laboratory manual organic chemistry 241napavalley.edu/people/sfawl/documents/chem 241/chem 241 lab -...
TRANSCRIPT

LABORATORY MANUALORGANIC CHEMISTRY 241
THIRD EDITION
Dr. Steven Fawl

LABORATORY MANUAL
ORGANIC CHEMISTRY 241
THIRD EDITION
Dr. Steven Fawl
Mathematics and Science DivisionNapa Valley College
Napa, California

N A P A V A L L E Y C O L L E G E
COURSE: Chemistry 241
INSTRUCTOR: Dr. Steven Fawl, Room 1843, 253-3195 LECTURES: MW 9:30 to 10:50 LABS: MT 2:00 to 4:50OFFICE HRS: MTW 12:20 to 1:20 Room 1843 EXAM DATES:
The following are tentative dates for your exams and the material each exam will cover.
Exam #1 - Wednesday, February 24 - Spectroscopyth
Exam #2 - Wednesday, March 24 - Aromatics, Diels-Alder, MO Theoryth
Exam #3 - Wednesday, May 12 - Carbonyl Chemistryth
Final Exam - Comprehensive - Monday, May 24 , 8:00-10:00th
DESCRIPTION: A continuation of CHEM 240. Introduction to NMR, IR, and MassSpectroscopy. Chemical reactions and syntheses of aromatic, carbonyl, and amine compounds. Special topics in carbohydrate, amino acid, and lipid chemistry. Lab work includes simple andmulti-step synthesis and spectral identification.
COURSE CONTENT:
1. SpectroscopyNuclear Magnetic Resonance Infrared SpectroscopyMass Spectrometry
2. Aromatics I: AromaticityReactions of benzene, Kekules structure, stability, and modern theories of the structurefor benzene.Huckel's Rule and other aromatics.Nomenclature of benzene derivatives.Heterocyclic aromatics and aromatics in biochemistry.
3. Aromatics II: Reactions with ElectrophilesElectrophilic aromatic substitutions and their mechanisms.Halogenation, nitration, sulfonation and alkylation of benzene.Effects of substituents-reactivity and directing influences.Alkyl and alkenyl benzenes and their reactions.Carbenes.
4. Aldehydes and KetonesStructure, nomenclature and physical properties.

Reactions of carbonyls - acetals, hemi-acetals, ammonia derivatives, and Schiff basereactons.
Keto-enol tautomerism, and the Cannizzaro Reaction.5. Carboxylic Acids and their Derivatives
Nomenclature and physical properties.Preparation and reactions at acyl carbon.Synthesis and reactions of acyl halides, acid anhydrides, esters and amides.Hell-Volhard-Zelinski and decarboxylation reactions.Claisen, Michael, and Aldol reactions and synthetic pathways.
6. AminesNomenclature, physical properties and their basicities.Some biologically important amines.Preparation and reactions of amines with nitrous acid, diazonium salts.Analysis of amines.
7. CarbohydratesMonosaccharides, mutarotation and glycoside formation.Oxidation and reduction, osazone formation.Synthesis and degradation of monosaccharides.The D-family of aldoses.Methylation of monosaccharides.Di- and poly- saccharides.Nitrogen containing sugars.
8. LipidsFatty acids and Triacylglycerols.Steroids and prostaglandins.Phospholipids, waxes and terpenes.
9. Amino Acids and Proteins Important amino acids.
Laboratory synthesis and analysis of amino acids.Amino acid sequence of proteins and polypeptides.Primary structure of polypeptides and their synthesis.Secondary and tertiary structure of proteins.
COURSE OBJECTIVES
1. Solve complex reaction mechanisms.2. Synthesize compounds starting with simple ingredients.3. Determine the structure of organic compounds from spectrographic data.4. Name organic compounds based on their structure.

STUDENT LEARNING OUTCOMES
1. Communicate chemical and physical processes at the molecular level and how they relate to themacroscopic environment.
2. Solve synthetic reaction pathways and mechanisms while demonstrating the reasoning clearlyand completely.
3. Implement laboratory techniques correctly using appropriate safety procedures and express themclearly in written laboratory reports.
GRADING POLICY: Three exams and a final, quizzes, plus laboratory scores will count towardthe final grade according to the following schedule,
3 Exams = 300pts (100pts each) Final = 200pts Quizes = 100pts (10 @ 10pts each) Lab = 120pts Total = 720pts
Grading is based on the class average = B-. The approximate breakdown of grades is,
100-85% A / 84-70% B / 69-60% C / 59-50% D / <50% F
ALL of the labs must be completed to pass the course regardless of the overall performance of thestudent or else an "F" will be given.
LABS ARE CONSIDERED LATE IF THEY ARE TURNED IN ANY TIME AFTER THEFRIDAY THAT THEY ARE DUE. LABS THAT ARE TWO WEEKS LATE WILL RECEIVENO CREDIT. Special arrangements must be made if a lab must be missed!

SAFETY AND TECHNIQUE RULES
Safety in the laboratory is extremely important. It is expected that you know laboratory safetyrules. It is important that if you feel uncomfortable with your knowledge of these rules thatyou take the time to learn them. The following list is NOT complete. The Media Center,room 1028, there is a video tape available in which a Napa Valley College instructor explainsin detail safety and technique rules. There is NO excuse for not following safety rules.
1) Be attentive to instructions and follow them carefully. Read the board at the back of theclass room when you first come to class, any changes in procedure will be written there.
2) If you ever have any questions about the procedure, apparatus, or chemicals it isimportant that you ask the Instructor or Instructional assistant.
3) Do not perform any unauthorized experiments. Anyone found doing so faces permanent
expulsion from class.
4) Do not handle chemicals or materials not assigned to you or not called for in theexperiment.
5) Learn the location and proper use of the fire extinguisher, safety shower, eye and facewash. Keep the first aide sink area clear at all times
6) Coats, books, etc, should be kept in the space provided for them at the back of the lab. Many of the chemicals used in the lab can ruin or stain paper and clothing.
7) Never taste chemicals, nor pipet by mouth. Always use pipet bulbs or wheels.
8) Smell chemicals by fanning a little vapor towards you.
9) Experiments in which dangerous or obnoxious fumes are produced must be done in thefume hood. Be sure to stop these reactions as soon as possible.
10) No eating, drinking or smoking in the lab.
11) Never point test tubes at yourself or others.
12) In the event of any injury, spill or glass breakage inform the Instructor immediately.
13) Goggles must be worn at all times when in the lab.
14) Chemicals may not be taken out of the lab (not even to the I.A.'s desk.)
15) Chemicals may not be stored in lockers.

16) Avoid unnecessary contact with ALL chemicals.
17) Do not leave lit burners unattended.
18) Every time you use a chemical read it's label carefully. If any discrepancies inform theIA or instructor immediately.
19) All containers which contain a chemical or in which a reaction occurs must be labeled.
20) When labeling a storage container include name and/or formula of chemical, anyappropriate warnings, concentration, date and your name.
21) NEVER place anything inside a reagent bottle, no spatulas, droppers, nor pipets. If thereagent is a clumpy solid inform the IA. Proper technique is to "roll" containers fromside to side to remove solids and to pour liquids into smaller containers (such as abeaker) first.
22) NEVER return unused chemical (liquids or solids) back to the original container - offer
excess to another student or dispose of it appropriately.
23) Be conservative of reagents, place only the amount you need into a labeled container(such as a beaker). Do not take the reagent bottles to your work area - leave them whereevery one can find them.
24) Use tap water to wash glassware - you should rinse with DI water - please beconservative.
25) To dilute acids and bases, Add the Acid (or Base) to the Water.
26) Dispose of liquids and solids appropriately, read the board, or your experimentalprocedure for special instructions, otherwise dispose of liquids and soluble solids downthe sink with lots of water, insoluble materials (such as paper towels) should be put inthe waste basket. KEEP THE SINKS CLEAN
27) It is very important to keep the lab clean. Before you leave each time be sure to:
a) return equipment to its proper place b) clean up your workspace with the sponge c) put away your labwared) lock your locker
There is NO reason for a messy lab. Everything you need to keep your lab neat and cleanis available. Dirty counters, paper towels left in the sink or troughs, labware left out,messes left under the fume hood, chemical spills left on the balance, are BAD techniqueand as such will not be tolerated.

