neuronal uv-initiated apoptosis is prevented by 5- bromo … · neuronal uv-initiated apoptosis is...
TRANSCRIPT

Neuronal UV-Initiated Apoptosis Is Prevented By 5-Bromo-2’-Deoxyuridine (BrdU) Or A Deficiency in
Cockayne Syndrome B Or Xeroderma Pigmentosum A
by
Nishani Rajakulendran
A thesis submitted in conformity with the requirements for the degree of Master of Science
Graduate Department of Pharmacology and Toxicology University of Toronto
© Copyright by Nishani Rajakulendran 2012

ii
Neuronal UV-Initiated Apoptosis is Prevented by 5-Bromo-2’-
Deoxyuridine (BrdU) or a Deficiency in Cockayne Syndrome
B or Xeroderma Pigmentosum A
Nishani Rajakulendran
Master of Science
Graduate Department of Pharmacology and Toxicology University of Toronto
2012
ABSTRACT
This project addressed mechanisms of the neuronal DNA damage response after
treatment with the model DNA damaging agent ultraviolet light (UV). The thymidine
analogue, 5-bromo-2’-deoxyuridine (BrdU) protected against UV-initiated neuronal
apoptosis in a concentration-dependent manner (p<0.001). BrdU did not protect
proliferating mouse embryonic fibroblasts from UV-induced apoptosis. We assessed
whether the mechanism of BrdU neuroprotection was through a modification in the
neuronal DNA damage response. BrdU neuroprotection was independent of BrdU
incorporation into DNA, neuronal DNA repair, p53 activation or cell cycle re-entry, a
neuronal DNA damage response. Neurons deficient in Cockayne Syndrome B (CSB) or
Xeroderma Pigmentosum A (XPA) were paradoxically resistant to UV-initiated
apoptosis. Therefore, CSB and XPA play essential roles in the neuronal DNA damage
response.

iii
ACKNOWLEDGEMENTS
This thesis was made possible by the guidance and encouragement I received
from an incredible group of people. I would like to express my gratitude to everyone who
has been extremely supportive during the past two years.
First, I would like to thank my supervisor and mentor, Dr. Rebecca Laposa, for
her inspiration and support. Her enthusiasm and positive outlook on science has helped
shape my appreciation for scientific research. Also, her endless encouragement has
been instrumental to my success as a graduate student.
I would also like to thank past and present members of the Laposa lab for
creating a wonderful work environment. I am very grateful to Laura Tamblyn who has
trained me in most of the techniques I used in this project and has been incredibly
patient with my numerous questions. I would also like to thank Fernando Bralha who
was extremely helpful, especially on those days when I was working on too many
experiments, and for our engaging discussions. Sharanya and Temi have also become
good friends and it was a joy working with them.
I would also like to thank my advisor Dr. McPherson whose feedback has been
invaluable. I am also grateful to the McPherson lab members, who have allowed us to
use their equipment and for their assistance in technical problems.
Finally, I have to thank my family and friends. I am extremely grateful to my
brother and sister for their encouragement and motivation during my ups and downs.
Most importantly, I am deeply grateful to my parents for their unconditional support in
my academic pursuits and career goals. They are incredible people who have taught
me the value and importance of education and I would not have come this far without
their love and inspiration.

iv
TABLE OF CONTENTS
ABSTRACT .................................................................................................................... II
ACKNOWLEDGEMENTS ............................................................................................. III
TABLE OF CONTENTS ................................................................................................ IV
LIST OF TABLES ......................................................................................................... VII
LIST OF FIGURES ...................................................................................................... VIII
ABREVIATIONS ............................................................................................................. X
1. INTRODUCTION ....................................................................................................... 1
1.1 DNA damage in eukaryotic cells ............................................................................ 1
1.1.1 Endogenous and exogenous sources of DNA damage ...................................... 1
1.1.2 DNA damage response signaling ....................................................................... 1
1.1.3 UV DNA lesions .................................................................................................. 8
1.2 Nucleotide excision repair ................................................................................... 11
1.2.1 Overview of nucleotide excision repair ............................................................. 11
1.2.2 Diseases caused by defective nucleotide excision repair ................................. 14
1.2.3 Nucleotide excision repair in neurons ............................................................... 15
1.3 Neuronal cell cycle re-entry ................................................................................. 16
1.3.1 Overview of the cell cycle ................................................................................. 16
1.3.2 In vivo evidence of neuronal cell cycle re-entry ................................................ 19
1.2.3 In vitro evidence of neuronal cell cycle re-entry ................................................ 20
1.4 5-bromo-2’-deoxyuridine (BrdU) .......................................................................... 24
1.4.1 Overview of BrdU ............................................................................................. 24
1.4.2. The role of BrdU in DNA repair ........................................................................ 26
1.4.3. The role of BrdU in the cell cycle ..................................................................... 29
2. OVERVIEW............................................................................................................... 33
2.1 Hypothesis............................................................................................................. 34
2.2 Aims ....................................................................................................................... 34

v
3. MATERIALS AND METHODS ................................................................................. 35
3.1 Cortical neuron culture ........................................................................................ 35
3.2 Embryonic genotyping ......................................................................................... 36
3.3 Mouse embryonic fibroblast (MEF) isolation ...................................................... 36
3.4 Cortical neuron survival assays .......................................................................... 37
3.4.1 Neuronal immunocytochemistry ....................................................................... 37
3.4.2 Quantification of apoptosis with flow cytometry ................................................ 38
3.4.3 Quantification of apoptosis with immunocytochemistry .................................... 38
3.5 Mouse embryonic fibroblast survival assay ....................................................... 39
3.6 BrdU labeling using immunocytochemistry ....................................................... 39
3.7 Quantification of UV DNA lesions ....................................................................... 40
3.8 DNA content quantification using immunocytochemistry ................................ 41
3.9 Quantification of cell cycle distribution using flow cytometry ......................... 42
3.10 Cell cycle marker expression ............................................................................. 42
3.11 Quantification of global DNA methylation ........................................................ 43
3.12 Statistical analyses ............................................................................................. 43
4. RESULTS ................................................................................................................. 44
4.1 BrdU prevents mouse cortical neurons from UV-induced death ...................... 44
4.1.1 BrdU reduces UV-induced cleaved caspase-3 in cortical neurons ................... 47
4.2 Absence of detectable BrdU labeling in cortical neurons ................................. 49
4.3 Cell type specificity of BrdU protection .............................................................. 51
4.4 Chemical specificity of BrdU protection ............................................................. 53
4.5 The UV-initiated increase of p53 is unaltered by BrdU ...................................... 55
4.6 BrdU does not alter UV DNA lesion removal ...................................................... 57
4.7 Xpa-/- and Csb-/- neurons are resistant to UV-initiated death ............................. 59
4.7.1 BrdU does not alter caspase-3 in Xpa-/- and Csb-/- neurons ............................ 60

vi
4.8 Influence of BrdU on neuronal cell cycle ............................................................ 63
4.8.1 BrdU does not alter neuronal DNA content ...................................................... 63
4.8.2 Neither UV-irradiation nor BrdU incubation alter cell cycle distribution ............. 65
4.8.3 UV does not induce cell cycle markers in neurons ........................................... 65
4.9 BrdU does not alter neuronal global DNA methylation ..................................... 68
5. DISCUSSION............................................................................................................ 70
5.1 Characterization of BrdU protection ................................................................... 70
5.2 UV-induced cell signaling .................................................................................... 71
5.3 UV-induced DNA repair ........................................................................................ 71
5.4 UV-induced cell signaling in Xpa-/- and Csb-/- neurons ...................................... 73
5.5 Neuronal cell cycle re-entry ................................................................................. 75
5.6 Neuronal DNA methylation................................................................................... 78
6. CONCLUSIONS AND FUTURE DIRECTIONS ........................................................ 79
7. REFERENCES ......................................................................................................... 82

vii
LIST OF TABLES
INTRODUCTION:
TABLE 1.1 Evidence of neuronal cell cycle re-entry after DNA damage ....................... 23
TABLE 1.2 Evidence of cell cycle modulation by thymidine analogues ........................ 30
MATERIALS AND METHODS:
TABLE 3.1 Summary of Xpa primers, cycling conditions and band sizes ..................... 36
TABLE 3.2 Summary of Csb primers, cycling conditions and band sizes ..................... 36
RESULTS:
TABLE 4.1 Flow cytometric analysis of cell cycle phase in DNA-damaged cortical
neurons .......................................................................................................................... 65

viii
LIST OF FIGURES
INTRODUCTION:
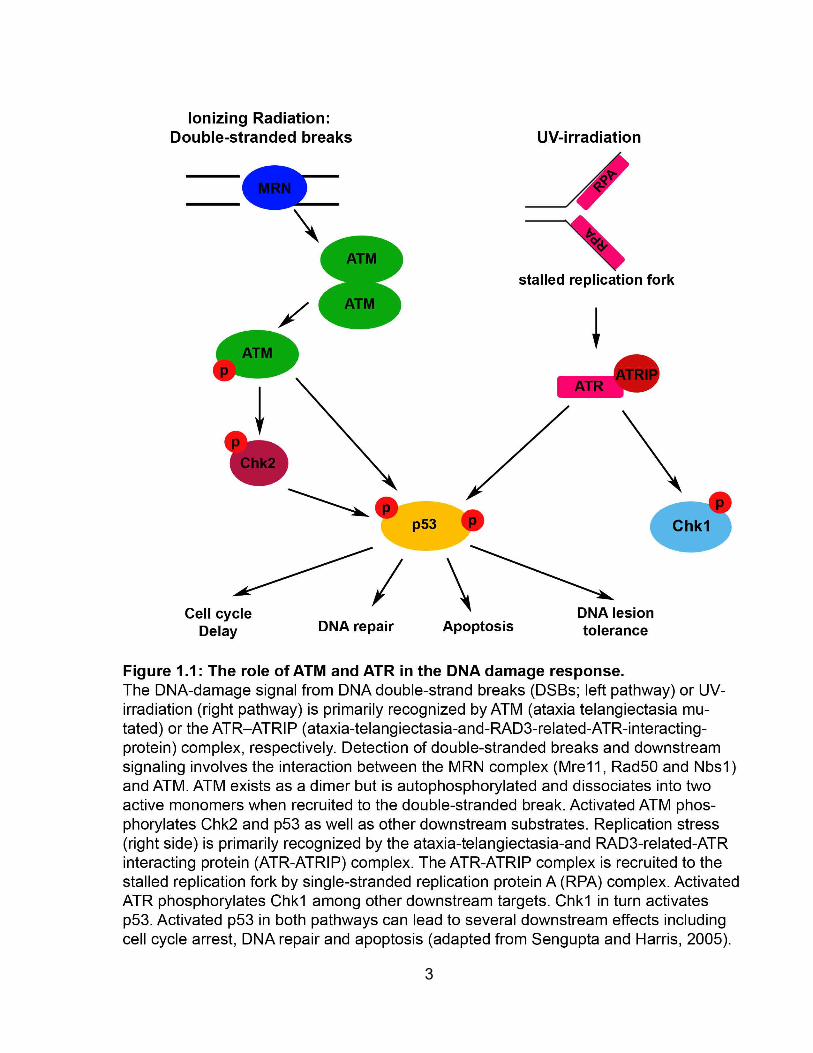
FIGURE 1.1 The role of ATM and ATR in the DNA damage response ............................ 3
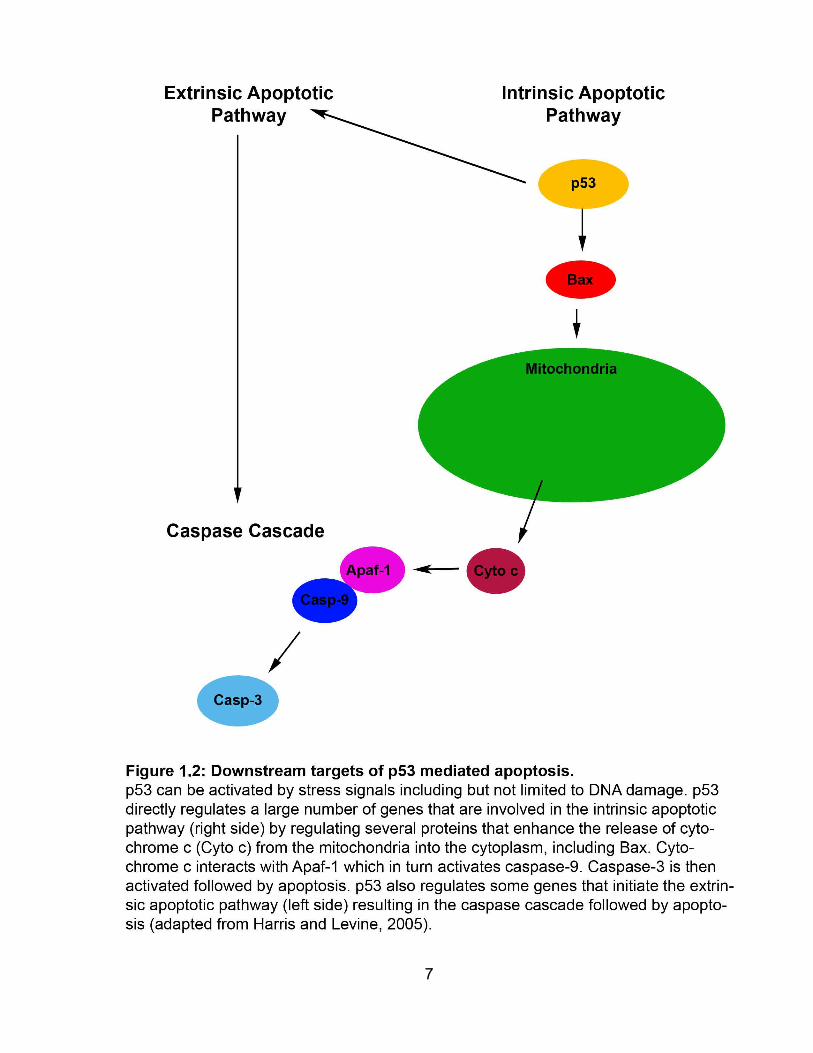
FIGURE 1.2 Downstream targets of p53 mediated apoptosis ......................................... 7
FIGURE 1.3 UV DNA lesions ......................................................................................... 10
FIGURE 1.4 The two sub-pathways of nucleotide excision repair (NER): global genome
repair (GGR) and transcription-coupled repair (TCR) .................................................... 13
FIGURE 1.5 Chemical structures of thymidine and 5-bromo-2’-deoxyuridine (BrdU) .... 25
RESULTS:
FIGURE 4.1 BrdU protects cortical neurons from UV-induced apoptosis ...................... 46
FIGURE 4.2 BrdU reduces UV-induced cleaved caspase-3 in neurons ........................ 48
FIGURE 4.3 Absence of detectable BrdU labeling in UV-irradiated and control cortical
neurons .......................................................................................................................... 50
FIGURE 4.4 In fibroblasts, BrdU does not reduce UV-induced apoptosis ..................... 52
FIGURE 4.5 Nucleosides and halogenated thymidine analogues prevent UV-initiated
apoptosis ....................................................................................................................... 54
FIGURE 4.6 UV-induced p53 up-regulation is unaltered by BrdU ................................. 56
FIGURE 4.7 Time course of disappearance of UV DNA lesions in cortical neurons ...... 58
FIGURE 4.8 Xpa-/- and Csb-/- cortical neurons are resistant to UV-initiated apoptosis ... 61
FIGURE 4.9 BrdU does not alter the proportion of cleaved caspase-3 + neurons after
UV in Xpa-/- and Csb-/- ................................................................................................... 62
FIGURE 4.10 DNA content of cortical neurons is unaltered by UV or BrdU .................. 64

ix
FIGURE 4.11 Neither UV nor BrdU alter neuronal Cdk4, Cdk6 or Ki67 expression ...... 67
FIGURE 4.12 Neither UV-irradiation nor BrdU alter neuronal global DNA methylation .. 69

x
ABREVIATIONS 6-4PP pyrimidine 6-4 pyrimidone photoproducts Apaf-1 apoptotic protease activating factor 1 APV 2-Amino-5-phosphonopentannoic acid ATM ataxia telangiectasia mutated ATR ATM and Rad3-related protein ATRIP ATR interacting protein βIIITub βIIITubulin Bak Bcl-2 homologous antagonist/killer Bax Bcl-2 associated X protein Bcl-2 B-cell lymphoma 2 Bxl-XL B-cell lymphoma-extra large BrdU 5-bromo-2’-deoxyuridine Cdk cyclin dependent kinase CDKI cyclin dependent kinase inhibitor Chk1 checkpoint kinase 1 Chk2 checkpoint kinase 2 CldU chlorodeoxyuridine CPD cyclobutane pyrimidine dimers cPu cyclopurines CS Cockayne Syndrome CSB Cockayne Syndrome B DAR domain associated repair DDR DNA damage response DNA-PK DNA-dependent serine/threonine protein kinase dNTP Deoxyribonucleotide triphosphates DS dissecting solution DSB double-stranded bread dTTP thymidine triphosphate EdU 5-ethynyl-2’-deoxyuridine FdU fluorodeoxyuridine GGR global genome repair IdU iododeoxyuridine i.p. intraperitoneal injection IR ionizing radiation MEF mouse embryonic fibroblast MRN Mre11, Rad50 and Nbs1 NER nucleotide excision repair NPC neural precursor NSC neural stem cell PAN-118 3-aminopyridine-2-carboxaldehyde thiosemicarbazone PARP poly (ADP-ribose) polymerase PI propidium iodide PIKK phosphatidylinositol 3-kinase-like protein kinase RPA replication protein A SSB single-stranded break s.q. subcutaneous injection

xi
TCR transcription-coupled repair TFIIH multi-subunit transcription factor-IIH TTD trichothiodystrophy ROS reactive oxygen species RR ribonucleotide reductase UV ultraviolet XP Xeroderma Pigmentosum XPA Xeroderma Pigmentosum A

1
1. INTRODUCTION
1.1 DNA damage in eukaryotic cells
1.1.1 Endogenous and exogenous sources of DNA damage
DNA damage can be caused by exogenous or endogenous sources.
Approximately 105 DNA adducts are estimated to be generated in every cell each day
(Hoeijmakers, 2009). Exogenous DNA damage can be caused by physical sources
such as ultraviolet (UV) light or ionizing radiation (IR) or by chemical sources such as
chemotherapeutic agents or cigarette smoke (Ciccia and Elledge, 2010). Different types
of damage can be caused by chemotherapeutic agents depending on their mode of
action. For example some drugs are alkylating agents (i.e. temozolomide) which attach
alkyl groups to DNA bases whereas other drugs are crosslinking agents (i.e. cisplatin).
which can induce the formation of covalent links between DNA bases (Ciccia and
Elledge, 2010). Endogenous DNA damage can be caused by reactive oxygen species
(ROS) which are produced during regular cellular metabolism. ROS can oxidize DNA
bases and cause DNA breaks (Ciccia and Elledge, 2010).
1.1.2 DNA damage response signaling
The ability of a cell to detect DNA damage and respond is a signal transduction
pathway that is referred to as the DNA damage response (DDR). At the molecular level,
key proteins involved in DDR are members of the phosphatidylinositol 3-kinase-like
protein kinase (PIKKs) family and of the poly (ADP-ribose) polymerase (PARP) family.
Members of PIKKs family include ATM (ataxia telangiectasia mutated), ATR (ATM and

Rad3 related) and DNA-PK (DNA-dependent serine/threonine protein kinase) (Ciccia
and Elledge, 2010).
ATM is primarily activated by DNA strand breaks that can be formed by ionizing
radiation (Caporali et al., 2004) whereas ATR signals after DNA replication fork stalling
by DNA lesions (Fig. 1.1) including but not limited to those caused by UV irradiation
(Caporali et al., 2004; Myers and Cortez, 2006; Vrouwe et al., 2011). Double-stranded
break (DSB) detection and downstream signaling following DSBs involves the
interaction between the MRN (Mre11, Rad50 and Nbs1) complex and ATM (Roos and
Kaina, 2012). The MRN complex recruits and activates ATM at the site of DSBs
(Bakkenist and Kastan, 2003; Bartek and Lukas, 2003; Sengupta and Harris, 2005).
ATM generally exists as a dimer but following its recruitment to the DSB, it
autophosphorylates which results in the dissociation of the dimer into two active
monomers (Bakkenist and Kastan, 2003). Once ATM is activated, it phosphorylates
downstream signaling cascades which may lead to DNA repair, cell cycle arrest and/or
apoptosis (Bartek and Lukas, 2003). The ATR-ATRIP (ATR-interactive protein) complex
is recruited to the site of DNA damage that causes replication stress by RPA (replication
protein A) (Byun et al., 2005). Several other proteins are also recruited to form a
complex which results in the phosphorylation of ATR (Kurz and Lees-Miller, 2004).
Similar to ATM, the activation of ATR by phosphorylation initiates phosphorylation of
downstream targets and can also at times involve ATM (Kurz and Lees-Miller, 2004).
Targets of ATR and ATM include checkpoint kinase 1 (Chk1) and checkpoint kinase 2
(Chk2) which are involved in the phosphorylation of p53 (tumor suppressor) (Latonen
and Laiho, 2005). Activation of p53 can result in several downstream effects including
DNA repair, cell cycle arrest and/or apoptosis.
2
3

