mir-205 targets pten and phlpp2 to augment akt...
TRANSCRIPT

1
miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant
phenotypes in non-small cell lung cancer
Junchao Cai1, 3, §, Lishan Fang1, 3, §, Yongbo Huang1, 3, Rong Li1, 3, Jie Yuan3, 4,
Yi Yang2, 3, Xun Zhu1, 3, Baixue Chen1, 3, Jueheng Wu1, 3, Mengfeng Li1, 3 *
Departments of 1Microbiology and 2Pharmacology, Zhongshan School of Medicine, Sun
Yat-Sen University; 3Key Laboratory of Tropical Disease Control (Sun Yat-Sen University),
Ministry of Education; 4Key Laboratory of Functional Molecules from Oceanic
Microorganisms (Sun Yat-sen University), Department of Education of Guangdong Province,
Guangzhou, Guangdong 510080, China
*Correspondence to:
Mengfeng Li, MD, Ph.D.
Departments of Microbiology, Zhongshan School of Medicine, Sun Yat-Sen University, 74
Zhongshan Road II, Guangzhou, Guangdong 510080, China. Voice: +86(20)87332748; Fax:
+86(20)87331209; E-mail: [email protected]
§ These authors contributed equally to this work.
Running Title: miR-205 activates AKT signaling by targeting PTEN and PHLPP2.
Keywords: miR-205; Lung cancer; Proliferation; Angiogenesis; NF-κB
The authors disclose no potential conflicts of interest.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

2
Abstract
AKT signaling is constitutively activated in various cancers, due in large part to loss of
function in the PTEN and PHLPP phosphatases that act as tumor suppressor genes. However,
AKT signaling is activated widely in non-small cell lung cancers (NSCLC) where genetic
alterations in PTEN or PHLPP genes are rare, suggesting an undefined mechanism(s) for their
suppression. In this study, we report upregulation of the oncomir miR-205 in multiple
subtypes of NSCLC, which directly represses PTEN and PHLPP2 expression and activates both
the AKT/FOXO3a and AKT/mTOR signaling pathways. miR-205 overexpression in NSCLC
cells accelerated tumor cell proliferation and promoted blood vessel formation in vitro and in
vivo. Conversely, RNAi-mediated silencing of endogenous miR-205 abrogated these effects.
The malignant properties induced by miR-205 in NSCLC cells were reversed by AKT inhibitors,
FOXO3a overexpression, Rapamycin treatment, or restoring PHLPP2 or PTEN expression.
Mechanistic investigations revealed that miR-205 overexpression was a result of
NF-κB-mediated transactivation of the miR-205 gene. Taken together, our results define a
major epigenetic mechanism for suppression of PTEN and PHLPP2 in NSCLC, identifying a
pivotal role for miR-205 in development and progression of this widespread disease.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

3
Introduction
Lung cancer remains a leading cause of cancer-related deaths worldwide, and among all lung
cancer cases, over 80% are non-small cell lung cancer (NSCLC), including squamous cell
carcinoma, adenocarcinoma, adenosquamous cell carcinoma, large cell carcinoma and several
other histologic types (1, 2). The overall five-year survival rate for NSCLC, with all stages
and subtypes combined, remains as low as 15% (1). Despite advances in surgical therapy,
radiotherapy, chemotherapy and recently developed targeting therapy, such as anti-epithelial
growth factor receptor (EGFR) and anti-vascular endothelial growth factor (VEGF) targeting
strategies, the mortality of the disease has not been markedly reduced thus far (2, 3). The
suboptimal efficacies of currently available therapeutic approaches to the treatment of lung
cancer is largely attributable to difficulties in dealing with the malignant phenotype of the
disease, namely, its highly uncontrolled characteristics of overactivated proliferation,
neovascularization, invasiveness and metastasis supported and/or promoted by deregulated
molecular signaling pathways within lung cancer cells (4).
Indeed, mounting evidence has demonstrated that the poor prognosis of NSCLC patients and
therapeutic failure are associated with a number of abnormally activated signaling pathways,
among which PI3K/AKT signaling represents one of the most important regulatory pathways
for the malignancy (5, 6). Notably, aberrant Akt activation is a poor prognostic factor for
NSCLC of all stages and contributes to resistance to first-generation single-agent targeting
therapy, such as gefitinib, a tyrosine kinase inhibitor clinically used for NSCLC patients with
EGFR overactivation (7, 8). Biologically, activated AKT confers NSCLC cells resistance to
chemotherapy and radiation, and promotes cancer cell survival (9, 10), and by contrast,
chemically synthetic compounds inhibiting AKT activation induce apoptosis of NSCLC cells in
vitro as well as in vivo (11). Moreover, AKT signaling contributes to oncogenesis through
activating multiple downstream effector molecules (12). Of note, activated AKT
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

4
phosphorylates tumor suppressor FOXO3a and impairs the transcription of its target genes
related to cell growth arrest such as p21, inactivation of which has also been implicated in the
promotion of tumor angiogenesis (13, 14). In addition, mTOR, another substrate subjected to
phosphorylation by AKT, enhances phosphorylation of S6K1 and 4E-BP1 (15) and plays crucial
roles in the regulation of ribosomal protein synthesis, for example, production of cyclin D1 and
VEGF-A at both transcriptional and translational levels (16, 17). It has been found that
mediated by the above molecular mechanisms, both AKT/FOXO3a and AKT/mTOR pathways
underlie lung cancer development and progression (18, 19). Thus, inhibitors targeting these
pathways might represent potentially applicable therapeutic agents against NSCLC.
Under physiological conditions, PI3K/AKT signaling is sophistically regulated by both
positive signals, such as signals emitted from growth factor-activated receptor tyrosine kinases
(RTK) (12, 20), and negative regulators, mainly including phosphatases PTEN and PHLPP.
PTEN, a well-known tumor suppressor, for instance, is essential for both normal lung
morphogenesis and the prevention of lung carcinogenesis (21). Lack of PTEN expression
represents a common event in a variety of tumor types. Unlike frequently observed mutations,
chromosomal deletion and/or loss of heterozygosity (LOH) in other cancer types, however, it
appears that loss of PTEN in NSCLC has not been associated with genomic alterations but
instead, more likely due to epigenetic silencing (22). PHLPP2, a recently characterized
member of the PHLPP family and suppressor of PI3K/AKT signaling, inhibits cell cycle
progression and promotes apoptosis in cells of various cancer types, including lung cancer cells
(23), albeit the expression profile of PHLPP2 in lung cancer tissues remains unclear.
Knowledge of how these negative regulators are suppressed in NSCLC so that AKT signaling is
aberrantly activated might provide a new basis for future targeting therapies against the disease.
In the framework of gene expression regulation, it is widely recognized that
miRNA-mediated post-transcriptional repression plays important roles, largely attributed to the
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

