nmda receptor-dependent long-term potentiation …bioslp/neural_plasticity/luscher_malenka.pdf ·...
TRANSCRIPT

published online April 17, 2012Cold Spring Harb Perspect Biol Christian Lüscher and Robert C. Malenka Depression (LTP/LTD)NMDA Receptor-Dependent Long-Term Potentiation and Long-Term
Subject Collection The Synapse
Synapses and Alzheimer's Disease
C. SüdhofMorgan Sheng, Bernardo L. Sabatini and Thomas Brain
Ultrastructure of Synapses in the Mammalian
Kristen M. Harris and Richard J. Weinberg
(LTP/LTD)Potentiation and Long-Term Depression NMDA Receptor-Dependent Long-Term
Christian Lüscher and Robert C. Malenka
Synapses and Memory Storage
KandelMark Mayford, Steven A. Siegelbaum and Eric R.
Synaptic Cell Adhesion
BiedererMarkus Missler, Thomas C. Südhof and Thomas
Calcium Signaling in Dendritic SpinesMichael J. Higley and Bernardo L. Sabatini
DisabilitiesIntellectualDisorders Associated with Autism and
Synaptic Dysfunction in Neurodevelopmental
Huda Y. Zoghbi and Mark F. Bear
Synaptic Neurotransmitter-Gated ReceptorsTrevor G. Smart and Pierre Paoletti
The Postsynaptic Organization of SynapsesMorgan Sheng and Eunjoon Kim
Synaptic Vesicle ExocytosisThomas C. Südhof and Josep Rizo
Inhibitory SynapsesPresynaptic LTP and LTD of Excitatory and
Pablo E. CastilloNeurotransmittersVesicular and Plasma Membrane Transporters for
Randy D. Blakely and Robert H. EdwardsCalcium Control of Neurotransmitter Release
Thomas C. Südhof Mechanisms for Stabilizing Neuronal FunctionHomeostatic Synaptic Plasticity: Local and Global
Gina Turrigiano
in Synapse Development and Cognitive FunctionNeuronal Activity-Regulated Gene Transcription
Anne E. West and Michael E. Greenberg
http://cshperspectives.cshlp.org/cgi/collection/ For additional articles in this collection, see
Copyright © 2012 Cold Spring Harbor Laboratory Press; all rights reserved
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

NMDA Receptor-Dependent Long-TermPotentiation and Long-Term Depression(LTP/LTD)
Christian Luscher1 and Robert C. Malenka2
1Department of Basic Neurosciences and Clinic of Neurology, University of Geneva and Geneva UniversityHospital, 1211 Geneva, Switzerland
2Nancy Pritzker Laboratory, Department of Psychiatry and Behavioral Sciences, Stanford University School ofMedicine, Palo Alto, California 94305-5453
Correspondence: [email protected] and [email protected]
Long-term potentiation and long-term depression (LTP/LTD) can be elicited by activatingN-methyl-D-aspartate (NMDA)-type glutamate receptors, typically by the coincident activityof pre- and postsynaptic neurons. The early phases of expression are mediated by a redistri-bution of AMPA-type glutamate receptors: More receptors are added to potentiate thesynapse or receptors are removed to weaken synapses. With time, structural changesbecome apparent, which in general require the synthesis of new proteins. The investigationof the molecular and cellular mechanisms underlying these forms of synaptic plasticity hasreceived much attention, because NMDA receptor–dependent LTP and LTD may constitutecellular substrates of learning and memory.
Long-term synaptic plasticity is a generic termthat applies to a long-lasting experience-de-
pendent change in the efficacy of synaptic trans-mission. Here we will focus on N-methyl-D-as-partate (NMDA) receptor–dependent synapticpotentiation (LTP) and depression (LTD), twoforms of activity-dependent long-term changesin synaptic efficacy that have been extensivelystudied. Because both LTP and LTD are believedto represent cellular correlates of learning andmemory, they have attracted considerable inter-est. In this article we will focus on the molecularand cellular mechanisms associated with LTPand LTD. As for other forms of long-term syn-aptic plasticity, a characterization of LTP andLTD involves describing the molecular mecha-nisms that are required to elicit the change (in-
duction), followed by an investigation of themechanism of expression (hours) and mainte-nance (days). The best-characterized form ofNMDA receptor (NMDAR)-dependent LTP oc-curs between CA3 and CA1 pyramidal neuronsof the hippocampus (Fig. 1). Throughout thechapter we will mostly refer to this specific formof LTP. At these CA3-CA1 Schaffer collateralsynapses, the loci of both induction and expres-sion are situated in the postsynaptic neuron.
AMPA-TYPE AND NMDA-TYPEGLUTAMATE RECEPTORS
After exocytotic release of the content of thesynaptic vesicle from the presynaptic specializa-tion (see Castillo 2012), the neurotransmitter
Editors: Morgan Sheng, Bernardo Sabatini, and Thomas C. Sudhof
Additional Perspectives on The Synapse available at www.cshperspectives.org
Copyright # 2012 Cold Spring Harbor Laboratory Press; all rights reserved.
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710
1
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from
rapidly diffuses across the synaptic cleft andreaches the postsynaptic membrane, where itbinds to its respective receptors. The neuro-transmitter concentration in the synaptic cleftremains high for only a very brief period. Iono-tropic receptors are direct ligand-gated ionchannels that respond rapidly to the brief pulseof neurotransmitter released from the synapticvesicle. The evoked synaptic currents are typi-cally believed to last only a few milliseconds.The most common excitatory transmitter is glu-tamate, whereas many inhibitory synapses useg-aminobutyric acid (GABA) for transmission.
Here we will first discuss the ionotropicmembers that are directly involved in synapticplasticity of excitatory transmission, NMDAand AMPA-type glutamate receptors (NMDARs,AMPARs) (Dingledine et al. 1999). Both are ion-otropic receptors that are permeable to Naþ and
Kþ. Activation of ionotropic glutamate recep-tors leads to strong influx of Naþ and only asmall efflux of Kþ, such that the net effect isthe depolarization of the postsynaptic neuron.
