaraschKM,WindhausenAN,Dorsey TH, LizardoMM,YfantisHG, et al. Nitric oxide synthase and breast cancer: role of TIMP-1 in NO-mediated Akt activation. PLoS One 2012;7:e44081.
7. Switzer CH, Glynn SA, Cheng RY, Ridnour LA, Green JE, Ambs S, et al.S-Nitrosylation of EGFR and Src activates an oncogenic signaling net-work in human basal-like breast cancer.Mol Cancer Res 2012;10:1203–15.
8. Switzer CH, Cheng RY, Ridnour LA, Glynn SA, Ambs S, Wink DA. Ets-1 is atranscriptional mediator of oncogenic nitric oxide signaling in estrogenreceptor-negative breast cancer. Breast Cancer Res 2012;14:R125.
9. Glynn SA, Boersma BJ, Dorsey TH, Yi M, Yfantis HG, Ridnour LA, et al.Increased NOS2 predicts poor survival in estrogen receptor-negative breastcancer patients. J Clin Invest 2010;120:3843–54.
10. Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J1994;298 (Pt 2):249–58.
11. Ridnour LA, Thomas DD, Donzelli S, Espey MG, Roberts DD, Wink DA,et al. The biphasic nature of nitric oxide responses in tumor biology.Antioxid Redox Signal 2006;8:1329–37.
12. Heinecke JL, Ridnour LA, Cheng RY, Switzer CH, Lizardo MM, Khanna C,et al. Tumormicroenvironment-based feed-forward regulation of NOS2 inbreast cancer progression. Proc Natl Acad Sci U S A 2014;111:6323–8.
13. Ekmekcioglu S, Ellerhorst J, Smid CM, Prieto VG, Munsell M, Buzaid AC,et al. Inducible nitric oxide synthase and nitrotyrosine in humanmetastaticmelanoma tumors correlate with poor survival. Clin Cancer Res 2000;6:4768–75.
14. Zhang W, He XJ, Ma YY, Wang HJ, Xia YJ, Zhao ZS, et al. Inducible nitricoxide synthase expression correlates with angiogenesis, lymphangiogen-
esis, and poor prognosis in gastric cancer patients. Hum Pathol 2011;42:1275–82.
15. Eyler CE,WuQ, Yan K,MacSwords JM, Chandler-Militello D,Misuraca KL,et al. Glioma stem cell proliferation and tumor growth are promoted bynitric oxide synthase-2. Cell 2011;146:53–66.
16. Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxideproduction contributes to the angiogenic properties of vascular endo-thelial growth factor in human endothelial cells. J Clin Invest 1997;100:3131–9.
17. Ng QS, Goh V, Milner J, Stratford MR, Folkes LK, Tozer GM, et al. Effect ofnitric-oxide synthesis on tumour blood volume and vascular activity: aphase I study. Lancet Oncol 2007;8:111–8.
18. Cuneo KC, Geng L, Fu A, Orton D, Hallahan DE, Chakravarthy AB.SU11248 (sunitinib) sensitizes pancreatic cancer to the cytotoxic effectsof ionizing radiation. Int J Radiat Oncol Biol Phys 2008;71:873–9.
19. Leach JK, Black SM, Schmidt-Ullrich RK, Mikkelsen RB. Activation ofconstitutive nitric-oxide synthase activity is an early signaling eventinduced by ionizing radiation. J Biol Chem 2002;277:15400–6.
20. Cardnell RJ, Mikkelsen RB. Nitric oxide synthase inhibition enhances theantitumor effect of radiation in the treatment of squamous carcinomaxenografts. PLoS One 2011;6:e20147.
21. Roses RE, Datta J, Czerniecki BJ. Radiation as immunomodulator:implications for dendritic cell-based immunotherapy. Radiat Res 2014;182:211–8.
22. Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination ofT-cell therapy and trigger of inflammation induces remodeling of thevasculature and tumor eradication. Cancer Res 2002;62:1462–70.
23. Kaur P, Asea A. Radiation-induced effects and the immune system incancer. Front Oncol 2012;2:191.
24. Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumorimmune escape and angiogenesis. Cancer Res 2012;72:2162–71.
25. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, WhitesideTL. A unique subset of CD4þCD25highFoxp3þ T cells secreting interleu-kin-10 and transforming growth factor-b1 mediates suppression in thetumor microenvironment. Clin Cancer Res 2007;13:4345–54.
26. Niedbala W, Wei XQ, Campbell C, Thomson D, Komai-KomaM, Liew FY.Nitric oxide preferentially induces type 1 T cell differentiation by selectively
Ridnour et al.