5
capability of a miRNA to inhibit multiple target mRNAs through binding to their 3’UTRs.
Previously, upregulated miR-205, which is located at lung cancer-associated genomic
amplification region 1q32.2, was implicated, respectively, in 104 cases of primary lung cancers
and 38 stage-I and -II NSCLC specimens (24, 25). Moreover, miR-205 was reported to be
expressed at higher level in squamous cell lung carcinoma than other types of NSCLC (26).
Nevertheless, whether miR-205 is mechanistically associated with NSCLC progression remains
unknown. In the current study, we identify that miR-205 is highly expressed in multiple
subtypes of NSCLC tissues and causes simultaneous downregulation of PHLPP2 and PTEN,
leading to activation of both AKT/FOXO3a and AKT/mTOR pathways, consequently leading to
accelerated proliferation and enhanced angiogenesis in NSCLC. We have also found that the
observed overexpression of miR-205, at least partly, is ascribed to transactivation by the NF-κB
transcription factor.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

6
Materials and Methods
Cell culture
NSCLC cell lines NCI-H460 (H460) (large cell carcinoma), NCI-H596 (H596)
(adenosquamous carcinoma), A549 (adenocarcinoma) and Calu-1(squamous cell carcinoma), as
well as NCI-H292 and NCI-H1299 were purchased from Shanghai Institutes of Biological
Sciences (Shanghai, China) and maintained in the DMEM medium (Invitrogen, Carlsbad, CA)
supplemented with 10% fetal bovine serum (HyClone, Logan, UT) and 1%
penicillin/streptomycin (Invitrogen). Primary normal lung epithelial cells (NLEs) were
obtained according to our previous report (27). In brief, surgically resected specimens of
normal lung tissue were promptly removed and transported aseptically in Hanks' solution
(Invitrogen, Carlsbad, CA) supplemented with 100 units/ml penicillin, and 100 µg/ml
streptomycin (Invitrogen) and 5 µg/ml gentamycin (Invitrogen). The tissue specimens were
then incubated with 1.5 units/ml dispase (Roche Molecular Biochemicals, Indianapolis, IN) at
4 °C overnight, and the epithelium was dissected and incubated with trypsin (Invitrogen). The
reaction was stopped with soybean trypsin inhibitor (Sigma, Saint Louis, MI) and centrifuged,
followed by resuspension in keratinocyte-SFM medium (KSFM) supplemented with 40 µg/ml
bovine pituitary extract, 1.0 ng/ml EGF, 100 units/ml penicillin, 100 µg/ml streptomycin, 5
µg/ml gentamycin and 100 units/ml nyastatin (Invitrogen). The BEAS-2B immortalized
human bronchial epithelial cell line (Shanghai Institutes of Biological Sciences, Shanghai,
China) was cultured in LHC-9 medium as instructed by the provider. All cell lines were
authenticated by short tandem repeat fingerprinting at IDEXX RADIL (Columbia, MO, USA)
and Services at SYSU Forensic Medicine Lab (Guangzhou, China).
Tissue specimens
All clinical NSCLC specimens conducted in this study were histopathologically and
clinically diagnosed at the Sun Yat-sen University Cancer Center during the period of 2009 to
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

7
2011. Clinicopathological classification of staging and subtypes of the tumor samples were
characterized according to the current International Union Against Cancer (UICC) standard.
Normal lung tissue specimens were taken from a standard distance (3cm) from the margin of
resected neoplastic tissues of NSCLC patients who underwent surgical lung resection. For the
use of these clinical materials for research purposes, prior patients’ consents and approval from
the Institutional Research Ethics Committee were obtained. Clinical information of 60 cases
of NSCLC patient specimens is presented in Supplemental Table 1.
Western blotting analysis
Western blotting analysis was performed using anti-phospho-AKT (ser473), anti-AKT,
anti-FOXO3a, anti-phospho-S6K1 (Thr389), anti-S6K1 and anti-4E-BP1(Epitomics,
Burlingame, CA), anti-phospho-FOXO3a (ser253), and phospho-4E-BP1 (Ser65) (Cell
signaling, Danvers, MA), anti-p21, anti-cyclinD1 and anti-PTEN (BD PharMingen, San Diego,
CA), anti-PHLPP2 (Abcam, Cambridge, MA) antibodies. Blotted membranes were stripped
and re-blotted with an anti-GAPDH, β–actin or p84 monoclonal antibody (Sigma, Saint Louis,
MO) as loading control. Surgically resected tumor tissues and corresponding adjacent normal
lung tissues were homogenized and the nuclear proteins were extracted using CelLytic Nuclear
Extraction Kit (Sigma) according to the manufacturer’s instructions. Relative protein levels
were quantified by densitometric scanning, and the relative gray values of protein were
calculated as Band Intensity of Protein of Interest / Band Intensity of Loading Control.
Xenografted tumor model in vivo and immunohistochemical analysis (IHC)
H460-vector and H460-miR-205 cells (2x106) were subcutaneously inoculated into the dorsal
thighs of individual female BALB/C nude mice (n = 5/group). Tumor lengths (L) and widths
(W) were measured every three days using a digital caliper, and tumor volumes were calculated
using the equation volume (mm3) = L*W2/2. After 19 days, mice were anesthetized and
sacrificed, and tumors were removed, photographed, weighed and sectioned (5µm in thickness),
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

8
followed by immunostaining. Following deparaffinization, sections were IHC analyzed using
antibodies for Ki-67, CD31, VEGF-A, PHLPP2 and PTEN, respectively, and subsequently were
pathologically confirmed for the tumor phenotype and specific immunostaining. The resultant
immunostaining images were captured using the AxioVision Rel.4.6 computerized image
analysis system (Carl Zeiss, Oberkochen, Germany). Briefly, the stained sections were
evaluated at 200x magnification, and 10 representative staining fields of each section were
analyzed to calculate the mean optical density (MOD), which denotes the strength of staining
signals as measured by positive pixels. The microvessel density (MVD) was measured
according to the Weidner’s method (28). Every endothelial cell cluster of positive CD31
immunoreactivity, clearly separated from tumor cells, was counted as an individual vessel.
The immunostaining of p65, PHLPP2 and PTEN in 60 cases of formalin-fixed,
paraffin-embedded sections was evaluated and scored by two independent observers as
described in supplementary information.
Statistical analysis
Comparative analysis between groups for statistical significance was performed with
two-tailed paired Student’s t test. The correlation between miR-205 expression and nuclear
p65 level or PHLPP2 and PTEN protein levels in frozen and paraffin-embedded NSCLC tumor
tissues was respectively calculated using chi-square test and Pearson test. All error bars
represent mean ± SD derived from three independent experiments. P < 0.05 was considered
statistically significant in all cases.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