The workhorse of glutamatergic transmis-sion is the AMPAR, which drives large and rapidsynaptic signaling (Fig. 2). There are four genesencoding AMPARs (GRIA1 to -4). Each AM-PAR is composed of four subunits, which canbe a homomeric or heteromeric mixture ofGluA1 to -4. Most AMPARs contain at leastone subunit of GluA2. Following transcriptionthis subunit undergoes RNA editing, wherebythe RNA coding for a glutamine residue in theion channel pore-forming region is exchangedfor the RNA codon for arginine. This process isan essential step in the quality control. Noned-ited GluA2 will be retained in the endoplasmicreticulum. The large arginine residue located in
2
2
1 1Dentate
C
A B
CA3
21
12
1 Hz for 10 min
0
200
150
100
50
EP
SP
slo
pe (
% b
asel
ine)
020 40 60 80 100
min
2 × 100 Hz
3
3
CA1
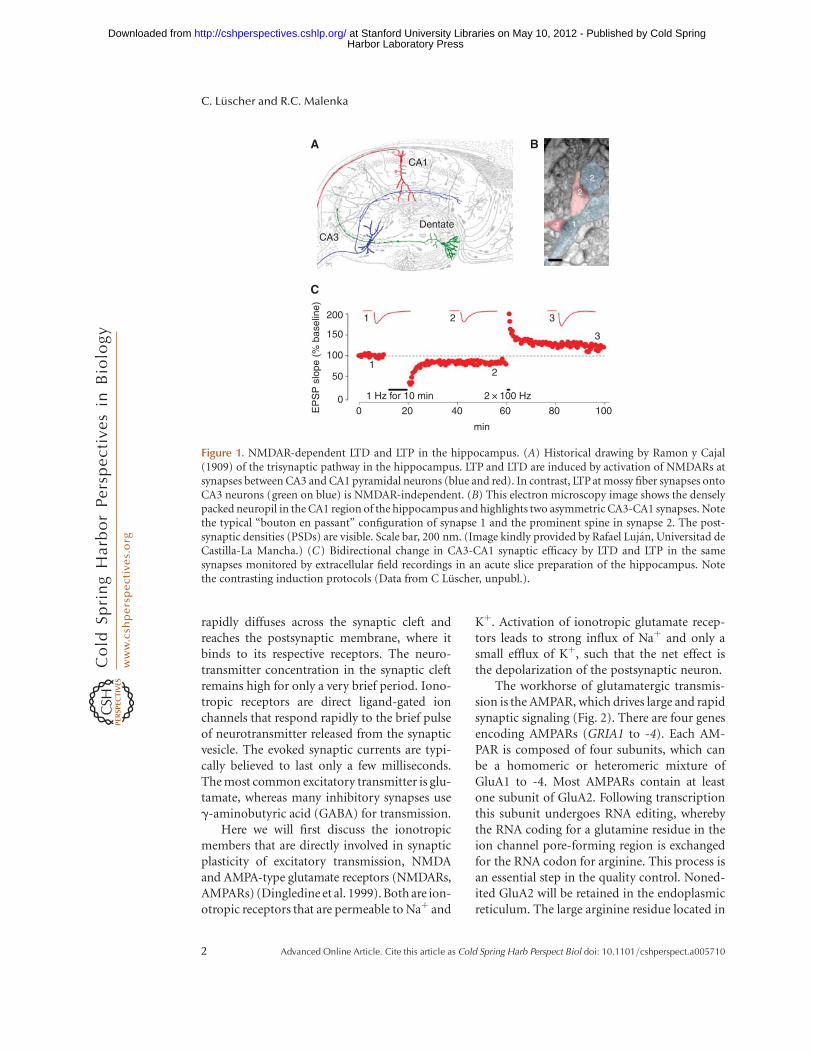
Figure 1. NMDAR-dependent LTD and LTP in the hippocampus. (A) Historical drawing by Ramon y Cajal(1909) of the trisynaptic pathway in the hippocampus. LTP and LTD are induced by activation of NMDARs atsynapses between CA3 and CA1 pyramidal neurons (blue and red). In contrast, LTP at mossy fiber synapses ontoCA3 neurons (green on blue) is NMDAR-independent. (B) This electron microscopy image shows the denselypacked neuropil in the CA1 region of the hippocampus and highlights two asymmetric CA3-CA1 synapses. Notethe typical “bouton en passant” configuration of synapse 1 and the prominent spine in synapse 2. The post-synaptic densities (PSDs) are visible. Scale bar, 200 nm. (Image kindly provided by Rafael Lujan, Universitad deCastilla-La Mancha.) (C) Bidirectional change in CA3-CA1 synaptic efficacy by LTD and LTP in the samesynapses monitored by extracellular field recordings in an acute slice preparation of the hippocampus. Notethe contrasting induction protocols (Data from C Luscher, unpubl.).
C. Luscher and R.C. Malenka
2 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

the pore region of the channel limits the flow ofNaþ and Kþ ions and prevents divalent ionsfrom entering the cell. This design renders AM-PARs that contain GluA2 calcium-impermeable(Liu and Zukin 2007).
AMPARs carry inward currents at negativepotentials and outward currents at positive po-tentials, and the reversal potential is 0 mV. Forreceptors that contain GluA2, this relationshipis symmetrical, i.e., the current–voltage rela-tionship is linear. In contrast, for receptorsthat lack GluA2 (e.g., GluA1 homomeric orGluA1/3 heteromeric channels), the current–voltage relationship shows an interesting non-linear voltage-dependent interaction. GluA2-lacking AMPARs have a glutamine residue inthe pore region instead of the arginine in the
GluA2. Such channels have a high conductancefor sodium and are even permeable for calcium.Because endogenous polyamines, which arenegatively charged, can also access a site closeto the cytoplasmic mouth of the pore, the chan-nels are inhibited at positive potentials. As aconsequence, GluA2-lacking AMPARs have aninward-rectifying current–voltage relationship(i.e., they conduct current more easily intothe cell than out of the cell; Fig. 2). Such calci-um-permeable AMPARs are expressed on manyGABAergic neurons.
The current–voltage relationship of NM-DARs is even more complex. At negative mem-brane potentials close to the resting membranepotential, magnesium ions enter the pore ofthe NMDAR, blocking the passage for all other
AMPA receptor
AMPA receptor
+GluA2
–GluA2–60 mV
I I
–60 mVV V
0 mV
A
B
–60 mV K+ K+
Na+
Glu GluMg2+
Ca2+ and Na+
NMDA receptor
NMDA receptor
Figure 2. Major ionotropic glutamate receptors involved in LTD and LTP. (A) When glutamate binds to AMPAreceptors, many sodium ions flow into the cell while only some potassium ions leave the neuron, causing a netdepolarization of the membrane. NMDA receptors are also permeable for calcium but only if the magnesium isexpelled by a slight depolarization of the neuron. (B) The current–voltage (I–V ) relationship provides abiophysical signature for the different receptors. AMPA receptors have a linear I–V relationship when theycontain the subunit GluA2, but are inward-rectifying (see text for definition) without GluA2. NMDA receptorshave a complex I–V curve because Mg blocks the pore at negative potentials.
NMDA Receptor-Dependent LTP/LTD
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710 3
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

ions. Upon depolarization the magnesium isexpelled from the pore, allowing sodium, potas-sium, and, importantly, calcium ions to pass. Atpositive potentials NMDARs then show maxi-mal permeability (i.e., large outward currentscan be observed under these circumstances;Fig. 2). NMDARs also have much slower kinet-ics than AMPARs. Following release of gluta-mate, NMDARs activate more slowly, having apeak conductance long after the AMPAR peakconductance. NMDARs can also remain openfor hundreds of milliseconds after presynapticrelease of glutamate, much longer than the AM-PARs, which typically open for only a few mil-liseconds. It is important to note that NMDARsconduct currents only when glutamate is boundand the postsynaptic neuron is depolarized. Inother words, both the pre- and postsynapticneurons need to be active to open NMDARs.Through this mechanism NMDARs play therole of molecular coincidence detectors, which,as we will see below, is essential for several formsof synaptic plasticity.
Ionotropic glutamate receptors do not workin isolation. They interact with many proteinsof the postsynaptic density (PSD) (Elias andNicoll 2007). Some of these proteins modulateglutamate receptor function, whereas otherscontrol their membrane insertion and removal.The number of glutamate receptors at a synapse(normally only a few dozens of receptors) cantherefore be regulated through these interac-tions. A particularly important interacting pro-tein is stargazin, as well as other members ofthe transmembrane AMPAR regulatory protein(TARP) family. Stargazin is in fact an auxiliarysubunit of most AMPARs that affects con-ductance, kinetics, and rectification. In otherwords, in the physiological situation in whichTARPs are present, the kinetics and pharmacol-ogy of neuronal AMPARs differ from those pre-dicted by classical in vitro expression studies ofAMPARs. PICK1 is another example of a pro-tein that regulates the number of receptors at thesynapse. Unlike the TARPs, which have littlesubunit specificity, PICK1 is believed to interactprimarily with GluA2. Interfering with PICK1and other interacting proteins has shown thatsynaptic AMPARs dynamically recycle within
tens of minutes using mechanisms that allowrapid removal from and insertion into the PSD.Although under baseline conditions the num-ber of receptors remains stable, rapid redistri-bution leading to a change in the number ofsynaptic receptors is a widely used mechanismfor synaptic plasticity (see below).
Although the major role of ionotropic glu-tamate receptors is postsynaptic, they have alsobeen found on presynaptic boutons, where theymay participate in the regulation of transmitterrelease. Such presynaptic receptors can be ac-tivated by strong release from the bouton onwhich they are located (homosynaptic modula-tion) or by glutamate released by neighboringsynapses (heterosynaptic modulation). Wheth-er this activation enhances or inhibits furtherrelease varies from synapse to synapse.
INDUCTION OF LTP AND LTD
LTP and LTD are induced by specific patternsof activity (Malenka 1994). For LTP inductionboth pre- and postsynaptic neurons need tobe active at the same time because the postsyn-aptic neuron must be depolarized when gluta-mate is released from the presynaptic bouton tofully relieve the Mg2þ block of NMDARs. As aconsequence of coincident depolarization andglutamate binding, calcium influx throughNMDARs is maximal, which activates intracel-lular signaling cascades that ultimately are re-sponsible for the altered synaptic efficacy.NMDAR-dependent LTP is therefore an associ-ative form of plasticity and fulfills the criteria forcorrelated activity as the origin of the strength-ening of the connection between two neuronsproposed by Donald Hebb more than 60 yearsago (Hebb 1949). Conversely, LTD can be in-duced by repeated activation of the presynap-tic neuron at low frequencies without postsyn-aptic activity (see below). Because the drivingforce for calcium entry is very large for a neuronat rest and the block of NMDARs by Mg2þ isincomplete even at resting potentials (Jahr andStevens 1993), significant Ca enters the cell inresponse to synaptic stimulation (Sabatini et al.2002; Bloodgood and Sabatini 2007; Blood-good et al. 2009) during low-frequency synaptic
C. Luscher and R.C. Malenka
4 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

stimulation. Presumably it is repeated occur-rence of this smaller NMDAR-dependent Ca in-flux that triggers LTD induction.
