methods for chemical analysis of soils (1981)
TRANSCRIPT

(
A. METHODS FOR CHEMICAL ANALYSIS OF SOILS
Bv L.C. BLAKEMOREJ P.L. SEARLEJ B.K. DALY
Soil Bureau, Department of Scientific and Industrial Research, Lower Hutt
N-EW ZEALAND SoIL BUREAU Sc1ENTIF1c REPORT lOA Department of Scientific and Industrial Research, New Zealand
1981

Bibliographic Reference:
First printed 1972 Revised 1977 Revised 1981
BLAKEMORE, L.C.; SEARLE, P.L.; DALY, B.K. 1981: A. Methods for Chemical Analysis of Soils. New Zealand Soil Bureau Scientific Report lOA.
ISSN 0304-1735
P.D. HASSELBERG, GOVERNMENT PRINTER, WELLINGTON, NEW ZEALAND - 1981
c
(
( ....

c:
CONTENTS
Introduction
Al
A2
A3
A4
AS
A6
Soil pH
A. pH in H20 and chloride salt solutions
B. pH in NaF solution
Moisture factor
Carbon
A. Determination of total carbon using high frequency induction furnace equipment
B. Colorimetric determination of organic carbon
Nitrogen
A. Total nitrogen determination where nitrate is present in trace amounts
B. Total nitrogen determination where nitrate content is high
Phosphorus
A. Truog-soluble phosphorus determination
B. 0.5 M H2S0 4 -soluble phosphorus determination
C. Phosphate retention determination
D. Organic phosphorus determination ignition method
E. Total phosphorus determination
Cation exchange properties
A. Leaching procedures
B. Determination of individual exchangeable bases
C. Determination of total exchangeable bases by titration (TEBtitn)
D. Calculation of total exchangeable bases by summation (TEBsum or ~ bases)
E. Determination of cation exchange capacity (CEC)
F. Calculation of percentage base saturation (%BS)
G. Semi-micro leaching
H. KCl-Extractable Aluminium
I. Exchange Acidity
page
Al. l
Al. l
Al.4
A2.l
A3. l
A3.2
A3. 7
A4.l
A4.l
A4.7
AS.l
AS.2
AS.6
AS.9
AS.11
AS.13
A6.l
A6.4
A6.6
A6.8
A6.9
A6.10
A6.12
A6.13
A6.14
A6.15

A7 Reserve potassium (Kc) and magnesium (Mgr)
A. Determination of Kc value
B. Determination of Mgr value
AS Extractable iron, aluminium and silicon
A. Oxalate-extractable iron, aluminium and silicon
B. Pyrophosphate extractable iron and aluminium
C. Dithionite-citrate extractable iron and aluminium
A9 Soluble salts
A. Measurement of conductivity
B. Determination of water-soluble cations
C. Determination of water-soluble anions
AlO Calcium carbonate
A. Gasometric carbon dioxide method
B. Differential method
C. Correcting exchangeable calcium for calcium carbonate-calcium
All Phosphate extractable sulphur
Al2 Analyses of plant litters, peats and compost
Al3 Presentation and rating of results
A. Ratings for chemical properties
A7.l
A7 .1
A7.3
AS.l
AS.S
AS. 7
AS.Cl
A9.l
A9.2
A9.3
A9.4
AlO.l
AlO.l
Al0.3
Al0.4
All. l
Al2.l
Al3. l
Al3.3
l
C.

c
c
INTRODUCTION
In 1956, "Methods of Chemical Analysis for Soil Survey Samples" by A.J. Metson was published. This comprehensive account of the chemical analytical methods that were being used by Soil Bureau at that time provides background information on the purpose of the determinations and on the technical details of the methods.
In common with all scientific disciplines, soil chemistry has made tremendous advances over recent years and many new analytical techniques have been developed. Not only have these new techniques of measuring soil properties improved the speed and accuracy of standard analytical procedures, but they have also facilitated measurement of a wider range of properties. It seems probable that this progress, with its consequent improvement in analytical methods, will continue.
These methods have been written with a minimum amount of explanatory information on the principles of the methods and application of the results. They describe new methods or new techniques that have been developed since 1956 and they may require modern and expensive equipment. If this equipment is not available the basic methods described in Metson (1956) may be used with confidence.
Mr A.J. Metson and the staff of the Soil Analysis Section, Soil Bureau are thanked for their help in developing several of the methods, and for their helpful criticism of the manuscript.
REFERENCE
METSON, A.J. 1956: Methods of chemical analysis for soil survey samples. New Zealand Soil Bureau Bulletin 12. 208 p.
1972
This 1977 revised edition incorporates recent developments in instrumental methods within the Soil Analysis Section. Also, some changes to procedures, and to the ratings for chemical properties have been made in light of recent knowledge. The opportunity has been taken to express most concentrations in moles/litre, except for standard solutions in cation exchange properties where measurements are expressed in terms of milliequivalents, as this unit is still used in most soil literature. The following expressions for cation exchange and related properties are used:
1 me.% = 1 m mol per 100 g oven dry (105°C) soil charge on free ion
1 mg% = 1 mg per 100 g oven dry (10S°C) soil

This second rev1s1on (1981) includes several changes to technique and terminology. Some are relatively minor but those concerning determination of extractable sulphur and oxalate extractable Al, Fe and Si, are quite major. Another area of important change is the determination of exchangeable (--bases - both in terminology (bases and cations) and in the extent of dilution before measurement. This is to allow a more convenient range to be used during flame spectrometry for most soils.
The use of 'Soil Taxonomy' for classification of soils by Soil Bureau, on a trial basis, has made it necessary for this laboratory to carry out several additional analyses to those listed in the first revision. These comprise pH NaF, KCl-extractable Al, exch. acidity, dithionite-citrate Fe and Al, pyrophosphate extractable Fe and Al. Details of methods for these determinations are given.
c

(
(
Al.l
Al. SOIL PH
Al.A PH IN H20 AND CHLORIDE SALT SOLUTIONS
INTRODUCTION
The measurement of soil pH is usually regarded as one of the simplest determinations used in soil chemistry. However, even a cursory inquiry into the factors involved will show that many complications and difficulties are involved.
Several of these difficulties are caused by the fact that the pH theory applies to dilute solutions of simple electrolytes (Bates 1964). Although measurements may be made quite easily on soils and soil suspensions, there is considerable doubt about the significance of the results obtained in terms of hydrogen-ion activities. Available evidence indicates that the apparent pH of soil obtained by use of a glass electrode pH meter reflects hydrogen-ion activities in the bulk solution surrounding the electrodes, rather than hydrogen-ion activities in the ion atmosphere around the soil particles. However, the values obtained by carrying out pH measurements on soils do provide useful correlations with other soil properties and it is important to give considerable thought to choice of conditions for making the measurements and to realise the limitations of these conditions when interpreting results. The use of glass electrodes with calomel half-cells is almost universal for measurement of pH and details of use of the instrument are adequately dealt with in the relevant literature and in manufacturers' brochures.
FACTORS INFLUENCING pH MEASUREMENT
Choice of conditions for measurement of pH in soils involves the following factors:
Moistness of soil to be tested,
Suspension medium,
Ratio of soil to suspension medium,
Degree of stirring,
Positioning of electrodes.
MOISTNESS OF SOIL TO BE TESTED. The choice is between air-dry soil (as prepared for chemical analysis) and soil in its natural state of moisture. The principal objection to using air-dry soil is that the drying process can cause changes in pH values (e.g., through oxidation of sulphides), while the main objection to using naturally moist soil is the added difficulties in obtaining a representative sample and weighing out the naturally moist material, and the effects of biological activity on pH during storage of moist soil. At Soil Bureau, we have found that these effects can be most important and now routinely use air-dry soil.

Al.2
SUSPENSION MEDIUM. Ideally, measurement of soil pH should be made in soil in its natural condition but for several reasons, e.g., fragility of the glass electrode, this is not possible and, in practice, the soil is usually suspended in sufficient liquid to enable measurement to be taken.
In New Zealand laboratories water has usually been the suspension medium for pH determination for both pedological and agricultural measurements. However, electrolyte solutions are often preferred because the results are more reproducible, they are not influenced by the salt content of the soil, and the values obtained are less dependent on the positioning of the electrodes. 1 M KCl and 0.01 M CaCl2 are the most commonly.used salts.
When 1 M KCl is used, extensive ion exchange takes place, including the release of aluminium. Hydronium and other proton donors are brought into solution, influencing the glass electrode and lowering the measured pH (Black 1968). It is not to be expected that the pH values obtained with 1 M KCl will represent those of soil solutions but Black has suggested that they may approach the pH values in the ion atmospheres of the original soil.
Peech (1965) advocated use of 0.01 M CaC1 2 as the suspension medium because it is similar in electrolyte composition to soil solutions found at optimum moisture conditions for plant growth in "non-saline" soils (i.e., soils of low salt concentration). He noted that this medium is independent of dilution over a wide range of soil:suspension medium ratios. At Soil Bureau, limited experience with 0.01 M CaC1 2 has shown that it has some advantage over water as a medium for measuring soil pH because results are more easily reproduced.
The overall effect of using salts is to lower the measured pH values. With KCl the effect varies considerably and the pH is often more than 1 unit lower than results obtained with water. For New Zealand soils, results with CaC1 2 are about 0.5 to 1 pH unit lower than with water.
RATIO OF SOIL TO SUSPENSION MEDIUM. Because soil pH values decrease with increasing electrolyte concentration, it is to be expected that the electrolyte effect would significantly change with different dilutions of soil with water. In practice it has been found that the dilution effect varies considerably from soil to soil, and in order to render measurements of different investigators comparable, the International Society of Soil Science committee on soil reaction measurements (1930) adopted a standard dilution ratio of 1 part (by weight) of soil to 2.5 parts of water. This ratio was chosen for convenience of pH measurement in mineral soils. Other ratios have from time to time been recommended but it has been decided at Soil Bureau that standardisation is important and that the international 1:2.5 ratio should be used. However in soils which are very high in organic
(
(_
matter content (peats), it has been found necessary to use wider ratios ( (1: 5 or 1: 10) in order to obtain a workable slurry. ,,

(
(
Al.3
DEGREE OF STIRRING. Because it has been found that pH levels for some soils vary with degree of stirring during preparation of soil suspensions, it has been made standard practice to stir the suspensions vigorously using a mechanical stirrer or homogeniser.
POSITIONING OF ELECTRODES. Electr0des can be positioned to be in contact with the supernatant liquid or with the sediment.
When water or 0.01 M CaC1 2 is used as the suspension medium, the position of the calomel electrode is most important. A potential difference is apparently set up at the junction of the soil and the KCl bridge. As the potential difference varies with the concentration and nature of the soil in the contact area, standardisation is better achieved by positioning the calomel electrode in the supernatant liquid.
Although the positioning of the glass electrode is not of such importance it is preferable to place it in the sediment zone where the system is more strongly buffered and hence less susceptible to minor disturbances such as carry-over from the previous sample.
PREP~RATION OF REAGENTS
pH 6.5 BUFFER: CONCENTRATED BUFFER. Dissolve 89.7 g disodium hydrogen phosphate (Na2HP04.2H20) A.R, and 158.4 g potassium dihydrogen phosphate (KH2P04) A.R.,in distilled water. Make up to one litre with distilled water.
WORKING BUFFER. Pipette 20 ml concentrated buffer into a 500 ml volumetric flask and make to volume with distilled water.
pH 4.0 .BUFFER: 0.05 M ~OTASSIUM HYDROGEN PHTHALATp. Dissolve 5.106 g COOH.C 6H4.COOK in distilled water and make up to 500 ml with distilled water.
1 M POTASSIUM CHLORIDE. distilled water.
Dissolve 74.56 g KCl, A.R., in one litre of
0.01 M CALCIUM CHLORIDE. Dry CaCh (anhydrous) at 110°C and dissolve 11.1 g in 10 litres distilled water.
PROCEDURE
PREPARATION OF SAMPLE. Weigh 10 g of air-dry soil into a 100-ml beaker and add 25 ml distilled water, or 0.01 M CaC1 2.
Stir vigorously with a homogeniser or high-speed stirrer.
Leave to stand overnight.
NOTE: When wider soil:suspension medium ratios are needed to achieve a workable slurry (e.g., for peats), record the ratio used.

Al.4
MEASUREMENT OF pH. Position the electrodes so that the contact point of the calomel electrode is approximately 1 cm higher than the centre-point of the sphere of the glass electrode (Fig. A.1.1). Combined electrodes are suitable providing the bulb is positioned in the sediment when the calomel outlet is in contact with the supernatant.
Adjust the pH meter so that correct pH readings are obtained for two buffer solutions of widely differing pH (e.g., pH 4.00 and 6.50). Thoroughly wash the electrodes with distilled water. Position the soil sample on the instrument so that both electrodes are well-covered. Without stirring, measure and record the pH. Carry out duplicate determinations on separate subsamples. Replicate determinations should give results within 0.1 pH unit.
Glass electrode
-Calomel electrode
~---. ..... Liquid level (water or O·OlM CaCl 2 ) _(or M KCI) -
Fig. Al.1. Positioning of electrodes.
Al.B PH IN NAF SOLUTION
INTRODUCTION
This method is based on that of Fieldes and Perrott (1966) and is used as an indication of the presence of active aluminium. It is the basis of a "Fluoride field test" in which a portion of soil is reacted with NaF and the resultant high pH (if 'allophane' is present) is shown by phenolphthalein soaked filter paper turning red. High pH NaF values are found in soils derived from volcanic ash and in the illuvial horizons of podzolised soils. The method depends ·on active aluminium sorbing fluoride ions with consequent release of hydroxyl ions.
PREPARATION OF REAGENT
SODIUM FLUORIDE, saturated (approximately 1 M). Add 4 R. distilled water to 180 g NaF, A.R., in a 4 R. plastic bottle. Shake well, and let stand for 2 days with occasional shaking. After excess NaF has settled, check that the pH is between 7.2 and 8.1. Take 50 ml aliquot, heat to boiling, add 5 drops of 0.25% phenolphthalein and titrate with 0.01 M NaOH to a pink end-point while hot. If the solution has a pH of more than 8.1 or if the titratable acidity exceeds 0.25 me. per litre (more than 1.25 ml 0.01 M NaOH for 50 ml aliquot), discard and try another sou:rce of NaF. This laboratory uses Fisons A.R. grade.
(
(
(

Al.5
(- PROCEDURE
(
Weight 1 g soil (air-dry and < 2 mm) into a 100-ml beaker, add 50 ml NaF reagent and stir vigorously for 1 min. Place pH electrode/s in suspension and swirl gently. Read pH exactly 2 min. after adding reagent, ensuring that the suspension is well stirred immediately prior to taking reading.
REFERENCES
BATES, R.G. 1964: "Determination of pH; Theory and Practice". Wiley, New York. 435 p.
BLACK, C.A. 1968: "Soil-Plant Relationships". 2nd ed. Wiley, New York. 792 p.
FIELDES, M.; PERROTT, K.W. 1966: The nature of allophane in soils. Part 3. Rapid field and laboratory test for allophane. N.Z. Journal of Science 9: 623-9.
INTERNATIONAL SOCIETY OF SOIL SCIENCE COMMITTEE ON SOIL REACTION MEASUREMENTS, 1930:
Part 1. Results of Comparative Investigations on the Quinhydrone Electrode Method. Soil Research 2: 77-139
PEECH, M. 1965: Hydrogen-ion activity. Agronomy 9: 914-26.

c
(_

( A2. MOISTURE FACTOR
INTRODUCTION
Most results should be reported on an oven-dry basis (see Section Al3), but as oven drying causes changes in several chemical properties of soils, analyses are carried out on air-dried samples (i.e. dried at tempo eratures of no more than 30 C). In order to convert results to an oven-dry basis, a moisture factor is applied (see Section Al3).
PROCEDURE
Weigh 10 g to 20 g soil (air-dry, < 2 mm) into a weighed dish with lid (a labelled, aluminium dish is recommended), dry in an oven at 10s0 c for 8-24 hours.
A2.1
Remove from oven, fit lid, cool and reweigh (all weighings should be made to the nearest 10 mg). Because oven-dry soil picks up water from the atmosphere very rapidly (even in some desiccators), it is necessary to reweigh without delay. In practice this can either be achieved by weighing with a top-loading balance as soon as the dish is cool enough to handle, or after cooling in an efficient desiccator.
CALCULATION OF RESULTS
1. weight air-dry soil (g) weight oven-dry soil (g) =Moisture Factor (M.F.)

c
c

c
c
(_
A3. CARBON
INTRODUCTION
At Soil Bureau, two methods for determination of carbon in soils are in current use.
The method more often used is the induction furnace method, in which
A3.1
the carbon is converted to carbon dioxide and this is measured volumetrically. This method has the advantages of being rapid, convenient for most soils, and accurate. Its main disadvantage is that it measures total soil carbon content which includes that of any carbonate, charcoal, or live root material present.
The alternative is a colorimetric method which has the advantage that it is more specific to humified organic carbon. This method involves a wet oxidation of the carbon by dichromate, and the measurement of the amount of reduced chromium present. The disadvantage of this method is that direct calibration is not possible. Therefore calibration is made against amounts of sucrose which have the same reducing power on dichromate (measured colorimetrically) as have known amounts of soil carbon. The relationship between sucrose carbon and soil carbon is calculated from figures found by previous experience with Metson's (1956) adaption of the Schollenberger (1927a, b, 1931, 1945) and Allison (1935) methods, and the dry combustion method.
Methods of determining organic carbon in soils have been reviewed by Metson et al. (1979).

