mart-1–specific melanoma tumor-infiltrating lymphocytes
TRANSCRIPT

of January 3, 2019.This information is current as Following Antigenic Restimulation In Vitro
CapabilityImproved Survival and Expansion Maintaining CD28 Expression HaveTumor-Infiltrating Lymphocytes
Specific Melanoma−MART-1
Patrick Hwu and Laszlo RadvanyiYufeng Li, Shujuan Liu, Jessica Hernandez, Luis Vence,
ol.0901101http://www.jimmunol.org/content/early/2009/11/30/jimmun
published online 30 November 2009J Immunol
MaterialSupplementary
1.DC1http://www.jimmunol.org/content/suppl/2018/08/07/jimmunol.090110
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved.1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on January 3, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
MART-1–Specific Melanoma Tumor-Infiltrating LymphocytesMaintaining CD28 Expression Have Improved Survival andExpansion Capability Following Antigenic RestimulationIn Vitro
Yufeng Li,*,† Shujuan Liu,* Jessica Hernandez,*,† Luis Vence,* Patrick Hwu,*,† and
Laszlo Radvanyi*,†
We determined how CD8+ melanoma tumor–infiltrating lymphocytes (TILs) isolated from two distinct phases of expansion in
preparation for adoptiveT cell therapy respond tomelanomaAg restimulation.We found thatTILs isolated after the rapid expansion
protocol (REP) phase, used to generate the final patient TIL infusion product, were hyporesponsive to restimulation with MART-1
peptide-pulsed dendritic cells, withmanyCD8+ T cells undergoing apoptosis. Telomere length was shorter post-REP, but of sufficient
length to support further cell division. Phenotypic analysis revealed that cell-surface CD28 expression was significantly reduced in
post-REPTILs, whereasCD27 levels remained unchanged. Tracking post-REPTILproliferation byCFSEdilution, aswell as sorting
for CD8+CD28+ and CD8+CD282 post-REP subsets, revealed that the few CD28+ TILs remaining post-REP had superior survival
capacity and proliferated after restimulation withMART-1 peptide. An analysis of different supportive cytokinemixtures during the
REP found that a combination of IL-15 and IL-21 facilitated comparable expansion of CD8+ TILs as IL-2, but prevented the loss of
CD28 expression with improved responsiveness to antigenic restimulation post-REP. These results suggest that current expansion
protocols using IL-2 formelanoma adoptive T cell therapy yields largelyCD8+T cells unable to persist and divide invivo followingAg
contact. The few CD8+CD28+ T cells that remain may be the only CD8+ TILs that ultimately survive to repopulate the host and
mediate long-term tumor control. A REP protocol using IL-15 and IL-21may greatly increase the number of CD28+ TILs capable of
long-term persistence. The Journal of Immunology, 2010, 184: 000–000.
Tumor vaccines against defined melanoma Ags, such asgp100 and MART-1, have not yet been successful in thetreatment of human metastatic melanoma. A critical
reason for this may be the inhibition of CD8+ CTL function andinduction of T cell energy in the tumor microenvironment or lackof adequate activation of tumor-reactive T cells in vivo. Adoptivecell therapy (ACT) is an alternative approach that removes tumor-infiltrating lymphocytes (TILs) from this suppressive tumor mi-croenvironment and expands and activates CD8+ and CD4+ T cellsin vitro, differentiating them into potent antitumor effector cellsthat can be reintroduced back into the patient (1, 2). In fact, in
recent clinical trials, combining TIL infusion with a single cycleof high-dose IL-2 therapy has demonstrated clinical response ratesas high as 51% in patients who underwent prior lymphodepletioninduced by cyclophosphamide and fludarabine (3–6). AlthoughACT using expanded TILs has shown great promise, there are stilla number of outstanding issues with this type of therapy that limitits widespread application. Furthermore, although clinical re-sponse rates are quite remarkable for Stage IV melanoma, thefrequency of actual complete response is still low (,10% oftreated patients) and, in many patients, tumor regression is tran-sient in nature (3).One of the key problems that may limit tumor regression and
long-term durable clinical responses in ACT is the persistence ofTILs following infusion (7, 8). Long-term TIL persistence hasbeen correlated with accumulation in vivo of T cells from theinfused TIL population having an effector memory phenotype,characterized by the expression (or re-expression) of CD27 andCD28 (7). However, the factors regulating or facilitating this TILsurvival and persistence are not known, and, moreover, the pos-sible fates of TILs shortly after infusion into patients, when T cellsmake contact with tumor Ags, reactivating cells through the TCR,has not been addressed systematically. The current approach forACT in melanoma involves expanding TILs from small tumorfragments or tumor digests by exposure to IL-2 for a period of 4–5wk. This is followed by acute stimulation of TILs by CD3 ligationwith anti-CD3 in the presence of an excess number of irradiatedfeeder cells and IL-2 for 2 wk, a process called the rapid expan-sion protocol (REP). The REP greatly expands the number ofTILs, usually by 1000- to 3000-fold, before infusion of this finalproduct into the patient. The REP not only expands TILs (up to100 billion or more), but also drives further T cell differentiation
*Department of Melanoma Medical Oncology and †The Immunology Program of theUniversity of Texas Health Science Center, Graduate School of Biomedical Sciences,The University of Texas M. D. Anderson Cancer Center, Houston, TX 77030
Received for publication April 6, 2009. Accepted for publication October 26, 2009.
This work was supported by National Institutes of Health/National Cancer InstituteGrants 1RO1 CA111999-01A2 and P50 CA093459 and by a grant from the Miriamand Sheldon Adelson Medical Research Foundation.
The sequences presented in this article have been submitted to GEO database underaccession number GSE16517.
Address correspondence and reprint requests to Dr. Laszlo G. Radvanyi, Departmentof Melanoma Medical Oncology, The University of Texas M. D. Anderson CancerCenter, Box 904, 1515 Holcombe Boulevard, Houston, TX 77030. E-mail address:[email protected]
The online version of this article contains supplemental material.
Abbreviations used in this paper: 7-AAD, 7-aminoactinomycin D;ACT, adoptive T celltherapy; DC, dendritic cell; FISH, fluorescence in situ hybridization; ITIP, mixture ofIL-1b, TNF-a, IL-6, and PGE2; KIR, killer inhibitory receptor; REP, rapid expansionprotocol; TIL, tumor-infiltrating lymphocyte; TIL-CM, TIL culture medium.
Copyright� 2009 by The American Association of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0901101
Published November 30, 2009, doi:10.4049/jimmunol.0901101 by guest on January 3, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

and phenotypic changes that can affect the survival and pro-liferative capacity of T cells in vivo shortly after infusion, as thesecells are reactivated by cytokines and through APCs presentingmelanoma Ags (3). How TILs respond to melanoma Ag restim-ulation after the REP and how this relates to specific phenotypicchanges during the REP has not been studied thoroughly.As reported in this study, we determined the response ofMART-1
peptide–specific CD8+ TILs to antigenic restimulation before andafter the REP and how this relates to changes in T cell phenotypeand effector function. MART-1 is a major Ag in melanoma ex-pressed in.95% of patients, and CD8+ T cells specific for this Agare commonly found in TIL preparations and can be reliablytracked using tetramers (9–11). Targeting MART-1 has also beenused in vaccine clinical trials and adoptive T cell therapy usingTCR-transduced PBMCs can mount antitumor responses againstmelanoma (9, 11). Thus, the fate of MART-1–specific CD8+ T cellscan have important ramifications for ACT. TILs from HLA-A2.1+
melanomas were stimulated under optimal conditions using HLA-A2.1+ MART-1 peptide–pulsed mature dendritic cells (DCs) andadded IL-2, mimicking a possible scenario in vivo in which infusedTILs can make contact with their cognate melanoma Ags. In-terestingly, post-REP TILs were found to be hyporesponsive topeptide restimulation, manifested as slow entry into the cell cycleand increased apoptosis. In contrast, under identical conditions,pre-REP MART-1–reactive TILs expanded well, with little in-duction of apoptosis. Phenotypic and functional analysis of the twoTIL populations using a number of different T cell differentiationmarkers found that CD28 was markedly downmodulated in post-REP cells, whereas CD27 and CD57 levels showed no statisticallyrelevant changes in expression. Sorting of CD28+ and CD282 post-REP CD8+ T cells revealed that only the few remaining CD28+
TILs retained proliferative capacity in response to MART-1 peptiderestimulation. Sorted CD282 post-REP TILs did not re-expressCD28. We also went on to study whether loss of CD28 during theREP can be prevented with different supportive cytokine mixturesusing IL-21 and/or IL-15. Interestingly, a synergy between IL-21and IL-15 was found with IL-15 driving TIL expansion and IL-21maintaining CD28 expression. Our results suggest that loss ofCD28 but not CD27 expression marks diminished proliferative andsurvival potential of post-REP melanoma TILs generated usingcurrent expansion protocols with IL-2. This has implications forhowTIL persistencemay be regulated in vivo and suggests that onlythe few remaining younger, less-differentiated CD28+ TILs arecapable of long-term survival and expansion in vivo. These resultsalso explain recent clinical observations in ACT patients showingthat CD28 expression predominantly marks a long-term persistentTIL population in patients that have a durable response to ACT (12).Expansion of TILs using IL-15 together with IL-21 instead of IL-2may help overcome these current shortcomings in ACT.
Materials and MethodsPatient TIL samples and initial TIL expansion
TILs for laboratory studies were obtained from patients with HLA-A0201+
(HLA-A2.1+) Stage IV melanoma who were undergoing surgery at TheUniversity of Texas M. D. Anderson Cancer Center according to an In-stitutional Review Board–approved protocol and following patient consent.The tumor samples were cut into 3- to 5-mm2 pieces and cultured in 2 mlof TIL culture medium (TIL-CM) consisting of RPMI 1640, 10% humanAb serum, 1 mM glutamine, 1 mM sodium pyruvate, 50 mM 2-mercap-toethanol, 13 penicillin-streptomycin, and 20 mg/ml gentamicin (In-vitrogen, Carlsbad, CA) in 24-well plates. IL-2 at a concentration of 6000U/ml was added to all wells. The dividing TIL lines were fed with freshTIL-CM containing 3000 U/ml IL-2 (Proleukin, Novartis, East Hanover,NJ) and subcultured 1:1 in TIL-CM with 6000 U/ml IL-2 to maintainviable cell density of 0.5–2 3 106/ml.
