mature b- and t-cell lymphoproliferative neoplasms · chronic lymphoproliferative neoplasms...
TRANSCRIPT

22[22.1] Chronic Lymphoproliferative Neoplasms 475
[22.2] Chronic Lymphocytic Leukemia 476
[22.3] B-Cell Prolymphocytic Leukemia 491
[22.4] Hairy Cell Leukemia 493
[22.5] Peripheralizing B-Cell Lymphomas 501
[22.6] T-Chronic Lymphoproliferative Neoplasms 504
[22.7] Components of the Diagnostic Interpretation in CLPN 514
[22.8] Clues and Caveats 514
[22.9] Acknowledgments 516
[22.10] References 516
C H A P T E R
Mature B- and T-Cell Lymphoproliferative
NeoplasmsKathryn Foucar, MD

475
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
L ymphoid neoplasms are broadly categorized into precursor lymphoid neoplasms and mature B-cell, T-cell, or natural killer (NK) cell neoplasms
[Swerdlow 2008] . For practical purposes, mature B-, T-/NK- neoplasms can be further segregated into disorders likely to exhibit primary manifestations in blood and bone marrow (ie, chronic leukemias) compared with mature neoplasms that predominate in extramedullary sites but may involve blood or bone marrow more typically as a secondary event (B-, T-/NK- lymphomas). Furthermore, some mature B-cell/plasma cell disorders exhibit dominant immunosecretory manifestations. To aid the diagnostician in a logical systematic approach, the mature lymphoid/plasma cell neoplasms are separated into 4 chapters in this text:1. chronic B-and T-cell leukemias, by convention termed
chronic lymphoproliferative disorders/neoplasms (CLPD/CLPN), are the focus of this chapter
2. myeloma and other immunosecretory disorders are comprehensively reviewed in Chapter 23
3. bone marrow involvement by B- and T-cell lymphomas is the focus of Chapter 24
4. indolent and aggressive neoplasms of true NK cells are the focus of Chapter 26
[22.1] Chronic Lymphoproliferative NeoplasmsChronic lymphoproliferative neoplasms represent clonal
proliferations of morphologically and immunophenotypically mature B or T cells characterized by a low proliferation rate and prolonged cell survival. t22.1 lists the specifi c subtypes of CLPNs, which are the focus of this chapter, based on the 2008 World Health Organization (WHO) criteria and defi nitions [Swerdlow 2008]. A systematic approach to CLPN generally begins with a morphologic review of blood smears t22.2. Key decision-making steps in blood smear review include monotony vs heterogeneity of the lymphoid cells, absolute lymphocyte count, nuclear features such as chromatin condensation, and nuclear contours, as well as the features of the cytoplasm. It is also important to assess all other lineages for qualitative and quantitative features. For many of the CLPNs discussed in this chapter, the index of suspicion for an overt neoplastic disorder can be very high based on just morphologic and CBC data. However, multicolor fl ow cytometric immunophenotyping is the “mainstay” for establishing stage of maturation for B- and T-cell disorders and clonality for B-CLPN. Molecular assessment for T-cell receptor gene rearrangement is the gold standard for establishing T-cell clonality.
t22.1 Chronic Lymphoproliferative Neoplasms with Characteristic Involvement of Blood and Bone Marrow at Presentation
Mature B-Cell Neoplasms Mature T-Cell Neoplasms*
Chronic lymphocytic leukemiaB-cell prolymphocytic leukemiaHairy cell leukemiaSplenic zone lymphomaSplenic B-cell leukemia including
hairy cell leukemia-variant
T-cell large granular lymphocytic leukemia
T-cell prolymphocytic leukemiaSézary syndromeAdult T-cell leukemia
*Chronic lymphoproliferative disorders of natural killer (NK) cells and aggressive NK cell leukemias are the focus of Chapter 26
t22.2 Approach to B- and T-Cell Chronic Lymphoproliferative Neoplasms (CLPN)
For Diagnosis
Morphologic review of blood smear with CBC data
Determine stage of maturation based on nuclear features and likelihood of neoplastic vs non-neoplastic process
Immuno pheno-typing
Delineate full IP profi le of lymphoid cells including patterns and intensity of surface/cytoplasmic antigen expression to confi rm lineage and stage of maturation
Assess for neoplasm-specifi c markersGenotyping Assess for recurrent cytogenetic aberrations
linked to specifi c CLPN subtypesMolecular assessment for B- or T-cell clonality
may be necessary for diagnosis of some CLPNFor Prognosis
Staging Largely based on clinical assessment, but cytopenias in blood are component of both Rai and Binet staging systems for CLL
Delineating other prognostic factors (major focus in CLL, not as relevant in other CLPN)
Major area of emphasis in CLLCD38 and ZAP-70 assessment by IP in CLLKaryotypic assessment by conventional
cytogenetic/FISH in CLL and other B-, T-CLPNSomatic mutation status by molecular
assessment for CLLOther specialized molecular/genetic testing
may be useful for prognostication or in defi ning pathophysiologic mechanisms of leukemogenesis
Numerous other serum and molecular/genetic factors linked to prognosis (most notably in CLL)
CLL = chronic lymphocytic leukemia; FISH = fl uorescence in situ hybridization

476
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
In addition, recurrent cytogenetic abnormalities may provide support for the diagnosis of some B- and T-CLPNs t22.2. Although a fi nal diagnosis has been the historical endpoint of the pathologic evaluation of neoplasms, the detection and delineation of numerous prognostic factors has become an area of much greater emphasis in CLPN, especially chronic lymphocytic leukemia (CLL).
Because fl ow cytometric immunophenotyping is readily available in most clinical practice sett ings, the discussions in this chapter will be segregated by B- or T-cell lineage, with the greatest emphasis on the most frequent CLPNs. Consequently, B-CLPN will be presented fi rst, followed by T-CLPN.
[22.2] Chronic Lymphocytic Leukemia
[22.2.1] General FeaturesAlthough it has a worldwide distribution, CLL is much
more frequent in Western countries, where it accounts for up to 30% of all leukemias [Montserrat 2008]. Th is disease characteris-tically aff ects middle-aged to elderly patients, especially men, with a median age of 70 years. CLL can occur in younger patients (≤55 years); a more aggressive disease course has been noted in these patients [Mauro 1999]. Th e incidence of CLL increases dramatically with age and may exceed 50 in 100,000 men >80 years of age [Dores 2007]. Most patients are asymptomatic, and the disease is detected by complete blood count (CBC) assessment. Symptomatic patients may manifest with lymphadenopathy, various autoimmune disorders such as hemolytic anemia or immune-mediated thrombo-cytopenia, or, less commonly, fatigue, fever, and weight loss [Dearden 2008, Fuller 2008, Montserrat 2008, Morton 2008, Visco 2008].
Some insights into the pathogenesis of CLL have been delineated from both the assessment of familial kindreds and sophisticated evaluations of CLL cells. Since CLL is the most common familial leukemia, both genetic linkage studies and IGHV mutation analyses of kindreds have helped to defi ne genomic linkages and an association of gene usage patt erns in familial CLL [Aoun 2007, Brown 2008, Crowther-Swanepoel 2008, Ng 2007]. Even the link between CLL and monoclonal B-lymphocytosis has been solidifi ed by family studies (see [22.2.9] “Diff erential Diagnosis,” p 485) [Fuller 2008]. Recent studies have provided numerous insights into the pathogenesis of CLL, but a clear-cut linear delineation of the precise, sequential pathogenesis of CLL has not been forthcoming. Factors at play in the pathogenesis of CLL include chronic antigenic stimulation, the B-cell surface receptor molecule, the bone marrow microenvironment, and numerous genomic alterations, especially microRNA mutations sidebar 22.1. [Caligaris-Cappio 2008, Chiorazzi 2005, Linet 2007]
[22.2.2] Blood FindingsTh e diagnosis of CLL requires the integration of absolute
monoclonal B lymphocyte count, lymphocyte morphology, and the immunophenotypic profi le of the neoplastic cells t22.3. An unexplained, sustained absolute monoclonal B lymphocytosis of >5.0 × 103/μL (≥5.0 × 109/L) or more for 3 months is required [Hallek 2008, Muller-Hermelink 2008]. Studies subsequent to the 2008 WHO classifi cation indicate that an absolute monoclonal B lymphocytosis of 11 × 103/μL (11 × 109/L) is highly predictive of outcome (see Prognostic Factors section). Consequently, requirements for diagnosis may diff er from outcome predictors. Similarly, the diagnostician needs to be aware that criteria used to distinguish overt low-stage B-CLL from monoclonal B lymphocytosis may be revised based on these recent studies (see [22.2.9] “Diff erential Diagnosis,” p 485) [Marti 2009, Rossi 2009, Shanafelt 2009].
t22.3 Prototypic Features of Chronic Lymphocytic Leukemia (CLL)
Blood† Absolute monoclonal B lymphocyte count ≥5.0 × 103/μL(≥5.0 × 109/L) with CLL immunophenotype for 3 months*
Mature, monotonous lymphocytes with round nuclei, condensed chromatin, inconspicuous nucleoli, and generally scant cytoplasm
Bone marrow Mature lymphocytosis generally >30% of total cellsFocal nonparatrabecular or diffuse infi ltrates on core
biopsy sectionsOnly rarely see proliferation foci on core biopsy; true
germinal centers very rarely noted
Immuno-pheno type (IP)
CD5+ monoclonal light chain-restricted B cell with dim CD20, dim CD22, CD23, and weak surface immunoglobulin
Antigens typically absent: CD10, FMC7, and CD79bKey prognostic
factors‡
(assessed at diagnosis)
Clinical stage (Rai, Binet)Absolute monoclonal B lymphocyte count of
≥11 × 103/μL (≥11 × 109/L)CD38 expression (IP)ZAP-70 expression (IP)Karyotype (FISH)IGHV mutation status (molecular)MicroRNA gene deletions or gene overexpression
(not yet utilized in routine clinical practice) (see sidebar 22.1)
*Criteria used to distinguish overt CLL from monoclonal B-cell lymphocytosis may be revised based on outcome studies†Distinction between absolute lymphocyte count and absolute monoclonal B-cell count is critical‡See later discussion regarding prognostic factorsFISH = fl uorescence in situ hybridization
References: [Hallek 2008, Kim 2000, Marti 2009, McCarron 2000, Montserrat 2008, Morice 2008, Muller-
Hermelink 2008, Shanafelt 2009, Viswanatha 2010]

477
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
In general, hematopoietic lineages are preserved in patients with CLL, especially in asymptomatic patients. If cytopenias are detected, this can refl ect either a more advanced stage of the disease with bone marrow eff acement or an immune-mediated destruction of cells secondary to autoantibody production [Montserrat 2008]. Likewise, rare cases of anemia secondary to red cell aplasia have been described in CLL patients, a presumed consequence of the T-cell aberrations that are well-described in this disorder [Lanasa 2009].
In CLL, a monotonous population of mature lympho cytes is generally identifi ed in the blood, although some hetero-geneity is noted occasionally i22.1, i22.2, i22.3. Th ese cells have round nuclei and highly condensed nuclear chromatin with separation of chromatin and parachromatin, creating a blocky patt ern i22.4. Cytoplasm is generally scant, but may be more moderate in some cases that exhibit otherwise typical morphologic features of CLL i22.5. In rare cases, the leukemic
i22.1 Th is peripheral blood smear fr om an 81-year-old female with chronic lymphocytic leukemia and preserved hematopoiesis shows a predominance of mature lymphocytes and multiple smudge cells, along with normal hemato-poietic cells. ( Wright)
[sidebar 22.20] MicroRNAs, the “miRNome,” & CLL
T he past decades have seen a fundamental shift in our understanding of hematologic malignancy, with an increasing emphasis on the centrality of genetic abnormalities and their consequences for protein
expression or function. Th e recently discovered and incompletely understood microRNA (miRNA) world, sometimes termed the miRNome, is yet another level at which cellular homeostasis in these neoplasms is disturbed. Just as genetic abnormalities provide an opportunity for diagnostic and prognostic investigations, so alterations at the miRNA level are proving to be a promising determinant of neoplastic pathogenesis and behavior. Among hematopoietic malignancies, CLL is the most extensively studied at this level.
As described in sidebar 1.1 (p 4), miRNAs are short RNA molecules, coded for by specifi c regions of the genome, which are not translated into a protein product, as is the fate of traditional messenger RNA (mRNA). Instead, miRNAs are processed and incorporated into the RNA-induced silencing complex (RISC), whereupon they suppress the translation of other target mRNAs. Th e nucleotide sequence of the miRNA is complementary to that of portions of the target mRNA, providing specifi city to the translational repression. Th us far, hundreds of miRNAs have been identifi ed in human cells, and their presence aff ords an additional level of control over gene expression beyond that achieved through strictly genetic and epigenetic means. Greater than 30% of the human genome may be under translational regulation by miRNA [Schickel 2008]. It is theorized that miRNA may have originated as a primitive cellular defense against RNA viruses or transposons [Zhang 2007].
Th ere is an unusual concentration of genes encoding miRNAs in regions of the genome known to be aff ected by copy number change or structural alterations in malignancy, suggesting that modulation of miRNA expression underlies at least some acquired chromosomal abnormalities in cancer [Calin 2004]. One of the most commonly aff ected chromosomal regions in CLL is 13q14.3, isolated deletions of which confer a relatively good prognosis. Th e frequency and consistency with which deletion of 13q14.3 arises in CLL indicates that a tumor suppressor gene is likely present at this location, but extensive evaluation of the known genes in the minimally deleted region did not reveal a convincing candidate [Aqeilan 2010]. However, aft er the discovery and early characterization of miRNAs, investigators identifi ed 22 sequences encoding miRNAs within the critical region [Calin 2002, Nicoloso 2007]. Interestingly, these miRNA sequences were located within an intron of another gene, DLEU2, which had been previously considered and largely excluded as a candidate tumor suppressor at this locus [Nicoloso 2007]. Th e specifi c miRNAs encoded, miR-15a and miR-16 - 1, were found to be highly expressed in CD5+ lymphocytes, and to be downregulated or deleted in 70% of CLL cases [Calin 2002]. miR-15a and miR-16 - 1 both target for interference and/or destruction mRNA transcribed from the anti-apoptotic gene BCL2, providing a plausible mechanism by which 13q14.3 deletion could confer a proliferative advantage [Cimmino 2005].
Recently, yet another common chromosomal abnormality in CLL has been functionally linked to miRNAs. Th e well-known tumor suppressor TP53, which is deleted in cases of CLL with loss at 17p, positively regulates the transcription of miR-34a [Calin 2009]. Low levels of miR-34a in CLL are associated with resistance to chemotherapy and to apoptosis, suggesting the participation of this miRNA in generating the adverse prognostic phenotype associated with TP53 deletion in CLL [Zenz 2009a].
While cytogenetic abnormalities are of great importance in CLL, other cellular characteristics, including ZAP70 expression and IGHV somatic hypermutation status, are known to be prognostically signifi cant. An initial study examining global miRNA expression in subsets of CLL delineated by such prognostic markers showed that favorable and unfavorable prognostic groups maintain largely distinct miRNA expression patt erns [Calin 2005]. Th is and similar studies have consistently demonstrated downregulation of miR-223 and members of the miR-29 family in CLL [Calin 2005, Fulci 2007, Marton 2008], with low levels specifi cally associated with disease progression, poor prognostic factors, and reduced treatment-free and overall survival [Stamatopoulos 2009].
Given that we are only beginning to unravel the processes by which miRNAs aff ect cellular function, it is highly likely that the success of these initial investigations will be replicated in the future, yielding further dramatic insight into the pathogenesis of CLL and of hematopoietic malignancies in general. One might even hazard a prediction that miRNA-based data could some day be incorporated into diagnostic classifi cation schemes, much as chromosomal abnormalities and alterations involving protein-coding genes have assumed a central place in the current 2008 WHO system.
i22.2 Th is peripheral blood smear fr om an 81-year-old female with preserved hematopoiesis and a white blood cell count of 68.3 × 103/μL (68.3 × 109/L) shows a striking monotonous mature lymphocytosis. Note homogeneous chromatin clumping , inconspicuous nucleoli, round nuclear contours, and scant cytoplasm, all classic features of chronic lymphocytic leukemia. ( Wright)

478
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
cells have cytoplasmic vacuoles or crystals derived from monotypic immunoglobulin i22.6, i22.7 [Peters 1984, Ralfk iaer 1982].
In addition to the monotonous lymphocytes that typify B-CLL, a range of nuclear aberrations may be noted in cases of otherwise straightforward CLL. For example, a small portion of these lymphoid cells (usually <5%) may exhibit prominent nuclear cleft ing reminiscent of that seen in mantle cell and follicular lymphomas. Also, the proportion of lymphocytes with prominent nucleoli (prolymphocytes), although generally low and stable over time, may sometimes be increased at presentation i22.8. A progressively increasing proportion of prolymphocytes on sequential specimens may indicate disease transformation (see Transformations section).
Th e designation “atypical” CLL has been applied under several circumstances. Th e more common usage relates to cases of CLL in which the morphologic features diff er from classic CLL, while other parameters (clinical, CBC, and immuno-phenotypic profi le) are characteristic of CLL. In these cases of so-called atypical CLL, the circulating lymphocytes are more heterogeneous, and both nuclear irregularity and nucleolated lymphocytes are more prominent than prototypic CLL i22.9, i22.10, i22.11. Associations between morphologic atypia and genotype have been described (see Cytogenetics section). Th e other circumstance in which atypical CLL has been used relates to cases in which the immunophenotypic profi le diff ers somewhat from prototypic CLL (see Immunophenotypic Analysis section). Caution should be exercised in using the term atypical CLL in that diff erential diagnostic considerations must be thoroughly assessed and excluded.
[22.2.3] Bone Marrow FindingsBone marrow examination should be considered in
patients with CLL to determine both the patt ern and extent of the leukemic infi ltrates and to evaluate the residual hemato-poietic cells. On bone marrow aspirate smears, lymphocytes generally account for >30% of cells, and they exhibit cytologic features similar to those of circulating lymphocytes i22.12, i22.13. Th e evaluation of good quality bone marrow core biopsy sections is especially useful in determining patt ern and extent of infi ltration in CLL. CLL infi ltrates are designated as nodular, interstitial, diff use, or mixed, which is a combination of either nodular and interstitial or nodular and diff use. Because of the scant cytoplasm and closely packed nuclei that typify CLL, these leukemic infi ltrates are characteristically dark blue on low magnifi cation of bone marrow biopsy sections.
Nodular infi ltrates of CLL are characteristically nonpara-trabecular in location and are typically composed of small lymphocytes with very closely packed nuclei exhibiting round nuclear contours and condensed chromatin. Litt le mitotic activity is evident i22.14, i22.15, i22.16, i22.17 [Hsi 2009, Viswanatha 2010]. Th ese nodules generally demonstrate infi ltrative margins with
i22.5 Blood smear depicts chronic lymphocytic leukemia cells with relatively abundant amounts of cytoplasm. ( Wright)
i22.4 Blood smear highlights exaggerated chromatin clumping with separation of chromatin and parachromatin in chronic lymphocytic leukemia cells. ( Wright)
a b
i22.3 Blood smears illustrate lymphocyte morphologic features in a patient with a striking leukocytosis fr om chronic lymphocytic leukemia. Rare prolym-phocytes are present. ( Wright)
a b

479
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
i22.6 Blood smear depicts cytoplasmic vacuoles in a chronic lymphocytic leukemia cell. ( Wright)
i22.8 Blood smears illustrate prolymphocytes fr om 3 cases of chronic lymphocytic leukemia. Note the more dispersed chromatin with central nucleoli that characterizes these more immature cells. ( Wright)
i22.7 Blood smear illustrates numerous immunoglobulin crystals in the cytoplasm of chronic lymphocytic leukemia cells. ( Wright)
i22.9 Th is peripheral blood smear fr om a 57-year-old male with atypical chronic lymphocytic leukemia shows a greater heterogeneity in cell size and nuclear confi gurations. Th is patient had concurrent cytopenias. ( Wright)
i22.10 Th is peripheral blood smear fr om a patient with atypical chronic lymphocytic leukemia shows a fairly signifi cant range in cell size. ( Wright)
i22.11 Large prolymphocytic cells are present in this peripheral blood smear fr om a patient with atypical chronic lymphocytic leukemia. ( Wright)
a b c
a b c

480
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
percolation of small lymphocytes into the surrounding hemato-poietic cells. Features that help to distinguish these neoplastic nodules from benign lymphoid aggregates include the relatively higher number of nodules, the infi ltrative margins, the monomorphic cellular composition, and the lack of a consistent perivascular distribution in CLL (see Volume 1, Chapter 21 for a discussion of benign lymphoid aggregates). In addition, immunoperoxidase staining can be used to document the predominance of B cells coexpressing either CD5 or CD43 in CLL, while T cells generally predominate in benign lymphoid aggregates i22.18, i22.19.
Interstitial infi ltrates of CLL consist of an admixture of CLL cells with residual fat and hematopoietic cells i22.20, i22.21. Th e “mixed” designation usually refers to an admixture of nodular and interstitial infi ltrates, while complete eff acement of both fat and hematopoietic cells defi nes diff use solid infi ltrates of CLL. Infi ltrates can also
occasionally involve bone marrow sinuses in conjunction with other patt erns [Kent 2000]. Th e cytologic features of both these interstitial and densely packed diff use cellular infi ltrates are similar to those found in the nodular lesions i22.22.
Unlike the situation in an involved lymph node, prolif-eration foci are only rarely identifi ed in bone marrow core biopsy sections, possibly a refl ection of the small sample size i22.16. Reports of non-neoplastic germinal centers in core biopsy sections from CLL patients are even more rarely encountered, at essentially the “case report” frequency [Kim 2000]. Occasional large transformed cells may be admixed with the small lymphocytes in CLL infi ltrates within the bone marrow. As long as the proportion of these larger cells is low, there is no adverse impact on survival. In rare cases, Reed-Sternberg-like cells exhibiting multinucleation and huge nucleoli are admixed with otherwise typical CLL infi ltrates i22.23 [Kanzler 2000]. Th e signifi cance of this fi nding
i22.14 Bone marrow biopsy section illustrates nodular infi ltrates of chronic lymphocytic leukemia that are primarily nonparatrabecular. ( H&E)
i22.15 Many focal nonparatrabecular nodules of chronic lymphocytic leukemia are present on this bone marrow biopsy section. ( H&E)
i22.12 Th is bone marrow aspirate smear shows extensive involvement by chronic lymphocytic leukemia, which accounts for >80% of cells on diff erential counts. ( Wright)
i22.13 High magnifi cation of bone marrow aspirate smear shows marked infi ltration by chronic lymphocytic leukemia. ( Wright)

