molecular basis of cancer non-lethal genetic damage a tumor is formed by the clonal expansion of a...
TRANSCRIPT

MOLECULAR BASISof CANCER
• NON-lethal genetic damage• A tumor is formed by the clonal expansion
of a single precursor cell (monoclonal)• Four classes of normal regulatory genes– PROTO-oncogenes– Oncogenes Oncoproteins– DNA repair genes– Apoptosis genes
• Carcinogenesis is a multistep process

TRANSFORMATION &PROGRESSION
• Self-sufficiency in growth signals• Insensitivity to growth-inhibiting signals• Evasion of apoptosis• Defects in DNA repair: “Spell checker”• Limitless replicative potential: Telomerase• Angiogenesis• Invasive ability• Metastatic ability
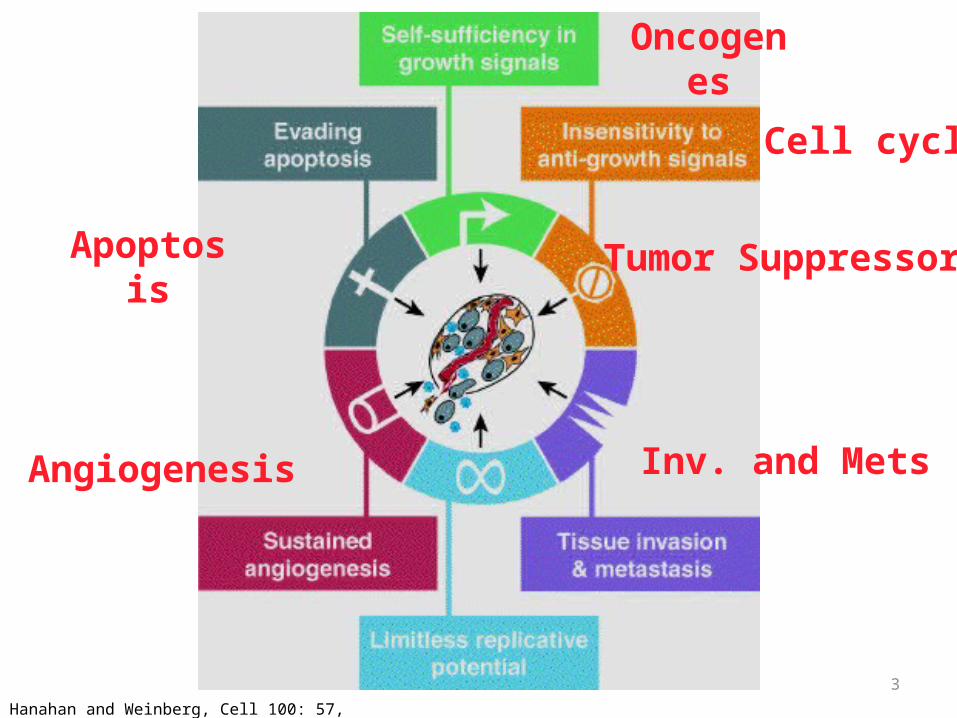
3Hanahan and Weinberg, Cell 100: 57, 2000
Apoptosis
Oncogenes
Tumor Suppressor
Inv. and MetsAngiogenesis
Cell cycle

4
ONCOGENES
• Oncogenes are mutated forms of cellular proto-oncogenes.
• Proto-oncogenes code for cellular proteins which regulate normal cell growth and differentiation.

5
Class I: Growth Factors
Class II: Receptors for Growth Factors and Hormones
Class III: Intracellular Signal Transducers
Class IV: Nuclear Transcription Factors
Class V: Cell-Cycle Control Proteins
Five types of proteins encoded by proto-oncogenes participate in control of cell growth:

6
4. NuclearProteins:
TranscriptionFactors
5. Cell GrowthGenes
3. CytoplasmicSignal Transduction
Proteins
1. Secreted Growth Factors
2. Growth Factor Receptors
Functions of Cellular Proto-Oncogenes

ONCOGENES• Are MUTATIONS of NORMAL genes
(PROTO-oncogenes)–Growth Factors–Growth Factor Receptors– Signal Transduction Proteins (RAS)–Nuclear Regulatory Proteins–Cell Cycle Regulators
• Oncogenes code for Oncoproteins