At the cellular level there are several consequences following DNA damage
including DNA repair, DNA lesion tolerance, cell cycle arrest and/or apoptosis. Not all
cell types are able to execute all of these functions. These four consequences will be
described briefly.
1.1.2.1 DNA repair
Several DNA repair pathways have evolved to remove a variety of DNA lesions.
When DNA bases are incorporated into DNA incorrectly during DNA replication, the
mispaired bases are excised and are replaced with the correct bases during mismatch
repair (Ciccia and Elledge, 2010; Friedberg, 2003). DNA lesions caused by hydrolytic
deamination, alkylating agents, ionizing radiation or intracellular metabolites are
repaired by base excision repair and involves DNA glycosylases (Rastogi et al., 2010).
A wide range of DNA lesions that result in the helical distortion of DNA, including lesions
caused by UV light, bulky chemical adducts and DNA intrastrand crosslinks, are
repaired by nucleotide excision repair (NER) (Hess et al., 1997). During NER, incisions
are created on either side of the lesion and a fragment, about 30 nucleotides, is
removed (Ciccia and Elledge, 2010; Friedberg, 2003). NER is described in more detail
in section 2.2. Double strand breaks are repaired by homologous recombination or non-
homologous end joining (Rastogi et al., 2010).
1.1.2.2 DNA lesion tolerance
Occasionally the DNA lesion is not repaired and the DNA strand break or adduct
remains but the cell tolerates the damage. For example, translesion DNA synthesis may
occur where the damaged DNA strand is passed over during DNA replication and gene
expression by low-fidelity DNA polymerases (Goodman, 2002). These DNA
4

polymerases are less stringent and can continue DNA replication in the presence of
DNA lesions which will often stall high-fidelity polymerases. Although these low-fidelity
polymerases are able to add nucleotides during replication of a region of DNA
containing a DNA lesion, mutations are more likely to occur in the recently synthesized
DNA strand since the inserted nucleotide may not be accurate (Goodman, 2002).
1.1.2.3 Cell cycle arrest
DNA damage may arrest the advancement of the cell cycle in order to provide
the cell an opportunity to repair the damage prior to DNA replication or the separation of
a damaged chromosome (Hartwell and Weinert, 1989). DSBs activate ATM and DNA-
PK whereas single-stranded breaks (SSBs) activates ATR. Once ATM and ATR are
activated, Chk2 and Chk1 are phosphorylated respectively. The DNA damage signal is
then passed to proteins involved in the cell cycle such as Cdc25 that is phosphorylated
by Chk1/Chk2. When Cdc25 is phosphorylated it is degraded by an ubiquitin-mediated
pathway resulting in G1-S phase arrest. It has also been suggested that the activation
of ATM/ATR may directly, or indirectly through Chk2, phosphorylate p53 which results in
the arrest of the G1/S or G2/M cell-cycle checkpoint (Harris and Levine, 2005; Lukas et
al., 2004; Rastogi et al., 2010).
1.1.2.4 Apoptosis
If a cell does not repair damaged DNA it is theorized that programmed cell death
(apoptosis) may be activated. Apoptosis is a controlled death mechanism in cells where
cellular characteristics include blebbing, cell shrinkage, chromatin condensation and
DNA and nuclear fragmentation (Munoz-Pinedo, 2012). Apoptosis can prevent
carcinogenesis following DNA damage (Rastogi et al., 2010). The extrinsic death
5

receptor apoptosis pathway and/or the intrinsic mitochondrial apoptosis pathways are
the most frequent programmed cell death pathways that are activated following DNA
damage (Fig. 1.2) (Roos and Kaina, 2006). p53 initiates the extrinsic apoptotic pathway
by regulating several genes including the Fas ligand (Harris and Levine, 2005). The
caspase cascade is then activated which results in the degradation of DNA by caspase-
activated DNase and caspases 3 and 7 inactivating cellular proteins (Roos and Kaina,
2012). p53 also activates the intrinsic apoptotic pathway including Bax (Bcl-2-
associated X protein) (Harris and Levine, 2005). Bax as well as pro-apoptotic factor Bak
(Bcl-2 homologous antagonist/killer) and anti-apoptotic factors such as Bcl-2 (B-cell
lymphoma 2) and Bxl-XL (B-cell lymphoma-extra-large) regulate the release of
cytochrome c from mitochondria. Cytochrome c interacts with Apaf-1 (Apoptotic
protease activating factor 1) to initiate a protease cascade which results in the activation
of caspase-9 followed by caspase-3 (Harris and Levine, 2005). After caspase-3
activation the anionic phospholipid phosphatidylserine is exposed on the cell surface
and apoptosis occurs (Niu and Chen, 2010).
6
7

1.1.3 UV DNA lesions
Oxidative DNA damage is likely the most common form of DNA damage found in
neurons (McMurray, 2005). NER can remove oxidized DNA bases but the majority of
DNA lesions caused by ROS is repaired by base excision repair (Bjelland and Seeberg,
2003). Severe neurodegeneration is found in patients without functional NER (Brooks,
2002). Since neurons are not exposed to UV light which produces DNA adducts
repaired by NER, there may be other types of endogenous DNA damage in neurons
which is primarily repaired by NER (Andrews et al., 1978; Robbins et al., 1983). There is
no conclusive evidence regarding an endogenous form of DNA damage found in
neurons which is primarily repaired by NER but there are several candidate DNA
lesions which may be involved. For example, it has been proposed that cyclopurines, a
class of oxidative lesions, may be repaired by NER (Brooks, 2002). Cyclopurine is a
substrate for NER and not measurably repaired by base excision repair or direct repair
(Brooks et al., 2000; Kuraoka et al., 2000).
UV light was utilized in the current study as the DNA damaging agent since it
induces the formation of DNA lesions that are substrates for NER. UV light was utilized
for its reliability, simplicity, and efficacy, lack of requirement for bioactivation and lack of
metabolism. UV light can be categorized by wavelength. UV light with short
wavelengths (high energy) correlates with DNA lesions such as cross-links, while UV
light with long wavelengths (low energy) correlates with DNA modifications, strand-
breaks and sites of base loss (Kielbassa et al., 1997). UVA (wavelength: 315-400nm)
has low energy and is inefficient in inducing DNA damage since it is not absorbed by
native DNA (Rastogi et al., 2010). UVA often causes the production of ROS which result
in oxidative DNA lesions (i.e. 8-oxoguanine) or strand breaks (Ananthaswamy and
8

Pierceall, 1990). UVB (280-315nm) and UVC (<280nm) have higher energy and can
induce chemical modifications in DNA and alter the molecular structure of DNA by
causing the formation of pyrimidine dimers (Rastogi et al., 2010). We used UVC as our
DNA damaging agent since it produces lesions that are substrates for NER.
The most frequent DNA lesions induced by UVC are cyclobutane pyrimidine
dimers (CPDs) and pyrimidine 6-4 pyrimidone photoproducts (6-4PPs) (Fig. 1.3)
(Rastogi et al., 2010). The structure of CPDs is a four-member ring structure which
includes two carbon atoms of neighboring pyrimidine bases. 6-4PPs are noncyclic
structures with a bond between adjacent pyrimidine bases (Rastogi et al., 2010).
Adjacent thymine-thymine or thymine-cytosine bases are more photoreactive than
cytosine-thymine or cytosine-cytosine bases (Rastogi et al., 2010). CPDs are more
frequent and are often 75% of UV-induced DNA adducts whereas 6-4PPs are 25% of
lesions (Sinha and Hader, 2002). Even though CPDs and 6-4PPs are both repaired by
NER, 6-4PPs are repaired at least 5-fold faster than CPDs (de Laat et al., 1999; van
Hoffen et al., 1995).
9

10

1.2 Nucleotide excision repair
1.2.1 Overview of nucleotide excision repair
The nucleotide excision DNA repair (NER) pathway, removes bulky DNA adducts
which distort the DNA helix. These lesions include intrastrand crosslinks, bulky chemical
lesions, cyclodeoxyadenosine, thymine glycol, cyclopurines and sometimes 8-oxo-G
(Friedberg, 2003). Bulky DNA adducts can arise from UV irradiation or the metabolism
of endogenous or exogenous compounds. During NER, over 30 proteins are involved in
the recognition, incision, and excision of an approximately 30 nucleotide long
oligonucleotide which is replaced and resealed by DNA polymerase and DNA ligase
respectively (Le May et al., 2010).
NER can be divided into two sub-pathways depending on how the DNA adduct is
initially recognized, global genome repair (GGR) and transcription-coupled repair (TCR)
(Fig.1.4). In GGR, DNA adducts are recognized by the XPC-hHR23B complex that
recruits other NER factors. The rate of GGR is dependent on the type of DNA lesion, for
example, 6-4PPs are repaired at a faster rate than CPDs even though both are UV
lesions. The XPC protein is restricted to GGR and not required in TCR (van Hoffen et
al., 1995). In TCR, DNA adducts are recognized by a stalled RNA polymerase which
recruits two proteins CSA and CSB. Following this initial DNA damage recognition,
GGR and TCR are very similar (Rastogi et al., 2010). The DNA at the location of the
damage is unwound by components of multi-subunit transcription factor-IIH (TFIIH).
Two helicases that are subunits of TFIIH, XPB and XPD, open the DNA double helix
(Tirode et al., 1999). Next, RPA and XPA are recruited to verify the damage (Hitomi et
al., 2007), XPG and ERCC1-XPF cleave the DNA strand on 3’ and 5’ ends of the
damage and an approximately 30 base oligonucleotide fragment is removed (Houtgraaf
11

et al., 2006). DNA polymerase δ and ε fill in the gap and DNA ligase seals it (Moser et
al., 2007).
12

13

1.2.2 Diseases caused by defective nucleotide excision repair
Inherited deficiency in NER has been linked to at least three diseases in humans:
xeroderma pigmentosum (XP), Cockayne syndrome (CS), and trichothiodystrophy
(TTD). Eleven out of the 30 proteins discovered to date to be involved in the NER
pathways have been connected to one or more of these three diseases. XP is caused
by mutations in one or more of the 7 complementation groups of XP, XPA to G,
whereas CS is caused by mutations in CSA or CSB. TTD is caused by mutations in
TFIIH subunits (XPB, XPD or TTDA) which results in dysfunction of the transcription
factor.
The frequency of XP is estimated to be approximately 1 in 105 and is autosomal
recessive. XP is characterized by photosensitivity, hyperpigmentation, dry scaly skin
caused by sun exposure and a 1000 fold increased risk of carcinomas and melanomas
of the skin and eyes (Kraemer et al., 1987). The symptoms vary depending on the
mutation and the degree to which the mutation disrupts NER. For example, patients
with defective XPC have a milder form of the disorder than patients with defective XPA
since XPC is only required for GGR while XPA is required for GGR and TCR
(Niedernhofer, 2008). Neurological symptoms occur in approximately 20% of XP
patients (Robbins et al., 1991) and characterized by mental retardation, microcephaly,
growth retardation, spasticity and ataxia (Brooks, 2002). The majority of neurological
symptoms are found in patients with defective XPA (Hentati et al., 1992). The source of
these neurological symptoms is due to progressive neurodegeneration (Niedernhofer,
2008).
Similarly to XP, CS is also an autosomal recessive condition. Although CS is less
common than XP it is a more severe disorder and a majority of the patients exhibit
14

progressive postnatal growth failure, neurological dysfunction and accelerated aging
(Nance and Berry, 1992). Neuropathological findings in the central nervous system
include atrophy of the white matter, enlarged ventricles and atherosclerosis
(Niedernhofer, 2008). Similar to XP, apoptosis in granule cells and loss of Purkinje cells
were found in several CS patients (Kohji et al., 1998).
1.2.3 Nucleotide excision repair in neurons
Apoptotic cell death of neurons found in XP and CS patients is suggested to be
caused by the lack of repair of endogenous DNA damage. As DNA adducts accumulate
in the neuron, defective proteins may be transcribed which may ultimately lead to cell
death (Lu et al., 2004; Taddei et al., 1997). Thus, functional NER appears to be
important in neurons.
NER is attenuated in neurons relative to proliferating cells (Nouspikel, 2007). In
human neuroblastoma cells (Jensen and Linn, 1988), NT2 neuroteratoma cells
(Nouspikel and Hanawalt, 2000) and primary neurons (Nouspikel and Hanawalt, 2002),
NER has been shown to be attenuated. For instance, when NT2 human neuroteratoma
cells were differentiated into neuron-like cells there was a loss of NER of CPD lesions.
CPDs were removed in active genes but were not removed from silent genes indicating
a lack of GGR (Nouspikel and Hanawalt, 2000). NER has been observed to be
attenuated in other differentiated cells such as striated muscle, macrophages and
keratinocytes (Nouspikel, 2008). A decrease in repair of silent genes was also seen in
other types of differentiated cells like myoblasts. In rat L8 myoblast cell line, UV-induced
CPD adducts were repaired in active genes but not in silent genes (Ho and Hanawalt,
1991). An explanation for functional TCR but attenuation of GGR in differentiated cells
is that since these cells do not divide and do not need to replicate DNA, they do not
15

need to expend energy in repairing DNA damage. However, repair of active genes
should be maintained since these genes are transcribed (Nouspikel, 2007). When DNA
repair of active genes was examined in detail, it was discovered that both the
transcribed and non-transcribed strands of transcribed genes are both repaired in
human neurons (Nouspikel and Hanawalt, 2000) and macrophages in a process called
domain associated repair (DAR) (Hsu et al., 2007). The lack of GGR in neurons has
been hypothesized to be connected to neurodegenerative diseases such as Alzheimer’s
and Parkinson’s.
1.3 Neuronal cell cycle re-entry
Neurons are considered to be post-mitotic cells. One stress response observed
in neurons is re-entry into the cell cycle, an event that is followed by cell death (Herrup
et al., 2004). The precise molecular mechanisms that initiate cell cycle re-entry and
couple it to a death response remain poorly understood and areas of active research.
1.3.1 Overview of the cell cycle
Cell proliferation is a tightly regulated process controlled by the cell cycle. For a
brief review, the cell cycle can be divided into four phases: the first gap phase (G1), the
DNA synthesis phase (S phase), the second gap phase (G2) and mitosis (M). Cells that
have exited the cell cycle are in a quiescent state (G0). The cell cycle manages the
transition of cells from G1 through M with several checkpoints to assess damage and
ensure successful completion of the previous phases. The progression of a cell through
the cell cycle is regulated by two classes of proteins: cyclins and cyclin-dependent
kinases (Cdks) (Malumbres and Barbacid, 2001). Cyclins can be divided into two main
families, mitotic cyclins (cyclin A and cyclin B) and G1 cyclins (cyclin C, cyclin D and
16

17
cyclin E) (Hess et al., 1997; Kurz and Lees-Miller, 2004). Cdks are a group of
serine/threonine kinases that bind to cyclins (Malumbres and Barbacid, 2001) and form
active heterodimer complexes and drive the cell through the cell cycle (Malumbres and
Barbacid, 2001). Nine structurally related Cdks (Cdk1-Cdk9) have been identified
(Schwartz and Shah, 2005). Several Cdks function together, primarily Cdk4, Cdk6,
Cdk2, Cdk1 and perhaps Cdk3, to ensure successful progression through the cell cycle
(Malumbres and Barbacid, 2001).
Cdks need to form a complex with cyclins in order for kinase activity to be
activated (Malumbres and Barbacid, 2001). The transition from G1 to S phase referred
to as the restriction point is controlled by growth factors (Coller, 2007). During G1
progression, levels of cyclin D isoforms (cyclin D1, D2 and D3) increase in the presence
of extracellular mitogens. Cyclin D1 interacts with Cdk2, 4 and 6 and phosphorylates
downstream targets (Coller, 2007; Schwartz and Shah, 2005). Cyclin E interacts with
Cdk2 in late G1 (Schwartz and Shah, 2005). S phase progression and the transition
from S phase to G2 are regulated by the cyclin A/Cdk2 complex (Schwartz and Shah,
2005). After the G2 phase, the cyclin B/Cdk1 complex shuttles from the cytoplasm into
the nucleus in order to initiate mitosis (Malumbres and Barbacid, 2001). For the cell to
exit mitosis, cyclin B needs to be degraded (Malumbres and Barbacid, 2001).
During G1 progression, several members of the retinoblastoma protein (Rb)
family are phosphorylated by cyclin D/Cdk4/6 followed by cyclin E/Cdk2 (Schwartz and
Shah, 2005). Rb is tumor suppressor gene and is hypophosphorylated in its active
state. An active Rb protein interacts with a group of transcription factors, E2F-DP
(E2F1, E2F2 and E2F3) to form an inhibitory complex that prevents the transcription of
S-phase proteins (Malumbres and Barbacid, 2001). The consecutive phosphorylation of

Rb by cyclin D/Cdk4/6 followed by cyclin E/Cdk2 alters Rb activity. First, Cyclin
D/Cdk4/6 partially phosphorylates Rb. While Rb continues to be bound to E2F-DP, it
does not completely inhibit E2F-DP and E2F-DP transcribes several genes including
cyclin E. Cyclin E, in turn, binds to Cdk2 and this complex (hyper)phosphorylates Rb
(pRb) (Malumbres and Barbacid, 2001). pRb releases E2F-DP and the E2F
transcription factors transcribe many S phase proteins (Schwartz and Shah, 2005).
Once the cell has entered the S phase, cyclins D and E are degraded and cyclin A/Cdk2
regulates the S phase. Several proteins required for DNA synthesis such as histones
and proliferating cell nuclear antigen (PCNA) are produced. At the same time, cyclin
A/Cdk2 phosphorylates E2F; this results in the inactivation and degradation of E2F
(Schwartz and Shah, 2005).
Cdk inhibitors (CDKIs) are able to control the cell cycle at several checkpoints by
preventing the formation or inhibiting the function of cyclin/Cdk complexes (Schwartz
and Shah, 2005). CDKIs can be divided into two families INK4 and Cip/Kip. The INK4
family of CDKIs prevents Cyclin/Cdk complexes from forming whereas the kinase
activity of already formed cyclin/Cdk complexes is inhibited by the Cip/Kip family (Sherr
and Roberts, 1999).
The differentiation of neurons occurs in the ventricular zone (VZ) and
subventricular zone (SVZ) of the central nervous system (CNS) where neuroblasts
become mature neurons and exit the cell cycle, becoming permanently post-mitotic
(Lennington et al., 2003; Yang and Herrup, 2007). Neurons cannot proliferate once they
have exited the cell cycle (Liu and Greene, 2001). However, recent evidence has
indicated that after neurons have entered the G0 phase they can still re-enter the cell
18

cycle but contrary to expectations, they undergo apoptosis instead of completing the cell
cycle (Kruman, 2004).
1.3.2 In vivo evidence of neuronal cell cycle re-entry
Increasing experimental evidence demonstrates a correlation between cell cycle
re-entry and neuronal loss in neurodegenerative diseases (Wang et al., 2009). Cell
cycle protein expression has been shown in several neurodegenerative disorders
including Alzheimer’s disease (AD), Parkinson’s disease, amyotrophic lateral sclerosis,
stroke and ataxia-telangiectasia. In addition, several animal models of diseases, such
as AD, amyotrophic lateral sclerosis, traumatic brain injury and cerebral hypoxia-
ischemia, have shown neuronal cell cycle re-entry (Copani et al., 2007; Wang et al.,
2009).
Alzheimer’s disease is a well-studied area of research in neuronal cell cycle re-
entry. One of the first pieces of evidence of neuronal cell cycle re-entry in AD was the
presence of active cell cycle molecules in AD postmortem brains shown by Vincent and
Davies, 1996. Neuronal cell cycle re-entry was primarily shown indirectly using
immunocytochemistry. In AD but not control postmortem brains, several cell cycle
proteins such as cyclin D, E, Cdk4, PCNA, cyclin B, cdc2, and Ki67 were found. CDKIs,
p16, p21 and p105 were also shown to be present in AD postmortem brains but not in
controls (Herrup et al., 2004). However, these findings did not confirm neuronal cell
cycle re-entry since these proteins may be involved in other unknown cellular functions.
One of the strongest pieces of direct evidence of neuronal cell cycle re-entry was
conducted by Yang et al who used fluorescent in situ hybridization (FISH) to illustrate
that DNA replication had actually occurred in neurons of AD brains. These findings
suggest that neurons may initiate the S phase of the cell cycle. Yang et al also showed
19

that neuronal cell cycle re-entry occurred in early stages of AD by looking at individuals
who died with mild cognition impairment (MCI) (Yang et al., 2001). MCI is considered to
be an early stage of AD since a large percentage of individuals with MCI are diagnosed
with AD several years later (Herrup et al., 2004).
Abnormal neuronal cell cycle may be an early indication of neuronal stress in
human and mouse AD (Herrup et al., 2004).There are also several transgenic mouse
models of AD. In four of these models the presence of abnormal cell cycle re-entry of
neurons in susceptible areas of the brain was shown (Herrup et al., 2004). Although
there is no significant loss of neurons in these models, amyloid plaques are formed
(Herrup et al., 2004). Interestingly, in one of the models, R1.40, Yang et al. showed that
neuronal cell cycle re-entry can be seen as early as 6 months, several months before
amyloid plaques begin forming (Herrup et al., 2004). This finding along with the
discovery from patients with MCI described above suggests that abnormal neuronal cell
cycle re-entry may be an early indication of neuronal stress in human and mouse AD
(Herrup et al., 2004)
1.2.3 In vitro evidence of neuronal cell cycle re-entry
Similar to the above in vivo findings, in vitro findings also noted neuronal cell
cycle re-entry (Table 1.1). There is indirect evidence suggesting that cerebellar granule
neurons, sympathetic neurons and cortical neurons re-enter the cell cycle after
conditions of stress and ultimately undergo apoptosis (Yang and Herrup, 2007). Many
DNA damaging agents including UV, etoposide, and camptothecin also cause abnormal
neuronal cell cycle re-entry (Table 1.1). The degree of neuronal progression through the
cell cycle prior to apoptosis depends on the stressor. For instance, neurons treated with
thrombin undergo apoptosis as soon as they transition from G0 to G1 (Liu et al., 2010).
20

