mithramycin depletes specificity protein 1 and activates...
TRANSCRIPT

Cancer Therapy: Preclinical
Mithramycin Depletes Specificity Protein 1 andActivates p53 to Mediate Senescence andApoptosis of Malignant Pleural MesotheliomaCellsMahadevRao1, ScottM. Atay1,Vivek Shukla1,YoungHong1,Trevor Upham1, R.Taylor Ripley1,Julie A. Hong1, Mary Zhang1, Emily Reardon1, Patricia Fetsch2, Markku Miettinen2,Xinmin Li3, Cody J. Peer4, Tristan Sissung4,William D. Figg4, Assunta De Rienzo5,Raphael Bueno5, and David S. Schrump1
Abstract
Purpose: Specificity protein 1 (SP1) is an oncogenic transcrip-tion factor overexpressed in various human malignancies. Thisstudy sought to examine SP1 expression in malignant pleuralmesotheliomas (MPM) and ascertain the potential efficacy oftargeting SP1 in these neoplasms.
Experimental Design: qRT-PCR, immunoblotting, and immu-nohistochemical techniques were used to evaluate SP1 expressionin cultured MPM cells and MPM specimens and normal meso-thelial cells/pleura. MTS, chemotaxis, soft agar, b-galactosidase,and Apo-BrdUrd techniques were used to assess proliferation,migration, clonogenicity, senescence, and apoptosis inMPM cellsfollowing SP1 knockdown, p53 overexpression, or mithramycintreatment. Murine subcutaneous and intraperitoneal xenograftmodels were used to examine effects of mithramycin on MPMgrowth in vivo. Microarray, qRT-PCR, immunoblotting, and chro-matin immunoprecipitation techniques were used to examinegene expression profilesmediated bymithramycin and combined
SP1 knockdown/p53 overexpression and correlate these changeswith SP1 and p53 levels within target gene promoters.
Results: MPM cells and tumors exhibited higher SP1 mRNAand protein levels relative to control cells/tissues. SP1 knockdownsignificantly inhibited proliferation,migration, and clonogenicityof MPM cells. Mithramycin depleted SP1 and activated p53,dramatically inhibiting proliferation and clonogenicity of MPMcells. Intraperitoneal mithramycin significantly inhibited growthof subcutaneous MPM xenografts and completely eradicatedmesothelioma carcinomatosis in 75% of mice. Mithramycinmodulated genes mediating oncogene signaling, cell-cycle regu-lation, senescence, and apoptosis in vitro and in vivo. The growth-inhibitory effects ofmithramycin inMPM cells were recapitulatedby combined SP1 knockdown/p53 overexpression.
Conclusions: These findings provide preclinical rationale forphase II evaluation of mithramycin in patients with mesotheli-oma. Clin Cancer Res; 1–14. �2015 AACR.
IntroductionMalignant pleural mesotheliomas (MPM) are highly lethal
neoplasms attributable primarily to occupational or environ-mental exposures to asbestos and other related mineral fiberssuch as erionite (1–3). Because of the long latency associatedwith these neoplasms, the global incidence of MPM continues
to increase, despite asbestos being banned in many countries(1, 3). Presently, in the United States, the incidence of MPM isapproximately 3,000 cases annually (4). Median overall sur-vival of patients with MPM undergoing aggressive multimod-ality therapy ranges from 14 to 22 months, depending ontumor stage and histology, extent of surgical resection, andresponse to chemotherapy (5–7).
Recent investigative efforts have provided new insights regard-ing the pathogenesis of MPM (11). For example, rare familialMPMs and approximately 65% of sporadic MPM exhibit muta-tions involving BRCA1-associated protein-1 (BAP1), whichencodes a nuclear ubiquitin hydrolase with diverse activitiesincluding DNA repair and deubiquitination of the repressivehistone mark H2AK119Ub (8–11). In addition, MPMs exhibitrecurrent cytogenetic abnormalities including allelic loss ofCDKN2A and p14 ARF (12) and amplification of MYC and PVT1oncogenes (13). Epigenomic perturbations including aberrantactivity of DNA methyltransferases and overexpression of poly-comb repressor complex-2, silence tumor suppressor genes andnoncoding RNAs (14, 15). To date, clinical efforts to specificallytarget oncogene signaling or reverse epigenomic derangements inMPM have had limited success (16).
1Thoracic Epigenetics Section, Thoracic and GI Oncology Branch,National Cancer Institute, Bethesda, Maryland. 2Laboratory of Pathol-ogy, National Cancer Institute, Bethesda, Maryland. 3Clinical Micro-arrayCore,UniversityofCalifornia, LosAngeles,California. 4MolecularPharmacologySection,GenitourinaryMalignanciesBranch,Center forCancer Research, National Cancer Institute, Bethesda, Maryland.5DivisionofThoracicSurgery,BrighamandWomen'sHospital, Boston,Massachusetts.
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
Corresponding Author: David S. Schrump, NCI, Building 10; Rm 4-3942, 10Center Drive, MSC 1201, Bethesda, MD 20892-1201. Phone: 301-496-2128; Fax:301-451-6934; E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-14-3379
�2015 American Association for Cancer Research.
ClinicalCancerResearch
www.aacrjournals.org OF1
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

Specificity protein 1 (SP1) is a zinc-finger transcription factorthat binds to GC-rich motifs (17) and mediates diverse physio-logic processes such as cell-cycle regulation, apoptosis, and angio-genesis, which are essential for normal embryonic developmentand tissue differentiation (18, 19). SP1 interacts with a variety oftranscription factors, histone acetyltransferases and deacetylases,as well as chromatin remodeling complexes to activate or represstranscription in a context-dependent manner (18, 20). SP1 isoverexpressed and contributes to the malignant phenotype of avariety of human cancers by upregulating genes that enhanceproliferation, invasion, and metastasis (18, 19), as well as stem-ness and chemoresistance (21–24). To date, the role of SP1 inMPM has not been defined. The present study was undertaken toexamine SP1 expression in MPM and ascertain the potentialclinical efficacy of targeting this transcription factor for mesothe-lioma therapy.
Materials and MethodsCell lines, tumor samples, and in vitro mithramycintreatments
H28 and H2452 human MPM lines were obtained from ATCCand maintained in RPMI plus Pen/Strep and 10% FBS (normalmedia). LP3 and LP9 normal mesothelial cell lines were obtainedfrom the Coriell Institute for Medical Research and cultured pervendor recommendations. MES1-9 cell lines were established inthe Thoracic Epigenetics Laboratory, Thoracic and GI OncologyBranch, NCI from primary or metastatic MPM, and cultured innormal media. Validation of these cell lines, which have beencontinuously passaged for over 3 years, was performed by HLAtyping as well as immunohistochemical and molecular profilingrelative to the primary tumors (Hong and colleagues, manuscriptin preparation). All tumor samples were obtained from resectedspecimens at the NCI in accordance with Institutional ReviewBoard (IRB)-approved protocols. Normal pleura RNA wasobtained from ProteoGenex. Mithramycin (Sigma-Aldrich) wasprepared as described previously (21). For in vitro experiments,cells were seeded in tissue culture plates. The next day,mediawerereplaced with fresh normal media with or without mithramycin.Twenty-four hours later, media were removed, cells were washedthree times with HBSS, and either harvested immediately ormaintained in normal media for analysis at later time points.Cell proliferation, migration, and clonogenicity assays aredescribed in Supplementary Methods.
RNA isolation, real-timequantitative reverse transcriptionPCR,and microarray analysis
RNA isolation, quantitative reverse transcription PCR(qRT-PCR) using comparative Ct method (25), and microarrayanalysis are described in Supplementary Methods.
ImmunoblottingImmunoblotting was done as previously described (26) using
antibodies listed in Supplementary Methods. Cells for immuno-blot analysis were harvested immediately after 24-hour mithra-mycin treatment in vitro and 3 days following the last intraper-itoneal mithramycin injection for xenograft experiments.
Immunohistochemistry of primary tumor samplesSee Supplementary Methods.
Murine xenograft experimentsAthymic nude mice were injected in bilateral flanks with
2 � 106 of MES1 or MES7 cells. After 10 to 15 days, mice weresorted by tumor size and randomly assigned to receive eithernormal saline or mithramycin (1 or 2 mg/kg) intraperitoneallyevery Monday, Wednesday, and Friday for 3 weeks. Tumor sizeand mouse weights were measured twice weekly, and mithramy-cin doses were held for weight loss >20% of initial body weight.After completion of 3 weeks of treatment (day 31, 3 days after lastmithramycin injection) or when control tumors reached maxi-mum allowable size, mice were euthanized, tumors were excisedand processed for additional studies. For the intraperitonealtumor model, 3 � 106 MES1 cells were injected intraperitoneallyinto athymic nude mice. Ten days later, mice were randomized toreceive saline or mithramycin (1 mg/kg) intraperitoneally everyMWF for 3 weeks. Approximately 1 week later, mice were eutha-nized and laparotomies were performed to assess tumor burden.All animal procedures were approved by the National CancerInstitute Animal Care and Use Committee, and were in accor-dance with the NIH Guide for the Care and Use of LaboratoryAnimals.
