machine learning in computational biology csc 2431goldenberg/csc2431/csc_2431... · machine...
TRANSCRIPT

Machine Learning in Computational Biology CSC 2431
Lecture 4: Missing Heritability Instructor: Anna Goldenberg

Heritability (of a trait)
� Fraction of phenotypic variability attributable to genetic variation
� NOT: how much genetics influences trait in one person
� Relative to specific population in a particular environment (since contribution of genetic factors is relative to contribution of other factors such as environment)

Heritability Phenotype P, Genotype G, Environment E G: Additive A, Dominant D, Epistatic J Var(P) = Var(G)+Var(E)+2Cov(G,E) Var(G) = Var(A) + Var(D) + Var(J)
Broad Sense Heritability: (includes additive, epistatic, dominant, maternal, paternal)
H2 =V ar(G)
V ar(P )
h2 =V ar(A)
V ar(P )Narrow Sense Heritability (only additive effects)

Regress offspring value on midparent value
← Slope = h2
Example of a height trait

Parent-offspring regression
Heritability = slope Problem: Parents and children share other factors besides genome

Heritability estimates from other regression analyses
Comparison Slope
Midparent-offspring h2
Parent-offspring 1/2h2
Half-sibs 1/4h2
First cousins 1/8h2
• as the groups become less related, the precision of the h2 estimate is reduced.
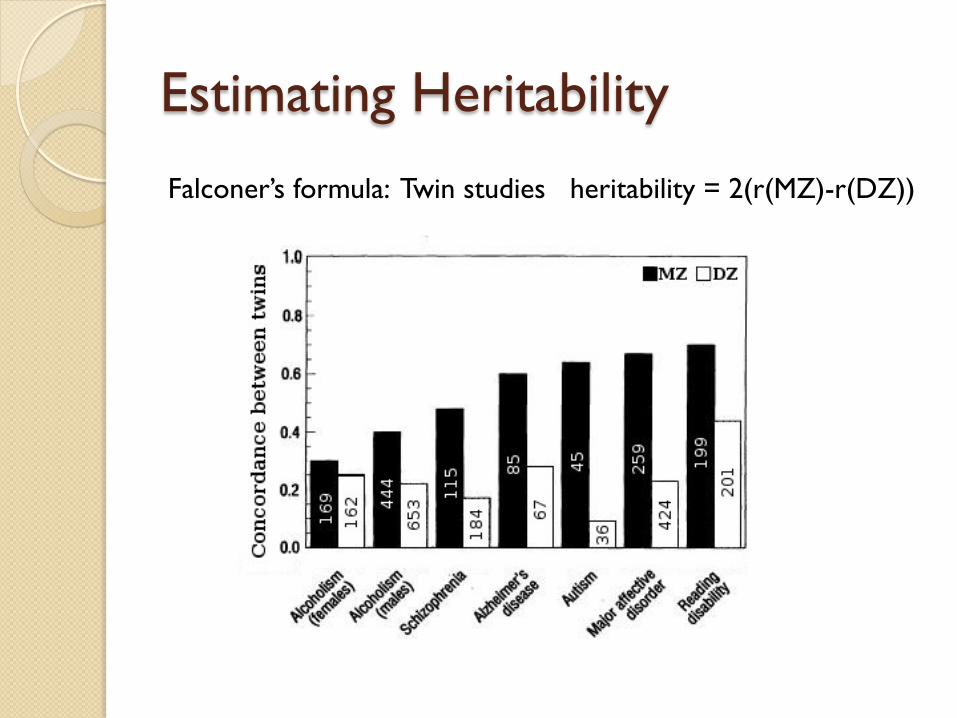
Estimating Heritability
Falconer’s formula: Twin studies heritability = 2(r(MZ)-r(DZ))

Estimating Heritability
� Tetrachoric correlation: correlation of disease among relatives of particular type vs random pair from population
� Twin Method: resemblance between MZ twins vs DZ twins
� Falconer’s method � Mixed Linear Models – uses Bayesian
method or MLE to estimate variances from families and pedigrees
Tenesa, Albert, and Chris S. Haley. "The heritability of human disease: estimation, uses and abuses." Nature Reviews Genetics 14.2 (2013): 139-149.

Examples of estimated heritability Trait/Disease Estimated heritability
Alcoholism 50-60%
Alzheimers 58-79%
Asthma 30%
Bipolar Disorder 70%
Depression 50%
Hair Curliness 85-95%
Lung Cancer 8%
Height 81%
Obesity 70%
Longetivity 26%
Sexual Orientation 60%
Schizophrenia 81%
Type 1 diabetes 88%
Type 2 diabetes 26%
http://snpedia.com/index.php/Heritability

Genetically Explained Heritability Disease # of
Loci Heritability Explained
Heritability Estimated
Measure of Heritability
Age related macular degeneration
5 50% 46-71% Sibling recurrent risk
Crohn’s Disease 32 20% 50-60% Genetic risk (liability)
Systemic Lupus Erithematosus
6 15% 44-66% Sibling recurrent risk
Type 2 diabetes 18 6% 26% “
HDL Cholesterol 7 5.2% “
Height 40 5% 81% Phenotypic Variance
Fasting glucose 4 1.5% “
Manolio, Teri A., et al. "Finding the missing heritability of complex diseases." Nature 461.7265 (2009): 747-753.

