koordinationschemie ii (ac7–9) · 1 eine vorübung aus dem qualitativ-analytischen praktikum...
TRANSCRIPT

Koordinationschemie II(AC7–9)wann und wo?
2 SWS, Fr 10–12, Willstätter-HörsaalBeginn: 18. Oktober 2013, Ende: 7. Februar 2014
Klausur
Die erste Klausur für das WS 2013/2014 findet am Freitag, dem 7. Februar 2014, von10:15–11:45 Uhr im Willstätter-Hörsaal statt.
Das Anmeldeskript für die Klausur wird ca. 1 Woche vor dem Klausurtermin geöffnet sein.
Alte Klausuren:
WS 2004/2005: Klausur LösungWS 2005/2006: Klausur LösungWS 2009/2010: Klausur Lösung
Klausur LösungWS 2010/2011: Klausur Lösung
Klausur LösungWS 2011/2012: Klausur Lösung
Klausur LösungWS 2012/2013: Klausur Lösung
Klausur Lösung
für wen?
Master-Studiengang Chemie: AC 7, 8 oder 9
was?
Der Vorlesung Koordinationschemie II vertieft den Stoff der Vorlesung Einführung indie Koordinationschemie in verschiedene Richtungen. Ziel ist es, unter besondererBerücksichtigung der verwendeten Methoden aktuelle Schwerpunkte der Koordinations-chemie anzusprechen.
Die aktuelle Koordinationschemie profitiert als sehr methodenreiche Disziplin vomenormen Fortschritt der technischen Möglichkeiten. Gerade bei den hier interessierenden

Methoden wie der Strukturaufklärung an Kristallen, den verschiedenen spektroskopischenVerfahren und, als verbindende Klammer, der Computerchemie – hier vor allem die DFT-Methoden an offenschaligen Spezies – hat es in der jüngeren Vergangenheit beachtlicheFortschritte gegeben. Dabei haben diese Methoden ihren Weg aus den spezialisiertenArbeitskreisen in die experimentell-synthetische Laborpraxis gefunden. Ziel der Vorlesungist es, genau solche Methoden und Konzepte vorzustellen, die heute zur Arbeit einersynthetisch orientierten Gruppe zum Standard gehören, … und daher Teil praktisch jederkoordinationschemischen Publikation sind.
Ausgangspunkt ist die Vorlesung Koordinationschemie des Bachelorstudienganges (AC3). Die dort formulierten Konzepte sind so ausgewählt, dass Ihnen dort kein zu starkvereinfachtes Scheinwissen vermittelt wurde, sondern, im Gegenteil, solcheModellvorstellungen eingeführt wurden, die sich ohne Abstriche erweitern lassen.
Technisches
Computerchemische Rechnungen sind entweder mit Gaussian ausgeführt, das Sie aufden Rechnern in den WAP-Räumen in der Version 03 vorfinden. Die Ergebnisse könnensie mit GaussView ansehen, das Sie ebenfalls dort vorfinden. Frei verfügbar ist Orca, zudessen Bedienung einschließlich der Darstellung der Ergebnisse Sie ebenfalls auf freieProgramme zugreifen können. Empfehlenswert ist zum Beispiel Gabedit.
Prüfen Sie hier, ob Ihr Browser das Skript korrekt darstellt.
Um das Skript auszudrucken, verwenden Sie am Besten die pdf-Version.
Zuletzt geändert: 17. Oktober 2013.
2

