manual gaussian
TRANSCRIPT

Gaussian Overview 09 Input09 entrada de Gauss consiste em uma série de linhas em um arquivo de texto ASCII. A estrutura de base de um ficheiro de entrada de Gauss inclui várias secções diferentes:
• Ligação 0 Comandos : Localize e arquivos nome scratch (linha em branco não terminado).
• Seção Rota ( # linhas ): Especifique o tipo de cálculo desejado, modelo química e outras opções (linha em branco terminado).
• Seção Título : Breve descrição do cálculo (linha em branco terminado). Esta secção é necessária na entrada, mas não é de forma alguma interpretados pelo programa Gaussian 09. Ele aparece na saída para fins de identificação e descrição.Normalmente, esta seção pode conter o nome composto, sua simetria, o estado eletrônico, e qualquer outra informação relevante. A seção de título não pode exceder cinco linhas e deve ser seguido por uma linha em branco terminar. Os seguintes caracteres devem ser evitados na seção de título: @ #! - _ \ caracteres de controle (especialmente Ctrl-G)
• Especificação Molecule : Especifique sistema molecular a ser estudado (linha em branco terminado).
• Opcional seções adicionais : entrada adicional necessário para tipos de trabalho específicos (normalmente linha em branco terminado).
Muitos Gaussian 09 postos de trabalho vai incluir apenas a segunda, terceira e quarta seções. Aqui está um exemplo de um arquivo, que solicita um cálculo da energia de ponto único na água:
# HF/6-31G (d) seção Rota
energia da água seção Título
0 1 especificação MoleculeO -0,464 0,177 0.0H -0.464 1.137 0,0H 0,441 -0,143 0,0
Neste trabalho, os troços e título cada um composto de uma única linha. A seção de especificação molécula começa com uma linha dando a carga e rotação multiplicidade para a molécula: 0 carga (molécula neutra) e rotação multiplicidade 1 (singlete) neste caso. A linha de carga e a multiplicidade de centrifugação é seguida por linhas descrevendo a localização de cada um dos átomos da molécula; este exemplo utiliza coordenadas cartesianas para fazê-lo. Especificações molécula são discutidos em mais detalhes mais adiante neste capítulo.
O seguinte arquivo de entrada ilustra o uso de ligação 0 comandos e uma seção de entrada adicional:
Chk% = pesado Ligação 0 seção # HF/6-31G (d) Opt = ModRedundant seção Rota
Trabalho Opte seção Título
0 1 seção Especificação Molecule coordenadas atômicas ...

3 8 Adicionar uma ligação e um ângulo para os internos 2 1 3 coordenadas utilizadas durante o geom. opt.
Esse trabalho requer uma otimização de geometria. A secção de entrada seguinte especificação molécula é usado pelo Opt = ModRedundant palavra-chave, e que serve para adicionar uma ligação e ângulo adicional nas coordenadas internas utilizadas na optimização da geometria. O trabalho também especifica um nome para o arquivo de verificação.
Por conveniência, a seguinte tabela de todos os setores possíveis que podem aparecer dentro de um arquivo de entrada Gaussian 09, juntamente com as palavras-chave associadas a cada um.
Gaussian 09 Input Seção de encomendaSeção Palavras-chave Final de
linha embranco?
Ligação 0 comandos % comandos não
Route Section ( # linhas) tudo sim
Sobreposições extra ExtraOverlays sim
Seção Título todos, exceto Geom = AllCheck sim
Especificação Molecule todos, exceto Geom = AllCheck sim
Especificações de conectividade
Geom = Ligar ou ModConnect sim
Alterações átomos congelados
Geom = ReadFreeze sim
Modificações coordenadas
Opt = ModRedundant sim
2 º título e especificação molécula
Opt = QST2 ou QST3 sim paraambos
Especificações de conectividade. para o 2 º conjunto de coordenadas
Geom = Ligar ou ModConnect e Opt = QST2 ou QST3
sim
2 Alterações átomos congelados
Geom = ReadFreeze sim
Modificações segundo conjunto de coordenadas
Opt = QST2 ou QST3 sim
3 º título e estrutura TS inicial
Opt = QST3 sim paraambos
Especificações de conectividade. para o 3 º conjunto de coordenadas
Geom = Ligar ou ModConnect Opt = (ModRedun, QST3)
sim
Alterações a 3 átomos congelados
Geom = ReadFreeze sim
Modificações terceiro conjunto de coordenadas
Opt = (ModRedun, QST3) sim

PDB informações estrutura secundária
automática se informações resíduo na especificação molécula
sim
Massas atômicas ReadIsotopes opção sim
Parâmetros de mecânica molecular
HardFirst, SoftFirst, SoftOnly, Modificar sim
Frequência de interesse CPHF = RdFreq sim
Distribuição de carga de fundo
Cobrar sim
Entrada BOMD / ADMP (1 ou mais seções)
ADMP e BOMD exigido entrada e ReadVelocity ,ReadMWVelocity opções
sim
Entrada PCM SCRF = (ExternalIteration, Ler) sim
Coordenadas para mesa de IRC
IRC = Relatório sim
Restrições harmônicas Geom = ReadHarmonic sim
Parâmetros semi-empíricos (formato Gaussian)
Entrada de opção, AM1 = Ambos sim
Parâmetros semi-empíricos (formato MOPAC)
MOPAC , Ambas as opções sim
Especificação conjunto de base
Gen, GenECP, ExtraBasis sim
Alterações conjunto de base
Massagem sim
Coeficientes de campo finito
Campo = Leitura sim
Especificação ECP Pseudo = Cartões, GenECP sim
Densidade especificação conjunto de base montagem
ExtraDensityBasis sim
Entrada do modelo PCM solvatação
SCRF = Leitura sim
Parâmetros DFTB DFTB sim
Fonte para estimativa inicial
Adivinha = Entrada sim
Tipos de simetria para combinar
Adivinha = LowSymm não
Especificações Orbital (α e β em separado)
Adivinha = Cartões sim
Alterações orbitais (α e β em separado)
Adivinha = Alter sim
Reordenação Orbital (α e β em separado)
Adivinha = Permuta sim

# Orbitais / par GVB GVB não
Pesos para CAS média estadual
CASSCF = StateAverage não
Unidos de interesse para acoplamento de spin órbita
CASSCF = SpinOrbit não
Informações congelamento Orbital
ReadWindow opções sim
Orbitais EPT para refinar EPT = ReadOrbitals sim
Lista Átomos para constantes de acoplamento spin-spin
NMR = ReadAtoms sim
Raios atômicos alternativos
Pop = ReadRadii ou ReadAtRadii sim
Os dados para as propriedades electrostáticas
Prop = Ler ou Opt sim
Entrada NBO Pop = NBORead não
Seleção Modo Normal Harmonic
Freq = SelectNormalModes sim
Rotor entrada impedida Freq = ReadHindered sim
Seleção Modo Normal anarmônicas
Freq = SelectAnharmonicNormalModes sim
Modos normais para FCHT
Freq = SelectFCHTNormalModes sim
Entrada para anarmônicas Freq = ReadAnharmonic sim
Entrada para FCHT Freq = ReadFCHT sim
Pickett arquivo de saída Output = Pickett não
PROAIMS arquivo de saída
Output = WFN não
Sintaxe de entradaEm geral, a entrada de Gauss está sujeita às seguintes regras de sintaxe:
• A entrada é livre de formato e case-insensitive.
• Espaços, tabulações, vírgulas ou barras podem ser usados em qualquer combinação de itens separados dentro de uma linha.Vários espaços são tratados como um único delimitador.
• Opções para as palavras-chave podem ser especificados em qualquer das seguintes formas:
o keyword = opção
o palavra-chave ( opção )
o keyword = ( opção 1, opção 2, ...)

o palavra-chave ( opção 1, opção 2, ...)
• Várias opções estão entre parênteses e separados por qualquer delimitador válido(vírgulas são convencionais e são mostrados acima). O sinal de igual antes do parêntese de abertura pode ser omitido, ou espaços podem, opcionalmente, ser incluído antes e / ou depois dela. Note que algumas opções também assumir valores, neste caso, o nome da opção é seguido por um sinal de igual: por exemplo, CBSExtrap (nmin = 6) .
• Todas as palavras-chave e opções pode ser reduzido à sua menor abreviatura única em todo o sistema Gaussian 09. Assim, oconvencional opção para o SCF palavra-chave pode ser abreviado para convenção , mas não para conv (devido à presença da convergência opção). Isto é verdade ou não, tanto convencional e Convergência acontecerá a ser opções válidas para qualquer palavra-chave.
• O conteúdo de um arquivo externo pode ser incluída dentro de um arquivo de entrada Gaussian 09 usando a seguinte sintaxe: @nome do arquivo . Isso faz com que todo o arquivo seja colocado na posição atual no fluxo de entrada. Anexando / N para tais comandos irá impedir o conteúdo do arquivo incluído de ser ecoado no início do arquivo de saída.
• Os comentários começam com um ponto de exclamação ( ! ), que pode aparecer em qualquer lugar em uma linha. Linhas de comentário separados podem aparecer em qualquer lugar dentro do arquivo de entrada.
Gauss Tipos 09 empregoA parte do percurso de um arquivo de entrada Gaussian 09 especifica o tipo de cálculo aser realizado. Existem três componentes principais para essa especificação:
• O tipo de trabalho
• O método
• O conjunto de base
A tabela a seguir lista os tipos de trabalho disponíveis no Gaussian 09:
• SP : a energia de ponto único.
• Opte : Otimização de Geometria.
• Freq : Freqüência e análise termoquímica.
• IRC : caminho de reação seguinte
• IRCMax : Encontre o máximo de energia ao longo de um caminho de reação específica.
• Digitalizar : Potencial exame de superfície de energia.
• Polar : polarizabilidades e hiperpolarizabilidades.
• ADMP e BOMD : Dinâmica diretas cálculo trajetória.
• Força : forças de computação sobre os núcleos.
• Estável : a estabilidade da função de onda de teste.

• Volume : o volume molecular Compute.
• Densidade = Guess Checkpoint = Somente : Recompute somente a análise da população.
• Adivinha = Somente : Imprimir apenas suposição inicial; gerar estimativa inicial baseada em fragmentos.
Em geral, deve ser especificado apenas um tipo de trabalho de palavras-chave. As exceções a essa regra são:
• Polar e Opt pode ser combinada com Freq . Neste último caso, a optimização da geometria é automaticamente seguido por um cálculo de frequência na estrutura optimizada.
• Opt podem ser combinados com os compostos método palavras-chave, a fim de especificar as opções para a parte de otimização do cálculo: por exemplo, Opt = (TS, ReadFC) CBS-QB3 .
Quando nenhum tipo de trabalho-chave é especificado na seção de rota, o tipo de cálculo padrão é geralmente um único cálculo de ponto de energia ( SP ). No entanto, uma parte do percurso da forma: método 2 / base 2 / / método 1 / base 1 pode ser usado para solicitar um cálculo de otimização (pelo método 1 / base 1) seguido por um cálculode ponto único da energia (pelo método 2 / base2) na geometria otimizada. Por exemplo, a parte do percurso a seguir solicita uma B3LYP/6-31G (d) otimização de geometria seguido por um cálculo da energia de ponto único usando o CCSD/6-31G (d) modelo de química:# CCSD/6-31G (d) / / B3LYP/6-31G (d) Teste
Neste caso, o Opt palavra-chave é opcional e é o padrão. Note-se que Opt Freq cálculos podem não usar essa sintaxe.
Prevendo Propriedades Moleculares
A tabela a seguir fornece um mapeamento entre as quantidades previstas comumente desejados e os Gaussian 09 palavras-chave que irá produzi-los:
• Acoplamento antiferromagnético: Adivinha = Fragmento , Estabilidade
• Cargas atômicas: Pop
• ÄG de hidratação: SCRF = SMD
• Momento de dipolo: Pop
• Afinidades eletrônicas: CBS-QB3 , CCSD , EPT
• Densidade de elétrons: cubegen
• Dicroísmo circular eletrônico: CIS , TD , MOE , SAC-CI
• Potencial eletrostática: cubegen , Prop
• Derivados de potenciais cargas eletrostáticas: Pop = Chelp , ChelpG ou MK
• Forma eletrônica banda de transição: Freq = FC , Freq = HT
• Polarizabilidades / hiperpolarizabilidades: Freq , Polar [ CPHF = RdFreq ], Polar = DCSHG
• Energias de alta precisão: CBS-QB3 , G2 , G3 , G4 , W1U , W1BD
• Constantes de acoplamento Hyperfine (anisotrópicos): Prop

• Tensores espectros Hyperfine (incluindo g tensores): Freq = (VCD, VibRot [ , anarmônicas ] )
• Potenciais de ionização: CBS-QB3 , CCSD , EPT
• Espectros IV e Raman: Freq [ = anarmônicas ]
• Os espectros de Raman pré-ressonância: Freq CPHF = RdFreq
• Orbitais moleculares: Pop = regular
• Momentos multipolares: Pop
• RMN de blindagem e químicas turnos: RMN
• RMN spin-spin constantes de acoplamento: NMR = Mista
• Rotações ópticas: Polar = OptRot
• Atividade óptica Raman: Freq = ROA , CPHF = RdFreq
• Análise termoquímica: Freq
• UV / espectros visível: CIS , ZINDO , TD , MOE , SAC-CI
• Acoplamento vibração-rotação: Freq = VibRot
• Dicroísmo circular Vibracional: Freq = VCD
Químicos modeloA combinação do método ea base set especifica um modelo de química para Gaussian, especificando o nível de teoria. Todo trabalho de Gauss deve especificar um método e um conjunto de base. Isso geralmente é realizado através de duas palavras-chave separadas dentro do troço do arquivo de entrada, apesar de algumas palavras-chave do método implica uma escolha de conjunto de base.Alguns trabalhos utilizando um método funcional da densidade pode também incluir um conjunto adequado de densidade (ver a seção de conjuntos de base para mais informações).
A tabela seguinte lista os métodos que estão disponíveis em Gauss, juntamente com os tipos de trabalho para as quais cada um pode ser utilizado. Um asterisco indica cálculos analíticos, enquanto numérica somente cálculos são indicados por n (veja a discussão sobre a palavra-chave específica em questão para detalhes).

Lista de métodos e seus tipos de emprego disponíveis
Se nenhuma palavra-chave método for especificado, HF é assumido. A maioria das palavras-chave do método pode ser prefaciado por R for-shell fechado wavefunctions restritos, U para irrestritos wavefunctions-shell aberto, ou RO para wavefunctions-shell aberto limitado: por exemplo, ROHF , UMP2 ou RQCISD . RO está disponível apenaspara Hartree-Fock energias e densidade métodos funcionais e AM1, PM3, PM3MM, PM6 e PDDG energias e gradientes, e MP2, MP3, MP4 e CCSD.
Em geral, deve ser especificado apenas um único método de palavras-chave , e incluindo mais de um deles irá produzir bizarrasresultados. No entanto, há exceções:
• CASSCF podem ser especificados junto com MP2 para solicitar um cálculo CASSCF incluindo correlação eletrônica dinâmica.
• ONIOM e IRCMax empregos exigem várias especificações do método. No entanto, eles são dadas como opções para a palavra-chave correspondente.
• A forma do modelo 2 / / modelo 1 anteriormente descrito pode ser utilizado para gerar uma optimização automática seguido por um cálculo de ponto único para ageometria optimizada.
Conjuntos de baseA maioria dos métodos requerem um conjunto de base ser especificada, se nenhuma palavra-chave conjunto de base está incluído na parte do percurso, então a base STO-3G

será usado. As excepções consistem de alguns métodos pelos quais o conjunto de base édefinida como uma parte integral do método, são listados a seguir:
• Todos os métodos semi-empíricos, incluindo ZINDO para estados excitados.
• Todos os métodos de mecânica molecular.
• Modelo composto químicos: tudo G n , métodos CBS e W1.
Os seguintes conjuntos de base são armazenadas internamente no programa Gaussian 09(ver referências citadas para obter descrições completas), listados abaixo pelo seu correspondente Gaussian 09 palavra-chave (com duas exceções):
• STO-3G [ Hehre69 , Collins76 ]
• 3-21G [ Binkley80a , Gordon82 , Pietro82 , Dobbs86 , Dobbs87 , Dobbs87a ]
• 6-21G [ Binkley80a , Gordon82 ]
• 4-31G [ Ditchfield71 , Hehre72 , Hariharan74 , Gordon80 ]
• 6-31G [Ditchfield71, Hehre72, Hariharan73, Hariharan74, Gordon80, Francl82, Binning90, Blaudeau97, Rassolov98, Rassolov01]
• 6-31G †: Gaussian 09 também inclui a 6-31G † e 6-31G ‡ conjuntos de bases deGeorge Petersson e colegas de trabalho, definido como parte dos métodos completos conjunto de base [ Petersson88 , Petersson91 ] . Estes são acedidos através da 6-31G (d ') e 6-31G (d ', p') palavra-chave, a qual também pode ser adicionada funções individuais ou duplas difusas; pode também ser adicionado funções f: por exemplo, 6-31G ( d'f ), e assim por diante.
• 6-311G : Especifica a base 6-311G para átomos de primeira linha ea McLean-Chandler (12s, 9p) conjuntos → (621111,52111) Base de átomos de segunda linha [ McLean80 , Raghavachari80b ] (note que os conjuntos de base para P, S e Cl são os chamados conjuntos de base de íons negativos por McLean e Chandler, estes foram considerados para dar melhores resultados para moléculas neutras também), o conjunto de base Blaudeau e colegas de trabalho para o Ca e K [ Blaudeau97 ] , os Wachters- Hay [ Wachters70 , Hay77 ] tudo base elétron definido para a primeira linha de transição, utilizando os fatores de escala de Raghavachari e Caminhões [ Raghavachari89 ] eo conjunto de base 6-311G de McGrath, Curtiss e colegas de trabalho para os outros elementos da terceira linha [ Binning90 , McGrath91 , Curtiss95 ] . Note-se que Raghavachari e Caminhões recomendar dimensionamento e incluindo funções difusas quando se utiliza o conjunto de base Wachters-Hay para elementos de linha de primeira transição, o 6-311 + G . formulário deve ser especificado para incluir as funções difusas MC-311G é um sinônimo para 6 -311G .
• D95V : Dunning / Huzinaga valência double-zeta [ Dunning76 ] .
• D95 : Dunning / Huzinaga completo zeta duplo [ Dunning76 ] .
• SHC : D95V em primeira linha, Goddard / Smedley ECP na segunda linha [ Dunning76 , Rappe81 ] . Também conhecida como SEC.
• CEP-4G : Stevens / Basch / Krauss ECP base mínima [ Stevens84 , Stevens92 , Cundari93 ] .
• CEP-31G : Stevens / Basch / Krauss ECP divisão saia [ Stevens84 , Stevens92 , Cundari93 ] .