Cancer Res; 75(14) July 15, 2015 Cancer Research2798
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

up-regulating IL-12 receptor b2 expression via cGMP. Proc Natl Acad SciU S A 2002;99:16186–91.
27. NiedbalaW, Cai B, LiuH, PitmanN, Chang L, Liew FY.Nitric oxide inducesCD4þCD25þ Foxp3 regulatory T cells from CD4þCD25 T cells via p53,IL-2, and OX40. Proc Natl Acad Sci U S A 2007;104:15478–83.
28. Davies KM,Wink DA, Saavedra JE, Keefer LK. Chemistry of the diazenium-diolates. 2. Kinetics and mechanism of dissociation to nitric oxide inaqueous solution. J Am Chem Soc 2001;123:5473–81.
29. Cox GW, Mathieson BJ, Gandino L, Blasi E, Radzioch D, Varesio L.Heterogeneity of hematopoietic cells immortalized by v-myc/v-raf recom-binant retrovirus infection of bone marrow or fetal liver. J Natl Cancer Inst1989;81:1492–6.
30. Chou MI, Hsieh YF, Wang M, Chang JT, Chang D, Zouali M, et al. In vitroand in vivo targeted delivery of IL-10 interfering RNA by JC virus-likeparticles. J Biomed Sci 2010;17:51–9.
31. Saito K, Matsumoto S, Yasui H, Devasahayam N, Subramanian S, Muna-singhe JP, et al. Longitudinal imaging studies of tumor microenvironmentin mice treated with the mTOR inhibitor rapamycin. PLoS One 2012;7:e49456.
32. Rozen S, Skaletsky H. Primer3 on the WWW for general users and forbiologist programmers. Methods Mol Biol 2000;132:365–86.
33. ReinholdMI,Green JM, Lindberg FP, TicchioniM, BrownEJ. Cell spreadingdistinguishes the mechanism of augmentation of T cell activation byintegrin-associated protein/CD47 and CD28. Int Immunol 1999;11:707–18.
34. Beccano-Kelly DA, Kuhlmann N, Tatarnikov I, Volta M, Munsie LN, ChouP, et al. Synaptic function is modulated by LRRK2 and glutamate releaseis increased in cortical neurons of G2019S LRRK2 knock-in mice.Front Cell Neurosci 2014;8:301.
35. Boer R, Ulrich WR, Klein T, Mirau B, Haas S, Baur I. The inhibitory potencyand selectivity of arginine substrate site nitric-oxide synthase inhibitorsis solely determined by their affinity toward the different isoenzymes.Mol Pharmacol 2000;58:1026–34.
36. Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into thecomplex roles of IL-2 as a broad regulator of T helper cell differentiation.Curr Opin Immunol 2011;23:598–604.
37. Stout RD, Watkins SK, Suttles J. Functional plasticity of macrophages: insitu reprogramming of tumor-associated macrophages. J Leukoc Biol2009;86:1105–9.
38. Connelly L, Jacobs AT, Palacios-Callender M, Moncada S, Hobbs AJ.Macrophage endothelial nitric-oxide synthase autoregulates cellular acti-vation and pro-inflammatory protein expression. J Biol Chem 2003;278:26480–7.
39. Thomas DD, Espey MG, Ridnour LA, Hofseth LJ, Mancardi D, Harris CC,et al.Hypoxic inducible factor 1a, extracellular signal-regulated kinase, andp53 are regulated by distinct threshold concentrations of nitric oxide. ProcNatl Acad Sci U S A 2004;101:8894–9.
40. Ridnour LA,WindhausenAN, Isenberg JS, YeungN, ThomasDD, VitekMP,et al.Nitric oxide regulatesmatrixmetalloproteinase-9 activity by guanylyl-cyclase-dependent and -independent pathways. Proc Natl Acad Sci U S A2007;104:16898–903.
41. Ridnour LA, Isenberg JS, Espey MG, Thomas DD, Roberts DD, Wink DA.Nitric oxide regulates angiogenesis through a functional switch involvingthrombospondin-1. Proc Natl Acad Sci U S A 2005;102:13147–52.
42. Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, RobertsDD. Thrombospondin-1 inhibits endothelial cell responses to nitric
oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A 2005;102:13141–6.
43. Zhang YH, Casadei B. Sub-cellular targeting of constitutive NOS inhealth and disease. J Mol Cell Cardiol 2012;52:341–50.
