nf-κb localization in multiple myeloma plasma cells and ... · nf- b localization in multiple...
TRANSCRIPT

N
CEAMa
b
c
d
e
f
a
ARRAA
KMNBM
1
imiai[[
ap
piS
tf
0d
Leukemia Research 35 (2011) 52–60
Contents lists available at ScienceDirect
Leukemia Research
journa l homepage: www.e lsev ier .com/ locate / leukres
F-�B localization in multiple myeloma plasma cells and mesenchymal cells
oncetta Conticelloa, Raffaella Giuffridab, Luana Adamob, Gabriele Anastasib, Daniela Martinettib,dvige Salomonec, Cristina Colarossi a, Gabriella Amatoa, Ausilia Gorgoned,lessandra Romanod, Gioacchin Iannolob, Ruggero De Mariae, Rosario Giustolisi a,assimo Gulisanoa,f, Francesco Di Raimondod,∗
Department of Experimental Oncology, Mediterranean Institute of Oncology (IOM), Viagrande, Catania, ItalyIOM (Mediterranean Institute of Oncology) Ricerca, Viagrande, Catania, ItalyPathology Unit, Azienda Ospedaliera V. Emanuele – Ferrarotto – S. Bambino, Catania, ItalyDepartment of Biomedical Sciences, Hematology Section, University of Catania, Catania, ItalyDepartment of Oncology and Molecular Medicine, Istituto Superiore di Sanità, Rome, ItalyDepartment of Physiological Sciences, University of Catania, Catania, Italy
r t i c l e i n f o
rticle history:eceived 26 January 2010eceived in revised form 5 May 2010ccepted 28 June 2010
a b s t r a c t
Several reports demonstrated that the activation of Nuclear Factor-kappa B NF-�B is essential for thepathogenesis of multiple myeloma (MM). We analyzed the nuclear localization of NF-�B in MM-cellsderived from 60 different patients with MM at presentation and in relapse, as well as in three myeloma
vailable online 31 July 2010
eywords:ultiple myelomaF-�Bortezomib
cell lines. Nuclear localization (the active form) of NF-�B was detected in only one MM-sample froma refractory patient and in two samples from relapsed patients, while all the other samples, includingthe MM-cell lines, almost exclusively express the cytoplasmic (inactive) form of NF-�B. In mesenchymalcells from MM-patients NF-�B was clearly present in the nucleus. In addition, the proteasome inhibitorBortezomib, which is described to antagonize NF-�B activity, had a consistent antitumor activity againstboth chemoresistant and chemosensitive MM-cells, regardless the NF-�B localization, thus suggesting
lecul
esenchymal cells the existence of other mo. Introduction
Multiple myeloma (MM) is a clonal B-cell malignancy character-zed by accumulation of terminally differentiated B-lymphocytes,
alignant plasma cells (PCs), within the BM. MM PCs localizento the BM where cell adhesion-mediated autocrine or paracrinectivation of different cytokines, such as interleukin 6 (IL-6) andnsulin-like growth factor 1 (IGF-1), results in PCs accumulation1]. A major role in the pathogenesis of MM is attributed to NF-�B
2].The NF-�B family of transcription factors is composed by anrray of homo- and heterodimers (containing p50, p52, c-Rel,65/RelA, and RelB). These proteins are held in the cytoplasm
Abbreviations: NF-�B, Nuclear Factor-kappa B; MM, multiple myeloma; PCs,lasma cells; IL-6, interleukin 6; BM, bone marrow; MCs, mesenchymal cells; IF,
mmunofluorescence; IHC, immunohistochemistry; EMSA, Electrophoresis Mobilityhift Assay; ELISA, Enzyme-Linked Immuno Sorbent Assay.∗ Corresponding author at: Department of Biomedical Sciences, Hematology Sec-
ion, University of Catania, Via Citelli 7, 95100 Catania, Italy. Tel.: +39 095 7435911;ax: +39 095 7435913.
E-mail address: [email protected] (F. Di Raimondo).
145-2126/$ – see front matter © 2010 Elsevier Ltd. All rights reserved.oi:10.1016/j.leukres.2010.06.023
ar targets of proteasome inhibitors in MM.© 2010 Elsevier Ltd. All rights reserved.
of most normal cells as an inactive latent form by specificproteins, the inhibitors of NF-�Bs (I�Bs). Several stimuli acti-vate NF-�B through kinase-dependent phosphorylation of I�Band its subsequent degradation by the 26S proteasome [3,4].This process leaves NF-�B free to translocate into the nucleus,where it activates the transcription of numerous genes, suchas cyclin D1 [5], Bcl-xL, and IAPs [6], and activates numeroustranscriptional processes. The active form of the transcriptionfactor plays an important role in inflammation, immune andstress responses, oncogenesis, cell migration, and angiogene-sis.
Two major pathways lead to the activation of NF-�B: a classicaland an alternative pathway. The classical pathway is defined byactivation of p50–p65 complexes upon degradation of associatedI�B. The alternative pathway is characterized by processing of aninactive p100-RelB dimer to active p52-RelB through proteosomaldegradation of inhibitory C-terminal I�B-like sequences of NF-�B2p100 [7,8].
In both pathways, NF-�B activation can be effectively blocked byproteasome inhibitors, which interrupt NF-�B translocation fromthe cytoplasm to the nucleus by inhibiting degradation of I�B pro-teins (Fig. 1). This mechanism has been demonstrated in differentmalignancies, where NF-�B is aberrantly activated and contributes

C. Conticello et al. / Leukemia R
Fig. 1. NF-�B activation pathways.The so-called classical/canonical NF-�B pathway is triggered by many inflammatorystimuli to induce IKK2-containing IKK complexes that specifically phosphorylatethe three canonical I�B proteins, thereby marking them for ubiquitination andproteasome-mediated proteolysis. Cytoplasmic RelA (A) as well as cRel-containingdimers are thereby released to translocate to the nucleus and activate genes mainlyinvolved in inflammatory processes. The alternative/non-canonical pathway ismediated by IKK1, is strictly dependent on IKK� homodimers and is activated bylymphotoxin � receptor (LT�R), B cell-activating factor belonging to the TNF family(BAFF), and CD40 ligand (CD40L). This pathway induces the release of RelB (B)-coCr
tpt
itaeacria(o(ttttB
2
2
n
ontaining dimers to the nucleus where NF-�B plays a central role in the expressionf genes involved in development and maintenance of secondary lymphoid organs.rosstalk between canonical and non-canonical signalling pathways is of currentesearch interest.
o the drug resistance, and where proteasome inhibitors block I�Broteasomal degradation, thus inhibiting NF-�B nuclear transloca-ion [9,10].