However, neurons treated with etoposide proceed as far as the S phase before they
undergo apoptosis (Kruman et al., 2004). Interestingly, the furthest the neurons have
progressed through the cell cycle before they die is the S phase. To date, there has
been no evidence of neurons in the G2 phase or M (Liu et al., 2010).
Since cell cycle proteins appear in neurons prior to apoptosis, several
investigators have tested whether inhibitors of these cell cycle proteins prevent neuronal
apoptosis. Previous studies using caspase inhibitors to prevent apoptosis slowed down
neuronal death but did not prevent it. Cdk inhibitors (CDKIs), on the other hand,
protected neurons longer than caspase inhibitors (Wang et al., 2009). By inhibiting
Cdks, there was not any cytochrome c release, loss of mitochondria transmembrane
potential and caspase activation. These findings indicated that cell cycle re-entry is
upstream of other apoptotic pathways (Wang et al., 2009). Pharmacological inhibition of
Cdks prevents the G1/S transition and protects cultured neurons from death initiated by
several DNA damaging agents: UV, camptothecin and cytosine arabinoside (Park et al.,
1998a; Park et al., 1998b). Furthermore, dominant-negative kinase-inactive forms of
Cdk4 and Cdk6 reduced UV-induced death (Park et al., 1998a; Park et al., 1998b).
Thus, there has been some indirect evidence of neuronal cell cycle re-entry in vitro and
correlation between DNA damage and neuronal cell cycle regulation (Kruman et al.,
2004). There has been relatively little direct evidence of cell cycle re-entry in neurons.
For example, one study reported a significant increase of S phase neurons from
cultures treated with genotoxic chemicals (etoposide, homocysteine, methotrexate and
AB1-42) using flow cytometry to measure DNA content (Kruman et al., 2004) which
indicate that neurons can initiate the S phase prior to apoptosis. The majority of studies
conducted suggest neuronal cell cycle re-entry based on evidence of cell cycle marker
21

expression or prevention of DNA-damage induced apoptosis using Cdk inhibitors
however these proteins may have cellular functions independent of the cell cycle in
neurons.
22
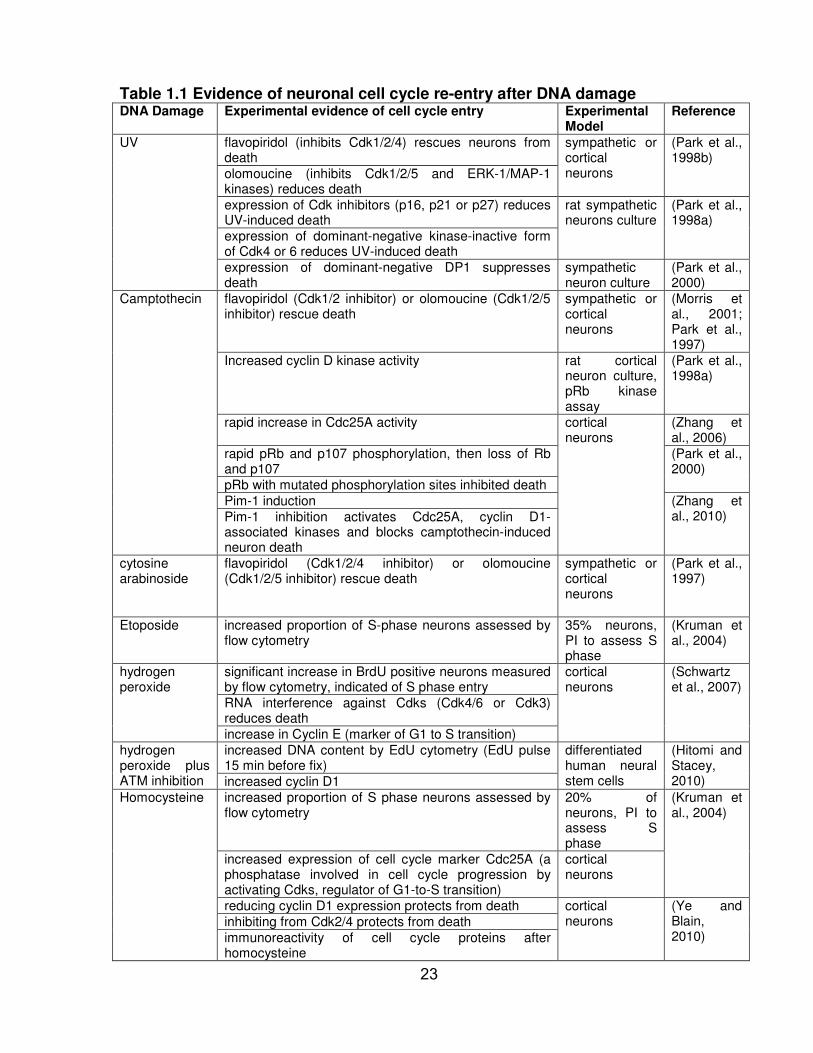
Table 1.1 Evidence of neuronal cell cycle re-entry after DNA damage DNA Damage Experimental evidence of cell cycle entry Experimental
Model Reference
UV flavopiridol (inhibits Cdk1/2/4) rescues neurons from death
sympathetic or cortical neurons
(Park et al., 1998b)
olomoucine (inhibits Cdk1/2/5 and ERK-1/MAP-1 kinases) reduces death
expression of Cdk inhibitors (p16, p21 or p27) reduces UV-induced death
rat sympathetic neurons culture
(Park et al., 1998a)
expression of dominant-negative kinase-inactive form of Cdk4 or 6 reduces UV-induced death
expression of dominant-negative DP1 suppresses death
sympathetic neuron culture
(Park et al., 2000)
Camptothecin flavopiridol (Cdk1/2 inhibitor) or olomoucine (Cdk1/2/5 inhibitor) rescue death
sympathetic or cortical neurons
(Morris et al., 2001; Park et al., 1997)
Increased cyclin D kinase activity rat cortical neuron culture, pRb kinase assay
(Park et al., 1998a)
rapid increase in Cdc25A activity cortical neurons
(Zhang et al., 2006)
rapid pRb and p107 phosphorylation, then loss of Rb and p107
(Park et al., 2000)
pRb with mutated phosphorylation sites inhibited death
Pim-1 induction (Zhang et al., 2010) Pim-1 inhibition activates Cdc25A, cyclin D1-
associated kinases and blocks camptothecin-induced neuron death
cytosine arabinoside
flavopiridol (Cdk1/2/4 inhibitor) or olomoucine (Cdk1/2/5 inhibitor) rescue death
sympathetic or cortical neurons
(Park et al., 1997)
Etoposide increased proportion of S-phase neurons assessed by flow cytometry
35% neurons, PI to assess S phase
(Kruman et al., 2004)
hydrogen peroxide
significant increase in BrdU positive neurons measured by flow cytometry, indicated of S phase entry
cortical neurons
(Schwartz et al., 2007)
RNA interference against Cdks (Cdk4/6 or Cdk3) reduces death
increase in Cyclin E (marker of G1 to S transition)
hydrogen peroxide plus ATM inhibition
increased DNA content by EdU cytometry (EdU pulse 15 min before fix)
differentiated human neural stem cells
(Hitomi and Stacey, 2010) increased cyclin D1
Homocysteine increased proportion of S phase neurons assessed by flow cytometry
20% of neurons, PI to assess S phase
(Kruman et al., 2004)
increased expression of cell cycle marker Cdc25A (a phosphatase involved in cell cycle progression by activating Cdks, regulator of G1-to-S transition)
cortical neurons
reducing cyclin D1 expression protects from death cortical neurons
(Ye and Blain, 2010)
inhibiting from Cdk2/4 protects from death
immunoreactivity of cell cycle proteins after homocysteine
23

DNA Damage Experimental evidence of cell cycle entry Experimental Model
Reference
ionizing radiation
increase in proportion of neurons double positive for DNA synthesis and apoptosis
cortical neurons
(Kruman et al., 2004)
Methotrexate increased proportion of S-phase neurons assessed by flow cytometry
20% of neurons, PI to assess S phase
1.4 5-bromo-2’-deoxyuridine (BrdU)
1.4.1 Overview of BrdU BrdU is an analogue of the nucleoside thymidine, where a methyl group is
replaced by bromine (Fig. 1.5). Since it is structurally similar to thymidine, BrdU can be
incorporated into DNA during DNA repair or DNA replication during the S-phase of the
cell cycle (Beisker and Hittelman, 1988; Gratzner, 1982; Nowakowski et al., 1989; Ross
et al., 2011). When BrdU enters cells it is phosphorylated into BrdU triphosphate and
this precursor is incorporated into DNA in the place of deoxythymidine triphosphate
(Jackson and Cook, 2008). Numerous studies suggest negative effects of BrdU
incorporation in DNA (Littlefield and Gould, 1960; Stellwagen and Tomkins, 1971), but
since BrdU is not believed to have any significant detrimental effects if cells are treated
for a short period of time, BrdU is often used to label cycling cells in vitro and in vivo
(Cameron and McKay, 2001; Lehner et al., 2011; Levkoff et al., 2008). In vitro,
mammalian cells are often supplemented with BrdU for one hour and BrdU
incorporation is measured using immunocytochemistry or flow cytometry. In vivo,
animals are injected with BrdU and immunohistochemistry is conducted. Using these
techniques BrdU can be used for cell birth dating, tracking cells following transplantation
and understanding cells which proliferate in the brain such as neural stem cells
(Cooper-Kuhn and Kuhn, 2002; Kuhn and Cooper-Kuhn, 2007; Lehner et al., 2011).
24

25

BrdU incorporation has been suggested to change DNA stability and increase the
possibility of sister-chromatid exchanges, mutations and double stranded breaks when
cells are also exposed to other stresses. For instance, BrdU has been used as a
radiosensitizer since it can cause chromosomal breakage and cause cells to become
more sensitive to ionizing radiation (Djordjevic and Szybalski, 1960; Erikson and
Szybalski, 1963; Hsu and Somers, 1961). Therefore, BrdU has been used in
conjunction with chemotherapy and radiation for treatment of different cancers in clinical
trials (Groves et al., 1999; Kinsella et al., 1984; Phillips et al., 1991; Prados et al., 2004;
Robertson et al., 1997a; Robertson et al., 1997b). The clinical benefits of BrdU as a
radiosensitizer has not been great but there may be other uses for BrdU (Levkoff et al.,
2008). The following two sub-sections will discuss the possible role of BrdU in DNA
repair and cell cycle progression.
1.4.2. The role of BrdU in DNA repair
Deoxyribonucleotide triphosphates (dNTPs) are required for DNA synthesis and
repair. As a cell transitions from the G1 phase to the S phase of the cell cycle dNTP
pools are increased approximately 5 to 10 fold and maintained at this level until DNA
replication is finished. When cells progress pass the S phase dNTP levels are reduced
(Reichard, 1988). Even though the dNTP pool sizes of neurons have not been
determined, examination of G0 phase 3T3 mouse fibroblasts indicated that thymidine
triphosphate (dTTP) pools are reduced by 20-fold in comparison with S phase cells.
Furthermore, dTTP turnover was 200-fold faster in S phase cells than G0 cells (Spyrou
and Reichard, 1988). Since G0 cells are not replicating their DNA, a small dNTP pool
may be sufficient for DNA repair and mitochondrial DNA synthesis. Yet, if there is a
great deal of DNA damage, neurons may not have enough dNTPs for repair. Thus,
26

increasing neuronal dNTP pool size may improve DNA repair. Thymidine supplement
does increase intracellular dTTP pool size in hepatocytes (Rottgen and Rabes, 1989)
and BrdU exposure to hamster melanoma cells formed a large BrdU triphosphate pool
which remained at a high concentration for several days (Ashman et al., 1981). At the
same time, deoxycytidine triphosphate levels dropped drastically after the addition of
BrdU (Ashman and Davidson, 1981). This finding supports the idea that increasing
extracellular concentrations of pyrimidines may increase intracellular concentrations of
dNTPs.
Terminally differentiated cells such as neurons are often relatively deficient in
DNA repair. During cellular differentiation, DNA repair is gradually reduced and cells
may be more vulnerable to DNA damage (Kruman, 2004; McCombe et al., 1976). In rat
neurons there is evidence of low NER (Subrahmanyam and Rao, 1991) and double-
strand break repair (Gobbel et al., 1998; Wang and Wheeler, 1978). NER was also
shown to be decreased in human neuroblastoma and NT2 neuroteratoma (Jensen and
Linn, 1988; Nouspikel and Hanawalt, 2000). DNA repair consumes a great deal of ATP
(Roca and Cox, 1997). Since neurons are metabolically active cells that require a large
amount of energy, it has been suggested that DNA repair is down-regulated in neurons
to save energy (Kruman, 2004). NER can be divided into two pathways, TCR which
repairs DNA lesions in transcribed genes and GGR which repairs DNA lesions
throughout the entire genome. Although repair of active genes in neurons by TCR has
been maintained in neurons (Kruman, 2004), GGR is significantly lower in neurons than
fibroblasts (Kruman, 2004; Nouspikel and Hanawalt, 2000; Yamamoto et al., 2007).
Removal of DNA adducts in transcribed genes is important in order for the cell to
maintain proper cellular function whereas GGR may not be essential since neurons
27

have exited the cell cycle and do not divide (Kruman, 2004). The attenuation of GGR
may be responsible for the higher sensitivity of neurons to DNA damage-initiated
apoptosis. Improving neuronal DNA repair could theoretically improve neuronal survival
following DNA damage.
NER is inducible. DNA repair in general was first discovered to be inducible in
1975 in bacteria and is referred to as the SOS response (Radman, 1975). NER has also
been shown to be inducible in mammalian cells as well (Gilchrest and Eller, 1999). For
example, single stranded DNA fragments such as thymidine dinucleotide (pTT) induce
protective responses in skin cells as well as intact guinea pig skin (Eller et al., 1994).
Furthermore, pTT partly increases repair of UV lesions by activating p53 (tumor
suppressor protein and transcription factor) and up-regulating several proteins involved
in DNA repair (Gilchrest and Eller, 1999). Interestingly, in vitro, pTT enhanced skin cells
ability to repair UV lesions by activating p53 and improving endogenous DNA repair
(Goukassian et al., 2004). pTT did not only improve removal of UV lesions but also
benzo(α)pyrene adducts which are also repaired by NER (Maeda et al., 1999).
Oligonucleotides similar to the mammalian telomere repeat sequence 5’-TTAGGG-3’ (T-
oligos) also improved DNA repair similar to pTT (Eller et al., 2002). Other compounds,
in addition to pTT, also improve DNA repair. For example, treatment with green tea
polyphenols improved the removal and/or repair of CPDs (DNA adducts caused by UV)
in mice skin exposed to UVB (Katiyar et al., 2010). The improvement in DNA repair of
DNA adducts in mouse skin could be due to increased levels of NER proteins XPA and
XPC. In order for green tea polyphenols to improve repair of UV-induced DNA adducts,
functional NER is required indicating that the effect of green tea polyphenols is through
the NER pathway (Katiyar et al., 2010).
28

1.4.3. The role of BrdU in the cell cycle
Although BrdU has been used to label proliferating cells and assumed that it
incorporates into DNA, there are other cellular effects of BrdU. Around the time BrdU
was initially introduced, cellular effects of BrdU were studied and it was discovered that
BrdU does not benignly substitute for thymidine. For instance, BrdU causes mouse
neuroblastoma C1300 cells to differentiate and structurally look like mature neurons
even without DNA synthesis (Schubert and Jacob, 1970). Thus, halogenated
pyrimidines do not need to be incorporated into DNA in order to change the function of
cells (Schubert and Jacob, 1970). While it has not been shown that BrdU alters cell
cycle re-entry in neurons, BrdU does alter cell cycle progression in proliferating cells
(Table 1.2). A single BrdU exposure to cancer cells impairs cell cycle progression.
Interestingly, these cells slowly accumulate in the G1 phase of the cell cycle and do not
die (Levkoff et al., 2008). BrdU treatment has also slowed down cell growth in vivo (Gyi
and Wrathall, 1983). Similarly, other thymidine analogues, CldU, IdU, FdU and EdU,
have all been shown to inhibit cell proliferation (Levkoff et al., 2008; Ross et al., 2011).
29

Table 1.2 Evidence of cell cycle modulation by thymidine analogues Nucleoside analogue
Cell type Effect of nucleoside analogue Experimental conditions
Reference
BrdU B-cell lines Synergistically cytostatic with cisplatin, bleomycin and chlorambucil
100-500 µM BrdU
(Poot et al., 1991)
Melanoma cells
Reduced growth in vivo 3 µg/mL BrdU (Gyi and Wrathall, 1983)
Neurogenic astrocytes
Caused rapid increase in proportion of cells in G1 and decrease in proportion of cells in S
Single 24h pulse of 50 µM BrdU;
(Ross et al., 2008)
Breast and bladder cancer lines
Reduced cell cycle progression but no increase in death
3.5, 5 and 60 µM BrdU
(Diermeier et al., 2004)
Gastric carcinoma lines
Caused G1 arrest and S phase delay 20 µM BrdU (Peng et al., 2001)
A549 lung cancer cells
Reduced proliferation 50 µM BrdU for 24 h, cells are p16-null
(Masterson and O'Dea, 2007)
MG-63 osteosarcoma cell line
Reduced proliferation 50 µM BrdU for 18 h
(Levkoff et al., 2008)
H9 T-cell lymphoma cell line
50 µM BrdU for 24 h
Saos-2 human osteosarcoma cell line
TT human thyroid tumour cell line
BJ human fibroblasts
RG2 rat glioma cell line
Reduced proliferation in a dose-response manner
1 – 50 µM BrdU
RG2 rat glioma cell line
Cells pretreated with BrdU then injected into mice showed suppressed proliferation in vivo relative to vehicle controls
50 µM BrdU for 24 h
RG2 rat glioma cell line
Mice inoculated with glioma cells and treated with BrdU showed improved survival relative to vehicle controls
6 i.p. injections of 300 mg/kg BrdU over 2 days
RG2 rat glioma cell line
Mice inoculated with glioma cells and treated with BrdU showed improved survival relative to vehicle controls
BrdU 0.8 mg/mL in drinking water
CldU H9 (human lymphoma cells)
Reduced proliferation in a dose-response manner similar to BrdU
1 – 50 µM CldU for 18 h
IdU H9 Reduced proliferation in a dose-response manner similar to BrdU
1 – 50 µM IdU for 18 h
FdU H9 Transiently reduced proliferation in a dose-response manner, but not as effective as BrdU
1 – 50 µM FdU for 18 h
30