Because NMDAR-dependent calcium in-flux induces both LTP and LTD, the cell musthave a way to decide whether to potentiate ordepress a synaptic connection. An early modelinvoked the level of activity or depolarization inthe postsynaptic cell as the critical variable dic-tating whether LTP or LTD was evoked (or nochange in synaptic strength). Indeed, it is nowfairly well accepted that modest activation ofNMDARs leading to modest increases in post-synaptic calcium are optimal for triggering LTD,whereas much stronger activation of NMDARsleading to much larger increases in postsynapticcalcium are required to trigger LTP (Malenka1994).
An important property of these NMDAR-dependent forms of LTP and LTD is that they areinput- or synapse-specific. That is, only syn-apses that actively contribute to the inductionprocess will undergo the plasticity. This is be-cause NMDARs on the synapses must be acti-vated by synaptically released glutamate duringthe induction protocol. The synapse specificityof LTP has been explicitly tested using photolyt-ic release of glutamate to bypass the presynapticterminal and directly stimulate a visualized den-dritic spine with glutamate. Experiments usingthis approach to induce NMDAR-dependentLTP have shown that LTP can be induced ata single synapse without causing postsynap-tic potentiation or depression of neighboringsynapses (Matsuzaki et al. 2004; Harvey andSvoboda 2007; but see Engert and Bonhoeffer1997). However, interactions between neigh-boring synapses do occur, such that the intra-dendritic spread of the activated Ras from anactive synapse briefly lowers the threshold forsubsequent LTP induction at neighboring syn-apses (Harvey et al. 2008).
In acute slices of the hippocampus, LTP isoften induced by tetanic stimulation of Schaffercollaterals. High-frequency stimulation proto-cols typically comprise delivery of one or severaltrains of pulses at 50–100 Hz for 1 sec. It maybe surprising that stimulation of the presynap-tic axons alone is sufficient for LTP induction.
However, this makes sense because high-fre-quency stimulation protocols cause a strongtemporal summation of the excitatory postsyn-aptic potentials (EPSPs), and the resultant largedepolarization of the postsynaptic cell is suffi-cient to relieve the Mg block of the NMDARand allow a large amount of calcium to enterthe postsynaptic cells during the induction pro-tocol. In contrast, low-frequency stimulation ofpresynaptic axons is commonly used to induceLTD. Typically such protocols involve stimula-tion at 1–3 Hz for 5–15 min. This causes only amodest amount of postsynaptic depolarization,resulting in a modest but prolonged increase inpostsynaptic calcium due to modest and repet-itive activation of NMDARs. A variant of theseinduction protocols is the so-called pairing pro-tocol, whereby during low-frequency activationof axons (0.1–1 Hz), individual cells are heldat depolarized membrane potentials by passingcurrent through the recording electrode. Thisprovides relief of the Mg-dependent block ofNMDARs, allowing calcium to enter the post-synaptic cell when the NMDARs are activatedby the presynaptically released glutamate. Con-sistent with original ideas (Bienenstock et al.1982), modest depolarization can elicit LTD,whereas stronger depolarization leads to LTP.
More recently studies have shown that LTPand LTD can be elicited if action potentials inthe pre- and postsynaptic neurons coincide withthe appropriate timing. From the perspectiveof the postsynaptic neuron (which is wherethe recordings are made), these manipulationslead to both a synaptic potential and an actionpotential that backpropagates into the den-drites, which presumably provides the addi-tional depolarization required to facilitate cal-cium influx through the activated NMDARs(Stuart et al. 1997; Waters et al. 2005). If a pre-synaptic spike is repetitively elicited slightly be-fore (e.g., 5 msec) the postsynaptic neuron isfired, the EPSP precedes the backpropagatingaction potential and such repetitive “pre-post”action potential firing can generate LTP. Con-versely, when the backprogating action poten-tial is repetitively elicited before the presynapticspike, “post-pre” firing, LTD is often observed.The time window for such pairings is critical,
NMDA Receptor-Dependent LTP/LTD
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710 5
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

such that the closer in time presynaptic andpostsynaptic spikes are fired, the larger the mag-nitude of the plasticity induced. If both stimu-lations, however, occur at once, an asymptoteis reached, in which the resultant long-termchange in synaptic strength can no longer bereliably predicted. As the direction of synapticplasticity depends on the timing between thepresynaptic and postsynaptic spikes, this phe-nomenon has been named spike-timing-depen-dent plasticity (Dan and Poo 2006; Caporaleand Dan 2008). It is often a considered a morephysiological method of inducing LTP or LTDbecause it is imagined that these patterns ofspiking may occur during real behavior.
EXPRESSION MECHANISM
During the last two decades of the last century,the locus of expression of NMDAR-dependentLTP was the focus of an intense debate (Ma-lenka and Nicoll 1999; Nicoll 2003). Althoughin hindsight it seems difficult to understandhow an apparently simple question could keepmany researchers in the field busy, the confu-sion could largely be attributed to the lack ofunderstanding of the basic physiology of excit-atory synapses in the mammalian central ner-vous system. Although a minority of researchersin the field would argue that the question is stillnot fully answered, there is general agreementthat the experiments that were performed toaddress this topic significantly furthered ourunderstanding of the basic properties of synap-tic transmission.
In theory, how LTP and LTD are expressed isa simple question. For LTP the increase in syn-aptic strength could be due either to more trans-mitter being released from the presynaptic ax-ons being activated during the course of theexperiment or to the same amount of transmit-ter being released but having a greater effectbecause the postsynaptic cell was more sensitiveto the same amount of released neurotransmit-ter. It is important to note that if LTP (or LTD) iscaused by enhanced (or decreased) transmitterrelease, it requires that the postsynaptic cellsomehow communicate to the presynaptic ter-minals and modify their function. This is re-
quired because it is well established thatNMDAR-dependent LTP and LTD are triggeredin the postsynaptic cell. Indeed, during the de-bate about the locus of expression for LTP (andLTD), there was much discussion about the pos-sible identity of so-called retrograde messen-gers, the substances that might be releasedfrom postsynaptic cells following appropriateactivation of NMDARs and modify presynapticfunction (Arancio et al. 1996; but see Williamset al. 1993).
To solve the pre- versus post- debate, whichprimarily focused on the locus of expression ofLTP, a large number of experiments were per-formed. To assess whether presynaptic functionchanged, the assays included:
† Using use-dependent open channel blockersof postsynaptic receptors to estimate the re-lease probability. Most importantly, dizocil-pine (MK-801) irreversibly blocks NMDARsonce they are activated by glutamate. Thus,once glutamate is released from a single pre-synaptic terminal and activates its correspond-ing postsynaptic NMDARs, those NMDARsare irreversibly blocked. This means thatapplication of MK-801 while activating apopulation of synapses leads to a gradual de-crease of the postsynaptic currents generatedby NMDARs, and the rate of this decreasedirectly correlates with the overall probabilityof release of the synapses being activated.Importantly, it was found that the rate ofthis decrease remains unaltered by LTP butis clearly affected by other manipulationsthat are known to influence the probabilityof transmitter release (Manabe and Nicoll1994).
† Monitoring short-term plasticity before andafter the induction of LTP. Paired-pulse ratiosand short-term facilitation or depression ofsynaptic responses during short, high-fre-quency bursts of stimulation reflect short-term changes in release probability. Theseubiquitous forms of short-term synapticplasticity are greatly influenced by the base-line release probability yet were unaffected bythe generation of LTP (McNaughton 1982;
C. Luscher and R.C. Malenka
6 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

Manabe et al. 1993; but see Schulz et al.1995).
† Monitoring glial glutamate transporter cur-rents before and after LTP. The reuptake ofglutamate by astrocytes is electrogenic, andthus currents can be measured from astro-cytes that reflect the amount of transmitterreleased. There is, however, no change in thesize of these currents after induction of LTPeven though other manipulations that affecttransmitter release have clear effects. Theseexperiments are particularly convincing, be-cause this approach relies on a readout thatis independent of glutamate receptors andtherefore unlikely to participate in the ex-pression of LTP (Diamond et al. 1998; Lu-scher et al. 1998).