Important question: how is the genetic heritability estimated from GWAS?
Typically: add up the estimated heritability contributed by each of the genetic variants that have achieved clear genome-wide statistical significance Problem: this is just a lower bound Solution: estimate common variant heritability without identifying the exact loci
Disease # of Loci
Heritability Explained
Heritability Estimated
Measure of Heritability
Type 2 diabetes 18 6% 26% “
Height 40 5% 81% Phenotypic Variance

Miscalculated heritability estimates 1
� Yang … Visscher, Nature Genetics, 2010: ◦ Problem: Given SNPs do not account for rare
variants, so genetic heritability is under computed ◦ Method: linear mixed models, REML ◦ Fix: model the extent to which the phenotypic
similarity across pairs of individuals in a sample is explained by their genotypic similarity at common variants. ◦ Results: using all SNPs found genetic estimate of
heritability of height to be 45% (compared to 5% before)

Miscalculated heritability estimates 2
� Golan, Lander and Rosset, PNAS, 2014: ◦ Problem: small sample size, small effect size,
true heritability, number of genotyped SNPs ◦ Method: Phenotype-correlation genotype-
correlation (PCGC) ◦ Fix: regress pairs of phenotypes to pairs of
genotypes

Miscalculated heritability estimates

Missing heritability continued � Much larger numbers of variants of smaller
effects � Rarer variants not present on arrays � Structural variants � Low power to detect gene-gene interactions � Inadequate accounting for shared environment
by twins
Manolio, Teri A., et al. "Finding the missing heritability of complex diseases." Nature 461.7265 (2009): 747-753.

Missing heritability continued � Much larger numbers of variants of smaller
effects � Rarer variants not present on arrays � Structural variants � Low power to detect gene-gene interactions � Inadequate accounting for shared environment
by twins Manolio, Teri A., et al. "Finding the missing heritability of complex diseases." Nature 461.7265 (2009): 747-753.
� Heterogeneity of phenotype in complex diseases (our inability to distinguish between multiple less common but similarly manifesting diseases)

Missing heritability continued Problem: Much larger numbers of variants of smaller effects Solution: Bigger cohorts (number of people)

Rare variants
Low: 0.5% < MAF < 5% of the population Rare: MAF < 0.5%
Example: 20 variants with MAF < 1% and risk of 3 would account for most variation in Type 2 diabetes! But they were not found yet.
Reason: Small sample sizes or insufficiently large arrays
Solution: pooling (collapsing)

Rare variants � CAST - cohort allelic sum test, collapses information on all rare variants within a
region (e.g., the exons of a gene) into a single dichotomous variable for each subject by indicating whether or not the subject has any rare variants within the region and then applies a univariate test (Morris and Zeggini, Gen Epi, 2010)
� Calpha - non-burden-based test, robust to the direction and magnitude of effect. For case-control data, it compares the expected variance to the actual variance of the distribution of allele frequencies (Neale et al, PloS Genetics, 2011)
� RWAS - Rare variant Weighted Aggregate Statistic –groups variants and computes a weighted sum of differences in mutation counts between case and control individuals. Weights of RWAS are estimated from data to achieve nearly optimal power under a disease model in which all variants make an equally small contribution to population disease risk (Sul et al, Genetics)
� SKAT – sequence kernel association test: supervised, flexible, computationally efficient regression method to test for association between genetic variants (common and rare) in a region and a continuous or dichotomous trait while easily adjusting for covariates (Wu et al, AJHG, 2011)
� SKAT+dmGWAS – SKAT + network aggregation (Jia, Bioinformatics, 2011)