28) You may not be in the laboratory at any time other than your scheduled laboratory periodunless you have the permission of the instructor in charge as well as your courseinstructor. Do not visit friends during their lab time and do not invite your friends orfamily to visit you.

TABLE OF CONTENTS
EXPERIMENT 1Synthesis of p-Nitroacetanilide 1
WORKSHEET 4
EXPERIMENT 2Synthesis of Aspirin - Ester Formation 6
WORKSHEET 9
EXPERIMENT 3 Reactions of Aromatic Aldehydes - Cannizaro Reaction 11
WORKSHEET 15
EXPERIMENT 4Synthesis of 9,10-Dihyroanthracene-9,10-endo-á,â-succinic Anhydride:A Diels-Alder Reaction 16
WORKSHEET 17
EXPERIMENT 5sec-Butylbenzene - Friedel Craft Alkylation 18
WORKSHEET 19
EXPERIMENT 6Sodium Borohydride Reduction of Acetophenone 20
WORKSHEET 23
EXPERIMENT 7Synthesis of 2-Methyl-2-Hexanol: A Grignard Reaction 24

EXPERIMENT ONE
SYNTHESIS OF P-NITROACETANILIDE
Discussion:
The aromatic nitration of acetanilide is an exothermic reaction ; the temperature must becarefully controlled by chilling, stirring, and the slow addition of reagents. Acetanilide is firstdissolved in the solvent, glacial acetic acid, by warming. Glacial acetic acid is used because itis a polar solvent capable of dissolving acetanilide and the acetate ion is a poor nucleophile sono substitution is possible. After the solution is cooled, sulfuric acid is added; however, evenwith cooling, the temperature of the solution rises almost 40E. Both the acetanilide solution
3 2 4and the nitrating solution (a mixture of HNO , and H SO ) must be chilled to about 10ECbefore the reaction is begun.
To prevent dinitration of the acetanilide, the nitrating mixture is added in small portions to the
3acetanilide solution (and not vice versa) so that the concentration of HNO , is kept at a
3 2 4minimum. After all the HNO , H SO solution has been added, the reaction mixture is allowedto warm slowly to room temperature. If the reaction mixture has been kept excessively cold
3during the addition, there will be a relatively large amount of unreacted HNO , present, whichmay cause the temperature to rise above room temperature. If this should happen, the mixturemust be rechilled.
The work-up procedure consists of removal of the acids and crystallization of the product.Every trace of acid must be removed because hydrogen ions catalyze the hydrolysis of theamide to p-nitroaniline or its protonated cation. Most of the acid is removed by pouring thereaction mixture onto ice and water, then filtering the flocculent yellow precipitate of p--nitroacetanilide. The last traces of acetic acid are removed by neutralization. Because basesalso catalyze the hydrolysis of amides, the neutralizing agent used is disodium hydrogen
2 4 2 4phosphate (Na HPO ). This reagent reacts with acids to yield NaH PO . The result is abuffered solution with a pH near neutral.
The crude product is air-dried before crystallization. If all of the acid was removed, theproduct will be light yellow. A deep yellow to yellow-orange product is indicative of thepresence of p-nitroaniline from hydrolysis. Unfortunately, p-nitroaniline is difficult to removefrom p-nitroacetanilide by crystallization.

Equipment
250-mL beakerdropper or disposable pipettwo 50-mL and one 125-mL Erlenmeyer flasks10-mL graduated cylinderhot plateice bathspatulastirring rodthermometervacuum filtration assemblywatch glassacetanilide, 6.5 gdisodium hydrogen phosphate, 15 g95% ethanol, 60 mLglacial acetic acid, 10 mLconc. nitric acid, 3.5 mLconc. sulfuric acid, 15 mL
Time Required: about two hours to crude product; 15-20 minutes for crystallization; twoovernight dryings; 15 minutes for melting-point determination
STOPPING POINTS: during either of the two drying periods or while the product iscrystallizing from ethanol
>>>>SAFETY NOTE 1: A mixture of concentrated nitric and sulfuric acids is used as thenitrating mixture. Use extreme caution when preparing and using this mixture.
>>>>SAFETY NOTE 2: Nitro compounds are toxic and can be absorbed through the skin.You may wish to wear disposable plastic gloves during portions of this experiment.
PROCEDURE
Place 6.5 g of acetanilide in a 125-mL Erlenmeyer flask, add 10 mL of glacial acetic acid(CAUTION: strong irritant), and warm the flask on a hot plate in a fume hood until theacetanilide dissolves. Cool the flask in an ice bath to about 20E; then add 10 mL of cold, conc.sulfuric acid. The temperature of the mixture will rise to about 60E. Chill the solution to about10E in an ice bath. (The solution will become very viscous.)
Mix 3.5 mL of conc. nitric acid and 5 mL of conc. sulfuric acid in a 50-mL flask, and chill the
3 2 4flask in an ice bath. When both solutions are cold, slowly add the HNO , H SO solution, 1mL at a time, to the acetanilide solution. Keep the reaction flask in an ice bath so that thetemperature of the reaction mixture is maintained between 10-20E. Stir the reaction mixturecarefully after each addition. The entire addition requires about 15 minutes.

After the addition is completed, allow the reaction flask to stand at room temperature for 30minutes. Monitor the temperature; if it rises above 25E, chill the flask in an ice bath. Shouldthe rechilling be necessary, allow the flask to stand for 30 minutes or more at roomtemperature after the rechilling.
Pour the reaction mixture into a 250-mL beaker containing 100 mL of water and 25 g ofcracked ice. Using a large Buchner funnel, filter the heavy lemon-yellow precipitate withvacuum. Press out as much aqueous acid from the filter cake as possible with a spatula orclean cork while suction is being applied (CAUTION: see Safety Note 2). The precipitate isvoluminous; use care in transferring it to the Buchner funnel or a substantial amount ofproduct will be lost.
Transfer the filter cake to a clean 250-mL beaker, and add 100 mL of 15% aqueous disodiumhydrogen phosphate. Stir the mixture to a paste-like consistency and refilter using vacuum.Wash the beaker with two 30-mL portions of cold water. Finally, wash the filter cake with anadditional 50 mL of cold water. Press the filter cake with a spatula or clean cork to remove asmuch water as possible, then dry the solid overnight on a watch glass. Determine the yield andmelting point.
The crude product can be purified by crystallization from 30-60 mL of 95% ethanol. (Thecrude product dissolves very slowly, even with heating; avoid using an excess of solvent.)

NAME DATE
SYNTHESIS OF P-NITROACETANILIDE WORKSHEET
Mass of Product Melting Point CRC MeltingPoint
TheoreticalYield (grams)
Actual Yield(Percent)
Problems
1. List at least two reasons for the choice of glacial acetic acid as the solvent for the nitrationof acetanilide.
2. What would be the effects of each of the following changes of reaction conditions in thisexperiment, assuming that all other conditions are held constant? Explain your answers.
(a) increasing the amount of glacial acetic acid from 10 mL to 20 mL
(b) increasing the amount of nitric acid from 3.5 mL to 7.0 mL
(c) decreasing the amount of sulfuric acid from 15 mL to 5 mL
3. Write equations for the hydrolysis of p-nitroacetanilide in (a) aqueous acid; (b) aqueoushydroxide. The products of the reation should be p-nitroaniline and acetic acid (or acetate ionin base).
4. Predict what would happen during the crystallization of p-nitroacetanilide from 95%ethanol if all the acidic material had not been neutralized previously? Use an equation in youranswer.

