mps iiia patients by the first quarter of 2018. lysogene ... · lysogene (lys.pa) initiation report...
TRANSCRIPT

Initiating CoverageApril 19, 2017
Lysogene (LYS.PA)Initiation Report
LifeSci Investment Abstract
Lysogene (Euronext Paris: LYS.PA) is a French biotechnology Company focused ondeveloping novel gene therapies for lysosomal storage disorders. The Company’s leadcandidate, LYS-SAF302, is an adeno-associated virus (AAV) capable of delivering a missinggene into patients with mucopolysaccharidosis type IIIA (MPS IIIA), an orphan indicationfor which there are no approved treatments. The Company recently went public throughan IPO on the Euronext Paris exchange and plans to initiate a pivotal Phase II/III trial inMPS IIIA patients by the first quarter of 2018. Lysogene is also pursing a treatment forGM1 gangliosidosis, another orphan indication, and may opt to leverage their platform andexpertise to move into additional rare CNS diseases.
Key Points of Discussion
■ Lysogene Plans to Initiate a Pivotal Study for MPS IIIA in the First Quarter of 2018.Lysogene’s lead program is a gene therapy for MPS IIIA, a lysosomal storage disorder withno approved therapies. Mucopolysaccharidosis type III is characterized by an inability tobreak down glycosaminoglycans (GAGs) in the lysosome due to an enzyme deficiency. Thetherapy uses an adeno-associated virus (AAV) to deliver the SGSH gene, which encodes themissing protein. The Company has demonstrated the safety and tolerability of the treatmentin a Phase I/II study and plans to launch a pivotal Phase II/III trial by the first quarterof 2018.
■ Developing Gene Therapies for Rare CNS Diseases. Lysogene intends to use its viralvector technology and expertise to address unmet needs in the treatment of lysosomalstorage disorders that have CNS involvement. The Company has chosen to first pursueMPS IIIA as well as GM1 gangliosidosis, but may expand their development pipeline intoother rare CNS diseases. The lack of approved therapies and potential for premium pricingin these orphan indications provide for an attractive market opportunity for the Company.
Expected Upcoming Milestones
■ Q4 2017 – IND submission for LYS-SAF302.■ Early 2018 – One-year data from natural history study for MPS IIIA.■ Q1 2018 – Launch of pivotal Phase II/III study for LYS-SAF302 in MPS IIIA.■ Q4 2018 – IND submission for LYS-GM101.■ Q1 2019 – Initiation of a Phase I/II study for LYS-GM101.■ Q1 2019 – Last patient enrolled into pivotal Phase II/III study for LYS-SAF302.■ Q2 2020 – Topline results from Phase II/III study for LYS-SAF302.■ Q3 2020 – Regulatory filings for LYS-SAF302 expected.
Analysts
David Sherman, Ph.D. (AC)(212) [email protected]
Market Data
Price $5.79Market Cap (M) $70EV (M) $42Shares Outstanding (M) 12.1Avg Daily Vol 2,09252-week Range: $5.78 - $8.01Cash (M) $28.9Net Cash/Share $2.33Annualized Cash Burn (M) $5.8Years of Cash Left >2.0Debt (M) $0.7
Financials
FY Dec 2014A 2015A 2016AEPS H1 NA NA NA
H2 NA NA NAFY (1.68)A (5.49)A NA
For analyst certification and disclosures please see page 33Page 1

▪ Lysogene Has Designed Pivotal Study Based on Feedback from US and EU Regulators. This pivotal Phase
II/III study is designed to evaluate the safety and efficacy of LYS-SAF302 in patients with MPS IIIA. The Company
has discussed this trial with US and EU regulators and it has been designed to satisfy the provided guidance. This
pivotal study will compare the disease progression of patients treated with LYS-SAF302 to a control cohort of patients
evaluated in the Company’s ongoing natural history study. The primary endpoint of this trial will be an assessment of
neurocognitive and motor development. Secondary endpoints are expected to include assessments of behavior, sleep,
quality of life, AEs, and MRI scans. The primary efficacy analysis will be conducted at 12 months with a secondary
efficacy analysis to follow at 24 months. Treated patients will be followed for a total of 5 years. Lysogene plans to
launch this study by the first quarter of 2018 and expects to enroll up to 20 patients. This could provide for a data
readout by the middle of 2020.
▪ Direct Delivery into the Brain May Be Critical. Lysogene has one primary competitor, Abeona Therapeutics
(NasdaqCM: ABEO), that is developing a competing AAV-based gene therapy to treat MPS IIIA. While the
companies have similar approaches, there are several key differences, particularly in terms of route of administration,
that may benefit Lysogene:
Direct Delivery into the Brain – Lysogene’s strategy involves the direct injection of an AAV encoding the
missing enzymes into the brain, compared with Abeona’s intravenous approach. Direct delivery is critical for
ensuring viral titers sufficient for strong transfection. Viral delivery using IV administration can be
problematic when trying to treat a CNS disorder, since viruses don’t readily cross the blood-brain barrier
(BBB). Abeona uses AAV9, which has an enhanced ability to cross the BBB. However, the doses required
for IV administration may increase the risk of systemic effects and other adverse events (AEs).
Potential for Off-Target Effects – Direct injection into the brain corrects the enzyme deficiency where the
toxicity occurs and minimizes the exposure of other cell types to the virus. In contrast, IV administration
exposes the whole body to the AAV. In the case of Abeona’s vector, AAV9 has broad tropisms for several
peripheral tissue types, including liver, heart, and skeletal muscle, in addition to neurons and glia.1 This
increases the likelihood of off-target effects or immune reactions. Since Abeona’s vector uses a non-specific
promoter, there would be transcription and translation of the SGSH gene in other peripheral tissues. While
IV administration is less invasive than intraparenchymal injection, it does also result in much broader viral
exposure in peripheral tissues.
Neutralizing Antibodies – AAV is a naturally-occurring virus, and up to 80% of humans have antibodies
against AAV serotype 2, 5, or 9.23 The presence of neutralizing antibodies can greatly reduce transduction
efficiency, since the immune system can clear out large quantities of virus. This is primarily an issue for IV
administration. Lysogene uses AAVrh10, an AAV serotype isolated from rhesus monkeys, which may be
more suitable for gene therapy due to the lack of endogenous anti-AAVrh10 antibodies found in humans.4
Proper Viral Titers – In their Phase I/II trial, Lysogene’s vector was delivered at a concentration of 7.21011
vg/mL, which is in the range of what is typically used for direct viral injections into the brain. Abeona is using
1 Inagaki, K, et al., 2006. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer
superior to that of AAV8. Molecular Therapeutics, 14(1), pp45-53. 2 Boutin, S, et al., 2010. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6,
8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Human Gene Therapy, 21(6), pp704–712. 3 Haas, MJ, 2013. MPS IIIA: gene therapy for brain and body. SciBX, 6(28). 4 Gao, GP, et al., 2002. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proceedings of
the National Academy of Sciences, 99, pp11854–11859.
April 19, 2017
Page 2

a low dose of 51012 vg/kg and a high dose of 11013 vg/kg. Considering the dilution in the blood, the
presence of the BBB, and uptake by cells in peripheral organs, this may not be sufficient to induce high levels
of SGSH expression in the brain. Due to the broad tropism of AAV9, Abeona is very likely to have better
transduction efficiency of the liver and other organs compared to the brain. This poses a substantial risk to
this clinical program, because they must select a dosage that gets sufficient virus into the brain while not
causing toxicity in the liver or other organs.
▪ Lysogene is Conducting IND-Enabling Studies to Support Development in GM1 Gangliosidosis. LYS-
GM101 is an AAVrh10-based gene therapy to treat GM1 gangliosidosis by direct delivery of the gene encoding the
beta-galactosidase enzyme into the CNS. Lysogene has collaborative agreements with investigators from the University
of Massachusetts Medical School (UMMS) and Auburn University to conduct IND-enabling preclinical studies. The
Company expects to launch a Phase I/II study to evaluate the safety and efficacy of LYS-GM101. Lysogene has
received Orphan Drug designation of FDA and EMA and Rare Pediatric Disease designation form the FDA for LYS-
GM101.
Financial Discussion
Initial Public Offering. In February, Lysogene announced the completion of an initial public offering (IPO) on the
Euronext Paris exchange. The Company sold 3.3 million new shares priced at €6.80 ($7.31) per share, and the deal
raised a total of €22.6 million ($24.3 million) in net proceeds. Immediately following the IPO, Lysogene had a market
capitalization of roughly €82.1 million ($88.3 million).
April 19, 2017
Page 3

Table of Contents
Company Description .................................................................................................................................................................... 5
LYS-SAF302: A Gene Therapy to Treat Mucopolysaccharidosis Type IIIA (MPA IIIA) ................................................. 5
Mechanism of Action ................................................................................................................................................................ 6
Comparison with Abeona Therapeutics’ Approach for MPSIIIA .................................................................................... 6
Preclinical Studies ...................................................................................................................................................................... 7
Safety Profile ............................................................................................................................................................................. 13
Mucopolysaccharidosis Type III ................................................................................................................................................ 13
Causes & Pathogenesis ........................................................................................................................................................... 14
Symptoms & Diagnosis .......................................................................................................................................................... 16
Treatment .................................................................................................................................................................................. 17
Disease Market Information ....................................................................................................................................................... 17
Epidemiology ............................................................................................................................................................................ 17
Market Size ................................................................................................................................................................................ 18
Pricing and Market Share Analysis ........................................................................................................................................ 18
Clinical Data Discussion .............................................................................................................................................................. 20
Planned Pivotal Phase II/III Trial ........................................................................................................................................ 24
Other Drugs in Development ..................................................................................................................................................... 24
LYS-GM101: A Gene Therapy to Treat GM1 Gangliosidosis ............................................................................................. 27
GM1 Gangliosidosis ..................................................................................................................................................................... 27
Causes & Pathogenesis ........................................................................................................................................................... 27
Symptoms & Diagnosis .......................................................................................................................................................... 28
Planned Clinical Program for LYS-GM101.............................................................................................................................. 29
Other Drugs in Development ..................................................................................................................................................... 29
Intellectual Property & Licensing ............................................................................................................................................... 30
Management Team ....................................................................................................................................................................... 30
Risk to an Investment .................................................................................................................................................................. 32
Analyst Certification ..................................................................................................................................................................... 33
Disclosures ..................................................................................................................................................................................... 33
April 19, 2017
Page 4