Rare variants
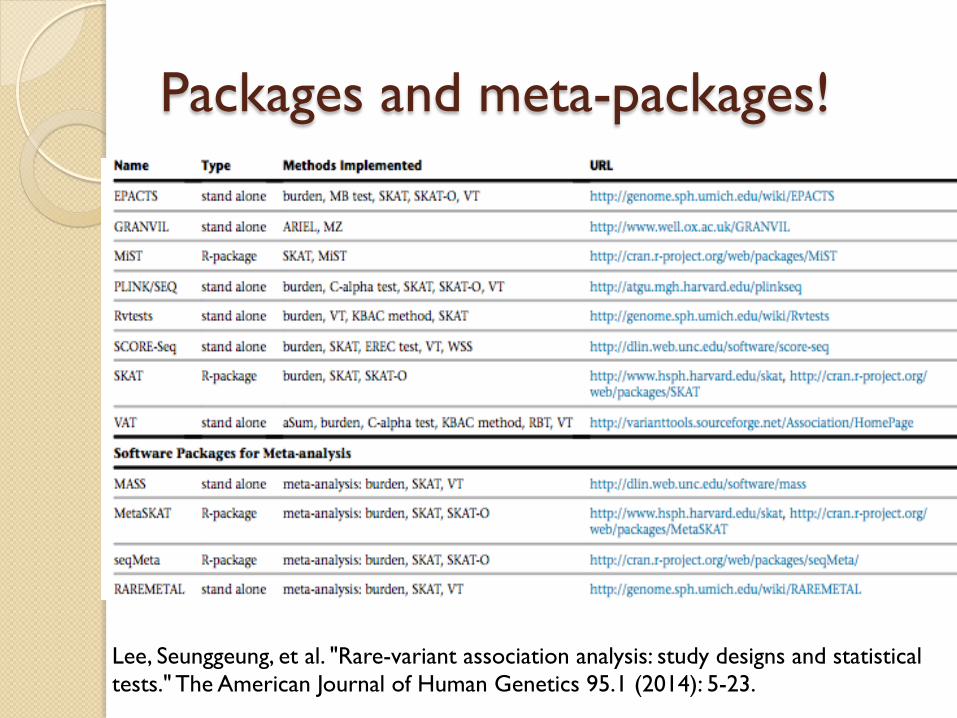
Packages and meta-packages!
Lee, Seunggeung, et al. "Rare-variant association analysis: study designs and statistical tests." The American Journal of Human Genetics 95.1 (2014): 5-23.

Structural Variations � Copy number variants (CNVs) – insertions
and deletions � Copy neutral variations – inversions and
translocations – largely unstudied with respect to complex diseases
� Common CNVs are large – 600kb-3Mb � Disease associated CNPs – 20-40kb � de novo CNVs are shown to be important in
neuropsychiatric and developmental conditions

Examples of identified CNVs
Similar to SNPs – rare variants have large effects common variants have small effects

Problem with studying structure
� Technical – several hundred genes that map to commonly duplicated regions are considered ‘inaccessible’ by most existing genotyping and sequencing technologies due to multicopy nature
� Need – characterize sequence content in highly variable regions
Evan Eichler et al. Nat. Rev. Gen. 2010. Missing Heritability and strategies for finding underlying causes of complex disease

Epistasis � The departure from the independence of the
effects of different genetic loci
� AB = Ab + aB – ab – no epistasis � AB > Ab + aB – ab – synergistic epistasis (SE) � AB < Ab + aB – ab – antagonistic epistasis
� E.g. Synthetic lethality – synergistic epistasis of harmful mutations (combined together they kill the organism)

Parent of origin effect
� Example – an allele if inherited from father – hurts, from mother – helps (T2D)
� Variants can increase a recombination rate for fathers and reduce – for mothers

Epistasis method review
Wei et al, Detecting epistasis in human complex traits, Nature Genetics, 2014


Interesting findings � Hemani et al. Nature 508, 249–253 (2014) Found: found 501 significant pairwise interactions between common SNPs influencing the expression of 238 genes (P < 2.91 × 10−16). Replication of these interactions in two independent data sets11, 12 showed both concordance of direction of epistatic effects (P = 5.56 × 10−31) � Wood et al: Another explanation for apparent epistasis Found: Using whole-genome sequencing data from 450 individuals we strongly replicated many of the reported interactions but, in each case, a single third variant captured by our sequencing data could explain all of the apparent epistasis.

Phenotypic heterogeneity

Phenotypic heterogeneity
0 0 0 0 0 0 1 1 1 0 1 0 0 0 1 0 2 0 0 0 0 0 0 1 0 1 0 0 0 1
0 0 2 0 0 0 1 2 1 0 1 0 0 2 0 0 2 0 2 2 0 0 0 1 2 1 0 0 0 2
CASES
CONTROL

Phenotypic heterogeneity
0 0 0 0 0 0 1 1 1 0 1 0 0 0 1 0 2 0 0 0 0 0 0 1 0 1 0 0 0 1
0 0 2 0 0 0 1 2 1 0 1 0 0 2 0 0 2 0 2 2 0 0 0 1 2 1 0 0 0 2
CASES
CONTROL
Age of onset
Warde-Farley, David, et al. ”Mixture model for subphenotyping in GWAS." Pac. Symp. Biocomput. Vol. 17. 2012.

Phenotypic heterogeneity

Next class presentations � Methods for Rare Variants - Liu, Dajiang J., and Suzanne M. Leal. "Estimating genetic effects and quantifying missing heritability explained by identified rare-variant associations." The American Journal of Human Genetics 91.4 (2012): 585-596.
� Methods for Epistasis – Schwarz, Daniel F., Inke R. König, and Andreas Ziegler. "On safari to Random Jungle: a fast implementation of Random Forests for high-dimensional data." Bioinformatics 26.14 (2010): 1752-1758.
REMINDER: Project Proposals are due by the end of the week