la filosofia del qbd applicata alla bioanalitica:...
TRANSCRIPT

La filosofia del QbD applicata alla bioanalitica: sviluppo di un
metodo per il monitoraggio terapeutico di elvitegravir
Unità di Farmacologia Clinica,"Luigi Sacco" University HospitalUniversità degli Studi di Milano
Sara Baldelli, Giorgio Marrubini
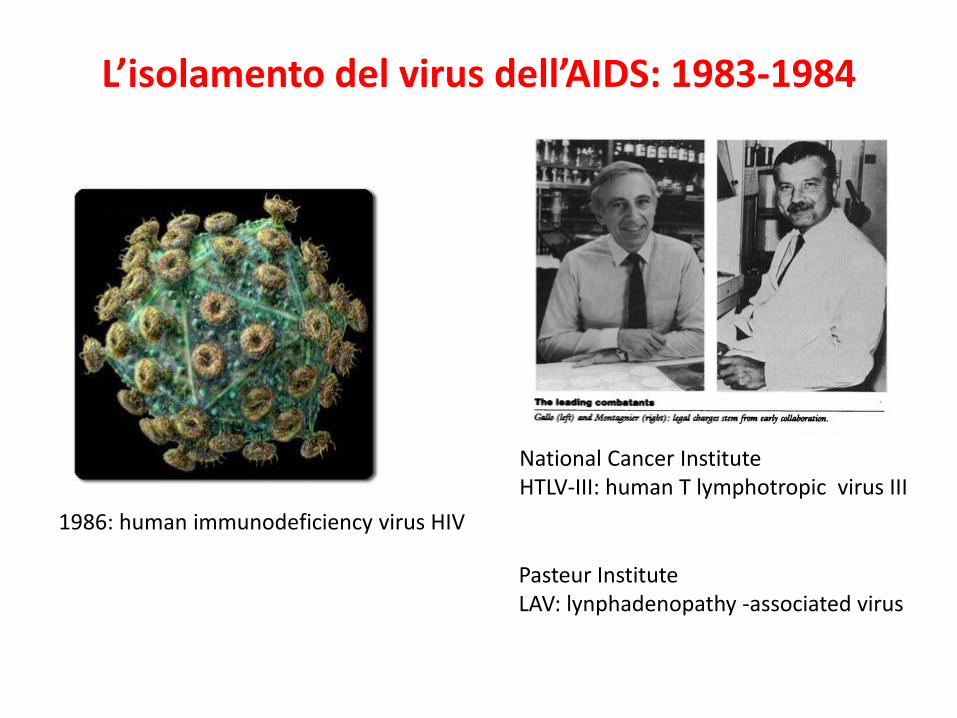
L’isolamento del virus dell’AIDS: 1983-1984
Pasteur InstituteLAV: lynphadenopathy -associated virus
National Cancer InstituteHTLV-III: human T lymphotropic virus III
1986: human immunodeficiency virus HIV
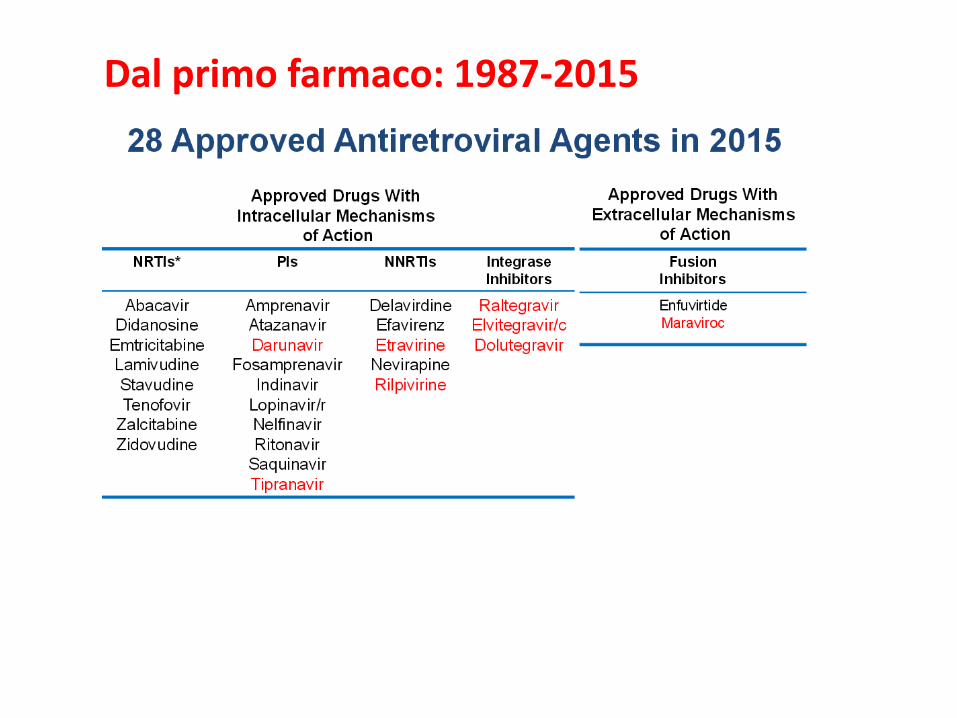
Dal primo farmaco: 1987-2015
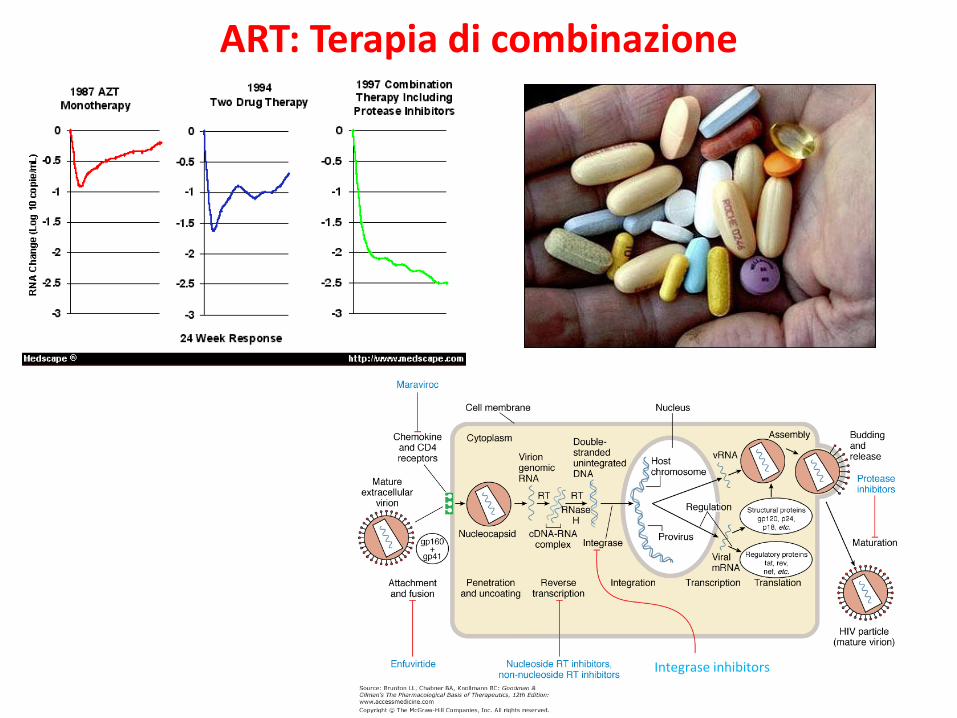
ART: Terapia di combinazione
Integrase inhibitors