481
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
i22.18 Immunohistochemical staining for CD20 on this bone marrow core biopsy section shows numerous B cells with weak to intermediate positivity (see i22.17). (immunoperoxidase for CD20)
i22.19 Immunohistochemical staining for CD3 on this lymphoid nodule fr om a patient with chronic lymphocytic leukemia shows only scatt ered T cells (see i22.17 and i22.18). (immunoperoxidase for CD3)
i22.16 Vague proliferation foci are present in a large chronic lymphocytic leukemia nodule on this bone marrow biopsy section. ( H&E)
i22.17 High magnifi cation of nodular lymphoid aggregate in the bone marrow core biopsy section fr om a patient with chronic lymphocytic leukemia shows relative uniformity in cell size and litt le, if any, mitotic activity. ( H&E)
i22.20 Th is low magnifi cation of a bone marrow core biopsy section fr om a patient with extensive diff use interstitial infi ltration of chronic lymphocytic leukemia shows normal bony trabeculae and relatively preserved fat. ( H&E)
i22.21 On high magnifi cation diff use interstitial infi ltrates of chronic lymphocytic leukemia show an admixture of the leukemia cells with preserved fat and occasional hematopoietic cells, most oft en megakaryocytes. ( H&E)

482
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
is controversial, because some of these patients eventually develop overt Hodgkin lymphoma, while others do not. Likewise, some cases exhibit an immunophenotype of true Reed-Sternberg cells, while these multinucleated cells exhibit a straight-forward B-cell phenotype in other cases i22.23 [Kanzler 2000, Momose 1992, Williams 1991, Ohno 1998]. Finally, in patients with known, longstanding CLL, foci of transformation to large cell lymphoma may be evident on either aspirate smears or biopsy sections (see Transformations section).
Recent studies provide insight into the association of a diff use patt ern of CLL infi ltration in bone marrow with progressive disease and an overall more aggressive disease course. Both ZAP-70 expression and unmutated IGHV genes are linked to a diff use core biopsy infi ltration patt ern [Schade 2006,
Zanott i 2007].
[22.2.4] Immunophenotypic AnalysisMulticolor fl ow cytometric immunophenotyping is
essential in establishing a diagnosis of CLL, in delineating specifi c prognostic factors (CD38 and ZAP-70 assessment), and in distinguishing CLL from other leukemic CLPN f22.1, f22.2 [Chiorazzi 2005, Hallek 2008, Morice 2008]. In addition to weak monotypic surface immunoglobulin, the classic immunophenotypic profi le of CLL includes expression of CD19, weak CD20, weak CD22, weak CD11c, CD5, and CD23 antigens. Cases of CLL characteristically lack CD25, CD10, and FMC7, while CD79b expression is weak or absent [McCarron 2000, Morice 2008]. Variations from this prototypic immunophenotypic profi le have been described including partial or absent CD23 expression, partial or absent CD5 expression, and bright CD20 expression [Morice 2008]. Th e designation “atypical CLL” is sometimes applied to cases with
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD38
CD19
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
ZAP-
70
CD3
CLL normal T
f22.2 Th ese fl ow cytometric histograms illustrate CD38 and ZAP-70 expression in chronic lymphocytic leukemia. Note comparison of ZAP-70 to normal T cells.
a b
For p
lace
men
t only
0
200
400
600
1000
800
4002000 600 800 1000
side
sca
tter
forward scatter
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD23
CD5
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD20
CD5
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD20
CD23
f22.1 Immunophenotypic features of chronic lymphocytic leukemia (CLL) (black) are illustrated in this composite fl ow cytometric histogram. Note coexpression of CD23, CD5, and CD20. Note normal T cells (green). CLL cells are small and agranular based on forward scatt er and side scatt er, respectively.
a b
c d
i22.22 Th is bone marrow core biopsy section fr om a 69-year-old male with chronic lymphocytic leukemia associated with severe cytopenias shows diff use solid eff acement. ( H&E)
i22.23 Th e morphologic and histochemical features of Reed-Sternberg-like cells in an infi ltrate of otherwise typical chronic lymphocytic leukemia are illustrated in this composite photomicrograph. Note membrane and Golgi staining of Reed-Sternberg-like cells for CD30 b and CD15 c. ( H&E plus immunoper-oxidase for CD30 and CD15)
a b c

483
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
“nonclassic” immunophenotype [Frater 2001]. Similar to cases of CLL with atypical morphology, cases of CLL with atypical immunophenotypic features have been linked to distinctive genetic and clinical features (see Cytogenetics section) [Frater 2001,
Schlett e 2003, Tam 2008]. When cell suspensions for fl ow cytometry are not available, paraffi n immunoperoxidase techniques can be used to assess CD20, cyclin D1, CD5, CD43, CD10, and CD23, further facilitating the distinction between CLL and other B-CLPN i22.24, i22.25, i22.26, i22.27 (see [22.2.9] “Diff erential Diagnosis,” p 485).
[22.2.5] Immunophenotypic Assessment of Prognostic Markers CD38 and ZAP-70
Numerous investigators have described the role of CD38 in the pathophysiology of CLL, the functional link between CD38 and ZAP-70 expression, and the adverse prognostic signifi cance of CD38 expression on ≥30% of CLL cells on fl ow cytometric immunophenotyping t22.4 [Chen 2007, Chiorazzi 2005, D’Arena 2007, Deaglio 2006, 2007,
i22.24 CD20 highlights discrete lymphoid aggregates on this bone marrow core biopsy section fr om a patient with chronic lymphocytic leukemia exhibiting exclusively nodular infi ltrates. (immunoperoxidase for CD20)
i22.25 Immunoperoxidase staining for CD3 shows only scatt ered T cells in this lymphoid nodule of chronic lymphocytic leukemia on this bone marrow core biopsy section. (immunoperoxidase for CD3)
i22.26 Immunoperoxidase staining for CD5 is strongly positive in this bone marrow core biopsy section fr om a patient with chronic lymphocytic leukemia ( CLL) with exclusively nodular infi ltrates (see i22.24, i22.25). Th e disparity between the number of CD5+ cells and the number of CD3+ cells is remarkable, indicating that the CLL cells coexpress CD5. (immunoperoxidase for CD5)
i22.27 Immunoperoxidase staining for BCL2 shows strong positivity in this bone marrow core biopsy section fr om a patient with chronic lymphocytic leukemia ( CLL) showing multiple nodular lymphoid infi ltrates. Th e BCL2 expression by CLL is linked to microRNA mutations; BCL2 positivity in CLL can result in diagnostic confusion with follicular lymphoma. (immunoperoxidase for BCL2)
t22.4 CD38 Expression in CLLSurface ADP-ribosyl cyclase adhesion molecule that supports B-cell
interactions and differentiationInduces proliferation and activation, and increases survival of CLL cellsKey element, in conjunction with ZAP-70, in coreceptor pathway that
may sustain B-cell receptor signalsSurface expression on ≥30% of CLL population independent prognostic
factorFunctionally linked with ZAP-70 and confers enhanced migratory
potential on CLL cellsLevel of expression may vary during disease course and by tissue site
CLL = chronic lymphocytic leukemia; ZAP-70 = zeta-chain-associated protein kinase
References: [Chen 2007, Chiorazzi 2005, D’Arena 2007, Deaglio 2006, 2007, Hamblin 2002, Montserrat 2008,
Patten 2008, Pittner 2005, Rassenti 2008, Schroers 2005, Van Bockstaele 2009]

484
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
Hamblin 2002, Montserrat 2008, Patt en 2008, Pitt ner 2005, Rassenti 2008, Schroers 2005, Van Bockstaele 2009]. Consequently, expression of CD38 should be assessed on all CLL cases in which immunophenotyping is performed. Reports vary in terms of the stability of CD38 expression over time and in diff erent disease sites [Chen 2007, Hamblin 2002, Patt en 2008].
Similarly, the independent prognostic signifi cance of ZAP-70 expression in CLL is well-established t22.5 [Crespo 2003, Del Principe 2006,
Gachard 2008, Rassenti 2008, Scielzo 2006, Sup 2004, Van Bockstaele 2009, Zanott i 2007]. ZAP-70 can be assessed with either fl ow cytometric techniques or immuno-histochemistry, but these assessments are highly problematic in many laboratories [Gachard 2008, Wilhelm 2006]. Nonetheless, several recent studies have shown that ZAP-70 expression appears to have the greatest weight in predicting outcome compared with other parameters [Del Principe 2006, Rassenti 2008].
[22.2.6] Cytogenetic and Molecular FeaturesWith modern cytogenetic techniques and enhanced
fl uorescence in situ hybridization (FISH) testing, 70% - 80% of CLL cases show cytogenetic abnormalities [Dicker 2006, Dohner 2000,
Haferlach 2007, Reddy 2006]. Likewise, multiplex ligation-dependent probe amplifi cation can be used to detect genomic aberrations in CLL [Coll-Mulet 2008]. Th e independent prognostic signifi cance of karyotype in CLL is well established [Dohner 2000, Grever 2007, Mayr 2006,
Van Den Neste 2007]. Th e key cytogenetic abnormalities in CLL include deletions of 6q, 13q, 11q, and17p, and trisomy 12 [Byrd 2006, Dohner 2000,
Grever 2007, Roos 2008]. Th e frequency of these cytogenetic abnormalities and key associations are listed in t22.6 [Cavazzini 2008, Cuneo 2004, Dicker 2006, 2009,
Dohner 2000, Grever 2007, Haferlach 2007, Kujawski 2008, Reddy 2006, Roos 2008, Saddler 2008, Tam 2008,
Zenz 2008]. In addition to these individual cytogenetic aberrations, cytogenetic complexity identifi es patients at risk for an aggressive disease course; in some reports genetic complexity is linked to short telomeres [Kujawski 2008, Roos 2008]. Th e best overall outcome is
associated with isolated del(13q14), which tracks with other biologically favorable features such as somatic hypermutation and absence of CD38 [Dicker 2006, Dohner 2000, Haferlach 2007, Viswanatha 2010].
Somatic Mutations of IGHV GenesAssessment for somatic mutations of IGHV genes in CLL is
the cornerstone in separating CLL into 2 biologic and prognostic groups. Cases of CLL with unmutated (germline) IGHV genes show a characteristic profi le consisting of CD38 and ZAP-70 expression, adverse karyotype, reduced time to therapy, and overall more aggressive disease course [Chiorazzi 2005, Montserrat 2008,
Morabito 2009, Murray 2008, Rassenti 2008]. In contrast, the biologically indolent subtype of CLL is characterized by somatic hypermutations, absence of CD38 and ZAP-70, and good risk karyotype. Recent evidence suggests that both of these biologic CLL subtypes are derived from an antigen-experienced B cell but diff er based on
t22.6 Cytogenetic Features of CLLCytogenetics % of Cases Comments
Normal 20% - 30% Linked to intermediate outcomedel(13q14) >40% Sole abnormality in 50% of untreated
patientsLinked to better outcome when sole
abnormalityLinked to mutated IGHV, absence of
CD38, typical morphologytrisomy 12 7% - 25% Linked to intermediate outcome, CD38
expressionLinked to atypical morphology, bright
CD20 expression, and high rate of response to rituximab
del(11q) 3% - 15% Linked to unmutated IGHV, adverse outcome, ATM gene deletions/alterations
del(17p) 5% - 12% TP53 mutations/inactivation may be independent of del(17p) and linked to low expression of miR-34a
Linked to rapid disease progression, adverse outcome
del(6q) 3% - 5% Linked to unmutated IGHVLinked to atypical morphology, CD38
expression, splenomegaly, and short interval to therapy
14q32 translocations
4% - 10% Multiple translocation partnersPoor outcome, CD38 expression
Complex karyotypes
10% - 40% Linked to unmutated IGHV, CD38 expression
Linked to short telomeres, short survival, and TP53 mutations
CLL = chronic lymphocytic leukemia
References: [Cavazzini 2008, Cuneo 2004, Dicker 2006, 2009, Dohner 2000, Grever 2007, Haferlach 2007, Kujawski 2008,
Reddy 2006, Roos 2008, Saddler 2008, Tam 2008, Viswanatha 2010, Zenz 2008, Zenz 2009b]
t22.5 ZAP-70 Expression in CLLZeta-associated protein of 70 kDaCytoplasmic tyrosine kinase essential for T-cell receptor signal
transductionExpressed in normal and neoplastic B cells at different maturation
stagesOriginally identifi ed by gene expression profi ling studies comparing
CLL with and without IGHV somatic mutationsAlthough linked to somatic mutation status some investigators report
that ZAP-70 expression has higher relative value in predicting outcome compared with CD38 expression and mutation status
May be assessed by fl ow cytometry and immunohistochemistry, but highly problematic in many laboratories
CLL = chronic lymphocytic leukemia; ZAP-70 = zeta-chain-associated protein kinase
References: [Crespo 2003, Del Principe 2006, Gachard 2008, Rassenti 2008, Scielzo 2006, Sup 2004,
Van Bockstaele 2009, Zanotti 2007]

485
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
the extent of somatic mutation [Chiorazzi 2005]. Consequently, the designation germline/unmutated, is not entirely correct, but by convention is used to delineate the biologically aggressive CLL subtype.
[22.2.7] Integration of Primary Prognostic FactorsIn patients with CLL in whom all of these immuno-
phenotypic and genetic prognostic factors are concordant and favorable, the median survival time may exceed 25 years [Shanafelt 2004]. In contrast, in patients with CLL in whom all of these prognostic factors are concordant and unfavorable, the median survival time is ≤8 years. Survival time for patients whose CLL cells show discordant CD38 and ZAP-70 is intermediate [Schroers 2005, Shanafelt 2004].
[22.2.8] Other Prognostic FactorsIn addition to traditional Rai and Binet staging and
the previously discussed biologic markers (CD38, ZAP-70 expression, somatic mutation status, and karyotype), other prognostic factors have been described in CLL [Moreno 2008,
Van Bockstaele 2009]. Th ese range from routine hematologic factors such as lymphocyte doubling time to various serum markers (soluble CD23 doubling time, LDH, β-2 microglobulin) to esoteric markers such as microRNA signature profi le (see sidebar 22.1, p 477), thrombopoietin levels, telomere length, and the expression of various genes (TCL1, MCL1, MDR1, and antiapoptotic genes) [Agrawal 2008, Delgado 2009, Koller 2006, Meuleman 2008, Moreno 2008, Pepper 2008, Ricca 2007,
Weinberg 2007, Wierda 2007, Zenz 2009b]. Of note, initial therapeutic modalities aff ect the prognostic signifi cance of many of these biologic factors [Lin 2009, Wierda 2009].
[22.2.9] Diff erential DiagnosisWhen an elderly patient presents with a markedly elevated
monotonous, mature lymphocytosis, the diagnosis of CLL is generally not diffi cult. However, the diagnosis of CLL is much more challenging in patients in whom the absolute lymphocyte count is only minimally elevated or in cases in which the morphologic and clinical features are not those associated with classic CLL. Disorders that can show morphologic overlap with CLL include reactive lymphocytoses, monoclonal B lympho-cytosis, B-cell prolymphocytic leukemia (B-PLL), hairy cell leukemia (HCL), splenic marginal zone lymphoma, and some other peripheralized non-Hodgkin lymphomas t22.7 [Delage 2001,
Himmelmann 2001, Karandikar 2002, Landgren 2009, Rawstron 2008, Troussard 1996]. Because most reactive lymphocytoses are composed of morphologically hetero-geneous T cells, their distinction from CLL is usually possible, oft en by morphologic review of the blood smears and clinical correlation. Both the pronounced morphologic heterogeneity and the association with viral syndromes in reactive transient, reactive lymphocytoses is generally suffi cient for their distinction from clonal disorders; immunophenotyping studies are not
generally required. However, there are rare reports of unexplained, sustained, mature lymphocytoses in young women (see Volume 1, Chapter 21) [Delage 2001, Himmelmann 2001, Troussard 1996]. Th e polyclonal B-cell surface antigen profi le of these processes can be used to exclude CLL from consideration.
For clonal B-cell disorders ranging from monoclonal B lymphocytosis to peripheralized lymphomas, the distinction from CLL may be more challenging, requiring the integration of clinical, morphologic, immunophenotypic, genetic, and outcome information. Several large studies of normal individuals confi rm that occult B-cell clones, termed monoclonal B lymphocytosis, are common in blood, especially in older patients [Landgren 2009, Nieto 2009, Rawstron 2008]. Th ese occult clones oft en, but not always, exhibit a CLL immunophe-notypic profi le and are typically detected in individuals with a normal absolute lymphocyte count. Although much more frequent than overt CLL, monoclonal B lymphocytosis is a likely precursor lesion because it was detected retrospectively in 98% of individuals who developed overt CLL in a cancer screening clinical trial [Landgren 2009]. Recent studies suggest that the criteria for distinguishing monoclonal B lymphocytosis from overt low-stage CLL may need refi nement, since the level of absolute monoclonal B lymphocyte count which is most signifi cantly correlated with outcome is 11 × 103/μL (11 × 109/L) [Marti 2009, Shanafelt 2009]. In addition, patients with small lymphocytic lymphoma may manifest minor blood involvement that may mimic monoclonal B lymphocytosis. Th e diagnostician must be aware of these potential pitfalls that may lead to either overdiagnosis or underdiagnosis of small monoclonal B-cell populations in blood.
Th e spectrum of features useful in distinguishing CLL from other overt B-cell neoplasms is delineated in various ways on t22.8, t22.9, t22.10, t22.11. Again, a systematic multipa-rameter approach is essential. Th e diagnostician must be aware, however, that some cases may be equivocal, even aft er extensive assessment. In these situations, a diagnosis of CLPN, not otherwise specifi ed (NOS), should be considered. Indeed, the diagnostic challenge of CD5+/CD10– leukemias has been well described, and lymphoid leukemias with a typical mantle cell lymphoma (MCL) phenotype may include cases of CLL by genetic assessment [Goldaniga 2008, Ho 2009].
[22.2.10] TransformationsTh e development of a second hematolymphoid neoplasm
in a patient with longstanding CLL represents a diagnostic challenge: is this neoplasm a transformation of the CLL or a therapy-related secondary disorder? For example, the alkylating agent therapy used to treat some patients with CLL can induce either secondary myelodysplasia or acute myeloid leukemia (see Volume 1, Chapters 16 and 18) i22.28, i22.29. In contrast, many

486
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
t22.8 Classic Immunophenotypic Profi le of B-Cell Chronic Lymphoproliferative Neoplasms (B-CLPN)Disorder SIg CD19 CD20 CD22 CD23* CD25 CD5 FMC7 CD11c CD10 CD79b CD103 Annexin 1 Cyclin D1 CD123
CLL/SLL w + w w + – + – w – –, w – – – –
B-PLL + + + + –, w +s v + +s – + – – – –
MCL + + + + –, w – + + – – + – – + –
FL + + + + – – – + – + + – – – –
SMZL + + + + –, w – v + v v + +s – – –
HCL + + + + –, w + – + + +s –, w + + +s +
LPL v, cIg+ + + + – – – – – – + – – – –
*Weakly to moderately expressed on many leukemia subtypesCLL/SLL = chronic lymphocytic leukemia/small lymphocytic leukemia; B-PLL = B-cell prolymphocytic leukemia; MCL = mantle cell lymphoma; FL = follicular lymphoma; SMZL = splenic marginal zone lymphoma; HCL = hairy cell leukemia; LPL = lymphoplasmacytic lymphomav = variable expression; w = weakly expressed; c = cytoplasmic; +s = subset of cases positive
References: [Dong 2009, Isaacson 2008, Matutes 2006, McCarron 2000, Morice 2008, Muller-Hermelink 2008, Viswanatha 2010]
t22.7 Differential Diagnosis of CLL
Disorder Key Distinguishing Feature in Clinical/Blood Findings
Reactive lymphocytosis* T cells predominateUsually virally induced; transientProminent morphologic spectrum
Transient stress lymphocytosis* Modest absolute lymphocytosis after severe stressTransient increase in helper and suppressor T cells as well as B and NK cells
Polyclonal B lymphocytosis* Familial link; HLA linkYoung females, heavy smokersBinucleated lymphocytes; sustained
Monoclonal B lymphocytosis CLL-like immunophentoypeDetected in up to 12% of normal individuals, especially elderlyIncreased frequency of detection with aging (100 times more frequent than overt CLL)Absolute lymphocyte count usually normalLikely precursor to overt CLL in some, but not all, casesIdentifi ed retrospectively in 98% of patients who developed overt CLL in a population-based cancer
screening clinical trialRisk of progression to treatment requirement linked to trisomy 12 or del(17p13)
B-cell prolymphocytic leukemia WBC strikingly increasedProlymphocytes predominateLacks both IP and genetic features of CLLUsually associated with marked hepatosplenomegaly
Hairy cell leukemia Cytopenias predominate, often pancytopeniaNeutropenia, pronounced monocytopeniaHairy cells usually rare in bloodVery distinctive morphology of hairy cellsVery distinctive IP profi le
Splenic marginal zone lymphoma Mild leukocytosis with spectrum with plasmacytoid cells; bipolar projectionsLacks both IP and genetic features of CLLSplenomegaly typical
Other peripheralized B-cell lymphomas
Often have clinical history of NHL in extramedullary sitesMay exhibit de novo leukemic manifestations (especially mantle cell lymphomas)Nuclear irregularities of lymphoid cells commonLack both IP and genetic features of CLL
*See Volume 1, Chapter 21 for more detailsCLL = chronic lymphocytic leukemia; IP = immunophenotype; NHL = non-Hodgkin lymphoma
References: [Dagklis 2009, Delage 2001, Himmelmann 2001, Karandikar 2002, Landgren 2009, Nieto 2009, Rawstron 2008, Rossi 2009, Troussard 1996]