A3.A : DETERMINATION OF TOTAL CARBON USING HIGH FREQUENCY INDUCTION FURNACE EQUIPMENT
INTRODUCTION
A3.2
The following method refers particularly to the equipment produced by the Laboratory Equipment Corporation (Leco), St Joseph, Michigan, U.S.A. The method involves the purification and measurement of carbon dioxide (C02) evolved when a sample is heated in a stream of oxygen. The heat is produced by a high frequency electrical flux induced in a mixture of the sample and of a conducting matrix of iron chips, and temperatures in excess of 1400°c can be attained. The carbon dioxide is purified by passing it in turn through a dust trap, through precipitated silver to remove chlorine, through a converter furnace to convert any carbon monoxide (CO) to carbon dioxide, and through manganese dioxide (Mn0 2) to remove sulphur dioxide (S02). The measurement of carbon dioxide is carried out by passing the remaining gases (C02 + 02) into a calibrated burette in which 5% H2SO~ is displaced. The gases (C02 + 02) are flushed through concentrated potassium hydroxide solution which absorbs the carbon dioxide. The difference between the original volume of gas in the burette and the volume of carbon dioxide-free gas is equal to the volume of carbon dioxide evolved from the sample. After correction for temperature and pressure,
c
the carbon content of the sample is calculated. ~
This method measures total carbon and the result obtained will be directly affected by the presence of calcium carbonate, undecomposed wood, charcoal, etc.
There are many details of operation of the induction furnace and associated equipment which are adequately described by the makers and are therefore not included here. The following method concentrates on details required for soil analysis.
REPARATION OF REAGENTS
IRON CHIPS. Leco cat. no. 501-077.
TIN METAL. Leco cat. no. 501-076.
SILICIC ACID A.R.
RED LEVELLING SOLUTION. see maker's manual.
CAUSTIC SOLUTION. see maker's manual.
MANGANESE DIOXIDE. Leco cat. no. 501-060.
PRECIPITATED SILVER METAL. precipitate from silver nitrate (AgN0 3 )
solution with sodium dithionite solution. Filter, wash precipitate with distilled water, and dry.
l

c
(
PROCEDURE
Use finely ground (< 0.25 mm) air-dry soil and take sample weights according to the following table:
Carbon Expected Sample Weight
% g
0.0 - 1.5 1.00
1.5 - 6 0.25
6 - 15 0.10
15 - 60 0.025
For loading the crucible use a scoop which delivers about 1 g iron chips and charge the crucible in the following order:
1 g Sample
1 scoop iron chips soil sample ~ scoop tin metal 2 scoops iron chips
less than 1 g Sample
1 scoop iron chips soil sample 1 scoop silicic acid ~ scoop tin metal 2 scoops iron chips
A3.3
Prepare blank crucibles, loaded as for samples, and run these until a zero reading is obtained (usually-after 2 or 3 blanks), before proceeding with samples.
Place the loaded crucible on the pedestal and adjust the rate of oxygen flow from a cylinder to 0.8 litres/minute.
Raise the pedestal into the induction field. This simultaneously closes the system and initiates induction, the combustion gases being passed through the dust and chlorine traps, the carbon monoxide converter furnace, and the sulphur trap before they reach the gasometric carbon analyser (Fig. A3.1).
When the gases have displaced most of the H2SO~ in the burette, stop the reaction and oxygen flow by lowering the pedestal.
Measure the percent carbon directly from the appropriate scale on the calibrated burette applying the appropriate corrections for sample weight, temperature and barometric pressure (see following table).

Factor Chart for Leco Carbon Furnace
Atmospheric Pressure (mm)
740 745 750 755 760 765 770 775
21 0.950 0.957 0 .963 0.970 0.976 0.983 0 .990 0.996 22 0.945 0.952 0.958 0.965 0.972 0.978 0.985 0.991 2J 0.941 0.947 0.954 0. 960 0.967 0.973 o .980 0.986 ,.......,.
o0 24 0.936 0.942 0.949 0.955 0.962 0.968 0.975 0.981
'-.-'
Q) 25 i-i
0.931 0.937 0.944 0.950 0.957 0.963 0 .970 0.976 E 26 0.926 0.932 0.939 0.945 0.952 0.958 0 .965 0.971 al
~ 27 P;
0.921 o. 927 0.934 0.940 0.946 0.953 0.959 0.966 ~ 28 0.916 0.922 0 .928
E-i 0.935 0.941 0.948 0.954 0.961
29 0.910 0.917 0.923 0.930 0.936 0.942 0.949 0.955 JO 0.905 0 .911 0.918 0.924 0.931 0.937 0.943 0.950
"Degrees C" is temperature of gas as read from thermometer in measuring burette.
A3.4
780
l.OOJ 0.998 0.993 o.9sa 0.983 0.978 0.972 0.967 0.962 0.956
"Atmospheric Pressure (mm)" is station pressure as read from a mercurial or aneroid barometer. As mercury and steel expand with temperature, it is necessary to correct the mm reading for surrounding air temperature. In an aneroid barometer, this is accomplished by using an instrument with a temperature compensated movement. With a mercurial barometer the correction is made simply by reference to the following chart, which brings the pressure to what it would be at 0 degrees C:
Room Subtract from Temperature Uncorrected (Degrees C) Reading
5 - 12 1 mm
13 - 20 2 mm
21 - 28 3 mm
29 - 35 4 mm
36 - 40 5 mm
(
c

c
(
02 + C02 ... ~ ...... - ... etc. from combustion tube
_.,. To gasometric ---- carbon analyser
1:~ lo~
Sulphur trap
Manganese dioxide
Carbon monoxide converter furnace
lo·°""..._+-Copper oxide chips
lo I~~ 0
Chlorine trap
Precipitated silver metal
Dust trap
Calico filter
Fig. AJ.1. Flow diagram for carbon determination using Leco induction furnace.
A3.5

A3.6
NOTES:
(1) Use of silicic acid in mixture. One effect of the silicic acid is to increase the viscosity of the molten material, thus decreasing crucible failures caused by the permeation of the melt through the crucible walls. While there is usually sufficient silica in soils to prevent this cause of failure, there is not sufficient in blanks or organic samples (e.g., litters, peats), and it is advisable always to include silicic acid in the crucible charge when less than 1 g of sample is used.
(2) Constant temperature. A supply of temperature-controlled water to the outer jacket of the carbon dioxide analyser is recommended, especially when the temperature of the water from the mains supply is variable or when it differs appreciably from room temperature.
(3) Cleaning of dust trap and gas delivery tube. The calico filter and delivery tube leading to the dust trap must be cleaned and dried regularly to prevent absorption of carbon dioxide by moisture and dust.
(4) Sulphur trap. Moist manganese dioxide has a tendency to absorb
(
carbon dioxide and cause low results. The effect of moisture on the manganese dioxide can be minimised by placing the sulphur trap immediately after the carbon monoxide converter furnace. It has been found that fresh manganese dioxide also absorbs carbon dioxide and it as necessary to counteract this effect by passing copious amounts of carbon dioxide through the trap and then flushing with oxygen to remove any excess carbon dioxide. (_ Experience has shown that as long as the sulphur trap is kept moisture-free, renewal of the manganese dioxide and subsequent carbon dioxide treatment are not frequently required.
(5) Use of variable transformer. Most furnaces are fitted with an automatic cut-out device which operates if the safe current for the induction coil is exceeded. To prevent this happening during an analysis it has been found necessary to use a variable transformer to limit the induction current to a safe level.
(_
(
c
A3. 7
A3.B COLORIMETRIC DETERMINATION OF ORGANIC CARBON
INTRODUCTION
In 1934, Walkley and Black published a colorimetric method for carbon determination. This was modified by Walkley in 1947, and the method given in Metson (1956) is derived from the two papers.
The following method is essentially that of Metson with several minor changes.
PREPARATION OF REAGENTS
CHROMIUM TRIOXIDE, about 3 M. 332.5 g chromium trioxide per litre. Dissolve in water, make to volume and filter through sintered glass before storing (the solution must be quite clear). The exact strength is unimportant.
SULPHURIC ACID, A.R., 98% w/w. This should be used fresh from the bottle, and not left standing in a burette or beaker as it rapidly picks up moisture from the air.
SUCROSE, A.R., powdered. If necessary grind, using a clean mortar and pestle, until particles are about 0.25 mm.
PREPARATION OF STANDARDS
FOR SOILS EXPECTED TO CONTAIN LESS THAN 10% C. Weigh out sucrose standards which are equivalent to 0, 20, 40, 60, 80 and 100 mg soil carbon according to the following table:
For 1 g Soil Samples
Soil Soil Sucrose Carbon Sucrose Carbon Carbon Equivalent (g) (%) (mg) (mg)
0 0 0 0
2 20 19.6 0.0465
4 40 39.0 0.0926
6 60 58.4 0.1386
8 80 77.9 0.1849
10 100 97.4 0.2312

FOR SOILS EXPECTED TO CONTAIN MORE THAN 10% C. Weigh out sucrose standards which are equivalent to 0, 20, 40, 60, 80 and 100 mg soil carbon according to the following table:
For 0.2 g Soil Samples
Soil Soil Sucrose Carbon Sucrose Carbon Carbon Equivalent (g) (%) (mg) (mg)
0 0 0 0
10 20 20.8 0.0494
20 40 41.6 0.0988
30 60 62.4 0.1481
40 80 83.2 0.1974
so 100 104.0 0.2469
PROCEDURE
FOR SOILS EXPECTED TO CONTAIN LESS THAN 10% C. Weigh out 1.00 g finely ground soil (air-dry, < 0.25 mm) into a 200-ml Taylor type flask which is calibrated to show the 200-ml level.
FOR SOILS EXPECTED TO CONTAIN MORE THAN 10% C. Weigh out 0.200 g finely ground soil (air-dry, < 0.25 mm) into a 200-ml Taylor type flask calibrated to show the 200-ml level.
A3.8
To all soil samples and sucrose standards, add 12 ml cone. H2 S04 (from a 250-ml rapid-flow dispensing burette) and swirl gently.
Stand for 10 minutes with occasional swirling.
Add 6 ml 3 M chromium trioxide solution (from a rapid automatic pipette) and mix well by swirling, washing down any particles that have lodged on the sides of the flask.
Stand for exactly 10 minutes and then dilute with water nearly to the 200-ml mark. It has been found that the addition of chromium trioxide and the subsequent dilution can be conveniently carried out at 30 second intervals.
Let the diluted solution stand overnight, preferably at a room temperature of about 20°c.
After 18 to 24 hours, make up to the 200-ml mark with distilled water and stir well.
Centrifuge for about 10 to 15 minutes at about 2000 rpm in a 40-ml centrifuge tube.
Read absorption at 600 nm using 10-mm cells and with the absorptiometer set to zero on water.
c
c

c
(
A3.9
CALCULATION OF RESULTS
Construct standard curves from the readings for the appropriate standards and calculate the % carbon.
For 1 g sample, (~~) = C in soil (%)
For 0.2 g sample,
REFERENCES
ALLISON, L.E. 1935:
(~) = C in soil (%) 2
Organic soil carbon by reduction of chromic acid. Soil Science 40: 311-20
METSON, A.J. 1956: Methods of chemical analysis for Soil Survey Samples. N.Z. Soil Bureau Bulletin 12: 208 p.
METSON, A.J.; BLAKEMORE, L.C.; RHOADES, D.A. 1979: Methods for the determination of soil organic carbon application to New Zealand so~ls. New Zealand Journal of Science 22: 205-28.
SCHOLLENBERGER, C.J. 1927a:
a review, and
A rapid approximate method for determining soil organic matter. Soil Science 24: 65-8.
1927b: Exchangeable hydrogen and soil reaction. Science 65: 552-3.
1931: Determination of soil organic matter. Soil Science 31: 483-6.
1945: Determination of soil organic matter. Soil Science 59: 53-6.
WALKLEY, A. 1947: A critical examination of a rapid method for determining organic carbon in soils - effect of variations in digestion conditions and of inorganic soil constituents. Soil Science 63: 251-64.
WALKLEY, A.; BLACK, I.A. 1934: An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Science 37: 29-38.

c
(

c
c
l
A4.l
A4. NITROGEN
INTRODUCTION
Method A4.A is a semi-micro method which employs a digestion to convert nitrogen present in the sample to ammonium sulphate. Aminonium-nitrogen is subsequently determined either by distillation and titration or by a colorimetric Autoanalyzer method.
For samples likely to have greater than 20 ppm N0 3 (e.g. litters, composts, plant materials, bioturbic soils) Method A4.B should be used as it incorporates a modified digestion mixture to reduce nitrates to ammonium sulphate.
A4.A : TOTAL NITROGEN DETERMINATION WHERE NITRATE IS PRESENT IN TRACE AMOUNTS
(<20 PPM (0.002%) N03)
INTRODUCTION
The method described is a modified semi-micro Kjeldahl method in which the digestion is carried out in 50 ml calibrated test tubes inserted in a drilled aluminium block on a hotplate. Two alternative determination steps are described, I, the traditional steam distillation and titration procedure and II, A colorimetric Autoanalyzer method described by Searle (1975).
The colorimetric determination is based on the rea~tion of ammonia. with a phenol (sodium salicylate) in alkaline oxidising conditions to form an indophenol which absorbs strongly at 660 nm. The reaction is catalysed by sodium nitroprusside, and a citrate-tartrate mixture is employed to chelate metals that would otherwise form insoluble hydroxides.
DIGESTION
APPARATUS
DIGESTION TUBES. 'Exelo' 50 ml calibrated test tubes cat. no. T2/30.
ALUMINIUM HEATING BLOCK. Aluminium block (30 cm x 30 cm x 5 cm) drilled with seven rows of six holes (2.7 cm diameter, 3.5 cm depth).

A4.2
HOT PLATE. Domestic single radiant element hotplate (element should be at least 20 cm diameter and have an output of about 2 kW).
PREPARATION OF REAGENTS
SULPHURIC ACID, CONC., A.R. not required for routine work).
(Special N-free or M.A.R. grades are
KJELDAHL CATALYST TABLETS. B.D.H. Cat. No. 33064. contains 1 g sodium sulphate and 0.1 g copper sulphate.
PROCEDURE
Each tablet
Weigh accurately 0.50 g finely ground (air-dry, < 0.25 mm) soil into a dry 50 ml calibrated test tube. A dry tube is desirable to prevent soil from sticking to the sides. For peaty soils use a smaller sample - 0.10 to 0.20 g - to avoid excessive frothing.
A reagent blank should be carried throughout the following procedure: Moisten the soil with a few drops of distilled water and allow the moisture to penetrate the soil. Lower results have been reported for nitrogen determinations on certain heavy clay soils where this slight wetting was omitted (Bal 1925, Walkley 1935, Alper 1938). Add a Kjeldahl catalyst tablet and 3.25 ml of cone. H2S04.
Significant errors will occur in the colorimetric Autoanalyzer method (II) if the final acid content of the digest falls outside the range 2.75 -3.25 ml of cone. H2S04. Some soils (particularly Pacific Island soils derived from andesitic ash) have been found to neutralise up to 0.5 ml cone. H2 S04 per 0.5 g sample, and consequently, the higher amount of acid (3.25 ml instead of 3.0 ml) has been adopted to eliminate this source of eTror.
Place the tubes in a preheated aluminium digestion block on a radiantelement hotplate. Boil the digestion mixture until it decolorises (usually 20-30 min.). Soils high in organic matter need careful watching as frothing occurs. The digestion is then carried on for another 20-30 min. to ensure conversion of all nitrogen to ammonium sulphate.
Remove tubes from block. For determination method I the complete sample may be transferred to the distillation apparatus. This procedure however may be difficult as the digest solidifies when cold and can be a problem to redissolve. It is preferable, therefore, to prepare the digests as for Method II and take an aliquot (usually 25 ml) for the distillation.
For determination method II, before the digests are completely cold (5-6 min. after removal) add carefully 10-15 ml of distilled water and swirl to hasten the solution of salts. When cool make to 50 ml mark with distilled water, stopper and shake vigorously. Allow solids to settle (usually overnight) before the colorimetric determination is carried out.
c
(_
(_

(
(
l. DETERMINATION USING MANUAL METHOD
PREPARATION OF REAGENTS
BORIC ACID, 1% SOLUTION. of distilled water.
Dissolve 100 g H3B0 3 , A.R., in 10 litres
HYDROCHLORIC ACID (HCl), 0.02 M.
A4.3
BROMOCRESOL GREEN - METHYL RED MIXED INDICATOR (Ma and Zuazaga 1942). Mix 5 parts 0.1% bromocresol green in 95% EtOH with 1 part 0.1% methyl red in 95% EtOH. · These proportions may have to be varied to obtain the neutralgrey transition colour of the indicator.
SODIUM HYDROXIDE, 1+2. Dissolve 1 kg NaOH pellets, A.R., in 2 litres distilled water, stirring constantly.
PROCEDURE
To transfer total sample digest to distillation apparatus, add, when cool, about 5 ml distilled water. Heat if necessary to hasten solution of the sodium sulphate, and transfer total content to the distilling flask, washing with two or three small portions of distilled water.
Add approximately 10 ml 1+2 NaOH (or more if extra H2S04 was added during the digestion). A little pH indicator will show whether sufficient NaOH has been added, but usually formation of a brown precipitate of ferric hydroxide, when the liquids are mixed, indicates neutralisation of the acid.
Rinse the funnel with a little distilled water.
Add 8 to 10 ml 1% boric acid* and 5-6 drops indicator to a 100 ml Erlenmeyer flask and place under the delivery tube of the condenser so that the tip is below the surface of the liquid.
Close sample inlet and drainage outlet and pass steam into the distillation flask. The liquid will soon boil, and the indicator in the boric acid solution will change colour as soon as ammonia begins to distil over.
NOTE: * At least 500 µg of N can be fixed by 10 ml 1% boric acid, with less than 0.5% error (Yuen & Pollard 1953). The more dilute the boric acid that can be used with complete retention of ammonia, the sharper is the end-point of the titration. The A.R. grade of boric acid is much superior to the 'pure' or L.R. grade.

A4.4
After a minute or two, lower the flask so that the tip of the delivery tube is clear of the liquid. No ammonia will be lost if the condenser is efficient. The temperature of the distillate should not rise above 40°c (Yuen & Pollard 1953).
When about 20 to 25 ml of distillate have collected, rinse the tip of the tube with a little distilled water and remove the flask.
Stop the entry of steam. The distilling flask will empty automatically and the vacuum can be used to rinse the apparatus by immersing the delivery tube in distilled water.
Titrate the distillate against 0.02 M HCl, to the neutral grey colour of the indicator.
Correct titration for re_age11t blank. 1 ml 0.02 M HCl = 280 µg N.
CALCULATION OF RESULTS
For total digests
0.5 g soil: 0.02 M HCl (ml) x 0.056 = 0.2 g soil: 0.02 M HCl (ml) x 0.140 =
For aliquot
0.5 g soil: 0.02 M HCl (ml) x 0.056 x
0.2 g soil: 0.02 M HCl (ml) x 0.140 x
N (%)
N (%)
50 N (%) aliquot (ml) =
50 N (%) aliquot (ml) =
(
(_
C.

c
c-
A4.5
II. DETERMINATION USING AUTOANALYZER
PREPARATION OF REAGENTS
DICHLORO-S-TRIAZINE - 2,4,6, TRIONE, SODIUM SALT (NaDTT),(Koch light Cat. No. 1615h). Dissolve 10 g NaOH in 400 ml distilled water, add 0.25 g NaDTT and make to 500 ml.
SODIUM SALICYLATE. Dissolve 15 g sodium salicylate A.R. in distilled water and make to 1 litre.
SODIUM NITROPRUSSIDE. Dissolve 1.2 g sodium nitroprusside A.R. in distilled water and make to 500 ml.
CITRATE-TARTRATE REAGENT. Dissolve 25 g NaOH in 800 ml distilled water, add 6 g sodium citrate, A.R. and 18 g sodium tartrate A.R., dissolve and make to 1 litre with distilled water.
WASH SOLUTION. Carefully add 120 ml cone. H2S04, A.R. to 1500 ml distilled water, add 40 g Na2S04, A.R. and 4 g CtiS04.5H20, A.R., dissolve and make to 2 litres with distilled water.
PREPARATION OF STANDARDS
STOCK SOLUTION (1 ml= 500 µg N). Dissolve 2.3585 g ammonium sulphate (M.A.R. or Aristar quality dried at 110°C) in approximately 800 ml distilled water. Carefully add 60 ml cone. H2S04, A.R.,20 g of Na2S04, A.R.,2 g CuS0 4.5H20,A.R.,dissolve and make to 1 litre with distilled water in a volumetric flask.
WORKING STANDARDS. Pipette 2.5, 5.0, 10.0, 15.0, 20.0, 25.0, 40.0, and 50.0 ml of stock solution into 250 ml volumetric flasks, and make each to volume with wash solution (see preparation of reagents). These standards contain 5, 10, 20, 30, 40, 50, 80, and 100 µg/ml N respectively.
PROCEDURE
Set ug Autoanalyzer as shown in flow diagram (Fig. A4.l) with heating bath at 45 C and 660 nm filters in the colorimeter.
Pump reagents and wash solution through system for about 10 min. to ensure complete flushing of the analytical system and wash receptacle.
Pour standard and sample solutions into cups on the sample tray, set the recorder baseline with the colorimeter baseline control. Sample top standard and when it reaches the colorimeter flow cell set recorder to read 100% absorption using the standard calibration control on colorimeter (a standard calibration of about 1 is usual). Reset baseline if necessary.
Sample standards and unknowns at a rate of 60/hr with a 5:1 sample:wash ratio.