EXPERIMENT TWO
SYNTHESIS OF ASPIRIN: ESTER FORMATION
Discussion
Although active salicylates are found in the bark of willow trees (trees of the genus Salix,from whence salicylic acid derives its name), all aspirin sold today is synthesized from phenol,which, in turn, is obtained from petroleum. In this experiment, you will conduct only the laststep of the commercial synthesis, the conversion of salicylic acid to aspirin using the Fisherester synthesis.
The reaction of salicylic acid with acetic anhydride occurs rapidly. The reactants and a sulfuricacid catalyst are mixed, then warmed in a hot water bath. Solid salicylic acid is insoluble inacetic anhydride. Aspirin is soluble in a hot mixture of acetic anhydride and acetic acid, whichis formed as the reaction proceeds. Thus, the course of the reaction can be followed by thedisappearance of the solid salicylic acid. The reaction is essentially complete when all thesalicylic acid has dissolved. Aspirin is only slightly soluble in a cold mixture of aceticanhydride and acetic acid. Therefore, as the mixture is cooled to room temperature, the aspirinprecipitates.
At the end of the reaction period, the mixture contains aspirin, acetic anhydride, and aceticacid. Acetic acid is miscible with water, and acetic anhydride reacts fairly rapidly with waterto yield acetic acid. By adding water to the reaction mixture and allowing the aqueous mixtureto stand at room temperature for a few minutes, we can achieve a reasonable separation-thecontaminants dissolve in the water, and the aspirin precipitates. Because of the presence ofacetic acid and because aspirin is slightly soluble in water, about 10% of the aspirin remains insolution. Therefore, the mixture is thoroughly chilled before filtration and crystallization.
An additional small amount of aspirin can be recovered from the mother liquor if it is allowedto stand overnight. Despite this fact, it is not good practice to leave the bulk of the aspirin inacidic solution for an extended period of time because it will undergo a slow hydrolysis toyield salicylic acid and acetic acid, a typical acid-catalyzed ester hydrolysis.

EQUIPMENT
400-mL beaker (for hot water bath) droppertwo 125-mL Erlenmeyer flasks10-mL and 100-mL graduated cylinders spatulathermometervacuum filtration assemblytwo watch glasses
CHEMICALS
acetic anhydride, 5.0 mLsalicylic acid, 2.8 gconc. sulfuric acid, 3-4 drops
TIME REQUIRED: 1 hour plus overnight drying and time for a melting-point determination
STOPPING POINTS: after the initial vacuum filtration; while the product is crystallizingfrom water; while the final product is being air-dried
>> SAFETY NOTE: Acetic anhydride is volatile and is a strong irritant.
PROCEDURE
Place 2.8 g of salicylic acid in a dry 125-mL Erlenmeyer flask, then add 5.0 mL aceticanhydride and 3-4 drops concentrated sulfuric acid. Mix the resultant white slurry thoroughlywith a spatula, and place the flask in a warm water bath (45-50EC) for 5-7 min. Swirl or stirthe mixture occasionally to dissolve all the solid material. Because the reaction is slightlyexothermic, a small temperature rise can be detected.
Allow the flask to cool. The aspirin begins to precipitate when the temperature of the solutionis about 35-40EC, and the mixture becomes semisolid. When this occurs, add 50 mL waterand break up any lumps with a spatula. Allow the mixture to stand for an additional 5 minutes,then chill the flask in an ice bath and remove the crystals by vacuum filtration.
Crystallize the crude aspirin from 25 mL of warm water not exceeding 80EC (seeExperimental Note). Allowing the mother liquor to sit overnight may produce a second cropof crystals. Air-dry the crystals and determine the percent yield and melting point.
EXPERIMENTAL NOTE
At temperatures exceeding 80EC, aspirin forms an oil that dissolves organic impurities fromthe water; in this case, it may be difficult to redissolve the aspirin in water. If the solid doesnot dissolve in 25 mL of water, add more water from a dropper. Let the mixture warm 2-4minutes between additions to allow the solid to dissolve.

NAME DATE
SYNTHESIS OF ASPIRIN WORKSHEET
Mass ofProduct
Melting Point CRC MeltingPoint
TheoreticalYield (grams)
Actual Yield(Percent)
PROBLEMS
1) Write an equation for the synthesis of aspirin from salicylic acid with an acid chlorideinstead of acetic anhydride.
2) The following compounds all have antipyretic and analgesic properties similar to aspirin.Suggest a synthesis for each from salicylic acid.
(a) sodium salicylate (b) methyl salicylate
(c) salicylamide (d) phenyl salicylate
(e) 2-0-acetyl-5-bromosalicylic acid

3a) Commercial aspirin sometimes has a distinct acetic acid odor. Why?
3b) Would ingestion of such aspirin be harmful? Explain.

EXPERIMENT THREE
THE CANNIZARO REACTION: THE DISPROPORTIONATION OFBENZALDEHYDE
DISCUSSION
In planning the laboratory schedule, it should be observed that this experiment requiresmaterials to be mixed and allowed to stand for 24 hr or longer.
In the presence of strong alkalis, benzaldehyde (like formaldehyde) undergoesdisproportionation to form the corresponding primary alcohol and a salt of the carboxylic acid: the Cannizzaro reaction.
The process involves addition of hydroxyl ion to the carbonyl group of one molecule andtransfer of hydride anion from the adduct to a second molecule of benzaldehyde, accompaniedby proton interchange to form the benzoate anion and benzyl alcohol. If the reaction iseffected under anhydrous conditions with the sodium derivative of benzyl alcohol as catalyst,the product is the ester, benzyl benzoate.
EQUIPMENT
18 g KOH20 mL Benzaldehyde125 mL Erlenmeyer/stopper100 mL methylene chlorideSteam bath125 mL separatory funnel10 mL 20% sodium bisulfite4 g anhydrous magnesium sulfate100 mL round bottom flaskThermometer (0-250EC)Bunsen burner40 mL conc. HClChipped iceBlue litmus paperSide arm flaskBuchner funnel

EXPERIMENT: In a small beaker dissolve 0.27 mole (18 g of 85% pure solid) of solidpotassium hydroxide in 18 mL of water and cool the solution to about 25EC. Place 0.2 mole(21 g, 20 mL) of benzaldehyde in a 125-mL Erlenmeyer flask (or narrow-mouth bottle) and toit add the potassium hydroxide solution. Cork the flask firmly and shake the mixturethoroughly until an emulsion is formed. Allow the mixture to stand for 24 hr or longer. At theend of this period, the odor of benzaldehyde should no longer be detectable.
Isolation of Benzyl Alcohol
To the mixture add just enough distilled water to dissolve the precipitate of potassiumbenzoate. Shake the mixture thoroughly to facilitate solution of the precipitate. Extract thealkaline solution with three or four 20-mL portions of methylene chloride to remove thebenzyl alcohol and traces of any unconverted benzaldehyde. Combine the methylene chlorideextracts for isolation of benzyl alcohol and reserve the aqueous solution to obtain the benzoicacid.
Concentrate the methylene chloride solution of benzyl alcohol by distillation on a steam bathusing a water-cooled condenser, until the volume of the residual liquid has been reduced to15-20 mL. A steam bath is just a beaker of boiling water into which the distillation flask isemersed. Cool the remaining liquid, transfer it to a small separatory funnel (using 2-3 mL ofmethylene chloride to rinse the distilling flask), and shake it thoroughly with two 5-mLportions of 20% aqueous sodium bisulfite to remove any benzaldehyde. Wash the methylenechloride solution finally with two 10-mL portions of water and dry it with 3-4 g of anhydrousmagnesium sulfate. Filter the solution into a small dry distilling flask and carefully distill offthe methylene chloride. Attach a short air-cooled condenser and distill the benzyl alcohol, byheating the flask directly with a luminous flame kept in motion. Collect the material boiling at200-206EC. The yield is 4-5 g.
Isolation of Benzoic Acid
To free the acid, pour the aqueous solution of potassium benzoate (from which the benzylalcohol has been extracted) into a vigorously stirred mixture of 40 mL of concentratedhydrochloric acid, 40 mL of water, and 40-50 g of chipped ice. Test the mixture with indicatorpaper to make sure that it is strongly acidic. Collect the benzoic acid with suction and wash itonce with cold water. Crystallize the product from hot water, collect the crystals, and allowthem to dry thoroughly. The yield is about 8g.