Moreover, it has been widely demonstrated that proteasomenhibitor Bortezomib induces apoptosis and reverses drug resis-ance of MM cells by affecting cytokine circuits, cell adhesion andngiogenesis in BM microenvironment. However, there are severalxperimental evidences that Bortezomib may also acts with mech-nisms different from the simple inhibition of NF-�B and it is notlear if there is a correlation between the NF-�B status and theesponse to Bortezomib. To better define the role of NF-�B statusn the pathogenesis of MM and the sensitivity to Bortezomib, wenalyzed the sub-cellular localization of the p65 subunit of NF-�Bclassical pathway) in 60 different samples from newly diagnosedr relapsed patients with MM, as well as in three myeloma cell linesXG1, RPMI 8226, KMS-18). In this study, we decided to analyze onlyhe sub-cellular localization of the p65 subunit of NF-�B becausehe majority of the studies reported on MM and NF-�B concernhe analysis of the classical pathway. Furthermore, we analyzedhe sensitivity of MM primary cells to the proteasome inhibitorortezomib.
. Materials and methods
.1. Primary cells/bone marrow cultures
37 BM biopsy and 22 marrow aspirates were obtained as part of routine exami-ation from patients with MM at diagnosis or at relapse who gave informed consent
esearch 35 (2011) 52–60 53
and were managed at the Division of Hematology, Ospedale Ferrarotto, Universityof Catania. One sample of MM PCs was obtained from the paracentesis of one MMpatient resistant to chemotherapy with neoplastic ascites. All samples were selectedfor PCs infiltration from 50 to 99%. BM biopsies were fixed with 10% buffered forma-lin or fixed and decalcified in Lowy’s solution. Four micron sections were dewaxedand antigen retrieval was carried out by treating sections for 30 min in pH 6 cit-rate buffer at 98 ◦C. BM aspirates were collected in tubes containing EDTA and cellswere isolated by Ficoll Hypaque (Cedarlane labs, Ontario, Canada) density gradi-ent centrifugation. The CD138+ cells were separated as described by Hata et al. [11].Cells were maintained in RPMI 1640 medium (Gibco, Invitrogen) supplemented with2 mM l-glutamine and 100 U/ml pennicillin–streptomycin and 10% heat-inactivatedfetal bovine serum (FBS) (Invitrogen, Carlsbad, CA). Cells were kept in a 5% CO2
atmosphere at a density of 5 × 105 cells/ml. BM mononuclear cells separated byFicoll-Hypaque density sedimentation were also used to establish long-term BMmesenchymal cells (MCs) cultures as described previously [12]. Briefly, BM cellswere cultured (1 × 105 cells/ml) for 24 h to obtain a confluent adherent cell mono-layer of BM MCs. After BM MCs formed a confluent adherent layer, remainingnon-adherent cells were washed with PBS.
2.2. Cell lines
The human MM cell lines XG-1, RPMI-8226 and KMS18 both plasmocytomasof B-cell origin, were obtained from the American Type Culture Collection (ATCC,Rockville, MD, USA). Cells were maintained in RPMI 1640 medium (Gibco, Invitro-gen) supplemented with 2 mM l-glutamine and 100 U/ml penicillin–streptomycinand 10% FBS (Invitrogen) and were kept in a 5% CO2 atmosphere at a density of5 × 105 cells/ml.
2.3. Reagents
Drugs were purchased from Sigma–Aldrich (St. Louis, MO) and resuspended inDMSO (Etoposide) or in water (Cytarabine, Daunorubicine and Bortezomib). Borte-zomib (also known as PS-341) was purchased from Millennium Pharmaceuticals(Cambridge, MA).
2.4. Cell viability assay
Cell viability was determined using the CellTiter 96 AQueous One SolutionCell Proliferation Assay (Promega, Madison, WI), according to the manufac-turer’s instructions. Cells were seeded into 3 wells of 96-well plates at5 × 103 cells/200 �l/well and treated with different combinations of the drugstested. After 24 and 48 h of incubation the culture medium was removed and thecells were washed with PBS (pH 7.4); 100 �l of fresh culture medium without drugsand 20 �l of assay solution were added to each well and the cells were incubatedfor 3 h. The plates were read on a Microplate Reader (Synergy HT, BIO-TEK). Survivalwas expressed as the percentage of viable cells in the treated sample versus theuntreated control cells.
2.5. Flow cytometry
105 BM aspirate cells were washed with cold PBS containing 1% BSA andincubated for 1 h at 4 ◦C with control or specific primary antibodies: goat anti-human CD138 (1:50, R&D Systems, Minneapolis, MN). After washing, the cells wereincubated for 40 min at 4 ◦C with phycoerythrin-conjugated anti-goat secondaryantibodies (1:100, Jackson Laboratories, West Grove, PA). Labeled cells were washedtwice with PBS/BSA and the fluorescence intensity was evaluated by a FACScan(Beckman Coulter, Fullerton, CA).
2.6. Immunostaining procedure
Immunohistochemical staining was performed on 2 �m thick paraffin-embedded BM biopsies. After deparaffination-hydration, sections were permeabi-lized with PBS containing 0.4% Triton X-100 for 30 min and blocked with PBScontaining 5% BSA for 30 min. The samples were incubated, over night at 4 ◦C, withrabbit anti-human NF-�B/p65 primary antibody (1:50, Santa Cruz). The day after,slides were first incubated for 1 h with biotinylated anti-goat secondary antibody,and then with streptavidin-HRP (Dako Corp., Carpintera, CA). Staining was detectedusing diaminobenzedine (DAB) as chromogen. Sections were counterstained withhematoxylin.