Company Description
Lysogene is a French biotechnology Company that was founded in 2009 to harness the potential of gene therapy to
address unmet needs in the orphan disease space and, in particular, lysosomal storage disorders. The Company has
also established a US presence with the launching of a subsidiary in Cambridge, Massachusetts. Lysogene completed
an initial public offering (IPO) in February 2017, raising net proceeds of €22.6 million ($24.3 million). Lysogene’s lead
program is a gene therapy for mucopolysaccharidosis type IIIA (MPS IIIA), a lysosomal storage disorder with no
approved therapies. The therapy uses an adeno-associated virus (AAV) to deliver the SGSH gene, which encodes a
protein that these patients are missing. The Company has discussed plans for a pivotal Phase II/III study with US and
EU regulators and expects to launch this trial by the first quarter of 2018.
Lysogene is also currently conducting an observational natural history study in up to 25 patients, which will provide
additional data on the natural disease progression and will serve as a control group for the pivotal study. The
Company’s development pipeline is shown in Figure 1. Lysogene is also developing a gene therapy treatment, LYS-
GM101, for patients with GM1 gangliosidosis, and is currently conducting IND-enabling preclinical studies for this
candidate. There are no approved treatments for either indication that Lysogene is pursuing, and there is the potential
for premium pricing for these orphan indications. In addition, the Company can leverage their existing viral delivery
technology to pursue additional lysosomal storage disorders (LSD).
Figure 1. Lysogene’s Development Pipeline
Source: LifeSci Capital
LYS-SAF302: A Gene Therapy to Treat Mucopolysaccharidosis Type IIIA (MPA IIIA)
LYS-SAF302 is a second-generation product candidate that uses the adeno-associated virus vector serotype rh10
(AAVrh10) to deliver the SGSH gene into the CNS of patients with MPS IIIA. AAVrh10 is able to efficiently transfect
neurons and glia. LYS-SAF302 is a more potent form of the previous candidate, LYS-SAF301, that was developed in
response to the results obtained in the Phase I/II trial. LYS-SAF301 encoded two genes, which may have diminished
the transduction efficiency of the virus and the rate of protein expression.
April 19, 2017
Page 5

Mechanism of Action. In MPS IIIA, there is a deficiency of the heparan-N-sulfatase enzyme. This enzyme is
responsible for the hydrolysis of the sulfate group attached to the glucosamine in heparan sulfate. The gene that
encodes heparan-N-sulfatase is SGSH. The catalytic site of the SGSH gene is activated by a sulfatase-modifying factor,
SUMF1.
By introducing the SGSH gene into individuals affected by MPS IIIA, there is the possibility to correct the deficiency
of the heparan-N-sulfatase enzyme. When this enzyme is present, the heparan sulfate breakdown process continues.
A decline in heparan sulfate build-up will slow neurological degeneration.
The SGSH gene can be delivered with a form of gene therapy using adeno-associated viruses (AAVs). AAVs are small
viruses (approximately 4.5 kilobases) with single-stranded DNA surrounded by non-enveloped capsids. To make the
complete AAV, the gene of interest is inserted into a plasmid as a transgene following a promoter. In the case of LYS-
SAF302, the promoter is the CAG promoter, a strong promoter capable of driving high levels of protein expression.
AAVs do not encode their own polymerases and therefore rely on the replication machinery in cells. The manufacture
of AAVs require two additional plasmids: a packaging plasmid containing the rep gene from AAV2 and cap gene from
AAVrh10, and a helper plasmid, which contains several other critical genes. The rep gene in AAVs recruits the cellular
machinery for translation. The cap gene encodes for structural proteins to make up the capsid. There are also segments
of the cDNA that code for the helping adenoviruses, which AAVs require. Once cells are transfected with these three
plasmids, the necessary genes are expressed and functional AAV particles are assembled.
A key advantage of using AAVs in gene therapy is the ability to infect post-mitotic cells, which lead to long-term gene
expression. When the AAV particles have been directly delivered to the central nervous system (CNS), they can infect
neurons and induce the expression of the missing enzyme. If sufficient expression of the protein is induced, the disease
progression may be slowed or even halted. Lysogene has developed an AAVrh10 virus that encodes the human SGSH
gene. To treat MPS IIIA, this viral vector is bilaterally injected directly into multiple sites in the brain to ensure broad
viral transfection and maximize the amount of enzyme that is translated and secreted.5
Comparison with Abeona Therapeutics’ Approach for MPSIIIA. Abeona Therapeutics is developing ABO-102,
an AAV9-based gene therapy, to treat MPS IIIA, taking a similar approach to Lysogene’s LYS-SAF302. Both ABO-
102 and LYS-SAF302 are AAVs that encode the SGSH gene. However, there are several important differences, most
important of which is in the route of administration, described below:
▪ Direct Delivery into the Brain – Lysogene’s strategy involves the direct injection of an AAV encoding the
missing enzymes into the brain. Direct delivery is critical for ensuring viral titers sufficient for strong
transfection. Viral delivery using intravenous administration can be problematic when trying to treat a CNS
disorder, since viruses don’t readily cross the blood-brain barrier (BBB). Abeona Therapeutics (NasdaqCM:
ABEO), Lysogene’s main competitor, uses this approach. However, they are using an AAV9, which has an
enhanced ability to cross the BBB. Higher local titers distributed over a larger region, the result of Lysogene’s
strategy, may induce greater overall protein expression than low titers across the entire brain. In addition, the
doses required for IV administration may increase the risk of systemic effects and other adverse events (AEs).
5 Tardieu M, Zerah M, Husson B, de Bournonville SP, Deiva K, Adamsbaum C, Vincent F, Hocquemiller M Broissand C,
Furlan V. 2014. Intracerebral administration of adeno-associated viral vector serotype rh. 10 carrying human SGSH and SUMF1
cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum Gene Ther 25:506-516.
April 19, 2017
Page 6

▪ Potential for Off-Target Effects – Direct injection into the brain corrects the enzyme deficiency where the
toxicity occurs and minimizes the exposure of other cell types to the virus. In contrast, IV administration
exposes the whole body to the AAV. In the case of Abeona’s vector, AAV9 has broad tropisms for several
peripheral tissue types, including liver, heart, and skeletal muscle, in addition to neurons and glia,6 which
increases the likelihood of off-target effects or immune reactions. In addition, since Abeona’s vector uses a
non-specific promoter, there would be transcription and translation of the SGSH gene in other peripheral
tissues. While IV administration is less invasive than intraparenchymal injection, it does also result in much
broader viral exposure in peripheral tissues.
▪ Neutralizing Antibodies – AAV is a naturally-occurring virus, and up to 80% of humans have antibodies
against AAV serotype 2, 5, or 9.78 The presence of neutralizing antibodies can greatly reduce transduction
efficiency, since the immune system can clear out large quantities of virus. This is primarily an issue for IV
administration; neutralizing antibodies are less of a problem with intracerebroventricular or intraparenchymal
delivery. Lysogene uses AAVrh10, an AAV serotype isolated from rhesus monkeys, which may be more
suitable for gene therapy due to the lack of anti-AAVrh10 antibodies found in humans.9
▪ Correct Viral Titers – In their Phase I/II trial, Lysogene’s vector was delivered at a concentration of 7.21011
vg/mL, which is in the range of what is typically used for direct viral injections into the brain. Abeona is using
a low dose of 51012 vg/kg and a high dose of 11013 vg/kg. Considering the dilution in the blood, the
presence of the BBB, and uptake by cells in peripheral organs, this may not be sufficient to induce high levels
of SGSH expression in the brain. Due to the broad tropism of AAV9, Abeona is very likely to have better
transduction efficiency of the liver and other organs compared to the brain. This poses a substantial risk to
this clinical program, because they must select a dosage that gets sufficient virus into the brain while not
causing toxicity in the liver or other organs.
Preclinical Studies
MPS IIIA Mouse Model. An AAVrh10 vector, SAF301, was used to deliver the human SGSH and SUMF1 genes
to mice affected with MPS IIIA.10 In this experiment, 5-week-old mice received viral injections into the region labeled
L2 in Figure 2. Following this procedure, measures of SGSH expression, heparan sulfate storage, ganglioside
accumulation, and neuroinflammation were assessed at 12, 23 and 30 weeks of age.
6 Inagaki, K, et al., 2006. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer
superior to that of AAV8. Molecular Therapeutics, 14(1), pp45-53. 7 Boutin, S, et al., 2010. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6,
8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Human Gene Therapy, 21(6), pp704–712. 8 Haas, MJ, 2013. MPS IIIA: gene therapy for brain and body. SciBX, 6(28). 9 Gao, GP, et al., 2002. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proceedings of
the National Academy of Sciences, 99, pp11854–11859. 10 Winner Leanne K., Beard Helen, Hassiotis Sofia, Lau Adeline A., Luck Amanda J., Hopwood John J., and Hemsley Kim M.
2016. A preclinical study evaluating AAVrh10-based gene therapy for Sanfillipo syndrome. Human Gene Therapy. 27(5): 363-375
April 19, 2017
Page 7

