materiały dodatkowe i uzupełniaj ą -...
TRANSCRIPT

1
Stanisław Krawczyk
Materiały dodatkowe i uzupełniające
do wykładu Bionanotechnologia
Spis treści
1. Plazmony w metalach – uwagi ogólne .................................................................... 2
2. Przenikalność elektryczna (funkcja dielektryczna) gazu elektronowego .................. 5
3. Plazmon – więcej szczegółów ............................................................................... 7
4. Elektrostatyka kuli dielektrycznej ............................................................................. 10
5. Teoria Mie rozpraszania i absorpcji światła przez obiekt kulisty ............................. 14
6. Rozszerzenia teorii Mie .......................................................................................... 25
7. Absorpcja światła w ośrodku materialnym ............................................................... 29
8. Podstawy fotofizyki molekuł ............................................................................... 33
9. Szybkość reakcji chemicznej i kataliza enzymatyczna ............................................ 37
10. Podstawy zjawisk bioelektrycznych .......................................................................... 44
11. Literatura .................................................................................................................... 48

2
1. Plazmony w metalach – uwagi ogólne
Plazmon jest zjawiskiem fizycznym występującym w metalu poddanym wpływowi fali
elektromagnetycznej lub innej przyczyny oddziałującej z elektronami metalu, np. elektronu o
stosunkowo wysokiej energii przelatującego przez sieć krystaliczną. Właściwości wzbudzanej
wówczas podłużnej fali gęstości elektronów – plazmonu – zależą od rodzaju metalu, a także
jego ukształtowania i rozmiarów. Ważne są tu rozmiary przede wszystkim w porównaniu z
długością fali światła, które wzbudza plazmony w metalu.
W ostatnich dekadach zjawisko to – znane od bardzo dawna i występujące w
rozlicznych konfiguracjach układów metal-dielektryk – znalazło szereg zastosowań i jest
nadal intensywnie badane. Dobrym przykładem możliwości, jakie oferuje odpowiednie
wykorzystanie plazmonów, jest ich zastosowanie w powierzchniowo wzmocnionej
spektroskopii ramanowskiej (ang. Surface-Enhanced Raman Spectroscopy, SERS).
Wzmocnione rozpraszanie ramanowskie światła przy powierzchni metali zostało odkryte w
połowie lat 70-tych. Uzyskiwane wzmocnienie rejestrowanego sygnału w takich
eksperymentach przekracza wartość 1010 i – jak wykazano już ponad 10 lat temu – przy
zastosowaniu nanocząstek metalicznych o odpowiednim kształcie może osiągać nawet
większe wartości. Zbadanie i wyjaśnienie tego efektu doprowadziło do tego, że przy jego
wykorzystaniu można obecnie uzyskać widma ramanowskiego rozproszenia światła –
zazwyczaj bardzo słabego w porównaniu z luminescencją – nawet dla pojedynczych molekuł.
Inne zastosowania plazmonów wynikające z ich czułości na właściwości dielektryczne
otoczenia polegają na ich zastosowaniu w mikro- i nanodetektorach różnych substancji.
Jeszcze inne polegają na wykorzystaniu ciepła wydzielanego w dużej ilości wokół bardzo
małych nanocząstek metalicznych (~10-100 nm) absorbujących promieniowanie
elektromagnetyczne. Wiele możliwych zastosowań jest obecnie w fazie badań i z pewnością
część z nich zostanie zrealizowana praktycznie w fizyce, technice, chemii, biologii,
medycynie. W zakres możliwych zastosowań wchodzi również tzw. elektronika molekularna
– hipotetyczna alternatywa dla makroskopowych układów elektronicznych polegająca na
manipulacji prądem elektrycznym przewodzonym przez pojedyncze molekuły.
Plazmon jest więc falą w zbiorze mobilnych elektronów, pojawiającą się gdy rozkład
gęstości elektronów zostaje zaburzony. Fale tego rodzaju występują często w plazmie
gazowej, stąd nazwa „plazmon”; oznacza ona również zjawiska w układach
skondensowanych, przede wszystkim w metalach, których objętość wypełniona jest gazem
swobodnych (w przybliżeniu) elektronów. Metale wyraźnie wykazujące właściwości plazmy
elektronowej to przede wszystkim metale alkaliczne (Na, K), Mg, Al, Cu oraz metale

3
szlachetne jak Ag, Au. Są to metale, których własności elektryczne i optyczne wynikają
głównie z właściwości elektronów przewodnictwa.
Plazmony mogą występować zarówno w objętości metalu jak i przy jego powierzchni,
a gdy rozmiary ciała metalicznego są porównywalne z długością fali światła, to mogą
wystąpić specyficzne efekty podobne do rezonansu.
Opis właściwości plazmonu jest zawarty w materiale wykładu. Tu opiszemy głównie
właściwości optyczne metalu wynikające z klasycznej (tj. niekwantowej) teorii elektronów
swobodnych, którą zazwyczaj określa się wspólnym mianem teorii Drudego. W klasycznym
opisie zjawiska plazmonu lokalne zaburzenie gęstości elektronowej w metalu powoduje
wystąpienie siły proporcjonalnej do tego zaburzenia i kierującej je w stronę przeciwną, co
stanowi podstawę opisu plazmonu jako rezonansu o częstotliwości własnej
2
0p
Ne
mω
ε= (1.1)
gdzie m – masa elektronu swobodnego w metalu, n – liczba elektronów w jednostce objętości.
W ogólnym przypadku fala elektromagnetyczna o częstości kołowej ω biegnąca w ośrodku
elektrycznie przewodzącym może być pochłaniana, wskutek czego jej energii stopniowo
ubywa. Jak wiadomo, lokalna gęstość prądu j i lokalna gęstość objętościowa wydzielanej
energii cieplnej w wyrażają się wzorami:
σ=j E 2w Eσ= ⋅ =j E , (1.2)
Należy tu mieć na uwadze, że rozważamy metal w polu elektrycznym szybkozmiennym, w
którym wektor polaryzacji opóźnia się względem natężenia pola elektrycznego, co oznacza,
że okres fali może być porównywalny z czasem relaksacji rozkładu elektronów w metalu.
Jeżeli rozpatrujemy pole elektryczne harmonicznie zmienne o określonej częstości, to
pojawiające się wówczas opóźnienie wektora polaryzacji P względem E oznacza po prostu
przesunięcie fazowe między tymi wielkościami, skutkujące wykonaniem pracy przez pole
elektryczne nad przesuwającymi się ładunkami. Relację między polem elektrycznym i
polaryzacją ośrodka, która powinna uwzględniać nie tylko amplitudy ale również fazy tych
powiązanych ze sobą wielkości, opisuje się w dogodny sposób wprowadzając zespoloną
przenikalność dielektryczną ośrodka ε (odpowiednik „stałej dielektrycznej” w przypadku
pola statycznego):
( ) ( ) ( )1 2ˆ iε ω ε ω ε ω= + (1.3)
Wielkość ta jest zazwyczaj nazywana funkcją dielektryczną ośrodka. Zarówno część
rzeczywista jak i część urojona ε są funkcjami częstości pola ω.

4
Sens fizyczny zespolonej funkcji dielektrycznej pokazuje następujące przekształcenie
wyrażenia przedstawiającego falę płaską biegnącą w ośrodku materialnym. Jeżeli fala biegnie
w kierunku osi x, to natężenie pola elektrycznego tej fali o amplitudzie E0 jest:
( ) ( )2 2
0 0 0,n
i t x i t xi t kx cTx t e e e
π πω ωω λ − − − − − − = = =E E E E (1.4)
Skorzystaliśmy tu ze związków: k=2π/λ, λ=cT/n ; n oznacza współczynnik załamania.
Oznaczając: cT=λ0 (długość fali w próżni) i wprowadzając zespolony współczynnik
załamania:
n̂ n iκ= + (1.5)
otrzymujemy:
( ) 0 0 0
ˆ2 2 2
0 0,n n
i t x i t x x
t e e eπ π πκω ωλ λ λ
− − − − − = = ⋅E r E E (1.6)
Pierwszy czynnik wykładniczy w (1.6) przedstawia nadal falę płaską, której długość zależy
od rzeczywistej części współczynnika załamania n (tj. λ=λ0/n), zaś drugi czynnik przedstawia
zanik amplitudy fali wzdłuż współrzędnej x. Współczynnik przy współrzędnej x w
wykładniku stanowi współczynnik absorpcji promieniowania. Natężenie promieniowania
wyraża się przez kwadrat natężenia pola elektrycznego, a więc:
( ) 0
42 2
0,x
I r t eπκλ
−= ⋅E E∼ czyli w skrócie: 0
xI I e γ−= (1.7a)
przy czym:
0
4 2
c
πκ κωγλ
= = (1.7b)
Wielkość γ jest to współczynnik absorpcji. Jak widać, urojona część współczynnika
załamania κ ma związek z intensywnością absorpcji fali. Współczynnik absorpcji ma związek
z funkcją dielektryczną, który wynika z uniwersalnej relacji:
2ˆ n̂ε = (1.8a)
a mianowicie:
2 211 2
1
2 2
εκ ε ε= − + + (1.8b)
Pełny opis propagacji fali elektromagnetycznej, zawierający również np. intensywność
absorpcji energii w funkcji jej częstości lub długości fali (widmo absorpcji), wymaga
znajomości parametrów optycznych materiału, tj. obu części przenikalności elektrycznej
(funkcji dielektrycznej), w optycznym zakresie częstości (widzialny oraz UV).

5
2. Przenikalność elektryczna (funkcja dielektryczna) gazu elektronowego
Poniżej podany jest podstawowy opis właściwości optycznych metalu w ramach klasycznej
teorii elektromagnetyzmu, tzw. teorii Drudego. Rozpatrujemy elektrony poruszające się w
sieci krystalicznej, w której węzłach znajdują się jony dodatnie. Elektrony poruszają się
praktycznie całkowicie swobodnie, z wyjątkiem krótkich momentów zderzeń z jonami, w
trakcie których zachodzi wymiana energii między elektronem i jonem. Efekt ciągle
występującej utraty energii, stale nabywanej przez elektrony przyspieszane polem
elektrycznym, można potraktować jako rezultat stale obecnej, efektywnej siły tarcia,
skierowanej przeciwnie do prędkości elektronu i do niej proporcjonalnej. Równanie ruchu
elektronu w polu elektrycznym E w obecności siły tarcia ma zatem postać:
m m eβ+ =r r Eɺɺ ɺ (2.1)
Sens fizyczny wielkości β charakteryzującej tarcie wynika z rozpatrzenia przypadku, gdy
istniejące w przewodniku pole elektryczne E, istniejące np. w czasie przepływu prądu
elektrycznego ze średnią prędkością dryfu v0, nagle znika: E=0. Mamy wtedy równanie
jednorodne 0β+ =r rɺɺ ɺ .
Rozwiązanie tego równania ma postać:
0 0
1exp( ),tβ
β= − −r r v (2.2a)
z której dla prędkości elektronu nadawanej przez lokalne pole elektryczne wynika:
0 exp( )tβ= = −v r vɺ (2.2b)
Jak widać, w nieobecności pola elektrycznego prędkość dryfu elektronu v (związana z
przepływem prądu) zmniejsza się wykładniczo, a szybkość zaniku zależy od wielkości β,
która stanowi czas relaksacji elektronów w metalu i odzwierciedla opór elektryczny metalu. Z
tą stałą czasową β będą również zanikać lokalne zagęszczenia elektronów tworzące plazmon.
Stała relaksacji β wiąże się ze średnią drogą swobodną elektronu l w następujący sposób:
vF
lβ = (2.2c)
gdzie vF jest prędkością Fermiego, tzn. prędkością elektronu o energii równej energii
Fermiego.
Rozpatrzmy pole harmonicznie poprzecznej fali elektromagnetycznej w pewnym
punkcie wewnątrz metalu, zależne od czasu według: 0 exp( )i tω= −E E . Rozwiązanie
równania (9) jest sumą dwu składników, z których jeden przedstawia wykładniczy zanik
początkowego położenia elektronu (odpowiada to omówionemu już rozwiązaniu jednorodnej

6
postaci równania (9)), . Drugi składnik rozwiązania odpowiada stacjonarnemu ruchowi
periodycznemu elektronu wymuszanemu przez pole elektryczne:
( )2
e
m iω βω= −
+r E (2.3)
Ruch periodyczny wnosi do wektora polaryzacji ośrodka wkład p=e·r . Jeżeli jednostka
objętości zawiera N swobodnych elektronów, to całkowity wektor polaryzacji ośrodka P
(będący sumą wektorów p poszczególnych elektronów, przypadającą na jednostkę objętości
ii
V = ∑P p na podstawie wzoru (2.3) jest:
( )2
2
NeN Ne
m iω βω= = = −
+P p r E (2.4)
Skorzystamy teraz z ogólnych związków między wektorami indukcji elektrycznej D,
natężenia pola elektrycznego E i polaryzacji P:
0ˆ , εε ε= = +D E D E P (2.5)
z których wynika:
( )0 ˆ 1ε ε= −P E (2.6)
Z porównania wzorów (2.4) i (2.6) otrzymujemy ostatecznie wyrażenie przedstawiające
zespoloną przenikalność elektryczną gazu elektronowego:
( )2
20
ˆ 1Ne
m iε
ε ω βω= −
+ (2.7)
Powyższe wyrażenie może posłużyć do obliczenia części rzeczywistej i urojonej zespolonego
współczynnika załamania na podstawie ogólnego związku między zespoloną przenikalnością
elektryczną 1 2ˆ iε ε ε= + i współczynnikiem załamania: 2ˆ n̂ε = . Wynikające stąd wartości
rzeczywistej i urojonej części współczynnika załamania n oraz κ służą do opisu propagacji
fali elektromagnetycznej w ośrodku materialnym, np. w przypadku fali płaskiej podlegającej
absorpcji można posłużyć się wzorami (1.7). Występujące tam wartości n i κ (patrz: wzory
(1.5) i (1.8)) można wyliczyć z następujących związków wynikających z podstawienia ε z
równania (1.5) do równania (1.8a). Otrzymujemy w ten sposób:
( )22
2 21 2 22 2
0
1 1 pNen
m
ωε κ
ω βε ω β= − = − = −
++ (2.8a)
( ) ( )22
2 2 2 2 20
22
pNen
m
ω ββε κε ω ω β ω ω β
= = =+ +
(2.8b)