9
Results
miR-205 inhibited expression of PHLPP2 and PTEN and bound to their 3’UTRs.
To identify miRNAs that might contribute to activation of Akt signaling, we began our study
with searching for those miRNAs that potentially target PTEN, a major negative regulator of the
pathway, using the TargetScan database. Consequently, miR-205 was predicted to target
putative binding sites in the 3’UTR of PTEN mRNA, as well as that of PHLPP2 (Fig. 1A). Of
particular interest, miR-205 is located in a lung cancer-associated amplification region and
previously found to be upregulated in NSCLC tissues as aforementioned. Furthermore, we
found that transfection of miR-205 mimic oligonucleotides drastically reduced, but
anti-miR-205 (Anti-miR) oligonucleotides increased, the protein levels of both PHLPP2 and
PTEN, as compared to those in control oligonucleotides (NC) transfection in H460 and H596
NSCLC cells, which showed moderately enhanced expression of miR-205 compared to primary
normal lung epithelial cells (NLEs) and immortalized human bronchial epithelial cell line
BEAS-2B. These results were also validated in two other subtypes of NSCLC cells, namely,
A549 and Calu-1, which expresses slightly and markedly higher, respectively, level of miR-205
than NLEs and BEAS-2B, in response to transfection of miR-205 mimic and Anti-miR
oligonucleotides, respectively. (Fig. 1B and Fig. S1). To further assess whether miR-205 and
mRNA of PHLPP2 or PTEN interact and form miRNP complexes, RNA-immunoprecipitation
(RIP) analysis performed by pulling down Ago1 showed that far more PHLPP2 and PTEN
transcripts were assembled into miR-205 mimic oligonucleotides-containing miRNPs than into
NC oligonucleotides-containing miRNPs, due to binding of miR-205 with the mRNA 3’UTR
regions (Fig. 1C). Moreover, we separately cloned 3’UTRs of PHLPP2 and PTEN into a
luciferase reporter plasmid to determine the direct inhibitory binding of miR-205 to the 3’UTRs.
As shown in Fig. 1, D and E, miR-205 mimic oligonucleotides transfection imposed a
dose-dependent reduction in the normalized luciferase activities of both reporters, which were
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

10
dose-dependently increased by antagonizing endogenous miR-205. The suppressive effects of
miR-205 mimic oligonucleotides on luciferase activities were completely deprived by
introduction of 4-nucleotide mutations in miR-205, suggesting an importance of appropriate
binding of miR-205 to the target 3’UTRs. The engineered levels of miR-205 expression in the
above transfection with miR-205 mimic or anti-miR oligonucleotides were confirmed (Fig. 1F).
Taken together, these data strongly suggested that miR-205 posttranscriptionally inhibited
protein expression of PHLPP2 and PTEN through directly binding to the 3’UTRs of their
mRNAs.
Ectopic miR-205 expression in NSCLC cells stimulated proliferation and angiogenesis.
To investigate whether miR-205 is biologically relevant to the development of malignant
phenotype of NSCLC cells, we stably overexpressed miR-205 in H460, H596 and A549
NSCLC cell lines (Fig. S2A) and performed gain-of-function experiments in vitro. As
analyzed by MTT assay and shown in Fig. 2A, miR-205 transduction significantly accelerated
the proliferation of H460, H596 and A549 cells by 58.3%, 48.5% and 44.6%, respectively, as
compared to control vector transduction. Additionally, clonogenic and anchorage-independent
growth assays revealed that H460-miR-205, H596-miR-205 and A549-miR-205 cells formed
more and larger-sized colonies in either 2-D or 3-D setting than their corresponding control
cells (Fig. 2, B and C). Furthermore, serum-free conditioned medium collected from cultured
miR-205-overexpressing cells of various subtypes stimulated HUVEC to form markedly longer
and more well-organized network of tubule structure than those from control vector cells (Fig.
2D), accompanied by formation of more secondary- and third-order vessels from preexisting
vessels as determined by chicken chorioallantoic membrane (CAM) assay (Fig. 2E). Taken
together, our data suggested a functionally important role of miR-205 in promoting the
proliferative and angiogenic phenotype of NSCLC cells.
Silencing endogenous miR-205 abrogated the abilities of NSCLC cells to proliferate and to
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

11
induce vascular growth.
To further investigate whether endogenous miR-205 contributes to maintenance of the
proliferative and pro-angiogenic properties of NSCLC cells, loss-of-function studies were
performed by employing anti-miR-205 oligonucleotides to silence miR-205 expression (Fig.
S2B). As shown in Fig. 3A, transfection with anti-miR-205 oligonucleotides suppressed cell
proliferation by 43.1%, 34.5% and 38.9%, respectively, in H460, H596 and Calu-1 cells. In
parallel, silencing miR-205 remarkably compromised the tumorigenic abilities of all cell lines
as demonstrated by clonogenic and anchorage-independent growth assays (Fig. 3, B and C).
Furthermore, knockdown of miR-205 completely abrogated the ability of NSCLC cells of
various subtypes to induce tube formation of cultured HUVEC and formation of lower-order
blood vessels within CAMs (Fig. 3, D and E). Together with the above overexpression
experiments, these data strongly suggested an essential contribution of endogenous miR-205 to
the tumor proliferation and angiogenesis in NCSLC.
miR-205 promoted NSCLC tumor growth and tumor blood vessels growth in vivo.
To confirm whether the biological effects of miR-205 observed in cultured cells is relevant to
NSCLC tumor growth in vivo, H460-vector and H460-miR-205 cells, respectively, were
subcutaneously inoculated into BALB/C athymic mice. As shown in Fig. 4A, tumors formed
by miR-205-overexpressing cells grew more rapidly than those by vector-control cells within
the first week following inoculation, and the difference in average tumor volume between
experimental and control animals continued to increase for 2.5 folds at the experimental
endpoint (19 days). In parallel, increases in sizes and weights of tumors excised from animals
of the miR-205 overexpression group were also observed as compared to those of the control
group (Fig. 4, B and C). Notably, high degrees of neovascularization were seen on the surface
of miR-205-overexpressing tumors, whereas control tumors displayed nearly avascular surface,
as shown in Fig. 4D, strongly suggesting that miR-205 enhanced tumor angiogenesis.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

