mechanisms contributing to malignant dysrhythmias induced by
TRANSCRIPT

Mechanisms Contributing to Malignant DysrhythmiasInduced by Ischemia in the Cat
PETER B. CoRR, FRANCIS X. WITKOWSKI, and BURTON E. SOBEL, The CardiovascularDivision, Washington University School of Medicine, St. Louis, Missouri 63110
A B S T R A C T Continuously recorded bipolar elec-trograms were obtained simultaneously from epi-,endo-, and mid-myocardial regions of the ischemic andnormal zones of cat left ventricle in vivo after coronaryocclusion, analyzed by computer, and compared to re-gional cyclic AMP levels. Regional cyclic AMP con-tent was used as an index of the combined local ef-fects of: (a) efferent sympathetic nerve discharge; (b)release of myocardial catecholamines due to ischemia;and (c) circulating catecholamines. Ischemia resultedin a progressive increase in pulse width and rise timeand a decrease in rate of rise ofvoltage (dV/dt) ofthe localelectrograms from ischemic zones reaching a maximumwithin 2.4+0.3 min (mean+SE) at the time of onset of se-vere ventricular dysrhythmias, all of which returnedtoward control before the cessation of the dysrhythmia(33.5±1.5 min after coronary occlusion). Increases incyclic AMP in ischemic zones preceded corresponding in-creases in the frequency of premature ventricularcomplexes (PVCs). Propranolol inhibited the increasesin cyclic AMP and reduced the frequency of PVCs inanimals without ventricular fibrillation. In animalswith ventricular fibrillation, cyclic AMP was signifi-cantly elevated in normal and ischemic zones com-pared to animals with PVCs only. Electrical inductionof PVCs or ventricular fibrillation in ischemic andnonischemic hearts failed to increase cyclic AMP.The results suggest that the changes in regional adren-ergic stimulation of the heart may contribute to per-petuation of ventricular dysrhythmia and the genesisof ventricular fibrillation early after the onset of myo-cardial ischemia.
INTRODUCTIONIn experimental animals, dysrhythmias occurring soonafter the onset of myocardial ischemia differ markedly
A preliminary report was presented at the 1977 AnnualMeeting of the American Society for Clinical Investigation,Washington, D. C.Receivedfor publication 11 May 1977 and in revisedform 6
September 1977.
from the relatively benign dysrhythmias occurringlater (1-5). Thus, elucidation of factors contributingto dysrhythmias associated with sudden death in manmay require analysis of processes occurring duringthe interval immediately after the onset of myocardialischemia.
Several observations implicate deleterious sympa-thetic nervous system influences on these early dys-rhythmias. Among patients seen within 30 min afterthe onset of acute myocardial ischemia (6), prominentalterations in the activity of the autonomic nervoussystem occur in 92%. Complete myocardial catechola-mine depletion (i.e., after mediastinal neural abla-tion) confers striking protection against ventricularfibrillation in animals (7). However, prior treatmentwith propranolol in doses sufficient to attenuate the re-sponse to maximal stellate ganglion stimulation doesnot confer protection against ventricular fibrillation (8,9). This may be due to incomplete obliteration ofneural as opposed to humoral sympathetic input to theheart by pharmacological blockade (10) or a-receptorcontributions to the malignant dysrhythmias inducedby ischemia (11).
Sympathetic nerve recordings during myocardial is-chemia may not reflect alterations in sympatheticinput to selected regions of myocardium because ofextensive arborization ofthe nerves distal to the record-ing site as well as additional influences of circulatingcatecholamines. Measurement of local myocardial cate-cholamines may not reflect regional efferent sympa-thetic activity because of the combined loci of nor-epinephrine in peripheral nerve terminals and cardiaceffector cells. Incompleteness of pharmacologicalblockade or intramyocardial release of norepinephrinemay cloud the interpretation of results based solely onchanged global function after fl-receptor blockade.For these reasons, in the present study we assayed
regional myocardial cyclic adenosine 3',5'-monophos-phate (cyclic AMP) content as one index of regionaladrenergic activity, based on the concept that transientchanges in myocardial cyclic AMP content may reflect
The Journal of Clinical Investigation Volume 61 January 1978 -109-119 109

regional postsynaptic influences of catecholamines(12-14). Accordingly, measurement of regional cyclicAMP content may permit assessment of the combinedneural, humoral, and intramyocardial effects of cate-cholamines in selected regions of the heart. Althoughfactors other than catecholamines may contribute tochanges in cyclic AMP, including alterations in phos-phodiesterase activity (15), effects of anoxia per se(16-18), or decreased utilization and (or) washoutof cyclic AMP, most of the increase in cyclic AMPin anoxic or ischemic hearts has been shown to reflectadrenergic stimulation rather than direct effects ofanoxia (17, 18).
In the present study, we utilized an experimentalanimal preparation exhibiting consistent ventriculardysrhythmia early after coronary occlusion to deter-mine: (a) whether specific changes in local electro-grams in ischemic tissue correlate with the onset orspontaneous termination of ventricular dysrhythmia;(b) whether characteristic changes occurred in re-gional adrenergic activity, reflected by increased cyclicAMP content in ischemic and nonischemic zones be-fore, during, and after termination of ventricular dys-rhythmia; and (c) whether alterations in cyclic AMPcontent after ischemia depend on regional adrenergicinfluences based on results after f8-adrenergicblockade.
METHODS
Animal preparation. Male and female adult cats (n- 157) (2.1-3.9 kg) were anesthetized with a-chloralose(75 mg/kg) to minimize depression of cardiac reflexes by anes-thesia. After endotracheal intubation, each animal wasmechanically ventilated with a mixture of oxygen and roomair to maintain normal arterial pH, Pco2 and Po2 monitoredevery 5 min in several animals. Adjustment of tidal volumes(-20 cm3/kg) resulted in maintenance of normal pH andblood gas values. Left thoracotomy was performed by ex-cision of ribs two through four, and body temperature wasmaintained at 37.50C with the use of a thermostatic esopha-geal probe controlling an infrared lamp. The pericardium wasopen and sutured to the chest wall to form a cradle, and theleft anterior descending coronary artery was isolated at itsbifurcation from the main left trunk, proximal to all branchpoints under microscope visualization. A Gould-Brushmodel 260 recorder (Gould, Inc., Instrument Systems Div.,Cleveland, Ohio) was used to monitor multiple lead surfaceelectrocardiograms and systemic arterial pressure via afemoral artery catheter.Bipolar electrogram recordings and automated analysis.