7
W powyższych wzorach wyrażenie (Ne2/mε0) zostało zastąpione przez częstość plazmonową
ωp zgodnie z wzorem (1.1). Powyższe wzory dotyczą fali w ośrodku zawierającym gaz
swobodnych elektronów, nie zawierają jednak efektów pochodzących od elektronów rdzeni
jonowych metalu ani od struktury pasmowej metalu, której istnienie implikuje dodatkowe
mechanizmy absorpcji energii wynikające z kwantowych przejść międzypasmowych i
wewnątrzpasmowych.
W szczególności, gdy tłumienie β jest odpowiednio małe, tj. β << ω, obie części
funkcji dielektrycznej we wzorach (17) mogą być przedstawione w postaci:
( )2
1 21 pω
ε ωω
≈ − (2.9a)
( )2
2 3
pω βε ω
ω≈ (2.9b)
Jak widać, dla częstości fali w metalu zbliżonej do częstości plazmonowej jest: ε1(ω)=0.
Model Drudego dla gazu elektronowego wyznacza dwa zakresy optycznych właśności metali.
Ich granicę stanowi tzw. częstość plazmonowa, nazywana też częstością odcięcia. Oddziela
ona dwa zakresy częstości fal elektromagnetycznych, w których:
■ poniżej pω rzeczywista część funkcji dielektrycznej jest ujemna, wektor falowy jest
urojony i fala nie może rozchodzić się w gazie elektronowym, jedynie płytko wnika w metal i
zostaje całkowicie odbita w powierzchniowej warstwie metalu o grubości zaledwie kilku
warstw atomów; metal wykazuje typowe, dobrze znane właściwości optyczne,
■ powyżej pω (gdy ε1>0) fala rozchodzi się w gazie elektronowym i nie jest tłumiona.
Długości fal elektromagnetycznych odpowiadających częstości odcięcia wynoszą np. dla litu
155 nm, dla potasu 315 nm. Równania (2.9) objaśniają istnienie dwu zakresów optycznych, w
których właściwości optyczne metali są całkiem odmienne. Poniżej częstości odcięcia metale
wykazują wysoki współczynnik odbicia światła (w zakresie widzialnym i w części
nadfioletu), zaś powyżej tej częstości metale są w znacznym stopniu przezroczyste dla światła
(nadfioletu). Przezroczystość metali w zakresie częstości powyżej częstości odcięcia dobrze
potwierdza się doświadczalnie szczególnie dla litowców, w których gaz elektronowy
wykazuje stosunkowo proste właściwości dobrze opisywane modelem Drudego.
3. Plazmon – więcej szczegółów
Gdy mamy do czynienia z odpowiednio małymi obiektami, których rozmiary są dużo
mniejsze od długości fali światła, to pole elektryczne fali elektromagnetycznej oddziałującej

8
na ten obiekt można uwazać w przybliżeniu za jednorodne. Jest to tzw. przybliżenie
elektrostatyczne, zaniedbujące fakt, że różne punkty obiektu znajdują się w różnych co do
wartości oraz fazy natężeniach pola elektrycznego. Rozpatrując nanocząstkę metaliczną
przyjmujemy też, że tylko elektrony przewodnictwa poruszają się pod wpływem pola, zaś
dodatnie ładunki rdzeni atomowych pozostają nieruchome.
W granicach przybliżenia elektrostatycznego w stosunkowo prosty sposób można
objaśnić właściwości optyczne obiektu o kształcie kuli (patrz uzupełnienie: Elektrostatyka
kuli dielektrycznej). Przy pomocy warunków granicznych dla pola elektrycznego na
powierzchni kuli można obliczyć polaryzację kuli. Jeżeli kula o przenikalności elektrycznej ε
zostanie umieszczona w jednorodnym, nieabsorbującym ośrodku o stałej dielektrycznej
(rzeczywistej) εm , w którym istnieje jednorodne pole elektryczne o natężeniu E0 , to natężenie
pola wewnątrz kuli również jest jednorodne i wynosi:
0
3
2m
im
E Eε
ε ε=
+ (3.1)
zaś jej polaryzowalność α zdefiniowana jako
0α=µ E (3.2)
jest:
304
2m
m
Rε εα πε
ε ε−=
+ (3.3)
Zauważmy, że w szczególnym przypadku kuli metalowej (ε = ∞) znajdującej się w polu
elektrostatycznym otrzymujemy:
304kuli Rα πε= . (3.4)
Powyższe wzory elektrostatyki stosują się też do małych kul metalowych w oscylującym polu
elektrycznym jednorodnym. Warunkowi jednorodności pola elektrycznego odpowiada w
przypadku pola zmiennego w czasie nierówność: R/λ << 1. W takim przypadku należy tylko
zastąpić statyczne stałe dielektryczne ε oraz εm odpowiednimi rzeczywistymi funkcjami
dielektrycznymi ε(ω) oraz εm(ω). Zarówno pole wewnętrzne Ei jak i polaryzowalność
wykazują rezonans gdy spełniony jest warunek (por. wzór 3.3):
2 minimummε ε+ = (3.5)
Gdy funkcja dielektryczna kuli metalowej jest zespolona, ten warunek ma postać:
( ) ( )2 2
1 22 minimummε ω ε ε ω+ + = (3.6)

9
Ostatnie z tych wyrażeń oznacza, że rezonans występuje przy ujemnej wartości ε1 , która
oznacza odpowiednią relację faz pola elektrycznego fali i wektora momentu dipolowego kuli
metalowej. Częstość rezonansowa jest nieco zależna od ε2(ω), ale gdy wartość ε2 jest dużo
mniejsza od 1 lub bardzo słabo zależy od ω, częstość rezonansowa wynika z równości ε1 =
−2εm. W szczególnym przypadku, dla kuli metalowej w próżni (εm=1) z wzoru (2.9a) wynika
rezonans przy częstości:
2 3 24
3p c
kuli kuli
Ne R N e
m m
ω πωα α⋅= = = (3.7)
gdzie Nc=4πR3·N jest całkowitą liczbą elektronów zawartych w metalowej kuli (symbol N
oznacza, jak w poprzednim rozdziale, liczbę elektronów w jednostce objętości).
Obiekty o kształcie innym niż kulisty wykazują częstości rezonansowe inaczej powiązane z
częstością plazmową materiału. Relacje te podsumowane są w poniższej Tabeli 3.1.
Tabela 3.1. Plazmonowe częstości rezonansowe obiektów metalowych o różnych kształtach,
znajdujących się w próżni.
Geometria Warunek rezonansu Częstość rezonansowa
lity metal ( )1 0ε ω = rez pω ω=
płaska powierzchnia ( )1 1ε ω = − 2p
rez
ωω =
cienka warstwa# ( )( ) [ ]1
exp1 xk d
ε ωε ω
+= ± − ⋅
− [ ]1 exp
2p
rez xk dω
ω = ± − ⋅
kula (przybliż. dipolowe) ( )1 2ε ω = − 3p
rez
ωω =
# kierunek x równoległy do warstwy

10
4. Elektrostatyka kuli dielektrycznej
Pole elektryczne wokół kuli (lub elipsoidy) wypełnionej ośrodkiem dielektrycznym o stałej
dielektrycznej ε2 , otoczonej przez rozległy (nieskończony) ośrodek o stałej dielektrycznej ε1
spolaryzowany polem elektrycznym jednorodnym E0, jest jednym z podstawowych zagadnień
elektrostatyki (Rys. 4.1). Gdy ε1 = ε2 , pole wewnątrz kuli i w jej otoczeniu powinno być
polem jednorodnym tak jak w dużej odległości; gdy zaś ε1 ≠ ε2 , polaryzacja kuli wprowadza
Rys. 4.1.
dodatkowe pole zaburzające jednorodność pola zewnętrznego. Naszym celem jest
wyznaczenie pola elektrycznego wewnątrz kuli i w jej otoczeniu. Zarówno wewnątrz jak i na
zewnątrz kuli potencjał elektryczny spełnia równanie Laplace’a
2 0ϕ∇ = (4.1)
które wynika z prawa Gaussa
0
divρ
ε ε≡ ∇ =E E (4.2)
w nieobecności ładunków swobodnych (ρ = 0), oraz ze związku natężenia z potencjałem
ϕ= −∇E . Potencjały elektryczne na zewnątrz i wewnątrz kuli, odpowiednio ϕ1 i ϕ2, powinny
spełniać równania:
( )1 0 0 cosr E z E rϕ θ→ ∞ = − = − (z dala od kuli pole jest jednorodne), (4.3a)
( ) ( )1 2r a r aϕ ϕ
= == (4.3b)
1 21 2
r a r ar r
ϕ ϕε ε= =
∂ ∂ = ∂ ∂ . (4.3c)
Ostatnie z tych równań jest warunkiem ciągłości składowej normalnej wektora D = ε0εE.
Równanie Laplace’a (4.1) we współrzędnych sferycznych ma zbiór rozwiązań:
( )1cosl l
l l ll
BA r P
rϕ θ+
= + ⋅
(4.4)
ε1
ε2
a
r θ
E0
oś Z

11
w których l jest liczbą naturalną lub zerem. Pl(x) oznacza tu wielomian Legendre’a
argumentu x. Ogólne rozwiązanie zatem, jako suma rozwiązań szczególnych, jest szeregiem:
( )10
cosl ll ll
l
BA r P
rϕ θ
∞
+=
= + ⋅
∑ (4.5)
Ponieważ ϕ nie może mieć osobliwości w środku kuli , wszystkie współczynniki Bl muszą
być zerowe. Z warunku (4.3a) wynika z kolei, że z wszystkich potęg r l w (4.5) musi pozostać
tylko pierwsza potęga r, zatem współczynniki Al muszą być zerowe dla l=0 oraz l > 1.
Pozostaje to również w zgodności z postacią P1, jako że P1(cosθ) = cosθ. Współczynnik a1
wynosi więc –E0. Rozwiązania (4.5) dla ϕ w obszarach na zewnątrz i wewnątrz kuli
przyjmują postać:
( )1 0 10
cos coslll
l
BE r P
rϕ θ θ
∞
+=
= − +∑ (4.6a)
( )20
cosll l
l
A r Pϕ θ∞
==∑
(4.6b)
Zastosowanie warunków (4.3b) i (4.3c) do powyższych wyrażeń na ϕ1 i ϕ2 prowadzi do
następujących równości dla każdego l ≠ 1:
1
llll
BA a
a + = oraz ( ) 11 22
1 llll
Bl l A a
aε ε −
+− + = (4.7)
z których wynika, że Al=0 oraz Bl=0 dla wszystkich l z wyjątkiem l=1. Te same warunki
(4.3b) i (4.3c) dla l =1 dają równości:
10 12
BaE aA
a− = oraz 1
1 0 2 13
2BE A
aε ε + = −
(4.8)
z których wynika:
32 11 0
2 12B a E
ε εε ε
−=+
oraz 11 0
1 2
3
2A E
εε ε
= −+
(4.9)
i ostateczna postać potencjałów ϕ1 i ϕ2 na zewnątrz i wewnątrz kuli:
3
2 11 03
2 1
12
aE z
r
ε εϕε ε −= ⋅ − +
(4.10)
12 0
2 1
3
2E z
εϕε ε
= −+
(4.11)
Wzór (4.10) ma tę właściwość, że na dużych odległościach od kuli (gdy r >> a) pierwszy
składnik znika i ϕ1 ≈ −E0z, czyli pole staje się polem jednorodnym. Wzór (4.11) wyraża zaś

12
bardzo istotną właściwość kuli spolaryzowanej polem jednorodnym: potencjał wewnątrz kuli
jest liniową funkcją współrzędnej z i zależy tylko od tej współrzędnej. Wynika stąd, że pole
elektryczne wewnątrz kuli jest jednorodne, a wektor natężenia ma składowe kartezjańskie:
2 2 2 10
2 1
3, , 0,0,
2E
x y z
ϕ ϕ ϕ εε ε
∂ ∂ ∂= − = ∂ ∂ ∂ + E (4.12)
co wynika wprost z wzoru (4.11).
Wzory (4.10) i (4.11) przedstawiają potencjał elektryczny na zewnątrz i wewnątrz
kuli, na który składa się potencjał zewnętrznego, jednorodnego pola polaryzującego E0 oraz
potencjał wytwarzany przez ładunki polaryzacyjne na powierzchni kuli. We wzorze (4.10)
wyodrębnijmy oba te składniki potencjału zapisując go w postaci:
3
2 11 0 03
2 12
aE z E z
r
ε εϕε ε
−= ⋅ −+
(4.13)
Drugi składnik (−E0z) jest potencjałem pola zewnętrznego, zatem pierwszy składnik
przedstawia pole na zewnątrz kuli wytwarzane przez jej ładunki polaryzacyjne. Można go
przekształcić do postaci:
3 3 3
2 1 2 1 2 11 0 0 03 2 2
2 1 2 1 2 1
cos2 2 2
a a z aE z E E
r r r r
ε ε ε ε ε εϕ θε ε ε ε ε ε
− − −= ⋅ = ⋅ = ⋅+ + +
(4.14)
Porównując ten wzór z wyrażeniem przedstawiającym potencjał wytwarzany przez dipol
elektryczny o momencie dipolowym µ:
2
0
1 cos
4dipola r
µ θϕπε
= (4.15)
zauważymy, że wzór (4.14) przedstawia pole dipola o momencie dipolowym:
3 2 10 0
2 1
42
a Eε εµ πε
ε ε−=
+ (4.16)
Tak więc kula z dielektryka o stałej dielektrycznej ε2 znajdująca się w otoczeniu o stałej
dielektrycznej ε1 i w jednorodnym polu o natężeniu E0 wytwarza pole:
− na zewnątrz takie, jak umieszczony w jej środku dipol o momencie dipolowym (4.16),
− wewnątrz swojej objętości pole jednorodne, które sumując się z polem zewnętrznym E0
stanowi nadal pole jednorodne, chociaż o innym już natężeniu danym wzorem (4.12).
Dipolowy charakter pola na zewnątrz spolaryzowanej kuli można scharakteryzować
definiując polaryzowalność kuli jako współczynnik proporcjonalności α między natężeniem
pola zewnętrznego i wyindukowanym momentem dipolowym kuli:
0Eµ α=

13
Z wzoru (4.16) wynika natychmiast polaryzowalność kuli dielektrycznej:
3 2 10
2 1
42
aε εα πε
ε ε−=
+ (4.17)
W szczególnym przypadku gdy kula jest metalowa (ε2 → ∞) otrzymujemy polaryzowalność o
wartości 304 aα π ε= . Wzory powyższe są analogiczne jak wzory (3.1)-(3.4) przytoczone w
części 3 niniejszego opracowania.
Warunkiem jednorodności pola elektrycznego wewnątrz kuli jest jednorodność pola
elektrostatycznego zewnętrznego E0, które tę kulę polaryzuje. W przypadku pól zmiennych w
czasie, np. gdy kula znajduje się w polu fali elektromagnetycznej, spełnienie warunku
jednorodności pola wymaga, aby rozmiary kuli były dużo mniejsze od długości fali. Ten
warunek jest dobrze spełniany przez obiekty takie jak kuliste cząstki metaliczne lub z innych
materiałów, o nanometrowych rozmiarach. W takim przybliżeniu można pominąć, jako
bardzo małe, różnice faz pola polaryzującego cząstkę w różnych jej punktach. Przestają być
wówczas istotne efekty retardacyjne i cząstka kulista ma właściwości punktowego dipola
elektrycznego. Ten szczególny przypadek jest rozpatrywany w teorii Mie jako przybliżenie
dla małych cząstek (patrz: część 5. niniejszego opracowania). Jednak pełny opis polaryzacji
kuli polem fali elektromagnetycznej, nie tylko niejednorodnym przestrzennie ale też szybko
zmieniającym się w czasie, wymaga znacznie bardziej złożonego opisu matematycznego,
również naszkicowanego w części 5.