12
Consistent with the above observations, as shown in Fig. 4E, the proportion of proliferative
Ki67-positive cells increased from 44.6% in control tumors to 89.4% in
miR-205-overexpressing tumors, and there were more intratumoral microvessels formed with
higher microvascular densities (MVD) in miR-205-overexpressing tumors, as assessed by
CD31 staining, than the control tumors. Moreover, significant upregulation of VEGF-A
expression was shown in miR-205-transduced tumors, which simultaneously presented lower
protein levels of PHLPP2 and PTEN than control tumors, as confirmed by quantifying staining
intensities of these proteins (Fig. 4E). Collectively, these in vivo data strongly demonstrated a
biological role of miR-205 as a promoter of tumor growth and inducer of angiogenesis in
NSCLC.
Both AKT/FOXO3a and AKT/mTOR signaling contributed to miR-205-mediated
malignant phenotype of NSCLC cells.
We next examined the role of miR-205-mediated inhibition of PTEN and PHLPP2 in the
development and maintenance of the malignant phenotype of NSCLC cells. Of note, ectopic
miR-205 expression in H460, H596 and A549 cells remarkably increased the level of
phosphorylated AKT, resulting in enhanced phosphorylation of FOXO3a, S6K and 4E-BP1,
while knockdown of miR-205 in H460, H596 and Calu-1 cells robustly suppressed
phosphorylation of AKT, FOXO3a, S6K and 4E-BP1 (Fig. 5A), indicating that miR-205 indeed
activated the AKT/FOXO3a and AKT/mTOR pathways. Furthermore, decreased p21 and
increased cyclin D1 expression could be caused by miR-205 overexpression, whereas opposite
effects on the regulation of p21 and cyclin D1 were found at both the mRNA and protein levels
when miR-205 was knocked down (Fig. 5B). In addition, as determined by real-time PCR and
ELISA, VEGF-A expression was shown to be upregulated in miR-205 overexpressing cells and
downregulated in miR-205 knocked-down cells (Fig. 5C), providing a molecular basis for the
regulatory effects of miR-205 on NSCLC cell proliferation and angiogenesis.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

13
We next separately tested the effect of specific blockage of AKT/FOXO3a and AKT/mTOR
pathways on the pro-proliferative and pro-angiogenic functions of miR-205. As shown in Fig.
5D, treatment with a specific PI3K inhibitor, LY294002, or an AKT inhibitor drastically
retarded the proliferation of miR-205-overexpressing cells, such as H460-miR-205 and
H596-miR-205 cells, and considerably weakened the abilities of these miR-205-transduced
cells to induce HUVEC tube formation. Similarly, potentiation of the proliferation and
pro-angiogenic ability of H460 and H596 cells by miR-205 were markedly eliminated in
response to Rapamycin inhibition of the mTOR pathway (Fig. 5E). Furthermore,
overexpression of FOXO3a repressed the growth of, and HUVEC tube formation induced by,
H460-miR-205 or H596-miR-205 cells (Fig. 5F). The effectiveness of treatment with
LY294002, AKT inhibitor, Rapamycin or FOXO3a overexpression was confirmed, as shown in
Fig. 5G. These data suggested that miR-205 promoted the proliferative and angiogenic
phenotype of NSCLC cells by simultaneously activating both AKT/FOXO3a and AKT/mTOR
pathways.
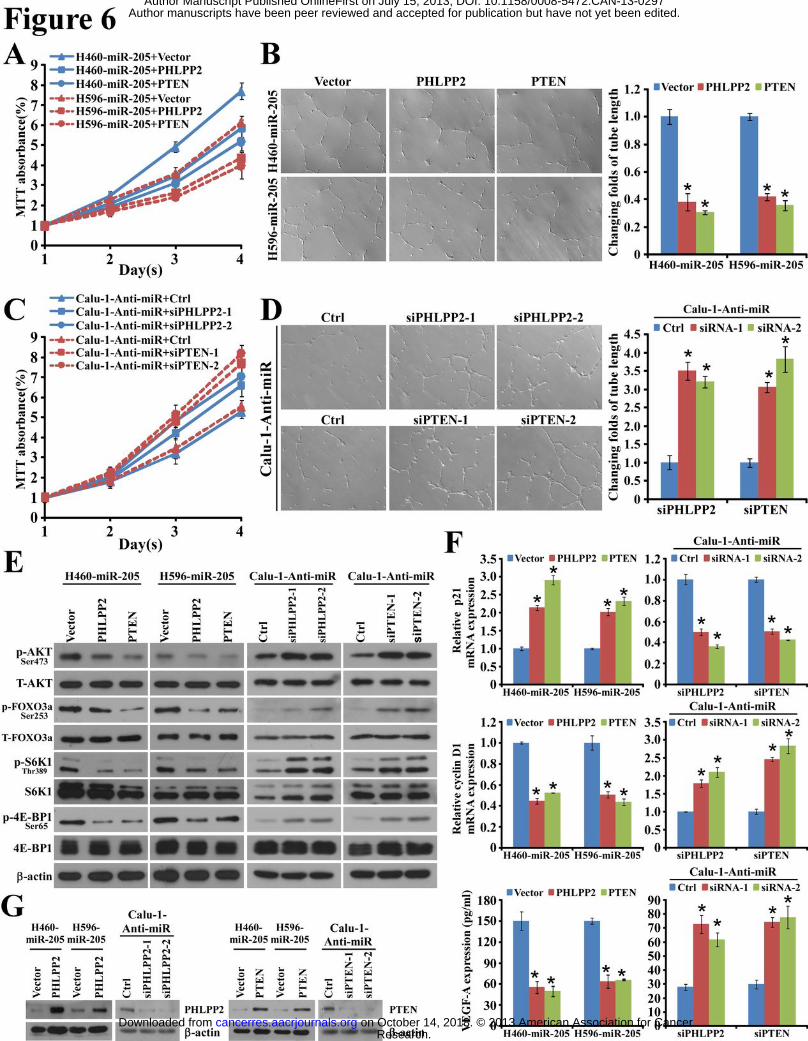
Repression of PHLPP2 and PTEN in NSCLC cells was essential for miR-205-induced
proliferation and neovascularization.
Based on the indispensable role of AKT activation in the biological functions of miR-205, we
then asked whether sustained repression of PHLPP2 and PTEN was essential. Indeed,
re-expression of the open reading frame (ORF, without 3’-UTR) of PHLPP2 or PTEN partially,
but significantly, impaired the abilities of miR-205-transduced cells to proliferate and to induce
tube formation of HUVEC as assessed by MTT assay and HUVEC/Matrigel assay, respectively
(Fig. 6, A and B). By contrast, in miR-205 knocked-down cells, namely, Calu-1-Anti-miR,
which expressed increased level of PHLPP2 and PTEN relative to its parental cells,
silencingPHLPP2 or PTEN significantly promoted their proliferation and induced tube
formation of HUVEC (Fig. 6, C and D). Unexceptionally, restoration of PHLPP2 or PTEN
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