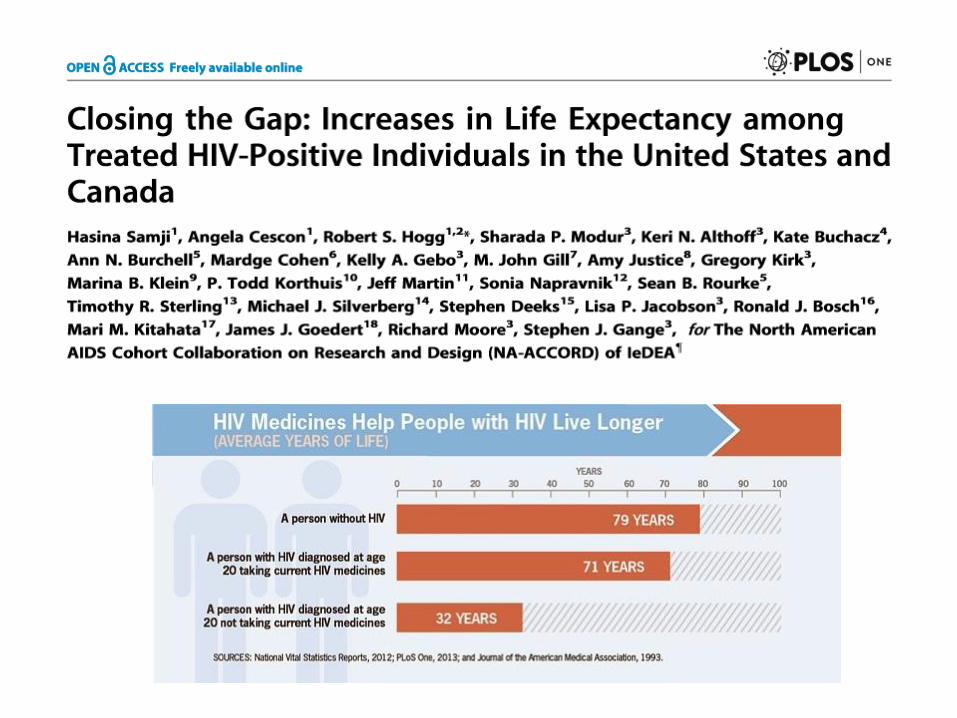
ART: aumento della sopravvivenza
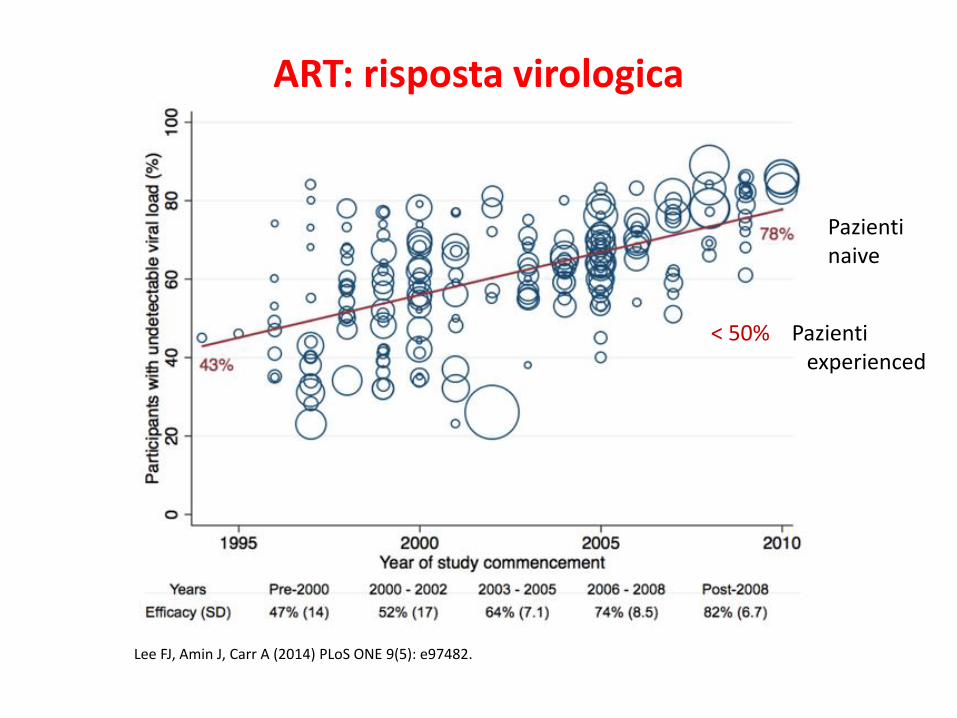
ART: risposta virologica
Lee FJ, Amin J, Carr A (2014) PLoS ONE 9(5): e97482.
Pazientinaive
< 50% Pazienti experienced
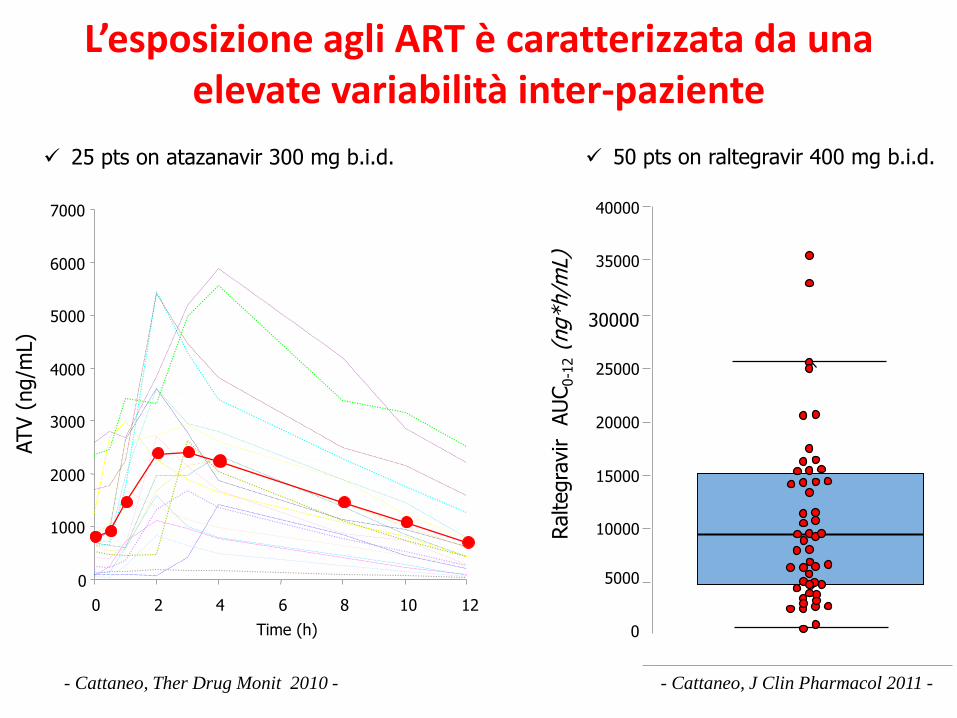
40000
35000
30000
25000
20000
15000
10000
5000
0
Raltegra
vir
AU
C0-1
2(n
g*h/m
L)
50 pts on raltegravir 400 mg b.i.d.
- Cattaneo, J Clin Pharmacol 2011 -
L’esposizione agli ART è caratterizzata da unaelevate variabilità inter-paziente
- Cattaneo, Ther Drug Monit 2010 -
0
1000
2000
3000
4000
5000
6000
7000
0 2 4 6 8 10 12
Time (h)
ATV (
ng/m
L)
25 pts on atazanavir 300 mg b.i.d.
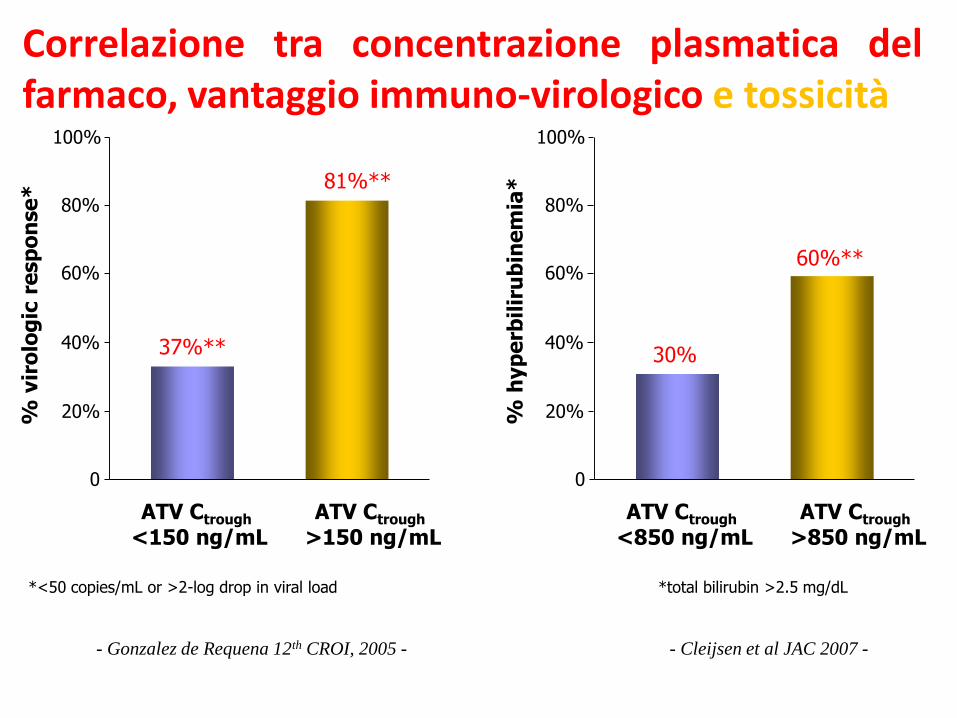
Correlazione tra concentrazione plasmatica delfarmaco, vantaggio immuno-virologico e tossicità
- Cleijsen et al JAC 2007 -
0
60%**
30%
ATV Ctrough
<850 ng/mL%
hyp
erb
ilir
ub
ine
mia
*ATV Ctrough
>850 ng/mL
- Gonzalez de Requena 12th CROI, 2005 -
0
20%
100%
37%**
81%**
ATV Ctrough
<150 ng/mL
% v
iro
log
ic r
esp
on
se
*
ATV Ctrough
>150 ng/mL
40%
60%
80%
*<50 copies/mL or >2-log drop in viral load
20%
100%
40%
60%
80%
*total bilirubin >2.5 mg/dL
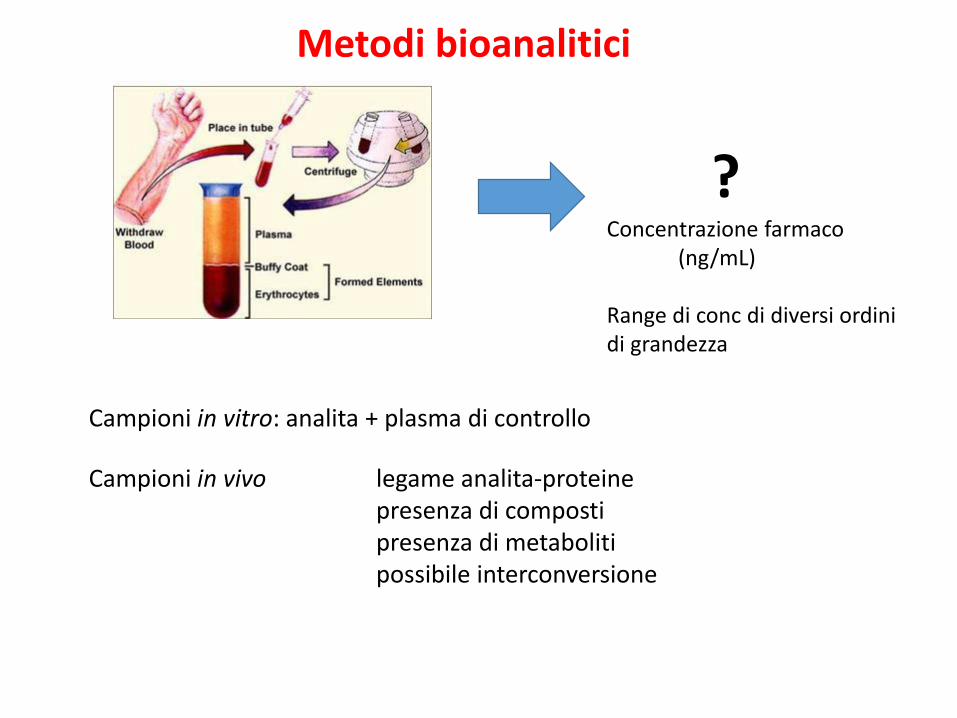
?Concentrazione farmaco
(ng/mL)
Range di conc di diversi ordini di grandezza
Campioni in vitro: analita + plasma di controllo
Campioni in vivo legame analita-proteinepresenza di composti presenza di metabolitipossibile interconversione
Metodi bioanalitici