Generation of stable cells expressing shRNA constructsMES1 and MES7 were transduced with commercially vali-
dated shRNA targeting SP1 or sham sequences (Sigma) accord-ing to manufacturer's instructions (Supplementary Methods).Cell lines were selected with puromycin (Sigma) and expandedafter confirmation of knockdown by qRT-PCR and immuno-blot techniques.
Chromatin immunoprecipitation experimentsQuantitative chromatin immunoprecipitation (ChIP) assays
were conducted as described (26)withminormodifications usingeither nonspecific immunoglobulin G (IgG) or ChIP-grade anti-bodies as well as PCR primers listed in Supplementary Methods.
Adenoviral transduction of p53 in SP1-knockdown cellsBoth sham control or SP1-knockdown MES1 and MES7 were
transduced with high titer Ad-GFP or Ad-GFP-p53 (Vector Labs)for 48 hours. Thereafter, the cells were collected for counting,RNA, and protein analysis. For in vivo studies, 2 � 106 cells wereinjected subcutaneously into the bilateral flanks of athymic nudemice and tumor volume was measured every 4 days until theexperiment was completed.
Translational Relevance
Pleural mesotheliomas are highly lethal neoplasms forwhich there are no effective treatments. Experimentsdescribed in this article demonstrate that under exposureconditions potentially achievable in clinical settings,mithramycin depletes SP1 and activates p53 signaling tomediate profound growth arrest associated with senescenceand subsequent apoptosis of pleural mesothelioma cells invitro and in vivo. These findings provide the preclinicalrationale for evaluation of mithramycin administered sys-temically or by regional perfusion techniques in patientswith mesothelioma.
Rao et al.
Clin Cancer Res; 2015 Clinical Cancer ResearchOF2
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

Senescence, cell-cycle, and apoptosis analysesA total of 3 � 104 MPM cells were seeded in each of four-
chamber slides overnight. The following day, cells were treated for24 hours withmithramycin (0–100 nmol/L), and senescence wasassessed using the SA-b-gal Staining Kit fromCell BioLabs. Similartechniqueswere used to detect senescence in sham control or SP1-knockdown MES1 and MES7 cells 48 hours following transduc-tionwith high titer Ad-GFPor Ad-GFP-p53. For cell-cycle analysis,1 � 106 cells were plated overnight in serum-containing media.The following day, normal media were replaced with serum-freeRPMI for 48hours. Thereafter, cellswere exposed tonormalmediawith or without mithramycin (0–100 nmol/L) for 24 hours. Cellswere harvested at indicated time points and processed for cell-cycle analysis using flow cytometric techniques (27). Apoptosiswas assessed by flow cytometry using reagents and protocolscontained in the Apo-BrdU Kit (BD Pharmingen).
Statistical analysisSD or SEM are indicated by bars on figures and were calculated
using GraphPad Prism 6.0. All experiments were conducted withat a minimum of triplicate samples, and all P values were calcu-lated with two-tailed t tests.
ResultsSP1 expression in MPM
qRT-PCR and immunoblot experiments were performed toexamine SP1 expression in cultured MPM cells and tumorsrelative to cultured normal mesothelial cells or normal pleura.Seven of nine MPM lines exhibited overexpression of SP1 mRNArelative to normal mesothelial cells (LP3 or LP9; Fig. 1A, top).Immunoblot experiments demonstrated markedly higher SP1protein levels in MPM cells relative to normal mesothelial cells(Fig. 1A, bottom). Additional qRT-PCR experiments demonstrat-ed overexpression of SP1 in 5 of 7 primary MPM specimens fromwhich the aforementioned cell lines were derived, although themagnitude of SP1 overexpression in cell lines and primary tumorsdid not exactly coincide (Fig. 1A, middle top). Immunohisto-chemical experiments demonstrated overexpression of SP1 in 18of 19 primary MPM specimens compared with normal pleura,including seven from which our cell lines were derived (Supple-mentary Fig. S1A). To extend these observations, IHC techniqueswere used to evaluate SP1 expression in commercial tissue arrayscontaining 59 primary mesotheliomas and 22 normal mesothe-lial tissues. A spectrum of SP1 expression was detected, withsignificantly increased SP1 staining in MPMs compared withnormal mesothelia (Fig. 1A, middle bottom).
Additional analysis was undertaken to ascertain whetherintratumoral SP1 expression detected by Illumina array tech-niques correlated with survival in 39 patients with locallyadvanced MPM undergoing potentially curative resections atBrigham and Women's Hospital. Twenty-four patients hadepithelial mesotheliomas, whereas seven patients had biphasicand eight patients had sarcomatoid malignancies. Increasedexpression of SP1 tended to be associated with shorter survivalof patients with MPM, although this was not statisticallysignificant (Fig. 1A, right).
Effects of SP1 depletion in MPM cellsAdditional experiments were performed to examine whether
SP1 expression modulates the malignant phenotype of pleural
mesothelioma cells. Briefly, shRNA techniques were used toknockdown SP1 in cultured MPM cells. qRT-PCR and immuno-blot experiments demonstrated significant decreases in SP1expression in MES1 and MES7 cells following transfection witheither of two shRNA sequences (shSP1 #1 and #2) relative tocontrols (Fig. 1B, left and right). Knockdown of SP1 significantlydiminished proliferation, migration, and soft agar clonogenicityof MES1 and MES7 cells (Fig. 1C and D).
Effect of mithramycin in MPM cellsSeeking potential translation of the aforementioned findings to
the clinic, additional experiments were performed to examinewhether mithramycin, an antineoplastic agent that inhibits bind-ing of SP1to DNA (17), could similarly inhibit the malignantphenotype of pleural mesothelioma cells. Briefly, MES1 andMES7 cells were cultured in normal media with or withoutmithramycin (25–100 nmol/L) for 24 hours. qRT-PCR andimmunoblot experiments (Supplementary Fig. S1B) demonstrat-ed dose-dependent decreases in SP1 expression in both cell linesfollowing mithramycin exposure. MTS assays demonstrated that24-hour mithramycin exposure dramatically inhibited prolifera-tion of MES1 and MES7 cells (Fig. 2A, left and middle). Cyto-toxicity was not evident until 24 hours following removal ofmithramycin, suggesting delayed effects of drug exposure.Although some recovery of cell growth was observed 3 to 5 daysfollowing mithramycin exposures of 25 or 50 nmol/L, treatmentwith doses of 100 nmol/L or more resulted in progressivedecreases in cell viability over the ensuing 4 days. Furthermore,24-hour exposure ofmithramycin significantly inhibited soft agarclonogenicity of MES1 and MES7 cells in a dose-dependentmanner (Fig. 2A, right).
Additional experiments were performed to examine whethermithramycin inhibited growth of MPM cells in vivo. As shownin Fig. 2B (left and middle), as well as Supplementary Fig. S1C,mithramycin significantly diminished growth of subcutaneousMES1 and MES7 xenografts in a dose-dependent manner. Thegrowth-inhibitory effects of mithramycin were readily apparentshortly after treatment was initiated, with statistically significantdifferences in xenograft volumes evident within 7 days of com-mencing treatment (3 intraperitoneal injections). The 1 mg/kgdose was well-tolerated with no significant weight loss, lethargy,or decreased activity; however, the 2 mg/kg dose group experi-enced moderate or statistically significant weight loss and somevisible toxicities (Fig. 2B, right, and Supplementary Fig. S1C).These latter findings were in contrast to our previously publishedexperiments pertaining to intraperitoneal mithramycin treatmentof lung cancer xenografts (21), in which 2 mg/kg mithramycinappeared to be well tolerated, possibly because clinical grademithramycin was used for the lung cancer studies but not thecurrent experiments.