A4.6
Digests containing more than 50 µg/ml N can be diluted using the wash solution as diluent. Soils with extremely high nitrogen contents should be redigested using a smaller weight of soil.
CALCULATION OF RESULTS
Prepare a standard curve of µg/ml N against % absorption.
For 0.5 g of soil made to 50 ml
3 ML HEATING BATH COIL
4S0c
COLORIMETER 660 NM
15 MM FLOW CELL
µg/ml N = 100
N (%)
AlO CONNECTORS
WASTE
FLOW RATE HUHIN
0.32
0.8
0.1
0.6
0.23
1.2
LO
2.0
REAGENT
AIR
COMPLEXING AGENT
SAMPLE
NAllTI
NITROPRUSS !DE
SALICYLATE
WASH
PULL FROM CO LOR I METER
Fig A4.l Flow diagram of Autoanalyzer II manifold for total nitrogen and CEC determinations.
TUBE COLOUR CODE
BLACK
RED
ORANGE/GREEN
WHITE
ORANGE/WHl"i E
YELLOW
GREY
GREEN
c
(~

c
(_'
A4.B TOTAL NITROGEN DETERMINATION WHERE NITRATE CONTENT IS HIGH (>20 PPM)
NOTE: The digestion used in this method is also suitable for the determination of total potassium and phosphorus in plant material.
DIGESTION
PREPARATION OF REAGENTS
KJELDAHL CATALYST TABLETS. See method A4.A.
A4.7
SULPHURIC ACID-SALICYLIC ACID MIXTURE. Dissolve 20 g salicylic acid, A.R. in 600 ml cone. H2S04, A.R.
SODIUM THIOSULPHATE, A.R. crystals.
PROCEDURE
Weigh a 0.1 g sample into a 50 ml calibrated tube (plant material needs to be dried for 2 hours at 105°C before weighing). Add 4.·o ml sulphuric acid-salicylic acid mixture and allow to stand with occasional shaking for 1 hour. Stopper tubes and leave overnight.
Add 0.25 g sodium thiosulphate through a long-necked funnel and heat gently for about 5 min.
Remove from heat, and when cool, add a Kjeldahl catalyst tablet and carry out the digestion as described in method A4.A.
A reagent blank should be carried throughout the procedure.
I, DETERMINATION USING MANUAL METHOD
Carry out digestion, distillation, and titration according to method A4.A, but use approximately 20 ml 1+2 NaOH for the distillation.
CALCULATION OF RESULTS
For total digest: 0.1 g sample:
For aliquot: 0.1 g sample:
0.02 M HCl (ml) x 0.280 = N (%) 50
0.02 M HCl (ml) x 0.280 x aliquot = N (%)

II. DETERMINATION USING AUTOANALYZER
PREPARATION OF REAGENTS
See method A4.AII.
PREPARATION OF STANDARDS
STOCK SOLUTION(l ml= 500 µg N, 500 µg Kand 50 µg P). Dissolve 2.3585 g anunonium sulphate (M.A.R. or Aristar quality, dried at llOOC), 0.9533 g potassium chloride(A.R.,dried at 110°c), and 0.2292 g disodium hydrogen orthophosphate(A.R.,dried at 110°c) in distilled water. Add 0.5 ml toluene as a preservative and make to 1 litre.
WORKING STANDARDS. Prepare 6 blank digests as described above but add approximately 20 ml distilled water instead of making to 50 ml.
Pipette O, 1.0, 2.5, S.O, 7.5 and 10.0 ml of stock solution into the blank digests. Make to 50 ml with distilled water.
A4.8
These standards contain O, 10, 2S, SO, 75 and 100 µg/ml N and K; and 0, 1.0, 2.5, 5.0, 7.S and 10.0 µg/ml P.
PROCEDURE
As for A4.A.
CALCULATION OF RESULTS
Prepare a standard curve of µg/ml N against % absorption.
For 0.1 g sample made to SO ml
µg/ml N 20 = N (%)
NOTE: Determination of potassium in digest.
Compare potassium concentration in samples with standards using flame emission spectrometry at 766.5 nm.
µg/ml K = 20 K (%)
c_-
c

c
c
. Determination of phosphorus in digest.
Prepare 1+1 dilutions of samples and standards with distilled water and determine P as for AS.All.
µg/ml P 20
REFERENCES
=
ALPER, Pauline 1938:
p (%)
An accurate wet-combustion method for the determination of carbon in soils. Journal of Agricultural Science 75: 173-80.
BAL, D.V. 1925: The determination of nitrogen in heavy clay soils. Journal of Agricultural Science 15: 454-9.
MA, T.S.; ZUAZAGA, G. 1942: Micro-Kjeldahl determination of nitrogen. A new indicator and an improved rapid method.
A4.9
Industrial and Engineering Chemistry. Analytical edition 14: 280-2.
SEARLE, P.L. 1975: Automated colorimetric determination of ammonium ions in soil extracts with 'TechniconAutoanalyzer II equipment". N.Z. Journal of Agricultural Research 18: 183-7.
WALKLEY, A. 1935: An examination of methods for determining organic carbon and nitrogen in soils. Journal of Agricultural Science 25: 598-609.
YUEN, S.H.; POLLARD, A.G. 1953: Determination of nitrogen in soil and plant materials: use of boric acid in the micro-Kjeldahl method. Journal of the Science of Food and Agriculture 4: 490-6.
c
(__

(
c
AS.l
A5. PHOSPHORUS
INTRODUCTION
At the present time (1977) the main phosphorus values determined by the New Zealand Soil Bureau Soil Analysis Section are:
AS.A TRUOG-SOLUBLE PHOSPHORUS. Determinations are normally carried out on topsoil samples only. The values measure readily soluble phosphorus and may be used, with some limitations, as a guide to soil fertility.
AS.B 0.5 M H2S04-SOLUBLE PHOSPHORUS. Determinations are carried out on the separate horizons of selected whole profile samples and are used to assess the amount of inorganic phosphorus which is present but not fixed. The values so obtained are useful in pedological studies as they provide an indication of the state of weathering and leaching.
AS.C PHOSPHATE RETENTION. An empirical measure of the ability of the soil to rapidly remove phosphorus from solution, a process which is considered to be a precursor to the much slower process of phosphorus fixation which renders phosphorus unavailable to plants. In acid soils of pH <6.5, compounds of iron and aluminium are considered to play an increasingly important role (Saunders 1968) . The method has been devised so that the concentration of phosphate used gives a high degree of differentiation between soils of high and low phosphorus-retention ability, and the pH used (4.6) is close to the point of maximum phosphate retention in many soils.
AS.D ORGANIC PHOSPHORUS. Determinations are normally carried out on all soil horizons and, when considered as a proportion of the total phosphorus present (particularly for B horizons), are useful in the study of soil-forming processes.
AS.E TOTAL PHOSPHORUS. Determinations are used in conjunction with other phosphorus fractions to elucidate the state of weathering and leaching of a soil.
DETERMINATION
Two colorimetric methods are given for the determination of phosphorus in the extracts. I - a manual procedure and I I - an Autoanalyzer procedure. These methods are applicable for all the phosphorus values determined except phosphate retention, for which a separate method is used.

A5.2
AS.A TRUOG-SOLUBLE PHOSPHORUS DETERMINATION
EXTRACTION
PREPARATION OF REAGENT
EXTRACTING REAGENT. 0.001 M H2S0 4 buffered to pH 3. Dissolve 15 g ammonium sulphate (NH 4 ) 2 S0 4 , A.R., in 10 ml 0.5 M H2S0 4 , and dilute to 5 litres.
PROCEDURE
The amount of phosphorus extracted depends on the extraction time which is relatively short (30 min.). Therefore it is necessary to arrange the work so that shaking time is kept as close to the 30 min. as possible, with minimum time spent in adding the extracting reagent to the soils, placing flasks on shaker and filtering extracts after shaking.
Place 1 g soil (air-dry, < 2 mm) into a shaking bottle, add 200 ml extracting reagent and shake for 30 min. on an end-over-end shaker. Filter as soon as possible through No. 1 (Whatman) paper until at least 100 ml have been collected, if using determination method I. If using method II filter (_ about 18 ml into a sampler test tube. (Fill to about 1 cm from top). A reagent blank should be carried throughout the determination.
I, DETERMINATION USING MANUAL METHOD
INTRODUCTION
This colorimetric procedure is based on the method of Murphy and Riley (1962) as adapted by Watanabe and Olsen (1965). The procedure involves reduction of the phospho-molybdate complex by ascorbic acid in a reaction catalysed by antimony.
PREPARATION OF REAGENTS
MURPHY AND RILEY REAGENT A. 1.2% solution of ammonium molybdate ((NH4 ) 6Mo 7024 .4H20) with 0.1 mg/ml antimony in 2.5 M H2 S04 •
To prepare 5 litres of solution:
Dissolve 60 g ammonium molybdate A.R., in 1 litre distilled water. The (-rate of solution may be increased by warming, but do not warm above 60°C. -Cool the solution.
In 250 ml distilled water dissolve 1.3343 g antimony potassium tartrate. Add both of the dissolved reagents to 2.5 litres of 5 M H2 S0 4 (705 ml cone.

(
c
c
H2SO~, A.R., made to 2.5 litres with distilled water). make to 5 litres and store in dark bottles.
Mix thoroughly,
A5.3
MURPHY AND RILEY REAGENT B. In each 100 ml of reagent A dissolve 1.056 g ascorbic acid and mix. This reagent must be prepared as required as it does not keep for more than 24 hours.
PREPARATION OF STANDARDS
STOCK SOLUTION (100 µg/ml P). Dissolve 0.220 g potassium dihydrogen phosphate (KH2 PO~), A.R., in distilled water, add 0.5 ml toluene as a preservative, and make up to 500 ml.
WORKING STOCK (1 µg/ml P). Pipette out 5 ml stock solution and dilute to 500 ml in a volumetric flask.
WORKING STANDARDS. Pipette O, 5, 10, 15, 20 and 25 ml of the 1 µg/ml P solution into 100 ml volumetric flasks (preferably of the mixing variety) and dilute to approximately 80 ml with distilled water. These standards contain O, 5, 10, 15, 20 and 25 µg P respectively. Treat these standards as for samples.
PROCEDURE
Pipette out a 50 ml aliquot of the Truog extract into a 100 ml volumetric flask (mixing variety if available) and dilute to approximately 80 ml with distilled water.
To standards and unknowns add 8 ml of Reagent B, make to 100 ml, and mix well. The colour produced is stable for 24 hours and maximum intensity is reached in 10 min.
Maximum absorption occurs at 880-885 nm. However an absorptionmeter (e.g. Spekker) may be used, using 4 cm cells and 608 filter (660 nm). When values of greater than 8 mg % P are encountered, smaller aliquots should be taken and the same procedure followed.
CALCULATION OF RESULTS
Construct a standard curve. This should be near-linear. For a 50 ml aliquot (= 0.25 g soil) of the extract (from 2 g of soil and 400 ml extracting reagent) made up to 100 ml:
P concentration in final solution (µg) x 0.40 =
Truog-soluble P (mg %)

A5.4
II. DETERMINATION USING AUTOANALYZER
INTRODUCTION
The Autoanalyzer method is a modification of that proposed by Colwell (1965) who utilised the ascorbic acid reduction of the phosphomolybdate complex.
PREPARATION OF REAGENTS
ACID AMMONIUM MOLYBDATE. 0.5% aIIDnonium molybdate (NH4) 6 Mo 7024 .4H20 in 1.0 M H2S04. Dissolve 5 g ammonium molybdate in 800 ml distilled water; add carefully 56 ml cone. H2S04 A.R., cool and make up to 1 litre. Add about 2 ml Wetting Agent A when cool.
ASCORBIC ACID. Dissolve 2.25 g ascorbic acid A.R. in distilled water to make to 250 ml. This solution will keep for about a week if kept in a refrigerator.
WASH SOLUTION. 0.5 M H2S04.
PREPARATION OF STANDARDS
STOCK SOLUTION (100 µg/ml P). Dissolve 0.220 g potassium dihydrogen phosphate (KH2P0 4), A.R. in distilled water, add 14 ml cone. H
2S0 4 and make
to 500 ml.
WORKING STANDARDS. Pipette 1.0 and 2.0 ml aliquots of stock solution (100 µg/ml P) into 200 ml volumetric flasks and make to volume with 0.5 M H2S0 4 • These standards contain 0.5 and 1.0 µg/ml P.
PROCEDURE
To 18 ml of filtered extract add 0.5 ml of cone. H2S04 with a dispenser, stopper and shake. Treat 18 ml of extracting solution in the same manner to use as a blank. Cone. H2S04 is added to make the final solution 0.5 M H2S04 and this enables the Truog solutions to be analysed with the same manifold and reagents as for the other phosphate measurements. The error introduced by the addition of the small volume of acid is insignificant.
Set up Autoanalyzer as shown in manifold diagram (Fig. A5.1), with heating bath at 9o0 c and 880 nm filters in the colorimeter. Pump reagents and wash solution for about 10 min. to ensure complete flushing of the analytical system and wash receptacle. Pour standard and sample solutions into tubes on the sample tray. Set recorder baseline with baseline control on colorimeter.
c
(_

(
c
Sample top standard and when it reaches the colorimeter flowcell adjust recorder to read 100% transmission with the standard calibration control on the colorimeter. Reset baseline if necessary, and repeat
A5.5
this process to check calibration. Sample standards followed by unknowns, at a rate of 60/hr with a 5:1 sample:wash ratio.
CALCULATION OF RESULTS
Prepare a standard curve of µg/ml P against % absorption.
Using a 1:200 soil:solution ratio:
µg/ml P x 20 = mg % P
AlO CONNECTOR
10 TURN 5 TURN
3 ML HEATING MIXING COIL MIXING COIL
BATH COIL 9o0c
COLORIMETER 880 NM
15 MM FLOW-CELL
WASTE
FLOW RATE REAGENT MVMIN
0.23 AIR
0.6 MOLYBDATE
0.23 SAMPLE
0.6 ASCORBIC
0.8 WASH
1.0 PULL FROM COLORIMETER
TUBE COLOUR CODE
ORANGE/WH lTE
WHITE
ORANGE/WH lTE
WHITE
RED
GREY
Fig A5.l Flow diagram of autoanalyzer II manifold for phosphorus detennination.

AS.6
AS.B 0.5 M H2S04 - SOLUBLE PHOSPHORUS DETERMINATIOI~
EXTRACTION
PREPARATION OF REAGENTS
SULPHURIC ACID, O.S M. Make up 280 ml cone. H2S04 A.R.,to 10 litres (add the acid carefully and cool). Standardise to O.S M with standard alkali.
PROCEDURE
Place O.S g of finely ground soil (air-dry, < 0.2S mm) into a 2SO ml shaking jar and add 100 ml O.S M H2S04 . Shake on an end-over-end shaker for 16 hr (overnight) under temperature-controlled conditions (200 C). Filter through a No. 42 (Whatman) 12.S cm filter paper and collect from 40 to SO ml clear extract for determination method I (Manual). If using method II (automated), collect about 18 ml in a sampler tube. Include a reagent blank.
I, DETERMINATION USING MANUAL METHOD
PREPARATION OF REAGENTS
p-NITROPHENOL INDICATOR. distilled water.
Dissolve 0.S g p-nitrophenol in 2S ml
AMMONIUM HYDROXIDE, 1 + 1. One part of cone. NH40H, A.R., (sp.gr. 0.88 -· 0.90) plus one part distilled water.
MURPHY AND RILEY REAGENT A, see Method AS.AI.
MURPHY AND RILEY REAGENT B, see Method AS.AI.
PREPARATION OF STANDARDS
WORKING STOCK SOLUTION (1 µg/ml P) . See Method AS.AI.
WORKING STANDARDS. Prepare standards by pipetting 0, S, 10, lS, 20, and 2S ml aliquots of working stock phosphate solution (1 µg/ml P) into 100 ml volumetric mixing flasks and make to about 80 ml with distilled water. These solutions contain 0, S, 10, lS, 20 and 2S µg P respectively. Treat these standards as for samples.
c
c
c

c
(
A5.7
PROCEDURE
Pipette an aliquot of the filtrate containing not more than 25 µg P into a 100 ml volumetric (mixing) flask; a 10 ml aliquot will be suitable for most soils.
Dilute with water to about 80 ml and then adjust the pH to about 4.5 to 5.5 as follows: add 1 drop of p-nitrophenol indicator and then add 1 + 1 NH40H drop by drop until the solution just turns yellow. For a 10 ml aliquot, this should take from 1.5 to 2 ml (depending on the strength of the NH40H).
Add 0.5 M H2 S0 4 drop by drop until the solution just loses its yellow colour. When organic matter causes a yellow tinge in the solution, it is advisable to add 2 drops of p-nitrophenol indicator and to make the pH adjustment according to the change in indicator colour only.
Some soils give a precipitate of iron and aluminium hydroxide, but this can be disregarded as it redissolves when the acid molybdate is added during colour development.
Develop and measure the molybdenum blue colour using Murphy and Riley molybdate reagent according to the procedure described under Method AS.AI.
CALCULATION OF RESULTS
Construct a standard curve.
For 0.5 g sample:
(µg P in final 100 ml) x 20 Aliquot (ml)
This should be near-linear.
= 0. 5 M HiS0 4 - soluble P (mg %)
II. DETERMINATION USING AUTOANALYZER
PREPARATION OF REAGENTS
ACID AMMONIUM MOLYBDATE. As for Method A5.AII.
ASCORBIC ACID. As for Method A5.AII.
WASH SOLUTION. 0.5 M H2 S0 4 extraction solution.
PREPARATION OF STANDARDS
STOCK SOLUTION (100 µg/ml P). See Method A5.AII.