Bipolar electrograms from epi-, myo-, and subendocardialareas of ischemic and normal zones of the left ventricle wereobtained as follows: The epicardial electrodes consisted oftwo gold disks (1.5 mm diameter and 0.004 inch thickness)soldered to fine stainless steel wires coated with polyure-thane, imbedded in a pliable silicone mat and sutured to theepicardial surface. The myocardial and subendocardial elec-trodes were made from four polyurethane-coated stainlesssteel wires which were bonded together with an additionalcoat of polyurethane. The two subendocardial leads were
turned at right angles to form a hook and the tips were cut toprovide electrical contact. The two myocardial leads werecut 3.5 mm proximal to the beveled end of the subendo-cardial electrode to provide a mid-myocardial recording. Theentire electrode bundle was checked for continuity and lackof crossover between leads with an ohmmeter and subse-quently placed in a 25-gauge needle and inserted through theleft ventricular wall immediately adjacent to the epicardialelectrode. The needle was withdrawn so that the subendo-cardial leads engaged the inner wall and the myocardialelectrode engaged the myocardium at the mid wall level.Each of the six bipolar electrode contacts (in both ischemicand normal zones) were placed in multiple silver-contactelectrode holders, amplified by a constant gain AC amplifierand variable gain DC amplifier-universal coupler (GouldInc.) with low frequency cutoff (3 dB) at 1.0 Hz and highfrequency cutoff (3 dB) at 1.0 kHz before storage on FM-magnetic tape and display on an eight-channel monitor os-cilloscope. Zero level (short-circuit of the amplifier inputlines) and calibration level (constant 1 mV) were also re-corded on FM-magnetic tape for subsequent calibration ofthe waveforms. The location of each electrode was verifiedat the termination of each experiment. Variance in elec-trode placement from experiment to experiment was mini-mized by placing the ischemic zone electrodes 3.0 mmlateral to the left anterior descending coronary arteryjust distal to the first medial branch (mid-ischemic zone)and placing the normal zone electrode on the lateral leftventricular wall adjacent to the first major branch of theleft circumflex coronary artery.Automated analysis of the bipolar electrograms was per-
formed in real time with a PDP-12 computer system andinteractive controlling software written in LINC/PDP-8Assembly language. The system digitizes the analog signalat a 10-kHz sampling rate. When a signal equal to or in excessof threshold is identified, the program retains the 200samples before the time when the threshold requirement isfulfilled along with the 300 subsequent samples providing atotal of 500 data points (50 ms) per pulse. The followingparameters are then obtained from each captured bipolarelectrogram: (a) maximum (MAX = 100%) and minimum(MIN = 0%)-the highest absolute voltage detected afterthreshold and the lowest detected within the 20-ms preced-ing threshold; (b) the difference (millivolt) between MAXand MIN; (c) the rise time (the interval (millisecond) duringwhich voltage increases from 25 to 75% of MAX-MIN);(d) the pulse width at 50% amplitude (millisecond); and(e) the rate of rise of voltage (millivolt/millisecond) from25 to 75% of MAX-MIN/(dV/dt).
Acquisition of data occurs during each systole and analysisduring diastole. Thus, as soon as each electrogram has beenanalyzed, acquisition is reinstituted automatically. The systemcalculates the mean and SD for each derived parameter anddisplays all parameters graphically as a function of timeutilizing a Houston Instruments (Div. Bausch & Lomb, Inc.,Austin, Tex.) DP1-5H incremental plotter.Biochemical procedures. Cyclic AMP content in normal
and central ischemic zones of the left ventricle was assessedat selected intervals after coronary occlusion in fast-frozenbiopsies (-200 mg) obtained with a suction-drill through theleft ventricular wall from normal and ischemic zones. Onepair of samples was obtained from each cat after which theexperiment was terminated. Biopsy samples, stored at -80°C(REVCO, Inc., West Columbia, S.C.) were pulverized in asteel mortar frozen in liquid nitrogen, quickly transferredto a cold homogenizer cup containing 0.75 ml of 6% tri-chloroacetic acid, thoroughly homogenized (Omni Mini-
110 P. B. Corr, F. X. Witkowski, and B. E. Sobel

SURFACE ECGLEAD I[
EPICARDIAL EPICARDIALISCHEMIC ZONE NORMAL ZONE
CONTROLBefore Occlusion AA.AJ.A.
2.2 Min afterOcclusion at Onset
of Dysrhythmia ALWYL
34.8 Min afterOcclusion at Termination
of Dysrhythmia
10 20 30 40 50 10- 20 30 40 50
time in msFIGURE 1 Computer-generated digital reconstruction of typical epicardial wave forms obtainedfrom the ischemic (center panels) and normal zones (right panels) ofthe left ventricle both beforeand 2.2 min after occlusion and immediately before the termination of the dysrhythmia (34.8 minafter occlusion). The surface ECG shown depicts the rhythm at the moment when the wave
forms were obtained. PW = pulse width, RT = rise time, and dV/dt = rate of change of voltagewith time. The wave forms shown are digital reproductions obtained with a high-speed plotterwith each pulse shown as 500 data points (50 ms at 10 KHz sampling rate).