487
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
t22.9 Defi ning Immunophenotypic Features* in Chronic B-Cell Neoplasms
CLL B-PLL HCL MCL SMZL FL
Grading System†
CD20 DimBright
+0
0+
0+
0+
0+
0+
CD22 Dim/absentBright
+–
0+
–+
0+
0+
0+
CD23 + 0 0 0 0 0
Light chain DimBright
+–
0+
0+
0+
0+
0+
CD79b – 0 – 0 0 0
CD11c Weak/moderateBright
+0
00
–+
00
+0
00
CD10 – 0 0 – 0 +CD25 0 0 + 0 – 0CD5 + 0 0 + 0 +FMC7 – 0 0 + 0 0*Only characteristic immunophenotypic features given; many exceptions reported†Grading system: + = Typical feature of the disease
0 = Not a defi ning feature of disease; may be present but does not have diagnostic specifi city– = Feature which does not support diagnosis of disease (can be used to refute diagnosis)
CLL = chronic lymphocytic leukemia; B-PLL = B-cell prolymphocytic leukemia; HCL = hairy cell leukemia; MCL = mantle cell lymphoma; SMZL = splenic marginal zone lymphoma; FL = follicular lymphoma
t22.10 Pattern and Morphology of Bone Marrow Involvement in B-Chronic Lymphoproliferative Neoplasms (B-CLPN)*
Disorder Aspirate Morphology Core Biopsy Features
CLL/SLL Monotonous small, round lymphocytes with scant cytoplasm
Focal nonparatrabecular infi ltrates predominate
Other patterns include diffuse interstitial or diffuse solid infi ltratesB-PLL Intermediately sized lymphocytes exhibiting round
nuclei with relatively condensed nuclear chromatin and prominent central nucleoli
Usually diffuse
HCL Distinctive cell with reniform nuclei, spongy “checkerboard” nuclear chromatin, and moderate to abundant amounts of slightly basophilic cytoplasm exhibiting circumferential shaggy contours
Diffuse interstitial and sinusoidal infi ltrates characteristic; can be very subtle and best appreciated by immunohistochemical assessment
Discrete, nodular lesions distinctly uncommonMCL Small to intermediately sized lymphocytes with
variably condensed chromatin and irregular nuclear contours; a variable number of cells may exhibit either prolymphocytic or blastic features
Usually focal nonparatrabecular and/or paratrabecular infi ltrates evident, although interstitial and diffuse lesions also described
FL Variable, but small cleaved lymphoid cells typically predominate
Focal paratrabecular lesions predominate
MZL (including splenic MZL)
Variable with admixture of plasmacytoid forms
Some cells may exhibit shaggy cytoplasmic contours that tend to be bipolar
Discrete nodular lesions very characteristic
May be associated with sinusoidal infi ltrates
May see “naked” germinal centers*See Chapter 24 for more detailsCLL/SLL = chronic lymphocytic leukemia/small lymphocytic leukemia; B-PLL = B-cell prolymphocytic leukemia; HCL = hairy cell leukemia; MCL = mantle cell lymphoma; FL = follicular lymphoma; MZL = marginal zone lymphoma including splenic MZL
References: [Campo 2008b, Foucar 2008, Muller-Hermelink 2008, Schlette 2003, Viswanatha 2010]

488
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
subsequent lymphoid lesions that develop in patients with CLL likely represent transformations of the original disease t22.12.
Th e most common transformation, occurring in 5% - 15% of cases of CLL, is the development of increased numbers of prolymphocytes, so-called prolymphocytoid transfor-mation of CLL. Th is transformation may be associated with progressive disease, loss of responsiveness to chemothera-peutic agents, and decreased survival time i22.30, i22.31, i22.32, i22.33. Th e increasing proportion of prolymphocytes with prominent central nucleoli may be linked to clonal progression on cytogenetic studies and to changes in the immunophentoypic profi le [Kroft 2001, Schlett e 2003, Viswanatha 2010]. If increased prolymphocytes are a feature at the time of initial presentation, disease course cannot be predicted, in that
some cases demonstrate a prolonged stable disease course, while others exhibit disease progression. In contrast, the development of a progressively rising prolymphocyte count in patients with established CLL is more compatible with a prolymphocytoid transformation. Th ese prolymphocytoid cells retain many of the immunophenotypic features of the original CLL, though both surface immunoglobulin and CD22 expression may be brighter, while CD23 and CD5 coexpression may diminish f22.3.
Th e development of a large cell lymphoma in a patient with CLL, so-called Richter syndrome, is a more dramatic event that occurs in 2% - 12% of patients with established CLL [Aydin 2008, Mao 2007, Richter 1928, Rossi 2008, Swords 2007]. Th is phenomenon is relatively more common in younger patients with CLL (≤55 years) [Mauro 1999]. Th ese patients experience
i22.28 Th is bone marrow core biopsy section is fr om an 86-year-old male with a long history of chronic lymphocytic leukemia ( CLL). Th is patient received chemotherapy and developed secondary acute myeloid leukemia ( AML) with complex cytogenetic abnormalities. Th e bone marrow core biopsy shows persistent CLL (upper half), while the therapy-related AML (lower half) is highlighted by atypical megakaryocytes. ( H&E)
i22.29 Immunoperoxidase staining for CD34 fr om an 86-year-old male with longstanding chronic lymphocytic leukemia ( CLL) who developed therapy-related acute myeloid leukemia ( AML) (see i22.28) shows markedly increased CD34+ cells in the area of AML (left ), while the CLL (right) is CD34–. (immunoperoxidase for CD34)
t22.11 Morphologic Features of Bone Marrow in Chronic B-Cell NeoplasmsCLL B-PLL HCL MCL SMZL FL
Grading System*Pattern Nodular
Diffuse, interstitial++
++
–+
+0
+0
+0
Cytology Closely packed nucleiWidely spaced nucleiRound nucleiIrregular nuclei (clefted, folded, indented)
+–+0
00+–
–+0+
+––+
0+00
+––+
Associated features
Fibrosis 0 0 + 0 0 0
*Grading System: + = Typical feature of the disease0 = Not a defi ning feature of disease; may be present but does not have diagnostic specifi city– = Feature which does not support diagnosis of disease (can be used to refute diagnosis)
CLL = chronic lymphocytic leukemia; B-PLL = B-cell prolymphocytic leukemia; HCL = hairy cell leukemia; MCL = mantle cell lymphoma; SMZL = splenic marginal zone
lymphoma; FL = follicular lymphoma

489
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
the abrupt onset of severe symptoms, oft en related to large, extramedullary lymphomatous mass lesions composed of pleomorphic, large immunoblastic cells. Foci of large cell transformation may be evident in the bone marrow of some patients, but in most patients the bone marrow is uninvolved at the time of extramedullary large cell
t22.12 Types of Transformation of CLLType of Transformation Comments
Prolymphocytoid Occurs in 5% - 15% of cases of CLLGradual increase in prolymphocytes in
blood and bone marrowMay be associated with progressive
symptoms, refractoriness to therapyPhenotype similar to original CLL but minor
changes describedDiffuse large B-cell
lymphoma or rarely Hodgkin-like transformation (Richter syndrome)
Occurs in 2% - 15% of CLL patients (15% at 10 years)
Abrupt onset of severe symptomsLarge mass lesions develop that may be
extranodalOften immunoblastic and pleomorphic;
rarely Hodgkin-likeVery aggressive disease with median
survival <1 yearClonal association with underlying CLL
established in most cases (about 80%)Other putative
transformationsRare reports of acute lymphoblastic
leukemia, Burkitt lymphoma, myeloma, hairy cell leukemia, and even aggressive T-cell disorders have been described as possible “transformations” of CLL
Supporting data marginal; may be secondary neoplasms
CLL = chronic lymphocytic leukemia
References: [Aydin 2008, Mao 2007, Richter 1928, Rossi 2008, Swords 2007]
i22.30 Th is cytospin preparation of a peripheral blood specimen fr om a 62-year-old female with chronic lymphocytic leukemia ( CLL) in prolympho-cytoid transformation highlights the spectrum of lymphoid cells, more abundant cytoplasm, and distinct nucleoli that are characteristic of this type of transfor-mation of CLL. Cytogenetic studies revealed 17p13 (TP53) deletion. ( Wright)
i22.31 Extensive diff use infi ltrates with substantial numbers of large lymphoid cells are illustrated on this bone marrow biopsy section fr om a patient with atypical chronic lymphocytic leukemia. ( H&E)
i22.32 Th is peripheral blood smear is fr om a 75-year-old female with a rising white blood cell count and no evidence of adenopathy. Th e fl ow cytometric immunophenotyping was classic for chronic lymphocytic leukemia ( CLL). A presumptive diagnosis of CLL in prolymphocytoid transformation was made. ( Wright)
i22.33 Th e corresponding bone marrow biopsy section (see i22.32) shows a prominent diff use chronic lymphocytic leukemia ( CLL)-like infi ltrate with individually dispersed large atypical cells with prominent nucleoli compatible with either prolymphocytoid transformation of CLL or early Richter transfor-mation. ( H&E)

490
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
lymphomatous transformation. Rarely, both blood and bone marrow involvement by large cell lymphoma may be evident i22.34, i22.35, i22.36, i22.37, i22.38. In some cases the large cell tranformation has a Hodgkin lymphoma appearance. Most recent evidence suggests a clonal relationship between the large cell lymphoma of Richter syndrome and CLL, but some cases, including Hodgkin-like Richter syndrome, represent secondary immunodefi ciency-associated disorders, oft en Epstein-Barr virus (EBV) induced [Mao 2007]. A link to fl udarabine therapy and subsequent Hodgkin lymphoma has been suggested in one study [Fong 2005].
Chronic lymphocytic leukemia factors linked to the subsequent development of Richter syndrome include unmutated IGHV, lymph node size, and absence of del(13q14) [Rossi 2008]. Other studies propose an association of Richter syndrome with CD38 gene polymorphisms, high levels of activation-induced cytidine deaminase, or aberrant somatic mutations [Aydin 2008, Reiniger 2006].
Other putative types of transformation of CLL are rare and include the development of either a B lymphoblastic leukemia or plasmablastic (myelomatous) blood and bone marrow picture, while both nodal paraimmunoblastic or Burkitt lymphomas have also been described t22.12, i22.39, i22.40. Data supporting a clonal association with the underlying CLL are limited in these largely older case reports.
i22.34 Th is peripheral blood smear is fr om a 66-year-old female with a long-term history of chronic lymphocytic leukemia who developed Richter transformation involving blood and bone marrow. ( Wright)
i22.35 A touch prep of a bone marrow core biopsy section fr om a 66-year-old female with long-term chronic lymphocytic leukemia who developed Richter syndrome shows a striking infi ltrate of immature-appearing large lymphoid cells (see i22.34). ( Wright)
i22.36 Imprint smear illustrates chronic lymphocytic leukemia along with large immunoblasts in a patient with Richter syndrome. ( Wright)
For p
lace
men
t only
0
200
400
600
1000
800
4002000 600 800 1000
side
sca
tter
forward scatter
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD23
CD5
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD20
CD5
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD20
CD23
f22.3 Transformation of CLL (black and yellow populations) to a more prolympho cytic process is characterized immunophenotypically by reduced expression of CD23 and increased overall cell size (red population on forward scatt er) in this composite fl ow cytometric histogram.

491
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
[22.3] B-Cell Prolymphocytic LeukemiaB-cell prolymphocytic leukemia is a very rare disorder
that is essentially defi ned by the morphologic features of the circulating monoclonal B cells, lacking both defi nitive immunophenotype and molecular/genetic features. Prolymphocytes, defi ned as cells with round nuclei, moderately condensed chromatin, and central prominent nucleoli, must exceed a threshold of 55% of lymphocytes, but in practice most cases exceed 90% [Campo 2008a]. Cases fulfi lling these morphologic criteria are oft en linked to a clinical/hematologic profi le that consists of age >60 years, marked leukocytosis, splenomegaly, and B symptoms [Campo 2008a, Del Giudice 2006]. Th e white blood cell (WBC) count generally exceeds 100 × 103/μL (100 × 109/L), consisting of a relatively homogeneous population of large cells with
huge central nucleoli and moderate amounts of pale blue, agranular cytoplasm i22.41,i22.42, i22.43. Bone marrow examination generally reveals extensive eff acement of normal hematopoietic cells and fat cells by the leukemic infi ltrate, which oft en exhibits either a diff use or a mixed nodular and diff use patt ern of infi ltration on biopsy sections i22.44 [Campo 2008a, Schlett e 2003]. Th ese leukemic infi ltrates are characterized by fairly widely spaced nuclei, prominent central nucleoli, and generally moderate mitotic activity; reticulin fi brosis may be present.
B-cell prolymphocytes exhibit a characteristic, though nonspecifi c, immunophenotypic profi le, including strong monoclonal surface immunoglobulin expression, as well as expression of CD19, CD20, CD22, CD79a, CD79b, and FMC7, while either CD5 or CD23 expression is present in
i22.38 Bone marrow biopsy sections show a focus of Richter syndrome in a patient with chronic lymphocytic leukemia. Note the immunoblastic appearance of these cells. ( H&E)
i22.37 Imprint smear depicts immunoblasts eff acing bone marrow in a case of Richter syndrome. ( Wright)
i22.39 Peripheral blood smear is fr om a patient with a 6-year history of chronic lymphocytic leukemia. Note prolymphocyte and blast-like forms with associated profound hematopoietic failure. ( Wright)
i22.40 Blast-like forms predominate in this peripheral blood smear fr om a patient with long-term chronic lymphocytic leukemia who developed an acute leukemic blood and bone marrow picture. ( Wright)
a b

492
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
only a minority of cases (10% - 30%) f22.4 [Campo 2008a]. Both CD38 and ZAP-70 are commonly expressed [Campo 2008a,
Del Giudice 2006]. A diagnosis of MCL or CLL with prolympho-cytoid transformation should be excluded in CD5+ cases. Indeed, one main focus of both immunophenotyping and cytogenetic assessment in cases of putative B-PLL is to exclude either leukemic MCL or CLL with prolym-phocytoid transformation [Matutes 2004, Ruchlemer 2004, Schlett e 2003]. In addition to karyotype, cyclin D1 staining is helpful in identifying cases of MCL that mimic B-PLL.
Th ere are no recurrent cytogenetic abnormalities in B-PLL, and complex cytogenetic abnormalities are common. Unlike CLL, neither genetics nor CD38/ZAP-70 expression is linked to survival [Campo 2008a, Del Giudice 2006]. Similarly, the presence or absence of somatic hypermutation is not predictive of outcome. As anticipated by the absence of either clear-cut molecular/
i22.41 Th is peripheral blood smear is fr om an 88-year-old male with a striking leukocytosis. Flow cytometry confi rmed a B-cell phenotype. Based on the nucleoli, the rapidly rising white blood cell count, and the clinical features, a diagnosis of B-cell prolymphocytic leukemia was made. ( Wright)
i22.42 Classic features of prolymphocytes are evident on this peripheral blood smear fr om a 76-year-old male with marked leukocytosis and moderate anemia. Note single distinctive nucleolus in each nuclei. ( Wright)
i22.43 Blood smears illustrate the morphologic spectrum of prolymphocytes in 2 cases of B-cell prolymphocytic leukemia. ( Wright)
i22.44 Th is bone marrow core biopsy section shows an extensive diff use infi ltrate of B-cell prolymphocytic leukemia. Note prominent nucleoli. ( H&E)
a b
For p
lace
men
t only
For p
lace
men
t only
For p
lace
men
t only
For p
lace
men
t only
For p
lace
men
t only
For p
lace
men
t only
0
256
512
768
1024
0 256 512 768 1024
100
101
102
103
104
100 101 102 103 104
100
101
102
103
104
100 101 102 103 104
100
101
102
103
104
100 101 102 103 104
100
101
102
103
104
100 101 102 103 104
100
101
102
103
104
100 101 102 103 104
side
sca
tter
forward scatter
CD22
CD5
λλ
κ
CD23
FMC7
CD20
κ
CD19
CD22
f22.4 Th ese fl ow cytometric histograms show a typical immunophenotypic profi le of B-cell prolymphocytic leukemia. Note absence of distinctive immuno-phenotypic features of this clonal B-cell leukemia, both CD23+ and FMC7+.

493
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
genetic or immunophenotypic features, the cell of origin of B-PLL is an unknown mature B cell [Campo 2008a].
Patients presenting with striking leukocytosis and marked organomegaly generally experience an aggressive disease course with median survival times ranging from 30 - 50 months. Unlike CLL, optimal staging and treatment strategies are not clear-cut, and treatment responses are generally poor, possibly the consequence of consistently detected p53 abnormalities [Campo 2008a].
[22.4] Hairy Cell Leukemia
[22.4.1] Defi nition and General FeaturesHairy cell leukemia is an indolent mature B-cell neoplasm
derived from cells with distinctive morphology (circum-ferential hairy projections) which exhibit a propensity for diff use bone marrow and splenic red pulp infi ltration [Foucar 2008]. Evidence supports an activated mature memory B cell of origin [Forconi 2004]. HCL is rare, accounting for <5% of chronic leukemias.
Hairy cell leukemia typically aff ects middle-aged to elderly patients (median age, 50 years), usually men (male-to-female ratio of 4:1), who present with splenomegaly and pancytopenia [Robak 2006]. HCL can occur as early as the third decade of life, but it is extremely unusual in patients <20 years of age [Foucar 2008] . Splenomegaly is a typical clinical feature in patients with HCL; however, because of earlier detection, the incidence of marked splenomegaly has declined. Hepatomegaly is variable but common, while most patients with HCL lack signifi cant lymphadenopathy. Patients with HCL are oft en symptomatic, either from splenomegaly, producing left upper quadrant pain, or from the consequences of cytopenias (fever, fatigue, infection) [Hoff man 2006].
Th e clinical course of patients with HCL is generally indolent. Asymptomatic patients do not require therapy. In symptomatic patients, remarkable hematologic responses have been achieved using purine analogy therapy (eg, 2-chlorodeoxyadenosine, cladribine, or 2-CDA) and other agents [Belani 2006, Robak 2006]. Th e overall 10-year survival rate exceeds 90%; median relapse-free survival times of 16 years have been reported [Chadha 2005, Else 2009].
[22.4.2] Diagnosis of HCLAlthough HCL lacks a defi ning recurrent cytogenetic
abnormality, this disease has numerous distinctive clinical and pathologic features. A list of prototypic clinical, hematologic, cyto/morphologic, and immunophenotypic features is provided in t22.13. Th e successful diagnosis of HCL requires a high index of suspicion in conjunction with the integration of these diverse parameters.
[22.4.3] Blood FindingsEither single or multilineage cytopenias dominate
blood fi ndings in patients with HCL [Sharpe 2006]. Leukopenia, characterized by both neutropenia and marked monocy-topenia, is the most common abnormality. Anemia is also a frequent fi nding in these patients. Erythrocytes are generally normocytic and normochromic, and the cause of anemia is multifactorial, including bone marrow production failure, splenic sequestration, and dilutional anemia secondary to increased plasma volume. Although characteristically inconspicuous, low numbers of hairy cells can oft en be identifi ed on peripheral blood smears. A high index of suspicion is essential, and areas to include in scanning of the blood smear include the feather edge and thicker regions on the slides. Aside from reduced numbers, there are no qualitative abnormalities of neutrophils, monocytes, or platelets, a useful fi nding in distinguishing HCL from myelodysplasia.
t22.13 Prototypic Features of HCL
Parameter Comments
Clinical Older, male, splenomegaly, possible recurrent infections
Blood Pancytopenia, profound monocytopenia, and neutropenia
Rare circulating hairy cells (inconspicuous)
Cytology Round to oval nuclei with “spongy” chromatin; inconspicuous nucleoli
Abundant pale cytoplasm with circumferential projections
Cytochemistry Strong tartrate-resistant acid phosphatase positivity (TRAP) (can also be assessed by immunohistochemistry)
Bone marrow Inaspirable in most cases due to diffuse reticulin fi brosis directly associated with hairy cell infi ltrates
Subtle, patchy infi ltrates on core biopsy (may be inconspicuous)
Leukemic cells show wide collar of cytoplasm (fried egg appearance)
Flow cytometric immunophenotype
Monoclonal B cells with bright SIg, bright CD20, bright CD22, bright CD11c, bright CD103, and CD25
Subset CD10+Immuno histo-
chemical featuresPositive for CD20, annexin-1, T-bet, CD123,
DBA.44, TRAP; subset weakly cyclin D1+HCL = hairy cell leukemia
References: [Del Giudice 2004, Dong 2009, Falini 2004, Foucar 2008, Johrens 2007, Matutes 2006, Sharpe 2006,
Went 2005]

494
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
[22.4.4] Morphologic and Cytochemical Features on Smear Preparations
Th e nuclear and cytoplasmic features of classic hairy cells on smear preparations are well-delineated t22.13 [Foucar 2008,
Sharpe 2006]. Th e nuclei of prototypic hairy cells range from round to oval, containing homogeneous, spongy, ground-glass chromatin with absent or inconspicuous nucleoli i22.45, i22.46. Th e cytoplasm is abundant to voluminous, slate blue, and contains poorly delineated, patchy variations in the intensity of cytoplasmic staining sometimes described as fl occulent. Occasional vacuoles, granules, and rod-shaped inclusions may be identifi ed. Th ese rod-shaped inclusions correspond to the ribosome lamellar complexes identifi ed in ultrastructural studies [Cawley 2006]. Th e cytoplasmic membrane is characterized by numerous hairy projections, which are generally uniformly distributed around the circumference.
When aspiration is successful, abundant mast cells are oft en admixed with hairy cell infi ltrates i22.47 [Macon 1993].
Th e disorder formerly designated as HCL-variant is categorized by the 2008 WHO criteria as a type of splenic diff use red pulp B-cell lymphoma, and its relationship to HCL has been questioned (see [22.4.8] “Diagnostic Criteria and Diff erential Diagnosis,” p 499) [Piris 2008] . However, several other bona fi de morphologically distinctive variants of HCL have been described. Despite the unusual morphologic character-istics, the diagnosis of HCL can be established in these cases by integrating cytochemical stain reactions, immunophe-notypic features, clinical fi ndings, and patt erns of either bone marrow or splenic infi ltration. Th ese morphologic variants of HCL include convoluted, multilobulated, blastic, and spindled
i22.45 Blood smears highlight some nuclear variability in 3 hairy cells. ( Wright)
i22.46 Blood smears illustrate 3 characteristic cells fr om hairy cell leukemia. ( Wright)
i22.47 Increased mast cells are present on this bone marrow aspirate smear fr om a patient with extensive bone marrow eff acement by hairy cell leukemia. ( Wright)
i22.48 Rare unusual circulating cells are evident in the peripheral blood specimen of a patient with confi rmed hairy cell leukemia based on the splenic features and immunophenotype. Th e complete blood count showed a low white blood cell count with monocytopenia. Note large nuclear size and irregularity with subtle circumferential hairy projections. ( Wright)
a b ca b c