Mutations that confer these properties fall into two categories
• Oncogene• : a cancer-causing gene that has been mutated to cause an
increase in• activity, or the activity becomes constitutive, or a new
activity is acquired.• -a mutation in a single allele is sufficient to transform cells
(dominant).• -originally identified as viral proteins that resembled
normal human proteins.• -the term "proto-oncogene" refers to the normal protein
that has not been mutated

• tumor Suppressor gene• : cancer-causing gene that has been mutated
to cause a loss of activity.• -mutations are required in both alleles to
transform cells (recessive)

1
2
3
4
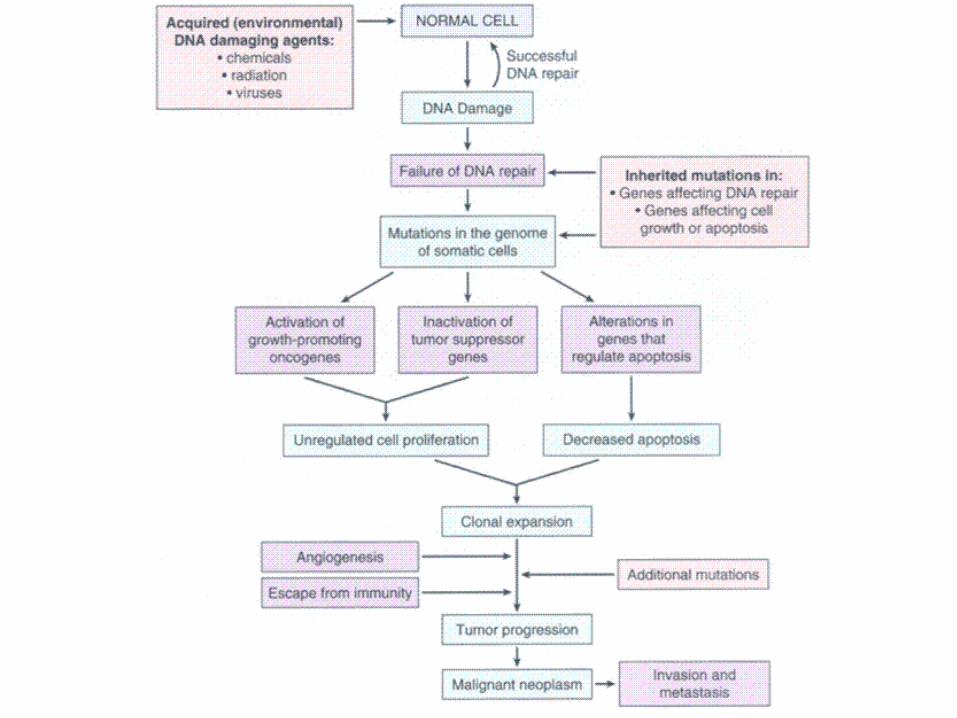
4 types of genetic mutations that contribute to cancer

• Categories of oncogenes• A. Growth factors• -generally not directly involved
transformation, but increased expression seen as part of
• an autocrine loop due to changes in other steps in the same pathway

growth factor receptors
• -They are transmembrane proteins with an external ligand binding domain and an
• internal tyrsosine kinase domain.• -oncogenic mutations can result in
dimerization and activation in the absence of • ligand• -more commonly, increased activity is a result
of overexpression of receptors

Growth factor receptors
• They are transmembrane proteins with an external ligand binding domain and an
• internal tyrsosine kinase domain.• -Oncogenic mutations can result in
dimerization and activation in the absence of • ligand• -More commonly, increased activity is a result
of overexpression of receptors.

signal transducers
• -Activated directly or indirectly by growth factor receptors
• -Activation of signal transducers triggers a phosporylation cascade that ultimately
• results in changes in gene expression at the transcriptional level.
• -mutations in RAS• , a GTPase, are the most common oncogenic
abnormality in tumors• -failure to hydrolyze GTP locks RAS in its active form.