14
protein expression greatly diminished phosphorylation of AKT, FOXO3a, S6K1 and 4E-BP1 in
both H460-miR-205 and H596-miR-205 cells (Fig. 6E), accompanied by elevated p21, reduced
cyclin D1 transcription, and decreased VEGF-A secretion (Fig. 6, F and G). Conversely,
silencing PHLPP2 or PTEN in Calu-1-Anti-miR cells exhibited opposite effects (Fig. 6, E, F
and G). These data supported that downregulation of PHLPP2 and PTEN was essential for
miR-205-induced increase of cell-proliferative activity and angiogenesis-inducing ability in
NSCLC.
NF-κB-transactivated miR-205 expression correlated with PHLPP2 and PTEN expression.
Previous array-based miRNA profiling has demonstrated that miR-205 expression is
upregulated in human NSCLC tissues. Indeed, we confirmed the upregulation of miR-205 in
22 frozen NSCLC tumor lesions consisting of 6 cases of adenocarcinoma (AD), 6 cases of
squamous cell carcinoma (SCC), 6 cases of large cell carcinoma (LCC) and 4 cases of
adenosquamous carcinoma (ASC) paired with non-cancerous adjacent tissues, among which
miR-205 was markedly highly expressed in SCC compared to other subtypes (Fig. 7A). To
elucidate the mechanism underlying the upregulation of miR-205 in NSCLC, we employed the
UCSC genome browser tool and identified that putative binding sequences for NF-κB p65
subunit were present at a ENCODE H3K4Me1 site, an indicator for the presence of active
chromatin region and transcription enhancer(s), located 3.5kb upstream to the miR-205 gene
locus (Fig. 7B). Moreover, overexpression of p65 or treatment with TNF-α to activate NF-κB
transcriptional activity robustly elevated pri-miR-205 expression, and in contrast, p65 siRNA or
a dominant-negative mutant IκBα, which blocked NF-κB activation, abated pri-miR-205
expression (Fig. 7B), suggesting that NF-κB signaling contributed to the modulation of
miR-205 transcription. In further support of this notion, ChIP analysis revealed enhanced
binding of p65 with the putative binding site located in the miR-205 upstream region, as
compared to that with the control sequence, when detected by primers spanning irrelevant
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

15
sequences (Fig. 7C). Of note, when a 500bp DNA fragment covering the putative p65 binding
site was cloned upstream to luciferase reporter gene and transfected into cells overexpressing
p65, significantly increased luciferase activity was achieved, whereas luciferase activity was not
changed when linked to mutated putative p65 binding sequence or irrelevant control sequence
(Fig. 7D), suggesting that miR-205 could be directly transactivated by the NF-κB transcription
factor in NSCLC cells.
In an effort to reveal the clinical relevance of above identified correlation, we found that in 8
frozen primary NSCLC tissues, a high miR-205 level was linked to diminished expression of
PHLPP2 and PTEN and increased nuclear p65 abundance as compared with those in 2
non-cancerous lung tissue specimens (Fig. 7E). Furthermore, examination of miR-205
expression and PHLPP2 and PTEN expression, as well as p65 localization was conducted in a
cohort of 60 cases of paraffin-embedded archived human NSCLC specimens (Supplementary
Table 1). The high (> median, n=30) and low (< median, n=30) miR-205 expression group
were identified. Of note, 63.3% of 30 specimens with high miR-205 expression exhibited
strong nuclear localization of p65, in contrast to the mainly cytoplasmic p65 staining in 73.3%
of 30 specimens expressing low level of miR-205 (P = 0.004, r = 0.369). Moreover, in the
high miR-205 expression group, 63.3% and 66.7% of cases, respectively, showed low levels of
PHLPP2 and PTEN, which were highly expressed, respectively, in 63.3% and 70.0% of
specimens with low miR-205 expression (P = 0.039, r = -0.265 and P = 0.004, r = -0.367,
respectively) (Fig. 7, F and G). Taken together, these data suggest that the experimental
demonstration that NF-κB-transactivated miR-205 overexpression negatively regulates PHLPP2
and PTEN is clinically relevant in NSCLC.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

16
Discussion
In the current study, we have demonstrated a tumor-promoting role of miR-205 in NSCLC,
overexpression of which robustly promotes cell proliferation and vascular growth both in vitro
and in vivo. In contrast, inhibition of endogenous miR-205 remarkably abrogates the abilities
of NSCLC cells of various tissue subtypes to proliferate and to induce angiogenesis. At the
molecular level, both the AKT/FOXO3a and AKT/mTOR pathways contribute to
miR-205-mediated malignant phenotype of NSCLC cells, likely mediated by suppressing
PHLPP2 and PTEN expression. Of note, the close correlation between high miR-205
expression and low expression of PHLPP2 and PTEN, as well as with the malignant properties
of NSCLC tumors, were also confirmed in xenoplanted tumors and in clinical NSCLC samples,
suggesting a possible role of miR-205 in the development and progression of NSCLC.
It has been previously indicated that miR-205 might represent a specific biomarker in lung
squamous cell carcinoma (SCC), a subtype of NSCLC (26). Interestingly, our current study on
one hand confirms a higher level of miR-205 expression in SCC tissues than that in other
NSCLC subtypes, and on the other hand verifies that the non-SCC NSCLC subtypes also
overexpress miR-205 as compared with their adjacent non-cancerous tissues, suggesting that
miR-205 overexpression might indeed be an indicator for the presence and/or progression of
NSCLC. Notably, the enhanced expression of miR-205 in multiple subtypes of NSCLC may
be attributed, at least in part, to direct transactivation of NF-κB. It is widely recognized that
NF-κB signaling plays a key role in stimulating oncogenesis, and that its activation can be
triggered by carcinogenic or tumor-promoting factors (29, 30). Our study uncovers a novel
mechanism, mediated by miR-205, linking NF-κB overactivation to supporting the malignant
phenotype in NSCLC, highlighting the importance of miRNA in the NF-κB-promoted
oncogenic cascades, and such a finding is of particular significance in the interest of developing
interventional strategies against NF-κB signaling, as direct NF-κB inhibitors may cause severe
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