† Visualization of presynaptic exocytosis withstyryl dyes such as FM1-43. These dyes stainpresynaptic vesicles and are washed out oncethe vesicles fuse with the membrane in anactivity-dependent fashion. Just as with MK-801, the destaining curves were superimpos-able before and after tetanic induction ofNMDAR-dependent LTP in CA1 neurons at50–100 Hz (Zakharenko et al. 2001).
Taken together, these findings (and addi-tional experiments that are not covered becauseof space limitations) make it very unlikely thatLTP is associated with an increase in releaseprobability or the amount of glutamate releasedfrom a presynaptic vesicle. But they say nothingabout what postsynaptic mechanisms contrib-ute to LTP (and LTD). An important clue to thisquestion came from the observation that in thehippocampi of very young animals, some syn-apses contain only NMDARs and no, or veryfew, AMPARs. Thus, these synapses are func-tionally silent under baseline conditions. Afterthe application of an LTP induction protocol,these synapses “wake up” and become function-al because of the insertion of AMPARs into theirpostsynaptic membrane (Isaac et al. 1995; Liaoet al. 1995). This result immediately raised thepossibility that at both silent synapses and syn-apses that already contain AMPARs, LTP in-volves the insertion of more AMPARs into the
synapse, whereas conversely, LTD may involvethe removal or endocytosis of synaptic AMPARs(Fig. 3) (Lledo et al. 1998; Luscher et al. 1999).
A large body of experimental evidence nowsupports this hypothesis (Luscher et al. 2000;Luscher and Frerking 2001; Malinow and Mal-enka 2002; Nicoll 2003; Collingridge et al. 2004;Malenka and Bear 2004). In fact, AMPARs canbe quite mobile and recycle between the cyto-plasm and the cell membrane even under base-line conditions within tens of minutes. This canbe shown, for example, by interfering with en-docytosis, which leads to a run-up of synapticresponses. It is presumably this mobile pool ofAMPARs that allows for rapid but sustainedchanges in synaptic efficacy. The insertionand removal of AMPARs during LTP and LTD,respectively, is believed to involve classical me-chanisms of SNARE protein–mediated exocy-tosis and dynamin-dependent endocytosis viaclathrin-coated vesicles (Luscher et al. 1999;Carroll et al. 2001; Kennedy and Ehlers 2011).Current evidence favors the idea that the endo-cytosis and exocytosis of AMPARs during LTDand LTP happens not directly at the synapse butat slightly perisynaptic locations, from wherethe receptors reach the postsynaptic density bylateral diffusion.
A large family of proteins associates withthe AMPARs to regulate their mobility and bio-physical properties as well as their stabilizationwithin the PSD (see Sheng and Kim 2012). Dif-ferent AMPAR subunits may play distinct rolesin this redistribution process. HeteromericGluA1/GluA2 receptors seem to be the primarysubtype of AMPAR that is inserted into syn-apses during LTP (Adesnik and Nicoll 2007).In addition, there are also forms of synapticpotentiation that are expressed by an exchangeof GluA2-containing AMPARs in the synapsefor GluA2-lacking AMPARs (Liu and Zukin2007). Because the latter have a higher conduc-tance, this will potentiate the synapse even if thetotal number remains the same. It has also beensuggested that during the very early phase ofLTP (its first 10–20 min), there is a transientappearance of GluA2-lacking AMPARs at thesynapse and this is required for the maintenanceof LTP (Plant et al. 2006). However, this finding
NMDA Receptor-Dependent LTP/LTD
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710 7
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from
is controversial (Adesnik and Nicoll 2007). Ofparticular interest are members of the TARPfamily, which interact with all AMPA receptorsubunits and control not only membrane inser-tion but also lateral redistribution (Chen et al.2000; Tomita et al. 2005; see Blakely and Ed-wards 2012). If interactions with scaffoldingproteins are manipulated through genetic inter-ventions or the perfusion of dominant-negativeproteins, LTP and LTD can be blocked (Luscher
et al. 1999; Luthi et al. 1999; Collingridge andIsaac 2003).
SIGNALING CASCADES FOR TRIGGERINGLTP AND LTD
We have briefly reviewed the mechanisms un-derlying the induction of LTP and LTD, as wellas their expression mechanisms. Appropriatecoincident activity of the pre- and postsynaptic
AMPA
NMDA
Endocytosis
Exocytosis
Strong depolarizationWeak depolarizationLTD
Blocking LTD by disruption of dynamin–amphiphysin interaction (D15 peptide )
Blocking LTP with tetanus toxin
120 3001+2
1 2
1+2
200
100
2 × 100 Hz, 0 mV
80
40
0
0 10 20 30min min
0 10 20 30 40
5 Hz, 3 min, –40 mV
1 2
1 2
2
EP
SC
(%
bas
elin
e)
EP
SC
(%
bas
elin
e)
A
B
LTP
Weak Strong
CaMKllPP2BPP1
PP
Ca2+
Ca2+
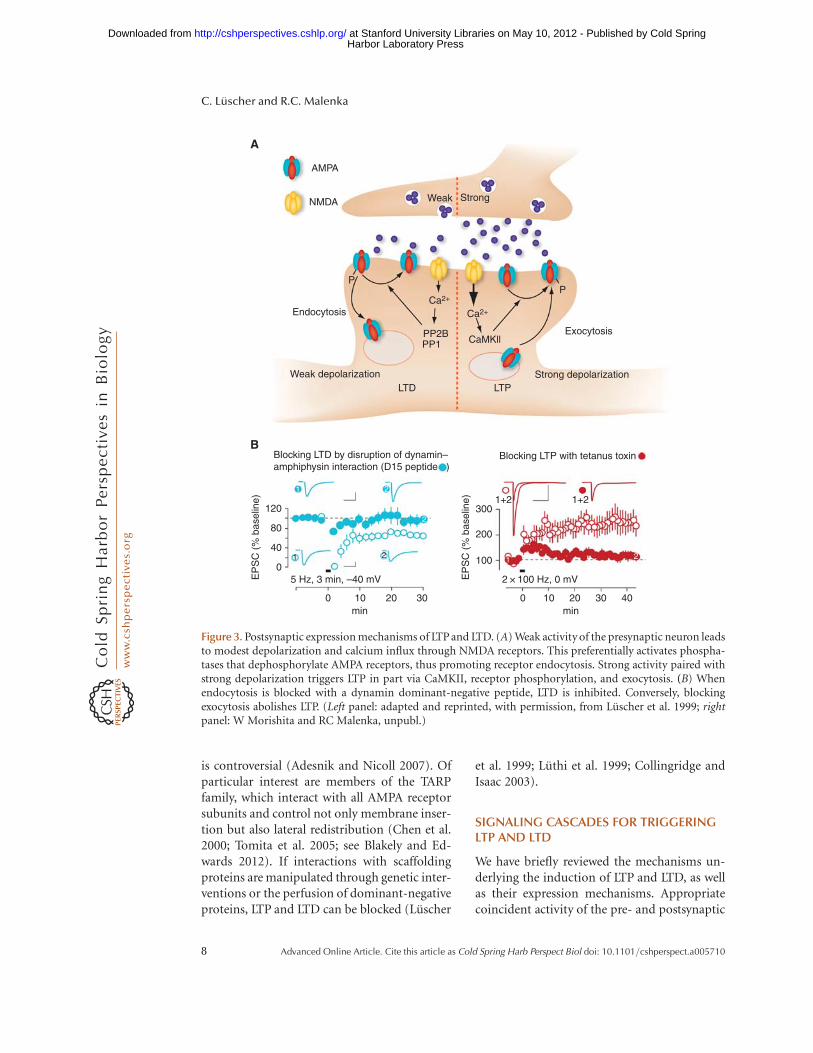
Figure 3. Postsynaptic expression mechanisms of LTP and LTD. (A) Weak activity of the presynaptic neuron leadsto modest depolarization and calcium influx through NMDA receptors. This preferentially activates phospha-tases that dephosphorylate AMPA receptors, thus promoting receptor endocytosis. Strong activity paired withstrong depolarization triggers LTP in part via CaMKII, receptor phosphorylation, and exocytosis. (B) Whenendocytosis is blocked with a dynamin dominant-negative peptide, LTD is inhibited. Conversely, blockingexocytosis abolishes LTP. (Left panel: adapted and reprinted, with permission, from Luscher et al. 1999; rightpanel: W Morishita and RC Malenka, unpubl.)