Immunofluorescence staining was performed on cells in suspension seeded onglass slides. Cells were washed in PBS buffer and fixed with 4% paraformaldheyde
for 10 min. After rinsing, cells were permeabilized with 0.2% Triton X-100 in PBSfor 5 min and nonspecific staining was blocked in 4% BSA in PBS for 30 min. Cellswere then incubated with rabbit anti-human NF-�B/p65 (1:50, Santa Cruz, CA)for 1 h at room temperature. Subsequently, cells were washed and incubated withCy3-conjugated mouse anti-rabbit immunoglobulins (1:500 Jackson Laboratories).Nuclei were stained with DAPI (Sigma, St. Louis, MO).
5 mia Re
2
t((c4lmpntBTwow
F(mNrm(fl
4 C. Conticello et al. / Leuke
.7. Western Blotting
Western Blot analysis was carried out on proteins isolated after cellular frac-ionation. Cytoplasmic proteins were obtained by lysing cells in hypotonic buffer10 mmol/l Trizma Base, 10 mmol/l KCl, 2 mmol/l phenylmethylsulfonyl fluoridePMSF), 2× Protease Inhibitor Cocktail, 0.2% NP40). After 2 min incubation in ice,ytosolic proteins were recovered by centrifugation at 2000 rpm for 10 min at◦C. Nuclear proteins were extracted resuspending the residual pellet in nuclear
ysis buffer (10 mmol/l trizma base, 10 mmol/l KCl, 100 mmol/l NaCl, 7 mmol/l �-ercaptoethanol, 2× protease inhibitor cocktail), and subsequently harvesting the
roteins by centrifugation at 14,000 rpm for 15 min at 4 ◦C. 30 �g cytoplasmic anduclear proteins were loaded onto 10% SDS-polyacrylamide gels. Gels were elec-
roblotted onto nitrocellulose membranes (Hybond C-extra, Amersham Biosciences,uckinghamshire, UK). Membranes were blocked for 1 h in 5% nonfat dry milk inBS-T (2.5 mM Tris–HCl, 15 mM NaCl, 0.05% Tween 20) and then incubated for 1 hith the primary antibody rabbit anti-human NF-�B/p65 (1:50, Santa Cruz), mon-clonal anti-Tubulin and polyclonal anti-Histone 2B antibodies (Santa Cruz). Filtersere then rinsed and incubated for 1 h with the corresponding secondary antibod-
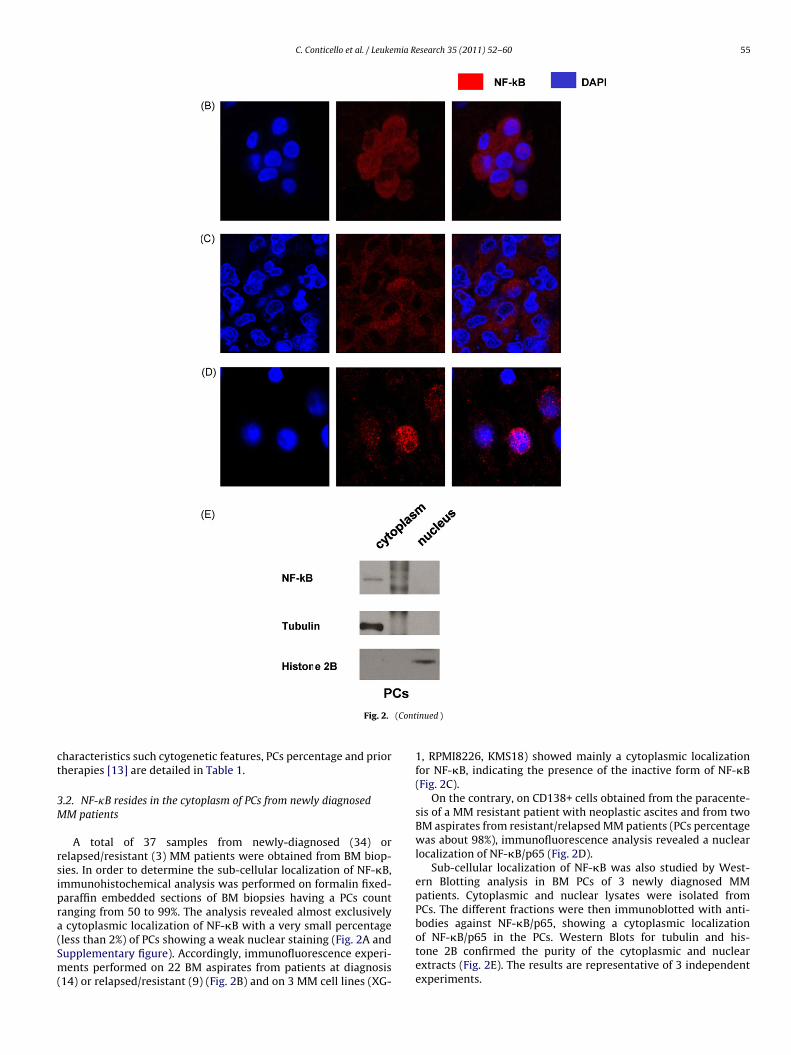
ig. 2. NF-�B cellular localization in MM patients and cell lines.A) NF-�B/p65 immunohistochemical analysis on 8 BM specimen from newly diagnosed
agnification. Small square represents a picture with an higher magnification.F-�B/p65 immunofluorescence staining on MM primary cells from a patients (out of 20 a
elapsed/resistant patient (one out of 3 analyzed) (D). Blue stain indicates the position of terged in the third picture. Original magnification 63×.
E) Cellular fractionation analysis for NF-�B/p65 lysates isolated from BM PCs of 3 newly dor the cytoplasmic and nuclear extracts purity. The results are representative of 3 indeegend, the reader is referred to the web version of the article.)
search 35 (2011) 52–60
ies conjugated with peroxidase (Amersham Biosciences). After washing, proteinswere detected by the enhanced chemiluminescent method (ECL plus; AmershamBiosciences).