Transcription factors
• -Transcription factors contain DNA binding domains.
• Sequences• Regulate expression of genes essential for
passage through the cell cycle, or• regulation of apoptosis.
• -

Normal CELL CYCLE Phases
INHIBITORS: Cip/Kip, INK4/ARF
Tumor (really growth) suppressor genes: p53

cyclins and cyclin-dependent kinases
• -cyclins are only expressed at specific stages of the cell cycle
• -cyclin-dependent kinases are expressed constitutively, but must bind cyclins for
• activation; phosphorylation of target proteins essential for progression through
• cell cycle
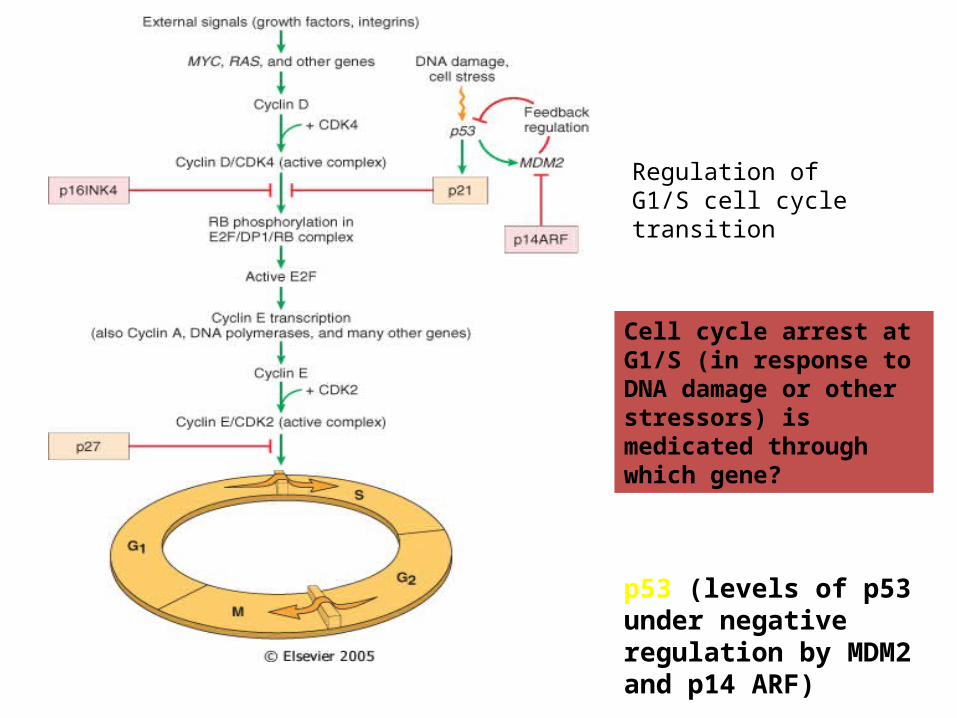
Regulation of G1/S cell cycle transition
Cell cycle arrest at G1/S (in response to DNA damage or other stressors) is medicated through which gene?
p53 (levels of p53 under negative regulation by MDM2 and p14 ARF)

• a second level of control is achieved by CDK inhibitors
• -p21 family (broad specificity) and the INK4 (p16) family (CDK4/6
• specific)• -overexpression of cyclin D and CDK4 common.• -phosphorylate and inactivate • Rb

CategoryPROTO- Oncogene
Mode of Activation
Associated Human Tumor
GFsPDGF-β chain SIS Overexpression Astrocytoma
OsteosarcomaFibroblast growth factors
HST-1 Overexpression Stomach cancer
INT-2 Amplification Bladder cancer
Breast cancerMelanoma
TGFα TGFα Overexpression Astrocytomas
Hepatocellular carcinomas
HGF HGF Overexpression Thyroid cancer

CategoryPROTO- Oncogene
Mode of Activation
Associated Human Tumor
GF ReceptorsEGF-receptor family
ERB-B1 (ECFR)
Overexpression Squamous cell carcinomas of lung, gliomas
ERB-B2 Amplification Breast and ovarian cancers
CSF-1 receptor FMS Point mutation Leukemia
Receptor for neurotrophic factors
RET Point mutation Multiple endocrine neoplasia 2A and B, familial medullary thyroid carcinomas
PDGF receptor PDGF-R Overexpression Gliomas
Receptor for stem cell (steel) factor
KIT Point mutation Gastrointestinal stromal tumors and other soft tissue tumors