Figure 2. Injection Site of SAF301 into the Mouse Brain
Source: Winner, 2016
In mice affected with MPS IIIA and treated with SAF301, there was a mean 185-fold increase in SGSH compared to
control mice. At 30 weeks of age, mice were observed to have detectable SGSH levels at sites distant from the injection
site, including the L4, R2 and R4 regions, indicating broad spread of the enzyme. Figure 3 shows the relative levels
of a derivative of heparan sulfate, known as glucosamine-N-sulfate [α-1,4] hexuronic acid (GlcNS-UA). Injection of
SAF301 into L2 results in lower levels of GlcNS-UA, suggesting that this strategy of viral delivery may be able to
correct the underlying enzyme deficiency in MPS IIIA.
Figure 3. SGSH Concentrations in Various Brain Slices in Treated and Control Mice
Source: Winner et al., 2016 11
Figure 4 shows the quantities of GlcNS-UA remaining following treatment with SAF301. In the L2 region where the
injection was administered, the GlcNS-UA levels were 14% of that observed in control mice at 30 weeks. While the
reduction was less pronounced outside of the point of injection, there is a general downward trend for all areas,
11 Winner Leanne K., Beard Helen, Hassiotis Sofia, Lau Adeline A., Luck Amanda J., Hopwood John J., and Hemsley Kim M.
2016. A preclinical study evaluating AAVrh10-based gene therapy for Sanfillipo syndrome. Human Gene Therapy. 27(5): 363-
375
April 19, 2017
Page 8

especially after 23 weeks of age, indicating that the virus spread a considerable distance from the original injection site.
The spread also increased with time.
Figure 4. Percentage of GlcNS-UA Remaining in SAF-301 Treated vs Control Mice
Source: Winner, 2016
Figure 5 illustrates the reduction in ganglioside accumulation in the rostral cortex between the treated and untreated
mice at 23 weeks. It is evident that the gene therapy strongly reduced ganglioside levels in the treated mice (panel G)
compared to untreated MPS IIIA mice (panel F), approximating levels observed in unaffected control mice (panel H).
Similar reductions were observed for mice at 30 weeks of age.
Figure 5. GM3 Ganglioside Staining in Ipsilateral Rostral Cortex at 23 Weeks
Source: Winner et al., 201612
AAVrh.10 was effective in treating a variety of neuropathologies in a mouse model of MPS IIIA, suggesting its
therapeutic potential in human patients with MPS IIIA. However, the results also suggest that using multiple sites of
injection may be essential to ensure sufficient coverage.
12 Winner Leanne K., Beard Helen, Hassiotis Sofia, Lau Adeline A., Luck Amanda J., Hopwood John J., and Hemsley Kim M.
2016. A preclinical study evaluating AAVrh10-based gene therapy for Sanfillipo syndrome. Human Gene Therapy. 27(5): 363-
375
April 19, 2017
Page 9

AAV Delivery in MPS IIIA Mouse Model
AAV5 was used to deliver SUMF1 and SGSH directly into the central nervous system via an intraventricular injection
in newborn mice affected with MPSIIIA.13 Infection with GFP allowed for visualization of SGSH enzyme activity in
the mice. The importance of SUMF1 to SGSH activity was first established by comparing the effects of vectors either
encoding SGSH alone or together with SUMF1. Figure 6 shows how enzyme activity increases when SUMF1 is
encoded in the AAV vector. The results show that inclusion of SUMF1, connected with an internal ribosome entry
site (iRES) linker, more than doubles the amount of enzyme activity.
Figure 6. SGSH activity for MPS IIIA affected mice
Source: Fraldi, 200714
Behavioral tests also showed an improvement in activity levels following gene therapy. In Figure 7, hind-limb gait
was measured in MPSIIIA and control mice that either received an injection of SAF301 or a GFP-expressing vector
as a sham control. MPSIIIA mice treated with SAF301 (thin diagonal lines) had a significant improvement in gait
length and width relative to MPS IIIA mice receiving sham injections (black).
13 Fraldi, A., Hemsley, K., Crawley, A., et al. (2007). Functional correction of CNS lesions in an MPS-IIIA mouse model by
intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum. Mol. Genet. 16, 2693–2702. 14 Fraldi, A., Hemsley, K., Crawley, A., et al. (2007). Functional correction of CNS lesions in an MPS-IIIA mouse model by
intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum. Mol. Genet. 16, 2693–2702.
April 19, 2017
Page 10

Figure 7. Gait Length and Width for MPSIIA and Normal Mice Receiving SAF301 or Sham Treatment
Source: Fraldi et al, 2007 15
Routes of Delivery for AAVrh10 into the CNS. Investigators evaluated the use of AAVrh10 to treat a lysosomal
storage disorder via a variety of routes of delivery. In these experiments, AAVrh10 carrying human cDNA was
delivered to primates.16 Five different routes of delivery were compared, including:
▪ Delivery into white matter.
▪ Delivery to deep gray matter and overlying white matter.
▪ Convection-enhanced delivery to deep gray matter.
▪ Intraventricular delivery to the lateral cerebral ventricles.
▪ Intraarterial delivery to the middle cerebral artery.
Figure 8 shows activity in the central nervous system following viral delivery of arylsulfatase A (ARSA) cDNA via
multiple routes of administration. The percentage of brain cubes with increased ARSA activity was highest for white
matter delivery. These results suggest that viral delivery into the white matter can achieve the highest transfection
efficiency.
15 Fraldi, A, et al., 2007. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated
delivery of sulfamidase and SUMF1 genes. Human Molecular Genetics, 16(22), pp2693-2702. 16 Rosenberg JB, Sondhi D, Rubin DG, et al. 2014. Comparative Efficacy and Safety of Multiple Routes of Direct CNS
Administration of Adeno-Associated Virus Gene Transfer Vector Serotype rh.10 Expressing the Human Arylsulfatase A cDNA
to Nonhuman Primates. Human Gene Therapy Clinical Development. 25(3):164-177.
April 19, 2017
Page 11
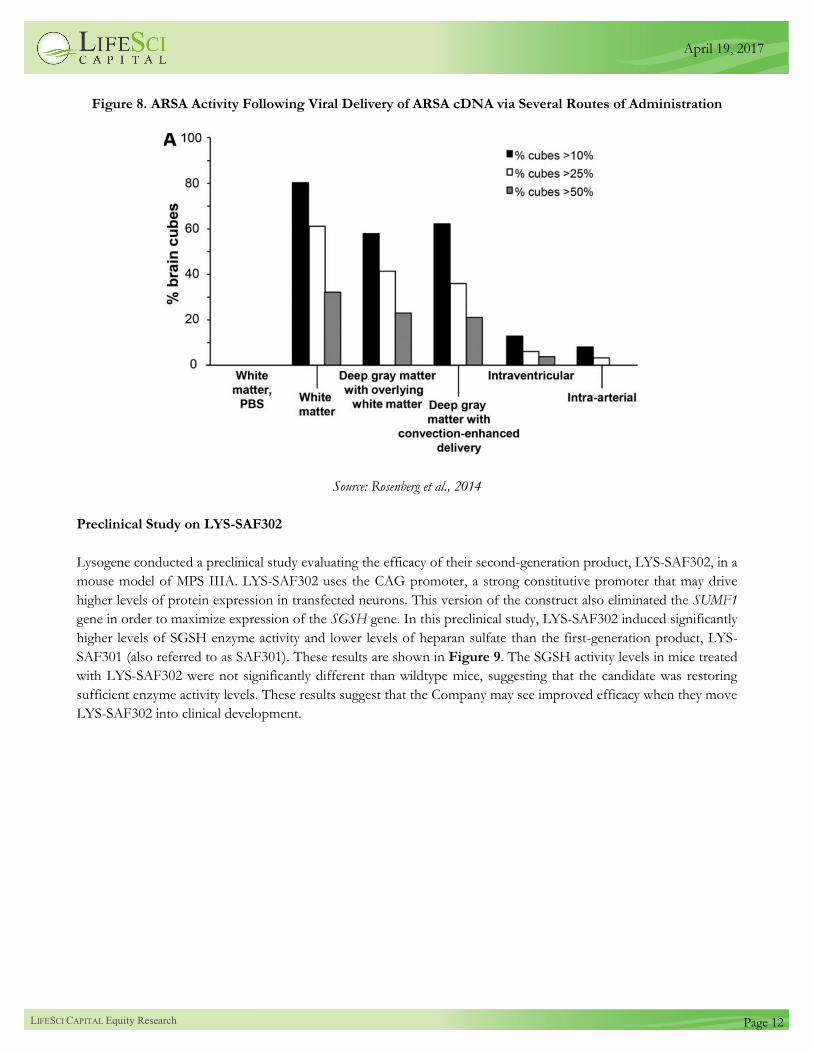
Figure 8. ARSA Activity Following Viral Delivery of ARSA cDNA via Several Routes of Administration
Source: Rosenberg et al., 2014
Preclinical Study on LYS-SAF302
Lysogene conducted a preclinical study evaluating the efficacy of their second-generation product, LYS-SAF302, in a
mouse model of MPS IIIA. LYS-SAF302 uses the CAG promoter, a strong constitutive promoter that may drive
higher levels of protein expression in transfected neurons. This version of the construct also eliminated the SUMF1
gene in order to maximize expression of the SGSH gene. In this preclinical study, LYS-SAF302 induced significantly
higher levels of SGSH enzyme activity and lower levels of heparan sulfate than the first-generation product, LYS-
SAF301 (also referred to as SAF301). These results are shown in Figure 9. The SGSH activity levels in mice treated
with LYS-SAF302 were not significantly different than wildtype mice, suggesting that the candidate was restoring
sufficient enzyme activity levels. These results suggest that the Company may see improved efficacy when they move
LYS-SAF302 into clinical development.
April 19, 2017
Page 12
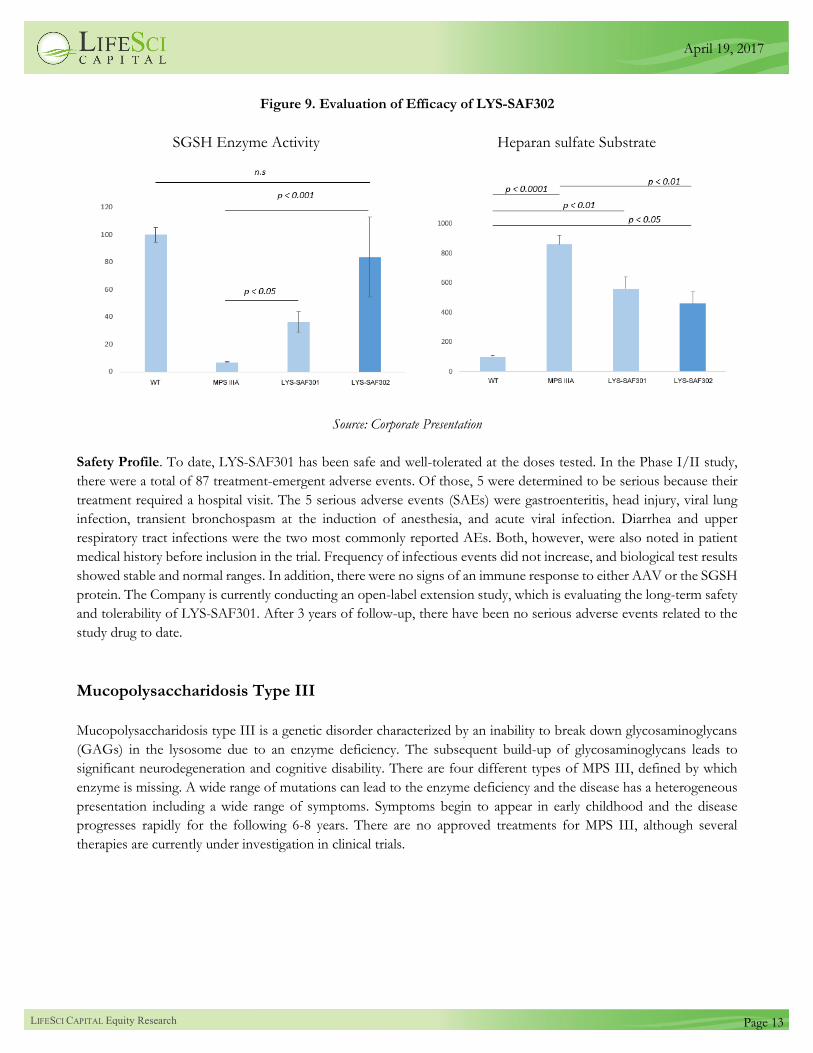
Figure 9. Evaluation of Efficacy of LYS-SAF302
SGSH Enzyme Activity Heparan sulfate Substrate
Source: Corporate Presentation
Safety Profile. To date, LYS-SAF301 has been safe and well-tolerated at the doses tested. In the Phase I/II study,
there were a total of 87 treatment-emergent adverse events. Of those, 5 were determined to be serious because their
treatment required a hospital visit. The 5 serious adverse events (SAEs) were gastroenteritis, head injury, viral lung
infection, transient bronchospasm at the induction of anesthesia, and acute viral infection. Diarrhea and upper
respiratory tract infections were the two most commonly reported AEs. Both, however, were also noted in patient
medical history before inclusion in the trial. Frequency of infectious events did not increase, and biological test results
showed stable and normal ranges. In addition, there were no signs of an immune response to either AAV or the SGSH
protein. The Company is currently conducting an open-label extension study, which is evaluating the long-term safety
and tolerability of LYS-SAF301. After 3 years of follow-up, there have been no serious adverse events related to the
study drug to date.
Mucopolysaccharidosis Type III
Mucopolysaccharidosis type III is a genetic disorder characterized by an inability to break down glycosaminoglycans
(GAGs) in the lysosome due to an enzyme deficiency. The subsequent build-up of glycosaminoglycans leads to
significant neurodegeneration and cognitive disability. There are four different types of MPS III, defined by which
enzyme is missing. A wide range of mutations can lead to the enzyme deficiency and the disease has a heterogeneous
presentation including a wide range of symptoms. Symptoms begin to appear in early childhood and the disease
progresses rapidly for the following 6-8 years. There are no approved treatments for MPS III, although several
therapies are currently under investigation in clinical trials.
April 19, 2017
Page 13