TIL REP
TILs, initially expanded from tumor fragments as above,were harvested after5 wk and stained for CD8 expression and recognition of the HLA-A2.1MART-1 peptide tetramer and other T cell differentiation markers (see be-low). These TILs were designated as pre-REP and were restimulated withMART-1 peptide as described below or subjected to continued expansionusing the REP originally designed byRiddell andGreenberg (13) that is usedcurrently for ACT for Stage IV melanoma. Briefly, 1.33 105 pre-REP TILsin TIL-CM were added to upright T-25 flasks containing 30 ng/ml OKT3(Abbott Labs, Abbott Park, IL) and 26 3 106 allogeneic irradiated (5000cGy) PBMC feeder cells obtained by pooling PBMC from four normaldonors (obtained from the Gulf Coast Regional Blood Bank, Houston, TX)in 20 ml of TIL-CM. On day 2, 10 ml of AIM V medium (Invitrogen) wasadded together with 6000 U/ml IL-2 (final concentration). The TILs wereexpanded for another 12 d and diluted as needed with AIM V and IL-2 tokeep the viable cell density in the range of 1–43 106/ml. The post-REPTILswere isolated and washed in TIL-CM and rested for 3–6 h before restim-ulation as described below. In some experiments, IL-2 was substituted withIL-15 at 100 ng/ml (R&DSystems,Minneapolis,MN) or IL-21 at 100 ng/ml(BioVision, Mountainview, CA) or a combination of IL-15 (100 ng/ml)together with and IL-21 (100 ng/ml), at all steps in the REP.
Flow cytometric Abs and analysis and sorting of expanded TILs
Expanded TILs were routinely stained for human T cell differentiationmarkers using fluorochrome-conjugated mAb recognizing CD3, CD4, CD8,CD27, CD28, CD57, and CD62L obtained from BD Biosciences (San Jose,CA) or eBiosciences (La Jolla, CA). TILs were stained with HLA-A2.1MART-1 peptide (ELAGIGILTV) tetramer (Beckman Coulter, Fullerton,CA) to track changes in the MART-1–specific CD8+ subpopulation. Insome experiments, pre-REP and post-REP TILs were also stained forCTLA-4, CD25, CD122, CD132, granzyme B, IFN-g, and Ki67 (all fromBD Biosciences) and PD-1 (BioLegend, San Diego, CA). Apoptosis wasmonitored by 7-amino-actinomycin D (7-AAD; Sigma-Aldrich, St. Louis,MO) and Annexin V (BD Biosciences) staining of CD8+ and CD8+MART-1 tetramer+ subpopulations. The stained cells were acquired by using a BDFACScanto II flow cytometry analyzer and FACSDiva software (BD Bi-osciences). Data were analyzed by FlowJo software (TreeStar, San Carlos,CA). In some experiments, the TILs were subjected to FACS usinga FACSAria sorter (BD Biosciences) to isolate CD8+CD28+ and CD8+
CD282 subpopulations and CD8+CD27+ from CD8+CD272 sub-populations.
Generation of DCs and TIL restimulation with MART-1peptide–pulsed mature DCs
Human DCs were generated from monocytes obtained from HLA-A2.1+
normal donors (obtained from the Gulf Coast Regional Blood Bank) in T-75 flasks after plastic adherence. The DCs were differentiated with 1000U/ml GM-CSF and 1000 U/ml IL-4 (R&D Systems) in IMDM withGlutamax, 2% human Ab serum, 13 penicillin-streptomycin, and 50 mM2-mercaptoethanol (designated as DC-CM). After 5 d, the DCs were ma-tured through exposure to a mixture of IL-1b TNF-a, IL-6, and PGE2
(ITIP) (14). The mature DCs were irradiated at 2000 cGy and pulsed with3 mg/ml of MART-1 peptide for 90 min. TILs (2 3 106 TIL in 24-wellplates) were restimulated by adding 2 3 105 peptide-pulsed mature DCs inTIL-CM with 100 U/ml IL-2 and culturing for 7 to 8 d. IL-2 (to 100 U/ml)was added on day 4.
Measurement of TIL cell division and effector cell activity
Expansion of TILs following restimulation was determined by countingviable cells after trypan blue staining using a hemocytometer. In someexperiments, cell division was determined by CFSE dilution in prelabeledpre-REP or post-REP TILs. For these CFSE experiments, TILs werewashedin Dulbecco’s PBS and resuspended in Dulbecco’s PBS with 1 mM ofCFSE (Molecular Probes-Invitrogen, Carlsbad, CA) for 5–7 min and thenwashed three times in TIL-CM. Cell division was monitored by using theFITC channel in a FACScanto II flow cytometer (BD Biosciences). CTLactivity of TILs was monitored by a flow cytometry-based assay measuringthe cleavage of caspase 3 in MART-1 peptide–pulsed T2 target cells asreadout (15). Briefly, the T2 cells were labeled with DDAO-SE (MoleculeProbes-Invitrogen), washed, and then pulsed with MART-1 peptide ora control HLA-A2.1-binding HIV rev peptide (SLYNTVATL), both at 5 mg/ml, for 1 h. The pulsed T2 cells were incubated with TILs at different ef-fector:target ratios for 3 to 4 h and then fixed, permeabilized, and stainedwitha PE-conjugated anticleaved caspase 3 rabbit mAb. Staining for IFN-gproduction by TILs coincubated withMART-1 peptide–pulsed T2 cells for 5
2 CONTINUED CD28 EXPRESSION IS ASSOCIATED WITH TIL PERSISTENCE
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

to 6 h was done by using an intracellular cytokine staining protocol. Gol-giStop (BD Biosciences) was added 1 h into the coincubation time. In eachcase, stained cells were analyzed in a FACScanto II flow cytometer (BDBiosciences).
Fluorescence telomere length assay
Quantitative flow-fluorescence in situ hybridization (FISH) analysis wasused to measure the average length of telomere repeats at chromosome endsin individual T cells, as previously described (8). FITC-conjugated telomereprobe (FITC-OO-CCCTAACCCTAACCCTAA, O indicating a moleculelinking FITC to DNA sequence) was obtained from Dako (Carpinteria,CA). FITC-labeled fluorescent calibration beads (Quantum TM-24; BangsLaboratories, Fishers, IN) were used to convert telomere fluorescence datato molecules of equivalent soluble fluorescence units (8). Length in basepairs was calculated by the equation: bp = molecules of equivalent solublefluorescence3 0.495 (8). Aliquots of the K562 cell line, which has a stabletelomere length between 4.0 and 4.5 kb, were used each time the assay wasrun to normalize telomere lengths. Nonspecific sequence probes were in-cluded as negative controls.
Microarray experiment and analysis
Post-REP TILs were freshly harvested from the flasks and sorted after anti-CD4 and anti-CD28 staining into two subsets, CD42CD28+ and CD42
CD282. After a 3-h rest period to shed staining Abs after sorting, TILswere processed with the RNeasy kit (Qiagen, Valencia, CA) to obtain RNAfor microarray analysis. Samples (1 mg) of RNAwere subjected to reversetranscription and probe blotting using an Illumina kit, which uses a humanRef6 chip (Illumina, San Diego, CA). Image acquisition and data pro-cessing, conducted with BeadStudio software (Illumina), generated a set ofgenes (detected with a p value , 0.01) as well as data from supervisedgroup analysis. Microarray data sets were explored further using a heatmapserver (http://noble.gs.washington.edu/prism/) and Onto-Tool (12; http://vortex.cs.wayne.edu/projects.htm). Real-time PCR was conducted to val-idate key genes of interest.
Statistical analysis
Data were analyzed by Microsoft Excel (Microsoft, Redmond, WA). TheStudent t test was used to analyze statistical differences.
ResultsPost-REP TILs are hyporesponsive to Ag restimulation
Post-REP melanoma TILs must survive and be able to undergo celldivision after infusion into lymphodepleted patients to repopulatethe empty lymphoid compartment. Thus, melanoma-specific CD8+
TIL subpopulations should have the capacity to selectively expandand persist in the host when restimulated by APC-presentingmelanoma Ags. These cells should have a proliferative advantageover the bulk T cell population if they are to effectively mediateantitumor effector cell responses. We established an in vitro sys-tem to address these possibilities by tracking the fate of theMART-1 peptide–specific CD8+ T cell population in pre-REP andpost-REP TILs after restimulation with MART-1 peptide–pulsedmature DCs. HLA-A2.1-matched DCs were generated with GM-CSF and IL-4, followed by 1 to 2 d of maturation using ITIP.Screening for CD83, CD86, CD80, CD70, and HLA-A2.1 re-vealed their expression at expected levels for DC matured by thismixture (Supplemental Fig. 1).We first tested a number of pre-REPHLA-A2.1+ TIL cultures and
confirmed that restimulation with the peptide-pulsed HLA-A2.1-matched DC selectively expanded the CD8+MART-1 tetramer+ Tcell population over a background level of expansion (Fig. 1A). Wethen tested a set of post-REPTIL lines in the same fashion and foundthat, in contrast, CD8+MART-1–specific T cells were hypores-ponsive during the 7-d restimulation period, as determined by tet-ramer staining (Fig. 1B). This was reflected by a low level of CFSEdilution (Fig. 1C) and a lack of increase of total viable CD8+MART-1 tetramer+ cells (Fig. 1D) in the different patient TIL lines shown.Alack of appreciable proliferation after DC restimulation was alsofound in the bulk (Ag-nonspecific) CD8+ TILs, as shown by CFSE
dilution and counts of total viable cells. Thus, this hypores-ponsiveness of post-REPMART-1–specificT cells was due not to anoverwhelming expansion of nonspecific T cells, but to the lack ofproliferative capacity of theMART-1–specific subpopulation. Fig. 2summarizes results of seven pre-REP and post-REP melanoma TILlines showing the fold changes in total T cells andMART-1 peptide–specific CD8+ T cells after the 7-d restimulation period. In bothcases, post-REP TILs had substantially lower levels of T cell ex-pansion than pre-REP TILs.
Post-REP restimulated TILs have stronger cytolytic ability
In addition to T cell expansion, the acquisition of cytolytic ability isalso crucial for effective antitumor T cell responses during ACT.CD8+ T cells exert their CTL function by inflammatory cytokinesecretion and direct killing by granzyme B and perforin release. Weassessedwhether post-REPTILs retained theirAg-specific cytolyticability despite the loss of proliferative capacity following antigenicrestimulation. Pre-REP and post-REP TILs were restimulated withpeptide-pulsed DCs as before and tested for CTL activity againstMART-1 peptide–pulsed T2 cells after 7 d using a previously de-scribed FACS-based caspase 3 cleavage assay (15). Post-REP TILshad a greater cytolytic ability after restimulation on a per-cell basisthan pre-REPTILs (Fig. 3A). Thiswas correlatedwith an increase inthe level of granzyme B expression in the MART-1 tetramer+ pop-ulation and ability to produce IFN-g in response to MART-1 pep-tide–pulsed targets (Fig. 3B). These data indicate that post-REPTILs, upon MART-1 restimulation, can differentiate into more po-tent CTL than pre-REP TILs, despite having a low proliferativepotential after restimulation with Ag.