• CEP-121G : Stevens / Basch / Krauss ECP base triplo-split [ Stevens84 , Stevens92 , Cundari93 ] .
Note-se que há apenas um conjunto de base CEP definido para além da segunda fila, e todas as três palavras-chave são equivalentes para esses átomos.
• LanL2MB : STO-3G [ Hehre69 , Collins76 ] em primeira linha, Los Alamos ECP mais MBS em Na-La, Hf-Bi [ Hay85 , Wadt85 , Hay85a] .
• LANL2DZ : D95V na primeira linha [ Dunning76 ] , Los Alamos ECP mais DZ no Na-La, Hf-Bi [ Hay85 , Wadt85 , Hay85a ] .
• SDD : D95 até AR [ Dunning76 ] e PAE Stuttgart / Dresden no restante do periódico O SDD , SHF , SDF , MHF , MDF ,MWB formas podem ser usadas para especificar estes conjuntos de base / potenciais dentro Gen entrada base. Note-se que o número de electrões de núcleo deve ser especificado a seguira forma (por exemplo, MDF28 para o potencial de substituir 28 MDF electrões do núcleo). OldSDD solicita o padrão anterior.
• SDDAll : Seleciona potenciais Estugarda para Z> 2.
• cc-pVDZ , cc-pVTZ , cc-pVQZ , cc-pV5Z , cc-pV6Z : correlação consistentes conjuntos de bases de Dunning [ Dunning89 ,Kendall92 , Woon93 , Peterson94 , Wilson96 ] (duplos, triplos, quádruplos, quíntuplos-zeta e sêxtuplo-zeta , respectivamente). Estes conjuntos de base ter funções redundantes removido e foram rodados [ Davidson96 ] , a fim de aumentar a eficiência computacional.
Estes conjuntos de bases incluem funções de polarização por definição. A tabela a seguir lista as funções de polarização de valência presentes para os vários átomos incluídos nestes conjuntos de base:
Átomos cc-pVDZ
cc-pVTZ
cc-pVQZ
cc-pV5Z
cc-pV6Z
H 2s, 1p 3s, 2p,1d
4s, 3p,2d, 1f
5s, 4p,3d, 2f,
1g
6s, 5p, 4d,3F, 2g, 1h
Ele 2s, 1p 3s, 2p,1d
4s, 3p,2d, 1f
5s, 4p,3d, 2f,
1g
nãodisponível
Li-Be 3s, 2p,1d
4s, 3p,2d, 1f
5s, 4p,3d, 2f,
1g
6s, 5p,4d, 3F,2g, 1h
nãodisponível
B-Ne 3s, 2p,1d
4s, 3p,2d, 1f
5s, 4p,3d, 2f,
1g
6s, 5p,4d, 3F,2g, 1h
7s, 6p, 5d,4f, 3G, 2h,
1iNa-Ar 4s, 3p,
1d 5s, 4p,
2d, 1f 6s, 5p,
3d, 2f,1g
7s, 6p,4d, 3F,2g, 1h
nãodisponível
Ca 5s, 4p,2d
6s, 5p,3d, 1-F
7s, 6p,4d, 2f,
1g
8s, 7p,5d, 3F,2g, 1h
nãodisponível
Sc-Zn 6s, 5p,3d, 1-F
7s, 6p,4d, 2f,
8s, 7p,5d, 3F,
9s, 8p,6d, 4f,
nãodisponível

1g 2g, 1h 3G, 2h,1i
Ga-Kr 5s, 4p,2d
6s, 5p,3d, 1-F
7s, 6p,4d, 2f,
1g
8s, 7p,5d, 3F,2g, 1h
nãodisponível
Estes conjuntos de base pode ser aumentada com funções difusas, adicionando a -AUG prefixo para a palavra-chave conjunto de base (em vez de usar a + e + +notação veja abaixo).
• SV , SVP , TZV , TZVP [ Schaefer92 , Schaefer94 ] , QZVP [ Weigend05 ] de Ahlrichs e colegas de trabalho.
• MIDI! de Truhlar e colaboradores [ Easton96 ] . O MidiX palavra-chave é usada para solicitar este conjunto de base.
• EPR-II e EPR-III : Os conjuntos de bases de Barone [ Barone96a ] que são otimizados para o cálculo de constantes de acoplamento hiperfinos por métodos DFT (particularmente B3LYP). EPR-II é uma base zeta double set com um único conjunto de funções de polarização e uma s parte reforçada: (6,1) / [4,1] para H e (10,5,1) / [6,2,1 ] para B a F. EPR-III é um conjunto de base triple-zeta incluindo funções difusas, D-polarizações de casal e um único conjunto de funções f-polarização. Também neste caso, a parte s-é melhorado para melhor descrever a região nuclear: (6,2) / [4,2] por H e (11,7,2,1) / [7,4,2,1] para B a F.
• UGBs : O conjunto de base Gaussian universal de Castro, Jorge e colaboradores [ Silver78 , Silver78a , Mohallem86 , Mohallem87 ,daCosta87 , daSilva89 , Jorge97 , Jorge97a ,
deCastro98 ] . Funções adicionais de polarização pode ser adicionado através da inclusão de um sufixo para esta palavra-chave:
• UGBs n P | V | O
onde n é um número inteiro que indica se deve adicionar 1, 2 ou 3 funções de polarização para cada função na normalidadeUGBs conjunto de base. O segundoitem é uma carta de código que indica qual função deve ser aumentada funções de polarização: P adiciona-los a todas as funções, V adiciona-los a todas as funções de valência, e O solicita o esquema usado no Gaussian 03 (veja abaixo). Por exemplo, os UGBS1P pedidos de palavra-chave nesta base definidacom uma função de polarização adicional a todos os orbitais, e UGBS2V acrescenta dois função adicional de polarização para todos os orbitais de valência.
O O sufixo acrescenta as mesmas funções que os UGBs n P . palavras-chave emGaussian 03 UGBS1O acrescenta função ap para cada s, a função do anúncio para cada p, e assim por diante; UGBS2O acrescenta ap e função d para cada s, anúncio e função f para cada p, e UGBS3O acrescenta ap, d e f para cada s, etc
Difusas funções podem ser adicionados como habitualmente com + ou + + , o primeiro deles pode ser especificado como 2 +para adicionar duas funções difusas por átomos pesados.
• MTSmall de Martin e de Oliveira, definida como parte de seu método W1 (veja a W1U palavra-chave) [ Martin99 ] .
• Os DGDZVP , DGDZVP2 e DGTZVP conjuntos de base utilizados na DGauss [ Godbout92 , Sosa92 ] .

• CBSB7 : Seleciona o 6-311G (2d, d, p) conjunto de base utilizado pelo método de energia de alta precisão CBS-QB3 [Montgomery99 ] . A notação especifica duas funções de polarização d adicionais no segundo átomos de linhas, uma função d em átomos de primeira linha e função ap em hidrogênios (note que este três campos sintaxe da função de polarização é nãosuportado pelo Gaussian 09).
Adicionando polarização e Difusos FunçõesFunções primeiro single de polarização também pode ser solicitado através do costume * ou ** notação. Note-se que ( d, p ) e ** são sinónimos- 6-31G ** é equivalente a 6-31G (d, p) , por exemplo, e que o 3-21G * conjunto base tem funções de polarização em átomos segunda fileira única . O+ e + + funções difusas [ Clark83 ] estãodisponíveis com alguns conjuntos de base, como são múltiplas funções de polarização [ Frisch84 ] . A sintaxe de palavra-chave é melhor ilustrado por exemplo: 6-31 + G ( 3df, 2p ) designa o conjunto de base 6-31G complementado por funções difusas, 3 conjuntos de funções D e um conjunto de funções M em átomos pesados, e complementados por 2 conjuntos de funções de p sobre hidrogénios.
Quando o -AUG prefixo é usado para adicionar funções difusas para os cc-PV * Z conjuntos de base, uma função difusa de cada tipo de função em uso por um determinado átomo é adicionado [ Kendall92 , Woon93 ] . Por exemplo, o AUG-cc-pVTZ base lugares One S, um d, e uma funções p difusas sobre os átomos de hidrogênio e um de d, um p, um d, e uma funções difusas de f sobre B através de Ne e Al através Ar.
Existem várias opções para aumentar as cc-PV * Z conjuntos de bases com funções difusas:
• spAug-cc-PV * Z aumenta com s e p apenas funções, incluindo s funções de H e He.
• DAUG-cc-pV * Z aumenta com duas conchas de cada movimento angular em vez de um.
• "Calendário" variações do conjunto de bases de Truhlar [ Papajak11 ] estão disponíveis. A nomeação desta série de conjuntos de base vêm do fato de que os cc-PV * Z conjuntos de bases com funções adicionais de polarização são conhecidos como Aug-cc-PV * Z . Truhlar observou que "Agosto" é também uma abreviação para o mês de agosto, em Inglês, então ele propôs novos esquemas de aumento para os cc-PV * Z conjuntos de base, também chamado depois de meses do ano. Eles são construídos através da remoção de funções difusas a partir dos agosto conjuntos de base. Por exemplo, os jul-cc-PV * Z conjuntos de bases remover a função difusa de H e ele a partir de ago-cc-PV * Z . Jun-cc-PV * Z também remove a maior função difusa momento angular de todos os outros átomos, Maio-cc-PV * Z remove as duas mais altas funções de momento angular, e Apr-cc-PV * Z remove as três mais altas funções momento angular.
No entanto, por padrão, pelo menos s e p difusas funções são sempre incluídos nestes conjuntos de base. Isso serve para evitar algumas inconsistências inerentes, mas difere da Truhlar e definições originais "colegas de trabalho. Use as formas TJul , Tjun , e assim por diante para especificar as versões originais em que o limite é aplicada incondicionalmente: por exemplo, FSV-cc-pVDZ inclui apenas uma função difusa s em Cl mas ambas s

difusos e funções p sobre Fé e Br, enquanto M ay-cc-pVDZ tem s difusos e funções p sobre todos esses átomos.
Adicionando uma única função de polarização para 6-311G (ou seja, 6-311G ( d )) resultará em uma função d para átomos de primeira e segunda linha e uma função f paraátomos de primeira linha de transição, uma vez que as funções d já estão presentes para os elétrons de valência neste último. Da mesma forma, a adição de uma função difusa para o 6-311G conjunto de base irá produzir um s, um p, e um d funções difusas para átomos da terceira fileira.
Quando um cálculo núcleo congelado é feito usando o D95 base, ambos os orbitais centrais ocupados e os orbitais virtuais correspondentes estão congelados. Assim, enquanto uma D95 ** cálculo sobre a água tem 26 funções de base, e uma ** 6-31Gcálculo no mesmo sistema tem 25 funções, haverá 24 orbitais usados em um núcleo de cálculo pós-SCF congelado envolvendo ou conjunto de base.
A tabela seguinte lista de polarização e difusa disponibilidade da função e da faixa de aplicabilidade para cada alto-definido no Gaussian 09 base:
Conjunto debase
Aplica-sea
Funções depolarização
Funções Difusos
3-21G H-Xe +6-21G H-Cl * ou **
4-31G H-Ne * ou **
6-31G H-Kr através ( 3df,3pd )
+ , + +
6-311G H-Kr através ( 3df,3pd )
+ , + +
D95 H-Cl ,exceptoNa e Mg
através ( 3df,3pd )
+ , + +
D95V H-Ne ( d ) ou ( d, p ) + , + +SHC H-Cl *
CEP-4G H-Rn * (Li-Ar somente)
CEP-31G H-Rn * (Li-Ar somente)
CEP-121G H-Rn * (Li-Ar somente)
LanL2MB H-La,Hf-Bi
LANL2DZ H, Li-La,Hf-Bi
SDD, SDDAll todos,mas o
padre eRa
cc-pVDZ H-Ar,Ca-Kr
incluídas nadefinição
adicionadovia AUG- prefixo (H-Ar,
Sc-Kr)cc-pVTZ H-Ar,
Ca-Kr incluídas na
definição adicionado
via AUG- prefixo (H-Ar,Sc-Kr)
cc-pVQZ H-Ar, incluídas na adicionado

Ca-Kr definição via AUG- prefixo (H-Ar,Sc-Kr)
cc-pV5Z H-Ar,Ca-Kr
incluídas nadefinição
adicionadovia AUG- prefixo (H-Na,
Al-Ar Sc-Kr)cc-pV6Z H, B-Ne incluídas na
definição adicionado
via AUG- prefixo (H,BO)
SV H-Kr
SVP H-Kr incluídas nadefinição
TZV e TZVP H-Kr incluídas nadefinição
QZVP H-Rn incluídas nadefinição
MidiX H, CF,S-Cl, I,
Br
incluídas nadefinição
EPR-II , III-EPR
H, B, C,N, O, F
incluídas nadefinição
UGBs H-LR UGBs ( 1,2,3 ) P + , + + , 2 + , 2 + +MTSmall H-Ar
DGDZVP H-Xe
DGDZVP2 HF,Al-Ar,Sc-Zn
DGTZVP H, CF,Al-Ar
CBSB7 H-Kr incluídas nadefinição
+ , + +
STO-3G e 3-21G aceitar um * sufixo, mas isso na verdade não adicionar quaisquer funções de polarização.
Base adicional Set-chave relacionadasAs seguintes palavras-chave adicionais são úteis em conjunto com essas palavras-chave do conjunto de bases:
• 5D e 6E : Utilização de 5 ou 6 funções d (puro vs cartesianas funções d), respectivamente.
• 7F e 10F : Use 7 ou 10 f funções (puros vs funções f cartesianas), respectivamente. Essas palavras-chave também se aplicam a todas as funções superiores (g) e além.
Outros conjuntos de base também pode ser entrada para o programa usando os ExtraBasis e Gen palavras-chave. O ChkBasispalavra-chave indica que o conjunto de base é a de ler a partir do arquivo de verificação (definido através da % Chk de comando).Veja as descrições individuais destas palavras-chave mais adiante neste capítulo para mais detalhes.

As questões decorrentes da Pure vs Funções de Base cartesianasGauss usuários devem estar cientes dos seguintes pontos relativos puro vs funções de base cartesianas:
• Todos os conjuntos de bases internas usar funções f puros. A maioria também usar puro funções d, as exceções são 3-21G, 6-21G, 4-31G, 6-31G, 6-31G †, 6-31G ‡, CEP-31G, D95 e D95V. As palavras-chave anteriores pode ser usado para substituir a configuração padrão puro / cartesiano. Note-se que as funções de base geralmente, são convertidos para o outro tipo automaticamente, quando necessário, por exemplo, quando uma função de onda é lido a partir do arquivo do ponto de verificação para o uso em um cálculo utilizando uma base constituída por outro tipo [ Schlegel95a ] .
• Dentro de um trabalho, todas as funções de d deve ser 5D ou 6D, e todas as funções f e superior deve ser puro ou cartesiano.
• Ao usar o ExtraBasis , Gen e GenECP palavras-chave, a base definida explicitamente especificado na seção rota sempre determina o formulário padrãodas funções de base (para Gen , estes são 5D e 7F ). Por exemplo, se você usar um conjunto de base geral, tendo algumas funções do 3-21G e conjuntos de base6-31G, funções puras será usada a menos que você especifique explicitamente 6D na parte do percurso, além de Gen . Da mesma forma, se você adicionar funções de base para um metal de transição da base 6-311G (d) definir via ExtraBasis a um trabalho que especifica a base 6-31G (d) definido na seção rota, cartesianas funções d será usado. Da mesma forma, se você quiseradicionar funções de base para Xe da base 3-21G definido para a base 6-311 definido através do ExtraBasis palavra-chave, as funções de base Xe será funções puras.
Conjuntos de bases de montagem de DensidadeGaussian 09 proporciona a aproximação adequada densidade para cálculos DFT puros [ Dunlap83 , Dunlap00 ] . Essa abordagem amplia a densidade de um conjunto de funçõescentrada no átomo ao calcular a interação de Coulomb em vez de computar todas as integrais de dois elétrons. Ele oferece ganhos significativos de desempenho para os cálculos DFT puros em sistemas de médio porte muito pequeno para tirar vantagem dos algoritmos de escalonamento linear sem uma degradação significativa na precisão das estruturas previstas, energias relativas e propriedades moleculares. Gaussian 09 pode gerar uma base encaixe apropriado automaticamente da base de AO, ou você pode selecionar um dos conjuntos de montagem embutidas.
O conjunto de base de montagem desejado é especificado como um terceiro componente do modelo de química, como neste exemplo:# BLYP / TZVP / TZVPFit
Note-se que cortes devem ser utilizados como caracteres de separação entre o método, conjunto de base e conjunto de montagem quando a densidade de montagem conjunto de base é especificado.
As seguintes palavras-chave conjuntos de montagem estão disponíveis no Gaussian 09:
• DGA1 e DGA2 [ Godbout92 , Sosa92 ] . DGA1 está disponível para H através de Xe, e DGA2 está disponível para H, He e B através de Ne.

