lymphoma studies in patients with sjögren's...
TRANSCRIPT

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2017
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1331
Lymphoma studies in patients withSjögren's syndrome
LILIAN VASAITIS
ISSN 1651-6206ISBN 978-91-554-9912-9urn:nbn:se:uu:diva-320220

Dissertation presented at Uppsala University to be publicly examined in Enghoffsalen, Ingång50 bv, Akademiska sjukhuset, Uppsala, Wednesday, 7 June 2017 at 13:00 for the degree ofDoctor of Philosophy (Faculty of Medicine). The examination will be conducted in Swedish.Faculty examiner: Associate Professor Thomas Mandl (Department of Clinical SciencesMalmö, Lund University).
AbstractVasaitis, L. 2017. Lymphoma studies in patients with Sjögren's syndrome. DigitalComprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1331.94 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-554-9912-9.
Patients with primary Sjögren’s syndrome (pSS) are at increased risk of developing malignantlymphoma. The studies in this thesis aim at broadening our understanding of the associationbetween these two conditions.
Germinal centre (GC)-like structures were found in minor salivary gland biopsies taken at thetime of pSS diagnosis in 25% of 175 studied patients. Lymphoma development was observed in86% of the GC-positive pSS patients and 14% of the GC-negative patients. GC-like structuresin salivary gland biopsies at pSS diagnosis might identify pSS patients at high risk for laterlymphoma development.
We used the National Patient Register and the Cancer Register to identify pSS patients withlymphoid malignancy for the following studies. The lymphoma tissues were reviewed andclassified according to the WHO classification.
In a study of 79 patients with available lymphoma tissues, we identified histopathologicaland clinical features compatible with IgG4-related disease (IgG4-RD) in one patient (1.3%).Histological features of IgG4-RD in lymphoma tissue in patients with an initial pSS diagnosisseem to be rare but, if present, may indicate underlying IgG4-RD.
We identified and compared pSS patients with (n=18/17%) and without (n=87) pre-existinglymphoma at pSS diagnosis and found similar pSS characteristics in both groups. Mucosa-associated lymphoid tissue (MALT) lymphoma in salivary glands was more common in patientswith pre-existing lymphoma. The findings support the removal of pre-existing lymphoma as ageneral exclusion criterion for a pSS diagnosis in classification criteria. Further, the findingssuggest an investigation for pSS in patients presenting with MALT lymphoma in salivary glands.
We compared the distribution of lymphoma subtypes with a general population reference.Both diffuse large B-cell lymphoma (DLBCL) (32%) and marginal zone lymphoma (MZL)(31%) were common, but only MZL (MALT lymphomas) occurred at an increased relativefrequency compared to the general population.
Men constituted 15% of 105 pSS patients with lymphoma. Men had a shorter time between thepSS and lymphoma diagnoses and more often had lymphoma in the salivary glands comparedwith women. Increased awareness of signs of lymphoma in salivary glands already during thefirst years after pSS diagnosis is justified in men with pSS.
Keywords: Sjögren's syndrome, primary Sjögren's syndrome, lymphoma, IgG4-related disease
Lilian Vasaitis, Department of Medical Sciences, Akademiska sjukhuset, Uppsala University,SE-75185 Uppsala, Sweden.
© Lilian Vasaitis 2017
ISSN 1651-6206ISBN 978-91-554-9912-9urn:nbn:se:uu:diva-320220 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-320220)

To Indra and Paulius


List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Theander E, Vasaitis L, Baecklund E, Nordmark G, Warfvinge G, Liedholm R, Brokstad K, Jonsson R, Jonsson MV. (2011) Lymphoid organisation in labial salivary gland biopsies is a possible predictor for the development of malignant lymphoma in primary Sjögren's syndrome. Annals of the Rheumatic Diseases, 70(8):1363-8
II Vasaitis L, Sundström C, Backlin C, Nordmark G, Baecklund E. (2016) Sporadic occurrence of non–diagnosed IgG4-related disease in lymphoma patients with a previous Sjögren’s syndrome diagnosis. Acta oncologica, 55(9-10):1139-44
III Vasaitis L, Nordmark G, Theander E, Backlin C, Smedby K E, Ask-ling J, Rönnblom L, Sundström C, Baecklund E. Support for remov-al of lymphoma as an exclusion criterion in classification criteria for primary Sjögren’s syndrome. Manuscript
IV Vasaitis L, Nordmark G, Theander E, Backlin C, Smedby K E, Ask-ling J, Rönnblom L, Sundström C, Baecklund E. Population-based study of primary Sjögren’s syndrome and lymphoma: gender differ-ences, clinical characteristics, and lymphoma subtypes. Manuscript
Reprints were made with permission from the respective publishers.

Related Papers
Vasaitis L (former Vasiliauskiene L), Wiik A, Høier-Madsen M. (2001) Prevalence and clinical significance of antikeratin antibodies and other sero-logical markers in Lithuanian patients with rheumatoid arthritis. Annals of the Rheumatic Diseases, 60(5):459-66 Nordmark G, Kristjansdottir G, Theander E, Appel S, Eriksson P, Vasaitis L, Kvarnström M, Delaleu N, Lundmark P, Lundmark A, Sjöwall C, Brun JG, Jonsson MV, Harboe E, Gøransson LG, Johnsen SJ, Söderkvist P, Elor-anta ML, Alm G, Baecklund E, Wahren-Herlenius M, Omdal R, Rönnblom L, Jonsson R, Syvänen AC. (2011) Association of EBF1, FAM167A (C8orf13)-BLK and TNFSF4 gene variants with primary Sjögren's syn-drome. Genes & Immunity, 12(2):100-9 Bolstad AI, Le Hellard S, Kristjansdottir G, Vasaitis L, Kvarnström M, SjöwallC, Johnsen SJ, Eriksson P, Omdal R, Brun JG, Wahren-Herlenius M, Theander E, Syvänen AC, Rönnblom L, Nordmark G, Jonsson R. (2012) Association between genetic variants in the tumour necrosis fac-tor/lymphotoxin α/lymphotoxin β locus and primary Sjögren's syndrome in Scandinavian samples. Annals of the Rheumatic Diseases, 71(6):981-8 Nordmark G, Wang C, Vasaitis L, Eriksson P, Theander E, Kvarnström M, Forsblad-d'Elia H, Jazebi H, Sjöwall C, Reksten TR, Brun JG, Jonsson MV, Johnsen SJ, Wahren-Herlenius M, Omdal R, Jonsson R, Bowman S, Ng WF, Eloranta ML, Syvänen AC; UK Primary Sjögren’s Syndrome Registry. (2013) Association of genes in the NF-κB pathway with antibody-positive primary Sjögren's syndrome. Scandinavian Journal of Immunology, 78(5):447-54 Vasaitis L. (2016) IgG4-related disease: A relatively new concept for clini-cians. European Journal of Internal Medicine, 27:1-9 Vasaitis L. (2016) Metal dust as a possible inducer of immunoglobulin G4-related skin nodules. British Journal of Dermatology, 175(5):871

Contents
Introduction ................................................................................................... 11Sjögren’s syndrome .................................................................................. 12Primary Sjögren’s syndrome .................................................................... 13
Classification criteria ........................................................................... 13ESSDAI ................................................................................................ 17ESSPRI ................................................................................................ 18Epidemiology ....................................................................................... 18Etiology and pathogenesis ................................................................... 19Haematological disturbances ............................................................... 24Autoantibodies ..................................................................................... 25Cellular infiltration in salivary glands ................................................. 25Germinal centre-like structures in minor salivary glands .................... 26Glandular manifestations ..................................................................... 27Extraglandular manifestations ............................................................. 27Constitutional features ......................................................................... 29Co-morbidity ........................................................................................ 29Risk factors and causes of mortality in pSS ......................................... 29Sex differences ..................................................................................... 29Diagnosis of pSS in clinical practice ................................................... 29Treatment ............................................................................................. 30
Lymphoma ................................................................................................ 31Risk and predictors for lymphoma in pSS ........................................... 31Possible mechanisms of lymphoma development in pSS .................... 34Lymphoma subtypes in pSS and lymphoma diagnosis ........................ 36Outcome and treatment of lymphoma in pSS ...................................... 37
IgG4-related disease ................................................................................. 37Pathogenesis of IgG4-RD .................................................................... 38Similarities and differences with pSS .................................................. 38Diagnosis of IgG4-RD ......................................................................... 40Lymphoma in IgG4-related disease ..................................................... 40
Aims of the thesis .......................................................................................... 42

Subjects and methods .................................................................................... 43Data sources .............................................................................................. 43National Registers used in the studies ...................................................... 43
The Patient Register ............................................................................. 43The Cancer Register ............................................................................. 44The Lymphoma Register ...................................................................... 44
PAPER I ................................................................................................... 45Patients and clinical information ......................................................... 45Salivary gland tissue re-evaluation ...................................................... 46The Swedish Cancer Register and lymphoma tissues .......................... 47
PAPERS II-IV .......................................................................................... 47Patients ................................................................................................. 47Tissue analyses ..................................................................................... 50Validation of pSS diagnosis ................................................................. 50
Statistics .................................................................................................... 53Ethical approval ........................................................................................ 53
Results ........................................................................................................... 54PAPER I ................................................................................................... 54PAPER II .................................................................................................. 55PAPER III ................................................................................................. 58
Underlying reasons for a SS diagnosis code ........................................ 58pSS patients with and without pre-existing lymphoma ....................... 59
PAPER IV ................................................................................................. 61Characteristics of pSS patients with a lymphoid malignancy .............. 61Sex differences ..................................................................................... 62Comparison of pSS-MALT lymphoma with pSS-DLBCL ................. 63Lymphoma subtypes in pSS patients and comparison with a general population reference ............................................................. 64
Discussion ..................................................................................................... 66Lymphoid formations in minor salivary gland biopsies ........................... 66IgG4-RD in patients with a previous pSS diagnosis and lymphoma ....... 68Underlying reasons behind a SS diagnosis code ...................................... 70Support for removal of pre-existing lymphoma as an exclusion criterion for pSS ........................................................................................ 71Sex differences in pSS patients with a lymphoid malignancy .................. 71Lymphoma characteristics in pSS patients and comparison with a general population reference .................................................................. 73
Concluding remarks ...................................................................................... 75
Acknowledgements ....................................................................................... 76
References ..................................................................................................... 77

Abbreviations
ANA Anti-nuclear antibody ACR American College of Rheumatology AECG American European Consensus Group DC Dendritic cell DLBCL Diffuse large B-cell lymphoma EBV Epstein-Barr virus ESSDAI EULAR Sjögren’s syndrome disease activity index EULAR European League Against Rheumatism GC Germinal centre H&E Haematoxylin and eosin HPF High power field HL Hodgkin lymphoma ICD International Classification of Diseases IFN Interferon Ig Immunoglobulin IgG4-RD IgG4-related disease IL Interleukin MALT Mucosa-associated lymphoid tissue MD Mikulicz’s disease MZL Marginal zone lymphoma NF-κB Nuclear factor kappa-light-chain-enhancer of activated
B cells NHL Non-Hodgkin lymphoma PC Plasma cell pSS Primary Sjögren’s syndrome RA Rheumatoid arthritis RF Rheumatoid factor RTA Renal tubular acidosis SIR Standardised Incidence Ratio SLE Systemic lupus erythematosus SS Sjögren’s syndrome sSS Secondary Sjögren’s syndrome SSA/Ro Sjögren’s syndrome antigen A=Ro SSB/La Sjögren’s syndrome antigen B=La Th T helper TLR Toll-like receptor

TNF Tumour necrosis factor WHO World Health Organisation

11
Introduction
Autoimmune diseases are mostly chronic disorders in which the immune system mistakenly attacks the body’s own organs and tissues. There are over 80 different autoimmune diseases and about 4.5% of the population are af-fected by them (1). Most autoimmune diseases predominantly affect women (2) indicating different etiopathogenetic mechanisms of immune response and development of autoimmunity in female and male individuals.
Typically, autoimmune chronic inflammation has a fluctuating course with periods of remission (no or little activity) and flare-ups (worsening of symptoms). Diagnosis of the autoimmune disease may often be delayed or mistakenly diagnosed as another disorder because of unspecific vague symp-toms and fluctuating course. Whence the autoimmune disease is diagnosed, it often requires continuing or long periods of immunosuppressive treatment.
Over time, continuous or periodical activation of the immune response can lead to the development of lymphoid malignancy. Chronic autoimmune conditions, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and particularly primary Sjögren’s syndrome (pSS) are associated with increased risk of lymphoma (3). It is, therefore, an important research field to study mechanisms and associated factors behind lymphoma and au-toimmunity.
In this thesis, the introduction first gives an overview of the hallmarks of Sjögren’s syndrome, lymphoma and IgG4-related disease (IgG4-RD), and then four studies of pSS and lymphoid malignancy are introduced. The first study presents lymphoid organisation in labial salivary gland biopsies taken at pSS diagnosis as a possible predictor for future lymphoma development. This study is performed on pSS patients from two hospitals. The remaining studies (II-IV) are nationwide and performed on non-selected pSS patients. The second study elucidates the occurrence of undiagnosed IgG4-RD in previously misdiagnosed pSS patients with lymphoma. In the third study, detailed analysis of the pSS patients’ characteristics with and without pre-existing lymphoma at pSS diagnosis was performed, and investigation of underlying causes of Sjögren’s syndrome (SS) code in the patient register is presented. The last study elucidates gender differences in pSS patients with lymphoid malignancy, compares the pSS patients with the two most com-mon subtypes of low-grade and high-grade lymphomas, and evaluates the distribution of the subtypes of lymphoma in pSS patients in comparison with a general population reference group.

12
Sjögren’s syndrome SS, also known as ‘sicca syndrome’, is a systemic chronic autoimmune dis-ease which affects the moisture-producing glands. Patients develop charac-teristic sicca symptoms, namely dry eyes (xerophthalmia) and/or dry mouth (xerostomia). In 1933, Henrik Sjögren was the first who described this syn-drome in 19 patients with RA and dry eyes, establishing that sicca symptoms may extend beyond glandular involvement (4). SS is currently divided in pSS, existing by itself, and in secondary SS (sSS), usually coexisting with other autoimmune diseases, such as RA (5), SLE (6) or systemic sclerosis (7, 8). Compared to RA and SLE, SS is a relatively mild disease but with signif-icant morbidity (9) that attracts both clinical and research interest.
Stress, smoking, drugs, and diabetes are also common causes for unspe-cific dryness symptoms. In about a quarter of the elderly individuals (above the age of 65 years) sicca complaints are present, which are largely due to medication. Moreover, aging also leads to diminished glandular function (10) and development of dry mucous membranes. Dryness of the eyes and mouth may also occur in the following conditions: after radiation therapy to the head and neck; after allogeneic stem cell transplantation, when graft ver-sus host disease develops; in patients with malignancy in the salivary or lac-rimal glands; in some cases of sarcoidosis; in tuberculosis or other infec-tions, such as hepatitis C or acquired immunodeficiency syndrome. Causes of sicca syndrome (other than pSS and sSS) are summarised in Table 1.
Table 1. Causes of sicca symptoms other than primary and secondary Sjögren’s syndrome*
Conditions Drug side effects Age-related glandular atrophy Alzheimer’s disease Amyloidosis Cystic fibrosis Dehydration Diabetes mellitus Fibromyalgia Graft versus host disease IgG4-related disease Infections: hepatitis C, HIV, tuberculosis Previous irradiation to the head and neck Psychological factors Salivary gland trauma or tumour Sarcoidosis Smoking
Anticholinergic drugs (atropine, scopolamine) Antihistamines α1-antagonists (prazosin, terazosin) α2-antagonists (clonidine) Benzodiazepines β-blockers (atenolol, propranolol) Diuretics Nicotine Phenothiazines Opioids Tricyclic antidepressants Selective serotonin reuptake inhibitors Sympathomimetic drugs (ephedrine)
*Adapted from Rischmueller et al. (11).
The World Health Organisation (WHO) International Classification of Dis-eases (ICD), does not, however reflect the diversity of the causes of sicca

13
symptoms (http://apps.who.int/classifications/icd10/browse/2016/en). In this system, the same code M35.0 (ICD-10 classification) is assigned for “Sjögren’s syndrome/sicca syndrome” regardless of the underlying reason for the dryness symptoms. From registers based on ICD codes, it is therefore not possible to identify whether patients have, for example, pSS or sSS, or other underlying causes of a sicca syndrome diagnosis code.
Primary Sjögren’s syndrome No diagnostic criteria and no single gold-standard test exist for the diagnosis of pSS. The typical patient with pSS exhibits sicca symptoms. However, clinically pSS extends from dryness symptoms due to lymphocytic infiltra-tion and diminished function of salivary and lacrimal glands to a systemic involvement of inflammation accompanied by fatigue, and occurrence of extraglandular manifestations.
The clinical symptoms, both sicca and systemic, as well as serological features, are not specific for pSS only. They can mimic or overlap with other rheumatic autoimmune systemic diseases, infections or fibromyalgia. In fact, no specific diagnosis can be made at the first presentation of a patient with sicca symptoms. In some cases, the difficulties of diagnosing pSS may lead to erroneous diagnosis, for example, fibromyalgia, SLE or RA (12). Patients with vague symptoms of sicca syndrome can also be undiagnosed or diag-nosed in the latter stages of the disease. Therefore, other causes of sicca syn-drome and systemic features need to be considered in diagnosing pSS through a detailed medical history with an accurate examination of the pa-tient, and in some cases, a multidisciplinary approach and exclusions of oth-er diagnoses.
Classification criteria The variability of clinical pSS presentation led to the development of differ-ent classification criteria for SS. It is important to note that classification criteria are not synonymous with diagnostic criteria. The ultimate goal for diagnosing a patient with SS is to be correct at the level of the individual patient even if the patient does not completely fulfil the criteria, whereas classification aims to maximally increase the homogeneity of the population for study purpose.
Before 2002, many different sets of classification criteria for SS were published (13-15). The numerous proposed criteria reflect the difficulties of defining this heterogeneous syndrome. During the 1980s-90s, the most known criteria in Sweden were the San Diego or Californian (13), the Co-penhagen (14), and the preliminary European (16) criteria.

14
The preliminary European criteria were proposed in 1993 and focused on the three characteristic features of SS: sicca symptoms, glandular dysfunc-tion, and systemic autoimmunity. These criteria could misclassify patients with non-autoimmune sicca syndrome as having pSS, based on subjective ocular and oral symptoms alone. To overcome this possible misclassifica-tion, the European criteria were revised in 2002 and published as the Ameri-can-European consensus group (AECG) criteria for the classification of SS (17) (Table 2).
Table 2. 2002 Revised American-European consensus group (AECG) criteria* I. Ocular symptoms: positive response to one of the following claims: 1) daily persistent trouble with dry eyes for more than 3 months; 2) recurrent sensation of sand or gravel in the eyes; 3) usage of tear substitutes more than three times per day II. Oral symptoms: positive response to one of the following claims: 1) daily feeling of dry mouth for more than three months; 2) recurrent or persistent swollen salivary glands as an adult; 3) need frequently drink liquids to aid swallowing dry food III. Ocular signs: positive Schirmer’s test performed without anesthesia (≤5 mm in 5 minutes) or positive rose Bengal or another ocular dye score (≥4 according to van Bijsterveld) IV. Histopathology: focal lymphocytic sialadenitis with a focus score ≥1 focus, defined as a number of lymphocytic foci per 4 mm2 of minor salivary gland tissue (one focus contains >50 lymphocytes) V. Salivary gland involvement: unstimulated whole salivary flow (≤1.5 mL in 15 minutes) VI. Autoantibodies: presence of anti-SSA/Ro or anti-SSB/La or both Rules: for pSS: in patients without any associated disease and the presence of four of the six items, including positive histopathology (IV) or serology (VI), or the presence of three of the four objective items (III, IV, V, VI); for sSS: in patients with associated other well-defined connective tissue disease and the pres-ence of item I or item II, plus any two from among items III, IV, and V. Exclusion criteria: pre-existing lymphoma, past head and neck radiation therapy, hepatitis C, acquired immunodeficiency disease, sarcoidosis, graft versus host disease, use of anticholin-ergic drugs *Adapted from Vitali et al. (17).
The 2002 AECG criteria have been commonly used in epidemiological SS studies and also as support for the diagnosis of SS in everyday clinical prac-tice. The criteria have 90% sensitivity and 95% specificity for pSS diagnosis. They distinguish between pSS and sSS and differ from previously used crite-ria in their requirements for either a positive serology (anti-SSA/Ro and/or anti-SSB/La) or a positive labial biopsy (a focal lymphocytic sialadenitis with a focus score of ≥1). According to the 2002 AECG criteria, pSS is de-fined as the presence of four of the six items, including positive histopathol-ogy (item IV) or serology (item VI), or the presence of three of the four ob-jective items (III, IV, V, VI).
In 2012, the Sjögren’s International Collaborative Clinical Alliances Co-hort (SICCA) and the American College of Rheumatology (ACR) (18) ap-proved solely objective criteria for SS (Table 3).

15
Table 3. 2012 American College of Rheumatology criteria for SS*
I. Autoantibodies: presence of anti-SSA/Ro and/or anti-SSB/La or RF and ANA titer ≥1:320 II. Histopathology: focal lymphocytic sialadenitis with a focus score ≥1 focus, defined as a number of lymphocytic foci per 4 mm2 of minor salivary gland tissue III. Ocular staining: keratoconjuctivitis sicca with ocular staining score ≥3 Rules for SS: in individuals with signs/symptoms that may be suggestive of SS who exhibited at least 2 out of 3 objective features Exclusion criteria: past head and neck radiation therapy, hepatitis C, acquired immunodeficiency disease, sar-coidosis, amyloidosis, graft versus host disease, IgG4-related disease *Adapted from Shiboski SC et al. (18).
These criteria were developed for enrolling patients with SS in clinical trials. The subjective sicca symptoms were considered as inclusion criteria in the subject population before applying these objective criteria. The criteria have only slightly higher sensitivity (93%) and the same specificity as the AECG criteria. The 2012 ACR criteria set does not distinguish between pSS and sSS. Furthermore, IgG4-RD, recognised as a new disease entity in 2012, has been added into the exclusions, while pre-existing lymphoma has been re-moved from the exclusion criteria. The rationale of removing the criterion of pre-existing lymphoma from the 2012 ACR criteria is unknown and this issue has not been discussed.
Recently, the international Sjögren’s syndrome criteria working group from both the European League Against Rheumatism (EULAR) and the ACR has developed and approved the new 2016 ACR/EULAR criteria (19), merging both the 2002 AECG and the 2012 ACR criteria (Table 4).
Subjective symptoms of ocular and oral dryness and isolated anti-SSB/La positivity without positivity for anti-SSA/Ro (20) were excluded by the ex-pert-panel, as these items had no independent diagnostic significance for pSS (19). Instead, sicca symptoms or suspicion of SS based on at least one of the domains of the EULAR Disease Activity Index (ESSDAI) (21) for patients with pSS are preliminary requirements for applying these 2016 ACR/EULAR criteria (19). The criteria are based on the weighted sum of five objective items/tests applicable to any patient with at least one symptom of ocular or oral dryness and individuals are classified as having pSS if they have a score of ≥4.
The last criteria from 2016 are improved compared to the previous criteria and are more practically applicable as simple tests, such as Schirmer’s and unstimulated salivary flow tests, which are already a part of common clinical practice, have been added to the items of the criteria. Additionally, the scores of these simple tests are equal to the ocular staining score. The ocular stain-ing, as it was required in 2012 ACR criteria, craves a specific evaluation performed by a trained ophthalmologist, an examination, which is not avail-able in all cases.

16
Table 4. 2016 ACR/EULAR criteria for pSS*
Item Weight/Score Focal lymphocytic sialadenitis with a focus score ≥1 focus, defined as a num-ber of lymphocytic foci per 4 mm2 of minor salivary gland tissue
3
Anti-SSA/Ro-positive 3 Ocular staining score ≥5 (or von Bijsterveld score ≥4) in at least one eye 1 Schirmer’s test ≤5 mm/5 min in at least one eye 1 Unstimulated whole saliva flow rate ≤0.1 mL/min 1 Inclusion criteria: The criteria are applicable to any individual with at least one sicca symp-tom, defined as a positive response to at least one of the following questions: (1) Have you had daily, persistent, troublesome dry eyes for more than 3 months? (2) Do you have a recurrent sensation of sand or gravel in the eyes? (3) Do you use tear substitutes more than three times a day? (4) Have you had a daily feeling of dry mouth for more than 3 months? (5) Do you frequently drink liquids to aid in swallowing dry food? Or in whom there is suspicion of SS from the ESSDAI questionnaire (at least one domain with a positive item). Exclusion criteria: past head and neck radiation therapy, active hepatitis C infection, acquired immunodeficiency disease, sarcoidosis, amyloidosis, graft versus host disease, IgG4-RD Rules for pSS: for individuals who meet inclusion criteria, does not have any of the conditions listed as exclusion criteria and has a score of ≥4 *Adapted from Shiboski CH et al. (19).
The 2016 ACR/EULAR criteria also consider a systemic disease at inclusion by evaluation of the ESSDAI and allow classification of patients with pSS at early stages of the disease. Moreover, these criteria seem to be more liberal and eligible to classify pSS patients than the 2002 AECG criteria. For in-stance, a patient with dry mouth symptoms (subjective sicca symptoms at the time of inclusion) or swollen salivary gland (the glandular domain in the ESSDAI at the time of inclusion) with a positive unstimulated salivary flow test (one weight point), and positive anti-SSA/Ro antibodies (three weight points) has four weight points and fulfils the 2016 ACR/EULAR criteria even if other items are not fulfilled. Meanwhile, a patient in the same situa-tion fulfils only three items of the 2002 AECG criteria and would not fulfil these criteria for pSS.
The latest criteria have the same specificity as the 2002 AECG and the 2012 ACR criteria, but the sensitivity is higher (96%). The criteria have been developed specifically for classification of pSS in clinical trials and studies. They do not define sSS, but the criteria can still be applicable to SS associat-ed with other autoimmune rheumatic diseases. The exclusion criteria are almost the same as in the 2012 ACR criteria, except for the addition of active hepatitis C infection confirmed by polymerase chain reaction. Pre-existing lymphoma is not an exclusion criterion in these criteria because according to the experts in the working groups, pSS can be diagnosed after lymphoma. However, no confirmative study on this issue has been addressed.
So far, the 2002 AECG criteria are the most widely used criteria in pSS studies. The 2012 ACR criteria are too strict and not so practical. The new

17
2016 ACR/EULAR criteria are simple to apply in pSS studies and possibly well suited as a support in everyday clinical practice.
ESSDAI In 2009, the EULAR committee developed the ESSDAI (21) to measure systemic disease activity in patients with pSS (Table 5). Like the SLEDAI (22) or BILAG (23) in lupus, the ESSDAI is a gold standard for evaluation of pSS activity in clinical studies (24). The index includes 12 domains: con-stitutional domain (fever, weight loss, sweating), nine organ-specific do-mains (lymph nodes, glandular, articular, cutaneous, pulmonary, renal, mus-cular, peripheral and central nervous system), haematological (cytopenia), and biological (clonal component, hypocomplementemia, hypergammag-lobulinemia or recent decrease of IgG level).
Depending on the degree of activity of the organ manifestation, 3-4 levels of activity with different weight points (ranging from one to six) are as-signed to each domain. The score of each domain is obtained by multiplying the level of activity by the domain weight. The final score is the sum of all domain scores ranging from zero to theoretically a max score that can be achieved, 123. Low activity has been defined as the ESSDAI score being <5, a moderately active disease if the ESSDAI score is ≥5 but ≤13, and a high activity if the index is ≥14. Threshold of minimal clinically important im-provement is a decrease of the initial score of at least by three points (25).
Some pSS patients can exhibit systemic disease or extraglandular mani-festations without sicca symptoms (26, 27). The ESSDAI measurement in some cases may help to diagnose pSS in early stages and differentiate pSS from other autoimmune diseases. Moreover, baseline ESSDAI is associated with the prognosis of pSS. Activity in the constitutional and lymphadenopa-thy domains is associated with lymphoma, and pulmonary involvement can be one of the main predictors of death (28). Thus, the ESSDAI can be used not only for measuring the disease activity at pSS diagnosis, but also during treatment as a tool to assess the efficacy of the treatment, and can also be useful for identifying patients with a systemic disease.