CategoryPROTO- Oncogene
Mode of Activation
Associated Human Tumor
Signal TransductionProteins
GTP-binding K-RAS Point mutation Colon, lung, and pancreatic tumors
H-RAS Point mutation Bladder and kidney tumors
N-RAS Point mutation Melanomas, hematologic malignancies
Nonreceptor tyrosine kinase
ABL Translocation Chronic myeloid leukemia
Acute lymphoblastic leukemia
RAS signal transduction
BRAF Point mutation Melanomas
WNT signal transduction
β-catenin Point mutation Hepatoblastomas, hepatocellular carcinoma
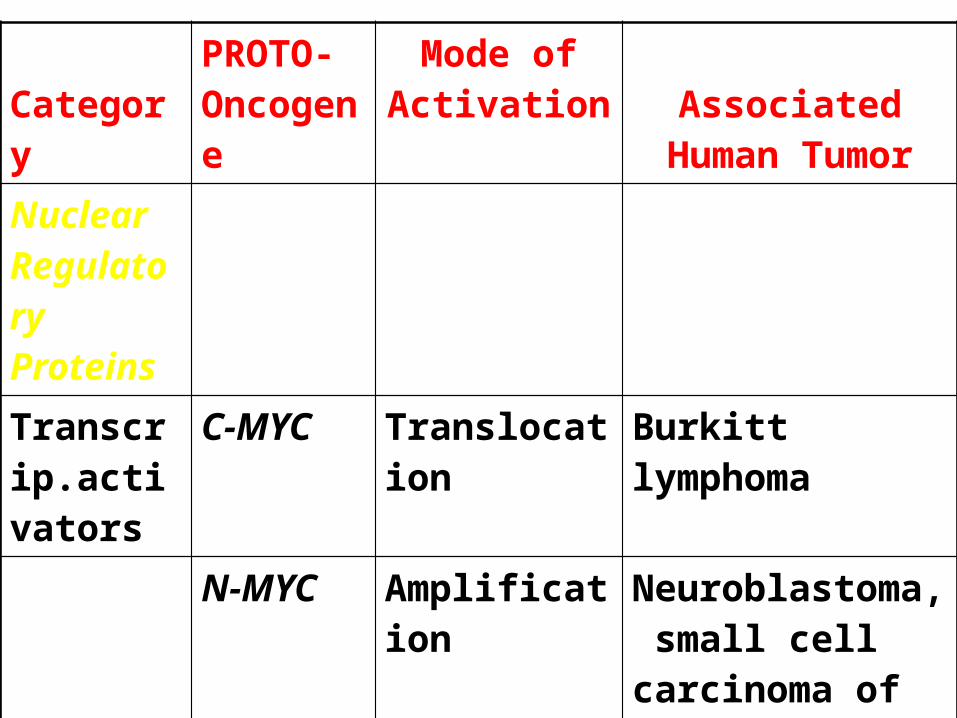
CategoryPROTO- Oncogene
Mode of Activation Associated Human
TumorNuclear Regulatory Proteins
Transcrip.activators
C-MYC Translocation Burkitt lymphoma
N-MYC Amplification Neuroblastoma, small cell carcinoma of lung
L-MYC Amplification Small cell carcinoma of lung
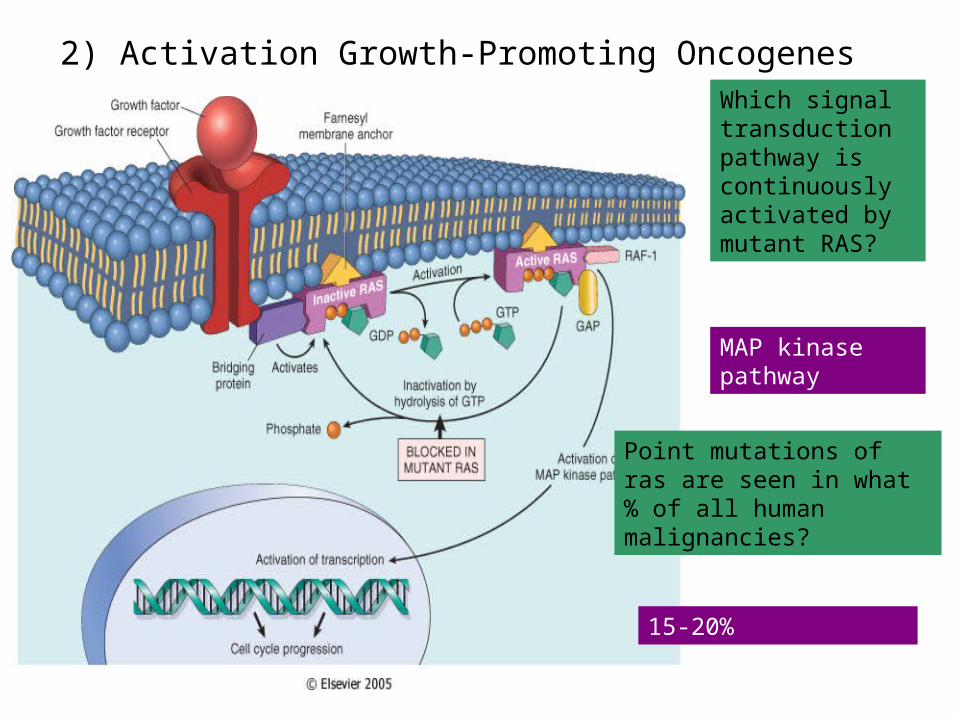
2) Activation Growth-Promoting OncogenesWhich signal transduction pathway is continuously activated by mutant RAS?
MAP kinase pathway
Point mutations of ras are seen in what % of all human malignancies?
15-20%