Massima sensibilità
Nessun/Minimo trattamento campione
Corse cromatografiche con tempi di ritenzione molto brevi
Multipli analiti analizzati contemporaneamente
La spettrometria di massa è considerata il goldstandard per la determinazioni di farmaci e deimetaboliti

Linee guida validazione metodi bioanalitici
Usare pool di plasma diversiStudiare l’effetto della matriceReincurrent sample reanalysis



Applicazione dei principi del QbD nello sviluppo diun metodo per il monitoraggio terapeutico dielvitegravir
Cos'è il Quality by design?
1. «Un approccio sistematico allo sviluppo»
2. «si attua prestabilendo obiettivi precisi»
3. Pone in rilievo primario la comprensione e l’approfondimento del processo e del prodotto
4. …e il controllo
5. fondato sulla migliore conoscenza scientifica
6. …e la stima del rischio per la qualità
Adattato da G. Boccardi, D. Fraioli, G. Marrubini, Il Quality by
Design nello sviluppo e convalida dei metodi analitici,
presentazione al 53° Simposio AFI, Rimini 2013.
Applicazione dei principi del QbDICH Q8 (R2) per lo sviluppo e
produzione di medicinali
Applicazione dei principi del QbD nello sviluppo diun metodo per il monitoraggio terapeutico dielvitegravir
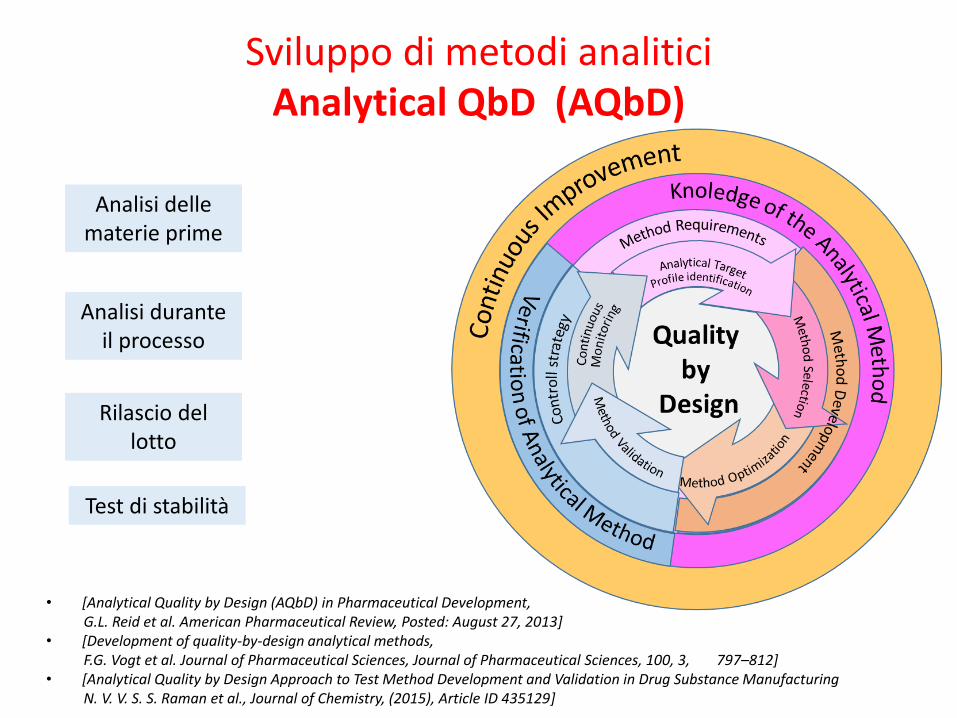
Analytical QbD (AQbD)
• [Analytical Quality by Design (AQbD) in Pharmaceutical Development, G.L. Reid et al. American Pharmaceutical Review, Posted: August 27, 2013]
• [Development of quality-by-design analytical methods, F.G. Vogt et al. Journal of Pharmaceutical Sciences, Journal of Pharmaceutical Sciences, 100, 3, 797–812]
• [Analytical Quality by Design Approach to Test Method Development and Validation in Drug Substance Manufacturing N. V. V. S. S. Raman et al., Journal of Chemistry, (2015), Article ID 435129]
Sviluppo di metodi analitici
Analisi delle materie prime
Analisi durante il processo
Rilascio del lotto
Test di stabilità