AS.8
WORKING STANDARDS. Pipette 2.0, 4.0, 6.0, 8.0, 10.0, 12.0, 16.0, and 20.0 aliquots of stock solution (100 µg/ml P) into 200 ml volumetric flasks c·· and make to 200 ml with 0.5 M H2 SO~. These standards then correspond to 1, 2, 3, 4, 5, 6, 8, and 10 µg/ml P.
PROCEDURE
Set up the Autoanalyzer as described in Method AS.All. Sample unknowns at a rate of 60/hr with a 5:1 sample:wash ratio, using the appropriate range of standards (1-5 µg/ml or 2-10 µg/ml).
CALCULATION OF RESULTS
Prepare a standard curve of µg/ml P against % absorption.
For a 1:200 soil:solution ratio:
µg/ml P x 20 = mg % P.
(
l

c
c
A5.9
AS.C PHOSPHATE RETENTION DETERMINATION
EXTRACTION
PREPARATION OF REAGENT
P-RETENTION SOLUTION (1 mg P/ml). Dissolve 8.80 g potassium dihydrogen phosphate (KH 2 P0 4), A.R., and 32.8 g anhydrous sodium acetate (CH 3COONa), A.R., in distilled water, add 23 ml glacial acetic acid, A.R., and dilute to 2 litres in a volumetric flask. The pH of this solution should be 4.6 ± 0.05.
PROCEDURE
Weigh 5 g soil (air dry, < 2 mm) into a stoppered 50 ml polypropylene centrifuge tube, and add 25 ml P-retention solution. Shake for 24 hr at about 20°c.
Centrifuge at 2000 rpm for 15 min.
DETERMINATION
PREPARATION OF REAGENT
NITRIC VANADOMOLYBDATE ACID REAGENT. Vanadate solution - dissolve 0.8 g ammonium vanadate, A.R., in 500 ml boiling distilled water, cool the solution, add 6 ml cone. HN03, A.R. and dilute to 1000 ml with distilled water. Mo16bdate solution - dissolve 16 g ammonium molybdate in distilled water at 50 C, cool and dilute to 1000 ml. Prepare dilute HN03 by diluting 100 ml cone. HN03 , A.R., to 1000 ml with distilled water. To this add first the vanadate solution and then the molybdate solution. Mix well.
PREPARATION OF STANDARDS
WORKING STOCK SOLUTIONS. Pipette 0, 10, 20, 30, 40, and 50 ml aliquots of P-retention solution (1 mg P /ml) into 50 ml flasks and make to volume with distilled water. These solutions contain O, 0.2, 0.4, 0.6, 0.8, and 1.0 mg P/ml and correspond to 100, 80, 60, 40, 20 and 0 percent retention respectively.

AS.10
PROCEDURE
Take 1 ml aliquots with an automatic dilutor from the samples in the centrifuge tubes, and from the working standards, and dispense into labelled 30 ml tubes with 19 ml of the nitric vanadomolybdate acid reagent. Shake well.
Wipe the delivery tip of the dilutor after each step, and carry out a complete step with distilled water between standards to avoid possible contamination of working stock solutions.
Read the absorbance, after at least 30 min., at 466 nm, setting the instrument to read zero on distilled water.
CALCULATION OF RESULTS
Prepare a standard curve of % phosphate retention against absorbance. Read off unknowns.
c
l_

c
(
A5.D
INTRODUCTION
ORGANIC PHOSPHORUS DETERMINATION IGNITION METHOD
This phosphorus fraction is determined from the increase in 0.5 M H2S04-soluble phosphorus caused by ignition of the soil, which converts organic phosphorus to inorganic phosphate. It has been found that in some soils with very low base status, phosphorus may be lost during ignition. To prevent this, calcium acetate is added prior to ignition.
AS.11
In strongly weathered soils ignition causes positive errors due to solubilisation of some forms of inorganic phosphorus (which were previously insoluble in 0.5 M H2S04). In some tropical soils (e.g. Cook Island soils) these errors are sufficient to render the results for organic phosphorus invalid.
EXTRACTION
PREPARATION OF REAGENTS
CALCIUM ACETATE, 20%. Dissolve 100 g anhydrous salt, A.R., by heating in 400 ml distilled water. Add about 5 ml glacial acetic acid to aid solution and make up to 500 ml with water.
0.5 M SULPHURIC ACID. See Method AS . B .
PROCEDURE
Weigh 0.5 g finely ground soil (air-dry, < 0.25 mm) into a 5 ml silica crucible, wet with 0.5 ml calcium acetate solution (20%) and dry in an oven (110°C). Ignite for 60 minutes at SS0°C, cool, and drop crucible into a 250 ml shaking jar containing 100 ml 0.5 M H2S04. Carry out extractions as for Method AS.B.
I DETERMINATION BY MANUAL METHOD
Carry out colour development and calculation as described in Method AS.BI. This calculation gives the amount of ignited 0.5 M H2S04 soluble phosphorus.
II DETERMINATION USING AUTOANALYZER
Carry out determination and calculation described in Method AS.BII. This calculation gives the amount of ignited 0.5 M H2S04 soluble phosphorus.

CALCULATION OF RESULTS
[Ignited o. s M H2SOa. -soluble P (mg%)l-Co. 5 M H2SOa. -soluble P (mg%)]=
Organic P (mg%)
AS.12
c

(
(
AS.E TOTAL PHOSPHORUS DETERMINATION
INTRODUCTION
The following method is based on the method recommended by Syers et al. (1968) who compared several extraction procedures for determining total phosphorus in soils and parent materials. They found that the sodium carbonate procedure of Muir (1952) modified by Jackson (1958) gave the most reliable results.
FUSION
PREPARATION OF REAGENTS
SULPHURIC ACID, 1 + 3. Carefully add 250 ml cone. H2S04, A.R., to 750 ml distilled water. Cool in running water.
SODIUM CARBONATE anhydrous Na2C0 3 , A.R.
PROCEDURE
Weigh 0.25 g finely ground soil (air-dry, < 0.25 mm) into a 60 ml platinum crucible. Add approximately half of a weighed 2 g amount of anhydrous Na2C03,A.R., and stir thoroughly with a platinum wire or glass rod. Cover the mixture with the rest of the Na2C0 3 •
Place the covered crucible at a slight angle on a silica triangle over a small diameter head Meker-type burner. Heat gently for about 5 min, until sample is molten, carefully lifting the lid to allow air in through a slit.
NOTE: In order to avoid reducing conditions during the fusion, it is most impoT ant to ensure that the flame from the burner does not completely envelope the crucible.
A5.13
Increase the heat gradually until the full heat of the flame is being used. The bottom of the· crucible should be a bright cherry red. Maintain this heat for 4-5 min. The crucible lid should be about ~ removed to allow the admission of air, and for the last 2 min. removed completely. Remove the crucible and swirl until the melt has solidified around the sides of the crucible. Cool, and transfer the crucible and lid to a 250 ml beaker. Holding the crucible lid with platinum tipped tongs, wash any residue from the underside, with approximately 2 ml 1 + 3 H2S04 from a full 15 ml pipette, into the beaker and set the lid aside. Cover the beaker with a watch-glass and carefully add the remainder of the 15 ml 1 + 3 H2S04 into the crucible through the gap between the beaker spout and the watch-glass. After the effervescence has completely subsided, tip the contents of the crucible into the beaker, and remove the crucible after washing it thoroughly with hot distilled water. Digest for 1 hour on a boiling water bath and filter through a No. 42 (Whatman) 12.5 cm filter paper into a 100 ml volumetric flask. Wash beaker and filter paper with hot distilled water, cool and make to volume. A Na2C0 3 blank should be treated in the same way as the samples.

I DETERMINATION. BY MANUAL METHOD
PREPARATION OF REAGENTS
MURPHY AND RILEY REAGENTS A AND B. see Method AS.AI.
PREPARATION OF STANDARDS
WORKING STOCK ( 1 ml = 1 µg P. ) See Method AS. AI.
WORKING STANDARDS. Pipette O, S, 10, lS, 20 and 2S ml aliquots of standard phosphate solution (1 ml = 1 µg P) into 100 ml volwnetric flasks and dilute to approximately 30 ml with distilled water. These solutions contain O, S, 10, lS, 20 and 2S µg P respectively. Treat these solutions as for samples as regards pH adjustment and colour development.
PROCEDURE
Pipette a suitable aliquot (usually 10 ml) of the sample solution into a 100 ml yolwnetric flask. Adjust the pH, develop and read the colour as described in Method AS.BI.
CALCULATION OF RESULTS
Prepare a standard curve from the readings for standards and read off the phosphorus concentrations for the unknowns in µg p per 100 ml:
AS.14
P in final 100 ml (µg) volume (ml) 10 x weight soil (g) x aliquot (ml) = Total phosphorus (mg %)
or, for a 0.2S g sample, making up to lOOml and taking a 10 ml aliquot:
P in final 100 ml (µg) x 4 ~ Total P (mg %)
II DETERMINATION USING AUTOANALYZER
Set up Autoanalyzer as described in Method AS.AII, using appropriate standards for the range expected.
CALCULATION OF RESULTS
Prepare a standard curve of µg/ml P against % absorption.
For 0.2S g made to 100 ml
µg/ml P x 40 = mg % P
Apply a blank correction.
c-

c
(
REFERENCES
COLWELL, J.D. 1965: Determination of phosphorus in sodium hydrogen carbonate extracts of soils. Chemistry and Industry 21: 893-5.
JACKSON, M.L. 1958: "Soil Chemical Analysis". Prentice-Hall. Jersey. 490 p.
MUIR, J.W. 1952:
Englewood Cliffs, New
AS.15
The determination of total phosphorus in soil with particular reference to the control of interference by soluble silica. Analyst, London 77: 313-7.
MURPHY, J.; RILEY, J.P. 1962: A modified single solution method for the determination of phosphate in natural waters. Analytica Chimica Acta, 27: 31-6.
SAUNDERS, W.M.H. 1968: Phosphorus. Chapter 7.7 pp. 95-102 in "Soils of New Zealand, Part 2". New Zealand Soil Bureau Bulletin 26(2): 221 p.
SYERS, J.K.; WILLIAMS, J.D.H.; WALKER, T.W. 1968: The determination of total phosphorus in soils and parent materials. N.Z. Journal of Agricultural Research 11: 757-62.
WATANABE, F.S.; OLSEN, S.R. 1965: Test of an ascorbic acid method for determing phosphorus in water and NaHC0 3 extracts from soil. Soil Science Society of America Proceedings 29: 677-8.

(

c
c
A6.l
A6. CATION EXCHANGE PROPERTIES
INTRODUCTION
Cation exchange properties in:lude cation exchange capacity (CEC), total exchangeable bases by titration (TEB), exchangeable bases, i.e., Ca, Mg, K, Na, and percentage base saturation(% BS).
The methods described here are suitable only for the usual soils of humid regions, that is, for soils in which calcium carbonate and soluble salts are both virtually absent. Where appreciable amounts of such substances are present, other more complex methods of analysis must be used for the determination of the cation exchange properties (e.g. Metson 1956).
The following methods enable the levels of individual exchangeable bases to be determined by atomic absorption spectrometry (AAS) or by flame emission spectrometry (FES). They also permit determination of CEC and of TEBt. on the same sample. 1tn
The exchangeable bases are leached from the soil with 1 M _ ammonium acetate, pH 7.0, the excess removed by washing with alcohol, leaving the exchange sites saturated with ammonium ions. These are removed by leaching with M sodium chloride and the CEC determined from the amount of ammonium ions in the extract (distillation or Autoanalyzer colorimetric determination of annnonia). Although this procedure involves an extra leaching step for the determination of CEC, it is preferable to the method in which the ammonia is replaced directly from the soil by distillation ( using magnesium oxide) because the results have been found to be more reliable and more convenient (Metson & Blakemore 1964).
TEB measurements are carried out on aliquots of the leachates. Although summation of exchangeable Ca, Mg, K, and Na (i.e., I bases, or TEBsum) should be used to calculate% BS (Blakemore 1964), the TEBt.t value is useful as a check on the individual base determinations 1 n and also gives a useful indication of the presence of adsorbed sulphate when compared with I bases.
Choice between AAS and FES will depend partly on the availability of instruments and partly on individual preferences. At Soil Bureau FES is preferred for Ca, K, and Na. It is essential that the instrument is of sufficiently good resolution to avoid spectral interferences such as those caused by calcium during sodium determination and strontium during calcium determination. Advantages of FES over AAS for these elements include greater sensitivity for K and Na, steadier base line and sample readings, and elimination of the recurring trouble and expense of failing hollow cathode lamps. However, FES is not sufficiently sensitive for the determination of exchangeable Mg, and AAS is more suitable.
For AAS analysis, strontium is added to the test solutions to overcome the depression of calcium absorption by phosphorus and suppression of magnesium absorption by aluminium and silicate. It is also necessary to

add strontium during FES analysis to overcome phosphate interference on calcium emission. Hydrochloric acid is added to ensure that there is adequate chloride present for optimum atomic absorption, especially for calcium determinations by AAS. Caesium is added to test solutions for both AAS and FES to eliminate ionization of the analyte in the flame.
PREPARATION OF REAGENTS see Method A6.E.II. for reagents for Autoanalyzer determination of CEC)
A6.2
ACID-WASHED SILICA SAND. Silica sand to be treated should be relatively pure (white) and should be sieved so that it is in the 0.25 -0.5 mm size range (to pass through a 30-60 mesh sieve). Place about 10 litres of this material into a suitable container (e.g. lixiviating beaker, chromatography tank), wash with tap water to disperse organic matter, silt, and clay, and after vigorous stirring, remove these impurities by decanting. This process should be.repeated until the supernatant water is clear. Add 2.5 litres cone. HCl (commercial grade) to sand and leave overnight. Stirring vigorously, wash out the acid from the sand with copious quantities of tap water. Using a large Buchner funnel (with a Whatman No. 542 filter paper) connected to a vacuum pump, wash portions of the sand with distilled water until the filtrate is chloride-free. Dry in an oven.
MACERATED FILTER PAPER. Macerate by means of a suitable machine (e.g. food blender), a quantity of fast-filtering acid-washed filter paper (e.g. Whatman No. 31 or 41) in distilled water. Keep moist in a closed jar.
AMMONIUM ACETATE (CH3COONH4 ), M,pH 7.0. This is best prepared from glacial acetic acid and strong ammonia solution as it is cheaper, and the results for blank solutions are usually lower than when it is prepared from the solid salt, even of A.R. grade .
. Prepare as follows: Add 1150 ml acetic acid (CH::iCOOH) (about 99%,A.R.) to about 15 litres distilled water, shake, and add 1500 ml ammonium hydroxide (NH 40H) solution (A.R. sp. gr. 0.91). Make up to ·20 litres and mix by shaking. Che.ck pH and adjust to pH 7.0 ± 0.05 with 2 M NH1tOH and 2 M CH 3COOH. Allow to cool before using.
WASH ETHANOL (WASH ALCOHOL). Dilute ethanol (L.R. or commercial grade) to 90% w/w with distilled wate.r, using a hydrometer to check strength. (N.B. water required= 9.7 ml water/litre alcohol/percentage change. e.g. 20 litres of 95% alcohol requires 970 ml water.) Add 0.25 ml of 2 M NH 40H per litre. Determine the ammonium content by distillation of a 10 ml aliquot with NaOH (steam distillation as for nitrogen determination), and titrate the distillate with 0.0200 M HCl. The titration should take between 0.18 and 0.33 ml, using bromocresol green - methyl red indicator.
SODIUM CHLORIDE, approximately lM. 20 litres distilled water.
Dissolve 1170 g NaCl, A.R., in
SODIUM HYDROXIDE 1 + 2. Dissolve 1 kg NaOH pellets, A.R., in 2 litres distilled water stirring constantly.
BORIC ACID, 1% solution, A.R.
c

(
(
HYDROCHLORIC ACID, 0.020 M.
HYDROCHLORIC ACID, 0.050 M.
HYDROCHLORIC ACID, 0.2 M.
HYDROCHLORIC ACID, 1 M.
SODIUM HYDROXIDE, 0.020 M.
BROMOCRESOL GREEN-METHYL RED MIXED INDICATOR (Ma and Zuazaga 1942). Mix 5 parts of 0.1% bromocresol green in 95% ethanol with 1 part of 0.1% methyl red in 95% ethanol. These proportions may have to be varied to obtain the neutral-grey transition colour of the indicator.
METHYL RED INDICATOR, 0 .1%. Weigh 1. 0 g finely ground methyl red powder into a 1500 ml beaker. Add 964 ml 95% ethyl alcohol and 36 ml 0.1 M. NaOH. Stir until all the methyl red has dissolved.
A6.3
STRONTIUM CHLORIDE - CAESIUM CHLORIDE SOLUTION. (7500 ppm Sr, 25000 ppm Cs). Weigh 46 g SrCh.6H20 A.R., and 6:1.6 g CsCl A.R., into a beaker, dissolve and make to 2· litres with distilled water. This solution contains strontium to eliminate possible phosphate interference during the flame emission determination of calcium. Caesium is added to eliminate ionisation of all exchangeable cations in the air/acetylene flam.e.
1 + 3 DILUENT SOLUTION. Carefully add 54 ml cone. HCl, A.R., to about 200 ml distilled water, add 213 ml SrC1 2 /CsCl solution and make to 2 litres with distilled water.
l + 9 DILUENT SOLUTION. Carefully add 45 ml cone. HCl, A.R., to 233 ml 1 M ammonium acetate, add 180 ml SrCl/CsCl solution and make to 2 litres with distilled water.