Pre-REP TILs exhibited better long-term persistence in cultureafter Ag restimulation
In addition to following pre-REP and post-REP TILs over a 7-d peptide restimulation assay, we also continued to track the ex-pansion of MART-1–specific CD8+ TILs for up to 28 d after re-stimulation to determine their long-term expansion or persistencecapacities. Both pre-REP and post-REP TILs were restimulatedfor 1 wk by MART-1 peptide–pulsed mature DCs. Following thisrestimulation, the TILs were replated in culture medium with IL-2and cultured for another 3 wk with IL-2 feeding every 4 d. Sup-plemental Table I shows the changes in total number and per-centage of CD8+MART-1 tetramer+ pre-REP and post-REP T cellsin five different patient TIL lines over 28 d after MART-1 peptiderestimulation. Overall, pre-REP, MART-1–specific TILs expandedmuch better during the first 7 d and cells from 3 of the linescontinued to expand up to the 28-d time point, with the other twohaving a drop in MART-1–specific CD8+ T cell recovery andpercentage. This is in contrast to pre-REP TILs in which a ex-pansion of MART-1–specific CD8+ T cells was found in only oneout of the five TIL lines tested, with most having undetectableMART-1–specific cells at 28 d (Supplemental Table I).
Most MART-1–reactive post-REP TILs fail to enter cell cyclenormally and undergo delayed apoptosis after restimulationwith MART-1 peptide
Wewent on to investigate possible reasons accounting for the lack ofMART-1 CD8+ T cell expansion in post-REP TILs following re-stimulation with peptide-pulsed DCs. We first measured the abilityof post-REP TILs to enter the cell cycle and determined the level ofapoptosis at different times following restimulation with MART-1peptide. Cell cycle progression was determined by costaining CD8+
MART-1 tetramer+ TILs for Ki67 and DNA content using 7-AAD.As shown in Fig. 4A, post-REP TILs mostly failed to enter the cellcycle after the 7-d restimulation period, whereas a significant
The Journal of Immunology 3
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

fraction of pre-REP TILs entered the cell cycle, as determined byKi67+ staining. Ki67+ cells in the S phase andG2/Mphase of the cellcycle, as determined by DNA content, could be clearly seen,whereas very few such cells were found among post-REP TILs.These results correlate with the differences in CFSE dilution de-scribed earlier. Measurement of apoptosis (Fig. 4C) in the MART-1peptide–specific CD8+ and bulk CD8+ T cell populations followingpeptide-restimulation found that post-REP TILs had an increase inAnnexinV+, 7-AAD+ cells (67.06%and 65.84%, respectively, in thetwo TIL lines shown) after 4 d in comparison with pre-REP TIL(3.93%and 12.98%, respectively, in the twoTIL lines shown). Thus,most post-REP TILs failed to enter the cell cycle after antigenicrestimulation, and many of these cells later underwent apoptosis(Fig. 4B, 4D). It must be noted that this cell death was not due tocytokine deprivation after day 4, because the cultures were fed with200 U/ml IL-2 after restimulation and again on day 4.
Post-REP TILs have decreased telomere length
Decreased telomere length has been observed in post-REP TILs,and the loss of persistence of many infused TILs in patients in vivohas been attributed to this decrease (8). We therefore measuredtelomere lengths by flow-FISH analysis in a panel of independentpatient TIL lines. This analysis did not determine telomere lengthsof the MART-1–specific CD8+ T cell population because notenough Ag-specific TILs could be isolated for the flow-FISH.Nevertheless, the results should reflect the changes in the MART-1–specific population due to the overall small variation of the
flow-FISH peaks in the samples (Fig. 5A), suggesting that most Tcell clones in the population had similar telomere lengths. As seenin Fig. 5B, telomere erosion occurred during the REP in most TILlines; the average telomere length of the pre-REP TIL was 5.7 kb,whereas that of the post-REP TILs had an average loss of 2 kbdown to 3.7 kb (p = 0.0004 using Student t test). Thus, a loss oftelomeres in CD8+ T cells occurs during the REP; however, suf-ficient telomere length (3.7 kb) was retained that would supportcontinued cell division.
Loss of CD28 expression in CD8+ MART-specific TILs after theREP
We went on to look at markers of CD8+ T cell differentiation todetermine whether any specific phenotypic changes correlated withthe loss of post-REP TIL proliferation potential. Decreased cellcycle progression can result from either an increase in T cell–suppressive signaling molecules and coreceptors or a loss of cos-timulatory or activating receptors. Staining for CD3 (TCR), IL-2Rb(CD122), IL-2Rg (CD132), and IL-2Ra (CD25) did not reveal anysignificant differences between pre-REP and post-REP TILs (datanot shown). We also looked at two negative costimulatory mole-cules, CTLA4 and PD1, and found no significant differences in thepercentage of bulk or MART-1–specific CD8+ T cells expressingthese molecules between pre-REP and post-REP cultures (data notshown). We stained for the costimulatory molecules CD27 andCD28, key markers of the state of CD8+ T cell differentiation (16,17). As shown in Fig. 6A, CD8+ TILs exhibited a marked
FIGURE 1. MART-1–specific TILs preferentially expand when restimulated with MART-1 peptide–pulsed DCs, and post-REP TILs are hyporesponsive
to this form of restimulation. Mature DCs were generated and pulsed with MART-1 peptide as described inMaterials and Methods and then used to activate
TILs (1:10 ratio). The cultures were incubated for 7 d. IL-2 (200 U/ml) was added to all cultures to maintain viability. A, Preferential increase in the
percentage of MART-1 tetramer+ TILs (pre-REP) by flow cytometry analysis before and after the 7-d restimulation with MART-1 peptide–pulsed normal
donor HLA-A2.1+ DCs. Changes in MART-1 tetramer staining (B) and CFSE dilution (C) in pre-REP versus post-REP TILs 7 d after restimulation with
MART-1 peptide–pulsed DCs. D, Total number of CD8+MART-1 tetramer+ T cells in post-REP melanoma TILs before and after restimulation with MART-
1 peptide–pulsed DCs.
4 CONTINUED CD28 EXPRESSION IS ASSOCIATED WITH TIL PERSISTENCE
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

downregulation of CD28 expression (average of 58% CD28+ in theCD8+ in pre-REP TILs versus 28.8% CD28+ in the same sub-population of post-REPTILs). CD27 expressionwas also somewhatdecreased in the MART-1–specific CD8+ T cell population, but thiswas not statistically significant over the 15 different TIL lines tested.CD28 expression was also markedly lost in the post-REP CD8+
MART-1 tetramer+ population, and only a minor population re-tained significant CD28 expression (Fig. 6B). CD27 expression,however, was retained in most of the CD8+ MART-1 tetramer+
population (Fig. 6B). CD57, a marker for end-stage effector CD8+
T cells shown to be hyporesponsive to activation, increased slightlyafter the REP (Fig. 6A). Thus, the most consistent change in T cellphenotype in CD8+ and the MART-1–specific CD8+ TILs after theREP was a loss of CD28 expression, with a slight increase in thepercentage of CD57+ cells and an insignificant change in CD27expression.
CD28+ post-REP TILs have improved persistence afterrestimulation with MART-1 peptide
The data presented so far show that, of the key effector memorycostimulatory markers (CD27 and CD28), melanoma TILs losemostly CD28 expression, and to a lesser extent CD27 expression,following the REP. Although CD28 has been known for many yearsasacriticalcostimulatorysignal for theproductiveactivationofnaiveT cells and to prevent activation-induced cell death, several recentstudies have shown that memory CD8+ T cells may still requireCD28 costimulation for optimal reactivation and expansion (18, 19).With this in mind, we hypothesized that the few remaining CD28+
MART-1–specificCD8+T cells in post-REPTILsmay exhibit betterpersistence and may retain the capacity to divide after restimulationwith MART-1 peptide. Although most CD28 expression was lost,some post-REPTILs still retained enoughCD28+ cells to allow us totrack the fate of CD28+ and CD282, as well as CD27+ and CD272,MART-1 tetramer+CD8+ post-REP TILs after restimulation withpeptide-pulsed DCs. For these experiments, we also used CD40L-activated and expanded HLA-A2.1+ human B cells as APCs, asthese cells express high levels ofCD70 (ligand forCD27) in addition
to CD86 and CD80 (Supplemental Fig. 1), allowing us to determinethe effects of both CD28 and CD27 costimulation on TIL restim-ulation. TheDCs (ITIP-matured) expressed high levels of CD86 andCD80, but low levels of CD70 (Supplemental Fig. 1). Post-REPTILs having MART-1 tetramer+CD8+ T cells were labeled withCFSE and restimulated by HLA-A2.1+ MART-1 peptide–pulsedDCs orCD40L-activatedB cells asAPCs.After 5–7 d, the cellswerestained for CD8, CD28, CD27, andMART-1 tetramer and analyzedby flow cytometry. As shown in Fig. 7A, no distinct difference wasobserved between the responses to the two different types of APCsused to stimulate the TILs. However, subset analysis (Fig. 7B) re-vealed a significant degree of CFSE dilution in the MART-1 tetra-mer+CD28+CD8+T cells, whereas CFSE remained largely undilutedin the corresponding CD28- population. Interestingly, when CD27+
versus CD272 CD8+ T cells were analyzed in the same manner, nodifference in CFSE dilution was observed within the MART-1 tet-ramer+ population. Annexin V staining of post-REP TILs after re-stimulation with MART-1 showed that CD282 TILs underwentsignificantlymore apoptosis upon stimulation thanCD28+TILs (datanot shown). Further tracking of post-REP MART-1–specific CD8+
TILs over a 10-d period after restimulation found that the CD28+
subpopulation lost CFSE staining and was the dominant sub-population remaining after 10 d, whereas the CD282 subpopulationdisappeared (Fig. 7C). Thus, the small fraction of CD8+CD28+ post-REP TILs exhibited a measurable proliferative advantage followingantigenic restimulation and were more resistant to apoptosis.To further explore whether maintenance of CD28 expression is
associated with persistence and/or expansion of post-REP TILsduring restimulation with Ag, we sorted CD8+CD28+ and CD8+
CD282T cells frompost-REPTILs using FACS (Supplemental Fig.
FIGURE 2. Pre-REP TILs have better proliferative ability than post-
REP TILs in response to restimulation with Ag-pulsed DCs. Multiple lines
of TILs were expanded from different patients and restimulated at the pre-
REP or post-REP stage with MART-1 peptide–pulsed DCs. The fold in-
crease in the bulk CD8+ TIL population (A) and the CD8+MART-1 tet-
ramer+ subset (B) 7 d after restimulation is shown for seven different TIL
lines. Each point represents a different TIL sample; the horizontal bars
show the averages. The p value was calculated using the Student t test.