1 Eine Vorübung aus dem qualitativ-analytischen Praktikum
Wegen des Bezugs zur AC3-Vorlesung beginnen wir zur Einstimmung mit einer Vorübung,bei der wir uns die verschiedenen Regeln (einschließlich der etwas langweiligenNomenklaturregeln) bewusst machen. Wir beginnen mit einem Experiment: Eisen(III)-nitrat-Nonahydrat wird in Wasser gelöst und eine schwache Braunfärbung mit wenigSalpetersäure beseitigt. Anschließend wird zuerst etwas, dann viel Natrium- oderKaliumthiocyanat zugegeben. Wir beobachten die Farbänderung und fragen uns nach derUrsache. Dabei diskutieren wir, welche Spezies bei der Reaktion entsteht, wie sie heißt,wie ihre Struktur ist, wie der Spinzustand des Zentralmetalls ist, welche Isomere denkbarsind, und ähnliche Fragen. (Am Schluss fügen wir der Lösung Fluorid hinzu und schauen,ob wir das Resultat verstehen.) Ganz zum Schluss wiederholen wir etwas Nomenklatur,indem wir die κ-Konvention benutzen und den Nutzen des IUPAC-Polyedersymbols wieOC-6 gegenüber einer Punktgruppenbezeichnung wie Oh herausstellen.
Materialien hierzu.
3

2 Methoden der Koordinationschemie
2.1 13C{1H}-NMR-Spektroskopie
Der coordination-induced shift
Die erste Lehreinheit knüpft an den Inhalt der Lehreinheit 2 der Einführung in dieKoordinationschemie an. Es geht um die Frage, wie ein Komplexgleichgewicht in wässrigerLösung analysiert werden kann. Im Mittelpunkt steht als wohlvertrauter Ligand Tartrat,und zwar das C2-symmetrische L-Tartrat, das in mehr oder weniger alkalischer Lösungeingesetzt wird. Um übersichtlich zu bleiben, wird ein Palladium(II)-Zentrum miteingeschränkter Funktionalität eingesetzt: zwei der vier (warum vier?) Bindungsstellendes Zentralmetalls sind durch einen zweizähnigen Stickstoff-Chelatliganden blockiert. Beider Umsetzung des Edukts [(R,R-chxn)PdII(OH)2] (chxn = 1,2-Diaminocyclohexan) mitWeinsäure (H2tart) in wässrig-alkalischer Lösung entstehen neue Komplexverbindungen.Da diese diamagnetisch sind (warum?), können 13C-NMR-Spektren mit Standard-methoden ausgewertet werden (1H-NMR-Spektren helfen nicht viel weiter; warum nicht?).
13C-NMR Spektren in D2O bei verschiedenen molaren Verhältnissen [(R,R-chxn)Pd(OH)2]:L-H2tart:OH− von 1:1:1, 3:1:0, 1:1:2 und 3:1:2. Die auf der Abszisseangegebene chemische Verschiebung bezieht sich auf TMS (δ = 0 ppm). Spektrum a:Dinatrium-L-tartrat in D2O; Komplexspezies sind farbig dargestellt:
4

5

Die Spektren werden auf folgende Aspekte hin untersucht:
• Welche Komplexspezies entstehen?
• Wodurch wird gesteuert, welche Spezies dominiert?
Die wichtigste und zur nächsten Lehreinheit überleitende Frage ergibt sich jedoch aus derBetrachtung der Spektren d und e: Wie ist es zu verstehen, dass ein Gleichgewicht wie
L2Pd2(tartH−2) + 2 OH− = LPd(tartH−2) + LPd(OH)2
nach dem 13C-NMR-Spektrum praktisch vollständig auf der rechten Seite liegt, dass alsoder hoch eingeschätzte Chelateffekt Schwächen zeigt (L = R,R-chxn)? Die oft als chaotischempfundene Chemie in wässriger Lösung zeigt hier eine wesentliche Besonderheit: dieerwarteten Komplexgleichgewichte spielen mit Protolysegleichgewichten zusammen.Liegt, wie im Beispiel, auf der einen Seite des Gleichgewichts starke Säure oder Basefrei vor, so gewinnt die Natur bei der Verschiebung des Gleichgewichtes auf die andereSeite Neutralisationsenthalpie. Dieser Beitrag kann entscheidend sein, wenn dieKomplexstabilität nicht sehr hoch ist – so wie hier: beachten Sie die großen Mengen anfreiem Ligand zum Beispiel in Spektrum b.
2.2 NMR-Spektroskopie diamagnetischer Komplexemit anderen Kernen
19F-, 29Si-, 31P-, 103Rh-, 195Pt-NMR-Spektroskopie
Preetz et al. berichten über die Synthese und Charakterisierung gemischter Chlorido-fluorido-platinate(IV) der allgemeinen Formel [PtFnCl6 − n]2− für n = 0–6. Wieviele Komplex-spezies erwarten Sie? Beachten Sie bei den folgenden Überlegungen, dass der I=½-Kern195Pt eine natürliche Häufigkeit von ca. 34% hat, während Fluor ein Reinelement ist; auchhier ist I = ½. Im 195Pt-NMR-Spektrum sind die Signale aller Spezies gut aufgelöst undnicht überlagert. Eine der Spezies ergibt das folgende Multiplett (ohne Berücksichtigungvon 35Cl-Satelliten):
6