Homogenizer, Sorvall, Ivan, Inc., Norwalk, Conn.) for 1 min,and transferred to a centrifuge tube with 0.25 ml trichloro-acetic acid used as a rinse. Extracts were assayed for pro-tein (19) and cyclic AMP by radioimmunoassay in triplicate(20) with succinyl cyclic AMP tyrosine methyl ester (1251)and cyclic AMP antiserum obtained commercially (Schwarz-Mann Div., Becton, Dickinson Co., Orangeburg, N. Y.).Results in recovery experiments were between 96 and 102%of expected, and all assays were verified with internalstandards.Physiological and pharmacological procedures. Because
both the incidence of spontaneous ventricular fibrillation andpremature ventricular complexes occurred simultaneous withthe changes in cyclic AMP (see Results), the influence oncyclic AMP of premature beats or ventricular fibrillationinduced experimentally was assessed. A bipolar epicardialgold disk electrode was sutured to the right ventricularsurface to induce premature beats or ventricular fibrillationwithout interfering with actcess to left ventricular sites forsubsequent biopsy and so that fibrillation could be inducedwith lower current intensities than those required to induceit from left ventricular sites (21). The stimulus (5 ms dura-
Malignant Dysrhythmias and Ischemia 111
PW 2.4 msRT : 0.8 msdV/dt- 9.9mV/ms
01 20 30 40 50
PW: 2.1 msRT : 0.9 msdV/dt - 15.6 mV/ms
I
D 10 20 30 40 5
time in ms
-I N4sf0PW : 8.8 msRT : 73 msdV/dtz 0.8 mV/ms
D 10 20 30 40 5
r
PW a 2.3 msRT : 0.9 msdV/dt a16.4 mV/ms
-A0 10 20 30 40 50
time in ms
PW 5.7msRT : 4.8 msdV/dt 2.3 mV/ms
PW: 2.2 msRT: 0.8 msdV/dt - 16.4mV/ms
I1

tion) obtained from an Anapulse stimulator (W-P Instruments,Inc., New Haven, Conn.) was triggered by the R wave of thesurface ECG (Gould amplifier, Gould Inc.). The output of theAnapulse stimulator was connected to a photo-opticallycoupled, battery powered, current generator which was de-signed to supply a constant current (programmable from 1to 99 mA) at a voltage up to 150 V, in 1 mA incrementsused to prevent ground-loop currents from affecting the cur-rent input at the electrode. The current generator was de-signed so that the current and voltage output delivered tothe electrode could be monitored simultaneously through abattery-powered Tektronix oscilloscope (Tektronix, Inc.,Beaverton, Oreg.). To induce premature beats, a 1-2-mApulse was used for capture, and to induce ventricularfibrillation, a single pulse (5 ms) was delivered duringthe upstroke of the T wave of the surface electro-cardiogram. Initial stimuli were 30 mA with increasesin 5-mA steps until ventricular fibrillation ensued. The amountof current required to induce ventricular fibrillation with asingle pulse is greater than that required when trains ofpulsesare utilized (21). However, trains of pulses may cause releaseof norepinephrine from the heart (22), and might thereforeinfluence cyclic nucleotide levels appreciably more.To ascertain whether the changes in cyclic AMP could be
attenuated by pharmacological 8-receptor blockade, d,l-propranolol (0.75 mg/kg) was administered intravenously,over 5 min, 15 min before coronary occlusion. This dose isthe minimum required to entirely antagonize the tachycardiainduced by right stellate ganglion stimulation (bipolarplatinum electrodes, Grass stimulator Grass Instruments Co.,Quincy, Mass.). Similar dosages of d,l-propranolol have alsobeen shown to be required to insure ,8-receptor blockade ofcirculating catecholamines (23).Because anoxia increases both myocardial cyclic AMP
(16-18) and adenosine release, two types of experimentswere performed to determine whether adenosine, a potentstimulant of cyclic AMP accumulation in brain tissue (24),would stimulate cyclic AMP accumulation in the mammalianheart. In the first set of experiments, adenosine (0.1 mM) wasinfused into the left anterior descending coronary artery viaa 29-gauge needle. 90 s later, samples were taken from myo-cardium perfused with adenosine and from the lateral myo-cardium perfused by the circumflex coronary arterial blood(adenosine-free) and assayed for cyclic AMP. The high con-centration of adenosine was chosen because adenosine israpidly converted to inosine largely by adenosine deaminasein crythrocytes (25). Despite this phenomenon, the dose wassufficient to slow the ventricular rate in this preparation.In other experiments, isolated rabbit hearts perfused retro-grade with Krebs-Henseleit bicarbonate at 37°C equilibratedwith 95% 02, 5% CG2 at 20 ml/min were studied sothat adenosine could be administered at more physiologicallevels (10 nM) in a perfusion medium without erythrocytes,before assay of cyclic AMP in fast-frozen samples.
RESULTS
The feline preparation utilized in the present experi-ments (26, 27) exhibits: (a) ventricular dysrhythmiawith a consistent onset time (2.4+±0.3 min), duration(33.5+1.5 min), and associated overall mortality (20%)due to ventricular fibrillation; (b) consistent frequencyof premature ventricular complexes within the 1st hafter occlusion (1,205±97); (c) a coronary arterial dis-tribution very similar to that ofman (27); (d) consistentalterations in heart rate, systemic arterial pressure,cardiac output, and total peripheral resistance; and (e)
c200 200 4/X 40
U~~~~~ ~o
P <t005&e<+0 P<0.05
400 Myo ,.400 Myo J80 Myo
200 p200 * o40
P< 0.05
400 Endo 400 Endo 80 Eno
200 - 200 _ __A4 40
t + _.
- C cC-o o
u _ u 0EO
FIGURE 2 The combined data from 11 animals subjected tocoronary occlusion. The figure illustrates changes in pulsewidth, rise time, and dV/dt at three times during the controlperiod before occlusion, the interval 1 min after occlusion,and immediately before the onset of the dysrhythmia, andthe interval immediately before the termination of the dys-rhythmia in epicardial (Epi), myocardial (Myo), and endo-cardial (Endo) layers of the ischemic zone. Values shownare percentage changes from average values in the controlperiod (100%) and are expressed as means±SD.
enhanced autonomic cardiac neural activity in theperiod immediately after coronary occlusion.Electrophysiological alterations. Striking altera-
tions were observed in electrograms from the ischemicarea within minutes after occlusion (Fig. 1). In theabsence of ischemia, electrograms remained stablewith no significant alterations in width, rise time,velocity of upstroke or amplitude for 7 h. Coronaryocclusion resulted in a progressive decline in thevelocity of upstroke with a corresponding increase inthe rise time and pulse width in electrograms fromischemic sites only. The maximal changes in electro-gram parameters in recordings from the ischemic zonecoincided with the onset of ventricular dysrhythmia.Before termination of the dysrhythmia, partial but sig-nificant regression of the abnormalities was evidentin all three parameters (Fig. 1).The combined data from 11 such experiments in
which animals recovered from the ventricular dys-rhythmia are shown in Fig. 2. All values were stand-ardized on the basis of 100% representing the initialcontrol value. Coronary occlusion resulted in consis-tent and progressive increases in pulse width and risetime and a decrease in velocity of upstroke in the is-chemic zones, with peak changes in each parameteroccurring immediately before the onset ofthe ventricu-lar dysrhythmia (Fig. 2). Myocardial and epicardialregions consistently demonstrated partial regression ofabnormalities in all three electrogram parametersbefore the termination of the dysrhythmia. In contrast,
112 P. B. Corr, F. X. Witkowski, and B. E. Sobel

endocardial regions of the ischemic zone exhibitedcontinued depression of these parameters at termina-tion of the dysrhythmia. Normal zone recordings failedto demonstrate any significant alterations in parametersafter ischemia and during normal sinus rhythm.A comparison of the changes in pulse width, rise
time, and dV/dt in the three levels of the ischemiczone at the onset of the dysrhythmia showed that thealterations in pulse width were greatest in epicardialareas and least prominent in endocardial regions (Fig.2). Likewise, rise time was increased to a greater ex-tent in epicardial compared to changes in myo- orendocardial areas. However, the velocity of upstrokewas depressed comparably in all three regions of theventricular wall at the time of onset ofthe dysrhythmia.Regional changes in cyclic AMP content. In initial
studies, simultaneous biopsies for assay of cyclic AMPwere obtained from normal and central ischemic zonesof the left ventricle from six cats at each selected timeinterval after coronary occlusion. Each experiment was"terminated immediately after" the biopsy was ob-tained. Coronary occlusion resulted in a significantincrease in cyclic AMP in the ischemic zone 5 minlater and cyclic AMP levels subsequently rose to peakvalue at 15 min, returning to preocclusion levels by30 min (Fig. 3). However, increases in cyclic AMPcontent in the normal zone were striking only duringthe 15-20 min after occlusion. Cyclic AMP in thecentral ischemic zones increased significantly com-pared to values in the normal zone 5, 15, 20, 25, 30, and35 min after occlusion (Fig. 3), suggesting a regionaldifference in localized adrenergic activity or possiblymarkedly decreased phosphodiesterase activity. Thedistribution of premature ventricular complexes(PVCs),' (n = 30 different animals) at 5-min intervalsafter occlusion exhibits a progressive increase in PVCfrequency which parallels closely the rise in cyclicAMP in the ischemic zone based on observations madein biopsies from other cats. It should be noted thatthe time of occurrence of peak level of cyclic AMPafter coronary occlusion in the ischemic and normalzone precedes the time of occurrence of the peak PVCfrequency (Fig. 3) suggesting not only that the twophenomena were temporally associated, but also thatincreased regional adrenergic activity reflected by in-creased cyclic AMP might have contributed to sus-taining the dysrhythmia. However, this does not implythat increased regional adrenergic activity is necessaryfor initiation of the dysrhythmia. Indeed, when biop-sies were obtained from seven other animals at the timeof onset of the first abnormal beat, levels of cyclicAMP were not increased (6.1+0.35 and 6.4+0.30pmol/mg protein in ischemic and normal zones, respec-
I Abbreviations used in this paper: PVCs, premature ven-tricular complexes; VF, ventricular fibrillation.