In the second series of experiments, mice with intraperito-neal mesothelioma carcinomatosis were randomly allocated toreceive saline or mithramycin (1 mg/kg) intraperitoneally everyMonday, Wednesday, and Friday for 3 weeks, followed byeuthanasia 3 days later. Preliminary experiments were per-formed to confirm the reproducibility and growth kinetics ofthe carcinomatosis model and timing of intervention. Resultsof three independent experiments totaling 60 mice are sum-marized in Fig. 2C. Whereas 29 of 30 control mice (97%)had extensive carcinomatosis when euthanized, 23 of 30mithramycin-treated mice (77%) had no evidence of disease
Mithramycin and MPM
www.aacrjournals.org Clin Cancer Res; 2015 OF3
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379
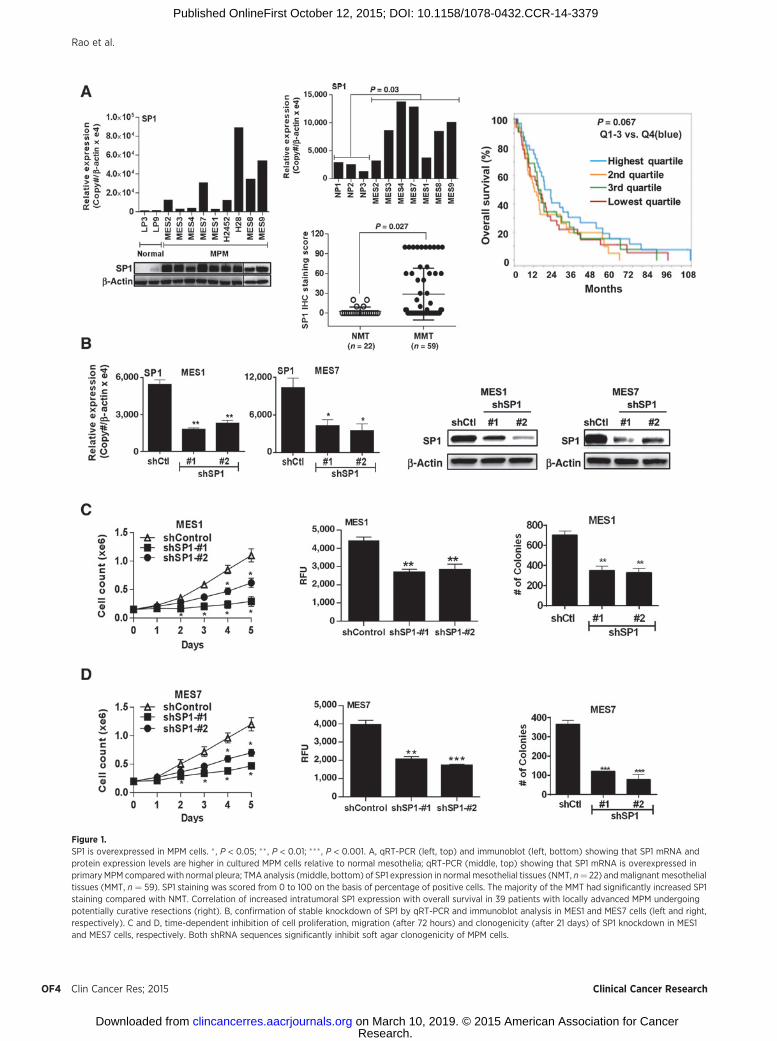
Figure 1.SP1 is overexpressed in MPM cells. � , P < 0.05; �� , P < 0.01; ��� , P < 0.001. A, qRT-PCR (left, top) and immunoblot (left, bottom) showing that SP1 mRNA andprotein expression levels are higher in cultured MPM cells relative to normal mesothelia; qRT-PCR (middle, top) showing that SP1 mRNA is overexpressed inprimary MPM compared with normal pleura; TMA analysis (middle, bottom) of SP1 expression in normal mesothelial tissues (NMT, n¼ 22) andmalignant mesothelialtissues (MMT, n ¼ 59). SP1 staining was scored from 0 to 100 on the basis of percentage of positive cells. The majority of the MMT had significantly increased SP1staining compared with NMT. Correlation of increased intratumoral SP1 expression with overall survival in 39 patients with locally advanced MPM undergoingpotentially curative resections (right). B, confirmation of stable knockdown of SP1 by qRT-PCR and immunoblot analysis in MES1 and MES7 cells (left and right,respectively). C and D, time-dependent inhibition of cell proliferation, migration (after 72 hours) and clonogenicity (after 21 days) of SP1 knockdown in MES1and MES7 cells, respectively. Both shRNA sequences significantly inhibit soft agar clonogenicity of MPM cells.
Rao et al.
Clin Cancer Res; 2015 Clinical Cancer ResearchOF4
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379
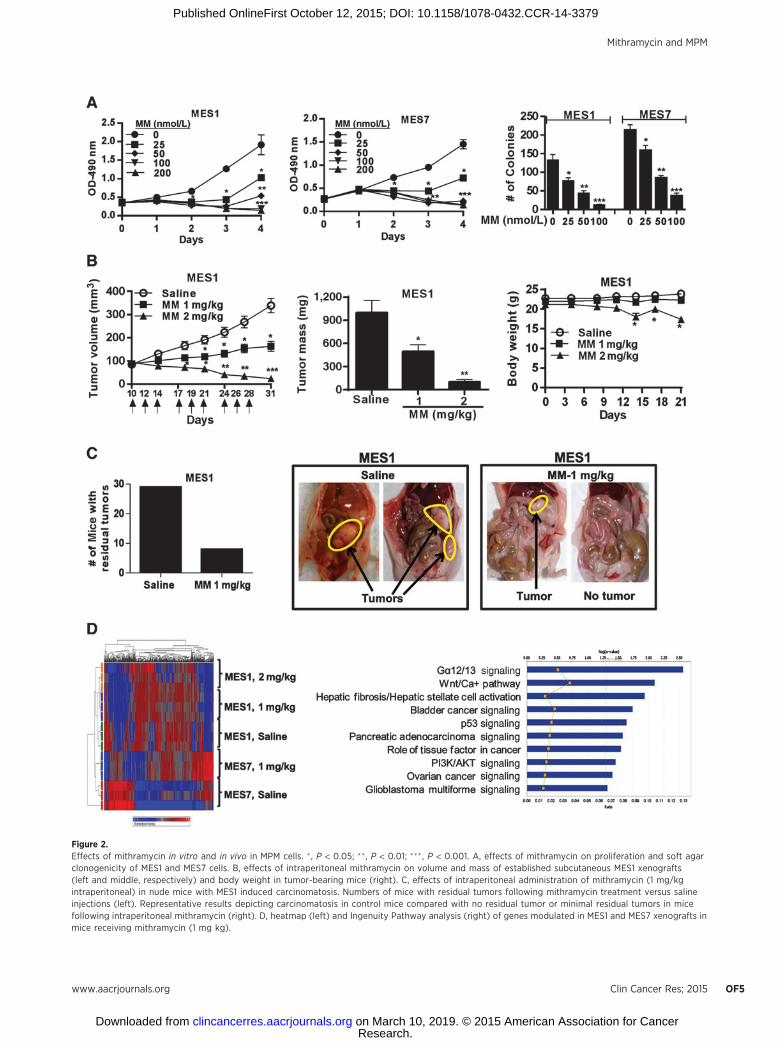
Figure 2.Effects of mithramycin in vitro and in vivo in MPM cells. � , P < 0.05; �� , P < 0.01; ���, P < 0.001. A, effects of mithramycin on proliferation and soft agarclonogenicity of MES1 and MES7 cells. B, effects of intraperitoneal mithramycin on volume and mass of established subcutaneous MES1 xenografts(left and middle, respectively) and body weight in tumor-bearing mice (right). C, effects of intraperitoneal administration of mithramycin (1 mg/kgintraperitoneal) in nude mice with MES1 induced carcinomatosis. Numbers of mice with residual tumors following mithramycin treatment versus salineinjections (left). Representative results depicting carcinomatosis in control mice compared with no residual tumor or minimal residual tumors in micefollowing intraperitoneal mithramycin (right). D, heatmap (left) and Ingenuity Pathway analysis (right) of genes modulated in MES1 and MES7 xenografts inmice receiving mithramycin (1 mg kg).
Mithramycin and MPM
www.aacrjournals.org Clin Cancer Res; 2015 OF5
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

(P < 0.02); the remaining seven mithramycin-treated mice hadminimal residual tumors.
Effects of mithramycin on global gene expression in MPM cellsAffymetrixmicroarray experiments were performed to examine
global gene expression profiles in cultured MES1 and MES7 cellsimmediately after 24-hour exposure to normal media with orwithout mithramycin (25 or 100 nmol/L) and their correspond-ing xenografts harvested three days after the last intraperitonealsaline ormithramycin injection.Mithramycin-mediated dramaticdose-dependent alterations in gene expression in cultured MPMcells. Using criteria of fold change greater than 2 for drug treat-ment versus control, 986 genes were commonly modulated inMES1 andMES7 cells following 25 and 100 nmol/L mithramycinexposures, respectively (Supplementary Fig. S2A); significantoverlapwas observed at both drug concentrations. Approximately65%of genesmodulated bymithramycin in culturedMPMcells ateither drug concentration were repressed by mithramycin. Topcanonical pathways affected by mithramycin exposure in vitro aredepicted in Supplementary Fig. S2B. Within each cell line, moreoverlap was observed with cells treated with 100 nmol/L ratherthan 25 nmol/L in vitro and tumor xenografts (SupplementaryFig. S2C). Using criteria of fold change equal to or greater than 2relative to appropriate controls, 27 genes were differentiallyregulated in cultured MES1 and MES7 cells following exposureto 100 nmol/L mithramycin as well as xenografts from micetreated with 1 mg/kg mithramycin (Supplementary Table S1).Top canonical pathways (depicted in Supplementary Fig. S2D)
included p53 signaling, G-alpha 12/13, and Rho signaling, as wellas pathways associated with immunologic destruction of cancercells.