17
adverse pharmacological effects due to the important roles of NF-κB in a broad array of
physiological processes (31). In this context, more experimental investigation regarding the
therapeutic efficacy of anti-miR-205 approaches is warranted.
It should be noted that miR-205 can be upregulated or downregulated in different cancer
types or subtypes, as previous studies have suggested. For example, miR-205 has been
reported to be highly expressed in normal mammary gland stem cells and triple-positive breast
cancer tissues but present low-level expression in metastatic samples (32-34). In addition,
miR-205 is markedly upregulated in specimens of cervical cancer but in contrast,
downregulated in those of prostate cancer (35, 36). In the scenario of lung cancer
development and progression, however, several previous studies have identified an upregulation
of miR-205, which is localized at 1q32.2, a lung cancer-associated genomic amplification
region (24-26). In support of the tumor-promoting role of miR-205 in NSCLC, our current
study provides comprehensive evidence for the functional, mechanical as well as clinical
significance of this molecule. These findings together indicate that the regulation of miR-205
expression, as well as its biological functions, might be tissue- and cancer-type dependent.
Thus, it remains important to thoroughly understand the molecular mechanisms mediating the
differential biological effects and targets of miR-205 in NSCLC and other cancer types.
Importantly, two bona fide target genes of miR-205, PTEN and PHLPP2, have been
identified by our study, both of which are phosphpatases essential for specific and effective
termination of the oncogenic AKT signaling. PTEN, of note, controls normal lung
morphogenesis and is essential for prevention of lung carcinogenesis in mice (21). Indeed,
low PTEN expression correlates with poor prognosis in NSCLC patients, and nearly 70% of
NSCLC cases have been detected to display reduced or lost PTEN expression (37, 38).
Genetic alterations, including LOH or inactivating mutation of PTEN, however, are rare in
NSCLC, identified in only approximately 10% of NSCLC tumors (39, 40), and promoter
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

18
methylation of PTEN is found in only 24%-26% of NSCLC tumors (22, 41), suggesting that
other suppressive mechanism(s) also plays important roles in the cancer type. Thus, our
current finding adds an important dimension of regulatory mechanism underlying the inhibition
of PTEN in NSCLC. On the other hand, PHLPP2, which has been postulated to act as a tumor
suppressor that inhibits cell growth and promotes apoptosis in cancer cell lines (23), is located
at 16q22.3, a region that undergoes LOH in breast and ovarian cancers, prostate cancer and
hepatocellular carcinoma (42). Nonetheless, expression status and biological function of
PHLPP2 in lung cancer remains unclear. Our data show that experimental overexpression of
PHLPP2 in NSCLC cells significantly antagonizes the stimulatory effect of miR-205 on cell
growth and vascularization, silencing of which rescues the compromised abilities of miR-205
knocked-down NSCLC cells to proliferate and to induce angiogenesis, and that PHLPP2 is
downregulated in NSCLC in a manner inversely correlated with high miR-205 expression,
underscoring the importance of this phosphatase in the cancer type. Interestingly, we also
observe that neither PTEN nor PHLPP2 restoration could completely reverse the malignant
properties of NSCLC cells induced by miR-205, suggesting that a simultaneous repression of
PTEN and PHLPP2 may be necessary for the resultant oncogenic effects of miR-205.
In summary, this study provides a miR-205-mediated mechanism for the frequent activation
of AKT signaling in NSCLC pathogenesis, causing uncontrolled proliferation and neoplastic
angiogenesis. It is well documented that AKT activation confers survival disadvantage in
NSCLC patients (37, 43), and that kinase inhibitors targeting PI3K/AKT signaling or/and
mTOR pathway exhibit promising antitumor activities in preclinical trials but probably induce
upstream RTK activity and reversely activate AKT signaling in a wide spectrum of tumor types,
including NSCLC, resulting in compromised therapeutic efficacy, suggesting that effective
therapy may require combined inhibitors targeting both the upstream signaling and the
downstream specific pathways (44-46). Therefore, the feature that miR-205 inhibition almost
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

19
shuts down upstream signaling of AKT activation and simultaneously suppresses both
AKT/mTOR and AKT/FOXO3a pathways makes it a reasonable candidate target for therapeutic
intervention in NSCLC.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

20
Grant support
This work was supported by the Ministry of Science and Technology of China grant (No.
973-2011CB11305); The Natural Science Foundation of China (No. 81071647, 81071762,
30900415, 81272339, 81272417); Guangdong Recruitment Program of Creative Research
Groups (No. 2009010058, 2010B030600003); National Science and Technique Major Project
(No. 201005022-2, 2012ZX10004213, 311030).The Key Science and Technique Research
Project of Guangdong Province (12B292060029).
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

21
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin.
2011;61:69-90.
2. Buyukcelik A, Yalcin B, Utkan G. Multidisciplinary management of lung cancer. N Engl J Med.
2004;350:2008-10; author reply -10.
3. Pallis AG, Fennell DA, Szutowicz E, Leighl NB, Greillier L, Dziadziuszko R. Biomarkers of clinical benefit for
anti-epidermal growth factor receptor agents in patients with non-small-cell lung cancer. Br J Cancer.
2011;105:1-8.
4. Petrosyan F, Daw H, Haddad A, Spiro T. Targeted therapy for lung cancer. Anticancer Drugs. 2012;23:1016-21.
5. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene.
2005;24:7455-64.
6. Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for
therapeutic targeting. Adv Cancer Res. 2005;94:29-86.
7. Tsurutani J, Fukuoka J, Tsurutani H, Shih JH, Hewitt SM, Travis WD, et al. Evaluation of two phosphorylation
sites improves the prognostic significance of Akt activation in non-small-cell lung cancer tumors. J Clin Oncol.
2006;24:306-14.
8. Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G. Response to epidermal growth factor receptor inhibitors in
non-small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with
persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res.
2003;9:2316-26.
9. Brognard J, Clark AS, Ni YC, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung
cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Research.
2001;61:3986-97.
10. Schuurbiers OC, Kaanders JH, van der Heijden HF, Dekhuijzen RP, Oyen WJ, Bussink J. The PI3-K/AKT-pathway
and radiation resistance mechanisms in non-small cell lung cancer. J Thorac Oncol. 2009;4:761-7.
11. Lee HY, Moon H, Chun KH, Chang YS, Hassan K, Ji L, et al. Effects of insulin-like growth factor binding
protein-3 and farnesyltransferase inhibitor SCH66336 on Akt expression and apoptosis in non-small-cell lung
cancer cells. J Natl Cancer Inst. 2004;96:1536-48.
12. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer.
2002;2:489-501.
13. Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, et al. Involvement of Foxo transcription
factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115:2382-92.
14. Dansen TB, Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol.
2008;18:421-9.
15. Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through
its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Molecular and Cellular Biology.
2004;24:200-16.
16. Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for
therapeutic invention. Mol Cancer. 2007;6:24.
17. Land SC, Tee AR. Hypoxia-inducible factor 1 alpha is regulated by the mammalian target of rapamycin (mTOR)
via an mTOR signaling motif. Journal of Biological Chemistry. 2007;282:20534-43.
18. Pisick E, Jagadeesh S, Salgia R. Receptor tyrosine kinases and inhibitors in lung cancer. ScientificWorldJournal.
2004;4:589-604.
19. Blake DC, Jr., Mikse OR, Freeman WM, Herzog CR. FOXO3a elicits a pro-apoptotic transcription program and
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