495
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
cell variants; all are infrequently encountered in clinical practice [Bartl 1983, Catovsky 1984, Diez Martin 1987, Hanson 1989, Nazeer 1997]. Th ese morphologically unique subtypes of HCL can usually be identifi ed on both smears and sections. In both the convoluted and multilobulated subtypes, nuclear irregularity is frequent with lobulated, folded, cerebriform confi gurations i22.48. Th e blastic variant is characterized by an oval or indented nuclear contour, a fi nely dispersed nuclear chromatin patt ern, and a central nucleolus i22.49. Elongated or spindled hairy cells may be evident on smears but tend to be more prominent on tissue biopsy sections (see next section). In all of these hairy cell variants, the cytoplasmic, cytochemical, and immunophe-notypic features of prototypic HCL are retained.
Th e strong, fairly uniform reaction of hairy cells with tartrate-resistant acid phosphatase (TRA P) stains is useful in the diagnosis of HCL, even though the cytochemical stain has been replaced by the immunohistochemical method in many laboratories [ Janckila 1995, Went 2005]. In most cases, a classic intense, diff use cytoplasmic reaction product is identifi ed in at least a subset of the leukemic cells i22.50. Cases of HCL that fail to exhibit any TRA P reactivity are very rare, ranging from 1% - 5% in published series. Both normal myeloid and histiocytic cells and other neoplasms including leukemias, lymphomas, and mast cell disease can all exhibit some degree of TRA P positivity [Hoyer 1997, Janckila 1995]. However, if morphologic features, the intensity of the TRA P reaction product, and immunophenotypic fi ndings are considered, cases of HCL can generally be distinguished from these other neoplastic and non-neoplastic TRA P+ cell types.
[22.4.5] Bone Marrow Biopsy FindingsBecause of the very high frequency of unsuccessful
aspiration, the bone marrow core biopsy is invaluable in establishing a diagnosis of HCL, despite the variation in
both the patt ern and extent of HCL infi ltrates evident on these sections [Foucar 2008, Sharpe 2006]. Th e patt erns of bone marrow involvement in HCL include subtle patchy infi ltrates, diff use interstitial infi ltrates, and diff use solid infi ltrates that completely eff ace fat and hematopoietic cells. Th e patchy infi ltrates are very poorly delineated from surrounding hematopoietic cells, oft en requiring both a high index of suspicion and very careful morphologic review for their identifi cation i22.51. Th ese subtle infi ltrates can be highlighted by immunoperoxidase staining using pan B-cell antibodies and many other more HCL-specifi c/characteristic antibodies (see next section). Th e readily identifi able discrete lesions that are characteristic of bone marrow involvement by many other lymphoproliferative neoplasms are not a typical feature of bone marrow involvement by HCL i22.52. Sinusoidal
i22.49 Blood smears illustrate blastic hairy cells. ( Wright) i22.50 Intense, uniform tartrate-resistant acid phosphatase( TRA P) positivity is evident in these hairy cells on a bone marrow imprint smear. Cytochemical staining for TRA P has largely been replaced by immunohistochemical staining. (cytochemical stain for TRA P)
i22.51 Th is bone marrow core biopsy section shows a subtle infi ltrate of hairy cell leukemia ( HCL). Note the monotonous appearance of these cells along with widely spaced nuclei, both typical features of HCL. ( H&E)
a b

496
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
infi ltration in HCL is fairly common, but this distinctive patt ern of infi ltration is admixed with the diff use infi ltrates and is oft en subtle.
Early reports on the morphology of bone marrow biopsy involvement by HCL emphasized the “fried egg” appearance of these infi ltrates. Th is appearance resulted from the wide spacing of the nuclei secondary to the abundant cytoplasm of the hairy cells, distinct cytoplasmic membranes, and cleared-out cytoplasm i22.53 [Bouroncle 1994, Hounieu 1992]. With the subsequent development of improved fi xation techniques, this “fried egg” appearance is much less conspicuous. Th e nuclei, however, are still widely spaced, round to oval, and exhibit very litt le mitotic activity. Th e cytoplasm is moderate to abundant i22.54.
A spectrum analogous to the variant morphologic forms of hairy cells described on aspirate smears can be identifi ed on well-fi xed and thinly sectioned bone marrow biopsy specimens i22.55, i22.56, i22.57. Good correlation
exists between the identifi cation of blastic and convoluted morphologic characteristics on both aspirate smears and biopsy sections. In addition, the spindled cell and osteosclerotic variants of HCL can be identifi ed exclusively on biopsy sections i22.56, i22.57.
Fibrosis in HCLTh ere is a consistent association between hairy cell
infi ltrates and reticulin fi brosis in virtually all organs studied, accounting for the high rate of aspiration failure in this disorder. Factors released by hairy cells such as fi bronectin and/or transforming growth factor β1 are the likely cause of this fi brosis i22.58 [Aziz 2003, Shehata 2004, Tiacci 2006]. Th e patt ern of fi brosis varies from case to case, paralleling the extent of hairy cell infi ltrates. For example, in cases of HCL with minimal patchy infi ltrates, the reticulin fi brosis is localized to these minute foci of bone marrow involvement, while diff use infi ltrates are associated with extensive diff use reticulin fi brosis. Rare cases of HCL are associated with both reticulin fi brosis and osteosclerosis; both lytic and blastic lesions have been noted radiographically [Filippi 2007, VanderMolen 1989].
Unusual Mast Cell and Vascular LesionsSeveral additional fi ndings noted on some bone marrow
biopsy sections from patients with HCL include increased numbers of mast cells and vascular lesions [Macon 1993]. Increased numbers of mast cells are consistently identifi ed on aspirate smears; these cells are also sometimes evident on biopsy sections. In rare cases, these mast cell aggregates are distinctive enough on biopsy sections to suggest concordant HCL and systemic mastocytosis i22.59. Even less commonly, increased numbers of dilated vascular spaces producing angiomatous lesions may also be evident on bone marrow biopsy sections from patients with HCL reminiscent of the vascular lakes described in the spleen i22.60.
i22.52 Low magnifi cation of a bone marrow core biopsy section shows extensive infi ltration by hairy cell leukemia ( HCL) with a diff use interstitial and partially solid infi ltrate without apparent nodular lesions. Th is low magnifi cation appearance is characteristic of HCL in which there is massive bone marrow involvement. ( H&E)
i22.53 Bone marrow biopsy sections illustrate the classic “fr ied-egg” appearance of hairy cells on biopsy sections. ( H&E)
i22.54 Well-fi xed bone marrow biopsy sections highlight the appearance of hairy cell leukemia at various magnifi cations. ( H&E)
a b
a b c

497
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
i22.55 Convoluted hairy cell leukemia is illustrated on this bone marrow biopsy section. ( H&E)
i22.56 Th is bone marrow core biopsy section is fr om a 64-year-old female with profound pancytopenia. Th e specimen shows osteosclerosis along with a relatively subtle infi ltrate of hairy cell leukemia, which was highlighted on immunohisto-chemical staining. ( H&E)
i22.59 A prominent perivascular infi ltrate containing abundant mast cells is present on this bone marrow biopsy section fr om a patient with hairy cell leukemia. ( H&E)
i22.60 Bone marrow biopsy sections illustrate the unique focal angiomatous patt ern present in this case of hairy cell leukemia. ( H&E)
i22.57 Bone marrow biopsy sections illustrate a case of spindled hairy cell leukemia, which can closely mimic systemic mastocytosis. ( H&E)
i22.58 Th e spectrum of reticulin fi brosis on bone marrow biopsy sections fr om patients with hairy cell leukemia ( HCL) is illustrated. When fi brosis is patchy, it tracks with patchy HCL infi ltrates a. (reticulin)
a b a b
a b

498
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
Hypocellular HCLAnother unique feature of HCL on bone marrow biopsy
sections is the marked variability of cellularity of residual hematopoietic cells, even when hairy cell infi ltration is minimal i22.61. Up to 1/3 of cases of HCL exhibit marked bone marrow hypocellularity mimicking aplastic anemia [Foucar 2008, Sharpe 2006]. In these cases of hypocellular HCL, the granulocytic lineage is more potently suppressed than either the erythroid or megakaryocytic cell lines. Several publications suggest that inhibition of normal hematopoiesis in these patients is the consequence of either excess cytokine production (notably tumor necrosis factor α or transforming growth factor β1) by the hairy cells or inadequate colony-stimulating factor production by the bone marrow microenvironment [Cawley 2006,
Tiacci 2006]. Immunoperoxidase staining for CD20 is an essential
screening stain used to highlight subtle hairy cell infi ltrates in these hypocellular specimens i22.62. Once the occult B-cell infi ltrate has been identifi ed, further immunohistochemical stains are warranted (see below and t22.14).
i22.61 Patchy infi ltrates of hairy cell leukemia are subtle on these markedly hypocellular bone marrow biopsy sections. ( H&E)
i22.62 Immunoperoxidase staining for CD20 highlights the subtle, interstitial infi ltrates that predominate in this case of hypocellular hairy cell leukemia. (immunoperoxidase for CD20)
a b t22.14 Immunohistochemical Studies in HCL
Antibody Reaction Pattern Comments
CD20 Surface membrane Best marker for screening specimens for occult B-cell infi ltrates
Expression lost in patients on rituximab
CD79a Cytoplasmic “Back-up” stain for CD20 in detecting occult B-cell infi ltrates
Also highlights plasma cells
Annexin 1 Cytoplasmic Antibody identifi ed by gene expression profi ling
Highly specifi c for HCL, but cross-reacts with myeloid cells limiting utility in partially involved bone marrow specimens
T-bet Nuclear T-cell associated transcription factorStrongly positive in HCL, but weak
positivity in many other B-cell neoplasms
Not coexpressed by hematopoietic cells
Useful in the detection of minimal residual disease
CD123 Surface membrane Expressed by variety of normal hematopoietic cells, acute leukemias, and HCL
Moderate to strong reactivity in HCLMost useful in distinguishing HCL
from SMZLDBA.44 Surface membrane
(highlights hairy projections)
Positive in HCL, but also positive in other B-CLPN
Most useful when combined with other antibodies (eg, TRAP)
TRAP Cytoplasmic granules
Expressed in variety of neoplasms and benign histiocytes
More strongly positive in HCL compared to other B-CLPN
Most useful when combined with other antibodies
B-CLPN = B-chronic lymphoproliferative neoplasms; HCL = hairy cell leukemia; SMZL = splenic marginal zone lymphoma; TRAP = tartrate-resistant acid phosphatase cytochemical stain
References: [Del Giudice 2004, Falini 2004, Johrens 2007, Went 2005]
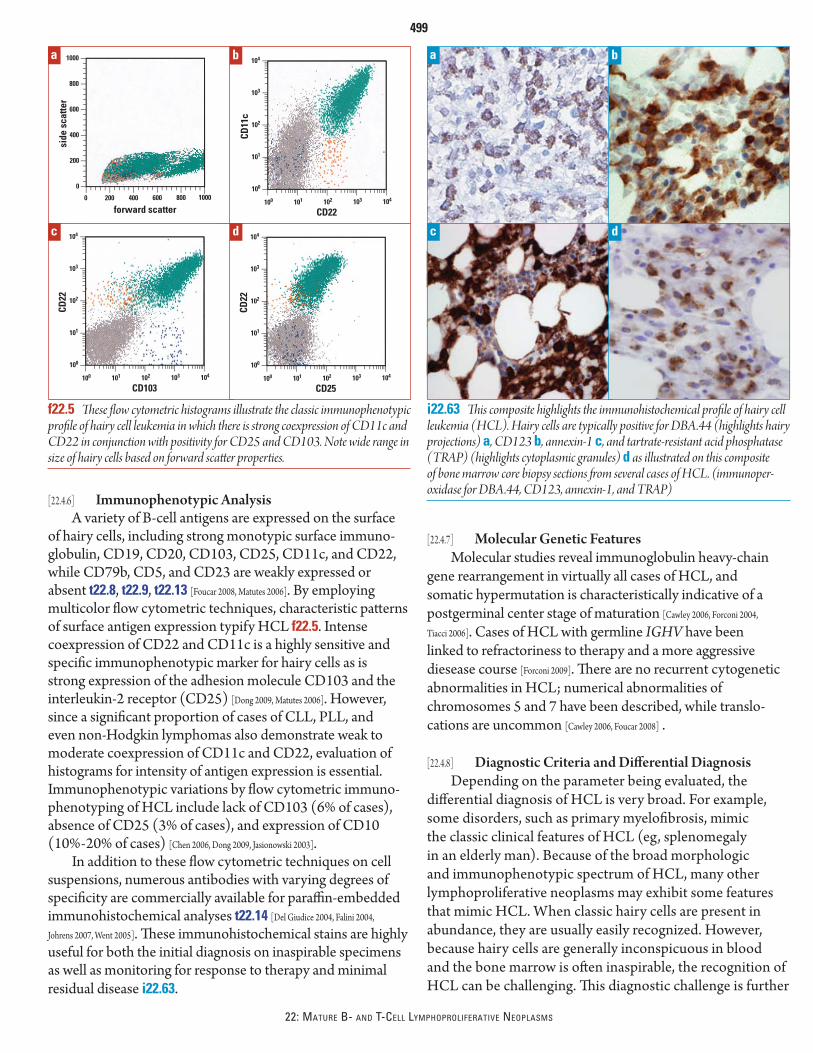
499
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
[22.4.6] Immunophenotypic AnalysisA variety of B-cell antigens are expressed on the surface
of hairy cells, including strong monotypic surface immuno-globulin, CD19, CD20, CD103, CD25, CD11c, and CD22, while CD79b, CD5, and CD23 are weakly expressed or absent t22.8, t22.9, t22.13 [Foucar 2008, Matutes 2006]. By employing multicolor fl ow cytometric techniques, characteristic patt erns of surface antigen expression typify HCL f22.5. Intense coexpression of CD22 and CD11c is a highly sensitive and specifi c immunophenotypic marker for hairy cells as is strong expression of the adhesion molecule CD103 and the interleukin-2 receptor (CD25) [Dong 2009, Matutes 2006]. However, since a signifi cant proportion of cases of CLL, PLL, and even non-Hodgkin lymphomas also demonstrate weak to moderate coexpression of CD11c and CD22, evaluation of histograms for intensity of antigen expression is essential. Immunophenotypic variations by fl ow cytometric immuno-phenotyping of HCL include lack of CD103 (6% of cases), absence of CD25 (3% of cases), and expression of CD10 (10% - 20% of cases) [Chen 2006, Dong 2009, Jasionowski 2003].
In addition to these fl ow cytometric techniques on cell suspensions, numerous antibodies with varying degrees of specifi city are commercially available for paraffi n-embedded immunohistochemical analyses t22.14 [Del Giudice 2004, Falini 2004,
Johrens 2007, Went 2005]. Th ese immunohistochemical stains are highly useful for both the initial diagnosis on inaspirable specimens as well as monitoring for response to therapy and minimal residual disease i22.63.
[22.4.7] Molecular Genetic FeaturesMolecular studies reveal immunoglobulin heavy-chain
gene rearrangement in virtually all cases of HCL, and somatic hypermutation is characteristically indicative of a postgerminal center stage of maturation [Cawley 2006, Forconi 2004,
Tiacci 2006]. Cases of HCL with germline IGHV have been linked to refractoriness to therapy and a more aggressive diesease course [Forconi 2009]. Th ere are no recurrent cytogenetic abnormalities in HCL; numerical abnormalities of chromosomes 5 and 7 have been described, while translo-cations are uncommon [Cawley 2006, Foucar 2008] .
[22.4.8] Diagnostic Criteria and Diff erential DiagnosisDepending on the parameter being evaluated, the
diff erential diagnosis of HCL is very broad. For example, some disorders, such as primary myelofi brosis, mimic the classic clinical features of HCL (eg, splenomegaly in an elderly man). Because of the broad morphologic and immunophenotypic spectrum of HCL, many other lymphoproliferative neoplasms may exhibit some features that mimic HCL. When classic hairy cells are present in abundance, they are usually easily recognized. However, because hairy cells are generally inconspicuous in blood and the bone marrow is oft en inaspirable, the recognition of HCL can be challenging. Th is diagnostic challenge is further
For p
lace
men
t only
0
200
400
600
1000
800
4002000 600 800 1000
side
sca
tter
forward scatter
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD22
CD103
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD11
c
CD22
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD22
CD25
f22.5 Th ese fl ow cytometric histograms illustrate the classic immunophenotypic profi le of hairy cell leukemia in which there is strong coexpression of CD11c and CD22 in conjunction with positivity for CD25 and CD103. Note wide range in size of hairy cells based on forward scatt er properties.
a ab b
c cd d
i22.63 Th is composite highlights the immunohistochemical profi le of hairy cell leukemia ( HCL). Hairy cells are typically positive for DBA.44 (highlights hairy projections) a, CD123 b, annexin-1 c, and tartrate-resistant acid phosphatase ( TRA P) (highlights cytoplasmic granules) d as illustrated on this composite of bone marrow core biopsy sections fr om several cases of HCL. (immunoper-oxidase for DBA.44, CD123, annexin-1, and TRA P)

500
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
complicated by the variant forms of HCL that overlap with other B-CLPNs. Splenomegalic disorders such as splenic marginal zone lymphomas, B-PLL, splenic B-cell leukemias, unclassifi able (including HCL-variant), and splenomegalic MCL show substantial overlap of both morphologic and immunophenotypic features with HCL (see later sections for images). Th ese disorders comprise the primary diff er-ential diagnostic considerations in HCL t22.15. However, rare cases of either plasma cell leukemia or myeloma can closely mimic HCL i22.64.
[22.4.9] Possible Clonal TransformationsTh ere is an increased incidence of second lymphoid
neoplasms in patients with HCL. Th ese include both B- and T-cell neoplasms, but B-cell disorders predominate. Although few molecular studies have been performed, an overlap of morphologic and immunophenotypic features suggest that at least some B-cell disorders identifi ed in patients with
i22.64 Th is composite of plasma cell leukemia a and myeloma b closely mimic hairy cell leukemia. Note prominent circumferential projections in circulating cells in plasma cell leukemia and fr ied egg appearance of bone marrow core biopsy section in a patient with extensive eff acement by myeloma. Th e plasma cells closely resemble hairy cells, with round nuclei and abundant cleared cytoplasm. Note Russell bodies. ( Wright, H&E)
t22.15 Comparison of Features of Splenomegalic B-Chronic Lymphoproliferative Neoplasms (B-CLPN)
Feature B-PLL SMZL* HCLSplenic B-Cell Leukemia, Unclassifi able (HCL Variant)
MCL (Splenomegalic Subtype)
WBC Very high Variable, but not markedly increased
Low (monocytopenia) Moderately increased (normal monocyte count)
Variable, often moderately increased
Morphology of lymphoid cells
Moderately sized, round nuclei, condensed chromatin, striking central nucleoli
Small to moderately sized, round nuclei, condensed chromatin, inconspicuous nucleoli
Scant to moderate basophilic cytoplasm, bipolar villous projections
Plasmacytoid features common
Moderately sized, round to reniform nuclei, dispersed “spongy” chromatin
Abundant pale cytoplasm, circumferential hairy projections
Several variants described
Moderately sized, condensed chromatin, prominent central nucleoli
Moderate to abundant cytoplasm, circumferential hairy projections
Spectrum of cell size, usually condensed, nuclear chromatin, irregular nuclear contours, scant to moderate cytoplasm
Blastoid variant may show prominent nucleoli
Bone marrow sections
Nodular and/or diffuse infi ltrates
Intrasinusoidal and nodular Interstitial (sometimes subtle) or diffuse infi ltrates, variable intrasinusoidal
30% hypocellular
Distinctly sinusoidal; also interstitial
Discrete nodules, usually non-paratrabecular, but may be paratrabecular
TRAP Negative to weak
Negative to moderate Strong (in at least a portion of cells)
Negative to weak Negative
IP Strong surface Ig, CD19, CD20, CD22, FMC7, CD79b
Variable CD5 expression
CD11, CD25, CD123, CD103 usually absent
Strong surface Ig, CD19, CD20, CD22, FMC7, CD79b, CD11c
CD5, CD23, CD25, CD123, and CD103 usually absent or present in <20% of cases
Bright surface Ig, CD19, CD20, CD22, CD11c, CD103, CD25, CD103, FMC7
Also express CD123, annexin-1, t-bet
Lack CD5, CD79b
Strong surface expression of Ig, CD20, CD11c, CD103, FMC7
Negative for CD123, annexin-1, CD25 , and TRAP
Strong surface expression of Ig, CD20, FMC7, CD79b, moderate CD5, variable CD23
Negative for CD10, CD11c
*See Chapter 24 for illustrationsB-PLL = B-cell prolymphocytic leukemia; HCL = hairy cell leukemia; IP = immunophenotype; MCL = mantle cell lymphoma; SMZL = splenic marginal zone lymphoma; TRAP = tartrate-resistant acid phosphatase cytochemical stain; WBC = white blood cell count
References: [Campo 2008a, Dong 2009, Falini 2004, Foucar 2008, Isaacson 2008, Johrens 2007, Matutes 2004, 2006, 2008, Piris 2008, Ruchlemer 2004]
a b

501
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
longstanding HCL may represent clonal transformations of the HCL i22.65 [Nazeer 1997, Sun 2004]. Likewise, morphologic features suggesting 2 distinct lymphoid processes are occasionally identifi ed on bone marrow biopsy sections of cases of HCL i22.66.
[22.5] Peripheralizing B-Cell Lymphomas
[22.5.1] Splenic Marginal Zone Lymphoma (SMZL)Even though SMZL is a recognized subtype of
non-Hodgkin lymphoma, this disorder is included in this chapter because a leukemic blood picture is typical at presen-tation (see Chapter 24 for a more detailed discussion). Th is
type of non-Hodgkin lymphoma aff ects elderly men who present with symptoms secondary to splenomegaly. Peripheral blood examination reveals mild anemia and/or thrombocy-topenia, while the WBC count is moderately elevated, rarely exceeding 40 × 103/μL (40 × 109/L) t22.15 [Isaacson 2008, Matutes 2006,
Matutes 2008]. Cytologic heterogeneity is evident on blood and bone marrow aspirate smears. Overall cell size and amount of cytoplasm are variable. Some cells are typically plasma-cytoid with bipolar cytoplasmic projections, while others exhibit HCL-like circumferential projections i22.67, i22.68. Th e immunophenotypic profi le of SMZL is provided in t22.8 and t22.15 [Campo 2008a, Foucar 2008, Isaacson 2008, Matutes 2004, 2006, 2008, Piris 2008,
Ruchlemer 2004]. On bone marrow biopsy sections, the infi ltrate is characteristically nodular with readily apparent infi ltrates at low
i22.65 A hairy cell leukemia-like blood picture preceded by 12 years the development of mantle cell lymphoma in lymph node and multiple lympho-matous polyposis in bowel. All 3 lesions are derived fr om the same clone. ( Wright)
i22.66 A large, dense lymphoid infi ltrate is admixed with extensive hairy cell leukemia infi ltrates on these bone marrow biopsy sections, suggesting a composite disorder. ( H&E)
i22.67 Low magnifi cation of a peripheral blood smear fr om a 54-year-old male with a modest leukocytosis (white blood cell count of 13.4 × 103/μL [13.4 × 109/L]) exhibits a spectrum of lymphocytes showing variable cytologic features in conjunction with mild rouleaux compatible with splenic marginal zone lymphoma. ( Wright)
i22.68 Th is composite of peripheral blood cells in several patients with splenic marginal zone lymphoma highlights the variability and degree of hairy projections that can be seen in this disorder. ( Wright)
a b a b
a b