• SVPFit [ Eichkorn95 , Eichkorn97 ] e Def2SV [ Weigend05 ] , correspondente ao SVP conjunto de base.
• TZVPFit [ Eichkorn95 , Eichkorn97 ] e DefTZV [ Weigend05 ] , correspondente ao TZVP conjunto de base.
• QZVP [ Weigend03 , Weigend05 ] , correspondente ao QZVP conjunto de base.
• O W06 set montagem de Ahlrichs e colegas de trabalho [ Weigend05 , Weigend06 ] .
• Fit : Selecione o conjunto de montagem correspondente ao conjunto de base especificado. Se não houver um conjunto de montagem tal, ocorrerá um erro.
• NoFit : Desligue o uso conjunto apropriado para este cálculo. Esta palavra-chave é usado para substituir o DensityFit palavra-chave com um Default.Route arquivo.
• Auto : Gerar um conjunto apropriado automaticamente (veja abaixo).
Conjuntos de montagem de densidade podem ser gerados automaticamente a partir dos primitivos AO dentro do conjunto de base.Este é solicitada usando o Auto -chave conjunto de montagem. O programa trunca automaticamente o conjunto em um momento angular razoável: o padrão é Max ( MaxTyp +1,2 * MaxVal ), onde MaxTyp é o mais alto momento angular na base AO, e MaxValé o mais alto momento angular de valência. Você pode solicitar que todas as funções geradas ser usado com Auto = Todos , ou solicitar aqueles com até um certo nível com Auto = N , onde N é o momentum angular máxima retida nas funções de ajuste.Finalmente, o PAuto forma gera todos os produtos de funções AO em um centro, em vez de apenas quadrados dos primitivos AO, mas este é geralmente mais funções que são necessárias.
Por padrão, nenhum conjunto de montagem é usado. Densidade de montagem conjuntosde base pode ser aumentada com oExtraDensityBasis palavra-chave, definida na íntegra com o Gen palavra-chave e, opcionalmente, recuperadas do arquivo de verificação (use ChkBasis a fazê-lo). As opções para o DensityFit palavra-chave pode ser usado para controlar alguns aspectos do conjunto de montagem usado dentro de cálculos.
Densidade montagem pode ser feita o padrão para trabalhos usando funcionais DFT puros, adicionando a DenFit palavra-chave para a parte do percurso ( - # - ) linha no Default.Route arquivo. Montagem é mais rápido do que fazer o termo de Coulomb exatamente para sistemas de até várias centenas de átomos (dependendo conjunto de base), mas é mais lento do que exata Coulomb usando técnicas de escala linear (que são ligados automaticamente com exata Coulomb) para sistemas muito grandes.
Visão geral das especificações moléculaEsta seção de entrada especifica as posições nucleares e o número de elétrons de α-e β-spin. Existem várias maneiras pelas quais a configuração nuclear pode ser especificados: como uma matriz-Z, como as coordenadas cartesianas, ou como uma mistura dos dois (note que as coordenadas cartesianas é apenas um caso especial da matriz-Z).
A primeira linha da seção de especificação molécula especifica a carga líquida elétrica (um inteiro assinado) ea multiplicidade de spin (geralmente um número inteiro positivo). Assim, para uma molécula neutra em um estado de singuleto, a entrada 1 0 é

apropriado. Para um anião radical, -1 2 seriam utilizados. Vários pares de carga / rotação pode / deve ser incluída para alguns tipos de cálculo.
A linha de carga e spin é a única entrada especificação molécula necessária se Geom = CheckPoint é usado. Toda a especificação molécula (e seção de título) pode ser omitido, incluindo Geom = AllCheck na seção rota.
O restante da molécula de especificação dá o tipo de elemento e posição nuclear para cada átomo do sistema molecular. O formato mais geral para a linha no seu interior é a seguinte:
Elemento-label [- Atom do tipo [- Carga ]] [( param = valor [, ...])] Atomposições parâmetros-
Cada linha contém o tipo de elemento, e, possivelmente, um tipo de átomo de mecânica molecular opcional e carga parcial. Parâmetros nucleares para este átomo são especificados na lista entre parênteses. O restante da linha contém informações sobre a localização do átomo, ou como as coordenadas cartesianas ou como uma definição de Z-matriz. Vamos começar por considerar os itens iniciais e finais e, em seguida, passar adiscutir os demais itens.
Aqui é o formato básico para especificar átomos dentro da molécula de especificação (omitindo todos os itens opcionais):
Elemento-label [ congelamento-code ] x y z
Embora estes exemplos usam os espaços para separar itens dentro de uma linha, qualquer separador de validade, pode ser utilizado. A posição do átomo é especificado em coordenadas cartesianas. congelar-code é um parâmetro opcional relacionado ao congelamento átomos durante otimizações usando ONIOM (ver ONIOM para detalhes).
Elemento-label é uma cadeia de caracteres que consiste em um ou outro o símbolo químico do átomo ou seu número atômico. Se o símbolo elementar é utilizado, ele pode ser opcionalmente seguido por outros caracteres alfanuméricos para criar uma etiqueta de identificação para esse átomo. Uma prática comum é a seguir o nome do elemento com um número inteiro identificando secundário: C1, C2, C3, e assim por diante; esta técnica é útil na seguinte ordem química convencional. O comprimento máximo do elemento da etiqueta é 4 caracteres.
Na primeira forma, os demais itens em cada linha são coordenadas cartesianas especificam a posição desse núcleo. Na segunda forma, o átomo de 1, átomo 2, átomo 3 são os rótulos dos átomos anteriormente especificadas, que serão utilizados para definir os átomos de correntes 'posição (em alternativa, os outros átomos' números de linha entre a secção de especificação molécula pode ser usada para os valores das variáveis, onde a linha de multiplicidade de carga e rotação é a linha 0).
A posição do átomo de corrente é então determinado mediante a extensão da ligação unindo-se a atom1, o ângulo formado por esta ligação e a ligação unindo átomo 1 e átomo de 2, e o (torção), ângulo diedro formado pela ligação unindo átomo 2 e átomo de 3 com o plano que contém o átomo de corrente, átomo e um átomo de 2.
Aqui está uma seção simples especificação molécula de etano, que utiliza etiquetas de elemento para os átomos de carbono e os tipos de elemento para os átomos de hidrogênio:0,1 C1 0.00 0.00 0.00

C2 0,00 0,00 1,52 H 1.02 0.00 -0.39 H -0.51 -0.88 -0.39 H -0.51 0.88 -0.39 H -1.02 0.00 1.92 H 0.51 -0.88 1.92 H 0,51 0,88 1,92
Especificações molécula Z-matriz também são aceitos. Veja o Apêndice C para mais detalhes.
Especificando Isótopos e outros parâmetros nuclearesIsótopos e outros parâmetros nucleares pode ser especificado dentro do campo de tipo de átomo usando palavras-chave e os valores entre parênteses, como no exemplo a seguir:C (ISO = 13, rotação = 3) 0,0 0,0 0,0
A linha especifica um 13 átomo de C, com um spin nuclear de 3/2 (3 * 1/2), localizado na origem. Os seguintes itens podem ser incluídos na lista de parâmetros:
• Iso = n : seleção Isotope. Se inteiros são usados para especificar as massas atômicas, o programa irá automaticamente usar a massa correspondente real exata isotópica (por exemplo, 18 especifica 18 O, e Gaussian utiliza o valor 17,99916).
• Rotação = n : spin nuclear, em unidades de 1/2.
• Zeff = n : carga efetiva. Este parâmetro é utilizado no acoplamento órbita rotação (ver CASSCF = SpinOrbit ), eo ESR gtensor eo eletrônico spin-molecular hiperfina rotação tensor ( RMN Output = Pickett ).
• QMom = n : momento de quadrupolo Nuclear.
• GFAC = n : momento magnético nuclear em magnetons nucleares.
Especificando fragmentosFragmentos dentro de um sistema molecular pode ser definido usando o fragmento parâmetro, que aparece entre parênteses após o rótulo átomo juntamente com qualquer isótopo e / ou valores de parâmetros nucleares. O valor de fragmento é um número inteiro; todos os átomos com o mesmo número do fragmento são definidas como um fragmento. Os fragmentos são úteis para o cálculo fragmento suposição, cálculos contrapeso, e assim por diante.
Por exemplo, a seguinte estrutura de bifenilo é dividido em dois fragmentos de pelo anelde benzeno:
0,1 0,1 0,1 rotação total e carga, seguidos por aqueles específicos do fragmento. C (Fragmento = 1) -3,05015529 -0,24077322 0,00000698C (Fragmento = 1) -1,64875545 -0,24070572 0,00067327C (Fragmento = 1) -,94811361 0,97297577 0,00020266C (Fragmento = 1) -1,64887160 2,18658975 -0,00093259C (Fragmento = 1) -3,05027145 2,18652225 -0,00159819C (Fragmento = 1) -3,75091329 0,97284076 -0,00112735H (Fragmento = 1) -3,58511088 -1,16744597 0,00036555H (Fragmento = 1) -1,11371117 -1,16732692 0,00154256H (Fragmento = 1) -1,11391601 3,11326250 -0,00129286H (Fragmento = 1) -3,58531573 3,11314346 -0,00246648H (Fragmento = 1) -4,82091317 0,97278922 -0,00163655C (Fragmento = 2) 0,59188622 0,97304995 0,00093742

C (Fragmento = 2) 1,29252806 2,18673144 0,00046795C (Fragmento = 2) 1,29264421 -0,24056403 0,00207466C (Fragmento = 2) 2,69392790 2,18679894 0,00113535C (Fragmento = 2) 2,69404405 -0,24049653 0,00274263C (Fragmento = 2) 3,39468590 0,97318496 0,00227326H (Fragmento = 2) 0,75768862 -1,16723678 0,00243403H (Fragmento = 2) 0,75748378 3,11335264 -0,00040118H (Fragmento = 2) 3,22888349 3,11347169 0,00077519H (Fragmento = 2) 3,22908834 -1,16711773 0,00360969H (Fragmento = 2) 4,46468577 0,97323650 0,00278063
Este exemplo também ilustra o uso de carga específico do fragmento e as especificaçõesde multiplicidade de spin. O formato da linha de entrada correspondente, neste caso, é a seguinte:
carga total , cisão total , carga fragment1 , rotação fragment1 , responsável fragment2 , rotação fragment2
Valores de multiplicidade de spin negativos têm um significado especial para Guess = Fragmento cálculos, indicando que os orbitais desemparelhados para o fragmento correspondente estão a tornar-se orbitais de spin β no conjunto combinado especificado.Multiplicidades de spin negativos irá gerar um erro em qualquer outro tipo de trabalho.
Para Guess = Fragmento e contrapeso cálculos, números de fragmentos deve começar em 1 e executar consecutivamente. Para outros tipos de cálculo, esta restrição não é imposta, mas sua violação pode resultar em alguns, seções de dados vazios estranhos na saída (por exemplo, análises de toda a população de zero fragmento).
GaussView fornece uma ferramenta gráfica para a definição de fragmentos.
Tipos Molecular Atom MecânicaEspecificações da molécula por cálculos de mecânica molecular pode também incluir digitação átomo e as informações de carga parcial. Aqui estão alguns exemplos:
C-CT Especifica um átomo de carbono SP3 alifático. C-CT-0.32 Especifica um átomo de carbono SP3 alifáticos com uma carga parcial de 0,32. OO - 0,5 Especifica um átomo de oxigênio grupo carbonila com uma carga parcial de -0,5.
Tipos Atom e cargas parciais opcionais podem ser especificados para cada átomo. Parâmetros nucleares também pode ser definida, como nos exemplos:C-CT (Iso = 13)C-CT - 0,1 (spin = 3)
Parâmetros arquivo PDBVários itens adicionais podem ser definidas, juntamente com os parâmetros nucleares e /ou definições de fragmento. Esses itens são projetados para uso com arquivos PDB parareter resíduos e outras informações estruturais que contêm e, como tal, não vai ser definido pelo usuário. No entanto, você pode vê-los em Gaussian 09 arquivos de entradacriados por GaussView usando estruturas originárias de arquivos PDB.
• RESNum especifica o resíduo em que está localizado o átomo. O valor toma a forma de n [ X [ Y ]], em que n é um número inteiro (que não precisa ser positivo), X é o código de inserção de um carácter opcional, e Y é a letra de cadeia facultativo. Se a corrente é especificado, mas não existe um código de inserção, então X pode ser um sublinhado: ResNum =-17_C para o resíduo com o número -17 em cadeia C.

• ResName especifica o nome do resíduo de três caracteres.
• PDBName especifica o nome atribuído ao átomo se ele não é apenas o nome do elemento.
Especificando Átomos fantasmaUm átomo com a mecânica digite Bq (por exemplo, O-Bq ) é configurado como um fantasma [ Macbeth ] do átomo correspondente, com as suas funções de base normais e de integração numérica pontos de grade, mas nenhuma acusação ou elétrons nuclear. Esta substituição exige um cálculo contrapeso. Tais cálculos diferir ligeiramente as solicitadas com Massagem nas versões anteriores do Gaussian na medida em que incluem os pontos de grade dos átomos fantasmas em DFT XC quadratura. A nova forma é uma correção de superposição mais consistente e também mais fácil de usar. Note-se que os cálculos contrapeso também podem ser solicitados com o contrapeso palavra-chave.
Especificar sistemas periódicosSistemas periódicas são especificados com uma especificação molécula normal para a célula unitária. A única entrada adicional necessário é um, dois ou três vetores de tradução acrescentados à especificação molécula (sem linha em branco interveniente), indicando a direção (s) de replicação. Por exemplo, a seguinte entrada especifica um cálculo da energia unidimensional PBC único ponto de neoprene:# PBEPBE/6-31g (d, p) / Auto SCF = apertado
neopreno,-CH2-CH = C (Cl)-CH 2-geometria optimizada
0 1C, -1.9267226529,0.4060180273,0.0316702826H, -2.3523143977,0.9206168644,0.9131400756H, -1.8372739404,1.1548899113, -,770750797C, -0,5737182157, -0.1434584477,0.3762843235H, -0,5015912465, -0.7653394047,1.2791284293C, 0.5790889876,0.0220081655, -,3005160849C, 1,9237098673, -0.5258773194,0.0966261209H, 1,772234452, -1.2511397907,0.915962512H, 2,3627869487, -1,0792380182, -,752511583Cl, 0.6209825739,0.9860944599, -1,7876398696TV, 4.8477468928,0.1714181332,0.5112729831
A última linha especifica o vetor de translação. Note-se que especifica TV como o símbolo do átomo.
O seguinte especificação molécula poderia ser utilizada para o cálculo PBC bidimensional sobre uma folha de grafite:0 1C 0.000000 0.000000 0.000000C 0.000000 1.429118 0.000000TV 2.475315 0.000000 0.000000TV -1,219952 2,133447 0,000000
Aqui é a especificação de molécula que pode ser utilizada para o cálculo PBC tridimensional sobre o arsenieto de gálio:0 1 Ga 0.000000 0.000000 0.000000 Ga 0.000000 2.825000 2.825000 Ga 2.825000 0.000000 2.825000

Ga 2.825000 2.825000 0.000000 Como 1.412500 1.412500 1.412500 Como 1.412500 4.237500 4.237500 Como 4.237500 1.412500 4.237500 Como 4.237500 4.237500 1.412500 TV 5.650000 0.000000 0.000000 TV 0.000000 5.650000 0.000000 TV 0.000000 0.000000 5.650000
Jobs multi-passoVários trabalhos de Gauss podem ser combinados em um único arquivo de entrada. A entrada para cada posto de trabalho sucessivas é separada daquela da etapa precedente trabalho por uma linha com a forma:- Link1 -
Aqui está um exemplo de arquivo de entrada que contém duas etapas de trabalho:Chk% = freq# HF/6-31G (d) Freq Frequências em STP Especificação Molecule - Link1 -Chk% = freq% Nosave# HF/6-31G (d) Geom = Verificar Guess = Leitura Freq = (ReadFC, ReadIsotopes) Frequências em 300 K carga e rotação
300,0 2,0Especificações Isotope
Este arquivo de entrada calcula freqüências vibratórias e realiza a análise termoquímica em duas temperaturas e pressões diferentes: primeiro a 298,15 K e 1 atmosfera, e depoisnovamente a 300 K e 2 atmosferas. Note-se que uma linha em branco deve preceder a - Link1 - linha.
Gaussian 09W ReferênciaEste trabalho serve como referência para Gaussian 09W . Ele documenta a interface do usuário para esta versão. Ele pressupõe o conhecimento dos conceitos básicos do Windows, técnicas e caixas de diálogo (por exemplo, abertura de arquivo e salvar). Consulte a documentação do Windows se precisar de assistência nessas áreas. Este documento está organizado em torno das várias janelas (caixas de diálogo) que compõem o Gaussian 09W interface, e seu associado menus, botões e campos.
Consulte a Referência do Usuário do Gaussian 09 para obter informações gerais sobre os recursos de Gauss, palavras-chave e utilitários.

