kuliah enzyme replacement therapy 2012 palangkaraya
DESCRIPTION
modul neurosainsTRANSCRIPT

Enzyme Replacement Therapy:
Lesson from Gaucher Diseases
Yoga Devaera, Damayanti Rusli SjarifDiv Pediatric Nutrition and Metabolic Diseases
Dept of Child Health – FKUI/RSCM Jakarta - Indonesia

After the lecture you should make a flowchart of how the
enzyme replacement therapy generated Collect it tomorrow to your chief of the class
Assignment

Introduction
Enzyme replacement therapy
Summary
Outlines

They are looking for the answers to three basic
questions: Is there a disease? What causes it? Can we prevent, treat, or cure it?
How do scientists investigate diseases?

Is there a disease ?

What’s wrong with this child?
Splenomegaly In 1882, the French
medical student Phillipe Charles Ernest Gaucher described a 32-year old woman whose spleen was very enlarged.
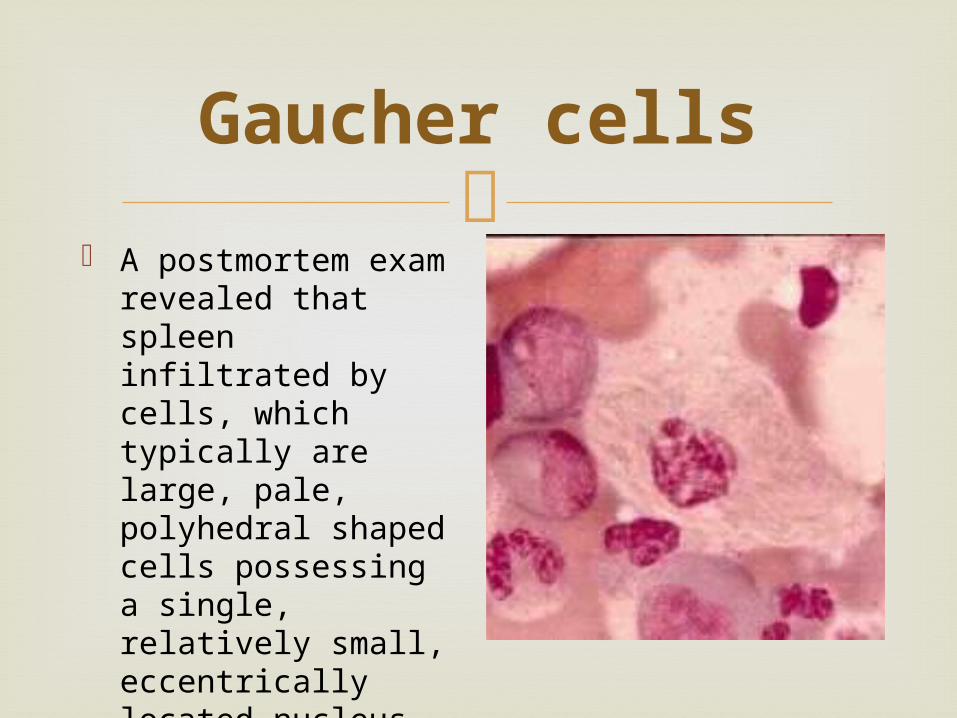
Gaucher cells
A postmortem exam revealed that spleen infiltrated by cells, which typically are large, pale, polyhedral shaped cells possessing a single, relatively small, eccentrically located nucleus


Phillipe Charles Ernest Gaucher

Gaucher disease

Did people with Gaucher disease exist before
1882 ?
Yes, they did. But because a set of symptoms wasn't identified with the condition, "Gaucher disease" as a disease diagnosis did not exist.
Question 1

What causes it?