., 12.C0cL
E
0E 8.0
6.0
0- <4.0u
U 2.0u
< 500ccui
400I
u 300uJ
O 200z
o 100
0-
---Ischemic ZoneNormal Zone
0 10 20 30 40
TIME AFTER CORONARYOCCLUSION
50
FIGURE 3 Changes in cyclic AMP (upper panel, n = 6 catsat each point) in the ischemic and normal zones of theleft ventricle at selected time intervals before and aftercoronary occlusion. The shaded areas indicate a significantdifference (P < 0.05) between values from the ischemic andnormal zones. The (*) indicates a significant difference fromthe control value. The frequency distribution of PVCs oc-curring during the same time intervals is illustrated in thelower panel (n = 30 cats). The vertical bars represent SE.
tively) suggesting that enhanced regional adrenergicactivity and (or) accumulation of cyclic AMP werenot necessary conditions for initiation of the dys-rhythmia.Effects of induced PVCs and induced ventricular
fibrillation on regional cyclic AMP. Premature beatsat selected coupling intervals were induced electricallyin six nonischemic hearts for 15 min and the myo-cardium was subsequently sampled and assayed forcyclic AMP (Table I). Cyclic AMP levels did not in-crease even with a frequency of induced PVCs (552+53) in a 15-min interval comparable to the spon-taneous frequency after occlusion (665±63). Likewise,electrical induction of PVCs for 15 min at a rate of742+98 in six animals, beginning 50 min aftercoronary occlusion when the spontaneous increase incyclic AMP had abated, did not increase the cyclicAMP in normal or ischemic myocardium (Table I).The data illustrated in Fig. 3 were obtained from 66
animals. An additional 13 animals succumbed to ven-tricular fibrillation within 2.5 min after coronary
Malignant Dysrhythmias and Ischemia 113
I
I
I -
I,-ir-,

TABLE IInfluence of Induction of PVCs on Cyclic AMP in
Ischemic and Nonischemic Hearts
Cyclic AMP
Anterior zone Lateral zone
pmol/mg protein
Before occlusion 6.4+0.5 6.0±0.2(6) (6)
After induced PVCs in 5.7±0.3 5.7±0.2nonischemic hearts (6) (6)
After induced PVCs in hearts 5.1+0.4 5.4±0.4ischemic for 50 min (6) (6)
Values are means±SE.Numbers in parentheses indicate numbers of animals in eachgroup.In the ischemic hearts, the zone of ischemia was anterior ineach case.
occlusion and biopsies were obtained immediatelythereafter (5-10 s). Results indicate that in thoseanimals developing ventricular fibrillation (VF), cyclicAMP in both the central ischemic and normal zoneswas significantly (P < 0.01) elevated compared tochanges in the corresponding zones at the same timeafter coronary occlusion (i.e. 2.0-min values in Fig. 3) inanimals exhibiting PVCs but not VF (Fig. 4). In thoseanimals developing VF, cyclic AMP in the ischemiczone was significantly elevated compared to values innormal zones in the same animals (Fig. 4). Althoughthese results suggest that VF induced by myocardial
2c0
QJ0)E0E
UL
UlU:
Ischemic Zone
O Normal
ischemia is associated with increased regional adrener-gic activity, VF per se might itself cause cyclic AMPto increase. However, when fibrillation was inducedwith a single pulse to the right ventricle, biopsies ob-tained within 5-10 s from normal (n = 5) and heartssubjected to coronary occlusion (n = 5) failed to exhibitincreased cyclic AMP levels in either ischemic or nor-mal zones (Table II). This lack of increase within thebrief interval indicates that accumulation secondary tosympathetic reflex discharge requires a long intervalafter the onset of fibrillation. In addition, despite themarked differences in cyclic AMP in spontaneouslyfibrillating hearts after coronary occlusion, hemo-dynamic changes in animals with and without spon-taneous fibrillation were comparable at the time cor-responding to the interval before the rhythm disturb-ances (Table III, top two rows).Effect ofpropranolol on cyclic AMP in ischemic and
normal zones. To determine whether changes in ef-ferent sympathetic activity accounted for the increasesin cyclic AMP observed, we administered propranolol(0.75 mg/kg i.v.) 15 min before ischemia in eight addi-tional animals, a dose found previously to entirelyblock the increase in heart rate induced by isopro-terenol injection (0.5 ,ug/kg) or right stellate ganglionstimulation in the cat (9, 23). Because the maximalincrease in regional cyclic AMP occurred 15 min aftercoronary occlusion (Fig. 3), biopsies were obtained atthis interval after occlusion in propranolol-treated ani-mals. In the five of eight treated animals who survivedfor 15 min, significantly fewer PVCs occurred in thefirst 15 min after occlusion compared to control ani-mals. The reduction in PVC frequency may be due in
TABLE IIThe Influence of Induction of VF on Cyclic AMP
in Ischemic and Nonischemic Hearts
Cyclic AMP
P<0.01
T
6
No VF
FIGURE 4 Cyclic AMP levels in ischemic and normal zones ofthe left ventricle in animals with and without spontaneous VF.The vertical bars represent the SEM and the numbers withinthe histograms indicate the numbers of animals in each group.