A tabela a seguir lista algumas tarefas comuns que você pode querer realizar com Gaussian 09W , juntamente com o número da página onde a discussão das características e / ou técnicas relevantes começa assim:
Gaussian 09W ReferênciaA janela de processamento do trabalhoO Processamento de trabalho janela é o lugar a partir do qual Gaussian 09W empregos são controladas e executadas e onde sua produção é exibida. Suas principais peças são descritas na ilustração a seguir:
O restante desta seção discute os menus e botões disponíveis nesta janela.
O menu Arquivo
O arquivo menu permite-lhe criar e acessar Gaussian 09W arquivos de entrada e definir as preferências do programa.
NovoCriar nova Gaussian 09W entrada (que residem apenas na memória até que seja salva em disco).

Abrir ... Abra um existente Gaussian 09W arquivo de entrada. A extensão de um Gaussian 09W arquivo de entrada é. GJF. O Abrir ... item de menu também pode ser usado para carregar um arquivo de controle de lote existente. A instalação em lote é descrito mais adiante nesta seção. Finalmente, pode ser usada para abrir um ficheiro APO para a conversão de (este processo é discutido mais tarde).
Modificar ... Edite a entrada de corrente, através do Editar Job arquivo existente janela.
PreferênciasDefinir Gaussian 09W preferências. As preferências são descritas em uma seção separada, mais adiante neste documento.
Sair Sair do Gaussian 09W . Você será perguntado se deseja salvar todos os arquivos que nãoforam salvos novos ou modificados de entrada, bem como as alterações não salvas para as preferências.
O Menu de Processos
O Processo de menu permite que você manipule os trabalhos de execução. Todos os seus itens têm ícones equivalentes noProcessamento Job janela (descritos mais adiante nesta seção).
Comece Processamentocomeçar a executar a entrada atualmente carregado.
Pausaimediatamente suspender o trabalho atualmente em execução.
Pausa → Próximo Link suspender a execução do trabalho atualmente em execução após completar a ligação actual. (O Gaussian 09 programa é dividido em uma série de módulos conhecidos como ligações . Diferentes ligações executar diferentes partes do cálculo, e as várias ligações executar seqüencialmente, tornando-se o trabalho total.)
Resume Restart execução de um trabalho em pausa.
Morte Jobabortar imediatamente o trabalho atualmente em execução. Se um lote for executado, o próximo trabalho no lote (lotes são formalmente definidos mais adiante nesta seção) vai começar a executar (a menos que o Batch Run End em erro a preferência é definida).
Batch EndParar de executar o lote atual, quando o trabalho atual terminar.
Matar Batchabortar imediatamente o trabalho atualmente em execução e encerrar o processamento em lote, sem correr mais nenhum trabalho.
O Menu Utilitários
O Utilities menu dá acesso às instalações em lote e conversão de arquivos e outras utilidades fornecidas com Gaussian 09W . Vamos considerá-las em detalhe mais adianteneste manual.

Editar Lista BatchEditar o arquivo de controle de lote atualmente carregado (extensão . BCF ), através da Lista de lote Editar janela (descrito mais tarde). Se nenhum arquivo de controle de lotes é carregado, em seguida, uma nova lista de lote é criado e qualquer entrada carregado atualmente é apagada da memória.
NewZMatConverta arquivos usando o NewZMat utilidade. Após selecionar esta opção, você designa o arquivo a ser convertido a partir doArquivo Abrir caixa de diálogo. A Conversão NewZMat Arquivo janela aparece então (descrito posteriormente neste documento).
CubeGen Gerar um arquivo de cubo para uso em um programa de visualização. Você será solicitado para todas as informações necessárias.
CubManmanipular ou transformar um ou mais arquivos de cubo existentes. Você será solicitado para todas as informações necessárias.
FreqChkRecuperar freqüência e termoquímica de dados a partir de um arquivo de ponto de verificação. Após selecionar esta opção, você designa o arquivo de ponto de verificação para ser usado com o arquivo aberto caixa de diálogo.
FORMCHKConverter um arquivo binário para um posto de controle (ASCII) versão formatada. Após selecionar esta opção, você designa o arquivo de ponto de verificação para ser usado com o arquivo aberto caixa de diálogo.
UnFchkConverter um arquivo de verificação formatado para o formato binário G09W. Após selecionar esta opção, você designa o arquivo de ponto de verificação para ser usado com o arquivo aberto caixa de diálogo.
ChkChkExibir informações sobre o conteúdo de um arquivo de verificação. Após selecionar estaopção, você designa o arquivo de ponto de verificação para ser usado com o arquivo aberto caixa de diálogo.
C8609 Converta arquivos de ponto de verificação entre a versão atual e as de versões anterioresdo Gaussian para Windows. Após selecionar esta opção, você designa o arquivo de ponto de verificação para ser usado com o arquivo aberto caixa de diálogo.
External PDB VisualizadorVer a estrutura molecular atual com um programa de visualização PDB externo. O programa a ser usado é especificado nas preferências (descritos mais adiante neste documento).
O Menu Exibir
O Ver menu controla a aparência da janela e permite invocar um editor de texto externo. As configurações padrão das várias opções de exibição também podem ser controladas através das preferências. As opções de edição também têm equivalentes ícone (descritos mais adiante nesta seção).

Barra de ferramentasAlterna a exibição da parte da barra de ferramentas da janela. Quando a barra de ferramentas é visível, este item é verificado.
Saída de ProcessamentoAlterna a exibição da exibição de saída área da janela. Quando a exibição de saída área é visível, este item é verificado.
Barra de statusAlterna a exibição da parte da barra de status da janela, o que mostra uma breve descrição do item de menu atual. Quando a barra de status é visível, este item é verificado.
Editor de chamar o editor externo (que editor é usado é definido nas preferências).
Editor de arquivo de saída →Chame o editor externo no arquivo de saída atual. Note-se que um trabalho de execução deve ser interrompida antes de invocar um editor em seu arquivo de saída.
O Menu Ajuda
A Ajuda cardápio segue as convenções padrão do Windows.
ConteúdoMostrar a tabela de conteúdos para a ajuda on-line.
Sobre ...Mostra uma janela de informações sobre esta versão e cópia do Gaussian 09W , incluindo a versão do programa eo número de série deste exemplar:
Ajuda também pode ser alcançado a qualquer momento, pressionando o F1 chave.
Ícones
Os seguintes ícones estão disponíveis no processamento do trabalho da janela (o item demenu é equivalente entre parênteses):
Comece o processamento do arquivo de entrada ou lote atual ( Processo: Run ).
Imediatamente uma pausa no trabalho ( processo: Pausa ).

Pausa após a ligação atual ( Processo: Pausa → Próximo Link ).
Retomar a execução de uma tarefa interrompida ( Processo: Resume ).
Terminar o trabalho atual ( Processo: Morte Job ).
Edite o arquivo de controle de lote atual, ou criar um novo, se nenhum está atualmente carregado ( Utilities: Editar lista de lote).
Acabar com o lote em execução após o trabalho atual é concluída ( Processo: BatchEnd ).
Abortar imediatamente o lote atual, matando o trabalho atual ( Processo: Mate Batch ).
Abra o editor externo ( Ver: Editor ).
Edite o arquivo de saída de corrente. Certifique-se de dar uma pausa no trabalho emexecução em primeiro lugar! ( Ver: Editor → arquivo de saída ).
Execução de arrastar-e-soltar arquivos
Gaussian 09W empregos também pode ser iniciada usando a técnica de execução de arrastar-e-soltar. Aqui estão os passos envolvidos:
• Iniciar Gaussian 09W (se necessário).
• Navegue até o diretório que contém o arquivo de entrada desejado (s).
• Selecione e arraste os arquivos para o Gaussian 09W ícone (se o programa é minimizado) ou em qualquer lugar dentro doProcessamento Job janela. Se mais de um arquivo for arrastado e solto, um arquivo de lote que consiste nos arquivos de entrada selecionados serão criados. O arquivo de controle de lote resultante será denominada DropList.BCF , e será colocado no diretório atual.
• Escolha iniciar o processamento do Processo menu (ou clique no ícone equivalente). Você pode direcionar Gaussian 09Wpara iniciar automaticamente o processamento de arquivos que são descartados no Preferências do Processo janela (descrito mais tarde).
Gaussian 09W ReferênciaA janela Editar JobEsta janela é utilizada para criar e editar Gaussian 09 arquivos de entrada. Ele tem dois títulos: trabalho de entrada , quando usado para criar novos entrada e arquivo existente Editar Job , quando utilizado para modificar um arquivo de entrada existente. Observe que a nova entrada e mudanças para arquivos de entrada existentes são armazenados na memória como eles são feitos e, portanto, será usado quando a execução do trabalho começa, mas eles devem ser explicitamente salvas no disco.
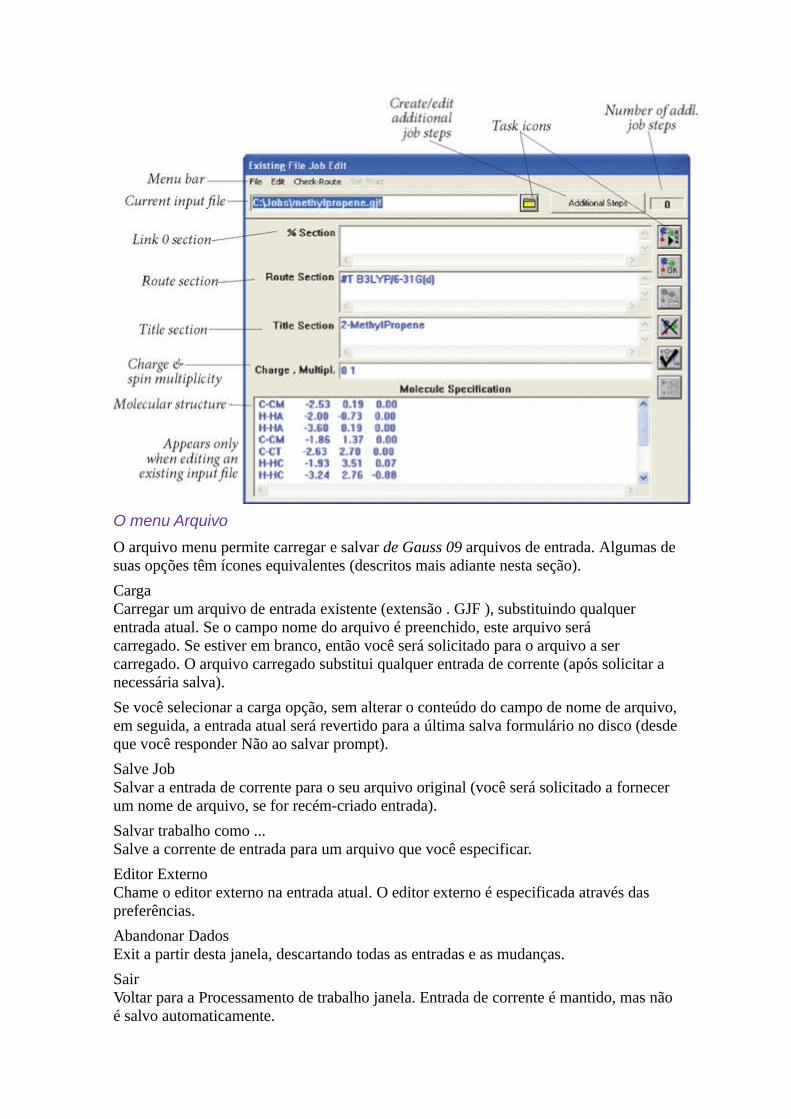
O menu Arquivo
O arquivo menu permite carregar e salvar de Gauss 09 arquivos de entrada. Algumas desuas opções têm ícones equivalentes (descritos mais adiante nesta seção).
CargaCarregar um arquivo de entrada existente (extensão . GJF ), substituindo qualquer entrada atual. Se o campo nome do arquivo é preenchido, este arquivo será carregado. Se estiver em branco, então você será solicitado para o arquivo a ser carregado. O arquivo carregado substitui qualquer entrada de corrente (após solicitar a necessária salva).
Se você selecionar a carga opção, sem alterar o conteúdo do campo de nome de arquivo,em seguida, a entrada atual será revertido para a última salva formulário no disco (desdeque você responder Não ao salvar prompt).
Salve JobSalvar a entrada de corrente para o seu arquivo original (você será solicitado a fornecer um nome de arquivo, se for recém-criado entrada).
Salvar trabalho como ...Salve a corrente de entrada para um arquivo que você especificar.
Editor ExternoChame o editor externo na entrada atual. O editor externo é especificada através das preferências.
Abandonar DadosExit a partir desta janela, descartando todas as entradas e as mudanças.
Sair Voltar para a Processamento de trabalho janela. Entrada de corrente é mantido, mas não é salvo automaticamente.

Saia & RunVoltar para a Processamento de trabalho janela e começar a executar a entrada de corrente (não salvos automaticamente para o disco).
O Menu Editar
O Editar menu inclui o padrão do Windows Editar opções de menu Desfazer , Recortar , Copiar , Colar e Apagar . Ele também tem essa opção adicional:
Limpar formulárioapagar todas as informações em todas as seções da janela. Nenhum aviso é dado sobre as alterações não salvas. Você pode criar um novo arquivo de entrada a partir deste formulário selecionando Limpar formulário , entrando na entrada desejada, e depois salvá-la.
A opção Check-Route
Este item é executado a Route check utilidade na entrada de corrente (descrito mais adiante neste documento). Existe um ícone equivalente para esta opção (descrito mais tarde).
A opção Set-Start
Esta opção permite-lhe definir a etapa de trabalho de partida para este arquivo de entrada (etapas de trabalho adicionais são discutidos mais adiante nesta seção). O padrão é o principal (primeiro) passo;
Selecione o passo de iniciar clicando duas vezes sobre o passo desejado. Sair da janela, escolhendo Fechar a partir da janela do Sistema menu (alcançado através da estreita barra no canto superior esquerdo). Existe um ícone equivalente para esta opção (descritomais tarde).
Ícones
Os seguintes ícones estão disponíveis no Job Editar janela (o item de menu é equivalente entre parênteses):
Voltar para o menu principal e iniciar a execução do trabalho ( Arquivo → Sair & Run ). Volte ao menu principal ( Arquivo → Sair ). Salve todo entrada de corrente para o disco ( File → Save Job ). Descartar todas as entradas e retornar ao menu principal (Arquivo → Abandonar Dados ). Execute o utilitário Route Check ( Check-Rota ). Especifique o passo de iniciar trabalho ( Set-Start ). Carregue um arquivo de entrada, sempre pedindo o nome do arquivo, substituindo qualquer entrada de corrente (após solicitar para qualquer necessidade salva) . Apenas apresentar ao editar um arquivo existente ( Arquivo → Carga ).

O Passos botão adicional
Este botão leva você para a etapas de trabalho adicionais janela, descrito na próxima seção. O campo de exibição de seu direito indica o número de etapas de trabalho adicionais dentro deste trabalho.
O posto de trabalho adicional Passos JanelaA etapas de trabalho adicionais janela é usada para criar e editar multicomponentes Gaussian 09 postos de trabalho. Note-se que nenhuma economia de arquivo ocorre a partir desta janela; salva deve ser feita a partir do Job Editar janela.
Aspecto inicial Janela
Quando você pressiona o adicional Passos botão para um arquivo de entrada com apenas uma única etapa de trabalho, a etapas de trabalho adicionais janela consiste unicamente em sua barra de menus:

As várias áreas de entrada não são visíveis até que você crie uma nova etapa, selecionando Adicionar Etapa da Etapa menu.
O Menu de Passo
A Etapa de menu é usado para criar, remover e reorganizar a ordem das etapas de trabalho.
Adicionar Passo Crie uma nova etapa de trabalho após o atual. O conteúdo da Seção% , Seção Título e Carga & Multipl. áreas da actividade principal são automaticamente copiados para a nova etapa. Eles podem ser editadas como desejado como as áreas adicionais são preenchidos
Excluir PassoRetire a etapa atual do trabalho.
Reordenar Alterar a ordem das etapas de trabalho utilizando o re-ordenação de dados janela (descrito em uma seção separada, mais adiante neste documento).
Carga De ArquivoSubstitua a etapa atual com o trabalho armazenado em um arquivo externo (você será solicitado a informar o nome do arquivo). Se o arquivo contém mais de uma etapa de trabalho em si ea etapa atual é a última etapa do trabalho, então todos os passos do arquivo será carregado em sua ordem atual.
Se o arquivo contém várias etapas de trabalho ea etapa atual não é a última etapa do trabalho, então apenas o primeiro passo a partir do arquivo será carregado, como o passo atual, e uma mensagem de erro será exibida.
SairVoltar para a Job Editar janela. Existe um ícone equivalente para este item de menu (descrito mais adiante nesta seção).
O Menu Editar
O Editar menu contém os itens de série: Desfazer , Cut , Copiar , Colar e Apagar .