Nucleoside analogue
Cell type Effect of nucleoside analogue Experimental conditions
Reference
EdU hGBM, primary human glioblastoma cell lines
Suppressed proliferation in culture better than BrdU
1 – 50 µM EdU for 24 h
(Ross et al., 2011)
MG63 osteosarcoma cell line
Suppressed proliferation in culture better than BrdU
1 – 10 µM EdU for 24 h
Mouse embryonic fibroblasts
Suppressed proliferation in culture better than BrdU
1 – 10 µM EdU for 24 h
hGBM glioblastoma cells
Reduced tumour volume in vivo in xenograft model
50 mg/kg/day EdU i.p. or s.q. long term (up to 100 days)
hGBM glioblastoma cells
Improved survival of xenografted mice 50 mg/kg/day EdU i.p. or s.q. long term (up to 100 days)
BrdU inhibits ribonucleotide reductase (RR). RR catalyzes the rate-limiting step
of the synthesis of deoxyribonucleotides (dNTPs) from ribonucleotide precursors (Cory
and Sato, 1983; Jiang et al., 2006; Thelander and Reichard, 1979). In the 1970s, BrdU
was discovered to be an allosteric inhibitor RR (Meuth and Green, 1974). Thus,
functional RR is required for sustaining dNTP supply necessary for DNA repair and
synthesis (Finch et al., 2000). 3-aminopyridine-2-carboxaldehyde thiosemicarbazone
(PAN-118) is an inhibitor of RR and was developed for cancer treatment (Jiang et al.,
2006). PAN-811 has some neuroprotective effects and prevents ischemic neurotoxicity
as well as hypoxic toxicity and also prevents neuronal death from neurotoxic agents
such as staurosporine, veratridine and glutamate (Jiang et al., 2006). Although Jiang et
al suggest that PAN-118 may be neuroprotective by altering excitatory neurotoxic
pathways (Jiang et al., 2006), PAN-118 may also be neuroprotective by inhibiting RR.
Recently, the effect of BrdU on neural stem cells (NSCs) has also been studied
in the mammalian brain. NSCs are found in the subependymal zone of the lateral
31

ventricles and in the hippocampus and can differentiate into neurons and glia
(astrocytes and oligodendrocytes) (Doetsch, 2003). BrdU prevents NSC cell cycle
progression, induces them to differentiate into astrocytes and causes DNA
demethylation (Schneider and d'Adda di Fagagna, 2012). Methylation (adding a methyl
group) to cytosines in DNA occurs in regions called CpG islands and is highly regulated
in stem cells (Hermann et al., 2004; Wu and Zhang, 2010). Particularly, DNA
methylation of specific genes affects differentiation (Meissner et al., 2008). DNA
methyltransferases (DNMT) methylate either de novo (DNMT3a and DNMT3b) or newly
replicated DNA (DNMT1) (Hermann et al., 2004). DNMT1 localizes to replication foci
during S phase of the cell cycle and is down-regulated in proliferating cells whereas
DNMT3a and DNMT3b may be independent of the cell cycle (Hermann et al., 2004;
Schneider and d'Adda di Fagagna, 2012). Demethylation of DNA can either occur by
removing methylated cytosines or inhibition of DNMTs (Wu and Zhang, 2010). Other
thymidine analogues, CldU and IdU, also have effects similar to BrdU in neural stem
cells (Schneider and d'Adda di Fagagna, 2012). Studies on NSC have also shown that
BrdU treatment of neurosphere culture caused cellular senescence (Lehner et al., 2011;
Ross et al., 2008).
32

2. OVERVIEW The limited capacity to replace neurons stresses the importance of the neuronal
DNA damage response. Neuronal DNA damage can lead to apoptosis. To prevent
neuronal DNA damage-induced apoptosis, understanding the neuronal DNA damage
response is essential.
In this project, it was observed that BrdU prevented UV-induced neuronal death.
This is the first report of the neuroprotective action of BrdU and the mechanism of BrdU
neuroprotection is unknown. Therefore, we investigated dose-response relationships as
well as determined whether proliferating cells were also protected by BrdU. Since BrdU
is a thymidine analogue, we determined whether other thymidine analogues and DNA
nucleosides protect neurons. p53 is a central stress response protein involved in many
neuronal death pathways (Harris and Levine, 2005). To determine whether BrdU altered
the UV-induced neuronal DNA-damage response we investigated p53 expression after
UV and its potential modulation by BrdU. Theoretically, the repair of DNA damage
should be neuroprotective, thus we also interrogated whether BrdU improves neuronal
DNA repair. Indirect evidence indicates that UV induces neuronal cell cycle re-entry
which ultimately leads to apoptosis (Park et al., 1998a). Since BrdU has been shown to
impair cell cycle progression in proliferating cells (Levkoff et al., 2008; Schneider and
d'Adda di Fagagna, 2012) we examined whether BrdU prevented neuronal cell cycle re-
entry.
33

2.1 Hypothesis
We hypothesized that BrdU prevented UV-induced neuronal apoptosis by
improving the DNA damage response.
2.2 Aims
This work has four specific aims:
1. Characterize dose-response relationships, cell type specificity and chemical
specificity of BrdU protection.
2. Determine whether BrdU modulates UV-induced cell signaling by p53
expression.
3. Assess the role of nucleotide excision repair in UV-initiated neuronal apoptosis.
4. Determine whether UV or BrdU modulate neuronal cell cycle re-entry.
34

3. MATERIALS AND METHODS
3.1 Cortical neuron culture
CD1 males and females were mated and the plug date used was E0.5. The
cortex was dissected in cold dissecting solution (DS) from embryo brains (E16.5). DS
contains 137 mM NaCl, 5.4 mM KCl, 0.17 mM Na2HPO4, 0.22 mM KH2PO4, 9.9 mM
Hepes, 33.3 mM D (+)-Glucose and 43.8 mM sucrose in ultra-filtered water. The freshly
dissected embryonic cortices were digested in DS solution containing 9 units/ml Papain
(Worthington), 10 ng/ml 2-Amino-5-phosphonopentannoic acid (APV, Sigma) and 20
ng/ml L-cysteine at 37oC for 30 mins. The tissue was then transferred to an enzyme
inhibitor solution containing 20 mg/ml BSA (Bovine Albumin, Sigma), 20 mg/ml Trypsin
Inhibitor and 10 ng/ml of APV in DS using a Pasteur pipette. The tissue was rinsed once
with cortical neuron media before dissociation using micropipetting to single cell
suspensions in cortical neuron media. Cortical neuron media consisted of neurobasal
medium, 2% B-27 serum-free supplement, 0.25% glutaMAXTM-I Supplement and 0.2%
Penicillin-Streptomycin from Life Technologies. Culture ware (BD) was coated with 50
µg/ml poly-L-lysine (Sigma). 2.3 x 102 cells/mm2 were plated on 6 cm dishes and 2.9 x
102 cells/mm2 on 18 by 18 mm coverslips. For coverslips, cells were diluted to a
concentration of 1.36 x 105 and 700 ul was seeded on the coverslip. The media was
replaced in coverslips and dishes after approximately 3 hours after the cells had
adhered to the surface. The cultures were maintained at 37oC and 5% CO2.
For Xpa-/- or Csb-/- cortical neuron isolation, Xpa+/- or Csb+/- males and females
were mated respectively. Similar to CD1 cultures, the date used was E16.5 and the
cortex was dissected in cold DS. Embryonic tails were also removed for genotyping of
each embryo. The tissue was stored in Hibernate-E with 2% B-27 serum-free
35

supplement (Life Technologies) for approximately 3 hours while genotyping was
completed. The processing of the tissue was identical to the above protocol.
3.2 Embryonic genotyping
Embryonic tails were removed from each embryo from mixed Xpa+/- and Csb+/-
litters during dissections and labeled to correspond to the culture samples from that
embryo. The tails were digested, neutralized and amplified using the REDExtract-N-
AmpTM Tissue PCR (XNAT) kit (Sigma) according to manufacturer’s instructions. Table
3.1 and 3.2 summarizes the primer sequences, PCR conditions and band sizes of XPA
and CSB respectively. PCR products were run on a 2% agarose gel at 120 Volts for 1 h
and visualized using ethidium bromide and a UV transilluminator.
Table 3.1 Summary of Xpa primers, cycling conditions and band sizes Primers Cycling
Conditions Band Sizes
XP26: 5’-GTGTCAGGCATAAGATCTATGACAA-3’
1. 95oC for 5min 2. 94oC for 1min 3. 58oC for 1min 4. 72oC for 2min Repeat 2-4 34x 5. 72oC for 10min
WT: 300bp Xpa-/-: 200bp
XP47: 5’-AGGCAAGCACCTGCAGCTGT-3’ PGK2: 5’-GGCCACTTGTGTAGCGCCAA-3’
Table 3.2 Summary of Csb primers, cycling conditions and band sizes Primers Cycling
Conditions Band Sizes
CSB4: 5’-GCTGCTTATAATAATCCTCATC-3’
1. 95oC for 5min 2. 94oC for 30s 3. 58oC for 45s 4. 72oC for 1min Repeat 2-4 34x 5. 72oC for 10min
WT: 200bp Csb-/-: 500bp
CSB5: 5’-ATCTGGGTGTTCGAATTCGCCAATG-3’ CSB6: 5’-GTCTTCTGATGACGTTAGCTATGAG-3’
3.3 Mouse embryonic fibroblast (MEF) isolation
CD1 males and females were mated and the plug date used was E0.5. Embryos
were collected at E14.5 and the head and all internal organs were removed. The
36

embryo body was transferred to microfuge tube on ice. The embryo was cut into small
pieces in the microfuge tube and then transferred to a 6-well plate. The tissue was
incubated at 37oC with 0.05% Trypsin-EDTA (Gibco) for 30 mins. The tissue was
dissociated with a pipette and incubated again with 0.05% Trypsin-EDTA for 20mins.
The trypsin was inactivated with fetal bovine serum (FBS, Life Technologies). The cells
were centrifuged at 524 x g at room temperature for 10 mins and the top layer was
removed. The cultures were maintained in Dulbecco’s Modified Eagle Medium (DMEM)
containing 25 mM glucose, 10% FBS, 1% L-glutamine, 1% Penicillin-Streptomycin and
55 µM B-mercaptoethanol (all reagents from Life Technologies). The cultures were
maintained at 37oC and 5% CO2.
3.4 Cortical neuron survival assays
3.4.1 Neuronal immunocytochemistry
CD1 cortical neurons were seeded on coverslips. At 5 days post isolation, media
was removed from coverslips and using the CL-1000 UVP Crosslinker, wells were
individually treated with 0, 10 or 50 J/m2 of UVC at a wavelength of 254 nm. Cortical
neuron media was added with vehicle or BrdU (30 µM, Sigma) and cells were fixed
using 4% paraformaldehyde (PFA) from Sigma 24 hours later. Cells were washed with
PBS, permeabilized with 0.4% TritonX-100 in PBS for 15 mins and blocked with 1%
goat serum and 0.2% TritonX-100 in PBS (blocking solution) for 30 mins. The cells were
incubated with the primary antibody, mouse anti-tubulin, beta III isoform (1:1000,
StemCell Technologies, 01409) overnight at 4oC in blocking solution. This antibody
reacts with the neurons specific beta III isoform of tubulin (βIIITub) and is commonly
used as a neuronal marker. The cells were washed the following day with PBS and
37

incubated with the secondary antibody goat anti-mouse Alexa-568 (Invitrogen) for an
hour and then mounted onto slides using Vectashield Mouting Media (Vector)
containing DAPI (nuclear stain). Cells were imaged using AxioImager.M2
Epifluorescence Microscope from Zeiss.
3.4.2 Quantification of apoptosis with flow cytometry
CD1 cortical neurons were seeded on 6 cm dishes at the density 2.3 x 102
cell/mm2. At 5 days post isolation, media was removed from dishes and UVC-irradiated
using the CL-1000 UVP Crosslinker (0, 5 or 10 J/m2) and media was added with vehicle
or BrdU (30 µM) for the UV dose-response. For the BrdU dose-response, cells were
irradiated with 0 or 10J/m2 and supplemented with media containing 0 to 10 µM BrdU.
24 hours post-irradiation, media was collected (to ensure dead cells were harvested as
well), and the cells were incubated with 0.05% trypsin-EDTA (Gibco) for 7 mins at 370C.
Trypsin was deactivated with FBS (Life Technologies) and cells were dissociated and
harvested into 15 ml falcon tubes (BD). Cells were centrifuged at 524 x g; supernatant
aspirated, washed with PBS and transferred to 5 ml round-bottom tubes (BD). The cells
were centrifuged at 524 x g for 5mins, supernatant removed and cells resuspended in
150ul binding buffer containing 5 µl of FITC Annexin V (apoptotic marker) and 50ng/ml
propidium iodide (PI). The binding buffer and FITC Annexin V were from BD
Pharmingen and PI was from Sigma. The cells were incubated at room temperature and
analyzed using BD FACSCalibur flow cytometer (BD) and FLOJO software (Tree Star).
3.4.3 Quantification of apoptosis with immunocytochemistry
Cortical neurons were UVC-irradiated (0 or 10J/m2) using the CL-1000 UVP
Crosslinker 5 days post isolation and supplemented with media containing vehicle or
38

BrdU as before. CD1 cortical neurons were treated with 0 or 10 J/m2 UV, Xpa-/- and
Csb-/- cortical neurons were treated with 0, 2, 10 or 50 J/m2 UV and Xpa+/+ and Csb+/+
(wild type controls) were treated 0, 10 or 50 J/m2 UV. Cells were fixed using 4% PFA
and the staining was identical to 2.2.1 but with different antibodies. The primary
antibodies were mouse βIIITub (1:1000) and rabbit cleaved caspase-3 (1:100, Cell
signaling, Asp175) as an apoptotic marker. The secondary antibodies used were goat
anti-mouse Alexa-568 and goat anti-rabbit Alexa 488 (Invitrogen). AxioImager.M2
Epifluorescence Microscope (Zeiss) was used to take sequential individual channel
images at 10x magnification using the same exposure times. Metamorph Software
(Olympus) was used to quantify the percentage of apoptotic neurons (cells that were
positive for cleaved caspase-3 and βIIITub) and the total number of neurons (cells that
were positive for βIIITub).
3.5 Mouse embryonic fibroblast survival assay
CD1 passage 0 MEFs were seeded at the density 1.12 x 102 cells/mm2. 24 hours
later the media was removed and the cells were UVC-irradiated (0 or 25J/m2) using the
CL-1000 UVP Crosslinker and supplemented with media containing vehicle or BrdU (0-
30 µM). The cells were harvested 24 hours later for flow cytometry with FITC Annexin V
(BD Pharmingen) and PI (Sigma) following the same protocol that was used for cortical
neurons described above.
3.6 BrdU labeling using immunocytochemistry
Cortical neurons were UVC-irradiated (0 or 50J/m2) using the CL-1000 UVP
Crosslinker 5 days post isolation and supplemented with media containing 10 µM BrdU
and fixed with 4% PFA at 24 h. MEFs were used as a positive control and incubated
39

with 30 µM BrdU for 1 h and fixed with 4% PFA. Cells were washed with PBS and
permeabilized with 0.4% TritonX-100 in PBS for 15mins. The cells were than washed
with PBS and incubated with 10U/ml DNase (Thermo Scientific) in 10mM Tris, 0.1mM
CaCl2 and 2.5mM MgCl2 for 1h at 37oC. The cells were than washed with PBS and
blocked similar to above and incubated with the primary antibodies mouse βIIITub
(1:1000) and rat BrdU (1:500, Abcam, ab6326) and then secondary antibodies goat
anti-mouse 568 and goat anti-rat Alexa-488 (Invitrogen) for an hour and then mounted
onto slides using Vectashield Mounting Media (Vector) containing DAPI (nuclear stain).
Cells were imaged using AxioImager.M2 Epifluorescence Microscope from Zeiss.
3.7 Quantification of UV DNA lesions
UV DNA lesions were quantified using an enzyme-linked immunosorbant assay
(ELISA). CD1 cortical neurons were seeded on 10cm dishes and UVC-irradiated (0, 50
or 100J/m2) using the CL-1000 UVP Crosslinker 5 days post isolation. Cortical neuron
media with vehicle or BrdU (10 µM) supplemented was added to the dishes and cells
were harvested 0, 2, 4 or 8 h after UV-irradiation similar to cells harvested for flow
cytometry described above. The pellet was washed with PBS and flash frozen. Genomic
DNA was extracted using GenEluteTM Mammalian Genomic DNA Miniprep kit (Sigma)
according to manufacturer’s instructions and stored at 4oC. The DNA was quantified
using Nanodrop 1000 (Thermo Scientific) and diluted to a concentration of 4.0 ng/µl for
the 6-4PP lesion and 0.2 ng/µl for the CPD lesion. In order to denature the DNA, the
DNA solution was incubated at 100oC for 10mins and then rapidly chilled on ice.
In order for the DNA to adhere to the plate, 96 well microtiter plates were coated
with 50 µl of 0.003% protamine sulfate solution overnight at 37oC in order to dry
completely and washed with distilled water the following day. 50 µl of the diluted DNA
40

was added per well and dried completely overnight at 37oC. The following day, the
DNA-coated plates were washed with PBS-T (0.05% Tween-20) and blocked with 2%
FBS in PBS for 30 mins. The wells were washed again with PBS-T and either the 64M-2
antibody (1:1500, Cosmo Bio Co.) for the 6-4PP lesion or the TDM-2 antibody (1:1000,
Cosmo Bio Co.) for the CPD lesion diluted in PBS was added to each well and the plate
was incubated at 37oC for 30mins. The plates were then washed with PBS-T and then
incubated with Biotin-F(ab’)2 fragment of anti-mouse IgG (H+L) (1:2000) in PBS for
30mins at 37oC. The plates were washed again with PBS-T and incubated with 1:10000
Peroxidase-Streptavidin in PBS for 30mins at 37oC. The wells were washed with PBS-T
and then washed with Citrate-phosphate buffer at a pH of 5. Then 100 µl of o-
Phenylene diamine and H2O2 in Citrate-phosphate buffer was added to each well for 2-3
mins. In order to stop the enzyme reaction 50 µl of 2M H2SO4 was added to each well
and the absorbance at 492 nm was measured using a plate reader.
3.8 DNA content quantification using immunocytochemistry
CD1 cortical neurons were UVC-irradiated (0 or 50J/m2) using the CL-1000 UVP
Crosslinker 5 days post isolation and supplemented with media with vehicle or BrdU (10
µM). Cells were fixed with 4% PFA at 12 or 18 h after UV-irradiation. Similar to 3.4.1
immunocytochemistry protocol, cells were washed with PBS, permeabilized with 0.4%
TritonX-100 and blocked with 0.2% TritonX-100 and 1% goat serum in PBS. Neurons
were incubated with mouse βIIITub (1:1000) overnight at 4oC and washed with PBS the
following day. The cells were then incubated with the secondary antibody goat anti-
mouse Alexa-568 (Invitrogen) for one hour. The cells were washed with PBS and
incubated with Hoechst 33342 (1 µg/ml) for 15 mins at room temperature and washed
with PBS five times. The coverslips were then mounted onto slides using Vectashield
41

Mounting Media (Vector) without DAPI. The cells were imaged using AxioImager.M2
Epifluorescence Microscope from Zeiss.
3.9 Quantification of cell cycle distribution using flow cytometry
CD1 cortical neurons were UVC-irradiated (0 or 50J/m2) using the CL-1000 UVP
Crosslinker 5 days post isolation and supplemented with vehicle or BrdU (10 µM).
Control neurons were incubated with 1 µM etoposide. 12 hours post-irradiation cells
were trypsinized (0.05% Trypsin-EDTA, Gibco) for 7 mins at 37oC and collected as a
single cell suspension in a 15 ml falcon tube (BD). The cells centrifuged at 524 x g for 5
mins at room temperature and the supernatant was removed. The cells were
resuspended and fixed with ice cold 70% ethanol and stored at -20oC for at least 20
mins. The cells were transferred to 5 ml round-bottom tubes (BD), centrifuged and the
supernatant removed. The cells were then washed with PBS and permeabilized with
0.4% TritonX-100 in PBS for 15 mins. The cells were washed again with PBS and then
incubated with the primary antibody βIIITub (1:200, StemCell Technologies) with 0.2%
TritonX-100 and 1% donkey serum for 3 h. The cells were washed with PBS and
incubated with the secondary antibody, donkey anti-mouse-FITC (1:200), in PBS for 30
mins at room temperature. The cells were finally washed with PBS and resuspended
with PI (50ng/ml, Sigma) and analyzed using BD FACSCalibur flow cytometer (BD) and
FLOJO software (Tree Star).
3.10 Cell cycle marker expression
Immunocytochemistry was similar to the protocol described above for βIIITub
only. The cells were incubated with primary antibodies, mouse anti-tubulin, beta III
isoform (1:1000, StemCell Technologies) and one of the following cell cycle markers:
42

rabbit anti-Cdk4 (1:100, Santa Cruz Technology, sc-260), rabbit anti-Cdk6 (1:100, Santa
Cruz Technology, sc-177) or Ki67 (1:100, abcam, ab66155). The secondary antibodies
were goat anti-mouse Alexa-568 (Invitrogen) and anti-rabbit Alexa-568 (Invitrogen).
Cells were imaged using AxioImager.M2 Epifluorescence Microscope from Zeiss.
3.11 Quantification of global DNA methylation
CD1 cortical neurons were seeded at a density of 2.3 x 102 in 10 cm dishes and
UVC-irradiated (0 or 50 J/m2) using the CL-1000 UVP Crosslinker 5 days post isolation.
Cortical neuron media with vehicle or BrdU (10 µM) supplemented was added to the
dishes and cells were harvested 0, 6, 12 or 18 h after UV-irradiation. The cells were
harvested and genomic DNA extracted was similar to protocol described above for
quantification of UV lesions. Global DNA methylation was quantified using the
MethylampTM Global DNA Methylation Quantification Kit (Epigentek) according to
manufacturer’s instructions. The assay consists of binding the genomic DNA to assay
well. The methylated portion of DNA was quantified using an anti-5-methylcytosine
antibody with an enzyme with an ELISA-like reaction. The following formula was used to
calculate the percent methylation.
% Methylation = (OD (sample-blank)/2)/ (OD (positive control-blank)) x 100
3.12 Statistical analyses
All statistical analysis was performed with GraphPad Prism software where data
was expressed with error bars indicating standard deviation of the mean (+/- S.D.). Two
way analysis of variance (two-way ANOVA) where p<0.05 was considered statistically
significant was used to determine the effect of BrdU on UV-induced apoptosis followed
by Bonferroni analysis.
43