Sviluppo di metodi analitici mediante approccio AQbD
1.Obiettivo della analisi
2.Selezione della metodo
3.Analisi del rischio
4.Sviluppo del metodo
5.Strategia di controllo
6.Miglioramento continuo
Analytical Target Profile: cosa, quando e in quale matrice, misurare con quali criteri di accettabilità Critical QualityAttributes (CQAs)
Selezione della tecnica analitica appropriata. Risk assessment
Analisi dei rischi connessi ai parametri operativi del metodoCritical Process Parameters (CPPs) e della variazione delcampione. Risk assessment
[Quality by Design Approaches to Analytical Methods, Y. Tang, AAPS Meeting 2011]
Identificazione dei fattori e dello spazio/dominiosperimentale (Knowledge space /design space)
Validazione del metodo
Monitoraggio delle performance del metodo eaggiornamento, se necessario.

1. Selezione degli Analytical Target Profile (ATP)
A. Analita da quantificare ELVITEGRAVIRB. Specifiche di performance del metodo (previste dalle linee
guida)
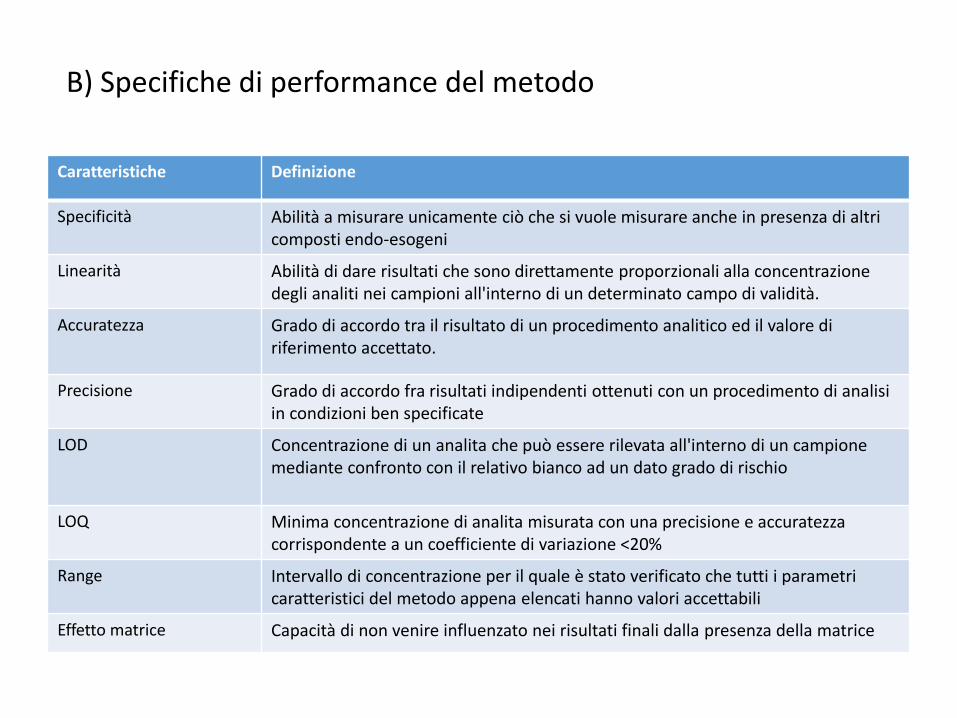
B) Specifiche di performance del metodo
Caratteristiche Definizione
Specificità Abilità a misurare unicamente ciò che si vuole misurare anche in presenza di altri composti endo-esogeni
Linearità Abilità di dare risultati che sono direttamente proporzionali alla concentrazione degli analiti nei campioni all'interno di un determinato campo di validità.
Accuratezza Grado di accordo tra il risultato di un procedimento analitico ed il valore di riferimento accettato.
Precisione Grado di accordo fra risultati indipendenti ottenuti con un procedimento di analisi in condizioni ben specificate
LOD Concentrazione di un analita che può essere rilevata all'interno di un campione mediante confronto con il relativo bianco ad un dato grado di rischio
LOQ Minima concentrazione di analita misurata con una precisione e accuratezza corrispondente a un coefficiente di variazione <20%
Range Intervallo di concentrazione per il quale è stato verificato che tutti i parametri caratteristici del metodo appena elencati hanno valori accettabili
Effetto matrice Capacità di non venire influenzato nei risultati finali dalla presenza della matrice

A. Analita da quantificare ELVITEGRAVIRB. Specifiche di performance del metodo (previste dalle linee
guida)C. La procedura deve essere in grado di quantificare l’analita
accuratamente e precisamente in campioni di plasma dipazienti in terapia con il farmaco [con una concentrazioneda 50 a 10000 ng/mL con una accuratezza e precisionecomprese in un intervallo di ± 15%; (LOQ 20%)]
1. Selezione degli Analytical Target Profile (ATP)

Elementi da considerare:
• Potenzialità di raggiungere Analytical Target Profile (ATP)• Disponibilità presso il laboratorio• Esperienza del personale nello sviluppo, impiego e
mantenimento delle metodica• Sicurezza per gli operatori• Tempo di lavoro• Specificità• Range• Costi di funzionamento
2. Selezione del metodo analitico
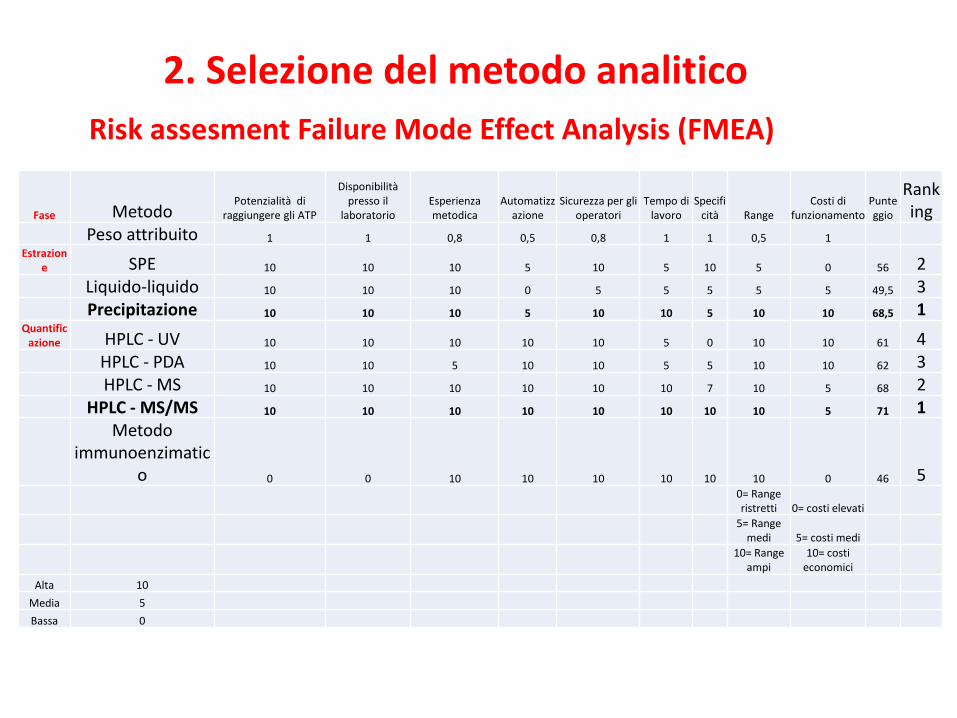
Fase MetodoPotenzialità di
raggiungere gli ATP
Disponibilità presso il
laboratorioEsperienza metodica
Automatizzazione
Sicurezza per gli operatori
Tempo di lavoro
Specificità Range
Costi di funzionamento
Punteggio
Ranking
Peso attribuito 1 1 0,8 0,5 0,8 1 1 0,5 1
Estrazione SPE 10 10 10 5 10 5 10 5 0 56 2
Liquido-liquido 10 10 10 0 5 5 5 5 5 49,5 3Precipitazione 10 10 10 5 10 10 5 10 10 68,5 1
Quantificazione HPLC - UV 10 10 10 10 10 5 0 10 10 61 4
HPLC - PDA 10 10 5 10 10 5 5 10 10 62 3HPLC - MS 10 10 10 10 10 10 7 10 5 68 2
HPLC - MS/MS 10 10 10 10 10 10 10 10 5 71 1Metodo
immunoenzimatico 0 0 10 10 10 10 10 10 0 46 5
0= Range ristretti 0= costi elevati
5= Range medi 5= costi medi
10= Range ampi
10= costi economici
Alta 10
Media 5
Bassa 0
2. Selezione del metodo analitico
Risk assesment Failure Mode Effect Analysis (FMEA)