Causes & Pathogenesis. MPS III has four different types (A, B, C, D), shown in Figure 10, each with a different
underlying mutated gene. All four mutated genes affect function of a specific enzyme involved in the breakdown of
GAGs.17
Figure 10. Types of MPS IIIA
MPS III Type Mutated gene Affected enzyme
A SGSH heparin N-sulfatase
B NAGLU alpha-N-acetylglucosaminidase
C HGSNAT acetyl-CoA-glucosaminide acetyltransferase
D GNS N-acetylglucosamine-6-sulfatase
Source: LifeSci Capital
GAGs, also known as mucopolysaccharides, are long unbranched sugar chains made up of a repeating disaccharide
units. The strong polarity of GAGs allow them to function as lubricants in joints. They are also involved in the
formation of bones, cartilage, skin and other tissues in the body. The specific GAG of interest in MPS III is heparan
sulfate.18
Heparan sulfate is a carbohydrate chain that contains sulfated residues. Heparan sulfates interact with various proteins
on the surface of the cell and in the extracellular matrix. While the function of heparan sulfates is not fully understood,
they are believed to play a role in growth factor signaling pathways. The degradation of heparan sulfate is carried out
by a series of enzymes and ends with the formation of heparan sulfate fragments and oligosaccharides.19 The enzymes
involved are three exoglycosidases, at least three sulfatases, and an acetyltransferase. Figure 11 outlines the degradation
process of heparan sulfate and the figure is labeled with which enzyme is deficient in each type of MPS III (A-D).
17 Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA (2008). Sanfilippo syndrome: A mini-review. J Inherit
Metab Dis. 31:240–52. 18 Fedele AO (2015). Sanfilippo syndrome: causes, consequences, and treatments. The Application of Clinical Genetics. 8:269-281. 19 Bame KJ (2001). Heparanases: endoglycosidases that degrade heparan sulfate proteoglycans. Glycobiology 11: 91R–98R.
April 19, 2017
Page 14

Figure 11. Degradation of Heparan sulfate
Source: Wijburg et al., 200820
Heparan sulfate can’t be broken down if an individual has deficiencies in any of the four enzymes described above.
Instead, heparan sulfate will build-up in the lysosomes of cells, as shown in Figure 12. Lysosomes are cellular
compartments where molecules are digested and recycled. A build-up in the lysosome interferes with other proteins
and lysosomal functions. Decline in lysosomal functioning can affect the overall functioning and health of the cell.
20 Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA (2008). Sanfilippo syndrome: A mini-review. J Inherit
Metab Dis. 31:240–52.
April 19, 2017
Page 15

Figure 12. Build-Up in Lysosomes Resulting from Enzyme Deficiency
Source: Pathology Outlines21
The link between accumulation of heparan sulfate and neurodegeneration in the CNS is not fully understood.
However, it is known that monosialic gangliosides build up in lysosomes following the build-up of heparan sulfate.
Ganglioside build-up occurs either through the inhibition ganglioside degradation enzymes or deregulated ganglioside
synthesis. The exact effect of build-up of heparan sulfate and gangliosides remains unclear.22
When a molecule cannot be broken down in the lysosomes, it builds up and can interfere with normal cellular
functioning. Animal model studies have suggested that both gangliosides and heparan sulfate build-up play a role in
the brain’s inflammatory response. When heparan sulfate is exocytosed from lysosomes and when ganglioside-filled
neurons are phagocytosed, the cell may elicit an immune response that can worsen damage to the CNS. Microglial
cells may become activated in response to the build-up, leading to the release of pro-inflammatory cytokines that can
contribute to neurodegeneration.23 Animal studies have also shown the role of heparan sulfate in interfering with
lysosomal membrane composition. When the membrane cannot associate with target membranes, there is a decline
in the trafficking of proteins, which can affect the function of organelles such as the mitochondria. A decline in
mitochondria function has widespread consequences.24
Symptoms & Diagnosis. Symptoms of MPS III begin to appear during early childhood, usually at around one year
of age. At first, behavior problems and delayed speech are most evident. An affected child may become restless and
21 http://pathologyoutlines.com/topic/thyroidlsd.html. 22 McGlynn R, Dobrenis K, Walkley SU (2004) Differential subcellular localization of cholesterol, gangliosides, and
glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J Comp Neurol 480: 415–426. 23 Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF (2003). Activated microglia in cortex of mouse models
of mucopolysaccharidoses I and IIIB. 100(4):1902-1907. 24 Fedele AO (2015). Sanfilippo syndrome: causes, consequences, and treatments. The Application of Clinical Genetics. 8:269-281.
April 19, 2017
Page 16

