localized peeling skin syndrome: case report with ultrastructural study
TRANSCRIPT

Localized peeling skin syndrome: case report withultrastructural study
A.BRUSASCO, S.VERALDI, G.TADINI AND R.CAPUTOCentre for Inherited Cutaneous Diseases, Institute for Dermatological Sciences, IRCCS, Ospedale Maggiore, University of Milan,Via Pace 9, 20122 Milan, Italy
Accepted for publication 7 April 1998
Summary We report a young woman in whom the history, clinical features, histopathological and ultra-structural findings led to a diagnosis of peeling skin syndrome (PSS). PSS is a rare and not wellclassified genodermatosis, mainly characterized by the spontaneous separation of the stratumcorneum from the stratum granulosum. The unusual feature in our patient was the strictlocalization to the palm. PSS has been described as a more generalized disease frequently sparingpalms and soles. We propose the diagnosis label of ‘localized PSS’ for this previously undescribedvariant of a rare keratinization defect.
Peeling skin syndrome (PSS) is a very rare and not wellcharacterized keratinization disorder, probably inher-ited as an autosomal recessive trait, characterized byspontaneous exfoliation of the stratum corneum and,histologically, by subcorneal or intracorneal splitting.1,2
PSS comprises cases with different clinical pictures anddifferent evolution.1–11 This clinical heterogeneitydetermined the recent distinction into two subgroupsbased mainly on the presence or absence of inflamma-tory changes and seasonal variation.12
Case report
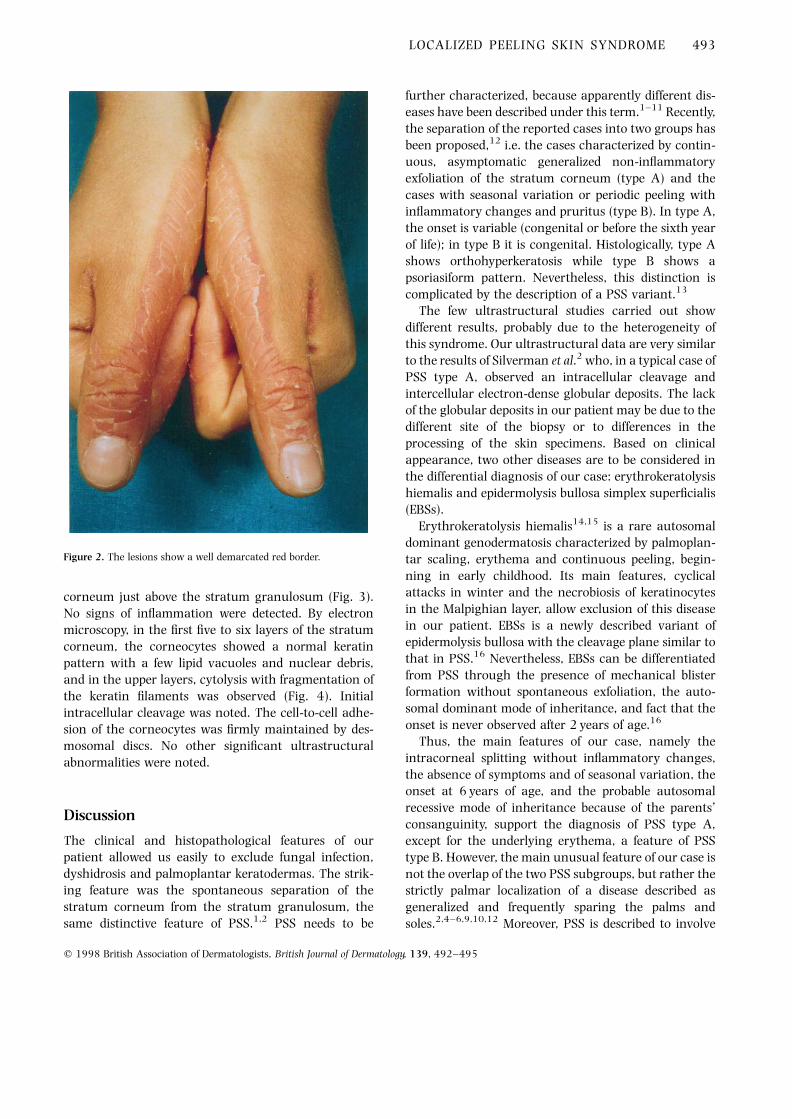
A 26-year-old woman has been affected by asympto-matic erythematous and thin-scaling patches with asharply demarcated border (Figs 1 and 2) localizedexclusively to the palms and dorsal side of the fingers.The scaling seems to occur spontaneously, and thepatient is able to peel off sheets of stratum corneumwithout pain. The patient stated that the dermatitisdeveloped at the age of 6 years and was stable and didnot worsen during hot or cold weather. She had usedseveral topical agents without benefit. Her parents wereconsanguineous but no family members showed similarlesions. Mycological examination was negative. Histo-pathological examination of a skin specimen from theborder of a palmar lesion showed only orthokeratotichyperkeratosis with an initial separation of the stratum
British Journal of Dermatology 1998; 139: 492–495.