Tumor supressor gene
• . Tumor suppressor were originally identified as inherited mutations that confer a
• predisposition to cancer (familial form).• -inheritance is dominant, meaning a single
defective allele is sufficient to confer• the predisposition

• Inactivation of tumor suppressors can occur • Sporadically• -sequential inactivation of both alleles in somatic cells• You may hear the term • haploinsufficiency• , which refers to inactivation of a single• allele contributing to malignancy.• -usually not the initiating event, but exacerbating.• Viral inactivation• -HPV expresses proteins that inhibit Rb and p53 function.

P53 and RASp53
• Activates DNA repair proteins
• Sentinel of G1/S transition
• Initiates apoptosis• Mutated in more than
50% of all human cancers
RAS• H, N, K, etc., varieties• Single most common
abnormality of dominant oncogenes in human tumors
• Present in about 1/3 of all human cancers

RB gene
• a.Loss of RB function confers a predisposition to retinoblastoma.
• occurs in both the familial form (early onset) and sporadic fromthe basis for tissue specificity of some tumor suppressors is unknown, but
• presumably is due to the transcriptional profile of the tissue, determined by tissue
• function

P53
• p53 is the most commonly mutated gene in tumors
• -over 50% of all tumors lack functional p53• -• Li-Fraumeni syndrome• : inheritance of a single defective copy of p53
results in a • predisposition to a wide spectrum of cancers.• -p53 is a transcription factor.

1: Failure of DNA Repair (acquired)
Normal function of p53 is to upregulate activity of which 2 genes to allow repair of DNA?
p21
GADD45

• Unlike Rb, p53 inhibits G1 progression only in response to DNA damage
• -normally p53 is very unstable, due to proteolytic degradation triggered by
• mdm2• .• -p53 is phosphorylated in response to DNA damage; mdm2 no
longer binds p53• -p53 upregulates expression of p21, which in turn inhibits G1/S
CDKs.• c. In response to excessive DNA damage, p53 can trigger
apoptosis

• Some other tumor suppressors found to be inactivated in tumors inhibit proliferation by
• various mechanisms:• -APC: degradation of • b• -catenin, a transcriptional activator anchored to E-cadherins• -NF-1: activates GTPase activity of ras• -TGF-• b• receptor: a tyrosine kinase that upregulates expression of CDK
inhibitors• -

• -PTEN: dephosphorylates inositol phospholipids, which act as docking sites for
• intracellular signaling proteins
• VHL: transcriptional elongation• -WT-1: transcriptional regulator

MYC• Encodes for transcription factors• Also involved with apoptosis

Tumor (really “GROWTH”) suppressor genes
• TGF-β COLON• E-cadherin STOMACH• NF-1,2 NEURAL TUMORS• APC/β-cadherin GI, MELANOMA• SMADs GI• RB RETINOBLASTOMA• P53 EVERYTHING!!• WT-1 WILMS TUMOR• p16 (INK4a) GI, BREAST• BRCA-1,2 BREAST• KLF6 PROSTATE

Evasion of APOPTOSIS
•BCL-2•p53•MYC

DNA REPAIR GENE DEFECTS
• DNA repair is like a spell checker
• HNPCC (Hereditary Non-Polyposis Colon Cancer [Lynch]): TGF-β, β-catenin, BAX
• Xeroderma Pigmentosum: UV fixing gene• Ataxia Telangiectasia: ATM gene• Bloom Syndrome: defective helicase• Fanconi anemia