Anterior zone Lateral zone
pmol/mg protein
At the time of spontaneous VF 10.7+0.8 8.4+0.6(13) (13)
After induced VF in 4.8+0.6* 4.9+0.4*nonischemic hearts (5) (5)
After induced VF in hearts 5.5+0.7* 6.1+0.2*ischemic for 50 min (5) (5)
Values are means+SE.Numbers in parentheses indicate the number of animals ineach group.In the ischemic hearts, the zone of ischemia was anteriorin each case.* P < 0.01 compared to results in animals with spon-taneous VF.
114 P. B. Corr, F. X. Witkowski, and B. E. Sobel

TABLE IIIHemodynamic Alterations
Before coronary occlusion 1.5 Min after coronary occlusionNumberof cats Heart rate Arterial pressure Heart rate Arterial pressure
beatslmin mm Hg beats/min mm Hg
Control* 66 198+7.9 105+8.2 173+5.51 88+9.1tVF§ 13 204+9.2 111±4.1 181±6.1t 92±10.2tPropranolol" 5 150±4.1 96±10.2 128±7.5t 78±10.1tPropranolol and VF§ 3 141±1.2 103±7.1 122±7.6t 84±11.2t
Numbers are mean±SE.Propranolol refers to animals treated with this drug as described in the text.* Animals with coronary occlusion who were not treated with propranolol.t P < 0.05 with paired comparisons between the pre- and postocclusion period.§ Animals with hearts biopsied at the moment of spontaneously occurring VF.11 Animals with hearts biopsied 15 min after coronary occlusion.
part to the corresponding decrease in sinus rate withpropranolol treatment. In addition, myocardial cyclicAMP content at this time was significantly reduced(Fig. 5). The control values were obtained 15 min afterocclusion (Fig. 3) and compared to values in biopsiesobtained at corresponding intervals after occlusion inanimals treated with propranolol. Nevertheless, threeanimals treated with propranolol did develop spon-taneous VF within 3 min after coronary occlusion. All
0 Ischemic Zone
EJ Normal Zone
12.0 -
*C- 10.0 .
a-
$ .Ec 8.0
E..a 6.0
< 4.0U.U) 2.0u
665 + 63 PVCsin first 15 min
(n' 30)
Control15 min
348±129 PVCsin first 1S min
(n=5)
Propranolol15 min
P<0.05
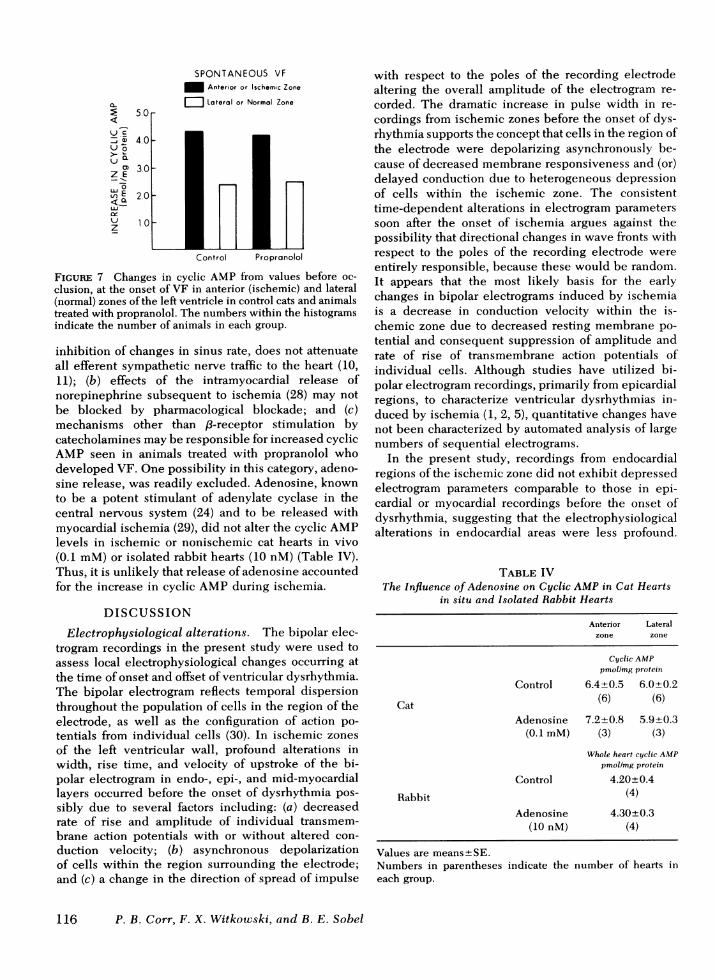
three exhibited a significant increase in cyclic AMPwithin ischemic zones, compared to values before oc-clusion in animals treated with propranolol (Fig. 6).Although the absolute levels of cyclic AMP were re-duced by propranolol, the increase from control valuesin animals who developed VF was not precluded by/3-receptor blockade (Fig. 7). Furthermore, althoughpropranolol significantly reduced the sinus rate be-fore coronary occlusion, the alterations in systemicarterial pressure and heart rate induced by ischemiawere similar in treated animals with and without VF(Table III).At least three possibilities might account for the
association of ventricular fibrillation with increasedcyclic AMP in animals treated with propranolol: (a)adequate pharmacological /-blockade, as assessed by
PREOCC LUS ION SPONTANEOUS VF
j20
E0Eca
CLi
uu
FIGURE 5 CyclicAMP levels in ischemic and normal zones ofthe left ventricle 15 min after coronary occlusion in controlanimals (left) and animals treated with propranolol (right) whosurvived for at least 15 min. The numbers of PVCs in thecontrol (top) and treated animals (bottom) are indicated. The(*) indicates a significant decrease in the cyclic AMP levels inpropranolol-treated compared to control animals (P < 0.01).Vertical bars represent SEM and the numbers within thehistograms indicate the number of animals in each group.
FIGURE 6 Cyclic AMP levels in anterior (ischemic zones) orlateral (normal zones) ofthe left ventricular wall in the intervalbefore occlusion (left side) and at the time of onset ofspontaneous VF (VF-right side) in control animals and catstreated with propranolol. Vertical bars represent SEM and thenumbers within the histograms indicate the numberofanimalsin each group.
Malignant Dysrhythmias and Ischemia 115
SPONTANEOUS VFAnterior or Ischemic Zone
E Lateral or Normal Zone5.0
_ 40
30-<E 220~
W 10
z
Control Propronolol
FIGURE 7 Changes in cyclic AMP from values before oc-clusion, at the onset of VF in anterior (ischemic) and lateral(normal) zones of the left ventricle in control cats and animalstreated with propranolol. The numbers within the histogramsindicate the number of animals in each group.
inhibition of changes in sinus rate, does not attenuateall efferent sympathetic nerve traffic to the heart (10,11); (b) effects of the intramyocardial release ofnorepinephrine subsequent to ischemia (28) may notbe blocked by pharmacological blockade; and (c)mechanisms other than 8-receptor stimulation bycatecholamines may be responsible for increased cyclicAMP seen in animals treated with propranolol whodeveloped VF. One possibility in this category, adeno-sine release, was readily excluded. Adenosine, knownto be a potent stimulant of adenylate cyclase in thecentral nervous system (24) and to be released withmyocardial ischemia (29), did not alter the cyclic AMPlevels in ischemic or nonischemic cat hearts in vivo(0.1 mM) or isolated rabbit hearts (10 nM) (Table IV).Thus, it is unlikely that release of adenosine accountedfor the increase in cyclic AMP during ischemia.