4. RESULTS
4.1 BrdU prevents mouse cortical neurons from UV-induced death
Since BrdU labeling is a common technique to examine the S phase of the cell
cycle we decided to treat cortical neurons with UV and supplement the cells with BrdU
to determine if these neurons entered the S phase following DNA damage. DNA repair
and DNA replication involve DNA synthesis but DNA replication likely represents the
majority of BrdU labeling (Cooper-Kuhn and Kuhn, 2002). In proliferating cells, BrdU
supplementation for an hour is often sufficient for BrdU to become phosphorylated,
incorporated into DNA and label S phase (Cavanagh et al., 2011; Taupin, 2007).
However, since neurons are post-mitotic and it is unknown at what time neurons might
enter the S phase following UV treatment, neurons were UV treated and immediately
supplemented with BrdU (30 µM) for a full 24 hours. This BrdU concentration is routinely
used to label dividing cells in culture (Tamblyn et al., 2009). If neurons entered the S
phase at any point during this 24 hour time period, they should incorporate BrdU. The
approach of utilizing BrdU to mark neurons entering the S phase after stress has been
shown in other studies (Kruman et al., 2004). 50 J/m2 UVC-irradiation generates up to 5
DNA lesions per 10 kB DNA (van Hoffen et al., 1995). UV treated neurons died by
apoptosis. Surprisingly, UV treated neurons with BrdU for 24 h survived. UV irradiation
at 10 and 50 J/m2 caused extensive neuronal death by 24 h (Fig. 4.1A-C). UV-irradiated
neurons cultured with BrdU immediately after UV did not die. Instead, qualitative
assessment of neuronal morphology using the structural marker βIIITubulin revealed
that BrdU-treated neurons (Fig. 4.1D-F) appeared healthy and similar to unirradiated
neurons (Fig. 4.1A).
44

In order to assess whether BrdU protected neurons from UV-initiated apoptosis,
neurons were stained with the apoptotic marker Annexin V (Niu and Chen, 2010) and
the fraction of Annexin V positive cells was assessed by flow cytometry 24 h after
treatment. UV (5 and 10 J/m2) caused a significant increase in Annexin V positive cells
(p<0.05, Fig. 4.1G). Treatment with 30 µM BrdU immediately after UV significantly
reduced the proportion of Annexin V positive cells. Therefore, BrdU can prevent UV-
induced apoptosis in cortical neurons confirming our qualitative observations.
In order to determine whether BrdU concentrations lower than the 30 µM utilized
above were neuroprotective, the effect of 0.1 – 10 µM BrdU on UV-initiated neuronal
apoptosis was assessed (Fig. 4.1H). A concentration dependent neuroprotective effect
of BrdU was observed, with BrdU concentrations as low as 0.1 µM significantly reducing
UV-initiated neuronal apoptosis.
In order to determine the toxicity of BrdU on its own, unirradiated neurons were
incubated with a range of BrdU concentrations (0.1-10 µM) for 24 hours and apoptosis
was quantified using flow cytometry with Annexin V (Fig. 4.1H). BrdU (0-10 µM) did not
significantly increase the basal levels of neuronal apoptosis. Since 10 µM BrdU reduced
UV-induced apoptosis but was not toxic, 10 µM BrdU was used for future experiments.
45

46

4.1.1 BrdU reduces UV-induced cleaved caspase-3 in cortical neurons
The Annexin V flow cytometry experiments described above assess apoptosis in
all cell types present in our cultures. Since cortical neuron cultures derived from
embryonic mice produce predominantly but not exclusively neurons, cleaved caspse-3
staining (marker of late apoptosis) was conducted on neurons (immunocytochemistry)
taking advantage of double labeling with a neuronal marker. βIIITub was used as a
neuronal marker to restrict the analysis to neurons. Approximately 1500 neurons were
scored for each treatment group. Cortical neurons were UV-irradiated (0 or 10 J/m2) and
incubated with vehicle or BrdU (10 µM) for 18 h (Fig. 4.2A-L). Among βIIITub positive
neurons, cells were classified as cleaved caspase-3 positive using automated image
analysis. BrdU significantly reduced UV-induced neuronal apoptosis (Fig. 4.2M).
47
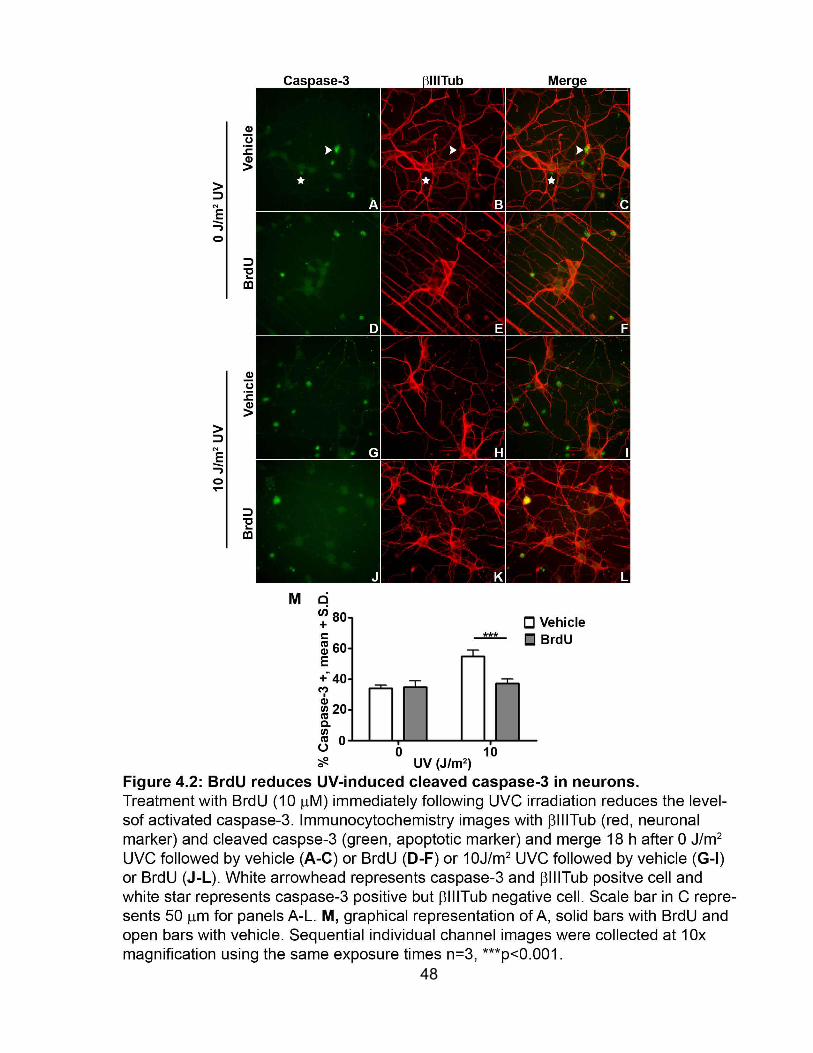
48

4.2 Absence of detectable BrdU labeling in cortical neurons
BrdU was initially utilized as a DNA synthesis marker. To assess whether the
neuroprotective effects of BrdU resulted from BrdU incorporation into newly synthesized
DNA, immunocytochemistry with an anti-BrdU antibody was conducted. Proliferating
mouse embryonic fibroblasts (MEFs) were used as a positive control. BrdU labeling was
readily detectable in MEFs (Fig. 4.3A). MEFs were labeled with BrdU (30 µM) for one
hour. The intensity (green) of labeling varied among MEFs indicating that these cells
were at different phases of the cell cycle. Absence of BrdU labeling is seen in control
MEFs (Fig. 4.3B) not incubated with BrdU. Detectable BrdU labeling was absent in UV-
irradiated (50 J/m2) neurons incubated with BrdU (30 µM) for 24 hours (Fig. 4.3C) as
well as negative control neurons: in the absence of BrdU (Fig. 4.3D) and unirradiated
neurons incubated with BrdU (Fig. 4.3E). βIIITub (red) was used as a neuronal marker.
BrdU neuroprotection is independent of global DNA synthesis and UV-irradiation did not
appear to trigger cell cycle re-entry in our model system. However, this methodology
does not have the sensitivity to detect BrdU incorporation during DNA repair when small
patches of new DNA are synthesized.
49
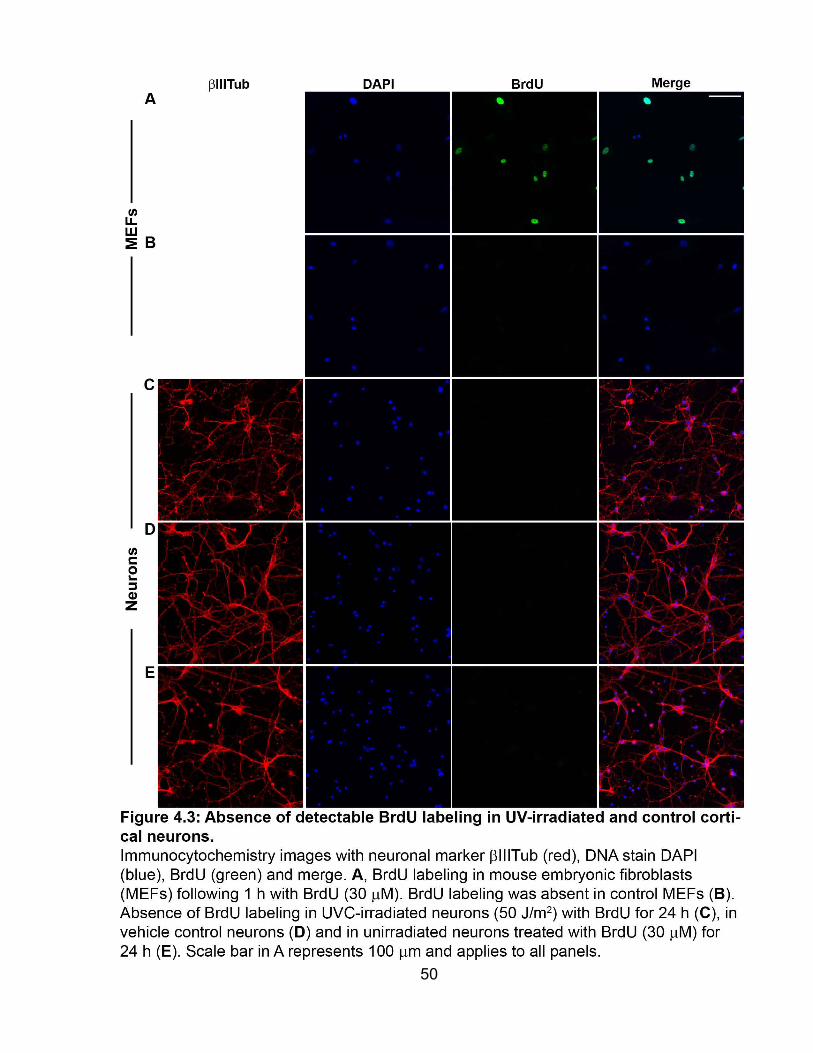
50

4.3 Cell type specificity of BrdU protection
To determine whether BrdU protection was specific to neurons the cell type
specificity of BrdU protection was analyzed. MEFs were utilized to determine whether
BrdU reduces UV-induced apoptosis in proliferating cells. MEFs were UV-irradiated (25
J/m2) and incubated with 0 – 30 µM BrdU for 24 hours. Apoptosis was quantified using
flow cytometry with Annexin V. UV-irradiation significantly increased apoptosis in MEFs
and BrdU did not reduce UV-induced apoptosis (Fig. 4.4).
51
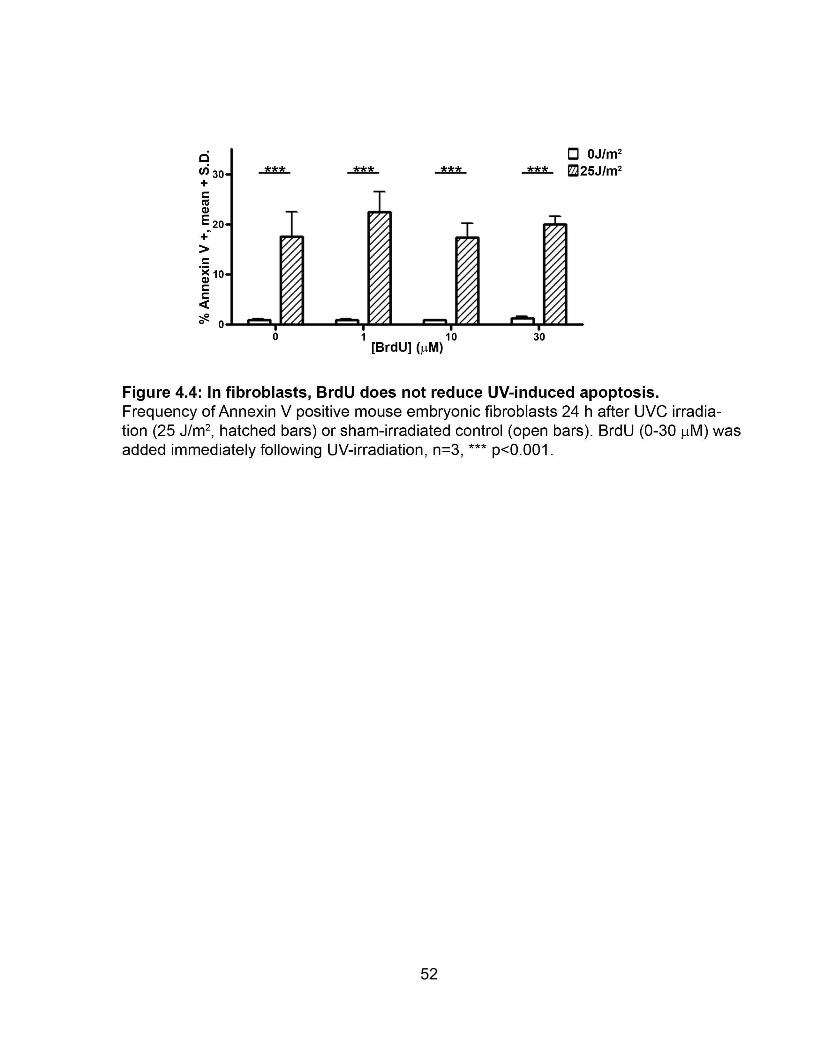
52

4.4 Chemical specificity of BrdU protection
To determine if the protective effect of BrdU could be extended to structurally or
functionally related molecules the potential neuroprotective effect of DNA nucleosides
and halogenated thymidine analogues was assessed. In order to evaluate the chemical
specificity of BrdU protection, UV-irradiated neurons (10 J/m2) were incubated with
equimolar concentrations (30 µM) of thymidine or its halogenated analogues (CldU or
IdU) for 24 h. Immunocytochemistry with TUNEL and βIIITub was used to evaluate
neuronal apoptosis. Qualitatively, there appears to be a reduction in TUNEL positive
neurons when UV-irradiated cells were incubated with thymidine or its analogues, BrdU,
CldU or IdU (Fig. 4.5A-E). Since thymidine is a DNA nucleoside, UV-irradiated neurons
were also incubated with equimolar concentrations (30 µM) of the other DNA
nucleosides (adenosine, cytidine or guanosine) as well as a mix of the four DNA
nucleosides (dNT, 30 µM each). Qualitatively, the other nucleosides as well as a mix of
the four nucleosides also appeared to reduce UV-induced apoptosis to varying degrees
(Fig. 4.5F-I) but none were as potent as thymidine and its analogues. Although Fig.
4.5H indicates very few apoptotic neurons when UV-irradiated neurons are incubated
with guanosine, the number of neurons at 24 h of guanosine treatment is lower
suggesting that some neurons have lost adherence and washed away which may occur
when neurons are undergoing cell death.
53
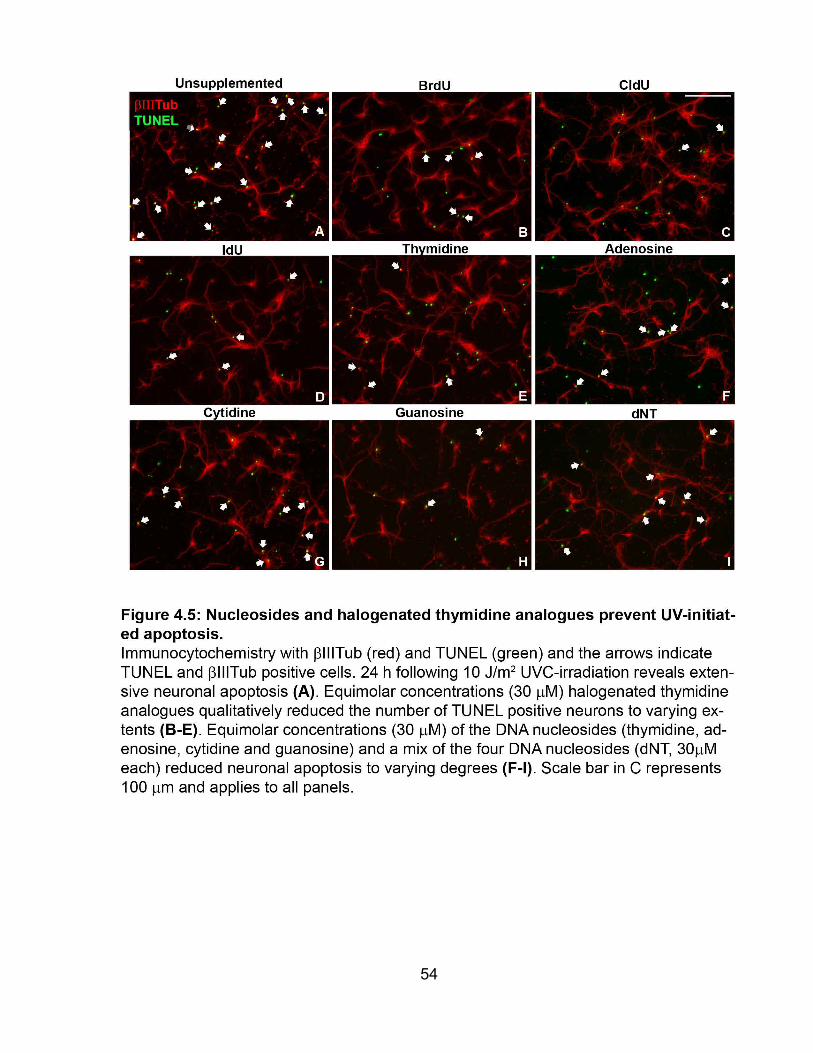
54