What Causes It? In 1934, the French
chemist A. Aghion discovered the chemical cause of the enlarged spleens and liver: a buildup of a lipid (fatty substance) called "glucocerebroside."

did people with Gaucher disease make too much of
the lipid for their bodies to handle? Or did their bodies not break it down and dispose of
it?
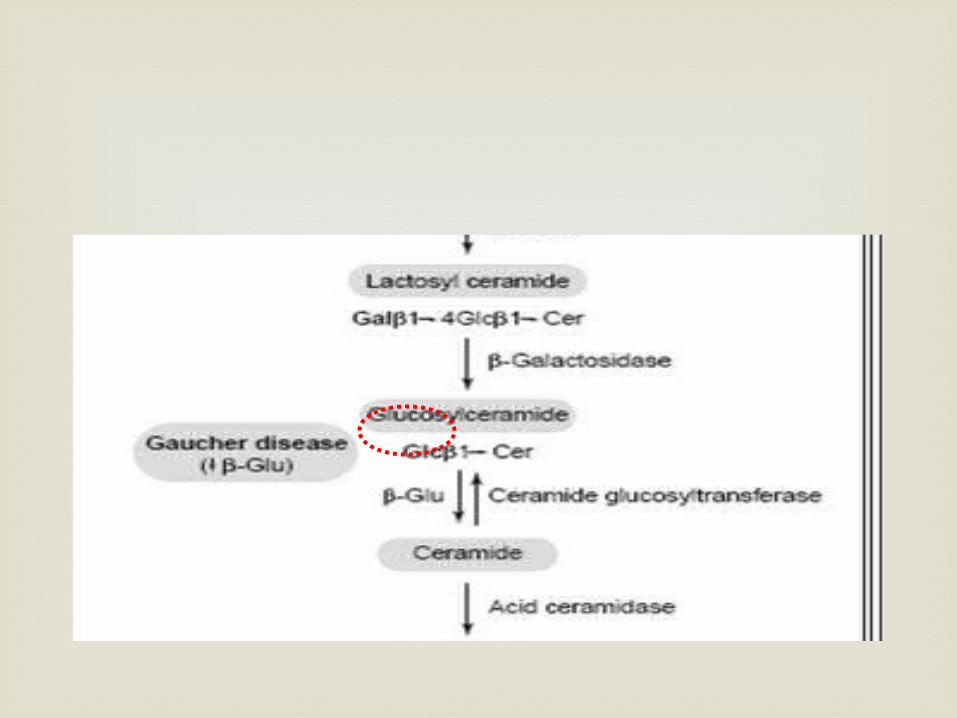
Why there was too much lipid in Gaucher cell ?

The answer to this question came during the
early 1960s, when Dr. Roscoe Brady's group showed that people with Gaucher disease made the lipid normally
but did not make enough of the enzyme "glucocerebrosidase" to break it down and clear it out of the body.

Development of a Gaucher cell

Gaucher’s disease is resulting from either
severely decreased functioning or a complete lack of lysosomal acid β-glucosidase or
glucocerebrosidase
Causes?

A single mutation in the gene coding for GCase can result
in partial (60-70%) or complete lack of enzyme activity (Lieberman et al. 2007).
In affected cells, glucosylceramide accumulates and is difficult to remove (glucosylceramide forms deposits within macrophages called Lewy bodies that these cells can't break down)
Buildup of glucosylceramide within macrophages can lead to simultaneous enlargement of the spleen and liver, abnormal bone turnover, and diseases of the central nervous system in more severe cases (Lieberman et al. 2007).
Next findings


In 1967, Brady's group developed a convenient
diagnostic test for Gaucher disease which works by measuring the activity of the enzyme glucocerebrosidase in white blood cells.
The amount of enzyme directly relates to how severe a case of Gaucher disease
The enzyme activity is also one way that may help to distinguish the three types of Gaucher disease described in this chart.
Enzyme activity

The Three Types of Gaucher Disease Type 1 Type 2 Type 3Whom it Strikes young adults
/ adultsinfants children/
young adults
Distinguishing symptom
no nervous system problems
early nervous system problems
later onset of nervous system problems
Effects of disease varies from mild to severe
dies in infancy
becomes severe
Glucocerebrosidase Activity
some activity but much less than normal
very little activity
little activity




"Why do some people make too little enzyme?"
The answer to this question came in 1987, when the first gene mutation that causes Gaucher disease was discovered by Dr. Shoji Tsuji and coworkers.
chromosome 1

How Gaucher disease is passed on ?
Autosomal recessive

In the early 1970s, Dr. Brady's group devised
an enzymatic test based on the enzyme's activity to tell people if they were carriers or not, and
a procedure for prenatal diagnosis. These tests give people information about their
genetic status so that they can prepare for the future.
Enzymatic based diagnosis

Can We Prevent, Treat, or Cure It?