I.R. SPECTROSCOPY
Setup Instructions
1. Remove the cover from the machine.
2. The "ON" switch is located to the rear on the right side of the machine. Switch the machineon. The spectrometer will take about a minute to warm-up. When it is ready for use it willbeep twice.
3. Running the machine involves several simple steps. The machine can scan at two rates, athree minute and a twelve minute scan. A three minute scan will give you all of the majorspectroscopic peaks, but none of the details. The twelve minute scan will give you the detailsthat the three minute scan missed. Set the machine to a three minute scan.
4. There are several kinds of chart paper that can be used for plotting the output. We use theshortest sheets. Set the machine for short paper output.
5. Before a plot can be made a pen must be placed into the slide bar located near the center ofthe machine. The pens are located on a shelf next to the spectrometer (several colors areavailable). Slide one of the pens (any color) into the pen holder on the slide bar.
6. Occasionally the edge of the chart paper becomes misaligned with the pen position. Thechart can be moved by pressing the Chart button (either the UP or the DOWN button) to movethe chart into the proper position.
The spectrometer is now ready to accept a sample.
Sample Preparation
Infrared spectroscopy can be done on either liquid or solid samples. The preparation of thesesamples differ dramatically. Follow these general guidelines.
Liquid Samples
Liquid samples are loaded into "cells". What this means is that the liquid will be sandwichedbetween two plates. Each plate has a shallow indentation in it's surface that holds a smallamount of sample. When the plates are put together the sample is trapped in this indentationproducing a thin film of sample which can be analyzed by the spectrophotometer.
The process is very simple. Locate the cell and it's holder. It should be found in a small whitebox on a shelf adjacent to the machine. The cell holder is a round piece of white plastic with ahole in the center and should be found together with small piece of protective cloth whichcontains the cells themselves. The cells are small round disks of what appear to be plastic butthey are really made of solid AgCl so be careful with them.

You will notice that the cell holder unscrews. Unscrew the cell holder and inside you will seea black rubber "O" ring. Take one of the two cells and place it, shallow side up, on top of the"O" ring. Place two or three drops of sample onto the cell surface and quick place the otherhalf cell and place it on top (shallow side down). Now screw the top of the cell holder backonto the cell. The cell should be snug but not overly tight. Overtightening can warp or evenbreak the AgCl cells. Please be careful.
Now that the sample is in the cell you are ready to mount the cell into the spectrometer andtake a spectrum of the sample.
Solid Samples
Solid samples can be prepared by mixing (actually grinding) the solid together with KBr. Youwill do this using an agate morter and pestle. It is usually wise to use about two to three timesmore KBr than the amount of solid sample (eye-ball it). Use very small amounts of each, 1gram of KBr and 0.3 grams of solid are more than enough (usually). After the sample hasbeen well mixed place a small amount in the KBr wafer press and make a thin, nearlytransparent wafer of this mixture. Small cracks in the sample are alright. Mount the pellet inthe IR and run the sample.
Problem
Write equations for the preparation of benzaldehyde from
(a) benzene(b) toluene(c) benzoic acid
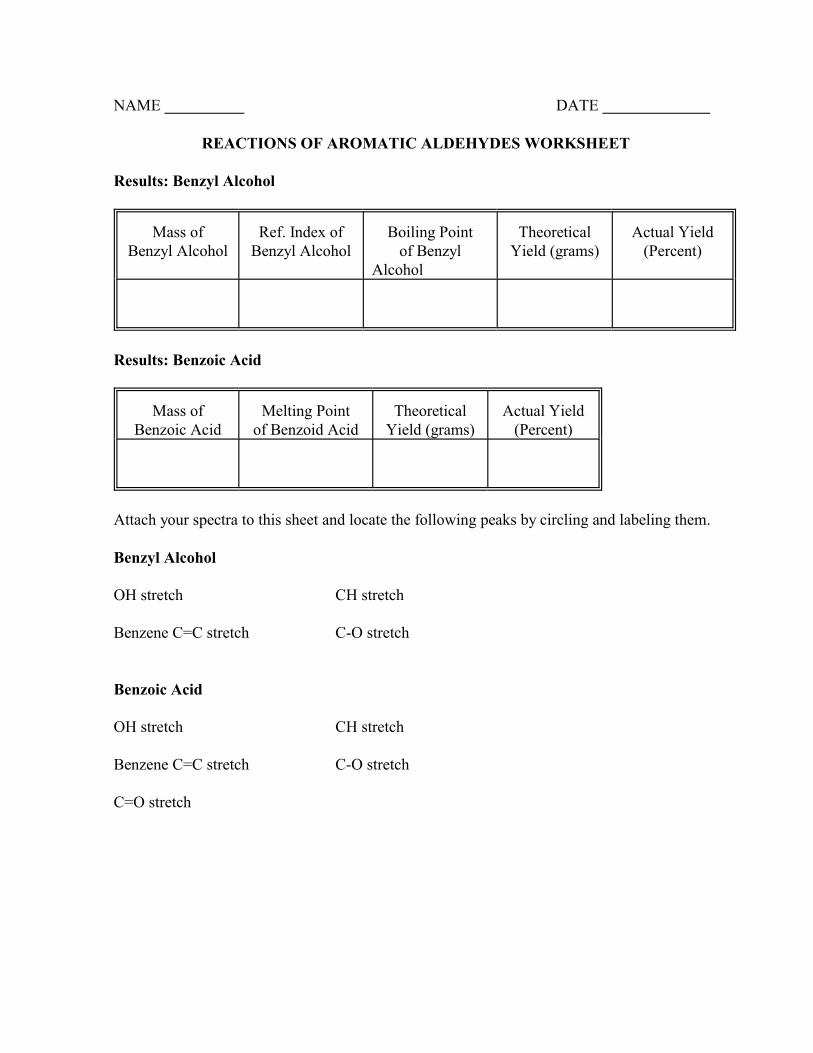
NAME DATE
REACTIONS OF AROMATIC ALDEHYDES WORKSHEET
Results: Benzyl Alcohol
Mass ofBenzyl Alcohol
Ref. Index ofBenzyl Alcohol
Boiling Pointof Benzyl
Alcohol
TheoreticalYield (grams)
Actual Yield(Percent)
Results: Benzoic Acid
Mass ofBenzoic Acid
Melting Pointof Benzoid Acid
TheoreticalYield (grams)
Actual Yield(Percent)
Attach your spectra to this sheet and locate the following peaks by circling and labeling them.
Benzyl Alcohol
OH stretch CH stretch
Benzene C=C stretch C-O stretch
Benzoic Acid
OH stretch CH stretch
Benzene C=C stretch C-O stretch
C=O stretch