2.8. Statistical analysis
A two-tailed paired t-test was used to analyze the statistical significance of theresults. Values of p < 0.05 was considered statistically significant. One asterisk indi-cates p < 0.05, two asterisks p > 0.05. Data are presented as mean values ± SD of themean.
3. Results
3.1. Samples
A total of 48 samples from patients with MM at diagnosis and12 samples from resistant/refractory MM were examined. Patients’
MM patients. NF-�B/p65 staining in brown, nuclear staining in blue. 40× Original
nalyzed) at diagnosis (B), on KMS18 MM cell line (C) and on MM primary cells fromhe nuclei (DAPI), red stain indicates the specific staining for NF-�B/p65, signals are
iagnosed MM patients. Western Blots for tubulin and histone 2B are used as controlpendent experiments. (For interpretation of the references to color in this figure
C. Conticello et al. / Leukemia Research 35 (2011) 52–60 55
(Cont
ct
3M
rsipra(Sm(
Fig. 2.
haracteristics such cytogenetic features, PCs percentage and priorherapies [13] are detailed in Table 1.
.2. NF-�B resides in the cytoplasm of PCs from newly diagnosedM patients
A total of 37 samples from newly-diagnosed (34) orelapsed/resistant (3) MM patients were obtained from BM biop-ies. In order to determine the sub-cellular localization of NF-�B,mmunohistochemical analysis was performed on formalin fixed-araffin embedded sections of BM biopsies having a PCs countanging from 50 to 99%. The analysis revealed almost exclusively
cytoplasmic localization of NF-�B with a very small percentageless than 2%) of PCs showing a weak nuclear staining (Fig. 2A andupplementary figure). Accordingly, immunofluorescence experi-ents performed on 22 BM aspirates from patients at diagnosis
14) or relapsed/resistant (9) (Fig. 2B) and on 3 MM cell lines (XG-
inued )
1, RPMI8226, KMS18) showed mainly a cytoplasmic localizationfor NF-�B, indicating the presence of the inactive form of NF-�B(Fig. 2C).
On the contrary, on CD138+ cells obtained from the paracente-sis of a MM resistant patient with neoplastic ascites and from twoBM aspirates from resistant/relapsed MM patients (PCs percentagewas about 98%), immunofluorescence analysis revealed a nuclearlocalization of NF-�B/p65 (Fig. 2D).
Sub-cellular localization of NF-�B was also studied by West-ern Blotting analysis in BM PCs of 3 newly diagnosed MMpatients. Cytoplasmic and nuclear lysates were isolated fromPCs. The different fractions were then immunoblotted with anti-
bodies against NF-�B/p65, showing a cytoplasmic localizationof NF-�B/p65 in the PCs. Western Blots for tubulin and his-tone 2B confirmed the purity of the cytoplasmic and nuclearextracts (Fig. 2E). The results are representative of 3 independentexperiments.
56 C. Conticello et al. / Leukemia Research 35 (2011) 52–60
Table 1MM samples characteristics.
Samples (BM biopsy) Immunoglubulin heavy chain type Durie –Salmon stage %PCs Cytogenetics Status of disease
1 IgA� III 65 Nd Newly diagnosed2 IgA� I 55 Nd Newly diagnosed3 IgA� III 50% Nd Newly diagnosed4 IgG� III 50 Nd Newly diagnosed5 IgA� III 50 del 13 Newly diagnosed6 IgA� III 78 Nd Newly diagnosed7 IgG� III 60 Nd Newly diagnosed8 IgG� III 50 Nd Newly diagnosed9 IgA� III 75 del 13 Newly diagnosed
10 IgG� III 70 del13q Newly diagnosed11 IgG� III 65 del 13 Newly diagnosed12 Light chain only, Bence Jones protein III 60 Nd Newly diagnosed13 IgG� III 70 del13q Newly diagnosed14 IgA� III 90 Nd Newly diagnosed15 IgA� III 55 Nd Newly diagnosed16 IgA� II 70 Nd Newly diagnosed17 IgA� III 90 Nd Resistant18 IgA� III 80 Del13q Newly diagnosed19 IgA� III 65 Nd Newly diagnosed20 IgA� III 50 Nd Newly diagnosed21 IgA� III 50 Nd Newly diagnosed22 Light chain only, Bence Jones protein III 90 Nd Newly diagnosed23 IgA� III 90 Nd Newly diagnosed24 IgA� II 60 Nd Relapsed25 IgGK III 80 Nd Newly diagnosed26 IgGK III 55 Nd Newly diagnosed27 IgGK II 90 del13q Relapsed28 IgGK III 50 Nd Newly diagnosed29 IgGK III 80 Nd Newly diagnosed30 IgGK III 90 Nd Newly diagnosed31 IgGK III 55 Nd Newly diagnosed32 IgGK III 65 Nd Newly diagnosed33 IgGK III 70 46 XX, 45 X, 13 (1 metaphase) Newly diagnosed34 IgA� III 80 Nd Newly diagnosed35 Light chain only, Bence Jones protein III 80 Nd Newly diagnosed36 IgG� III 65 Nd Newly diagnosed37 IgGK II 55 Nd Newly diagnosed
Samples (BM cells) Immunoglubulin heavy chain type Durie–Salmon stage %PCs Cytogenetics Status of disease
1 IgG� III 50 Nd Newly diagnosed2 Light chain only, Bence Jones protein II 72 Nd Newly diagnosed3 IgA� III 90 Nd Newly diagnosed4 IgA� III 80 Nd Newly diagnosed5 IgG� III 75 Nd Newly diagnosed6 IgG� III 70 Nd Newly diagnosed7 IgG� III 75 Nd Newly diagnosed8 IgG� III 85 Nd Newly diagnosed9 IgG� III 65 Nd Newly diagnosed
10 IgG� III 78 Nd Newly diagnosed11 IgA� III 90 Nd Newly diagnosed12 IgG� III 86 Nd Newly diagnosed13 IgG� II 58 Nd Newly diagnosed14 IgA� III 63 Nd Newly diagnosed15 IgA� III 75 Nd Relapsed/resistant16 IgA� III 65 Nd Relapsed/resistant17 IgG� III 80 Nd Relapsed/resistant18 IgG� III 95 Nd Relapsed/resistant19 Light chain only, Bence Jones protein III 80 Nd Relapsed/resistant20 IgA� III 90 Nd Relapsed/resistant21 IgG� II 74 Nd Relapsed/resistant22 IgGK III 50 Nd Relapsed/resistant23 (neoplastic ascites) IgGK III 98 Nd Relapsed/resistant
R and dR imenN
3f
mci
elapsed patients: relapsed to the first line of chemotherapy including thalidomideelapsed/resistant: resistance to 2 or more line of chemotherapy including VAD regd, not determined.