The third question that medical researchers try to
answer is the most important, and often depends on the answer to the question what the cause of it ?
To be truly successful, a treatment would have to address the cause of the disease, not just the symptoms.

For Gaucher disease, physicians initially attempted to
address the symptoms that accompany the disease. They removed enlarged spleens, and performed liver
transplants, blood transfusions, and orthopedic procedures.
Only bone marrow transplantation for people with Type I Gaucher disease was sometimes successful.

mutation in gene coding enzyme
mutated enzyme
loss ofenzyme activity
accumulation of substrate
cell dysfunction / biochemical and pathological change
clinical symptoms
The Pathophysiology of IEMs

The Principle of Treatment
gene mutation
mutated lysosome enzyme
loss ofenzyme activity
accumulation of substrate
cell dysfunction / biochemical and pathological change
clinical symptoms
modification ofmutated protein
ChemicalChaperone
inhibition ofsubstrate synthesis
SubstrateDeprivation
supplementationof enzyme protein
ERT
BMT
Gene Therapy
ERT

In 1966, Dr. Roscoe Brady suggested a therapy for
Gaucher disease based on replacing the enzyme. Using human placentas, Dr. Peter Pentchev of Dr.
Brady's team isolated a tiny sample of purified glucocerebrosidase.
Enzyme Replacement Therapy

In 1973, Brady put that enzyme into two splenectomised
patients with Gaucher disease. The first patient was a 15 year old boy with Type 3 Gauchers
disease. a liver biopsy was obtained, the enzyme at ½ u/kg/bw (unit
per kilo of bodyweight) was given and two days later performed another liver biopsy → a 26% reduction of the accumulation of glucocerebroside in the liver biopsy.
The second patient also showed a 26% decrease. The third patient received 2½ u/kg/bw but the reduction was
only 8%
Development ERT

A large-scale purification method was completed in 1977.
The enzyme had to be treated with an alcohol to make it stick to the purifying columns.
But this preparation of the enzyme produced inconsistent results during clinical trials. No reduction of hepatic (liver) glucocerebroside occurred in 4 out
7 patients who received this preparation The alcohol had removed a lipid (fat) that activates the
enzyme and targets it to the affected cells, called macrophages.
Development ERT

The problem was
the glucocerebrosidase was not going into the macrophages, the cells in the liver, spleen and bone marrow
that accumulate glucocerebroside: 95% of the infused enzyme was going to other cells, primarily hepatocytes in the liver, and being wasted.
Dr. Brady's challenge was now to get the purified enzyme into the targeted cells.
Development ERT

John Barranger, Clifford Steer and Scott Furbish removed
oligosaccharides from the enzyme so that the mannose (a sugar at the end of the sugar side chains) would attach to the macrophage.
Modified Enzyme



In the first clinical trial with macrophage-targeted
glucocerebrosidase, eight people with Gaucher disease received a fixed dose of the modified enzyme. Only the smallest one - a child - experienced beneficial
effects. He was given 13 u/kg/bw every week.His hemoglobin and platelet counts increased; the size of his spleen and liver decreased; and the damage to his bones lessened.
Then deliberately stopped the enzyme infusions and his haemoglobin and platelets gradually decreased to pre-infusion values.
Development ERT

When re-instated his enzyme infusions at 30 u/kg/bw, his blood
counts rose to normal range, there was a reduction in the size of his spleen and liver and the damage to his bones improved.
The other seven people were adults and had not received enough of the enzyme to improve their condition.

It was at this point that Henri Termeer (now President
and Chief Executive Officer of Genzyme Corporation) learned about the work.
Henri raised 10 million dollars to produce enough enzyme for a clinical efficacy trial.
Brady's team then carried out a dose response study and elected to give patients 60 u/kg/bw every two weeks. In this clinical efficacy trial, all 12 patients improved.
All of these people had strikingly good clinical responses within a few months. For example, their height and weight increased; their anemia improved; their liver and spleen sizes decreased; and their bone damage lessened.
Dose Response Study

Hemoglobin Chart

Before and after X-
Rays

Macrophage-targeted glucocerebrosidase was
approved as a specific treatment for Gaucher disease by the Federal Drug Administration on April 5, 1991.
Enzyme replacement therapy worked. Ceredase worked and the recombinant enzyme Cerezyme is just as good.’
Enzyme replacement therapy is an effective treatment for most people with Type 1 Gaucher disease.
FDA approval

FDA approval

ERT for Gaucher Diseases

PretreatmentFemale; Age 8 Years, 8 Months
Post-treatmentFemale; Age 10 Years, 10 Months
Gaucher’s patient Response to Enzyme Therapy
Courtesy of NW Barton. Developmental and Metabolic Neurology Branch of the NINDS.