More than 1,200 genes and nearly 640 genes were signifi-cantly modulated by mithramycin treatment in subcutaneousMES1 and MES7 xenografts, respectively. More than 700 geneswere commonly regulated in MES1 cells exposed to mithra-mycin (100 nmol/L) in vitro and MES1 xenografts from 1 and 2mg/kg treated mice (Supplementary Fig. S2C). Similar analysiscould not be performed for MES7 because of the limitedquantity and poor quality of the RNA from xenografts from2 mg/kg treated mice. Fifty-six genes were commonly regulatedin MES1 and MES7 xenografts from mice treated with 1 mg/kgmithramycin intraperitoneally (heatmap is depicted in Fig.2D, left); 55% of these genes were downregulated (Table 1).Notably, KIAA1199, which recently has been implicated inmodulating metabolism, EGFR and Wnt signaling in cancercells (28, 29) was markedly repressed in MES1 as well asMES7 xenografts from mithramycin-treated mice. In addition,the oncofetal receptor tyrosine kinase ROR1, which is over-expressed in a variety of human malignancies but to datehas not been studied in pleural mesotheliomas, was signifi-cantly downregulated in MPM xenografts from mithramycin-treated mice. Top canonical pathways included G-alpha 12/13signaling, which mediates cytoplasmic b-catenin levels (30),Wnt/Ca2þ signaling, axonal guidance signaling, which hasrecently been implicated recently in pluripotency and meta-static potential of cancer cells (31), p53 signaling, as well as
Table 1. Genes induced or repressed by mithramycin in MES1 as well as MES7 xenografts
Downregulated Upregulated
Probeset ID Gene symbolIn vivo 1 mg/kg
Probeset ID Gene symbolIn vivo 1 mg/kg
MES1 MES7 MES1 MES7
1 212942_s_at KIAA1199 �12.6 �10.5 1 232165_at EPPK1 9.1 5.22 209183_s_at C10orf10 �5.2 �6 2 236646_at C12orf59 2.4 3.33 202688_at TNFSF10 �3.8 �4.5 3 1560683_at BCL8 6.6 3.34 213931_at ID2/ID2B �6.8 �3.7 4 221577_x_at GDF15 5.3 3.15 227955_s_at EFNA5 �2.6 �3.5 5 207761_s_at METTL7A 4.9 36 201438_at COL6A3 �3.2 �3.5 6 228293_at DEPDC7 2.5 2.97 205199_at CA9 �4 �3.2 7 235230_at PLCXD2 3.6 2.98 224771_at NAV1 �2.2 �3.1 8 237737_at LOC100289026 8.5 2.89 210601_at CDH6 �2.8 �2.9 9 224566_at NEAT1 2.2 2.710 205681_at BCL2A1 �3 �2.8 10 1556194_a_at LOC100507455 2.3 2.711 46142_at LMF1 �2.2 �2.7 11 205543_at HSPA4L 2.4 2.612 232060_at ROR1 �3.4 �2.7 12 204286_s_at PMAIP1 2 2.513 216222_s_at MYO10 �3.6 �2.6 13 202672_s_at ATF3 2 2.414 1561615_s_at SLC8A1 �2.8 �2.5 14 234303_s_at GPR85 2.9 2.215 221029_s_at WNT5B �2 �2.5 15 215440_s_at BEX4 2.2 2.216 1554452_a_at C7orf68 �3.8 �2.5 16 226103_at NEXN 2.7 2.217 200632_s_at NDRG1 �8.3 �2.4 17 1554020_at BICD1 2.7 2.218 234994_at TMEM200A �3 �2.3 18 202704_at TOB1 2 2.119 203397_s_at GALNT3 �4.4 �2.2 19 230748_at SLC16A6 2.3 2.120 202821_s_at LPP �2.1 �2.2 20 206066_s_at RAD51C 2.2 221 204298_s_at LOX �4.8 �2.1 21 205386_s_at MDM2 2.9 222 228570_at BTBD11 �2.1 �2.1 22 228855_at NUDT7 2.5 223 213506_at F2RL1 �2.1 �2.1 23 219179_at DACT1 2 224 220227_at CDH4 �4.6 �2 24 209481_at SNRK 2 225 210512_s_at VEGFA �2.9 �2 25 1557765_at LOC643401 5.3 226 43544_at MED16 �2.1 �227 224646_x_at H19 �3.8 �228 242301_at CBLN2 �2.1 �229 204348_s_at AK4 �5.6 �230 33778_at TBC1D22A �8.2 �231 226899_at UNC5B �2.8 �2
Rao et al.
Clin Cancer Res; 2015 Clinical Cancer ResearchOF6
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

Mithramycin and MPM
www.aacrjournals.org Clin Cancer Res; 2015 OF7
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

pathways associated with bladder, pancreas, ovarian, andbrain cancer (Fig. 2D, right).
Because p53 signaling was observed to be a top pathwayaffected by mithramycin in vitro and in vivo, genes from Table 1and Supplementary Table S1 were screened for SP1- and p53-binding sites within their respective promoters using softwareguided analysis (DECODE: DECipherment Of DNA Elements orUCSC Genome Browser). Approximately 50% of these genes hadrecognition elements for either SP1, p53, or both within theirpromoters.
qRT-PCR and immunoblot techniques were used to validateAffymetrix array results using selected genes modulated bymithramycin in vitro and/or in vivo. qRT-PCR analysis (Fig. 3A)confirmed that mithramycin decreased ROR1 expression in vitroand in vivo. Furthermore, although not identified as a commonlyregulated gene bymicroarray analysis, HDAC4, awell-establishedSP1 target (32), was repressed by mithramycin in vitro and in vivoin MES1 and MES7 cells. mithramycin also decreased expressionof SP1 as well as EZH2, which previously has been shown to be anepigenetic driver of malignancy in pleural mesotheliomas (14;Supplementary Fig. S3A); this phenomenon was not seen incorresponding xenografts possibly due to the timing and inter-mittent nature of the intraperitoneal drug treatments and intervalfrom last drug treatment to euthanasia. In addition, mithramycinmediated dose-dependent increases in p53, p21, PMAIP1, andPRDM1 in vitro and in vivo (Fig. 3A). Subsequent immunoblotexperiments (Fig. 3B) confirmed results of qRT-PCR experiments.
Mithramycin activates p53 in MPM cellsFollowing stress or DNA damage, p53 undergoes various
posttranslational modifications such as acetylation or phosphor-ylation, mechanisms which are known to activate and stabilizethis transcription factor (33). To examine this issue, MES1 andMES7 cells, which express wt p53 were treated with mithramycinat various doses for 24 hours. Immunoblot experiments demon-strated dose-dependent increases in gH2AX in MES1 and MES7cells indicative ofDNAdamage,which coincidedwith increases intotal p53 as well as acetylated p53-K320, K373, K382, andphosphorylated p53-S15 levels in MES7 cells, and to a lesserextent, MES1 cells following mithramycin exposure (Supplemen-tary Fig. S3B–S3D). Collectively, these data suggest that DNAdamage contributes to mithramycin-mediated activation of p53in MPM cells.
Effects of mithramycin on target gene promotersChIP experiments were conducted to further investigate the
mechanisms by which mithramycin regulates oncogene andtumor suppressor gene expression in MPM cells. ChIP wasperformed using primers flanking p53 or SP1 response ele-ments to quantitate occupancy of p53, SP1, or RNA Pol II, aswell as levels of H3K4Me3 and H3K27Me3 (euchromatin and
heterochromatin histone marks, respectively) within the pro-moter regions of p21, PMAIP1, HDAC4, ROR1, DACT1, andPRDM1. As shown in Fig. 3C and Supplementary Fig. S4A andS4B, activation of p53 or repression of SP1 by mithramycinincreased p53 and decreased SP1 occupancy, respectively, with-in the p21, PMAIP1, and PRDM1 promoters, which coincidedwith increased occupancy of RNA Pol II, increased H3K4Me3,and decreased levels of H3K27Me3 in the respective promotersin MES1 and MES7 cells. mithramycin appeared to decreaseoccupancy of p53 as well as SP1 and RNA Pol II and diminishH3K4Me3 levels, while increasing H3K27Me3 levels within theROR1 promoter. Furthermore, mithramycin decreased occu-pancy of SP1 and RNA PolII, coinciding with decreasedH3K4Me3 and increased H3K27Me3 within the HDAC4 pro-moter. Finally, mithramycin increased occupancy of RNAPolIIand increased H3K4Me3 levels while decreasing H3K27Me3within the DACT1 promoter region, which contains no proto-typic SP1 or p53 recognition elements (Supplementary Fig. S4Aand S4B), suggesting an indirect mechanism of activation ofthis tumor suppressor gene by mithramycin.