4.5 The UV-initiated increase of p53 is unaltered by BrdU
A common cellular response to stress including DNA damage is an up-regulation
of p53 (Lanni et al., 2012). UV-induced cell signaling in cortical neurons was determined
by examining p53 expression using immunocytochemistry. There was no p53
expression in unirradiated neurons (Fig. 4.6A) and 1h after UV-irradiation in the
absence or presence of BrdU (10 µM, Fig. 4.6B-C) but there was an increase in p53
expression as early as 4 hours following UV-irradiation (Fig. 4.6D-E) in both the
absence and presence of BrdU. p53 expression continued to increase 8 (Fig. 4.6F-G)
and 12 (Fig. 4.6H-I) hours in UV-irradiated neurons regardless of BrdU incubation.
There appears to be no difference in p53 expression following UV-irradiation with or
without BrdU. Thus, the neuroprotective activation of BrdU is not due to a change in p53
accumulation.
55
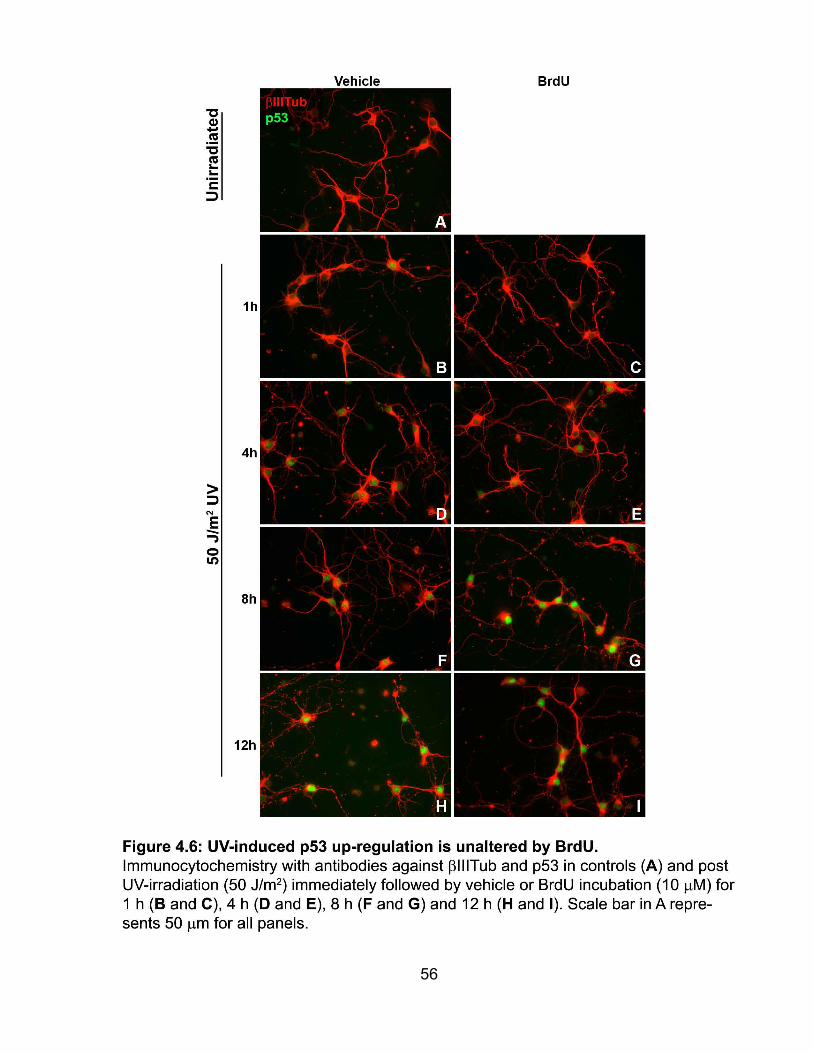
56

4.6 BrdU does not alter UV DNA lesion removal
The removal of UV DNA lesions over time was examined in cortical neurons to
determine whether BrdU incubation alters DNA repair. DNA lesions (6-4PP and CPD)
were measured using an ELISA. Figure 4.7A and B show the removal of the 6-4PP
lesion over time following 50 or 100 J/m2 UV-irradiation respectively whereas figures
4.7C and D show the removal of the CPD lesion over time following 50 or 100 J/m2 UV-
irradiation respectively. There was a significant reduction in 6-4PP lesions 4 hours after
UV-irradiation. Although BrdU appears to delay or stop repair of 6-4PP and CPD
lesions, the removal of 6-4PP and CPD lesions following UV-irradiation was not
statistically altered with BrdU incubation. Thus, BrdU does not protect UV-irradiated
neurons by improving the global genome repair (GGR) of UV lesions.
57

58

4.7 Xpa-/- and Csb-/- neurons are resistant to UV-initiated death
DNA lesions created by UVC-irradiation are primarily repaired by the NER
pathway. To determine the role of NER in UV-initiated neuronal death, two mouse
models of NER were used. Xpa-/- neurons are deficient in NER throughout the genome
while Csb-/- neurons lack NER of transcribed genes. Since MEFs from Xpa-/- and Csb-/-
mice and pluripotent embryonic stem cells from Xpa-/- mice are hypersensitive to UV (de
Vries et al., 1995; de Waard et al., 2008; Nakane et al., 1995), we hypothesized that
Xpa-/- and Csb-/- neurons would also be hypersensitive to UV. Wild type, Xpa-/- and Csb-
/- neurons were UV-irradiated at several doses and neuronal apoptosis was quantified
18 hours later using immunocytochemistry with βIIITub and cleaved caspase-3. Xpa-/-
and Csb-/- neurons were UV-irradiated with a lower dose (2 J/m2 UV) as well as higher
doses since other cell types of Xpa-/- and Csb-/- mice are hypersensitive to DNA damage
(de Waard et al., 2008). Immunocytochemistry images of UV-irradiated (0, 2, 10 and 50
J/m2) Xpa-/- and Csb-/- neurons are shown in figures 4.8A-D and 4.8E-H respectively.
Cells were classified as apoptotic neurons if they were positive for both cleaved
caspase-3 and βIIITub staining using automated image analysis software Metamorph.
As shown previously, UV-irradiation (50 J/m2) significantly increased apoptosis in wild
type neurons (Fig. 4.8I). In stark contrast to wild type, UV did not increase apoptosis in
Xpa-/- or Csb-/- neurons. Neurons deficient in NER, the DNA repair pathway that repairs
UV lesions are paradoxically protected from UV-initiated apoptosis at this 18 h time
point.
As a complementary measure of neuronal death following UV-irradiation, the
number of neurons remaining per unit area 18 h after UV was calculated since neurons
lose their adherence to coverslips when they undergo cell death. Figure 4.8J represents
59

the number of neurons remaining per mm2 18 hours following UV treatment for wild
type, Xpa-/- and Csb-/- neurons. The number of wild type neurons per unit area
significantly decreased following UV-irradiation (10 and 50 J/m2). However, the number
of Xpa-/- neurons per unit area following UV-irradiation is unaltered and the number of
Csb-/- neurons is significantly decreased at the highest UV dose (50J/m2) in comparison
to unirradiated control. When the number of neurons per unit area is compared among
the three genotypes, there was a significant reduction in the number of wild type
neurons but not Xpa-/- and Csb-/- neurons remaining at both 10 and 50 J/m2 UV. These
results further support the resistance of Xpa-/- and Csb-/- neurons to UV-induced
apoptosis.
4.7.1 BrdU does not alter caspase-3 in Xpa-/- and Csb-/- neurons
In wild type neurons, BrdU protected against UV-induced apoptosis (Fig. 4.9A-
B). BrdU incubation (10 µM) did not change the fraction of cleaved caspase-3 positive
neurons or the number of cells per unit area for both Xpa-/- and Csb-/- neurons (Fig.
4.9C-F) following UV irradiation. Therefore, BrdU prevents UV-induced apoptosis in wild
type neurons but is ineffective in Xpa-/- and Csb-/- neurons at this time point since they
are already UV-resistant.
60

61

62

4.8 Influence of BrdU on neuronal cell cycle
4.8.1 BrdU does not alter neuronal DNA content
To study whether UV-irradiation induces neuronal cell cycle re-entry, neuronal
DNA content was quantified. UV-irradiated cortical neurons were incubated with or
without BrdU and harvested at 0, 8 and 12 h. Neurons were stained with βIIITub and
Hoechst (DNA stain), imaged at identical exposure times and Hoechst intensity was
quantified using Metamorph. There was a statistically significant increase in Hoechst
intensity of cortical neurons 12 hours following UV-irradiation (50 J/m2) in the absence
and presence of BrdU (10 µM) and a significant increase 18 hours following UV-
irradiation without BrdU in comparison to unirradiated neurons (Fig. 4.10). However,
there was a significant reduction in the Hoechst intensity of UV-irradiated neurons
incubated with BrdU for 18 hours in comparison to unirradiated neurons as well as
neurons 12 hours following UV-irradiation (with and without BrdU) and neurons 18
hours after UV-irradiation. Furthermore, Hoechst intensity was variable following UV-
irradiation and there was a significant reduction in the coefficient of variation of Hoechst
staining 18 hours after UV-irradiation with BrdU in comparison to 12 hours after UV-
irradiation. Although there are statistically significant changes in DNA intensity of
cortical neurons after UV-irradiation the increase is only 5% which may not be
biologically significant.
63

64

4.8.2 Neither UV-irradiation nor BrdU incubation alter cell cycle distribution
As an alternative measure of neuronal cell cycle distribution, neuronal DNA
content was measured using flow cytometry with propidium iodide (DNA content) and
βIIITub. Cortical neurons were harvested 12 hours following treatment and analyzed
using flow cytometry. DNA content of unirradiated neurons, etoposide treated neurons
(1 µM, positive control), and UV-irradiated neurons (50 J/m2) in the absence and
presence of BrdU (10 µM) was analyzed. The percentage of neurons in the G0/G1
phase, S phase and G2 phase of the cell cycle was determined using the Watson
Pragmatic model (Table 4.1). The majority of the neurons in all treatments were in the
G0/G1 phase and only a very small percentage (less than 3%) were in the S phase or G2
phase. There was no significant difference in the percentage of neurons in G0/G1 phase,
S phase or G2 phase in comparison to unirradiated control neurons. These results
indicate that cortical neurons do not re-enter the S phase of the cell cycle following UV-
irradiation in our model.
Table 4.1: Flow cytometric analysis of cell cycle phase in DNA-damaged cortical neurons Sample G0/G1 mean (S.D.) S phase mean
(S.D.) G2 mean (S.D.)
Unirradiated 92% (3.1) 2.2% (1.0) 2.8% (0.4) Etoposide (1µM) 89% (1.2) 1.8% (0.4) 1.7% (0.5) 50J/m2 UV 97% (2.0) 1.5% (0.5) 1.3% (1.0) 50J/m2 UV + BrdU 98% (1.9) 1.1% (0.1) 1.3% (0.2)
4.8.3 UV does not induce cell cycle markers in neurons
UV-induced neuronal death can be rescued by using the Cdk inhibitors
flavopiridol and olomoucine (Roos and Kaina, 2012). Flavopiridol inhibits Cdk1/2/4
whereas olomoucine inhibits Cdk1/2/5 and ERK-1/MAP-1 kinases (extracellular signal-
regulated-1/mutagen-activated protein kinases) (Bakkenist and Kastan, 2003; Bartek 65

and Lukas, 2003; Byun et al., 2005; Roos and Kaina, 2012). In addition, the dominant-
negative kinase-inactive forms of both Cdk4 and Cdk6 reduced UV-induced neuronal
death (Park et al., 1998a). Since Cdk4 and Cdk6 are involved in the cell cycle
progression through the G1 phase (Liu et al., 2010), we analyzed Cdk4 and Cdk6
expression following UV-irradiation in the presence and absence of UV. Ki67 expression
was also analyzed since it is expressed during G1, S, G2 and mitosis but is absent
during G0 (Sinha and Hader, 2002). Furthermore, Ki67 expression has been used to
verify neuronal cell cycle re-entry after hydrogen peroxide treatment (Schwartz et al.,
2007). In order to verify that UV-irradiation does not induce neuronal cell cycle re-entry
in our model, we determined whether UV-irradiation or BrdU incubation changes cell
cycle marker expression. MEFs were used as a positive control to determine the quality
of Cdk4 and Cdk6 antibodies (Fig. 4.11A and B respectively). Neither UV-irradiation nor
BrdU altered Cdk4 (Fig. 4.11C-H), Cdk6 (Fig. 4.11I-N) and Ki67 (Fig. 4.11O-T)
expression. These results support our previous findings that neither UV-irradiation nor
BrdU induce neuronal cell cycle re-entry in our model.
66

67

4.9 BrdU does not alter neuronal global DNA methylation
BrdU can cause global DNA demethylation in neural stem cells (Schneider and
d'Adda di Fagagna, 2012). To investigate whether BrdU caused neuronal DNA
demethylation, global DNA methylation, using a 5-methylcytosine antibody against
isolated genomic DNA, was measured at 6, 8 and 12 h. The positive control is
proprietary and was provided by the manufacturer of the global DNA methylation kit.
There was no change in neuronal global DNA methylation levels in UV-irradiated
neurons in the absence or presence of BrdU (Fig. 4.12).
68

69

5. DISCUSSION
5.1 Characterization of BrdU protection
The initial goal of this project was to study the neuronal DNA damage response,
in particular the DNA damage response to bulky DNA adducts that are primarily
repaired by the nucleotide excision repair pathway. An interesting neuronal death
mechanism and response to other forms of DNA damage (e.g. double stranded breaks)
involves neuronal cell cycle re-entry (Kruman et al., 2004) that precedes apoptosis. In
order to determine whether bulky DNA adducts trigger neuronal cell cycle re-entry,
cortical neurons were UVC-irradiated and incubated with the cell cycle marker BrdU for
24 hours. Since BrdU is a thymidine analogue, it is incorporated into DNA during
replication and can be used as a maker for the S phase of the cell cycle. Interestingly,
UV-irradiated neurons incubated with BrdU as low as 100 nM had significantly lower
levels of apoptosis than UV-irradiated neurons. The most common application of BrdU
is as an S phase marker but the absence of detectable BrdU labeling in UV-irradiated
cortical neurons suggested that the protective mechanism of BrdU is not due to its
incorporation into new DNA during DNA replication. Thus, we concluded that BrdU
prevents UV-induced apoptosis through an alternative mechanism.
Neurons are post-mitotic cells that have exited the cell cycle. In order to
determine whether BrdU protection was specific to neurons or could also be extended
to proliferating cell types, we assessed BrdU protection in primary MEFs and observed
a lack of protection. These observations support the concept that BrdU protection is
specific to post-mitotic cells and thus the mechanism of protection may involve features
of the DNA damage response that are unique to post-mitotic cells. Qualitative
observations indicated that UV induced neuronal apoptosis could be reduced by
70

thymidine or halogenated thymidine analogues and not only BrdU. Thus, it appears that
several chemical structures similar to thymidine may reduce UV-induced neuronal
apoptosis, which may provide clues to the mechanism of BrdU neuroprotection.
5.2 UV-induced cell signaling
One of the initial questions regarding the protective effect of BrdU was whether
BrdU prevented neurons from responding to UV-induced DNA damage through
canonical signaling. p53 expression was analyzed since it is generally required for
neuronal apoptosis following DNA damage (Lanni et al., 2012). Although p53 is
expressed at low levels in unstressed cells, when cells are stressed, p53 degradation is
reduced and p53 accumulates (Essmann and Schulze-Osthoff, 2012). Qualitative
observations indicated that after UV, BrdU-treated neurons increased p53 levels after
UV similar to vehicle-treated neurons. This suggests that the mechanism of BrdU
protection occurs after p53 activation or independent of p53 activation. p53 is activated
by one or more post-translational modifications including phosphorylation, acetylation,
methylation, ubiquitination or sumolation depending on the stress response (Harris and
Levine, 2005). However, our methodology looked at total p53 levels and did not
distinguish between post-translationally modified forms of p53.
5.3 UV-induced DNA repair
A rational approach to preventing UV-initiated apoptosis is to repair UV-initiated
DNA damage. Interestingly, a molecule related to thymidine, thymidine dinucleotide
(pTT) has been shown to improve the repair of UV lesions in part by activating p53 and
up-regulating gene products involved in DNA repair (Gilchrest and Eller, 1999). Since
the chemical structure of pTT is related to that of thymidine and BrdU, we postulated
71

that the neuroprotective action of BrdU may be similar to pTT. To measure the impact of
BrdU on NER, an ELISA was used to measure the abundance of UV DNA damage in
the form of 6-4PP and CPD lesions in DNA extracted from UV-irradiated neurons. A
limitation of this methodology is that a decrease in 6-4PP or CPD lesion level only
provides information about initial steps in the multistep NER pathway (Fig. 2.4).
Furthermore, the ELISA used to measure the removal of UV lesions does not
distinguish between UV lesions on transcribed genes and non-transcribed genes. Since
the transcribed genes are a small percentage of the genome (1-2%) (Elgar and Vavouri,
2008), GGR is measured by this methodology. If GGR is silenced due to differentiation,
there is a possibility that this assay may be reporting the repair of transcribed genes.
Whether, GGR is attenuated in our cultures and the sensitivity of this assay to report
TCR is unknown.
There was a significant reduction in 6-4PP lesions 4 hours following UV-
irradiation compared to the amount of lesions immediately after UV-irradiation indicating
that post-mitotic neurons can repair this form of damage. This is in agreement with
previous results that showed a 40% removal of 6-4PP lesions in rat neurons four hours
after UV-irradiation using immunofluorescence (Yamamoto et al., 2007). Our results
were also consistent with these results and there was about a 40% reduction in 6-4PP
lesions in cortical neurons 4 hours after UV-irradiation. However, the level of CPD
lesions did not decline during the 8 hour time period. These findings are consistent with
previous reports that CPDs are repaired at a slower rate than 6-4PPs (de Laat et al.,
1999; van Hoffen et al., 1995). Although both UV lesions are repaired at the same rate
by TCR, 6-4PPs are repaired faster than CPDs by GGR in human skin (Nijhof et al.,
2007). In addition, GGR of CDPs is very low or absent in rodent epidermal cells (Nijhof
72

et al., 2007). Our results also confirmed that CPD lesions were repaired at a slower rate
or not repaired at all in murine cells.
There was no significant difference in the removal of UV lesions (6-4PP and
CPD) at 8 hours following UV-irradiation in the absence or presence of BrdU. However,
there appears to be trend toward inhibition or slowing of repair with BrdU. BrdU may be
preventing UV-induced apoptosis by inhibiting repair. Experiments with higher sensitivity
are required to determine whether BrdU inhibits DNA repair and if BrdU alters the repair
of transcribed genes. We concluded from these findings that BrdU does not improve
DNA repair in neurons and thus BrdU is preventing UV-induced neuronal apoptosis
through an alternative mechanism.
5.4 UV-induced cell signaling in Xpa-/- and Csb-/- neurons
To determine the role of NER in BrdU neuroprotection we utilized NER-deficient
neurons. We hypothesized that Xpa-/- and Csb-/- cortical neurons would be
hypersensitive to UV compared to wild type neurons since neurological dysfunction is
characteristic of patients with defective XPA or CSB (Nance and Berry, 1992; Robbins
et al., 1991). Furthermore, XPA and CSB patient fibroblasts, Xpa-/- MEFs and
pluripotent embryonic stem cells as well as Csb-/- MEFs and Xpa-/- and Csb-/- neural
stem cells are hypersensitive to UV in comparison to wild type controls (de Vries et al.,
1995; de Waard et al., 2008; Nakane et al., 1995; Sacco et al., (submitted)). Xpa-/-
cerebellar neurons are also more vulnerable to UV than wild type at 48 h (Enokido et
al., 1997). Surprisingly, our findings show that Xpa-/- and Csb-/- cortical neurons were
less sensitive to UV than their wild type counterparts at 18h. In light of the
neurodegeneration phenotype of patients with defective NER, our results suggest that
Xpa-/- neurons may have a delayed response to UV compared to wild type neurons.
73

Differences that might account for the contrasting findings between our study and
Enokido et al are the age of mice (embryonic vs. postnatal), brain region (cortical vs.
cerebellar neurons), media conditions and assessment of UV-induced apoptosis (18h
vs. 48h). A longer time course is necessary to determine whether Xpa-/- cortical neurons
have a delayed response to UV-irradiation relative to wild type neurons.
One possible explanation for the UV resistance observed in Xpa-/- neurons is that
these cells have defective DNA damage response signaling. UV-irradiation activates
ATR (Paulsen and Cimprich, 2007) which in turn phosphorylates Chk1 and p53 (Guo et
al., 2000; Hsu et al., 2007; Nouspikel, 2007). Expression of p53 increases after UV DNA
damage (Lanni et al., 2012) as we observed. In support of the importance of functional
NER in signaling, NER is required for UV-induced Chk1 and p53 phosphorylation in
non-proliferating fibroblasts (Marini et al., 2006). Chk1 and p53 are phosphorylated after
UV in fibroblasts from normal donors and CS patients (defective CSA or CSB) but are
not phosphorylated in fibroblasts from patients defective in XPA (Marini et al., 2006). A
limitation of this study is that phosphorylated Chk1 and p53 were only measured at one
time point (1 h) after UV in non-proliferating Xpa-/- human fibroblasts (Marini et al.,
2006). These results suggest that non-proliferating cells require NER to recognize and
start processing the UV DNA lesion in order to activate a cellular stress response that
results in the activation of Chk1 and p53 (Marini et al., 2006). Similarly, it is possible that
in Xpa-/- cortical neurons, Chk1 and p53 are not phosphorylated after UV, resulting in a
delay or absence of UV-induced apoptosis and this is currently being assessed
experimentally. It is also possible that UV-induced Chk1 and p53 activation could occur
at a later time point through an alternative pathway that is NER-independent.
74