Therapeutic Goals and Monitoring for Type 1
Gaucher Disease
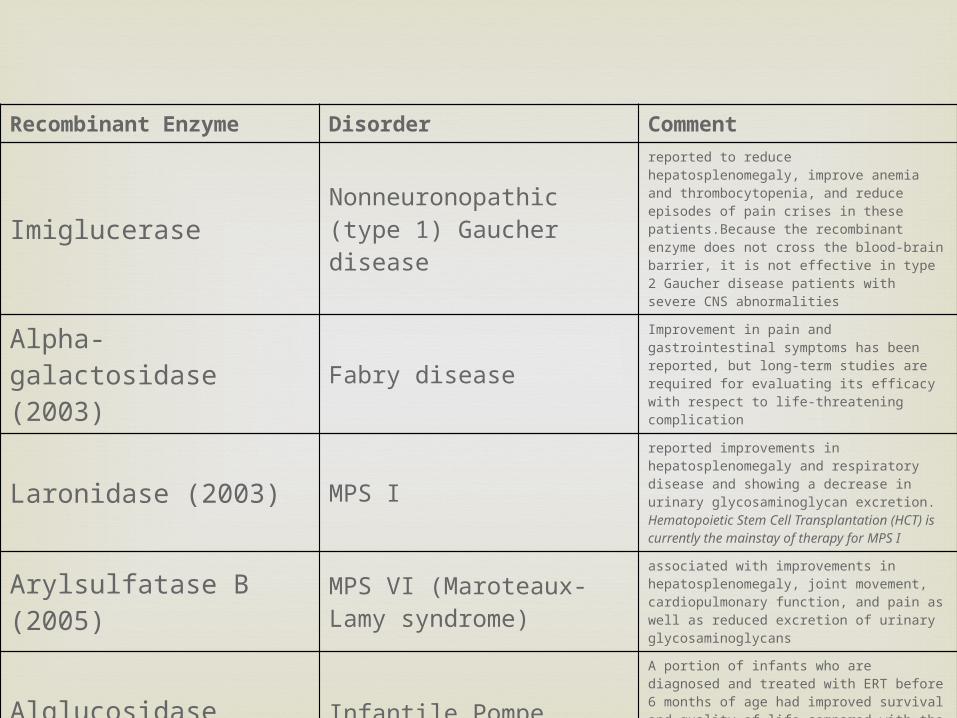
Recombinant Enzyme Disorder Comment
Imiglucerase Nonneuronopathic (type 1) Gaucher disease
reported to reduce hepatosplenomegaly, improve anemia and thrombocytopenia, and reduce episodes of pain crises in these patients.Because the recombinant enzyme does not cross the blood-brain barrier, it is not effective in type 2 Gaucher disease patients with severe CNS abnormalities
Alpha-galactosidase (2003) Fabry disease
Improvement in pain and gastrointestinal symptoms has been reported, but long-term studies are required for evaluating its efficacy with respect to life-threatening complication
Laronidase (2003) MPS Ireported improvements in hepatosplenomegaly and respiratory disease and showing a decrease in urinary glycosaminoglycan excretion. Hematopoietic Stem Cell Transplantation (HCT) is currently the mainstay of therapy for MPS I
Arylsulfatase B (2005) MPS VI (Maroteaux-Lamy syndrome)
associated with improvements in hepatosplenomegaly, joint movement, cardiopulmonary function, and pain as well as reduced excretion of urinary glycosaminoglycans
Alglucosidase alpha Infantile Pompe diseaseA portion of infants who are diagnosed and treated with ERT before 6 months of age had improved survival and quality of life compared with the observed natural history of the disease, yet, for unknown reasons, a subset of the treated cohort did not show effects from ERT
Idursulfase MPS II (Hunter syndrome)
Clinical trials showed improvement in cardiopulmonary disease and hepatosplenomegaly and decreased excretion of urinary glycosaminoglycans