have frequent sleep disturbances. Between the ages of 2-6, there is often a progression of intellectual disability and a
loss of previously learned skills. In the later stages, affected children become unsteady with frequent falls and eventually
lose the ability to walk. Seizures and movement disorders may also occur.25
There will also be notable differences in physical features for those affected. Coarse facial features, while less
prominent in individuals with MPS III compared to individuals with other types of MPS, may be present. Individuals
with MPS III will most likely have macrocephaly, mild hepatomegaly (enlarged liver), and umbilical or inguinal hernia.
It is also possible that individuals will have short stature, joint stiffness, and mild skeletal deformities. Chronic diarrhea,
respiratory problems, and difficulties with hearing and vision may also develop with age. MPS III type A will lead to
the most severe symptoms out of the four types of the disorder.26
To diagnose MPS III, a urine test will initially be used to detect GAG since most individuals with MPS will have
elevated levels. Another important test is measurement of enzymatic activity in blood or skin cells. To diagnose
different types of MPS III, tests need to be run to examine the specific enzymatic activity that varies between A, B, C
and D. ELISA is combined with a more sensitive assay that makes use of radiolabeled oligosaccharides. The
combination of these two assays allows for accurate prediction of the disease and its severity.27 Genetic testing can
also be performed to identify the disease-causing mutation. Prenatal diagnosis is also possible through screening of
cells from chorionic villi at 9-10 weeks of gestation or cells from amniocentesis at 15-16 weeks gestation.28 This is
typically performed when there is a family history of the disease.
Treatment. Currently, there are no effective treatments for individuals affected by MPS IIIA. Neither enzyme
replacement therapies (ERT) nor hematopoietic stem cell transplantation have been shown to be effective in MPS
IIIA. Attempts have been made to develop ERTs that can cross the blood-brain barrier (BBB); however, this work
has not resulted in the approval of any therapies for MPS IIIA. Current treatments for MPS IIIA are palliative and
intended to improve quality of life. These can include anticonvulsants to treat seizures, as well as sedatives and
melatonin to addresses sleep problems.
Disease Market Information
Epidemiology. Lysosomal storage disorders (LSD) are a collection of more than 50 inherited diseases resulting from
lysosomal dysfunction. LSDs have a collective prevalence of 1 per 7,000 live births.29 MPS IIIA is the most common
of the four types of MPS.30 There are an estimated 0.5 to 1.2 cases per 100,000 live births,31 which translates to an
annual incidence of roughly 82 MPS IIIA cases combined in the US and Europe. The worldwide prevalence is thought
25 Fedele, A, 2015. Sanfilippo syndrome: causes, consequences, and treatments. Applied Clinical Genetics, 8, pp269-281. 26 Valstar MJ, et al., 2008. Sanfilippo syndrome: A mini-review. Journal of Inherited Metabolic Disorders, 31, pp240–252. 27 Perkins, KJ, et al., 2001. Prediction of Sanfilippo phenotype severity from immunoquantification of heparan-N-sulfamidase in
cultured fibroblasts from mucopolysaccharidosis type IIIA patients. Mol Genet Metab, 73, pp306-312. 28 Hopwood JJ (2005). Prenatal diagnosis of Sanfilippo syndrome. Prenat Diagn. 25:148–150. 29 Hocquemiller, M, et al., 2016. Adeno-Associated Virus-Based Gene Therapy for CNS Diseases. Human Gene Therapy, 27(7),
pp478-496. 30 Meyer, A., et al., 2007. Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type
A). Pediatrics, 120, e1255– e1261. 31 Heron, B, et al., 2011. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United
Kingdom and Greece. American Journal of Medical Genetics, 155A(1), pp58-68.
April 19, 2017
Page 17

to be roughly 3,000. Because MPS IIIA is an autosomal recessive disease that does not affect sex chromosomes, it
affects males and females equally.32 MPS IIIA is most prevalent in northwestern Europe. Frequency and progression
of the disease may differ by country depending on the types of mutations present in certain regions. This suggests that
different mutations confer varying degrees of disease severity, although no firm correlations between genotype and
phenotype have been established.33
Market Size. The current market for drugs to lysosomal storage disorders is approximately $5.4 billion. However,
there are no approved treatments for lysosomal storage disorders (LSD) that involve the CNS, even though these
types of disorders make up 60% of the total cases of LSDs. A new entrant addressing LSDs with CNS involvement
can expect to capture a substantial portion of the market. The lysosomal disorders drug market is dominated by
enzyme replacement therapies (ERT), which account for roughly 90% of the market. Substrate reduction therapy
(SRT) and cysteine depletors together make up about 7% of the market.
Pricing and Market Share Analysis. Given the lack of currently approved drugs for MPS IIIA, the best indicators
for pricing are the cost and revenues of treatments for other lysosomal storage disorders. Figure 13 shows the sales
of ERTs and SRTs achieved in 2015, as well as the incidence of the targeted disease. Sales of drugs to treat MPS types
I, II, and IV generate annual sales between $200 million and $550 million. MPS type I, which has a similar incidence
to MPS IIIA, may provide some guidance for the market opportunity that may exist in MPSIIIA. Genzyme’s
Aldurazyme (iduronidase) for MPS I reached sales of $218 million in 2015, and we note that pricing for a gene therapy
may be substantially higher.
32 Blanch L, et al., 1997. Molecular defects in Sanfilippo syndrome type A. Hum Mol Genet, 6(5), pp787-91. 33 Heron, B., Mikaeloff, Y., Froissart, R., et al. (2010). Incidence and natural history of mucopolysaccharidosis type III in France
and comparison with United Kingdom and Greece. American Journal of Medical Genetics (Part A), 155, pp58–68
April 19, 2017
Page 18

Figure 13. 2015 Revenue and Disease Incidence for Drugs Treating Lysosomal Storage Disorders
Disease Drug Company 2015 Sales Incidence*
Gaucher I
Cerezyme Genzyme $848 M 1/100,000
Cerdelga Genzyme $73 M 1/100,000
Elelyso Protalix $26 M 1/100,000
Vpriv Shire $342 M 1/100,000
Zavesca Actelion $96 M 1/100,000
Fabry Fabrazyme Genzyme $657 M 1/80,000
Replagal Shire $441 M 1/80,000
Pompe Myozyme/
Lumizyme Sanofi $721 M 1/57,000
MPS I Aldurazyme Genzyme $218 M 1/100,000
MPS II Elaprase Shire $553 M 1/166,000
MPS IVA Vimizim Biomarin $228 M 1/250,000
MPS IV Naglazyme Biomarin $303 M 1/250,000
Others $929 M
Total $5,435 M
* Incidence in terms of live births
Source: Corporate Presentation
Figure 14 shows the annual pricing of treatments for lysosomal storage disorders. The average annual cost is roughly
$321,000, reflecting the substantial premium pricing that is possible for ultra-orphan indications. Pricing for orphan
indications is often a function of the disease prevalence. We have explored the relationship between pricing and disease
prevalence in a previous note.
April 19, 2017
Page 19
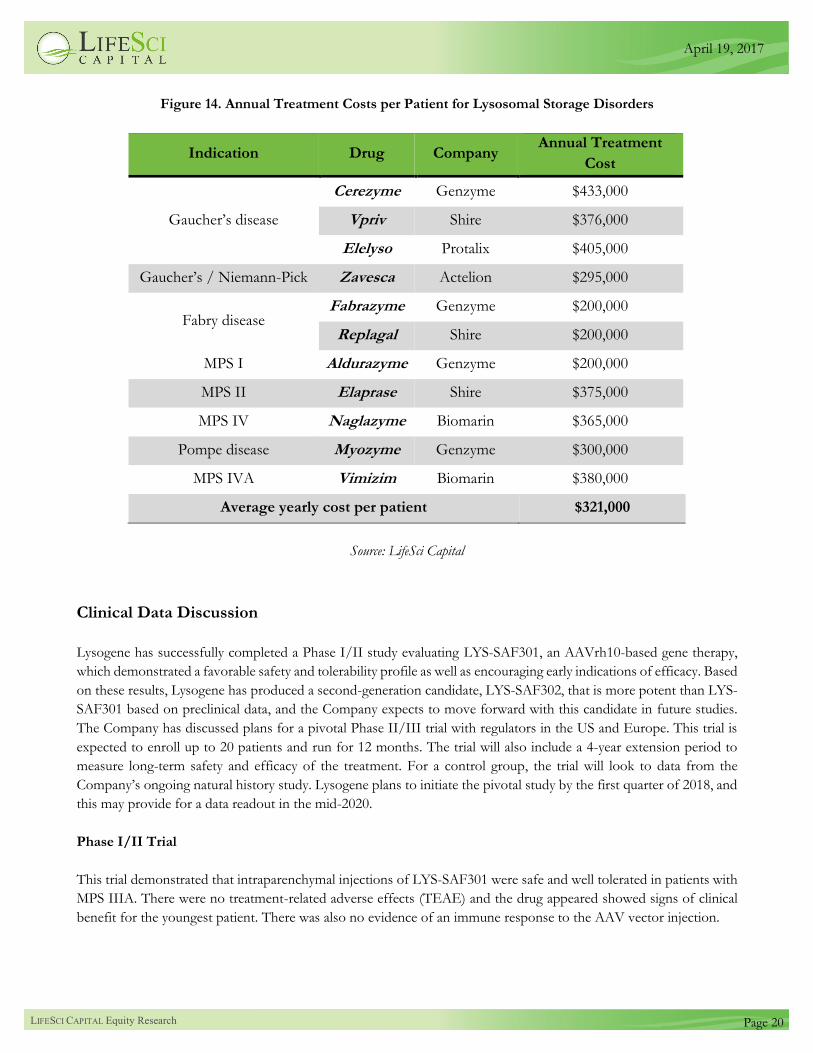
Figure 14. Annual Treatment Costs per Patient for Lysosomal Storage Disorders
Indication Drug Company Annual Treatment
Cost
Gaucher’s disease
Cerezyme Genzyme $433,000
Vpriv Shire $376,000
Elelyso Protalix $405,000
Gaucher’s / Niemann-Pick Zavesca Actelion $295,000
Fabry disease Fabrazyme Genzyme $200,000
Replagal Shire $200,000
MPS I Aldurazyme Genzyme $200,000
MPS II Elaprase Shire $375,000
MPS IV Naglazyme Biomarin $365,000
Pompe disease Myozyme Genzyme $300,000
MPS IVA Vimizim Biomarin $380,000
Average yearly cost per patient $321,000
Source: LifeSci Capital
Clinical Data Discussion
Lysogene has successfully completed a Phase I/II study evaluating LYS-SAF301, an AAVrh10-based gene therapy,
which demonstrated a favorable safety and tolerability profile as well as encouraging early indications of efficacy. Based
on these results, Lysogene has produced a second-generation candidate, LYS-SAF302, that is more potent than LYS-
SAF301 based on preclinical data, and the Company expects to move forward with this candidate in future studies.
The Company has discussed plans for a pivotal Phase II/III trial with regulators in the US and Europe. This trial is
expected to enroll up to 20 patients and run for 12 months. The trial will also include a 4-year extension period to
measure long-term safety and efficacy of the treatment. For a control group, the trial will look to data from the
Company’s ongoing natural history study. Lysogene plans to initiate the pivotal study by the first quarter of 2018, and
this may provide for a data readout in the mid-2020.
Phase I/II Trial
This trial demonstrated that intraparenchymal injections of LYS-SAF301 were safe and well tolerated in patients with
MPS IIIA. There were no treatment-related adverse effects (TEAE) and the drug appeared showed signs of clinical
benefit for the youngest patient. There was also no evidence of an immune response to the AAV vector injection.
April 19, 2017
Page 20