.3. NF-�B localization is nuclear in BM mesenchymal cells (MCs)rom newly diagnosed MM patients
The bone-marrow microenvironment plays a key role inyeloma cells growth and survival and the interactions of myeloma
ells with the microenvironment are widely believed to be criticaln the pathophysiology of MM. Several studies have investigated
esametazone.and/or thalidomide and desametazone and/or MP regimen.
mesenchymal cells from multiple myeloma patients with par-ticular attention to cell surface antigens, cytokines, and growth
factors expression, concluding that MM mesenchymal cells arephenotypically and functionally distinguishable from normal donormesenchymal cells.On the basis of these studies and the studies that describe arole of new drugs such as Bortezomib counteracting the inter-

C. Conticello et al. / Leukemia Research 35 (2011) 52–60 57
Fig. 3. NF-�B localization in BM MCs from newly diagnosed MM patients.(A) NF-�B/p65 fluorescence immunostaining on MCs from one (out of three) newly diagnosed MM patients. Blue stain indicates the position of the nuclei (DAPI), red staini re. Ori n andr referea
awc
BwBciirmf�(ilne
3B
opo(rwo8
ea
ndicates the specific staining for NF-�B/p65, signals are merged in the third pictusolated from BM MCs of 3 newly diagnosed MM patients. Western Blots for tubuliesults are representative of 3 independent experiments. (For interpretation of therticle.)
ction between MM plasma cells and mesenchymal cells [14],e decide to investigate the NF-�B status of MM mesenchymal
ells.Sub-cellular localization of NF-�B was therefore studied in
M MCs from 3 newly diagnosed MM patients. The studyas performed, either by immunocytochemistry and Westernlotting experiments, on MCs. Immunocytochemistry analysisarried out on BM MCs, isolated from cell culture by their abil-ty to adhere to the plate within 48–72 h, showed NF-�B/p65n the active status by nuclear localization (Fig. 3A). Similaresults were obtained by Western Blotting analysis. Cytoplas-ic and nuclear lysates were isolated from MCs. The different
ractions were then immunoblotted with antibodies against NF-B/p65, showing a nuclear localization of NF-�B/p65 in the MCs
Fig. 3B) compared to PCs where the signal was clearly visiblen the cytoplasmic fraction (Fig. 2E). Western Blots for tubu-in and histone 2B confirmed the purity of the cytoplasmic anduclear extracts. The results are representative of 3 independentxperiments.
.4. MM PCs exhibit a great dose-dependent sensitivity toortezomib
We next studied the effect of proteasome inhibitor Bortezomibn MM PCs. For this purpose, MM PCs were incubated in vitro in theresence of 0.01 �M Bortezomib for 48 h (Fig. 4A) or in the presencef decreasing concentrations (10–0.01 �M) of Bortezomib for 24 hFig. 4B). These biologically active concentrations are fully compa-able with those achieved in vivo in the clinical setting. Cell viabilityas measured by MTS assay. The results represent the means ± SD
f 3 independent experiments for each MM sample (15 at diagnosis,resistant/relapsed).
Despite the cytoplasmic localization of NF-�B, with the onlyxception for the sample obtained from the paracentesis and BMspirates of 2 relapsed/resistant patients (nuclear localization),
iginal magnification, 20×. (B) Cellular fractionation analysis for NF-�B/p65 lysateshistone 2B are used as control for the cytoplasmic and nuclear extracts purity. Thences to color in this figure legend, the reader is referred to the web version of the
24 h of treatment with low dose (0.01 �M) Bortezomib triggeredto a great reduction of viable cells in both newly diagnosed andresistant/relapsed patient without any significant difference in thesensitivity to Bortezomib. Furthermore, the cytotoxic effect wasdose- and time-dependent in both early and resistant/relapsedsamples. In addition, although some of the samples in our studyharboured the del(13), they showed the same sensitivity to Borte-zomib as other samples, thus confirming recent published data [15],that Bortezomib cytotoxic activity overcomes this poor prognosticfeature.
CD138+ cells from the patient with neoplastic ascites weretreated with increasing concentrations of Bortezomib (from 1 to0.01 �M) and different chemotherapeutic drugs at concentrationscomparable with those administrated in vivo in the clinical setting.Cell viability was measured by MTS assay. After 24 h of treatment,Bortezomib induced, even at lowest doses (0.01 �M), reduction of50% of cell viability. At this concentration, Bortezomib in combina-tion with Melphalan induced 85% of cytotoxicity, while Melphalanalone reached only 15% of reduction of viability (Fig. 4C) thus con-firming a recent study on synergistic effect of alkylating substancestogether with Bortezomib [16].
4. Discussion
In the present study, we have demonstrated, in contrast to otherreports, that at least the p65 (RelA) member of the NF-�B family,that belongs to canonical pathway, resides mainly in the cytoplasm(the inactive form) of PCs from all newly diagnosed MM patients,the majority of relapsed patients and some MM cell lines. We havefound a nuclear localization of NF-�B/p65 (active form) only in
three samples from refractory patients and in BM mesenchymalcells from MM patients. However, we do not exclude that othermembers of the NF-�B family belonging to noncanonical pathwaymight be found in the active form (nuclear localization) in the sameMM patients.