A6.A LEACHING PROCEDURES
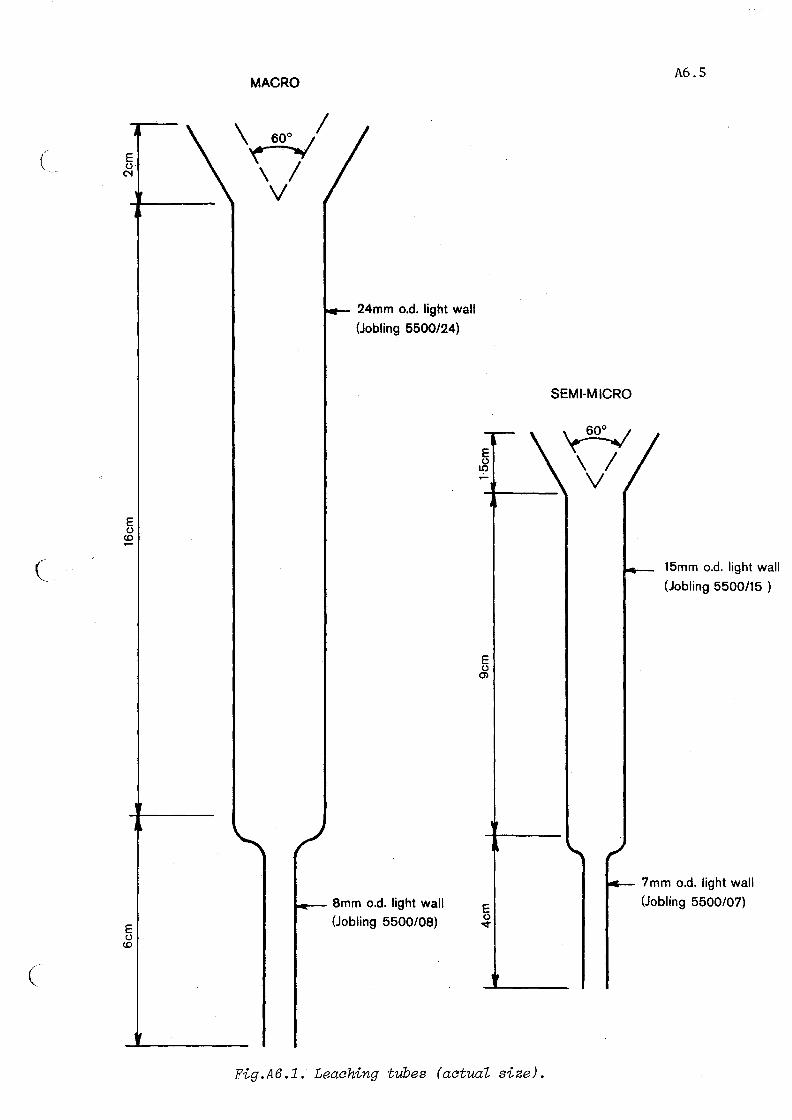
Mix 5 g soil (air-dry, < 2 mm) with 10 g acid-washed silica sand and pack into a leaching tube (see Fig. A6.1) which has a macerated filter paper plug.
A6.4
Percolate the soil with 230 ml 1 M ammonium acetate, pH 7.0 into~ 250 ml volumetric flask and make to volume (250 ml) with distilled water. Leaching time should be at least one hour, or low results may be obtained.
Carry out a blank determination with each set on 10 g acid washed silica sand.
NOTE: It is essential that formation of channels and air-locks in the colum.i1 of soil and sand are avoided during leaching. This may be ensured by application of suction to the outlet of the leaching tube when the ammonium acetate is first added. The suction must be gentle, and may be generated by attaching a short length of rubber tube to the outlet, squeezing the whole tube and,while blocking off the end, releasing the squeezed portion. The suction so obtained is applied until the ammonium acetate just reaches the filter paper plug, at which time the end of the rubber tube is released and the rubber tube quickly removed. The outlet of the leaching tube is then placed into the neck of the receiving volumetric flask during leaching.
Rinse the top of the leaching tube and the soil several times with smc-_ll ai-nounts of wash ethanol, allowing to drain between each washing. Percolate 200 ml ethanol through the tube and discard the percolate.
Place a 250 ml volumetric flask under the leaching tube, and leach the soil column with 230 ml 1 M NaCl solution. Make the solution to 250 ml with distilled water.
NOTE: ,--
1. In order to avoid contamination with sodium, it is advised that· a separate set of glassware be used for the sodium chloride leaching.
2. A further leaching of the sample may be made to extract oxalate soluble iron, aluminium, manganese and silicon (see Method AS).
I 1 •.
(
(
(
E (.)-
CN
E 0
<O
E 0
<O
MACRO
\~~ ... '!/ ' I v
24mm o.d. light wall
(Jobling 5500/24)
8mm o.d. light wall
(Jobling 5500/08)
E 0 ~
Fig.A6.1. Leaching tubes (actual size).
SEMI-MICRO
~ \ I v
A6.5
15mm o.d. light wall
(Jobling 5500/15 )
7mm o.d. light wall
(Jobling 5500/07)

A6.B DETERMINATION OF INDIVIDUAL EXCHANGEABLE BASES
PREPARATION OF STANDARDS
CONCENTRATED STOCK SOLUTIONS:
CALCIUM, 2.000 N solution: carefull6 dissolve 50.045 g calcium carbonate (CaC03) A.R. (dried at 110 C), in enough 1 + 1 HCl to just dissolve it (about 200 ml), and make up to 500 ml with 0.2 M HCl.
MAGNESIUM, 0.2000 N solution: carefully dissolve 1.216 g magnesium ribbon, A.R., in 1 + 1 HCl, as for calcium, and make to 500 ml with 0.2 M HCl.
POTASSIUM, 0.2000 N solution: dissolve 7.455 g potassium chloride (KCl) A.R. (dried at 110°C), in 0.2 M HCl, and make to 500 ml with 0.2 M HCl.
SODIUM0 0.2000 N solution: dissolve 5.845 g NaCl A.R. (dried at 110 C), in 0.2 M HCl, and make to 500 ml with 0.2 M HCl.
A6.6
MULTIPLE WORKING STOCK SOLUTION. Pipette 25 ml of each concentrated stock solution into a 1000 ml volumetric flask, and make to mark with deionised water. Store in a plastic (preferably teflon) bottle.
WORKING STANDARDS. Pipette out aliquots of the multiple working stock solution to 500 ml volumetric flasks according to the following table:
Working Volume of Multiple Concentration of Working Standard Working Stock Standards (10- 4 N)
(ml per 500 ml) Ca Mg K Na
1 0.0 0.0 0.0 0.0 0.0
2 2.5 2.5 0.25 0.25 0.25
3 5.0 5.0 0.50 0.50 0.50
4 10.0 10.0 1.00 1.00 1.00
5 15.0 15.0 1.50 1.50 1.50
6 25.0 25.0 2.50 2.50 2.50
7 50.0 50.0 5.00 5.00 5.00
(
c
(

(
(
A6.7
To each flask add 10 ml cone. HCl, 40 ml strontium-caesium solution (7500 ppm Sr, 25000 ppm Cs) and 125 ml 1 M ammonium acetate. Make up to 500 ml with deionised water and mix well. Store in plastic bottles.
PROCEDURE
Dilute sample solutions using the 1 + 3 diluent solution.
Determine exchangeable bases by flame spectrometry using the working standards (previously prepared as above) for comparison. Normally standard number 5 is used as the top standard, however if sample concentrations exceed this standard, standards 6 and 7 may be used to avoid further dilution of the sample. If further sample dilution is necessary, perform a 1 + 9 dilution using the appropriate diluting solution.
CALCULATION OF RESULTS
Prepare a standard curve from the readings for the standards in each case (i.e. Ca*, Mg, Na and K) and read off concentrations as N x 10- 4 •
For a 1 + 3 dilution; N x 10- 4 x 2 = me.%
For a 1 + 9 dilution; N x 10- 4 x 5 = me.%
Apply blank corrections in terms of me.%.
* See Section AlO (p. Al0.4) for correction needed when free CaC03
present.

A6.C
PROCEDURE
DETERMINATION OF TOTAL EXCHANGEABLE
BASES BY TITRATION <TEBtitn)
Pipette a 100 ml aliquot of the ammonium acetate leachate into a silica basin, evaporate to dryness, and muffle for 30 min. at 50o0 c.
Take up in a known amount of 0.050 M HCl, (usually 10 ml), digest on a waterbath for 30 min., and back titrate with 0.020 M NaOH, while still hot, using methyl red as indicator.
This procedure is basically that described by Metson (1956) except that the reagents are weaker and therefore NaOH is preferable to NH 40H.
CALCULATION OF RESULTS
0.0200 M HCl used (ml) = TEB (me.%)
or
(0.0500 M HCl added (ml) x 2.5) - (0.0200 M NaOH required (ml)) =
TEB (me.%).
Apply a blank correction in terms of me.%.
A6.8
(
c
c

( A6.D : CALCULATION OF TOTAL EXCHANGEABLE
BASES BY SUMMATION <TEB sum. or E Bases)
CALCULATION
This valUe represents the sum of the individual exchangeable bases
(i.e., Ca, Mg, K, Na) when each is expressed as me.%. For the usual
soils of humid regions, TEB .t closely corresponds to TEB However, ti n sum. when soluble salts are present and are not removed prior to determination,
A6.9
TEBsum. will exceed TEBtitn by approximately the equivalent of the metal
cations present as neutral salts. When calcium carbonate is present some
is dissolved by the ammonium acetate leaching solution, but this is titratable
by the TEBtitn procedure, .so that both the apparent TEBtitn and the apparent
TEB are increased by an amount equivalent to the amount of calcium sum. carbonate dissolved.
In very acid soils much aluminium, replaceable by aIIUllonium acetate,
may be present. As this aluminium is associated with the sulphate ion,
an acid reaction is developed on ignition of the aluminium sulphate formed
on evaporation, reducing the apparent TEBtitn figure and, in extreme cases,
a negative value for TEBtitn may even be obtained (Blakemore 1964) .
. Disregarding such exceptions, a comparison of TEBt.t with TEB 1 n sum.
provides a useful guide to the accuracy of the analysis as a whole.

A6.E : DETERMINATION OF CATION EXCHANGE CAPACITY <CEC)
I DETERMINATION BY DISTILLATION
PROCEDURE
Transfer a 20 ml aliquot of the 1 M NaCl leachate to a suitable ammonia distillation apparatus, using about 5 ml 1 + 2 NaOH to drive off the ammonia. See Section A4.Al (p. A4.3 ) for details.
Collect the ammonia in about 10 ml 1% boric acid and titrate with 0.0200 M HCl using bromocresol green-methyl red indicator.
CALCULATION OF RESULTS
For a 5 g sample and 20 ml aliquot:
0.0200 M HCl used (ml) x 5 = CEC (me.%)
Apply a blank correction in terms of me.%.
II DETERMINATION USING AUTOANALYZER
INTRODUCTION
The colorimetric determination of ammonium-nitrogen as described in Method A4.A is used to determine the ammonia present in the 1 M NaCl leachates obtained by Method A6.A. This procedure is an alternative to the distillation method.
PREPARATION OF REAGENTS
NaDTT, see Method A4.A.
SODIUM NITROPRUSSIDE, see Method A4.A.
SODIUM SALICYLATE, see Method A4.A.
A6.10
WASH SOLUTION distilled water plus a few drops Brij 35 Wetting Agent.
CITRATE-TARTRATE, see Method A4.A.
PREPARATION OF STANDARDS
STOCK SOLUTION (1 ml= 700 µg N). Dissolve 3.3011 g (NH4)zS04 (M.A.R. or Aristar quality dried at ll0°C) in 1 M NaCl and make to 1 litre with 1 M NaCL
c

(
(~
l
A6.ll
WORKING STANDARDS. Pipette 5.0, 10.0, 20.0, 30.0, and 40.0 ml aliquots of stock solution into 250 ml volumetric flasks and make to mark with 1 M NaCl. These solutions then correspond to 14.0, 28.0, 56.0, 84.0 and 112.0 µg/ml N respectively.
PROCEDURE
Prepare sample solutions as in Method A6.A.
Set up manifold as in Method A4.A (see figure A4.1 but use 1 M NaCl as wash solution). Set the recorder baseline using the colorimeter baseline control.
Sample 112.0 µg/ml N standard and when it reaches the colorimeter flowcell, set the recorder to read 100%_absorption using the standard calibration control on colorimeter. Reset baseline if necessary. Sample standards and unknowns at 60/hr with a 5:1 sample:wash ratio. Samples containing more than 112 µg/ml N can be diluted using 1 M NaCl as diluent.
CALCULATION OF RESULTS
With 5 g soil made to 250 ml the standards are equivalent to the following CEC (me.%) values.
Standard CEC µg/ml N me. %.
112.0 40.0 84.0 30.0 56.0 20.0 28.0 10.0 14.0 5.0
Construct a standard curve of CEC (me.%) against % absorption for . the standards (this should be near-linear).
Read off unknowns directly as me.%.
Apply a blank correction in terms of me.%.

CALCULATION
A6.F : CALCULATION OF PERCENTAGE BASE SATURATION C%BS)
A6.12
This value is derived from the CEC and TEB (or sometimes TEBtitn) figures, when both are expressed as me.%, thus, sum.
TEB (me.%) x 100 sum. = %BS CEC (me.%)
(__
c

A6.13
A6.G SEMI-MICRO LEACHING
PROCEDURE
Mix 1 g of soil (air-dry, < 2 mm) with 2 g of acid-washed silica sand and pack into a semi-micro leaching tube (see Fig. A6.l) which has a macerated filter paper plug. Percolate the soil with SO ml 1 M ammonium acetate, pH 7.0 into a SO ml flask. Make to volume with distilled water. Dilute 1 + 3 as for Method A6.B.
The solution is then equivalent to the diluted macro-leachate and ready for flame determination of exchangeable bases. Cation exchange capacity is determined by washing the soil with SO ml of 90% ethanol, leaching with 4S ml of 1 M NaCl, and making to volume in a SO ml flask. The NaCl leachate is equivalent in concentration to that obtained in the macro determination of CEC.
Leachings may be carried out on smaller amounts of soil with suitable adjustment of reagent volumes and glassware.

A6.14
A6.H KCL-EXTRACTABLE ALUMINIUM
INTRODUCTION
This property is sometimes referred to as "exchangeable" aluminium and is used together with the sum of exchangeable bases to estimate the Effective Cation Exchange Capacity (ECEC).
i.e., KCl extractable Al (me.%) + ~bases (me.%) = ECEC (me.%)
PREPARATION OF REAGENTS
POTASSIUM CHLORIDE 1 M. Dissolve 373 g potassium chloride, A.R. in S litres of distilled water.
PREPARATION OF STANDARDS
STOCK SOLUTION (200 µg/ml Al). Dissolve 0.200 g pure aluminium wire in about S ml cone. HCl with the addition of a trace (one single crystal) of a mercury salt to catalyse the reaction. Make to 1 litre with distilled water.
WORKING STANDARDS. Pipette 0, S, 10, 2S and SO ml of stock solution (200 µg/ml Al) into 200 ml vol~etric flasks, add SO ml of 1 M KCl, 1 ml cone. H
2S0
4, and then make to 200 ml with distilled water. These solutions then
contain 0, S, 10, 2S and SO µg/ml Al respectively.
PROCEDURE
Weigh S.O g of soil (air-dried, < 2 mm) into a 12S ml conical flask. Add 2S ml of 1 M KCl and swirl gently to saturate soil. Allow to stand for about 16 h. (overnight). Filter through a No. 42 (Whatman) filter paper into a 200 ml volumetric flask. Wash the remaining soil in the conical flask and filter paper with a further 2S ml of 1 M KCl. Wash soil and filter paper twice with distilled water. When filtered add 1 ml cone. H2 S0 4 and make volumetric flask to volume with distilled water. Include a reagent blank in the procedure.
Determine aluminium in extracts directly by high-temperature flame spectrometry.
N.B. H2S0 4 is added to extracts to prevent biological growths.
CALCULATION OF RESULTS
Prepare a standard curve of µg/ml Al against % transmission or absorbance.
µg/ml Al x 0.444 =Al (me.%).
( '
c

A6.15
A6.I EXCHANGE ACIDITY
INTRODUCTION
This property refers to the acidity measured by releasing aluminium and displaced hydrogen at pH 8.2, neutralising the resulting acidity with triethanolamine (TEA) and titrating the excess TEA with standard acid. The value is an indicator of pH dependent charge, and when added to total exchangeable bases, gives a value for CEC at pH 8.2
There are a number of methods available for the determination of this property, including leaching and shaking methods. The method described here has been found to offer advantages in reproducibility and convenience. Main features are the use of overnight shaking rather than leaching and the titration of an aliquot of the equiliberated buffer solution rather than the whole solution. This avoids the need to recover the remaining triethanolamine completely by leaching with replacement solution, or with further portions of buffer solution.
PREPARATION OF REAGENTS
BUFFER SOLUTION. To 61.07 g BaC1 2 .2H20 add 28 ml triethanolamine and make to 1 litre with distilled water. Adjust the pH of the solution to 8.2 with 1 + 1 HCl (usually about 10 ml per litre). Protect from C0 2 by attaching a drying tube containing a C0
2 absorbant (e.g. soda lime) to the
air intake.
HYDROCHLORIC ACID. 0.10 M
BROMOCRESOL GREEN. 0.1% aqueous solution.
MIXED INDICATOR. Dissolve 1.250 g methyl red and 0.825 g methylene blue in 1 litre 90% ethanol.
PROCEDURE
Weigh 2.5 g soil (air-dry, < 2 mm) (1.0 g for soils high in organic matter or amorphous oxides) into so ml, screw-cap, polypropylene centrifuge tubes. Add 25 ml of buffer solution and shake overnight (16 h.) on an endover-end shaker. Centrifuge at 2000 r.p.m. for 15 min. Also shake at least two blanks (i.e. 25 ml buffer solution).
Transfer 10 ml aliquot to 125 ml conical flask and add about 20 ml distilled water. Add 1 drop bromocresol green and 5 drops mixed indicator. Titrate with 0.10 M HCl until solution reaches first full purple colour. Titrations on blanks should be carried out first and unknowns titrated to same point of colour change.

A6.16
CALCULATION OF RESULTS
Using 2.5 g soil and 25 ml buffer solution,
[ml 0.10 M HCl, (Blank) - ml 0.10 M HCl (Sample)] x 10 = Exchange Acidity (me.%).
Using 1.0 g soil and 25 ml buffer solution,
[ml 0.10 M HCl, (Blank) - ml 0.10 M HCl (Sample)] x 25 = Exchange Acidity (me.%).
REFERENCES
BLAKEMORE, L.C. 1964: Effect of adsorbed sulphate on the determination of total exchangeable bases in soils. N.Z. Journal of Agricultural Research 7: 435-8.
MA, T.S.; ZUAZAGA, G. 1942: Micro-Kjeldahl determination of nitrogen. A new indicator and an improved rapid method. Industrial and Engineering Chemistry. Analytical Edition 14: 280-2.
METSON, A.J. 1956: Methods of chemical analysis for soil survey samples. N.Z. Soil Bureau Bulletin 12: 208 p.
METSON, A.J.; BLAKEMORE, L.C. 1964: Determination of cation-exchange capacity of soils by ammonium absorption: comparison of methods for determination of absorbed ammonium ion. N.Z. Soil Bureau Report 6/1964: 12 p.