EXPERIMENT FOUR
Synthesis of 9,10-Dihyroanthracene-9,10-endo-á,â-succinic AnhydrideA Diels-Alder Reaction
EQUIPMENT
2.0 g Anthracene50 mL round bottom flask55 mL mixed xylenes1.0 g maleic anhydrideReflux condenserIce bathBuchner funnelSide-arm flask10 mL iced xyleneWatch glassParafilm
EXPERMENT
Place 2.0 g of anthracene in a 50 mL round bottom flask and add 25 mL of mixed xylenes. Add 1.0 g of well ground maleic anhydride, fit the flask with a reflux condenser, and heat themixture at reflux for 30 minutes. Allow the flask to cool to room temperature, then chill it inan ice bath. The product if fairly insoluble in xylene. Once crystallization begins, it cannot bereversed by heating the solution. Filter the solid using a Buchner funnel and wash the productwith 10 mL of of ice-cold mixed xylene. Dry the solid on a watch glass next to a small beakerof paraffin wax, both covered by a larger beaker. A typical yield is 2.5 g of colorless productmp 262-264EC. If the sample is tinged with yellow and has a depressed melting point,recrystallize the product from xylene (20 mL per gram of product).
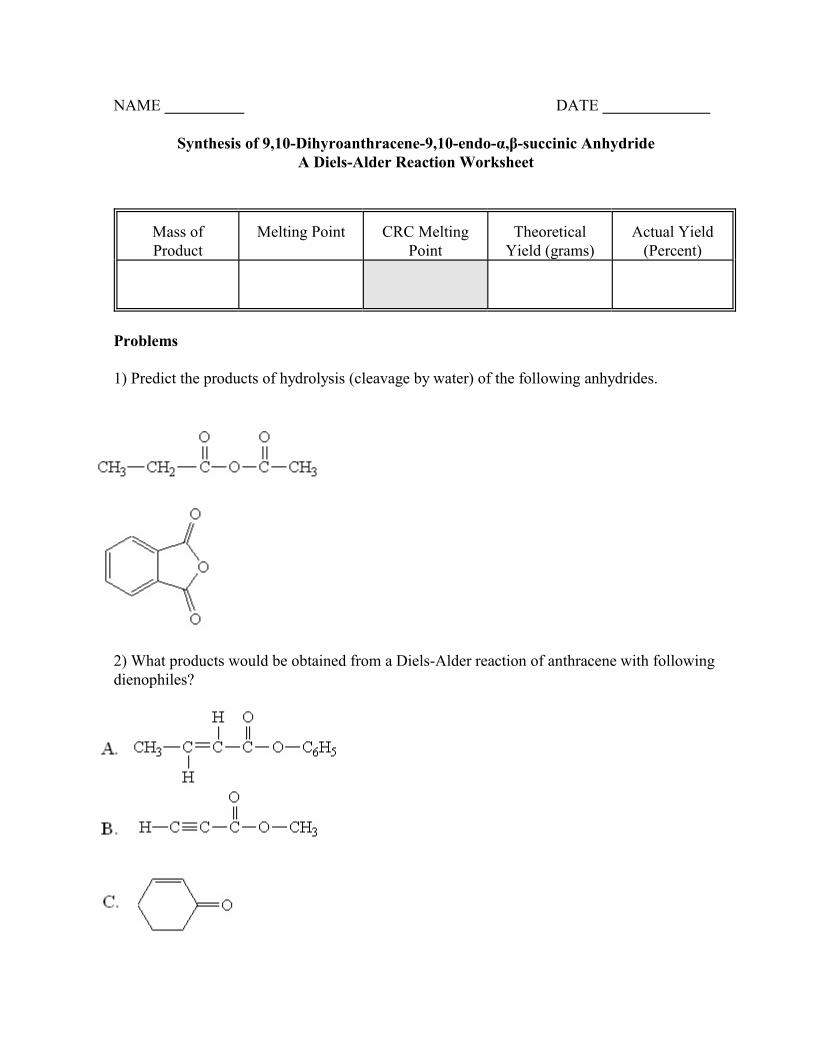
NAME DATE
Synthesis of 9,10-Dihyroanthracene-9,10-endo-á,â-succinic AnhydrideA Diels-Alder Reaction Worksheet
Mass ofProduct
Melting Point CRC MeltingPoint
TheoreticalYield (grams)
Actual Yield(Percent)
Problems
1) Predict the products of hydrolysis (cleavage by water) of the following anhydrides.
2) What products would be obtained from a Diels-Alder reaction of anthracene with followingdienophiles?

EXPERIMENT FIVE
Friedel Craft Alkylation: Formation of sec-Butylbenzene
DISCUSSION
3The reaction between bromobutane and benzene in the presence of AlCl is a classic Friedel-
3Craft alkylation reaction. The reaction begins with the removal of the bromine by the AlCl toproduce a bromobutane carbocation. This cation rearranges to place the positive charge on thesecondary carbon of butane. Thus, when benzene attacks the butane carbocation theattachment is on the secondary rather than the primary carbon. The only product formed inthis reaction is sec-butylbenzene. No butylbenzene is formed.
EQUIPMENT
250 mL round bottom flaskcondenser7.8 mL n-butyl bromide50 mL benzene3.25 g Anhydrous Aluminum chloride6 mL conc. HCl40 g icesteam bath250 mL separatory funnel
23-4 g CaClthermometer (0-200EC)
EXPERIMENT
In a dry 250 mL round-bottomed flask fitted with an upright condenser, place 11g (7.8 mL) ofn-butyl bromide and 50 mL of benzene. To provide for the acid vapors evolved during thereaction do the experiment in the hood.
Add 0.025 mole (3.25g) of pulverized anhydrous aluminum chloride and allow the reaction toproceed in the cold, shaking the flask occasionally on a steam bath, and finally reflux themixture on a steam bath for about an hour. Cool the reaction mixture and pour it with stirringinto a mixture of 40 g ice, 25 g water, and 6 mL of concentrated HCl. Stir thoroughly todissolve the excess aluminum compounds and transfer the mixture to a large separatoryfunnel. Separate the benzene layer and dry it with 3-4 g anhydrous calcium chloride, and

decant the dried liquid into a 250 mL round bottom flask. Fit the flask with a fractionatingcolumn and distill the mixture. Distill very slowly but at a regular rate. Save all the samplethat distills above 130EC and put this into a 50 mL round bottom flask and refractionate,keeping the ethylbenzene fraction which boils from 173-178EC.

NAME DATE
sec-Butyl Benzene Worksheet
Mass ofProduct
Boiling Point CRC BoilingPoint
TheoreticalYield (grams)
Actual Yield(Percent)
Questions
1) Why is such a large excess of benzene used in this experiment?
2) What products would be formed by the reaction of the following alkenes with benzene, inthe presence of aluminum chloride: ethylene? isobutylene? cyclohexene?
3) Explain why n-propyl bromide reacts with benzene in the presence of aluminum chloride toform mainly isopropylbenzene. How could you make propylbenzene?
4) Write the reactions for the formation of;
diphenylmethane
triphenylmethane
p-chloroethylbenzene

EXPERIMENT SIX
SODIUM BOROHYDRIDE REDUCTION OF ACETOPHENONE
DISCUSSION
The reduction of an aldehyde or ketone with sodium borohydride is straight forward andusually affords a high yield of the alcohol. The usual procedure (and the one employed in thisexperiment) involves dissolving the borohydride in 95% ethanol and adding the carbonylcompound to this solution. To ensure complete reaction, an excess of sodium borohydride isused.
The reaction between sodium borohydride and acetophenone is exothermic. Therefore, it isimportant to add the acetophenone drop-wise and to control the reaction temperature with anice bath. After the reaction has been completed, the excess borohydride and theethoxyborohydrides are destroyed with aqueous acid. Because hydrogen gas is evolved, thistreatment with acid must be carried out in a fume hood or a very well-ventilated room.
Because ethanol, the reaction solvent, is water-soluble, a clean separation of organic andinorganic products cannot be achieved by a simple extraction with water and diethyl ether atthis point. (Too much product would be lost in the aqueous ethanol layer.) To circumvent thisproblem, the first step in the work-up is to boil off much of the ethanol. In a larger-scalereaction, the ethanol would be distilled and collected. In a small-scale reaction such as in thisexperiment, the ethanol can be boiled away in the fume hood. When most of the ethanol hasbeen removed, the product 1-phenylethanol oils out.
Water and diethyl ether are then added to the residue for the extraction of the organiccompounds from the inorganic salts. The ether extract is dried with either sodium sulfate ormagnesium sulfate. The crude product is obtained by distilling the ether.
Because of its high boiling point, 1-phenylethanol cannot be distilled at atmospheric pressure.Although it could be vacuum-distilled, distillation could not separate it from unreacted startingmaterial (if any) because the two compounds have boiling points only 1E apart. (An infraredspectrum can be used to determine if any ketone is present in the product.)