ERT in Pompe diseases

Pompe: patient response to enzyme therapy
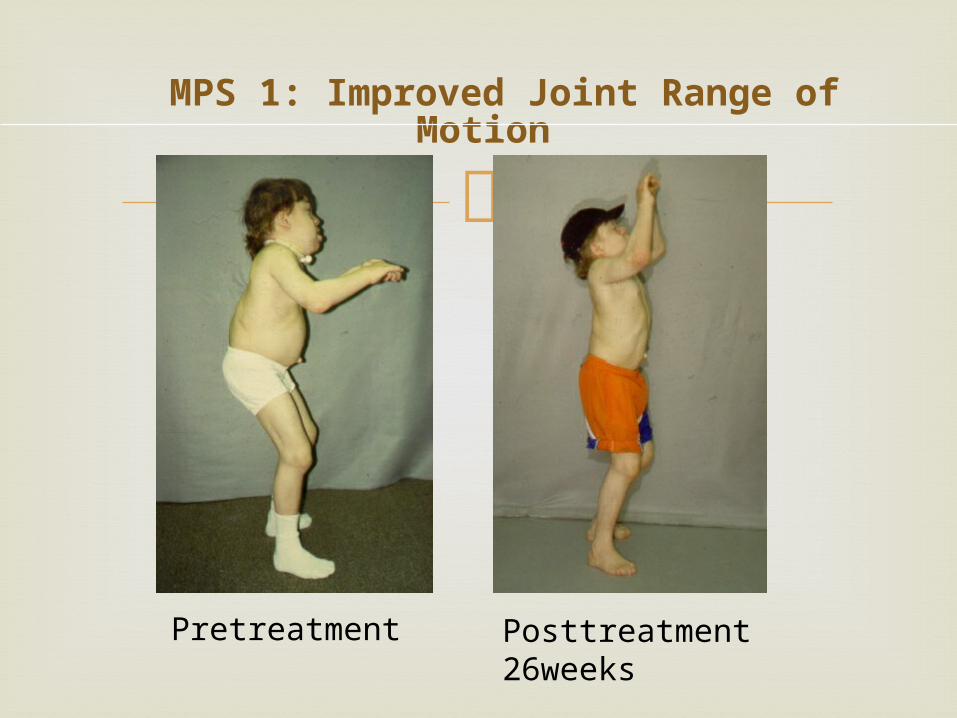
MPS 1: Improved Joint Range of Motion
Pretreatment Posttreatment 26weeks

Mean Changes in the Restriction of Range of Motion in
Patients with Mucopolysaccharidosis I during -l-Iduronidase Therapy

ERT has little effect on the brain, skeletal tissue, and
valvular heart disease in LSDs. ERT treats only the symptoms and not the underlying
disease Lifelong frequent infusions (weekly to monthly depending
on the protocol) The estimated cost of ERT is between
$90,000 to $565,000 (US), depending on the disease (therapy) and patient size.
Intravenous infusions of GCase are administered weekly and it can cost a patient from $100,000 to $750,000 per year (Sawkar et. al, 2002).
Infusion reactions including fatal anaphylaxis have been reported
Limitations of enzyme replacement therapy

Other therapy options are
highly desirable !!!!


How about Indonesia ?

K, female, 8 years old Suspected Fabry since 3 years old Diagnosed Fabry at 8 years old ERT cost
Rp 180.000.000,-/month Lifelong Donation ?
Fabry Diseases : Waiting for ERT

Fabry disease progression

Clinical staging of Fabry disease in the kidney. (Reproduced
with permission from Branton MH, et al. Medicine. 2002;81:122-38.)

F, 3 years old boy diagnosed as MPS IVA (Morquio
Syndrome) No ERT available yet Trial of ERT beginning 2012 in Taiwan, refused because of
nationality
MPS IVA : Waiting for ERT (trial)

Thank you

Gaucher disease - massive
splenomegaly
splenomegaly

The enlarged cells (now called "Gaucher cells") and spleen became signs of the disease.
Gaucher's description of the them enabled other physicians to diagnose people with Gaucher disease, and introduce the term into medical literature

Lysosomal glucosylceramide accumulation in Gaucher stabilizes α-synuclein oligomers ► α-synuclein inhibits
lysosomal trafficking of glucocerebrosidase in synucleinopathies