DISCUSSION
Electrophysiological alterations. The bipolar elec-trogram recordings in the present study were used toassess local electrophysiological changes occurring atthe time of onset and offset of ventricular dysrhythmia.The bipolar electrogram reflects temporal dispersionthroughout the population of cells in the region of theelectrode, as well as the configuration of action po-tentials from individual cells (30). In ischemic zonesof the left ventricular wall, profound alterations inwidth, rise time, and velocity of upstroke of the bi-polar electrogram in endo-, epi-, and mid-myocardiallayers occurred before the onset of dysrhythmia pos-sibly due to several factors including: (a) decreasedrate of rise and amplitude of individual transmem-brane action potentials with or without altered con-duction velocity; (b) asynchronous depolarizationof cells within the region surrounding the electrode;and (c) a change in the direction of spread of impulse
with respect to the poles of the recording electrodealtering the overall amplitude of the electrogram re-corded. The dramatic increase in pulse width in re-cordings from ischemic zones before the onset of dys-rhythmia supports the concept that cells in the region ofthe electrode were depolarizing asynchronously be-cause of decreased membrane responsiveness and (or)delayed conduction due to heterogeneous depressionof cells within the ischemic zone. The consistenttime-dependent alterations in electrogram parameterssoon after the onset of ischemia argues against thepossibility that directional changes in wave fronts withrespect to the poles of the recording electrode wereentirely responsible, because these would be random.It appears that the most likely basis for the earlychanges in bipolar electrograms induced by ischemiais a decrease in conduction velocity within the is-chemic zone due to decreased resting membrane po-tential and consequent suppression of amplitude andrate of rise of transmembrane action potentials ofindividual cells. Although studies have utilized bi-polar electrogram recordings, primarily from epicardialregions, to characterize ventricular dysrhythmias in-duced by ischemia (1, 2, 5), quantitative changes havenot been characterized by automated analysis of largenumbers of sequential electrograms.
In the present study, recordings from endocardialregions of the ischemic zone did not exhibit depressedelectrogram parameters comparable to those in epi-cardial or myocardial recordings before the onset ofdysrhythmia, suggesting that the electrophysiologicalalterations in endocardial areas were less profound.
TABLE IVThe Influence of Adenosine on Cyclic AMP in Cat Hearts
in situ and Isolated Rabbit Hearts
Anterior Lateralzone zone
Cyclic AMPpmollmg protein
Control 6.4+0.5 6.0+0.2
Cat (6) (6)Adenosine 7.2±+0.8 5.9±+ 0.3
(0.1 mM) (3) (3)
Whole heart cyclic AMPpmollmg protein
Control 4.20+0.4
Rabbit (4)Adenosine 4.30+0.3
(10 nM) (4)
Values are means+SE.Numbers in parentheseseach group.
indicate the number of hearts in
116 P. B. Corr, F. X. Witkowski, and B. E. Sobel

Oxygenation, substrate supply, or washout of noxiousmetabolites in intraventricular cavitary blood may re-duce the coronary perfusion requirements of Purkinjefibers in endocardial regions and may contribute totheir protection (31).Delay in activation within the ischemic zone has
been correlated with the occurrence of ventriculardysrhythmia (1, 2, 5). The present study was designedto characterize the nature of depolarization of cells inthe ischemic zone by analyzing quantitatively the ini-tial deflections in electrograms recorded at the time ofoccurrence and cessation of dysrhythmia rather thanfocusing on the temporal pattern of activation. At thetime of onset of dysrhythmia, the abnormalities de-tected in regional electrograms reached a maximum,and this, coupled with their regression before cessa-tion of dysrhythmia suggests that the regional elec-trophysiological changes detected contributed to thedysrhythmia.Regional adrenergic activity. In the present study,
regional alterations in cyclic AMP content wereutilized as a potential index of local adrenergicinput. Catecholamine stimulation of the heart is ac-companied by increases in cyclic AMP (12-14).Furthermore, administration of dibutyryl cyclic AMP(32, 33) and ionotophoretic injection of cyclic AMPintracellularly (34) produce electrical alterations in iso-lated cardiac tissue similar to those evoked by cate-cholamines. Nevertheless, factors such as local ac-cumulation due to decreased washout or phos-phodiesterase inhibition may directly or indirectlyincrease cyclic AMP levels. Decreased washout seemsunlikely to account for the increase because the half-life of cyclic AMP in tissues is so short and becausepersistent flow (10-20% of normal) is typical in zonesof infarction. In the present study, we observed com-plete attenuation by ,8-receptor blockade ofthe large in-crease in cyclic AMP 15 min after coronary occlusion.Furthermore, the dose of propranolol utilized wassufficient for f8-adrenergic blockade assessed byattenuation ofthe tachycardia induced by isoproterenolor right stellate ganglion but insufficient to exertquinidine-like membrane effects (35). Thus, it appearslikely that the changes in cyclic AMP did reflectcatecholamine activation of cardiac ,8-receptors. Otherpossibilities such as inhibition of phosphodiesteraseappear to be less likely because catecholamines in-hibit phosphodiesterase only at very high levels (15)and because propranolol blocks the increase of cyclicAMP so effectively.
Results of this study implicate an increase in re-gional adrenergic activity in the perpetuation but notthe initiation of dysrhythmia after coronary occlusionbased on: (a) the progressive increase of cyclicAMP in the ischemic zone preceding the progressiveincrease in frequency of PVCs; (b) the failure of in-
duced PVCs to alter regional cyclic AMP; and (c)effective attenuation by propranolol ofthe rise in cyclicAMP 15 min after occlusion with simultaneous depres-sion of PVCs. This interpretation is compatible withfindings in pharmacological studies with propranolol(9) with results in dogs exhibiting diminished dys-rhythmia induced by ischemia after ganglionectomy(36).