Effects of SP1 knockdown and p53 overexpression inMPM cellsAdditional experiments were undertaken to ascertain whether
SP1 knockdown (shSP1) and p53 overexpression (p53-OEX)could mimic the effects of mithramycin in MPM cells. MES1 andMES7 cells exhibiting either knockdown of SP1, or respectivevector controls, were transduced with adenovirus GFP-p53-WT orcontrol adenovirus. Cell count assays for 48 hours post-adeno-viral transduction demonstrated a significant additive effect ofshSP1 and p53-OEX in MPM cells (representative results pertain-ing toMES7 cells are depicted in Fig. 4A, left). Further experimentsdemonstrated that shSP1 and p53-OEX significantly diminishedgrowth of subcutaneous MPM xenografts (Fig. 4A, middleand right); the in vivo growth-inhibitory effects of combinedshSP1/p53-OEX were more pronounced than either shSP1 orp53-OEX alone in MES7 cells. Similar results were observed inMES1 cells (Supplementary Fig. S5A).
Gene expression profiles were obtained from additionalmicroarray experiments in MPM cells following combinedshRNA mediated knockdown of SP1 and adenoviral-mediatedoverexpression of p53 (shSP1/p53-OEX). As shown in Fig. 4B(left), 706 and 806 genes were commonly regulated by com-bined shSP1/p53-OEX in MES1 and MES7 cells, respectively.Additional analysis was performed to compare gene expressionprofiles in mithramycin-treated and shSP1/p53-OEX MPMcells. The heatmap for this analysis is shown in Fig. 4B, middle.Fifty-three genes were differentially regulated by mithramycinand shSP1/p53-OEX in either MES1 or MES7 (SupplementaryTable S2). Subsequent ingenuity pathway analysis demonstrat-ed that these commonly regulated genes mediated p53 signal-ing, G1–S, G2–M and DNA checkpoint regulation, ATM and
Figure 3.Validation of microarray data. � , P < 0.05; �� , P < 0.01; ��� , P < 0.001. A, qRT-PCR showing the effects of mithramycin on ROR1, HDAC4, p53, p21, PMAIP1,and PRDM1 in MES1 and MES7 cells harvested immediately after 24-hour exposure, as well as corresponding xenografts harvested 3 days following the lastintraperitoneal saline or mithramycin injection. B, immunoblot analysis (left) showing the dose-dependent effects of mithramycin for 24 hours on SP1, ROR1,HDAC4, EZH2, p53, p21, PMAIP1, PRDM1 protein levels in culturedMPM cells; representative immunoblot analysis (right) demonstrating the effects ofmithramycin onSP1, ROR1, HDAC4, EZH2, p53, p21, PMAIP1, PRDM1 protein levels in subcutaneous MPM xenografts frommithramycin-treated and control mice. Three tumors (T1, T2,and T3) were selected from saline control versus 1 mg/kg mithramycin. Tumors were harvested 3 days following the last intraperitoneal saline or mithramycininjection. C, quantitative ChIP analysis of various gene promoters in MES1 cells cultured in normal media with or without mithramycin (100 nmol/L, 24 hours).
Rao et al.
Clin Cancer Res; 2015 Clinical Cancer ResearchOF8
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379
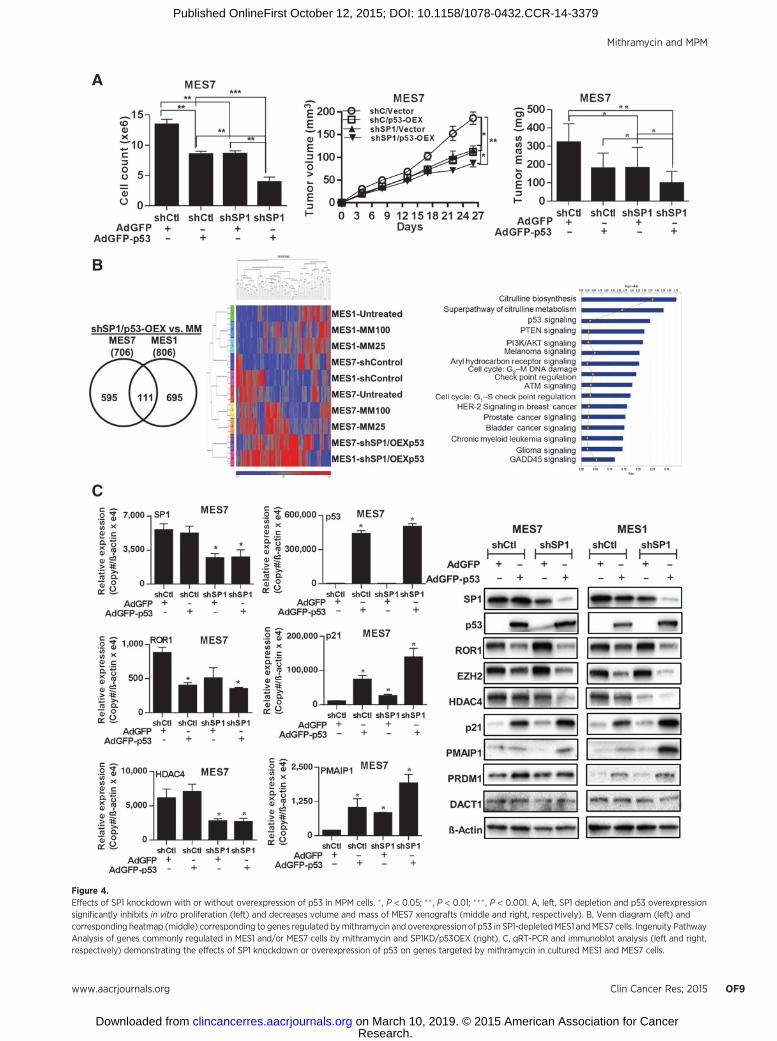
Figure 4.Effects of SP1 knockdown with or without overexpression of p53 in MPM cells. � , P < 0.05; �� , P < 0.01; ���, P < 0.001. A, left, SP1 depletion and p53 overexpressionsignificantly inhibits in vitro proliferation (left) and decreases volume and mass of MES7 xenografts (middle and right, respectively). B, Venn diagram (left) andcorresponding heatmap (middle) corresponding to genes regulated bymithramycin and overexpression of p53 in SP1-depletedMES1 andMES7 cells. Ingenuity PathwayAnalysis of genes commonly regulated in MES1 and/or MES7 cells by mithramycin and SP1KD/p53OEX (right). C, qRT-PCR and immunoblot analysis (left and right,respectively) demonstrating the effects of SP1 knockdown or overexpression of p53 on genes targeted by mithramycin in cultured MES1 and MES7 cells.
Mithramycin and MPM
www.aacrjournals.org Clin Cancer Res; 2015 OF9
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379
Rao et al.
Clin Cancer Res; 2015 Clinical Cancer ResearchOF10
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

GADD45 signaling, as well as citrulline, glutamine, and argi-nine metabolism (Fig. 4B, right). Results of this analysis as wellas other aforementioned experiments were further validated byqRT-PCR and immunoblot experiments (Fig. 4C; Supplemen-tary Fig. S5B and S5C). Knockdown of SP1 had no effecton endogenous levels of p53 in MES1 or MES7 cells (Supple-mentary Fig. S5D) but modestly induced expression of p21(Fig. 4C). Overexpression of p53 markedly increased p21 levelsin these cells. These findings suggest that upregulation of p21 inMPM cells by mithramycin occurs via p53-dependent as well as-independent mechanisms. Modulation of other targets such asROR1 and EZH2 appeared to be determined by SP1 depletionas well as activation of p53.