14
5. Teoria Mie rozpraszania i absorpcji światła przez obiekt kulisty
W badaniach i zastosowaniach zjawisk specyficznych dla obiektów fizycznych o rozmiarach
nanometrowych (tj. poniżej 1 µm) szczególną rolę odgrywają zjawiska z udziałem cząstek
metalicznych lub dielektrycznych o kształcie kulistym. Pierwszy opis matematyczny
pochłaniania i rozpraszania światła na zawiesinie małych kulistych cząstek, z zastosowaniem
dobrze zdefiniowanych przybliżeń, podał w roku 1908 Gustav Mie. Jego artykuł na ten temat
opublikowany w Annalen der Physik (25 (1908), str. 377) należy do dziesiątki najczęściej
cytowanych w dziedzinie fizyki, a w minionych dwudziestu latach liczba cytowań przyrasta o
kilkaset rocznie. Teoria Mie zawdzięcza tę niezwykłą popularność szerokiej stosowalności w
opisie nie tylko właściwości optycznych kulistych nanocząstek metalicznych, ale też np.
właściwości optycznych aerozoli (mgła, smog, pyły), zawiesin substancji w stanie silnego
rozdrobnienia w cieczy (w tym koloidów). Stanowi też podstawę działania takich urządzeń
badawczych jak lidar czy nefelometr. Opis rozpraszania i absorpcji światła na cząstkach
kulistych został już dawno rozszerzony na podobne obiekty, np. cząstki wydłużone
elipsoidalnie, cząstki kuliste z równomierną powłoką (także wielowarstwową) z innego
materiału.
Przy założeniu, że zawiesina jest na tyle rzadka, że obserwacji podlega światło tylko
jednokrotnie rozproszone na cząstkach zawiesiny, można opisać absorpcję i rozpraszanie
światła jako addytywnie złożone z efektów pochodzących od oddzielnych cząstek. Wystarczy
wówczas opisać wpływ pojedynczej cząstki na falę padającą, która ulega częściowo
rozproszeniu, a częściowo absorpcji. Mie podał taki opis w roku 1908 w ramach wyłącznie
elektrodynamiki klasycznej, rozpatrując oddziaływanie fali z cząstką kulistą o funkcji
dielektrycznej εs (w ogólności zespolonej) w ośrodku niepochłaniającym światła, czyli o
funkcji dielektrycznej εm rzeczywistej. Nie zakładał też ograniczeń co do promienia
Rys. 5.1. Orientacja przestrzenna i układy współrzędnych do opisu rozpraszania światła. Materiał kuli charakteryzuje funkcja dielektryczna εs (w ogólności zespolona); w ośrodku otaczającym funkcja dielektryczna εm jest rzeczywista.
padająca fala płaska
x
y
z θ
ϕ
r
εm εs

15
cząstki, który może być zarówno mniejszy jak i dużo większy od długości fali światła.
Schemat układu fizycznego oraz orientację układu współrzędnych przedstawia Rys. 5.1.
Problem sprowadza się do wyznaczenia wektorów pola elektrycznego w dowolnym punkcie r
(transmisja i światło rozproszone), gdy na cząstkę pada fala o określonej częstości, w
ogólności nie będąca falą płaska, a więc mająca pewien niejednorodny rozkład intensywności
w przestrzeni (przypadek fali płaskiej jest przybliżeniem ogólnych rozwiązań, p. poniżej).
Pole elektryczne i magnetyczne zakłada się w ogólnej postaci fali biegnącej, o zmodulowanej
przestrzenie amplitudzie:
( ) ( ) ( ) ( )0 0, ( ) , , ( )i t i tt e t eω ω− −= =kr krE r E r H r H r (5.1)
Amplitudę stanowi czynnik zależny od współrzędnych. Jest on poszukiwanym rozwiązaniem
równań Maxwella, które w ośrodku niemagnetycznym (µ = 1) bez prądów i bez ładunków
swobodnych mają postać:
0 tµ ∂∇ × = −
∂H
E (5.2)
0 0 tεε µ ∂∇ × =
∂E
H (5.3)
Korzystając z tożsamości:
( ) ( ) 2∇ × ∇ × = ∇ ∇ − ∇A A Ai (5.4)
oraz z przemienności różniczkowania względem współrzędnych (∇) i czasu, dla założonych
w postaci (5.1) pól otrzymuje się dwa analogiczne równania, np. dla pola elektrycznego:
( ) ( )2 20 0 0 0 0εε µ ω∇ + =E r E r (5.5)
Uwzględniając związki:
0 0 2
1 , k
c c
ωε µ = = (5.6)
otrzymujemy równania dla obu pól takie jak dla pola elektrycznego:
2 2 0kε∇ + =E E
gdzie dla uproszczenia postaci równań pominięty został indeks dolny oraz jawna zależność od
współrzędnych E(r ). Jest to podstawowe równanie, stanowiące punkt wyjścia dla teorii
podanej przez Mie, jej późniejszych modyfikacji rachunkowych, a także teorii opisujących
rozpraszanie i absorpcję światła przez obiekty niesferyczne, wielowarstwowe, etc.
Metoda rachunkowa Mie polega na przedstawieniu pola elektromagnetycznego w
całym rozpatrywanym obszarze (kula + otoczenie) w postaci fali rozproszonej Esca (scattered,

16
na zewnątrz kuli) oraz pola wewnętrznego Eint (internal, wewnątrz kuli), nałożonych na falę
padającą Einc (incident) oraz analogicznie dla pola magnetycznego H. Rozwiązania dla pól
otrzymuje się metodą potencjału skalarnego podaną przez Hertza. W metodzie potencjału
skalarnego ogólna postać poszukiwanych rozwiązań dla pól elektrycznego i magnetycznego
obierana jest w postaci iloczynu pewnych funkcji skalarnych ψe (r,θ,ϕ) , ψm
(r,θ,ϕ) i wektora
położenia r . Zakłada się przy tym, że oba pola są rezultatem rotacji z iloczynu r ·ψ (który
pełni rolę potencjału wektorowego), np. dla pola elektrycznego powinno być:
( ) ( )e mikψ ψ= ∇ × ∇ × + ∇ ×E r r (5.7)
Oba pola, elektryczne i magnetyczne, można wtedy wyrazić jako kombinację liniową
pewnych funkcji wektorowych M i N,
( ), ,rψ θ ϕ= ∇ × ⋅ M r (5.8)
( )1 1, ,
m m
rk k
ψ θ ϕ= ∇ × = ∇ × ∇ × ⋅ N M r (5.9)
gdzie km oznacza tu wartość wektora falowego w ośrodku otaczającym kulę:
0
2 2m mk n
π πλ λ
= =
zaś nm jest współczynnikiem załamania ośrodka otaczającego.
W dalszym ciągu naszkicowanych tu obliczeń pominiemy pole magnetyczne, dla
którego rozwiązania mają analogiczną postać jak dla pola elektrycznego. Ponieważ z
równania falowego wynikają zbiory rozwiązań dla funkcji ,l kψ (l, k – liczby całkowite), to
rozwiązania dla pola elektrycznego fali padającej (Einc), pola elektrycznego fali rozproszonej
(Esca) i pola wewnętrznego (Eint) stanowią szeregi wyrażeń zawierających funkcje wektorowe
M l,k oraz Nl,k :
, , , ,2,
minc l k l k l k l k
l km
kA B
ω ε µ = + ∑E M N (5.10a)
, , , ,2,
msca l k l l k l k l l k
l km
kA a B b
ω ε µ = + ∑E M N (5.10b)
, , , ,2,
mint l k l l k l k l l k
l km
kA c B d
ω ε µ = + ∑E M N (5.10c)
Współczynniki Al,k oraz Bl,k są wyrażeniami algebraicznymi o postaci funkcji wymiernej
liczb całkowitych l, k. Funkcje wektorowe M l,k i Nl,k w przypadku symetrii sferycznej
przedstawiają wzory:

17
( ) ( )
,
1 1ˆ ˆ
sinl k
d r d r
r d r dθ ϕψ ψ
θ ϕ θ= −M e e (5.11)
( ) ( ) ( ), , , ,,
1ˆ ˆ ˆ1
l m l m
l k r
m
d rM d rMl l
k r dr drϕ θ
θ ϕψ = + + +
N e e e (5.12)
w których skalarna funkcja ψ wyraża się przez wielomian Bessela Zl i funkcję Legendre’a Plk:
( ) ( ) ( ),
2, , cosk ik
l k l m lr Z k r P e ϕψ θ ϕ θπ
= (5.13)
Współczynniki al, bl, cl, dl wynikają z warunków granicznych dla pola fali na powierzchni
kuli. Warunki graniczne dla składowej stycznej wektora pola elektrycznego na granicy kula-
otoczenie można przedstawić w postaci równości:
[ ] 0inc sca int+ − × =E E E r (5.14)
Warunek ten powinien być spełniony dla |r | ≡ r = a. Bardziej szczegółowo, warunek ten
oznacza spełnienie następujących równań przez transwersalne składowe pola elektrycznego:
, , , , , , inc sca int inc sca intE E E E E Eθ θ θ ϕ ϕ ϕ+ = + = (5.15)
Analogiczne wzory jak powyżej podane wyrażają natężenie pola magnetycznego fali oraz
warunki graniczne dla tego pola. Z warunków granicznych wynikają następujące ogólne
wyrażenia dla współczynników Mie:
( ) ( ) ( ) ( )( ) ( ) ( ) ( )
2
2
l l l l
l
l l l l
n j n j j n j na
n j n h h n j n
χ χ χ χ χ χ
χ χ χ χ χ χ
′ ′− =′ ′−
(5.16a)
( ) ( ) ( ) ( )( ) ( ) ( ) ( )
l l l l
l
l l l l
j n j j n j nb
j n h h n j n
χ χ χ χ χ χ
χ χ χ χ χ χ
′ ′− =′ ′−
(5.16b)
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )l l l l
l
l l l l
j h h jc
j n h h n j n
χ χ χ χ χ χ
χ χ χ χ χ χ
′ ′− =′ ′−
(5.16c)
( ) ( ) ( ) ( )( ) ( ) ( ) ( )2
l l l l
l
l l l l
nj h nh jd
n j n h h n j n
χ χ χ χ χ χ
χ χ χ χ χ χ
′ ′− =′ ′−
(5.16d)
Wielkość s
m
nn n= , przy czym n oznacza tu (zespolony) współczynnik załamania materiału
kuli w stosunku do otoczenia, zaś nm jest współczynnikiem załamania ośrodka. Występujące
tu funkcje j l oraz hl są funkcjami cylindrycznymi Riccatiego-Bessela. Znaki „prim”

18
oznaczają w tych wzorach pochodne względem argumentu χ. W argumencie funkcji j l oraz
hl występuje parametr χ będący iloczynem wektora falowego światła i promienia kuli:
0
2 2 ma anπ πχλ λ
= = . (5.17)
We wzorze tym nm oznacza bezwzględny współczynnik załamania (rzeczywisty) ośrodka
otaczającego kulę, zaś λ0 długość fali światła w próżni. Wartość tego wyrażenia stanowi
podstawowy parametr w całej teorii Mie, jako że stosunek promienia kuli do długości fali
determinuje liczbę oraz położenia rezonansów występujących w widmie ekstynkcji cząstki, a
w przypadku a << λ stanowi podstawę przyjęcia uproszczonych wzorów (p. poniżej).
Pole elektryczne fali rozproszonej zależy od odległości od kuli rozpraszającej. W bliskiej
odległości zawiera składową radialną, która szybko maleje na większych odległościach i
znika w strefie falowej (dalekiego pola, r >> a), gdzie istnieją już tylko fale poprzeczne
biegnące radialnie. Rys. 5.2 przedstawia rozkłady przestrzenne linii sił pola elektrycznego i
pola magnetycznego odpowiadające poszczególnym składnikom szeregów (5.10a-c),
obrazujące tzw. fale parcjalne. Linie te obrazują kierunki wektorów pola na powierzchni kuli
o promieniu
Rys. 5.2. Linie sił pól elektrycznego (u góry) i magnetycznego (u dołu) pochodzących od pierwszych czterech składników w szeregu (5.4b) w projekcji na przekrój kuli. Wg: M.Born, E. Wolf – Principles of Optics, Pergamon Press, 1965 (tłum. ros. 1973).
dużo większym od promienia kuli oddziałującej z falą padającą. Rys. 5.2 przedstawia linie sił
na powierzchni półkuli znajdującej się ponad płaszczyzną YZ na Rys. 5.1, zrzutowane na
płaszczyznę YZ. Dostrzegalne jest tu podobieństwo do promieniowania multipolowego.
Względne znaczenie poszczególnych składników zależy od parametru χ , wyrażającego
stosunek promienia kuli rozpraszającej do długości fali. Dla małych wartości χ w szeregach

19
(5.10b,c) dominuje składnik odpowiadający l = 1, który odpowiada przybliżeniu dipolowemu
(p. poniżej).
Skutkiem zerowania się wyrażeń w mianownikach wzorów przedstawiających al, bl,
cl, dl są obserwowane doświadczalnie rezonanse w rozproszeniu i absorpcji światła.
Rezonanse obserwowane są zarówno w funkcji długości fali rozpraszanego światła jak i w
zależności od rozmiarów cząstek rozpraszających (p. poniżej).
Wyznaczenie całkowitego pola oraz pól składowych fali elektromagnetycznej na
zewnątrz kuli w sposób skrótowo naszkicowany powyżej pozwala na wyznaczenie
przekrojów czynnych przy użyciu wektora Poyntinga, przedstawiającego strumień energii
niesionej przez pole fali w kierunku wektora falowego. Wektor Poyntinga np. dla fali
rozproszonej, scałkowany po wszystkich kierunkach w dużej odległości od kuli (przybliżenie
długofalowe), umożliwia wyznaczenie całkowitego strumienia energii fali rozproszonej.
Porównanie mocy niesionej przez pole fali rozproszonej ze strumieniem energii fali padającej,
przypadającym na przekrój kuli (πa2) umożliwia zdefiniowanie przekroju czynnego na
rozpraszanie. Ponieważ nie dysponujemy „polem absorbowanym”, moc absorbowaną można
wyrazić jako różnicę między mocą fali padającej i całkowitą mocą fali zmodyfikowanej przez
obecność kuli na jej drodze. Daje to podstawę do zdefiniowania przekroju czynnego dla
całkowitej ekstynkcji, tj. całkowitego ubytku energii fali w rezultacie jej oddziaływania z
pojedynczą cząstką kulistą. Z kolei, po odpowiednim zsumowaniu przekrojów czynnych dla
zbioru cząstek (z założenia nieoddziałujących między sobą) można obliczyć całkowity ubytek
energii w wiązce światła oddziałującej z badaną próbką. Ta właśnie wielkość jest mierzona
eksperymentalnie, zazwyczaj przy użyciu równoległej wiązki światła. Można też, oczywiście,
zmierzyć strumień światła rozproszonego w funkcji kątów θ oraz ϕ w celu np. określenia
średniego rozmiaru cząstek, od którego rozkład kątowy jest silnie zależny). Ten ostatni
pomiar jest podstawą nefelometrii – metody wyznaczania rozmiarów, kształtu i własności
dielektrycznych małych cząstek (np. w zawiesinie lub koloidzie).
Głównym celem teorii jest zdefiniowanie i podanie wzorów określających wielkości
mierzalne doświadczalnie. W przypadku małego obiektu rozpraszającego i absorbującego
światło takimi wielkościami, które mają bezpośredni związek z obserwowanymi
doświadczalnie zjawiskami są przekroje czynne na rozpraszanie i absorpcję światła. Pojęcie
przekroju czynnego w spektrofotometrii jest bardziej szczegółowo omówione w rozdziale o
absorpcji światła. Przekrój czynny na rozpraszanie Cscat oraz całkowity przekrój czynny
wyrażający ekstynkcję fali (wskutek rozpraszania i absorpcji) Cext są definiowane
następująco:

20
, scat extscat ext
inc inc
W WC C
I I= = (5.18)
gdzie I inc oznacza strumień energii światła padającego (incident) w W/m2. Moc rozpraszaną i
moc pochłanianą, Wscat (od: scatter) i Wext (od: extinct) (w watach) można wyznaczyć przez
całkowanie strumieni energii (wat/m2) wyrażonych wektorem Poyntinga, w pełnym kącie
bryłowym wokół kuli:
2
2
0 0
1Re sin
2scat scat scatW r d dπ π
θ θ ϕ= ×∫ ∫ E H (5.19)
Z kolei I inc jako moc padająca na koło o średnicy cząstki można wyznaczyć całkując strumień
energii fali padającej wprost na cząstkę kulistą (lub mnożąc go przez pole przekroju cząstki w
przypadku gdy fala padająca jest falą płaską). Powyższy wzór może też być użyty w formie
zmodyfikowanej tak, aby obliczyć moc promieniowania niesioną przez pole fali o określonej
polaryzacji, np. z wektorem pola elektryczngo równoległym do osi X lub Y na Rys. 5.1. Z
obliczenia mocy całkowitej (tj. niezależnie od polaryzacji, przedstawionej powyższym
wzorem) wynikają przekroje czynne wyrażające się wzorami:
( )( )2 2
21
22 1scat l l l l
lm
C l a H b Fk
π ∞
=
= + +∑ (5.20)
( )( )2 2
21
2Re 2 1ext l l l l
lm
C l a H b Fk
π ∞
=
= + +∑ (5.21)
w których:
( )
( )( )
( ) 2! cos2
1 !
kll
lk l
l k dPH
l l l k d
θθ=−
−= + +
∑ (5.22)
( )
( )( )
( ) 2! cos2
1 ! sin
kll
lk l
l k PF
l l l k
θθ=−
−= + +
∑ (5.23)
Powyższe wzory dotyczą rozpraszania i absorpcji fali padającej o dowolnym, niejednorodnym
rozkładzie intensywności w przestrzeni. Są one stosowalne np. w przypadku, gdy cząstki
rozpraszające znajdują się w polu fali zanikającej w pobliżu granicy dwu ośrodków,
występującej w zjawisku całkowitego wewnętrznego odbicia. Osadzone na granicy ośrodków
cząstki znajdują się wówczas w polu, którego amplituda szybko zmienia się w kierunku
prostopadłym do granicy i może niemal całkowicie zanikać na odległości znacznie mniejszej
od długości fali. Silnie niejednorodne natężenie fali świetlnej (laserowej) występuje również
w mikroskopie konfokalnym i w pułapkach optycznych służących do manipulacji cząstkami
(optical tweezers). W przypadku prostszym i znacznie częściej spotykanym, gdy fala padająca

21
jest falą płaską o jednorodnym rozkładzie energii w przestrzeni, w powyższych bardzo
złożonych wzorach zerują się wszystkie składniki z wyjątkiem wskaźnika k = 1. Wtedy
wyrażenia na przekroje czynne przyjmują znacznie prostszą formę:
( )( )2 2
21
22 1scat l l
lm
C l a bk
π ∞
=
= + +∑ (5.24)
( )( )21
2Re 2 1ext l l
lm
C l a bk
π ∞
=
= + +∑ (5.25)
Ta właśnie postać wzorów jest najczęściej stosowana gdy badane są widma ekstynkcji w
typowej, równoległej wiązce światła w spektrofotometrze. Ma ona postać sumy, która – jak to
wzmiankowano powyżej – wynika z rozwinięcia rozwiązań w szereg analogiczny do
rozwinięcia multipolowego.
W przypadku cząstek metalicznych jak i dielektrycznych o szczególnie małych
rozmiarach określanych jako nanocząstki (∼10-9 m), obiekty rozpraszające światło mają
zwykle rozmiary od 2-3 nm do 50-100 nm. 2 nm to minimalny rozmiar, przy którym
nanocząstka traci właściwości wieloatomowego klastra (molekuły) uzyskując właściwości
objętościowe (bulk) materiału. W takim zakresie rozmiarów, dla światła z zakresu bliskiego
ultrafioletu, widzialnego i bliskiej podczerwieni, parametr χ jest znacznie mniejszy od jedynki
i w sumach (5.24) i (5.25) istotny pozostaje jedynie składnik dla l = 1, który odpowiada
elektrycznemu momentowi dipolowemu indukowanemu w małej kuli przez pole fali płaskiej
(por. Rys. 5.2 powyżej). Wynika to z zaniku różnic fazowych fali w różnych punktach kuli;
polaryzacja kuli staje się wówczas polaryzacją statyczną, jak w opisie elektrostatycznym
(patrz: część 4 niniejszego opracowania). Całkowity przekrój czynny dla ekstynkcji przyjmuje
wtedy postać:
( ) ( ) ( )( ) ( )
3 220 2 2
1 2
92
ext m
m
C Vc
ε ωωω εε ω ε ε ω
=+ +
(5.26)
gdzie V0 = (4/3)πa3 jest objętością cząstki, ε1 jest częścią rzeczywistą a ε2 częścią urojoną
funkcji dielektrycznej cząstki kulistej. Jest to najczęściej używana postać wzoru
przedstawiającego kształt widma ekstynkcji nanocząstek, szczególnie metalicznych, dla
których oba składniki funkcji dielektrycznej ε1(ω), ε2(ω) są dobrze znane w pełnym zakresie
widm optycznych. Powyższe wyrażenie przedstawia całkowity przekrój czynny pojedynczej

22
nanocząstki. Aby otrzymać absorbancję zawiesiny takich nanocząstek, należy uwzględnić
liczbę nanocząstek w jednostce objętości (patrz: część 7 niniejszego opracowania).
Rysunki poniżej ilustrują efekty interferencyjne pojawiające się przy pewnych
rozmiarach cząstek rozpraszających. Przekrój czynny pokazany w lewej części Rys. 5.3
wzrasta wraz z rozmiarem cząstki rozpraszającej, a na tym tle widoczne są oscylacje o
różnym okresie, odzwierciedlające rezonanse fali w objętości cząstki kulistej. Należy mieć na
uwadze, że zakres rozmiarów cząstki objęty poziomą skalą wykresu jest dość szeroki: od zera
do 3λ0 (p. wzór 5.17). Szerokie maksima ekstynkcji przy χ wynoszącym kolejno 5, 10, 15,
20 i 25 są rezultatem kolejnych interferencji gdy kolejne połowki fali mieszczą się na jej
średnicy, co bezpośrednio wskazuje skala w kolorze niebieskim. Jest to efekt analogiczny do
interferencji światła w cienkiej płytce płaskorównoległej. Na tym tle występują bardzo wąskie
rezonanse pochodzące od poszczególnych składników multipolowych, wzmiankowanych
powyżej w odniesieniu do sum we wzorach (5.10b,c) i zilustrowanych na Rys. 5.2. Prawa
część Rys. 5.3 przedstawia te same dane co w części lewej, podzielone przez pole przekroju
cząstki kulistej. Przekrój czynny, z oczywistych przyczyn wzrastający wraz z rozmiarem
cząstki, jest tu przedstawiony w zmodyfikowany sposób jako „efektywny przekrój czynny”
Q:
2 2
, sca extsca ext
C CQ Q
a aπ π= = (5.27)
Rys. 5.3. Po lewej – obliczony całkowity przekrój czynny Cext na rozpraszanie światła przez kuliste cząstki dielektryczne o rzeczywistym współczynniku załamania ns=1.59, w zależności od ich rozmiaru w postaci parametru χ (p. wzór 5.11 powyżej). Po prawej – te same dane przedstawione w postaci przekroju efektywnego Qext = Cext / πa 2. Obliczenia wykonano dla ustalonej długości fali rozpraszanej λ0 = 1000 nm. Według: http://www.orc.soton.ac.uk
0 a=λ0 a=2λ0 a=3λ0

23
Tak przedstawiony przekrój czynny pokazuje, że wąskie rezonanse są najbardziej znaczące w
zakresie małych rozmiarów cząstek, lecz przy wartości χ mniejszej niż 2 znikają zupełnie.
Ten zakres, poniżej 0.2 długości fali, charakteryzuje się brakiem rezonansów optycznych w
cząstkach dielektrycznych nieabsorbujących. Jest to zakres rozmiarów cząstek, w którym
stosowalny jest opis rozpraszania światła przez cząstki dużo mniejsze od długości fali. Został
on sformułowany przez Rayleigha; objaśnia m.in. dobrze znaną proporcjonalność
efektywności rozpraszania do odwrotności czwartej potęgi długości fali: Q ∼ λ-4. Teoria
Rayleigha objaśnia m.in. niebieski kolor nieba i czerwony kolor słońca nad horyzontem.
Teoria Mie obejmuje ten zakres wielkości cząstek, a jej wzory w odpowiednich warunkach
przechodzą we wzór Rayleigha. Stosowalna jest jednak w dowolnie szerokim zakresie
rozmiarów cząstek kulistych, i, co istotne, stosuje się również do cząstek absorbujących
światło, np. metalicznych, których funkcja dielektryczna jest zespolona.
Rys. 5.4. Obliczone widma ekstynkcji nanocząstek złota o różnych rozmiarach, wraz ze składnikami tych widm wnoszonymi przez poszczególne wzbudzenia multipolowe odpowiadające kolejnym wartościom liczby l (por. wzory (5.18) 9 (5.19)). Promień nanocząstki jest podany na poszczególnych panelach. Według: http://www.orc.soton.ac.uk
a) 10 nm
Suma
b) 40 nm
c) 100 nm
d) 250 nm

24
Podstawową charakterystyką spektralną zjawisk rozpraszania i absorpcji światła przez
małe cząstki jest widmo ekstynkcji, tj. zależność przekroju czynnego od długości fali. Rys.
5.4 pokazuje obliczone widma ekstynkcji dla kulistych cząstek złota o różnych rozmiarach,
wraz z wkładami wnoszonymi przez poszczególne składniki multipolowe. Podobnie jak w
przypadku kulki dielektrycznej, tylko jeden składnik – dipolowy – jest istotny gdy rozmiar
kulki jest odpowiednio mały (Rys. 5.4a). Jak widać, również i w tym przypadku metalicznej
cząstki absorbującej światło, gdy promień nanocząstki nie przekracza ≈50 nm, jej widmo
ekstynkcji jest bardzo dobrze opisywane tylko składnikiem z l = 1 (dipolowym). Gdy promień
nanocząstki staje się porównywalny z długościami fal w badanym zakresie, w widmie mogą
pojawić się wielokrotne maksima odpowiadające rezonansom multipolowym.
Rozpraszanie światła charakteryzuje nie tylko całkowity przekrój czynny;
intensywność światła rozproszonego wykazuje również silną zależność od kąta rozproszenia
wynikającą z istnienia rezonansów w polaryzacji rozpraszającej kulki wzbudzanej falą
swietlną. Również tu powtarza się tu regularna zmiana charakteru rozpraszania jak te opisane
powyżej. Ilustruje to Rys. 5.5, pokazujący rozkład intensywności światła rozproszonego w
Rys. 5.5. Kątowe rozkłady intensywności światła rozproszonego przez kuliste cząstki złota o różnych rozmiarach. Długość fali padającej 550 nm; współczynniki załamania: ośrodka nm = 1.33 (woda), złota ns = 0.57 + i·2.45 (dla częstości fali padającej). [M. Born, E. Wolf – Principles of Optics]
płaszczyźnie polaryzacji światła padającego. Intensywność światła rozproszonego na tych
wykresach polarnych (we współrzędnych biegunowych) odpowiada radialnej współrzędnej
punktu na krzywej. Krzywa wewnętrzna pokazuje rozkład intensywności światła
rozproszonego gdy światło padające jest spolaryzowane liniowo w płaszczyźnie rysunku, zaś
a → 0 a = 80 nm
a = 90 nm

25
krzywa zewnętrzna odpowiada rozpraszaniu światła niespolaryzowanego. Jak widać, gdy
rozmiary kuli rozpraszającej są znikome, cząstka rozpraszająca ma charakter promieniującego
dipola. Gdy jej rozmiary są porównywalne z λ, przeważa rozproszenie do przodu pod
niewielkimi kątami, a także pojawiają się ostre maksima światła rozproszonego wstecz,
których liczba wzrasta przy większych rozmiarach cząstki.
6. Rozszerzenia teorii Mie
Teoria Mie rozpraszania i absorpcji światła dotyczy obiektów o kształcie kulistym z
dowolnego materiału – dielektryka lub przewodnika. Przytoczona powyżej wersja
oryginalnego rozwiązania tego zagadnienia nie jest jedyną. Już w rok po opublikowniu teorii
przez Gustava Mie, Peter Debye w 1909 r. rozważał zagadnienie ciśnienia światła na kulistą
cząstkę, również używając metody obliczeniowej wykorzystującej potencjały skalarne
(funkcje ψ w przytoczonych w cz. 5 obliczeniach). W połowie XX w. pojawiły się inne
matematyczne metody rozwiązywania zagadnienia Mie dla cząstek kulistych, a wraz z
rozwojem komputerowej techniki obliczeniowej także wiele wersji programów do obliczania
pól i intensywności rozproszonego światła oraz absorbowanej mocy promieniowania. Od lat
1970 datują się metody analityczne pozwalające opisać zjawiska także dla kuli w ośrodku
absorbującym (tj. z zespoloną funkcją dielektryczną w miejsce stałej εm).
Nanocząstki mogą składać się z atomów więcej niż jednego pierwiastka, tworzących
bądź stopy, bądź też odrębne warstwy ułożone wokół kulistego rdzenia. Własności
nanocząstek utworzonych ze stopów metali opis właściwości nie odbiega od przypadku
prostszego – cząstek z czystego metalu. Natomiast cząstki stanowiące rdzeń z
wielowarstwowym pokryciem (metalicznym, dielektrycznym lub na przemian) zawierają w
swej objętości dodatkowe granice, na których pole elektromagnetyczne musi spełniać
dodatkowe warunki. Jako że opis układów o symetrii sferycznej jest stosunkowo nietrudny,
rozwiązane zostało również zagadnienie rozpraszania i absorpcji światła przez cząstki kuliste
pokryte warstwami. W takich układach mogą powstawać odrębnie i sprzęgać się ze sobą
rezonanse w rdzeniu i w powłokach, prowadząc w rezultacie do złożonych widm optycznych.
Ich opis wzorami analitycznymi został sformułowany zarówno dla prostszego przypadku
kwazistatycznego (stosowalnego gdy a << λ), jak i ogólnego, dla większych cząstek, w tym
wielowarstwowych, gdy należy wziąć pod uwagę różnice faz pola w różnych punktach
cząstki. Polaryzowalność kwazistatyczną cząstki o promieniu a (funkcja dielektryczna εs(ω)),