502
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
magnifi cation i22.69, i22.70. In addition, intrasinusoidal invasion is a common and distinctive feature of SMZL that can be highlighted by immunohistochemical staining i22.71 [Isaacson 2008,
Matutes 2006, 2008]. Th e disease course of SMZL is indolent, and some patients may not initially require therapy.
[22.5.2] Hairy Cell Leukemia-Variant (HCL-v)Although the acronym HCL-v implies a relationship to
classical HCL, this rare disorder is currently considered to be a subtype of splenic B-cell leukemia, unclassifi able [Piris 2008]. In this rare, poorly defi ned disorder, the cytologic features of the circulating cells in the blood typically show hybrid features between classical HCL and B-PLL t22.15 i22.72 [Matutes 2003,
Piris 2008] . In addition to splenomegaly, leukocytosis is common, and, unlike classical HCL, monocytes are present in normal
numbers. Cytologic features on blood smears may be quite variable with a range in cell types. Nuclei are typically round, but may be convoluted, and nucleoli are generally prominent. Cytoplasmic projections are usually present, but variable i22.73. Bone marrow infi ltrates may be subtle with a predilection for sinusoidal infi ltration i22.74 [Dong 2009, Matutes 2003, Ya-In 2005]. Th e immunophenotype is also variable, but the classical HCL “profi le” is lacking in that CD25, annexin-1, CD123, and TRA P are characteristically negative [Cessna 2005, Dong 2009]. VH4-34 gene usage is common in cases classifi ed as HCL-v and is linked to unmutated IGHV and adverse outcomes [Arons 2009].
[22.5.3] Other NHLs with Leukemic PictureIn addition to SMZL, peripheralizing lymphoma cells can
be identifi ed in a substantial proportion of patients with other
i22.69 Low magnifi cation of a bone marrow core biopsy section of a patient with splenic marginal zone lymphoma ( SMZL) highlights discrete lymphoid aggregates that characterize this disease. Of note is the germinal center formation in the center of the larger aggregate, another characteristic feature of bone marrow infi ltrates of SMZL. ( H&E)
i22.70 Intermediate and high magnifi cation of a bone marrow core biopsy section fr om a patient with splenic marginal zone lymphoma highlights the widely spaced nuclei, generally round nuclear contours, and lack of mitotic activity that characterize this disorder. ( H&E)
i22.71 CD20 immunoperoxidase staining highlights both discrete aggregates and sinusoidal infi ltrates in splenic marginal zone lymphoma on this bone marrow core biopsy section. (immunoperoxidase for CD20)
i22.72 Th is circulating monoclonal B cell highlights the hybrid features of this hairy cell leukemia ( HCL) variant in which the nucleus resembles that of prolymphocytic leukemia, while the cytoplasm shows circumferential projections characteristic of HCL. ( Wright)
a
a
b
b

503
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
types of NHL. However, in contrast to SMZL, the peripheral blood involvement in other types of NHL does not usually produce a de novo leukemic blood picture, except for the splenomegalic subtype of MCL t22.15 [Matutes 2004, Ruchlemer 2004]. Likewise, peripheralization of MCL can also occur later in the disease course in a fair proportion of cases. In both de novo leukemic MCL and cases in which peripheralization is a late feature, a morphologic spectrum has been described t22.15. Some cases exhibit a mature nuclear chromatin confi gu-ration with moderate nuclear irregularity, while other cases have a distinctly blastic appearance with variably prominent nucleoli; overlap with prolymphocytic leukemia and even acute leukemia is common i22.75 [Ruchlemer 2004, Schlett e 2003].
For most other types of NHL, the identifi cation of substantial numbers of circulating lymphoma cells occurs in association with longstanding NHL and extensive bone marrow replacement. In these patients, the blood-bone marrow barrier is thought to be disrupted by the neoplastic infi ltrate, resulting in the release of these cells into the blood. Circulating lymphoma cells are generally morphologically similar to those seen on bone marrow aspirate smears. Although small cell lymphomas such as follicular and mantle cell lymphomas account for the majority of peripheralized lymphomas, both lymphoblastic and large cell lymphoma cells may also circulate (see also Chapter 24). In ~10% of patients with NHL, a frank leukemic peripheral blood picture develops in which a striking leukocytosis is evident i22.76. On immunophenotyping studies, the majority of peripheralized lymphomas are of B-cell type, and follicular lymphomas demonstrating coexpression of CD10 predominate t22.8, t22.9. Peripheralized MCLs exhibit
i22.73 Th is peripheral blood smear is fr om an elderly patient with a longstanding monoclonal B-cell neoplasm characterized by moderate leukocytosis (white blood cell count of 24.9 × 103/μL [24.9 × 109/L]) with preserved hematopoiesis and a normal absolute monocyte count. Th e spectrum of morphology of the circulating monoclonal B cells is highlighted. Some cells have shaggy cytoplasmic contours, while others have more prominent nucleoli. ( Wright)
i22.74 CD20 staining on this bone marrow clot section highlights the prominent intrasinusoidal patt ern of infi ltration in this bone marrow specimen fr om a patient with hairy cell leukemia variant. (immunoperoxidase for CD20)
i22.75 Composite of peripheral blood smears fr om 2 patients with periph-eralized mantle cell lymphoma illustrates the spectrum of cytologic features that characterizes this disorder, ranging fr om circulating cells with condensed chromatin a to lymphoblast-like circulating forms b. ( Wright)
a b
i22.76 Th is composite of peripheral blood smears fr om 2 patients with peripheralized non-Hodgkin lymphoma shows a mantle cell lymphoma with a leukemic blood picture very reminiscent of chronic lymphocytic leukemia a and a peripheralized follicular lymphoma b in which 2 highly cleft ed mature-appearing lymphoid cells are present. Note normal lymphocyte for comparison. ( Wright)
a b

504
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
immunophenotypic and genotypic features analogous to the more common nodal forms of this disease t22.8, f22.6 [Matutes 2004]. See t22.8, t22.9, t22.10, t22.11 for comparisons of B-CLPN in blood, bone marrow, and on immunophenotyping.
[22.6] T-Chronic Lymphoproliferative Neoplasms
Analogous to B-CLPN, there are several types of T-chronic lymphoproliferative neoplasms (T-CLPN) with leukemic manifestations, including T-cell large granular lymphocytic leukemia (T-LGL), T-cell prolymphocytic leukemia (T-PLL), Sézary syndrome (SS), and adult T-cell leukemia lymphoma (ATLL). Th ese mature T-CLPN oft en manifest as systemic disorders with splenomegaly with or without hepatomegaly, skin lesions, and variable lymphade-nopathy in conjunction with hematologic abnormalities [Foucar 2007]. Th e focus of this chapter will be the blood and bone marrow manifestations of these disorders. Note that both non-neoplastic and neoplastic disorders of true NK cells are the focus of Chapter 26. Mature T-cell leukemias can be broadly defi ned as clonal proliferations of morphologically and immunophenotypically mature T cells of either helper (CD4+) or cytotoxic/suppressor (CD8+) type.
Clinical, hematologic, morphologic, immunophe-notypic, and selected genetic features are essential in the subclassifi cation of T-CLPN t22.16. Based on the integration
t22.16 Key Features of Mature T-Cell LeukemiasFeature T-LGL T-PLL SS ATLL (acute)
Clinical Wide age range, no sex predilection
Symptoms linked to cytopenias, splenomegaly common
Association with rheumatoid arthritis, recurrent infections and red cell aplasia†
Elderly, male predominanceHepatosplenomegaly
with generalized lymphadenopathy
Skin lesions and pleural effusions in subset
Middle age to elderly, male predominance
Cutaneous disease, especially generalized erythroderma
Generalized lymph- adenopathy common
Endemic HTLV-1 associated leukemia
Occurs in minority of carriers after long latency period
Aggressive multisystem disease
HypercalcemiaCBC fi ndings Absolute lymphocyte count
2 - 20 × 103/μL (2 - 20 × 109/L; sustained >6 months)
Variably severe anemia and/or neutropenia
Marked lymphocytosis (>100 × 103/μL [>100 × 109/L])
Rapidly rising WBCAnemia and
thrombocytopenia common
Variable lymphocytosisVariable cytopeniasEosinophilia common
Variable lymphocytosis, often striking
Variable cytopenias
Morphology of lymphocytes
Round nuclei, condensed chromatin, moderate amounts of cytoplasm with distinct granules
Granules contain granzyme and perforin
Spectrum, variably prominent nucleoli, variable nuclear irregularity
Spectrum in cell sizeSubtle nuclear convolutions
Spectrum in size and nuclear confi gurations
Pronounced nuclear lobulation (cloverleaf)
Key immuno-pheno typic features
Pan-T+*, TCR α/β, CD8, CD57 Pan-T+*, TCR α/βUsually CD4, but CD4/CD8
coexpression in 1/4TCL-1 (nuclear & cytoplasmic)
Pan-T+*, TCR α/βCD4, variable CD25, reduced
or absent CD7
Pan-T+*, TCR α/βCD4+, CD25+Reduced or absent CD7
Genetic features TCR gene rearrangement + (may be useful in distinguishing from reactive LGLs)
No recurrent cytogenetic fi nding
inv(14)(q11q23) results in TCL-1 translocation to promoter region of TCR genes (other translocations described)
No recurrent cytogenetic fi ndings
No recurrent cytogenetic fi ndings
*Some pan T-cell antigens may exhibit aberrant reduced expression†Other less common associations include myelodysplasia and aplastic anemiaT-LGL = T-cell large granular lymphocytic leukemia; T-PLL = T-cell prolymphocytic leukemia; SS = Sézary syndrome; ATLL = adult T-cell leukemia/lymphoma; CBC = complete blood count; TCL = T-cell leukemia; HTLV = human T-cell leukemia virus; TCR = T-cell receptor; WBC = white blood cell
References: [Catovsky 2008, Chan-WC 2008, Foucar 2007, Go 2003, Huh 2009, O’Malley 2007, Ohshima 2008, Osuji 2006, Pise-Masison 2009, Ralfkiaer 2008b]
0
256
512
768
1024
0 256 512 768 1024
100
101
102
103
104
100 101 102 103 104
100
101
102
103
104
100 101 102 103 104
100
101
102
103
104
100 101 102 103 104
100
101
102
103
104
100 101 102 103 104
side
sca
tter
forward scatter
CD23
FMC7
CD3
CD20
CD5
CD19
λ
κ
f22.6 Peripheralizing mantle cell lymphoma characterized by bright CD20 and CD5 coexpression in addition to CD23 expression is evident on this fl ow cytometric histogram composite fr om peripheral blood. Neoplastic cells are circled.

505
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
of multiple parameters, most cases of neoplastic T-cell disorders in blood can be subclassifi ed into the 4 disorders encompassed in this chapter. However, the diagnostician must be aware that other peripheral T-cell lymphomas may occasionally manifest in blood (see “Diff erential Diagnosis,” p 507 and Chapter 24).
[22.6.1] T-Cell Large Granular Lymphocytic Leukemia
General FeaturesNormally, only a small proportion (~15%) of peripheral
blood lymphocytes contain coarse cytoplasmic azurophilic granules, the so-called large granular lymphocytes i22.77. Cells exhibiting this distinct morphologic appearance consist largely of either T-cytotoxic/suppressor or true NK cells. Although NK cells were originally defi ned by a functional activity, major histocom-patibility complex-nonrestricted cytotoxicity, subsequent studies have successfully delineated both the immunophenotypic charac-teristics and the diverse functional activities useful in defi ning these 2 subtypes of large granular lymphocytes t22.17 [Caligiuri 2008,
Chen 2009, Farag 2006, Morice 2007, Verheyden 2008]. Indolent and aggressive disorders of true NK cells are reviewed in Chapter 26.
Numbers of large granular lymphocytes are transiently (rarely chronically) increased in a wide variety of disorders, especially in viral infections or immune aberrations (see Volume 1, Chapter 21) [O’Malley 2007]. In contrast to these transient disorders, clonal T-LGL disorders are sustained (>6 months) and linked to clinically signifi cant manifestations though the disease course may be stable for decades t22.16.
Morphologic FeaturesIn most patients with T-LGL, the blood contains
increased numbers of morphologically mature, generally normal-appearing LGLs i22.78, i22.79 [Chan 2008, O’Malley 2007]. Occasionally the clonal cells of T-LGL exhibit more pleomorphism and/or immaturity [Alekshun 2007]. Other common blood features include neutropenia and anemia.
Although bone marrow examination is oft en not necessary, especially in patients with stable indolent disease, bone marrow fi ndings in T-LGL are well delineated [Chan 2008,
Evans 2000, O’Malley 2007, Osuji 2007]. It is important for the diagnostician to be aware that T-LGL infi ltrates in the bone marrow are subtle, poorly delineated, and patchy i22.80, i22.81, i22.82. Consequently, immunohistochemical assessment is critical [Morice 2002, Osuji 2007]. In patients with a more aggressive T-LGL, the bone marrow may be eff aced by a diff use infi ltrate of leukemic cells [Alekshun 2007]. Overall bone marrow cellularity is oft en increased, especially in patients with myeloid expansion as a response to neutropenia [Burks 2006, Osuji 2007].
Although lymphoid aggregates may be readily apparent on H&E-stained sections, these lymphoid cells are charac-teristically non-neoplastic based on morphology and immunohistochemistry [Osuji 2007]. Other nonspecifi c features include reticulin fi brosis, eosinophilia, and megakaryocytic hyperplasia. Assessment for T-LGL-associated red cell aplasia and myelodysplasia is essential i22.83 [Epling-Burnett e 2007,
Huh 2009]. Consequently, each hematopoietic lineage must
i22.77 A typical large granular lymphocyte is evident in the blood. ( Wright)
t22.17 Functions of Large Granular LymphocytesT Lymphocytes with Large Granular Lymphocytic Morphology
Cytotoxic/suppressor subset (CD8+, CD3+, CD57+)
Role in regulation of hematopoiesis
Regulation of B lymphocytes
Variable, often low, natural killer cell activity (major histocompatibility complex-nonrestricted cell lysis and destruction of normal tissue) (cytoplasmic granules contain perforin and granzyme)
Variable antibody-dependent cytotoxicity
Contrasuppressor activity, ie, inhibition of other T-suppressor cells from inhibiting T-helper cell activity
True Natural Killer Cells with Large Granular Lymphocytic Morphology
High natural killer cell activity (major histocompatibility complex-nonrestricted cell lysis) (cytoplasmic granules contain perforin and granzyme)
Multiple classes of stimulatory or inhibitory NK-associated major histocompatibility complex receptors defi ned (eg, KIRs)
Lack of cell surface CD3; expression of CD16 and CD56
Key role in innate immunity
Immunosurveillance for spontaneously occurring neoplasms
Regulation of hematopoiesis
Resistance to viral infections
Antibody-dependent cytotoxicity
Graft vs leukemia effect after bone marrow transplantationKIR = killer cell inhibitory receptors; NK = natural killer
References: [Caligiuri 2008, Chen 2009, Farag 2006, Morice 2007, Verheyden 2008]

506
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
i22.78 Th is peripheral blood smear fr om a 73-year-old female with T-cell large granular lymphocytic leukemia shows a marked increase in circulating mature-appearing large granular lymphocytes. ( Wright) (courtesy Q-Y Zhang)
i22.79 Th is peripheral blood smear fr om a 73-year-old patient with T-cell large granular lymphocytic leukemia shows prominent cytoplasmic granules in the majority of circulating neoplastic cells. Note binucleation and occasional shaggy cytoplasmic contours. ( Wright)
i22.80 Th is bone marrow aspirate smear fr om this patient with T-cell large granular lymphocytic leukemia shows an increase in mature-appearing lymphoid cells, which exhibit nondescript morphologic features. ( Wright)
i22.81 Low magnifi cation of a bone marrow core biopsy section fr om a patient with T-cell large granular lymphocytic leukemia shows subtle areas of increased cellularity, but no discrete lymphoid infi ltrates are appreciated. ( H&E)
i22.82 On high magnifi cation, the subtle lymphoid infi ltration in this case of T-cell large granular lymphocytic leukemia is more apparent (see i22.81). Th e areas of increased cellularity noted at low magnifi cation consist largely of mature, lymphoid-appearing cells with widely spaced nuclei compatible with moderate amounts of cytoplasm. ( H&E)
i22.83 Immunoperoxidase staining for hemoglobin A on this bone marrow core biopsy section shows only widely scatt ered erythroblasts in this patient with T-cell large granular lymphocytic leukemia and associated red cell aplasia. (immunoperoxidase for hemoglobin A)

507
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
be systematically assessed for quantitative and qualitative abnormalities.
Immunoperoxidase staining of the bone marrow biopsy sections for T-cell antigens (CD3, CD8, TIA-1, CD57) can highlight subtle infi ltrates i22.84. Immunohistochemical staining is particularly useful in highlighting sinusoidal infi ltrates of T-LGL [Morice 2002, O’Malley 2007, Osuji 2007]. TIA-1 stains the intracytoplasmic granules of LGLs and is thus much less conspicuous on immunohistochemical staining i22.85.
Immunophenotypic and Molecular AnalysesTh e classic fl ow cytometric T-LGL profi le consists
of expression of surface CD3, CD2, CD5, CD8, CD16, and CD57 f22.7, but anomalies in the intensity of antigen expression are common [Chan-WC 2008, Lundell 2005]. Clonal T-cell receptor gene rearrangement is characteristically identifi ed. Although various numerical and structural cytogenetic abnormalities have been described, there are no known recurrent cytogenetic abnormalities in T-LGL [Chan-WC 2008]. Rare reports describe CD4+, CD56+, CD57+ clonal prolif-erations of large granular lymphocytes [Olteanu 2008].
Diff erential DiagnosisTh e distinction between chronic LGL disorders and other
mature T-cell leukemias is generally not a problem, since both granulated lymphocyte morphology and the cytotoxic suppressor immunophenotype are unique to T-LGL. More problematic is the distinction between reactive and neoplastic T-LGL proliferations; molecular assessment for T-cell receptor gene rearrangement is particularly useful in this
i22.84 Th is composite of immunohistochemical staining for CD3 a and CD8 b in 2 bone marrow biopsy sections fr om patients with T-cell large granular lymphocytic leukemia shows the diff use and focally patchy increase in CD3+ T cells that characterizes this disorder a. Most of the T cells are CD8+ and are sometimes sinusoidal b. (immunoperoxidase for CD3 and CD8)
i22.85 Th is bone marrow core biopsy section fr om a patient with T-cell large granular lymphocytic leukemia highlights increased numbers of TIA-1+ cells characteristic of this disorder. Because these cytoplasmic granules are relatively sparse, staining with TIA-1 is much less conspicuous (especially at low magnifi -cation) than staining for either CD3 or CD8 (see i22.84). (immunoperoxidase for TIA-1)
a b
For p
lace
men
t only
0
200
400
600
1000
800
4002000 600 800 1000
side
sca
tter
forward scatter
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD8
CD4
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD3
CD5
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD3
CD7
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD2
CD5
For p
lace
men
t only
100
101
102
103
104
100 101 102 103 104
CD2
CD7
f22.7 Th ese fl ow cytometric histograms fr om a patient with T-cell large granular lymphocytic leukemia highlight many of the characteristic immunophe-notypic features. Note reduced expression of CD5 and CD7 in conjunction with coexpression of CD57 on the CD8+ T-cell population.
a b
c
c
d
d

508
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
sett ing. Similarly, immunophenotypic assessment is essential in the distinction between T-LGL and chronic lymphoprolif-erative disorders of NK cells, which can be morphologically identical (see Chapter 26 for images) [Chan 2008, Villamor 2008].
[22.6.2] T-Cell Prolymphocytic Leukemia
General FeaturesAlthough controversial, the current recommendation is
to combine disorders previously termed T-PLL and T-CLL into a single disease category. Because of similarities in both clinical course and cytogenetic abnormalities, T-CLL may be best viewed as a small cell variant of T-PLL [Catovsky 2008]. Like patients with B-PLL, most patients with T-PLL are elderly men who present with marked leukocytosis and pronounced splenomegaly t22.16. However, patients with T-PLL more commonly exhibit lymphadenopathy, cutaneous involvement, and pleural eff usions than those with B-PLL [Catovsky 2008,
Foucar 2007].
Blood FindingsAlthough the morphologic features tend to be
homogeneous within individual cases, a spectrum of morphologic features has been noted in T-PLL. In some cases the cells are similar to those seen in B-PLL, exhibiting round nuclear contours with huge central nucleoli. In other cases the leukemic cells exhibit greater nuclear irregularities, oft en appearing knobby with cytoplasmic protrusions and blebs i22.86, i22.87, i22.88. In occasional T-PLL cases, the nuclear irregularity approaches that seen in SS or ATLL. Th e nuclear chromatin is generally condensed, and nucleoli
are variably prominent but sometimes inconspicuous. Th e relatively scant cytoplasm is basophilic and agranular and may exhibit prominent blebs. One distinctive hematologic feature in T-PLL is a rapidly rising WBC [Herling 2004]. Other common hematologic features include anemia and thrombocytopenia.
Bone marrow infi ltrates of T-PLL are characteristically diff use with extensive replacement of hematopoietic cells and fat cells i22.89 [Catovsky 2008, Viswanatha 2003]. Occasional cases exhibit a mixed nodular and diff use patt ern of infi ltration, and fi brosis has sometimes been associated with these leukemic infi ltrates [Nieto 1989]. Despite this mild fi brosis, numerous prolymphocytes are generally evident on bone marrow aspirate smears, and these atypical cells exhibit the same cytologic features as the circulating neoplastic cells.
Immunophenotypic and Genetic FeaturesT-prolymphocytic leukemia is derived from immuno-
phenotypically mature T cells that express the pan T-cell antigens, CD2, CD3, CD5, and CD7 t22.16. Th e majority of cases exhibit a helper cell phenotype, although one fourth of cases of T-PLL demonstrate coexpression of both CD4 and CD8, while others exclusively express CD8 [Catovsky 2008, Foucar 2007]. Despite morphologic heterogeneity, a consistent cytogenetic abnormality has been identifi ed in 80% of cases of T-PLL. Th is abnormality is inversion (14)(q11q32) f22.8; this translocation juxtaposes TCL1 oncogene into the promoter region of the T-cell receptor α/δ gene. Constitutive dysregulation of TCL1 is leukemogenic [Catovsky 2008, Pekarsky 2004]. Consequently, immunohisto-chemical staining for TCL1 shows both nuclear and cytoplasmic positivity in T-PLL and is absent in other T-CLPN i22.90 [Herling 2004, 2008]. Other cytogenetic abnormalities in T-PLL have
i22.86 Th is peripheral blood smear fr om a 36-year-old male with a white blood cell count higher than 350 × 103/μL (350 × 109/L) shows a striking mature lymphocytosis that on immunophenotyping was immunologically mature, but coexpressed CD4 and CD8 as oft en seen in T-cell prolymphocytic leukemia. ( Wright)
i22.87 Th is peripheral blood smear is fr om an 89-year-old female with T-cell large granular lymphocytic leukemia who presented with a skin rash. Complete blood count showed a white blood cell count of 530 × 103/μL (530 × 109/L). Th ese cells were confi rmed to be mature T cells by immunophenotyping. Note generally prominent central nucleoli resembling B-cell prolymphocytic leukemia. ( Wright)