18
Table 5. EULAR proposed Sjögren’s syndrome Disease Activity Index (ESSDAI)*
Domain Weight Characteristics of the domain Activity levels**
1. Constitutional 3 Fever, night sweats, weight loss 0-2 2. Lymphadenopa-thy and lymphoma
4 Swollen lymph nodes, splenomegaly, current B-cell proliferative malignancy
0-3
3. Glandular 2 Swollen salivary and/or lacrimal glands 0-2 4. Articular 2 Arthralgias with morning stiffness, or synovitis
among 28 joints 0-3
5. Cutaneous 3 Erythema multiforme, cutaneous, including urticari-al, vasculitis, or purpura, or subacute cutaneous lupus, ulcers related to vasculitis
0-3
6. Pulmonary 5 A persistent cough, bronchial involvement, or radio-logical evidence of ILD
0-3
7. Renal 5 Tubular acidosis, glomerular involvement with pro-teinuria >0,5 g/L, or haematuria, or renal failure, or histological evidence of glomerulonephritis, intersti-tial nephritis, or cryoglobulinemia-related renal involvement
0-3
8. Muscular 6 Active myositis proven by abnormal EMG or biopsy with or without weakness or elevated creatine kinase
0-3
9. Peripheral nerv-ous system
5 Evidence of active peripheral nerve involvement proven by nerve-conductive studies, trigeminal neuralgia, or cranial peripheral nerve involvement
0-3
10. Central nervous system
5 Cranial nerve involvement, optic neuritis, multiple sclerosis-like syndrome with pure sensory, or cogni-tive impairment, or motor deficit, cerebral vasculitis with cerebrovascular accident, seizures, transverse myelitis, lymphocytic meningitis
0-2
11. Haematological 2 Cytopenia of autoimmune origin with neutropenia, anaemia, thrombocytopenia or lymphopenia
0-3
12. Biological 1 Clonal component, cryoglobulinemia, or hypocom-plementemia, or hypergammaglobulinemia with IgG>15 g/L, or recent onset of hypogammaglobu-linemia (IgG<5 g/L)
0-2
*Adapted from Seror et al. 2010 (21). **0=no activity, 1=low activity, 2=moderate activity, 3=high activity. ILD=interstitial lung disease; EMG=electromyogram.
ESSPRI The EULAR SS Patients Reported Index (ESSPRI) has been proposed in 2011. It uses numerical scales of 0 to 10 and assesses the patients’ symptoms in three domains: dryness, fatigue, and musculoskeletal pain over the preced-ing two weeks (29). The ESSPRI score is the mean of the three scales.
Epidemiology Estimated incidence and prevalence of pSS varies in different studies and depends on studied population and the classification criteria used. Similar to

19
most autoimmune diseases, pSS is more common in women than in men. The female-to-male ratio varies from 9:1 to 20:1 (30-34). An estimated inci-dence of pSS is 3.9-11 per 100,000 inhabitants with a significantly higher incidence in women compared to men (31, 32, 35-38).
When the 2002 AECG criteria (17) are applied in epidemiological studies, the prevalence of pSS is 0.01-0.7%, (31, 39-46). It seems that those with non-European background have two times higher prevalence of pSS (46).
The onset of sicca symptoms may be exhibited at all ages and many years before diagnosis. The mean age of established pSS diagnosis is usually in the fourth or fifth decades of life (31, 34, 47, 48).
Etiology and pathogenesis
The etiology of pSS is largely unknown as in other autoimmune diseases. It is supposed that a combination of multiple environmental and hormonal factors with a genetic predisposition can contribute to pSS development (49). Innate and adaptive hyperactive immune responses occur locally, in salivary and lacrimal glands, and systematically. Different cells, chemokines, and cytokines are involved in the complex interactions and pathways, which are not yet fully elucidated. A trigger inducing the cascade of the autoreactive inflammatory response in pSS has not yet been identified. The initial signal, viral or non-viral, may lead to a process ending in tissue damage and cell death or apoptosis (Fig. 1).
Ribonucleoprotein particles Ro and La autoantigens, expressed on blebs at the surface of apoptotic epithelial cells of the exocrine glands (50), that have also been identified as antigens in lupus (51), are supposed to lead to chronic inflammation and dysfunction of the glands (autoimmune epithe-litis). Another possible autoantigen in pSS can be the cytoskeletal protein α-fodrin also found in apoptotic cells (52). Type I interferon (IFN) is activated in both blood and salivary glands in pSS patients, so-called ‘IFN signature’. IFN activation is seen in virus infection, therefore, viruses have been sug-gested as a possible trigger of pSS, but so far this has not been proven (53). Recent data indicate that gut-commensal bacteria may be relevant to the development of pSS (54, 55).
pSS occurs most commonly in middle-aged women. Diminished oestro-gen production in post-menopausal women could contribute to pSS (10). Some studies have suggested that not only sex hormones but also X-chromosome dosage may play a role in pSS development (56, 57).

20
Figure 1. A potential explanation of autoimmune epithelitis in primary Sjögren’s syndrome. The first step is tissue damage by an unknown trigger leading to apopto-sis with subsequent expression of the SSA/Ro and/or SSB/La proteins. The next step is T and B cell activation, autoantibody production by B cells, and dysfunction of dendritic cells in the exocrine glands, the formation of germinal-centre (GC)-like structures and development of histopathological lesions. The third step is perpetua-tion, in which cytokines and chemokines promote migration of lymphocytes and dendritic cells and further secretion of the cytokines. PDC=plasmacytoid dendritic cell, Tfh=T follicular helper, Th17=T helper 17, IC=immune complexes, IL=interleukin, IFN=interferon, TLR=toll-like receptor, BAFF=B-cell activating factor, FDC=follicular dendritic cell. The figure was produced by the author using Servier Medical Art and adapted from Brito-Zeron et al. (4).
Environmental factors Virus It has been proposed that viruses from the Herpesviridae family, such as Epstein-Barr virus (EBV), human herpesvirus 6 (HHV6), and retrovirus human T cell lymphotropic virus (53), may be involved in pSS pathogenesis, but so far this hypothesis has not been confirmed by studies (58). However, by increasing understanding of the genetic factors and the central role that type I and II IFN play in autoimmunity (59), it is of interest how viruses may trigger autoimmunity a long time before manifestation of a disease (4). This interest of the “pre-pSS” phase is based on reports that antibodies linked to pSS are present in sera many years before clinical manifestations of the dis-ease (26, 60).

21
Smoking Smoking is a risk factor for SLE, myositis (61), and RA (62), whereas, in some conditions, such as ulcerative colitis, there is a negative correlation between current smoking and disease activity (63).
There is limited and inconsistent information about smoking and pSS. A lower rate of smoking with a higher frequency of antinuclear antibody (ANA) positivity in smokers has been reported in pSS (64). However, some other studies have reported lower frequencies of sialadenitis and SSA/Ro positivity in active smokers with pSS (65-67). Thus, current smoking may have a protective effect against pSS disease-associated humoral and cellular autoimmunity (67), but former smoking may increase risk for pSS (66).
Innate immune system dysfunction in pSS Cells and cytokines Plasmacytoid dendritic cells (DCs), one of the many innate immune system cells infiltrating glandular tissue, are potent producers of type I IFN (68). It is supposed that an activated type I IFN system plays a significant role in the pathogenesis of SLE and pSS (69-71). The role of IFN in pSS is discussed in a separate chapter (see below).
Other cells, for example, macrophages and salivary gland epithelial cells, are involved in the pathogenesis as well. Macrophages produce proinflam-matory cytokines, such as interleukin (IL)-18 (72) and IL-12, and contribute to glandular enlargement (73). The epithelial cells in the glandular tissue may act as antigen-presenting cells (74), express chemokines, promoting glandular localisation of T cells, and produce proinflammatory cytokines, such as IL-6, IL-7, and B-cell activating factor (BAFF) that are important in B-cell pathophysiology (72, 75).
Toll-like receptors There are ten different Toll-like receptors (TLRs) or pattern-recognition receptors expressed in cellular membranes or intracellularly in the endo-somes. They are involved in the first defence against infections by recognis-ing a variety of pathogen-associated molecular patterns (PAMPs), such as viral nucleic acid or bacterial DNA. TLRs can also be activated by so-called damage-associated molecular pattern (DAMP) molecules from apoptotic cells (76). The interaction between BAFF and TLR3 on epithelial cells, and TLR7 and TLR9 on plasmacytoid DCs and epithelial cells promote B-cell maturation and production of self-reactive antibodies (77, 78). This inflam-matory process has a vicious circle-like mechanism (Fig. 1) with increased autoantibody production and formation of more endogenous IFN producers (59). Further, different expression of TLRs has been reported in peripheral blood cells in pSS and healthy controls (79) supporting that TLRs may play a role in pSS pathogenesis.

22
Adaptive immune system dysfunction in pSS T cells In 1983, it was published that T cells are a predominate component of the lymphocytic infiltrate in the salivary and lacrimal glands in pSS and the ma-jority of them are CD4+ T cells (80). Further, a predominant T helper 1 (Th1) expression has been reported in pSS including increased levels of pro-inflammatory cytokines, responsible for Th1 response, such as IL-1β, IL-6, tumour necrosis factor (TNF)-α, and INF-γ in salivary glands (81). Moreo-ver, increased expression of IL-17, IL-22, and IL-23, products of mucosal natural killer cells and Th17 cells, has been found in the inflamed salivary glands of patients with pSS (82). The related cytokines to IL-17, for exam-ple, TGF-β, IL-6, IL-12, and IL-23, have been found in increased levels in plasma from pSS patients (83, 84). Moreover, dysfunction of T regulatory (Treg) cells, expressing Foxp3+, with impaired function of suppression on Th17 cells, may play a role in pSS development (85).
It is supposed that T cells, activated by unknown autoantigen, may acti-vate B cells. It is known that CD4+ T follicular helper (Tfh) cells arise from activated T cells (86). The Tfh cells produce Bcl-6, IL-6, and IL-21, and assist B cells during germinal centre (GC) formation in lymphoid organs (86). In this way, T cells control antigen-specific B-cell immunity.
B cells B cells play a critical role in pSS pathogenesis. B cells are involved in auto-antibody and cytokine production, antigen presentation and regulatory func-tions (87). High levels of autoantibodies that target the self-antigens SSA (Ro52 and Ro60) and SSB (La) are common in pSS (88). B cells also pro-duce other antibodies in pSS, inclusive rheumatoid factor (RF), ANA, and anti-muscarinic acetylcholine M3 receptor antibodies (89, 90). This hyperac-tivity of B cells may result in hypergammaglobulinemia (91) and formation of GC-like structures in salivary glands (92).
Moreover, alterations in B cell subsets have been reported. Decreased numbers of CD27+ memory B cells in peripheral blood (93-95) and salivary glands (96) have been found. In contrast, increased numbers of marginal zone B cells have been reported in peripheral blood and salivary glands (97). These disturbances in B cell subset distribution can be important in lym-phoma development in pSS patients.
BAFF and APRIL BAFF, also known as BLys or TNFSF18, and a proliferation-inducing ligand (APRIL) or TNFSF13A, are members of the TNF-superfamily described in 1999. BAFF and APRIL are expressed by T cells, DCs, monocytes, and macrophages. The overexpression of BAFF and APRIL by these cells acti-vate B cells and may in this way contribute to autoimmunity (98, 99). More-

23
over, BAFF that is generated from memory B cells has emerged as a potent survival factor for plasmablasts and may promote B-cell lymphomas (99). BAFF is found in increased levels in pSS and plays a significant role for GC-like structure development and establishment of follicular DC networks (92, 100).
NF-κB signalling and A20 regulation Abnormalities in nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) signalling mechanisms play a central role in pSS inflamma-tion, cell differentiation, and apoptosis (101, 102). Several studies have re-ported polymorphisms in genes associated with the NF-κB pathways in auto-immune diseases and pSS (103-105).
The ubiquitin-editing enzyme A20 (tumour necrosis factor-α-induced pro-tein 3, TNFAIP3) inhibits NF-κB signalling (106-108). Impaired function in A20 regulation may enhance activation of the NF-κB pathway and lead to sustained autoimmune inflammation and oncogenic mutations (103).
Interferon Induction of transcription of type I IFN genes may be triggered by a virus or by immune complexes containing nucleic acids. Plasmacytoid DCs synthe-sise IFN-α that may have a major role in the development of inflammation in pSS. IFN-α activates infiltrating DCs, T, and B cells. The B cells produce autoantibodies against SSA/SSB, which bind to autoantigens on the epitheli-um cells or form immune complexes. These immune complexes may bind to TLR7 and TLR9 in endosomes of the plasmacytoid DCs, and in this way, may further enhance IFN-α production and perpetuation of the autoimmune process in pSS (70) (Fig. 1). Type I IFN also induces upregulation of the transcription of several genes, ‘IFN signature’, encoding antiviral and im-munomodulatory proteins. Such ‘IFN signature’ has been described in both the minor salivary glands (71) and peripheral blood of patients with pSS (109).
Histopathological studies have shown that epithelial cells in glandular tis-sue express a type I IFN signature (mostly IFNβ) and type II signature (pre-dominately IFNγ), and lymphocytes express a type II IFN (IFNγ). This ‘IFN signature’ in the exocrine glands lesions and high expression of TLR3 in epithelial cells may support the implications of viral infection in pSS patho-genesis (59, 110, 111).
Genetics The genetic factors are important to the pathogenesis of pSS and constitute an active area of research. Familial clustering studies have revealed that ap-proximately 35% of patients with pSS have relatives with pSS or another autoimmune disease (112). It is known that the HLA class II system has genetic influence on susceptibility to autoimmunity and pSS. A meta-

24
analysis of 23 worldwide studies has shown a significantly increased risk for pSS in association with the alleles DQA1*05:01, DQB1*02:01 and DRB1*03:01, while DQA1*02:01, DQA1*03:01 and DQB1*05:01 alleles have been shown to be protective for pSS (113).
Several significant non-HLA gene associations with pSS have been re-ported in Scandinavian studies by the candidate gene approach. Polymor-phisms in IRF5 (encoding interferon regulatory factor 5), STAT4 (encoding signal transducer and activator of transcription 4) (114, 115), in four signal-ling proteins from the NF-κB pathway (104), and polymorphisms in lympho-toxin (LTA/LTB)/TNF have shown strong association with pSS (116).
Two large genome-wide association studies (GWAS) of patients with pSS have shown the strongest associations with the HLA-II locus, followed by type I and II IFN signalling such as STAT4, IRF5, IL-12A, B lymphocyte kinase (BLK) and chemokine receptor type 5 (CXCR5), which are important for B-cell function. A strong association with pSS has also been noted with the TNFAIP3 interacting protein 1 (TNIP1) gene that is involved in the NF-κB pathway. These studies have also uncovered several new immunity-related genes and multiple different single nucleotide polymorphisms for each gene (117, 118) which can be important in pSS pathogenesis.
Epigenetics Genetic regulatory mechanisms are also of interest in studies of the pathoge-netic mechanisms underlying the development of pSS. One of these mecha-nisms is epigenetic factors or biological mechanisms that can switch genes on and off. Defective patterns of DNA methylation may lead to different gene expressions in pSS. For example, hypomethylation of IFN-regulated genes in B cells can increase the genes’ expression (119) and promote pSS development.
Haematological disturbances B-cell hyperactivity in pSS may result in hypergammaglobulinemia, which can be detected in about half of the patients with pSS (91, 120). Cytopenia, which includes anaemia, leucopenia, and thrombocytopenia, may be present in one-third of pSS patients. Leucopenia is more common than anaemia and thrombocytopenia and occurs in about 40% of pSS patients (121).
Reduced concentrations of complement factors (C3 and/or C4) have been reported in 3-16% of the patients with pSS and can be associated with in-creased lymphoma risk (30, 47, 120).
Cryoglobulins (the most common type being type III-mixed cryoglobuli-naemia followed by type II-mixed cryoglobulinaemia) can be detected in 5-15% of pSS patients (30, 47, 120, 122).

25
Autoantibodies Autoantibodies against ribonucleoproteins SSA/Ro and SSB/La are among the most frequently detected autoantibodies in sera of patients with pSS. Anti-SSA/Ro is found in 33-74% and anti-SSB/La in 23-52% of patients with pSS (122). Moreover, anti-SSA/Ro and anti-SSB/La can be detected many years before sicca symptom onset (26). These autoantibodies are a useful diagnostic marker for pSS and are included in the classification crite-ria for pSS (17-19). However, anti-SSA/Ro and anti-SSB/La antibodies are not specific for pSS as they also can be found in sera of patients with other systemic autoimmune diseases e.g., SLE, systemic sclerosis, and myositis, and the pathologic significance of these antibodies is still poorly understood.
Anti-SSA/Ro and anti-SSB/La antibodies are associated with parotid gland damage (decreased unstimulated salivary flow and higher focus scores on minor salivary gland biopsy), and a higher prevalence of extraglandular manifestations (122). In addition, the presence of anti-SSA/Ro antibodies in the mother increases the risk of neonatal lupus (subacute cutaneous lupus, photosensitivity, cytopenias, hepatosplenomegaly, myocarditis, and pneu-monitis) and complete heart block in the fetus. The risk is 1–2% in the first pregnancy and increases significantly to 17% in the subsequent pregnancy if a previous child has been affected (123).
ANA is also one of the most frequent autoantibodies found in 59-85% of pSS patients, whereas RF is reported in 36-74% of the patients with pSS (122). ANA positivity is associated with a higher prevalence of RF, hyper-gammaglobulinemia, a higher risk of cutaneous vasculitis, as well as articu-lar and renal involvement (123).
Cellular infiltration in salivary glands A positive histopathological examination of the minor salivary gland biop-sies in pSS demonstrates lymphocytic infiltration containing ≥50 cells per 4 mm2, so-called focus score (Fig. 2) (124). A focus score ≥1 is considered as a pathological finding and is included in the criteria for pSS diagnosis (17-19). The sensitivity and specificity of the test are around 85% and it has a high predictive value (125). Approximately 40-70% of the patients with pSS have pathological minor salivary gland biopsy findings. The infiltrating cells are able to organise themselves into B and T cell areas in minor salivary glands. The majority of these cells are T cells, and only 20% are B cells (94, 126).

26
Figure 2. Lymphocytic infiltration containing ≥50 cells per 4 mm2 (focus score) in a biopsy of the minor salivary gland. The figure kindly provided by Dr. Gunnel Nordmark.
Germinal centre-like structures in minor salivary glands At the sites of inflammation in minor salivary glands, infiltrating T and B cells can organise themselves into tertiary lymphoid structures, referred to as ectopic GC-like structures (127-134). GC-like structures appear as mononu-clear cell infiltrates consisting of a dark zone and a light zone located within normal salivary gland epithelium and can be seen in minor salivary gland biopsies in approximately 25-30% of the patients with pSS (130, 135) (ref. 135 is part of this thesis).
The precise mechanism of formation of GC-like structures is unknown. Many cells, chemokines, and cytokines are involved in this process. Th1 cells and IL-17, CD4+ lymphoid tissue inducer cells, CXC-chemokine lig-and 13 and CC-chemokine ligand 21, including pro-inflammatory mediators IL-7 and lymphotoxin-α1β2, are necessary for recruitment of B and T cells and formation of GC-like structures (134).
GC-like structures share not only morphological features with secondary lymphoid organs (lymph nodes and spleen), but also similar mechanisms controlling their induction and maintenance. B cells in these structures un-dergo somatic hypermutations and antigen-driven selection of the variable region genes of immunoglobulins.
Formation of the GC-like structures may point to a more aggressive pSS disease course. The presence of these structures correlates with a higher fo-cus score, higher concentrations of autoantibodies, hypergammaglobuline-mia, and high levels of BAFF and IFN in the serum of the patients with pSS (100).

27
Glandular manifestations Lymphocytic infiltration of the exocrine glands causes primarily sicca symp-toms in the mouth and eyes due to destruction and impaired secretory func-tion of the glands.
Xerostomia is typically noticed as it becomes necessary to drink frequent-ly liquids to aid in the chewing and swallowing of dry food. Severe dryness in the mouth feels like a burning sensation in the mouth, may be complicated by bad breath, cracking and soreness in the lips, inflammation of the gums, and may lead to an increased risk to develop dental caries.
Patients with xerophthalmia complain of irritation, sensation of grit or sand in the eyes, and itching. It is common to get a conjunctival injection, often complicated with a bacterial infection, and the eyes may become sensi-tive to light. Chronically dry-eyes can lead to the destruction of epithelium in the cornea and conjunctiva, so-called keratoconjuctivitis sicca.
Inflammation in other secretory glands producing moisture predisposes to impaired glandular function and dryness symptoms as well. Dryness in the mucosa of the nose results in rhinitis sicca. Dryness in the throat and the upper and lower airway results in a hoarse voice, cough, tracheitis sicca, recurrent bronchitis sicca, and pneumonitis. Impaired secretion of the mucus of glandular in the gastric mucosa causes gastrointestinal symptoms. Vaginal dryness leads to painful vaginal intercourse (vaginitis sicca) and recurrent urinary tract infections. Abnormally dry skin (xerosis) may cause discomfort and itching.
Chronic or recurrent, uni- or bilateral swelling of the major salivary glands is observed in 18-31% of patients with pSS (30, 31, 47, 120).
Extraglandular manifestations Extraglandular involvement is crucial for pSS prognosis (136). About 20-50% of patients with pSS may develop extraglandular manifestations (120, 137), but only 15% of the patients have severe extraglandular manifestations (120). Most of the extraglandular manifestations are listed in domains of the ESSDAI (Table 5) (25, 138).
Arthralgia has been reported in more than 50% of patients with pSS (136). Arthritis is characterised by inflammation/synovitis in one or more joints or defined as active articular involvement, that is, joint pain accompa-nied by morning stiffness for more than 30 minutes, and has been described in approximately 16% of pSS patients (136).
Muscle pain, especially fibromyalgia is common in pSS (up to 27%). His-topathological signs of polymyositis or subclinical myositis have been de-tected in up to 47% of the patients. However, clinical and histopathological findings of myositis appearing at the same time are uncommon in pSS pa-tients (up to 5%) (139).

28
Central nervous system involvement may occur in up to 20% of pSS pa-tients and can manifest as a combination of migraine-like symptoms, sen-sorimotor deficits, neuropsychiatric diseases, cognitive disturbances, and unspecific subcortical lesions (137, 140, 141).
There can be diverse symptoms of the peripheral neurologic involvement. Motor, sensory, or autonomic nerve system, alone or in combination can be affected, but sensory, sensorimotor and particularly small fibre neuropathy (SFN) are the most common features and are reported in up to 64% of the patients with pSS (142, 143). In SFN, patients complain of painful or burn-ing paraesthesias, but an objective examination, inclusive nerve conduction studies, are normal. SFN is diagnosed by the counting of nerve fibres in a skin biopsy. No small fibres are detected in positive cases.
Pulmonary manifestations, including both bronchial and parenchymal in-volvement, have been reported in up to 16% of the patients with pSS. Com-puter tomography (CT) findings include bronchiectasis/bronchiolar abnor-malities (50%) and ground glass opacities/interstitial changes (49%), being the most frequent abnormalities. The pulmonary function tests disclose de-creased diffusing capacity for carbon monoxide and/or abnormal forced vital capacity. The most frequent histopathological diagnoses are: non-specific interstitial pneumonia (45%), bronchiolitis (25%), usual interstitial pneumo-nia (16%), and lymphocytic interstitial pneumonia (15%) (136). Lung in-volvement is more frequent in older (>70 years of age) patients and is one of the main causes of death in pSS (47).
Annular erythema or subacute cutaneous lupus erythematosus is a photo-sensitive rash, most frequently localised in the face, the upper arms, and the neck, characterised by a wide elevated border and central pallor. It is report-ed in up to 9% of pSS patients. Cutaneous vasculitis has been reported in approximately 10% of pSS patients. It may present as cutaneous purpura (88%), nodules, digital lesions, cutaneous ulcers, urticarial vasculitis or maculopapular rash. The biopsy usually shows cutaneous leukocytoclastic vasculitis (90%) (136). Severe cryoglobulinemic vasculitis is associated with increased rate of mortality in patients with pSS (47).
Raynaud’s phenomenon has a prevalence of 10-37% in pSS. Its clinical course is milder than in systemic sclerosis (11) and it may precede the onset of sicca symptoms (144).
Chronic tubulointerstitial nephritis is the main renal involvement associ-ated with pSS. Renal tubular acidosis (RTA) occurs in approximately 9% of patients with pSS. It is caused by a generalised dysfunction of the distal re-nal tubules leading to renal acid retention and bicarbonate loss. Clinically, RTA is seen in conjunction with hypokalaemic weakness/paralysis (69%), renal colic (12%), radiologic nephrocalcinosis (17%), osteomalacia (13%), and polyuria/polydipsia (diabetets insipidus) (4%). Renal failure in RTA is reported in up to 24% of the patients (136, 145) and is associated with excess mortality in pSS patients (47).