58 C. Conticello et al. / Leukemia Research 35 (2011) 52–60
Fig. 4. Comparation of Bortezomib with standard chemotherapeutics on MM PCs.MM PCs were incubated in vitro for 48 h in the presence of Bortezomib 0.01 �M (A) or in the presence of decreasing concentrations (10–0.01 �M) of Bortezomib for 24 h (B).Cell viability was measured by MTS assay. The results represent the means ± SD of 23 independent experiments for each MM sample (15 at diagnosis, 8 resistant/relapsed).Data are presented as mean values ± SD of the mean.(C) CD138+ cells from neoplastic ascites of a resistant patient were treated with increasing concentrations of Bortezomib [1–0.01 �M] and standard chemotherapeuticdrugs: Melphalan (Mel) 10 �g/ml, Melphalan plus Bortezomib (Mel + Bort), Cytarabin (Cyt) 1 �M, Cisplatin (Cispl) 300 ng/ml, Etoposide (Eto) 0.5 �g/ml, Mitoxantrone (Mito)1 in (Epe
dcMctmht
bcnen
�g/ml, Metotrexate (MTX) 10 �M, Taxol 5 �M, Doxorubicin (Doxo) 5 �M, Epirubicxperiments for each drug.
On the other side, we found a consistent dose- and time-ependent antitumor activity of Bortezomib against both myelomaell lines and all the PCs from both relapsed and at diagnosisM patients, independently of NF-�B/p65 localization, thus indi-
ating that Bortezomib is able to induce cell death regardless tohe NF-�B/p65 localization and suggesting the existence of other
olecular targets of proteasome inhibitors in MM. In addition, weave confirmed the synergism of Bortezomib with Melphalan inhe cytotoxicity of MM cells.
Constitutive nuclear localization (the active form) of NF-�B has
een reported in different solid and haematological malignan-ies suggesting that targeting the NF-�B pathway may provideovel therapeutic strategies, particularly in haematological dis-ases [17,18]. We and others have demonstrated that constitutiveuclear localization of NF-�B has been associated with lympho-i) 5 �M for 24 h. MTS assay. The results represent the means ± SD of 3 independent
proliferative diseases such as B cell chronic lymphocytic leukaemia[19], a subgroup of B-cell non-Hodgkin’s lymphoma [20,21], acutemyeloid leukaemia [22,23], and it is also likely that NF-�B activa-tion contributes to malignant transformation of myelodysplasticsyndromes [24].
Several experiments have clearly demonstrated that the activa-tion of NF-�B contributes to the pathogenesis of MM by regulatingthe expression of many proteins that act as growth factors for MMplasma cells, of several antiapoptotic genes, of proteins that areinvolved in angiogenesis and in the interaction between myeloma
cells and bone marrow mesenchymal cells. In addition, the clinicalsuccess of the proteasome inhibitor [25], Bortezomib, consideredto act by inhibiting I�B proteasomal degradation [26], has fur-ther highlighted the relevance of NF-�B pathway in MM. However,the mechanisms that activate the NF-�B pathway in MM are not
C. Conticello et al. / Leukemia Research 35 (2011) 52–60 59
Table 2Literature on NF-kB localization in MM cells.
MM cells Methods Results References
Dexa-resistant cell lines EMSA Nuclear Feinman et al. [2](ARP-1, RPMI8226, AR1177)MM primary cells Nuclear (2/4)
MM primary cells IHC Nuclear (2/13) Ni et al. [35]U266, HS-Sultan, K620 EMSA NuclearRPMI 8226 IF Cytoplasmic
MM1.S, RPMI8226 EMSA Weakly nuclear Hideshima et al. [33]MM1.S ELISA Nuclear Mitsiades et al. [6]
U266, RPMI8226 EMSA, IF Weakly nuclear Ma et al. [30]ARH77 NuclearU266/doxR, RPMI82 Nuclear26/doxRMM primary cells
MM primary cells IF, EMSA Nuclear (4/22) Bharti [36]
C
yaaiita[falcwOomalsNnwlPdiaeno
faisatcss
Nap
U266, RPMI8226, RPMI8226LR5, MM.1/MM.1R
MM primary cells RPMI8226, U266 EMSA
ell lines are undersigned.
et clear. Recently, it has been shown that primary MM samplesnd MM cell lines may have mutations in genes encoding positivend negative regulators and effectors of NF-�B signaling, lead-ng to chronic NF-�B target gene expression. However mutationsnvolved in the activation of NF-�B pathway were found in no morehan 20% of MM samples and the level of NF-�B signature in MMnd MGUS are comparable, on average, to those in normal PCs27–29]. Ma et al. demonstrated that mutations in the I�B generom two MM cell lines result in enhanced phosphorylation of I�Bnd, consequently, reduced NF-�B/I�B binding and nuclear activeocalization of NF-�B [30]. Activation of the NF-�B pathway in MMells by mutation has been described only in a single MM patientith an activating mutation of NF-�B2 by Fracchiolla et al. [31].ther authors have examined the potential structural alterationsf the RelA gene in 50 MM patients but they found a single pointutation in just one patient, concluding that RelA gene alterations
re involved only rarely in MM pathogenesis [32]. Indeed, nuclearocalization of NF-�B has been shown in MM resistant cell lines andeveral studies correlated the expression of constitutively activeF-�B in MM primary or cell lines with chemoresistance of malig-ant MM cells [33,34]. In the majority of these studies experimentsere performed with MM drug-resistant or cytokine-treated cell
ines. Ni et al. have reported a nuclear localization of NF-�B in MMCs but they have examined 13 biopsies of MM patients withoutescribing the phase of disease and the nuclear localization shown
n the manuscript is a rare staining in comparison with the percent-ge of malignant PCs infiltration indicated [35]. Bharti et al. havexamined 4 MM cell lines and 22 MM samples in which a clearuclear localization of p65 was found just in 4 cases by immunoflu-rescence and 3 by EMSA [36].