3. Identificazione dei parametri critici del metodo analitico selezionato
Suddivisione del metodo in unità operative Per ciascuna unità individuazione delle variabili critiche Costruzione di uno schema ordinato
(Cause & Effect, Ishikawa, Fishbone diagram)
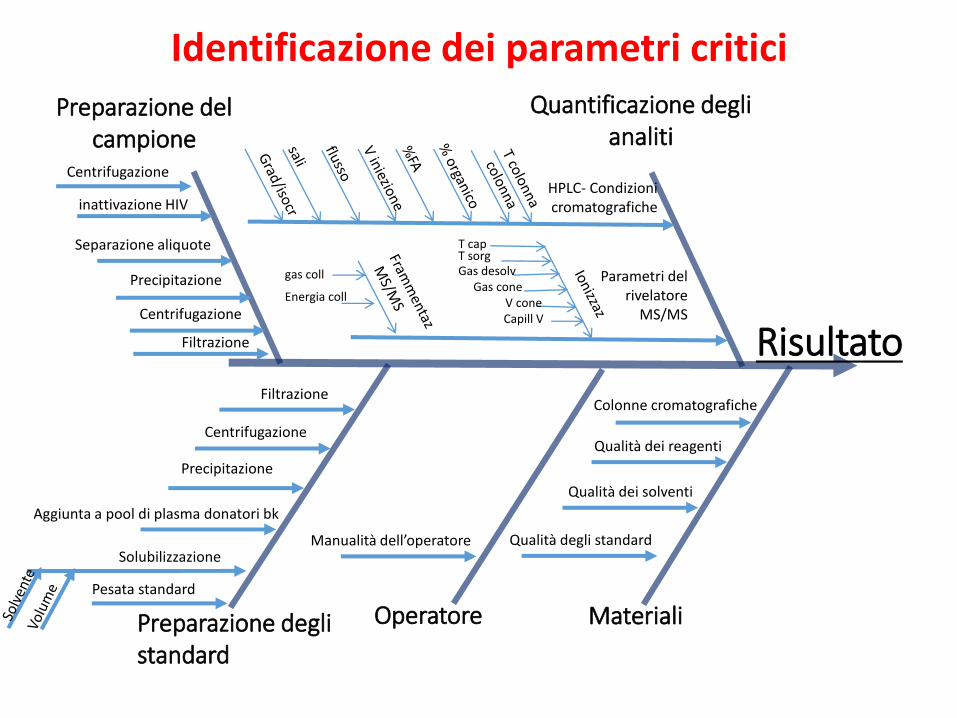
Identificazione dei parametri critici
Preparazione del campione
Preparazione degli standard
Operatore Materiali
Quantificazione degli analiti
Risultato
inattivazione HIV
Separazione aliquote
Precipitazione
Filtrazione
Centrifugazione
Precipitazione
Aggiunta a pool di plasma donatori bk
Filtrazione
Solubilizzazione
Pesata standard
Manualità dell’operatore
HPLC- Condizioni cromatografiche
Parametri del rivelatore
MS/MS
Qualità degli standard
Qualità dei solventi
Qualità dei reagenti
Colonne cromatografiche
Centrifugazione
Centrifugazione
Gas desolvGas cone
V coneCapill V
T sorgT cap
Energia coll
gas coll
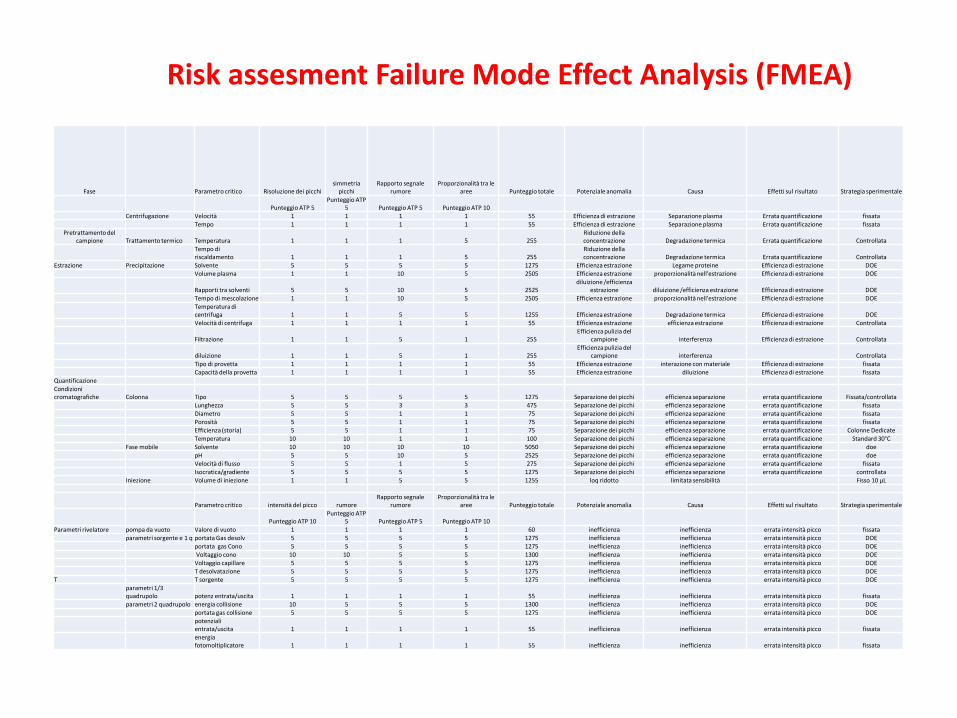
Fase Parametro critico Risoluzione dei picchisimmetria
picchiRapporto segnale
rumoreProporzionalità tra le
aree Punteggio totale Potenziale anomalia Causa Effetti sul risultato Strategia sperimentale
Punteggio ATP 5Punteggio ATP
5 Punteggio ATP 5 Punteggio ATP 10Centrifugazione Velocità 1 1 1 1 55 Efficienza di estrazione Separazione plasma Errata quantificazione fissata
Tempo 1 1 1 1 55 Efficienza di estrazione Separazione plasma Errata quantificazione fissataPretrattamento del
campione Trattamento termico Temperatura 1 1 1 5 255Riduzione della concentrazione Degradazione termica Errata quantificazione Controllata
Tempo di riscaldamento 1 1 1 5 255
Riduzione della concentrazione Degradazione termica Errata quantificazione Controllata
Estrazione Precipitazione Solvente 5 5 5 5 1275 Efficienza estrazione Legame proteine Efficienza di estrazione DOEVolume plasma 1 1 10 5 2505 Efficienza estrazione proporzionalità nell'estrazione Efficienza di estrazione DOE
Rapporti tra solventi 5 5 10 5 2525diluizione /efficienza
estrazione diluizione /efficienza estrazione Efficienza di estrazione DOETempo di mescolazione 1 1 10 5 2505 Efficienza estrazione proporzionalità nell'estrazione Efficienza di estrazione DOETemperatura di centrifuga 1 1 5 5 1255 Efficienza estrazione Degradazione termica Efficienza di estrazione DOEVelocità di centrifuga 1 1 1 1 55 Efficienza estrazione efficienza estrazione Efficienza di estrazione Controllata
Filtrazione 1 1 5 1 255Efficienza pulizia del
campione interferenza Efficienza di estrazione Controllata
diluizione 1 1 5 1 255Efficienza pulizia del
campione interferenza ControllataTipo di provetta 1 1 1 1 55 Efficienza estrazione interazione con materiale Efficienza di estrazione fissataCapacità della provetta 1 1 1 1 55 Efficienza estrazione diluizione Efficienza di estrazione fissata
QuantificazioneCondizioni cromatografiche Colonna Tipo 5 5 5 5 1275 Separazione dei picchi efficienza separazione errata quantificazione Fissata/controllata
Lunghezza 5 5 3 3 475 Separazione dei picchi efficienza separazione errata quantificazione fissataDiametro 5 5 1 1 75 Separazione dei picchi efficienza separazione errata quantificazione fissataPorosità 5 5 1 1 75 Separazione dei picchi efficienza separazione errata quantificazione fissataEfficienza (storia) 5 5 1 1 75 Separazione dei picchi efficienza separazione errata quantificazione Colonne DedicateTemperatura 10 10 1 1 100 Separazione dei picchi efficienza separazione errata quantificazione Standard 30°C
Fase mobile Solvente 10 10 10 10 5050 Separazione dei picchi efficienza separazione errata quantificazione doepH 5 5 10 5 2525 Separazione dei picchi efficienza separazione errata quantificazione doeVelocità di flusso 5 5 1 5 275 Separazione dei picchi efficienza separazione errata quantificazione fissata Isocratica/gradiente 5 5 5 5 1275 Separazione dei picchi efficienza separazione errata quantificazione controllata
Iniezione Volume di iniezione 1 1 5 5 1255 loq ridotto limitata sensibilità Fisso 10 µL
Parametro critico intensità del picco rumoreRapporto segnale
rumoreProporzionalità tra le
aree Punteggio totale Potenziale anomalia Causa Effetti sul risultato Strategia sperimentale
Punteggio ATP 10Punteggio ATP
5 Punteggio ATP 5 Punteggio ATP 10Parametri rivelatore pompa da vuoto Valore di vuoto 1 1 1 1 60 inefficienza inefficienza errata intensità picco fissata
parametri sorgente e 1 q portata Gas desolv 5 5 5 5 1275 inefficienza inefficienza errata intensità picco DOEportata gas Cono 5 5 5 5 1275 inefficienza inefficienza errata intensità picco DOEVoltaggio cono 10 10 5 5 1300 inefficienza inefficienza errata intensità picco DOEVoltaggio capillare 5 5 5 5 1275 inefficienza inefficienza errata intensità picco DOET desolvatazione 5 5 5 5 1275 inefficienza inefficienza errata intensità picco DOE
T T sorgente 5 5 5 5 1275 inefficienza inefficienza errata intensità picco DOEparametri 1/3 quadrupolo potenz entrata/uscita 1 1 1 1 55 inefficienza inefficienza errata intensità picco fissataparametri 2 quadrupolo energia collisione 10 5 5 5 1300 inefficienza inefficienza errata intensità picco DOE
portata gas collisione 5 5 5 5 1275 inefficienza inefficienza errata intensità picco DOEpotenziali entrata/uscita 1 1 1 1 55 inefficienza inefficienza errata intensità picco fissataenergia fotomoltiplicatore 1 1 1 1 55 inefficienza inefficienza errata intensità picco fissata
Risk assesment Failure Mode Effect Analysis (FMEA)