29
Constitutional features Constitutional symptoms, such as fatigue, night sweats, low-grade fever, and weight loss may develop in 50-70% of the patients with pSS during the dis-ease course (146, 147). Swelling of lymph nodes may also occur in patients with pSS. Fatigue has a serious negative impact on patients’ quality of life and is often associated with anxiety, depression, and sleep disturbance (148).
Co-morbidity Oral dryness increases the risk of caries, and the dry mucosa is more prone to bacterial infections and candidiasis. Prevalence of candidiasis has been reported to be up to 37% of cases in pSS patients (149). Autoimmune chron-ic hepatitis and primary biliary cirrhosis can be associated diseases in 2-9% of patients with pSS (150, 151). Hypothyroidism and Grave’s thyrotoxicosis can be found in 7-14% and 2-3%, respectively, in pSS (151, 152).
Cardiovascular risk factors, such as diabetes mellitus have been found in 27%, while hypertriglyceridemia has been found in 22% (153) and hyperten-sion in 28-50% (154) of pSS patients.
Risk factors and causes of mortality in pSS Lymphoid and solid organ malignancies, cardiovascular disease, and infec-tions have been reported as predominant causes of death in pSS (28, 155). Older age at pSS diagnosis, male sex, parotid swelling, extraglandular in-volvement, vasculitis, low complement (C3 and C4), anti-SSB/La positivity, and cryoglobulinemia have been reported as risk factors associated with increased mortality. However, pSS is not associated with an all-cause mor-tality compared with the general population (155).
Sex differences There are very limited data about sex differences in pSS. A couple of studies (47, 156) showed that men might have a more severe glandular involvement and less-pronounced systemic and immunologic disease than women. One study reported that pSS in men is more common among individuals 65 years of age or older compared to women (11). A systematic review of 7,888 pa-tients with pSS has shown that male gender can be associated with increased mortality (155), but the reason for this is not known.
Diagnosis of pSS in clinical practice The diagnosis of pSS is complex and requires a stepwise approach for the evaluation of: (i) ocular and oral dryness symptoms, which can precede pSS

30
diagnosis by several years and is present in 98% of pSS patients (157), (ii) objective measures of lacrimal and salivary gland dysfunction, (iii) evidence of autoimmunity with positivity for SSA/Ro (and/or SSB/La) antibodies, and (iv) evidence of inflammation in labial salivary gland biopsy.
Essentially, if a patient presents with sicca and/or systemic features of pSS accompanied by laboratory abnormalities, such as increased erythrocyte sedimentation rate (ESR) in the setting of normal CRP, polyclonal hyper-gammaglobulinemia, and/or cytopenia, that patient should undergo a Schirmer’s test and a 15-minute unstimulated whole saliva collection. Fur-ther, the patient should be tested for SSA/Ro antibodies. The patient needs to have either a positive test result for anti-SSA/Ro or a positive labial biopsy in combination with objective glandular tests for a final pSS diagnosis. Labi-al biopsy of the minor salivary glands is performed under local anaesthesia with a collection of 4-5 minor salivary glands by blunt dissection via an inci-sion through the normal-appearing mucosa (158).
Classification criteria for pSS can be used as support of the diagnosis. It is essential to eliminate other causes of sicca syndrome and evaluate for sys-temic features and extraglandular manifestations (11).
Treatment Most patients with pSS are treated symptomatically with tear and saliva sub-stitution, chewing gum and other moisture replacement therapies, and mois-ture stimulating products taken to prevent dental caries and oral infections. Proper dental care and regular visits to a dentist and dental hygienist are important. Cholinergic drugs (muscarinic agonists), such as pilocarpine or salagen and cevimeline hydrochloride sometimes have an effect on both dry eyes and dry mouth by increasing glandular secretion, but side effects impair their usefulness.
Patients experiencing joint or muscular pain respond well to non-steroid anti-inflammatory drugs and analgesics. In some patients, particularly with arthritis, disease modifying drugs (e.g., hydroxychloroquine, methotrexate or azathioprine) and corticosteroids are used.
Patients with extraglandular manifestations are treated as similar condi-tions in other autoimmune diseases (corticosteroids in combination with immunosuppressive drugs). Biological agents with B-cell depletion (Rituxi-mab®) (159-161), anti-BAFF (Belimumab®) (162), and drugs targeting T cells (Orencia®) (163) may be used in selected cases of moderate-severe forms of pSS.

31
Lymphoma Lymphoma is a type of malignancy which develops from malignant trans-formed immune cells. Lymphomas are usually divided into non-Hodgkin lymphoma (NHL) and Hodgkin lymphoma (HL). NHLs account for about 90% of all lymphomas. According to the WHO classification, NHLs are subdivided according to cell type (B, T and NK cells), relation to the lymph node area, and morphology (164). Recently, a revision of the classification of blood malignancies was published, which clarifies the diagnosis and man-agement of lymphoid neoplasms at the very early stages of their develop-ment, refines the diagnostic criteria for some entities, and details the expand-ing genetic/molecular features of different lymphoid neoplasms with their clinical correlates (165).
In the general population, most NHLs are of B-cell origin, and less than 10% are derived from T or NK cells. The most frequent subtypes of B-cell lymphoma in the general population are diffuse large B-cell lymphoma (DLBCL) (25-35%) and follicular lymphoma (20%). Marginal zone lym-phoma (MZL) is subdivided into three subtypes according to the sites in-volved: extranodal marginal zone of mucosa-associated lymphoid tissue (MALT) lymphoma, splenic MZL, and nodal MZL (164).
DLBCL is an aggressive lymphoma of large B lymphoid cells with nucle-ar size more than twice the size of a normal lymphocyte and has a diffuse growth pattern. By using immunohistochemistry and antibodies against CD10, BCL6, and IRF4/MUM1 and/or genetic expression analyses (166), DLBCL can be divided into two subsets with different pathologies, treatment outcomes, and prognoses: the germinal centre B-cell-like (GCB) type with better prognosis and the activated B-cell-like (ABC) type (also referred to as the non-GC type) with worse prognosis (166-168).
DLBCL is more common in the elderly. The median age at diagnosis is in the seventh decade and DLBCL is slightly more common in males than fe-males (164).
MALT lymphoma comprises 7-8% of all B-cell lymphomas, whereas up to 50% are gastric MALT lymphoma (164). In most cases, MALT lympho-ma occurs in adults with a median age of 61 and a slight female preponder-ance (male: female ratio is 1:1.2) (164).
Risk and predictors for lymphoma in pSS An increased risk of malignant B-cell NHL in patients with pSS was first reported in 1978 and the risk for lymphoma in pSS was estimated to be 44 times greater compared to age, sex, and race-matched controls (169). The patients with pSS have the highest risk of lymphoma development reported in autoimmune diseases. A meta-analysis of 20 studies showed a standard-

32
ised incidence ratio (SIR) of 18.8 for NHL in pSS (Table 6), whereas in SLE, the SIR was 7.4 and in RA, 3.9 (170).
Table 6. Prevalence of lymphoma in primary Sjögren’s syndrome
Study period, country
Follow-up time, years
pSS patients
n
Lympho-mas
n (%)
Standardised incidence ratio
(95% confidence interval)
Reference
1978-2001 meta-analysis of
5 studies
18.4 1300 30 (2) 18.8 (9.5-37.3) Zintzaras et al. (170)
1984-2002 Sweden
8 286 11 (3.8) 15.6 (7.8-27.9) Theander et al. (171)
1979-2003 Britain
10.8 112 11 (9.8) 37.5 (20.7-67.6) Lazarus et al. (172)
1990-2005 China
4.4 1320 8 (0.6) 48 (20.7-94.8) Zhang et al. (173)
2005-2007 Taiwan
6911 23 (0.3) 7.1 (4.3-10.3) Weng et al. (38)
1980-2009 Norway
3,813 per-son-years
443 7 (1.6) 9.0 (7.1-25.3) Johnsen et al. (174)
1964-2010 Sweden
9.4 14,570 143 (0.9) 4.9 (4.2-5.8) Fallah et al. (175)
A Swedish study has shown a relative risk of 16 for the development of lymphoma in pSS patients compared to the general population (171). The risk of NHL in Norwegian patients with pSS was increased up to nine times compared to the general population (174). A nationwide Swedish study re-ported an SIR of 4.9 of NHL in 14,570 SS patients (175). The prevalence of lymphoma is 4-5% in patients with pSS (9, 120, 175) and risk for lymphoma development increases during the course of pSS. The cumulative risk of developing lymphoma may reach 3.5% during the first five years and 9.8% after 15 years since the established pSS diagnosis (176).
Lymphoma development in pSS is associated with certain risk factors. Significant predictors reported for the development of lymphoma are low complement (C3 and C4), the presence of cryoglobulins, a low CD4:CD8 ratio, the persistence of unilateral or bilateral parotid gland swelling, sple-nomegaly, lymphadenopathy, and palpable purpura (171, 177, 178) (Table 7).
By contrast, ANA, anti-SSA/Ro, anti-SSB/La, RF, and hypergammag-lobulinemia have not been associated with lymphoma development (178). Parotid gland swelling, haematological manifestations, lymphadenopathy, and disturbances in the biological parameters (low complement, hypergam-maglobulinemia, and the presence of cryoglobulinemia), are very common (>70%) in the patients with pSS developing subsequent lymphoma (185).

33
Table 7. Risk and predictive factors of lymphoma development in pSS
Risk factors References • Clinical features
Persistent swelling of major sali-vary glands
Anaya et al. (179), Ioannidis et al. (9), Nishishinya et al. (178), Sutcliffe et al. (177)
Swollen lymph nodes Anaya et al. (179), Nishishinya et al. (178), Sutcliffe et al. (135, 177)
Paplpable purpura Ioannidis et al. (9), Nishishinya et al. (178), Theander et al. (171)
• Laboratory Cryoglobulinemia Nishishinya et al. (178), Baimpa et al. (180) Lymphopenia Soalns-Laque et al. (176), Theander et al. (135, 171) Low C4 Ioannidis et al. (9), Nishishinya et al. (178), Soalns-Laque
et al. (176), Theander et al. (135, 171) Monoclonal component Anaya et al. (179) New predictors
• Histopathology of salivary glands Presence of GC-like structures Theander et al. (135)* Focus score ≥3 Risselada et al. (181, 182)
• Cytokines High levels of BAFF (TNFSF13B) Nezos et al. (183) High levels of Flt3-ligand Tobon et al. (184)
• Genetic predictors Impairment of TNFAIP3 (A20) Nocturne et al. (108) *This reference is part of the current thesis.
All these suggested clinical and laboratory risk factors of lymphoma are easy to check up and follow up in daily practice. However, there is no consensus on the monitoring of pSS patients with risk factors.
Regarding the risk of development of lymphoma, it has been suggested to divide the patients with pSS into two types based on two identified risk fac-tors, i.e., low C4 levels and/or palpable purpura (9): 1) Type 1 (80-85%) with a low risk of lymphoma, patients without the pre-dictors for lymphoma development; 2) Type 2 (15-20% of the patients) with a high risk of lymphoma, those hav-ing one or both risk factors.
New predictors of lymphoma have also been proposed, such as GC-like structures (ref. 135 is part of this thesis), high focus score in labial biopsy (181, 182), high BAFF (183), Flt3-ligand (184) levels, and genetic impair-ment of TNFAIP3 (108, 186) (Table 7).
Data concerning male gender and lymphoma risk in pSS are inconsistent. A meta-analysis of 18 studies could not reveal that male gender was associ-ated with the occurrence of lymphoma (178).

34
Possible mechanisms of lymphoma development in pSS It is presumed that chronic antigenic stimulation by an as-yet-unknown anti-gen may be associated with the development of pSS-related lymphoprolif-eration. Epigenetic mechanisms may play a role in the disturbed function of Th17 or Treg cells (187), and dysregulation and hyperactivity of B cells. This imbalance and dysfunction of the T and B cells contribute to both auto-immunity and haematologic malignancies associated with autoimmune dis-ease (188). Additional oncogenic events, loss of the B-cell cycle control, and overproduction of B-cell specific stimulators and defective apoptosis of B cells may contribute to lymphomagenesis (179, 189-191).
Somatic mutations of the TNFAIP3 (A20) protein, which plays a key role in controlling NF-κB activation, have been observed in pSS associated MALT lymphoma (127). Another pathway with signalling abnormalities in NOTCH system (a highly conserved cell signalling system present in most multicellular organisms), that also mediate autoimmune diseases, may con-tribute to lymphoma development as well (192).
EBV is associated with some lymphoproliferative diseases, such as HL, post-transplant lymphoma, subsets of DLBCL, and immunodeficiency-related lymphoproliferation (164). However, it is presumed that pSS-lymphomas are not associated with EBV infection (193).
After contact with antigen, activated B cells migrate to secondary lym-phoid organs (e.g., lymph nodes or the spleen) and form GCs (Fig. 3), which are important for B-cell maturation and lymphoma development (194, 195). The GC is divided into two parts: the dark zone and the light zone. The dark zone contains rapidly proliferating B cells, so-called centroblasts. Centro-blasts divide and undergo somatic hypermutation to adapt antibodies to anti-gen. This process introduces mutations, which mainly are single nucleotide polymorphisms in the light chain of immunoglobulin genes. The light zone contains non-dividing centrocytes. The centrocytes bear high-affinity B-cell antigen receptor and have the capacity to bind the antigen, which is present-ed by follicular DCs. The centrocytes undergo a selection of beneficial muta-tions, which enhance the affinity for the original antigen. Follicular DCs and Th lymphocytes assist centrocytes during this selection process. Those cells with disadvantageous mutations die by apoptosis. Further, centrocytes un-dergo class switching, which is the second mechanism to adapt an antibody to an antigen. The class switching changes Ig heavy chains from IgM to IgG, IgA, or IgE. Finally, selected GC B-cells differentiate into plasmablasts and plasma cells or memory B cells expressing mutated Ig with increased affinity for the immunising antigen (196).
DLBCLs may originate from centrocytes of the GC B-cells (GCB sub-type) or outside GC from activated B-cells (ABC subtype). The GCB-DLBCL has overexpression of GC-related genes and a higher rate of BCL2

35
translocations. In contrast, the ABC-DLBCL has overexpression of the NF-κB pathway (167, 168).
Some naïve B-cells migrate to the marginal zone, which is located at the periphery of the lymphoid follicles and accommodates a marginal zone B-cell population of varied maturation stages (197). MALT lymphoma and MZL arise from these monocytic marginal zone B cells. Some subtypes of B-cell lymphoma origin are shown in Fig. 3.
Figure 3. Origin of lymphomas and the main oncogenic pathways. Germinal centre (GC)-derived lymphomas are blocked at different stages of maturation. Diffuse large B-cell lymphoma (DLBCL) of GCB-subtype and follicular lymphoma originate from the light zone B cells. Marginal zone lymphomas (MZL) and MALT-type lymphomas originate from the marginal zone B cells. Activated B cell-like (ABC)-DLBCL originates from late GC B cells (plasmablasts). PDC=plasmacytoid dendrit-ic cell; Tfh=T follicular helper; FDC=follicular dendritic cell. The figure was pro-duced by the author using Servier Medical Art.
Recurrent genetic abnormalities, such as translocations t(11;18) (q21;21), t(14;18) (q32;q21), t(1;14) (p22;q32) are reported in MALT lymphoma in the general population. These translocations may lead to activation of the NF-κB pathway, which results in cell survival and proliferation. However, in pSS-related MALT lymphoma, other lymphomagenesis mechanisms may be involved compared to the general population. These translocations have been reported only in small proportions of pSS-MALT lymphoma patients (198). On the other hand, a possible genetic aberration correlating to pSS-MALT

36
lymphomas could be the deletion of the long arm of chromosome 6(6q23) leading to the deletion of TNFIAP3 (A20), which is required for termination of NF-κB activation (199). To confirm A20 molecular aberration in pSS-related MALT lymphoma development, a larger cohort of patients to study is required.
Lymphoma subtypes in pSS and lymphoma diagnosis Extranodal MALT lymphoma is the most common histological subtype of lymphoma encountered in patients with pSS (169, 177, 180, 185, 189, 200). These lymphomas are generally characterised by an indolent course and good performance status in patients. MALT lymphomas in pSS are mostly localised in the salivary glands, which are the major sites of inflammation in pSS, but other mucosal sites such as lacrimal glands, the naso-pharynx, the stomach, the thyroid, and the lung can be affected. A rapidly enlarging major salivary gland may herald the emergence of a malignant lymphoma. The parotid gland enlargement is often unilateral and the tumour affection is fixed and hard. Magnetic resonance imaging (MRI) (201) or ultrasound with Doppler may help to differentiate between focal salivary gland inflammation or suspected lymphoma, and gives the possibility of performing fine-needle- aspiration or biopsy of the gland during the same diagnostic procedure (202).
The histology of MALT lymphoma displays diffuse lymphoid infiltrates with small lymphocytes with slightly irregular nuclei, cells with pale cyto-plasm resembling those of centrocytes, or plasmacytic differentiation. In glandular tissues, the epithelium is destroyed by aggregates of lymphoma cells resulting in the so-called lymphoepithelial lesions. The lymphoma cells may colonise the GCs. Transformed centroblasts or immunoblasts may be present in MALT lymphoma. The phenotype of tumour cells of MALT lym-phoma is CD20+, CD79+, CD5-, CD10-, CD23-, CD43+/-, and CD11+/-. They express the marginal zone cell-associated antigens CD21 and CD35. However, there is no specific marker for MALT lymphoma at present. Im-munoglobulin light chain restriction is relevant in the differentiation from benign lymphoid infiltration. Additionally, to distinguish from other B-cell lymphomas, a confirmation of the absence of CD5, cyclin D1, and CD10 is important (164).
High-malignant DLBCL is the other lymphoma subtype reported in high frequency in pSS (169, 171, 203, 204). In some cases (up to 10%), a histo-logic transformation of a previous low-grade lymphoma has been observed in pSS (193, 200, 204).
An aggressive clinical picture can suggest high-malignant lymphoma or transformation. The clinical criteria of a transformation are as follows: a rise in the lactate dehydrogenase level, a rapid localised nodal growth, new ex-tranodal sites of disease, the presence of B symptoms (e.g., fever, weight loss, and severe night sweats) or hypercalcemia (205). Imaging with CT,

37
MRI, ultrasound and biopsy with immunohistochemistry, and molecular biology studies are helpful for diagnosis. Several studies have reported on the clinical utility of an 18F(fluorodeoxyglucose (FDG) positron emission tomography (PET) scan to detect sites of transformation or to diagnose DLBCL de novo (206, 207).
Other mature B-cell malignancies, for example, HL (208) and plasma cell neoplasms/myeloma (171, 173, 209), have rarely been studied in pSS.
Outcome and treatment of lymphoma in pSS MALT lymphoma in pSS is usually associated with a good prognosis. A retrospective study in Greece reported a 3-year overall survival (OS) and event-free (EFS) survival of 97% and 78%, respectively, in pSS-MALT lymphomas (210).
For some asymptomatic MALT lymphoma patients with a localised dis-ease in the salivary glands, a ‘watch and wait’ policy may be adopted (211). Patients with symptomatic MALT lymphoma localised in the salivary glands may be treated with low-dose radiotherapy or, in some cases, with surgery or combination of both, and combined with or without chemotherapy (212). According to a recent large multicentre international study, salivary gland MALT lymphoma associated with pSS has a good prognosis regardless of the initial treatment and a median overall survival can be achieved up to 20 years after diagnosis (212).
Disseminated MALT lymphoma and pSS-DLBCL are treated with chem-otherapy with or without rituximab as in other patients with DLBCL. A suc-cessful outcome (100% OS and EFS at the three-year mark) in eight patients with pSS-DLBCL was reported when R-CHOP (rituximab-cyclophosphamide, doxorubicin, vincristine, prednisone) or CHOP was used (210).
IgG4-related disease IgG4-RD is a chronic fibrotic inflammation, characterised by tissue infiltra-tion by lymphocytes, mostly IgG4-positive plasma cells (PCs), development of fibrosis, and often elevated IgG4 levels in serum (213, 214). The disease can affect nearly all organs. A wide variety of different diseases (Mikulicz’s disease, type 1 autoimmune pancreatitis, lymphocytic hypophysitis, Riedel’s thyroiditis, Kuttner’s tumour, retroperitoneal fibrosis, inflammatory aortic aneurysm and aortitis, interstitial nephritis and pneumonitis, and inflamma-tory pseudotumour) are now recognised as IgG4-RD manifestations. The nomenclature of the disease and an international pathology consensus state-ment have been published in 2012 when IgG4-RD had been established (215, 216). However, IgG4-RD, as a recently recognised disease still re-

38
mains unknown or is a very new entity for many clinicians. Therefore, it may first come to clinical attention as visible organ swelling or detected organ dysfunction due to fibrosis. Sometimes it can be identified incidentally by imaging and specific biopsy findings when patients undergo an investiga-tion of suspected tumour disease, infection, or another immunity mediated disease (217). Most studies about this disease come from Japan and, there-fore, the epidemiology of the disease is not yet described in Western coun-tries.
Pathogenesis of IgG4-RD Immunopathogenesis is not yet fully understood. It is supposed that oligo-clonal B and T cells develop during chronic stimulation by unknown anti-gen(s). Oligoclonal expansion of plasmablasts positive for IgG4 arises after somatic hypermutation from naïve B cells in GCs (Fig. 3). T follicular helper cells can be involved in the differentiation of B cells during their develop-ment in the GCs and contribute to class switching (86, 218). The oligoclonal plasmablasts are identified by flow cytometry as CD19+CD20-
CD27+CD138+ and correlate with activity of the disease (219, 220). Plas-mablasts differentiate into antibody producing short- and long-lived PCs producing IgG4 (Fig. 3). Short-lived PCs can be depleted by rituximab (219). However, the plasmablasts may reemerge from naïve B cells again during relapses with an expression of other oliglonalities by rearrangements of V-J repertoires (219). It is supposed that the chronic disease may be en-hanced by long-lived memory PCs (221), which are not depleted by rituxi-mab.
T cells, in particular Th2 immune response cells (IL-4, IL-5, and IL-13) in atopic individuals, may be important in the pathogenesis of IgG4-RD (222, 223). Recent studies have shown that effector memory CD4+ T cells with cytotoxic function are expanded in the peripheral blood and in affected or-gans. They produce IL-1β cytokine and transforming growth factor (TGF)- β1, which are both important for inflammation and fibrosis (224).
Type 1 autoimmune pancreatitis is associated with IgG4-RD and is the best studied of all clinical manifestations of the disease so far. Interestingly, as in pSS, plasmacytoid DCs producing type I IFN-α have been shown to be important in activating B cells to produce IgG4 and promoting the develop-ment of this type of pancreatitis (225).
Similarities and differences with pSS Patients with IgG4-RD in salivary and/or lacrimal glands share many simi-larities with pSS patients (Table 8) (217). Therefore, some patients with sicca symptoms previously diagnosed as incomplete or atypical pSS could instead have undiagnosed IgG4-RD (226).

39
Mikulicz’s disease (MD), which is now recognised as one manifestation of IgG4-RD, is characterised by symmetrical and painless swelling of the lacrimal, parotid, and submandibular glands and was first described in 1892. For a long time, MD has been considered to be a subtype of SS because of clinical and histopathological similarities (227). However, many differences between MD and SS have been reported in Japan. In contrast to pSS, MD usually manifests in the form of bilateral and persistent swelling in at least two sites of the lacrimal and salivary glands, with a higher frequency of males represented. Lymphoplasmacytic infiltration with lymphoid follicle formation causes swollen glands but, differently from pSS, duct destruction is uncommon in MD. Serology is usually negative for anti-SS-A/SS-B anti-bodies, and in contrast to SS, dryness symptoms, which are mild compared to SS, improve within days of starting glucocorticoids in MD patients (228). In addition, a minor salivary gland biopsy reveals an intact structure of sali-vary ducts despite lymphocytic infiltration and formation of lymphoid folli-cles, and immunostaining shows IgG4-positive PCs (214, 228, 229).
Table 8. Similarities and differences between pSS and IgG4-RD with salivary gland involvement
Characteristics pSS IgG4-RD Mean age at onset, years 40-50 50-60 Female: male 1:9-20 1:3, if a head region
affected 1:1 Swelling of the major salivary glands Recurrent Persistent or recurrent Major salivary glands affected Parotid glands Submandibular and/or
parotid glands Sicca symptoms Moderate - severe No or mild Serum IgG level Often high Often high Serum IgG4 level Normal Often elevated Serum IgE level Normal Often elevated ANA positivity Positive in 59-85% Positive in approx. 20% SSA positivity Positive in 33-74% Absent RF positivity Positive in 36-74% Positive in 70-80% Low complement levels Present in 3-16% Present in approx. 50% Lymphocytic infiltration in the minor salivary gland biopsies with focus score ≥1
Present in 40-70% May be present
IgG4+ plasma cells in the salivary gland biopsies
Not present Present
Storiform fibrosis in the biopsies Not present Present Response to steroids Sometimes Good in most cases Increased risk of MALT lymphoma Present May be present pSS=primary Sjögren’s syndrome; IgG4-RD=IgG4-related disease.

40
Diagnosis of IgG4-RD The disease usually affects middle-to-older age men and is characterised by increased IgG4 levels in serum, clinical symptoms of affected organs, typical histopathology and immunohistochemistry and/or radiology features (Fig. 4).
Figure 4. Flow chart for workup of diagnosis of IgG4-related disease. Adapted from Vasaitis (217).
Early diagnosis of IgG4-RD is difficult because of vague unspecific symp-toms. (217). Increasing experience with IgG4-RD has led to the recognition that the gold standard to diagnose IgG4-RD is a typical histopathology (216) and compatible clinical features and/or radiology (217). High levels of IgG4 in serum support the diagnosis.
Several sets of diagnostic criteria have been proposed for IgG4-RD (230-233). However, these criteria have not been approved internationally, since they are not sufficiently sensitive and specific. The ACR/EULAR working group is now developing classification criteria for IgG4-RD on the basis of histopathology, clinical, and radiological features.
Lymphoma in IgG4-related disease Malignancies have been reported in 7-10% of the patients with IgG4-RD (234, 235). An association with lymphoma development has also been de-
Suspicion of IgG4-related disease Clinical and/or radiological findings (swelling of organs or soft tissue masses), history of ongoing atopic disease
Alternative diagnosis (malignancy, other inflammatory disease, infection) Yes
Laboratory tests (may help diagnosis): Serum-IgG4 > 1.35 g/L (high specificity if increased twice or more over the upper the cut-off), hypergammaglobulinemia, mild-moderate eosinophilia, increased serum IgE
30-50% of patients have normal serum IgG4
No Yes Ye Biopsy with characteristic histopathology (H&E): 1) dense lymphoplasmacytic infiltrates 2) storiform-type fibrosis 3) obliterative phlebitis
Yes
No • Repeat larger biopsies • Review biopsy samples obtained earlier
• 18F-FDG PET/CT helps diagnosis and may guide biopsy decision F FDG PET/CT helps diagnosis and may gui
Compatible clinical findings and typical histopathology Diagnosis of IgG4-related disease
es
bora
),
eses
psy w
) obli
es
s
No ••
o y d
G4
c o )
o
Immunostaining for IgG4+ and IgG+ plasma cells: 1) > 10 IgG4+ plasma cells/ high power field 2) Ratio of IgG4+/IgG+ > 0.4
IgG+

41
scribed. Similar to pSS, MALT lymphoma has been reported in ocular ad-nexa, salivary glands, or meningeal dura within five years after diagnosis of IgG4-RD (236-241). It has also been described that cancer tissue may be infiltrated by IgG4+ plasma cells without other signs of IgG4-RD (242-244).