(
c
INTRODUCTION
A7. RESERVE POTASSIUM <Kc) AND MAGNESIUM (MGR)
Al.A DETERMINATION OF Kc VALUE
This value, which was proposed by Metson et al. (1956), represents
A7.l
the long-term potassium-supplying power of the soil. The method disregards the more readily extractable fraction of the non-exchangeable potassium (which would be obtained in a single extraction or a small number of 1 M HN03 extractions) and represents the nearly constant rate of release after several extractions. This rate of release is a function of the mineral reserves present in the soil (Metson 1968) and is not affected by organic matter return, fertiliser applications, urine patches, etc.
In order to determine this value, the more readily soluble potassium is removed by a single extraction with 1 M HN03 at a wide acid:soil ratio. This fraction is discarded. Two or three successive extractions with 1 M HN03 at a narrower acid:soil ratio are then made and K concentration in these extracts is determined. The results of these two or three extractions, which should be fairly similar, are averaged to give the K value. ·
c
PREPARATION OF REAGENTS
NITRIC ACID, approximately 1 M. Dilute 318 ml cone. HN0 3, A.R., sp. gr. 1.42, to 5 litres with distilled water.
CAESIUM CHLORIDE (20000 ppm Cs). Weigh 25.4 g CsCl A.R., into a beaker, dissolve and make to 1 litre with distilled water. This solution is added to samples and standards to suppress potassium ionisation in the air/acetylene flame.
PREPARATION OF STANDARDS
STOCK SOLUTION 0.200N K, see Method A6.B.
WORKING STOCK SOLUTION 0.002 N K. Pipette 10.0 ml of stock solution (0.200 N K) into a 1 litre flask and make to volume with distilled water.
WORKING STANDARDS. Pipette O, 5, 10, 15, 20, 30 and 50 ml aliquots of the working stock solution (0.002 N K ) into 200 ml volumetric flasks add 20 ml caesium chloride solution (20000 ppm Cs) 100 ml of 1 M HN0 3 and make to volume. These solutions then correspond to 0, 0.5, 1.0, 1.5, 2.0, 3.0 and 5.0 N x 10~ 4 with respect to potassium.

A7.2
PROCEDURE
Weigh out 2.00 g soil (air-dry, < 2 mm) into a 400 ml extraction flask, or wide-mouth conical flask.
Add 200 ml 1 M HN03 and boil for 20 min. with a smaller conical flask inverted in the neck to act as a reflux condenser.
Allow solution to stand until supernatant is clear (about 30 min.). Remove the bulk of the supernatant with a suitable suction device taking care not to remove any soil. Alternatively if solution does not clear on standing, transfer to a 250 ml centrifuge bottle using distilled water and centrifuge for 15 min. at about 2000 rpm. Paur off and discard the liquid. With approximately 25 ml 1 M HN0 3, transfer soil to a 40 ml centrifuge tube, centrifuge, and discard liquid.
Transfer soil to a 100 ml conical flask with 819/26 Socket, using exactly 25 ml 1 M HN03 in approximately five portions (dispensed from a 25 ml burette).
Fit a 819/26 cone joint (about 110 mm in length) in the neck of the flask to act as an air condenser, and gently boil on a hot plate for 10 min. (actual boiling time).
Transfer liquid to the 40 ml heavy-duty centrifuge tube (coarse sand etc. may be left in flask) with a very small amount of distilled water, centrifuge, and pour liquid into a 50 ml volumetric flask. Add 5 ml of caesium chloride solution and make to volume with distilled water.
Transfer soil to original 100 ml conical flask with a further 25 ml of 1 M HN03 and repeat this boiling and centrifuging process.
Repeat, again, with a further 25 ml 1 M HN03, if a third extraction is to be made (normally two extractions give a sufficiently accurate result).
Determine potassium concentrations in the separate extractions by flame emission spectrometry (FES) (cf. p. A6.l). Include a blank determination throughout the procedure.
CALCULATION OF RESULTS
Prepare a standard curve of N x 10""4 K against % transmission.
For 2 g made to 50 ml
mean N x 10- 4 K 4
= Kc (me.%)
c
(

c
(
A7.3
A7.B DETERMINATION OF MGR VALUE
INTRODUCTION
This value represents the acid soluble 'reserve' magnesium of the soil (Metson & Brooks 1975) and is calculated by determining the magnesium soluble in boiling 1 M HCl (Mg ) and subtracting the exchangeable magnesium (Mg) obtained by leaching witRs 1 M ammonium acetate (see method A6.B). e
PREPARATION OF REAGENTS
HYDROCHLORIC ACID 1 M. Carefully add 430 ml of cone. HCl A.R.,to 2 litres of distilled water and make to 5 litres. Standardise with standard alkali of comparable strength.
1 + 9 DILUENT SOLUTION. Add 175 ml SrC12 /CsCl solution (see method A6) and 16 ml cone. HCl A.R.,to 1100 ml 1 M ammonium acetate and make to 2 litres with distilled water.
PREPARATION OF STANDARDS
WORKING STANDARDS. As for method A6.B.
PROCEDURE
Weigh 2.5 g of soil (air dry, < 2 mm) into a 400 ml extraction flask, add 100 ml 1 M HCl and place an inverted 50 ml conical flask in the neck of the extraction flask to act as a condenser. Place the extraction flasks on a preheated hotplate (the liquid should boil after approximately 10 min and then the hotplate heat should be reduced). Boil for 15 min (use timer) with occasional swirling. Cool for 10 min after removing from hotplate and rinse the condenser flask into the extraction flask. Filter extract into a 250 ml volumetric flask through a No. 42 (Whatman) filter paper and wash four times with hot distilled water. Allow the filtrate to cool before making to volume with distilled water. Include a reagent blank throughout the procedure.
Dilute the sample solutions 1 + 9 with the diluent solution and determine magnesium by atomic absorption spectrometry (AAS) at 285.2 nm
(_ using the exchangeable base standards.

A7.4
CALCULATION OF RESULTS
Prepare a standard curve of N x 10- 4 Mg against absorbance.
For 1 + 9 dilution
Mg (N x 10- 4) x 10 = Mgas (me.%)
Mg (moisture factored)(me.%)- Mg (moisture factored)(me.%)= Mg (me.%) as e r
REFERENCES
METSON, A.J.; ARBUCKLE, R.H.; SAUNDERS, M.L. 1956: The potassium-supplying power of New Zealand soils as determined by a modified normal-nitric-acid method. Transactions of the 6th International Congress of Soil Science B: 619-27.
METSON, A.J. 1968: The long-term potassium-supplying power of New Zealand soils. Transactions of the 9th International Congress of Soil Science 2: 621-9.
METSON, A.J.; BROOKS, Jean M. 1975: Magnesium in New Zealand soils. II. Distribution of exchangeable and reserve magnesium in the main soil groups. N.Z. Journal of Agricultural Research 18: 317-35.
c
(
l

c:
c
(
A8.l
A8. EXTRACTABLE IRON, ALUMINIUM AND SILICON
A8.A
INTRODUCTION
ACID OXALATE-EXTRACTABLE IRON, ALUMINIUM AND SILICON
The acid oxalate extraction method (Tamm 1922) depends mainly on the complexing affinity of acid oxalate to extract colloid complexes. The reagent dissolves amorphous oxides and hydrous oxides (its attack on crystalline oxides and crystalline clay minerals is very limited (Tamm 1932) and, because the forms which it dissolves play a major part in cation and anion retention and other surface phenomena, extraction with this reagent is useful in soil studies (Saunders 1965).
Two extraction procedures are used:
(1) A shaking method.
(2) A leaching method (Daly & Binnie 1974), following the leachings for cation exchange properties in the soil, thus avoiding .. weighings, shakings and filterings.
Historically at Soil Bureau the extraction solution proposed by Tamm (1922) has been used. This was a 0.175 M solution of ammonium oxalate buffered to pH 3.25 with oxalic acid, giving a total oxalate strength of 0.275 M. To be consistent with what most workers in soil science use, and perhaps to avoid extracting more Al, Fe and Si than they would, it has been decided to change to the weaker extracting solution described by McKeague and Day (1966). This is a 0.2 M solution of ammonium oxalate buffered to pH 3 with 0.2 M oxalic acid giving a total oxalate strength of 0.2 M. It has been found that the stronger reagent (0.275 M) used at a 1:40 soil reagent ratio for extraction by leaching gives very similar values to a 1:100 ratio extraction by shaking, for a wide range of Al, Fe and Si values. However, when using the weaker reagent (0.2 M) for samples with relatively large amounts of amorphous (> about 2% Al) materials, it has been found that neither a 1:40 leaching nor a 1:40 shaking extract as much as the stronger reagent or a 1:100 shaking.
Therefore it is recommended that if high values (> about 2% Al) are obtained or expected, the extraction be carried out by shaking at a 1:100 ratio. For lower values, the 1:40 ratio leaching is preferred for analytical convenience.
· One of the main factors which has enabled greater use to be made of the acid oxalate method is the development of atomic absorption and flame emission techniques by which iron, aluminium and silicon·determinations may be carried out directly on the extracts much more easily and quickly than previously.

A8.2
Choice of the flame technique to be used and of the conditions under which this technique is employed will be governed by the types of instruments available. However, there are some considerations which should be taken into account and the following notes are based on experience at Soil Bureau using both absorption and emission techniques (Searle & Daly 1977).
Iron concentration may be determined by either atomic absorption spectrometry (AAS) or flame emission spectrometry (FES). The use of the nitrous oxide-acetylene flame for both techniques is recommended.
Aluminium concentrations may be determined either by AAS or by FES with a nitrous oxide-acetylene flame. The FES method is more sensitive and convenient and should be used whenever there is available a good quality instrument equipped for the nitrous oxide-acetylene flame.
Silicon concentrations are determined only-by AAS with acetylene flame because FES is insufficiently sensitive. is not highly sensitive and a good quality instrument with capabilities is required for successful determinations.
a nitrous oxideThe AAS method
scale expansion
NOTE: For maximum sensitivity, in both AAS and FES it is necessary to suppress-ionisation of the species being determined, and this can be achieved by adding an excess of a readily ionisable salt, e.g., caesium chloride.
PREPARATION OF REAGENTS
ACID OXALATE REAGENT. A. 0.2 M Ammonium Oxalate. Dissolve 284 g (NH~) 2C 20~.H20 in 10 litres distilled water.
B. 0.2 M Oxalic Acid. Dissolve 252 g H2C 20~.2H20 in 10 litres distilled water.
Mix 1 part solution A with 0.75 parts solution B, (e.g., 5700 ml A and 4300 ml B to give 10 litres). Check and adjust pH to 3 by adding either A or B.
CAESIUM CHLORIDE, 20 OOO µg/ml Cs. 500 ml distilled water.
Dissolve 12.65 g CsCl A.R., in
1 + 4 DILUENT SOLUTION. 1 litre with distilled water.
Make 1.6 g CsCl A.R., and 25 ml cone. HCl to
1 + 19 DILUENT SOLUTION. Make 1.35 g CsCl A.R., 190 ml acid oxalate reagent and 21 ml cone. HCl to 1 litre with distilled water.
1 SUPERFLOC 1 SOLUTION, 0.4%.
PREPARATION OF STANDARDS
STOCK SOLUTION (1000 µg/ml Fe, Al, Si). Dissolve 1.000 g pure aluminium wire in 20 ml cone. HCl with the addition of a trace (one small crystal) of a mercury salt to catalyse the reaction.
Dissolve 1.000 g pure iron wire in 40 ml 1:1 HCl. The dissolution is hastened if the wire is cut up into small pieces and the solution heated on a water bath.
c
c
l

A8.3
C-. NOTE: It may be necessary to clean the iron before weighing. This can be done by immersing 1.5-2.0 g in cone. HCl until clean, washing with distilled water and drying quickly.
c
Weigh 2.139 g pure silica sand (finely ground in an agate motar and pestle and dried at 450°C) into a platinum crucible and add 12 g Na2 C0 3 A.R. Heat over a burner till molten and transfer to a muffle furnace at 1000°C for half an hour. Cool, and dissolve the melt in about 200 ml of distilled water. If the melt solution is cloudy, discard and refuse with finer ground sand.
Add Al and Fe solutions to a 1000 ml volumetric flask, add approximately 500 ml H20, then add Si solution slowly, while stirring and make to volume with distilled water. Add about 1 ml of toluene as a preservative.
WORKING STANDARDS (Fe, Al and Si). Pipette 0, l, 2, 6, 10, 15 and 20 ml stock solution (1000 µg/ml Fe, Al and Si) into 200 ml flasks, add 10 ml CsCl solution (20 OOO µg/ml), 40 ml acid oxalate reagent, 4 ml cone. HCl, and make to volume with distilled water.
EXTRACTION
SHAKING METHOD. Shake 1.0 g soil (air-dry, < 2 mm) with 100 ml acid oxalate reagent in a 250 ml centrifuge bottle on an end-over-end shaker for 4 h. (The extraction should be carried out in the dark.) Add 5 drops 0.4% superfloc and shake vigorously.
Centrifuge for 5 min at about 2000 r.p.m., filter through a No. 42 (Whatman) filter paper and collect about 50 ml. Include a reagent blank throughout the procedure.
LEACHING METHOD. Leach 190 ml acid oxalate reagent through the soil retained in leaching tube following the various leachings for cation exchange properties (Method A6.A), into a 200 ml volumetric flask.
NOTE: If cation exchange properties are not required on the samples, the same results are obtained by leaching acid oxalate reagent through the soil without any pre-treatment by the cation exchange leachings (Daly & Binnie 1974) .
Make to volume with acid oxalate reagent.
PROCEDURE
Dilute samples 1 + 4 using the 1 + 4 diluent. Using the 0-100 µg/ml standards this dilution ratio gives a range of 0-2% (Fe, Al and Si) in the soil for the leaching method, and 0-5% for the shaking method. Samples containing higher amounts should either be diluted with the 1 + 19 diluent or read with higher standards (up to 300 µg/ml).
Measure Al and Fe by AAS or FES using a lean nitrous oxide-acetylene flame and Si by AAS using a moderately rich nitrous oxide-acetylene flame.

A8.4
NOTE: A fuel-rich nitrous oxide-acetylene flame has a strong emission band r-near the Fe and Al wavelengths (370.0 nm to 390.0 nm) so that it is necessary \_ to use a lean flame to obtain a suitable signal:noise ratio for FES. However, a very lean flame is relatively cool and will cause a decrease in sensitivity. The ideal flame has a "red feather" zone 1 to 2 cm high.
CALCULATION OF RESULTS
Prepare standard curves for µg/ml (Fe, Al or Si) against % transmission or absorbance.
(1) Shaking method (1:100)
µg/ml in final solution x dilution factor (5 or 20) 100
= % (Fe, Al or Si)
i.e., for 5 x dilution (1 + 4) µg/ml
20 = % (Fe, Al or Si)
and for 20 x dilution (1 + 19)
(2) Leaching method (1:40)
µg/ml 5
= % (Fe, Al or Si)
µg/ml in final solution x dilution factor (5 or 20) 250 = % (Fe, Al or Si)
i.e.' for 5 x dilution (1 + 4) µg/ml 50 = % (Fe, Al or Si)
and for 20 x dilution (1 + 19) µg/ml 12.5 = % (Fe, Al or Si)
c
c

(_
(_
A8.5
A8.B PYROPHOSPHATE EXTRACTABLE IRON AND ALUMINIUM
INTRODUCTION
This method extracts Fe and Al complexed to organic matter. The pyrophosphate has only a slight attack on amorphous inorganic forms (McKeague 1967). Results are used in a number of soil classification systems to separate podzolised soils.
In other published methods, a choice is offered for ways of cleaning the extracts prior to measurement. Some use high speed tabout 20 OOO r.p.m.) centrifugation while others use 'superfloc' followed by low speed centrifugation (up to 2 OOO r.p.m.). It has been found that the latter method, when used on soils which have large amounts of amorphous material or finely divided secondary iron oxides, does not provide suitably clear extracts. This causes erroneously high values to be obtained by flame spectrometry. This problem has also been noted in Canada (Ballantyne et al. 1980). The procedure used at Soil Bureau combines high speed centrifugation (20 OOO r.p.m.) and the addition of 'superfloc'. The presence of 'superfloc' helps avoid the resuspension of the solid material after centrifuging.
PREPARATION OF REAGENTS
SODIUM PYROPHOSPHATE, 0.1 M. water and make to 5 litres.
1 SUPERFLOC 1 SOLUTION, 0.4%.
PREPARATION OF STANDARDS
Dissolve 223 g Na~P 20 7 .lOH20 in distilled
STOCK SOLUTION (1000 µg/ml Fe, Al and Si) (see method AS.A).
WORKING STANDARDS. Pipette 0, 2, 5, 10, 15 and 20 ml stock solution into 200 ml volumetric flasks, add 40 ml 0.1 M sodium pyrophosphate solution and make to volume with distilled water. These solutions contain O, 10, 25, SO, 75 and 100 µg/ml Fe and Al (and Si).
PROCEDURE
Shake 1 g soil (air-dry, < 2 mm) with 100 ml 0.1 M sodium pyrophosphate reagent in a 250 ml centrifuge bottle on an end-over-end shaker overnight (16 h).

Add 5 drops 0.4% superfloc and shake vigorously. r.p.m. for 30 min.
A8.6
Centrifuge at 20 OOO
Dilute cleaned extracts 1 + 4 with distilled water and read concentration of Fe and Al by high temperature FES or AAS. Include a reagent blank throughout the procedure.
CALCULATION OF RESULTS
Prepare standard curves for µg/ml (Fe or Al) against % transmission or absorbance.
µg/ml (Fe or Al) 20 = % Fe or Al
(
l

c
A8.7
A8.C DITHIONITE-CITRATE EXTRACTABLE IRON AND ALUMINIUM
INTRODUCTION
Dithionite-citrate reagent extracts 'free' (non-silicate)iron, which includes some inorganic crystalline forms, plus the fractions extracted by pyrophosphate and acid-oxalate (organic complexes and amorphous inorganic forms).
The amounts of aluminium extracted by this reagent are not easy to interpret however, and usually approximate the amount removed by acid-oxalate. In s9me podzols and volcanic ash derived soils, acid-oxalate extracts larger amounts of aluminium than does dithionite-citrate.
The method described is an adaption of that of Holmgren (1967) and involves an overnight shaking at room temperature. This is more convenient than the traditional methods in which the sample is extracted a number of
(' times, with heating.
PREPARATION OF REAGENTS
SODIUM CITRATE, 22%. Dissolve 1100 g sodium citrate (Na 3C6H50 7 .2H20) in distilled water and make to 5 litres. This solution should be made on the day required.
SODIUM DITHIONITE, Na2S20 4 , A.R.
SUPERFLOC SOLUTION, 0.4%
PREPARATION OF STANDARDS
STOCK SOLUTION (1000 µg/ml Fe, Al and Si). See method AS.A.
DITHIONITE-CITRATE SOLUTION. Dissolve 55 g sodium citrate and 5 g sodium dithionite in distilled water and make to 250 ml.
WORKING STANDARDS
Pipette 0, 2, 5, 10, 15 and 20 ml of stock solution in 200 ml volumetric flasks; add 20 ml dithionite-citrate solution and make to volume with distilled water. These solutions contain 0, 10, 25, 50, 75 and 100 µg/ml Fe and Al (and Si).