In the present study, cyclic AMP was consistentlyelevated in the ischemic compared to the correspond-ing normal zone. Prior treatment with propranololabolished not only the rise in cyclic AMP 15 minafter occlusion but also the disparity between cyclicAMP levels in ischemic and normal zones. This dis-parity may be important because heterogeneous,adrenergic neural stimulation of the heart is morearrhythmogenic than uniform, humoral stimulation(37). The heterogeneity of cyclic AMP levels in thepresent study may reflect many factors including: (a)local intramyocardial, non-neurally mediated re-lease of norepinephrine confined to ischemic regions;or (b) persistence of catecholamines in ischemic zonesbecause ofimpaired metabolism, reuptake, or washout.The first possibility is supported by findings in
isolated perfused hearts devoid ofefferent sympatheticinput, demonstrating that anoxia results in rapid in-creases in cyclic AMP (13, 16-18) that can be in-hibited by ,-receptor blockade (13, 17). Increases inextracellular potassium secondary to ischemia (38) mayrelease intramyocardial catecholamines (39). The re-leased norepinephrine may originate from peripheralsympathetic nerve endings since administration oftyramine 30 min before anoxia precludes the increasein catecholamines seen with anoxia (40). The secondpossibility, regarding washout, would not account forthe decreased cyclic AMP seen relatively late afterischemia, but no direct evidence is yet available tosupport or refute it.Among the animals in whom ventricular fibrillation
developed after coronary occlusion, cyclic AMP inthe ischemic and normal zones was significantly ele-vated compared to corresponding values in hearts withonly premature ventricular beats. Because electricalinduction of ventricular fibrillation failed to alter thecyclic AMP levels in either normal or ischemic hearts,it appears likely that the increased regional efferentsympathetic activity elicited the increased cyclicAMP and contributed to, rather than resulted from, VF.No demonstrable differences were noted in hemo-dynamics after coronary occlusion in animals whodeveloped VF compared to those who did not.
,8-Receptor blockade with propranolol did not attenu-ate the incidence of VF, the magnitude of the increasein cyclic AMP in normal or ischemic zones of the leftventricle (despite the lower initial values), or the dis-parity in cyclic AMP levels between the two zones in
Malignant Dysrhythmias and Ischemia 117

those hearts that did fibrillate. In animals with fibrilla-tion despite treatment with propranolol, the increase incyclic AMP was large, even though the baseline valuewas depressed and the peak absolute value was alsodepressed (Fig. 6). These observations suggest that fac-tors other than adrenergic stimulation could be re-sponsible for the increases in cyclic AMP in these par-ticular hearts, or that ,8-receptor blockade does notattenuate all efferent sympathetic nerve traffic to theheart (10). Ephaptic transmission, independent ofchemical neurotransmitters, may be one contributingfactor. Furthermore, because VF in animals not treatedwas preceded by large increases in cyclic AMP (Fig.4), it seems probable that in propranolol-treated ani-mals developing VF, complete blockade would notoccur due to the competitive nature of the antagonist.
Studies have demonstrated that clamping or transec-tion of the aorta in dogs (41, 42) or in rats (18, 43)increases cyclic AMP, an effect abolished by ,B-receptor blockade (18, 41-43); and anoxia increases ratheart cyclic AMP (16-18) also blocked by 1-receptorblockade (17, 18). However, these changes have notbeen related previously to electrophysiological eventssubsequent to ischemia. A recent editorial (44) hasimplicated cyclic AMP itself in the genesis of VF. Thepresent study presents direct evidence supporting thehypothesis that cyclic AMP or the processes re-sponsible for its accumulation such as regionally en-hanced adrenergic activity in ischemic zones contrib-ute to the maintenance of ventricular dysrhythmiaafter myocardial ischemia and to the occurrence of VF.
ACKNOWLEDGMENTS
The authors wish to thank Mr. Gerard Clarke and Ms.Elaine Carlson for their expert technical assistance and Ms.Carolyn Lohman for preparation of the manuscript.This work was supported in part by National Institutes of
Health grant HL 17646, Specialized Center of Research inIschemic Heart Disease; and Missouri Heart Associationgrant-in-aid.
REFERENCES1. Scherlag, B. J., R. H. Helfant, J. I. Haft, and A. N.
Damato. 1970. Electrophysiology underlying ventriculararrhythmias due to coronary ligation. Am. J. Physiol.219: 1665-1671.
2. Scherlag, B. J., N. El-Sherif, R. Hope, and R. Lazzara.1974. Characterization and localization of ventriculararrhythmias resulting from myocardial ischemia and in-farction Circ. Res. 35: 372-383.
3. Lazzara, R., N. El-Sherif, and B. J. Scherlag. 1973.Electro-physiological properties of canine Purkinje cellsin one-day-old myocardial infarction. Circ. Res. 33:722-734.
4. Lazzara, R., N. El-Sherif, and B. J. Scherlag. 1974.Early and late effects of coronary artery occlusion oncanine Purkinje fibers. Circ. Res. 35: 391-399.
5. Corr, P. B., and B. E. Sobel. 1977. Automated dataprocessing. An essential decision-making aid in the treat-ment of acute myocardial infarction. Adv. Cardiol.20: 54-71.
6. Webb, S. W., A. A. J. Adgey, and J. F. Pantridge.1972. Autonomic disturbance at onset of acute myo-cardial infarction. Br. Med. J. 3: 89-92.
7. Ebert, P. A., R. B. Vanderbeek, R. J. Allgood, andD. C. Sabiston, Jr. 1970. Effect ofchronic cardiac denerva-tion on arrhythmias after coronary artery ligation. Cardio-vasc. Res. 4: 141-147.
8. Khan, M. I., J. T. Hamilton, and G. W. Manning. 1972.Protective effect ofbeta adrenoceptor blockade in experi-mental coronary occlusion in conscious dogs. Am. J.Cardiol. 30: 832-837.
9. Corr, P. B., and R. A. Gillis. 1975. Effect of autonomicneural influences on the cardiovascular changes inducedby coronary occlusion. Am. Heart J. 89: 766-774.
10. Gillis, R. A., D. L. Pearle, and T. Hoekman. 1974.Failure of beta-adrenergic receptor blockade to preventarrhythmias induced by sympathetic nerve stimulation.Science (Wash. D. C.). 185: 70-72.
11. Govier, W. C. 1968. Myocardial alpha adrenergic recep-tors and their role in the production of positive inotropiceffect by sympathomimetic agents. J. Pharmacol. Exp.Ther. 159: 82-90.
12. Sobel, B. E., and S. E. Mayer. 1973. Cyclic adenosinemonophosphate and cardiac contractility. Circ. Res. 32:407-413.
13. Wollenberger, A. 1975. The role of cyclic AMP in theadrenergic control ofthe heart. In Contraction and Relaxa-tion in the Myocardium. W. G. Nayler, editor. AcademicPress, Inc., New York. 113-190.
14. Drummond, G. I., and S. J. Hemmings. 1973. Role ofadenylate cyclase-cyclic AMP in cardiac actions ofadrenergic amines. Recent Adv. Stud. Card. Struct.Metab. 3: 213-222.