C. Luscher and R.C. Malenka
8 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

neurons causes an influx of calcium throughNMDARs, and depending on the quantitativecharacteristics of this calcium signal, AMPARsare either inserted into or removed from thesynapses, resulting in LTP or LTD, respectively.But how are the triggering of LTP and LTD andtheir expression linked? What are the interme-diate signaling events that translate the increasein postsynaptic calcium into receptor redistri-bution? For LTP there is strong evidence that theopening of NMDARs increases calcium concen-tration sufficiently in the dendritic spine to ac-tivate calcium/calmodulin-dependent kinase II(CaMKII), which is found at very high concen-trations in spines and which is clearly requiredfor LTP (Lisman et al. 2002). This leads to thephosphorylation of a number of proteins in-cluding AMPARs themselves (Derkach et al.1999). The phosphorylation of AMPAR sub-units can cause an increase in the conductanceof the AMPAR channel (Benke et al. 1998), an-other postsynaptic mechanism that contributesto at least the early phase of LTP. In addition, inways that remain to be determined, the increasein CaMKII activity contributes to the insertionof AMPARs (Ehlers 2000). This can be shown byperfusing the postsynaptic neurons with acti-vated CaMKII, which not only leads to an in-crease of synaptically evoked currents but alsoenhances responses to iontophoretically appliedAMPA. Importantly, this manipulation oc-cludes further LTP, suggesting a shared mech-anism between the ways of increasing synapticstrength (Malenka and Nicoll 1999; Lismanet al. 2002).
Although CaMKII is well accepted to be onemajor requisite trigger for LTP, like many othercell biological phenomena, the signaling cas-cades underlying the induction and mainte-nance (see below) of LTP are extremely complex.A host of additional protein kinases, such ascAMP-dependent protein kinase (PKA), pro-tein kinase C (PKC), mitogen-activated proteinkinases, and tyrosine kinases, have all been sug-gested to contribute to LTP in various ways(Bliss and Collingridge 1993; Malenka and Nic-oll 1999; Salter and Kalia 2004; Sweatt 2004).However, many of the details of their precisefunction in LTP remain to be worked out.
If LTP involves the activation of CaMKII(and other kinases) and LTD represents the in-verse of LTP, then a logical hypothesis is thatLTD involves the preferential activation of pro-tein phosphastases. Indeed, a very influentialmodel proposed that NMDAR-dependent LTDdepends on the calcium/calmodulin-depen-dent protein phosphatase calcineurin as well ason protein phosphatase 1 (PP1) (Lisman 1989).This is a very attractive model because calci-neurin has a much higher affinity for calci-um/calmodulin than does CaMKII and thuswill be preferentially activated by a modest in-crease in calcium, the exact trigger for LTD.There is now strong evidence that these twophosphatases do indeed play a role in LTD(Mulkey et al. 1993, 1994: Carroll et al. 2001),perhaps in part by influencing the phosphory-lation state of AMPARs. As was the case for LTP,the intracellular signaling cascades underlyingLTD are certainly more complex than simply theactivation of two phosphatases. For example, ithas been suggested that apoptotic mechanismsincluding activation of caspase 3 via mitochon-dria are critical for LTD (Li et al. 2010). Further-more, the AMPARs that have internalized mayneed to be degraded via lysosomal or proteaso-mal pathways so that they are not returned tothe plasma membrane.
MAINTENANCE OF LTP AND LTD
AMPARs are tightly anchored in the PSD bya large number of scaffolding proteins linkingthem to cytoskeletal elements including actin.Insertion of additional receptors therefore islikely to affect the ultrastructure of the synapse,and in fact, spines associated with synapsesthat underwent LTP enlarge (Matsuzaki et al.2004; Harvey and Svoboda 2007; Holtmaatand Svoboda 2009; Kasai et al. 2010). Further-more, at the ultrastructural level in electronmicrographs, synapses that underwent LTP hadenlarged PSDs that were often discontinuous(Toni et al. 2001). Such perforated synapsesalso contain a higher proportion of smoothendoplasmic reticulum and a spine apparatus(Luscher et al. 2000). A second morphologicalcorrelate of LTP besidesthe physical enlargement
NMDA Receptor-Dependent LTP/LTD
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710 9
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

of spines is the appearance of new spines fromthe shaft within minutes of the induction pro-tocol (perhaps also by division of an existingspine). Consistent with this idea, the spine den-sity increases following LTP induction, and asignificantly higher frequency of multiple-spinesynapses (Toni et al. 1999) is observed, as well as areduction of their turnover (De Roo et al. 2008).Conversely, the triggering of LTD is associatedwith the shrinkage of dendritic spines and per-haps even their disappearance (Nagerl et al.2004; Zhou et al. 2004; Wang et al. 2007; Kasaiet al. 2010). These observations raise the ques-tion of the interdependence of the mechanismsunderlying structural and functional plasticityduring LTPand LTD, a topic that is being activelyinvestigated.
Not only does long-term plasticity causestructural changes in synapses, but the mainte-nance of the change in synaptic strength dur-ing LTP is protein synthesis–dependent. PKA,CaMKIV, protein kinase M-z, and extracellularsignal–regulated kinase (ERK), as well as othersignaling molecules, initiate protein synthesiseither locally in the dendrites from prefabricat-ed mRNA or by nuclear transcription (Sacktor2008). The latter involves interactions with tran-scription factors, including cAMP response el-ement–binding protein (CREB). Both localdendritic and nuclear transcription and somatictranslation together are believed to synthesizethe proteins required for the maintenance offunctional and structural plasticity followingthe triggering of LTP. In reality, the events ofLTP expression and maintenance do not occursequentially, and the first structural changes,such as the increase of the size of the PSD andspine growth, can be observed rapidly after in-duction. Through these mechanisms LTP mayguide the selective stabilization of synaptic in-puts that show coincident activity, whereas non-activated inputs may be removed and replacedby new spines. Arc is one immediate early genethat may orchestrate the translation of dendriticmRNA required for actin polymerization andstable expansion of dendritic spines during LTP(Bramham et al. 2010). Although such struc-tural changes in spines have been studied insome detail, they are not absolutely required
for LTP, as synapses made directly on the den-drites (i.e., shaft synapses) are capable of ex-pressing and maintaining LTP. Moreover, selec-tive interference with the structural changes thataccompany LTD do not seem to affect the in-crease in synaptic strength, at least not in earlystages, up to 1–2 hr following induction (Fig. 4)(Wang et al. 2007; Redondo and Morris 2011).
NMDAR-DEPENDENT LTP OF INHIBITORYTRANSMISSION
Although the vast majority of work on themechanisms and functions of LTP and LTDhave focused on excitatory synapses, potentia-tion and depression of inhibitory transmission(I-LTP/I-LTD) can also be observed in the brain(Castillo et al. 2011). The underlying molecularmechanisms of I-LTP and I-LTD are variable,but one common feature is that they are oftenheterosynaptic. Synaptic plasticity of GABAergic
LTPA
B
LTD
5 m
V
10 min
12 h
Figure 4. Structural changes associated with LTP andLTD. (A) Synaptic strength correlates with spine vol-ume and the area of the postsynaptic density (or-ange). Note that the PSD in potentiated synapses isoften perforated. (B) LTP can also lead to the appear-ance of new spines. Within 30 min of triggering LTP(30 stimuli applied to the presynaptic axon at 10-secintervals paired with depolarizing current injectioninto the postsynaptic neuron; black bar) of a synapticconnection in the hippocampus, new spines appear.(Panel created from data adapted from Engert andBonhoeffer 1999.)
C. Luscher and R.C. Malenka
10 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

transmission needs the activation of glutama-tergic synapses and a specific messenger thatpasses the signal from one type of synapses tothe other. Here we will focus on the forms ofI-LTP/I-LTD that are induced by NMDARs,which, as for the plasticity of excitatory trans-mission, are classified according to the locus ofinduction and expression.