44. Zhao Y, Xiong Z, Lechner EJ, Klenotic PA, Hamburg BJ, Hulver M, et al.Thrombospondin-1 triggers macrophage IL-10 production and promotesresolution of experimental lung injury. Mucosal Immunol 2014;7:440–8.
45. Morcos PA, Li Y, Jiang S. Vivo-Morpholinos: a non-peptide transporterdelivers Morpholinos into a wide array of mouse tissues. Biotechniques2008;45:613–4, 6, 8 passim.
46. Soto-Pantoja DR, Ridnour LA, Wink DA, Roberts DD. Blockade of CD47increases survival of mice exposed to lethal total body irradiation. Sci Rep2013;3:1038.
47. Sonveaux P, Brouet A, Havaux X, Gregoire V, Dessy C, Balligand JL, et al.Irradiation-induced angiogenesis through the up-regulation of the nitricoxide pathway: implications for tumor radiotherapy. Cancer Res 2003;63:1012–9.
48. Nagane M, Yasui H, Yamamori T, Zhao S, Kuge Y, Tamaki N, et al.Radiation-induced nitric oxide mitigates tumor hypoxia and radioresis-tance in a murine SCCVII tumor model. Biochem Biophys Res Commun2013;437:420–5.
49. Howard-Flanders P. Effect of nitric oxide on the radiosensitivity of bacteria.Nature 1957;180:1191–2.
50. Wardman P, Rothkamm K, Folkes LK, Woodcock M, Johnston PJ. Radio-sensitization by nitric oxide at low radiation doses. Radiat Res 2007;167:475–84.
51. Coulter JA, McCarthy HO, Worthington J, Robson T, Scott S, Hirst DG.The radiation-inducible pE9 promoter driving inducible nitric oxidesynthase radiosensitizes hypoxic tumour cells to radiation. Gene Ther2008;15:495–503.
52. Kashiwagi S, Tsukada K, Xu L, Miyazaki J, Kozin SV, Tyrrell JA, et al.Perivascular nitric oxide gradients normalize tumor vasculature. Nat Med2008;14:255–7.
53. Thoday JM, Read J. Effect of oxygen on the frequency of chromosomeaberrations produced by a-rays. Nature 1949;163:133–4.
54. Schwentker A, Vodovotz Y, Weller R, Billiar TR. Nitric oxide and woundrepair: role of cytokines? Nitric Oxide 2002;7:1–10.
55. Yamasaki K, Edington HD, McClosky C, Tzeng E, Lizonova A, Kovesdi I,et al. Reversal of impaired wound repair in iNOS-deficient mice by topicaladenoviral-mediated iNOS gene transfer. J Clin Invest 1998;101:967–71.
56. Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 toregulate vascular radiosensitivity in tumors: role of reoxygenation, freeradicals, and stress granules. Cancer Cell 2004;5:429–41.
57. Bernstein MB, Garnett CT, Zhang H, Velcich A, Wattenberg MM, GameiroSR, et al. Radiation-inducedmodulation of costimulatory and coinhibitoryT-cell signaling molecules on human prostate carcinoma cells promotesproductive antitumor immune interactions. Cancer Biother Radiopharm2014;29:153–61.
58. Gerber SA, Sedlacek AL, Cron KR,Murphy SP, Frelinger JG, Lord EM. IFN-gmediates the antitumor effects of radiation therapy in a murine colontumor. Am J Pathol 2013;182:2345–54.
59. Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effectorresponses, tolerance, and immunotherapy. Immunity 2013;38:13–25.
60. Nozaki S, Sledge GW Jr., Nakshatri H. Cancer cell-derived interleukin 1acontributes to autocrine and paracrine induction of pro-metastatic genes inbreast cancer. Biochem Biophys Res Commun 2000;275:60–2.
www.aacrjournals.org Cancer Res; 75(14) July 15, 2015 2799
NOS Inhibition Alters Post-IR Tumor Immune Response
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011

2015;75:2788-2799. Published OnlineFirst May 19, 2015.Cancer Res Lisa A. Ridnour, Robert Y.S. Cheng, Jonathan M. Weiss, et al. Radiation-Induced Tumor Growth DelayNOS Inhibition Modulates Immune Polarization and Improves
Updated version
10.1158/0008-5472.CAN-14-3011doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2015/05/19/0008-5472.CAN-14-3011.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/75/14/2788.full#ref-list-1
This article cites 60 articles, 17 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/75/14/2788.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/75/14/2788To request permission to re-use all or part of this article, use this link
on May 20, 2020. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 19, 2015; DOI: 10.1158/0008-5472.CAN-14-3011