Trial Design. Lysogene conducted an open-label, single-arm Phase I/II study to assess the safety and tolerability of
LYS-SAF301 in four children diagnosed with MPSIIIA.34 All four patients were between the age of 18 months and 6
years. All patients received intracerebral injections of LYS-SAF301 at a dose of 7.2 x 1011 viral copies/patient directly
into the brain. Injections were made along 6 tracks at 2 different depths, resulting in a total of 12 injection sites during
a single neurosurgical procedure. The primary endpoint was safety and tolerability through a one-year follow up. Safety
and tolerability were measured by the following: the type and severity of adverse events (AE), clinical parameters
including fever, seizure, headache, lethargy, and new neurological symptoms, radiological parameters based on MRI
results, and measurements of liver function. The secondary endpoint was a series of exploratory tests, including brain
MRI, neurocognitive and behavioral tests, and biomarkers in blood, urine, and cerebrospinal fluid (CSF). Cognitive
tests in this trial include the PEP-3, Vineland Adaptive Behavior-II, and Toddler Behavioral Assessment Questionnaire
(TBAQ) tests, which test the following:
▪ PEP-3 – a test used in children with autism or other communicative disorders.
▪ Vineland Adaptive Behavior II – a test of a child’s personal and social skills.
▪ TBAQ – a parent assessment tool to evaluate temperament.
The exploratory tests were intended to provide potential surrogate markers upon which to evaluate patients in
subsequent trials.35
Trial Results. Overall, this trial demonstrated that LYS-SAF301 was safe and well-tolerated at the dose tested. At the
one year follow up, the number of adverse events (AE) was similar to that of other protocols.36 There were a total of
87 emergent adverse events. Of those 87, 5 were determined to be serious, because their treatment required a hospital
visit. The 5 serious adverse events (SAE) were gastroenteritis, head injury, viral lung infection, transient bronchospasm
at the induction of anesthesia, and acute viral infection. Diarrhea and upper respiratory tract infections were the two
most commonly reported AEs. Both, however, were also noted in patient medical history before inclusion in the trial.
Figure 15 shows the levels of anti-AAV (top) or anti-SGSH (bottom) antibodies following treatment, as a percentage
of baseline levels. Immune response to AAV vector injection is often a cause of concern in gene therapy, particularly
since AAV is a wildtype virus and a substantial portion of the population naturally possess antibodies against the virus.
However, levels of both antibodies remained stable through the one year follow up, suggesting the absence of an
immune response.
34 https://clinicaltrials.gov/ct2/show/NCT01474343 35 Tardieu, M, et al., 2014. Intracerebral administration of adeno-associated viral vector serotype rh. 10 carrying human SGSH
and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Human Gene
Therapies, 25, pp506-516. 36 Leone, P et al., 2012. Long-term follow-up after gene therapy for Canavan disease. Science Translational Medicine, 4, pp163-165.
April 19, 2017
Page 21

Figure 15. Anti-AAVrh10 and Anti-SGSH Antibody Titers as a Percentage of Baseline for Patients 1-4
Anti-AAVrh10 Titers
Anti-SGSH Titers
Source: Tardieu et al., 2014.
Efficacy of treatment was the secondary endpoint for the current trial. Researchers looked at brain atrophy in MRI
images. Atrophy levels were high in both patients #1 and #3, and patients #2 and #4 showed increases over the
follow-up period.
Figure 16 shows changes in performances on cognitive tests, graphed geometrically according to the domains in each
test. Data points within the shaded region indicate decreases, while points outside of the shaded region represent
improvements. The PEP-3 results showed that cognitive and motor skills were low but stable throughout the follow-
up period in patients 1, 2, and 3, although patients 2 and 3 did show declines in fine motor skills. Patient 4 experienced
improvements in motor abilities, language and cognition. Based on the TBAQ test, all four patients showed
improvements in social interactions and ability to perform focused behaviors.
April 19, 2017
Page 22

Figure 16. Changes in Cognitive and Behavioral Performance Measured by PEP-3, Vineland and TBAQ
Source: Tardieu et al., 2014.
Long-Term Open-Label Extension (OLE) Study
Lysogene is currently conducting an open-label extension study to evaluate the long-term safety and tolerability of
LYS-SAF301. This study will follow patients for 5 years post-treatment. Three-year results have been reported from
the study, suggesting that the AAV-based gene therapy is safe and well-tolerated over the long-term in patients affected
with MPS IIIA. The trial is still ongoing and we await 5 year data.
Trial Design. Lysogene’s long-term follow up study is an open-label extension (OLE) study for patients who have
already been treated with LYS-SAF301.37 The primary endpoint is an evaluation of long-term safety and tolerability
of intracerebral LYS-SAF301 administered during the Phase I/II trial. Safety and tolerability are measured through
37 https://clinicaltrials.gov/ct2/show/NCT02053064
April 19, 2017
Page 23

assessment of AEs. The secondary endpoints are an assessment of potential biomarkers for drug efficacy and
collection of further data on the use of LYS-SAF301 to treat neurological and psychological impairments.
Planned Pivotal Phase II/III Trial
This pivotal Phase II/III study is designed to evaluate the safety and efficacy of LYS-SAF302, Lysogene’s second-
generation product candidate, in patients with MPS IIIA. The Company has discussed this trial with US and EU
regulators and it has been designed to satisfy the provided guidance. Lysogene plans to launch this study by the first
quarter of 2018 and expects to enroll up to 20 patients. This could provide for a data readout by the middle of 2020.
Trial Design. This pivotal study will compare the disease progression of patients treated with LYS-SAF302 to a
control cohort of patients evaluated in the Company’s ongoing natural history study. Lysogene expects to enroll up
to 20 MPS IIIA patients with a developmental quotient greater than 50%. The developmental quotient is the ratio of
the age of child into which test scores put the child to the actual age of the child. Enrolled subjects will receive direct
CNS delivery of LYS-SAF302 into multiple brain sites. LYS-SAF302 is more potent than its predecessor product and
is expected to be used in this trial at a higher dose than was used in the Phase I/II study. The primary endpoint of this
trial will be an assessment of neurocognitive and motor development. Secondary endpoints are expected to include
assessments of behavior, sleep, quality of life, AEs, and MRI scans. The primary efficacy analysis will be conducted at
12 months with a secondary efficacy analysis to follow at 24 months. Treated patients will be followed for a total of 5
years.
Natural History Study
Lysogene is currently conducting a natural history study (NHS) in up to 25 MPS IIIA patients who remain untreated.
The purpose of this study is to add to the published data on the course of MPS IIIA and to use the data as a control
group for the pivotal Phase II/III trial. The first patient was enrolled in June 2016 and first-year data are expected in
early 2018. Second-year data from the study will follow in the first quarter of 2019.
Trial Design. This observational natural history study will recruit up to 25 patients under the age of 10 with an
emphasis on patients under the age of 5.38 The study will provide details on the course of MPS IIIA through clinical,
biochemical, neurocognitive, developmental, and behavioral measures. The primary endpoint is the measurement of
cognitive function on the Bayley scales of Infant and Toddler Development (3rd edition). The secondary endpoints
are measures of change in behavior, sleep disturbance, quality of life, and cortical grey matter volume.
Other Drugs in Development
Aside from Lysogene, Abeona Therapeutics is the only other company with a program in clinical development.
Abeona is also developing an AAV9-based gene therapy to treat MPS IIIA and is Lysogene’s closest competitor.
Abeona is currently conducting a Phase I/II trial for ABO-102 and reported interim data in October 2016. There are
4 preclinical programs as well, including 2 gene therapies. The current development landscape for MPS IIIA is shown
in Figure 17.
38 https://clinicaltrials.gov/ct2/show/NCT02746341
April 19, 2017
Page 24

Figure 17. Other Drugs in Development for MPS IIIA
Treatment Company Type Stage Next Milestone
LYS-SAF302 Lysogene Gene therapy Phase II/III
ready
Initiation of Phase
II/III in Q1 2018
ABO-102 Abeona Therapeutics Gene therapy I/II Updated results in
Q1 2017
EGT-101 Esteve S.A. Gene therapy Preclinical
Initiation of Phase
I/II study in H1
2017
AGT-184 ArmaGen
Therapeutics
Enzyme
replacement Preclinical -
Lentivector Orchard Therapeutics Gene therapy Preclinical -
SOBI003 Swedish Orphan
Biovitrum
Enzyme
replacement
therapy
Preclinical Clinical trials in 2018
Source: LifeSci Capital
ABO-102 – Abeona Therapeutics (NasdaqCM: ABEO)
Abeona Therapeutics is developing ABO-102, an AAV9-based gene therapy, to treat MPS IIIA, taking a similar
approach to Lysogene’s LYS-SAF302. Both ABO-102 and LYS-SAF302 are AAVs that encode the SGSH gene.
However, there are several important differences, most important of which is in the route of administration. ABO-
102 is given intravenously, while LYS-SAF302 is administered through a series of injections directly into the brain.
Abeona has received Orphan Drug designation from the FDA and EMA, as well as Rare Pediatric Disease designation
and Fast Track status from the FDA. While Abeona has presented interim data showing encouraging signs of efficacy,
it is not yet clear what the regulatory path for ABO-102 will be.
Phase I/II Trial. In Abeona’s first-in-human, open-label Phase I/II trial, MPS IIIA patients are given a single
intravenous injection of ABO-102.39 The delivery approach has been described in another manuscript on the use of
viral vectors to treat MPS IIIB.40 Patients are separated into two cohorts: one cohort (n=3) receiving a low-dose of
ABO-102 (5x1012 vg/kg) and another (n=3-6) receiving a high dose (1x1013 vg/kg). The following assessments will
be taken at baseline, 6 and 12 months: neurocognitive tests, motor tests, standard lab test, SGSH activity levels,
heparan sulfate levels, and quality of life. MRIs of the liver, spleen, and brain are also taken at baseline and 12 months.
The primary endpoint of this Phase I/II trial is safety and tolerability, determined by whether the patient develops
unacceptable toxicities. Secondary endpoints include measuring heparan sulfate and urine GAG levels, SGSH activity
39 https://clinicaltrials.gov/ct2/show/NCT02716246 40 Murrey, DA, et al., 2014. Feasibility and Safety of Systemic rAAV9-hNAGLU Delivery for Treating Mucopolysaccharidosis
IIIB: Toxicology, Biodistribution, and Immunological Assessments in Primates. Human Gene Therapy Clinical Development, 25(2),
pp72-84.
April 19, 2017
Page 25