LIMITLESS REPLICATIVE POTENTIAL
• TELOMERES determine the limited number of duplications a cell will have, like a cat with nine lives.• TELOMERASE, present in >90% of
human cancers, changes telomeres so they will have UNLIMITED replicative potential

TUMOR ANGIOGENESIS• Q: How close to a blood vessel must a cell be?• A: 1-2 mm
• Activation of VEGF and FGF-b
• Tumor size is regulated (allowed) by angiogenesis/anti-angiogenesis balance

TRANSFORMATIONGROWTH
BM INVASIONANGIOGENESISINTRAVASATIONEMBOLIZATION
ADHESIONEXTRAVASATION
METASTATIC GROWTHetc.

Invasion Factors
• Detachment ("loosening up") of the tumor cells from each other • Attachment to matrix components • Degradation of ECM, e.g.,
collagenase, etc. • Migration of tumor cells


METASTATIC GENES?
• NM23• KAI-1• KiSS

CHROMOSOME CHANGESin CANCER
• TRANSLOCATIONS and INVERSIONS
• Occur in MOST Lymphomas/Leukemias• Occur in MANY (and growing numbers) of NON-
hematologic malignancies also

Malignancy Translocation Affected Genes
Chronic myeloid leukemia (9;22)(q34;q11) Ab1 9q34
bcr 22q11
Acute leukemias (AML and ALL) (4;11)(q21;q23) AF4 4q21
MLL 11q23
(6;11)(q27;q23) AF6 6q27
MLL 11q23
Burkitt lymphoma (8;14)(q24;q32) c-myc 8q24
IgH 14q32
Mantle cell lymphoma (11;14)(q13;q32) Cyclin D 11q13
IgH 14q32
Follicular lymphoma (14;18)(q32;q21) IgH 14q32
bcl-2 18q21
T-cell acute lymphoblastic leukemia (8;14)(q24;q11) c-myc 8q24
TCR-α 14q11
(10;14)(q24;q11) Hox 11 10q24
TCR-α 14q11
Ewing sarcoma (11;22)(q24;q12) Fl-1 11q24
EWS 22q12

Carcinogenesis is “MULTISTEP”• NO single oncogene causes cancer
• BOTH several oncogenes AND several tumor suppressor genes must be involved
• Gatekeeper/Caretaker concept–Gatekeepers: ONCOGENES and TUMOR
SUPPRESSOR GENES
–Caretakers: DNA REPAIR GENES
• Tumor “PROGRESSION”– ANGIOGENESIS– HETEROGENEITY from original single cell

Carcinogenesis: The USUAL (3) Suspects
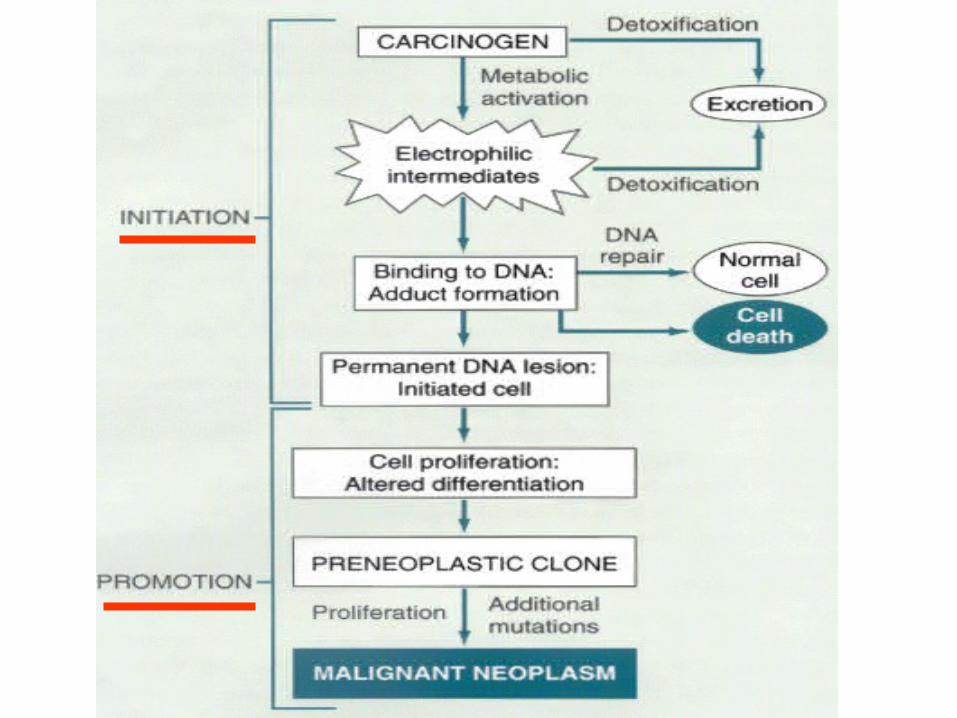
• Initiation/Promotion concept:– BOTH initiators AND promotors are needed– NEITHER can cause cancer by itself
–INITIATORS (carcinogens) cause MUTATIONS– PROMOTORS are NOT carcinogenic by themselves,
and MUST take effect AFTER initiation, NOT before
–PROMOTORS enhance the proliferation of initiated cells