26
pokrytej pojedynczą powłoką o grubości d (funkcja dielektryczna ε(ω)), znajdującej się w
otoczeniu o stałej dielektrycznej εm przedstawia wzór:
( )( ) ( )( )
( )( ) ( )( )( )
3
3
3
2 24
32 2 2
s m s s m s
s m s s s m
a
a da d
a
a d
ε ε ε ε ε ε ε επα
ε ε ε ε ε ε ε ε
− + + − + + = + + + + − − +
(6.1)
Ponieważ funkcje dielektryczne rdzenia i powłoki mogą być zespolone, to ze względu na
złożoną postać wzorów oblicza się odrębnie zespoloną polaryzowalność cząstki, a następnie
używa się jej do obliczenia przekroju czynnego dla absorpcji i rozproszenia światła. Dla
cząstek wielowarstwowych istnieją rozwiązania analogiczne do wzorów (5.24) i (5.25), w
których współczynniki al i bl wynikają z wzorów rekurencyjnych o stopniu zagnieżdżenia
zależnym od liczby warstw.
Istnieją również rozwiązania umożliwiające obliczenie ekstynkcji dla cząstek z
materiałów o bardziej zróżnicowanych właściwościach, np. z materiału dwójłomnego
optycznie, aktywnego optycznie, a także dla cząstek o określonych właściwościach
magnetycznych, które w innych wariantach teorii rozpraszania były zawsze pomijane przez
założenie przenikalności magnetycznej µ = 1.
Osobnego opracowania doczekało się zagadnienie rozpraszania krótkotrwałego
impulsu pola w formie femtosekundowego impulsu światła lub impulsu pola elektrycznego.
W takich przypadkach pole fali rozpraszanej zmienia się w czasie zanim zdąży ustalić się
stacjonarny obraz interferencyjny wokół cząstki. Sposób obliczeń przy takiej dodatkowej
komplikacji trudności polega na rozłożeniu impulsu rozpraszanej fali na składowe
fourierowskie (które mają postać nieskończonego, a więc stacjonarnego ciągu fal). Po
oddzielnym rozpatrzeniu oddziaływania tych składowych z cząstką, składowe fourierowskie
pola należy złożyć (z uwzględnieniem stosunków fazowych) w pole będące rezultatem ich
interferencji.
Metodą podobną do rachunku zaburzeń (używanego w mechanice kwantowej, a
wywodzącego się z opisu zaburzeń ruchu planet w mechanice klasycznej) opisane zostały
właściwości optyczne cząstek o kształcie nieco odbiegającym od kulistego, a w przypadku
cząstek o kształcie elipsoidy obrotowej otrzymano ścisłe wartości przekroju czynnego dla
ekstynkcji. W ich obliczaniu wyznaczana jest najpierw zespolona polaryzowalność α wzdłuż
osi elipsoidy. Polaryzowalność elipsoidy dla k-tej osi wyraża się wzorem:
( )( ) 0
mk
m m k
VL
ε ω εα ε
ε ε ω ε−
=+ −
(6.2)

27
przy czym V jest objętością, V = (4/3)πabc (por. wzory (3.3) i (3.4) dla kuli). Współczynniki
Lk wyrażają kształt elipsoidy, przy czym kL∑ =1. Dla kuli jest: La = Lb = Lc =1/3, dla
elipsoidy obrotowej La ≠ Lb = Lc. Następnie ekstynkcja jest uśredniana dla różnych orientacji
cząstki w polu fali padającej. Dla wydłużonej elipsoidy obrotowej, której półoś A jest większa
od półosi B i C, przy czym B=C, współczynnik ekstynkcji uśredniony po wszystkich
orientacjach wynosi:
( ) ( )3
232 2
21
21 2
12
3 1
jm
jj
mj
PNV
P
P
επ εκ
λε ε ε
=
= −
+ +
∑ (6.3)
Pj oznaczają tu tzw. współczynniki depolaryzacji dla osi elipsoidy zdefioniowane jako:
2
2
1 1 1ln 1
2 1A
e eP
e e e
− + = − − (6.4)
( ) ( )21 2 e= 1B C AP P P B A= = − − (6.5)
W przypadku, gdy cząstki oddziałujące z falą elektromagnetyczną mają kształt, dla
którego nie jest możliwe analityczne rozwiązanie równań Maxwella, niezbedne jest użycie
metod numerycznych. W ostatnim ćwierćwieczu opracowano kilka metod rozwiązywania
tego zagadnienia, wśród których najbardziej efektywne jest tzw. przybliżenie dyskretnych
dipoli (Discrete Dipole Approximation, DDA). W metodzie DDA opisywany obiekt jest
reprezentowany w kubicznej sieci zawierającej N polaryzowalnych elementów (np.
10x10x10). Następnie wybiera się część z tych elementów jako „obsadzone”, tak aby
tworzyły one opisywany obiekt o dowolnym kształcie przestrzennym (metoda jest stosowalna
również do układu złożonego z wielu oddzielnych obiektów, np. dwu odrębnych kul).
Każdemu obsadzonemu elementowi przypisywana jest polaryzowalność dipolowa, np.
elementowi i-temu polaryzowalność αi , oraz współrzędne (wektor położenia r i). Nie
uwzględnia się przy tym polaryzowalności wyższych rzędów (kwadrupolowej), jako że z
założenia elementy reprezentują na tyle małe fragmenty obiektu, że do każdego z nich
stosowalne jest przybliżenie dipolowe. Polaryzacja każdego pojedynczego elementu jest
rezultatem jego oddziaływania z lokalnym polem elektrycznym Eloc :
( )i i loc iα= ⋅p E r (6.6)
Pomijając czynnik zależności od czasu eiωt można zapisać pole lokalne działające na i-ty
element jako:

28
( ) ( ), , , 0 exploc i loc i inc i dip i i ij jj i
i≠
≡ = + = ⋅ −∑E r E E E E k r A p (6.7)
gdzie E0 i k są amplitudą i wektorem falowym fali padającej, zaś macierz oddziaływania A
ma następującą postać (dla i≠j):
( ) ( ) ( ) ( )2 2
3 2
exp 13
ij ij
ij j ij ij j ij j ij ij j
ij ij
i ik r
r r
⋅ − ⋅ = × × + − ⋅
k r k rA p r r p p r r p (6.8)
W powyższych wyrażeniach funkcje dielektryczne cząstki (ε) jak i otaczającego ją ośrodka
(εm) występują niejawnie w polaryzowalności αi. Kwadrat wektora falowego w ośrodku
otaczającym ma we wzorze (6.8) wartość
2
22 mk
c
ω ε=
Postać wyrażenia dla αi jest taka, że przy jej użyciu dla poszczególnych elementów uzyskuje
się dla nieskończonego ośrodka złożonego z takich elementów funkcję dielektryczną taką,
jaką ma funkcja dielektryczna materiału w dużej objętości. Relacje fazowe w
oddziaływaniach między poszczególnymi elementami uwzględnione są poprzez czynnik
exp(ik·r ij), który po pomnożeniu przez exp(-iωt) nadaje odpowiednią fazę sile oddziaływania
między elementami i-tym oraz j-tym. Wstawiając wyrażenia z (6.8) i (6.7) do (6.6) otrzymuje
się równanie
′ ⋅ =A P E (6.9)
w którym macierz A’ wyraża się przez A ij (6.8). Dla układu złożonego z N elementów
równanie (6.9) przedstawia układ 3N złożonych równań liniowych dla 3N składowych
wszystkich wektorów Ei oraz pi. Rozwiązanie daje wektor P, który służy do obliczenia
przekroju czynnego i innych własności cząstek i pola elektrycznego. Teoria DDA nie jest
całkowicie ścisła, ale w praktyce odtwarza widma ekstynkcji nanocząstek metalicznych z
błędem nie przekraczającym 10%, niezależnie od kształtu, wielkości i składu cząstki. Można
np. stosunkowo łatwo wyznaczyć natężenie pola elektrycznego fali świetlnej przy
powierzchni cząstek, uzyskując informacje o bardzo szerokim zakresie wartości natężenia
pola przy krawędziach nanocząstek o różnych geometrycznych kształtach, a także w
szczelinie między np. dwiema blisko położonymi cząstkami kulistymi. Znajomość pola
elektromagnetycznego w takich układach, dla których niemożliwy jest opis analityczny a
jedynie obliczenia numeryczne, jest kluczowa dla wyjaśnienia zjawisk takich jak
wzmocnienie ramanowskiego rozpraszania światła lub wpływ nanostruktur metalicznych na
fluorescencję molekuł.

29
Osobny rodzaj zagadnień związanych z oddziaływaniem mikro- i nanocząstek z
promieniowaniem elektromagnetycznym wynika z konieczności opisu efektów
specyficznych, jakim podlegają molekuły zaadsorbowane na powierzchni nanocząstek. Efekt
specyficzny oznacza tu oddziaływanie zależne od budowy elektronowej molekuł, ich
struktury przestrzennej i składu chemicznego, a także analogiczne własności materiału z
jakiego utworzone są nanocząstki. Rezultatem oddziaływań specyficznych są zmiany w
optycznym i oscylacyjnym widmie absorpcji molekuł, zmiany w widmie i intensywności
fluorescencji (gaszenie fluorescencji lub, zależnie od geometrii takiego układu, wzmocnienie
fluorescencji), oraz silny efekt wzmocnienia ramanowskiego rozpraszania światła. Efekty te
mają i potencjalnie mogą mieć wiele zastosowań o znaczeniu praktycznym. Ich objaśnienie
wymaga dobrego opisu pola elektromagnetycznego przy powierzchni nanocząstek, co
zasadniczo odróżnia tego rodzaju zagadnienia od teorii Mie skoncentrowanej na dalekim polu
promieniowania. Pole przy powierzchni cząstki np. metalicznej wnika nieco w głąb cząstki w
skali atomowej, zatem powierzchnia cząstki nie stanowi tak ostrej granicy dla sformułowania
warunków granicznych w opisie pól wewnętrznego i zewnętrznego, jak to się zwykle
przyjmuje w elektrodynamice klasycznej. Pojawiają się tu kwestie wykraczające poza
klasyczną teorię Mie. Także zupełnie nowe zagadnienia występują w przypadku nanocząstek
o średnicy poniżej 2-3 nm, gdy właściwości elektryczne i optyczne – dobrze określone dla
nanoczastek o „dużej” objętości – zmieniają się, ponieważ istotna staje się ziarnista, atomowa,
nieciągła struktura materii. Obiekty zawierające od ∼10 do około 500 atomów mają
właściwości wymagające uwzględnienia oddziaływań tworzących je atomów i należą już do
innej kategorii, tzw. klastrów atomowych.
7. Absorpcja światła w ośrodku materialnym
Światło lub inne fale elektromagnetyczne biegnące w ośrodku materialnym niezupełnie
przezroczystym mogą ulegać absorpcji i rozpraszaniu. Intensywność każdego z tych zjawisk
zależy na ogół dość silnie od częstości światła, a jej rozkład spektralny stanowi odpowiednie
widmo – absorpcji lub rozpraszania. Często bywa, że zjawisko rozpraszania powoduje
usuwanie z biegnącej fali stosunkowo nieznacznej części jej energii, znacznie mniejszej niż
energia przekazywana ośrodkowi poprzez absorpcję fotonów. Intensywność pochłaniania
energii fali przedstawia wówczas w pełni widmo absorpcji (a w przeciwnym wypadku –
widmo rozpraszania). Jeżeli jednak ubytek energii fali wynika w porównywalnym stopniu z

30
obu zjawisk, to spektralna zależność intensywności tego ubytku przedstawia tzw. widmo
ekstynkcji.
Niezależnie od fizycznej natury zjawiska osłabiającego falę biegnącą w ośrodku
materialnym, opis ilościowy ma prostą postać matematyczną, jednak formułowany jest przy
użyciu różnych wielkości fizycznych, jak: współczynnik absorpcji, indeks absorpcji,
absorbancja, przekrój czynny, współczynnik ekstynkcji. Poniżej opisane są podstawowe
pojęcia i prawa jakim podlegają rozpraszanie i absorpcja światła, oraz ograniczenia w ich
stosowalności.
Gdy płaska fala biegnie w ośrodku, to, jak to już opisano w części 1 niniejszego
opracowania (wzory (1.5-1.7), natężenie pola elektrycznego zanika wzdłuż geometrycznej
drogi x w tym ośrodku:
( ) 0 0
2 2
0,n
x i t x
t e eπκ πωλ λ
− − − = ⋅E r E (7.1)
a zatem zanika również strumień energii fali:
0 00 0
4 4exp , =xI I x I e γπκ πκγ
λ λ−
= − =
(7.2)
Najczęściej są używane miary absorpcji światła opisane poniżej. Absorpcję (wraz z
rozpraszaniem) substancji w postaci ciała stałego (kryształu) charakteryzuje współczynnik
absorpcji. Światło o długości fali λ na krótkiej drodze dx w ośrodku materialnym doznaje
ubytku intensywności proporcjonalnego do intensywności światła padającego i do grubości
tej warstwy. Przyjmując współczynnik proporcjonalności k, mamy równanie:
dI k I dx= − ⋅ ⋅ (7.3)
Całkując powyższe równanie otrzymujemy zależność I(x):
( ) 0kxI x I e−= (7.4)
Współczynnik k o wymiarze cm-1 jest współczynnikiem absorpcji; zależy on od długości fali
światła. Zależność k(λ) przedstawia widmo absorpcji.
Gdy światło przechodzi przez roztwór substancji absorbującej w przezroczystym
rozpuszczalniku, intensywność jego pochłaniania jest również funkcją długości fali, ale też
stężenia c substancji absorbującej. Równanie analogiczne do (7.3) ma wówczas postać:
( )dI a I c dxλ= − ⋅ ⋅ ⋅ (7.5)
Powyższy wzór zakłada proporcjonalność absorpcji do stężenia, która nie zawsze jest
zachowana. Wzór (7.5) oparty na takim założeniu (lub wynikający z niego wzór (7.6)) nosi

31
nazwę prawa Lamberta-Beera. Rozwiązanie tego równania, analogiczne do (7.4), w postaci
stosowanej w odniesieniu do roztworów jest przedstawiane w postaci potęgi dziesiętnej:
( ) ( ) ( )log0 0 010 10a c x e a c x c xI I e I Iλ λ ε λ− ⋅ ⋅ − ⋅ ⋅ ⋅ − ⋅ ⋅= ⋅ = ⋅ = ⋅ (7.6)
Występujący tu współczynnik ε(λ), podobnie jak k i a powyżej, charakteryzuje intensywność
absorpcji światła przez substancję rozpuszczoną. Jeżeli stężenie c tej substancji jest wyrażone
w molach na litr (mol/L), to liczbowa wartość ε(λ) stanowi dziesiętny molowy współczynnik
ekstynkcji tej substancji dla długości fali λ. Ta właśnie wielkość jako funkcja długości fali,
częstości lub liczby falowej, przedstawia właściwą miarę absorpcji światła przez substancję
rozpuszczoną i może być użyta do obliczenia stopnia pochłaniania światła przez jej roztwór o
dowolnym innym stężeniu. Przyjęte jest podawanie wartości ε o wymiarze odpowiadającym
standardowej grubości kuwetek fotometrycznych 1 cm; wymiar ε wynosi wówczas
L/(mol·cm).
Pomiar absorpcji światła (a w zasadzie ekstynkcji, bo pomiar wraz z rozpraszaniem)
polega na porównaniu intensywności przed i po przejściu przez badaną próbkę. Na podstawie
tych dwu wartości intensywności I0 oraz I przyrząd pomiarowy dostarcza wartość
absorbancji A próbki:
( ) ( )( )
0logI
AI
λλ
λ= (7.7)
Z porównania z wzorem (7.6) wynika, że:
( ) ( )A c xλ ε λ= ⋅ ⋅ (7.8)
Powyższy wzór stanowi podstawę metod analitycznych opartych na pomiarze absorpcji
światła przez roztwory – spektrofotometrii absorpcyjnej. Zwykle służy do określania stężenia
substancji na podstawie pomiaru absorbancji A warstwy roztworu o grubości x, lecz wymaga
znajomości molowego współczynnika ekstynkcji w użytym rozpuszczalniku i przy określonej
długości fali, zwykle odpowiadającej maksimum absorpcji. Wartości ε wielu substancji są
skatalogowane; można też posługiwać się własną kalibracją przy pomocy roztworów
wzorcowych.
Inna, rzadziej używaną miarą absorpcji światła jest przekrój czynny dla absorpcji.
Gdy roztwór, przez który biegnie światło o określonej długości fali, pochłania część mocy
wiązki światła, to na każdą molekułę przypada średnio ten sam udział w całkowitej ilości
zaabsorbowanej mocy. Molekuły w roztworze stanowią więc zbiór czarnych „tarcz” dla
fotonów, takich, że trafiający w taką tarczę foton zostaje pochłonięty. Oznaczmy efektywne