EQUIPMENT
150-mL beakerdistillation apparatusdropperthree 50-mL Erlenmeyer flasks10-mL and 50-mL graduated cylindershot plate or steam bath with magnetic stirrer (optional)ice bathtwo 50-mL round-bottom flasks125-mL separatory funnelthermometer
CHEMICALS
acetophenone, 12.0 g
2 4anhydrous magnesium sulfate (or Na SO ) (1 g)diethyl ether, 40 mL95% ethanol, 30 mL3M hydrochloric acid, 10 mLsodium borohydride, 1.5 g
TIME REQUIRED: about 4 hours
STOPPING POINTS: after the acetophenone has been added; afterthe excess ethanol has been boiled off ; while the ether solution is drying
>>>> SAFETY NOTE: Sodium borohydride is caustic. Do not let it come into contact withyour skin. If accidental contact should occur, wash immediately and thoroughly.
PROCEDURE:
Place 1.5 g of sodium borohydride (see Safety Note) in a 150-mL beaker. Add 30 mL of 95%ethanol and stir until the solid is dissolved (see Experimental Note). Weigh 12.0 g ofacetophenone into a 50-mL Erlenmeyer flask, and prepare an ice bath.
Add the acetophenone dropwise to the borohydride solution while stirring the mixturecontinuously, preferably with a magnetic stirrer. Keep the temperature of the reaction mixturebetween 30-50EC by controlling the rate of addition and by cooling the beaker in the ice bathas necessary. As the acetophenone is added, a white precipitate forms. The addition shouldtake about 45 minutes. After the addition is complete, allow the reaction mixture to stand atroom temperature for 15 minutes with occasional stirring (or continuous stirring if you areusing a magnetic stirrer).
(Stopping Point)

In the fume hood, add about 10 mL of 3M HCl to the reaction mixture. After the reaction hassubsided, heat the mixture to boiling on a hot plate or steam bath in the fume hood until themixture separates into two layers.
(Stopping Point)
Cool the reaction mixture in an ice bath, then transfer it to a separatory funnel. Wash theresidual material in the beaker into the separatory funnel with 20 mL of diethyl ether(flammable). If the inorganic salts precipitate, add 20-40 mL of water, as necessary, todissolve them. Extract the aqueous layer with this ether, then with a second 20-mL portion ofether; combine the ether extracts; wash them with an equal volume of water; and dry themwith anhydrous magnesium sulfate or sodium sulfate.
(Stopping Point)
Decant or filter the dried solution into a tared flask, and distil the ether slowly, using a heatingmantle or steam bath and an efficient condenser. Do not overheat the flask. You are notkeeping the ether that is being removed, keep what is left in the tared flask. A typical crudeyield is 10 g (82%). Measure the refractive index, and run the infrared spectrum (thin film).From these two pieces of data, estimate the purity of the crude product.
EXPERIMENTAL NOTE
Fresh sodium borohydride dissolves in 95% ethanol, but partially hydrolyzed sodiumborohydride will not all dissolve. This should not affect the experiment because an excess ofborohydride is used. (If your borohydride is extremely poor quality, your instructor maysuggest that you use a greater excess than is called for.)

Name Date
SODIUM BOROHYDRIDE REDUCTION WORKSHEET
Mass ofProduct
Ref. Index CRC Ref.Index
TheoreticalYield (grams)
Actual Yield(Percent)
Attach the I.R. Spectrum of the Product
Locate the position of the following peaks in your spectrum;
-OH peak
-phenyl peak (benzene stretch)
-CH stretch
-C-OH stretch
PROBLEMS
4 2 3 21) Write an equation for the hydrolysis of NaBH . The products should be NaH BO and Hgas.
2) Under what circumstances would you expect to find unreacted acetophenone in the productin this experiment?
3) Which compound would you expect to undergo borohydride reduction more rapidly?Explain.
3 2 3 2 2 3(a) CH CH CHO or (b) CH CH COCH CH

Figure 10: Grignard apparatus
EXPERIMENT SEVEN
SYNTHESIS OF 2-METHYL-2-HEXANOL:A GRIGNARD REACTION
DISCUSSION
A standard Grignard synthesis is carried out in three steps: (1) preparation of RMgX; (2) the reactionof RMgX with the carbonyl compound or other reactant; and (3) the acidic hydrolysis. The first twosteps (and often all three steps) are generally carried out in the same reaction vessel. The intermediateproducts (the Grignard reagent,and the alkoxide) are rarely isolated.
PREPARATION OF THE GRIGNARD REAGENT (STEP 1)
In this experiment, the Grignard reagent is prepared by slowly adding a solution of 1-bromobutane inanhydrous diethyl ether (not solvent ether, which is wet)to Mg turnings. Unfortunately, the reaction leading to theformation of a Grignard reagent is often difficult toinitiate. Difficulties can usually be traced to contaminants,primarily water. Therefore, scrupulous care must be takento dry all glassware and to use only dry reagents andsolvents. The techniques used to start the reaction arediscussed in Experimental Note 3.
Once started, the formation of a Grignard reagent isexothermic; therefore, excess 1-bromobutane should notbe added to initiate the reaction. If a large excess of 1-bromobutane is present in the reaction vessel, the reactionmay be difficult to control. Once the reaction has begun,the 1-bromobutane is added at a rate that will maintain agentle reflux of the ether in the reaction flask.
At the end of the reaction, some of the magnesium mayremain unconsumed. The reason for this is that some 1-bromobutane is destroyed by undergoing a couplingreaction with the Grignard reagent to yield octane. Otherthan providing a mechanical inconvenience in theextraction steps, the residual magnesium metal does notinterfere with the remainder of the experiment.
An ether solution of a Grignard reagent has a translucentgray-to-black tint. The color arises from impurities in themagnesium metal, rather than from the Grignard reagentitself. Once formed, the Grignard reagent must be carriedon to Step 2 (the reaction with acetone) immediately. It cannot be saved until the next laboratoryperiod because it reacts with oxygen and moisture from the air.
REACTION WITH ACETONE (STEP 2)