Our results diverge also from those of Markovina et al. [37] whoound evidence of NF-�B activation in most plasma cell samples. Inddition, such activation was insensitive to Bortezomib treatmentn the majority of samples. The authors indicate the PIR (protea-ome inhibitor-resistant) pathways as an alternative mechanismctivating NF-�B in the samples studied. However, disease stage,reatment history and/or response, and other identifying patientharacteristics are not available for the samples analyzed in thistudy. Reports from the literature on NF-�B localization in MM are
ummarized in Table 2.Therefore, the outcome from these and other papers, was thatF-�B pathway is constitutively active in MM and that Bortezomibnti-MM activity is strictly due to inhibition of NF-�B by preventingroteasome degradation of I�B�.
Weakly nuclear
Nuclear (14/14) Markovina et al. [37]
However, our results and a recently published paper ofHideshima strongly argue against this dogma.
Hideshima et al. have demonstrated that Bortezomib in MMcell lines and primary cells, while exerts an high cytotoxic effect,actually activates 2 upstream NF-�B-activating kinases (RIP2 andIKK�), promotes down-regulation of NF-�B’s inhibitor (I�B�), andincreases NF-�B DNA binding in vitro, thus improving NF-�B activ-ity [38–40].
Activation of NF-�B is a dynamic phenomenon that can beinduced by cytokines, chemokines, growth factors, and contact ofcells belonging to the microenvironment and we do not doubt ofits role in the pathogenesis of MM. However, in agreement withthe new current dogma, we demonstrated that in a steady state,especially in patients at diagnosis, NF-�B resides mainly in the cyto-plasm (the inactive form) but the PCs maintain the sensitivity toBortezomib. Indeed we cannot exclude that a small amount of p65(RelA) in the nucleus of MM cells it is not detected by the meth-ods we have used in our experiments and this may contribute tothe Bortezomib activity we observed. However, our results pro-vide believable evidence that blocking of I�B degradation is notthe only mechanism responsible for proteasome inhibitor-inducedapoptosis and for the proven clinical activity of Bortezomib. Inparticular several studies demonstrated that NF-�B activation can-not be the unique mechanisms by which the proteasome inhibitorBortezomib performs its anti-tumor activity. Indeed, Bortezomibas a proteasome inhibitor is able to inhibit the degradation ofmultiubiquitinated target proteins, i.e., cell cycle regulatory pro-teins such as cyclins and cyclin-dependent kinase inhibitors, thusregulating cell cycle progression. In addition, several possibleBortezomib-induced proteasome-independent mechanisms havebeen described. Among these mechanisms, Landowsky et al. [41]have shown that Bortezomib treatment induces the expression ofgene products associated with the endoplasmic reticulum secre-tory pathway with deregulation of intracellular Ca2+, leading tocaspase activation and cell death. Pei et al. [42] and more recentlyKikuchi et al. [43], indicate that Bortezomib target the HDAC in MMcells and can be used in association with HDAC inhibitors. Qin etal. [44] described the involvement of p53-independent BH3-onlyprotein NOXA in mitochondrial-based apoptotic pathway induced
by Bortezomib. Different studies [45] demonstrate that Bortezomibboth directly induces apoptosis of human MM cells and abrogatesparacrine growth of MM cells in the BM via altering cellular inter-actions and cytokine secretion in the BM milieu. The antiangiogeniceffect of Bortezomib represents another mechanism of its anti-MM
6 mia Re
ae
C
A
t
e
A
t
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[[
[
[
[
0 C. Conticello et al. / Leuke
ctivity [46] that should be considered in its possible therapeuticmployment.
onflict of interest
The authors do not have any conflict of interest.
cknowledgments
The authors thank the Fondazione Giuseppe Alazio for fundinghis work.
Contributions. Conticello Concetta and Raffaella Giuffridaqually contributed to this work.
ppendix A. Supplementary data
Supplementary data associated with this article can be found, inhe online version, at doi:10.1016/j.leukres.2010.06.023.
eferences
[1] Hussein MA, Juturi JV, Lieberman I. Multiple myeloma: present and future. CurrOpin Oncol 2002;14:31–5.
[2] Feinman R, Koury J, Thames M, et al. Role of NF-kappaB in the rescue of mul-tiple myeloma cells from glucocorticoid-induced apoptosis by bcl-2. Blood1999;93:3044–52.
[3] Hoffmann A, Baltimore D. Circuitry of nuclear factor �B signaling. Imm Rev2006;210:171–86.
[4] Wan F, Lenardo MJ. The nuclear signaling of NF-kappaB: current knowledge,new insights, and future perspectives. Cell Res 2010;20(January (1)):24–33.
[5] Panwalkar A, Verstovsek S, Giles F. Nuclear factor-kappaB modulation asa therapeutic approach in hematologic malignancies. Cancer 2004;100:1578–89.
[6] Mitsiades N, Mitsiades CS, Poulaki V, et al. Biologic sequelae of nuclearfactor-kappaB blockade in multiple myeloma: therapeutic applications. Blood2002;99:4079–86.
[7] Senftleben U, Cao Y, Xiao G, et al. Activation by IKKalpha of a sec-ond, evolutionary conserved, NF-kappa B signaling pathway. Science2001;293(5534):1495–9.
[8] Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processingof NF-kappaB2 p100. Mol Cell 2001;7(2):401–9.
[9] An WG, Hwang SG, Trepel JB, Blagosklonny MV. Protease inhibitor-inducedapoptosis: accumulation of wt p53, p21WAF1/CIP1, and induction ofapoptosis are independent markers of proteasome inhibition. Leukemia2000;14:1276–83.
10] Adams J, Palombella VJ, Elliott PJ. Proteasome inhibition: a new strategy incancer treatment. Invest New Drugs 2000;18:109–21.
11] Hata H, Xiao H, Petrucci MT, et al. Interleukin-6 gene expression in multiplemyeloma: a characteristic of immature tumor cells. Blood 1993;81:3357–64.
12] Chauhan D, Uchiyama H, Akbarali Y, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involvesactivation of NF-kappa B. Blood 1996;87:1104–12.
13] Barlogie B, Shaughnessy J, Tricot G, et al. Treatment of multiple myeloma. Blood2004;103:20–32.
14] Mitsiades CS, Mitsiades NS, Richardson PG, et al. Multiple myeloma: a pro-totypic disease model for the characterization and therapeutic targeting ofinteractions between tumor cells and their local microenvironment. J CellBiochem 2007;101(July (4)):950–68.