492 q 1998 British Association of Dermatologists
Some of this material was presented at the 23rd Annual Meeting of theSociety for Cutaneous Ultrastructure Research, Gardone Riviera, Italy,11–13 April 1996.
Figure 1. Scaling patches with underlying erythema are seen, strictlylocalized to the palms and dorsal aspect of the fingers.
corneum just above the stratum granulosum (Fig. 3).No signs of inflammation were detected. By electronmicroscopy, in the first five to six layers of the stratumcorneum, the corneocytes showed a normal keratinpattern with a few lipid vacuoles and nuclear debris,and in the upper layers, cytolysis with fragmentation ofthe keratin filaments was observed (Fig. 4). Initialintracellular cleavage was noted. The cell-to-cell adhe-sion of the corneocytes was firmly maintained by des-mosomal discs. No other significant ultrastructuralabnormalities were noted.
Discussion
The clinical and histopathological features of ourpatient allowed us easily to exclude fungal infection,dyshidrosis and palmoplantar keratodermas. The strik-ing feature was the spontaneous separation of thestratum corneum from the stratum granulosum, thesame distinctive feature of PSS.1,2 PSS needs to be
further characterized, because apparently different dis-eases have been described under this term.1–11 Recently,the separation of the reported cases into two groups hasbeen proposed,12 i.e. the cases characterized by contin-uous, asymptomatic generalized non-inflammatoryexfoliation of the stratum corneum (type A) and thecases with seasonal variation or periodic peeling withinflammatory changes and pruritus (type B). In type A,the onset is variable (congenital or before the sixth yearof life); in type B it is congenital. Histologically, type Ashows orthohyperkeratosis while type B shows apsoriasiform pattern. Nevertheless, this distinction iscomplicated by the description of a PSS variant.13
The few ultrastructural studies carried out showdifferent results, probably due to the heterogeneity ofthis syndrome. Our ultrastructural data are very similarto the results of Silverman et al.2 who, in a typical case ofPSS type A, observed an intracellular cleavage andintercellular electron-dense globular deposits. The lackof the globular deposits in our patient may be due to thedifferent site of the biopsy or to differences in theprocessing of the skin specimens. Based on clinicalappearance, two other diseases are to be considered inthe differential diagnosis of our case: erythrokeratolysishiemalis and epidermolysis bullosa simplex superficialis(EBSs).
Erythrokeratolysis hiemalis14,15 is a rare autosomaldominant genodermatosis characterized by palmoplan-tar scaling, erythema and continuous peeling, begin-ning in early childhood. Its main features, cyclicalattacks in winter and the necrobiosis of keratinocytesin the Malpighian layer, allow exclusion of this diseasein our patient. EBSs is a newly described variant ofepidermolysis bullosa with the cleavage plane similar tothat in PSS.16 Nevertheless, EBSs can be differentiatedfrom PSS through the presence of mechanical blisterformation without spontaneous exfoliation, the auto-somal dominant mode of inheritance, and fact that theonset is never observed after 2 years of age.16
Thus, the main features of our case, namely theintracorneal splitting without inflammatory changes,the absence of symptoms and of seasonal variation, theonset at 6 years of age, and the probable autosomalrecessive mode of inheritance because of the parents’consanguinity, support the diagnosis of PSS type A,except for the underlying erythema, a feature of PSStype B. However, the main unusual feature of our case isnot the overlap of the two PSS subgroups, but rather thestrictly palmar localization of a disease described asgeneralized and frequently sparing the palms andsoles.2,4–6,9,10,12 Moreover, PSS is described to involve
LOCALIZED PEELING SKIN SYNDROME 493
q 1998 British Association of Dermatologists, British Journal of Dermatology, 139, 492–495
Figure 2. The lesions show a well demarcated red border.