Welche? (Quelle: E. Parzich, G. Peters, W. Preetz, Z. Naturforsch. B 1993, 48,1169–1174.) Welches 19F-Spektrum erwarten Sie, wenn Sie von einer 19F-19F-Kopplungausgehen, die merklich kleiner als die 195Pt-19F-Kopplung ist (vernachlässigen Sie auchhier Chlor-Satelliten)?
2.3 Paramagnetische NMR-Spektroskopie
Die Verkürzung der Relaxationszeiten in der Umgebung eines paramagnetischenTeilchens führt dazu, dass die Signale NMR-aktiver Kerne innerhalb eines charakterischenRadius' im Untergrundrauschen verschwinden. Der Radius dieser verblindeten Regionum ein paramagnetisches Atom hängt vom paramagnetischen Element und dessenSpinzustand ab, außerdem von der Art des NMR-Kerns. Der Umstand, dass in Großteil derLiteratur zur paramagnetischen NMR-Spektroskopie den Eisen(III)-Porphyrinen gewidmetist, hängt mit dem ungewöhnlich geringen Radius des blinden Bereichs zusammen.Typisch ist dagegen zum Beispiel in der 13C-NMR-Spektroskopie, dass alle Speziesin einer ca.-10-Å-Umgebung um ein Zentralmetall herum im Spektrum keine Signaleergeben (ca. 15 Å in der 1H-NMR-Spektroskopie). Dies heißt, dass die Liganden einesparamagnetischen Komplexes üblicherweise nicht zu Signalen führen.
Trotz dieser Einschränkung gibt es eine Standardanwendung der Methode in derKoordinationschemie, nämlich die Evans-Methode. Hierbei wird die molare Suszeptibilitätχm aus der Differenz der chemischen Verschiebungswerte einer nicht koordinierendenSubstanz und der molaren Konzentration des Metallkomplexes errechnet:
3 Δ δχm =
cM
Die meist interessierende Zahl der Bohrschen Magnetonen (deren Berechnung nachder Spin-only-Formel kam in der Koordinationschemie I vor), ergibt sich dann aus der
7