Effects of mithramycin on senescence and apoptosis in MPMcells
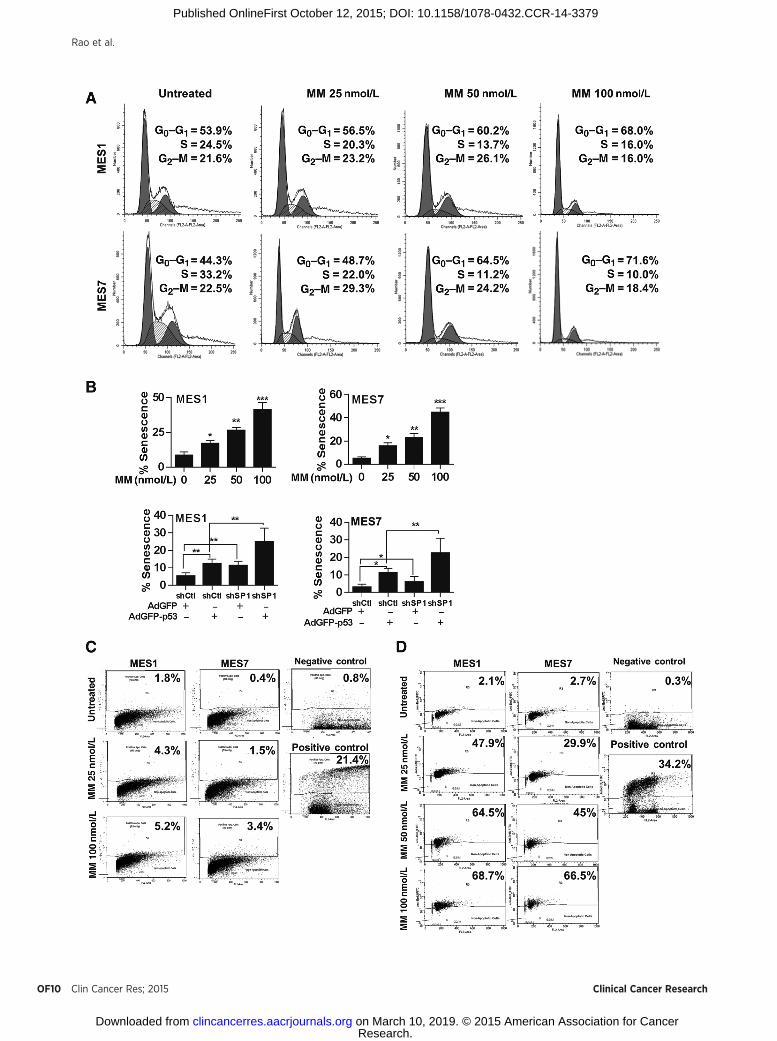
Because mithramycin modulated a variety of genes includ-ing p53, p21, PMAIP1, PRDM1 SP1, HDAC4, and EZH2 thateither promote or inhibit cell-cycle arrest, senescence, andapoptosis in cancer cells, additional experiments were per-formed to further characterize the mechanisms by whichmithramycin mediated cytotoxicity in MPM cells. Flow cyto-metric experiments (Fig. 5A) demonstrated dose-dependentaccumulation of MPM cells in G0–G1 without a sub-G0 frac-tion consistent with a G0–G1 arrest immediately following24-hour mithramycin exposure. Histochemical experimentsperformed at this time point demonstrated that mithramycinmediated dose-dependent increases in b-galactosidase expres-sion indicative of senescence in MPM cells; similarly, shSP1and p53-OEX had additive effects on senescent phenotype inMPM cells (Fig. 5B; additional data available upon request).Apo-BrdUrd experiments demonstrated no significant increasein apoptosis in MPM cells immediately following 24-hourmithramycin treatment (Fig. 5C). However, significant dose-dependent apoptosis was observed 48 hours following com-pletion of mithramycin treatment in MES1 as well as MES7cells (Fig. 5D). These findings were consistent with results ofMTT assays depicted in Fig. 2A.
DiscussionDespite being relatively rare, pleural mesotheliomas continue
to challenge clinicians due to relentless growth and resistance toconventional treatment modalities as well as novel therapeutics(1). As such, there is an urgent need for innovative treatmentregimens targeting specific genetic/epigenetic drivers in pleuralmesotheliomas and a more thorough appreciation of drug deliv-ery to these neoplasms in clinical settings.
In the present study, we sought to examine the potentialefficacy of targeting SP1 expression in MPM. We observedoverexpression of SP1 in the majority of cultured MPM linesand primary pleural mesotheliomas relative to cultured normalmesothelial cells or normal pleura. Knockdown of SP1 inhib-
ited growth, migration, and tumorigenicity of MPM cells,strongly suggesting that SP1 functions as an oncogene in MPM.Whereas SP1 overexpression has been associated withdecreased survival of patients with lung and esophageal cancers(34, 35), our microarray experiments did not reveal an asso-ciation between SP1 mRNA expression and survival in patientswith MPM, possibly due to the limited number of samplesanalyzed, as well as potential discrepancies between mRNA andprotein levels detected in MPM specimens using various tech-niques. Additional experiments demonstrated that mithramy-cin markedly diminished growth of MPM cells in vitro andin vivo via induction of DNA damage, cell-cycle arrest, andsenescence with subsequent apoptosis. To the best of ourknowledge, these experiments are the first to demonstrateoverexpression of SP1 in MPM and the potential efficacy ofmithramycin for mesothelioma therapy.
Mithramycin is a naturally occurring polyauroleic acid iso-lated from Streptomyces, which was originally evaluated as achemotherapeutic agent in patients with a variety of malig-nancies during the 1960s and 1970s; although completeresponses were observed in approximately 10% to 15% ofpatients with sarcomas and germ cell tumors, the drug wasdiscontinued because of excessive systemic toxicities that werepoorly characterized (36, 37). Recently, there has beenrenewed interest in clinical development of mithramycin andits analogues because of their ability to specifically inhibitbinding of SP1 to GC-rich DNA resulting in repression ofnumerous genes mediating proliferation, invasion, and metas-tasis of cancer cells (21, 24, 38–40). In an ongoing phase IItrial at the NCI using drug of higher purity than previouslyavailable, mithramycin has been surprisingly well-tolerated inpatients with cancer when administered at the previouslyrecommended dose and schedule (25–30 mg/kg i.v. over 6hours � 7 days every 4 weeks). Specifically, no nausea, vomit-ing, bleeding, or myelosuppression have been observed in 12adult patients with various malignancies; however, nine ofthese individuals developed dose-limiting transaminitis,which resolved spontaneously following cessation of drug.Affymetrix Drug Metabolizing Elimination and Transport(DMET) microarray experiments demonstrated that mithra-mycin-induced hepatotoxicity correlated with SNP in severalgenes encoding transporter proteins regulating bile flow(Schrump and colleagues, manuscript in preparation). On thebasis of these findings as well as review of pharmacokineticdata from this trial, the protocol has been amended to enrollonly those patients with favorable genotypes while intensify-ing the treatment regimen to recapitulate drug exposure con-ditions achieved in our preclinical studies.
Although we initially used mithramycin to target SP1 expres-sion, our microarray and gene manipulation experimentsrevealed that activation of p53 is a major mechanism bywhich mithramycin inhibits growth of MPM cells. These findingsare consistent with recent studies demonstrating that the
Figure 5.Effects of mithramycin on cell-cycle progression, senescence, and apoptosis in MPM cells. � , P < 0.05; �� , P < 0.01; ��� , P < 0.001. A, propidium iodidestaining demonstrating that 24-hour mithramycin treatment induces dose-dependent G0–G1 arrest in MPM cells. B, b-galactosidase staining assaysdemonstrating that 24-hour mithramycin treatment induces dose-dependent senescence in MES1 and MES7 cells. C and D, Apo-BrdUrd analysis demonstratingminimal apoptosis in MPM cells immediately following 24-hour mithramycin exposure (C) but significant dose-dependent apoptosis 48 hours following drugtreatment (D).
Mithramycin and MPM
www.aacrjournals.org Clin Cancer Res; 2015 OF11
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

antiangiogenic effects of mithramycin inmyeloma cells in vivo aremediated not by inhibition of SP1 signaling, but rather byactivation of p53 (41), and that knockdown of p53 significantlyattenuates mithramycin-mediated cytotoxicity in endometrialcarcinoma cells (38).
Themajority of pleuralmesotheliomas retainwt p53 expressionand exhibit functional disruption of p53 activity via allelic loss ofp14ARF (1); as such, our experiments didnot specifically delineatethe effects of mithramycin in MPM cells bearing p53 mutations.However, our recent studies have demonstrated dramatic cytotoxiceffects of mithramycin irrespective of p53 mutation status in lungand esophageal cancer cells (21); these latter observations suggestthat p53 activation may not be essential for growth arrest inducedby mithramycin in MPM cells. Nevertheless, p53 status may affectextent of cell-cycle arrest and propensity for apoptosis in cancercells following SP1 depletion bymithramycin (42). Recent elegantstudies have demonstrated that highly complex interactionsbetween p53, SP1, and mdm2 regulate cell-cycle arrest and apo-ptosis in cancer cells following exposure to chemotherapeuticagents which activate p53 (43). SP1 is a critical determinant ofapoptosis but not cell-cycle arrest mediated by p53. Furthermore,although dispensable for induction of proapoptotic genes, SP1 isrequired for proapoptotic transcriptional repression by p53 (43).Mdm2, which is upregulated by p53, facilitates proteosomaldegradation of SP1, and negatively regulates p53 (44, 45). Con-ceivably, early cell-cycle arrest/senescence and subsequent apopto-sis ofMPMcells followingmithramycin exposure is attributable, atleast in part, to time- anddose-dependentfluctuationsof SP1, p53,and mdm2. The fact that combined SP1 knockdown/p53 over-expression did not fully recapitulate growth inhibition and geneexpression profiles observed in mithramycin-treated MPM cellssuggests that additional as yet uncharacterized mechanisms con-tribute to mithramycin-mediated cell-cycle arrest, senescence, andapoptosis in MPM cells. For example, recently described interac-tions ofmithramycin with core histones could account, in part, forrepression of genes lacking prototypic SP1 or p53 recognitionelements within their respective promoters (46). Experiments areunderway to examine these issues and to ascertain whether small-molecule mdm2–p53 inhibitors (47) or agents which inhibit p21expression (48) can augment mithramycin-mediated apoptosis inMPM cells.