levels, behavioral changes, quality of life, and neurocognitive assessments using the Leiter International Performance
Scale and the Mullen Scales of Early Learning.
Trial Results. Abeona has reported interim results from the trial on 3 subjects who were treated with the low-dose
of ABO-102. The trial is ongoing and enrollment for the high-dose cohort has commenced. To date, ABO-102 has
been considered safe and well-tolerated, although the data come from a small number of patients. At 6 months,
Abeona has reported a 63.1% reduction in heparan sulfate fragments in the cerebrospinal fluid (CSF). The company
noted a 25.8% reduction in HS fragments at 1 month, suggesting that there is a sustained improvement over time. In
addition, there decreases in liver and spleen volumes were observed. There are no accepted surrogate markers for
MPS IIIA, so it is difficult to gauge the impact on the disease.
The cognitive effects induced with ABO-102 are not easily interpreted, in light of the mixed results and small sample
size. On the Vineland Adaptive Behavior Scale (n=2), one patient improved, while the other worsened, and neither
change was meaningful relative to the natural history data. On another cognitive measure, the Mullen Scales of Early
Learning, one of the patients showed declines on 3 of the subscores relative to the natural history of MPS IIIA.
Preclinical Programs for MPSIIIA
There are currently 4 preclinical programs of interest: Esteve S.A. ( private), Orchard Therapeutics (private), ArmaGen
(private), and Swedish Orphan Biovitrum (Stockholm: SOBI.ST).
Esteve S.A. and Orchard Therapeutics both have preclinical research on gene therapy in models of MPS IIIA. Esteve
S.A.’s EGT-101 program uses the AAV9 vector to increase SGSH enzyme activity levels.41 Orchard Therapeutics, in
combined efforts with Oxford BioMedica (LSE: OXB.L), used lentiviral vectors in gene therapy to restore SGSH
activity.42 Lentiviral vectors are able to transfer larger amounts of genetic information to cells than AAV, making them
a good candidate for use in gene therapy.
ArmaGen and Swedish Orphan Biovitrum carried out preclinical research on enzyme replacement therapy in models
of MPS IIIA. ArmaGen’s AGT-184 enzyme has similar activity to SGSH with the ability to cross the blood brain
barrier.43 Swedish Orphan Biovitrum’s SOBI003 is a chemically modified variant of recombinant human SGSH. 44 Its
increased half-life allows for it to cross the blood brain barrier and act in the brain.
41 Haas, Michael J. 2013. "MPS IIIA: gene therapy for brain and body." SciBX: Science-Business eXchange 6.28. 42 "Stem Cell Gene Therapy for Fatal Childhood Disease Ready for Human Trial." Stem Cell Gene Therapy for Fatal Childhood
Disease Ready for Human Trial. 11 May 2016. 43 Urayama, A.; Grubb, J. H.; Sly, W. S.; Banks, W. A. 2008. Mannose 6-phosphate receptor-mediated transport of sulfamidase
across the blood−brain barrier in the newborn mouse. Mol. Ther. 1261−1266. 44 SOBI. European Commission Grants SOBI003 Orphan Designation for the Treatment of MPS IIIA. N.p., 19 Oct. 2016.
<http://mb.cision.com/Main/14266/2104284/577060.pdf>.
April 19, 2017
Page 26

LYS-GM101: A Gene Therapy to Treat GM1 Gangliosidosis
LYS-GM101 is an AAVrh10-based gene therapy to treat GM1 gangliosidosis by direct delivery of the gene encoding
the beta-galactosidase enzyme into the CNS. Lysogene has collaborative agreements with investigators from the
University of Massachusetts Medical School (UMMS) and Auburn University to conduct IND-enabling preclinical
studies. The Company expects to launch a Phase I/II study to evaluate the safety and efficacy of LYS-GM101.
Lysogene has received Orphan Drug designation of FDA and EMA and Rare Pediatric Disease designation form the
FDA for LYS-GM101.
GM1 Gangliosidosis
GM1 gangliosidosis is an inherited disorder that leads to degeneration of neurons in the central and peripheral nervous
systems. While there are three subtypes of this disease depending on age of onset, all arise from mutations in the
GLB1 gene. The GLB1 gene codes the beta-galactosidase enzyme in lysosomes, which breaks down gangliosides;
however, when the enzyme is mutated gangliosides accumulate in lysosomes. This build-up leads to neuronal cell
death and pathogenesis. GM1 gangliosidosis is an autosomal recessive disease. According to the NIH, incidence is
roughly 1 in 100,000-200,000 newborns. Symptoms are most severe for infantile GM1 gangliosidosis, which is the
most common form of the disease, and those affected with the disease usually do not survive past early childhood.
There are currently no effective treatments for the disease.
Causes & Pathogenesis. GM1 gangliosidosis is caused by mutations to the GLB1 gene. The three subtypes of GM1
gangliosidosis are infantile, juvenile and adult, and all have similar symptoms and are caused by mutations to the same
gene. Thus far, 102 mutations have been reported in the GLB1 gene, with the majority being missense or nonsense
mutations.45 This gene is responsible for making the beta-galactosidase enzyme, which has a key function in the brain.
Beta-galactosidase acts in lysosomes, where it degrades GM1 gangliosides, as shown in Figure 18. It does this by
catalyzing the hydrolysis of beta-galactosidases into monosaccharides through breaking glycosidic bonds.46
45 Brunetti-Pierri N, Scaglia F. 2008. GM1 gangliosidosis: review of clinical, molecular and therapeutic aspects. Mol Genet Metab.
94: 391-396. 46 Sandhoff K., Harzer K. 2013. Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis. J.
Neurosci. 33: 10195–10208.
April 19, 2017
Page 27

Figure 18. Pathway of Lysosomal Ganglioside Degradation
Source: Brunetti-Pierri et al, 2008
With mutations in the gene encoding beta-galactosidase, GM1 ganglioside accumulates in the lysosomes. Gangliosides
are the primary glycolipids in neuronal plasma membranes, and they play an important role in multiple signaling
pathways. In high levels, however, GM1 gangliosides are toxic, especially in the brain. Ganglioside accumulation leads
to neurodegeneration, which eventually can disrupt normal function.
It is unknown exactly how ganglioside accumulation leads to neuronal cell death. However, it is hypothesized that
ganglioside buildup may impair synaptic transmission. Other possible mechanisms include disturbed neuron-
oligodendrocyte interactions, stress response, and abnormal axoplasmic transport leading to myelin deficiency.47
Symptoms & Diagnosis. GM1 patients can be separated into three subtypes based on the age of disease onset. The
infantile form (Type I) is the most severe and is usually diagnosed by 6 months of age. Symptoms become apparent
in the juvenile form (Type II) between 18 months to 5 years of age. The least common adult form (Type III) has a
wide onset range of 3 to 30 years of age.48
Symptoms are the most severe for infantile GM1 gangliosidosis, also referred to as Type I. By 6 months, development
begins to slow and muscles begin to weaken. There is eventually a developmental regression. The children may develop
an enlarged liver and spleen, skeletal abnormalities, seizures, severe intellectual disabilities, and clouding of the cornea.
The retina will deteriorate and vision loss occurs. Some individuals may also have coarse facial features, enlarged gums,
and cardiomyopathy. Most individuals affected by Type I GM1 gangliosidosis do not survive past early childhood.
47 Brunetti-Pierri N, Scaglia F. 2008. GM1 gangliosidosis: review of clinical, molecular and therapeutic aspects. Mol Genet Metab.
94: 391-396. 48 Weismann C, Ferreira J, Keeler A, Linghua Q, Shaffer S, Xu Z, Gao G, Sena-Esteves M; 2015. Systemic AAV9 gene transfer
in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum Mol Genet. 24 (15): 4353-4364.
April 19, 2017
Page 28

Patients with juvenile GM1 gangliosidosis can experience a wide variety of symptoms. Meanwhile, those affected with
adult GM1 gangliosidosis usually have normal early neurological development and no physical abnormalities. 49 Adult
GM1 gangliosidosis patients will also undergo a slow progression of dementia with certain aspects of Parkinson’s
disease.50
To diagnose GM1 gangliosidosis, patients may undergo enzyme analysis or genetic testing of the GLB1 gene. Prenatal
diagnosis may also be done through enzyme activity analysis or genetic testing of chorionic villus or amniotic fluid
cells.51 There are currently no approved therapies for individuals affected by GM1 gangliosidosis. While some
symptoms may be treated, there are no effective means for slowing or stopping the progression of the disease.
Planned Clinical Program for LYS-GM101
Lysogene plans to conduct a Phase I/II study to evaluate the safety, tolerability, and efficacy of LYS-GM101 in the
treatment of GM1 patients. The trial will consist of two parts: a 14-month dose-escalation safety study and a 12-month
evaluation of efficacy with an additional 12 month follow-up. In the dose-escalation portion of the study, 3 subjects
will be enrolled per dose tested for a total of 6 subjects. The efficacy portion of the study will enroll 6 new subjects.
The trial will follow an adaptive design, allowing for adjustments to the study design as the trial progresses. The
primary endpoint will assess motor function, and the secondary endpoints will assess speech ability, plus safety and
tolerability. The analysis of these endpoints will compare these data to natural history data. Lysogene expects to launch
this trial in the first quarter of 2019 with sites in both the US and Europe.
Other Drugs in Development
Aside from Lysogene’s LYS-GM101, there is only other gene therapy in development for GM1. However, unlike
Lysogene’s direct delivery into the brain, the other gene therapy, in development by UMass, uses IV administration.
This may not be the ideal route of administration and may not provide for sufficient viral titers in the brain. The
development pipeline for GM1 is shown in Figure 19.
49 Muthane U, Chickabasaviah Y, Kaneski C, et al. 2004. Clinical features of adult GM1 gangliosidosis: report of three Indian
patients and review of 40 cases. Mov Disord. 11:1334-41. 50 Suzuki K. 1991. Neuropathology of late onset gangliosidosis. A review. Dev Neurosci. 13(4-5): 205-10. 51 Regier D, Tifft C. 2013. GLB1-related disorders. GeneReviews. University of Washington, Seattle.
April 19, 2017
Page 29