FIGURE 3. Restimulated post-REP TILs exhibit stronger cytolytic ac-
tivity than pre-REP TILs. A, CTL activity of restimulated pre-REP and
post-REP TILs against MART-1 peptide–pulsed T2 cells using a caspase 3
cleavage CTL assay. The data shown are normalized by dividing the
percentage caspase 3 cleavage in the targets by the percentage of CD8+
MART-1 tetramer+ T cells added to the CTL assay (1:1 effector:target
ratio) to calculate killing per added TIL. Statistical difference was calcu-
lated by using Student t test. B, Granzyme B staining of a representative
TIL line shows a higher level of staining (MFI for granzyme B) in post-
REP TILs. Post-REP TILs synthesized more IFN-g in response to MART-1
peptide–pulsed DC restimulation as determined by an intracellular cyto-
kine staining assay. Results in B are representative of a minimum of three
separate TIL lines tested.
The Journal of Immunology 5
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

2). The sorted CD8+ T cells were CFSE-labeled and restimulatedwith peptide-pulsed APCs, as before. After 7 d, CFSE dilution andthe total number of recoveredMART-1 tetramer+CD8+ T cells weredetermined. Analysis of five independent post-REP TIL lines foundthat the numbers of sorted MART-1 tetramer+CD28+CD8+ T cellsremained stable or in other cases increased after the restimulationperiod, whereas the numbers of sorted CD282 cells decreased (Fig.7D). Both the CD28+ and CD282 subpopulations had similar levelsof CD27 expression after the sort (Supplemental Fig. 2); therefore,differences in CD27 costimulation or CD27 expression did not ac-count for these differences in post-REP CD8+ T cell persistence orexpansion. To confirm this, we sorted pre-REP TILs (which expandwell after restimulation) into CD27+ andCD272 subsets using FACSand analyzed their capacity to expand following restimulation withMART-1 peptide–pulsed DCs or B cells. The sorted CD27+ andCD272 CD8+ pre-REP TILs both expressed CD28 at similar levels(Supplemental Fig. 2). In this case, however, both sorted cell pop-ulations expanded similarly in response to restimulationwithMART-1 peptide (Supplemental Fig. 3). Thus, sustained and high-levelCD28expression, but notCD27expression, delineatesTILs that havea capacity to continue dividing after restimulation with cognate Ag.We went on to test whether inhibiting CD28 costimulation by
using anti-CD80 and anti-CD86 Abs, or by addition of CTLA4-Fcfusion protein, affected the persistence and/or further expansion ofthe CD28+ post-REP TILs after restimulation. The activity ofthese reagents was confirmed by their ability to block proliferationin a mixed lymphocyte response (data not shown). Blocking CD28costimulation by either of these techniques did not, however, in-hibit expansion of CD28+MART-1–specific TILs (data notshown). Thus, CD28 seems to be a marker for post-REP CD8+
TILs with better persistence or expansion capability, but may notbe required or function as a costimulatory molecule at this stage.
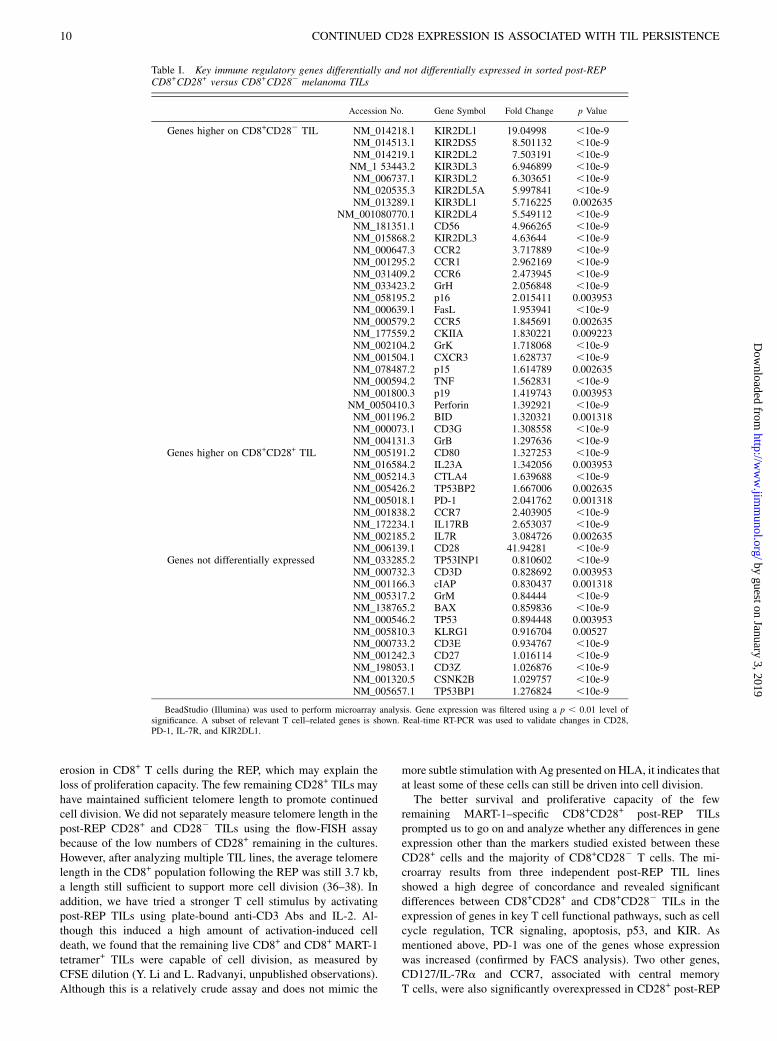
Differential gene expression analysis by cDNA microarrayanalysis reveals significant differences between sorted CD8+
CD282 and CD8+CD28+ post-REP TILs
To investigate the mechanism of why post-REP CD8+ TILs loseproliferation and survival capacity in relation to a loss of CD28 cell-surface expression, we performed cDNA microarray analysis onthree distinct post-REP TIL lines. Each TIL sample was sorted forCD8+CD28+ and CD8+CD282 immediately after the REP andsubjected to gene expression analysis using the Illumina human
Ref6 chip after RNA isolation (Illumina). The microarray data hasbeen deposited into the GEO database (Accession #GSE16517;http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE16517).The microarray results among the three different sorted post-REPTIL samples were highly concordant, with 12,430 genes expressed(overall p value, 0.01). Among these genes, thosewith a change inexpression of .2-fold were clustered together (Fig. 8A). Thisanalysis showed that a number of different genes coclustered withCD282 TILs and could be clearly separated from CD28+ TILs.Analysis of the most differentially expressed genes in the CD282
subset according to specific functional subsets (Fig. 8B) usingpathway analysis software (Onto-Tools; http://vortex.cs.wayne.edu/projects.htm) revealed genes in eight specific pathways thatsignificantly differed between CD28+ and CD282 TILs, includinggenes involved in Ag processing and presentation (28 of 88 genesdifferentially expressed), TCR signaling (38 of 93 genes differen-tially expressed), cell cycle (43 of 112 genes differentially ex-pressed), p53 pathway (29 of 68 genes differentially expressed), andkiller inhibitory receptor (KIR) pathway (47 of 131 genes differ-entially expressed). Interestingly, the KIR pathway genes exhibitedthe greatest difference between CD282 and CD28+ TILs (Fig. 8B).Table I shows a subset of the most differentially expressed genes inthis analysis. Overall, the CD282 TILs had higher expression ofcytolytic genes (granzyme family members, perforin, FasL, TNF)and cell cycle inhibitors (p15, p16, and p19). On the other hand,CD28+ TILs showed higher expression of a combined set of earlyeffector and central memory markers, such as IL-7R and CCR7.Interestingly, PD-1, TP53BP2, IL-17RB, and IL-23A were morehighly expressed on CD28+ TILs. Staining of post-REP TILs forCD8, CD28, and PD-1 confirmed this difference in PD-1 gene ex-pression at the protein level. PD-1 expression was significantlyhigher in the CD8+CD28+MART-1 tetramer+ subset, with 26% ofthis subset staining positive for PD-1 after the REP, whereas only14% of the post-REP CD8+CD282MART-1 tetramer+ cells ex-pressed PD-1 (Fig. 8C). Five days after MART-1 peptide restim-ulation, this difference in PD-1 expression between the two subsetspersisted (Fig. 8C). Another striking difference betweenCD282 andCD28+ TILs was a markedly greater expression of inhibitory KIRreceptors (20) on CD282 TILs, such as KIR2DL family members 1through 5 (Table I). Interestingly, CD28+ TILs expressed more ofa p53-binding protein, TP53BP2. As expected, the CD28 gene washighly overexpressed in the sorted CD28+ population (Table I). This
FIGURE 4. Cell cycle entry is reduced and apo-
ptosis increased in post-REP TILs after restimulation
with Ag. Pre-REP and post-REP TILs including
MART-1 tetramer+ CD8+ T cells were stimulated with
peptide-pulsed DCs in the presence of IL-2 as already
described. The cultures were assayed for cell cycle
entry and progression using staining for Ki67 expres-
sion and 7-AAD for DNA content after fixation and
permeabilization on day 7 of stimulation (A and C). A
shows flow cytometry plots of Ki67 versus 7-AAD
performed on day 7 after restimulation of two repre-
sentative TIL lines at the pre- and post-REP stages. B
shows the time course of changes in Ki67 staining in
these two pre-REP or post-REP CD8+ TILs after re-
stimulation. C, Post-REP CD8+MART-1 tetramer+
TILs underwent apoptosis after restimulation with
peptide, as measured by uptake of 7-AAD and Annexin
V staining of unfixed cells. D, Time course of changes
in 7-AAD+ and Annexin V+ CD8+MART-1 tetramer+
T cells in pre-REP and post-REP TILs after restim-
ulation in two TIL lines.
6 CONTINUED CD28 EXPRESSION IS ASSOCIATED WITH TIL PERSISTENCE
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

also served as an indicator for the purity of the sorted populationsused in the microarray analysis.