Our studies demonstrated that 24-hourmithramycin treatmentwas sufficient to mediate progressive decreases in MPM cellviability in vitro; furthermore, repeated intraperitoneal adminis-tration of mithramycin significantly inhibited growth of subcu-taneous MPM xenografts and eradicated established intraperito-neal mesothelioma xenografts in nearly 75% of mice. Thesefindings strongly suggest that the antiproliferative effects ofmithramycin are not primarily due to acute cytotoxicity but areinstead related to transcriptional reprogramming, which persistslong after mithramycin exposure. Our microarray experimentsdemonstrated considerable overlap between in vitro and in vivomithramycin exposures, which have been helpful regarding delin-eation of the mechanisms of mithramycin-mediated cytotoxicityin MPM cells. Some of the genes modulated by mithramycin
(particularly KIAA1199, ROR1, HDAC4, p21, PMAIP1, andPRDM1) may be relevant pharmacodynamic endpoints for clin-ical trials evaluatingmithramycin in patients withmesothelioma.
Although mithramycin was administered to approximately1,500 patients with cancer during the 1960's and early 1970's,no patients with mesothelioma were included in these trials.Furthermore, no pharmacokinetic data were available from anyof the initial trials, as methods for analyzing mithramycin levelshave only recently been developed (49). Data presented in thisarticle, as well as our previously published studies (21), indicatethat mithramycin-mediated cytotoxicity occurs in vitro following24-hour mithramycin exposure at concentrations > 50 nmol/L;mithramycin tissue concentrations in mice with regressing tumorxenografts range from 50 to 100 nmol/L for >24 hours followingintraperitoneal mithramycin injections (50). These exposure con-ditions are potentially achievable in clinical settings by systemicinfusions or regional perfusion techniques. Collectively, theseobservations support evaluation of mithramycin for mesotheli-oma therapy using precision medicine techniques to identifypatients for treatment andpharmacokineticmodeling tooptimizedose and scheduling to maximize clinical efficacy.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: M. Rao, V. Shukla, Y. Hong, R.T. Ripley, W.D. Figg,D.S. SchrumpDevelopment of methodology: M. Rao, V. Shukla, Y. Hong, R.T. Ripley,P. Fetsch, X. Li, D.S. SchrumpAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): M. Rao, S.M. Atay, V. Shukla, Y. Hong, R.T. Ripley,J.A. Hong,M. Zhang, E. Reardon,M.Miettinen, X. Li, A.D. Rienzo, D.S. SchrumpAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis):M. Rao, S.M. Atay, V. Shukla, Y. Hong, M. Miettinen,X. Li, C.J. Peer, T. Sissung, W.D. Figg, R. Bueno, D.S. SchrumpWriting, review, and/or revision of the manuscript: M. Rao, S.M. Atay,T. Upham, J.A. Hong, E. Reardon, M. Miettinen, T. Sissung, W.D. Figg,D.S. SchrumpAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): M. Rao, T. Upham, J.A. Hong, D.S. SchrumpStudy supervision: M. Rao, D.S. SchrumpOther (providing analysis of unpublishedmicroarray data for specific genes):R. Bueno
AcknowledgmentsThe authors express their gratitude to Jan Pappas for assistance regarding
article preparation.
Grant SupportThis work was supported by NCI Intramural grants ZIA BC 011122 (to D.S.
Schrump) and ZIA BC 011418 (to D.S. Schrump).The costs of publication of this articlewere defrayed inpart by the payment of
page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received January 7, 2015; revised September 22, 2015; accepted September27, 2015; published OnlineFirst October 12, 2015.
References1. Bononi A,NapolitanoA, PassHI, YangH, CarboneM. Latest developments
in our understanding of the pathogenesis of mesothelioma and the designof targeted therapies. Expert Rev Respir Med 2015;9:633–54.
2. Carbone M, Baris YI, Bertino P, Brass B, Comertpay S, Dogan AU, et al.Erionite exposure in North Dakota and Turkish villages with mesotheli-oma. Proc Natl Acad Sci USA 2011;108:13618–23.
Rao et al.
Clin Cancer Res; 2015 Clinical Cancer ResearchOF12
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

3. International Agency for Research on Cancer. IARC monographs on theevaluation of carcinogenic risks of humans: Volume 100C: a review ofhuman carcinogens: arsenic, metals, fibres, and dust. Lyon, France: Inter-national Agency for Research on Cancer; 2011.
4. Henley SJ, Larson TC, Wu M, Antao VC, Lewis M, Pinheiro GA, et al.Mesothelioma incidence in 50 states and the District of Columbia, UnitedStates, 2003–2008. Int J Occup Environ Health 2013;19:1–10.
5. Leuzzi G, Rea F, Spaggiari L, Marulli G, Sperduti I, Alessandrini G, et al.Prognostic score of long-term survival after surgery for malignantpleural mesothelioma: a multicenter analysis. Ann Thorac Surg 2015;100:890–7.
6. Opitz I, Friess M, Kestenholz P, Schneiter D, Frauenfelder T, Nguyen-Kim DL, et al. A new prognostic score supporting treatment allocationfor multimodality therapy for malignant pleural mesothelioma- Areview of 12 years' experience. J Thorac Oncol. 2015 Aug 27. [Epubahead of print].
7. Shersher DD, Liptay MJ. Multimodality treatment of pleural mesothelio-ma. Surg Oncol Clin N Am 2013;22:345–55.
8. Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, et al. The nucleardeubiquitinase BAP1 is commonly inactivated by somatic mutations and3p21.1 losses in malignant pleural mesothelioma. Nat Genet 2011;43:668–72.
9. NasuM, EmiM, Pastorino S, TanjiM, Powers A, LukH, et al.High incidenceof somatic BAP1 alterations in sporadicmalignantmesothelioma. J ThoracOncol 2015;10:565–76.
10. Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. GermlineBAP1 mutations predispose to malignant mesothelioma. Nat Genetics2011;43:1022–1025.
11. Scheuermann JC, de Ayala Alonso AG,Oktaba K, Ly-HartigN,McGinty RK,Fraterman S, et al. Histone H2A deubiquitinase activity of the Polycombrepressive complex PR-DUB. Nature 2010;465:243–7.
12. Jean D, Daubriac J, Le Pimpec-Barthes F, Galateau-Salle F, Jaurand MC.Molecular changes in mesothelioma with an impact on prognosis andtreatment. Arch Pathol Lab Med 2012;136:277–93.
13. Riquelme E, Suraokar MB, Rodriguez J, Mino B, Lin HY, Rice DC, et al.Frequent coamplification and cooperation between C-MYC and PVT1oncogenes promote malignant pleural mesothelioma. J Thorac Oncol2014;9:998–1007.
14. Kemp CD, Rao M, Xi S, Inchauste S, Mani H, Fetsch P, et al. Polycombrepressor complex-2 is a novel target for mesothelioma therapy. ClinCancer Res 2012;18:77–90.
15. Kubo T, Toyooka S, Tsukuda K, Sakaguchi M, Fukazawa T, Soh J, et al.Epigenetic silencing of microRNA-34b/c plays an important role in thepathogenesis of malignant pleural mesothelioma. Clin Cancer Res2011;17:4965–74.
16. Christoph DC, Eberhardt WE. Systemic treatment of malignant pleuralmesothelioma: new agents in clinical trials raise hope of relevant improve-ments. Curr Opin Oncol 2014;26:171–81.
17. Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. Mithra-mycin inhibits SP1 binding and selectively inhibits transcriptional activityof the dihydrofolate reductase gene in vitro and in vivo. J Clin Invest1991;88:1613–21.
18. Li L, Davie JR. The role of Sp1 and Sp3 in normal and cancer cell biology.Ann Anat 2010;192:275–83.
19. Safe S, Imanirad P, Sreevalsan S, Nair V, Jutooru I. Transcription factor Sp1,also known as specificity protein 1 as a therapeutic target. Expert Opin TherTargets 2014;18:759–69.