Estrazione del campione per precipitazione: definizione del dominio sperimentale
Screening dei fattori importanti
FATTORI PRESI IN ESAME LIVELLI
X1. Solvente precipitante
X2. Volume di plasma (µl)
X3. Rapporto in volume plasma:agente precipitante
X4. Tempo di miscelazione (sec)
X5. Temperatura centrifugazione (°C)
ALTRI PARAMETRI FISSATI
Velocità centrifugazione (rpm)
Tempo centrifugazione (min)
Filtrazione centrifugato
Diluizione 1:1 con eluente HPLC
Volume di iniezione in HPLC-MS (µl)
Valori pratici
Lo studio sistematico di tutte le combinazioni dei 5 fattori a 2 livelli richiederebbe 32
esperimenti

RISPOSTE
R1. Sensibilità (A, area del picco)R2. Sensibilità (h, altezza del picco)R3. Rapporto altezza/area del picco (h/A)R4. Rapporto segnale:rumore (S/N)R5. Simmetria del picco
Per analizzare le risposte abbiamo utilizzato la regressione multipla e icoefficienti ottenuti dalla regressione sono riportati in un grafico abarre per illustrare gli effetti e dare visivamente e in modo semplicel’informazione desiderata.

30
R2
R3
R4
R1
R5
Le 5 risposte non sono tutte ottimizzabili con un’unica scelta di fattori.
Il migliore compromesso è quello dato dalla scelta del precipitante
Da questa scelta deriva la selezione dei rapporti in V tra plasma e precipitante:
V di plasma:fisso
Il fattore b5 (T centrif.) sembra ininfluente tranne su R4.
Risultati

FATTORI PRESI IN ESAME LIVELLI (min-max, centro)
Solvente
Contenuto di HCOOH (%, v/v)
Contenuto di NH4OAc (mM)
IN QUESTA FASE SONO STATI FISSATI TUTTI GLI ALTRI PARAMETRI strumentali
cromatografici e di massa a valori obbligati (colonna HPLC, flusso, T colonna)
o “pratici” non definitivi (parametri della MS quali potenziali ionizzazione,
potenziali lenti, elettromoltiplicatore, ecc.)
Separazione cromatografica dai componenti della matrice biologica coestratti/coprecipitati

RISPOSTE
R1. Area del picco (obiettivo massimizzare sensibilità)
R2. Simmetria del picco (quanto più possibile vicina a) 1
R3. Effetto matrice% = obiettivo minimizzare effetto matrice se
presente
MF=area del picco in presenza della matrice (bk estratto e poi
spikato) / area del picco in assenza di matrice (soluzione pura
dell’analita)

RISPOSTE
R1. Intensità dello ione molecolare (proporz h picco)
R2. rapporto IC dello ione molecolare /TIC (rapporto S/N)
Iniezione diretta in massa di una soluzione standard di elvitegravir alla conc di 1000 ng/mL ricerca della massa m/z=448

intensità dello ione molecolare
il modello lineare con interazioni non funziona.La migliore % di varianza spiegata ottenibile è minore del 30% e sembra chiaro che esista una curvatura nella risposta e che serva quindi un modello quadratico per trovare l'optimum della intensità dello ione molecolare. In ogni caso è possibile individuare le tendenze dei fattori nel determinare la risposta, e queste sono le seguenti.
Risultati