42
Aims of the thesis
The overall aim of this thesis was to expand the knowledge of the associa-tion between primary Sjögren’s syndrome (pSS) and lymphoma develop-ment using a population-based approach to identify the study subjects. An-other overall aim was to establish a basis for future biologic studies by col-lecting a well-characterized population-based cohort of pSS patients with lymphoma with detailed clinical data, access to lymphoma tissues and diag-nostic salivary gland biopsies. The specific aims of the thesis were:
Paper I - To determine whether the formation of GC-like structures in a low-er labial salivary gland biopsy taken at pSS diagnosis predicts the subsequent development of lymphoma. Paper II - To identify the occurrence of histopathological features of IgG4-RD in lymphoma tissue in a large population-based cohort of patients with an initial pSS diagnosis complicated by lymphoma development and in posi-tive cases, to describe clinical and lymphoma characteristics in these pa-tients. Paper III - To explore the causes of an SS diagnosis in the patients registered with both SS and lymphoma diagnoses in the Swedish Patient and the Can-cer Registers; to assess whether patients with a lymphoma diagnosis before pSS diagnosis differ from pSS patients with a subsequent lymphoma diagno-sis. Paper IV - To explore sex differences in pSS patients with lymphoid neo-plasm, to assess patient characteristics of the most common subtypes of lymphoma in pSS and to assess the subtype distribution of lymphoid neo-plasms in pSS patients in a population-based setting in comparison with the general population.

43
Subjects and methods
Data sources We used the Swedish National Health Care Registers to identify the patients in the studies II-IV. In Sweden, each resident has a unique personal identifi-cation number (245). This number is used in all health care registers and enables linkages across the different registers. In connection with hospital discharges from inpatient care and visits in non-primary outpatient care, diagnoses are coded according to the International Classification of Diseases (ICD) codes (http://apps.who.int/classifications/icd10/browse/2016/en) and recorded in the National Patient Register.
The validity of the ICD codes for diagnoses in the Swedish National Pa-tient Register has been investigated by several studies by using chart data and validating the register-based diagnoses against classification criteria of the diseases and clinical diagnoses. It has been shown that the validity var-ies between different diagnoses and it is about 90% for RA (246) and about 70% for ankylosing spondylitis (247). There are no published reports of val-idations of the diagnosis code for SS, but as the code is assigned for all caus-es of sicca symptoms, we recognised at the start of this project that the valid-ity for the SS code to identify pSS patients could be a problem and included validation of the ICD code for SS as part of the project.
National Registers used in the studies The Patient Register The Swedish Patient Register was initiated in 1964 by the National Board of Health and Welfare and consists of two parts. One part contains information from all patients discharged from hospitals in Sweden (The Inpatient Regis-ter). The second part has been introduced since 2001 and contains data from specialist (non-primary) outpatient care (The Outpatient Register, e.g., rheumatology open care).
The coverage of the Inpatient Register became nationwide in 1987 (http://www.socialstyrelsen.se/Lists/Artikelkatalog/Attachments/18152/2010-10-20.pdf, http://www.socialstyrelsen.se/register/halsodataregister/patient registret/inenglish) and since then includes all inpatient care in Sweden. The

44
coverage of the Outpatient Register is lower (about 80%) than for the Inpa-tient Register as there are missing data from some private caregivers. Prima-ry care is not included in the Outpatient Register.
The medical data collected in the Patient Register include primary and secondary (up to eight) diagnoses/ICD codes, injuries and surgical proce-dures. The administrative data include the personal identification number of the patient, age, sex, dates of admission and discharge, hospital, department, and county.
The Swedish Population Register Patients were followed up for vital status and date of death by using the Swedish Population Register. The Population Register holds data since 1961 and is maintained by the Swedish Tax Agency. This register contains infor-mation of personal identity numbers of residents, their addresses, place of birth, citizenship, moves to and from Sweden, and date of death.
The Cancer Register The Swedish Cancer Register was founded in 1958 (http://www.socialstyrel-sen.se/register/halsodataregister/cancerregistret/inenglish). It covers the whole population of Sweden. The reporting rate is almost 100% through a system of mandatory reporting by both clinicians and pathologists of a newly detected malignancy. The information available in the register includes pa-tient data (personal identification number, sex, age, and place of residence), medical data (site of tumour, histological type, stage, basis and date of diag-nosis, reporting hospital and department, reporting pathology/cytology de-partment, identification number for the tissue specimen) and follow-up data (date and cause of death, date of migration, and whether a patient was regis-tered as a resident in Sweden at the end of a specific year). From data in the register, it is possible to localise available tumour tissue and retrieve it from pathology departments for reanalysis if needed.
Both the Swedish Patient Register and the Cancer Register are maintained by the National Board of Health and Welfare.
The Lymphoma Register The Lymphoma Register was started as a complement to the Cancer Regis-ter. The registration of malignant lymphomas in the Cancer Register was regarded as insufficient because of the complexity of lymphomas combined with frequent changes in their classification. Therefore, a national manage-ment committee formed the Swedish Lymphoma Group for establishing of the Lymphoma Register with the purpose to optimise the quality of care of patients with malignant lymphoma.

45
The Lymphoma Register was set up in 2000 and includes all patients ≥18 years of age (http://www.swedishlymphoma.com/rapporter) diagnosed with lymphoma in Sweden, and holds more detailed information on lymphoma subtypes according to the WHO classification than the Swedish Cancer Reg-ister. Information in the register is collected directly from the oncology cen-tres. The coverage of the register during the current project (2000-2010) is estimated to be >90% of all lymphomas in the Cancer Register. National reports are presented annually. Additionally, each clinic has the opportunity to obtain own data from the regional lymphoma register. From 2007, the Lymphoma Register, together with other blood malignancies, has a web-based platform with the electronic input of data and also contains data of response to the treatment.
Since 2008, all cases of multiple myeloma, plasmacytoma and plasma cell leukaemia are registered separately from the Cancer Register in the Myelo-ma Register. The coverage of the Myeloma Register is 91% in relation to the Cancer Register (https://www.cancercentrum.se/glbalassets/cancer-diagnoser/blod-lymfom-myelom/myelom/arsrapport-myelom-2008.pdf).
Definitions used for blood malignancies in the thesis Blood malignancies involving mature lymphoid cells are called lymphoid neoplasms or lymphoid malignancies in the thesis and include both lym-phoma and multiple myeloma/plasmacytoma.
The International Classification of Diseases The first classification of diseases was created by Bertillon for comparable cause-of-death statistics in 1885. This classification was the basis for the development of the ICD. Since 1900, the ICD classification has been revised 10 times until now. From the seventh revision, the WHO advisory group is involved in revising proposals for every new edition of the ICD classifica-tion (https://www.cdc.gov/nchs/data/misc/classification_diseases2011.pdf).
In studies II-IV, for identifying the patients with SS diagnoses and lym-phoid malignancies, the following ICD classifications have been used: ICD-7 (in use 1958-1967), ICD-8 (in use 1968-1978), ICD-9 (in use 1979-1994), and ICD-10 (in use 1995 to present).
PAPER I Patients and clinical information In this study, patients with pSS fulfilling the 2002 AECG criteria (17) were primarily selected from two Swedish centres with pSS research cohorts (Uppsala and Malmö University Hospitals) participating in a Nordic collabo-ration study on lymphoma and genetics. Of 241 consecutive pSS patients,

46
175 (Uppsala, n=49, Malmö, n=125, and Linköping, n=1) had haematoxylin and eosin (H&E) stained paraffin-embedded minor salivary gland tissue biopsies taken at the time of pSS diagnosis and were included in the study.
Of 175 pSS patients, 161 (92%) were women and 14 (8%) men. The mean age at pSS diagnosis was 51.3 years. Their clinical features, such as salivary gland swelling, vasculitis, lymphadenopathy, internal organ in-volvement, and laboratory parameters such as autoantibodies, blood status, and immunoglobulin values were regularly registered during the follow-up. T-cell subsets and complement function were studied only in the Malmö cohort. Antibody levels varied during the study period and were detected by different assays. Therefore, for serum antibodies against SSA/Ro-60, Ro-52, and SSB/La, a bead-based multiplex immune assay was performed (xMap technology; Luminex, Austin, Texas USA). For the remaining antibodies (ANA and RF), the local reference levels at the time of analysis were used. ANA was positive in 81%, anti-SSA/Ro in 64%, anti-SSB/La in 39%, and RF in 55% of the patients. The internal organ involvement was assessed according to the ESSDAI (21).
Salivary gland tissue re-evaluation The available H&E stained paraffin-embedded biopsies of 175 patients taken from minor salivary glands at the time of pSS diagnosis were re-evaluated by light microscopy at the Broegelmann Research Laboratory in Bergen, Norway. The investigator (MVJ) was blinded from the original biopsy eval-uation results.
Tissues were evaluated for the presence of periductal focal lymphocytic infiltration (124) (Fig. 5A, B) and counting of foci containing at least 50 mononuclear cells per 4 mm2. The presence of ectopic GC-like structures (GC-positive) was defined as well-circumscribed chronic inflammatory cell infiltrate consisted of at least 50 mononuclear cells presented with features indicative of the lymphoid organisation, such as a densely packed dark zone and a light zone within otherwise normal salivary gland epithelium (Fig. 5C, D).

47
Figure 5. Minor salivary gland tissue with periductal focal mononuclear cell infil-trates (focal sialadenitis) but no germinal centre-like structures (A and B) and focal sialadenitis with germinal centre-like structures (C and D). The figure reprinted with permission of the publisher from Theander et al. (135).
The Swedish Cancer Register and lymphoma tissues A linkage using the unique national identification number (245) was per-formed with the Swedish Cancer Register (248). In patients for whom lym-phoma was diagnosed, paraffin-embedded tissue blocks were retrieved, re-evaluated and lymphoma was classified according to the WHO classification (164) by a senior pathologist (GW).
PAPERS II-IV Patients A national population-based cohort of individuals with an ICD diagnosis code for “Sjögren’s syndrome/sicca syndrome” was identified using the Swedish Patient Register 1964-2007 (Fig. 6).

48
Figure 6. Identification of the patients with Sjögren’s syndrome and lymphoid neo-plasm diagnoses from the Patient and the Cancer Registries.
All individuals registered with a SS diagnosis code as the main or secondary diagnosis were identified using the following ICD codes: 374,06 (ICD-7), 734,90 (ICD-8), 710 C (ICD-9) and M35.0 (ICD-10). For M35.0, the addi-tional codes H19.3 (SS with keratoconjuctivitis), J199.0 (SS with pulmonary involvement), G73.7 (SS with myopathy), and N16.4 (SS with the renal tu-bulointerstitial disease) were added. Through linkage with the Cancer Regis-ter (which covers >95% of all incident cancer in Sweden) (249), we identi-fied SS patients diagnosed with lymphoma or myeloma (ICD-7 codes 200-203) at ≥18 years of age between 1990 and 2007, irrespective of the lym-phoid malignancy was diagnosed before or after the first registered SS code in the Patient Register (n=224).
As the diagnosis code for SS has not been used for pSS exclusively, the basis for the SS diagnosis was evaluated from the medical records of all patients (L.Vasaitis) (Fig. 7).
Clinical data was collected from medical records according to a compre-hensive questionnaire. Cases whose medical records could not be identified or were too sparse for diagnostic evaluation were excluded (n=6). Patients were divided into pSS and non-pSS patients. Those with a clear explanation for the SS-diagnosis code other than pSS and/or no support for a pSS diag-nosis in the medical records were considered to be non-pSS patients (n=99). The remaining patients, the pSS group (n=119), had been diagnosed as pSS by the treating physician and all available information in the medical records supported this diagnosis.

49
Figure 7. Flow chart of the validation of Sjögren’s syndrome and lymphoid malig-nancy diagnoses. SS=Sjögren’s syndrome; pSS=primary Sjögren’s syndrome; MGUS=Monoclonal gammopathy of undetermined significance.
We defined the date of lymphoid neoplasm diagnosis as the date of the first diagnosis of the lymphoid neoplasm in the Cancer Register and the date of pSS diagnosis as the diagnosis date documented in the medical records by the treating physician. The reference values of the local analysing laborato-ries were used for ANA, SSA/Ro, SSB/La antibodies, rheumatoid factor, cryoglobulins, and complement C3 and C4. Cytopenias were defined as haemoglobin <120 g/L, leukocytes <4.0 x10 9/L, platelets <150 x109/L, and hypergammaglobulinemia as immunoglobulin (Ig) G >15 g/L. Disease activ-ity at pSS diagnosis was retrospectively assessed in cases with available information using the ESSDAI (21).
To avoid a possible influence of the lymphoid malignancy, clinical and laboratory data of the pSS disease was collected from the medical records from the patient-reported onset of sicca symptoms until six months before lymphoid neoplasm diagnosis in all patients. Four patients formally diag-nosed with pSS after lymphoma diagnosis were excluded from the assess-ment of time period from pSS diagnosis to lymphoid neoplasm diagnosis. As the evaluation of salivary gland biopsies is part of the 2002 (17) and 2016 (19) classification criteria for pSS, we reviewed (C.Sundström, L.Vasaitis) all available H&E stained slides of the diagnostic minor salivary glands bi-opsies for focus score (124).

50
Patients were followed up for vital status and date of death in the Swedish Population Register until January 15th, 2017.
Tissue analyses The lymphoma tissues were reviewed by a senior haematopathologist (C.Sundström) to confirm, and if needed, to reclassify the lymphoma diag-nosis according to the WHO classification of tumours of haematopoietic and lymphoid tissues from 2008 (164). DLBCL were divided into the GCB and ABC subtypes by immunohistochemical staining for CD10, BCL6, and IRF4/MUM1 according to the algorithm by Hans et al. (166). Stainings for different lymphoma markers if needed and analyses for the presence of EBV using in situ hybridisation for EBV-encoded small RNAs in available lym-phoma tissues and minor salivary gland biopsies were also performed. In three cases, the lymphoma diagnosis was based on the original pathology reports, as specimens were not available for re-evaluation. Eleven patients were excluded, as the lymphoma/myeloma diagnosis could not be confirmed (Fig. 7).
Validation of pSS diagnosis The patients with pSS diagnosis according the treating physician and a confirmed lymphoid neoplasm were evaluated for the fulfillment of the 2002 AECG criteria for pSS (17) (lymphoma as an exclusion criterion was not employed) and the 2016 ACR/EULAR criteria for pSS (19).
Paper II Patients In this study, we included all patients with an initial diagnosis of pSS made by the treating physician, a confirmed lymphoid neoplasm by review, and available lymphoma biopsy materials for further analyses (n=79). Of the 79 patients, 44 fulfilled the 2002 AECG criteria for pSS (17), and 35 did not as most of them were diagnosed before 2002 and were not fully examined ac-cording to the criteria. Detailed clinical data was retrieved from the medical records of the 79 patients.
Histopathology and immunohistochemistry All available lymphoma tissues (n=79) were immunostained for IgG4 (clone HP6025, Genetex, CA). Specimens with identified IgG4-positive PCs were investigated for other histopathological features of IgG4-RD (the presence of lymphoplasmacytic infiltration, storiform fibrosis, obliterative phlebitis, and positive immunostaining for IgG4-positive PCs). Further stainings for IgG

51
and CD138, as a PC marker, were performed in tissues with >10 IgG4-postive PCs/high power field (HPF).
In cases with features of IgG4-RD in the lymphoma tissue, other archived tissue specimens were collected and examined for IgG4, IgG, CD138 posi-tive PCs, and histopathological features of IgG4-RD. Additionally, all avail-able minor salivary gland biopsies (n=11) obtained as part of the initial pSS investigation were stained for IgG4. Sections of tonsil were used as positive controls.
The immunostains were evaluated by using a light microscopy with a 40x field objective. The counting of IgG4, IgG, and CD138-positive PCs was performed in the three HPFs with the highest density of stained cells, and the average numbers were calculated.
Definitions pertaining to IgG4-RD were based on the consensus statement on the pathology of IgG4-RD, including the categorisation into histological features “probable” or “highly suggestive” of IgG4-RD (Table 9) (216).
Table 9. Overview of histopathological and immunohistochemistry features of IgG4-related disease based on the consensus statement* (216)
Features of IgG4-related disease Highly suggestive
Probable features
Histological 1. Dense lymphoplasmacytic infiltrate 2. Storiform fibrosis 3. Obliterative phlebitis
≥ 2 patholo-gy features
1 pathology feature
Immunohistochemical Number of IgG4+ plasma cells/high power field
Lacrimal glands >100** Salivary gland >100 Aorta, pleura >50 Surgical specimen of bile duct, liver, lung or pancreas >50 Retroperitoneum, surgical specimen of kidney >30 Biopsy of kidney or pancreas >10 Ratio IgG4+/IgG+ plasma cells >0.4
*Combination of histological and immunohistochemical features are needed for the diagnoses histologically highly suggestive and probable histological features of IgG4-RD; **In lacrimal glands only one pathology feature is required in combination with immunohistochemical features to be histologically highly suggestive of IgG4-RD.
Paper III Study population In this study, we included patients with pSS diagnosis according to the treat-ing physician and confirmed lymphoid neoplasm diagnosis (n=107), and patients with non-pSS but with ICD codes for SS and lymphoid neoplasm diagnoses (n=100) (Fig. 8).
The pSS patients were categorised into those with or without pre-existing lymphoma or myeloma at pSS diagnosis. We defined pre-existing lympho-ma/myeloma as a lymphoid neoplasm diagnosed before pSS diagnosis, or

52
within six months after pSS diagnosis. This definition was chosen, as signs of lymphoid neoplasm were present at pSS diagnosis in these patients, alt-hough the final diagnoses were delayed. In this comparison between the pSS groups with pre-existing and subsequent lymphoma, we excluded two pa-tients with verified DLBCLs, which occurred shortly after organ transplanta-tion (two and nine months after lung and kidney transplantation, respective-ly). It is well known that organ transplantation is associated with EBV-positive post-transplant lymphoma and, in addition, these two patients were included and described in another study about post-transplant lymphoma (250).
Figure 8. Study population in study III: patients with pSS and confirmed lymphoid neoplasm (n=107) and non-pSS patients with lymphoid neoplasm diagnosis (n=100). *Two pSS patients had confirmed diffuse large B-cell lymphoma development after solid organ transplantation and were excluded from the final comparison between the groups.
Paper IV Study cohort This study included 105 patients with pSS diagnosis according to the treating physician and a confirmed lymphoid malignancy (two patients with pSS and post-transplant lymphoma excluded, Fig. 8). Of these, 55 fulfilled the 2002 AECG criteria (17) (lymphoma as exclusion criteria not employed) and 57 fulfilled the 2016 ACR/EULAR criteria for pSS (19). The patients who did not fulfil these criteria were typically not fully examined according to the criteria or this information could not be found in the medical records, but as

53
the available information supported the pSS diagnosis and no other diagnosis, all 105 patients were kept in the study as pSS patients.
Cohort of the general population with lymphoma for comparison To compare the distribution of lymphoma subtypes in the pSS patients with the distribution in the general population, we used the Swedish National Quality Register of Lymphoma 2000-2010 as a reference population (http://www.swedishlymphoma.se/rapporter). The coverage of the register during the period in question is estimated to be about 95% of all lymphomas in the Cancer Register. Plasma cell neoplasms are not systematically includ-ed in the Lymphoma Register. Therefore, identified cases of plasma cell neoplasms were presented separately from the comparison of lymphoma subtype distribution in pSS patients with the distribution in the Lymphoma Register.
Statistics Demographic, serological, and clinical features were descriptively analysed (Paper I-IV). The risk of developing lymphoma in GC-positive and GC-negative patients was evaluated by Kaplan-Meier statistics/log-rank test (Pa-per I). For continuous variables, non-parametric (two-tailed) Mann-Whitney U-test to compare two independent groups was applied (Papers II-IV). For categorical variables, the Chi-square (two-tailed) test or Fisher’s exact test (if <5 observations) was used to compare frequencies between groups (Pa-pers I-IV). A two-tailed value of p<0.05 was considered statistically signifi-cant. The statistical analyses were performed using the Statistica version 13 software (StatSoft, Inc., Tulsa, Oklahoma, USA).
Ethical approval Ethical approval from the Regional Ethical Review Board, Uppsala (Paper I-IV) and approvals of the local ethics committees at the universities of Lund and Bergen were obtained (Paper I).

54
Results
PAPER I Of the 175 salivary gland biopsies, 25% had GC-like structures. Focal si-aladenitis with a focus score of ≥1 was detected in 136 (78%) patients and, among them, 43 (32%) showed GC-like structures. The remaining 39 pa-tients had normal biopsies.
Of the 175 patients with pSS, seven patients had developed lymphoma during the average follow-up time of 10.6 years. Six of seven patients with lymphoma had GC-like structures in their minor salivary glands at pSS diag-nosis, performed a median of seven years before the occurrence of lympho-ma. Thus, among 43 patients with GC-like structures, six patients (14%) developed lymphoma in contrast to one patient (0.8%) among the GC-negative patients (p=0.001). There was a significant difference in lympho-ma-free survival between GC-positive and GC-negative patients (a log-rank test of p=0.001) with worse survival for the GC-positive patients. The short-est interval from salivary gland biopsy until lymphoma development was two years and four months, and the longest interval was 12 years and seven months. The positive predictive value for GC positivity was 16%, whereas the negative predictive value was 99%. Lymphoma subtypes in the GC-positive group were the following: MALT lymphoma in salivary gland with transformation into mantle cell lymphoma (n=1), follicular lymphoma in salivary gland (n=1), anaplastic large T-cell lymphoma in lymph nodes and liver (n=1), and DLBCLs localised in the internal organs and lymph nodes (n=3).
Extraglandular manifestations using domains from the disease activity in-strument ESSDAI (21) occurred with different frequencies in GC-positive and GC-negative patients. Lymphadenopathy, haematological manifesta-tions, mainly leucopenia and hypergammaglobulinemia, systemic disease affecting a higher number of organs, lower C4 levels, positivity for anti-SSA/Ro, anti- SSB/La, ANA, and RF were more frequently observed among GC-positive than GC-negative patients (Table 10). The presence of GC-like structures in minor salivary gland biopsies and CD4 T-cell lymphocytopenia showed significant associations with lymphoma (p=0.001 and p=0.003 re-spectively). Complement C4 low levels were also more frequently found in the lymphoma patients. However, cryoglobulinemia and low C3 levels were not associated with lymphoma (p>0.05).

55
Table 10. Clinical variables in GC-positive and GC-negative patients
Clinical variables GC-positive GC-negative p value /% of available or mean (SD) Lymphoma 6/14 1/0.8 0.001 Lymphadenopathy 13/31 15/12 0.006 Leucopenia (<4000/mm3) 13/37 17/17 0.019 CD4 T lymphopenia* 8/26 11/14 0.16 Cryoglobulinemia 5/18 10/16 1 Systemic disease 30/74 57/51 0.007 Involved organ systems, n 2.56 (1.62) 1.58 (1.37) <0.001 Anti-SSA/Ro positivity 24/77 48/54 0.022 Anti-SSB/La positivity 19/61 32/36 0.014 ANA positivity 38/91 103/79 0.10 RF positivity 27/68 63/51 0.07 p-IgG (g/L) 18.3 (6.46) 15.0 (5.17) 0.003 C4 (g/L) 0.98 (0.26) 1.05 (0.25) 0.015 *CD4 T cells <300 cells/mL or CD4 T cells <30% of total lymphocyte count or low CD4/CD8 ratio: ≤0.8. GC=germinal centre; ANA=antinuclear antibody; RF=rheumatoid factor; p-IgG=plasma immunoglobulin.
One GC-negative lymphoma patient diagnosed with MALT lymphoma of a tear gland was ascertained to have a borderline positive focus score in the minor salivary gland biopsy and lacked positivity for anti-SSA/Ro and anti-SSB/La. Therefore, this patient could be classified as borderline-SS and the lymphoma might represent the background population risk of lymphoma.
The results of the present study show that the assessment of GC-like structures in minor salivary gland biopsies taken as part of the diagnostic procedure in pSS can be a useful tool for risk assessment of lymphoma de-velopment.
PAPER II Immunostainings of the 79 cases with lymphoma and an initial pSS-diagnosis revealed seven cases with infiltration by IgG4+PCs (8.9%) but only one (1.3%) patient showed “probable histological features of IgG4-RD” in the lymphoma tissue (Table 11). The biopsy material of this patient was taken from the submandibular gland. A review of the original lymphoma tissue slides and additional immunohistochemistry showed that the tumour cells expressed the B-cell markers CD20, CD79a, CD23 and monoclonal kappa light chain. Immunohistochemistry was consistent with MALT lym-phoma, but the present extensive patchy fibrosis was not. Therefore, the final lymphoma diagnosis was unspecified low-grade B-cell lymphoma.

56
The further assessment of H&E stainings of lymphoma tissue from this patient showed the following features: dense lymphoplasmacytic infiltrates and storiform fibrosis (Fig. 9a). Immunostaining for IgG4+ and IgG+ PCs showed a dense infiltration by IgG4+ PCs (Fig. 9b) and IgG+ PCs (Fig. 9c) giving a count of IgG4+ PCs 60/HPF and the IgG4+/IgG ratio of 0.62. Im-munostaining for PC marker CD138+ showed also a rich infiltration (Fig. 9d) with the ratio of IgG4+/CD138+ 0.60.
Figure 9. Features of IgG4-related disease in the surgical resection of the subman-dibular gland in the patient with unspecified low-grade B-cell lymphoma and a pre-viously suspected pSS diagnosis. (a) H&E staining (×100) shows abundant infiltra-tion by small lymphocytic and plasma cells, and features of storiform fibrosis; (b) Staining for IgG4+ plasma cells (×400); (c) Staining for IgG+ plasma cells (×400); (d) Staining for CD138+ plasma cells (×400). The figure reprinted with permission of the publisher from Vasaitis et al. (251).
In addition, this patient also had histological findings “highly suggestive of IgG4-RD” (dense lymphoplasmacytic infiltrate, storiform fibrosis, IgG4+ PCs 167/HPF, the IgG4+/IgG+ ratio 0.92, and the ratio IgG4+/CD138+ 0.94) in a surgical lung tissue specimen (Fig. 10) performed five years be-fore lymphoma diagnosis. A review of the medical records revealed that the patient did not fulfil the 2002 AECG criteria for pSS, but instead had a med-ical history compatible with IgG4-RD (sialadenitis with sicca symptoms and

57
interstitial lung disease with fibrosis). Altogether, this patient fulfilled the proposed histological and clinical criteria for IgG4-RD.
Figure 10. The lung tissue biopsy performed before lymphoma diagnosis with a rich infiltration by IgG4+ plasma cells (PCs), features of storiform fibrosis and germinal centre (GC) formation, ×100. The figure reprinted with permission of the publisher from Vasaitis et al. (251).
We observed occasional IgG4+ PCs (<10 cells/HPF) in another six lympho-ma cases (Table 11). Of these, two were situated in lymph nodes, two in parotid glands and one each in bone marrow and stomach. None of them had any other histopathological features of IgG4-RD in the lymphoma tissue. Five of these six patients fulfilled the 2002 AECG criteria for pSS, and all six patients had one or more extraglandular pSS manifestations (Raynaud, arthritis/arthralgia, rash, skin vasculitis, interstitial lung disease, and lym-phadenopathy).
None of the 44 patients who fulfilled the 2002 AECG criteria for pSS had any specific finding of IgG4-RD in the lymphoma tissue, and similarly, no specific findings and no IgG4+ PCs were found in the available minor sali-vary gland biopsies from 11 of these 44 patients.
Except for swellings of salivary glands and lymph nodes, none of the 79 patients had any other typical clinical manifestations of IgG4-RD (e.g., type 1 autoimmune pancreatitis, cholangitis or retroperitoneal fibrosis).