A8.8
PROCEDURE
Weigh 1 g soil (air-dry, < 0.25 nun) into a 250 ml centrifuge bottle. Add 1 g sodium dithionite and 50 ml 22% sodium citrate solution.
Shake overnight (16 h) on an end-over-end shaker. Add 50 ml distilled water and 5 drops 0.4% superloc. Shake vigorously for a few seconds. Centrifuge and filter through a No. 42 (Whatman) filter paper. Dilute 1 + 9 with distilled water and leave loosely stoppered for at least 2 days. Include a reagent blank throughout the procedure.
Read Fe and Al concentration by AAS.
NOTE: It has been found necessary to use AAS rather than FES for measuring Fe and Al levels in these extracts because a significant background interference occurs. This presumably is caused by dissolved S0 2 and the amount will vary from sample to sample and also between samples and standards. Severe interference has also been noted when determining iron by AAS. This would probably be overcome by digesting samples on a water bath, but it has been found that leaving the samples loosely stoppered for at least 2 days is a convenient and effective means of avoiding the interference.
CALCULATION OF RESULTS
Prepare standard curves for µg/ml (Fe or Al) against absorbance.
Then, µg/ml (i~ or Al) = % Fe or Al
REFERENCES
BALLANTYNE, A.K.; ANDERSON, D.W.; STONEHOUSE, H.B. 1980: Problems associated with extracting Fe and Al from Saskatchewan soils by pyrophosphate and low speed centrifugation. Canadian Journal of Soil Science 60: 141-3.
DALY, B.K.; BINNIE, H.J. 1974: A leaching method for the extraction of acid oxalate soluble aluminium and iron from soil in conjunction with cation exchange leachings. Communications in Soil Science and Plant Analysis 5: 507-14.
HOLMGREN, G.G.S. 1967: A rapid citrate-dithionite extractable iron procedure. Soil Science Society of America Proceedings 31: 210-1.
McKEAGUE, J.A. 1967: An evaluation of 0.1 M pyrophosphate and pyrophosphate-dithionite in comparison with oxalate as extractants of the accumulation products of podzols and some other soils. Canadian Journal of Soil Science 47: 95-9.
c
(~
(_

c·
(
A8.9
McKEAGUE, J.A.; DAY, J.H. 1966: Dithionite- and oxalate-extractable Fe and Al as aids in differentiating various classes of soils. Canadian Journal of Soil Science 46: 13-22.
SAUNDERS, W.M.H. 1965: Phosphate retention by New Zealand soils and its relationship to free sesquioxides, organic matter, and other soil properties. N.Z. Journal of Agricultural Research 8: 30-57.
SEARLE, P.L.; DALY, B.K. 1977: The determination of aluminium, iron, manganese and silicon in acid oxalate soil extracts by flame emission and atomic absorption spectrometry. Geoderma 19: 1-10.
TAMM, 0. 1922: Eine Methode Zur Bestimmung de anorganischen Komponente des Gelkomplexes im Boden.
0 Meddelanden fran Statens skogsforsoksanstalt Stockholm 19: 387~404.
TAMM, .0. 1932: Uber die Oxalatmethode in der chemischen Bodenanalyse.
0 Meddelanden fran Statens skogsforsoksanstalt Stockholm 27: 1-20.

(
c
(_

(
c
A9.1
A9. SOLUBLE SALTS
INTRODUCTION
For chemical analyses of soils from humid regions, determinations of amounts of soluble salts present is seldom required and is usually confined to soils from areas affected by sea water or by abnormally high amounts of cyclic salts, and to soils which have received large dressings of fertilisers or have been affected by animal excretion. Usually, the presence of salts will be suspected from knowledge of the nature of the sample, but the unexpected presence of salts is usually noted when results for total exchangeable bases by titration are compared with results for total exchangeable bases by swnmation. If significant amounts of salts are present, the result for TEBt.t will be lower than TEB because there is incomplete conversion of 1 n these cations to basicsum.oxides during ignition. Low TEBt.t values are also obtained when ammonium acetate displaces adsorbed sulpfia¥e from strongly acid soils during leaching (Blakemore 1964), and it is necessary to differentiate between the effects of soluble salts and of displaceable (adsorbed) sulphate. This is usually done by carrying out a conductivity test on a water extract, the result of which will not only confirm or disprove the presence or absence of soluble salts but will also give an approximate measure of the amount present.
Methods for extraction of soluble salts, measurement of conductivity of the extracts and determination of the anions and cations present are described in detail by Metson (1956). The only significant changes which have been made to these methods are the use of atomic absorption spectrometry (AAS) or flame emission spectrometry (FES) for the determination of concentrations of calcium, magnesium, potassium, and sodium, and the use of the method of Johnson and Nishita (1952) for the determination of the amounts of sulphate ions. However, for the sake of completeness the conductivity method, determination of cations, and determination of the common anions (chloride and sulphate) are all described here.
PREPARATION OF EXTRACT
Shake 20 g soil (air-dry, < 2 mm) with 100 ml distilled water for 30 minutes at about 20vC, and centrifuge. Alternative weights may be taken provided a 1:5 soil:water ratio is retained. For soils high in salts, centrifuging will suffice to provide an acceptable separation of extract (owing to flocculation of clay), but for less salty soils it may be necessary to filter through a retentive paper, a membrane filter, or a Pasteur-Chamberland filter candle.
Conductivity measurements should be made on the extract whatever its state of clarity, as soon as possible, because of possible changes in
(_ ionic content due to microbiological activity (Piper 1942).

A9.A MEASUREMENT OF CONDUCTIVITY
PROCEDURE
Measure the specific conductivity of the water extract in millimhos after first rinsing the cell with de-ionised water.
Note the temperature.
Correct the reading for cell constant and temperature (see table below) to obtain conductivity at 25°C (K25o).
Table A9 Temperature Factors, for Correcting Conductivity Data on Soil Extracts to 25°c
From data of Whitney and Means, as given in L.A. Richards (1947)
Temperature Correction Temperature Correction (oC) Factor, ft (oC) Factor, f
t
8 1.499 21 1.092
10 1.421 22 1.067
12 1.350 23 1.044
14 1.284 24 1.021
15 1.254 25 1. OOO
16 1.224 26 0.979
17 1.196 28 0.941
18 1.168 30 0.906
19 1.142 32 0.873
20 1.118 34 0.843
CALCULATION OF RESULTS
A9.2
The following equations give approximate values for total soluble salts in soil using 1:5 soil:water ratio:
K25o (millimho/cm) x 0.350 = Total Soluble Salts (%)
and K25o (millimho/cm) x 5 = Total Soluble Salts (me.%)
c·
(
C.

(
(
(
A9.3
A9.B DETERMINATION OF WATER-SOLUBLE CATIONS
INTRODUCTION
As with exchangeable base analysis, either AAS or FES is suitable for determination of Ca, K, and Na, but AAS is used for Mg. The standards used for exchangeable base determinations are suitable for soluble salt analysis, providing the extracts contain approximately the same amounts of CsCl, HCl, ammonium acetate (CH3COONH4) and SrC1 2 as the exchangeable base standards.
PREPARATION OF REAGENT
1 + 9 DILUENT SOLUTION. See Method A6.
PREPARATION OF STANDARDS
Although the proportions of cations present in soil/water extracts are usually different from the proportions in the standards used for exchangeable base analysis (sodium and magnesium usually predominate in soil/water extracts), it has been found convenient and sufficiently accurate to use the exchangeable base standards for determining concentrations of soluble salts.
PROCEDURE
Dilute sample solutions 1 + 9 with diluent solution. Determine Ca, Mg, Na and K concentrations as for Method A6.B.
CALCULATION OF RESULTS
Prepare standard curves of N x 10- 4 against % transmission or % absorbance.
For 1:5 soil:solution ratio and 1 + 9 dilution
= me.%

A9.C DETERMINATION OF WATER-SOLUBLE ANIONS
INTRODUCTION
The-only change in the methods for determination.of water-soluble anions described by Metson (1956) is in the method for.sulphate (described below) which is now (1977) carried out by the colorimetric method of Johnson and Nishita (1952).
SULPHATE (JOHNSON AND NISHITA 1952)
This method employs a special distillation apparatus in which a suitable aliquot of the unknown solution is taken to dryness and then heated with a strong reduction mixture (hydriodic, hypophosphorus, and formic acids) in a stream of nitrogen. The hydrogen sulphide evolved is trapped in zinc acetate and the resulting zinc sulphide is then used to form methylene blue from the "amino reagent", (NN'-dimethyl-p-phenylenediamine sulphate, Eastman Cat. No. 1333). The method is suitable for the range 0 to 100 µg S (0 to 300 µg S04 ) in a 2 ml aliquot and it
A9.4
is often necessary to dilute the soil/water extract fo.r this determination. Sulphur standards for calibration are treated similarly to the samples.
Details of the method and apparatus are given under "Phosphate extractable Sulphur", Section All.
Construct a standard curve.
For a 1:5 soil:water extract and 2 ml aliquot:
µg/ml (final solution) x 1.56 = so4 2-cme.%)
c
(

c
l
CHLORIDE (SCHALES AND SCHALES 1941)
The extract is titrated with mercuric nitrate (Hg(N0 3) 2 ) and forms the very slightly dissociated mercuric chloride (HgC1 2). The end point is recognised by the formation of a blue complex of the excess mercuric ions and the indicator, s-diphenylcarbazone.
PREPARATION OF REAGENTS
MERCURIC NITRATE SOLUTION (approximately 0.02 N). Add 20 ml 2 M HN0 3 , A.R.,to approximately 700 ml distilled water and then about 3.0 g of mercuric nitrate, A.R. Stir to dissolve, and make to 1 litre.
A9.5
DIPHENYLCARBAZONE INDICATOR, 0.1%. Dissolve 0.1 g s-diphenylcarbazone in 100 ml 95% ethyl alcohol, store in a refrigerator.
CHLORIDE STANDARD, 0.02 N. Dry NaCl, A.R., at 120°c for 4 hr, dissolve 1.169 g in distilled water, and make to 1 litre.
PROCEDURE
Standardise the mercuric nitrate solution by titrating against 10 ml 0.02 N NaCl solution using 10 drops of s-diphenylcarbazone indicator. The end point is recognised by the firs~ appearance of a faint violet-blue colour.
Transfer a suitable aliquot of the extract to a conical flask and add 10 drops of indicator. If a faint colour appears before the addition of mercuric nitrate, add a few drops of dilute HN0 3.
CALCULATION OF RESULTS
on the 1:5 soil:water extract
Hg(N03)2 (ml)x normality x 500 · aliquot taken (ml) = Cl- (me.%)

A9.6
REFERENCES
BLAKEMORE, L.C. 1964: Effect of adsorbed sulphate on the determination of total exchangeable bases in soils. N.Z. Journal of Agricultural Research 7: 435-8.
JOHNSON, C.M.; NISHITA, H. 1952: Microestimation of sulfur in plant material, soils, and irrigation waters. Analytical Chemistry 24: 736-42.
1 METSON,A.J. 1956:
Methods of chemical analysis for Soil Survey Samples. N.Z. Soil Bureau Bulletin 12: 208 p.
PIPER, C.S. 1942: "Soils and Plant Analysis". University of Adelaide, Adelaide. 368 p.
RICHARDS, L.A. (Ed.) 1947: Diagnosis and Improvement of Saline and Alkali Soi.ls. U.S. Department of Agriculture Regional Salinity Lalx>ratory, Riverside, California. 149 p.
SCHALES, O.; SCHALES, SELMA S. 1941: A Simple and Accurate Method for the Determination of Chloride in Biological Fluids. Journal of Biological Chemistry 140: 879-84.

(_
c
AlO. CALCIUM CARBONATE
INTRODUCTION
If a pH of 7.0 or more is obtained the soil should be analysed for carbonate because the presence of calcium carbonate (CaC03) must be allowed for in determinations of exchangeable cations. It occurs in soils from arid regions, soils derived from limestone, soils affected by lime-bearing ground waters and heavily limed soils.
AlO.A GASOMETRIC CARBON DIOXIDE METHOD
INTRODUCTION
The method described below involves treating a known weight of soil with 20% perchloric ac1d and measuring the C02 evolved. The procedure
Al0.1
is similar to that of Tinsley et al. (1951); as described in Metson (1956). However, the acid treatment is not carried out under vacuum and the C02 evolved is measured volumetrically instead of by titration.
PREPARATION OF REAGENT
PERCHLORIC ACID, 20% w/w. 2 parts of distilled water.
PROCEDURE
Dilute 1 part of 60% HC104 , A.R., with
Weigh an appropriate amount of finely ground soil (air-dry, < 0.25 mm) into a 125 ml conical flask. (5 g covers the range 0-2.5% CaC03 and 1 g, 0-12.5% CaC03).
Fit the special rubber stopper containing oxygen inlet, acid inlet, and gas outlet to the conical flask (Fig.Al0.1 ).
Flush the flask with oxygen to remove atmospheric C02 and then close the gas inlet.
Add enough perchloric acid through the acid inlet to cover the soil and gently shake the flask to ensure adequate mixing.
After about a minute, flush the flask with oxygen, transferring the gases (C02 + 02 ) into the gasometric analyser where the percentage carbon value is read as in method A3.A using the 1 g scale. Because of the possibility of inefficient flushing or prolonged evolution of C02 from the sample, it is advisable to repeat the flushing and gasometric determination step (about three times) until a zero reading is obtained.

Al0.2
The total of the individual readings is then corrected for temperature and pressure (see Method A3.A).
CALCULATION OF RESULTS
Total reading (corrected for temperature and pressure) x 12
lOO = x weight of
sample
CONTROLLED BY NEEDLE VALVE
20% w/w PERCHLORIC ACID RESERVOIR
~STOPCOCK --·
CaC03%
TO GASOMETRIC CARBON AN~LYSER (02 + C02)
-tt----RUBBER STOPPER
f "--r---GAS OUTLET
OXYGEN ___ --+---+
INLET ..,__--CONICAL FLASK
---+--ACID INLET
- . SAMPLE
Fig Al0.1 Apparatus for Gasometric CaC03 determination.
c
(
l_

(
(
(
AlO.B DIFFERENTIAL METHOD
INTRODUCTION
Although lack of sensitivity limits the accuracy of this method, especially when CaC0 3 contents are low (<5%), speed and simplicity makes it useful in assessing the CaC0 3 content of soils.
CaC0 3 is determined from the difference between total carbon % (Method A3.A) and the carbon content of the sample after pretreatment with acid to remove carbonate carbon as carbon dioxide (Wimberly 1969).
PREPARATION OF REAGENTS
HYDROCHLORIC ACID, A.R.,1 + 4.
IRON CHIPS. See Method A3.A.
SILICIC ACID. See Method A3.A.
. PROCEDURE
Al0.3
Weigh into art empty Leco combustion crucible the same weight of sample as used for total carbon method A3.A.
Add 1 + 4 HCl drop-wise until all visible C02 evolution ceases. with high carbonate content may require several additions.
Dry the crucible at loo0 c for 30 min.
Samples
Remove from oven, add one scoop of silicic acid (if weight of soil <1 g) followed by 2 scoops of iron chips.
Determine carbon content of the sample as for Method A3.A.
CALCULATION OF RESULTS
Total C% - Acid treated C% = carbonate carbon %
Assuming all carbonate to be CaC03, then:
Carbonate carbon x ~~O = CaC03%

Al0.4
CORRECTING EXCHANGEABLE CALCIUM FOR CALCIUM CARBONATE - CALCIUM
WHEN % CAC0 3 IS GREATER THAN 1
CaC03 at this level is not extracted quantitatively by 1 M ammonium acetate in the leaching process 'and values obtained for exchangeable Ca, TEB(titn)' ~bases and% BS are incorrect and therefore are not quoted. The words 'free lime' are inserted under these headings on the analysis card.
WHEN % CAC03 IS LESS THAN 1
Where less than 1% CaC03 occurs in New Zealand soils it is usually completely extracted by 1 M ammonium acetate during the leaching process and, for these soils, extractable calcium should be corrected using the relationship:
1% CaC03 = 20 me.% Ca
However, this relationship may not be valid for soils containing hard CaC0 3 fragments, such as shells, where complete solubility of the CaCOa .is not obtained for levels of less than 1%.
WHEN % CAC0 3 IS LESS THAN 0.1
Exchangeable Ca should not be corrected because the accuracy of the CaC03 determination at this level is inadequate for useful correction and the figures '<0.1%' are entered under 'CaC0 3' on the analysis card.
REFERENCES
METSON, A.J. 1956: Methods of chemical analysis for soil survey samples. N.Z. Soil Bureau Bulletin 12: 208 p.
TINSLEY, J.; TAYLOR, T.G.; MOORE, J.H. 1951: The determination of carbon dioxide derived from carbonates in agricultural and biological materials. Analyst, London, 76: 300-10.
WIMBERLY, J.W. 1969: A rapid method for the analysis of total and organic carbon in shale with a high-frequency combustion furnace. Analytica Chimica Acta 48: 419-23.
c

(
c
(
All.I
All. PHOSPHATE EXTRACTABLE SULPHUR
INTRODUCTION
In 1954 Ensminger showed the presence of soil sulphate which is insoluble in water but replaceable by other strongly adsorbed anions such as hydroxyl, acetate, phosphate and fluoride. This sulphate was termed "Adsorbed sulphate" and accumulations of it were found particularly in subsoils containing clays high in aluminium, iron oxides and 1:1 (kaolinitic) clay minerals. Ensminger measured this sulphate turbidimetrically as BaSO • Since then it has generally been accepted that although smaller amounts are ~sually found in topsails, measurement of such sulphate affords a good guide to available sulphur status and replacement by phosphate solutions is now widely used to measure available sulphur status.
In 1952 Johnson and Nishita developed a very sensitive distillation method for measurement of sulphate. This involves reduction of sulphate and measurement of the H S produced, by conversion to methylene blue. These authors noted that as well ~s sulphate, sulphides, sulphites and possibly some very labile forms of organic sulphur, would be included. For several reasons (e.g., high sensitivity) the Johnson and Nishita method became widely used to measure sulphur in soil extracts and a looseness of terminology crept in. Both "adsorbed sulphate" and "adsorbed sulphur" have been commonly used even though the Johnson and Nishita method is known to include some organic forms of sulphur.
Other measurement techniques available include Johnson and Nishita distillation with charcoal pre-treatment, turbidimetry with various pre-treatments including dialysis, oxidation and charcoal. Some of these techniques will measure varying amounts of extracted organic sulphur while others will measure only extracted inorganic sulphate. For this reason the following nomenclature is now used at Soil Bureau:
Phosphate-extractable sulphur: as determined by Johnson and Nishita distillation or by turbidimetry on pre-oxidised extracts.
Phosphate-extractable sulphate: as determined by Johnson and Nishita distillation of charcoal treated extracts.
Adsorbed sulphate: calculated by subtracting H20-soluble sulphate from phosphate-extractable sulphate.
The distillation apparatus used at Soil Bureau (Fig. All.l) is a modification of that described by Johnson and Nishita (1952). A bank of 6 individual distillation units forms a convenient group for simultaneous operation.
Differences in the method from that described in the previous edition (1977) include a narrower extraction ratio (1:5 instead of 1:20) and a more concentrated extraction solution (0.04 ~instead of 0.01 ~(Searle 1979).
PREPARATION OF REAGENTS
EXTRACTING SOLUTION 0.04 M Ca(H2P0 4 ) 2 • Add 44.6 g H3P0 4 , cone., A.R., (88%) to about 2.5 litres distilled water in a 5 litre flask. Slowly add with stirring a suspension containing 20 g CaC0 3 , A.R., in about a litre of

Condenser---------'•
814 joint---------'
Nitrogen carrier gas attachment ----------l
814 joint-------~
814 joint
t-tt----- Splash head
All .2
111&+----- Rubber joining tube r-----HH------Gas washer
>---100ml volumetric ...__.;:..._, flask
----------Nitrogen inlet
Electric heating mantle or gas - heated graphite block
Fig. All.l. Modified Johnson and Nishita apparatus. Single distillation unit.