22
cellular response to human lung carcinogen nicotine-derived nitrosaminoketone (NNK). Lung Cancer.
2010;67:37-47.
20. Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905-27.
21. Yanagi S, Kishimoto H, Kawahara K, Sasaki T, Sasaki M, Nishio M, et al. Pten controls lung morphogenesis,
bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J Clin Invest. 2007;117:2929-40.
22. Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, et al. Lack of PTEN expression in non-small cell lung cancer
could be related to promoter methylation. Clinical Cancer Research. 2002;8:1178-84.
23. Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the
amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917-31.
24. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in
lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189-98.
25. Vosa U, Vooder T, Kolde R, Fischer K, Valk K, Tonisson N, et al. Identification of MiR-374a as a Prognostic
Marker for Survival in Patients with Early-Stage Nonsmall Cell Lung Cancer. Gene Chromosome Canc.
2011;50:812-22.
26. Lebanony D, Benjamin H, Gilad S, Ezagouri M, Dov A, Ashkenazi K, et al. Diagnostic assay based on
hsa-miR-205 expression distinguishes squamous from nonsquamous non-small-cell lung carcinoma. J Clin Oncol.
2009;27:2030-7.
27. Cai J, Wu J, Zhang H, Fang L, Huang Y, Yang Y, et al. miR-186 downregulation correlates with poor survival in
lung adenocarcinoma, where it interferes with cell-cycle regulation. Cancer Res. 2013;73:756-66.
28. Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, et al. Lack of PTEN expression in non-small cell lung cancer
could be related to promoter methylation. Clin Cancer Res. 2002;8:1178-84.
29. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7-11.
30. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344-62.
31. Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat
Rev Drug Discov. 2004;3:17-26.
32. Ibarra I, Erlich Y, Muthuswamy SK, Sachidanandam R, Hannon GJ. A role for microRNAs in maintenance of
mouse mammary epithelial progenitor cells. Genes Dev. 2007;21:3238-43.
33. Mattie MD, Benz CC, Bowers J, Sensinger K, Wong L, Scott GK, et al. Optimized high-throughput microRNA
expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol
Cancer. 2006;5:24.
34. Sempere LF, Christensen M, Silahtaroglu A, Bak M, Heath CV, Schwartz G, et al. Altered MicroRNA expression
confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 2007;67:11612-20.
35. Wang XH, Tang S, Le SY, Lu R, Rader JS, Meyers C, et al. Aberrant Expression of Oncogenic and
Tumor-Suppressive MicroRNAs in Cervical Cancer Is Required for Cancer Cell Growth. PLoS One. 2008;3.
36. Schaefer A, Jung M, Mollenkopf HJ, Wagner I, Stephan C, Jentzmik F, et al. Diagnostic and prognostic
implications of microRNA profiling in prostate carcinoma. Int J Cancer. 2010;126:1166-76.
37. Tang JM, He QY, Guo RX, Chang XJ. Phosphorylated Akt overexpression and loss of PTEN expression in
non-small cell lung cancer confers poor prognosis. Lung Cancer. 2006;51:181-91.
38. Bepler G, Sharma S, Cantor A, Gautam A, Haura E, Simon G, et al. RRM1 and PTEN as prognostic parameters
for overall and disease-free survival in patients with non-small-cell lung cancer. Journal of Clinical Oncology.
2004;22:1878-85.
39. Kohno T, Takahashi M, Manda R, Yokota J. Inactivation of the PTEN/MMAC1/TEP1 gene in human lung
cancers. Genes Chromosomes Cancer. 1998;22:152-6.
40. Forgacs E, Biesterveld EJ, Sekido Y, Fong K, Muneer S, Wistuba, II, et al. Mutation analysis of the
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

23
PTEN/MMAC1 gene in lung cancer. Oncogene. 1998;17:1557-65.
41. Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, et al. PTEN expression in non-small-cell lung
cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol.
2005;36:768-76.
42. Brognard J, Newton AC. PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol Metab.
2008;19:223-30.
43. David O, Jett J, LeBeau H, Dy G, Hughes J, Friedman M, et al. Phospho-Akt overexpression in non-small cell
lung cancer confers significant stage-independent survival disadvantage. Clin Cancer Res. 2004;10:6865-71.
44. O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor
tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500-8.
45. Gridelli C, Maione P, Rossi A. The potential role of mTOR inhibitors in non-small cell lung cancer. Oncologist.
2008;13:139-47.
46. Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition
relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58-71.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