509
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
been described, some of which result in homologous oncogene dysregulation, while others are linked to deletion of ATM tumor suppressor gene [Catovsky 2008]. Th e disease course of T-PLL is typically aggressive but improved response rates have been achieved with anti-CD52 therapy [Catovsky 2008].
Diff erential DiagnosisTh e primary diff erential diagnostic challenge is the
distinction of T-PLL from other mature T-CLPNs t22.16. Features useful in identifying cases of T-PLL include the marked and rapidly rising leukocytosis, massive spleno-megaly, strong expression of CD7, TCL-1 expression, and the unique genetic features, all features absent in other CD4 positive T-CLPN.
[22.6.3] Sézary Syndrome
General FeaturesSézary syndrome and mycosis fungoides (MF) are
closely related, but pathophysiologically distinct, disorders that compose the bulk of cutaneous T-cell lymphomas [Ralfk iaer 2008a, 2008b, van Doorn 2009]. Both disorders are derived from mature T lymphocytes that express the helper antigen, CD4. Th ese cells are morphologically unique in that they exhibit a high nuclear-cytoplasmic ratio with marked nuclear convolutions resulting in a cerebriform appearance. Th e distinction between SS and MF is based largely on the patt ern of cutaneous disease. Patients categorized as having SS have generalized exfoliative erythroderma with characteristic blood involvement, while patients with MF tend to have
i22.88 Th is blood smear is fr om a patient with T-cell prolymphocytic leukemia. ( Wright) (courtesy T Keith, MD)
i22.89 Th is bone marrow core biopsy section fr om a patient with T-cell prolymphocytic leukemia shows a moderate to extensive diff use interstitial infi ltrate by mature-appearing lymphoid cells. ( H&E)
i22.90 Immunoperoxidase staining for TCL-1 shows both nuclear and cytoplasmic positivity in this patient with T-cell prolymphocytic leukemia associated with inv(14). (immunoperoxidase for TCL-1)
1 2 3 mar 4 5
67
8 9 10 11 12
13 14 15 16 17 18
19 20 21 10 X Y
f22.8 Th is karyotype fr om a patient with T-cell large granular lymphocytic leukemia (T-PLL) shows numerous marker chromosomes and other cytogenetic abnormalities. However, the key feature is a subtle inv(14)(q11q31) (arrow) characteristic of T-PLL

510
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
prolonged cutaneous disease with a very wide spectrum of cutaneous lesions ranging from plaques to tumor nodules. Consequently, this chapter focuses largely on the blood and bone marrow fi ndings in SS, the more aggressive variant of cutaneous T-cell lymphoma [Ralfk iaer 2008b].
Blood FindingsAlthough nonspecifi c fi ndings such as eosinophilia
and anemia of chronic disease may be evident, the major focus of blood examination in patients with SS is to identify circulating malignant T cells. By convention, these circulating cells are oft en referred to as Sézary cells, and these cells may be identifi ed by either morphologic appearance or immuno-phenotypic properties, although immunophenotype is much more reliable [Morice 2006, Vonderheid 2006]. Th e incidence of blood
involvement in patients with generalized erythroderma (SS) approaches 100%.
Despite the prominent variation in the size of these circulating atypical lymphoid cells, the nuclei of these cells are cerebriform with variably condensed nuclear chromatin, while nucleoli are absent or inconspicuous i22.91, i22.92, i22.93, i22.94. A predominance of large Sézary cells is linked to adverse outcome. Th e nuclear-cytoplasmic ratio is generally high, and the cytoplasm is typically basophilic and agranular. Occasional cytoplasmic vacuoles may be identifi ed.
Because it may be diffi cult to morphologically identify circulating Sézary cells with certainty, especially when present in low numbers, many routine and specialized techniques have been applied to facilitate this process (see later section for specialized methods). Cytochemical staining may highlight globules of cytoplasmic periodic acid-Schiff -positive
i22.91 Blood smears illustrate circulating Sézary syndrome cells fr om 3 patients. Note condensed chromatin, subtle nuclear convolutions, and size range. ( Wright)
i22.92 Th is intermediate magnifi cation of a peripheral blood smear fr om a patient with Sézary syndrome highlights the distinctly dark nuclear chromatin confi guration that characterizes this disorder. Note broad size variation of circulating Sézary cells including small forms of approximately the size of a normal lymphocyte (lower left ). ( Wright) (courtesy B Nelson, MD)
i22.93 Because of the subtle nuclear irregularities, Sézary cells can be confused with monocytes. Th e diff erences between a Sézary cell (center) and a monocyte (lower right) is highlighted in this peripheral blood smear fr om a patient with Sézary syndrome. ( Wright)
i22.94 Th e subtle cerebriform nuclear confi guration of circulating Sézary cells is evident in this composite fr om 2 patients with Sézary syndrome. ( Wright) (a courtesy B Nelson, MD).
a b c
a b

511
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
material, while the absence of nonspecifi c esterase activity can be used to distinguish Sézary cells from monocytes. Although electron microscopy is not utilized in routine practice, the cerebriform nuclear confi guration of circulating Sézary cells is best appreciated by this method i22.95.
Bone Marrow FeaturesDespite the high frequency of peripheral blood
involvement, overt bone marrow infi ltration by SS is much less frequent [Ralfk iaer 2008b]. Although some investigators report a 22% incidence of bone marrow infi ltration by SS, these infi ltrates are oft en very subtle and easily missed, identi-fi able only by a careful examination under oil immersion or immunohistochemical assessment i22.96, i22.97. In cases with more extensive bone marrow involvement, SS cells can be identifi ed on aspirate smears i22.98. However, bone marrow biopsy sections are superior both for the identifi cation of cells with markedly cerebriform nuclei and for assessing the patt ern of infi ltration (either small inconspicuous collections or interstitial infi ltrates of isolated cerebriform cells) i22.96. Immunohistochemical stains for CD3, CD4, CD7, and CD8 may be very helpful in highlighting CD3+, CD4+, CD7–, and CD8– interstitial infi ltrates i22.97.
Flow Cytometric Immunophenotypic FeaturesCirculating Sézary cells exhibit the same mature helper
T-cell phenotype demonstrated in cutaneous infi ltrates t22.16. In addition to CD4, these cells also express pan T-cell antigens such as CD2, CD3, and CD5, while CD7 is generally absent or weak f22.9 [Ralfk iaer 2008b]. Although multicolor fl ow cytometric immunophenotyping is being increasingly utilized to identify low numbers of circulating lymphoma cells in SS patients, the diagnostician must be aware that normal blood control samples and specimens from patients
i22.95 Th e cerebriform confi guration of a Sézary cell is well illustrated in this electron photomicrograph fr om peripheral blood.
i22.96 Th is bone marrow core biopsy section fr om a patient with Sézary syndrome shows a subtle interstitial diff use infi ltrate of lymphoid cells. Th e larger Sézary cells have dark nuclear chromatin, which makes them more readily apparent on H&E sections (lower portion). ( H&E)
i22.97 Immunohistochemical staining for CD3 on this bone marrow clot section fr om a patient with Sézary syndrome shows an extensive interstitial T-cell infi ltrate. Th e number of T cells is oft en much greater on immunohistochemical staining than is apparent on H&E (see i22.96). (immunoperoxidase for CD3)
i22.98 Bone marrow aspirate smear contains numerous atypical lymphocytes in a patient with Sézary syndrome. ( Wright)

512
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
with benign dermatoses contain a low proportion of CD4+ CD7– T cells [Ginaldi 1996, Harmon 1996]. More recent authors cite the utility of dim CD3 expression in identifying low numbers of circulating Sézary cells [Edelman 2000]. Flow cytometric assessment of Vβ chain antibodies may enhance the detection of blood involvement [Feng 2010].
Cytogenetic and Molecular StudiesIn stimulated peripheral blood specimens, clonal
cytogenetic abnormalities are common, but there are no recurrent cytogenetic features in SS [Ralfk iaer 2008b]. Molecular studies, whether performed on involved skin, lymph node, or peripheral blood, document consistent T-cell receptor gene rearrangements in patients with SS.
Diff erential DiagnosisTh e diff erential diagnosis of SS logically includes
the other T-CLPNs. Subclassifi cation among these uncommon leukemias depends on the integration of clinical, morphologic, and immunophenotypic data t22.16. Distinction between non-neoplastic circulating T cells and minimal involvement by SS is problematic [Olsen 2007]. Parameters utilized to defi ne blood involvement include fl ow cytometric immunophenotyping, molecular genetic analyses, and morphologic assessment [Olsen 2007]. Of these methodologies,
morphologic assessment is the most subjective and shows the greatest interobserver variation [Olsen 2007]. Th e detection of a signifi cant population of CD4+, CD7– T cells (exceeding laboratory normal reference ranges) is the main modality currently used in clinical practice to delineate the number of circulating SS [Morice 2006, Olsen 2007, Vonderheid 2006]. Loss of CD26 expression is also useful, as is a helper to suppressor ratio >10 [Morice 2006]. T-cell clonality is of most utility when combined with clonality assessment of other sites of disease (usually skin), since T-cell clones can be present in blood from patients with non-neoplastic disorders [Olsen 2007, Vonderheid 2006]. Rare reports of pseudoleukemic picture with features of SS following drug therapy have been published in patients without cutaneous T-cell lymphoma [ Jabbar 1998].
[22.6.4] Adult T-Cell Leukemia/Lymphoma
General FeaturesAdult T-cell leukemia/lymphoma is a distinctive mature
T-cell neoplasm originally described in Japanese patients in 1977 [Ferreira 1997, Franchini 1995, Uchiyama 1977, 1988]. Subsequent studies have documented its unique worldwide geographic distri-bution, with the vast majority of cases occurring in Japan, Africa, the southeast United States, and the Caribbean region [Ohshima 2008, Pise-Masison 2009]. However, nonendemic cases have been described, and it is possible that ATLL has a more widespread distribution than generally recognized.
Th e etiologic agent causing endemic ATLL is the human T-cell leukemia virus type 1 (HTLV-1), which can be identifi ed by serologic, immunologic, or molecular studies in virtually all endemic cases [Nicot 2005, Ohshima 2008]. However, only 2.5% of chronic HTLV-1 carriers develop overt malignancies, so infection alone is insuffi cient to induce neoplasia [Nicot 2005,
Ohshima 2008, Pise-Masison 2009].Although originally recognized as a leukemic disorder,
the range of HTLV-1-related disease is broad and even includes a neurodegenerative disorder. For that subgroup of HTLV-1-infected patients who develop leukemia/lymphoma-type manifestations, a spectrum of clinical and pathologic fi ndings has also been identifi ed, ranging from asymptomatic patients with sustained mild lymphocytosis to those with fl orid leukemic or lymphomatous manifestations [Ohshima 2008,
Yokote 2005]. Th e progression from an asymptomatic carrier state to a leukemic phase generally takes decades. Recent evidence suggests that gene expression profi ling may be useful in distinguishing biologic subtypes of HTLV-1-related disorders [Pise-Masison 2009].
Th e most aggressive type of HTLV-1-associated leukemia/lymphoma has been designated as acute ATL, and only this leukemic variant will be presented in this chapter. Patients suff ering from acute ATL present with a leukemic
100
101
102
103
104
100 101 102 103 104
100
101
102
103
104
100 101 102 103 104
0
200
400
600
1000
800
0
200
400
600
1000
800
100 101 102 103 104100 101 102 103 104
CD8
CD2
CD4 CD7
side
sca
tter
side
sca
tter
CD3 CD4
f22.9 Th is composite of fl ow cytometric immunophenotyping histograms is fr om a patient with Sézary syndrome in which the CD4+ helper T cells show greatly diminished expression of CD7.

513
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
blood picture, clinical manifestations of leukemia, oft en in conjunction with prominent extramedullary disease. Th ese patients oft en have lymphadenopathy, hepatospleno-megaly, skin lesions, and lytic bone lesions; ~1/3 also have hypercalcemia [Ohshima 2008]. Th e clinical course is very aggressive, with short median survival times.
Th e focus of this chapter discussion is on the peripheral blood fi ndings, bone marrow morphologic features, and immunophenotypic features of this acute type of ATL. Both endemic and nonendemic cases of acute ATL have been described in the United States. Because many of the nonendemic cases fail to demonstrate evidence of HTLV-1 infection, it is possible that another agent is responsible for this disease [Head 1987]. However, both the morphology and immunophenotype of these nonendemic cases are identical to those described in endemic acute ATL.
Peripheral Blood FindingsTh e most remarkable peripheral blood abnormality in
acute ATL is the striking leukocytosis, with a predominance of morphologically abnormal lymphoid cells. Th ese lymphoid cells generally show pronounced nuclear irregularity with coarse lobulations and cloverleaf forms, though cases resembling either CLL or PLL have been described i22.99, i22.100 [Foucar 2007]. Heterogeneity of both overall cell size and nuclear confi gurations is common. Th e chromatin of these cells tends to be coarsely clumped, and nucleoli are either absent or inconspicuous. Th e amount of cytoplasm varies from scant to moderate and is generally slightly basophilic and agranular. Both an associated eosinophilia and neutrophilia have been occasionally described in these patients, possibly the consequence of cytokines released from the leukemic cells. Hemoglobin and platelet counts vary, depending on the degree of bone marrow replacement by the leukemia.
Bone Marrow FindingsPatt erns of bone marrow infi ltration by acute ATL vary
from patchy to focal paratrabecular to extensive, diff use replacement of the entire hematopoietic space. Occasionally, the bone marrow involvement is subtle, despite prominent blood involvement. On aspirate smears and tissue sections, these cells exhibit the same striking nuclear irregularities identifi ed in the peripheral blood. Mitotic activity is generally not prominent. Eosinophilia may be associated with acute ATL infi ltrates in bone marrow and other tissues [Ogata 1998]. In addition, striking bony abnormalities, consisting predomi-nantly of increased numbers of osteoclasts and osteoblasts, are very frequently identifi ed on bone marrow biopsy sections from these patients i22.101. Increased bone resorption is
i22.99 A marked leukocytosis composed of highly atypical lymphoid cells characterizes this blood smear in a case of nonendemic adult T-cell leukemia (see i22.100). ( Wright)
i22.100 Note the marked nuclear lobulations on these blood smears in a case of adult T-cell leukemia. ( Wright)
i22.101 Prominent bone resorption with increased osteoclastic activity is evident on this bone marrow core biopsy section fr om a patient with adult T-cell leukemia. An associated hypercellularity of this bone marrow and a prominent diff use interstitial lymphoid infi ltrate are present. ( H&E)
a b

514
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
the cause of the hypercalcemia that is commonly identifi ed in these patients. Constitutive production of parathyroid hormone-related protein by the leukemia cells is the primary cause of this pathologic fi nding [Nadella 2007].
Immunophenotypic FindingsIn both endemic and nonendemic cases, ATLL cells
demonstrate a consistent immunophenotypic profi le [Ohshima 2008]. Th ese cells represent mature T cells, lacking both terminal deoxynucleotidyl transferase and CD1 expression. Several pan T-cell antigens are expressed on ATLL cells, including CD3, CD2, and CD5, while CD7 expression is absent or weak t22.16 [Foucar 2007, Ohshima 2008]. Reduced expression of CD3 was noted in about half of the cases in one study [Yokote 2005]. ATLL cells also commonly express activation antigens, most notably CD25. Most cases of ATLL demonstrate a helper cell (CD4+) phenotype; although small numbers of cases demonstrate coexpression of both CD4 and CD8, rare cases lack either CD4 or CD8, and other rare cases demonstrate only CD8 expression [Foucar 2007].
Cytogenetic and Molecular StudiesAlthough almost all case of ATLL exhibit cytogenetic
abnormalities, none is specifi c [Ohshima 2008]. Molecular studies of ATLL cells consistently demonstrate clonal T-cell receptor gene rearrangement. In addition, HTLV-1 is clonally integrated in the neopastic cells [Kamihira 2005, Ohshima 2008].
Diff erential DiagnosisDespite the disorder’s rarity in the United States, the striking
morphologic abnormalities of circulating acute ATL cells are
so unique that cases can generally be suspected. Although both mature and blastic T-cell leukemias can exhibit circulating cells with nuclear irregularities, these disorders can generally be distinguished from acute ATL by integrating morphologic, clinical, viral, and immunophenotypic fi ndings t22.16.
[22.7] Components of the Diagnostic Interpretation in CLPN
Th e goal in the assessment of a patient for a possible B- or T-CLPN is to fi rst exclude any reactive process, followed by an eff ort to delineate the hematologic, morphologic, and immunophenotypic profi le of the neoplastic clone t22.18. Th is complex, multifaceted endeavor facilitates the identifi cation of cases that fulfi ll diagnostic criteria for well-defi ned clinico-pathologic entities. For cases that exhibit hybrid features, the pathologist should acknowledge diagnostic uncertainty and recommend additional tests that may allow defi nitive subclas-sifi cation. For purposes of treatment, clinicians may require a “best fi t” label, with the caveat that the case exhibits some aberrant properties. It is also useful to discuss those features known to have prognostic signifi cance.
For both B- and T-cell neoplasms, peripheralized lymphomas are a key diff erential diagnostic consideration. Although lymphomas with typical “leukemic” features at presentation have been included in this chapter, other B- and T-cell lymphomas may manifest rarely with a leukemic blood picture. B-cell lymphomas to be considered include mantle cell, follicular, lymphoplasmacytic, and diff use large B-cell subtypes. T-cell lymphomas that can occasionally manifest a leukemic blood picture include hepatosplenic T-cell, anaplastic large cell lymphoma, angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, not otherwise specifi ed (NOS) (see Chapter 24).
[22.8] Clues and Caveats1. Current classifi cation proposals for B- and T-cell CLPNs
are based on the identifi cation of distinct clinico-pathologic entities by integrating clinical, morphologic, immunophenotypic, and oft en genotypic properties.
2. A leukemic blood and bone marrow picture is by defi nition a de novo feature of chronic leukemias, while a similar picture is noted occasionally in primarily lympho-matous disorders.
3. A spectrum of morphologic and immunophenotypic fi ndings characterizes all CLPNs, and overlap among these disorders is common. As a consequence, precise subclassifi cation may sometimes be problematic and arbitrary.
t22.18 Components of Diagnostic Interpretation in B-, T-Chronic Lymphoproliferative Neoplasms
Hematologic and morphologic features in bloodMorphologic features in bone marrowPattern and extent of bone marrow effacementFeatures of uninvolved bone marrowImmunophenotypic profi le of the neoplastic cloneProvide delineation of prognostic features assessed and explain
signifi canceIntegration of clinical, hematologic, morphologic features into
a fi nal diagnostic “label” that segregates cases into distinct clinicopathologic entities when feasible
Recognition of “overlap” disorders that exhibit hybrid features and discuss signifi cance
Exclusion of B- and T-cell lymphomas with leukemic manifestationsRecommendations for additional tests that may either resolve
diagnostic uncertainty or provide additional prognostic information on an individual case basis

515
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
4. A “classic” immunophenotypic profi le has been delineated for all B- and T-cell CLPNs, which takes into account patt ern and intensity of antigen coexpression as well as antigens that are characteristically absent.
5. Immunophenotype and genotype provide both diagnostic and prognostic information in CLL.
6. Monoclonal B lymphocytosis is a precursor lesion to CLL.7. Th e distinction between monoclonal B lymphocytosis
and low-stage CLL is problematic; current recommen-dations for CLL require a sustained absolute monoclonal B-cell count of ≥5 × 103/μL (≥5 × 109/L) for 3 months.
8. Th e diagnosis of CLL based exclusively on fl ow cytometric immunophenotyping can be problematic when the absolute lymphocyte count by CBC is not known.
9. Most cases of CLL are composed of small monotonous lymphocytes with round nuclei and scant cytoplasm; “atypical” CLL is characterized by greater cytologic heterogeneity, including nuclear cleft ing and increased prolymphocytes.
10. In CLL, atypical morphologic features, aberrant immunophenotype, and disease progression/ transfor-mation are oft en linked to specifi c genetic abnormalities.
11. In addition to disease stage, many other independent prognostic factors have been described in CLL, including CD38 expression, ZAP-70 expression, cytogenetic features, and IGHV mutation status.
12. Potential new prognostic factors such as microRNA profi les may become clinically useful.
13. Numerous T-cell and NK cell abnormalities have been noted in patients with B-CLL; even rare clonal T-cell defects have been described in these patients.
14. Th e distinction among CLL, prolymphocytoid transfor-mation of CLL, and B-PLL is based primarily on the percentage of prolymphocytes, and the monotony/hetero-geneity of the neoplastic cells. Despite some immuno-phenotypic and genetic diff erences, there may be overlap.
15. Although prolymphocytes typically exhibit prominent nucleoli, nuclear chromatin is more condensed than that of blasts.
16. Many types of transformation have been described in CLL, but prolymphocytoid transformation and Richter syndrome (overt large cell lymphoma) are the most common.
17. Th e majority of large cell lymphomas that develop in patients with underlying CLL represent clonal transformations; a minority represent secondary, oft en EBV-associated neoplasms.
18. Rare cases of CLL with scatt ered Reed-Sternberg-like cells must be distinguished from overt Hodgkin-like transformation of CLL.
19. Th e nuclear chromatin of CLL cells is densely and coarsely clumped; chromatin is less condensed in prolymphocytes, while the nuclear chromatin of hairy cells is dispersed with a spongy, ground-glass appearance.
20. Discrete, focal, nodular bone marrow infi ltrates are common in patients with CLL, B-PLL, and many lymphomas, but distinctly uncommon in HCL.
21. Strong, diff use tartrate-resistant acid-phosphatase positivity is characteristic of HCL, while less intense reactivity may be present in many other disorders.
22. Other relatively HCL-specifi c markers include annexin-1, CD123, and t-bet.
23. Patchy infi ltrates of hairy cells can be very subtle and are not sharply delineated from adjacent hematopoietic cells. Immunoperoxidase studies on bone marrow biopsy sections using pan B-cell antibodies can highlight these cells.
24. Hypocellular HCL is common and is probably the result of HCL-associated cytokine suppression of hematopoiesis.
25. Convoluted, multilobulated, blastic, and spindled variants of hairy cells have been described.
26. Distinctive features of B-PLL and T-PLL include predominance in elderly men, marked leukocytosis, generally homogeneous leukemic cells on blood smears, and massive splenomegaly.
27. Peripheralizing MCL should be excluded in cases of possible B-PLL, especially cases with CD5 coexpression; staining for cyclin D1 or FISH for CCDN/IgH is useful in confi rming MCL.
28. Although a fair proportion of T-PLLs are morphologically similar to B-PLLs, other cases exhibit “knobby” nuclear irregularity with inconspicuous or absent nucleoli.
29. Cases of T- PLL can be identifi ed by nuclear and cytoplasmic staining for TCL-1 or by cytogenetic detection of inv(14)(q11q32) or other T-PLL associated karyotype.
30. SMZL with villous lymphocytes by defi nition exhibits blood involvement at presentation; prominent clinical morphologic and immunophenotypic overlap between SMZL and HCL is evident.
31. Th e patt ern of bone marrow infi ltration can generally be used to distinguish SMZL from HCL; discrete nodules and intrasinusoidal lesions characterize SMZL, while HCL typically exhibits subtle, patchy interstitial infi ltrates early in the disease course with progression to diff use lesions in advanced cases.
32. Increased cytologically normal-appearing large granular lymphocytes are more common in T-cytotoxic/suppressor cell neoplasms, while true NK cell neoplasms are much less common and may exhibit greater cytologic immaturity and pleomorphism.