58
Table 11. IgG4+ plasma cells in the lymphoma tissue of 79 patients with an initial primary Sjögren’s syndrome diagnosis IgG4+ PCs Lymphoma subtype
n=79 ≤10/HPF
n=6 >10/HPF
n=1 I. B-cell neoplasms Unspecified low-grade B-cell lymphoma 5 1 1* Diffuse large B-cell lymphoma 28 2 0 MALT lymphoma 25 2 0 Nodal marginal zone lymphoma 3 1 0 Follicular lymphoma 2 0 Lymphoplasmacytic lymphoma 2 0 Mantle cell lymphoma 1 0 Plasma cell myeloma 3 0 Lymphomatoid granulomatosis 1 0 Unspecified high-grade B-cell lymphoma 2 0 II. T- and NK-cell neoplasms 0 Unspecified high-grade T-cell lymphoma 1 0 III. Hodgkin lymphoma 5 0 IV. Histiocytic and dendritic cell neoplasms 0 Histiocytic sarcoma 1 0 PCs=plasma cells; HPF=high power field; MALT= mucosa-associated lymphoid tissue; *Tissue with mean value of IgG4+ PCs/HPF 60 (range 40-70), IgG4+/IgG+ ratio 0.62 (60/97), IgG4+/CD138+ ratio 0.6 (60/100), dense lymphoplasmacytic infiltrate, and storiform fibrosis.
PAPER III Underlying reasons for a SS diagnosis code Out of 207 patients, 107 (52%) had pSS according to the treating physician. Of them, 57 fulfilled the 2002 AECG criteria and 59 the 2016 ACR/EULAR criteria for pSS (Table 12). In the non-pSS group, the most common reason for a SS code in the Patient Register was sSS in association with another rheumatic disease (n=54). Various other causes were identified in 46 patients with registered SS and lymphoid neoplasm diagnoses’ codes. Of them, 27 were well investigated by the treating physician (with autoimmune serology and minor salivary gland biopsies) without confirmation of a pSS diagnosis but were instead diagnosed with isolated keratoconjuctivitis sicca, or another final explanation for sicca syndrome (e.g., drugs, diabetes or old age). The SS diagnosis code was registered in the Patient Register while the patients had undergone different investigations of suspected pSS, which was not confirmed. Other reasons for a registered SS code in the non-pSS patients are shown in Table 12. One patient had previously been reported by us to have IgG4-RD in an analysis of archived tissues (Study II) (251) and was, therefore, included in this study in the non-pSS group. We could not detect

59
that lymphoid neoplasm was the underlying reason for the sicca symptoms leading to the SS diagnosis in any of the patients.
Table 12. Underlying conditions in 207 patients with “Sjögren’s syndrome/sicca syndrome” ICD code and lymphoid malignancy identified through cross-linkage between the Swedish Patient Register (1964-2007) and the Cancer Register (1990-2007)
Underlying condition Number of patients Primary Sjögren’s syndrome diagnosis1 107 (52%) Fulfilling the 2002 AECG criteria for pSS 57 (28%) Fulfilling the 2016 ACR/EULAR criteria for pSS 59 (28.5%) Non-primary Sjögren’s syndrome diagnosis 100 (48%) I. Secondary Sjögren’s syndrome and overlap: 54 (26%) Rheumatoid arthritis and SS 35 (17%) Systemic lupus erythematosus and SS2 15 (7%) Systemic sclerosis and SS3 4 (2%) II. Other causes of sicca diagnosis: 46 (22%) Sicca symptoms, various reasons4 15 (7%) Keratoconjunctivitis sicca only5 12 (6%) Radiation therapy head-neck before sicca onset 11 (5%) Graft versus host disease with sicca symptoms 5 (2%) Sarcoidosis with sicca symptoms 2 (1%) IgG4-related disease6 1 (0.5%) 1Include two patients with pSS with possibly post-transplantation-associated lymphoma; 2 Include one patient with overlap systemic lupus erythematosus and pSS (fulfil criteria for both diseases); 3Include one patient with overlap systemic sclerosis and pSS (fulfil criteria for both diseases); 4Investigation according to the 2002 AECG criteria excluded pSS, reasons include drugs, age, diabetes; 5Investigation according to the 2002 AECG criteria excluded pSS; 6Described in Study II (251).
pSS patients with and without pre-existing lymphoma Of the 105 patients, 18 (17%) had a pre-existing lymphoid malignancy: four patients with lymphoma diagnosis 1-5 years before pSS diagnosis and 14 patients diagnosed with a lymphoid neoplasm (one patient with myeloma and 13 with lymphoma) within six months after pSS diagnosis. Comparison of the features of the 18 pSS patients with pre-existing lymphoid neoplasm with the 87 pSS patients with lymphoid neoplasm diagnosis after pSS diag-nosis showed no significant differences in most demographic, clinical, and laboratory pSS findings (Table 13). The median ages at sicca onset and pSS diagnosis, the median time from patient-reported sicca onset until pSS diag-nosis, and the presence of extraglandular pSS manifestations and autoanti-bodies were similar in the groups. However, the relative frequency of men was higher among the patients with pre-existing lymphoid neoplasm. More-over, enlarged lymph nodes were also more common among the patients with pre-existing lymphoid malignancy.
Some differences were noted between the two groups regarding the lym-phoma characteristics. Patients with pre-existing lymphoid neoplasm at pSS
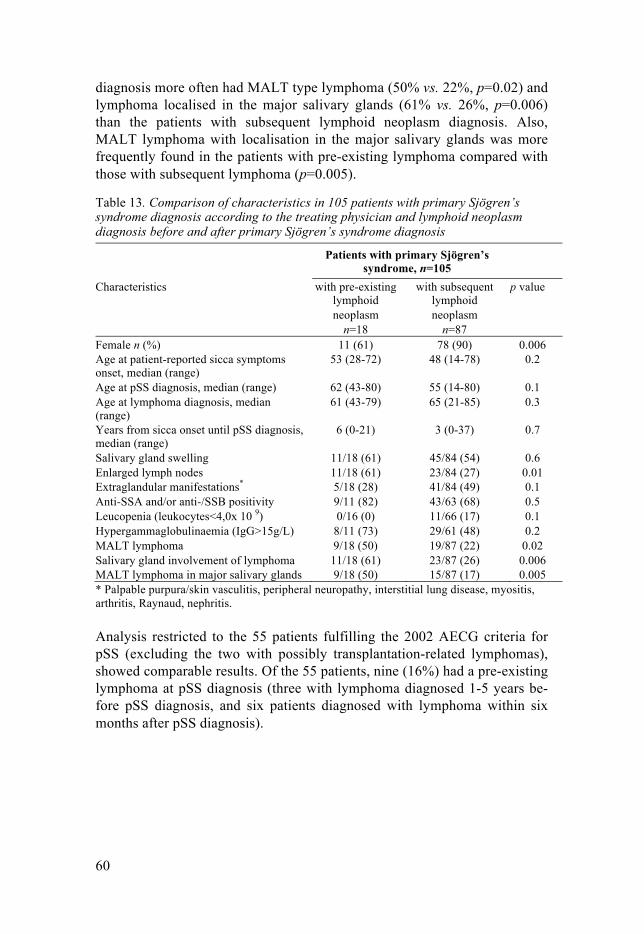
60
diagnosis more often had MALT type lymphoma (50% vs. 22%, p=0.02) and lymphoma localised in the major salivary glands (61% vs. 26%, p=0.006) than the patients with subsequent lymphoid neoplasm diagnosis. Also, MALT lymphoma with localisation in the major salivary glands was more frequently found in the patients with pre-existing lymphoma compared with those with subsequent lymphoma (p=0.005).
Table 13. Comparison of characteristics in 105 patients with primary Sjögren’s syndrome diagnosis according to the treating physician and lymphoid neoplasm diagnosis before and after primary Sjögren’s syndrome diagnosis
Patients with primary Sjögren’s syndrome, n=105
Characteristics with pre-existing lymphoid neoplasm
n=18
with subsequent lymphoid neoplasm
n=87
p value
Female n (%) 11 (61) 78 (90) 0.006 Age at patient-reported sicca symptoms onset, median (range)
53 (28-72) 48 (14-78) 0.2
Age at pSS diagnosis, median (range) 62 (43-80) 55 (14-80) 0.1 Age at lymphoma diagnosis, median (range)
61 (43-79) 65 (21-85) 0.3
Years from sicca onset until pSS diagnosis, median (range)
6 (0-21) 3 (0-37) 0.7
Salivary gland swelling 11/18 (61) 45/84 (54) 0.6 Enlarged lymph nodes 11/18 (61) 23/84 (27) 0.01 Extraglandular manifestations* 5/18 (28) 41/84 (49) 0.1 Anti-SSA and/or anti-/SSB positivity 9/11 (82) 43/63 (68) 0.5 Leucopenia (leukocytes<4,0x 10 9) 0/16 (0) 11/66 (17) 0.1 Hypergammaglobulinaemia (IgG>15g/L) 8/11 (73) 29/61 (48) 0.2 MALT lymphoma 9/18 (50) 19/87 (22) 0.02 Salivary gland involvement of lymphoma 11/18 (61) 23/87 (26) 0.006 MALT lymphoma in major salivary glands 9/18 (50) 15/87 (17) 0.005 * Palpable purpura/skin vasculitis, peripheral neuropathy, interstitial lung disease, myositis, arthritis, Raynaud, nephritis.
Analysis restricted to the 55 patients fulfilling the 2002 AECG criteria for pSS (excluding the two with possibly transplantation-related lymphomas), showed comparable results. Of the 55 patients, nine (16%) had a pre-existing lymphoma at pSS diagnosis (three with lymphoma diagnosed 1-5 years be-fore pSS diagnosis, and six patients diagnosed with lymphoma within six months after pSS diagnosis).

61
PAPER IV Characteristics of pSS patients with a lymphoid malignancy The most important patient characteristics are summarized in Table 14.
Table 14. Characteristics of the patients with pSS diagnosis according to the treat-ing physician and confirmed lymphoid neoplasm diagnosis
Demographic characteristics
All pa-tients n=105
DLBCL n=32
MALT type n=28
p value
Female n=89
Male n=16
p value
Female, n (%) 89 (85) 30 (94) 24 (86) 0.4 - - - A median age, years at: sicca onset 48 47 45 0.9 48 48 0.8 pSS diagnosis 55 55 50 0.1 55 60 0.9 lymphoma diagnosis 64 67 55 0.0001 65 60 0.2 A median time, years from: sicca onset until lymphoid neoplasm diagnosis
11 18.5 7 0.0001 11 10 0.03
pSS diagnosis until lymphoid neoplasm1
7 9 2 0.0005 8 1 0.0003
Clinical characteristics, n/available (%)2
Anti-SSA/Ro and/or SSB/La positivity
52/74 (70)
17/26 (65)
17/20 (85)
0.2 43/63 (68)
9/11 (82)
0.5
Salivary gland swelling 56/102 (55)
17/32 (53)
21/27 (78)
0.053 46/86 (53)
10/16 (63)
0.6
ILD 6/105 (6)
2/32 (6)
1/28 (3.6)
1 3/89 (3)
3/16 (19)
0.04
Current or previous smoking
26/87 (30)
9/30 (30)
4/20 (20)
0.5 19/75 (25)
7/12 (58)
0.04
Extraglandular mani-festations3
46/102 (45)
17/32 (53)
7/28 (25)
0.035 41/87 (47)
5/16 (31)
0.3
Hypergammaglobu-linemia (p-IgG>15g/L)
37/72 (51)
9/23 (39)
10/16 (63)
0.2 30/60 (50)
7/12 (58)
0.8
Any treatment given for pSS4
38/103 (37)
14/31 (45)
11/28 (39)
0.8 36/87 (41)
2/16 (13)
0.04
Lymphoma in salivary glands
34/105 (32)
4 /32 (12.5)
23/28 (82)
<0.0001 26 /89 (29)
9/16 (56)
0.04
pSS=primary Sjögren’s syndrome; MALT=mucosa associated lymphoid tissue; DLBCL=Diffuse Large B-cell Lymphoma; ILD=interstitial lung disease; 1Four patients with formal pSS diagnosis after lymphoma diagnosis at a median time of 4 years (range 1-5 years) were excluded from this assessment; 2Clinical manifestations evaluated from the onset of sicca symptoms until 6 months before diagnosis of the lymphoid neoplasm; 3In all 105 pSS patients: arthritis (n=20/102; 20%), skin vasculitis (n=11/102; 11%), intersti-tial lung disease (n=6/102; 6%), peripheral polyneuropathy (n=6/102; 6%), myositis (n=5/102; 5%), nephritis (n=2/102; 2%); some patients with >1 manifestation; 4Prednisolone (≥4 weeks), antimalarials, azathioprine, methotrexate, or cyclophosphamide.

62
Of the 105 patients with pSS diagnosis according to the treating physician and confirmed lymphoid neoplasm diagnosis, 89 (85%) were women and 16 men (15%) resulting in a male-to-female ratio of 1:6. The median age at the first patient-reported sicca symptoms was 48 years (range 14-78); pSS was diagnosed at a median age of 55 years (range 14-80), and lymphoid neo-plasm at 64 years (range 21-85). Anti-SSA/Ro and/or anti-SSB/La antibod-ies were present in 70% and extraglandular manifestations occurred in 45% of the patients.
The most common lymphoma site in the pSS patients was the major sali-vary glands, observed in 34 (32%) of the patients (Table 14) (parotid glands in 32, submandibular glands in two). As expected, the most common lym-phoma subtype in salivary glands was MALT lymphoma (n=23), but in 13 (12%) of the patients other subtypes were present in the glands (unspecified low-grade B-cell lymphomas in six, DLBCL in four, whereof two cases showed signs of transformation from MALT lymphoma, follicular lympho-ma in two and histiocytic sarcoma in one). Occasional MALT lymphomas were present in lung (n=2), lacrimal gland (n=1), breast (n=1), spleen (n=1), and stomach (n=1), but without notions of specific pSS involvement in these organs.
EBV was detected in the lymphoma tissue in 20% of investigated cases, most frequently in DLBCLs (22%) and HL (80%). One of the three DLBCLs with signs of transformation from MALT lymphoma was EBV-positive. Of the 28 MALT lymphomas, 22 were investigated for EBV, and, notably, all were EBV-negative.
In 27 DLBCL patients with available lymphoma tissues for further anal-yses, similar proportions were found of the ABC subtype (n=13) and the GCB subtype (n=13) (p=1.0). In one case, this subtype was unclassifiable. Further, no differences were found between the frequencies of the ABC and BCG subtype occurrence after stratification by age at lymphoma diagnosis (the age groups: <60, 61-69, 70-79, and 80+ years of age) (p>0.05). The overall median survival was similar in the ABC-DLBCL and GCB- DLBCL subgroups (7 years; range 0-18 years and 6 years; range 0-13 years, respec-tively; p=0.8).
During a median follow-up of 10 years after lymphoma diagnosis, 54 of the 105 (51%) patients had progression or relapse of the lymphoid malignan-cy. Further, three MALT lymphomas transformed into DLBCL localised in the same organ as the primary MALT lymphoma. The median overall sur-vival after lymphoma diagnosis was 10 years (0-24 years) and 5-year overall survival after lymphoma diagnosis 69% in the pSS patients.
Sex differences Some differences in patient characteristics between men and women were noted (Table 14). The time between patient-reported sicca symptom onset

63
and diagnosis of the lymphoid malignancy was shorter for men than women (10 vs. 11 years; p=0.03), and the difference was even more evident for the time between pSS diagnosis and diagnosis of the malignancy (one vs. eight years; p=0.0003).
The men more often had interstitial lung disease (ILD) (19% vs. 3%, p=0.04) and more often were, or had been, smokers than the women (58% vs. 25%, p=0.04). No men with ILD had lymphoma in the lung, but one of the women with ILD was diagnosed with DLBCL in the lung.
Women were more likely to have been treated with immune-modifying drugs than men (p=0.04), but there were no differences in the specific drug therapies for pSS between the groups. Other pSS manifestations and labora-tory test results were largely similar in men and women.
There was no statistically significant difference in the proportions of MALT lymphoma and DLBCL in men and women, but men more often had lymphoma involvement of the salivary glands (p=0.04) (Table 14). The outcome after lymphoma diagnosis was also similar in men and women, with no significant differences in the median overall survival (eight years in men and 10 years in women) (p=0.5).
Comparison of pSS-MALT lymphoma with pSS-DLBCL Main clinical characteristics and a comparison between patients with the two most frequent lymphoma subtypes, MALT lymphoma and DLBCL are pre-sented in Table 14. There were no significant differences in most pSS char-acteristics between the groups. The median age at the first patient-reported sicca symptoms, age at pSS diagnosis, autoimmune serology, the presence of focus score in diagnostic salivary gland biopsies, the ESSDAI at diagnosis, and results of the reported blood tests were similar.
Two differences were identified. Overall extraglandular manifestations during the course of pSS were more common in patients with DLBCL (53%) than in those with MALT lymphoma (25%) (p=0.04). On the other hand, there was a tendency of more major salivary gland swelling during the pSS disease in patients who developed MALT lymphoma (78%), than in those with DLBCL (53%) (p=0.05). There were no differences in the occurrence of specific extraglandular manifestations between the groups.
Regarding lymphoma characteristics, MALT lymphoma was diagnosed at a younger age (55 vs. 67 years, p=0.0001) and earlier after patient-reported sicca onset (seven vs. 18 years, p=0.0001) and pSS diagnosis (two vs. nine years, p=0.0005) than DLBCL. There were no differences between the DLBCL and MALT lymphoma patients for lymphoma relapse/progression and the median time to relapse (p=0.2). However, pSS-DLBCL patients had a significantly shorter median survival time after lymphoma diagnosis in comparison to pSS-MALT lymphoma patients (six vs. 13 years, p=0.00008).

64
Lymphoma subtypes in pSS patients and comparison with a general population reference
Of the 105 pSS patients with a lymphoid neoplasm, 99 (94%) had lymphoma and six (6%) had plasma cell neoplasms (Table 15).
Table 15. Subtypes of mature lymphoid neoplasms in 105 primary Sjögren’s syn-drome patients with lymphoid malignancy (1990-2007) and comparison with lym-phomas in the general Swedish population (Swedish Lymphoma Register 2000-2010)
Lymphoid neoplasms, WHO classification Primary SS Current study
1990-2007
Swedish Lym-phoma Register
2000-2010
p value
n/available (%) I. All lymphomas 99 18,058 1. B-cell lymphomas 91 (92) N/A Diffuse large B cell lymphoma: 32 (32) 5,789 (32) 1 ABC-type 13/27 (48)* N/A With signs of transformation from MALT lymphoma
3/32 (9) N/A
Marginal zone lymphoma: 31 (31) 887 (5) <0.0001 MALT lymphoma 28 (28) N/A Nodal marginal zone lymphoma 3 (3) N/A Follicular lymphoma 2 (2) 4,617 (26) <0.0001 Lymphoplasmacytic lymphoma 2 (2) 1,028 (6) 0.2 Mantle cell lymphoma 1 (1) 844 (5) 0.1 Lymphomatoid granulomatosis 1 (1) 4 (0.02) 0.004 Unspecified high-grade B-cell lymphoma 10 (10) 433 (2) <0.0001 Unspecified low-grade B-cell lymphoma 12 (13) 499 (3) <0.0001 2. T-cell lymphomas 1 (1) N/A Peripheral T-cell lymphoma, unspecified 1 (1) 287 (2) 1 3. Hodgkin lymphoma 5 (5) 1,757 (10) 0.2 Nodular sclerosis classical Hodgkin lymphoma 2 (2) 1,007 (5) 0.2 Mixed cellularity classical Hodgkin lymphoma 3 (3) 315 (2) 0.6 4. Other lymphomas 2 (2) N/A Histiocytic sarcoma 1 (1) N/A Unspecified lymphomas 1 (1) N/A II. Plasma cell neoplasms 6 N/A Multiple myeloma 5 (83) N/A Plasmacytoma 1 (17) N/A MALT=mucosa-associated lymphoid tissue; N/A= not applicable; *DLBCL subtypes: acti-vated B-cells (ABC) n=13, GC B-cells (GCB) n=13, and unclassifiable n=1.
Almost all lymphomas were of B-cell type (91/99; 92%). The two most common lymphoma subtypes, diagnosed in similar proportions in the pSS patients, were DLBCL in 32 patients (32%) and MZL in 31 of the patients (31%). Of the MZLs, all except three were MALT lymphomas (n=28; 90% of the MZLs). In three of the DLBCLs, signs of transformation from MALT lymphoma were observed at tissue review. In comparison with the Lym-

65
phoma Register in which MZL constitute 5% of all lymphomas (Table 15), MZL was significantly increased in the pSS patients (31%) (p<0.0001).
DLBCLs occurred with the same frequency in the pSS patients (32%) as in the general population (32%) (p=1). Other lymphoma subtypes occurred at much lower frequencies in the pSS patients. HL was not increased in the pSS patients (n=5; 5%) compared to the general population (10%) (p=0.2). Follicular lymphoma, common in the general population (26%), was present in only two (2%) pSS patients (p<0.0001).
Plasma cell neoplasms consisted of one patient diagnosed with plasmacy-toma and five with plasma cell myeloma, representing 6% of all the lym-phoid malignancies. In comparison, plasma cell neoplasms constitute 10-15% of haematological malignancies in the general population. Plasma cell neoplasms are not included in the Lymphoma Register and the frequency, therefore, could not formally be compared with the reference of the general population. Their characteristics did not differ from what is reported in plasma cell neoplasms in general, and there were no sex differences. All patients with myeloma had bone marrow involvement. Hypergammaglobu-linemia and a clonal component in blood were present already at pSS diag-nosis, and none of the patients had salivary gland swelling during the course of pSS.

66
Discussion
Lymphoid formations in minor salivary gland biopsies The finding in this study of the association between GC-like structure for-mation in labial salivary glands and lymphoma development later on is im-portant in the clinical management of pSS. Patients with pSS are at increased risk of lymphoma development, and lymphoma is one of the risk factors for premature mortality in pSS (155). The performance of biopsies of minor salivary glands is a routine procedure in pSS diagnostics and is also required within the 2002 AECG (17), the 2012 ACR (18), and the new 2016 ACR/EULAR (19) classification criteria for pSS.
GC-like structures were present in 25% of the pSS patients, and six out of seven (86%) of them developed malignant lymphoma. It is known that GCs in lymphoid organs are places where B cells undergo maturation and give origin to B-cell lymphomas (194, 195). However, not all our patients with GC-like structures developed lymphoma. This shows that many other factors may play a role in lymphoma development in pSS patients. Moreover, we did not study the occurrence of GCs in other organs, yet five of seven lym-phoma patients had another lymphoma localisation than salivary glands. It is not known if GC-formation is present systematically in pSS patients prone to lymphoproliferation. On the other hand, the study showed that GC-positive patients had more active pSS disease with swollen lymph nodes, leucopenia, hypergammaglobulinemia, and systemic disease with a higher amount of organs involved. Systemic findings of an upregulated type I IFN signature (71, 252), increased levels of BAFF (92, 100) and reduced levels of memory B cells in peripheral blood in pSS patients (94, 95), support that B cells are affected beyond the salivary glands. A single case of pSS has been reported (253) showing that partly the same B-cell clone was detected in parotid pro-liferation and pulmonary lymphoma. Thus, one might speculate that the for-mation of GC-like structures can be a multi-focal process in a subset of pSS patients prone to lymphoproliferation.
A trend was observed for lower levels of C3 and CD4 T-cell in the GC-positive patients. The statistical significance was not reached possibly due to few cases in the cohort or due to the independent expression of these predic-tors. On the other hand, it is not necessary that the peripheral situation in the blood mirrors the situation in the glands (129).

67
The present study showed association with lymphoma of other well-known markers for lymphoma development, such as C4 levels (9, 176, 178) and leucopenia (176). In addition, we present a possible new marker for lymphoma development, that is, GC-like structure formation in minor sali-vary glands at pSS diagnosis. The negative predictive value of the proposed marker is 99%. Thus, it allows for a reliable negative selection of a large number of patients spearing them from unnecessary investigations and sub-jective anxiety for lymphoma.
Our findings could have practical implications in the management of pSS patients. GCs can be detected in the routine procedure evaluating the biop-sies of minor salivary glands by a pathologist. Aware of that that up to 86% of GC-positive patients may develop lymphoma in the future and that these patients have a more active disease, allows the identification of high-risk patients already at pSS diagnosis. These selected pSS patients could be sub-ject to appropriate monitoring for lymphoma, close follow-up, and possibly selected for advanced interventions of treatment. Treatment in these cases could be directed to systemic manifestations of the pSS disease and for the prevention of lymphoma.
It should be admitted that we studied a limited number of pSS patients with lymphoma. Moreover, evaluation of the GC-like structures in labial salivary gland biopsies was a subjective assessment by an oral pathologist. A genetic expression analysis or immunohistochemical testing for CD10, BCL6, and IRF4/MUM1 used for differentiating DLBCL subtypes (166) allows for the detection of B cells originating from GCs. However, there is no sophisticated approach so far for objective evaluation of B cells in GC-like structures in minor salivary gland biopsies. Lymphocytes in salivary glands may organise themselves in B-cell rich and T-cell rich zones in a subset of pSS patients (134). These zones are associated with ectopic pro-duction of lymphoid chemokines (CXCL13 and CCL21 and expression of the activation-induced cytidine deaminise (AID) enzyme (132, 254, 255). AID is important in the local maturation of autoimmune responses and is expressed in association with CD21+ follicular DC networks (256). Howev-er, it is not clear how cellular infiltrates in minor salivary glands are associ-ated with B cell clonality and lymphoma development (257). Thus, further studies are desirable for the development of objective histopathological markers for more sophisticated diagnosis of GC-like structures in salivary glands.
Previous studies have shown that ANA, RF, and anti-SSA/Ro and/or anti-SSB/La positivity are not associated with lymphoma development (178). Two of our seven-lymphoma patients were negative for anti-SSA/Ro and/or anti-SSB/La autoantibodies. One of these two lymphoma patients had an only borderline positive biopsy for focus score in the minor salivary gland and could be categorised as borderline-SS and might represent the back-ground population risk of lymphoma.