20. Davie JR, He S, Li L, Sekhavat A, Espino P, Drobic B, et al. Nuclearorganization and chromatin dynamics–Sp1, Sp3 and histone deacetylases.Adv Enzyme Regul 2008;48:189–208.
21. Zhang M, Mathur A, Zhang Y, Xi S, Atay S, Hong JA, et al. Mithramycinrepresses basal and cigarette smoke-induced expression of ABCG2 andinhibits stem cell signaling in lung and esophageal cancer cells. Cancer Res2012;72:4178–92.
22. Saha S, Mukherjee S, Mazumdar M, Manna A, Khan P, Adhikary A, et al.Mithramycin A sensitizes therapy-resistant breast cancer stem cells towardgenotoxic drug doxorubicin. Transl Res 2015;165:558–77.
23. Chen DQ, Huang JY, Feng B, Pan BZ, De W, Wang R, et al. Histonedeacetylase 1/Sp1/microRNA-200b signaling accounts for maintenanceof cancer stem-like cells in human lung adenocarcinoma. PLoS One2014;9:e109578.
24. Yang WJ, Song MJ, Park EY, Lee JJ, Park JH, Park K, et al. Transcriptionfactors Sp1 and Sp3 regulate expression of human ABCG2 gene andchemoresistance phenotype. Mol Cells 2013;36:368–75.
25. AarskogNK, Vedeler CA. Real-time quantitative polymerase chain reaction.A new method that detects both the peripheral myelin protein 22 dupli-cation in Charcot-Marie-Tooth type 1A disease and the peripheral myelinprotein 22 deletion in hereditary neuropathy with liability to pressurepalsies. Hum Genet 2000;107:494–8.
26. RaoM,ChinnasamyN,Hong JA, ZhangY, ZhangM,Xi S, et al. Inhibition ofhistone lysine methylation enhances cancer-testis antigen expression inlung cancer cells: implications for adoptive immunotherapy of cancer.Cancer Res 2011;71:4192–204.
27. Thenappan A, Shukla V, Abdul Khalek FJ, Li Y, Shetty K, Liu P, et al. Loss oftransforming growth factor beta adaptor protein beta-2 spectrin leads todelayed liver regeneration in mice. Hepatology 2011;53:1641–50.
28. Jami MS, Hou J, Liu M, Varney ML, Hassan H, Dong J, et al. Functionalproteomic analysis reveals the involvement of KIAA1199 in breast cancergrowth, motility and invasiveness. BMC Cancer 2014;14:194.
29. Evensen NA, Kuscu C, Nguyen HL, Zarrabi K, Dufour A, Kadam P, et al.Unraveling the role of KIAA1199, a novel endoplasmic reticulum protein,in cancer cell migration. J Natl Cancer Inst 2013;105:1402–16.
30. Meigs TE, Fields TA, McKee DD, Casey PJ. Interaction of Galpha 12 andGalpha 13 with the cytoplasmic domain of cadherin provides a mech-anism for beta -catenin release. Proc Natl Acad Sci U S A 2001;98:519–24.
31. TangH,Wei P, Duell EJ, RischHA,Olson SH, Bueno-de-MesquitaHB, et al.Axonal guidance signaling pathway interacting with smoking inmodifyingthe risk of pancreatic cancer: a gene- and pathway-based interactionanalysis of GWAS data. Carcinogenesis 2014;35:1039–45.
32. Liu F, Pore N, Kim M, Voong KR, Dowling M, Maity A, et al. Regulation ofhistone deacetylase 4 expression by the SP family of transcription factors.Mol Biol Cell 2006;17:585–97.
33. Marouco D, Garabadgiu AV, Melino G, Barlev NA. Lysine-specificmodifications of p53: a matter of life and death? Oncotarget 2013;4:1556–71.
34. Sun Z, Wang L, Eckloff BW, Deng B, Wang Y, Wampfler JA, et al. Conservedrecurrent gene mutations correlate with pathway deregulation and clinicaloutcomes of lung adenocarcinoma in never-smokers. BMCMedGenomics2014;7:32.
35. Cao HH, Zheng CP, Wang SH, Wu JY, Shen JH, Xu XE, et al. A molecularprognosticmodel predicts esophageal squamous cell carcinomaprognosis.PLoS One 2014;9:e106007.
36. Curreri AR, Ansfield FJ. Mithramycin-human toxicology and preliminarytherapeutic investigation. Cancer Chemother Rep 1960;8:18–22.
37. Sewell IA, Ellis H. A trial of mithramycin in the treatment of advancedmalignant disease. Br J Cancer 1966;20:256–63.
38. Ohgami T, Kato K, Kobayashi H, Sonoda K, Inoue T, Yamaguchi S, et al.Low-dose mithramycin exerts its anticancer effect via the p53 signalingpathway and synergizes with nutlin-3 in gynecologic cancers. Cancer Sci2010;101:1387–95.
39. Zhao Y, Zhang W, Guo Z, Ma F, Wu Y, Bai Y, et al. Inhibition of thetranscription factor Sp1 suppresses colon cancer stem cell growth andinduces apoptosis in vitro and in nude mouse xenografts. Oncol Rep2013;30:1782–92.
40. Choi ES, Chung T, Kim JS, Lee H, Kwon KH, Cho NP, et al. Mithramycin Ainduces apoptosis by regulating the mTOR/Mcl-1/tBid pathway in andro-gen-independent prostate cancer cells. J Clin Biochem Nutr 2013;53:89–93.
41. Otjacques E, Binsfeld M, Rocks N, Blacher S, Vanderkerken K, Noel A, et al.Mithramycin exerts an anti-myeloma effect and displays anti-angiogeniceffects through up-regulation of anti-angiogenic factors. PLoSOne 2013;8:e62818.
42. Enge M, Bao W, Hedstrom E, Jackson SP, Moumen A, Selivanova G.MDM2-dependent downregulation of p21 and hnRNP K provides a switchbetween apoptosis and growth arrest induced by pharmacologically acti-vated p53. Cancer Cell 2009;15:171–83.
43. Li H, Zhang Y, Strose A, Tedesco D, Gurova K, Selivanova G. Integratedhigh-throughput analysis identifies Sp1 as a crucial determinant of p53-mediated apoptosis. Cell Death Differ 2014;21:1493–502.
44. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis andcancer therapy. Nat Rev Cancer 2013;13:83–96.
Mithramycin and MPM
www.aacrjournals.org Clin Cancer Res; 2015 OF13
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

45. LinRK,WuCY,Chang JW, Juan LJ,HsuHS,ChenCY, et al.Dysregulationofp53/Sp1 control leads to DNAmethyltransferase-1 overexpression in lungcancer. Cancer Res 2010;70:5807–17.
46. Banerjee A, Sanyal S, Kulkarni KK, Jana K, Roy S, Das C, et al. Anticancerdrug mithramycin interacts with core histones: An additional mode ofaction of the DNA groove binder. FEBS Open Bio 2014;4:987–95.
47. Zhang B, Golding BT, Hardcastle IR. Small-molecule MDM2-p53 inhibi-tors: recent advances. Future Med Chem 2015;7:631–45.
48. Nguyen DM, Schrump WD, Chen GA, Tsai W, Nguyen P, Trepel JB, et al.Abrogation of p21 expression by flavopiridol enhances depsipeptide-
mediated apoptosis in malignant pleural mesothelioma cells. Clin CancerRes 2004;10:1813–25.
49. Roth J, Peer CJ, Widemann B, Cole DE, Ershler R, Helman L, et al.Quantitative determination of mithramycin in human plasma by anovel, sensitive ultra-HPLC-MS/MS method for clinical pharmacoki-netic application. J Chromatogr B Analyt Technol Biomed Life Sci2014;970:95–101.
50. Kennedy BJ, Sandberg-Wollheim M, Loken M, Yarbro JW. Studieswith tritiated mithramycin in C3H mice. Cancer Res 1967;27:1534–8.
Clin Cancer Res; 2015 Clinical Cancer ResearchOF14
Rao et al.
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379

Published OnlineFirst October 12, 2015.Clin Cancer Res Mahadev Rao, Scott M. Atay, Vivek Shukla, et al. Mesothelioma Cellsto Mediate Senescence and Apoptosis of Malignant Pleural Mithramycin Depletes Specificity Protein 1 and Activates p53
Updated version
10.1158/1078-0432.CCR-14-3379doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2015/10/10/1078-0432.CCR-14-3379.DC1Access the most recent supplemental material at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://clincancerres.aacrjournals.org/content/early/2016/01/08/1078-0432.CCR-14-3379To request permission to re-use all or part of this article, use this link
Research. on March 10, 2019. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 12, 2015; DOI: 10.1158/1078-0432.CCR-14-3379