32
pole powierzchni tarczy jako σ. Jeżeli wiązka światła ma przekrój o powierzchni S0 , to
molekuły w cienkiej warstwie roztworu o grubości dx prezentują sumaryczną powierzchnie
absorbującą równą
0 AS cS dxNσ= (7.9)
Iloczyn S0dx jest objętością, a cS0dx jest liczbą moli molekuł absorbujących znajdujących się
w wiązce światła. NA oznacza stałą Avogadro. Stosunek powierzchni S do przekroju wiązki S0
określa ułamek liczby fotonów, które ulegają absorpcji. Mamy więc równanie:
0
0 0
AA
S cS dxNdI I I I N c dx
S S
σ σ= − = − = − (7.10)
Porównując to wyrażenie z wzorem (7.5) oraz biorąc pod uwagę, że ε = loge, otrzymujemy:
2,3
logA AN e N
ε εσ = = (7.11)
Przykład:
dla cząsteczki chlorofilu a, który w eterze etylowym wykazuje (przy λ = 662 nm) molowy
współczynnik ekstynkcji ε = 8,5·105 L/(mol·cm) otrzymujemy przekrój czynny
(odpowiadający długości fali 662 nm):
( )5
16 2 223
2,3 8,5 10 /2,332,6 10 32,6 (angstrom)
6 10 /A
L mol cmcm
N mol
εσ −⋅ ⋅ ⋅= = = ⋅ =
⋅.
Interesujące jest spostrzeżenie, że absorbujący światło fragment cząsteczki chlorofilu (tzw.
chromofor) jest płaskim pierścieniowym układem atomów o średnicy ok. 6 angstremów.
Stosowalność prawa Lamberta-Beera nie jest ograniczona do roztworów
rzeczywistych i obejmuje również koloidy złożone z nanocząstek. Warunkiem
ograniczającym stosowalność tego prawa jest niezmienność własności molekuł lub
nanocząstek od ich stężenia, a więc niewystępowanie ich wzajemnych oddziaływań. W
stężonych roztworach i koloidach, gdy średnia odległość między obiektami absorbującymi
jest mała, dochodzi do tworzenia tzw. statystycznych kompleksów zderzeniowych, które,
nawet w nieobecności oddziaływań specyficznych, skutkują zmianami intensywności i
przesunięciami pasm w widmie absorpcji. Dodatkowo mogą też tworzyć się dimery i wyższe
oligomery molekuł, które odznaczają się dużymi zmianami spektralnymi. Wynikające stąd
odchylenia od stosowalności prawa L-B, ograniczające jego stosowalność jako podstawy
metod analitycznych, niosą zarazem informacje o oddziaływaniach występujących w skali
nanometrowej, i z tego punktu widzenia są istotnym źródłem informacji o oddziaływaniach
nanocząstek i molekuł.

33
8. Podstawy fotofizyki molekuł
Każda trwała molekuła charakteryzuje się zbiorem wzbudzonych stanów elektronowych o
określonej energii całkowitej (elektronowej + oscylacyjnej + rotacyjnej). Stany wzbudzone
mogą być osiągane w różny sposób; fenomenologiczny opis wzbudzenia molekuł przez
absorpcję światła przedstawia rozdział poprzedni. Stan wzbudzony, niezależnie od sposobu
jego wytworzenia, jest stanem niestabilnym i molekuła w tym stanie podlega kilku procesom
fotofizycznym, które różnymi drogami sprowadzają ją do stanu podstawowego. Procesy te
ilustruje schemat Jabłońskiego przedstawiony na rys. 8.1. Molekuła w stanie podstawowym,
który z nielicznymi wyjątkami jest stanem singletowym (S0), może być przeprowadzona w
jeden ze stanów wzbudzonych (S1, S2 itd.) poprzez absorpcję fotonu (lub dwu i większej
liczby fotonów), w drodze reakcji chemicznej, przez krótkotrwałe oddziaływanie z
przelatującą w jej otoczeniu cząstką naładowaną itp. Pochłonięta energia wyznacza stan
elektronowy, a jej naddatek jest deponowany w postaci wewnątrzcząsteczkowego ruchu
oscylacyjnego (ponieważ rozważamy molekuły w cieczy lub w ciele stałym, udział ruchu
obrotowego pomijamy). Pierwszymi procesami, jakim podlega molekuła po nagłej
przebudowie powłoki elektronowej, jest szybka utrata nadmiaru energii oscylacyjnej na rzecz
Rys. 8.1. Schemat Jabłońskiego − procesy, jakim podlega molekuła wzbudzona na drodze do stanu podstawowego.
otoczenia, zachodzaca w czasie kilku okresów ruchu oscylacyjnego (∼10-13 s). Ustala się stan
równowagi statystycznej, w którym prawdopodobieństwo posiadania pewnej energii
oscylacyjnej jest zgodne z rozkładem Boltzmanna. Utracie energii przez molekułę może
towarzyszyć również zmiana stanu elektronowego np. z S2 do S1, czyli proces konwersji

34
wewnętrznej (stała szybkości kic – od internal conversion). Proces konwersji wewnętrznej
ulega znacznemu spowolnieniu gdy molekuła osiąga stan S1, a to ze względu na duży odstęp
energii między S1 i S0. Jest on dużo większy niż między S1 , S2 i wyższymi stanami
wzbudzonymi, co zmniejsza prawdopodobieństwo konwersji energii elektronowej w przejściu
S1 → S0 na energię oscylacyjną. Proces utraty energii przez molelułę zachodzi z
jednoczesnym wzbudzeniem ruchów cieplnych otoczenia molekuły. Silne zahamowanie tego
procesu w stanie S1 umożliwia emisję fotonu fluorescencji (ze stałą szybkości kf) lub zmianę
całkowitego momentu pędu molekuły (stała szybkości kix – od intersystem crossing), której
konsekwencją jest jej przejście do innego zbioru stanów energetycznych – stanów
trypletowych (T1, T2, ...). W zbiorze stanów trypletowych molekuła relaksuje do T1, z którego
może powoli przejść do singletowego stanu podstawowego w drodze konwersji wewnętrznej
lub poprzez emisję fotonu fosforescencji ze stałą szybkości kph.
Stałe szybkości poszczególnych procesów, jakim podlega molekuła w określonym
stanie, wyznaczają wydajności kwantowe tych procesów. Wydajność kwantowa jest to
stosunek liczby molekuł podlegających określonemu procesowi do łącznej liczby aktów
dezaktywacji molekuł wzbudzonych. Dla fluorescencji ze stanu S1, z którą konkurują procesy
konwersji wewnętrznej (S1 → S0) oraz przejścia międzysystemowe (S1 → T1), liczbę molekuł
fluoryzujących w pewnym czasie (dużo krótszym od czasu życia stanu wzbudzonego) wyraża
iloczyn kf · nwzb , zaś łączna liczba molekuł ubywających ze stanu S1 wynosi kf · nwzb + kic ·
nwzb + kix · nwzb. Zatem wydajność kwantowa fluorescencji wynosi
f wzb ff
f wzb ic wzb ix wzb f ic ix
k n kq
k n k n k n k k k= =
+ + + + (8.1)
Podobnie np. wydajność kwantowa przejść międzysystemowych (decydująca następnie o
intensywności fosforescencji) wyniesie
ix wzb ixix
f wzb ic wzb ix wzb f ic ix
k n kq
k n k n k n k k k= =
+ + + + (8.2)
Wydajności kwantowe fluorescencji molekuł zwykle są rzędu 0,01 do 0,5 a w przypadku
barwników używanych w laserach barwnikowych nawet bliskie 1. Wartość kix, zazwyczaj
niewielka, zwiększa się znacznie gdy molekuły barwnika zawierają podstawniki w postaci
atomów ciężkich pierwiastków, zazwyczaj jodu.
Jak wynika z powyzszych wzorów, zmiany wydajności kwantowej fluorescencji mogą
mieć różne przyczyny; np. zmniejszenie wartości qf (intensywności fluorescencji) może być
skutkiem:

35
■ zmniejszenia stałej szybkości kf ,
■ zwiększenia stałej szybkości kic, kix ,
■ wystąpienia nowego procesu polegającego na bezpromienistym (bez pośrednictwa fotonu)
przekazie energii wzbudzenia na inne, niefluoryzujące molekuły lub inne obiekty (np.
nanocząstki metaliczne lub półprzewodnikowe). W mianowniku we wzorze (8.1) wystąpi
wówczas dodatkowy składnik odpowiadający temu procesowi dezaktywacji, zmniejszając
wartość qf.
Istotne informacje o procesach zachodzących z udziałem molekuły w stanie
wzbudzonym niesie pomiar czasu zaniku fluorescencji po jej wzbudzeniu krótkim impulsem
światła. Wytworzona w ten sposób populacja molekuł wzbudzonych ulega stopniowo
zmniejszeniu w rezultacie zarówno fluorescencji jak i innych procesów dezaktywacji.
Szybkość zaniku populacji molekuł wzbudzonych n*( t) wyraża się wzorem analogicznym do
prawa rozpadu promieniotwórczego i jest sumą szybkości wszystkich procesów dezaktywacji:
*
* * *f ic ix
dnk n k n k n
dt= − ⋅ − ⋅ − ⋅ (8.3)
Z tego równania wynika zależność n* od czasu w postaci:
( ) ( )0 0
1* exp exp , f ic ix
f ic ix
tn t n k k k t n
k k kτ
τ = − + + = − = + +
(8.4)
Populacja molekuł wzbudzonych zanika wykładniczo ze stałą czasową τ. Ponieważ szybkość
fluorescencji jest kf · n*( t) , to τ jest czasem zaniku fluorescencji, lub inaczej średnim czasem
życia stanu wzbudzonego.
Schemat Jabłońskiego nie w pełni oddaje naturę procesów jakim podlegają molekuły
w stanach wzbudzonych. Na energię oscylacyjną molekuł składa się energia kinetyczna ruchu
atomów wokół położeń równowagi oraz energia potencjalna odkształcanych przez ten ruch
wiązań międzyatomowych. Energia potencjalna w przybliżeniu jest kwadratową funkcją
wychyleń atomów, wobec czego do opisu oscylacji molekularnych dobrze stosuje się model
oscylatora harmonicznego zilustrowany na rys. 8.2 dla stanów S0 i S1. O względnych
intensywnościach linii odpowiadających przejściom oscylacyjnym (0→0), (0→1), (0→2),...
decydują tzw. czynniki Francka-Condona, będące całkami z iloczynów odpowiednich
oscylacyjnych funkcji falowych w stanach S0 i S1. Ponieważ struktury molekuły w obu
stanach elektronowych są niemal identyczne, to intensywności analogicznych przejść
zarówno w absorpcji jak i w emisji są prawie jednakowe, co uwidacznia się w widmach
absorpcji i emisji jako ich zwierciadlana symetria. Jest to prawidłowość pozwalająca odróżnić

36
Rys. 8.2. Schemat przejść elektronowo-oscylacyjnych z absorpcją i emisją światła (po lewej) oraz widma absorpcji (A) i fluorescencji (F) (po prawej). Linie kropkowane odpowiadają tzw. poszerzeniu niejednorodnemu widm, które powoduje zanik ich struktury oscylacyjnej.
stany elektronowe aktywne w absorpcji i emisji światła, lub np. wyraźny brak takiej symetrii
może wskazywać na istnienie dwu form fluoryzujących molekuł o różnych widmach emisji.
Inną regułą w spektroskopii elektronowo-oscylacyjnej molekuł jest przesunięcie
maksimów widm absorpcji i fluorescencji, którego przyczyną jest częściowa utrata
zaabsorbowanej energii. W rezultacie ustalania się równowagi termicznej między molekułą i
jej otoczeniem, zarówno absorpcja jak i emisja zachodzą z niskich stanów oscylacyjnych, w
zasadzie odpowiadających oscylacyjnej liczbie kwantowej równej zeru. Po przejściu
absorpcyjnym np. (0-2) molekuła relaksuje w stanie S1 z powrotem do zerowego stanu
oscylacyjnego, z którego po pewnym czasie emituje fluorescencję. W ten sposób następuje
rozproszenie części zaabsorbowanej energii na ciepło. Co więcej, jeżeli emisja zachodzi w
przejściu np. (0-1) lub (0-2), tj. ze wzbudzeniem oscylacyjnym, to rozproszenie energii
jednego lub dwu kwantów oscylacyjnych powtórnie wystąpi już w stanie S0. Przesunięcie
widma fluorescencji względem widma absorpcji w stronę długofalową (niższych energii) nosi
nazwę przesunięcia stokesowskiego (od G.G. Stokesa, jego odkrywcy). W przypadku, gdy
widma są silnie poszerzone tak, że znika ich struktura oscylacyjna (p. rys. 8.2) przesunięcie
stokesowskie jest również widoczne jako różnica położeń maksimów obu widm.
Kolejna reguła spektroskopii molekularnej stwierdza niezależność widma emitowanej
fluorescencji od energii fotonów światła wzbudzającego tę fluorescencję. Widmo
fluorescencji zachowuje swój kształt i położenie na skali częstości niezależnie od tego, czy
molekuły są wzbudzane do stanu S1, S2 lub do wyższego stanu elektronowego. Ta reguła
3 2 1 0
3 2 1 0
abso
rpcj
a
fluorescencja
S0
S1
ν
A F
0-1
0-2
0-3
0-0
0-0
0-1
0-2
0-3
λ

37
Kashy (Michael Kasha) wynika z dużej szybkości procesu konwersji wewnętrznej, który
polega na przetwarzaniu energii powłoki elektronowej na wzbudzenia oscylacyjne w trakcie
relaksacji np. ze stanu S3 do S2, a następnie z S2 do S1. Proces ten jest zazwyczaj dużo
szybszy od fluorescencji, która prawie nie występuje z wyższych stanów wzbudzonych, a
jedynie z S1; w rezultacie widmo fluorescencji jest niezależne od warunków wzbudzenia.
9. Szybkość reakcji chemicznej i kataliza enzymatyczna
Szybkość reakcji chemicznej
Reakcje chemiczne przedstawiamy zazwyczaj w postaci informującej o ilościach
(proporcjach) poszczególnych reagentów, czyli w postaci sumarycznego równania
stechiometrycznego, np.:
aA + bB → cC + dD (9.1)
Gdy stanem początkowym rozważanego procesu chemicznego jest mieszanina zawierająca
jedynie substraty A i B, to z upływem czasu ich stężenia będą się zmniejszać, a wzrastają
stężenia produktów C i D. Prowadzi to do stopniowego wyczerpywania się substratów, a
wskutek istnienia procesu odwrotnego dochodzi ostatecznie do ustalenia się końcowego stanu
dynamicznej równowagi, w którym tyle samo moli produktów powstaje z substratów, ile
ulega rozpadowi. Spełnione jest wówczas równanie równowagi chemicznej
[ ] [ ][ ] [ ]C D
A B
c d
a b K= . (9.2)
Stała równowagi K zależy od warunków (temperatura, ciśnienie) oraz od termodynamicznych
wielkości charakteryzujących poszczególne reagenty, jak entalpie i entropie molowe
tworzenia tych substancji. Stała równowagi wyraża proporcje stężeń, jakie ustalą się w stanie
równowagi, nie mówi jednak nic o dynamicznym charakterze stale zachodzących reakcji „w
przód” oraz „wstecz”. Do ilościowego opisu przebiegu reakcji w czasie służy szybkość
reakcji przedstawionej równaniem (1) zdefiniowana w sposób następujący:
1 [A] 1 [B] 1 [C] 1 [D]
vd d d d
a dt b dt c dt d dt= − = − = = (9.3)
Wielkości w nawiasach kwadratowych oznaczają tu stężenia molowe reagentów. Szybkość
reakcji jest pojęciem bliskim innemu, również charakteryzującemu stopień zaawansowania
reakcji, mianowicie pojęciu współrzędnej reakcji. Szybkość reakcji jest stała w określonej
temperaturze, jest jednak funkcją aktualnie istniejących stężeń reagentów i w ogólności
zmienia się z upływem czasu w miarę postępu reakcji.