Effects of alternative cytokines during the REP on CD28expression and maintenance of TIL responsiveness to antigenicrestimulation
We went on to test whether alternative cytokines other than IL-2,such as IL-15 and IL-21, can support TIL expansion during the REPand yield TILs with a different phenotype, especially in terms ofpreventing CD28 loss in the CD8+ T cells. First, we tested IL-15,known for its ability to support T cell proliferation and effectorcell differentiation (21), in comparison with IL-2 in the REP.However, in six different TIL lines, no major differences wereobserved when IL-2 (IL-2 REP) or IL-15 (IL-15 REP) was used inthe REP. The IL-15 REP expanded TILs to a similar extent as IL-2(data not shown) with a similar CD8+ T cell phenotype observedafter staining for CD27, CD28 (Fig. 9A), and CD57 expression(data not shown). Next, we tested IL-21 alone, or together with IL-15, in comparison with IL-2 and IL-15 alone in the REP. IL-21 hasbeen shown to maintain CD28 expression in activated PBMCs andmaintain a younger T cell phenotype (22). However, although IL-21 used alone in the REP preserved CD28 expression on CD8+
TILs, it drove much lower levels of TIL expansion than IL-2 orIL-15 with a 50-fold lower T cell yield (Fig. 9B, 9C). In contrast,when IL-21 was combined with IL-15, TIL expansion was com-parable to IL-2 or IL-15 alone, but a significant increase in thepercentage of CD28+ CD8+ TILs post-REP was found (Fig. 9B,9C). Thus, a combination of IL-21 and IL-15 seems to preserveCD28 expression in a significant fraction of TILs while stilldriving high rates of TIL expansion.We then determined how TILs subjected to the REP using these
different cytokine mixtures responded to MART-1 peptide restim-ulation using TIL lines containing MART-1 tetramer+ CD8+ T cellsfrom HLA-A2.1+ patients. As shown in Fig. 10A, TILs from IL-21plus IL-15 REP cultures had an improved Ag-specific CD8+ T cellresponse to restimulation with MART-1 peptide–pulsed DCs overcells from the IL-2 REP and the IL-15 REP. In two out of three TILlines shown, TILs isolated from IL-21 plus IL-15 REP cultures hadmarkedly higher increase in CD8+ MART-1 tetramer+ T cells 7d after restimulation. Although IL-21 used alone preserved CD28expression during the REP, in most cases the isolated post-REPCD8+ T cells did not have an improved Ag-specific restimulation
response over cells from the IL-2 REP cultures (Fig. 10A). On fur-ther analysis, we found that CD8+ TILs isolated from IL-21 REPcultures had significantly higher levels of PD-1 expression thanTILs isolated after the REP with IL-2, IL-15, or IL-15 plus IL-21(Fig. 10B). Thus, although IL-21 preserves CD28 expression on theCD8+T cells, the rate of expansion during theREP is poor after TCRstimulation, and PD-1 levels are maintained at a higher level.
DiscussionThe critical goal of ACT using expanded TILs for melanoma andother cancers is generating a large enough population ofAg-reactiveT cells that not only mediates immediate antitumor killing afterinfusion, but also persists to mediate a longer-term control ofmetastatic tumor deposits. A major problem in current TIL ACTprotocols is the relatively rapid disappearance (within 14–21 d) ofmost of the infused TILs, which yield only transient control oftumor growth. The reason for the transience of infused TILs isa matter of current debate in the field. TILs infused into the body ofthe patient with melanoma are faced with a number of factors af-fecting their continued cell division, effector function, and survival.A key challenge for these cells is reactivation with tumor Ags andother self-Ags in addition to IL-2 signaling and contact with ad-ditional homeostatic cytokines in the body. These environmentalfactors, in context to the state of effector T cell differentiation, willultimately regulate long-term TIL persistence. At present, however,
FIGURE 5. Telomere length is shortened after the REP. Telomere
lengths in nine independent TIL lines were determined before and after the
REP using flow cytometric FISH assay. A shows how the telomere length
was determined using flow-FISH analysis on a representative TIL line. The
telomere lengths were measured in gated G1 phase cells using 7-AAD
staining. Flow-FISH assay of K652 cells (stable telomere length) was used
to calculate the telomere lengths. Telomere length for nine independent
pre-REP versus post-REP TIL lines, with p value, is shown in B. Each
point represents a different TIL sample; the heavy bar shows the average.
FIGURE 6. Cell-surface CD28 expression is lost after the REP. Pre-REP
and post-REP TILs from over 15 different lines were analyzed for CD27,
CD28, and CD57 cell-surface expression in the CD8+ subset using flow
cytometry. A, Percentage of positively staining CD8+ cells for each marker
is shown for multiple matched pre- and post- REP TILs. Each point rep-
resents a different TIL sample; the heavy bar shows the average. The p
values were calculated by the Student t test. B, Profound loss of cell-
surface CD28 expression in the CD8+MART-1 tetramer+ TIL subset after
the REP. Dot plots in B show analysis of CD27 versus CD28 staining in the
gated CD8+MART-1 tetramer+ subset in two different matched pre- and
post-REP TIL lines.
The Journal of Immunology 7
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

very little is known about how TILs generated for ACT via theclassical REP (13) respond to antigenic restimulation and whatphenotypic markers are associated with TILs capable of longer-term persistence and expansion after the REP.In thecurrent study,weaddressed thesequestions inawell-defined
in vitro assay system using expanded MART-1–specific TILs froma number of different melanoma patient tumors. Although othershared melanoma Ags and unique patient-specific Ags are alsoimportant in the antitumor response (23), MART-1 is the mostwidely expressed and most immunogenic melanoma Ag (10, 24).MART-1–specific CD8+ T cells are readily detected in most mela-noma TILs and can be reliably tracked using a number of methodssuch as tetramer staining, allowing us to track the phenotype and fateof these T cells in multiple patient samples (10, 24, 25). MART-1–reactive T cells can also mount effective antitumor responses duringACT in patients (9, 11, 24). Paradoxically, we found that post-REPCD8+MART-1–specific TILs were hyporesponsive to restimulation,even when stimulated in an optimal setting using mature peptide-pulsedDCs.Most TILs failed to enter the cell cycle,withmany of thecells undergoing delayed apoptosis 5 d after restimulation with Ag.TILs rested in lower doses of IL-2 (300 U/ml) for a period of 3 to 4 dafter the REP also exhibited this hyporesponsiveness. In contrast,TILs isolated from the initial phase of expansion from melanomatumor fragments (before the REP) were able to enter the cell cycleand proliferated markedly better in response to MART-1 peptiderestimulation. Thus, large-scale expansion in the REP induces a setof functional and phenotypic changes in TILs, limiting their con-tinued survival and responsiveness rechallenge with Ag, a situationthat may explain the lack of persistence observed in patients. Wesought to further understand the nature of this acquired hypores-
ponsiveness by tracking changes in a number of key effector-memorymarkers at both the pre-REPand post-REP stages, aswell asexamining anumberof other parameters suchas telomere erosion andgene expression.One of the key issues during T cell expansion in vitro and in vivo
is the induction of differentiation during multiple cell divisionsassociated with appearance of end-stage effector cells and de-creased proliferative potential. TILs isolated from melanomas arepreactivated T cells with mainly an effector-memory phenotype.We and others have found that freshly isolated CD8+ TILs havemostly a CD27+CD28+CD572GB+Perf2/low phenotype, indicatingan early effector-memory stage of differentiation (26, 27; Y. Liand L. Radvanyi, unpublished observations) before ex vivo ex-pansion for ACT. CD27 and CD28 are critical costimulatorymolecules for T cell activation and survival, and their expressionlevels are used to track the effector-memory and terminal-effectorstages of CD8+ T cell differentiation (17, 28–30). Long-term celldivision of viral-specific CD8+ T cells due to periodic or chronicviral restimulation eventually downmodulates CD27 and CD28associated with terminal CTL differentiation, high cytotoxic ac-tivity, IFN-g secretion, and loss of proliferative potential. CD57increases on fully differentiated CTLs and is another marker thathas been associated with hypoproliferative T cells. A classic caseis CMV-reactive CD8+CD272CD282CD57+GB+Perforin+ cellsthat are hypoproliferative, yet highly cytotoxic, and increase withage (26). In this context, a rapid and acute expansion of T cells(like the REP of TILs used for ACT) may mimic these long-termchanges and generate T cells reminiscent of such senescent anti-viral T cells. In our experiments, we stained for CD27, CD28,CD57, and other T cell phenotypic markers known to changeduring CD8+ T cell differentiation. We found that the most con-sistent change between pre-REP and post-REP TILs was a pro-found loss of cell-surface CD28 expression with a slight increasein the percentage of CD57+ cells. In studying over a dozen dif-ferent TIL lines from different patients, however, we did not ob-serve a statistically significant change in CD27 expression,especially in the MART-1–specific CD8+ subset. Our resultssupport previous studies (e.g., 12, 31, 32) on melanoma TIL ex-pansion where losses of CD28 and CD27 expression were foundafter extensive expansion of T cells in the REP used for ACT.However, these studies also noted a loss of CD27 expression afterthe REP that we did not observe in this study. The reason for thisdifference is unclear, but it is important to note that the other studydid not specifically look at a melanoma Ag-specific subpopulationand only tracked changes in the bulk TIL population after the REP.Moreover, subtle differences in protocols and feeders, as well asthe timing of IL-2 addition to the REP cultures, may account forthis difference. High IL-2 signaling (.3000 U/ml) can down-modulate cell surface CD27 expression through ligation by CD70on CD8+ TILs, whereas lower IL-2 doses led to CD27 being de-tected again on the cell surface (33). Ligation of CD27 by CD70expressed on the CD8+ T cells themselves was found to be themechanism involved in the CD27 downmodulation (33). Althoughwe did not track CD70 expression on our TILs during the REP, itis possible that the CD8+ TILs used in our study did not expressthese high levels of CD70 or that the cell density in our culturesdid not promote enough contact between CD70+ and CD27+
T cells to mediate this downmodulation. Despite these differencesin CD27 modulation between our study and previous studies, thekey finding in our study was that CD27 expression status did notaffect the proliferation or survival potential of post-REP TILswhen restimulated with melanoma Ag. Similar results were ob-tained in our experiments with pre-REP TILs sorted for CD27+
and CD272 subpopulations before restimulation. Rather, what
FIGURE 7. Only CD28+ TILs remaining after the REP are capable of
further cell division in response to restimulation with Ag. Post-REP TILs
from an HLA-A2.1+ patient were restimulated with either MART-1
peptide–pulsed HLA-A2.1+ DCs (CD70low/2) or MART-1 peptide–pulsed
CD40L-activated HLA-A2.1+ B cells (CD70+). A, FACS dot plots show the
change in CD27+ versus CD28+ cells in the CD8+MART-1 tetramer+
population before and after restimulation with the DCs or B cells. B,
Differential dilution of CFSE in the gated CD28+ versus CD282 and
CD27+ versus CD272MART-1 tetramer+ T cell subset 7 d after restim-
ulation with peptide-pulsed B cells. C, CFSE dilution versus CD28 staining
in the TIL line in B was tracked on days 5, 7, and 10 after restimulation,
showing the preferential outgrowth of CD28+ post-REP T cells during
culture. D, CD8+CD28+ and CD8+CD282 T cell subsets were sorted from
four independent HLA-A2.1+ MART-1–reactive patient TIL lines using
FACS followed by restimulation with MART-1 peptide–pulsed DCs and
measurement of the fold-change in CD8+MART-1 tetramer+ T cell num-
bers after 7 d using viable cell counting and FACS staining.