Another explanation for the UV resistance of Xpa-/- neurons could be an
alternative role of NER factors. Recent studies have shown that XPA, XPC, XPG and
XPF are recruited to initiate gene expression by RNA polymerases I and II-dependent
genes in the absence of DNA damage (Le May et al., 2010). Therefore, the absence of
gene expression in NER-null neurons that requires XPA may prevent UV-induced
apoptosis.
5.5 Neuronal cell cycle re-entry
CDK inhibitors have been shown to prevent UV-induced death suggesting that
UV-irradiation may cause neuronal cell cycle re-entry (Park et al., 1998a). BrdU inhibits
ribonucleotide reductase (Meuth and Green, 1974) which may prevent the production of
dNTPs that may be necessary for neuronal cell cycle re-entry. Since BrdU has been
shown to impair cell cycle progression in proliferating cells (Levkoff et al., 2008), BrdU
may be preventing UV-induced neuronal apoptosis by inhibiting neuronal cell cycle re-
entry. In contrast to previous studies which showed that DNA damage increased
neuronal DNA content (Kruman et al., 2004), UV-irradiation did not increase neuronal
DNA content in our cultures. We used two methodologies to analyze neuronal DNA
content. In the first methodology we analyzed neuronal DNA intensity using Hoechst
staining with immunocytochemistry. Hoechst is a DNA stain that binds to the minor
groove of the DNA helix (Garner, 2009). Although there was a significant increase in
neuronal Hoechst intensity 12 to 18 hours after UV-irradiation the increase was
approximately 5% which may not be biologically significant. However, the change in
neuronal Hoechst intensity could suggest chromatin remodeling after UV-irradiation.
Chromatin remodeling involving decondensation occurs early during the DNA damage
response. DNA damage appears to initiate chromatin decondensation by decreasing
75

the DNA double helix torsional strain (Kielbassa et al., 1997). On the other hand, UV-
induced chromatin condensation is a late DNA damage response and results in
apoptosis. For example, UV-induced chromatin condensation resulting in apoptosis was
found in both mouse microglial cells and human keratinocyte cell line (Bjelland and
Seeberg, 2003; Breen and Murphy, 1995; Friedberg, 2003). Chromatin condensation
results in higher intensity Hoechst staining and is often used to examine chromatin
structure. (Cheng et al., 1978; Kuraoka et al., 2000; Robbins et al., 1983).
UV-induced chromatin remodeling is important for DNA repair enzymes to
access the lesions that need to be repaired (Jiang et al., 2010). In chromatin, DNA is
wrapped around an octomer of histones which blocks DNA repair enzymes from UV-
induced DNA lesions. There are several mechanisms in place to make DNA more
accessible including ATP-dependent chromatin remodeling complexes, incorporation of
histone variants into the nucleosome and covalent histone modifications (Bjelland and
Seeberg, 2003). There is a possibility that the change in neuronal DNA intensity we
observed is caused by UV-induced chromatin remodeling. However, the changes we
observed were small and additional experiments are required to determine whether UV-
irradiation and/or BrdU changes chromatin remodeling.
In the second methodology we examined neuronal cell cycle distribution using
flow cytometry with PI. We examined the cell cycle distribution 12 hours after UV-
irradiation because the Hoechst methodology indicated that changes in DNA content
may occur as early as 12 hours after UV and remains consistent 18 hours after UV-
irradiation. Over 97% of the neurons were in the G0/G1 phase of the cell cycle and there
was no significant difference between any of our treatments. Etoposide was used as a
positive control because it was shown to increase the percentage of S-phase cortical
76

neurons (from ~3% to over 30%) (Kruman et al., 2004). Surprisingly, etoposide
treatment did not induce neuronal cell cycle re-entry and increase the number of
neurons in the S phase of the cell the cycle in our cultures. Although we used the same
concentration of etoposide (1 µM) as the previous study, we analyzed the cells after 12
hours whereas the previous study analyzed the cells 20 hours after treatment.
Furthermore, the previous study used rat cortical neurons whereas we used mouse
cortical neurons. Thus, additional experiments are required to analyze cell cycle re-entry
in neurons 20 hours after etoposide treatment. Nevertheless, these results further
confirm that neither UV-irradiation nor BrdU incubation alters neuronal DNA content and
that in our cultures, UV-irradiation does not induce neurons to enter the S phase of the
cell cycle.
Cell cycle markers were unaltered in cortical neurons following UV-irradiation
suggesting that UV-irradiation does not induce neuronal cell cycle re-entry in our
cultures. Ki67 is a nuclear antigen that is expressed during G1, S, G2 and mitosis but is
absent during G0 (Sinha and Hader, 2002). Neuronal cell cycle re-entry has been
verified using Ki67 in previous studies where neurons were treated with hydrogen
peroxide (Schwartz et al., 2007; Tomashevski et al., 2010). Although there were several
Ki67 positive neurons in all treatments, the majority of the neurons were Ki67 negative
at several time points following UV-irradiation. Cdk4 and Cdk6 expression in cortical
neurons was also analyzed because they are involved in the progression of the G1
phase in the cell cycle (Caporali et al., 2004). In addition, dominant-negative kinase-
inactive forms of Cdk4 and Cdk6 expression have reduced UV-induced neuronal death
as well as B-amyloid induced neuronal death (Giovanni et al., 1999; Park et al., 1998a).
Similarly to Ki67 findings, there were no differences in expression of Cdk4 or Cdk6 with
77

UV-irradiation or with BrdU incubation. These findings confirmed that neuronal cell cycle
re-entry does not occur following UV-irradiation in our cultures.
5.6 Neuronal DNA methylation
A recent study showed that proliferation of neural precursors (NPCs) decreases
when NPCs are incubated with BrdU (Schneider and d'Adda di Fagagna, 2012). BrdU-
treated neural stem cells (NSCs) lose expression of stem cell markers including Nestin,
Sox2 and Pax6. Furthermore, after BrdU treatment there was an up-regulation of GFAP,
an astrocytic marker (Schneider and d'Adda di Fagagna, 2012). Interestingly, the
increase in GFAP expression coincided with a decrease in DNA methyltransferases as
well as a reduction in global DNA CpG methylation (Schneider and d'Adda di Fagagna,
2012). However, in our cultures, global DNA methylation was unaltered after UV-
irradiation as well as with BrdU incubation. Since differentiation has been associated
with DNA demethylation (Singh et al., 2009), cortical neurons may already have low
levels of DNA methylation in comparison to stem cells. There is a possibility that BrdU
incubation after UV-irradiation does alter DNA methylation in cortical neurons but the
change may be small and/or gene-specific that it is undetectable with the methodology
we used. Therefore, a more in depth study is required to determine if BrdU changes
neuronal DNA methylation as well as whether it changes expression of DNA
methyltransferases.
78

6. CONCLUSIONS AND FUTURE DIRECTIONS UV damage causes neuronal apoptosis. Although neurons are not exposed to
UV radiation, UV-irradiation creates bulky DNA lesions that are representative of a
wide-range of lesions including chemotherapeutic agents. UV-induced neuronal
apoptosis can be prevented with BrdU concentrations as low as 100 nM, two orders of
magnitude below concentrations routinely used to label DNA synthesis. However, BrdU
does not protect proliferating cells such as fibroblasts from UV-induced apoptosis.
Therefore, BrdU likely protects cells from DNA damage-induced death through a
mechanism that is specific to neurons and perhaps other post-mitotic cells.
Furthermore, thymidine and other thymidine analogues may also be neuroprotective
suggesting that other structures similar to thymidine may also be neuroprotective. Since
thymidine may also be protective, the neuroprotective mechanism of BrdU may be an
interaction with a protein or a pathway that interacts with endogenous thymidine; the
bromine in BrdU is not essential for neuroprotection. In the current study, UV-irradiation
did not cause mouse cortical neurons to re-enter the cell cycle and the majority of
neurons remained in the G0 phase of the cell cycle regardless of the absence or
presence of BrdU.
BrdU reduces cleaved caspase-3 as well as phosphatidylserine exposure on the
cell surface in neurons after UV-irradiation. Since phosphatidylserine is exposed on the
cell surface after caspase-3 is cleaved (Niu and Chen, 2010), BrdU protects neurons
from apoptosis upstream of caspase-3 cleavage. UV-induced p53 expression occurs
after UV-irradiation in the absence and presence of BrdU. Therefore, BrdU likely
protects neurons downstream of p53 accumulation and up stream of caspase-3
activation.
79

Many aspects of BrdU neuroprotection remain to be clarified. Future investigation
is required to determine if BrdU following UV-irradiation delays neuronal apoptosis or if
neurons are protected indefinitely. Furthermore, the mechanism of BrdU protection
remains to be elucidated. Further examination of UV-induced cell signaling pathways,
specifically expression of proteins downstream of p53 and upstream of caspase-3 such
as activation of Bax and release of cytochrome c from the mitochondria, should provide
insight into the mechanism of BrdU protection.
The most surprising and definitive finding in the current study is that NER-null
neurons are resistant to UV-initiated apoptosis. UV-induced apoptosis occurs by UV
DNA damage activating ATR (Paulsen and Cimprich, 2007) which phosphorylates Chk1
and p53 (Guo et al., 2000) resulting in apoptosis. In non-proliferating fibroblasts,
functional NER is required for Chk1 and p53 phosphorylation (Marini et al., 2006). It has
been suggested that in proliferating cells the stalling of the replication fork and RPA on
single-stranded DNA recruits ATR and is an early trigger for the DNA damage response
(Byun et al., 2005). In neurons and post-mitotic cells, replication fork stalling cannot be
the trigger for ATR. Instead functional NER may be required to activate the DNA
damage response. Additional experiments are required to investigate the protective
mechanism of UV-resistance in NER-null neurons. Specifically, whether Chk1 and p53
are phosphorylated in NER-null neurons is an important area of future study.
About 20% percent of patients without functional NER have neurological
symptoms. Therefore, functional NER is important in neurons. Most of the neurological
symptoms in patients can be credited to progressive neurodegeneration (Rapin et al.,
2000). Thus, additional experiments are required to determine whether UV-irradiated
80

neurons without functional NER have a delayed response to DNA damage and undergo
death through an alternative mechanism.
Understanding the neuroprotective mechanism of BrdU can provide insight into
designing drugs to prevent neuronal DNA-damage induced apoptosis. Contrary to
expectations, NER deficient neurons are resistant to UV-induced apoptosis.
Understanding the role of NER in UV-induced apoptosis should provide insight into the
neuronal DNA damage response.
81

7. REFERENCES
Ananthaswamy, H.N., and W.E. Pierceall. 1990. Molecular mechanisms of ultraviolet radiation carcinogenesis. Photochemistry and photobiology. 52:1119-1136.
Andrews, A.D., S.F. Barrett, and J.H. Robbins. 1978. Xeroderma pigmentosum neurological abnormalities correlate with colony-forming ability after ultraviolet radiation. Proceedings of the National Academy of Sciences of the United States of America. 75:1984-1988.
Ashman, C.R., and R.L. Davidson. 1981. Bromodeoxyuridine mutagenesis in mammalian cells is related to deoxyribonucleotide pool imbalance. Molecular and cellular biology. 1:254-260.
Ashman, C.R., G.P. Reddy, and R.L. Davidson. 1981. Bromodeoxyuridine mutagenesis, ribonucleotide reductase activity, and deoxyribonucleotide pools in hydroxyurea-resistant mutants. Somatic cell genetics. 7:751-768.
Bakkenist, C.J., and M.B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 421:499-506.
Bartek, J., and J. Lukas. 2003. DNA repair: Damage alert. Nature. 421:486-488. Beisker, W., and W.N. Hittelman. 1988. Measurement of the kinetics of DNA repair
synthesis after uv irradiation using immunochemical staining of incorporated 5-bromo-2'-deoxyuridine and flow cytometry. Experimental cell research. 174:156-167.
Bjelland, S., and E. Seeberg. 2003. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutation research. 531:37-80.
Breen, A.P., and J.A. Murphy. 1995. Reactions of oxyl radicals with DNA. Free radical biology & medicine. 18:1033-1077.
Brooks, P.J. 2002. DNA repair in neural cells: basic science and clinical implications. Mutation research. 509:93-108.
Brooks, P.J., D.S. Wise, D.A. Berry, J.V. Kosmoski, M.J. Smerdon, R.L. Somers, H. Mackie, A.Y. Spoonde, E.J. Ackerman, K. Coleman, R.E. Tarone, and J.H. Robbins. 2000. The oxidative DNA lesion 8,5'-(S)-cyclo-2'-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. The Journal of biological chemistry. 275:22355-22362.
Byun, T.S., M. Pacek, M.C. Yee, J.C. Walter, and K.A. Cimprich. 2005. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes & development. 19:1040-1052.
Cameron, H.A., and R.D. McKay. 2001. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol. 435:406-417.
Caporali, S., S. Falcinelli, G. Starace, M.T. Russo, E. Bonmassar, J. Jiricny, and S. D'Atri. 2004. DNA damage induced by temozolomide signals to both ATM and ATR: role of the mismatch repair system. Molecular pharmacology. 66:478-491.
Cavanagh, B.L., T. Walker, A. Norazit, and A.C. Meedeniya. 2011. Thymidine analogues for tracking DNA synthesis. Molecules. 16:7980-7993.
Cheng, W.S., R.E. Tarone, A.D. Andrews, J.S. Whang-Peng, and J.H. Robbins. 1978. Ultraviolet light-induced sister chromatid exchanges in xeroderma pigmentosum and in Cockayne's syndrome lymphocyte cell lines. Cancer research. 38:1601-1609.
82

Ciccia, A., and S.J. Elledge. 2010. The DNA damage response: making it safe to play with knives. Molecular cell. 40:179-204.
Coller, H.A. 2007. What's taking so long? S-phase entry from quiescence versus proliferation. Nature reviews. Molecular cell biology. 8:667-670.
Cooper-Kuhn, C.M., and H.G. Kuhn. 2002. Is it all DNA repair? Methodological considerations for detecting neurogenesis in the adult brain. Brain Res Dev Brain Res. 134:13-21.
Copani, A., F. Caraci, J.J. Hoozemans, M. Calafiore, M.A. Sortino, and F. Nicoletti. 2007. The nature of the cell cycle in neurons: focus on a "non-canonical" pathway of DNA replication causally related to death. Biochimica et biophysica acta. 1772:409-412.
Cory, J.G., and A. Sato. 1983. Regulation of ribonucleotide reductase activity in mammalian cells. Molecular and cellular biochemistry. 53-54:257-266.
de Laat, W.L., N.G. Jaspers, and J.H. Hoeijmakers. 1999. Molecular mechanism of nucleotide excision repair. Genes & development. 13:768-785.
de Vries, A., C.T. van Oostrom, F.M. Hofhuis, P.M. Dortant, R.J. Berg, F.R. de Gruijl, P.W. Wester, C.F. van Kreijl, P.J. Capel, H. van Steeg, and et al. 1995. Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature. 377:169-173.
de Waard, H., E. Sonneveld, J. de Wit, R.E. Lange, J.H. Hoeijmakers, H. Vrieling, and G.T. van der Horst. 2008. Cell-type-specific consequences of nucleotide excision repair deficiencies: Embryonic stem cells versus fibroblasts. DNA repair.
Diermeier, S., E. Schmidt-Bruecken, M. Kubbies, L.A. Kunz-Schughart, and G. Brockhoff. 2004. Exposure to continuous bromodeoxyuridine (BrdU) differentially affects cell cycle progression of human breast and bladder cancer cell lines. Cell proliferation. 37:195-206.
Djordjevic, B., and W. Szybalski. 1960. Genetics of human cell lines. III. Incorporation of 5-bromo- and 5-iododeoxyuridine into the deoxyribonucleic acid of human cells and its effect on radiation sensitivity. The Journal of experimental medicine. 112:509-531.
Doetsch, F. 2003. The glial identity of neural stem cells. Nature neuroscience. 6:1127-1134.
Elgar, G., and T. Vavouri. 2008. Tuning in to the signals: noncoding sequence conservation in vertebrate genomes. Trends in genetics : TIG. 24:344-352.
Eller, M.S., N. Puri, I.M. Hadshiew, S.S. Venna, and B.A. Gilchrest. 2002. Induction of apoptosis by telomere 3' overhang-specific DNA. Experimental cell research. 276:185-193.
Eller, M.S., M. Yaar, and B.A. Gilchrest. 1994. DNA damage and melanogenesis. Nature. 372:413-414.
Enokido, Y., N. Inamura, T. Araki, T. Satoh, H. Nakane, M. Yoshino, Y. Nakatsu, K. Tanaka, and H. Hatanaka. 1997. Loss of the xeroderma pigmentosum group A gene (XPA) enhances apoptosis of cultured cerebellar neurons induced by UV but not by low-K+ medium. Journal of neurochemistry. 69:246-251.
Erikson, R.L., and W. Szybalski. 1963. Molecular Radiobiology of Human Cell Lines. V. Comparative Radiosensitizing Properties of 5-Halodeoxycytidines and 5-Halodeoxyuridines. Radiation research. 20:252-262.
Essmann, F., and K. Schulze-Osthoff. 2012. Translational approaches targeting the p53 pathway for anti-cancer therapy. British journal of pharmacology. 165:328-344.
83

Finch, R.A., M. Liu, S.P. Grill, W.C. Rose, R. Loomis, K.M. Vasquez, Y. Cheng, and A.C. Sartorelli. 2000. Triapine (3-aminopyridine-2-carboxaldehyde- thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochemical pharmacology. 59:983-991.
Friedberg, E.C. 2003. DNA damage and repair. Nature. 421:436-440. Garner, D.L. 2009. Hoechst 33342: the dye that enabled differentiation of living X-and
Y-chromosome bearing mammalian sperm. Theriogenology. 71:11-21. Gilchrest, B.A., and M.S. Eller. 1999. DNA photodamage stimulates melanogenesis and
other photoprotective responses. The journal of investigative dermatology. Symposium proceedings / the Society for Investigative Dermatology, Inc. [and] European Society for Dermatological Research. 4:35-40.
Giovanni, A., F. Wirtz-Brugger, E. Keramaris, R. Slack, and D.S. Park. 1999. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. The Journal of biological chemistry. 274:19011-19016.
Gobbel, G.T., M. Bellinzona, A.R. Vogt, N. Gupta, J.R. Fike, and P.H. Chan. 1998. Response of postmitotic neurons to X-irradiation: implications for the role of DNA damage in neuronal apoptosis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 18:147-155.
Goodman, M.F. 2002. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annual review of biochemistry. 71:17-50.
Goukassian, D.A., E. Helms, H. van Steeg, C. van Oostrom, J. Bhawan, and B.A. Gilchrest. 2004. Topical DNA oligonucleotide therapy reduces UV-induced mutations and photocarcinogenesis in hairless mice. Proceedings of the National Academy of Sciences of the United States of America. 101:3933-3938.
Gratzner, H.G. 1982. Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: A new reagent for detection of DNA replication. Science. 218:474-475.
Groves, M.D., M.H. Maor, C. Meyers, A.P. Kyritsis, K.A. Jaeckle, W.K. Yung, R.E. Sawaya, K. Hess, J.M. Bruner, P. Peterson, and V.A. Levin. 1999. A phase II trial of high-dose bromodeoxyuridine with accelerated fractionation radiotherapy followed by procarbazine, lomustine, and vincristine for glioblastoma multiforme. International journal of radiation oncology, biology, physics. 45:127-135.
Guo, Z., A. Kumagai, S.X. Wang, and W.G. Dunphy. 2000. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes & development. 14:2745-2756.
Gyi, K.K., and J.R. Wrathall. 1983. Histopathology and host response associated with reduced tumorigenicity of 5-bromodeoxyuridine--treated murine melanoma cells. The American journal of pathology. 110:135-147.
Harris, S.L., and A.J. Levine. 2005. The p53 pathway: positive and negative feedback loops. Oncogene. 24:2899-2908.
Hartwell, L.H., and T.A. Weinert. 1989. Checkpoints: controls that ensure the order of cell cycle events. Science. 246:629-634.
Hentati, F., C. Ben Hamida, M. Zeghal, M. Kamoun, B. Fezaa, and M. Ben Hamida. 1992. Age-dependent axonal loss in nerve biopsy of patients with xeroderma pigmentosum. Neuromuscular disorders : NMD. 2:361-369.
84

Hermann, A., H. Gowher, and A. Jeltsch. 2004. Biochemistry and biology of mammalian DNA methyltransferases. Cellular and molecular life sciences : CMLS. 61:2571-2587.
Herrup, K., R. Neve, S.L. Ackerman, and A. Copani. 2004. Divide and die: cell cycle events as triggers of nerve cell death. The Journal of neuroscience : the official journal of the Society for Neuroscience. 24:9232-9239.
Hess, M.T., U. Schwitter, M. Petretta, B. Giese, and H. Naegeli. 1997. Bipartite substrate discrimination by human nucleotide excision repair. Proceedings of the National Academy of Sciences of the United States of America. 94:6664-6669.
Hitomi, K., S. Iwai, and J.A. Tainer. 2007. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA repair. 6:410-428.
Hitomi, M., and D.W. Stacey. 2010. The checkpoint kinase ATM protects against stress-induced elevation of cyclin D1 and potential cell death in neurons. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 77:524-533.
Ho, L., and P.C. Hanawalt. 1991. Gene-specific DNA repair in terminally differentiating rat myoblasts. Mutation research. 255:123-141.
Hoeijmakers, J.H. 2009. DNA damage, aging, and cancer. The New England journal of medicine. 361:1475-1485.
Houtgraaf, J.H., J. Versmissen, and W.J. van der Giessen. 2006. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovascular revascularization medicine : including molecular interventions. 7:165-172.
Hsu, P.H., P.C. Hanawalt, and T. Nouspikel. 2007. Nucleotide excision repair phenotype of human acute myeloid leukemia cell lines at various stages of differentiation. Mutation research. 614:3-15.
Hsu, T.C., and C.E. Somers. 1961. Effect of 5-bromodeoxyuridine on mamalian chromosomes. Proceedings of the National Academy of Sciences of the United States of America. 47:396-403.
Jackson, D., and P.R. Cook. 2008. Analyzing DNA Replication I: Labeling Animals, Tissues, and Cells with Bromodeoxyuridine (BrdU). CSH protocols. 2008:pdb prot5031.
Jensen, L., and S. Linn. 1988. A reduced rate of bulky DNA adduct removal is coincident with differentiation of human neuroblastoma cells induced by nerve growth factor. Molecular and cellular biology. 8:3964-3968.
Jiang, Y., X. Wang, S. Bao, R. Guo, D.G. Johnson, X. Shen, and L. Li. 2010. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proceedings of the National Academy of Sciences of the United States of America.
Jiang, Z.G., M.S. Lebowitz, and H.A. Ghanbari. 2006. Neuroprotective activity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (PAN-811), a cancer therapeutic agent. CNS Drug Rev. 12:77-90.
Katiyar, S.K., M. Vaid, H. van Steeg, and S.M. Meeran. 2010. Green tea polyphenols prevent UV-induced immunosuppression by rapid repair of DNA damage and enhancement of nucleotide excision repair genes. Cancer prevention research. 3:179-189.
Kielbassa, C., L. Roza, and B. Epe. 1997. Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis. 18:811-816.
85