The reaction of n-butylmagnesium bromide with acetone is extremely vigorous. The acetone must beadded very slowly; otherwise, the reaction mixture will boil over. The product is a magnesiumalkoxide of an alcohol and thus insoluble in diethyl ether. This alkoxide sometimes forms a crustyprecipitate that must be broken up by swirling the flask so that the unreacted acetone can becomemixed with the Grignard reagent.
After the reaction of acetone and the Grignard reagent is completed, it is no longer necessary toprotect the reaction mixture from air or moisture. This mixture can be stored until the next laboratoryperiod.
HYDROLYSIS OF THE ALKOXIDE (STEP 3)
The alkoxide product of the Grignard reaction is converted to 2-methyl-2-hexanol by treatment with
4aqueous NH Cl instead of with a dilute mineral acid. The reason is that the final product is a tertiary
3alcohol (R COH) and is easily dehydrated to an alkene by a strong acid. When the magnesium
4 4alkoxide is poured into aqueous NH Cl, the alkoxide ion (a strong base) reacts with water or NH to+
extract a proton. Water alone is not used as a hydrolyzing agent for two reasons. First, the producthydroxide ion is only a slightly weaker base than the alkoxide ion. The addition of an acid results in amore favorable equilibrium.
Second, in alkaline solution, the magnesium ions are converted to a gelatinous precipitate of
2Mg(OH) , which is difficult to remove from the product. In a neutral or acidic medium, themagnesium ions remain in solution. The product alcohol is extracted from the aqueous layer withdiethyl ether (solvent grade). The aqueous layer, which contains the magnesium salts, is discarded.The ether solution is washed with sodium carbonate solution to ensure alkalinity prior to distillation.(Any acid remaining in the ether layer would cause dehydration of the alcohol during the distillation.)
2Because diethyl ether can dissolve a considerable amount of water (1.2 g H O in 100 g of ether), theether extract is partially dried by extraction with saturated NaCl solution before an inorganic dryingagent is used. The final drying is accomplished by allowing the ether solution to stand over anhydrous
4MgSO . The bulk of the ether is removed by simple distillation. Before the alcohol is distilled, theresidue is transferred to a smaller distillation flask; otherwise, a considerable amount of productwould be lost as vapor filling the large flask.
EQUIPMENT:
400-mL beakercalcium chloride drying tubeClaisen headcondenserdisposable pipet25-mL or 50-mL tared distillation receiving flask125-mL dropping funnel50-mL, 125-mL, and two 250-mL Erlenmeyer flasksheating mantle and rheostat (or steam bath)ice bath50-mL (or 100-mL) and 250-mL round-bottom flasks250-mL or 400-mL separatory funnelsimple distillation apparatusstirring rodwarm water bath

CHEMICALS:ammonium chloride, 25 ganhydrous acetone, 5.8 ganhydrous diethyl ether, 50 mLanhydrous magnesium sulfate or potassium carbonate, 5 g10% aqueous sodium carbonate, 25 mL1-bromobutane, 13.7 gdiethyl ether (for extraction), about 75 mLmagnesium turnings, 2.4 gsaturated aqueous NaC1, 25 mL
TIME REQUIRED: 3½ hours, plus 1½ hours for the distillation. The Grignard reagent must be usedimmediately after its formation. Therefore, enough time should be allotted to carry out Steps 1 and 2(about 1 hour each) in a single laboratory period. IMPORTANT: If anhydrous acetone is not
4available, then reagent acetone must be dried with anhydrous MgSO (5 g for each 50 mL) for at least24 hours prior to the Grignard reaction (see Experimental Note 4).
STOPPING POINTS: after the acetone has been added to the Grignard reagent (and reaction hassubsided); when the ether extracts are drying
>>SAFETY NOTE I Diethyl ether (bp 34.6 is used as a reaction solvent and as an extractionE)
solvent. It is very volatile and extremely flammable. There must be no flames in the laboratory. Anefficient condenser must be used for both the reaction and the distillation. The distillation should becarried out slowly to minimize ether vapors escaping into the room.
>> SAFETY NOTE 2 Because the formation of a Grignard reagent and a Grignard reaction are bothexothermic, there is the danger of a runaway reaction. Keep an ice bath handy at all times in case thereaction flask needs rapid cooling.
>>SAFETY NOTE 3 The heavy caked precipitate that sometimes forms makes thorough mixing ofacetone and the Grignard reagent difficult and can allow unreacted acetone to accumulate in one spot.When this acetone eventually contacts the Grignard reagent, the reaction may become impossible tocontrol. Therefore, swirl the reaction flask gently, but frequently and thoroughly.
PROCEDURE
Step 1, Preparation of n-Butylmagnesium Bromide. Heat a 250-mL round-bottom flask, a Claisenhead, a condenser, and a dropping funnel in a drying oven until they are hot to the hand. Thenassemble them as shown in Figure 10. Fit the reflux condenser with a drying tube containinganhydrous calcium chloride (see Experimental Note 1). To prevent atmospheric moisture fromcondensing inside the condenser, do not turn on the condenser water until the reaction is initiated.
Place 2.4 g of oven-dried magnesium turnings in the round-bottom flask. To the dropping funnel, adda well-mixed solution of 13.7 g of 1-bromobutane and 50 mL of anhydrous diethyl ether. (CAUTION:flammable! See Safety Note 1; see also Experimental Note 2.)
To initiate the reaction, add 10-15 mL of the ether solution from the dropping funnel to the reactionflask. Loosen the clamp holding the round-bottom flask and gently swirl the flask to mix the contents.When the Grignard reagent begins to form, the ether solution will become cloudy and then begin toboil. Turn on the condenser water at this time. If your Grignard reagent does not start to form within

5-10 minutes, follow the procedure outlined in Experimental Note 3. Because the reaction isexothermic once initiated, do not add an excessive amount of 1-bromobutane to the magnesium at anyone time (see Safety Note 2).
After the reaction has been initiated, add the remaining ether solution dropwise at a rate thatmaintains a gentle reflux. After all the solution has been added, close the stopcock of the droppingfunnel and heat the mixture at a gentle reflux for 15 minutes in a warm (50EC) water bath. As themagnesium is consumed, the mixture will become darker colored. At the end of the reflux period,proceed immediately to Step 2.
Step 2, Reaction of n-Butylmagnesium Bromide with Acetone. Chill the flask containing theGrignard reagent with an ice bath. Pour 5.8 g of anhydrous acetone (not ordinary reagent grade) in thedropping funnel, and add it a few drops at a time to the reaction mixture. After each addition, loosenthe clamps to the reaction assembly and gently swirl the reaction flask. (CAUTION: See Safety Note3.)
When the addition of the acetone is completed, allow the reaction mixture to stand at roomtemperature for 30 minutes or longer before going on to the hydrolysis step. If the mixture will bestanding for more than an hour, stopper the reaction flask with a glass stopper or a cork (not a rubberstopper) to prevent the solvent from evaporating.
Step 3, Hydrolysis and Purification. Prepare 100 mL of 25% aqueous ammonium chloride. Mix 75mL of this solution with 50 g of crushed ice in a 400-mL beaker. Transfer the remaining 25 mL of theammonium chloride solution to a 50-mL Erlenmeyer flask, and chill it in an ice bath. Slowly pour theGrignard reaction mixture into the ice mixture in the beaker, stirring vigorously (see Experimental
4Note 4). Rinse the reaction vessel into the ice mixture, first with the 25 mL of chilled NH C1solution, then with 25 mL of solvent ether. Transfer the contents of the beaker to a 400-mLseparatory funnel. (If a 250-mL separatory funnel must be used, divide the mixture into two batchesand process each separately.) Add solvent ether to bring the volume of the upper ether layer to about50 mL, shake the funnel, and allow the layers to separate. Drain the lower aqueous layer into a 250-mL Erlenmeyer flask or a second separatory funnel and drain the ether layer into a separateErlenmeyer flask. (Draining instead of pouring minimizes evaporation of the ether.) Return theaqueous layer to the separatory funnel, add 25 mL of fresh solvent ether, and shake the mixture again.Drain and discard the lower, aqueous layer.
Add the first 50-mL ether extract to the second extract in the separatory funnel. Rinse the flask thatcontained the original extract into the separatory funnel with a few mL of ether. Wash the combinedether extracts by shaking them with 25 mL of water, then with 25 mL of 10% sodium carbonatesolution. Finally, wash the ether solution with 20-25 mL of saturated sodium chloride solution.
Pour the ether solution into a clean, dry 250-mL Edenmeyer flask, add 5 g of anhydrous magnesiumsulfate or potassium carbonate, cork the flask tightly, and allow it to stand for at least one hour(overnight is better).
Carefully decant (or filter through a small plug of glass wool) the dried solution into a 250-mL round-bottom flask for distillation of the ether. Add a few boiling chips and slowly distil the bulk of theether (bp 34.6EC), using a steam bath or heating mantle and an efficient condenser. Stop thedistillation when there is about 25 mL remaining in the distillation flask. Cool the flask and, using adisposable pipet, transfer the residue to a 50-mL round-bottom flask for distillation of the product.