Intensità ione molecolare/TICil modello lineare con interazioni spiega bene i risultati.
In particolare, la % di varianza spiegata è del 90% con un errore piccolo e una convalida soddisfacente (quasi coincidenza, a meno della varianza sperimentale, tra dato previsto al centro e dato sperimentale misurato nelle condizioni centrali del dominio sperimentale).I fattori principali sono tutti importanti ad eccezione di Capillary voltage e Cone gas che possono essere impostati ai valori che convengono di più
Risultati
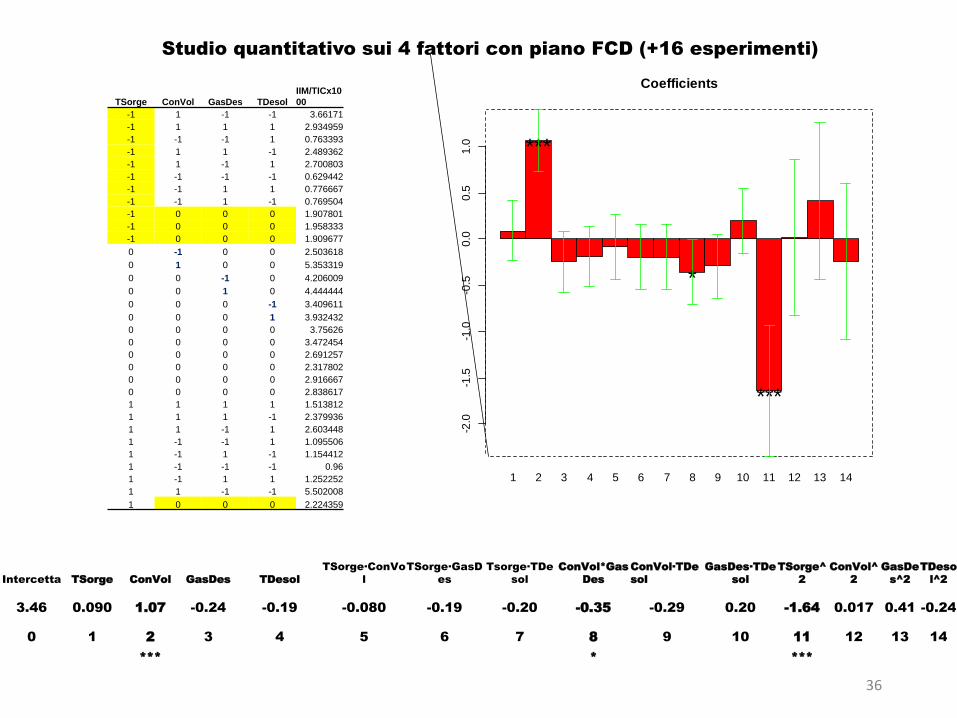
36
Intercetta TSorge ConVol GasDes TDesol
TSorge·ConVo
l
TSorge·GasD
es
Tsorge·TDe
sol
ConVol*Gas
Des
ConVol·TDe
sol
GasDes·TDe
sol
TSorge^
2
ConVol^
2
GasDe
s^2
TDeso
l^2
3.46 0.090 1.07 -0.24 -0.19 -0.080 -0.19 -0.20 -0.35 -0.29 0.20 -1.64 0.017 0.41 -0.24
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
*** * ***
TSorge ConVol GasDes TDesol
IIM/TICx10
00
-1 1 -1 -1 3.66171
-1 1 1 1 2.934959
-1 -1 -1 1 0.763393
-1 1 1 -1 2.489362
-1 1 -1 1 2.700803
-1 -1 -1 -1 0.629442
-1 -1 1 1 0.776667
-1 -1 1 -1 0.769504
-1 0 0 0 1.907801
-1 0 0 0 1.958333
-1 0 0 0 1.909677
0 -1 0 0 2.503618
0 1 0 0 5.353319
0 0 -1 0 4.206009
0 0 1 0 4.444444
0 0 0 -1 3.409611
0 0 0 1 3.932432
0 0 0 0 3.75626
0 0 0 0 3.472454
0 0 0 0 2.691257
0 0 0 0 2.317802
0 0 0 0 2.916667
0 0 0 0 2.838617
1 1 1 1 1.513812
1 1 1 -1 2.379936
1 1 -1 1 2.603448
1 -1 -1 1 1.095506
1 -1 1 -1 1.154412
1 -1 -1 -1 0.96
1 -1 1 1 1.252252
1 1 -1 -1 5.502008
1 0 0 0 2.224359
1 2 3 4 5 6 7 8 9 10 11 12 13 14
Coefficients
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0 ***
***
*
Studio quantitativo sui 4 fattori con piano FCD (+16 esperimenti)

37
Studio quantitativo dei fattori influenzantil’esperimento MRM
Esperimenti sulla transizione 448 -> 344
1) energia di collisione , intervallo 10-50 eV.
2) portata del gas di collisione , intervallo 0.25-0.35 mL/min.
8 esperimenti + 4 repliche del punto al centro del dominio sperimentale (30 eV; 0.30 mL/min).
Sono state condotte 3 serie di 12 esperimenti
Piano Face Centered Design dei 2 fattori (12 esperimenti)
Modello ipotizzato
Y = b0
+ b1·X
1+ b
2·X
2+ b
12·X
1·X
2+ b
11·X
1
2+ b
22·X
2
2

Y1: Intensità dello ione figlio (IM·106)
Y2: il rumore, N, rappresentata dalla TIC·108
Y3: il rapporto segnale/rumore, calcolato dal quoziente
IM/TIC*100
RISPOSTE
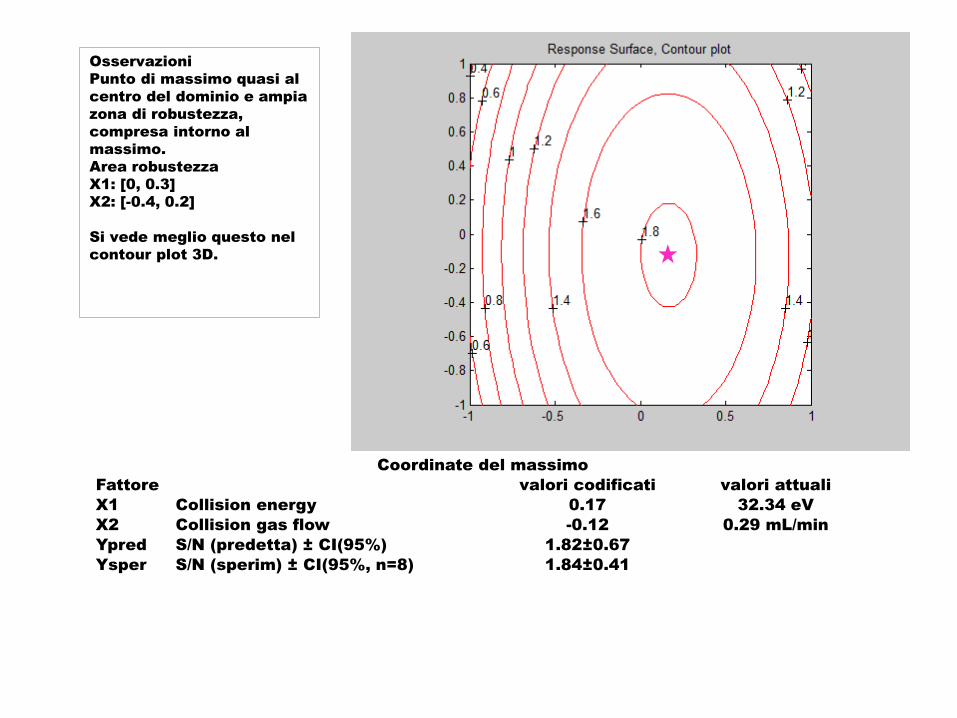
Osservazioni
Punto di massimo quasi al
centro del dominio e ampia
zona di robustezza,
compresa intorno al
massimo.
Area robustezza
X1: [0, 0.3]
X2: [-0.4, 0.2]
Si vede meglio questo nel
contour plot 3D.
Coordinate del massimo
Fattore valori codificati valori attuali
X1 Collision energy 0.17 32.34 eV
X2 Collision gas flow -0.12 0.29 mL/min
Ypred S/N (predetta) ± CI(95%) 1.82±0.67
Ysper S/N (sperim) ± CI(95%, n=8) 1.84±0.41