68
Further studies are needed to assess GC-like structures with objective methods and evaluate their significance in lymphoma development in pSS patients. We encourage to include minor salivary gland biopsy in the routine work-up in diagnosing pSS even if the patient is seropositive for anti-SSA/Ro and/or anti-SSB/La and formally fulfil the criteria for pSS. The value of ectopic GC-like structures can be evaluated prospectively by col-lecting minor salivary gland tissues in pSS patients.
IgG4-RD in patients with a previous pSS diagnosis and lymphoma In the population-based cohort of patients with pSS and lymphoid neoplasm diagnoses, we retrospectively revealed one male patient in his 60s with his-topathological features of IgG4-RD and lymphoma. Specific histopathologi-cal findings of IgG4-RD were present both in the submandibular gland with co-existing lymphoma and also in the previously performed surgical lung tissue biopsy without lymphoma. At chart review, we could not confirm that the patient fulfilled the 2002 AECG criteria for pSS (17). He had ocular and oral mild sicca symptoms confirmed by objective tests with unstimulated saliva flow test and Schirmer’s test, which normalised after one-year follow-up without any specific treatment. He was ANA positive but had no specific anti-SSA/Ro or anti-SSB/La antibodies. Minor salivary gland biopsy was not performed. He was diagnosed with suspected pSS by the treating rheumatol-ogist in the mid-1990s. Formally, the patient fulfilled the preliminary Euro-pean classification criteria for pSS (16). His medical history revealed that this patient had symptoms of recurrent nasal congestion, joint pain, swelling of submandibular lymph node from which a needle aspiration showed signs of a reactive lymph node. He had a high ESR (>90 mm/h), hypergammag-lobulinemia (IgG 38 g/L), and mild anaemia. Before the suspected pSS diag-nosis, the patient was diagnosed with severe idiopathic lung fibrosis con-firmed by surgical lung tissue biopsy. At the age of 64, the patient developed swelling of the right submandibular gland and low-grade B-cell lymphoma was diagnosed after the biopsy. Lymphoma was localised to only this gland and therefore no active lymphoma treatment was initiated. The lung fibrosis was treated with Prednisone 15-20 mg/d, cyclophosphamide, followed by azathioprine, and for respiratory insufficiency by oxygen therapy. The pa-tient died one year after lymphoma diagnosis from respiratory and heart failure.
Histological re-evaluation of the submandibular gland confirmed low-grade B-cell lymphoma and also showed highly suggestive features of IgG4-RD. The consensus statement on the pathology of IgG4-RD from 2012 (216) defines the requirements for a histological diagnosis of IgG4-RD in different

69
organs and tissues. Our patient fulfilled almost all requirements for the histo-logical diagnosis except the number of required IgG4+ PCS (>100/HPF in salivary gland) and therefore was categorised as probable histological fea-tures of IgG4-RD in the submandibular gland. The proposed cutoff for IgG4+ PCs varies from organ to organ (>10 - >200/HPF) and it is acknowl-edged that the extent of fibrosis may affect the cutoff point. However, our patient had co-existing lymphoma, which might influence the lower number of total IgG4+ PCs seen in the biopsy. On the other hand, the patient had been treated with steroids and cyclophosphamide for lung fibrosis, when he underwent the biopsy. Thus, the treatment might also influence the minor infiltration by IgG4+ PCs in the submandibular gland. Fortunately, we could re-evaluate the surgical lung biopsy performed five years before lymphoma diagnosis when the patient was not yet diagnosed with pSS and had no treatment for lung fibrosis. The findings in the lung tissue (>50 IgG+PCs/HPF required for surgical biopsy) fulfilled all criteria of “histolog-ically highly suggestive of IgG4-RD”.
The current knowledge of the clinical picture of IgG4-RD is rapidly ex-panding. Our described patient with histopathological features of IgG4-RD had a medical history, and clinical and laboratory signs well in line with what is described in IG4-RD. The disease mainly affects middle-to-older age males. Common symptoms are arthralgia, atopic symptoms, eosinophilia, lymphadenopathy, swelling of lacrimal and salivary (particular submandibu-lar) glands, and usually mild sicca symptoms (217). In about 70% of pa-tients, elevated IgG4 levels in serum may be detected (217, 235, 258, 259). Since our study was retrospective and we had no access to the patient’s se-rum, we could not test IgG4 levels in serum or perform other relevant la-boratory tests. On the other hand, elevated serum IgG4 levels are present in only a proportion of patients and are not a part of the gold standard for diag-nosis of IgG4-RD (216). Thus, the information about IgG4 serology levels would not have changed the results of the study.
Lung involvement in IG4-RD occurs in 12-54% of patients and may man-ifest as solid lung nodules, ground-glass opacities, lung infiltrates, an inter-stitial lung disease with honeycombing ad fibrosis, bronchiectasis, bronchial thickening, central airway stenosis, nodular pleural lesions and pleural effu-sion (217, 260, 261). The fibroinflammatory disease in the lungs may pro-gress to severe respiratory insufficiency and lead to death as in our case (262).
In the remaining 78 analysed patients, no or sporadic IgG4+ PCs without specific histopathological findings for IgG4-RD were detected in lymphoma tissue. Available diagnostic minor salivary gland biopsies (n=11) taken at the time of pSS diagnosis showed neither IgG+ PC nor other histological fea-tures of IG4-RD. Thus, our results suggest that infiltration by IgG4+ PCs can be found in the lymphoma tissue in patients with pSS diagnosis, but underly-

70
ing misdiagnosed IgG4-RD with specific histological features and clinical manifestations of this disease are rare (1.3%).
Occasional IgG4+ PCs (<10 cells/HPF) may occur in lymphoma tissue without underlying IgG4-RD (236). In these cases, IgG4+ PCs occur sporad-ically as in our other seven patients, and this spare infiltration by IgG4+ PCs is probably unspecific. Unspecific infiltration by sporadic IgG4+ PCs may be found in other inflammatory diseases, infections, and malignancies (217).
Underlying reasons behind a SS diagnosis code This is the first study reporting the utility of the SS diagnosis code for pSS and generally for SS diagnosis in clinical practice. In this population-based cohort of patients with a diagnosis of both SS and a lymphoid neoplasm, we found that not more than approximately half of the patients had pSS as the underlying cause of the SS diagnosis code, around 25% had sSS, and 25% had various other reasons for the SS diagnosis. Many different underlying causes of the sicca diagnoses have several implications for the interpretation of register-based information and poses particular problems to studies of pSS. The lack of an independent and unique ICD code for pSS when identi-fying patients in registries based on ICD codes, may affect the number of correctly included cases in an epidemiological study. Moreover, if ICD codes of SS are not validated, coding bias for pSS may lead to poor accuracy of the study. Thus, register-based information regarding SS diagnoses must be interpreted with caution and diagnoses should be properly validated. In the near future, however, epidemiological pSS studies may be facilitated. A release of ICD-11 is planned for 2018, and the ICD-11 Beta draft is available online (http://apps.who.int/classifications/icd11/browse/l-m/en). In this ver-sion of the ICD, each disorder will contain a definition, a set of inclusion and exclusion terms, and many other features, making ICD codes more applica-ble for epidemiological studies. In this ICD draft, Sjögren’s syndrome (GA63.3) is divided in primary (GA63.31), secondary SS (GA63.32), pediat-ric onset SS (GA63.33), and SS vasculitis (GA63.34).
Some limitations need to be discussed. The retrospective design of our study resulted in some patients having sparse historical data. Furthermore, the different classification criteria for pSS during the study period resulted in an incomplete assessment for the 2002 AECG and the 2016 ACR/EULAR criteria for pSS in a subset of the patients due to missing items in the medical records. Sicca onset was recorded at the time of subjective dryness symp-toms as noted in the medical records, and recall bias cannot be excluded. We also cannot exclude that oncologists may have underreported, or not regis-tered sicca symptoms, in patients with lymphoma.

71
Support for removal of pre-existing lymphoma as an exclusion criterion for pSS This report was the first study analysing why pre-existing lymphoma should or should not be an exclusion criterion in pSS patients. The results of this study support the decision to remove pre-existing lymphoma as a general exclusion criterion for pSS classification as it has been done in the newly published 2016 ACR/EULAR criteria for pSS (19). Most pSS characteristics were similar, irrespective, if the patients had a pre-existing lymphoma at pSS diagnosis or if pSS was diagnosed long before the lymphoma. A relatively large proportion of patients with pSS (in this study 17%) would not be cor-rectly classified as pSS if lymphoma had been employed as an exclusion criterion. It seems that the patients with pre-existing lymphoma before pSS diagnosis in general, neglected long-standing sicca symptoms due to pSS, and instead sought medical care for the lymphoma-related symptoms, that is, the swollen parotid glands caused by a MALT lymphoma.
Interestingly, there was a higher frequency of men with pre-existing lym-phoma compared with pSS patients with a subsequent lymphoma. One may speculate that men are less prone to seek medical attention for sicca symp-toms and will only be diagnosed with an underlying pSS once a lymphoma is present.
Obviously, a missed diagnosis of pSS is disadvantageous for the patient, as the disease will not be properly treated and extraglandular pSS manifesta-tions may be misinterpreted as lymphoma related. Highly active pSS is per se a prognostic factor for progression of lymphoma in pSS (211). There is also some support that lymphoma in pSS patients may need another lym-phoma treatment compared to lymphomas in non-pSS patients. Several re-ports indicate that rituximab, a treatment option for MALT lymphoma, is not as efficient in pSS patients with MALT lymphoma in the salivary glands compared with other sites or lymphoma subtypes (263-265).
Sex differences in pSS patients with a lymphoid malignancy In this population-based setting of pSS patients with lymphoid malignancy, the proportion of men was higher than in general pSS cohorts, indicating that men with pSS can be at a higher risk of lymphoma than women with pSS. In this study, the men-to-women ratio was 1:6, but typically it ranges between 1:10-20 in general pSS cohorts (4, 37, 47). Previous studies have not report-ed a clear sex difference in risk of lymphoid malignancy in pSS. In one study of 419 patients with pSS (42 men, 377 women), lymphoma development was slightly, but not significantly, more common in men than women based on

72
the finding of four men and seven women with lymphoma development (p=0.06) (266). In general, in the pSS-lymphoma studies, the power to esti-mate sex-specific risks for lymphoma in pSS has been too low due to low numbers of included men.
In some other autoimmune diseases, a higher incidence of lymphoma has been reported in men compared to women, including RA (175, 267), SLE, and autoimmune haemolytic anaemia (175). It has been suggested that this could be due to a more severe type of autoimmune disease in men than women, but there are also reports of a hormonal influence on prognosis and tumour growth in lymphoma. In DLBCL, it has been shown that prognosis is more favourable in fertile women than men (268) and that tumour growth in males is faster (269), indicating more complex sex-specific differences in lymphomagenesis in men and women.
We noted a significant difference in time from pSS diagnosis to lymphoid malignancy diagnosis in men compared to women. Men were diagnosed with lymphoid malignancy already after a median time of one year after pSS diagnosis while the corresponding time in women was eight years (p=0.0003). This could support sex differences in lymphomagenesis in pa-tients with pSS with a more aggressive process leading to lymphoid malig-nancy in men. Moreover, men more often had lymphoma involvement of the salivary glands than women (56% vs. 29%; p=0.045), which could be indica-tive of a more aggressive local process in the glands in men. In pSS, it is well described that chronic antigenic stimulation due to persistent inflamma-tion and continuous B cell activation with high levels of BAFF may lead to the clonal proliferation and development of MALT lymphoma in the salivary glands (190, 270). GC-like formations in minor salivary glands taken at pSS diagnosis also support the association between degree of local B-cell activa-tion and lymphoma risk (ref. 135 is part of the current thesis).
In the pSS patients with lymphoid malignancy, most clinical characteris-tics, pSS manifestations, and laboratory parameters were similar in men and women, with two observed differences. ILD and smoking were more com-mon in men than women. It is well known that ILD is more common in men than women with pSS (271), and ILD has been described as one of the ex-traglandular manifestations linked to increased lymphoma risk in pSS in general. Smoking in pSS is less well studied and there are no reports of sex differences (67). In lymphoma, in general, smoking has not been linked to an increased overall lymphoma risk, but associations with the risk of certain lymphoma subtypes, such as follicular lymphoma and HL, has been suggest-ed (272). To date, smoking has not been clearly linked to risk of MALT lymphoma in salivary glands.

73
Lymphoma characteristics in pSS patients and comparison with a general population reference In this population-based cohort of the pSS with lymphoid malignancies, a significant six-fold increase of the relative frequency of occurrence of MZL (31%), compared to that reported in the general population (5%), was found. In this study, MZL consisted of 28 MALT lymphomas and three nodal MZLs, whereas subgroups of the MZLs were not reported in the Lymphoma Register, which was used as a general population reference for comparisons.
The proportion of DLBCL was the same (32%) as in the general popula-tion, suggesting that the DLBCL cases in the pSS patients may represent the background population risk of lymphoma, which is present in all individuals in general. In addition, the pSS patients were diagnosed with DLBCL at a similar age (around 70 years) as DLBCL in the general population (268). Moreover, the GCB and ABC subtypes of DLBCL were equally distributed in our patients as in the general DLBCLs (164), which also supports that our DLBCL cases may represent the background lymphoma occurrence. On the other hand, in RA, a strong association with risk of DLBCL has been demonstrated and the distribution of the ABC-DLBCL has been reported to predominate (273). This subtype of DLBCL has a poorer prognosis and also, is associated with a higher frequency of elderly patients (≥80 years) (274) than the GCB type.
In MALT lymphoma, well-known for a long time to be associated with pSS, the characteristics differed between the pSS-MALT and the general MALT lymphoma patients. MALT lymphoma in our patients was diagnosed at a younger age (median 55 vs. 61 years reported in the general population) and the preferred site of lymphoma in the salivary glands was different from the most common site (i.e., stomach in 50%), in general MALT lymphoma patients (164). Additionally, the proportion of MALT lymphoma was similar in men and women in our pSS patients, whereas in general, there is a slight female preponderance in patients with MALT lymphoma (164).
Previous reports about lymphoma subtypes in pSS patients have been conflicting. Some studies have reported high proportions (9, 180, 193, 210, 275, 276) and up to 100% of marginal zone lymphomas or MALT lympho-ma in pSS populations (174, 177). Other studies have identified similar pro-portions of MALT lymphoma and DLBCL (173, 176, 204), and some have revealed a predominance of DLBCL (169, 171, 203). Many of the studies, though, represent selected populations and lack a valid general population comparator.
To be noted, there was a long median time from the patient-reported sicca onset (18 years) as well as from pSS diagnosis (nine years) to diagnosis of DLBCL. The corresponding times to diagnosis of MALT lymphoma were much shorter (seven years from sicca onset and two years from pSS diagno-

74
sis). In studies with a short follow-up (< 10 years) from pSS diagnosis, MALT lymphoma may, therefore, be falsely overrepresented.
In this setting, we confirm that the presence of EBV is not associated with MALT lymphoma in pSS (193). None of the investigated MALT lympho-mas expressed EBV; although, one case that was diagnosed with an EBV-positive DLBCL as the first lymphoma, showed signs of transformation from MALT lymphoma at tissue review. We identified transformation of MALT lymphoma to the high-grade DLBCL subtype in six of the cases (three cases at the first lymphoma diagnosis and three cases during the follow-up because of relapse or lymphoma progression), but as only one was EBV-positive, we cannot link the presence of EBV to a general increased risk of transfor-mation in these cases. Transformation to DLBCL in 19% of MALT lym-phomas is, however, higher than reported in MALT lymphoma in general, suggesting an increased risk of transformation in pSS patients with MALT lymphoma. In one study, 4% of general MALT lymphoma transformed to a high-grade subtype (277), whereas in another study, 7% of nodal MZLs transformed (278).
Strengths of the study include the population-based setting, the careful validation of the pSS and the lymphoid malignancy diagnoses, including the review of tissues, analyses of EBV and cell of origin, a uniform classifica-tion of the subtypes according to the WHO classification, and also the long follow-up. The population-based approach enabled us to compare the distri-bution of lymphoma subtypes to a valid general population reference and enhances generalizability. Most previous studies of lymphoma in pSS have included patients referred to specialised centres for pSS patients, which may lead to a selection of patients with a more severe pSS disease in such studies.
A weakness of the register-based approach to identify pSS patients is that not all patients had been investigated by rheumatologists and were not fully examined according to classification criteria for pSS. We, therefore, included comparisons between those fulfilling the 2002 AECG criteria for pSS and those who did not formally fulfil the criteria, as items included in the criteria were absent in all analyses. Patient characteristics and distribu-tion of lymphoma subtypes were almost identical supporting that these pa-tients were correctly included as pSS patients in this study. Another limita-tion when relying on retrospective data from medical records is missing clin-ical data. Although this study includes the largest number of men with pSS and lymphoma to date, the sixteen patients in this study is still a limited number, and further studies of this patient group are needed.

75
Concluding remarks
Paper I We present GC-like structures in minor salivary gland biopsies as a new potential marker of lymphoma development in pSS patients. This marker is detectable already at pSS diagnosis by routine light microscopy and allows identifying high-risk patients.
Paper II The presence of IgG4+ PCs and specific histopathological findings of IgG4-RD in lymphoma tissue in patients with an initial pSS diagnosis are rare, but if present may be associated with underlying undiagnosed IgG4-RD. Moreo-ver, IgG4-RD may co-exist with lymphoma in the same tissue.
Paper III In register-based studies using the ICD code for SS, validation of the under-lying diagnoses must be considered. Further, we found no support for pre-existing lymphoma as a general exclusion criterion for pSS. MALT or an-other lymphoma found in the major salivary glands should instead trigger adequate pSS investigation.
Paper IV Lymphoma may be more common in men than in women with pSS, and the findings support special awareness of lymphoma signs and a liberal attitude towards biopsy of major salivary glands in men already in the first years after pSS diagnosis. Only MALT lymphoma is relatively increased in pa-tients with pSS compared to a general population reference. Our findings strengthen the previously described strong association between pSS and MALT lymphoma in salivary glands and a history of major salivary gland swelling as a predictor of lymphoma development.

76
Acknowledgements
The work was carried out at the Department of Medical Sciences, Faculty of Medicine, Uppsala University. I would like to thank all who have supported me during this long journey until I finished the thesis. In particular, I would like to acknowledge: Eva Baecklund, my main supervisor, colleague, and friend. Thank you for encouraging, helping me and being so positive! Gunnel Nordmark and Lars Rönnblom, my co-supervisors, co-authors, and colleagues at Rheumatology department, for scientific discussions and help with doctoral studies. My co-authors: Carin Backlin, for helping me with pathology material collection and im-munostaining of tissues. Christer Sundström, for evaluating lymphoma and salivary gland tissues and valuable discussions about IgG4 staining. Karin E. Smedby, for valuable discussions about lymphoma. Elke Theander, for interesting discussions about Sjögren’s syndrome and lymphoma and fruitful collaboration. Johan Askling, for support in epidemiologic and register matters. My co-authors and colleagues from Norway, Roland Jonsson, Malin V Jons-son, and Karl Brokstad. Heriberto Vélëz, for reading my thesis and manuscripts, and valuable lin-guistic suggestions. Fredrik Granath, for register matters. Colleagues in the Sjögren network. All colleagues and friends at the department of Rheumatology. My much-loved family and friends. The studies in this thesis were supported by the ALF funding from County Council of Uppsala, the Lions Cancer Research Foundation, Uppsala Uni-versity Hospital, the Swedish Rheumatism Association, the Swedish Society of Medicine, the Gustav V:s 80-year foundation, and scholarships from Ag-nes & Mac Rudbergs and Gustav Prims Reumatikerfond foundations.

77
References
1. Hayter SM, Cook MC. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev. 2012;11(10):754-65.
2. Oertelt-Prigione S. The influence of sex and gender on the immune response. Autoimmun Rev. 2012;11(6-7):A479-85.
3. Leandro MJ, Isenberg DA. Rheumatic diseases and malignancy--is there an association? Scand J Rheumatol. 2001;30(4):185-8.
4. Brito-Zeron P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, et al. Sjogren syndrome. Nat Rev Dis Primers. 2016;2:16047.
5. Antero DC, Parra AG, Miyazaki FH, Gehlen M, Skare TL. Secondary Sjogren's syndrome and disease activity of rheumatoid arthritis. Rev Assoc Med Bras. 2011;57(3):319-22.
6. Baer AN, Maynard JW, Shaikh F, Magder LS, Petri M. Secondary Sjogren's syndrome in systemic lupus erythematosus defines a distinct disease subset. J Rheumatol. 2010;37(6):1143-9.
7. Avouac J, Sordet C, Depinay C, Ardizonne M, Vacher-Lavenu MC, Sibilia J, et al. Systemic sclerosis-associated Sjogren's syndrome and relationship to the limited cutaneous subtype: results of a prospective study of sicca syndrome in 133 consecutive patients. Arthritis Rheum. 2006;54(7):2243-9.
8. Baldini C, Mosca M, Della Rossa A, Pepe P, Notarstefano C, Ferro F, et al. Overlap of ACA-positive systemic sclerosis and Sjogren's syndrome: a distinct clinical entity with mild organ involvement but at high risk of lymphoma. Clin Exp Rheumatol. 2013;31(2):272-80.
9. Ioannidis JP, Vassiliou VA, Moutsopoulos HM. Long-term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjogren's syndrome. Arthritis Rheum. 2002;46(3):741-7.
10. Bouma HR, Bootsma H, van Nimwegen JF, Haacke EA, Spijkervet FK, Vissink A, et al. Aging and Immunopathology in Primary Sjogren's Syndrome. Curr Aging Sci. 2015;8(2):202-13.
11. Rischmueller M, Tieu J, Lester S. Primary Sjogren's syndrome. Best Pract Res Clin Rheumatol. 2016;30(1):189-220.
12. Rasmussen A, Radfar L, Lewis D, Grundahl K, Stone DU, Kaufman CE, et al. Previous diagnosis of Sjogren's Syndrome as rheumatoid arthritis or systemic lupus erythematosus. Rheumatology (Oxford). 2016;55(7):1195-201.
13. Fox RI, Robinson CA, Curd JG, Kozin F, Howell FV. Sjogren's syndrome. Proposed criteria for classification. Arthritis Rheum. 1986;29(5):577-85.
14. Manthorpe R, Oxholm P, Prause JU, Schiodt M. The Copenhagen criteria for Sjogren's syndrome. Scand J Rheumatol Supplement. 1986;61:19-21.
15. Homma M, Tojo T, Akizuki M, Yamagata H. Criteria for Sjogren's syndrome in Japan. Scand J Rheumatol Suppl. 1986;61:26-7.

78
16. Vitali C, Bombardieri S, Moutsopoulos HM, Balestrieri G, Bencivelli W, Bernstein RM, et al. Preliminary criteria for the classification of Sjogren's syndrome. Results of a prospective concerted action supported by the European Community. Arthritis Rheum. 1993;36(3):340-7.
17. Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjogren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002;61(6):554-8.
18. Shiboski SC, Shiboski CH, Criswell L, Baer A, Challacombe S, Lanfranchi H, et al. American College of Rheumatology classification criteria for Sjogren's syndrome: a data-driven, expert consensus approach in the Sjogren's International Collaborative Clinical Alliance cohort. Arthritis Care Res (Hoboken). 2012;64(4):475-87.
19. Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjogren's syndrome: A consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis. 2017;76(1):9-16.
20. Baer AN, McAdams DeMarco M, Shiboski SC, Lam MY, Challacombe S, Daniels TE, et al. The SSB-positive/SSA-negative antibody profile is not associated with key phenotypic features of Sjogren's syndrome. Ann Rheum Dis. 2015;74(8):1557-61.
21. Seror R, Ravaud P, Bowman SJ, Baron G, Tzioufas A, Theander E, et al. EULAR Sjogren's syndrome disease activity index: development of a consensus systemic disease activity index for primary Sjogren's syndrome. Ann Rheum Dis. 2010;69(6):1103-9.
22. Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35(6):630-40.
23. Isenberg DA, Rahman A, Allen E, Farewell V, Akil M, Bruce IN, et al. BILAG 2004. Development and initial validation of an updated version of the British Isles Lupus Assessment Group's disease activity index for patients with systemic lupus erythematosus. Rheumatology (Oxford). 2005;44(7):902-6.
24. Seror R, Bowman SJ, Brito-Zeron P, Theander E, Bootsma H, Tzioufas A, et al. EULAR Sjogren's syndrome disease activity index (ESSDAI): a user guide. RMD Open. 2015;1(1):e000022.
25. Seror R, Bootsma H, Saraux A, Bowman SJ, Theander E, Brun JG, et al. Defining disease activity states and clinically meaningful improvement in primary Sjogren's syndrome with EULAR primary Sjogren's syndrome disease activity (ESSDAI) and patient-reported indexes (ESSPRI). Ann Rheum Dis. 2014.
26. Jonsson R, Theander E, Sjostrom B, Brokstad K, Henriksson G. Autoantibodies present before symptom onset in primary Sjogren syndrome. JAMA. 2013;310(17):1854-5.
27. Brito-Zeron P, Ramos-Casals M, group E-Stf. Advances in the understanding and treatment of systemic complications in Sjogren's syndrome. Curr Opin Rheumatol. 2014;26(5):520-7.
28. Brito-Zeron P, Kostov B, Solans R, Fraile G, Suarez-Cuervo C, Casanovas A, et al. Systemic activity and mortality in primary Sjogren syndrome: predicting survival using the EULAR-SS Disease Activity Index (ESSDAI) in 1045 patients. Ann Rheum Dis. 2016;75(2):348-55.