these regions with different features from the rest of thebody, in particular erythema without scales1,8 or hyper-keratosis only.1 Only in two cases did the erythema withscaling involve the palmoplantar regions.7,11
We propose a diagnosis of localized PSS for ourpatient. This seems to be a new variant to be differen-tiated from the type A and B subgroups of PSS. In arecent report, another case of localized PSS was giventhe name ‘acral PSS’,17 confirming the existence of a
localized form of the disease. This case is very similar toour patient, except for a few differences, i.e. the involve-ment of arms and legs and the worsening with water,heat and friction. We prefer to call this new subgroup‘localized PSS’, because this term underlines the mainfeature and does not exclude the possibility of othertypes of partial localization of the disease. The identifi-cation of gene defects may enable investigators to over-come the semantics of classification in this disorder.
494 A.BRUSASCO et al.
q 1998 British Association of Dermatologists, British Journal of Dermatology, 139, 492–495
Figure 3. Histopathological examinationreveals hyperkeratosis with an initialseparation of the stratum corneum justabove the stratum granulosum(haematoxylin and eosin; originalmagnification × 100).
Figure 4. Electron microscopy: thecorneocytes show a normal keratin patternwith a few lipid vacuoles and nuclear debris(asterisks) in the first five to six layers. Inthe upper layers, cytolysis of thecorneocytes (white arrows) with a initialcleavage is found (black arrows) (originalmagnification × 1650).

References1 Levy SB, Goldsmith LA. The peeling skin syndrome. J Am Acad
Dermatol 1982; 7: 606–13.2 Silverman AK, Ellis CN, Beals TF, Woo TY. Continual skin peeling
syndrome. Arch Dermatol 1986; 122: 71–5.3 Fox H. Skin shedding (keratolysis exfoliativa congenita): report of a
case. (Letter.) Arch Dermatol 1921; 3: 202.4 Kurban AK, Azar HA. Familial continual skin peeling. Br J
Dermatol 1969; 81: 191–5.5 Abdel-Hafez K, Safer AM, Selim MM, Rehak A. Familial continual
skin peeling. Dermatologica 1983; 166: 23–31.6 Heid E, Harbit RB, Lazrak B. Desquamation familiale continue
(familial continual skin peeling). Ann Dermatol Venereol 1983;110: 141–3.
7 Dicken CH. Peeling skin syndrome. J Am Acad Dermatol 1985; 13:158–60.
8 Hacham-Zadeh S, Holubar K. Skin peeling syndrome in a Kurdishfamily. Arch Dermatol 1985; 121: 545–6.
9 Mevorah B, Frenk E, Saurat JH, Siegenthaler G. Peeling skinsyndrome: a clinical, ultrastructural and biochemical study. Br JDermatol 1987; 116: 117–25.
10 Aras N, Sutman K, Tastan HB et al. Peeling skin syndrome. J AmAcad Dermatol 1994; 30: 135–6.
11 Tolat SN, Gharpuray MB. Skin peeling syndrome. Cutis 1994; 53:255–7.
12 Traupe H. The Ichthyoses: a Guide to Clinical Diagnosis, GeneticCounseling, and Therapy. New York: Springer-Verlag, 1989.
13 Mevorah B, Salomon D, Siegenthaler G et al. Ichthyosiformdermatosis with superficial blister formation and peeling:evidence for desmosomal anomaly and altered epidermalvitamin A metabolism. J Am Acad Dermatol 1996; 34:379–85.
14 Findlay GH, Morrison JGL. Erythrokeratolysis hiemalis—kerato-lytic winter erythema or Oudtshoorn skin. Br J Dermatol 1978; 98:491–5.
15 Krahl D, Sigwart A, Hartschuh W et al. Erythrokeratolysis hiema-lis. Hautarzt 1994; 45: 776–9.
16 Fine JD, Johnson L, Wright T. Epidermolysis bullosa simplexsuperficialis. Arch Dermatol 1989; 125: 633–8.
17 Shwayder T, Conn S, Lowe L. Acral peeling skin syndrome. ArchDermatol 1997; 133: 535–6.
LOCALIZED PEELING SKIN SYNDROME 495
q 1998 British Association of Dermatologists, British Journal of Dermatology, 139, 492–495