38
Dla reakcji przebiegającej według prostego mechanizmu równanie kinetyczne ma
również prostą postać. Gdy molekuły dwu substratów zderzając się w ruchu cieplnym tworzą
produkt (chociaż nie w każdym zderzeniu; należy przyjąć, że tylko z pewnym
prawdopodobieństwem), to liczba molekuł produktu powstająca w jednostce czasu jest
proporcjonalna do liczby zderzeń, a ta do stężenia każdego z substratów, a więc do iloczynu
stężeń. Przykładowo, dla reakcji tworzenia jodowodoru bezpośrednio z jodu i wodoru w
gazowej mieszaninie (podobny jest też opis reakcji zachodzących w roztworach) mamy
sumaryczne równanie:
H2 + J2 → 2HJ (9.4)
któremu odpowiada równanie kinetyczne o postaci:
2 2v [H ][J ]k= (9.5)
Współczynnik k jest to stała szybkości reakcji – fundamentalna wielkość charakteryzująca
szybkość reakcji niezależnie od stężeń substratów, wyznaczana doświadczalnie, obliczana na
gruncie modeli teoretycznych, służąca do porównywania przebiegu różnych reakcji. Szybkość
reakcji wg wzoru (3) wyraża się jako:
[ ] [ ]2 2H J 1 [HJ]v
2
d d d
dt dt dt= − = − = (9.6)
Z porównania (5) i (6) otrzymujemy równanie tworzenia produktu HJ:
[ ] [ ][ ]2 2
HJ1H J
2
dk
dt= (9.7)
a ponieważ ilości aktualnie reagujących substratów można wyrazić przez ich stężenia
początkowe [H2]0 i [J2]0 , to powyższe równanie można zapisać w formie zawierającej już
tylko zależne od czasu stężenie produktu
[ ] [ ] [ ] [ ] [ ]2 20 0
HJ1 1 1H HJ J HJ
2 2 2
dk
dt = − −
(9.8)
Rozwiązanie tego równania różniczkowego wyznacza zależność stężenia produktu [HJ] od
czasu. Zakładając ustalone warunki fizyczne (np. stałą temperaturę) bez większych trudności
można otrzymać rozwiązania tego równania dla różnych warunków przebiegu reakcji, np. w
zależności od tego, czy oba substraty ulegają stopniowo wyczerpaniu, czy jeden lub oba są
stopniowo uzupełniane w mieszaninie reakcyjnej.
Na podstawie tylko równania stechiometrycznego przedstawiającego proporcje
reagujących substratów i produktów nie zawsze można napisać równania przedstawiające
szybkość reakcji, ponieważ nie każdej reakcji sumarycznej odpowiada proste równanie
kinetyczne. Gdy reakcja przebiega z udziałem różnych produktów pośrednich, to równania
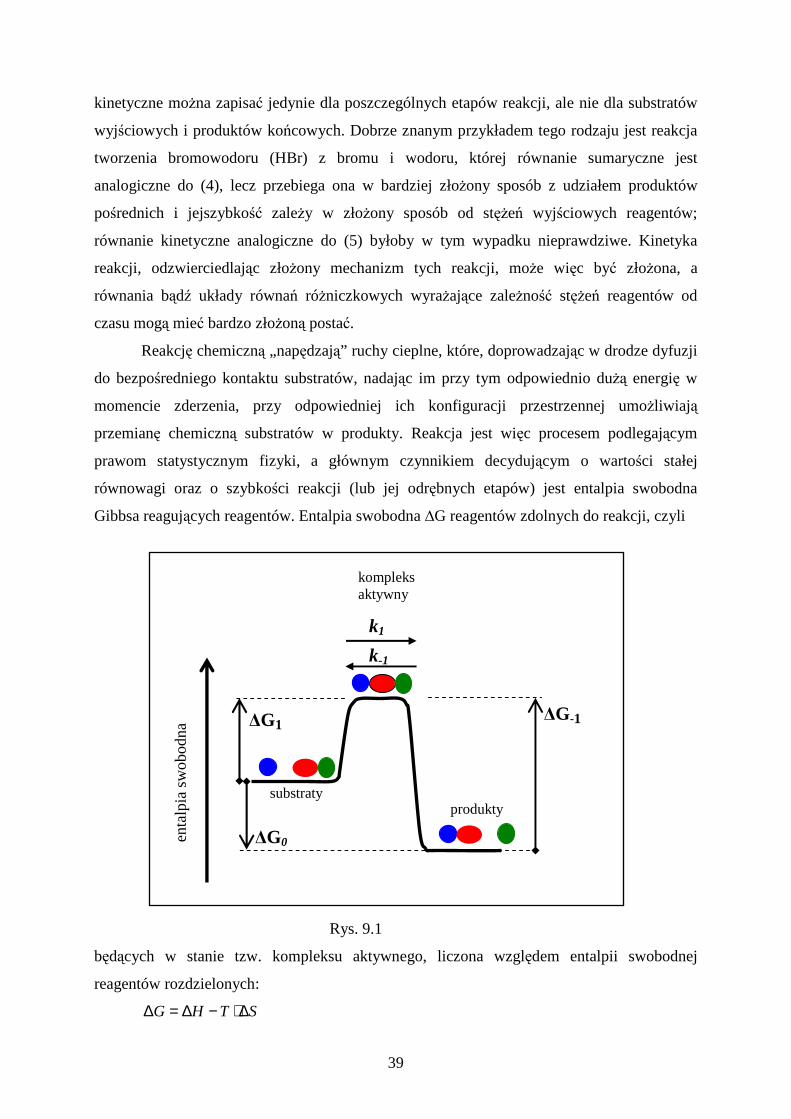
39
kinetyczne można zapisać jedynie dla poszczególnych etapów reakcji, ale nie dla substratów
wyjściowych i produktów końcowych. Dobrze znanym przykładem tego rodzaju jest reakcja
tworzenia bromowodoru (HBr) z bromu i wodoru, której równanie sumaryczne jest
analogiczne do (4), lecz przebiega ona w bardziej złożony sposób z udziałem produktów
pośrednich i jejszybkość zależy w złożony sposób od stężeń wyjściowych reagentów;
równanie kinetyczne analogiczne do (5) byłoby w tym wypadku nieprawdziwe. Kinetyka
reakcji, odzwierciedlając złożony mechanizm tych reakcji, może więc być złożona, a
równania bądź układy równań różniczkowych wyrażające zależność stężeń reagentów od
czasu mogą mieć bardzo złożoną postać.
Reakcję chemiczną „napędzają” ruchy cieplne, które, doprowadzając w drodze dyfuzji
do bezpośredniego kontaktu substratów, nadając im przy tym odpowiednio dużą energię w
momencie zderzenia, przy odpowiedniej ich konfiguracji przestrzennej umożliwiają
przemianę chemiczną substratów w produkty. Reakcja jest więc procesem podlegającym
prawom statystycznym fizyki, a głównym czynnikiem decydującym o wartości stałej
równowagi oraz o szybkości reakcji (lub jej odrębnych etapów) jest entalpia swobodna
Gibbsa reagujących reagentów. Entalpia swobodna ∆G reagentów zdolnych do reakcji, czyli
Rys. 9.1
będących w stanie tzw. kompleksu aktywnego, liczona względem entalpii swobodnej
reagentów rozdzielonych:
G H T S∆ = ∆ − ⋅ ∆
substraty produkty
kompleks aktywny
enta
lpia
sw
obod
na
k1
k-1
∆G1
∆G0
∆G-1

40
zawiera zarówno energię oddziaływania reagujących molekuł (entalpię H) jak i składnik
entropijny, zależny od orientacji molekuł. Energetykę i przebieg reakcji przedstawia schemat
na rys. 9.1. Zaznaczone tu szybkości reakcji, k1 , oraz reakcji wstecznej, k2 są funkcjami
entalpii swobodnej kompleksu aktywnego względem stanu wyjściowego (tj. względem
wolnych substratów lub rozpadających się w reakcji wstecznej produktów):
11 1 exp
Gk a
RT
∆ = ⋅ −
(9.9)
11 1 exp
Gk a
RT−
− −∆ = ⋅ −
(9.10)
W najprostszym przypadku, gdy reakcja polega na przemianie jedynego substratu w jedyny
produkt, można zapisać szybkość reakcji w formie równania:
1 1
[S] [P]v [S] [P]
d dk k
dt dt −= − = = − (9.11)
W stanie równowagi, gdy szybkość reakcji wynosi zero, otrzymujemy:
( )( ) [ ]( ) ( )1 11
1 1 01 2 1
exp[P]exp exp
[S] exp
a Gka G G a G K
k a G −− −
−∆= = = ⋅ − ∆ − ∆ = ⋅ −∆ =
−∆
(9.12)
Jak widać, stała równowagi reakcji K, której wartość decyduje o ilości utworzonego produktu,
zależy od zmiany entalpii swobodnej układu reagującego przy przejściu od produktu do
substratu, co jest naturalne w świetle praw termodynamiki (np. rozkładu Boltzmanna).
Powyższy wzór mówi jednak więcej. Zawiera on wskazówkę, że osiągany stan równowagi
zależy od stałych szybkości (k) reakcji prowadzących do powstania produktu. Tak więc
zwiększenie ilości produktu powstającego w prostej reakcji można osiągnąć poprzez wpływ
na wartości stałych szybkości k1 lub k-1 w tej reakcji. Na tej możliwości sterowania
wydajnością reakcji chemicznych zasadza się proces katalizy, w tym katalizy enzymatycznej.
Kataliza
Kataliza polega na zwiększeniu szybkości reakcji w rezultacie oddziaływania substratów w
stanie kompleksu aktywnego z katalizatorem. Wskutek tego oddziaływania obniża się energia
kompleksu aktywnego, skutkując zwiększeniem szybkości reakcji. Sposoby działania
katalizatorów są różne. Niektóre katalizatory jeszcze przed utworzeniem kompleksu
aktywnego tworzą kompleks z jednym z substratów.
Jak wynika z rysunku 9.1, obniżenie energii aktywacji ∆G1 spowoduje wzrost stałej
szybkości k1, a jednocześnie wzrost stałej szybkości k-1. Sprzyja to szybkiemu ustalaniu się

41
stanu równowagi między substratami i produktami, lecz nie zmienia samego stanu
równowagi, tj. równowagowych stężeń substratów i produktów; te są wyznaczone przez
termodynamikę i zależą tylko od ∆G0. Na czym więc polega korzyść z zastosowania
katalizatora? Na tym, że o ile należałoby długo czekać na osiągnięcie znacznego postępu
reakcji bez katalizatora, ten sam skutek w jego obecności można uzyskać w nieporównanie
krótszym czasie. Np. katalizator samochodowy powoduje szybkie dopalanie niekompletnie
spalonego paliwa; bez jego zastosowania, w tej samej temperaturze resztki paliwa i sadza też
uległyby spaleniu, ale znacznie wolniej, uniemożliwiając tym samym efektywne oczyszczenie
spalin z szybko pracującego silnika.
Ogólnie rozróżnia się katalizę heterogeniczną, w której katalizator znajduje się w
odrębnej fazie niż substraty i produkty (np. jest ciałem stałym, a reagenty gazami lub
cieczami), oraz katalizę homogeniczną, gdy katalizator i reagenty tworzą jedną fazę (np. gaz
lub ciecz). Katalizatorami homogennymi są biokatalizatory – enzymy. Decydującą rolę w
funkcjonowaniu katalizatorów odgrywają pewne szczegóły ich budowy molekularnej lub
krystalicznej. Wiadomo na przykład, że w katalizie heterogenicznej decydującą rolę odgrywa
specyficzna struktura defektów na powierzchni kryształów i istotne są stosunkowo nielicznie
występujące tzw. centra aktywne, podobnie też w strukturze molekularnej enzymów.
Kataliza enzymatyczna
Niemal wszystkie reakcje biochemiczne w żywych organizmach wymagają współudziału
wyspecjalizowanych katalizatorów – enzymów, by osiągnąć pożądaną szybkość reakcji.
Reakcje enzymatyczne charakteryzują się wysoką specyficznością; enzymy zazwyczaj
katalizują proste reakcje syntezy lub rozkładu ściśle określonych substratów, np. enzym
trawienny trypsyna rozszczepia wiązanie peptydowe w białkach, ale działa selektywnie na
wiązania lizyny lub argininy, nie działa zaś na wiązania peptydowe utworzone przez inne
aminokwasy.
Fragment molekuły enzymu, który bezpośrednio wiąże się i oddziałuje z substratem
oraz zawiera kluczowe do przebiegu reakcji reszty aminokwasowe lub inne grupy
prostetyczne, stanowi tzw. miejsce aktywne (centrum aktywne). Związanie substratu przez
enzym w miejscu aktywnym powoduje zmianę właściwości kilku atomów substratu,
ułatwiające zajście kattalizowanej reakcji. Formowanie kompleksu enzym-substrat jest więc
kluczowym elementem procesu katalizy na drodze od substratu do produktu.
Opis teoretyczny kinetyki reakcji enzymatycznych podali w 1913 r. L. Michaelis i M.
Menten. W tej teorii zasadniczą rolę odgrywa kompleks enzym-substrat (ES)*, który jest