15. Goren, E. N., and 0. M. Rosen. 1972. Inhibition of acyclic nucleotide phosphodiesterase from beef heartby catecholamines and related compounds. Mol. Pharma-col. 8: 380-384.
16. O'Brien, J. A., and R. C. Strange. 1975. The release ofadenosine 3':5'-cyclic monophosphate from the isolatedperfused rat heart. Biochem. J. 152: 429-432.
17. Shahab, L., A. Wollenberger, E. G. Krause, and S.Genz. 1972. Effect of Acute Ischaemia on MyocardialFunction. M. F. Oliver, D. G. Julian, and K. W. Donald,editors. Churchill-Livingstone, Edinburgh.
18. Dobson, J. G., and S. E. Mayer. 1973. Mechanisms ofactivation of cardiac glycogen phosphorylase in ischemiaand anoxia. Circ. Res. 33: 412-420.
19. Lowry, 0. H., N. J. Rosebrough, A. L. Farr, and R. J.Randall. 1951. Protein measurement with the folin phenolreagent.J. Biol. Chem. 193: 265-275.
20. Steiner, A. L., R. E. Wehmann, C. W. Parker, and D. M.Kipnis. 1972. Radioimmunoassay for the measurement ofcyclic nucleotides. Adv. Cyclic Nucleotide Res. 2: 51-61.
21. Tamargo, J., B. Moe, and G. K. Moe. 1975. Interactionofsequential stimuli applied during the relative refractoryperiod in relation to determination of fibrillationthreshold in the canine ventricle. Circ. Res. 37: 534-541.
22. Vincenzi, F. F., and T. C. West. 1963. Release ofautonomic mediators in cardiac tissue by direct sub-threshold electrical stimulation. J. Pharmacol. Exp. Ther.141: 185-194.
118 P. B. Corr, F. X. Witkowski, and B. E. Sobel

23. Nayler, W. G., and J. Tay. 1972. Effect of o-2-hydroxy-3-(tertbutylamino) propoxybenzonitrile HCl (KO 1366) onbeta adrenergic receptors in cardiovascular system. J.Pharmacol. Exp. Ther. 180: 302-316.
24. Schultz, J. 1975. Cyclic adenosine 3',5'-monophosphate inguinea-pig cerebral cortical slices: studies on the role ofadenosine. J. Neurochem. 24: 1237-1242.
25. Caen, J. P., C. S. P. Jenkins, H. Michel, J. J. Chivot,S. Levy-Toledano, and F. Rendu. 1973. Adenosine metab-olism in platelets and plasma. Ser. Haematol. 6: 317-332.
26. Corr, P. B., and R. A. Gillis. 1974. Role of the vagusnerves in the cardiovascular changes induced by coronaryocclusion. Circulation. 49: 86-97.
27. Corr, P. B., D. L. Pearle, J. R. Hinton, W. C. Roberts,and R. A. Gillis. 1976. Site of myocardial infarction. Adeterminant ofthe cardiovascular changes induced in thecat by coronary occlusion. Circ. Res. 39: 840-847.
28. Braasch, W., S. Gudbjamason, P. S. Puri, K. G. Ravens,and R. J. Bing. 1968. Early changes in energy metabolismin the myocardium following acute coronary artery oc-clusion in anesthetized dogs. Circ. Res. 23: 429-438.
29. Beme, R. M. 1964. Regulation of coronary blood flow.Physiol. Rev. 44: 1-29.
30. Brooks, C. M., B. F. Hoffman, E. E. Suckling, and 0.Orias. 1955. Excitability ofthe Heart. Grune and Stratton,Inc., New York. 124-130.
31. Wit, A. L., and P. L. Friedman. 1975. The basis forventricular arrhythmias accompanying myocardial infarc-tion: alterations in electrical activity ofventricular muscleand Purkinje fibers after coronary artery occlusion. Arch.Intern. Med. 135: 459-472.
32. Tsien, R. W., W. Giles, and P. Greengard. 1972. CyclicAMP mediates the effects of adrenaline on cardiacPurkinje fibres. Nat. New Biol. 240: 181-183.
33. Kobayashi, T., R. Nakayama, and K. Kimura. 1971.Effects ofglucagon, prostaglandin E, and dibutyryl cyclic3',5'-AMP upon the transmembrane action potential ofguinea pig ventricular fiber and myocardial contractileforce. Jpn. Circ. J. 35: 807-819.
34. Tsien, R. W. 1973. Adrenaline-like effects of intracellularionotophoresis of cyclic AMP in cardiac Purkinje fibres.Nat. New Biol. 245: 120-122.
35. Barrett, A. M. 1969. A comparison of the effect of (±)-propranolol and (+)-propranolol in anesthetized dogs; 13-receptor blocking and haemodynamic action. J. Pharm.Pharmacol. 21: 241-247.
36. Fowlis, R. A. F., C. T. M. Sang, P. M. Lundy, S. P.Ahuja, and H. Colhoun. 1974. Experimental coronaryartery ligation in conscious dogs six months after bilateralcardiac sympathectomy. Am. Heart J. 88: 748-757.
37. Han, J., P. Garcia de Jalon, and G. K. Moe. 1964.Adrenergic effects on ventricular vulnerability. Circ. Res.14: 516-524.
38. Harris, A. S. 1966. Potassium and experimental coronaryocclusion. Am. Heart J. 71: 797-802.
39. Borda, L., R. Shuchleib, and P. D. Henry. 1977. Effects ofpotassium on isolated coronary arteries: Modulation ofadrenergic responsiveness and release ofnorepinephrine.Circ. Res. 41: 778-786.
40. Wollenberger, A., and L. Shahab. 1965. Anoxia-inducedrelease ofnoradrenaline from the isolated perfused heart.Nature (Lond.). 207: 88-89.
41. Wollenberger, A., E. G. Krause, and G. Heier. 1969.Stimulation of 3',5'-cyclic AMP formation in dog myo-cardium following arrest of blood flow. Biochem. Bio-phys. Res. Commun. 36: 664-670.
42. Rabinowitz, B., W. W. Parmley, M. Kligerman, J. Norman,S. Fujimura, S.'Chiba, and J. M. Matloff. 1975. Myo-cardial and plasma levels of adenosine 3':5'-cyclic phos-phate. Studies in experimental myocardial ischemia.Chest. 68: 69-74.
43. Limas, C. J., D. Ragan, and E. D. Fries. 1974. Effectof acute cardiac overload on intramyocardial cyclic 3',5'-AMP: relation to prostaglandin synthesis. Proc. Soc. Exp.Biol. Med. 147: 103-105.
44. Podzuweit, T., W. F. Lubbe, and L. H. Opie. 1976.Cyclic adenosine monophosphate, ventricular fibrillationand antiarrhythmic drugs. Lancet. I: 341-342.
Malignant Dysrhythmias and Ischemia 119