O Menu Exibir
O Ver menu permite-lhe mover-se entre os trabalhos de passos adicionais dentro do trabalho atual. Seus itens também têm ícones equivalentes (descritos mais adiante nesta seção).
Próximo PassoMover para a próxima etapa (maior numerada) no trabalho.
Prev PassoVá para o passo anterior neste trabalho.
Escolha PassoMover para o número etapa de trabalho que você especificar.
O Check-Route item
Este item é executado a Route check facilidade na etapa de entrada de corrente (descritoem uma seção separada, mais adiante neste documento).
Ícones
Os seguintes ícones aparecem na etapas de trabalho adicionais janela (o item de menu é equivalente entre parênteses):
Mover para a próxima etapa de trabalho ( Ver: Próximo Passo ). Mover-se para a etapa anterior de trabalho ( Ver: Prev Etapa ). Mover-se para um número determinado passo ( Ver: Escolha Etapa ). Execute o utilitário de verificação de rotas ( Entrada Route ). Volte para a janela de Edição de trabalho ( Etapa: Exit ).
A Rota Facility VerifiqueA Rota do check instalação verifica a parte do percurso da corrente de entrada (ou etapa de trabalho adicional se correr a partir daetapas de trabalho adicionais janela). Ele exibe uma janela que contém a parte do percurso, o link mapa correspondente, e mensagens de erro. Isto caracteriza corresponde ao testrt utilitário fornecido com outros Gaussian versões.

A ausência de qualquer mensagem de erro indica uma parte do percurso válido (embora Verifique Route não pode determinar se é o que você pretende ou não). Você pode sair a partir desta janela, pressionando o Feito botão quando tiver terminado de examinar ocheck rota de saída. Consulte a Referência do Usuário do Gaussian 09 para mais informações sobre esta facilidade.
Gaussian 09 Palavras-chavePalavras-chave são geralmente dispostos em ordem alfabética, com as seguintes exceções:
• Conjunto de base palavras-chave não estão presentes, ver a seção sobre conjuntos de base para obter informações sobre conjuntos de bases disponíveis eas suas palavras-chave associadas. Note, entretanto, que os ChkBasis , ExtraBasis , Gen ePseudo palavras-chave são discutidos em suas próprias seções.
• Todas as palavras-chave relacionadas com a DFT são coletados sob o título Métodos DFT .
• Ligação 0 comandos são colocados depois de todas as palavras-chave do alfabeto (ou seja, após a discussão de ZINDO ), formando a penúltima seção .
• Palavras-chave relacionadas com a especificação de alternativas rotas ExtraLinks , ExtraOverlays , NonStd , Skip , e Use -são discutidos no Rotas Especificando Nonstandard seção. Informação relacionada também éapresentado na discussão dotestrt utilidade.
Dentro da discussão de uma determinada palavra-chave, as opções são listadas em ordem de importância e freqüência de uso, em vez de em ordem estritamente alfabética. Listas de opções grandes são frequentemente divididos em grupos com funções semelhantes.
Programas Utilitários

Esta seção descreve vários programas utilitários incluídos no Gaussian 09. Os utilitáriossão discutidos em ordem alfabética dentro deste capítulo.
A maioria dos utilitários estão disponíveis para ambas as versões do UNIX e Windows do Gauss. No entanto, certifique-se de consultar as notas de lançamento que acompanham o programa para obter informações referentes a sistemas operacionais específicos.
A seguir lista os serviços públicos disponíveis e suas funções (itens com estrela estão incluídos no 09W Gaussian Utilities menu):
c8609Converte arquivos de ponto de verificação a partir de versões anteriores do programa para o formato Gaussian 09.
chkchk *Apresenta o percurso e as seções de título de um arquivo de ponto de verificação.
cubegen *utilitário de geração de cubo Standalone.
cubman *Manipula cubos de Gauss-produzido de densidade de elétrons e potencial eletrostática (que permite que sejam adicionados, subtraídos, e assim por diante).
FORMCHK *Converte um arquivo de verificação binária em uma forma ASCII adequados para uso com programas de visualização e para mover arquivos de ponto de verificação entre diferentes tipos de sistemas de computador.
freqchk *Prints freqüência e termoquímica de dados de um arquivo de ponto de verificação. Fatorisótopos alternativos, temperatura, pressão e escala podem ser especificados para a análise termoquímica.
freqmemDetermina requisitos de memória para cálculos de freqüência.
gauoptExecuta otimizações de diferentes coordenadas moleculares variáveis.
ghelpajuda on-line para Gauss.
mmStandalone programa mecânica molecular.
newzmat *Conversão entre uma variedade de formatos de especificação de geometria molecular.
testrt *verificador de sintaxe seção Rota e geração de rota não-padrão.
unfchk *Converter um arquivo de checkpoint formatado de volta à sua forma binária (por exemplo, após o mover de um tipo diferente de sistema de computador).
GAUSS_MEMDEF variável de ambiente

O GAUSS_MEMDEF variável de ambiente pode ser usado para aumentar a memória disponível para os utilitários que não oferecem essa opção se. Seu valor deve ser ajustado para a quantidade desejada de memória em palavras.
Correndo GaussianEsta seção descreve os comandos do sistema operacional necessárias para executar Gaussian em sistemas de computadores baseados em Unix. Consulte as instruções adicionais que acompanham o programa de informações equivalentes para outros sistemas operacionais. Esta discussão pressupõe que o programa já foi instalado.
Correndo Gaussian envolve as seguintes atividades:
• Criando entrada Gaussian descrevendo o cálculo desejado.
• Especificando os locais dos vários arquivos de zero.
• Especificar os requisitos de recursos.
• Iniciando a execução do programa, ou em modo interativo ou batch.
Nesta seção, vamos supor que um arquivo de entrada Gaussian básico foi criado, e nossa discussão irá examinar os restantes três itens da lista.
Especificando Handling risco de arquivo e localGaussian utiliza vários arquivos de zero no curso de sua computação. Eles incluem:
• O arquivo Checkpoint: nome . chk
• O Read-Write file: nome . rwf
• O arquivo Integral Two-Electron: nome . int (vazio por padrão)
• O arquivo Integral Derivativo Two-Electron: nome . d2e (vazio por padrão)
• O arquivo de zero: nome . SKR
Por padrão, esses arquivos recebem um nome gerado a partir do ID de processo do processo de Gauss, e eles são armazenados no diretório raiz, designado pelo GAUSS_SCRDIR variável de ambiente (UNIX). Você também pode ver os arquivos da forma nome .inp neste diretório. Estes são os arquivos de entrada internos usados pelo programa. Se a variável de ambiente é definida, os padrões de localização para o diretório de trabalho atual do processo de Gauss.
Por padrão, esses arquivos são apagados no final de uma temporada de sucesso. No entanto, você pode querer salvar o arquivo de ponto de verificação para uso posterior em um outro trabalho de Gauss, para uso de um programa de visualização, para reiniciarum trabalho falhou, e assim por diante. Isto pode ser conseguido por nomear o arquivo de verificação, fornecendo um nome e / ou localização explícita para que, através de uma % de Chk comando no ficheiro de entrada de Gauss. Aqui está um exemplo:Chk% = água
Este comando, que é colocado no início do arquivo de entrada (antes da parte do percurso) dá ao arquivo de verificação do nomewater.chk , substituindo o nome gerado habitual e fazendo com que o arquivo seja salvo na conclusão do trabalho. Neste caso, oarquivo irá residir no diretório atual. No entanto, um comando como este irá especificar um local de diretório alternativo, bem como nome do arquivo:

Chk% = / chem/scratch2/water
Se o espaço em disco no diretório do zero é limitado, mas o espaço está disponível em outra parte do sistema, você pode querer dividir os arquivos scratch entre vários locais de disco. Os seguintes comandos permitem que você especifique os nomes e localizações dos outros arquivos scratch:
% RWF = caminho Arquivo de gravação de leitura
% Int = caminho Arquivo Integral
% D2E = caminho Arquivo Integral Derivativo
Em geral, o arquivo de leitura e escrita é de longe o maior, e por isso é o único para o qual um local alternativo é mais freqüentemente especificado.
Arquivos Divisão de raspadinhas em discos
Gaussian 09 pode resolver arquivos scratch únicas de até 16 GB em sistemas operacionais de 32 bits, como o Windows e Linux IA32.Não há necessidade de dividir arquivos de zero em 2 arquivos GB. O limite de espaço de rascunho total de 16 GB é inerente inteiros de 32 bits, no entanto, e dividir o arquivo de rascunho será não superá-lo.
Uma sintaxe alternativa é fornecida para dividir o arquivo de leitura e escrita, o arquivo Integral, e / ou o arquivo Integral Derivativo entre dois ou mais discos (ou sistemas de arquivos). Aqui está a sintaxe para o % RWF comando:
% RWF = loc 1, tamanho 1, loc 2, tamanho 2, ...
onde cada loc é um local de diretório ou um caminho de arquivo, e cada tamanho é o tamanho máximo para o segmento de arquivo naquele local. Gaussian irá gerar automaticamente nomes de arquivos originais para toda a loc que especifica apenas um diretório. Em sistemas UNIX, especificações de diretório (sem nomes) deve incluir umabarra terminal.
Por padrão, os tamanhos estão em unidades de 8 bytes palavras. Este valor também pode ser seguido por KB , MB , GB , TB , KW, MW , GW ou TW (sem intervir espaços) para especificar unidades de quilo-, mega-, giga-tera-bytes ou ou palavras. Note-se que 1 MB = 1024 2 bytes = 1.048.576 bytes (e não 1.000.000 bytes).
Um valor de -1 para qualquer parâmetro de tamanho indica que todo e qualquer espaço disponível pode ser usado, e um valor de 0 diz para usar o tamanho atual de um segmento existente. -1 é útil apenas para o último arquivo especificado, para a qual é o padrão.
Por exemplo, a seguinte diretiva divide o arquivo de leitura e escrita através de três discos:% RWF = / dalton/s0 /, 4GB, / scratch /, 3GB, / temp/s0/my_job, -1
Os tamanhos máximos para os segmentos de arquivo são 4 GB, 3 GB, e ilimitado, respectivamente. Gaussian irá gerar nomes para os dois primeiros segmentos, ea terceiraserá dado o nome my_job . Note-se que as especificações de diretório incluem barras determinais.
Devido a limitações no implementações atuais UNIX, -1 deve ser usado com cautela, uma vez que irá tentar estender um segmento de arquivo além de toda a capacidade do disco restante nesses sistemas; usá-lo também terá o efeito colateral de manter todos os segmentos de arquivos adicionais incluídos no a lista de nunca ter sido usado.

Salvando e Exclusão de arquivos de Risco
Por padrão, os arquivos são apagados scratch sem nome no final da corrida de Gauss, e arquivos nomeados são salvos. O % nosavecomando pode ser usado para alterar esse comportamento padrão. Quando esta directiva está incluído em um arquivo de entrada, chamado arquivos scratch cujas directivas aparecer no arquivo de entrada antes nosave% serão apagados no final de uma corrida (assim como todos os arquivos do zero sem nome). No entanto, se o % directiva nomear o arquivo é exibido após o nosave%directiva, o arquivo será mantido. Por exemplo, esses comandos especificar um nomepara o arquivo de verificação, e um nome alternativo e local do diretório para o arquivo de leitura e escrita, e fazer com que apenas o arquivo de ponto de verificação para ser salvo na conclusão do trabalho de Gauss:
% RWF = / chem/scratch2/water arquivos a ser excluída, clique aqui.% NosaveChk% = água arquivos sejam salvos aqui.
Observe que todos os arquivos são salvos quando um trabalho termina de forma anormal.
Arquivos de Inicialização
O sistema Gaussian inclui arquivos de inicialização para configurar o ambiente de usuário para executar o programa. Esses arquivos são:
$ G09root/g09/bsd/g09.login C shell $ g09root/g09/bsd/g09.profile Bourne shell
Note que o g09root variável de ambiente deve ser configurado pelo usuário. Assim, é habitual incluir linhas como o seguinte dentro dologon. ou perfil. arquivo para usuários de Gauss:
. arquivos de login: g09root setenv localizaçãofonte $ g09root/g09/bsd/g09.login
. Perfil arquivos: g09root = localizaçãog09root exportação. $ G09root/g09/bsd/g09.profile
Uma vez que as coisas estão configuradas corretamente, o G09 comando é usado para executar Gaussian 09 (veja abaixo).
Controlando o uso da memóriaA % Mem comando controla a quantidade de memória dinâmica a ser utilizado por Gaussian. Por padrão, 256 MB (32MW) são utilizados. Isso pode ser alterado para n palavras de dupla precisão, especificando:
% Mem = n
Por exemplo, o comando a seguir define o uso de memória para 320 milhões de bytes:% Mem = 40000000
Este valor também pode ser seguido por KB , MB , GB , TB , KW , MW , GW ou TW (sem intervir espaços) para especificar unidades de quilo-, mega-, giga-tera-bytes ou ou palavras. Por exemplo, o seguinte comando também define a quantidade de memória dinâmica de 1 GB:% = 1GB Mem

Pode ser necessário alocações ainda maiores para grandes cálculos SCF directos, pelo menos 3 N 2 palavras, onde N é o número de funções de base.
Aviso: Solicitando mais memória do que a quantidade de memória física realmente disponível em um sistema de computador vai levar a um desempenho muito pobre.
Se Gauss está sendo usado em uma máquina com memória física limitada, de modo que o padrão de 256 MB não está disponível, os algoritmos padrão, bem como a alocação dememória padrão deve ser definida de forma adequada durante a instalação.
Correndo Gaussian em Sistemas UNIXUma vez que todas as especificações de entrada e de recursos são preparados, você está pronto para executar o programa. Gaussian 09 de maio ser executado interativamente usando um dos dois estilos de comando:
G09 -nome de trabalho G09 <input-file> output-file
Na primeira forma, o programa lê a entrada de nome de trabalho . COM e escreve sua saída para -nome do trabalho . log . Quando-nome do trabalho não for especificado, o programa lê da entrada padrão e grava na saída padrão, e estes podem ser redirecionados ou canalizada na moda UNIX habitual. De qualquer forma de comando pode ser forçado no fundo da mesma forma como qualquer comando shell usando o E comercial.
Scripts e Gaussian
Scripts projetados para funcionar Gaussian 09 também podem ser criados de várias maneiras (vamos usar o shell C nestes exemplos).Primeiro, G09 comandos como aqueles acima pode ser incluído em um script shell. Em segundo lugar, a entrada efectiva de Gauss pode ser incluído no script usando o << construto:# / Bin / cshG09 << FIM> water.logChk% = água# RHF/6-31G (d) energia da água 0 1OH 1 1,0H 1 1,0 2 120,0 ENDecho "trabalho".
Todas as linhas anteriores a corda seguindo os << símbolos são tomados como entrada para o G09 de comando.
Finalmente, laços podem ser criados para executar diversos trabalhos de Gauss em sucessão. Por exemplo, o seguinte script executa todos os arquivos de entrada de Gauss especificados como argumentos de linha de comando, e mantém um registro de suas atividades no arquivo de Status :# / Bin / cshecho "Estado atual do trabalho:"> Estadoarquivo foreach ($ argv) echo "Iniciando $ file arquivo em` date `" >> Estado G09 <$ file> $ file: r.log

echo "$ file Feito com o status $ estado" >> Estadofinalecho "tudo feito." Estado >>
O seguinte script mais complexo cria arquivos de entrada de Gauss on-the-fly a partir daentrada parcial nos arquivos de dados como argumentos de linha de comando do script. O último faltam seções completas de rotas, os seus troços consistem simplesmente um #sinal ou # linha que contém palavras-chave especiais necessários para que o sistema molecular, mas nenhum método, conjunto de base, ou tipo de cálculo.
O script cria um trabalho de dois passos para cada arquivo-a otimização de Hartree-Fock entrada parcial seguido por um dos dois comandos literais incluídas no roteiro e os conteúdos de cada arquivo especificado em tempo de execução do script-consistindo cálculo MP2 energia de ponto único. Ele inclui o último explorando oGaussian 09 @ incluem mecanismo de arquivo:# / Bin / cshecho "Estado atual do trabalho:"> Estadoarquivo foreach ($ argv)echo "Iniciando $ file arquivo em` date `" >> EstadoG09 << FIM> $ file: r.logChk% = $ file: r# HF/6-31G (d) FOpt@ $ File / N - Link1 -Chk% = $ file: r% Nosave# MP2/6-31 + G (d, p) SP Guess = Leitura Geom = AllCheckENDecho "$ file Feito com o status $ estado" >> Estadoend # fim do foreachecho "tudo feito." Estado >>
Execução em lote com SNQ
Gaussian pode ser executado usando a facilidade lote SNQ nesses sistemas UNIX que suportam. O subg09 comando, definida nos arquivos de inicialização, envia um arquivode entrada para uma fila de lote. Ele tem a seguinte sintaxe:
subg09 fila-name-nome do trabalho [ -scrdir dir1 ] [ -exedir dir2 ] [ -pn ]
Os dois parâmetros necessários são os nomes de filas e de emprego. A entrada é tirado de -nome do trabalho . com e saída vai para-nome do trabalho . log , assim como para corridas interativas. O arquivo de log SNQ é enviado para trabalho de nome batch-log. .Os parâmetros opcionais -scrdir e -exedir são usados para substituir o zero defeito e diretórios executáveis, respectivamente. Todos os outros parâmetros estãoa ser tomadas opções SNQ. Em particular, p- n pode ser usado para definir a prioridade na fila de n . Esta é a prioridade para a iniciação (1 sendo o mais baixo), e não afeta a prioridade de tempo de execução.
Para submeter um trabalho SNQ de uma sessão interativa, um arquivo como o seguinte deve ser criado (com nome do arquivo nomedo trabalho. ):
# QSUB-r nome -o nome . fora-eo# QSUB-lt 2000-LT 2100# QSUB-lm 34mw 34mw-LMG09 < nome . com