8 CONTINUED CD28 EXPRESSION IS ASSOCIATED WITH TIL PERSISTENCE
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

correlated with the ability of TILs to continue dividing in responseto TCR restimulation was the state of CD28 expression.A key observation was that the few CD28+ TILs remaining after
the REPwere capable of continued proliferation after restimulation,whereas CD27+ and CD272 TILs reactivated with MART-1 pep-tide–pulsed APCs (mature DCs and CD40L-activated B cells) re-sponded similarly regardless ofwhether costimulationwas providedby CD70 on the APC (in our case with CD40L-activated B cells).Thus, sustained cell-surface CD28 expression and not CD27 ex-pression delineates TILs capable of continued expansion and per-sistence. This may explain why so few TIL clonotypes survive longterm in ACT patients and why long-term persistent clonotypesfound in patients with durable remission after ACT are associatedmost consistently with high CD28 expression (7, 12). Interestingly,we also found that, despite being a marker for TILs capable ofpersisting after the REP, CD28 itself did not seem to play a signalingor costimulatory role in driving cell division after restimulationwiththe peptide, despite the high levels of CD80 and CD86 on the APCsused. This was surprising to us considering recent reports demon-strating a role for CD28 costimulation beyond the naive T cell stageduring reactivation of effector memory cells (18). CD28 thereforeseems to be phenotypic marker rather than a functional marker onTILs capable of long-term persistence.
An alternative explanation for our inability to detect a signaling/costimulatory role for CD28 in inducing cell division of post-REPMART-1–specific CD8+CD28+ T cells is that a dominant negativesignaling pathway may have inhibited the effect of any CD28costimulation. FACS staining of post-REP TILs from patients withmelanoma unexpectedly revealed a relatively high level of cell-surface PD-1 expression on CD8+CD28+ post-REP TILs, whereassignificantly less PD-1 expression was found in CD8+CD282
post-REP TILs (Fig. 8C; Y. Li and L. Radvanyi, unpublishedobservations). We are currently addressing the possibility that PD-1 may play a dominant negative costimulatory role, masking anycostimulatory effect of the CD28 pathway during the restimulationof CD8+CD28+ post-REP TILs with MART-1 peptide–pulsedDCs. Increased expression of cell-surface PD-1 on CD8+CD28+
post-REP TILs was supported by our microarray study on sortedpost-REP CD8+CD28+ versus CD8+CD282 T cells that found 2-fold higher PD-1 gene expression in the CD8+CD28+ sorted post-REP TILs from all three patients analyzed.Another issue we addressed is the potential loss of telomere
length during T cell division. CD282 T cells have been found tohave decreased telomere lengths, and this has been associated withlack of proliferative ability and persistence in vivo (34, 35). In ourexperiments, we did observe a significant amount of telomere
FIGURE 8. CD8+CD28+ and CD8+CD282 post-REP TILs exhibit different gene expression profiles, as determined by cDNA microarray analysis. Post-
REP TILs from three patients were sorted for CD8+CD282 and CD8+CD282 T cells and gene expression analyzed using cDNA microarray as described in
Materials and Methods. A, Heat map showing genes differentially expressed (.2-fold) and having statistically significant differences in expression between
CD28+ and CD282 samples (p, 0.01). Onto-Tool software (http://vortex.cs.wayne.edu/projects.htm) was used to determine differences in gene expression
in major cellular signaling or functional pathways. B, The signaling pathways or functions that had statistically significant differences between CD282 and
CD28+ cells (impact factor .20) are shown, together with the number of genes with each difference out of the total number of genes represented in each
pathway. C, Flow cytometry analysis of post-REP TILs for CD28 versus PD-1 staining in gated CD8+MART-1 tetramer+ T cells after the REP and 5 d after
restimulation. The calculated percentage of PD-1+ and PD-12 cells as a fraction of the CD28+ or CD282 subset is depicted in the dot plots.
The Journal of Immunology 9
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
erosion in CD8+ T cells during the REP, which may explain theloss of proliferation capacity. The few remaining CD28+ TILs mayhave maintained sufficient telomere length to promote continuedcell division. We did not separately measure telomere length in thepost-REP CD28+ and CD282 TILs using the flow-FISH assaybecause of the low numbers of CD28+ remaining in the cultures.However, after analyzing multiple TIL lines, the average telomerelength in the CD8+ population following the REP was still 3.7 kb,a length still sufficient to support more cell division (36–38). Inaddition, we have tried a stronger T cell stimulus by activatingpost-REP TILs using plate-bound anti-CD3 Abs and IL-2. Al-though this induced a high amount of activation-induced celldeath, we found that the remaining live CD8+ and CD8+ MART-1tetramer+ TILs were capable of cell division, as measured byCFSE dilution (Y. Li and L. Radvanyi, unpublished observations).Although this is a relatively crude assay and does not mimic the
more subtle stimulation with Ag presented on HLA, it indicates thatat least some of these cells can still be driven into cell division.The better survival and proliferative capacity of the few
remaining MART-1–specific CD8+CD28+ post-REP TILsprompted us to go on and analyze whether any differences in geneexpression other than the markers studied existed between theseCD28+ cells and the majority of CD8+CD282 T cells. The mi-croarray results from three independent post-REP TIL linesshowed a high degree of concordance and revealed significantdifferences between CD8+CD28+ and CD8+CD282 TILs in theexpression of genes in key T cell functional pathways, such as cellcycle regulation, TCR signaling, apoptosis, p53, and KIR. Asmentioned above, PD-1 was one of the genes whose expressionwas increased (confirmed by FACS analysis). Two other genes,CD127/IL-7Ra and CCR7, associated with central memoryT cells, were also significantly overexpressed in CD28+ post-REP
Table I. Key immune regulatory genes differentially and not differentially expressed in sorted post-REPCD8+CD28+ versus CD8+CD282 melanoma TILs
Accession No. Gene Symbol Fold Change p Value
Genes higher on CD8+CD282 TIL NM_014218.1 KIR2DL1 19.04998 ,10e-9NM_014513.1 KIR2DS5 8.501132 ,10e-9NM_014219.1 KIR2DL2 7.503191 ,10e-9NM_1 53443.2 KIR3DL3 6.946899 ,10e-9NM_006737.1 KIR3DL2 6.303651 ,10e-9NM_020535.3 KIR2DL5A 5.997841 ,10e-9NM_013289.1 KIR3DL1 5.716225 0.002635
NM_001080770.1 KIR2DL4 5.549112 ,10e-9NM_181351.1 CD56 4.966265 ,10e-9NM_015868.2 KIR2DL3 4.63644 ,10e-9NM_000647.3 CCR2 3.717889 ,10e-9NM_001295.2 CCR1 2.962169 ,10e-9NM_031409.2 CCR6 2.473945 ,10e-9NM_033423.2 GrH 2.056848 ,10e-9NM_058195.2 p16 2.015411 0.003953NM_000639.1 FasL 1.953941 ,10e-9NM_000579.2 CCR5 1.845691 0.002635NM_177559.2 CKIIA 1.830221 0.009223NM_002104.2 GrK 1.718068 ,10e-9NM_001504.1 CXCR3 1.628737 ,10e-9NM_078487.2 p15 1.614789 0.002635NM_000594.2 TNF 1.562831 ,10e-9NM_001800.3 p19 1.419743 0.003953NM_0050410.3 Perforin 1.392921 ,10e-9NM_001196.2 BID 1.320321 0.001318NM_000073.1 CD3G 1.308558 ,10e-9NM_004131.3 GrB 1.297636 ,10e-9
Genes higher on CD8+CD28+ TIL NM_005191.2 CD80 1.327253 ,10e-9NM_016584.2 IL23A 1.342056 0.003953NM_005214.3 CTLA4 1.639688 ,10e-9NM_005426.2 TP53BP2 1.667006 0.002635NM_005018.1 PD-1 2.041762 0.001318NM_001838.2 CCR7 2.403905 ,10e-9NM_172234.1 IL17RB 2.653037 ,10e-9NM_002185.2 IL7R 3.084726 0.002635NM_006139.1 CD28 41.94281 ,10e-9
Genes not differentially expressed NM_033285.2 TP53INP1 0.810602 ,10e-9NM_000732.3 CD3D 0.828692 0.003953NM_001166.3 cIAP 0.830437 0.001318NM_005317.2 GrM 0.84444 ,10e-9NM_138765.2 BAX 0.859836 ,10e-9NM_000546.2 TP53 0.894448 0.003953NM_005810.3 KLRG1 0.916704 0.00527NM_000733.2 CD3E 0.934767 ,10e-9NM_001242.3 CD27 1.016114 ,10e-9NM_198053.1 CD3Z 1.026876 ,10e-9NM_001320.5 CSNK2B 1.029757 ,10e-9NM_005657.1 TP53BP1 1.276824 ,10e-9
BeadStudio (Illumina) was used to perform microarray analysis. Gene expression was filtered using a p , 0.01 level ofsignificance. A subset of relevant T cell–related genes is shown. Real-time RT-PCR was used to validate changes in CD28,PD-1, IL-7R, and KIR2DL1.
10 CONTINUED CD28 EXPRESSION IS ASSOCIATED WITH TIL PERSISTENCE
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

TILs. We are presently using FACS to determine which fractionsof post-REP CD8+CD28+ TILs express the cell-surface proteinproducts of these two genes as well as other similar markers, suchas CD62L. It could be that a small subset of MART-1 tetramer+
CD8+CD28+ post-REP TILs has central-memory–like character-istics with an even higher proliferation and survival potential,especially when IL-7 is added. This needs to be determined infuture experiments. Two other genes that exhibited increased ex-pression in CD8+CD28+ post-REP TILs were IL-17RB (IL-25R)and IL-23. Both of these genes play a role in the activation and
expansion of Th17 cells (39), but the role of IL-17 cells in CD8+
TILs is unknown at present. It is important to note, however, thatCD8+ IL-17–producing T cells have been found in breast andcolon cancers (40).A striking difference between CD8+CD282 and CD8+CD28+
sorted post-REP TILs in our microarray study was the high degreeof KIR overexpression in the CD282 subset. KIR2DL familymembers 1 to 5 were all considerably overexpressed in CD282
cells, at levels ranging from 6-fold (KIR2DL5A) to 19-fold(KIR2DL1). Interestingly, the activating NK receptor, NKG2D,
FIGURE 9. Effects of the REP done with IL-15 and IL-21 on TIL yield and CD8+ T cell phenotype. TILs from the indicated patient lines were subjected
to the REP using IL-2 (6000 U/ml), IL-15 (100 ng/ml), IL-21 (100 ng/ml), or IL-15 plus IL-21 (both at 100 ng/ml). Media changes with the indicated
cytokines were done as for the REP with IL-2. A, TILs harvested after the REP using IL-2 versus IL-15 alone were analyzed for the expression of CD28 and
CD27 in the CD8+ T cell subset by FACS. The percentage of CD27+ and CD28+ CD8+ T cells isolated from six different TIL REPs are shown with the
average percentage for each parameter shown as a black bar. No statistical differences in CD27 or CD28 expression were found. B, A representative
experiment showing the extent of CD28 expression in CD8+ MART-1 tetramer+ TILs after the REP using IL-2 alone, IL-15 alone, IL-21 alone, or
a combination of IL-15 and IL-21. Data are representative of three experiments with similar results. C, Five TIL lines were subjected to the REP with the
different cytokines indicated. On day 14 of the REP, the percentage of CD28+ cells in the CD8+ subset was determined in comparison with the fold
expansion of the CD8+ T cells under the different conditions. In the case of IL-15 alone, only three TIL lines were tested, with ND denoting “not de-
termined” for the other two lines.