In the hippocampus, visual cortex, and op-tic tectum, a form of I-LTP dependent on brain-derived neurotrophic factor (BDNF) has beendescribed (Inagaki et al. 2008). It is induced byactivation of NMDARs (and sometimes voltage-gated calcium channels) and modulated byGABAB receptors, which together drive an in-crease in cytoplasmic calcium, in part by trig-gering the release of calcium from intracellularstores. In this model NMDAR activation leadsto postsynaptic release of BDNF, which func-tions as a retrograde messenger and causes anincrease in GABA release through activation ofpresynaptic TrkB receptors.
In the ventral tegmental area, a similar formof plasticity called LTPGABA is induced by strongactivation of NMDARs on dopamine neurons(Nugent et al. 2007). This leads to activation of aCa2þ-dependent nitric oxide synthase, whichgenerates nitric oxide, which acts as a retrogrademessenger by diffusing back to presynaptic neu-rons. Nitric oxide in turn causes activation ofguanylate cyclase and synthesis of cGMP in thesynaptic terminals of inhibitory afferents ontodopamine neurons. The release probability forGABA then increases through a still unknownmechanism involving activation of the cGMP-dependent protein kinase, PKG.
The slow GABAB receptor–mediated inhib-itory postsynaptic potential (IPSP) can alsobe potentiated if the postsynaptic neuron isstrongly depolarized (Huang et al. 2005). Theincrease of this IPSP also depends on NMDARactivation and CaMKII, thus sharing two keyproperties with hippocampal LTP of AMPARs.
SYNAPTIC PLASTICITY AND DISEASE
Altered LTP and LTD has been implicated as amechanism that may contribute to brain dis-eases as diverse as dementia, movement disor-
ders, depression, addiction, posttraumatic stresssyndrome, neuropathic pain, and anxiety disor-ders. To illustrate this emerging field and to em-phasize the point that there are multiple ways bywhich LTP and LTD can be involved in diseasepathophysiology, we will focus on two contrast-ing conditions: the loss of synaptic plasticityassociated with Alzheimer disease’s (AD) andexcessive plasticity observed after exposure toaddictive drugs.
A definitive diagnosis of AD requires thevisualization of amyloid plaques and neurofi-brillary tangles in histological sections of thebrain. The cognitive decline, however, startswell before this stage, and there is growing evi-dence that one of the toxic protein species be-lieved to be etiologically related to AD, solubleAb oligomers, causes early memory problemsby disrupting LTP and LTD mechanisms (Walshet al. 2002; Tanzi 2005; Shankar et al. 2008; Cisseet al. 2011). Direct application or overproduc-tion of Ab oligomers both inhibits LTP andtriggers LTD-like changes (Fig. 5). The net resultis weaker synapses that have difficulty generat-ing LTP. Furthermore, the toxic Ab also de-creases synaptic NMDARs, a change that con-tributes to the impaired LTP (Kamenetz et al.2003). Based on these findings, there is greatinterest in finding compounds that prevent thesynaptic effects of Ab oligomers with the hopethat such compounds will be therapeuticallybeneficial if given to patients early enough dur-ing disease progression. See Sheng et al. (2012)for a comprehensive discussion of synapticchanges associated with AD.
Addictive drugs also have profound effectson synaptic transmission that may influenceLTP and LTD (Wolf 2003; Kauer and Malenka2007; Luscher and Malenka 2011). For example,a single dose of a drug of abuse such as cocaineenhances excitatory transmission onto dopa-mine neurons of the ventral tegmental area(Ungless et al. 2001). This drug-evoked plastic-ity requires activation of type 1 dopamine re-ceptors along with NMDAR activation and isexpressed by a redistribution of AMPARs andNMDARs. Calcium-permeable, GluA2-lackingAMPARs appear (Bellone and Luscher 2006),while NMDAR function is decreased. As a result,
NMDA Receptor-Dependent LTP/LTD
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710 11
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from
the primary source for synaptic calcium entryshifts from NMDARs in naıve animals toGluA2-lacking AMPARs after a dose of an ad-dictive drug. As a consequence, the rules forsubsequent activity-dependent plasticity areinverted (Mameli and Luscher 2011). Theseearly drug-evoked changes in the propertiesof excitatory synapses on dopamine cells canbe very long-lasting and are followed by manyadditional synaptic modifications in relatedstructures. For example, in the nucleus accum-bens chronic (5-d) administration of cocainetriggers an internalization of AMPARs in medi-um spiny neurons, a change that occludes thesubsequent synaptic induction of NMDAR-dependent LTD (Thomas and Malenka 2003;Kauer and Malenka 2007; Wolf and Ferrario2010).
CONCLUSIONS
Because of a major effort by a large numberof investigators, the mechanisms underlyingNMDAR-dependent LTP and LTD are under-stood in reasonable molecular detail. Coinci-
dent activity in pre- and postsynaptic neuronsresulting in calcium influx through synapticNMDARs is well established to be necessaryfor the triggering of both LTP and LTD. Thiscauses the activity-dependent redistribution ofAMPARs, a general mechanism for modifyingsynaptic strength in many neuronal cell types.Elucidating the detailed molecular mechanismsby which AMPARs traffic to and away from syn-apses is an active topic of current investigations(see West and Greenberg 2012). An equally im-portant topic is the identification of the pro-teins that need to be synthesized to maintainLTP and LTD. Through coordination of func-tional changes in synaptic efficacy to structuralchanges in synapses, the reorganization of neu-ral networks can be structurally maintained forlong periods of time. Certainly, more completeunderstanding of the mechanisms underlyingLTP and LTD will continue to contribute toour understanding of the mechanisms of learn-ing and memory (see Mayford et al. 2012), aswell as provide insight into the pathophysiologyof a broad spectrum of brain diseases (see Shenget al. 2012).
AMPAR
mGluRs
NMDAR
Healthy synapse
Aβ oligomers
Early stage AD
Thinning and lossof synapses
Late stage AD
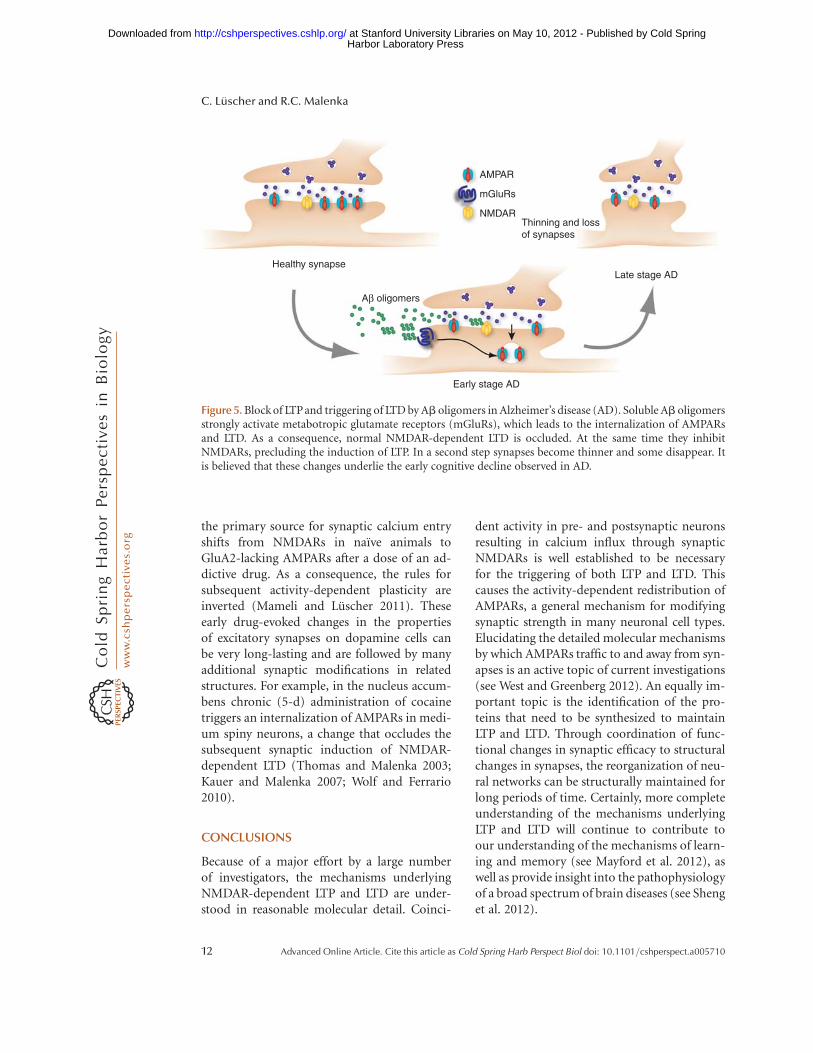
Figure 5. Block of LTPand triggering of LTD by Ab oligomers in Alzheimer’s disease (AD). Soluble Ab oligomersstrongly activate metabotropic glutamate receptors (mGluRs), which leads to the internalization of AMPARsand LTD. As a consequence, normal NMDAR-dependent LTD is occluded. At the same time they inhibitNMDARs, precluding the induction of LTP. In a second step synapses become thinner and some disappear. Itis believed that these changes underlie the early cognitive decline observed in AD.