24
Figure Legends
Figure 1. miR-205 directly targets PHLPP2 and PTEN genes in NSCLC cells. (A)
Predicted target sequences in 3’UTRs of PHLPP2 and PTEN bound to miR-205. (B) Western
blotting analysis of PHLPP2 and PTEN expression in indicated cells. (C) Quantitation of
recruited mRNAs of PHLPP2 and PTEN to miRNP complex immunoprecipitated with Ago 1,
examined by RIP analysis. IgG immunoprecipitation was used as a negative control. (D and
E) Luciferase assays of pGL3-PHLPP2-3’UTR or pGL3-PTEN-3’UTR reporter in indicated
cells transfected with increasing amounts (20 and 50 nM) of indicated oligonucleotides.
Sequence of miR-205 mutant is shown. (F) Validation of the levels of miR-205 expression in
indicated cells examined by real-time RT-PCR. *: P<0.05; **: P<0.01.
Figure 2. Ectopic miR-205 expression in NSCLC cells accelerates proliferation and
induces angiogenesis. (A) MTT assay reveals cell growth curves of H460, H596 and A549
cells. (B) Representative micrographs (left) and relative quantification (right) of crystal
violet-stained cell colonies analyzed by colony formation assay for 12 or 15 days. (C)
Representative micrographs (left) and quantification (right) of colonies larger than 50µm or
containing more than 100 cells. (D) Representative micrographs (left) and relative
quantification (right) of tube length by HUVEC tube formation assay. (E) Representative
micrographs of induced neovessels by CAM assay. *: P<0.05; **: P<0.01.
Figure 3. Silencing endogenous miR-205 abrogates the abilities of NSCLC cells to
proliferate and to induce vascular growth. (A) MTT assay reveals cell growth curves of
H460, H596 and Calu-1 cells. (B) Representative micrographs (left) and relative
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

25
quantification (right) of colonies analyzed by colony formation assay for 10 or 15 days. (C)
Representative micrographs (left) and quantification (right) of colonies that larger than 25µm or
containing more than 50 cells. (D) Representative micrographs (left) and relative
quantification (right) of tube length by HUVEC tube formation assay. (E) Representative
micrographs of induced neovessels by CAM assay. *: P<0.05; **: P<0.01.
Figure 4. Overexpressing miR-205 in NSCLC cells promotes tumor growth and blood
vessel formation in vivo. (A) Tumor growth curves in mice (n = 5/group) inoculated with
indicated cells at indicated days. (B) At the experimental endpoint, tumors were dissected and
photographed as indicated. (C) Each tumor formed by indicated cells was weighed. (D)
Stereoscopic microscope revealed surface blood vessels of indicated tumors. (E) H&E
staining confirms tumor cells in slices of indicated tumor sections and IHC stained for Ki-67,
CD31, VEGF, PHLPP2, and PTEN are quantified by staining intensity. Levels of miR-205
expression in indicated tumor tissues are confirmed by real-time PCR. For A and E, data are
presented as mean ± SD (n = 5 mice/group). *: P<0.05.
Figure 5. miR-205 activates both AKT/FOXO3a and AKT/mTOR pathways to exert its
biological effects on NSCLC cells. (A) Western blotting analysis of phospho-AKT, total
AKT (T-AKT), phospho-FOXO3a, total FOXO3a (T- FOXO3a), phospho-S6K1, total S6K1,
phospho-4E-BP1 and total 4E-BP1 in indicated cells. (B) Protein expression and relative
mRNA quantitation of p21 and cyclin D1 in indicated cells. (C) Relative VEGF-A mRNA
expression examined with real-time RT-PCR and VEGF-A protein secretion measured with
ELISA for indicated cells. (D) miR-205-transduced cells were pretreated with PI3K inhibitor
LY294002 or AKT inhibitor III, and their abilities to proliferate and induce HUVEC tube
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

26
formation were suppressed. (E) Suppressive effect of Rapamycin treatment on the malignant
phenotype of miR-205-transduced cells. (F) Suppressive effect of Foxo3a overexpression in
miR-205-transduced cells on cell growth and HUVEC tube formation. (G) Western blotting
analysis shows the effect of LY294002, AKT inhibitor III, Rapamycin or Foxo3a
overexpression. *: P<0.05.
Figure 6. Restoration of PHLPP2 and PTEN inverses miR-205-induced proliferation
and tube formation. (A) Growth rates of indicated miR-205-overexpressing cells transduced
with PHLPP2, PTEN or control vector. (B) Representative micrographs (left) and relative
quantification (right) of lengths of tubes by HUVEC tube formation assay. (C) Growth rates
of indicated miR-205 knocked-down cells, silenced with two different siRNAs against PHLPP2
or PTEN, respectively. (D) Representative micrographs (left) and relative quantification (right)
of lengths of tubes by HUVEC tube formation assay. (E) Western blotting analysis of
indicated proteins in indicated cells. (F) Measurement of p21 and cyclin D1 mRNAs and level
of secreted VEGF-A in indicated cells. (G) Western blotting confirmation of re-expression of
PHLPP2 or PTEN, as well as depletion of PHLPP2 or PTEN in indicated cells. *: P<0.05.
Figure 7. miR-205 overexpression by NF-κB transactivation correlates with low
expression of PHLPP2 and PTEN in NSCLC tumor tissues. (A) Relative expression of
miR-205 in twenty-two NSCLC tumor tissue specimens compared to their corresponding
adjacent non-cancerous tissues. (B) Schematic diagram of predicted p65 binding site within a
3.5kb region upstream to the miR-205 gene locus and relative expression of pri-miR-205 in
response to NF-κB activation (p65 overexpression or TNFα treatment) or inactivation (p65
siRNA or IκB α-mut transfection). (C) ChIP enrichment assay confirms that NF-κB subunit
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

27
p65 binds to the predicted promoter site of miR-205. (D) Luciferase activity of reporter
constructs spanning putative p65 binding site or control irrelevant sequences. (E) Correlation
of miR-205 overexpression with nuclear p65 abundance and PHLPP2 and PTEN
downregulation in indicated NSCLC tumor tissues. p84 and GAPDH were used as loading
controls. AD, adenocarcinoma; LCC, large cell carcinoma; ASC, adenosquamous carcinoma;
SCC, squamous cell carcinoma. (F) miR-205 expression level correlates with p65 subcellular
localization and expression of PHLPP2 and PTEN. Two representative cases (Low and High
miR-205) are shown. (G) Percentage of specimens showing cytoplasmic/nuclear p65 staining,
low- or high expression of PHLPP2 and PTEN in patient specimens, respectively, with low and
high miR-205 expression. *: P<0.05.
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297

Published OnlineFirst July 15, 2013.Cancer Res Mengfeng Li, Junchao Cai, Lishan Fang, et al. and drive malignant phenotypes in non-small cell lung cancermiR-205 targets PTEN and PHLPP2 to augment AKT signaling
Updated version
10.1158/0008-5472.CAN-13-0297doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2013/07/16/0008-5472.CAN-13-0297.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/early/2013/07/13/0008-5472.CAN-13-0297To request permission to re-use all or part of this article, use this link
Research. on October 14, 2018. © 2013 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0297