The Journal of Immunology 11
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

did not exhibit higher expression on the CD282 TILs. Other NKmarkers, such as CD56, known to be expressed on highly differ-entiated CD8+ CTLs, were also overexpressed. These resultscorrelate with the increased cytolytic activity noted in post-REPTILs and the reduced proliferative potential. The relatively highexpression of these KIRs in the post-REP CD282 subset furtherunderscores the great drive toward terminal CD8+ T cell differ-entiation taking place during the REP. KIRs have been shown tolimit the cytolytic activity of differentiated CTLs through ligationof self-HLA molecules such as HLA-C and HLA-A (20). Thismay have an effect on the tumor-killing function of infused post-REP TILs in patients with melanoma and may also inhibit T cellactivation during antigenic restimulation. In the future, it will beimportant to track the expression of these and other inhibitory NKreceptors (e.g., CD94/NKG2A) in patients after TIL infusion invivo to determine whether any correlation exists between clinicalresponses and TIL persistence and the quality and quantity ofKIRs. If so, blocking KIRs in vivo may enhance ACT success.A key question emerging from our studies is whether using other
methods of performing the REP can yield post-REP T cells witha “younger” phenotype associated with maintenance of CD28expression and other effector-memory markers that are capable ofbetter persistence in vivo during ACT. In other words, can we havethe best of both worlds by generating high numbers of tumor-reactive cytotoxic T cells while maintaining a memory phenotypefavorable for continued cell division and long-term survival invivo? One possibility is changing the culture technology used inthe REP (using bioreactors or other large-scale systems), but thisis unlikely to affect the resulting phenotype of the TILs after rapidexpansion due to the fact that, ultimately, cell division is the majorfactor driving further T cell differentiation and the phenotypicchanges observed. In fact, a recent study with melanoma TILs
rapidly expanded with anti-CD3 and IL-2 in a new closed con-tinuous perfusion bioreactor system versus the traditional REP,used by us in this study as well, found no significant difference inthe resulting TIL phenotype in terms of the expression of CD28,CD27, and other major phenotypic markers of T cell differentia-tion (41).The other obvious alternative is to test the effects of other
cytokines besides IL-2 in the REP, such as IL-15 or IL-21. IL-2used in traditional TIL expansion protocols has been shown to morerapidly drive effector cell differentiation than IL-15 or IL-21 (21,22). IL-15 may be a good alternative to IL-2, as it has been shownto sustain CD8+ T cells with a younger effector-memory pheno-type and facilitates maintenance of a central-memory (CD62L+
and CCR7+) T cell phenotype (42–45). IL-21 has been shown tohave an opposing role to IL-2 in driving CTL differentiation (22,46) and has been found to prevent the loss of CD28 expressionafter multiple rounds of stimulation of peripheral blood CD8+
T cells with MART-1 peptide (22). We tested the effects of IL-15and IL-21 versus IL-2 in the REP, but in each case found that it didnot improve the situation. IL-15 drove equivalent levels of CD8+ Tcell expansion, and the resulting phenotype of the cells wassimilar to that obtained with IL-2. IL-21 was remarkably good atmaintaining a younger phenotype with high CD28 expression, butwas poor at expanding the TIL, resulting in low T cell yields post-REP that may not be optimal for clinical application where highnumbers (billions) of Ag-specific TILs are needed for infusion. Inaddition, in most cases, TILs isolated from REP cultures grownwith IL-21 did not exhibit an improved response to restimulationwith Ag (MART-1) despite the higher fraction of CD28+ CD8+
TILs recovered. A possible explanation for this is that in additionto the increased levels of CD28 and a “younger” phenotype, CD8+
TILs from IL-21 REP cultures also had significantly higher PD-1
FIGURE 10. A combination of IL-15 and IL-21 in
the REP improves the responses of post-REP CD8+
TILs to restimulation with MART-1. TILs from HLA-
A2.1+ patients having a significant fraction of MART-
1–reactive T cells were subjected to the REP under the
cytokine conditions indicated. The isolated post-REP
TILs were restimulated for 1 wk with HLA-A2.1-
matched DCs pulsed with MART-1 peptide, as de-
scribed before. A, The fold expansion of CD8+ MART-
1 tetramer+ T cells is shown for three different TIL
lines. B, The levels of PD-1 expression in CD8+ TILs
isolated after rapid expansion under the cytokine con-
ditions indicated were for analyzed by FACS. A rep-
resentative experiment from one TIL line is shown.
12 CONTINUED CD28 EXPRESSION IS ASSOCIATED WITH TIL PERSISTENCE
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

expression that may have inhibited the activation and further ex-pansion of the cells (the DCs used as APCs in these experimentsall expressed B7H1 and B7DC). Although IL-15 or IL-21 alonedid not seem to be viable alternatives to IL-2 in the REP, sur-prisingly we found that in combination they supported botha significant preservation of CD28+ expression on the post-REPTILs together with high levels of TIL expansion. Thus, IL-15 andIL-21 may have synergistic effects acting through different re-ceptors, with IL-15 strongly driving cell division and having someeffects at inhibiting further differentiation and CD28 loss and IL-21 more strongly inhibiting differentiation and preserving CD28expression while itself being a poor inducer of cell division. It willbe interesting to determine how IL-15 and IL-21 signaling is in-tegrated in the CD8+ TILs and whether any unique gene expres-sion signatures exist in TILs expanded with both of thesecytokines together. Although we did not test the effects of IL-2and IL-21 together in the REP, we predict that this combinationwill have similar effects because both IL-2 and IL-15 signalthrough the IL-2Rb chain. However, this needs to be formallyaddressed.In conclusion, we have found a profound loss of melanoma Ag–
reactive proliferative and survival potential in TILs following theclassic REP protocol used for years for melanoma ACT. Loss ofCD28 expression in post-REP TILs was the most consistent ef-fector-memory marker loss during the REP, whereas CD27 levelsdid not change significantly. This observation had functional rel-evance in that the few remaining post-REP CD28+ Ag-specificCD8+ TILs were the only cells capable of continued cell divisionfollowing restimulation with APCs. Thus, our data offer an ex-planation for why CD28+ T cells dominate the repopulating T cellpool in lymphodepleted patients during ACT and why CD28+
T cells dominate the long-term persistent T cell population inpatients with more durable responses. Our microarray data onCD28+ and CD282 post-REP TILs also revealed potential targetsfor further therapeutic intervention (e.g., PD-1 blockade and KIRblockade) in vivo after TIL infusion, or during the REP, that mayfurther enhance the success of current ACT protocols. Finally, ourdata point out some of the limitations with current ACT protocolsusing TILs and the need to develop methods propagating TILs thatmaintain a “younger” phenotype associated with higher survivaland expansion capabilities in vivo after infusion into lymphode-pleted patients. This may be achieved by using pre-REP instead ofpost-REP TILs as the ACT infusion product. In addition, our re-sults suggest that IL-15 in synergy with IL-21 may be superior toIL-2 in driving TIL expansion during the classical REP used togenerate ACT products and may maintain a more Ag-responsive Tcell phenotype associated with the preservation of CD28 expres-sion. More detailed examination of the changes in gene expressionprofiles during the REP is another approach that can identify keygenes that can be modulated to enhance post-REP TIL survivaland improve Ag-induced cell division.
AcknowledgmentsWe thank Natalia Martin-Orozco for help in preparation of the figures and
tables. We thank Priscilla Miller for help in obtaining tissue samples and
Victor Prieto, Hafeez Diwan, and Alex Lazar of the Melanoma Tissue Bank
at M. D. Anderson Cancer Center. The following surgeons at M. D. Ander-
son Cancer Center generously helped in obtaining tissues for our ACT clin-
ical trial and for in vitro studies: Jeffrey Lee, Jeffrey Gershenwald, Merrick
Ross, Janice Cormier, Anthony Lucci, and Paul Mansfield. The following
technicians from M. D. Anderson Cancer Center helped process tumor ma-
terial to obtain TILs for in vitro studies: Kathryn Bushnell, Rahmatu Man-
saray, Orenthial Fulbright, Marissa Gonzalez, Chris Toth, and Renjith
Ramachandran.
DisclosuresThe authors have no financial conflicts of interest.
References1. June, C. H. 2007. Adoptive T cell therapy for cancer in the clinic. J. Clin. Invest.
117: 1466–1476.2. June, C. H. 2007. Principles of adoptive T cell cancer therapy. J. Clin. Invest.
117: 1204–1212.3. Rosenberg, S. A., N. P. Restifo, J. C. Yang, R. A. Morgan, and M. E. Dudley.
2008. Adoptive cell transfer: a clinical path to effective cancer immunotherapy.Nat. Rev. Cancer 8: 299–308.
4. Dudley, M. E., and S. A. Rosenberg. 2003. Adoptive-cell-transfer therapy for thetreatment of patients with cancer. Nat. Rev. Cancer 3: 666–675.
5. Rosenberg, S.A. 2005. Cancer immunotherapy comes of age. Nat. Clin. Pract.Oncol. 2: 115.
6. Rosenberg, S. A., J. C. Yang, and N. P. Restifo. 2004. Cancer immunotherapy:moving beyond current vaccines. Nat. Med. 10: 909–915.
7. Robbins, P. F., M. E. Dudley, J. Wunderlich, M. El-Gamil, Y. F. Li, J. Zhou,J. Huang, D. J. Powell Jr., and S. A. Rosenberg. 2004. Cutting edge: persistenceof transferred lymphocyte clonotypes correlates with cancer regression in pa-tients receiving cell transfer therapy. J. Immunol. 173: 7125–7130.
8. Zhou, J., X. Shen, J. Huang, R. J. Hodes, S. A. Rosenberg, and P. F. Robbins.2005. Telomere length of transferred lymphocytes correlates with in vivo per-sistence and tumor regression in melanoma patients receiving cell transfertherapy. J. Immunol. 175: 7046–7052.
9. Benlalam, H., V. Vignard, A. Khammari, A. Bonnin, Y. Godet, M. C. Pandolfino,F. Jotereau, B. Dreno, and N. Labarriere. 2007. Infusion of Melan-A/Mart-1specific tumor-infiltrating lymphocytes enhanced relapse-free survival of mela-noma patients. Cancer Immunol. Immunother. 56: 515–526.