(_
(
All.3
distilled water. Dilute to S litres with distilled water. Adjust to pH 4 (pH will usually be slightly lower than this) with calcium hydroxide (made by shaking Cao with distilled water and filtering).
CHARCOAL. Boil 2SO g decolourizing charcoal (e.g., Norit NK) with 2SO ml 1 + 1 HCl for 10 to lS min. Allow charcoal to settle and decant off clear liquid. Wash charcoal thoroughly in a Buchner funnel with distilled water and dry at lOS°C.
REDUCTION MIXTURE. Mix together: either SOO ml hydriodic acid, A.R., sp. gr. 1.7,
125 ml hypophosphorous acid, SO% w/w, and 200 ml formic acid, A.R., 90%.
or SOO ml hydriodic acid, A.R., sp. gr. 1.94, 171 ml hypophosphorous acid, SO% w/w, 274 ml formic acid, A.R., 90%, and 18S ml distilled water.
Heat this mixture to llS to 117°C for 10 min and bubble nitrogen through it to remove any hydrogen sulphide evolved, and to prevent bumping.
The reduction mixture may be re-used after regeneration as follows: to 200 ml used mixture, add: 20 ml hydriodic acid (sp. gr. 1.7 or 1.94)
4 ml hypophosphorous acid, SO% w/w, and 36 ml formic acid, A.R., 90%.
Heat, with nitrogen bubbling through to remove water, until the temperature reaches llS to 117°C, and then reflux (with nitrogen bubbling through) for l~ to 2 h. Johnson and Nishita (19S2) report that the mixture may be regenerated as many as four times provided excessive amounts of perchloric acid have not been introduced with the samples.
SULPHIDE-ABSORBING SOLUTION (zinc acetate-sodium acetate). Weigh out SO g zinc acetate, A.R., and 12.S g sodium acetate, A.R., hydrated, and dissolve in distilled water. Filter through a No. S31 (Whatman) filter paper and make up to 1 litre with distilled water.
COLOUR REAGENT ("amino reagent", Gustafsson 1960). This reagent is prepared by acidifying the amino compound, NN'-dimethyl-p-phenylenediamine sulphate (Eastman Cat. No. 1333) with H2 S0 4 as follows:
Dissolve 2.0 g of the amino compound in lSOO ml distilled water, add 400 ml H2 S0 4 , cone., A.R., and dilute to 2 litres with distilled water.
AMMONIUM FERRIC SULPHATE SOLUTION. To SO g ammonium ferric sulphate, A.R., ((NH4 ) 2S0 4 .Fe 2 (S0 4 ) 3 .24H20), add 10 ml H2S0 4 , cone., A.R., and 390 ml distilled water. Filter through a No. 42 (Whatman) filter paper.
NOTE: The methylene blue molecule contains an atom of S and the colour reaction depends on the formation of the dye methylene blue from the solution containing sulphide ions (equivalent in amount to the sulphate present in the original extract), amino reagent, and ferric iron. The reaction is very sensitive and entirely specific for sulphide (Gustafsson 1960). It is particularly suitable for the determination
of the trace amounts of sulphate present in many soils.

All.4
PREPARATION OF STANDARDS
STOCK SULPHATE SOLUTION (100 µg/ml S). Weigh 0.5436 g K2S0 4 A.R. (dried at 105°C) and dissolve in 0.04 M Ca (H2 P0 4 ) 2 extracting solution. Make to 1 litre with the extracting solution. Add a few drops of chloroform as preservative.
WORKING STANDARDS. Pipette 0, 2.5, 5, 10, 20, 30, 40 and 50 ml aliquots of stock solution (100 µg/ml S) into 100 ml volumetric flasks and make to volume with extracting solution. Add 2 or 3 drops of chloroform as a preservative. These solutions contain 0, 2.5, 5, 10, 20, 30, 40 and 50 µg/ml S. 2 ml aliquots will then contain 0, 5, 10, 20, 40, 60, 80 and 100 µg s.
PROCEDURE
PREPARATION OF EXTRACTS. Weigh 5 g soil (air-dried, < 2 mm) into stoppered 50 ml polypropylene centrifuge tubes and add 25 ml of extracting solution. Shake on an end-over-end shaker for 16 h (overnight) at 20°C and then centrifuge at 2000 r.p.m. for 15 min. Include reagent blanks.
PRETREATMENT OF EXTRACTS. 1. Phosphate-extractable sulphur: Transfer 2 ml aliquots of extract
to 50 ml flask (Fig. All.l). 2. Phosphate-extractable sulphate: Transfer 10 ml of extract to
25 ml tube containing 0.1 g charcoal. Stopper and shake for a few seconds, then allow to stand for 30 min. with occasional shaking. Filter through No. 42 (Whatman) filter paper and transfer 2 ml aliquot to 50 ml flask for distillation.
STANDARDS. Pipette 2 ml of each standard into 50 ml flasks and treat as for sample extracts.
DISTILLATION AND DETERMINATION. At the beginning of a set of determinations, flush out the apparatus with copious quantities of distilled water (using a vacuum to suck the water through) and suck air through the apparatus to remove the water. With a filter paper, remove any water adhering to the inner surface of the Bl4 cone. Turn on nitrogen supply. Wash carrier gas attachment and dry thoroughly with filter paper. Connect to apparatus. Add deioniseq water to the gas washer (about 1/3 full) and connect this to the apparatus. Fit a washed glass delivery tube to the rubber on the side arm of the gas washer.
Prepare collecting solution by measuring out 70 ml distilled water into a 100 ml volumetric flask and pipetting into this 10 ml zinc acetate-sodium acetate reagent. Fit the collecting flask to the distillation apparatus so that the glass delivery tube nearly reaches to the bottom. Add 6 ml reduction mixture to the distillation flask containing the sample extract, connect the flask to the apparatus. Turn on the water supply to the condensers and adjust the nitrogen flow so that the rate through each unit is about 150 ml/min.
c
c
Turn on heat source and carry out the distillation for 20 min. Disconnect ( the delivery tubes and collecting flasks from the apparatus, leaving the -delivery tube inside the flask. Stopper the collecting flask and set aside for colour development. Turn off the heat source. Disconnect the digestion flasks and retain the conents for regeneration of the reduction mixture.

(
(
All.5
Fit another delivery tube and the next rece1v1ng flask containing zinc acetate-sodium acetate reagent and distilled water. Carry out the distillation on this artd subsequent runs as for the first run. Change the water in the gas washer after 6 runs.
When all the distillations have been completed and the contents of the receiving flasks have been equilibrated with room temperature, carry out colour development and measurement as follows:
Add 10 ml colour reagent (using a rapid-delivery safety pipette, or an automatic dispensing pipette), re-stopper and shake gently. Add 2 ml ammonium ferric sulphate reagent, re-stopper flask and shake vigorously. Rinse the glass delivery tube into the flask and remove. Make up the contents of the flask to the 100 ml mark with distilled water. After at least 30 min. read absorbance at 670 nm on a suitable spectrophotometer (the colours are reported to be stable for several days if stored in the dark). The absorbance maximum is rather sharp at 670 nm (Johnson and Nishita 1952) and should be tested for the particular spectrophotometer in use.
CALCULATION OF RESULTS
Construct a graph of µg S against absorbance (670 nm) and read off unknowns as µg S.
µg S x 2.5 = S(ppm) (in soil).
REFERENCES
ENSMINGER, L.E. 1954: Some factors affecting the adsorption of sulphate by Alabama soils. Soil Science Society of America Proceedings 18: 259-64.
GUSTAFSSON, LILY 1960: Determination of ultramicroamounts of sulphate as methylene blue. Part II. The reduction. Talanta 4: 236-43.
JOHNSON, C.M.; NISHITA, H. 1952: Microestimation of sulfur in plant material, soil, and irrigation waters. Analytical Chemistry 24: 736-42.
SEARLE, P.L. 1979: Measurement of adsorbed sulphate in soils - effect of varying soilextractant ratios and methods of measurement. New Zealand Journal of Agricultural Research 22: 287-90.

c
c

Al2. ANALYSES OF PLANT LITTERS, PEATS AND COMPOST
INTRODUCTION
In the analytical work associated with soil surveys a sample should be treated as a litter when the loss on ignition exceeds 80%. Peat and compost samples are treated in a similar way to litters. The sample should be air-dried and pulverised in a Wiley mill, or similar apparatus, in order to obtain a homogeneous sample. The methods described below are basically those outlined by Metson (1956) but have been updated for modern instrumentation.
MOISTURE
(a) Moisture on sample as received. Compost samples only. Weigh out a sample of about 50 g and dry overnight in an oven at about 105°c. Cool in a desiccator and reweigh.
Al2.1
( CALCULATION OF RESULTS
(
(weight, moist sample - weight; oven dry sample) x 100 weight, moist sample
(b) Moisture factor. See Method A2.
LOSS ON IGNITION
= Moisture %
Weigh 10.00 g of air-dry material into an ignited weighed silica basin. Heat on a buns en burner until the material has c·ompletely charred, but do not allow it to burst into flame (smother flames with watch glass if necessary). Ignite at 50o0 c in a muffle furnace until stirring with a fine wire shows that all organic matter has disappeared (i. e. no glowing particles remaining). Cool in a desiccator and reweigh. Keep the ash for analysis.
CALCULATION OF RESULTS
Weight of ash = weight Qf basin + ash - weight of basin
Weight 0£ ash x ·10 = ash %
100 - (% ash x moisture factor) = loss on ignition.
Carry out determinations of insoluble ash, total phosphorus and total bases only if the loss on ignition exceeds 80%. If not, samples are treated as soils and analysed in the normal manner.

Al2.2
pH See Method Al.
CARBON See Method A3.
NITROGEN See Method A4.B.
CALCIUM CARBONATE See Method AlO.
INSOLUBLE ASH
To the ash from the loss on ignition determination add 20 ml 1 + 4 HCl, and about 20 ml distilled water. Cover with watch glass and digest for 30 min.on waterbath. Filter through No. 542 filter paper into a 250 ml volumetric flask and wash residue with hot water. Make filtrate to 250 ml with distilled water for total phosphorus and total bases determination. Place filter paper and residue in an ignited, weighed, silica crucible and char slowly over a bunsen burner. After charring, heat in muffle at 5oo0 c for 30 min, cool in desiccator and re-weigh.
CALCULATION OF RESULTS (~
Weight of residue (g) x 10 = Insoluble ash %
TOTAL PHOSPHORUS
Dilute an aliquot from the insoluble ash filtrate 1 + 4 with 0.55 M H2S04 (31 ml cone. H2S04/litre). Determine phosphorus (µg/ml in extract) by the automated method described in Method A5.B.II.
CALCULATION OF RESULTS
µg/ml P x 12.5 = Total P mg.%
TOTAL BASES (Ca, Mg, K and Na)
Dilute an aliquot of the insoluble ash filtrate 1 + 9 (using the 1 + 9 bases diluent (see Method A6).· Carry out the determination of individual bases, using the standards and procedure as outlined in Method A6.B.
CALCULATION OF RESULTS
N x 10- 4 x 2.5 =me.% (Ca, Mg, K and Na)
(_.

(
(
(
Al3. PRESENTATION AND RATING OF RESULTS
CONVERSION OF VALUES TO OVEN-DRY BASIS
As results are reported on an oven-dry basis, results of analyses of air-dry soil must be corrected using an appropriate moisture factor (see Method A2).
The moisture factor is to be applied to results of the following determinations:
Carbon and Nitrogen
Truog, 0.5 M H2SO~. Organic P, and Total P
CEC, TEB, Exchangeable bases (Ca, Mg, K, Na)
K , Mg c r
Acid oxalate-extractable. Al, Fe and Si
Soluble salts
Phosphate extractable sulphur and sulphate
Calcium carbonate
Insoluble ash
The moisture factor is not applied to:
(%)
(mg%)
(me.%)
(me.%)
(%)
(me.%) (%)
(ppm)
(%)
(%)
pH (values are not expressed on basis of a weighed amount of soil)
C/N (ratio of values that are already corrected)
% BS (calculated from corrected results)
Conductivity (property of an extract from soil)
P Retention (empirical method based on air""'..dry soil)
REPORTING OF RESULTS
or
Al3.l
Generally, results should be reported so that the uncertainty is restricted to the last figure given and should be rounded off to the recommended figure after the moisture factor has been applied. On this basis the following should be used as a guide to the number of significant decimal places reported.

Property Determined
pH c % >15
<15
N %
C/N
Truog-soluble P (mg.%) >l
<l
0.5 M H2 S0 4 -Soluble P (mg.%) >l
<l
Organic P (mg.%)
Total P (mg.%)
P retention (%)
CEC (me.%)
TEB (titn) (me.%)
L: bases (me.%)
BS (%)
Exchangeable Ca (me.%) >30
<30
Exchangeable Mg (me.%) >10
<10
Exchangeable K
Exchangeable Na
KCl extractable Al
Exchangeable Acidity
Acid Oxalate Al, Fe & Si (%) >l
<l
As for Mg
As for Mg
Pyrophosphate Al & Fe
Citrate dithionite Al &· Fe
As for acid oxalate
Phosphate extractable sulphur (ppm)
Kc (me.%) >l
<l
Mgr (me.%) >10
<10
As for acid oxalate
Water-soluble cations
Conductivity K25 oC (millimhos/cm)
Total Soluble Salts (%) (from conductivity)
Calcium Carbonate (%) <0.1
<l ..... ,
Al3.2
Recormnendation
1 decimal place
whole number
1 decimal place
2 decimal places
whole number
whole number
1 decimal place
whole number
1 decimal place
whole number
whole number
whole number
1 decimal place
1 decimal place
1 decimal place
whole number
whole number
1 decimal place
1 decimal place
2 decimal places
1 decimal place
1 decimal place
1 decimal place
2 decimal places
whole number
1 decimal place
2 decimal places
whole number
1 decimal place
(
(
as for exchangeable bases (
2 decimal places
2 decimal places
quote as <0.1%
1 decimal place
who1 e number

.....,;,,;:--...
f' ,,..--'
RATINGS FOR CHEMICAL PROPERTIES
The following ratings of chemical properties are used by Soil Bureau for New Zealand soils:
Phosphorus
RATING pH (1:2.5 soil:water) Organic C Total N C/N 0.5M (%) (%) Truog H2 S0 4 Inorg. Org. Total
(mg %) (mg %) (mg %) (Mg %) (mg %)
Very high ) >9.0 (extremely alkaline) >20 >1.0 >24 >5 >40 >50 >70 >120 ) 8.4-9.0 (strongly alkaline) ) 7.6-8.3 (moderately alkaline)
High ) 7.1-7.5 (slightly alkaline) 10-20 o. 6-1. 0 16-24 3-5 20-40 30-50 50-70 80-120 ) 6.6-7.0 (near neutral)
Medium ) 6.0-6.5 (slightly acid) 4-10 0.3-0.6 12-16 2-3 10-20 20-30 20-50 40-80 ) 5.3-5.9 (moderately acid)
Low 4.5-5.2 (strongly acid) 2-4 0.1-0.3 10-12 1-2 5-10 10-20 10-20 20-40
Very low <4.5 (extremely acid) <2 <0.1 <10 <l <5 <10 <10 <20
Cation-Exchange Properties RATING Exchangeable
CEC (me.%) TEB (me.%) BS (%) Ca (me.%) Mg (me.%) K (me.%) Na (me.%)
Very high >40 >25 80-100 >20 >7 >1.2 >2 High 25-40 15-25 60-80 10-20 3-7 o. 8-1.2 0.7-2 Medium 12-25 7-15 40-60 5-10 1-3 0.5-0.8 0.3-0.7 Low 6-12 3-7 20-40 2-5 0.5-1 0.3-0.5 0.1-0. 3 Very low <6 <3 <20 <2 <0.5 <0.3 <0.1
P retention %
90-100
-f;
60-90 00
30-60 7'() /
10-30 ,r:; 0-10
I
;i:.. ~
VI
VI

RATINGS FOR CHEMICAL PROPERTIES (cont)
Acid oxalate-extractable Phosphate- Soluble Salts RATING Al Fe Si extractable C d t. . t * s 1 h on UC 1V1 y % salt
(%) (%) (%) (~p~ ~~ (millirnhos/cm)
Very high >3.0 >2.0 >150 >2 >0.7 High 1.0-3.0 1.0-2.0 0.5 50-150 0.8 -2 0.3 -0.7 Medium 0.5-1.0 0.5-1.0 0.15-0.5 15-50 0.4 -0.8 0.15-0.3 Low 0.2-0.5 0.2-0.5 0.05-0.15 5-15 0.15-0.4 0.05-0.15 Very low <0.2 <0.2 <0.05 < 5 <0.15 <0.05
I ,r---, "
K c (me.%)
>0.5 0.35-0.5 0.20-0.35 0.10-0.20
<0.10
Mg r (me.%)
>30 15-30
7-15 3-7
< 3
~
;i:.. ~
VI
.j::.