79
29. Seror R, Ravaud P, Mariette X, Bootsma H, Theander E, Hansen A, et al. EULAR Sjogren's Syndrome Patient Reported Index (ESSPRI): development of a consensus patient index for primary Sjogren's syndrome. Ann Rheum Dis. 2011;70(6):968-72.
30. Garcia-Carrasco M, Ramos-Casals M, Rosas J, Pallares L, Calvo-Alen J, Cervera R, et al. Primary Sjogren syndrome: clinical and immunologic disease patterns in a cohort of 400 patients. Medicine (Baltimore). 2002;81(4):270-80.
31. Alamanos Y, Tsifetaki N, Voulgari PV, Venetsanopoulou AI, Siozos C, Drosos AA. Epidemiology of primary Sjogren's syndrome in north-west Greece, 1982-2003. Rheumatology (Oxford). 2006;45(2):187-91.
32. Qin B, Wang J, Yang Z, Yang M, Ma N, Huang F, et al. Epidemiology of primary Sjogren's syndrome: a systematic review and meta-analysis. Ann Rheum Dis. 2014.
33. Ramos-Casals M, Brito-Zeron P, Kostov B, Siso-Almirall A, Bosch X, Buss D, et al. Google-driven search for big data in autoimmune geoepidemiology: analysis of 394,827 patients with systemic autoimmune diseases. Autoimmun Rev. 2015;14(8):670-9.
34. Brito-Zeron P, Acar-Denizli N, Zeher M, Rasmussen A, Seror R, Theander E, et al. Influence of geolocation and ethnicity on the phenotypic expression of primary Sjogren's syndrome at diagnosis in 8310 patients: a cross-sectional study from the Big Data Sjogren Project Consortium. Ann Rheum Dis. 2016.
35. Pillemer SR, Matteson EL, Jacobsson LT, Martens PB, Melton LJ, 3rd, O'Fallon WM, et al. Incidence of physician-diagnosed primary Sjogren syndrome in residents of Olmsted County, Minnesota. Mayo Clin Proc. 2001;76(6):593-9.
36. Plesivcnik Novljan M, Rozman B, Hocevar A, Grmek M, Kveder T, Tomsic M. Incidence of primary Sjogren's syndrome in Slovenia. Ann Rheum Dis. 2004;63(7):874-6.
37. Kvarnstrom M, Ottosson V, Nordmark B, Wahren-Herlenius M. Incident cases of primary Sjogren's syndrome during a 5-year period in Stockholm County: a descriptive study of the patients and their characteristics. Scand J Rheumatol. 2015;44(2):135-42.
38. Weng MY, Huang YT, Liu MF, Lu TH. Incidence and mortality of treated primary Sjogren's syndrome in Taiwan: a population-based study. J Rheumatol. 2011;38(4):706-8.
39. Goransson LG, Haldorsen K, Brun JG, Harboe E, Jonsson MV, Skarstein K, et al. The point prevalence of clinically relevant primary Sjogren's syndrome in two Norwegian counties. Scand J Rheumatol. 2011;40(3):221-4.
40. Birlik M, Akar S, Gurler O, Sari I, Birlik B, Sarioglu S, et al. Prevalence of primary Sjogren's syndrome in Turkey: a population-based epidemiological study. Int J Clin Pract. 2009;63(6):954-61.
41. Kabasakal Y, Kitapcioglu G, Turk T, Oder G, Durusoy R, Mete N, et al. The prevalence of Sjogren's syndrome in adult women. Scand J Rheumatol. 2006;35(5):379-83.
42. Miyasaka N. [Epidemiology and pathogenesis of Sjogren's syndrome]. Nihon Rinsho. 1995;53(10):2367-70.
43. Bowman SJ, Ibrahim GH, Holmes G, Hamburger J, Ainsworth JR. Estimating the prevalence among Caucasian women of primary Sjogren's syndrome in two general practices in Birmingham, UK. Scand J Rheumatol. 2004;33(1):39-43.
44. Trontzas PI, Andrianakos AA. Sjogren's syndrome: a population based study of prevalence in Greece. The ESORDIG study. Ann Rheum Dis. 2005;64(8):1240-1.

80
45. Anagnostopoulos I, Zinzaras E, Alexiou I, Papathanasiou AA, Davas E, Koutroumpas A, et al. The prevalence of rheumatic diseases in central Greece: a population survey. BMC Musculoskelet Disord. 2010;11:98.
46. Maldini C, Seror R, Fain O, Dhote R, Amoura Z, De Bandt M, et al. Epidemiology of primary Sjogren's syndrome in a French multiracial/multiethnic area. Arthritis Care Res (Hoboken). 2014;66(3):454-63.
47. Ramos-Casals M, Solans R, Rosas J, Camps MT, Gil A, Del Pino-Montes J, et al. Primary Sjogren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine (Baltimore). 2008;87(4):210-9.
48. Theander E, Manthorpe R, Jacobsson LT. Mortality and causes of death in primary Sjogren's syndrome: a prospective cohort study. Arthritis Rheum. 2004;50(4):1262-9.
49. Hansen A, Lipsky PE, Dorner T. New concepts in the pathogenesis of Sjogren syndrome: many questions, fewer answers. Curr Opin Rheumatol. 2003;15(5):563-70.
50. Tzioufas AG, Kapsogeorgou EK, Moutsopoulos HM. Pathogenesis of Sjogren's syndrome: what we know and what we should learn. J Autoimmun. 2012;39(1-2):4-8.
51. Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179(4):1317-30.
52. Haneji N, Nakamura T, Takio K, Yanagi K, Higashiyama H, Saito I, et al. Identification of alpha-fodrin as a candidate autoantigen in primary Sjogren's syndrome. Science. 1997;276(5312):604-7.
53. Mariette X. Sjogren's syndrome and virus. Ann Med Interne (Paris). 1995;146(4):243-6.
54. Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011;10(4):311-23.
55. de Paiva CS, Jones DB, Stern ME, Bian F, Moore QL, Corbiere S, et al. Altered Mucosal Microbiome Diversity and Disease Severity in Sjogren Syndrome. Sci Rep. 2016;6:23561.
56. Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. 2010;10(8):594-604.
57. Liu K, Kurien BT, Zimmerman SL, Kaufman KM, Taft DH, Kottyan LC, et al. X Chromosome Dose and Sex Bias in Autoimmune Diseases: Increased Prevalence of 47,XXX in Systemic Lupus Erythematosus and Sjogren's Syndrome. Arthritis Rheumatol. 2016;68(5):1290-300.
58. Iakimtchouk K, Myrmel H, Jonsson R. Serological screening for hepatitis B and C and human herpesvirus 6 in Norwegian patients with primary Sjogren's syndrome. J Rheumatol. 1999;26(9):2065-6.
59. Hall JC, Baer AN, Shah AA, Criswell LA, Shiboski CH, Rosen A, et al. Molecular Subsetting of Interferon Pathways in Sjogren's Syndrome. Arthritis Rheumatol. 2015;67(9):2437-46.
60. Theander E, Jonsson R, Sjostrom B, Brokstad K, Olsson P, Henriksson G. Prediction of Sjogren's Syndrome Years Before Diagnosis and Identification of Patients With Early Onset and Severe Disease Course by Autoantibody Profiling. Arthritis Rheumatol. 2015;67(9):2427-36.
61. Klareskog L, Gregersen PK, Huizinga TW. Prevention of autoimmune rheumatic disease: state of the art and future perspectives. Ann Rheum Dis. 2010;69(12):2062-6.

81
62. Kallberg H, Ding B, Padyukov L, Bengtsson C, Ronnelid J, Klareskog L, et al. Smoking is a major preventable risk factor for rheumatoid arthritis: estimations of risks after various exposures to cigarette smoke. Ann Rheum Dis. 2011;70(3):508-11.
63. Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015;12(4):205-17.
64. Karabulut G, Kitapcioglu G, Inal V, Kalfa M, Yargucu F, Keser G, et al. Cigarette smoking in primary Sjogren's syndrome: positive association only with ANA positivity. Mod Rheumatol. 2011;21(6):602-7.
65. Manthorpe R, Benoni C, Jacobsson L, Kirtava Z, Larsson A, Liedholm R, et al. Lower frequency of focal lip sialadenitis (focus score) in smoking patients. Can tobacco diminish the salivary gland involvement as judged by histological examination and anti-SSA/Ro and anti-SSB/La antibodies in Sjogren's syndrome? Ann Rheum Dis. 2000;59(1):54-60.
66. Olsson P, Turesson C, Mandl T, Jacobsson L, Theander E. Cigarette smoking and the risk of primary Sjogren's syndrome: a nested case control study. Arthritis Res Ther. 2017;19(1):50.
67. Stone DU, Fife D, Brown M, Earley KE, Radfar L, Kaufman CE, et al. Effect of Tobacco Smoking on The Clinical, Histopathological, and Serological Manifestations of Sjogren's Syndrome. PLoS One. 2017;12(2):e0170249.
68. Bave U, Nordmark G, Lovgren T, Ronnelid J, Cajander S, Eloranta ML, et al. Activation of the type I interferon system in primary Sjogren's syndrome: a possible etiopathogenic mechanism. Arthritis Rheum. 2005;52(4):1185-95.
69. Ronnblom L, Eloranta ML, Alm GV. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 2006;54(2):408-20.
70. Nordmark G, Eloranta ML, Ronnblom L. Primary Sjogren's syndrome and the type I interferon system. Curr Pharm Biotechnol. 2012;13(10):2054-62.
71. Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren's syndrome. Proc Natl Acad Sci U S A. 2006;103(8):2770-5.
72. Bombardieri M, Barone F, Pittoni V, Alessandri C, Conigliaro P, Blades MC, et al. Increased circulating levels and salivary gland expression of interleukin-18 in patients with Sjogren's syndrome: relationship with autoantibody production and lymphoid organization of the periductal inflammatory infiltrate. Arthritis Res Ther. 2004;6(5):R447-56.
73. Manoussakis MN, Boiu S, Korkolopoulou P, Kapsogeorgou EK, Kavantzas N, Ziakas P, et al. Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren's syndrome: correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum. 2007;56(12):3977-88.
74. Manoussakis MN, Dimitriou ID, Kapsogeorgou EK, Xanthou G, Paikos S, Polihronis M, et al. Expression of B7 costimulatory molecules by salivary gland epithelial cells in patients with Sjogren's syndrome. Arthritis Rheum. 1999;42(2):229-39.

82
75. Bikker A, van Woerkom JM, Kruize AA, Wenting-van Wijk M, de Jager W, Bijlsma JW, et al. Increased expression of interleukin-7 in labial salivary glands of patients with primary Sjogren's syndrome correlates with increased inflammation. Arthritis Rheum. 2010;62(4):969-77.
76. Hurst J, von Landenberg P. Toll-like receptors and autoimmunity. Autoimmun Rev. 2008;7(3):204-8.
77. Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, et al. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med. 2007;204(8):1959-71.
78. Guerrier T, Le Pottier L, Devauchelle V, Pers JO, Jamin C, Youinou P. Role of Toll-like receptors in primary Sjogren's syndrome with a special emphasis on B-cell maturation within exocrine tissues. J Autoimmun. 2012;39(1-2):69-76.
79. Karlsen M, Jakobsen K, Jonsson R, Hammenfors D, Hansen T, Appel S. Expression of Toll-like receptors in peripheral blood mononuclear cells of patients with primary Sjogren's syndrome. Scand J Immunol. 2016.
80. Fox RI, Adamson TC, 3rd, Fong S, Young C, Howell FV. Characterization of the phenotype and function of lymphocytes infiltrating the salivary gland in patients with primary Sjogren syndrome. Diagn Immunol. 1983;1(3):233-9.
81. Boumba D, Skopouli FN, Moutsopoulos HM. Cytokine mRNA expression in the labial salivary gland tissues from patients with primary Sjogren's syndrome. Br J Rheumatol. 1995;34(4):326-33.
82. Ciccia F, Guggino G, Rizzo A, Ferrante A, Raimondo S, Giardina A, et al. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren's syndrome. Ann Rheum Dis. 2012;71(2):295-301.
83. Katsifis GE, Rekka S, Moutsopoulos NM, Pillemer S, Wahl SM. Systemic and local interleukin-17 and linked cytokines associated with Sjogren's syndrome immunopathogenesis. Am J Pathol. 2009;175(3):1167-77.
84. Hillen MR, Ververs FA, Kruize AA, Van Roon JA. Dendritic cells, T-cells and epithelial cells: a crucial interplay in immunopathology of primary Sjogren's syndrome. Expert Rev Clin Immunol. 2014;10(4):521-31.
85. Christodoulou MI, Kapsogeorgou EK, Moutsopoulos NM, Moutsopoulos HM. Foxp3+ T-regulatory cells in Sjogren's syndrome: correlation with the grade of the autoimmune lesion and certain adverse prognostic factors. Am J Pathol. 2008;173(5):1389-96.
86. King C. New insights into the differentiation and function of T follicular helper cells. Nat Rev Immunol. 2009;9(11):757-66.
87. Jonsson R, Nginamau E, Szyszko E, Brokstad KA. Role of B cells in Sjogren's syndrome--from benign lymphoproliferation to overt malignancy. Front Biosci. 2007;12:2159-70.
88. Jonsson R, Vogelsang P, Volchenkov R, Espinosa A, Wahren-Herlenius M, Appel S. The complexity of Sjogren's syndrome: novel aspects on pathogenesis. Immunol Lett. 2011;141(1):1-9.
89. Appel S, Le Hellard S, Bruland O, Brun JG, Omdal R, Kristjansdottir G, et al. Potential association of muscarinic receptor 3 gene variants with primary Sjogren's syndrome. Ann Rheum Dis. 2011;70(7):1327-9.
90. Elagib KE, Borretzen M, Jonsson R, Haga HJ, Thoen J, Thompson KM, et al. Rheumatoid factors in primary Sjogren's syndrome (pSS) use diverse VH region genes, the majority of which show no evidence of somatic hypermutation. Clin Exp Immunol. 1999;117(2):388-94.
91. Youinou P, Devauchelle-Pensec V, Pers JO. Significance of B cells and B cell clonality in Sjogren's syndrome. Arthritis Rheum. 2010;62(9):2605-10.

83
92. Jonsson MV, Szodoray P, Jellestad S, Jonsson R, Skarstein K. Association between circulating levels of the novel TNF family members APRIL and BAFF and lymphoid organization in primary Sjogren's syndrome. J Clin Immunol. 2005;25(3):189-201.
93. Bohnhorst JO, Bjorgan MB, Thoen JE, Jonsson R, Natvig JB, Thompson KM. Abnormal B cell differentiation in primary Sjogren's syndrome results in a depressed percentage of circulating memory B cells and elevated levels of soluble CD27 that correlate with Serum IgG concentration. Clin Immunol. 2002;103(1):79-88.
94. Hansen A, Lipsky PE, Dorner T. B cells in Sjogren's syndrome: indications for disturbed selection and differentiation in ectopic lymphoid tissue. Arthritis Res Ther. 2007;9(4):218.
95. Hansen A, Daridon C, Dorner T. What do we know about memory B cells in primary Sjogren's syndrome? Autoimmun Rev. 2010;9(9):600-3.
96. Aqrawi LA, Brokstad KA, Jakobsen K, Jonsson R, Skarstein K. Low number of memory B cells in the salivary glands of patients with primary Sjogren's syndrome. Autoimmunity. 2012;45(7):547-55.
97. Daridon C, Pers JO, Devauchelle V, Martins-Carvalho C, Hutin P, Pennec YL, et al. Identification of transitional type II B cells in the salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. 2006;54(7):2280-8.
98. Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189(11):1747-56.
99. Mackay F, Sierro F, Grey ST, Gordon TP. The BAFF/APRIL system: an important player in systemic rheumatic diseases. Curr Dir Autoimmun. 2005;8:243-65.
100. Szodoray P, Alex P, Jonsson MV, Knowlton N, Dozmorov I, Nakken B, et al. Distinct profiles of Sjogren's syndrome patients with ectopic salivary gland germinal centers revealed by serum cytokines and BAFF. Clin Immunol. 2005;117(2):168-76.
101. Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11(9):372-7.
102. Sisto M, Lisi S, Lofrumento DD, Ingravallo G, De Lucro R, D'Amore M. Salivary gland expression level of IkappaBalpha regulatory protein in Sjogren's syndrome. J Mol Histol. 2013;44(4):447-54.
103. Musone SL, Taylor KE, Nititham J, Chu C, Poon A, Liao W, et al. Sequencing of TNFAIP3 and association of variants with multiple autoimmune diseases. Genes Immun. 2011;12(3):176-82.
104. Nordmark G, Wang C, Vasaitis L, Eriksson P, Theander E, Kvarnstrom M, et al. Association of genes in the NF-kappaB pathway with antibody-positive primary Sjogren's syndrome. Scand J Immunol. 2013;78(5):447-54.
105. Sun F, Li P, Chen H, Wu Z, Xu J, Shen M, et al. Association studies of TNFSF4, TNFAIP3 and FAM167A-BLK polymorphisms with primary Sjogren's syndrome in Han Chinese. J Hum Genet. 2013;58(7):475-9.
106. Pujari R, Hunte R, Khan WN, Shembade N. A20-mediated negative regulation of canonical NF-kappaB signaling pathway. Immunol Res. 2013;57(1-3):166-71.
107. Sisto M, Barca A, Lofrumento DD, Lisi S. Downstream activation of NF-kappaB in the EDA-A1/EDAR signalling in Sjogren's syndrome and its regulation by the ubiquitin-editing enzyme A20. Clin Exp Immunol. 2016;184(2):183-96.

84
108. Nocturne G, Tarn J, Boudaoud S, Locke J, Miceli-Richard C, Hachulla E, et al. Germline variation of TNFAIP3 in primary Sjogren's syndrome-associated lymphoma. Ann Rheum Dis. 2016;75(4):780-3.
109. Hjelmervik TO, Petersen K, Jonassen I, Jonsson R, Bolstad AI. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjogren's syndrome patients from healthy control subjects. Arthritis Rheum. 2005;52(5):1534-44.
110. Hall JC, Casciola-Rosen L, Berger AE, Kapsogeorgou EK, Cheadle C, Tzioufas AG, et al. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proc Natl Acad Sci U S A. 2012;109(43):17609-14.
111. Kyriakidis NC, Kapsogeorgou EK, Gourzi VC, Konsta OD, Baltatzis GE, Tzioufas AG. Toll-like receptor 3 stimulation promotes Ro52/TRIM21 synthesis and nuclear redistribution in salivary gland epithelial cells, partially via type I interferon pathway. Clin Exp Immunol. 2014;178(3):548-60.
112. Reveille JD, Wilson RW, Provost TT, Bias WB, Arnett FC. Primary Sjogren's syndrome and other autoimmune diseases in families. Prevalence and immunogenetic studies in six kindreds. Ann Intern Med. 1984;101(6):748-56.
113. Cruz-Tapias P, Rojas-Villarraga A, Maier-Moore S, Anaya JM. HLA and Sjogren's syndrome susceptibility. A meta-analysis of worldwide studies. Autoimmun Rev. 2012;11(4):281-7.
114. Nordmark G, Kristjansdottir G, Theander E, Eriksson P, Brun JG, Wang C, et al. Additive effects of the major risk alleles of IRF5 and STAT4 in primary Sjogren's syndrome. Genes Immun. 2009;10(1):68-76.
115. Nordmark G, Kristjansdottir G, Theander E, Appel S, Eriksson P, Vasaitis L, et al. Association of EBF1, FAM167A(C8orf13)-BLK and TNFSF4 gene variants with primary Sjogren's syndrome. Genes Immun. 2011;12(2):100-9.
116. Bolstad AI, Le Hellard S, Kristjansdottir G, Vasaitis L, Kvarnstrom M, Sjowall C, et al. Association between genetic variants in the tumour necrosis factor/lymphotoxin alpha/lymphotoxin beta locus and primary Sjogren's syndrome in Scandinavian samples. Ann Rheum Dis. 2012;71(6):981-8.
117. Li Y, Zhang K, Chen H, Sun F, Xu J, Wu Z, et al. A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjogren's syndrome at 7q11.23. Nat Genet. 2013;45(11):1361-5.
118. Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjogren's syndrome. Nat Genet. 2013;45(11):1284-92.
119. Imgenberg-Kreuz J, Sandling JK, Almlof JC, Nordlund J, Signer L, Norheim KB, et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjogren's syndrome reveals regulatory effects at interferon-induced genes. Ann Rheum Dis. 2016;75(11):2029-36.
120. Baldini C, Pepe P, Quartuccio L, Priori R, Bartoloni E, Alunno A, et al. Primary Sjogren's syndrome as a multi-organ disease: impact of the serological profile on the clinical presentation of the disease in a large cohort of Italian patients. Rheumatology (Oxford). 2014;53(5):839-44.
121. Manoussakis MN, Georgopoulou C, Zintzaras E, Spyropoulou M, Stavropoulou A, Skopouli FN, et al. Sjogren's syndrome associated with systemic lupus erythematosus: clinical and laboratory profiles and comparison with primary Sjogren's syndrome. Arthritis Rheum. 2004;50(3):882-91.
122. Bournia VK, Vlachoyiannopoulos PG. Subgroups of Sjogren syndrome patients according to serological profiles. J Autoimmun. 2012;39(1-2):15-26.

85
123. Friedman DM, Rupel A, Buyon JP. Epidemiology, etiology, detection, and treatment of autoantibody-associated congenital heart block in neonatal lupus. Curr Rheumatol Rep. 2007;9(2):101-8.
124. Chisholm DM, Mason DK. Labial salivary gland biopsy in Sjogren's disease. J Clin Pathol. 1968;21(5):656-60.
125. Guellec D, Cornec D, Jousse-Joulin S, Marhadour T, Marcorelles P, Pers JO, et al. Diagnostic value of labial minor salivary gland biopsy for Sjogren's syndrome: a systematic review. Autoimmun Rev. 2013;12(3):416-20.
126. Jonsson R, Moen K, Vestrheim D, Szodoray P. Current issues in Sjogren's syndrome. Oral Dis. 2002;8(3):130-40.
127. Nocturne G, Boudaoud S, Miceli-Richard C, Viengchareun S, Lazure T, Nititham J, et al. Germline and somatic genetic variations of TNFAIP3 in lymphoma complicating primary Sjogren's syndrome. Blood. 2013;122(25):4068-76.
128. Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J, et al. Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjogren's syndrome. Arthritis Rheum. 2001;44(11):2633-41.
129. Salomonsson S, Jonsson MV, Skarstein K, Brokstad KA, Hjelmstrom P, Wahren-Herlenius M, et al. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren's syndrome. Arthritis Rheum. 2003;48(11):3187-201.
130. Jonsson MV, Skarstein K, Jonsson R, Brun JG. Serological implications of germinal center-like structures in primary Sjogren's syndrome. J Rheumatol. 2007;34(10):2044-9.
131. Jonsson MV, Skarstein K. Follicular dendritic cells confirm lymphoid organization in the minor salivary glands of primary Sjogren's syndrome. J Oral Pathol Med. 2008;37(9):515-21.
132. Bombardieri M, Barone F, Humby F, Kelly S, McGurk M, Morgan P, et al. Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in Sjogren's syndrome. J Immunol. 2007;179(7):4929-38.
133. Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. 2012;33(6):297-305.
134. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. 2014;14(7):447-62.
135. Theander E, Vasaitis L, Baecklund E, Nordmark G, Warfvinge G, Liedholm R, et al. Lymphoid organisation in labial salivary gland biopsies is a possible predictor for the development of malignant lymphoma in primary Sjogren's syndrome. Ann Rheum Dis. 2011;70(8):1363-8.
136. Ramos-Casals M, Brito-Zeron P, Seror R, Bootsma H, Bowman SJ, Dorner T, et al. Characterization of systemic disease in primary Sjogren's syndrome: EULAR-SS Task Force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology (Oxford). 2015.
137. Ienopoli S, Carsons SE. Extraglandular manifestations of primary Sjogren's syndrome. Oral Maxillofac Surg Clin North Am. 2014;26(1):91-9.

86
138. Seror R, Theander E, Brun JG, Ramos-Casals M, Valim V, Dorner T, et al. Validation of EULAR primary Sjogren's syndrome disease activity (ESSDAI) and patient indexes (ESSPRI). Ann Rheum Dis. 2015;74(5):859-66.
139. Lindvall B, Bengtsson A, Ernerudh J, Eriksson P. Subclinical myositis is common in primary Sjogren's syndrome and is not related to muscle pain. J Rheumatol. 2002;29(4):717-25.
140. Escudero D, Latorre P, Codina M, Coll-Canti J, Coll J. Central nervous system disease in Sjogren's syndrome. Ann Med Interne (Paris). 1995;146(4):239-42.
141. Soliotis FC, Mavragani CP, Moutsopoulos HM. Central nervous system involvement in Sjogren's syndrome. Ann Rheum Dis. 2004;63(6):616-20.
142. Pavlakis PP, Alexopoulos H, Kosmidis ML, Stamboulis E, Routsias JG, Tzartos SJ, et al. Peripheral neuropathies in Sjogren syndrome: a new reappraisal. J Neurol Neurosurg Psychiatry. 2011;82(7):798-802.
143. Pavlakis PP, Alexopoulos H, Kosmidis ML, Mamali I, Moutsopoulos HM, Tzioufas AG, et al. Peripheral neuropathies in Sjogren's syndrome: a critical update on clinical features and pathogenetic mechanisms. J Autoimmun. 2012;39(1-2):27-33.
144. Garcia-Carrasco M, Siso A, Ramos-Casals M, Rosas J, de la Red G, Gil V, et al. Raynaud's phenomenon in primary Sjogren's syndrome. Prevalence and clinical characteristics in a series of 320 patients. J Rheumatol. 2002;29(4):726-30.
145. Evans R, Zdebik A, Ciurtin C, Walsh SB. Renal involvement in primary Sjogren's syndrome. Rheumatology (Oxford). 2015;54(9):1541-8.
146. Barendregt PJ, Visser MR, Smets EM, Tulen JH, van den Meiracker AH, Boomsma F, et al. Fatigue in primary Sjogren's syndrome. Ann Rheum Dis. 1998;57(5):291-5.
147. Abrol E, Gonzalez-Pulido C, Praena-Fernandez JM, Isenberg DA. A retrospective study of long-term outcomes in 152 patients with primary Sjogren's syndrome: 25-year experience. Clin Med. 2014;14(2):157-64.
148. Theander L, Strombeck B, Mandl T, Theander E. Sleepiness or fatigue? Can we detect treatable causes of tiredness in primary Sjogren's syndrome? Rheumatology (Oxford). 2010;49(6):1177-83.
149. Hernandez YL, Daniels TE. Oral candidiasis in Sjogren's syndrome: prevalence, clinical correlations, and treatment. Oral Surg Oral Med Oral Pathol. 1989;68(3):324-9.
150. Lindgren S, Manthorpe R, Eriksson S. Autoimmune liver disease in patients with primary Sjogren's syndrome. J Hepatol. 1994;20(3):354-8.
151. Lazarus MN, Isenberg DA. Development of additional autoimmune diseases in a population of patients with primary Sjogren's syndrome. Ann Rheum Dis. 2005;64(7):1062-4.
152. Biro E, Szekanecz Z, Czirjak L, Danko K, Kiss E, Szabo NA, et al. Association of systemic and thyroid autoimmune diseases. Clin Rheumatol. 2006;25(2):240-5.
153. Perez-De-Lis M, Akasbi M, Siso A, Diez-Cascon P, Brito-Zeron P, Diaz-Lagares C, et al. Cardiovascular risk factors in primary Sjogren's syndrome: a case-control study in 624 patients. Lupus. 2010;19(8):941-8.
154. Juarez M, Toms TE, de Pablo P, Mitchell S, Bowman S, Nightingale P, et al. Cardiovascular risk factors in women with primary Sjogren's syndrome: United Kingdom primary Sjogren's syndrome registry results. Arthritis Care Res (Hoboken). 2014;66(5):757-64.