10. Derre, L., M. Ferber, C. Touvrey, E. Devevre, V. Zoete, A. Leimgruber,P. Romero, O. Michielin, F. Levy, and D. E. Speiser. 2007. A novel population ofhuman melanoma-specific CD8 T cells recognizes Melan-AMART-1 im-munodominant nonapeptide but not the corresponding decapeptide. J. Immunol.179: 7635–7645.
11. Morgan, R. A., M. E. Dudley, J. R. Wunderlich, M. S. Hughes, J. C. Yang,R. M. Sherry, R. E. Royal, S. L. Topalian, U. S. Kammula, N. P. Restifo, et al.2006. Cancer regression in patients after transfer of genetically engineeredlymphocytes. Science 314: 126–129.
12. Powell, D. J., Jr., M. E. Dudley, P. F. Robbins, and S. A. Rosenberg. 2005.Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effectormemory T cells in humans after adoptive cell transfer therapy. Blood 105: 241–250.
13. Riddell, S. R., and P. D. Greenberg. 1990. The use of anti-CD3 and anti-CD28monoclonal antibodies to clone and expand human antigen-specific T cells.J. Immunol. Methods 128: 189–201.
14. Berger, T. G., B. Feuerstein, E. Strasser, U. Hirsch, D. Schreiner, G. Schuler, andB. Schuler-Thurner. 2002. Large-scale generation of mature monocyte-deriveddendritic cells for clinical application in cell factories. J. Immunol. Methods 268:131–140.
15. He, L., J. Hakimi, D. Salha, I. Miron, P. Dunn, and L. Radvanyi. 2005. A sen-sitive flow cytometry-based cytotoxic T-lymphocyte assay through detection ofcleaved caspase 3 in target cells. J. Immunol. Methods 304: 43–59.
16. Hamann, D., P. A. Baars, M. H. Rep, B. Hooibrink, S. R. Kerkhof-Garde,M. R. Klein, and R. A. van Lier. 1997. Phenotypic and functional separation ofmemory and effector human CD8+ T cells. J. Exp. Med. 186: 1407–1418.
17. Sallusto, F., J. Geginat, and A. Lanzavecchia. 2004. Central memory and effectormemory T cell subsets: function, generation, and maintenance. Annu. Rev. Im-munol. 22: 745–763.
18. Borowski, A. B., A. C. Boesteanu, Y. M. Mueller, C. Carafides, D. J. Topham,J. D. Altman, S. R. Jennings, and P. D. Katsikis. 2007. Memory CD8+ T cellsrequire CD28 costimulation. J. Immunol. 179: 6494–6503.
19. Fang, M., and L. J. Sigal. 2006. Direct CD28 costimulation is required for CD8+T cell-mediated resistance to an acute viral disease in a natural host. J. Immunol.177: 8027–8036.
20. Vivier, E., and N. Anfossi. 2004. Inhibitory NK-cell receptors on T cells: witnessof the past, actors of the future. Nat. Rev. Immunol. 4: 190–198.
21. Rochman, Y., R. Spolski, and W. J. Leonard. 2009. New insights into the reg-ulation of T cells by gamma(c) family cytokines. Nat. Rev. Immunol. 9: 480–490.
22. Li, Y., M. Bleakley, and C. Yee. 2005. IL-21 influences the frequency, pheno-type, and affinity of the antigen-specific CD8 T cell response. J. Immunol. 175:2261–2269.
23. Michaeli, Y., G. Denkberg, K. Sinik, L. Lantzy, C. Chih-Sheng, C. Beauverd,T. Ziv, P. Romero, and Y. Reiter. 2009. Expression hierarchy of T cell epitopesfrom melanoma differentiation antigens: unexpected high level presentation oftyrosinase-HLA-A2 Complexes revealed by peptide-specific, MHC-restricted,TCR-like antibodies. J. Immunol. 182: 6328–6341.
24. Jager, E., M. Maeurer, H. Hohn, J. Karbach, D. Jager, Z. Zidianakis,A. Bakhshandeh-Bath, J. Orth, C. Neukirch, A. Necker, et al. 2000. Clonal ex-pansion of Melan A-specific cytotoxic T lymphocytes in a melanoma patientresponding to continued immunization with melanoma-associated peptides. Int.J. Cancer 86: 538–547.
25. Speiser, D. E., P. Baumgaertner, C. Barbey, V. Rubio-Godoy, A. Moulin,P. Corthesy, E. Devevre, P. Y. Dietrich, D. Rimoldi, D. Lienard, et al. 2006. Anovel approach to characterize clonality and differentiation of human melanoma-
The Journal of Immunology 13
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

specific T cell responses: spontaneous priming and efficient boosting by vacci-nation. J. Immunol. 177: 1338–1348.
26. Sacre, K., G. Carcelain, N. Cassoux, A. M. Fillet, D. Costagliola, D. Vittecoq,D. Salmon, Z. Amoura, C. Katlama, and B. Autran. 2005. Repertoire, diversity,and differentiation of specific CD8 T cells are associated with immune pro-tection against human cytomegalovirus disease. J. Exp. Med. 201: 1999–2010.
27. Takata, H., and M. Takiguchi. 2006. Three memory subsets of human CD8+T cells differently expressing three cytolytic effector molecules. J. Immunol.177: 4330–4340.
28. Kaech, S. M., and E. J. Wherry. 2007. Heterogeneity and cell-fate decisions ineffector and memory CD8+ T cell differentiation during viral infection. Immu-nity 27: 393–405.
29. Appay, V., P. R. Dunbar, M. Callan, P. Klenerman, G. M. Gillespie, L. Papagno,G. S. Ogg, A. King, F. Lechner, C. A. Spina, et al. 2002. Memory CD8+ T cellsvary in differentiation phenotype in different persistent virus infections. Nat.Med. 8: 379–385.
30. Appay, V., and S. L. Rowland-Jones. 2004. Lessons from the study of T-celldifferentiation in persistent human virus infection. Semin. Immunol. 16: 205–212.
31. Tran, K. Q., J. Zhou, K. H. Durflinger, M. M. Langhan, T. E. Shelton,J. R. Wunderlich, P. F. Robbins, S. A. Rosenberg, and M. E. Dudley. 2008.Minimally cultured tumor-infiltrating lymphocytes display optimal character-istics for adoptive cell therapy. J. Immunother. 31: 742–751.
32. Huang, J., H. T. Khong, M. E. Dudley, M. El-Gamil, Y. F. Li, S. A. Rosenberg,and P. F. Robbins. 2005. Survival, persistence, and progressive differentiation ofadoptively transferred tumor-reactive T cells associated with tumor regression. J.Immunother. 28: 258–267.
33. Huang, J., K. W. Kerstann, M. Ahmadzadeh, Y. F. Li, M. El-Gamil,S. A. Rosenberg, and P. F. Robbins. 2006. Modulation by IL-2 of CD70 andCD27 expression on CD8+ T cells: importance for the therapeutic effectivenessof cell transfer immunotherapy. J. Immunol. 176: 7726–7735.
34. Monteiro, J., F. Batliwalla, H. Ostrer, and P. K. Gregersen. 1996. Shortenedtelomeres in clonally expanded CD28-CD8+ T cells imply a replicative historythat is distinct from their CD28+CD8+ counterparts. J. Immunol. 156: 3587–3590.
35. Palmer, L. D., N. Weng, B. L. Levine, C. H. June, H. C. Lane, and R. J. Hodes.1997. Telomere length, telomerase activity, and replicative potential in HIVinfection: analysis of CD4+ and CD8+ T cells from HIV-discordant monozygotictwins. J. Exp. Med. 185: 1381–1386.
36. Ahmed, A., and T. Tollefsbol. 2001. Telomeres and telomerase: basic scienceimplications for aging. J. Am. Geriatr. Soc. 49: 1105–1109.
37. Brummendorf, T. H., and S. Balabanov. 2006. Telomere length dynamics innormal hematopoiesis and in disease states characterized by increased stem cellturnover. Leukemia 20: 1706–1716.
38. Roth, A., G. M. Baerlocher, M. Schertzer, E. Chavez, U. Duhrsen, andP. M. Lansdorp. 2005. Telomere loss, senescence, and genetic instability in CD4+T lymphocytes overexpressing hTERT. Blood 106: 43–50.
39. Dong, C. 2008. Regulation and pro-inflammatory function of interleukin-17family cytokines. Immunol. Rev. 226: 80–86.
40. Nam, J. S., M. Terabe, M. J. Kang, H. Chae, N. Voong, Y. A. Yang, A. Laurence,A. Michalowska, M. Mamura, S. Lonning, et al. 2008. Transforming growthfactor b subverts the immune system into directly promoting tumor growththrough interleukin-17. Cancer Res. 68: 3915–3923.
41. Klapper, J. A., A. A. Thomasian, D. M. Smith, G. C. Gorgas, J. R. Wunderlich,F. O. Smith, B. S. Hampson, S. A. Rosenberg, and M. E. Dudley. 2009. Single-pass, closed-system rapid expansion of lymphocyte cultures for adoptive celltherapy. J. Immunol. Methods 345: 90–99.
42. Bodnar, A., E. Nizsaloczki, G. Mocsar, N. Szaloki, T. A. Waldmann,S. Damjanovich, and G. Vamosi. 2008. A biophysical approach to IL-2 and IL-15 receptor function: localization, conformation and interactions. Immunol. Lett.116: 117–125.
43. Daudt, L., R. Maccario, F. Locatelli, I. Turin, L. Silla, E. Montini, E. Percivalle,R. Giugliani, M. A. Avanzini, A. Moretta, and D. Montagna. 2008. Interleukin-15 favors the expansion of central memory CD8+ T cells in ex vivo generated,antileukemia human cytotoxic T lymphocyte lines. J. Immunother. 31: 385–393.
44. Klebanoff, C. A., L. Gattinoni, P. Torabi-Parizi, K. Kerstann, A. R. Cardones,S. E. Finkelstein, D. C. Palmer, P. A. Antony, S. T. Hwang, S. A. Rosenberg, et al.2005. Central memory self/tumor-reactive CD8+ T cells confer superior antitumorimmunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 102:9571–9576.
45. Mueller, K., O. Schweier, and H. Pircher. 2008. Efficacy of IL-2- versus IL-15-stimulated CD8 T cells in adoptive immunotherapy. Eur. J. Immunol. 38: 2874–2885.
46. Hinrichs, C. S., R. Spolski, C. M. Paulos, L. Gattinoni, K. W. Kerstann,D. C. Palmer, C. A. Klebanoff, S. A. Rosenberg, W. J. Leonard, andN. P. Restifo. 2008. IL-2 and IL-21 confer opposing differentiation programs toCD8+ T cells for adoptive immunotherapy. Blood 111: 5326–5333.
14 CONTINUED CD28 EXPRESSION IS ASSOCIATED WITH TIL PERSISTENCE
by guest on January 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from