Q: WHO are the usual suspects?• Inflammation?• Teratogenesis?• Immune
Suppression?• Neoplasia?• Mutations?

A: The SAME 3 that are ALWAYS blamed!
•1) Chemicals•2) Radiation•3) Infectious Pathogens
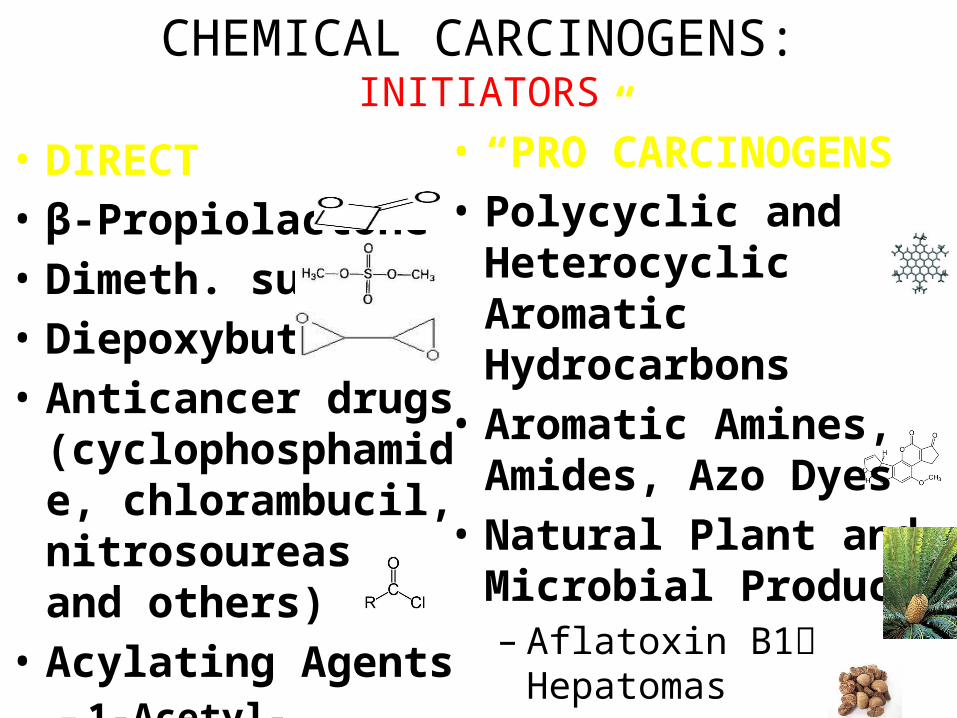
CHEMICAL CARCINOGENS:INITIATORS
• DIRECT• β-Propiolactone• Dimeth. sulfate• Diepoxybutane• Anticancer drugs
(cyclophosphamide, chlorambucil, nitrosoureas, and others)
• Acylating Agents– 1-Acetyl-imidazole– Dimethylcarbamyl
chloride
• “PRO”CARCINOGENS• Polycyclic and
Heterocyclic Aromatic Hydrocarbons
• Aromatic Amines, Amides, Azo Dyes
• Natural Plant and Microbial Products– Aflatoxin B1 Hepatomas– Griseofulvin Antifungal– Cycasin from cycads– Safrole from sassafras– Betel nuts Oral SCC

CHEMICAL CARCINOGENS:INITIATORS
•OTHERS• Nitrosamine and amides (tar, nitrites)• Vinyl chloride angiosarcoma in Kentucky• Nickel• Chromium• Insecticides• Fungicides• PolyChlorinated Biphenyls (PCBs)