42
tworzony ze stałą szybkości k1. Kompleks ten rozpadając się ze stałą szybkości k2 uwalnia
produkt, ale czasem też wolny substrat (k-1). Relacje te ujmuje następujący schemat reakcji:
( )1 2
-1 2
*
kE S ES E P
k k
k−
+ +� � (9.13)
Ze względu na znikome powinowactwo enzymu do produktu (tj. bardzo słabe wiązanie
enzymu z produktem, notabene powodujące szybkie uwalnianie utworzonego produktu i dużą
wartość k2), należy przyjąć, że k2 >> k -2, czyli efektywnie k -2 = 0. W stanie stacjonarnym
szybkości tworzenia kompleksu (ES)* z E i S (wynosząca k1[E][S]) oraz szybkość jego
rozpadu na E + S czyli (k-1[(ES)*]), oraz szybkość rozpadu na E + P (k2[(ES)*]) zrównują
się, mamy zatem równanie:
1 1 2[E][S] [(ES)*] [(ES)*] 0k k k−⋅ − ⋅ − ⋅ = (9.14)
z którego wynika:
[ ][ ]1
1 2
E S[(ES)*]
k
k k−
=+
(9.15)
Oznaczając [E]0 całkowite stężenie enzymu, które spełnia też warunek: [E]0 = [E] + [ES], i
podstawiając [E] do równania (15), po przekształceniach otrzymujemy:
[ ] [ ] [ ][ ]
1 0
1 2 1
E S(ES)*
S
k
k k k−
=+ +
(9.16)
a stąd szybkość tworzenia produktu:
[ ] [ ] [ ][ ]
1 2 02
1 2 1
E Sv ES
S
k kk
k k k−
= =+ +
(9.17)
Dzieląc licznik i mianownik przez k1 i oznaczając stałą Michaelisa KM:
1 2
1M
k kK
k− += (9.18)
otrzymujemy równanie Michaelisa i Menten wyrażające szybkość reakcji enzymatycznej:
[ ] [ ]
[ ]2 0
E Sv
SM
k
K=
+ (9.19)
Szybkość reakcji enzymatycznej jest więc proporcjonalna do całkowitego stężenia enzymu
[E]0. Przy małych stężeniach substratu ([S] << KM) jest też proporcjonalna do stężenia
substratu: v = k2[E]0[S], lecz ulega nasyceniu przy dużych stężeniach substratu; gdy [S] >>
KM, jest: v = vmax = k2[E]0. Ta maksymalna szybkość reakcji stanowi maksymalną aktywność
enzymu i nazywana jest maksymalną liczbą obrotów, tj. maksymalną liczbą cykli reakcji jakie

43
wykonują molekuły enzymu w jednostce czasu. Maksymalna liczba obrotów jest obiektywną
miarą efektywności enzymu w przetwarzaniu danego substratu.
Do wyznaczania parametrów charakteryzujących enzym służy równanie M-M w
postaci zlinearyzowanej. Odwrotności obu stron równania (19) przedstawiają liniową relację
między 1/v oraz 1/[S]:
[ ] [ ] [ ]2 20 0
1 1 1
v E E SMK
k k= + ⋅ (9.20)
Wykres tej liniowej relacji przedstawiony na rys. 9.2 służy do wyznaczania stałej Michaelisa
KM oraz maksymalnej liczby obrotów vmax = k2[E]0, a przy znajomości całkowitego stężenia
enzymu [E]0 – także wartości k2. Liniowość tego wykresu jest zarazem wskaźnikiem
kontrolnym czystości preparatu enzymu, substratu, oraz innych czynników mogących
naruszać prawidłowy przebieg eksperymentu.
Rys. 9.2. Wykres zależności szybkości reakcji enzymatycznej od stężenia substratu odpowiadający zlinearyzowanej postaci równania Michaelisa-Menten.
Istotnym warunkiem katalizy jest utrzymywanie przez molekuły enzymu ściśle
określonej struktury molekularnej, ponieważ nawet niewielkie zmiany struktury przestrzennej
w miejscu aktywnym enzymu silnie wpływają na jego funkcję katalityczną. Niektóre enzymy
zawierają też miejsca wiązania kofaktorów – regulujących ich aktywność, oraz dodatkowe
miejsca wiązania małych cząsteczek, na przykład pośrednich lub bezpośrednich produktów
lub substratów, które mogą dodatkowo regulować aktywność enzymu na zasadzie sprzężenia
zwrotnego. Istotnym elementem regulacji aktywności enzymu jest inhibicja allosteryczna –
zmniejszenie aktywności lub wyłączenie enzymu wskutek związania inhibitora poza
miejscem aktywnym. Powoduje to zmiany konformacyjne w strukturze molekularnej enzymu
i w ten sposób silnie wpływa na jego aktywność.
1/[S]
1/v
-1/KM
1/vmax

44
10. Podstawy zjawisk bioelektrycznych
Zjawiska elektryczne odgrywają bardzo ważną rolę w żywych organizmach, zarówno
roślinnych jak i zwierzęcych. Służą jako szybki przenośnik informacji między odległymi
tkankami i regulator niektórych procesów fizjologicznych, jak skurcz mięśni lub innych
efektorów mechanicznych. U zwierząt podstawowa rola zjawisk elektrycznych polega na
dostarczaniu informacji do mózgu oraz przetwarzaniu informacji w centralnym układzie
nerwowym. Informację tę stanowią sygnały słuchowe, wzrokowe i pochodzące z innych
zmysłów. Istnieją też przypadki zupełnie innego wykorzystania zjawisk elektrycznych jako
narzędzia walki, np. u węgorza elektrycznego, lub do wykrywania ukrytych organizmów
stanowiących pożywienie w trakcie polowania.
W każdym z tych przypadków mechanizm wytwarzania napięcia elektrycznego jest
taki sam i wynika ze zjawisk fizykochemicznych powstających z udziałem jonów niektórych
pierwiastków jak potas, sód, wapń, chlor. Zasadniczą rolę w generacji napięć pełnią błony
biologiczne – cienkie (ok. 7-10 nm grubości) warstwy lipidowo-białkowe pokrywające każdą
komórkę roślinną i zwierzęcą. Występują również we wnętrzu komórek, które dzielą na
odrębne przedziały, stanowiąc zarazem rusztowanie, na którym zachodzą procesy
biochemiczne (np. synteza białek) oraz służą jako bariera odgraniczająca wewnętrzne tzw.
organelle komórkowe (mitochondria, chloroplasty) od pozostałej zawartości wnętrza
komórki.
Podstawowa funkcja błony to rola bariery nieprzepuszczalnej dla określonych
substancji, a przepuszczalnej dla innych. Ten transport przez błonę jest – w ogólnym podziale
– dwojakiego rodzaju: transport bierny i transport aktywny. Transport aktywny jest
mechanizmem fizykochemicznym zużywającym część energii produkowanej w organizmie
na przemieszczanie przez błony substancji, które powinny znaleźć się wewnątrz komórki w
odpowiednio dużym stężeniu, lub odwrotnie – powinny zostać usunięte na zewnątrz aż do
osiągnięcia odpowiednio niskiego ich stężenia wewnątrz komórki. Oprócz przemieszczania
substancji organicznych, transport aktywny służy do gromadzenia lub pompowania na
zewnątrz jonów, które – mając ładunek elektryczny – w rezultacie nagromadzenia ładują
elektrycznie dwa „przewodniki” jakimi są elektrolity wypełniające wnętrze i obecne w
pokaźnych stężeniach w otoczeniu komórki. Transport ten, jeżeli ma ustanowić różnice stężeń
jonów po obu stronach błony, zachodzi wbrew gradientom stężeń i wymaga stałego dopływu
energii. Tym elektrogennym transportem aktywnym zajmować się nie będziemy. Stanowi on
tylko proces, który stwarza warunki do generacji napięć elektrycznych (stałych i

45
szybkozmiennych), które zachodzą już bez dopływu energii, a ich przebieg sterowany jest
wyłącznie prawami fizyki i chemii i napędzany jest energią wytworzonych gradientów stężeń
i potencjału elektrycznego (tj. polem elektrycznym). Są to więc – w odróżnieniu od transportu
aktywnego – zjawiska polegające na transporcie biernym.
Nagromadzeniu określonej substancji o molekułach elektrycznie obojętnych w
pewnym stężeniu towarzyszy potencjał chemiczny, który stanowi entalpię swobodną
przypadającą na 1 mol substancji, i w przybliżeniu (polegającym na zastosowaniu praw
analogicznych do praw gazu doskonałego) wyraża się wzorem:
0 lnRT aµ µ= + (10.1)
W dalszym ciągu, dla pominięcia komplikacji, będziemy uważać aktywność a za identyczną
ze stężeniem c. W przypadku molekuł posiadających ładunki elektryczne do energii
chemicznej 1 mola należy dodać energię elektryczną, zależną od potencjału elektrycznego w
którym znajdują się te jony. Wzór (10.1) przybiera postać potencjału elektrochemicznego:
0 lnRT c zFµ µ ϕ= + + (10.2)
(F jest stałą Faradaya, zaś z liczbą ładunków elementarnych danego jonu).
Gdy roztwory o różnych stężeniach zdysocjowanych elektrolitów, np. chlorku potasu
KCl, rozdzielone są błoną przepuszczającą tylko jeden rodzaj jonów, np. K+, a
nieprzepuszczalną dla anionów Cl−, to wypadkowy strumień jonów K+, przenikając przez
błonę na stronę niższego ich stężenia (w kierunku od C1 do C2), naładuje elektrycznie
Rys. 10.1. Schemat rozmieszczenia ładunków elektrycznych na błonie rozdzielającej elektrolity o różnych stężeniach i selektywnie przepuszczającej jeden rodzaj jonów.
dodatnio elektrolit po przeciwnej stronie, jak pokazuje to rys. 10.1. W rezultacie potencjał
elektryczny elektrolitu po prawej stronie staje się dodatni względem elektrolitu po stronie
lewej, wytwarzając wewnątrz błony pole elektryczne zmniejszające strumień jonów. W stanie
C1 > C2
K+ KCl KCl
+
+ + +
− −
− −

46
stacjonarnym ustala się równowaga strumienia dyfuzyjnego napędzanego różnicą stężeń (z
lewa na prawo) i strumienia napędzanego polem elektrycznym (z prawa na lewo). Odpowiada
to zrównaniu potencjałów elektrochemicznych jonów K+ po obu stronach błony:
0 1 1 0 2 2ln lnRT c zF RT c zFµ ϕ µ ϕ+ + = + + (10.3)
przy czym różnica potencjałów elektrycznych wynosi:
12 1
2
lnRT c
zF cϕ ϕ− = (10.4)
Jest to tak zwany potencjał równowagi jonu przepuszczanego przez błonę, identyczny z
wzorem Nernsta używanym w elektrochemii. Gdy c1 > c2 jak założono powyżej, oraz z = +1
jak dla jonów potasu, potencjał elektrolitu po prawej stronie jest bardziej dodatni a jego
wartość odpowiadająca dla przykładu e-krotnej różnicy stężeń wyniesie 25 mV (taka jest
wartość wyrażenia RT/zF w temperaturze normalnej 25 °C). Dziesięciokrotnej różnicy stężeń
odpowiada różnica potencjałów 59 mV. Jak wskazują dane liczbowe w tabeli poniżej, tego
rzędu są typowe stosunki stężeń jonów decydujących o potencjałach elektrycznych.
Stężenia wewnątrz- i zewnątrzkomórkowe jonów dla komórek mięśnia szkieletowego ssaków (mM oznacza stężenie milimolowe, tj. 0,001 mola/litr).
Jon stężenie
zewnętrzne (mM/l)
stężenie wewnętrzne
(mM/l)
potencjał równowagi (mV)
K+ 4 140 -89
Na+ 144 10 +68
Cl− 114 4 -85
Gdy błona nie jest doskonale selektywna, lub z innych powodów jednocześnie przepuszczalna
dla kilku różnych jonów, to ustalająca się różnica potencjałów po obu jej stronach jest
wypadkową, która zależy od stopnia przepuszczalności dla poszczególnych jonów. W
teoretycznym opisie tych zjawisk wprowadza się tzw. współczynnik przepuszczalności, który
jest stosunkiem stałej dyfuzji danego jonu wewnątrz błony do jej grubości δ:
D
Pδ
= (10.5)
Ustalającą się w takich warunkach różnicę potencjałów przedstawia szeroko stosowany w
elektrofizjologii wzór Goldmana. W realistycznej sytuacji, gdy błona rozdziela złożone

47
elektrolity wewnątrzkomórkowy (W) i zewnątrzkomórkowy (Z), zawierające stężenia jonów
K+, Na+ i Cl−, to różnica potencjałów wynosi:
lnZ Z W
K K Na Na Cl ClW Z W W Z
K K Na Na Cl Cl
RT P C P C P C
F P C P C P Cϕ ϕ + +− =
+ + (10.6)
(Ci są to stężenia poszczególnych jonów rozróżnione indeksem górnym W lub Z, zaś Pi są
współczynnikami przepuszczalności). Z reguły w stanie stacjonarnym jest: PK >> PNa > PCl i
o napięciu błony decydują jony potasu, których stężenia według danych w tabeli wynoszą
odpowiednio 140 mM wewnątrz i 4 mM na zewnątrz komórki. W tych warunkach napięcie
błony, tzw. potencjał spoczynkowy, wynosi -89 mV. Znak ujemny oznacza niższy potencjał
wewnątrz komórki.
Jak wskazuje wzór Goldmana, gdyby silnie wzrósł współczynnik przepuszczalności
np. dla jonów sodu, to decydujące o potencjale błony w takiej sytuacji jony Na+
wytworzyłyby potencjał dodatni wewnątrz komórki wynoszący +68 mV. Takie zjawisko
zachodzi w trakcie generacji tzw. potencjału czynnościowego, potocznie nazywanego
impulsem nerwowym. Napięcie elektryczne o odwrotnym znaku powstaje lokalnie i,
powodując zmianę ładunku pojemności elektrycznej błony, rozprzestrzenia się z dużą
prędkością, typowo kilkudziesięciu m/s, dochodzącą w niektórych komórkach nerwowych do
100 m/s. Ten biegnący impuls odwróconego napięcia błony przebiega przez ustalony punkt w
czasie 1 milisekundy. Mechanizm niezbędny do generacji i rozprzestrzeniania się potencjału
czynnościowego zapewniają kanały jonowe wbudowane w błonę komórki nerwowej (oraz
mięśniowej i in.). Kanały są to makrocząsteczki białkowe odpowiedzialne za selektywną
przepuszczalność dla różnych jonów i, co bardzo ważne, zmieniające swoje przewodnictwo
elektryczne pod wpływem pola elektrycznego jakie towarzyszy potencjałowi
czynnościowemu. Kanały jonowe są więc przykładem prostych, naturalnie występujących,
sterowanych polem elektrycznym maszyn molekularnych.

48
11. Literatura
Plazmon, własności klastrów i nanocząstek metalicznych, zarys teorii Mie:
U. Kreibig, M. Vollmer − Optical Properties of Metal Clusters. Springer, Berlin 1995 D.L. Feldheim, C.A. Foss, Jr., (Editors) − Metal Nanoparticles. Synthesis, Characterization and Applications. Marcel Dekker, New York 2002.
Kompletna teoria Mie:
M. Born, E. Wolf − Principles of Optics. Pergamon Press, 1968, tłum. ros.: Osnowy optiki, Nauka, Moskwa 1973.
Elektrostatyka, polaryzacja elektryczna:
C.J.F. Böttcher − Theory of Electric Polarisation. Elsevier, Amsterdam, wyd. I 1952, wyd. II 1973.
Elektrodynamika:
M. Piłat − Wykłady z elektrodynamiki. Wydawnictwo UMCS, Lublin 1985.
Absorpcja światła i fotofizyka:
Z. Kęcki − Podstawy spektroskopii molekularnej. PWN, Warszawa 1998.
Kataliza i enzymy:
L. Stryer − Biochemia. PWN Warszawa 1999. A.G. Whittaker, A.R. Mount, M.R. Heal − Chemia fizyczna. PWN, Warszawa 2006.
Biopotencjały:
R. Glaser − Wstęp do biofizyki. PZWL, Warszawa 1974. R. Glaser − Biophysics. An Introduction. Wyd. II: Springer, Heidelberg 2012. S. Miękisz, A. Hendrich − Wybrane zagadnienia z biofizyki. Volumed, Wrocław 1998.