5. Validazione del metodo analitico: Specificità
• Valutazione della performance in presenza di elevate
concentrazioni di altri antivirali, antifungini, antibiotici,
immunosoppressori
• Valutazione dell’EFFETTO MATRICE
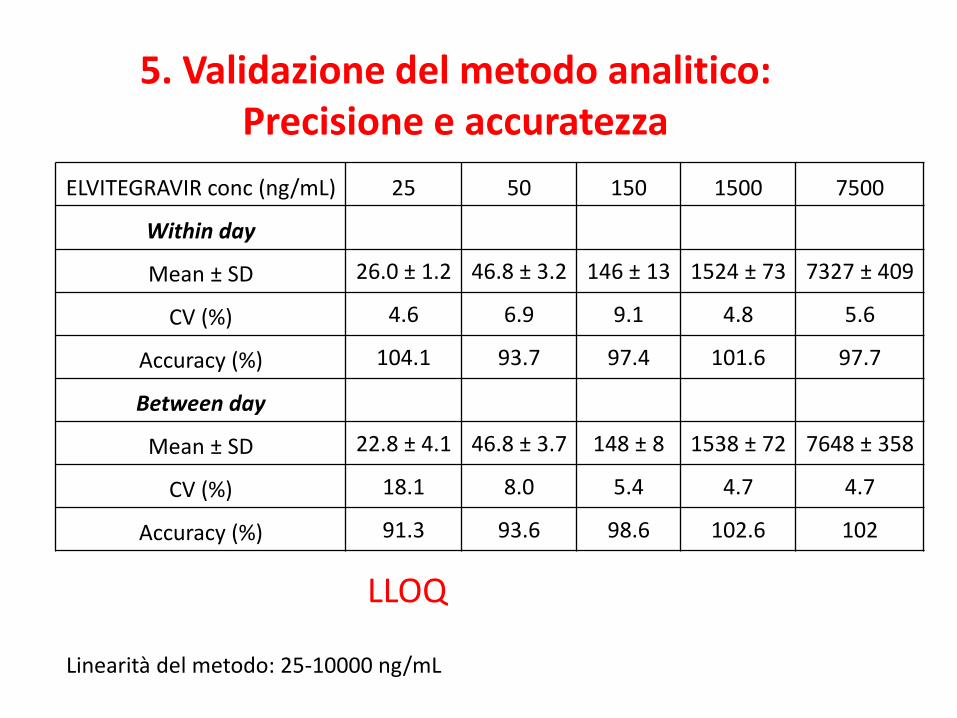
ELVITEGRAVIR conc (ng/mL) 25 50 150 1500 7500
Within day
Mean ± SD 26.0 ± 1.2 46.8 ± 3.2 146 ± 13 1524 ± 73 7327 ± 409
CV (%) 4.6 6.9 9.1 4.8 5.6
Accuracy (%) 104.1 93.7 97.4 101.6 97.7
Between day
Mean ± SD 22.8 ± 4.1 46.8 ± 3.7 148 ± 8 1538 ± 72 7648 ± 358
CV (%) 18.1 8.0 5.4 4.7 4.7
Accuracy (%) 91.3 93.6 98.6 102.6 102
5. Validazione del metodo analitico: Precisione e accuratezza
LLOQ
Linearità del metodo: 25-10000 ng/mL
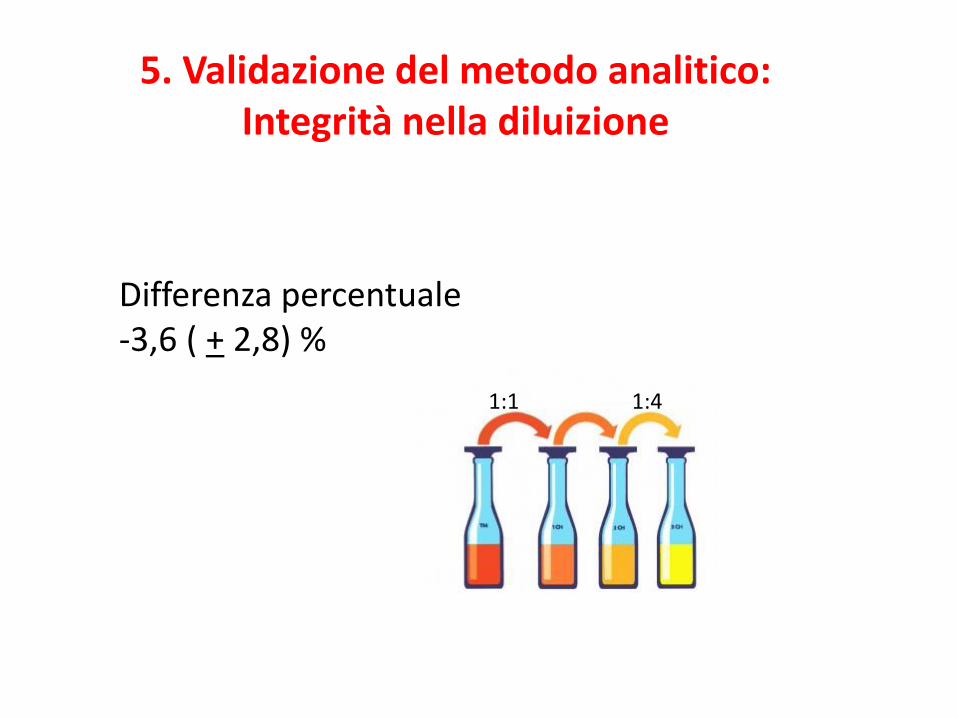
5. Validazione del metodo analitico:Integrità nella diluizione
Differenza percentuale-3,6 ( + 2,8) %
1:1 1:4
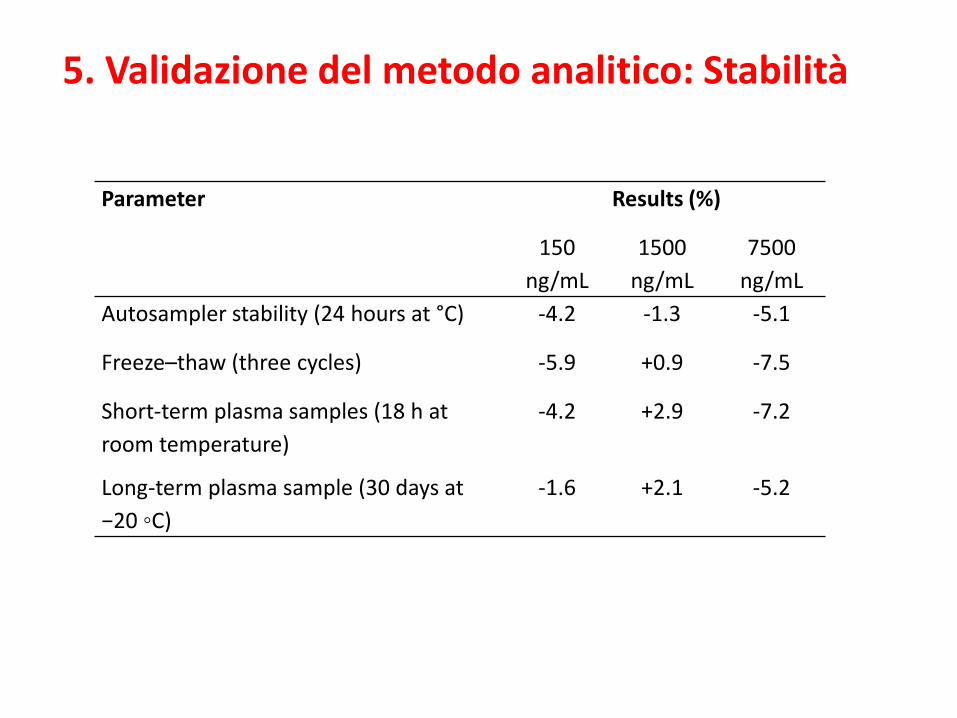
Parameter Results (%)
150
ng/mL
1500
ng/mL
7500
ng/mL
Autosampler stability (24 hours at °C) -4.2 -1.3 -5.1
Freeze–thaw (three cycles) -5.9 +0.9 -7.5
Short-term plasma samples (18 h at
room temperature)
-4.2 +2.9 -7.2
Long-term plasma sample (30 days at
−20 ◦C)
-1.6 +2.1 -5.2
5. Validazione del metodo analitico: Stabilità

0
20000
40000
60000
80000
100000
120000
140000
160000
180000
0 500 1000 1500 2000 2500 3000 3500
Patient chromatogram before and after reaction with enzyme

6. Controllo del metodo
• Quando avremo raccolto un numero significativo di dati potremo
prevedere una eventuale variazione del metodo affinchè venga
se necessario migliorata la metodica

Conclusioni
• Il QbD può effettivamente aiutare a mettere a punto metodi bioanaliticirobusti
• I numeri di metodi non performanti sono destinati a diminuire notevolmente
• Anche in questo campo è necessario che se ne comprenda l’importanza