C. Luscher and R.C. Malenka
12 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

REFERENCES�Reference is also in this collection.
Adesnik H, Nicoll RA. 2007. Conservation of glutamatereceptor 2-containing AMPA receptors during long-term potentiation. J Neurosci 27: 4598–4602.
Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, KandelER, Hawkins RD. 1996. Nitric oxide acts directly in thepresynaptic neuron to produce long-term potentiation incultured hippocampal neurons. Cell 87: 1025–1035.
Bellone C, Luscher C. 2006. Cocaine triggered AMPA recep-tor redistribution is reversed in vivo by mGluR-depen-dent long-term depression. Nat Neurosci 9: 636–641.
Benke TA, Luthi A, Isaac JT, Collingridge GL. 1998. Modu-lation of AMPA receptor unitary conductance by synapticactivity. Nature 393: 793–797.
Bienenstock EL, Cooper LN, Munro PW. 1982. Theory forthe development of neuron selectivity: Orientation spe-cificity and binocular interaction in visual cortex. J Neu-rosci 2: 32–48.
� Blakely RD, Edwards RH. 2012. Vesicular and plasma mem-brane transporters for neurotransmitters. Cold SpringHarb Perspect Biol doi: 10.1101/cshperspect.a005595.
Bliss TV, Collingridge GL. 1993. A synaptic model of mem-ory: Long-term potentiation in the hippocampus. Nature361: 31–39.
Bloodgood BL, Sabatini BL. 2007. Nonlinear regulation ofunitary synaptic signals by CaV2.3 voltage-sensitive calci-um channels located in dendritic spines. Neuron 53:249–260.
Bloodgood BL, Giessel AJ, Sabatini BL. 2009. Biphasic syn-aptic Ca influx arising from compartmentalized electricalsignals in dendritic spines. PLoS Biol 7: e1000190.
Bramham CR, Alme MN, Bittins M, Kuipers SD, Nair RR,Pai B, Panja D, Schubert M, Soule J, Tiron A, et al. 2010.The Arc of synaptic memory. Exp Brain Res 200: 125–140.
Caporale N, Dan Y. 2008. Spike timing-dependent plasticity:A Hebbian learning rule. Annu Rev Neurosci 31: 25–46.
Carroll RC, Beattie EC, von Zastrow M, Malenka RC. 2001.Role of AMPA receptor endocytosis in synaptic plasticity.Nat Rev Neurosci 2: 315–324.
� Castillo PE. 2012. Presynaptic LTP and LTD of excitatory andinhibitory synapses. Cold Spring Harb Perspect Biol doi:10.1101/cshperspect.a005728.
Castillo PE, Chiu CQ, Carroll RC. 2011. Long-term plastic-ity at inhibitory synapses. Curr Opin Neurobiol 21:328–338.
Chen L, Chetkovich DM, Petralia RS, Sweeney NT, KawasakiY, Wenthold RJ, Bredt DS, Nicoll RA. 2000. Stargazinregulates synaptic targeting of AMPA receptors by twodistinct mechanisms. Nature 408: 936–943.
Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B,Orr A, Lotz G, Kim DH, Hamto P, et al. 2011. ReversingEphB2 depletion rescues cognitive functions in Alz-heimer model. Nature 469: 47–52.
Collingridge GL, Isaac JT. 2003. Functional roles of proteininteractions with AMPA and kainate receptors. NeurosciRes 47: 3–15.
Collingridge GL, Isaac JT, Wang YT. 2004. Receptor traffick-ing and synaptic plasticity. Nat Rev Neurosci 5: 952–962.
Dan Y, Poo MM. 2006. Spike timing-dependent plasticity:From synapse to perception. Physiol Rev 86: 1033–1048.
De Roo M, Klauser P, Muller D. 2008. LTP promotes a se-lective long-term stabilization and clustering of dendriticspines. PLoS Biol 6: e219.
Derkach V, Barria A, Soderling TR. 1999. Ca2þ/calmodulin-kinase II enhances channel conductance of a-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type gluta-mate receptors. Proc Natl Acad Sci 96: 3269–3274.
Diamond JS, Bergles DE, Jahr CE. 1998. Glutamate releasemonitored with astrocyte transporter currents duringLTP. Neuron 21: 425–433.
Dingledine R, Borges K, Bowie D, Traynelis SF. 1999. Theglutamate receptor ion channels. Pharmacol Rev 51:7–61.
Ehlers MD. 2000. Reinsertion or degradation of AMPA re-ceptors determined by activity-dependent endocyticsorting. Neuron 28: 511–525.
Elias GM, Nicoll RA. 2007. Synaptic trafficking of glutamatereceptors by MAGUK scaffolding proteins. Trends CellBiol 17: 343–352.
Engert F, Bonhoeffer T. 1997. Synapse specificity of long-term potentiation breaks down at short distances. Nature388: 279–284.
Harvey CD, Svoboda K. 2007. Locally dynamic synapticlearning rules in pyramidal neuron dendrites. Nature450: 1195–1200.
Harvey CD, Yasuda R, Zhong H, Svoboda K. 2008. Thespread of Ras activity triggered by activation of a singledendritic spine. Science 321: 136–140.
Hebb DO. 1949. The organization of behavior: A neuropsy-chological theory. John Wiley & Sons, New York.
Holtmaat A, Svoboda K. 2009. Experience-dependent struc-tural synaptic plasticity in the mammalian brain. Nat RevNeurosci 10: 647–658.
Huang CS, Shi SH, Ule J, Ruggiu M, Barker LA, Darnell RB,Jan YN, Jan LY. 2005. Common molecular pathways me-diate long-term potentiation of synaptic excitation andslow synaptic inhibition. Cell 123: 105–118.
Inagaki T, Begum T, Reza F, Horibe S, Inaba M, Yoshimura Y,Komatsu Y. 2008. Brain-derived neurotrophic factor-me-diated retrograde signaling required for the induction oflong-term potentiation at inhibitory synapses of visualcortical pyramidal neurons. Neurosci Res 61: 192–200.
Isaac JTR, Nicoll RA, Malenka RC. 1995. Evidence for silentsynapses. Implications for the expression of LTP. Neuron15: 427–434.
Jahr CE, Stevens CF. 1993. Calcium permeability of the N-methyl-D-aspartate receptor channel in hippocampalneurons in culture. Proc Natl Acad Sci 90: 11573–11577.
Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D,Iwatsubo T, Sisodia S, Malinow R. 2003. APP processingand synaptic function. Neuron 37: 925–937.
Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Nogu-chi J. 2010. Structural dynamics of dendritic spines inmemory and cognition. Trends Neurosci 33: 121–129.
Kauer JA, Malenka RC. 2007. Synaptic plasticity and addic-tion. Nat Rev Neurosci 8: 844–858.
NMDA Receptor-Dependent LTP/LTD
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710 13
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

Kennedy MJ, Ehlers MD. 2011. Mechanisms and function ofdendritic exocytosis. Neuron 69: 856–875.
Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, ShengM. 2010. Caspase-3 activation via mitochondria is re-quired for long-term depression and AMPA receptor in-ternalization. Cell 141: 859–871.
Liao DZ, Hessler NA, Malinow R. 1995. Activation of post-synaptically silent synapses during pairing induced LTPin CA1 region of hippocampal slice. Nature 375: 400–404.
Lisman J. 1989. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. ProcNatl Acad Sci 86: 9574–9578.
Lisman J, Schulman H, Cline H. 2002. The molecular basisof CaMKII function in synaptic and behavioural memo-ry. Nat Rev Neurosci 3: 175–190.
Liu SJ, Zukin RS. 2007. Ca2þ-permeable AMPA receptors insynaptic plasticity and neuronal death. Trends Neurosci30: 126–134.