516
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
33. Because of morphologic and genetic overlap, cases formerly termed “T-chronic lymphocytic leukemia” are currently viewed as a subtype (small cell variant) of T-PLL.
34. Because ATLL is rare in nonendemic regions, other mature helper T-cell disorders such as T-PLL and Sézary syndrome (SS) should be systematically excluded.
35. Th e cerebriform appearance of Sézary cells in blood and bone marrow is best appreciated under high magnifi -cation, while the coarse nuclear lobulations of acute adult T-cell leukemia cells are more readily apparent on low magnifi cation.
36. Bone marrow infi ltrates by SS range from small, extremely subtle collections of cerebriform cells to frank lymphomatous infi ltrates. Only the conspicuous lesions are linked to adverse outcome.
37. By periodic acid-Schiff stain on biopsy sections, subtle infi ltrates of Sézary cells (periodic acid-Schiff negative) can be distinguished from diff usely positive myeloid precursor cells, but immunohistochemical detection is clearly the preferred modality to highlight occult infi ltrates.
38. T-PLL, ATLL, and SS are all mature T-cell neoplasms that characteristically express CD4; both ATLL and SS are typically CD7–, while CD25 expression is more characteristic in ATLL.
39. Caution should be exercised in the fl ow cytometric assessment of blood for evidence of minimal involvement by SS, since low numbers of CD4+/CD7– T cells are physiologic.
[22.9] Acknowledgments Th e author acknowledges the photographic contributions of T
Keith, MD, B Nelson, MD, and Q-Y Zhang.
[22.10] ReferencesAgrawal SG, Liu FT, Wiseman C, et al [2008] Increased proteasomal degradation of
Bax is a common feature of poor prognosis chronic lymphocytic leukemia. Blood 111:2790 - 2796.
Alekshun TJ, Tao J, Sokol L [2007] Aggressive T-cell large granular lymphocyte leukemia: a case report and review of the literature. Am J Hematol 82:481 - 485.
Aoun P, Zhou G, Chan WC, et al [2007] Familial B-cell chronic lymphocytic leukemia: analysis of cytogenetic abnormalities, immunophenotypic profi les, and immunoglobulin heavy chain gene usage. Am J Clin Pathol 127:31 - 38.
Aqeilan RI, Calin GA, Croce CM [2010] miR-15a and miR-16 - 1 in cancer: discovery, function and future perspectives. Cell Death Diff er 17: 215-220.
Arons E, Suntum T, Stetler-Stevenson M, Kreitman RJ [2009] VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood 114:4687-4695.
Aydin S, Rossi D, Bergui L, et al [2008] CD38 gene polymorphism and chronic lymphocytic leukemia: a role in transformation to Richter syndrome? Blood 111:5646 - 5653.
Aziz KA , Till KJ, Chen H, et al [2003] Th e role of autocrine FGF-2 in the distinctive bone marrow fi brosis of hairy-cell leukemia (HCL). Blood 102:1051 - 1056.
Bartl R, Frisch B, Hill W, et al [1983] Bone marrow histology in hairy cell leukemia. Identifi cation of subtypes and their prognostic signifi cance. Am J Clin Pathol 79:531 - 545.
Belani R, Saven A [2006] Cladribine in hairy cell leukemia. Hematol Oncol Clin North Am 20:1109 - 1123.
Bouroncle BA [1994] Th irty-5 years in the progress of hairy cell leukemia. Leuk Lymphoma 14:1 - 12.
Brown JR, Neuberg D, Phillips K, et al [2008] Prevalence of familial malignancy in a prospectively screened cohort of patients with lymphoproliferative disorders. Br J Haematol 143:361 - 368.
Burks EJ, Loughran TP, Jr [2006] Pathogenesis of neutropenia in large granular lymphocyte leukemia and Felty syndrome. Blood Rev 20:245 - 266.
Byrd JC, Gribben JG, Peterson BL, et al [2006] Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fl udarabine and rituximab in chronic lymphocytic leukemia: justifi cation for risk-adapted therapy. J Clin Oncol 24:437 - 443.
Caligaris-Cappio F, Ghia P [2008] Novel insights in chronic lymphocytic leukemia: are we gett ing closer to understanding the pathogenesis of the disease? J Clin Oncol 26:4497 - 4503.
Caligiuri MA [2008] Human natural killer cells. Blood 112:461 - 469.Calin GA [2009] MiR-sensing chemotherapy resistance in CLL. Blood
113:3652 - 3653.Calin GA, Dumitru CD, Shimizu M, et al [2002] Frequent deletions and
down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 99:15524 - 15529.
Calin GA, Ferracin M, Cimmino A, et al [2005] A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 353:1793 - 1801.
Calin GA, Liu CG, Sevignani C, et al [2004] MicroRNA profi ling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA 101:11755 - 11760.
Campo E, Catovsky D, Montserrat E, et al [2008a] B-cell prolymphocytic leukaemia. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 183 - 184.
Campo E, Pileri S, Jaff e E, et al [2008b] Nodal marginal zone lymphoma. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 218 - 219.
Catovsky D, Muller-Hermelink H, Ralfk iaer E [2008] T-cell prolymphocytic leukaemia. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 270 - 271.
Catovsky D, O’Brien M, Melo JV, et al [1984] Hairy cell leukemia (HCL) variant: an intermediate disease between HCL and B prolymphocytic leukemia. Semin Oncol 11:362 - 369.
Cavazzini F, Hernandez JA, Gozzett i A, et al [2008] Chromosome 14q32 translocations involving the immunoglobulin heavy chain locus in chronic lymphocytic leukaemia identify a disease subset with poor prognosis. Br J Haematol 142:529 - 537.
Cawley JC [2006] Th e pathophysiology of the hairy cell. Hematol Oncol Clin North Am 20:1011 - 1021.
Cessna MH, Hartung L, Tripp S, et al [2005] Hairy cell leukemia variant: fact or fi ction. Am J Clin Pathol 123:132 - 138.

517
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
Chadha P, Rademaker AW, Mendiratt a P, et al [2005] Treatment of hairy cell leukemia with 2-chlorodeoxyadenosine (2-CdA): Long-term follow-up of the Northwestern University experience. Blood 106:241 - 246.
Chan WC, Foucar K, Morice W, Catovsky D [2008] T-cell large granular lymphocyte leukaemia. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Tumours of Haematopoietic and Lymphoid Tissues. 2nd ed. Lyon, France: IARC Press; 272 - 273.
Chen X, Bai F, Sokol L, et al [2009] A critical role for DAP10 and DAP12 in CD8+ T cell-mediated tissue damage in large granular lymphocyte leukemia. Blood 113:3226 - 3234.
Chen YH, Peterson LC, Ditt mann D, et al [2007] Comparative analysis of fl ow cytometric techniques in assessment of ZAP-70 expression in relation to IgVH mutational status in chronic lymphocytic leukemia. Am J Clin Pathol 127:182 - 191.
Chen YH, Tallman MS, Goolsby C, Peterson L [2006] Immunophenotypic variations in hairy cell leukemia. Am J Clin Pathol 125:251 - 259.
Chiorazzi N, Rai KR, Ferrarini M [2005] Chronic lymphocytic leukemia. N Engl J Med 352:804 - 815.
Cimmino A, Calin GA, Fabbri M, et al [2005] miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 102:13944 - 13949.
Coll-Mulet L, Santidrian AF, Cosialls AM, et al [2008] Multiplex ligation-dependent probe amplifi cation for detection of genomic alterations in chronic lymphocytic leukaemia. Br J Haematol 142:793 - 801.
Crespo M, Bosch F, Villamor N, et al [2003] ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 348:1764 - 1775.
Crowther-Swanepoel D, Wild R, Sellick G, et al [2008] Insight into the pathogenesis of chronic lymphocytic leukemia (CLL) through analysis of IgVH gene usage and mutation status in familial CLL. Blood 111:5691 - 5693.
Cuneo A, Rigolin GM, Bigoni R, et al [2004] Chronic lymphocytic leukemia with 6q- shows distinct hematological features and intermediate prognosis. Leukemia 18:476 - 483.
D’Arena G, Tarnani M, Rumi C, et al [2007] Prognostic signifi cance of combined analysis of ZAP-70 and CD38 in chronic lymphocytic leukemia. Am J Hematol 82:787 - 791.
Dagklis A, Fazi C, Sala C, et al [2009] Th e immunoglobulin gene repertoire of low-count chronic lymphocytic leukemia (CLL)-like monoclonal B lymphocytosis is diff erent from CLL: diagnostic implications for clinical monitoring. Blood 114:26 - 32.
Deaglio S, Vaisitt i T, Aydin S, et al [2007] CD38 and ZAP-70 are functionally linked and mark CLL cells with high migratory potential. Blood 110:4012 - 4021.
Deaglio S, Vaisitt i T, Aydin S, et al [2006] In-tandem insight from basic science combined with clinical research: CD38 as both marker and key component of the pathogenetic network underlying chronic lymphocytic leukemia. Blood 108:1135 - 1144.
Dearden C, Wade R, Else M, et al [2008] Th e prognostic signifi cance of a positive direct antiglobulin test in chronic lymphocytic leukemia: a benefi cial eff ect of the combination of fl udarabine and cyclophosphamide on the incidence of hemolytic anemia. Blood 111:1820 - 1826.
Del Giudice I, Davis Z, Matutes E, et al [2006] IgVH genes mutation and usage, ZAP-70 and CD38 expression provide new insights on B-cell prolym-phocytic leukemia (B-PLL). Leukemia 20:1231 - 1237.
Del Giudice I, Matutes E, Morilla R, et al [2004] Th e diagnostic value of CD123 in B-cell disorders with hairy or villous lymphocytes. Haematologica 89:303 - 308.
Del Principe MI, Del Poeta G, Buccisano F, et al [2006] Clinical signifi cance of ZAP-70 protein expression in B-cell chronic lymphocytic leukemia. Blood 108:853 - 861.
Delage R, Jacques L, Massinga-Loembe M, et al [2001] Persistent polyclonal B-cell lymphocytosis: further evidence for a genetic disorder associated with B-cell abnormalities. Br J Haematol 114:666 - 670.
Delgado J, Pratt G, Phillips N, et al [2009] Beta2-microglobulin is a bett er predictor of treatment-free survival in patients with chronic lymphocytic leukaemia if adjusted according to glomerular fi ltration rate. Br J Haematol 145:801 - 805.
Dicker F, Herholz H, Schnitt ger S, et al [2009] Th e detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia 23:117 - 124.
Dicker F, Schnitt ger S, Haferlach T, et al [2006] Immunostimulatory oligonucleotide-induced metaphase cytogenetics detect chromosomal aberrations in 80% of CLL patients: a study of 132 CLL cases with correlation to FISH, IgVH status, and CD38 expression. Blood 108:3152 - 3160.
Diez Martin JL, Li CY, Banks PM [1987] Blastic variant of hairy-cell leukemia. Am J Clin Pathol 87:576 - 583.
Dohner H, Stilgenbauer S, Benner A, et al [2000] Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 343:1910 - 1916.
Dong HY, Weisberger J, Liu Z, Tugulea S [2009] Immunophenotypic analysis of CD103+ B-lymphoproliferative disorders: hairy cell leukemia and its mimics. Am J Clin Pathol 131:586 - 595.
Dores GM, Anderson WF, Curtis RE, et al [2007] Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol 139:809 - 819.
Edelman J, Meyerson HJ [2000] Diminished CD3 expression is useful for detecting and enumerating Sezary cells. Am J Clin Pathol 114:467 - 477.
Else M, Dearden CE, Matutes E, et al [2009] Long-term follow-up of 233 patients with hairy cell leukaemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol 145:733 - 740.
Epling-Burnett e PK, Painter JS, Rollison DE, et al [2007] Prevalence and clinical association of clonal T-cell expansions in myelodysplastic syndrome. Leukemia 21:659 - 667.
Evans HL, Burks E, Viswanatha D, Larson RS [2000] Utility of immunohis-tochemistry in bone marrow evaluation of T-lineage large granular lymphocyte leukemia. Hum Pathol 31:1266 - 1273.
Falini B, Tiacci E, Liso A, et al [2004] Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). Lancet 363:1869 - 1870.
Farag SS, Caligiuri MA [2006] Human natural killer cell development and biology. Blood Rev 20:123 - 137.
Feng B, Jorgensen JL, Jones D, et al [2010] Flow cytometric detection of peripheral blood involvement by mycosis fungoides and Sézary syndrome using T-cell receptor Vbeta chain antibodies and its application in blood staging. Mod Pathol 23:284-295.
Ferreira OC, Jr [1997] Planelles V, Rosenblatt JD. Human T-cell leukemia viruses: epidemiology, biology, and pathogenesis. Blood Rev 11:91 - 104.
Filippi AR, Franco P, Marinone C, et al [2007] Treatment options in skeletal localizations of hairy cell leukemia: a systematic review on the role of radiation therapy. Am J Hematol 82:1017 - 1021.
Fong D, Kaiser A, Spizzo G, et al [2005] Hodgkin’s disease variant of Richter’s syndrome in chronic lymphocytic leukaemia patients previously treated with fl udarabine. Br J Haematol 129:199 - 205.
Forconi F, Sahota SS, Raspadori D, et al [2004] Hairy cell leukemia: at the crossroad of somatic mutation and isotype switch. Blood 104:3312 - 3317.
Forconi F, Sozzi E, Cencini E, et al [2009] Hairy cell leukemias with unmutated IGHV genes defi ne the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood 114:4696-4702.
Foucar K [2007] Mature T-cell leukemias including T-prolymphocytic leukemia, adult T-cell leukemia/lymphoma, and Sézary syndrome. Am J Clin Pathol 127:496 - 510.
Foucar K, Falini B, Catovsky D, Stein H [2008] Hairy cell leukaemia. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 188 - 190.

518
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
Franchini G [1995] Molecular mechanisms of human T-cell leukemia/lympho-tropic virus type I infection. Blood 86:3619 - 3639.
Frater JL, McCarron KF, Hammel JP, et al [2001] Typical and atypical chronic lymphocytic leukemia diff er clinically and immunophentypically. Am J Clin Pathol 116:655 - 664.
Fulci V, Chiarett i S, Goldoni M, et al [2007] Quantitative technologies establish a novel microRNA profi le of chronic lymphocytic leukemia. Blood 109:4944 - 4951.
Fuller SJ, Papaemmanuil E, McKinnon L, et al [2008] Analysis of a large multi-generational family provides insight into the genetics of chronic lymphocytic leukemia. Br J Haematol 142:238-245.
Gachard N, Salviat A, Boutet C, et al [2008] Multicenter study of ZAP-70 expression in patients with B-cell chronic lymphocytic leukemia using an optimized fl ow cytometry method. Haematologica 93:215 - 223.
Ginaldi L, Farahat N, Matutes E, et al [1996] Diff erential expression of T cell antigens in normal peripheral blood lymphocytes: a quantitative analysis by fl ow cytometry. J Clin Pathol 49:539 - 544.
Go RS, Lust JA, Phyliky RL [2003] Aplastic anemia and pure red cell aplasia associated with large granular lymphocyte leukemia. Semin Hematol 40:196 - 200.
Goldaniga M, Ferrario A, Cortelazzo S, et al [2008] A multicenter retrospective clinical study of CD5/CD10– chronic B cell leukemias. Am J Hematol 83:349 - 354.
Grever MR, Lucas DM, Dewald GW, et al [2007] Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol 25:799 - 804.
Haferlach C, Dicker F, Schnitt ger S, et al [2007] Comprehensive genetic character-ization of CLL: a study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia 21:2442 - 2451.
Hallek M, Cheson BD, Catovsky D, et al [2008] Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute—Working Group 1996 guidelines. Blood 111:5446 - 5456.
Hamblin T, Orchard J, Ibbotson R, et al [2002] CD38 expression and immuno-globulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 99:1023 - 1029.
Hanson CA, Ward PC, Schnitzer B [1989] A multilobular variant of hairy cell leukemia with morphologic similarities to T-cell lymphoma. Am J Surg Pathol 13:671 - 679.
Harmon CB, Witzig TE, Katzmann JA, Pitt elkow MR [1996] Detection of circulating T cells with CD4+CD7- immunophenotype in patients with benign and malignant lymphoproliferative dermatoses. J Am Acad Dermatol 35:404 - 410.
Head DR, Kjeldsberg CR, Kadin ME, et al [1987] Childhood T-cell malignancy resembling adult T-cell leukemia/lymphoma. Hematol Pathol 1:15 - 25.
Herling M, Khoury JD, Washington LT, et al [2004] A systematic approach to diagnosis of mature T-cell leukemias reveals heterogeneity among WHO categories. Blood 104:328 - 335.
Herling M, Patel KA , Teitell MA, et al [2008] High TCL1 expression and intact T-cell receptor signaling defi ne a hyperproliferative subset of T-cell prolym-phocytic leukemia. Blood 111:328 - 337.
Himmelmann A, Gautschi O, Nawrath M, et al [2001] Persistent polyclonal B-cell lymphocytosis is an expansion of functional IgD+CD27+ memory B cells. Br J Haematol 114:400 - 405.
Ho AK, Hill S, Preobrazhensky SN, et al [2009] Small B-cell neoplasms with typical mantle cell lymphoma immunophenotypes oft en include chronic lymphocytic leukemias. Am J Clin Pathol 131:27 - 32.
Hoff man MA [2006] Clinical presentations and complications of hairy cell leukemia. Hematol Oncol Clin North Am 20:1065 - 1073.
Hounieu H, Chitt al SM, al Saati T, et al [1992] Hairy cell leukemia. Diagnosis of bone marrow involvement in paraffi n-embedded sections with monoclonal antibody DBA.44. Am J Clin Pathol 98:26 - 33.
Hoyer JD, Li CY, Yam LT, et al [1997] Immunohistochemical demonstration of acid phosphatase isoenzyme 5 (tartrate-resistant) in paraffi n sections of hairy cell leukemia and other hematologic disorders. Am J Clin Pathol 108:308 - 315.
Hsi ED [2009] Th e leukemias of mature lymphocytes. Hematol Oncol Clin North Am 23:843 - 871.
Huh YO, Medeiros LJ, Ravandi F, et al [2009] T-cell large granular lymphocyte leukemia associated with myelodysplastic syndrome: a clinicopathologic study of 9 cases. Am J Clin Pathol 131:347 - 356.
Isaacson P, Piris M, Berger F, et al [2008] Splenic marginal zone lymphoma. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 185 - 187.
Jabbar A, Siddique T [1998] A case of pseudolymphoma leukaemia syndrome following cefi xime. Br J Haematol 101:209.
Janckila AJ, Cardwell EM, Yam LT, Li CY [1995] Hairy cell identifi cation by immunohistochemistry of tartrate-resistant acid phosphatase. Blood 85:2839 - 2844.
Jasionowski TM, Hartung L, Greenwood JH, et al [2003] Analysis of CD10+ hairy cell leukemia. Am J Clin Pathol 120:228 - 235.
Johrens K, Stein H, Anagnostopoulos I [2007] T-bet transcription factor detection facilitates the diagnosis of minimal hairy cell leukemia infi ltrates in bone marrow trephines. Am J Surg Pathol 31:1181 - 1185.
Kamihira S, Sugahara K, Tsuruda K, et al [2005] Proviral status of HTLV-1 integrated into the host genomic DNA of adult T-cell leukemia cells. Clin Lab Haematol 27:235 - 241.
Kanzler H, Kuppers R, Helmes S, et al [2000] Hodgkin and Reed-Sternberg-like cells in B-cell chronic lymphocytic leukemia represent the outgrowth of single germinal-center B-cell- derived clones: potential precursors of Hodgkin and Reed-Sternberg cells in Hodgkin’s disease. Blood 95:1023 - 1031.
Karandikar NJ, Hotchkiss EC, McKenna RW, Kroft SH [2002] Transient stress lymphocytosis: an immunophenotypic characterization of the most common cause of newly identifi ed adult lymphocytosis in a tertiary hospital. Am J Clin Pathol 117:819 - 825.
Kent S, Malhotra R, Variakojis D, Peterson L [2000] Comparative study of bone marrow involvement in splenic marginal zone lymphoma (SMZL) and extranodal marginal zone lymphoma (EMZL). Mod Pathol 13:153A.
Kim YS, Ford R, Faber J, et al [2000] B-cell chronic lymphocytic leukemia/small lymphocytic lymphoma involving bone marrow with an interfollicular patt ern. Am J Clin Pathol 114:41 - 46.
Koller C, Bekele BN, Zhou X, et al [2006] Plasma thrombopoietin compared with immunoglobulin heavy-chain mutation status as a predictor of survival in chronic lymphocytic leukemia. Blood 108:1001 - 1006.
Kroft SH, Dawson DB, McKenna RW [2001] Large cell lymphoma transformation of chronic lymphocytic leukemia/small lymphocytic lymphoma: a fl ow cytometric analysis of 7 cases. Am J Clin Pathol 115:385 - 395.
Kujawski L, Ouillett e P, Erba H, et al [2008] Genomic complexity identifi es patients with aggressive chronic lymphocytic leukemia. Blood 112:1993 - 2003.
Lanasa MC, Allgood SD, Bond KM, et al [2009] Oligoclonal TRBV gene usage among CD8(+) T cells in monoclonal B lymphocytosis and CLL. Br J Haematol 145:535 - 537.
Landgren O, Albitar M, Ma W, et al [2009] B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med 360:659 - 667.