Wash the last traces of crude product from the 250-mL flask into the 50-mL flask with a few mL ofanhydrous diethyl ether.
Add fresh boiling chips and distil the product, collecting the fraction boiling at 135-143EC in a taredreceiver. A typical yield is 5.0 g (43%). (The yield may vary considerably, depending on the degree ofdryness of the anhydrous ether used in Step 1.) Determine the refractive index of the product. Placethe distilled product in a correctly labeled vial, and hand it in to your instructor.
EXPERIMENTAL NOTES
1) The purpose of the drying tube is to prevent atmospheric moisture from entering the reaction vesselvia the condenser and yet allow the reaction vessel to be open to the atmosphere so that gas pressuredoes not build up. There are two types of drying tubes: curved (better) and straight (less expensive). Astraight drying tube must not be connected directly to the top of the condenser because the dessicantcan liquefy and drain into the condenser. Connect the straight tube to the condenser by a short lengthof heavy-walled rubber tubing, as shown in Figure 10. In either type of drying tube, the dessicant isheld in place with loose plugs of glass wool. A one-hole rubber stopper may be used as a secondaryplug at the wide end of the drying tube.
2) Solvent ether contains an appreciable amount of water (up to 1-2%) and is totally unsuitable as aGrignard solvent. Anesthesia ether contains ethanol, which makes it also unsuitable. Commercialanhydrous ether is adequate only if a freshly opened can is used. Anhydrous ether must not be leftopen to the air because it absorbs both oxygen and moisture. (Oxygen and ethers yield peroxides,which can explode if the ether is distilled. Absorbed moisture will ruin a Grignard reagent.) Yourinstructor will probably provide anhydrous ether for this experiment. In many laboratories, storeroompersonnel prepare anhydrous ether by passing solvent ether through a column containing molecular
2sieves, which are adsorbents with pores that trap molecules of a certain size (in this case, H Omolecules). Another procedure for the preparation of anhydrous ether from solvent ether and aprocedure for the testing of peroxides in anhydrous ether follow. If you find it necessary to prepareyour own anhydrous ether, allot an additional laboratory period.
Preparation of Anhydrous Diethyl Ether. With cooling, mix a 2: ! ratio of solvent ether and
2 4conc. H SO in a large round-bottom flask, and distill about two-thirds of the ether. (Do notdistill all the ether.) Any water and ethanol contaminating the ether will remain with thesulfuric acid. To discard the residue, pour it onto cracked ice, allow the residual ether toevaporate in the hood, then dilute the aqueous acid with water and pour it down the hood drainwith additional water. Add freshly prepared sodium wire or ribbon to the distilled ether, thenallow the ether to stand at least overnight in the fume hood with the fan on. Stopper thecontainer with a very loose-fitting cork or drying tube to allow the hydrogen gas to escape.
Sodium wire or ribbon is prepared by pressing sodium metal through a die, using a press. If asodium press is not available, the ether can be dried with finely diced sodium; however, diced
2sodium is inferior to wire or ribbon. Another method is to add a few grams of CaCl to theether and allow the mixture to stand until hydrogen has ceased to be evolved. The dried ethercan then be decanted or (better) pipetted, using a rubber bulb, as needed. Commercialanhydrous ether can be further dried with sodium wire without the sulfuric acid purificationstep.

Peroxide Test. Shake 5 mL of ether with a solution of 1 mg of sodium dichromate and one
2 4drop of dilute H SO in 1 mL of water in a corked test tube. If the ether layer turns blue (fromperchromate ion), peroxides are present and must be removed.
Peroxide Removal. Shake the peroxide-contaminated ether with 5% aqueous ferrous sulfate
4 2 4.(FeSO ) solution acidified with H SO The iron(II) ions are oxidized with concurrent
2 3reduction of the peroxide. Aqueous sodium sulfite (Na SO ) can be substituted for the ferroussulfate solution.
3) The most common cause of failure of initiation of the reaction leading to the Grignard reagent ismoisture (in the apparatus, in the ether, or on the magnesium turnings). In addition, in a humidatmosphere, water will collect on the sides of a cold condenser. If the initial cloudiness becomes a
2white precipitate, then the Mg is being converted to Mg(OH) by the water, and not to RMgX. Ifexcessive moisture is present, it is best to begin anew with dry equipment and reagents.
Sometimes, Grignard reagents are reluctant to form because of a magnesium oxide coating on themetal turnings. The following procedure can often overcome this difficulty. First, warm the reactionflask with a pan of warm water (about 50-60EC). This warming will cause the ether to boil (not a signof initiation, in this case). Remove the warm water bath and watch for the signs of initiation(spontaneous boiling of the ether). This warming may be repeated if initiation does not occur.
If repeated warming does not initiate Grignard-reagent formation, add an additional 5 mL of the 1-bromobutane solution from the dropping funnel and warm the flask again.
As a last resort, another reagent may be added to activate the surface of the magnesium and/or
2indirectly complex with any water present. A number of reagents are useful: a crystal of I , a few
2drops of Br , 1.0 mL of iodomethane (methyl iodide) or dibromomethane (methylene bromide). (Onlyone, not all, of these should be added.) Add the reagent directly to the reaction mixture withoutswirling, then warm the flask in the water bath.
The two inorganic halogen compounds function by reacting with the magnesium to yield ananhydrous magnesium halide, which complexes with any water present. Iodomethane anddibromomethane are reactive organohalogen compounds that react with the magnesium in slightlydifferent ways. For example, iodomethane first forms a Grignard reagent (even when a less reactivealkyl halide does not react), which then reacts with any water present and thus removes it fromsolution.
4) The reaction mixture may contain small pieces of unreacted magnesium metal. If possible, avoidtransferring these bits of metal to the ice mixture. However, a tiny piece of magnesium that cannot beremoved easily from the ice mixture will do no harm.

PROBLEMS
1) Write equations for the three standard steps in a Grignard synthesis in which the principal reactantsare cyclohexanone and ethylmagnesium bromide.
2) A student oven-dries the glassware needed for a Grignard reaction, then stores them in a lockeruntil the next laboratory period. Will the glassware still be dry when the Grignard reaction is begun?Explain.
3) Suggest a reason for using magnesium turnings instead of magnesium powder or chunks in aGrignard reaction.
4) In which of the following steps in a Grignard synthesis is anhydrous ether (instead of solventether) necessary? Explain.
(a) Preparation of the Grignard reagent.
(b) Addition of an ether solution of a ketone (instead of pure ketone, as in this experiment).
(c) Extracting the product from the hydrolysis mixture.
(d) Washing the dried product into a distillation flask.
5) Are diethyl ether vapors lighter or heavier than air? What are the safety implications of youranswer?

6) As an alternative to drying tubes to protect a Grignard reaction, some chemists carry out thesereactions under a dry nitrogen atmosphere. Which of the following techniques could also be used tokeep a Grignard reagent dry? Explain.
(a) a dry argon atmosphere(b) a dry carbon dioxide atmosphere(c) a gentle stream of dried air passed over the surface of the mixture.