Lledo PM, Zhang X, Sudhof TC, Malenka RC, Nicoll RA.1998. Postsynaptic membrane fusion and long-term po-tentiation. Science 279: 399–403.
Luscher C, Frerking M. 2001. Restless AMPA receptors: Im-plications for synaptic transmission and plasticity. TrendsNeurosci 24: 665–670.
Luscher C, Malenka RC. 2011. Drug-evoked synaptic plas-ticity in addiction: From molecular changes to circuitremodeling. Neuron 69: 650–663.
Luscher C, Malenka RC, Nicoll RA. 1998. Monitoring glu-tamate release during LTP with glial transporter currents.Neuron 21: 435–441.
Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M,Malenka RC, Nicoll RA. 1999. Role of AMPA receptorcycling in synaptic transmission and plasticity. Neuron24: 649–658.
Luscher C, Nicoll RA, Malenka RC, Muller D. 2000. Synapticplasticity and dynamic modulation of the postsynapticmembrane. Nat Neurosci 3: 545–550.
Luthi A, Chittajallu R, Duprat F, Palmer MJ, Benke TA, KiddFL, Henley JM, Isaac JT, Collingridge GL. 1999. Hippo-campal LTD expression involves a pool of AMPARs reg-ulated by the NSF–GluR2 interaction. Neuron 24:389–399.
Malenka RC. 1994. Synaptic plasticity in the hippocampus:LTP and LTD. Cell 78: 535–538.
Malenka RC, Bear MF. 2004. LTP and LTD: An embarrass-ment of riches. Neuron 44: 5–21.
Malenka RC, Nicoll RA. 1999. Long-term potentiation—adecade of progress? Science 285: 1870–1874.
Malinow R, Malenka RC. 2002. AMPA receptor traffickingand synaptic plasticity. Annu Rev Neurosci 25: 103–126.
Mameli M, Luscher C. 2011. Synaptic plasticity and addic-tion: Learning mechanisms gone awry. Neuropharmacol-ogy 61: 1052–1059.
Manabe T, Nicoll RA. 1994. Long-term potentiation: Evi-dence against an increase in transmitter release probabil-ity in the CA1 region of the hippocampus. Science 265:1888–1892.
Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. 1993. Modula-tion of synaptic transmission and long-term poten-
tiation: Effects on paired pulse facilitation and EPSCvariance in the CA1 region of the hippocampus. J Neuro-physiol 70: 1451–1459.
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. 2004.Structural basis of long-term potentiation in single den-dritic spines. Nature 429: 761–766.
� Mayford M, Siegelbaum SA, Kandel ER. 2012. Synapses andmemory storage. Cold Spring Harb Perspect Biol doi:10.1101/cshperspect.a005751.
McNaughton BL. 1982. Long-term synaptic enhancementand short-term potentiation in rat fascia dentata actthrough different mechanisms. J Physiol 324: 249–262.
Mulkey RM, Herron CE, Malenka RC. 1993. An essentialrole for protein phosphatases in hippocampal long-termdepression. Science 261: 1051–1055.
Mulkey RM, Endo S, Shenolikar S, Malenka RC. 1994. In-volvement of a calcineurin/inhibitor-1 phosphatase cas-cade in hippocampal long-term depression. Nature 369:486–488.
Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. 2004.Bidirectional activity-dependent morphological plastici-ty in hippocampal neurons. Neuron 44: 759–767.
Nicoll RA. 2003. Expression mechanisms underlying long-term potentiation: A postsynaptic view. Philos Trans R SocLond B Biol Sci 358: 721–726.
Nugent FS, Penick EC, Kauer JA. 2007. Opioids block long-term potentiation of inhibitory synapses. Nature 446:1086–1090.
Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A,McBain CJ, Collingridge GL, Isaac JT. 2006. Transientincorporation of native GluR2-lacking AMPA receptorsduring hippocampal long-term potentiation. Nat Neuro-sci 9: 602–604.
Redondo RL, Morris RG. 2011. Making memories last: Thesynaptic tagging and capture hypothesis. Nat Rev Neuro-sci 12: 17–30.
Sabatini BL, Oertner TG, Svoboda K. 2002. The life cycle ofCa2þ ions in dendritic spines. Neuron 33: 439–452.
Sacktor TC. 2008. PKMz, LTP maintenance, and the dynam-ic molecular biology of memory storage. Prog Brain Res169: 27–40.
Salter MW, Kalia LV. 2004. Src kinases: A hub for NMDAreceptor regulation. Nat Rev Neurosci 5: 317–328.
Schulz PE, Cook EP, Johnston D. 1995. Using paired-pulsefacilitation to probe the mechanisms for long-term po-tentiation (LTP). J Physiol Paris 89: 3–9.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, ShepardsonNE, Smith I, Brett FM, Farrell MA, Rowan MJ, LemereCA, et al. 2008. Amyloid-b protein dimers isolated di-rectly from Alzheimer’s brains impair synaptic plasticityand memory. Nat Med 14: 837–842.
� Sheng M, Kim E. 2012. The postsynaptic organization ofsynapses. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005678.
� Sheng M, Sabatini B, Sudhof TC. 2012. Synapses and Alz-heimer’s disease. Cold Spring Harb Perspect Biol doi:10.1101/cshperspect.a005777.
Stuart G, Spruston N, Sakmann B, Hausser M. 1997. Actionpotential initiation and backpropagation in neurons ofthe mammalian CNS. Trends Neurosci 20: 125–131.
C. Luscher and R.C. Malenka
14 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from

Sweatt JD. 2004. Mitogen-activated protein kinases in syn-aptic plasticity and memory. Curr Opin Neurobiol 14:311–317.
Tanzi RE. 2005. The synaptic Ab hypothesis of Alzheimerdisease. Nat Neurosci 8: 977–979.
Thomas MJ, Malenka RC. 2003. Synaptic plasticity in themesolimbic dopamine system. Philos Trans R Soc Lond BBiol Sci 358: 815–819.
Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. 2005.Bidirectional synaptic plasticity regulated by phosphor-ylation of stargazin-like TARPs. Neuron 45: 269–277.
Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D. 1999.LTP promotes formation of multiple spine synapses be-tween a single axon terminal and a dendrite. Nature 402:421–425.
Toni N, Buchs PA, Nikonenko I, Povilaitite P, Parisi L, MullerD. 2001. Remodeling of synaptic membranes after induc-tion of long-term potentiation. J Neurosci 21: 6245–6251.
Ungless MA, Whistler JL, Malenka RC, Bonci A. 2001. Sin-gle cocaine exposure in vivo induces long-term potenti-ation in dopamine neurons. Nature 411: 583–587.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R,Wolfe MS, Rowan MJ, Selkoe DJ. 2002. Naturally secretedoligomers of amyloid b protein potently inhibit hip-pocampal long-term potentiation in vivo. Nature 416:535–539.
Wang XB, Yang Y, Zhou Q. 2007. Independent expression ofsynaptic and morphological plasticity associated withlong-term depression. J Neurosci 27: 12419–12429.
Waters J, Schaefer A, Sakmann B. 2005. Backpropagatingaction potentials in neurones: Measurement, mecha-nisms and potential functions. Prog Biophys Mol Biol87: 145–170.
� West AE, Greenberg ME. 2012. Neuronal activity-regulatedgene transcription in synapse development and cognitivefunction. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005744.
Williams JH, Li YG, Nayak A, Errington ML, Murphy KP,Bliss TV. 1993. The suppression of long-term potentia-tion in rat hippocampus by inhibitors of nitric oxidesynthase is temperature and age dependent. Neuron 11:877–884.
Wolf ME. 2003. LTP may trigger addiction. Mol Interv 3:248–252.
Wolf ME, Ferrario CR. 2010. AMPA receptor plasticity in thenucleus accumbens after repeated exposure to cocaine.Neurosci Biobehav Rev 35: 185–211.
Zakharenko SS, Zablow L, Siegelbaum SA. 2001. Visualiza-tion of changes in presynaptic function during long-termsynaptic plasticity. Nat Neurosci 4: 711–717.
Zhou Q, Homma KJ, Poo MM. 2004. Shrinkage of dendriticspines associated with long-term depression of hippo-campal synapses. Neuron 44: 749–757.
NMDA Receptor-Dependent LTP/LTD
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a005710 15
Harbor Laboratory Press at Stanford University Libraries on May 10, 2012 - Published by Cold Springhttp://cshperspectives.cshlp.org/Downloaded from