519
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
Lin KI, Tam CS, Keating MJ, et al [2009] Relevance of the immunoglobulin VH somatic mutation status in patients with chronic lymphocytic leukemia treated with fl udarabine, cyclophosphamide, and rituximab (FCR) or related chemoimmunotherapy regimens. Blood 113:3168 - 3171.
Linet MS, Schubauer-Berigan MK, Weisenburger DD, et al [2007] Chronic lymphocytic leukaemia: an overview of aetiology in light of recent developments in classifi cation and pathogenesis. Br J Haematol 139:672 - 686.
Lundell R, Hartung L, Hill S, et al [2005] T-cell large granular lymphocyte leukemias have multiple phenotypic abnormalities involving pan-T-cell antigens and receptors for MHC molecules. Am J Clin Pathol 124:937 - 946.
Macon WR, Kinney MC, Glick AD, Collins RD [1993] Marrow mast cell hyperplasia in hairy cell leukemia. Mod Pathol 6:695 - 698.
Mao Z, Quintanilla-Martinez L, Raff eld M, et al [2007] IgVH mutational status and clonality analysis of Richter’s transformation: diff use large B-cell lymphoma and Hodgkin lymphoma in association with B-cell chronic lymphocytic leukemia (B-CLL) represent 2 diff erent pathways of disease evolution. Am J Surg Pathol 31:1605 - 1614.
Marti GE [2009] Th e changing defi nition of CLL. Blood 113:4130 - 4131.Marton S, Garcia MR, Robello C, et al [2008] Small RNAs analysis in CLL reveals
a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia 22:330 - 338.
Matutes E [2006] Immunophenotyping and diff erential diagnosis of hairy cell leukemia. Hematol Oncol Clin North Am 20:1051 - 1063.
Matutes E, Oscier D, Montalban C, et al [2008] Splenic marginal zone lymphoma proposals for a revision of diagnostic, staging and therapeutic criteria. Leukemia 22:487 - 495.
Matutes E, Parry-Jones N, Brito-Babapulle V, et al [2004] Th e leukemic presen-tation of mantle-cell lymphoma: disease features and prognostic factors in 58 patients. Leuk Lymphoma 45:2007 - 2015.
Matutes E, Wotherspoon A, Catovsky D [2003] Th e variant form of hairy-cell leukaemia. Best Pract Res Clin Haematol 16:41 - 56.
Mauro FR, Foa R, Giannarelli D, et al [1999] Clinical characteristics and outcome of young chronic lymphocytic leukemia patients: a single institution study of 204 cases. Blood 94:448 - 454.
Mayr C, Speicher MR, Kofl er DM, et al [2006] Chromosomal translocations are associated with poor prognosis in chronic lymphocytic leukemia. Blood 107:742 - 751.
McCarron KF, Hammel JP, Hsi ED [2000] Usefulness of CD79b expression in the diagnosis of B-cell chronic lymphoproliferative disorders. Am J Clin Pathol 113:805 - 813.
Meuleman N, Stamatopoulos B, Dejeneff e M, et al [2008] Doubling time of soluble CD23: a powerful prognostic factor for newly diagnosed and untreated stage A chronic lymphocytic leukemia patients. Leukemia 22:1882 - 1890.
Momose H, Jaff e ES, Shin SS, et al [1992] Chronic lymphocytic leukemia/small lymphocytic lymphoma with Reed- Sternberg-like cells and possible transformation to Hodgkin’s disease. Mediation by Epstein-Barr virus. Am J Surg Pathol 16:859 - 867.
Montserrat E, Moreno C [2008] Chronic lymphocytic leukaemia: a short overview. Ann Oncol 19Suppl7:vii320 - 325.
Morabito F, Cutrona G, Gentile M, et al [2009] Defi nition of progression risk based on combinations of cellular and molecular markers in patients with Binet stage A chronic lymphocytic leukaemia. Br J Haematol 146:44 - 53.
Moreno C, Montserrat E [2008] New prognostic markers in chronic lymphocytic leukemia. Blood Rev 22:211 - 219.
Morice WG, Jevremovic D, Hanson CA [2007] Th e expression of the novel cytotoxic protein granzyme M by large granular lymphocytic leukaemias of both T-cell and NK-cell lineage: an unexpected fi nding with implications regarding the pathobiology of these disorders. Br J Haematol 137:237 - 239.
Morice WG, Katzmann JA, Pitt elkow MR, et al [2006] A comparison of morphologic features, fl ow cytometry, TCR-Vbeta analysis, and TCR-PCR in qualitative and quantitative assessment of peripheral blood involvement by Sezary syndrome. Am J Clin Pathol 125:364 - 374.
Morice WG, Kurtin PJ, Hodnefi eld JM, et al [2008] Predictive value of blood and bone marrow fl ow cytometry in B-cell lymphoma classifi cation: comparative analysis of fl ow cytometry and tissue biopsy in 252 patients. Mayo Clin Proc 83:776 - 785.
Morice WG, Kurtin PJ, Teff eri A, Hanson CA [2002] Distinct bone marrow fi ndings in T-cell granular lymphocytic leukemia revealed by paraffi n section immunoperoxidase stains for CD8, TIA-1, and granzyme B. Blood 99:268 - 274.
Morton LM, Wang SS, Cozen W, et al [2008] Etiologic heterogeneity among non-Hodgkin lymphoma subtypes. Blood 112:5150 - 5160.
Muller-Hermelink H, Montserrat E, Catovsky D, et al [2008] Chronic lymphocytic leukaemia/small lymphocytic lymphoma. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 180 - 182.
Murray F, Darzentas N, Hadzidimitriou A, et al [2008] Stereotyped patt erns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood 111:1524 - 1533.
Nadella MV, Dirksen WP, Nadella KS, et al [2007] Transcriptional regulation of parathyroid hormone-related protein promoter P2 by NF-kappaB in adult T-cell leukemia/lymphoma. Leukemia 21:1752 - 1762.
Nazeer T, Burkart P, Dunn H, et al [1997] Blastic transformation of hairy cell leukemia. Arch Pathol Lab Med 121:707 - 713.
Ng D, Toure O, Wei MH, et al [2007] Identifi cation of a novel chromosome region, 13q21.33-q22.2, for susceptibility genes in familial chronic lymphocytic leukemia. Blood 109:916 - 925.
Nicoloso MS, Kipps TJ, Croce CM, Calin GA [2007] MicroRNAs in the pathogeny of chronic lymphocytic leukaemia. Br J Haematol 139:709 - 716.
Nicot C [2005] Current views in HTLV-I-associated adult T-cell leukemia/lymphoma. Am J Hematol 78:232 - 239.
Nieto LH, Lampert IA, Catovsky D [1989] Bone marrow histological patt erns in B-cell and T-cell prolymphocytic leukemia. Hematol Pathol 3:79 - 84.
Nieto WG, Almeida J, Romero A, et al [2009] Increased frequency (12%) of circulating chronic lymphocytic leukemia-like B-cell clones in healthy subjects using a highly sensitive multicolor fl ow cytometry approach. Blood 114:33 - 37.
O’Malley DP [2007] T-cell large granular leukemia and related proliferations. Am J Clin Pathol 127:850 - 859.
Ogata M, Ogata Y, Kohno K, et al [1998] Eosinophilia associated with adult T-cell leukemia: role of interleukin 5 and granulocyte-macrophage colony-stimulating factor. Am J Hematol 59:242 - 245.
Ohno T, Smir BN, Weisenburger DD, et al [1998] Origin of the Hodgkin/Reed-Sternberg cells in chronic lymphocytic leukemia with “Hodgkin’s transformation.” Blood 91:1757 - 1761.
Ohshima K, Jaff e E, Kikuchi M [2008] Adult T-cell leukaemia/lymphoma. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 281 - 284.
Olsen E, Vonderheid E, Pimpinelli N, et al [2007] Revisions to the staging and classifi cation of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 110:1713 - 1722.
Olteanu H, Karandikar NJ, Eshoa C, et al [2008] Laboratory fi ndings in CD4(+) large granular lymphocytoses. Int J Lab Hematol 32:e9-16.

520
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
Osuji N, Beiske K, Randen U, et al [2007] Characteristic appearances of the bone marrow in T-cell large granular lymphocyte leukaemia. Histopathology 50:547 - 554.
Osuji N, Matutes E, Tjonnfj ord G, et al [2006] T-cell large granular lymphocyte leukemia: a report on the treatment of 29 patients and a review of the literature. Cancer 107:570 - 578.
Patt en PE, Buggins AG, Richards J, et al [2008] CD38 expression in chronic lymphocytic leukemia is regulated by the tumor microenvironment. Blood 111:5173 - 5181.
Pekarsky Y, Zanesi N, Aqeilan R, Croce CM [2004] TCL1 as a model for lymphomagenesis. Hematol Oncol Clin North Am 18:863 - 879.
Pepper C, Lin TT , Pratt G, et al [2008] Mcl-1 expression has in vitro and in vivo signifi cance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood 112:3807 - 3817.
Peters O, Th ielemans C, Steenssens L, et al [1984] Intracellular inclusion bodies in 14 patients with B cell lymphoproliferative disorders. J Clin Pathol 37:45 - 50.
Piris M, Foucar K, Mollejo M, et al [2008] Splenic B-cell lymphoma/leukaemia, unclassifi able. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 191 - 193.
Pise-Masison CA, Radonovich M, Dohoney K, et al [2009] Gene expression profi ling of ATL patients: compilation of disease-related genes and evidence for TCF4 involvement in BIRC5 gene expression and cell viability. Blood 113:4016 - 4026.
Pitt ner BT, Shanafelt TD, Kay NE, Jelinek DF [2005] CD38 expression levels in chronic lymphocytic leukemia B cells are associated with activation marker expression and diff erential responses to interferon stimulation. Leukemia 19:2264 - 2272.
Ralfk iaer E, Cerroni L, Sander C, et al [2008a] Mycosis fungoides. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 296 - 298.
Ralfk iaer E, Hou-Jensen K, Geisler C, et al [1982] Cytoplasmic inclusions in lymphocytes of chronic lymphocytic leukaemia. A report of 10 cases. Virchows Arch [Pathol Anat] 395:227 - 236.
Ralfk iaer E, Willemze R, Whitt aker S [2008b] Sezary syndrome. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 299.
Rassenti LZ, Jain S, Keating MJ, et al [2008] Relative value of ZAP-70, CD38, and immunoglobulin mutation status in predicting aggressive disease in chronic lymphocytic leukemia. Blood 112:1923 - 1930.
Rawstron AC, Bennett FL, O’Connor SJ, et al [2008] Monoclonal B-cell lympho-cytosis and chronic lymphocytic leukemia. N Engl J Med 359:575 - 583.
Reddy KS [2006] Chronic lymphocytic leukaemia profi led for prognosis using a fl uorescence in situ hybridisation panel. Br J Haematol 132:705 - 722.
Reiniger L, Bodor C, Bognar A, et al [2006] Richter’s and prolymphocytic transfor-mation of chronic lymphocytic leukemia are associated with high mRNA expression of activation-induced cytidine deaminase and aberrant somatic hypermutation. Leukemia 20:1089 - 1095.
Ricca I, Rocci A, Drandi D, et al [2007] Telomere length identifi es 2 diff erent prognostic subgroups among VH-unmutated B-cell chronic lymphocytic leukemia patients. Leukemia 21:697 - 705.
Richter MN [1928] Generalized reticular cell sarcoma of lymph nodes associated with lymphatic leukemia. Am J Pathol 4:285 - 292.
Robak T [2006] Current treatment options in hairy cell leukemia and hairy cell leukemia variant. Cancer Treat Rev 32:365 - 376.
Roos G, Krober A, Grabowski P, et al [2008] Short telomeres are associated with genetic complexity, high-risk genomic aberrations, and short survival in chronic lymphocytic leukemia. Blood 111:2246 - 2252.
Rossi D, Cerri M, Capello D, et al [2008] Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol 142:202 - 215.
Rossi D, Sozzi E, Puma A, et al [2009] Th e prognosis of clinical monoclonal B cell lymphocytosis diff ers from prognosis of Rai 0 chronic lymphocytic leukaemia and is recapitulated by biological risk factors. Br J Haematol 146:64 - 75.
Ruchlemer R, Parry-Jones N, Brito-Babapulle V, et al [2004] B-prolymphocytic leukaemia with t(11;14) revisited: a splenomegalic form of mantle cell lymphoma evolving with leukaemia. Br J Haematol 125:330 - 336.
Saddler C, Ouillett e P, Kujawski L, et al [2008] Comprehensive biomarker and genomic analysis identifi es p53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood 111:1584 - 1593.
Schade U, Bock O, Vornhusen S, et al [2006] Bone marrow infi ltration patt ern in B-cell chronic lymphocytic leukemia is related to immunoglobulin heavy-chain variable region mutation status and expression of 70-kd zeta-associated protein (ZAP-70). Hum Pathol 37:1153 - 1161.
Schickel R, Boyerinas B, Park SM, Peter ME [2008] MicroRNAs: Key players in the immune system, diff erentiation, tumorigenesis and cell death. Oncogene 27:5959 - 5974.
Schlett e E, Medeiros LJ, Keating M, Lai R [2003] CD79b expression in chronic lymphocytic leukemia. Association with trisomy 12 and atypical immuno-phenotype. Arch Pathol Lab Med 127:561 - 566.
Schroers R, Griesinger F, Trumper L, et al [2005] Combined analysis of ZAP-70 and CD38 expression as a predictor of disease progression in B-cell chronic lymphocytic leukemia. Leukemia 19:750 - 758.
Scielzo C, Camporeale A, Geuna M, et al [2006] ZAP-70 is expressed by normal and malignant human B-cell subsets of diff erent maturational stage. Leukemia 20:689 - 695.
Shanafelt TD, Geyer SM, Kay NE [2004] Prognosis at diagnosis: integrating molecular biologic insights into clinical practice for patients with CLL. Blood 103:1202 - 1210.
Shanafelt TD, Kay NE, Jenkins G, et al [2009] B-cell count and survival: diff eren-tiating chronic lymphocytic leukemia from monoclonal B-cell lympho-cytosis based on clinical outcome. Blood 113:4188 - 4196.
Sharpe RW, Bethel KJ [2006] Hairy cell leukemia: diagnostic pathology. Hematol Oncol Clin North Am 20:1023 - 1049.
Shehata M, Schwarzmeier JD, Hilgarth M, et al [2004] TGF-β1 induces bone marrow reticulin fi brosis in hairy cell leukemia. J Clin Invest 113:676 - 685.
Stamatopoulos B, Meuleman N, Haibe-Kains B, et al [2009] MicroRNA-29c and microRNA-223 down-regulation has in vivo signifi cance in chronic lymphocytic leukemia and improves disease risk stratifi cation. Blood 113:5237 - 5245.
Sun T, Grupka N, Klein C [2004] Transformation of hairy cell leukemia to high-grade lymphoma: a case report and review of the literature. Hum Pathol 35:1423 - 1426.
Sup SJ, Domiati-Saad R, Kelley TW, et al [2004] ZAP-70 expression in B-cell hematologic malignancy is not limited to CLL/SLL. Am J Clin Pathol 122:582 - 587.
Swerdlow S, Campo E, Harris N, et al [2008] WHO Classifi cation of Tumours: Pathology & Genetics: Tumours of Haematopoietic and LymphoidTissues. Lyon, France: IARC.
Swords R, Bruzzi J, Giles F [2007] Recent advances in the diagnosis and therapy of Richter’s syndrome. Med Oncol 24:17 - 32.
Tam CS, Otero-Palacios J, Abruzzo LV, et al [2008] Chronic lymphocytic leukaemia CD20 expression is dependent on the genetic subtype: a study of quantitative fl ow cytometry and fl uorescent in-situ hybridization in 510 patients. Br J Haematol 141:36 - 40.
Tiacci E, Liso A, Piris M, Falini B [2006] Evolving concepts in the pathogenesis of hairy-cell leukaemia. Nat Rev Cancer 6:437 - 448.

521
22: MATURE B- AND T-CELL LYMPHOPROLIFERATIVE NEOPLASMS
Troussard X, Flandrin G [1996] Chronic B-cell lymphocytosis with binucleated lymphocytes (LWBL): A review of 38 cases. Leuk Lymphoma 20:275 - 279.
Uchiyama T [1988] Adult T-cell leukemia. Blood Rev 2:232 - 238.Uchiyama T, Yodoi J, Sagawa K, et al [1977] Adult T-cell leukemia: clinical and
hematologic features of 16 cases. Blood 50:481 - 492.Van Bockstaele F, Verhasselt B, Philippe J [2009] Prognostic markers in chronic
lymphocytic leukemia: a comprehensive review. Blood Rev 23:25 - 47.Van Den Neste E, Robin V, Francart J, et al [2007] Chromosomal translocations
independently predict treatment failure, treatment-free survival and overall survival in B-cell chronic lymphocytic leukemia patients treated with cladribine. Leukemia 21:1715 - 1722.
van Doorn R, van Kester MS, Dijkman R, et al [2009] Oncogenomic analysis of mycosis fungoides reveals major diff erences with Sezary syndrome. Blood 113:127 - 136.
VanderMolen LA, Urba WJ, Longo DL, et al [1989] Diff use osteosclerosis in hairy cell leukemia. Blood 74:2066 - 2069.
Verheyden S, Demanet C [2008] NK cell receptors and their ligands in leukemia. Leukemia 22:249 - 257.
Villamor N, Morice W, Chan W, Foucar K [2008] Chronic lymphoproliferative disorders of NK cells. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classifi cation of Tumours: Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 274 - 275.
Visco C, Ruggeri M, Laura Evangelista M, et al [2008] Impact of immune thrombo-cytopenia on the clinical course of chronic lymphocytic leukemia. Blood 111:1110 - 1116.
Viswanatha D, Foucar K [2003] Hodgkin and non-Hodgkin lymphoma involving bone marrow. Semin Diagn Pathol 20:196 - 210.
Viswanatha D, Montgomery K, Foucar K [2010] Mature B-cell neoplasms: chronic lymphocytic leukemia/small lymphocytic lymphoma, B-cell prolym-phocytic leukemia and lymphoplasmacytic lymphoma. In: Jaff e E, Harris N, Vardiman J, eds. Diagnostic Hematopathology 2010 (in press).
Vonderheid EC [2006] On the diagnosis of erythrodermic cutaneous T-cell lymphoma. J Cutan Pathol 33Suppl1:27 - 42.
Weinberg JB, Volkheimer AD, Chen Y, et al [2007] Clinical and molecular predictors of disease severity and survival in chronic lymphocytic leukemia. Am J Hematol 82:1063 - 1070.
Went PT, Zimpfer A, Pehrs AC, et al [2005] High specifi city of combined TRA P and DBA.44 expression for hairy cell leukemia. Am J Surg Pathol 29:474 - 478.
Wierda WG, O’Brien S, Wang X, et al [2009] Characteristics associated with important clinical end points in patients with chronic lymphocytic leukemia at initial treatment. J Clin Oncol 27:1637 - 1643.
Wierda WG, O’Brien S, Wang X, et al [2007] Prognostic nomogram and index for overall survival in previously untreated patients with chronic lymphocytic leukemia. Blood 109:4679 - 4685.
Wilhelm C, Neubauer A, Brendel C [2006] Discordant results of fl ow cytometric ZAP-70 expression status in B-CLL samples if diff erent gating strategies are applied. Cytometry B Clin Cytom 70:242 - 250.
Williams J, Schned A, Cotelingam JD, Jaff e ES [1991] Chronic lymphocytic leukemia with coexistent Hodgkin’s disease. Implications for the origin of the Reed-Sternberg cell. Am J Surg Pathol 15:33 - 42.
Ya-In C, Brandwein J, Pantalony D, Chang H [2005] Hairy cell leukemia variant with features of intrasinusoidal bone marrow involvement. Arch Pathol Lab Med 129:395 - 398.
Yokote T, Akioka T, Oka S, et al [2005] Flow cytometric immunophenotyping of adult T-cell leukemia/lymphoma using CD3 gating. Am J Clin Pathol 124:199 - 204.
Zanott i R, Ambrosett i A, Lestani M, et al [2007] ZAP-70 expression, as detected by immunohistochemistry on bone marrow biopsies from early-phase CLL patients, is a strong adverse prognostic factor. Leukemia 21:102 - 109.
Zenz T, Krober A, Scherer K, et al [2008] Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood 112:3322 - 3329.
Zenz T, Mohr J, Edelmann J, et al [2009a] Treatment resistance in chronic lymphocytic leukemia: the role of the p53 pathway. Leuk Lymphoma 50:510 - 513.
Zenz T, Mohr J, Eldering E, et al [2009b] miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood 113:3801 - 3808.
Zhang W, Dahlberg JE, Tam W [2007] MicroRNAs in tumorigenesis: a primer. Am J Pathol 171:728 - 738.