Figure 19. Development Pipeline for GM1
Company Program Mechanism Route Stage
Univ of
Minnesota
Miglustat
& ketogenic diet Chaperone Oral Phase III
Lysogene LYS-GM101 AAVrh10 gene
therapy
Neurosurgical
procedure Preclinical
UMass AAV9 AAV9 gene
therapy
Single IV
injection Preclinical
Dorphan MPS IVB
(Morquio disease type B) Chaperone Oral Preclinical
Biostrategies
LC
Lectin-assisted delivery of
corrective enzyme
Enzyme
replacement Transnasal Preclinical
Source: LifeSci Capital
Intellectual Property & Licensing
LYS-SAF302. Lysogene has signed a licensing agreement with RegenX Bio, a subsidiary of the University of
Pennsylvania, covering the use of AAVrh10 to treat MPS IIIA.
GM1 Gangliosidosis. Lysogene has signed a partnership agreement with the University of Massachusetts covering
the development therapies for GM1. Lysogene also sponsors research at UMass and Auburn University.
Management Team
Karen Aiach
Founder & CEO
Founder and CEO of Lysogene, Ms. Aiach is also the mother of a child with MPS IIIA. She has a strong business
background starting her career with Arthur Andersen specializing in audit and transaction services. Her entrepreneurial
experience includes founding and running a financial business consultancy. From 2008 to 2009, Ms. Aiach served as
a Member of the Pediatric Committee at the European Medicines Agency (EMA), established in accordance with the
European Pediatric Regulation, as a patient representative. In 2008, she also served on the French Ethical Review
Board CCPPRB at Ambroise Paré Hospital. Ms. Aiach has been involved with several not-for-profit organizations
engaged in advocacy and research in the field of rare diseases such as Alliance Sanfilippo and Eurordis, where she
served on the board as treasurer from 2010 to 2011. She is a founding executive member of the International Rare
Diseases Research Consortium (IRDiRC). Ms. Aiach graduated from the ESSEC Business School and majored in
Economics.
April 19, 2017
Page 30

Kimberley S. Gannon
Chief Scientific Officer
Kim is responsible for the continuing development of Lysogene’s scientific programmes. Prior to joining Lysogene,
Kim was Senior Vice President of Preclinical Research and Development at NeuroPhage Pharmaceuticals, Inc., where
she directed preclinical and nonclinical drug development activities. She has also held various leadership positions at
several early stage ventures, including CereMedix where she was VP of Research & Development, EPIX (Predix)
Pharmaceuticals where she served as Senior Director of Biology, and Eolas Biosciences where she was Head of US
Operations. Dr. Gannon received her PhD in Neuroscience from Florida State University and conducted her
postdoctoral training at the Roche Institute of Molecular Biology, Hoffmann-LaRoche Inc., in New Jersey and at
Mount Sinai School of Medicine in the Department of Physiology and Biophysics in New York.
Mark Plavsic
Chief Technology Officer
Mark is responsible for Lysogene’ s manufacturing strategy and CMC operational development. He has extensive
technical experience across a diverse range of functions, proven knowledge of quality compliance and regulatory
requirements and strong business and leadership skills gained from over 30 years of experience working in North
America, Europe and Australasia. Prior to joining Lysogene, Mark worked as Head of Process Development and
Manufacturing at Torque Therapeutics, leading multiple functions including process and analytics development and
ensuring quality controls and design across manufacturing. Prior to that, Dr. Plavsic held multiple positions with
Sanofi Genzyme, Astra Zeneca and Invitrogen Corporation. Dr. Plavsic received his Ph.D in Virology and Molecular
Cell Biology, M.S. in Virology and Immunology, and Doctor of Veterinary Medicine degree from the University of
Belgrade in Yugoslavia. He has a Board certification in Microbiology, subspecialty Virology from the American
College of Veterinary Microbiologists.
Soraya Bekkali
Senior Vice President and Chief Medical Officer
Soraya is responsible for Lysogene’s clinical development and regulatory affairs. Physician by background, Soraya
specialized in clinical research and biostatistics before starting her career in academia. Prior to joining Lysogene,
Soraya worked for Aventis Pharma, followed by Orphan Europe, a pharmaceutical company dedicated to the
development of treatments for rare diseases, before joining Sanofi in 2007, where she held successively increasing
leadership positions including Vice President and Global Ophthalmology Unit Head. Soraya’s extensive industry
experience spanning from early development to ensuring market access, with an important focus on viral and non-
viral gene therapy products, has led to a rich breadth of experience across a variety of therapeutic areas.
Samantha Parker
Chief Patient Access Officer
Samantha Parker is Head of Lysogene’s Patient and Policy Affairs. Ms. Parker has specialised in rare disease research
and public health since 2000. She has focused on expanding the expert disease community of healthcare professionals,
patients, industry and regulators; building disease registries and natural history studies; developing consensus care
guidelines; and developing strategies to improve the quality of diagnosis and patient care. Ms. Parker contributed, for
several years, to the area of independent professional education in rare diseases. She is a member of the European
April 19, 2017
Page 31

Group of Experts on Rare Diseases (EGRD) and the International Rare Diseases Research Consortium (IRDiRC).
Ms. Parker has a B.A. (Hons) from Edinburgh University and an M.B.A (with distinction) in Life Sciences.
Sean O’Bryan
Vice President of Regulatory Affairs and Quality Assurance
Sean is responsible for providing global regulatory strategic leadership and for the creation and maintenance of quality
systems assuring global compliance. Prior to joining Lysogene, he was the Director of Regulatory Affairs at bluebird
bio leading efforts on the treatment of the rare disease CCALD through LVV gene therapy where he initiated and led
the RA CMC group across all programs including CNS, hematology and oncology. Prior to this, he worked 8 years
at Genzyme/Sanofi serving as a global regulatory lead for the Cell Therapy and Regenerative Medicine division. Sean
has more than 20 years of regulatory experience across a range of categories including biologics, gene therapy,
chemistry, manufacturing and control (CMC) and medical device. Sean holds a BS in Biology and Analysis and Policy
from Boston University and is RAPs certified.
Sarah Ankri
Finance Director
Sarah is responsible for overseeing Lysogene’s financial operations, budget and financing strategy. Prior to joining
Lysogene, she was CFO at Theraclion where she successfully led the company’s IPO in 2014. She acquired her
financial knowledge related to Bio- and Medtech companies, including fundraising (Europe and USA) during the six
years she spent at Ernst & Young working on legal audit and advisory/acquisition missions in the healthcare sector.
Sarah is a graduate of the National Institute of Agronomy (INA P-G/AgroParisTech) and has a Masters in Economy
and Management.
Risk to an Investment
We consider an investment in Lysogene to be a high-risk investment. Lysogene has generated limited clinical data to
date, and early signs of safety and efficacy may not necessarily translate into late-stage success. There are clinical and
commercialization risks associated with their program as well. As with any company, Lysogene may be unable to
obtain sufficient capital to fund planned development programs. There are regulatory risks associated with the
development of any drug, and Lysogene may not receive FDA or EMA approval for its candidates despite significant
time and financial investments. Regulatory approval to market and sell a drug does not guarantee that the drug will
penetrate the market, and sales may not meet expectations.
April 19, 2017
Page 32

Analyst CertificationThe research analyst denoted by an “AC” on the cover of this report certifies (or, where multiple research analysts are primarily responsiblefor this report, the research analyst denoted by an “AC” on the cover or within the document individually certifies), with respect to eachsecurity or subject company that the research analyst covers in this research, that: (1) all of the views expressed in this report accuratelyreflect his or her personal views about any and all of the subject securities or subject companies, and (2) no part of any of the researchanalyst's compensation was, is, or will be directly or indirectly related to the specific recommendations or views expressed by the researchanalyst(s) in this report.
DISCLOSURESThis research contains the views, opinions and recommendations of LifeSci Capital, LLC (“LSC”) research analysts. LSC (or an affiliate)has received compensation from the subject company for producing this research report. Additionally, LSC expects to receive or intendsto seek compensation for investment banking services from the subject company in the next three months. LSC (or an affiliate) has alsoprovided non-investment banking securities-related services, non-securities services, and other products or services other than investmentbanking services to the subject company and received compensation for such services within the past 12 months. LSC does not makea market in the securities of the subject company.
Neither the research analyst(s), a member of the research analyst’s household, nor any individual directly involved in the preparation ofthis report, has a financial interest in the securities of the subject company. Neither LSC nor any of its affiliates beneficially own 1% ormore of any class of common equity securities of the subject company.
LSC is a member of FINRA and SIPC. Information has been obtained from sources believed to be reliable but LSC or its affiliates (LifeSciAdvisors, LLC) do not warrant its completeness or accuracy except with respect to any disclosures relative to LSC and/or its affiliates andthe analyst's involvement with the company that is the subject of the research. Any pricing is as of the close of market for the securitiesdiscussed, unless otherwise stated. Opinions and estimates constitute LSC’s judgment as of the date of this report and are subject to changewithout notice. Past performance is not indicative of future results. This material is not intended as an offer or solicitation for the purchaseor sale of any financial instrument. The opinions and recommendations herein do not take into account individual client circumstances,objectives, or needs and are not intended as recommendations of particular securities, companies, financial instruments or strategies toparticular clients. The recipient of this report must make his/her/its own independent decisions regarding any securities or financialinstruments mentioned herein. Periodic updates may be provided on companies/industries based on company specific developments orannouncements, market conditions or any other publicly available information. Additional information is available upon request.
No part of this report may be reproduced in any form without the express written permission of LSC. Copyright 2017.
April 19, 2017
Page 33