CHEMICAL CARCINOGENS:PROMOTORS
• HORMONES• PHORBOL ESTERS (TPA), activate kinase C• PHENOLS• DRUGS, many
“Initiated” cells respond and proliferate FASTER to promotors than normal cells

RADIATION CARCINOGENS
• UV: BCC, SCC, MM (i.e., all 3)
• IONIZING: photons and particulate– Hematopoetic and Thyroid (90%/15yrs) tumors in
fallout victims– Solid tumors either less susceptible or require a
longer latency period than LEUK/LYMPH– BCCs in Therapeutic Radiation

VIRAL CARCINOGENESIS
• HPV SCC• EBV Burkitt Lymphoma• HBV HepatoCellular Carcinoma (Hepatoma)• HTLV1 T-Cell Malignancies• KSHV Kaposi Sarcoma

H. pylori CARCINOGENESIS
• 100% of gastric lymphomas (i.e., M.A.L.T.-omas)
• Gastric CARCINOMAS also!

HOST DEFENSES
• IMMUNE SURVEILLENCE CONCEPT
• CD8+ T-Cells• NK cells• MACROPHAGES• ANTIBODIES

CYTOTOXIC CD8+ T-CELLS are the main eliminators of tumor cells

How do tumor cellsescape immune surveillance?
• Mutation, like microbes
•↓ MHC molecules on tumor cell surface• Lack of CO-stimulation molecules, e.g.,
(CD28, ICOS), not just Ag-Ab recognition• Immunosuppressive agents• Antigen masking• Apoptosis of cytotoxic T-Cells (CD8), i.e., the
damn tumor cell KILLS the T-cell!

Effects of TUMOR on the HOST
• Location anatomic ENCROACHMENT• HORMONE production• Bleeding, Infection• ACUTE symptoms, e.g., rupture, infarction• METASTASES

CACHEXIA• Reduced diet: Fat loss>Muscle loss• Cachexia: Fat loss AND Muscle loss• TNF (α by default)• IL-(6)• PIF (Proteolysis Inducing Factor)

PARA-Neoplastic Syndromes
•Endocrine (next)• Nerve/Muscle, e.g., myasthenia w. lung ca.• Skin: e.g., acanthosis nigricans,
dermatomyositis• Bone/Joint/Soft tissue: HPOA (Hypertrophic
Pulmonary OsteoArthropathy)• Vascular: Trousseau, Endocarditis• Hematologic: Anemias• Renal: e.g., Nephrotic Syndrome

ENDOCRINECushing syndrome Small cell carcinoma of lung ACTH or ACTH-like substance
Pancreatic carcinoma
Neural tumors
Syndrome of inappropriate antidiuretic hormone secretion
Small cell carcinoma of lung; intracranial neoplasms
Antidiuretic hormone or atrial natriuretic hormones
Hypercalcemia Squamous cell carcinoma of lungParathyroid hormone-related protein
(PTHRP), TGF-α, TNF, IL-1
Breast carcinoma
Renal carcinoma
Adult T-cell leukemia/lymphoma
Ovarian carcinoma
Hypoglycemia Fibrosarcoma Insulin or insulin-like substance
Other mesenchymal sarcomas
Hepatocellular carcinoma
Carcinoid syndrome Bronchial adenoma (carcinoid) Serotonin, bradykinin
Pancreatic carcinoma
Gastric carcinoma
Polycythemia Renal carcinoma Erythropoietin
Cerebellar hemangioma
Hepatocellular carcinoma

GRADING/STAGING
• GRADING: HOW “DIFFERENTIATED” ARE THE CELLS?• STAGING: HOW MUCH ANATOMIC
EXTENSION? TNM• Which one of the above do you
think is more important?

WELL?(pearls)
MODERATE?(intercellular bridges)
POOR?(WTF!?!)
GRADING for Squamous Cell Carcinoma

ADENOCARCINOMA GRADINGLet’s have some FUN!

LAB DIAGNOSIS• BIOPSY• CYTOLOGY: (exfoliative)• CYTOLOGY: (FNA, Fine Needle
Aspirate)

IMMUNOHISTOCHEMISTRY
• Categorization of undifferentiated tumors• Leukemias/Lymphomas• Site of origin• Receptors, e.g., ERA, PRA