onde nome deve ser substituído por um nome que é apropriado para o seu cálculo. Os primeiros nomes a linha de trabalho em execução, os nomes dos arquivos de saída e causa erros a serem incluídas no arquivo de saída. Os parâmetros de tempo são diferentes para permitir a adição de controle de trabalho para limpeza, (por exemplo, arquivar o arquivo de verificação no caso em que o trabalho excede o seu limite de tempo). Os parâmetros de memória são utilizados tanto para a programação inicial de seu trabalho para a execução e pelo programa para determinar o uso de memória dinâmica.
Este trabalho seria então submetido ao emitir o comando,
$ qsub nome . trabalho
ea saída seria colocado no seu diretório de trabalho atual.
Correndo GaussianEsta seção descreve os comandos do sistema operacional necessárias para executar Gaussian em sistemas de computadores baseados no Windows. Consulte as instruções adicionais que acompanham o programa de informações equivalentes para outros sistemas operacionais. Esta discussão pressupõe que o programa já foi instalado.
Correndo Gaussian envolve as seguintes atividades:
• Criando entrada Gaussian descrevendo o cálculo desejado.
• Especificando os locais dos vários arquivos de zero.
• Especificar os requisitos de recursos.
• Iniciando a execução do programa, ou em modo interativo ou batch.
Nesta seção, vamos supor que um arquivo de entrada Gaussian básico foi criado, e nossa discussão irá examinar os restantes três itens da lista.
Especificando Handling risco de arquivo e localGaussian utiliza vários arquivos de zero no curso de sua computação. Eles incluem:
• O arquivo Checkpoint: nome . chk
• O Read-Write file: nome . rwf
• O arquivo Integral Two-Electron: nome . int (vazio por padrão)
• O arquivo Integral Derivativo Two-Electron: nome . d2e (vazio por padrão)
• O arquivo de zero: nome . SKR
Por padrão, esses arquivos recebem um nome gerado a partir do ID de processo do processo de Gauss, e eles são armazenados no diretório raiz, designado em Preferências. Você também pode ver os arquivos da forma nome . inp neste diretório. Estes são os arquivos de entrada internos usados pelo programa. Se a preferência é definida, os padrões de localização para o diretório de trabalho atual do processo de Gauss.
Por padrão, esses arquivos são apagados no final de uma temporada de sucesso. No entanto, você pode querer salvar o arquivo de ponto de verificação para uso posterior em um outro trabalho de Gauss, para uso de um programa de visualização, para reiniciarum trabalho falhou, e assim por diante. Isto pode ser conseguido por nomear o arquivo

de verificação, fornecendo um nome e / ou localização explícita para que, através de uma % de Chk comando no ficheiro de entrada de Gauss. Aqui está um exemplo:Chk% = água
Este comando, que é colocado no início do arquivo de entrada (antes da parte do percurso) dá ao arquivo de verificação do nomewater.chk , substituindo o nome gerado habitual e fazendo com que o arquivo seja salvo na conclusão do trabalho. Neste caso, oarquivo irá residir no diretório atual. No entanto, um comando como este irá especificar um local de diretório alternativo, bem como nome do arquivo:Chk% = c: \ chem \ Scratch2 \ água
Se o espaço em disco no diretório do zero é limitado, mas o espaço está disponível em outra parte do sistema, você pode querer dividir os arquivos scratch entre vários locais de disco. Os seguintes comandos permitem que você especifique os nomes e localizações dos outros arquivos scratch:
% RWF = caminho Arquivo de gravação de leitura
% Int = caminho Arquivo Integral
% D2E = caminho Arquivo Integral Derivativo
Em geral, o arquivo de leitura e escrita é de longe o maior, e por isso é o único para o qual um local alternativo é mais freqüentemente especificado.
Arquivos Divisão de raspadinhas em discos
Gaussian 09 pode resolver arquivos scratch únicas de até 16 GB em sistemas operacionais de 32 bits, como o Windows e Linux IA32.Não há necessidade de dividir arquivos de zero em 2 arquivos GB. O limite de espaço de rascunho total de 16 GB é inerente inteiros de 32 bits, no entanto, e dividir o arquivo de rascunho será não superá-lo.
Uma sintaxe alternativa é fornecida para dividir o arquivo de leitura e escrita, o arquivo Integral, e / ou o arquivo Integral Derivativo entre dois ou mais discos (ou sistemas de arquivos). Aqui está a sintaxe para o % RWF comando:
% RWF = loc 1, tamanho 1, loc 2, tamanho 2, ...
onde cada loc é um local de diretório ou um caminho de arquivo, e cada tamanho é o tamanho máximo para o segmento de arquivo naquele local. Gaussian irá gerar automaticamente nomes de arquivos originais para toda a loc que especifica apenas um diretório. Em sistemas UNIX, especificações de diretório (sem nomes) deve incluir umabarra terminal.
Por padrão, os tamanhos estão em unidades de 8 bytes palavras. Este valor também pode ser seguido por KB , MB , GB , TB , KW, MW , GW ou TW (sem intervir espaços) para especificar unidades de quilo-, mega-, giga-tera-bytes ou ou palavras. Note-se que 1 MB = 1024 2 bytes = 1.048.576 bytes (e não 1.000.000 bytes).
Um valor de -1 para qualquer parâmetro de tamanho indica que todo e qualquer espaço disponível pode ser usado, e um valor de 0 diz para usar o tamanho atual de um segmento existente. -1 é útil apenas para o último arquivo especificado, para a qual é o padrão.
Por exemplo, a seguinte diretiva divide o arquivo de leitura e escrita através de três discos:

% RWF = c: \ dalton \ s0 \, 4GB, e: \ zero \, 3GB, f: \ temp \ s0 \ my_job, -1
Os tamanhos máximos para os segmentos de arquivo são 4 GB, 3 GB, e ilimitado, respectivamente. Gaussian irá gerar nomes para os dois primeiros segmentos, ea terceiraserá dado o nome my_job . Note-se que as especificações de diretório incluem barras determinais.
Salvando e Exclusão de arquivos de Risco
Por padrão, os arquivos são apagados scratch sem nome no final da corrida de Gauss, e arquivos nomeados são salvos. O % nosavecomando pode ser usado para alterar esse comportamento padrão. Quando esta directiva está incluído em um arquivo de entrada, chamado arquivos scratch cujas directivas aparecer no arquivo de entrada antes nosave% serão apagados no final de uma corrida (assim como todos os arquivos do zero sem nome). No entanto, se o % directiva nomear o arquivo é exibido após o nosave%directiva, o arquivo será mantido. Por exemplo, esses comandos especificar um nomepara o arquivo de verificação, e um nome alternativo e local do diretório para o arquivo de leitura e escrita, e fazer com que apenas o arquivo de ponto de verificação para ser salvo na conclusão do trabalho de Gauss:
% RWF = c: \ chem \ Scratch2 \ água arquivos a ser excluída, clique aqui.% NosaveChk% = água arquivos sejam salvos aqui.
Observe que todos os arquivos são salvos quando um trabalho termina de forma anormal.
Controlando o uso da memóriaA % Mem comando controla a quantidade de memória dinâmica a ser utilizado por Gaussian. Por padrão, 256 MB (32MW) são utilizados. Isso pode ser alterado para n palavras de dupla precisão, especificando:
% Mem = n
Por exemplo, o comando a seguir define o uso de memória para 320 milhões de bytes:% Mem = 40000000
Este valor também pode ser seguido por KB , MB , GB , TB , KW , MW , GW ou TW (sem intervir espaços) para especificar unidades de quilo-, mega-, giga-tera-bytes ou ou palavras. Por exemplo, o seguinte comando também define a quantidade de memória dinâmica de 1 GB:% = 1GB Mem
Pode ser necessário alocações ainda maiores para grandes cálculos SCF directos, pelo menos 3 N 2 palavras, onde N é o número de funções de base.
Aviso: Solicitando mais memória do que a quantidade de memória física realmente disponível em um sistema de computador vai levar a um desempenho muito pobre.
Se Gauss está sendo usado em uma máquina com memória física limitada, de modo que o padrão de 256 MB não está disponível, os algoritmos padrão, bem como a alocação dememória padrão deve ser definida de forma adequada durante a instalação.
Configurando Gaussian

Esta seção descreve os procedimentos gerais para a instalação e configuração de Gauss em sistemas UNIX. Não deixe de verificar as instruções e notas de versão que acompanham a sua versão do programa para obter instruções adicionais ou alternativos referentes ao seu sistema de computador particular.
Requisitos do sistema• Os diretórios de Gauss vai exigir cerca de 1,5-2 GB de espaço em disco para os
arquivos executáveis, dependendo do sistema de computador.
• A alocação de memória padrão no Gaussian 09 é de 256 MB. As grandes dimensões fixas do programa exigir um tamanho de espaço de troca de 1-2 GB. É claro que o espaço de swap adicional será necessário se mais memória é solicitada em um trabalho usando a % Mem Ligação 0 comando, ou através do -M- comando no Default.Route arquivo. Esses requisitos são para cada trabalho executado simultaneamente.
• Consulte a lista de plataforma que vem com o CD. A versão mais recente deste documento pode ser encontrado nowww.gaussian.com/g09_plat.htm .
Configurando o ambiente de execução de GaussGaussian localiza executáveis e cria arquivos de zero em diretórios especificados por várias variáveis de ambiente. No entanto, o usuário é responsável pela criação de dois deles:
• g09root : Indica o diretório onde o G09 diretório reside (ou seja, o diretório acima dela).
• GAUSS_SCRDIR : Indica o diretório que deverá ser usado para arquivos de zero.
Os arquivos de inicialização de Gauss é responsável por inicializar outros aliases e variáveis de ambiente, conforme necessário. Todos os usuários de Gauss precisa executar o arquivo de inicialização Gaussian apropriado dentro de seu arquivo de inicialização específica do shell UNIX. Consulte o Capítulo 2 para mais detalhes.
As variáveis de ambiente criadas pelo g09.login e g09.profile incluem:
• GAUSS_EXEDIR : Especifica os diretórios nos quais as imagens de Gauss são armazenados. Por padrão, ele inclui o principal diretório $ g09root/g09 e vários diretórios alternativos.
• GAUSS_ARCHDIR : Especifica o diretório no qual o principal arquivo de todo o site é mantido, e no qual arquivos de arquivo temporário deve ser colocado se o arquivo principal não está disponível. O padrão é $ g09root/g09/arch se desactivado.
• G09BASIS : O diretório que contém os arquivos que especificam os conjuntos de bases Gaussianas armazenados internamente padrão, bem como alguns conjuntos de bases adicionais na forma de entrada de conjunto de base geral. Esta variável de ambiente é fornecido por conveniência e é projetado para uso com o @ incluem mecanismo.
Rede / fragmentação de cálculos paralelos usando Linda também pode usar a GAUSS_LFLAGS variável de ambiente para passar as opções para o processo de Linda. Veja a discussão de executar trabalhos de Gauss com Linda para mais detalhes.

Considerações risco arquivo
Em sistemas UNIX, Gaussian gera nomes exclusivos de arquivo do zero com base na identificação do processo, quando nenhum nome foi especificado pelo usuário. Este mecanismo é projetado para permitir que vários trabalhos de Gauss para executar simultaneamente usando um diretório zero comum.
Raspadinha arquivos são apagados automaticamente quando uma tarefa é concluída com êxito ou morre de forma limpa por padrão.No entanto, os arquivos de zero não são apagados quando um trabalho é morto externamente ou não terminar de forma anormal.Conseqüentemente, os arquivos que sobraram podem acumular-se no diretório raiz.
Um método fácil para evitar a desordem excessiva é ter todos os usuários compartilham um diretório zero comum, e ter esse diretório zero apuradas no momento da inicialização do sistema, adicionando um rm comando para o script de inicialização do sistema apropriado (por exemplo, / etc / rc ou um dos os arquivos sob / etc/rc.d/rc3.d ). Se o sistema de lote SNQ está em uso, limpando o diretório do zero também deve ser feito antes SNQ é iniciado, garantindo que nenhum trabalho estiver usando o diretório em que está desmarcada.
Usando Gaussian 09 com LindaEsta seção descreve o processo de instalação do software Linda que você adquiriu através de Gaussian, Inc. ea construção de uma versão paralela de memória distribuída de Gauss. Ele assume que você já construiu e testou a versão regular do programa. Ele também assume que você leu as instruções de instalação normais e também que você tem acesso a Referência do Usuário do Gaussian 09.
Métodos Paralelos LindaHF, CIS = direta, e DFT em moléculas são Linda paralelo, incluindo energias, otimizações e freqüências. Energias TDDFT e gradientes e energias MP2 e gradientes são também Linda paralelo. Porções de freqüência MP2 e cálculos CCSD são Linda paralelo, mas outros são apenas SMP-paralelo, então eles vêem alguma aceleração do uso de alguns nós, mas nenhuma melhoria adicional de um maior número de nós.
É sempre melhor usar SMP-paralelismo dentro de nós e Linda só entre nós. Por exemplo em um cluster de 4 nós, cada um com um duplo quad-core EM64T, deve-se usar% NProcShared = 8% LindaWorkers = no1, no2, node3, node4
ao invés de usar mais de um trabalhador Linda por nó.
Instalando o software Linda e Compilando G09/LindaSe você comprou os binários de Gauss, Linda é distribuído no mesmo CD, como os binários de Gauss e nenhuma instalação adicional é necessária. Siga as instruções do arquivo README arquivo no CD de distribuição.
Se você tiver adquirido o código-fonte de Gauss, então Linda é distribuído em um CD separado. Siga as instruções do arquivoREADME.source arquivo no CD de distribuiçãofonte Gaussian instalar Linda e compilar Gaussian usar Linda.

Em ambos os casos, você deve executar o comando bsd / install conforme detalhado nosarquivos LEIA-ME e folhas de instruções de instalação.
Correndo Gaussian com LindaO modelo de programação paralela Linda envolve um processo principal, que é executado no processador atual, e uma série de processos de trabalho que podem ser executados em outros nós da rede. Assim, uma corrida Gaussian 09/Linda deve especificar o número de processadores a serem usados, a lista de processadores onde os trabalhos devem ser executados, e, ocasionalmente, outros parâmetros de trabalho. Umavariável de ambiente é geralmente a maneira mais fácil de especificar essas informações(como veremos).
Cada um destes nós precisa ter algum acesso ao diretório árvore Gaussian 09. A configuração recomendada é ter G09 em cada sistema que irá ser utilizado para o trabalho em paralelo. Note-se que os binários Linda precisa ter o mesmo caminho em cada máquina. Se isso não for viável, a árvore G09 pode ser acessível através de um disco NFS-montado que é montado em um local idêntico em todos os nós.
Para cálculos MP2, cada nó também deve ter algum disco local onde Gaussian 09 pode colocar arquivos temporários. Este é definido como de costume através do GAUSS_SCRDIR variável de ambiente, que deve ser definido no . cshrc ou . perfil da sua conta em cada nó.
Configurando Gaussian 09/LindaGaussian 09 recebe informações de configuração a partir de três fontes primárias
• O arquivo de entrada Gaussian via % link0 comandos.
• O Default.Route arquivo.
• A variável de ambiente GAUSS_LFLAGS .
Detalhes sobre % link0 comandos ea Default.Route arquivo também pode ser encontrada no manual de referência do usuário do Gaussian 09. Entradas específicas para Gaussian 09/Linda são descritos abaixo.
Especificação dos computadores TRABALHADORES
O % LindaWorkers directiva é usado para especificar os computadores em que os processos de trabalho Linda deve ser executado.Ele tem a seguinte sintaxe:
% LindaWorkers = no1 [: n1 ] [, node2 [: n2 ]] [...]
Esta lista o nome do nó de TCP para cada nó de usar. Por padrão, um trabalhador Linda é iniciado em cada nó, mas o valor opcional permite que isso seja variada. Um trabalhador é sempre iniciado no nó em que o trabalho é iniciado (o nó mestre) se é ou não aparece na lista de nós. % LindaWorkers pode ser combinada com NProcShared% . Neste caso, um ou mais processos de trabalho paralelas será executado em cada nó (o número ainda determinada pelos valores em % LindaWorkers ). O valor a % NProcShared especifica o número de processadores SMP / núcleos de usar em cada sistema na lista de nós do trabalhador.
Não use o obsoleto % NProcLinda directiva. G09 irá calcular o número total de trabalhadores Linda baseada na % LindaWorkersentrada.
A seguinte diretiva faz com que um trabalho paralelo de rede para ser executado através dos especificados 5 nós. Nodes Hamlet eOfélia vai cada executar dois processos de trabalho.