Kinsella, T.J., J.B. Mitchell, A. Russo, M. Aiken, G. Morstyn, S.M. Hsu, J. Rowland, and E. Glatstein. 1984. Continuous intravenous infusions of bromodeoxyuridine as a clinical radiosensitizer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2:1144-1150.
Kohji, T., M. Hayashi, K. Shioda, M. Minagawa, Y. Morimatsu, K. Tamagawa, and M. Oda. 1998. Cerebellar neurodegeneration in human hereditary DNA repair disorders. Neuroscience letters. 243:133-136.
Kraemer, K.H., M.M. Lee, and J. Scotto. 1987. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Archives of dermatology. 123:241-250.
Kruman, II. 2004. Why do neurons enter the cell cycle? Cell cycle. 3:769-773. Kruman, II, R.P. Wersto, F. Cardozo-Pelaez, L. Smilenov, S.L. Chan, F.J. Chrest, R.
Emokpae, Jr., M. Gorospe, and M.P. Mattson. 2004. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 41:549-561.
Kuhn, H.G., and C.M. Cooper-Kuhn. 2007. Bromodeoxyuridine and the detection of neurogenesis. Current pharmaceutical biotechnology. 8:127-131.
Kuraoka, I., C. Bender, A. Romieu, J. Cadet, R.D. Wood, and T. Lindahl. 2000. Removal of oxygen free-radical-induced 5',8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proceedings of the National Academy of Sciences of the United States of America. 97:3832-3837.
Kurz, E.U., and S.P. Lees-Miller. 2004. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA repair. 3:889-900.
Lanni, C., M. Racchi, M. Memo, S. Govoni, and D. Uberti. 2012. p53 at the crossroads between cancer and neurodegeneration. Free radical biology & medicine. 52:1727-1733.
Latonen, L., and M. Laiho. 2005. Cellular UV damage responses--functions of tumor suppressor p53. Biochimica et biophysica acta. 1755:71-89.
Le May, N., J.M. Egly, and F. Coin. 2010. True lies: the double life of the nucleotide excision repair factors in transcription and DNA repair. Journal of nucleic acids. 2010.
Lehner, B., B. Sandner, J. Marschallinger, C. Lehner, T. Furtner, S. Couillard-Despres, F.J. Rivera, G. Brockhoff, H.C. Bauer, N. Weidner, and L. Aigner. 2011. The dark side of BrdU in neural stem cell biology: detrimental effects on cell cycle, differentiation and survival. Cell and tissue research. 345:313-328.
Lennington, J.B., Z. Yang, and J.C. Conover. 2003. Neural stem cells and the regulation of adult neurogenesis. Reproductive biology and endocrinology : RB&E. 1:99.
Levkoff, L.H., G.P. Marshall, 2nd, H.H. Ross, M. Caldeira, B.A. Reynolds, M. Cakiroglu, C.L. Mariani, W.J. Streit, and E.D. Laywell. 2008. Bromodeoxyuridine inhibits cancer cell proliferation in vitro and in vivo. Neoplasia. 10:804-816.
Littlefield, J.W., and E.A. Gould. 1960. The toxic effect of 5-bromodeoxyuridine on cultured epithelial cells. The Journal of biological chemistry. 235:1129-1133.
Liu, D.X., and L.A. Greene. 2001. Neuronal apoptosis at the G1/S cell cycle checkpoint. Cell and tissue research. 305:217-228.
Liu, D.Z., B.P. Ander, and F.R. Sharp. 2010. Cell cycle inhibition without disruption of neurogenesis is a strategy for treatment of central nervous system diseases. Neurobiology of disease. 37:549-557.
86

Lu, T., Y. Pan, S.Y. Kao, C. Li, I. Kohane, J. Chan, and B.A. Yankner. 2004. Gene regulation and DNA damage in the ageing human brain. Nature. 429:883-891.
Lukas, J., C. Lukas, and J. Bartek. 2004. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA repair. 3:997-1007.
Maeda, T., M.S. Eller, M. Hedayati, L. Grossman, and B.A. Gilchrest. 1999. Enhanced repair of benzo(a)pyrene-induced DNA damage in human cells treated with thymidine dinucleotides. Mutation research. 433:137-145.
Malumbres, M., and M. Barbacid. 2001. To cycle or not to cycle: a critical decision in cancer. Nature reviews. Cancer. 1:222-231.
Marini, F., T. Nardo, M. Giannattasio, M. Minuzzo, M. Stefanini, P. Plevani, and M. Muzi Falconi. 2006. DNA nucleotide excision repair-dependent signaling to checkpoint activation. Proceedings of the National Academy of Sciences of the United States of America. 103:17325-17330.
Masterson, J.C., and S. O'Dea. 2007. 5-Bromo-2-deoxyuridine activates DNA damage signalling responses and induces a senescence-like phenotype in p16-null lung cancer cells. Anti-cancer drugs. 18:1053-1068.
McCombe, P., M. Lavin, and C. Kidson. 1976. Control of DNA repair linked to neuroblastoma differentiation. International journal of radiation biology and related studies in physics, chemistry, and medicine. 29:523-531.
McMurray, C.T. 2005. To die or not to die: DNA repair in neurons. Mutation research. 577:260-274.
Meissner, A., T.S. Mikkelsen, H. Gu, M. Wernig, J. Hanna, A. Sivachenko, X. Zhang, B.E. Bernstein, C. Nusbaum, D.B. Jaffe, A. Gnirke, R. Jaenisch, and E.S. Lander. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 454:766-770.
Meuth, M., and H. Green. 1974. Induction of a deoxycytidineless state in cultured mammalian cells by bromodeoxyuridine. Cell. 2:109-112.
Morris, E.J., E. Keramaris, H.J. Rideout, R.S. Slack, N.J. Dyson, L. Stefanis, and D.S. Park. 2001. Cyclin-dependent kinases and P53 pathways are activated independently and mediate Bax activation in neurons after DNA damage. The Journal of neuroscience : the official journal of the Society for Neuroscience. 21:5017-5026.
Moser, J., H. Kool, I. Giakzidis, K. Caldecott, L.H. Mullenders, and M.I. Fousteri. 2007. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Molecular cell. 27:311-323.
Munoz-Pinedo, C. 2012. Signaling pathways that regulate life and cell death: evolution of apoptosis in the context of self-defense. Advances in experimental medicine and biology. 738:124-143.
Myers, J.S., and D. Cortez. 2006. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. The Journal of biological chemistry. 281:9346-9350.
Nakane, H., S. Takeuchi, S. Yuba, M. Saijo, Y. Nakatsu, H. Murai, Y. Nakatsuru, T. Ishikawa, S. Hirota, Y. Kitamura, and et al. 1995. High incidence of ultraviolet-B-or chemical-carcinogen-induced skin tumours in mice lacking the xeroderma pigmentosum group A gene. Nature. 377:165-168.
Nance, M.A., and S.A. Berry. 1992. Cockayne syndrome: review of 140 cases. American journal of medical genetics. 42:68-84.
87

Niedernhofer, L.J. 2008. Nucleotide excision repair deficient mouse models and neurological disease. DNA repair. 7:1180-1189.
Nijhof, J.G., C. van Pelt, A.A. Mulder, D.L. Mitchell, L.H. Mullenders, and F.R. de Gruijl. 2007. Epidermal stem and progenitor cells in murine epidermis accumulate UV damage despite NER proficiency. Carcinogenesis. 28:792-800.
Niu, G., and X. Chen. 2010. Apoptosis imaging: beyond annexin V. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 51:1659-1662.
Nouspikel, T. 2007. DNA repair in differentiated cells: some new answers to old questions. Neuroscience. 145:1213-1221.
Nouspikel, T. 2008. Nucleotide excision repair and neurological diseases. DNA repair. 7:1155-1167.
Nouspikel, T., and P.C. Hanawalt. 2000. Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Molecular and cellular biology. 20:1562-1570.
Nouspikel, T., and P.C. Hanawalt. 2002. DNA repair in terminally differentiated cells. DNA repair. 1:59-75.
Nowakowski, R.S., S.B. Lewin, and M.W. Miller. 1989. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. Journal of neurocytology. 18:311-318.
Park, D.S., E.J. Morris, R. Bremner, E. Keramaris, J. Padmanabhan, M. Rosenbaum, M.L. Shelanski, H.M. Geller, and L.A. Greene. 2000. Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. The Journal of neuroscience : the official journal of the Society for Neuroscience. 20:3104-3114.
Park, D.S., E.J. Morris, L.A. Greene, and H.M. Geller. 1997. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 17:1256-1270.
Park, D.S., E.J. Morris, J. Padmanabhan, M.L. Shelanski, H.M. Geller, and L.A. Greene. 1998a. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. The Journal of cell biology. 143:457-467.
Park, D.S., E.J. Morris, L. Stefanis, C.M. Troy, M.L. Shelanski, H.M. Geller, and L.A. Greene. 1998b. Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress. The Journal of neuroscience : the official journal of the Society for Neuroscience. 18:830-840.
Paulsen, R.D., and K.A. Cimprich. 2007. The ATR pathway: fine-tuning the fork. DNA repair. 6:953-966.
Peng, D.F., H. Sugihara, and T. Hattori. 2001. Bromodeoxyuridine induces p53-dependent and -independent cell cycle arrests in human gastric carcinoma cell lines. Pathobiology : journal of immunopathology, molecular and cellular biology. 69:77-85.
Phillips, T.L., V.A. Levin, D.K. Ahn, P.H. Gutin, R.L. Davis, C.B. Wilson, M.D. Prados, W.M. Wara, and M.S. Flam. 1991. Evaluation of bromodeoxyuridine in glioblastoma multiforme: a Northern California Cancer Center Phase II study. International journal of radiation oncology, biology, physics. 21:709-714.
88

Poot, M., A. Schuster, and H. Hoehn. 1991. Cytostatic synergism between bromodeoxyuridine, bleomycin, cisplatin and chlorambucil demonstrated by a sensitive cell kinetic assay. Biochemical pharmacology. 41:1903-1909.
Prados, M.D., W. Seiferheld, H.M. Sandler, J.C. Buckner, T. Phillips, C. Schultz, R. Urtasun, R. Davis, P. Gutin, T.L. Cascino, H.S. Greenberg, and W.J. Curran, Jr. 2004. Phase III randomized study of radiotherapy plus procarbazine, lomustine, and vincristine with or without BUdR for treatment of anaplastic astrocytoma: final report of RTOG 9404. International journal of radiation oncology, biology, physics. 58:1147-1152.
Radman, M. 1975. SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic life sciences. 5A:355-367.
Rapin, I., Y. Lindenbaum, D.W. Dickson, K.H. Kraemer, and J.H. Robbins. 2000. Cockayne syndrome and xeroderma pigmentosum. Neurology. 55:1442-1449.
Rastogi, R.P., Richa, A. Kumar, M.B. Tyagi, and R.P. Sinha. 2010. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. Journal of nucleic acids. 2010:592980.
Reichard, P. 1988. Interactions between deoxyribonucleotide and DNA synthesis. Annual review of biochemistry. 57:349-374.
Robbins, J.H., R.A. Brumback, M. Mendiones, S.F. Barrett, J.R. Carl, S. Cho, M.B. Denckla, M.B. Ganges, L.H. Gerber, R.A. Guthrie, and et al. 1991. Neurological disease in xeroderma pigmentosum. Documentation of a late onset type of the juvenile onset form. Brain : a journal of neurology. 114 ( Pt 3):1335-1361.
Robbins, J.H., R.J. Polinsky, and A.N. Moshell. 1983. Evidence that lack of deoxyribonucleic acid repair causes death of neurons in xeroderma pigmentosum. Annals of neurology. 13:682-684.
Robertson, J.M., W.D. Ensminger, S. Walker, and T.S. Lawrence. 1997a. A phase I trial of intravenous bromodeoxyuridine and radiation therapy for pancreatic cancer. International journal of radiation oncology, biology, physics. 37:331-335.
Robertson, J.M., C.J. McGinn, S. Walker, M.V. Marx, M.L. Kessler, W.D. Ensminger, and T.S. Lawrence. 1997b. A phase I trial of hepatic arterial bromodeoxyuridine and conformal radiation therapy for patients with primary hepatobiliary cancers or colorectal liver metastases. International journal of radiation oncology, biology, physics. 39:1087-1092.
Roca, A.I., and M.M. Cox. 1997. RecA protein: structure, function, and role in recombinational DNA repair. Progress in nucleic acid research and molecular biology. 56:129-223.
Roos, W.P., and B. Kaina. 2006. DNA damage-induced cell death by apoptosis. Trends in molecular medicine. 12:440-450.
Roos, W.P., and B. Kaina. 2012. DNA damage-induced apoptosis: From specific DNA lesions to the DNA damage response and apoptosis. Cancer letters.
Ross, H.H., L.H. Levkoff, G.P. Marshall, 2nd, M. Caldeira, D.A. Steindler, B.A. Reynolds, and E.D. Laywell. 2008. Bromodeoxyuridine induces senescence in neural stem and progenitor cells. Stem cells. 26:3218-3227.
Ross, H.H., M. Rahman, L.H. Levkoff, S. Millette, T. Martin-Carreras, E.M. Dunbar, B.A. Reynolds, and E.D. Laywell. 2011. Ethynyldeoxyuridine (EdU) suppresses in vitro population expansion and in vivo tumor progression of human glioblastoma cells. Journal of neuro-oncology. 105:485-498.
89

Rottgen, V., and H.M. Rabes. 1989. Deoxyribonucleoside triphosphate pools in regenerating rat liver: effect of hydroxyurea and exogenous deoxypyrimidines. Biochimica et biophysica acta. 992:349-354.
Sacco, R., L. Tamblyn, N. Rajakulendran, F.N. Bralha, V. Tropepe, and R. Laposa. (submitted). Cockayne syndrome b maintains neural precursor function after replicative stress, againg and UV DNA damage. DNA repair.
Schneider, L., and F. d'Adda di Fagagna. 2012. Neural stem cells exposed to BrdU lose their global DNA methylation and undergo astrocytic differentiation. Nucleic acids research.
Schubert, D., and F. Jacob. 1970. 5-bromodeoxyuridine-induced differentiation of a neuroblastoma. Proceedings of the National Academy of Sciences of the United States of America. 67:247-254.
Schwartz, E.I., L.B. Smilenov, M.A. Price, T. Osredkar, R.A. Baker, S. Ghosh, F.D. Shi, T.L. Vollmer, A. Lencinas, D.M. Stearns, M. Gorospe, and Kruman, II. 2007. Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell cycle. 6:318-329.
Schwartz, G.K., and M.A. Shah. 2005. Targeting the cell cycle: a new approach to cancer therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 23:9408-9421.
Sengupta, S., and C.C. Harris. 2005. p53: traffic cop at the crossroads of DNA repair and recombination. Nature reviews. Molecular cell biology. 6:44-55.
Sherr, C.J., and J.M. Roberts. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & development. 13:1501-1512.
Singh, R.P., K. Shiue, D. Schomberg, and F.C. Zhou. 2009. Cellular epigenetic modifications of neural stem cell differentiation. Cell transplantation. 18:1197-1211.
Sinha, R.P., and D.P. Hader. 2002. UV-induced DNA damage and repair: a review. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology. 1:225-236.
Spyrou, G., and P. Reichard. 1988. Dynamics of the thymidine triphosphate pool during the cell cycle of synchronized 3T3 mouse fibroblasts. Mutation research. 200:37-43.
Stellwagen, R.H., and G.M. Tomkins. 1971. Differential effect of 5-bromodeoxyuridine on the concentrations of specific enzymes in hepatoma cells in culture. Proceedings of the National Academy of Sciences of the United States of America. 68:1147-1150.
Subrahmanyam, K., and K.S. Rao. 1991. Ultraviolet light-induced unscheduled DNA-synthesis in isolated neurons of rat brain of different ages. Mechanisms of ageing and development. 57:283-291.
Taddei, F., M. Vulic, M. Radman, and I. Matic. 1997. Genetic variability and adaptation to stress. Exs. 83:271-290.
Tamblyn, L., E. Li, H. Sarras, P. Srikanth, M.P. Hande, and J.P. McPherson. 2009. A role for Mus81 in the repair of chromium-induced DNA damage. Mutation research. 660:57-65.
Taupin, P. 2007. BrdU immunohistochemistry for studying adult neurogenesis: paradigms, pitfalls, limitations, and validation. Brain research reviews. 53:198-214.
90

Thelander, L., and P. Reichard. 1979. Reduction of ribonucleotides. Annual review of biochemistry. 48:133-158.
Tirode, F., D. Busso, F. Coin, and J.M. Egly. 1999. Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Molecular cell. 3:87-95.
Tomashevski, A., D.R. Webster, P. Grammas, M. Gorospe, and Kruman, II. 2010. Cyclin-C-dependent cell-cycle entry is required for activation of non-homologous end joining DNA repair in postmitotic neurons. Cell Death Differ.
van Hoffen, A., J. Venema, R. Meschini, A.A. van Zeeland, and L.H. Mullenders. 1995. Transcription-coupled repair removes both cyclobutane pyrimidine dimers and 6-4 photoproducts with equal efficiency and in a sequential way from transcribed DNA in xeroderma pigmentosum group C fibroblasts. The EMBO journal. 14:360-367.
Vrouwe, M.G., A. Pines, R.M. Overmeer, K. Hanada, and L.H. Mullenders. 2011. UV-induced photolesions elicit ATR-kinase-dependent signaling in non-cycling cells through nucleotide excision repair-dependent and -independent pathways. Journal of cell science. 124:435-446.
Wang, T.S., and K.T. Wheeler. 1978. Repair of X-ray-induced DNA damage in rat cerebellar neurons and brain tumor cells. Radiation research. 73:464-475.
Wang, W., B. Bu, M. Xie, M. Zhang, Z. Yu, and D. Tao. 2009. Neural cell cycle dysregulation and central nervous system diseases. Progress in neurobiology. 89:1-17.
Wu, S.C., and Y. Zhang. 2010. Active DNA demethylation: many roads lead to Rome. Nature reviews. Molecular cell biology. 11:607-620.
Yamamoto, A., Y. Nakamura, N. Kobayashi, T. Iwamoto, A. Yoshioka, H. Kuniyasu, T. Kishimoto, and T. Mori. 2007. Neurons and astrocytes exhibit lower activities of global genome nucleotide excision repair than do fibroblasts. DNA repair. 6:649-657.
Yang, Y., D.S. Geldmacher, and K. Herrup. 2001. DNA replication precedes neuronal cell death in Alzheimer's disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 21:2661-2668.
Yang, Y., and K. Herrup. 2007. Cell division in the CNS: protective response or lethal event in post-mitotic neurons? Biochimica et biophysica acta. 1772:457-466.
Ye, W., and S.W. Blain. 2010. S phase entry causes homocysteine-induced death while ataxia telangiectasia and Rad3 related protein functions anti-apoptotically to protect neurons. Brain : a journal of neurology. 133:2295-2312.
Zhang, Y., M. Parsanejad, E. Huang, D. Qu, H. Aleyasin, M.W. Rousseaux, Y.R. Gonzalez, S.P. Cregan, R.S. Slack, and D.S. Park. 2010. Pim-1 kinase as activator of the cell cycle pathway in neuronal death induced by DNA damage. Journal of neurochemistry. 112:497-510.
Zhang, Y., D. Qu, E.J. Morris, M.J. O'Hare, S.M. Callaghan, R.S. Slack, H.M. Geller, and D.S. Park. 2006. The Chk1/Cdc25A pathway as activators of the cell cycle in neuronal death induced by camptothecin. The Journal of neuroscience : the official journal of the Society for Neuroscience. 26:8819-8828.
91