87
155. Singh AG, Singh S, Matteson EL. Rate, risk factors and causes of mortality in patients with Sjogren's syndrome: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford). 2016;55(3):450-60.
156. Mathews PM, Hahn S, Hessen M, Kim J, Grader-Beck T, Birnbaum J, et al. Ocular Complications of Primary Sjogren Syndrome in Men. Am J Ophthalmol. 2015;160(3):447-52 e1.
157. Brito-Zeron P, Theander E, Baldini C, Seror R, Retamozo S, Quartuccio L, et al. Early diagnosis of primary Sjogren's syndrome: EULAR-SS task force clinical recommendations. Expert Rev Clin Immunol. 2016;12(2):137-56.
158. Daniels TE, Silverman S, Jr., Michalski JP, Greenspan JS, Sylvester RA, Talal N. The oral component of Sjogren's syndrome. Oral Surg Oral Med Oral Pathol. 1975;39(6):875-85.
159. Devauchelle-Pensec V, Mariette X, Jousse-Joulin S, Berthelot JM, Perdriger A, Puechal X, et al. Treatment of primary Sjogren syndrome with rituximab: a randomized trial. Ann Intern Med. 2014;160(4):233-42.
160. van Nimwegen JF, Moerman RV, Sillevis Smitt N, Brouwer E, Bootsma H, Vissink A. Safety of treatments for primary Sjogren's syndrome. Expert Opin Drug Saf. 2016;15(4):513-24.
161. Bowman SJ, Everett CC, O'Dwyer JL, Emery P, Pitzalis C, Ng WF, et al. Randomized Controlled Trial of Rituximab and cost-effectiveness analysis in treating fatigue and oral dryness in primary Sjogren's Syndrome. Arthritis Rheumatol. 2017.
162. De Vita S, Quartuccio L, Seror R, Salvin S, Ravaud P, Fabris M, et al. Efficacy and safety of belimumab given for 12 months in primary Sjogren's syndrome: the BELISS open-label phase II study. Rheumatology (Oxford). 2015.
163. Meiners PM, Vissink A, Kroese FG, Spijkervet FK, Smitt-Kamminga NS, Abdulahad WH, et al. Abatacept treatment reduces disease activity in early primary Sjogren's syndrome (open-label proof of concept ASAP study). Ann Rheum Dis. 2014;73(7):1393-6.
164. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: ARC Press; 2008.
165. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-90.
166. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103(1):275-82.
167. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503-11.
168. Bea S, Zettl A, Wright G, Salaverria I, Jehn P, Moreno V, et al. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood. 2005;106(9):3183-90.
169. Kassan SS, Thomas TL, Moutsopoulos HM, Hoover R, Kimberly RP, Budman DR, et al. Increased risk of lymphoma in sicca syndrome. Ann Intern Med. 1978;89(6):888-92.
170. Zintzaras E, Voulgarelis M, Moutsopoulos HM. The risk of lymphoma development in autoimmune diseases: a meta-analysis. Arch Intern Med. 2005;165(20):2337-44.

88
171. Theander E, Henriksson G, Ljungberg O, Mandl T, Manthorpe R, Jacobsson LT. Lymphoma and other malignancies in primary Sjogren's syndrome: a cohort study on cancer incidence and lymphoma predictors. Ann Rheum Dis. 2006;65(6):796-803.
172. Lazarus MN, Robinson D, Mak V, Moller H, Isenberg DA. Incidence of cancer in a cohort of patients with primary Sjogren's syndrome. Rheumatology (Oxford). 2006;45(8):1012-5.
173. Zhang W, Feng S, Yan S, Zhao Y, Li M, Sun J, et al. Incidence of malignancy in primary Sjogren's syndrome in a Chinese cohort. Rheumatology (Oxford). 2010;49(3):571-7.
174. Johnsen SJ, Brun JG, Goransson LG, Smastuen MC, Johannesen TB, Haldorsen K, et al. Risk of non-Hodgkin's lymphoma in primary Sjogren's syndrome: a population-based study. Arthritis Care Res (Hoboken). 2013;65(5):816-21.
175. Fallah M, Liu X, Ji J, Forsti A, Sundquist K, Hemminki K. Autoimmune diseases associated with non-Hodgkin lymphoma: a nationwide cohort study. Ann Oncol. 2014;25(10):2025-30.
176. Solans-Laque R, Lopez-Hernandez A, Bosch-Gil JA, Palacios A, Campillo M, Vilardell-Tarres M. Risk, predictors, and clinical characteristics of lymphoma development in primary Sjogren's syndrome. Semin Arthritis Rheum. 2011;41(3):415-23.
177. Sutcliffe N, Inanc M, Speight P, Isenberg D. Predictors of lymphoma development in primary Sjogren's syndrome. Semin Arthritis Rheum. 1998;28(2):80-7.
178. Nishishinya MB, Pereda CA, Munoz-Fernandez S, Pego-Reigosa JM, Rua-Figueroa I, Andreu JL, et al. Identification of lymphoma predictors in patients with primary Sjogren's syndrome: a systematic literature review and meta-analysis. Rheumatol Int. 2015;35(1):17-26.
179. Anaya JM, McGuff HS, Banks PM, Talal N. Clinicopathological factors relating malignant lymphoma with Sjogren's syndrome. Semin Arthritis Rheum. 1996;25(5):337-46.
180. Baimpa E, Dahabreh IJ, Voulgarelis M, Moutsopoulos HM. Hematologic manifestations and predictors of lymphoma development in primary Sjogren syndrome: clinical and pathophysiologic aspects. Medicine (Baltimore). 2009;88(5):284-93.
181. Risselada AP, Kruize AA, Goldschmeding R, Lafeber FP, Bijlsma JW, van Roon JA. The prognostic value of routinely performed minor salivary gland assessments in primary Sjogren's syndrome. Ann Rheum Dis. 2014;73(8):1537-40.
182. Risselada AP, Hair M, Kruize AA, Bijlsma JW, van Roon JA. Lymphocytic focus score as a prognostic tool. Ann Rheum Dis. 2015;74(4):e31.
183. Nezos A, Papageorgiou A, Fragoulis G, Ioakeimidis D, Koutsilieris M, Tzioufas AG, et al. B-cell activating factor genetic variants in lymphomagenesis associated with primary Sjogren's syndrome. J Autoimmun. 2014;51:89-98.
184. Tobon GJ, Saraux A, Gottenberg JE, Quartuccio L, Fabris M, Seror R, et al. Role of Fms-like tyrosine kinase 3 ligand as a potential biologic marker of lymphoma in primary Sjogren's syndrome. Arthritis Rheum. 2013;65(12):3218-27.
185. Papageorgiou A, Ziogas DC, Mavragani CP, Zintzaras E, Tzioufas AG, Moutsopoulos HM, et al. Predicting the outcome of Sjogren's syndrome-

89
associated non-hodgkin's lymphoma patients. PLoS One. 2015;10(2): e0116189.
186. Nocturne G, Mariette X. Sjogren Syndrome-associated lymphomas: an update on pathogenesis and management. Br J Haematol. 2015;168(3):317-27.
187. Ngalamika O, Zhang Y, Yin H, Zhao M, Gershwin ME, Lu Q. Epigenetics, autoimmunity and hematologic malignancies: a comprehensive review. J Autoimmun. 2012;39(4):451-65.
188. Hansen A, Lipsky PE, Dorner T. B-cell lymphoproliferation in chronic inflammatory rheumatic diseases. Nat Clin Pract Rheumatol. 2007;3(10):561-9.
189. Voulgarelis M, Skopouli FN. Clinical, immunologic, and molecular factors predicting lymphoma development in Sjogren's syndrome patients. Clin Rev Allergy Immunol. 2007;32(3):265-74.
190. Baecklund E, Smedby KE, Sutton LA, Askling J, Rosenquist R. Lymphoma development in patients with autoimmune and inflammatory disorders--what are the driving forces? Semin Cancer Biol. 2014;24:61-70.
191. Goldin LR, Landgren O. Autoimmunity and lymphomagenesis. Int J Cancer. 2009;124(7):1497-502.
192. Kuksin CA, Minter LM. The Link between Autoimmunity and Lymphoma: Does NOTCH Signaling Play a Contributing Role? Front Oncol. 2015;5:51.
193. Royer B, Cazals-Hatem D, Sibilia J, Agbalika F, Cayuela JM, Soussi T, et al. Lymphomas in patients with Sjogren's syndrome are marginal zone B-cell neoplasms, arise in diverse extranodal and nodal sites, and are not associated with viruses. Blood. 1997;90(2):766-75.
194. Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5(4):251-62.
195. Illes A, Varoczy L, Papp G, Wilson PC, Alex P, Jonsson R, et al. Aspects of B-cell non-Hodgkin's lymphoma development: a transition from immune-reactivity to malignancy. Scand J Immunol. 2009;69(5):387-400.
196. Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8(1):22-33.
197. Martin F, Kearney JF. Marginal-zone B cells. Nat Rev Immunol. 2002;2(5):323-35.
198. Joao C, Farinha P, da Silva MG, Martins C, Crespo M, Cabecadas J. Cytogenetic abnormalities in MALT lymphomas and their precursor lesions from different organs. A fluorescence in situ hybridization (FISH) study. Histopathology. 2007;50(2):217-24.
199. Chanudet E, Ye H, Ferry J, Bacon CM, Adam P, Muller-Hermelink HK, et al. A20 deletion is associated with copy number gain at the TNFA/B/C locus and occurs preferentially in translocation-negative MALT lymphoma of the ocular adnexa and salivary glands. J Pathol. 2009;217(3):420-30.
200. Voulgarelis M, Dafni UG, Isenberg DA, Moutsopoulos HM. Malignant lymphoma in primary Sjogren's syndrome: a multicenter, retrospective, clinical study by the European Concerted Action on Sjogren's Syndrome. Arthritis Rheumatol. 1999;42(8):1765-72.
201. Makula E, Pokorny G, Kiss M, Voros E, Kovacs L, Kovacs A, et al. The place of magnetic resonance and ultrasonographic examinations of the parotid gland in the diagnosis and follow-up of primary Sjogren's syndrome. Rheumatology (Oxford). 2000;39(1):97-104.
202. Theander E, Mandl T. Primary Sjogren's syndrome: diagnostic and prognostic value of salivary gland ultrasonography using a simplified scoring system. Arthritis Care Res (Hoboken). 2014;66(7):1102-7.

90
203. Smedby KE, Hjalgrim H, Askling J, Chang ET, Gregersen H, Porwit-MacDonald A, et al. Autoimmune and chronic inflammatory disorders and risk of non-Hodgkin lymphoma by subtype. J Natl Cancer Inst. 2006;98(1):51-60.
204. Ekstrom Smedby K, Vajdic CM, Falster M, Engels EA, Martinez-Maza O, Turner J, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood. 2008;111(8):4029-38.
205. Al-Tourah AJ, Gill KK, Chhanabhai M, Hoskins PJ, Klasa RJ, Savage KJ, et al. Population-based analysis of incidence and outcome of transformed non-Hodgkin's lymphoma. J Clin Oncol. 2008;26(32):5165-9.
206. Schoder H, Noy A, Gonen M, Weng L, Green D, Erdi YE, et al. Intensity of 18fluorodeoxyglucose uptake in positron emission tomography distinguishes between indolent and aggressive non-Hodgkin's lymphoma. J Clin Oncol. 2005;23(21):4643-51.
207. Noy A, Schoder H, Gonen M, Weissler M, Ertelt K, Cohler C, et al. The majority of transformed lymphomas have high standardized uptake values (SUVs) on positron emission tomography (PET) scanning similar to diffuse large B-cell lymphoma (DLBCL). Ann Oncol. 2009;20(3):508-12.
208. Hollander P, Rostgaard K, Smedby KE, Chang ET, Amini RM, de Nully Brown P, et al. Autoimmune and Atopic Disorders and Risk of Classical Hodgkin Lymphoma. Am J Epidemiol. 2015.
209. Tomi AL, Belkhir R, Nocturne G, Desmoulins F, Berge E, Pavy S, et al. Brief Report: Monoclonal Gammopathy and Risk of Lymphoma and Multiple Myeloma in Patients With Primary Sjogren's Syndrome. Arthritis Rheumatol. 2016;68(5):1245-50.
210. Voulgarelis M, Ziakas PD, Papageorgiou A, Baimpa E, Tzioufas AG, Moutsopoulos HM. Prognosis and outcome of non-Hodgkin lymphoma in primary Sjogren syndrome. Medicine (Baltimore). 2012;91(1):1-9.
211. Pollard RP, Pijpe J, Bootsma H, Spijkervet FK, Kluin PM, Roodenburg JL, et al. Treatment of mucosa-associated lymphoid tissue lymphoma in Sjogren's syndrome: a retrospective clinical study. J Rheumatol. 2011;38(10):2198-208.
212. Jackson AE, Mian M, Kalpadakis C, Pangalis GA, Stathis A, Porro E, et al. Extranodal Marginal Zone Lymphoma of Mucosa-Associated Lymphoid Tissue of the Salivary Glands: A Multicenter, International Experience of 248 Patients (IELSG 41). Oncologist. 2015.
213. Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38(10):982-4.
214. Masaki Y, Dong L, Kurose N, Kitagawa K, Morikawa Y, Yamamoto M, et al. Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis. 2009;68(8):1310-5.
215. Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64(10):3061-7.
216. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181-92.
217. Vasaitis L. IgG4-related disease: A relatively new concept for clinicians. Eur J Intern Med. 2016;27:1-9.

91
218. McHeyzer-Williams LJ, Pelletier N, Mark L, Fazilleau N, McHeyzer-Williams MG. Follicular helper T cells as cognate regulators of B cell immunity. Curr Opin Immunol. 2009;21(3):266-73.
219. Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134(3):679-87.
220. Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190-5.
221. Hiepe F, Dorner T, Hauser AE, Hoyer BF, Mei H, Radbruch A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol. 2011;7(3):170-8.
222. Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy. 2014;69(2):269-72.
223. Kanari H, Kagami S, Kashiwakuma D, Oya Y, Furuta S, Ikeda K, et al. Role of Th2 cells in IgG4-related lacrimal gland enlargement. Int Arch Allergy Immunol. 2010;152 Suppl 1:47-53.
224. Mattoo H, Mahajan VS, Maehara T, Deshpande V, Della-Torre E, Wallace ZS, et al. Clonal expansion of CD4(+) cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol. 2016;138(3):825-38.
225. Arai Y, Yamashita K, Kuriyama K, Shiokawa M, Kodama Y, Sakurai T, et al. Plasmacytoid Dendritic Cell Activation and IFN-alpha Production Are Prominent Features of Murine Autoimmune Pancreatitis and Human IgG4-Related Autoimmune Pancreatitis. J Immunol. 2015;195(7):3033-44.
226. Moriyama M, Tanaka A, Maehara T, Furukawa S, Nakashima H, Nakamura S. T helper subsets in Sjogren's syndrome and IgG4-related dacryoadenitis and sialoadenitis: a critical review. J Autoimmun. 2014;51:81-8.
227. Morgan WS, Castleman B. A clinicopathologic study of Mikulicz's disease. Am J Pathol. 1953;29(3):471-503.
228. Yamamoto M, Harada S, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, et al. Clinical and pathological differences between Mikulicz's disease and Sjogren's syndrome. Rheumatology. 2005;44(2):227-34.
229. Doe K, Nozawa K, Okada T, Tada K, Yamaji K, Tamura N, et al. Usefulness of minor salivary gland biopsy in the diagnosis of IgG4-related disease: a case report. International journal of clinical and experimental pathology. 2014;7(5):2673-7.
230. Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Modern rheumatology / the Japan Rheumatism Association. 2012;22(1):21-30.
231. Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, Hisano S, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15(5):615-26.
232. Ohara H, Okazaki K, Tsubouchi H, Inui K, Kawa S, Kamisawa T, et al. Clinical diagnostic criteria of IgG4-related sclerosing cholangitis 2012. J Hepatobiliary Pancreat Sci. 2012;19(5):536-42.
233. Goto H, Takahira M, Azumi A, Japanese Study Group for Ig GROD. Diagnostic criteria for IgG4-related ophthalmic disease. Jpn J Ophthalmol. 2015;59(1):1-7.

92
234. Yamamoto M, Takahashi H, Tabeya T, Suzuki C, Naishiro Y, Ishigami K, et al. Risk of malignancies in IgG4-related disease. Mod Rheumatol. 2012;22(3):414-8.
235. Yamamoto M, Yajima H, Takahashi H, Yokoyama Y, Ishigami K, Shimizu Y, et al. Everyday clinical practice in IgG4-related dacryoadenitis and/or sialadenitis: results from the SMART database. Mod Rheumatol. 2015;25(2):199-204.
236. Cheuk W, Yuen HK, Chan AC, Shih LY, Kuo TT, Ma MW, et al. Ocular adnexal lymphoma associated with IgG4+ chronic sclerosing dacryoadenitis: a previously undescribed complication of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32(8):1159-67.
237. Oyama T, Takizawa J, Nakamura N, Aoki S, Aizawa Y, Abe H. Multifocal mucosa-associated lymphoid tissue lymphoma associated with IgG4-related disease: a case report. Jpn J Ophthalmol. 2011;55(3):304-6.
238. Venkataraman G, Rizzo KA, Chavez JJ, Streubel B, Raffeld M, Jaffe ES, et al. Marginal zone lymphomas involving meningeal dura: possible link to IgG4-related diseases. Mod Pathol. 2011;24(3):355-66.
239. Kubota T, Moritani S, Yoshino T, Nagai H, Terasaki H. Ocular adnexal marginal zone B cell lymphoma infiltrated by IgG4-positive plasma cells. J Clin Pathol. 2010;63(12):1059-65.
240. Nakayama R, Matsumoto Y, Horiike S, Kobayashi S, Nakao R, Nagoshi H, et al. Close pathogenetic relationship between ocular immunoglobulin G4-related disease (IgG4-RD) and ocular adnexal mucosa-associated lymphoid tissue (MALT) lymphoma. Leuk Lymphoma. 2014;55(5):1198-202.
241. Naresh KN, Barwick T, Karadimitris A. IgG4 positive mucosa associated lymphoid tissue lymphoma of the orbit - lesson of the month. Histopathology. 2014;65(5):718-21.
242. Zhang L, Notohara K, Levy MJ, Chari ST, Smyrk TC. IgG4-positive plasma cell infiltration in the diagnosis of autoimmune pancreatitis. Mod Pathol. 2007;20(1):23-8.
243. Deshpande V, Chicano S, Finkelberg D, Selig MK, Mino-Kenudson M, Brugge WR, et al. Autoimmune pancreatitis: a systemic immune complex mediated disease. Am J Surg Pathol. 2006;30(12):1537-45.
244. Strehl JD, Hartmann A, Agaimy A. Numerous IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders. J Clin Pathol. 2011;64(3):237-43.
245. Ludvigsson JF, Otterblad-Olausson P, Pettersson BU, Ekbom A. The Swedish personal identity number: possibilities and pitfalls in healthcare and medical research. Eur J Epidemiol. 2009;24(11):659-67.
246. Waldenlind K, Eriksson JK, Grewin B, Askling J. Validation of the rheumatoid arthritis diagnosis in the Swedish National Patient Register: a cohort study from Stockholm County. BMC Musculoskelet Disord. 2014;15:432.
247. Lindstrom U, Exarchou S, Sigurdardottir V, Sundstrom B, Askling J, Eriksson JK, et al. Validity of ankylosing spondylitis and undifferentiated spondyloarthritis diagnoses in the Swedish National Patient Register. Scand J Rheumatol. 2015;44(5):369-76.
248. The National Board of Health and Welfare. Cancer incidence in Sweden 2009. http://www.socialstyrelsen.se/statistik/statistikefteramne/cancer (accessed 13 April 2011).

93
249. Barlow L, Westergren K, Holmberg L, Talback M. The completeness of the Swedish Cancer Register: a sample survey for year 1998. Acta Oncol. 2009;48(1):27-33.
250. Kinch A, Cavelier L, Bengtsson M, Baecklund E, Enblad G, Backlin C, et al. Donor or recipient origin of posttransplant lymphoproliferative disorders following solid organ transplantation. Am J Transplant. 2014;14(12):2838-45.
251. Vasaitis L, Sundstrom C, Backlin C, Nordmark G, Baecklund E. Sporadic occurrence of non-diagnosed IgG4-related disease in lymphoma patients with a previous Sjogren's syndrome diagnosis. Acta Oncol. 2016;55(9-10):1139-44.
252. Nordmark G, Alm GV, Ronnblom L. Mechanisms of Disease: primary Sjogren's syndrome and the type I interferon system. Nat Clin Pract Rheumatol. 2006;2(5):262-9.
253. Gasparotto D, De Vita S, De Re V, Marzotto A, De Marchi G, Scott CA, et al. Extrasalivary lymphoma development in Sjogren's syndrome: clonal evolution from parotid gland lymphoproliferation and role of local triggering. Arthritis Rheum. 2003;48(11):3181-6.
254. Barone F, Bombardieri M, Rosado MM, Morgan PR, Challacombe SJ, De Vita S, et al. CXCL13, CCL21, and CXCL12 expression in salivary glands of patients with Sjogren's syndrome and MALT lymphoma: association with reactive and malignant areas of lymphoid organization. J Immunol. 2008;180(7):5130-40.
255. Barone F, Bombardieri M, Manzo A, Blades MC, Morgan PR, Challacombe SJ, et al. Association of CXCL13 and CCL21 expression with the progressive organization of lymphoid-like structures in Sjogren's syndrome. Arthritis Rheum. 2005;52(6):1773-84.
256. Fisher BA, Brown RM, Bowman SJ, Barone F. A review of salivary gland histopathology in primary Sjogren's syndrome with a focus on its potential as a clinical trials biomarker. Ann Rheum Dis. 2015;74(9):1645-50.
257. Johnsen SJ, Berget E, Jonsson MV, Helgeland L, Omdal R, Jonsson R. Evaluation of germinal center-like structures and B cell clonality in patients with primary Sjogren syndrome with and without lymphoma. J Rheumatol. 2014;41(11):2214-22.
258. Inoue D, Yoshida K, Yoneda N, Ozaki K, Matsubara T, Nagai K, et al. IgG4-related disease: dataset of 235 consecutive patients. Medicine (Baltimore). 2015;94(15):e680.
259. Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2015;74(1):14-8.
260. Inoue D, Zen Y, Abo H, Gabata T, Demachi H, Kobayashi T, et al. Immunoglobulin G4-related lung disease: CT findings with pathologic correlations. Radiology. 2009;251(1):260-70.
261. Matsui S, Taki H, Shinoda K, Suzuki K, Hayashi R, Tobe K, et al. Respiratory involvement in IgG4-related Mikulicz's disease. Mod Rheumatol. 2012;22(1):31-9.
262. Brito-Zeron P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4-related disease. Autoimmun Rev. 2014;13(12):1203-10.
263. Quartuccio L, Fabris M, Salvin S, Maset M, De Marchi G, De Vita S. Controversies on rituximab therapy in sjogren syndrome-associated lymphoproliferation. Int J Rheumatol. 2009;2009:424935.
264. Covelli M, Lanciano E, Tartaglia P, Grattagliano V, Angelelli G, Atzeni F, et al. Rituximab treatment for Sjogren syndrome-associated non-Hodgkin's lymphoma: case series. Rheumatol Int. 2012;32(10):3281-4.

94
265. Quartuccio L, Salvin S, Fabris M, Maset M, Pontarini E, Isola M, et al. BLyS upregulation in Sjogren's syndrome associated with lymphoproliferative disorders, higher ESSDAI score and B-cell clonal expansion in the salivary glands. Rheumatology (Oxford). 2013;52(2):276-81.
266. Gondran G, Fauchais A, Lambert M, Ly K, Launay D, Queyrel V, et al. Primary Sjogren's syndrome in men. Scand J Rheumatol. 2008;37(4):300-5.
267. Ansell P, Simpson J, Lightfoot T, Smith A, Kane E, Howell D, et al. Non-Hodgkin lymphoma and autoimmunity: does gender matter? Int J Cancer. 2011;129(2):460-6.
268. Hedstrom G, Peterson S, Berglund M, Jerkeman M, Enblad G, Swedish Lymphoma Study G. Male gender is an adverse risk factor only in young patients with diffuse large B-cell lymphoma - a Swedish population-based study. Acta Oncol. 2015;54(6):924-32.
269. Hasni MS, Berglund M, Yakimchuk K, Guan J, Linderoth J, Amini RM, et al. Estrogen receptor beta1 in diffuse large B-cell lymphoma growth and as a prognostic biomarker. Leuk Lymphoma. 2017;58(2):418-27.
270. Daridon C, Devauchelle V, Hutin P, Le Berre R, Martins-Carvalho C, Bendaoud B, et al. Aberrant expression of BAFF by B lymphocytes infiltrating the salivary glands of patients with primary Sjogren's syndrome. Arthritis Rheum. 2007;56(4):1134-44.
271. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjogren's syndrome. Eur Respir Rev. 2016;25(140):110-23.
272. Sergentanis TN, Kanavidis P, Michelakos T, Petridou ET. Cigarette smoking and risk of lymphoma in adults: a comprehensive meta-analysis on Hodgkin and non-Hodgkin disease. Eur J Cancer Prev. 2013;22(2):131-50.
273. Baecklund E, Backlin C, Iliadou A, Granath F, Ekbom A, Amini RM, et al. Characteristics of diffuse large B cell lymphomas in rheumatoid arthritis. Arthritis Rheum. 2006;54(12):3774-81.
274. Thunberg U, Enblad G, Berglund M. Classification of diffuse large B-cell lymphoma by immunohistochemistry demonstrates that elderly patients are more common in the non-GC subgroup and younger patients in the GC subgroup. Haematologica. 2012;97(2):e3; author reply e4.
275. Risselada AP, Kruize AA, Bijlsma JW. Clinical features distinguishing lymphoma development in primary Sjogren's Syndrome--a retrospective cohort study. Semin Arthritis Rheum. 2013;43(2):171-7.
276. Fragkioudaki S, Mavragani CP, Moutsopoulos HM. Predicting the risk for lymphoma development in Sjogren syndrome: An easy tool for clinical use. Medicine (Baltimore). 2016;95(25):e3766.
277. Conconi A, Franceschetti S, Aprile von Hohenstaufen K, Margiotta-Casaluci G, Stathis A, Moccia AA, et al. Histologic transformation in marginal zone lymphomasdagger. Ann Oncol. 2015;26(11):2329-35.
278. van den Brand M, van der Velden WJ, Diets IJ, Ector GI, de Haan AF, Stevens WB, et al. Clinical features of patients with nodal marginal zone lymphoma compared to follicular lymphoma: similar presentation, but differences in prognostic factors and rate of transformation. Leuk Lymphoma. 2015:1-8.


Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1331
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofMedicine. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Medicine”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-320220
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2017