% LindaWorkers = aldeia: 2, Ophelia: 2, Laertes, Horácio, lear
As seguintes diretivas especificar que um trabalho paralelo será executado em hosts Noruega , Itália e Espanha . Nodes Noruega eItália vão cada executar um 4-way SMP trabalhador paralelo e Espanha vai executar dois desses trabalhadores:
% NProcShared = 4 Especifica quatro vias paralelismo SMP.% LindaWorkers = Noruega, Itália, Espanha: 2
Essas diretrizes fazem sentido quando Noruega e Itália são 4 computadores de processador / núcleo, e Espanha é um 8 processador / computador central.
Note que o nproc% diretriz usada em versões anteriores de Gauss é obsoleto.
Especificando a quantidade de memória para um CÁLCULO PARALELO
Memória é especificado usando o Mem% comando link0, assim como para cálculos de série.
UTILIZAÇÃO ssh ao invés do RSH
Por padrão, Linda usa rsh para a comunicação entre os nós. Você pode usar o ssh ao invés, incluindo a seguinte opção noGAUSS_LFLAGS variável de ambiente:% setenv GAUSS_LFLAGS '...-opt "Tsnet.Node.lindarsharg: ssh"'
Outra maneira de substituir esse padrão, você precisa criar um arquivo de configuração no seu diretório home no nó mestre chamadotsnet.config. que contém a seguinte linha:Tsnet.Node.lindarsharg: ssh
Isso fará com que o ssh para ser usado em seu lugar. Note-se que sem senha ssh logins já deve estar configurado com o mestre de todos os nós do trabalhador.
ESPECIFICAÇÃO DE OUTRAS OPÇÕES LINDA
Algumas opções Linda, que são por vezes úteis são os seguintes:
-V Mostrar detalhado mensagens -vv Mostrar mensagens muito detalhados
Use o GAUSS_LFLAGS variável de ambiente para defini-las.
Por exemplo, pode-se ativar a saída Linda muito detalhado usando:% setenv GAUSS_LFLAGS-vv
Existem muitas outras opções de Linda, mas a maioria deles não são utilizados por Gauss. Verifique o manual Linda na Internet emwww.lindaspaces.com / downloads / lindamanual.pdf . O opt- forma pode ser usado em GAUSS_LFLAGS para invocar qualquer válido tsnet.config. directiva arquivo. Note-se que Gauss 09/Linda não usa o nativo recursos Linda minworker e maxworker .
Começando paralelas Gaussian 09 JobsO G09 comando é usado como de costume para iniciar uma memória distribuída paralelamente um Gaussian 09 emprego. Para um trabalho paralelo Linda para executar com êxito, as seguintes condições devem ser verdadeiras:
• Você já executou o Gaussian 09 arquivo de inicialização apropriado ( $ g09root/g09/bsd/g09.login ou $ g09root/g09/bsd/g09.profile ). Teste isso executando um cálculo Gaussian série 09 no nó mestre.
• O diretório $ g09root/g09 é acessível em todos os nós.

• O LD_LIBRARY_PATH variável de ambiente é definida (ver o G09 notas de instalação) para localizar as bibliotecas compartilhadas Linda.
• Espaço de rascunho local está disponível em cada nó, se necessário (via GAUSS_SCRDIR ).
• Todos os nós em que o programa será executado são confiáveis por parte do anfitrião atual. Você deve ser capaz de fazer o login remotamente com o rlogin ou ssh comando sem ter que dar uma senha para cada um deles. Contacte o seu administrador do sistema sobre a configuração de segurança para os nós da rede.
Os cálculos podem então ser iniciado como se fosse para um cálculo de série:% G09 entrada &
e Gaussian 09 irá iniciar os processos de mestrado e de trabalho, conforme necessário.
Acompanhamento do CálculoVocê vai ver os processos iniciados nos nós dos trabalhadores para os links que foram paralelizados, por exemplo, eles terão a * . exelentrada no diretório G09 principal. Usando os principais comandos ou outros em nós trabalhadores permitirá que você veja lxxx.exelquando é iniciado.
A medida relevante do desempenho para um cálculo paralelo é o tempo decorrido do relógio ou na parede. A maneira mais fácil de verificar isso é usar um monitor externo como tempo , tempos , ou timex , por exemplo,% tempos de entrada e G09
que apresentará um relatório decorrido, CPU e os tempos do sistema. Note-se que os dois últimos são apenas no nó mestre e quantidades similares de CPU e sistema de tempo são gastas em cada nó. Assim, a aceleração é a razão entre o tempo decorrido executando um trabalho de série e o tempo decorrido para o trabalho paralelo.
Especificando Trabalhadores por nó em Macs baseados em PPC.A -mp n opção pode ser usada para executar Gaussian com Linda através de vários de energia baseada em PC Mac OS X e outros sistemas com múltiplos processadores. Ele especifica o número máximo de processos Linda a ser programadas por nó. Configure-a para 2 quando todos os nós individuais são processadores dual.

Personalização Site: O Arquivo Default.RouteDependendo das características de um sistema de computador particular, às vezes é necessário por motivos de desempenho para substituir alguns dos padrões embutidos noprograma. Isto pode ser feito através da criação de um arquivo de personalização do site.Em sistemas Unix, este arquivo é chamado Default.Route , residente em $ g09root/g09 . No Windows, o arquivo de padrões de Gauss é Default.Rou , e está localizado no subdiretório zero Gaussian 09W (por exemplo, C: \ G09W \ zero ). O formato do arquivo é o mesmo em todos os sistemas de computador.
As subseções a seguir descrevem os tipos de informação que podem ser fornecidos no arquivo de padrões.
Padrões Rota
Esses parâmetros são introduzidos por - # - e têm a mesma forma que os comandos normais do troço. Por exemplo, esta linha irá definir o algoritmo SCF padrão para o algoritmo convencional (não-direta):- # - SCF = Convencional
Pode haver mais do que uma - # - linha do ficheiro.
Comandos listados na Default.Route alterar apenas os padrões, eles são substituídos pornada especificado na seção de rota de um arquivo de entrada. Assim, se o Default.Route contém:- # - = MP2 NoDirect
e a parte do percurso que contém o MP2 palavra-chave, em seguida, o algoritmo MP2 convencional será utilizado. No entanto, se a parte do percurso que contém o MP2 = directa palavra-chave, em seguida, o algoritmo directa será usado.
Todos os sites vão querer especificar a quantidade de espaço em disco de trabalho disponível através do MaxDisk palavra-chave noDefault.Route arquivo. Por exemplo, aseguinte linha define MaxDisk para 800 MB:- # - MaxDisk = 800MB
A directiva -R- é um sinônimo para - # - .
A quantidade de espaço em disco disponível é dada em palavras de 8 bytes (padrão). Este valor também pode ser seguido por KB ,MB , GB , TB , KW , MW , GW ou TW (sem intervir espaços) para especificar unidades de quilo-, mega-, giga-tera-bytes ou ou palavras . O espaço em disco do zero é definido ilimitado ( -1 ) por padrão, ou seja, presume-se que o espaço em disco suficiente disponível para executar um determinado cálculo sem trabalho redundante. Assim, especificando a quantidade de memória disponível e do disco é de longe a mais importante forma de otimizar o desempenho de seus cálculos. Isso permiteque o programa para decidir entre os vários algoritmos disponíveis, selecionando o caminho ideal para a configuração do seu sistema particular. Tenha em mente que o espaço em disco disponível, o mais rápido da avaliação, especialmente para MP2.
Default.Route Limitações
Nem todas as palavras-chave parte do percurso são homenageados no Default.Route arquivo. Em geral, a regra é que apenas as opções que não afetam o resultado de um cálculo (ou seja, não altere os valores de quaisquer quantidades previstas) são permitidos no arquivo. Assim, SCF = Convenção , o que muda apenas o

algoritmo de armazenamento integral, será homenageado, enquanto Int (Grid = 3) , o que afeta os resultados de vários tipos de cálculos, será ignorado.
Padrões de memória
Trabalhos de Gauss, que imprudentemente usam memória excessivo pode causar dificuldades graves no sistema. A memória directiva -M- impõe um limite de memória dinâmica padrão. Por exemplo, a linha a seguir define o uso de memória padrão para 400 MB:-M-50000000
A quantidade de memória disponível é dada em palavras de 8 bytes (padrão). Este valortambém pode ser seguido por KB , MB ,GB , TB , KW , MW , GW ou TW (sem intervir espaços) para especificar unidades de quilo-, mega-, giga-tera-bytes ou ou palavras. O tamanho da memória padrão é de 256 MB. Note-se que esse limite pode serultrapassado com a Mem% Ligação 0 comando.
A memória directiva -U- memória conjuntos padrão a ser usado em utilitários como FORMCHK e freqchk . A -F- directiva é o argumento de tipo de arquivo padrãopara FORMCHK .
Execução paralela em memória compartilhada multiprocessadores
Se o seu sistema de computador tem vários processadores / núcleos e processamento paralelo é compatível com a sua versão do Gaussian, você pode especificar o número padrão de processadores a serem usados no Default.Route arquivo. Por exemplo, o comando a seguir define o número padrão de processadores para 4:-P-4
Normalmente, o programa padrão para execução em um único processador. O % NProcShared Ligação 0 comando pode ser usado para substituir o padrão para um trabalho específico. Claramente, o número de processadores solicitados não deve exceder o número de processadores disponíveis, ou a uma diminuição substancial no desempenho vai resultar.
Você também pode especificar as CPUs específicas sobre a qual são executados com o -C- directiva. Por exemplo, a seguinte diretiva especifica que o programa deve ser executado nos primeiros cinco núcleos de um sistema hexacore (reservando um núcleo para outro uso):-C-0,1,2,3,4
Execução paralela Rede / Cluster
Você pode especificar a lista de trabalhadores Linda em Default.Route via -W- directiva:-W-dalton, Lavoisier: 2, Priestley, Agassiz: 3, curie = 20
Este exemplo vai usar os cinco nós especificados para execução paralela, colocando dois processos de trabalho em Lavoisier , 3 trabalhadores em Agassiz , e um trabalhadorem cada um dos outros sistemas. Se o mestre nó-nó em que o trabalho não foi iniciado, é um desses sistemas, um trabalhador também será executado no sistema (perfazendo um total de seis nós).
Esta directiva corresponde ao e pode ser substituído por-do link 0 comando % LindaWorkers .
Com relação ao uso de Default.Route com Linda, se você tem diferentes filas de lote

em um cluster que correspondem a diferentes conjuntos de nós e cada tarefa em lote é executado em seu próprio diretório temporário padrão, a melhor abordagem é criar um script que copia um padronizada Default.Route apropriado para a fila especial para o diretório padrão atual.
Quando -W- é combinado com -P- n , então um n processo de trabalho SMP paralelo caminho é iniciado em cada nó na lista de nós (ou mais de um tal processo, quando mais trabalhadores são especificados para que o nó em -W- ) .
A -L opções conjuntos padrão directiva para Linda, que são então passadas para o GAUSS_LFLAGS variável de ambiente.
Nome do site
O nome do local pode ser definido com -S- , cujo valor especifica o nome do local a serutilizada em entradas de arquivo gerada por Gaussian. O nome do site padrão é GINC. Por exemplo, a seguinte linha define o nome do site para EXPCONS:-S-EXPCONS
Nome do host
O nome do host pode ser definido com -H- , o valor especifica o nome do host para ser usado em entradas de arquivos gerados por Gauss. O padrão é o nome do host atual.
Configurações padrão típicos
Aqui estão as configurações padrão razoáveis para várias configurações de máquina:
• Por uma pequena estação de trabalho com 2 GB de memória e 200 GB de disco,os algoritmos padrão e alocação de memória estão bem. MaxDisk é tudo o que precisa ser especificado.
• - # - MaxDisk = 50GB
• Em uma estação de trabalho poderosa com 8 processadores e 8 GB de memória, sendo usados para grandes trabalhos, todos os 8 processadores deve ser usado por padrão. Além disso, mais memória deve ser dado a cada trabalho:
• -M-4GB• -P-8
- # - MaxDisk = 100GB
Padrões do usuário Arquivos
Gauss usuários podem definir seus próprios padrões, criando a sua própria Default.Route arquivo. Gaussian verifica o diretório de trabalho atual para um arquivo com este nome quando um trabalho é iniciado. Configurações no arquivo local têm precedência sobre aquelas no arquivo de todo o site, e as opções especificadas na parte do percurso do trabalho têm precedência sobre os dois.
Variáveis de Ambiente
Todas essas diretrizes também pode ser definido através de variáveis de ambiente ou argumentos de linha de comando UNIX. A variável de ambiente GAUSS_ X DEF oferece uma linha equivalente a - X- no Default.Route arquivo. Da mesma forma, o argumento de linha de comando para G09 - x = "valor" especifica a mesma configuração. Por exemplo, todas as seguintes características têm o efeito equivalente: -M-4GB em Default.Route

exportação GAUSS_MDEF = 4GB G09-m = "4GB" ...
Ordem de Prioridade
A ordem de prioridade é: o argumento de linha de comando, variável de ambiente, Default.Route definição, padrão de programa interno.
Correndo Gaussian Teste JobsUm extenso conjunto de trabalhos de teste para Gaussian são fornecidos, juntamente com seus arquivos de saída correspondentes. Os arquivos de entrada são encontrados nodiretório $ g09root/g09/tests/com . Um ou mais subdiretórios $ g09root/g09/tests , nomeados para os tipos de arquitetura de computadores (por exemplo, ia64 ), conter a saída de referência para os vários trabalhos de teste (arquivos de log gzipped). Um arquivo de script que corre faixas de postos de trabalho de teste automaticamente também é fornecido (descrito abaixo).
Se você construir o programa a partir do código fonte sob uma arquitetura suportada e sistema operacional e usando o compilador especificado, bibliotecas e outros softwares (se houver), recomendamos que você executar algumas das tarefas de teste para verificar se o programa foi construído corretamente . No entanto, não é necessário executar todo o conjunto de testes. Vejawww.gaussian.com/g09_plat.htm para a atual lista de plataformas suportadas e software necessário.
Você não precisa executar todos os trabalhos de teste para distribuições binárias.
Arquivos de entrada trabalho de teste têm nomes da forma teste nnn . com . Testes 1, 28, 94, 155, 194, 296, 302 e cobrem uma gama de capacidades de Gauss. Note-se que alguns postos de trabalho de teste são destinados a hardware rápido e são bastante carosem menores, sistemas de computadores mais lentos. O arquivo $ g09root/g09/tests/tests.idx lista o que cada trabalho de teste faz.
Renomeie existente Default.Route arquivo antes de executar o teste de Jobs
Se você optar por executar alguns ou todos os trabalhos de teste de Gauss, você precisará ter certeza de que eles correm com as configurações padrão built-in do programa. Portanto, você vai precisar para mudar o nome de ambos os em todo o site Default.Routearquivo (localizado no $ g09root/g09 diretório), bem como qualquer versão individual do arquivo de padrões que você pode ter antes de executar qualquer trabalho de teste. Note que certas configurações neste arquivo podem causar alguns trabalhos de teste falhar.
Exemplos
O script submit.csh pode ser usado para executar tarefas de teste. Ele aceita dois parâmetros: os números do primeiro e últimos trabalhos a executar (por padrão, todos os testes são executados). Observe que você deve executar os trabalhos de teste a partir de um diretório separado para evitar que sobrepor a saída de referência.
Os comandos a seguir ilustram o procedimento recomendado para a execução de um trabalho de teste, usando o diretório / chem / newtests como área de trabalho do teste executor e trabalho de teste 28 como um exemplo:$ mkdir / chem / newtests; cd / chem / newtests $ . ln-s $ g09root/g09/tests/com