15] Jagannath S, Richardson PG, Sonneveld P, et al. Bortezomib appears to overcomethe poor prognosis conferred by chromosome 13 deletion in phase 2 and 3 trials.Leukemia 2007;21:151–7.
16] Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R. Alkylating agentsinduce activation of NFkappaB in multiple myeloma cells. Leuk Res2008;32:1144–7.
17] Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasomepathway is required for processing the NF-kappa B1 precursor protein and theactivation of NF-kappa B. Cell 1994;78:773–85.
18] Andela VB, Schwarz EM, Puzas JE, et al. Tumor metastasis and the reciprocal reg-ulation of prometastatic and antimetastatic factors by nuclear factor kappaB.Cancer Res 2000;60:6557–62.
[
[
search 35 (2011) 52–60
19] Endo T, Nishio M, Enzler T, et al. BAFF and APRIL support chronic lympho-cytic leukemia B-cell survival through activation of the canonical NF-kappaBpathway. Blood 2007;109:703–10.
20] Lam LT, Davis RE, Pierce J, et al. Small molecule inhibitors of IkappaB kinaseare selectively toxic for subgroups of diffuse large B-cell lymphoma defined bygene expression profiling. Clin Cancer Res 2005;11:28–40.
21] Houldsworth J, Mathew S, Rao PH, et al. REL proto-oncogene is frequentlyamplified in extranodal diffuse large cell lymphoma. Blood 1996;87:25–9.
22] Frelin C, Imbert V, Griessinger E, et al. Targeting NF-kappaB activation viapharmacologic inhibition of IKK2-induced apoptosis of human acute myeloidleukemia cells. Blood 2005;105:804–11.
23] Conticello C, Adamo L, Vicari L, et al. Antitumor activity of bortezomib alone andin combination with TRAIL in human acute myeloid leukemia. Acta Haematol2008;120:19–30.
24] Braun T, Carvalho G, Coquelle A, et al. NF-kappaB constitutes a poten-tial therapeutic target in high-risk myelodysplastic syndrome. Blood2006;107:1156–65.
25] Richardson PG, Anderson KC. Bortezomib: a novel therapy approved for mul-tiple myeloma. Clin Adv Hematol Oncol 2003;1:596–600.
26] Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibitionas a novel therapeutic target in human cancer. J Clin Oncol 2005;23:630–9.
27] Gilmore TD. Multiple myeloma: lusting for NF-kappaB. Cancer Cell2007;12:95–7.
28] Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activatethe noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell2007;12:131–44.
29] Annunziata CM, Davis RE, Demchenko Y, et al. Frequent engagement of theclassical and alternative NF-kappaB pathways by diverse genetic abnormalitiesin multiple myeloma. Cancer Cell 2007;12:115–30.
30] Ma MH, Yang HH, Parker K, et al. The proteasome inhibitor PS-341 markedlyenhances sensitivity of multiple myeloma tumor cells to chemotherapeuticagents. Clin Cancer Res 2003;9:1136–44.
31] Fracchiolla NS, Lombardi L, Salina M, et al. Structural alterations of theNF-kappa B transcription factor lyt-10 in lymphoid malignancies. Oncogene1993;8:2839–45.
32] Trecca D, Guerrini L, Fracchiolla NS, et al. Identification of a tumor-associatedmutant form of the NF-kappaB RelA gene with reduced DNA-binding and trans-activating activities. Oncogene 1997;14:791–9.
33] Hideshima T, Chauhan D, Richardson P, et al. NF-kappa B as a therapeutic targetin multiple myeloma. J Biol Chem 2002;277:16639–47.
34] Hideshima T, Neri P, Tassone P, et al. MLN120B, a novel IkappaB kinase betainhibitor, blocks multiple myeloma cell growth in vitro and in vivo. Clin CancerRes 2006;12:5887–94.
35] Ni H, Ergin M, Huang Q, et al. Analysis of expression of nuclear factor kappaB (NF-kappa B) in multiple myeloma: downregulation of NF-kappa B inducesapoptosis. Br J Haematol 2001;115:279–86.
36] Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloylmethane) inhibitsconstitutive and IL-6-inducible STAT3 phosphorylation in human multiplemyeloma cells. J Immunol 2003;171:3863–71.
37] Markovina S, Callander NS, O’Connor SL, et al. Bortezomib-resistantnuclear factor-kappaB activity in multiple myeloma cells. Mol Cancer Res2008;6:1356–64.
38] Hideshima T, Ikeda H, Chauhan D, et al. Bortezomib induces canonicalnuclear factor-kappaB activation in multiple myeloma cells. Blood 2009;114:1046–52.
39] Di Raimondo F, Conticello C. Captivating Bortezomib: an active but still mys-terious drug. Leuk Res 2010;34(4):411–2. Epub 2009 Oct 12. PubMed PMID:19819547.
40] McConkey DJ. Bortezomib paradigm shift in myeloma. Blood 2009;114:931–2.41] Landowsky TH, Megli CJ, Nullmeyer KD, et al. Mitochondrial-mediated dis-
regulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib)cytotoxicity in myeloma cell lines. Cancer Res 2005;65(9):3828–36.
42] Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosisin human multiple myeloma cells by the proteasome inhibitor bortezomib andhistone deacetylase inhibitors. Clin Cancer Res 2004;10(11):3839–52.
43] Kikuchi J, Wada T, Shimizu R, et al. Histone deacetylases are critical targets ofbortezomib-induced cytotoxicity in multiple myeloma. Blood 2010 (March).[Epub ahead of print].
44] Qin JZ, Ziffra J, Stennett L, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res 2005;65(14):6282–93.
45] Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341inhibits growth, induces apoptosis, and overcomes drug resistance in humanmultiple myeloma cells. Cancer Res 2001;61(7):3071–6.
46] Podar K, Shringarpure R, Tai YT, et al. Caveolin-1 is required for vascularendothelial growth factor-triggered multiple myeloma cell migration and istargeted by bortezomib. Cancer Res 2004;64(20):7500–6.