$ mkdir `gau-máquina`
$ $ g09root/g09/tests/submit.csh mn &
O comando final é executado teste m por n .
Após cada trabalho de teste terminar, verifique se ela foi concluída com êxito. Em seguida, compare a sua produção atual com a saída de referência usando o d1 script. Por exemplo:
$ $ g09root/g09/tests/d1 m n
A d1 roteiro filtra diferenças insignificantes dos arquivos de saída para trabalhos de teste estou através n e envia a saída restante através de mais . As diferenças que aparecem deve ser limitado a itens não-essenciais.
Construindo Z-MatrizesEsta seção apresenta um breve panorama das descrições tradicionais de sistemas moleculares Z-matriz. Há restrições quanto ao tamanho de uma matriz-Z: O número máximo de variáveis eo número máximo de átomos dentro de um cálculo. Estes são definidos de forma consistente para um máximo de 250 mil átomos de reais (incluindo fantasma, mas os átomos não fictícios), e um máximo de 250 mil centros de Z-matriz (átomos, átomos de fantasmas, e átomos dummy).
Usando coordenadas internasCada linha de uma matriz-Z dá as coordenadas internas de um dos átomos dentro da molécula. O formato Z-matriz mais usada usa a seguinte sintaxe:
Elemento-label , átomo 1, vínculo de comprimento , átomo 2, bond-ângulo , átomo 3, diedro angular [ formato de código ]
Embora estes exemplos use vírgulas para separar itens dentro de uma linha, qualquer separador válido pode ser usado. Elemento-label é uma cadeia de caracteres que consiste em um ou outro o símbolo químico do átomo ou seu número atômico. Se o símbolo elementar é utilizado, ele pode ser opcionalmente seguido por outros caracteresalfanuméricos para criar uma etiqueta de identificação para esse átomo. Uma prática comum é a de seguir o nome do elemento com um número inteiro de identificação secundário: C1, C2, etcquímico de símbolo para o átomo ou seu número atômico. Se osímbolo elementar é utilizado, ele pode ser opcionalmente seguido por outros caracteresalfanuméricos para criar uma etiqueta de identificação para esse átomo. Uma prática comum é a de seguir o nome do elemento com um número inteiro de identificação secundário: C1, C2, etc
Atom 1, átomo 2, átomo 3 são os rótulos dos átomos previamente especificados e são usados para definir a posição dos átomos atuais. Alternativamente, os números de linha dos demais átomos dentro da seção especificação molécula poderá ser utilizada para os valores das variáveis, onde a carga ea linha multiplicidade de spin é a linha 0.rótulos para átomos previamente especificados e são usados para definir a posição dos átomos atuais. Alternativamente, os números de linha dos demais átomos dentro da seção especificação molécula poderá ser utilizada para os valores das variáveis, onde a carga ea linha multiplicidade de spin é a linha 0.
A posição do átomo de corrente é então determinado mediante a extensão da ligação unindo-a ao átomo 1, o ângulo formado por esta ligação e a ligação unindo átomo 1

e átomo de 2, e o (torção), ângulo diedro formado pelo plano que contém átomo de um,átomo de 2 e átomo de 3 com o plano que contém o átomo de corrente, átomo e um átomo de 2. Note que os ângulos de ligação deve estar no intervalo de 0 ° < ângulo <180 °. Ângulos diedros pode assumir qualquer valor.
O parâmetro de formato de código opcional especifica o formato da entrada Z-matriz. Para a sintaxe que está sendo descrito aqui, este código é sempre 0 . Este código é necessário apenas quando os parâmetros adicionais seguir os dados normais deespecificação Z-matriz, como em um ONIOM cálculo.
Como um primeiro exemplo, considere o peróxido de hidrogénio. A matriz-Z para esta estrutura seria:HO 1 0,9O 2 1 1.4 105.0H 3 0,9 2 105,0 1 120,0
A primeira linha da matriz-Z especifica apenas um átomo de hidrogénio. A linha seguinte lista um átomo de oxigênio e especifica a distância internuclear entre ele eo hidrogênio como 0,9 Angstrom. A terceira linha define outra oxigénio com uma distância de 1,4 Angstrom OO (isto é, a partir de átomo de 2, o outro de oxigénio) e com um ângulo OOH (com átomos 2 e 1), de 105 graus. A quarta e última linha é o único para o qual devem ser prestadas todas as três coordenadas internas. Ele define o outro hidrogénio como ligado ao segundo de oxigénio com uma distância de 0,9 Angstrom HO, um ângulo de 105 graus de HOO e um ângulo diedro HOOH de 120 graus.
As variáveis podem ser utilizadas para especificar alguns ou todos os valores dentro da matriz-Z. Aqui está outra versão do Z-matriz anterior:HO 1 R1 O 2 R2 1 A H 3 R 1 2 A 1 D Variáveis:R1 0,9R2 1.4A 105,0D 120,0
Restrições de simetria na molécula são reflectidos nas coordenadas internas. As duas distâncias HO são especificados pela mesma variável, assim como os dois ângulos de ligação HOO. Quando uma tal matriz Z é usado para uma optimização da geometria emcoordenadas internas ( Opt = Z-matriz ), os valores das variáveis serão optimizados para localizar a estrutura de menor energia. Para uma optimização completa ( FOpt ), as variáveis são obrigados a ser linearmente independente e incluem todos os graus de liberdade na molécula. Para uma otimização parcial ( Popt ), variáveis em uma segundaseção (muitas vezes rotulado Constantes :) são mantidos fixos em valor, enquanto aqueles na primeira seção são otimizados: Variáveis:R1 0,9R2 1.4A 105,0 Constantes:D 120,0
Veja os exemplos na discussão do Opt palavra-chave para obter mais informações

sobre otimizações em coordenadas internas.
Mistura Interna e coordenadas cartesianas
Coordenadas cartesianas são realmente um caso especial da matriz Z, como neste exemplo:C 0,00 0,00 0,00C 0,00 0,00 1,52H 1.02 0.00 -0.39H -0.51 -0.88 -0.39H -0.51 0.88 -0.39H -1.02 0.00 1.92H 0.51 -0.88 1.92H 0,51 0,88 1,92
Também é possível a utilização de ambas as coordenadas cartesianas e internas dentro da mesma de Z-matriz, tal como neste exemplo:O 0 0 xo. zoC 0 0. yc 0.C 0 0. Yc-0.N 0 0 xn. 0.H 2 r1 3 1 a1 b1H 2 r2 3 a2 1 b2H 3 r1 2 a1 1-b1H 3 r2 2 1-a2 b2H 4 r3 2 a3 3 d3 Variáveis: -1 xo.zo 0.yc 1.xn 1.r1 1,08r2 1,08r3 1.02a1 125.a2 125.d3 160.b1 90.b2 -90.
A matriz Z tem vários recursos dignos de nota:
• Os nomes das variáveis para as coordenadas cartesianas são dadas, simbolicamente, do mesmo modo como para as variáveis de coordenadas internas.
• O inteiro 0 após o símbolo atômico indica coordenadas cartesianas simbólicos a seguir.
• Coordenadas cartesianas pode ser relacionado por uma mudança de sinal ângulos diedros assim como pode.
Alternate Z-matriz Formato
Um formato de Z-matriz alternativa permite posições nucleares a serem especificados usando dois ângulos de ligação, em vez de um ângulo de ligação e um ângulo diedro. Isso é indicado por um uma em um campo adicional a seguir ao segundo ângulo(este campo padrão a 0 , o que indica um ângulo diedro como o terceiro componente):C4 0.9 O1 O2 C2 120,3 180,0 0C5 1.0 O1 C2 C4 110,4 105,4 1

C6 O1 R C2 C3 A1 A2 1
A primeira linha utiliza um ângulo diedro enquanto as duas últimas usa um segundo ângulo de ligação.
Usando manequim Átomos
Esta secção ilustram a utilização de átomos de manequim dentro Z-matrizes, os quais são representados pela pseudo atómica símboloX . O exemplo seguinte ilustra o uso de um átomo de manequim para fixar o eixo de três vezes na C 3v amoníaco:NX 1 1.H 1 NH 2 HNXH 1 NH 2 3 120,0 HNXH 1 NH 2 HNX 3 -120,0 nh 1.0HNX 70,0
A posição do manequim no eixo é irrelevante, e a distância de 1,0 utilizado pode ter sido substituído por qualquer outro número positivo. HNX é o ângulo entre uma ligaçãoNH e o eixo triplo.
Aqui está uma matriz-Z para oxirane:XC1 X halfccOX boi C1 90.C2 X halfcc O 90. C1 180,0H1 C1 ch X HCC O hccoH2 C1 ch X HCC O-hccoH3 C2 ch X HCC O hccoH4 C2 ch X HCC O-hcco halfcc 0,75boi 1.0ch 1,08HCC 130,0hcco 130,0
Este exemplo ilustra dois pontos. Em primeiro lugar, um átomo de manequim é colocado no centro da ligação CC para ajudar a limitar o cco triângulo isósceles ser. boi é em seguida a distância perpendicular a partir de O para a ligação CC, e os ângulos OXC são mantidas a 90 graus. Em segundo lugar, algumas das entradas na matriz Z são representados pela negativa do variável ângulo diedrohcco .
Os exemplos seguintes ilustram a utilização de átomos fictícios para especificar ligações lineares. Otimizações de geometria em coordenadas internas são incapazes de lidar com ângulos de ligação de graus l80 que ocorrem em fragmentos moleculares lineares, como o acetileno ou o C 4 cadeia em butatriene. Dificuldades também pode serencontrado em situações quase linear, tais como os grupos etinilo em moléculas assimétricas. Estas situações podem ser evitadas através da introdução de átomos fictícios ao longo da bissectriz do ângulo e utilizando a metade do ângulo que a variávelou constante:NC 1 cnX 2 1. 1 90.H 2 ch 3 90. 1 180. cn 1,20

ch 1,06
Da mesma forma, nesta matriz Z destinados para uma otimização de geometria, metade representa a metade do ângulo NCO que é esperado para estar perto de linear. Note-se que um valor de metade menor do que 90 graus corresponde a uma disposição de cis:NC 1 cnX 2 1. 1 metadeO 2 CO 3 metade 1 180,0H 4 oh 2 coh 3 0.0 cn 1,20co 1.3oh 1.080,0 metadecoh 105.
Geometria Builder Modelo EspecificaçõesO construtor modelo é outra facilidade dentro Gaussian para especificar rapidamente certos tipos de sistemas moleculares [ Pople67a ] .É solicitado aos Modela ou ModelB opções para o Geom palavra-chave, e que exige entrada adicional em uma seção separada dentro do arquivo de trabalho.
O insumo básico para o construtor do modelo é chamado de matriz de fórmula curta , uma coleção de linhas, cada um dos quais define um átomo (por símbolo atômico) e suaconectividade, por até mais seis entradas. Cada um destes pode ser um inteiro, que representa o número da linha que define um outro átomo explicitamente especificada deque o átomo de corrente está ligado, ou um símbolo atómico (por exemplo, H, F), ao qual o átomo de corrente está ligado por uma ligação de terminal , ou um símbolo para um grupo funcional terminal que é ligado ao átomo de corrente. Os grupos funcionais disponíveis atualmente são OH, NH2, Me, Et, NPr, IPR, NBu, IBU, e UTB.
A matriz de fórmula curta também define implicitamente a geometria de rotação sobre cada elo da seguinte maneira. Suponha que os átomos X e Y são explicitamente especificado. Então X aparecerão na fila Y e Y aparecerão na fila de X. Deixe eu ser o átomo à direita de X na linha Y e J ser o átomo à direita de Y na linha X. Em seguida, os átomos I e J são colocados em a orientação trans sobre o vínculo XY. A matriz de fórmula curta podem ser seguidos por linhas opcionais que modificam a estrutura gerada. Há zero ou mais de cada uma das seguintes linhas, as quais devem ser agrupados juntos na ordem dada aqui:
AtomGeom, I , GeomNormalmente a geometria local sobre um átomo é definido pelo número e tipos de ligação sobre o átomo (por exemplo, carbono em metano é tetraédrico, em etileno é triangular, etc). Todos os ângulos de ligação em um centro deve ser são iguais. O AtomGeom linha muda o valor dos títulos no centro eu . Geom pode ser o ângulo como um número de ponto flutuante, ou uma das cordas TetR , Pyra, Trig , Bent , ou de linha .
BondRot, I , J , K , L , GeomIsso muda as orientações do I - J e K - L títulos sobre a J - K . vínculo Geom ou é o ângulo diedro ou uma das cordas Cis (≥ 0),Trans (≥ 180), Gaup (≥ 60), ou Gaum (≥ -60).
BondLen, I , J , Newlen

Isso define o comprimento do I - J vínculo com Newlen (um valor de ponto flutuante).
O construtor de modelo só pode construir estruturas com átomos em suas valências normais. Se um radical é desejada, sua valência extra pode ser efetivamente "amarrado"usando átomos fictícios, que são especificados por um sinal de menos antes do símbolo atômico (por exemplo,-H). Apenas os átomos terminais podem ser átomos de manequim.
Os dois modelos disponíveis (A e B) diferem em que o modelo A leva em conta o tipo (simples, duplos, triplos, etc) de uma ligação na atribuição de comprimentos de ligação,comprimentos de ligação, enquanto o modelo B depende apenas dos tipos de átomos envolvido. Modelo B está disponível para todos os átomos de H para Cl, exceto Ele e Ne. Se o modelo A é solicitada e um átomo é usado para os quais não Model A comprimento de ligação é definido, o comprimento de ligação Modelo B apropriado é usado em seu lugar.
Limitações do ProgramaEsta seção lista as diversas limitações de tamanho que existem dentro Gaussian 09.
• O programa integral tem as seguintes limitações:
o O número máximo de átomos é de 250.000.
o O número máximo total de conchas primitivas é 750.000.
o O número máximo de primitivas d-conchas e superior é 250.000.
o O número máximo de conchas contratados é 250.000.
o A contracção graus de máximo permitido é de 100.
• Opt = (EF, EnOnly) otimizações-úteis apenas para os métodos analíticos, sem gradientes estão limitados a 50 variáveis.
• O programa GVB é limitado a 100 pares de orbitais (que não é uma restrição, naprática).
• NBO está dimensionado para 250 mil átomos e 10.000 funções de base.
Gaussian 09 LigaçõesA tabela a seguir lista os programas que compõem Gaussian 09 conhecidos como links, juntamente com as suas principais funções:
L0 Inicializa programa e controles de sobreposição
L1 Seção Processos rota, lista de links para executar constrói e inicializa arquivos scratch
L101 Lê título e molécula especificação
L102 Otimizações Fletcher-Powell
L103 Otimizações Berny a minima e TS, pesquisas estaduais STQN transição
L105 Otimizações Murtaugh-Sargent

L106 Diferenciação numérica de forças / dipolos obter polarizabilidade / hiperpolarizabilidade
L107 Linear-síncrono trânsito (LST) de pesquisa do estado de transição
L108 Unrelaxed exame de superfície potencial de energia
L109 Otimização de Newton-Raphson
L110 Duplo diferenciação numérica de energias para produzir freqüências
L111 Duplo diferenciação numérica de energias para calcular polarizabilidades e hiperpolarizabilidades
L112 Executa o método Scaling Virial auto-consistente (SCVS), extensão de TA Keith [ Lowdin59 , Magnoli82 , Lehd91 ]
L113 Otimização EF usando gradientes analíticos
L114 EF otimização numérica (usando apenas as energias)
L115 Segue o caminho de reação usando o algoritmo GS3
L116 Campo de reação numérica auto-consistente (SCRF)
L117 Executa cálculos de solvatação IPCM.
L118 Cálculos BOMD
L120 Controles ONIOM cálculos
L121 Cálculos ADMP
L122 Cálculos contrapeso
L123 Segue o caminho da reacção usando o algoritmo de HPC (e outros)
L124 Executa ONIOM com PCM e externa-iteração PCM
L202 Reorienta coordenadas, calcula simetria, e verifica as variáveis
L301 Gera definir base de informação
L302 Calcula sobreposição, cinética e integrais potenciais
L303 Calcula integrais multipolares
L308 Calcula velocidade de dipolo e integrais Rx ∇

L310 Calcula SPDF integrais 2-elétrons de uma forma primitiva
L311 Calcula sp integrais 2-elétrons
L314 Calcula SPDF integrais 2-elétrons
L316 Imprime integrais 2-elétrons
L319 Calcula integrais 1-elétron para rotação aproximado de acoplamento orbital
L401 Formulários o palpite MO inicial
L402 Executa semi-empíricos e moleculares cálculos de mecânica
L405 Inicializa um cálculo MCSCF
L502 Iterativamente resolve as equações SCF (conven. UHF & ROHF, todos os métodos diretos, SCRF)
L503 Iterativamente resolve as equações SCF usando minimização direta
L506 Executa um cálculo ROHF ou GVB-PP
L508 Programa SCF quadraticamente convergente
L510 MC-SCF
L601 População e afins análises (incluindo momentos multipolares)
L602 Propriedades 1-elétron (potencial, campo e gradiente de campo)
L604 Avalia MOs ou densidade sobre uma grade de pontos
L607 Realiza análises NBO
L608 Energias DFT não-iterativos
L609 Átomos em Moléculas propriedades
L610 Integração numérica (para testar códigos integrais)
L701 1-elétron integrais primeiras ou segundas derivadas
L702 2-elétrons integrais primeiras ou segundas derivadas (sp)
L703 2-elétrons integrais primeiras ou segundas derivadas (spdf)
L716 Processos de informação para otimizações e freqüências
L801 Inicializa transformação de integrais de 2 elétrons

L802 Executa a transformação integral ( N 3 in-core)
L804 Transformação Integral
L811 Transforma derivados integrais e calcula suas contribuições para MP2 2 ª derivados
L901 Anti-symmetrizes integrais 2-elétrons
L902 Determina a estabilidade da função de onda de Hartree-Fock
L903 Old MP2 no interior do núcleo
L904 Conjunto de base completo (CBS) método de extrapolação de Petersson, et. ai.
L905 Complexo MP2
L906 MP2 Semi-direto
L908 Electron Programa Propagator
L913 Calcula energias pós-SCF e termos de gradiente
L914 CI-Singles, RPA e ZINDO estados excitados; estabilidade SCF
L915 Calcula quantidades quinta ordem (para MP5, QCISD (TQ) e BD (TQ))
L916 MP4 e CCSD Velho
L918 Reoptimizes a função de onda
L923 Programa SAC-CI
L1002 Iterativamente resolve as equações CPHF; calcula várias propriedades (incluindo RMN)
L1003 Iterativamente resolve as equações CP-MCSCF
L1014 Calcula analíticas CI-Singles segundas derivadas
L1101 Calcula-1 elétrons derivados integrais
L1102 Calcula dipolo integrais derivativos
L1110 2-elétron contribuição derivado integrante para F (x)
L1111 Matriz densidade de partículas 2 e pós-SCF derivados
L1112 Derivados MP2 segunda

L9999 Finaliza cálculo e de saída