Boltzmann-Konstante, der Temperatur, der Suszeptibilität, der Vakuumpermeabilität, derAvogadro-Konstante und dem Bohrschen Magneton:
3 kBT χmμeff2 =
μ0NA μB2
2.4 Röntgenstrukturanalyse
Die Röntgenstrukturanalyse an Einkristallen gilt als Methode, die wenig Interpretations-spielraum lässt: so, wie es herauskommt, so ist es halt. Dass trotz der hohenAussagesicherheit der Röntgenstrukturanalyse allerhand Unsinn publiziert wird, zeigen diefolgenden Beispiele. Das Lernziel ist, dass Sie zwei besonders verdächtige Fälle erkennen:wenn (1) die publizierten Daten selbst nicht konsistent sind, und wenn (2) das Ergebnisden Regeln der Koordinationschemie zuwiderläuft.
Übungsobjekte sind fünf Strukturanalysen an Komplexen mit dem Anion des Salicyl-aldehyds (saldH), das in der Literatur meist Salicylaldehydat oder Formylphenolat genanntwird, und einem zweiwertigen Zentralmetall. Die lösungsmittelfrei kristallisierendenKomplexe werden genauso formuliert wie ein schon länger bekannter Prototyp, dieKupfer(II)-Verbindung [CuII(sald)2], deren Ci-symmetrische Struktur (nicht C2h, da dasMolekül nicht vollständig planar ist) zuletzt 1995 bestätigt wurde (A. Elmali, Y. Elerman,I. Svoboda, H. Fuess, Z. Kristallogr. NCS 1995, 210, 612). Die Atomabstände imKupferkomplex zeigen eine kürzere Bindung zwischen dem Zentralmetall und dem Phenol-O-Atom (1.887 Å) und eine längere Bindung zwischen dem Kupferatom und dem Aldehyd-O-Atom (1.935 Å):
Alle neueren Arbeiten an homologen Verbindungen erschienen in Acta Crystallogr., Sect.E, was den Vorteil hat, dass die zugrundeliegenden Beugungsdaten mitpubliziert sind, mankann also alles nachrechnen. Im Einzelnen ist M:
8

MII Autoren Jahr Band ArtikelMn Q. Wang, X.-N. Fang 2006 E62 m1492Fe Y.-M. Yang, P.-C. Lu, T.-T. Zhu, C.-H. Liu 2007 E63 m1613Co X.-Y. Qiu 2006 E62 m1191Ni Y.-G. Li, H.-J. Chen 2006 E62 m1038Zn Z.-Y. Xiong, L.-J. Liu 2005 E61 m863
Wir diskutieren als Übung, wo jeweils das Problem sein könnte (Anmerkung: die hierdiskutierten Strukturanalysen wurden inzwischen von Acta Crystallogr. zurückgezogen;in einigen Fällen dürfte es sich um bewusste Betrugsversuche handeln); siehe[actae_retraction_2010]. Als Grundlage verwenden wir den Zusammenhangzwischen Atomformfaktoren und Temperaturparametern:
U = 0.05 Å 2
U = 0 Å 2
O
N
2
4
6
8
e −
sinθ/λ0.2 0.4 0.6
Der abgebildete Zusammenhang wir auch unmittelbar beim Betrachten vonBeugungsbildern deutlich:
9

2.5 Ab-initio- und first-principles-Rechnungen
Das Ergebnis einer Strukturanalyse stimmt – vor allem, wenn die intermolekularenWechselwirkungen klein sind – innerhalb von typischerweise 2–3 pm (0.02–0.03 Å) mitroutinemäßigen computerchemischen Rechnungen überein. Abweichungen sind umsogrößer, je weniger die im Computer behandelte Baueinheit und ihre Einbindung in dieUmgebung mit der experimentell untersuchten Situation übereinstimmt.
Über eine Bestätigung der stabilsten Struktur hinaus bieten die Rechnungen (1)Gesamtenergien, auch von nicht experimentell gefundenen instabilen Isomereneinschließlich Übergangszuständen, (2) Orbitale und Orbitalenergien, und (3) weitereabgeleitete Größen wie UV/Vis-Absorptionen und NMR-Verschiebungen.
Schauen wir uns diese Aspekte im Beispiel an: Unter den im Röntgenkapitel als verdächtigerkannten Strukturanalysen war die eines quadratisch-planaren Zink-Salicylaldehyd-Komplexes, [Zn(sald)2], saldH = Salicylaldehyd. Die Strukturanalyse ergab ein Ci-symmetrisches Molekül mit den angegebenen Zn-O-Abständen:
10
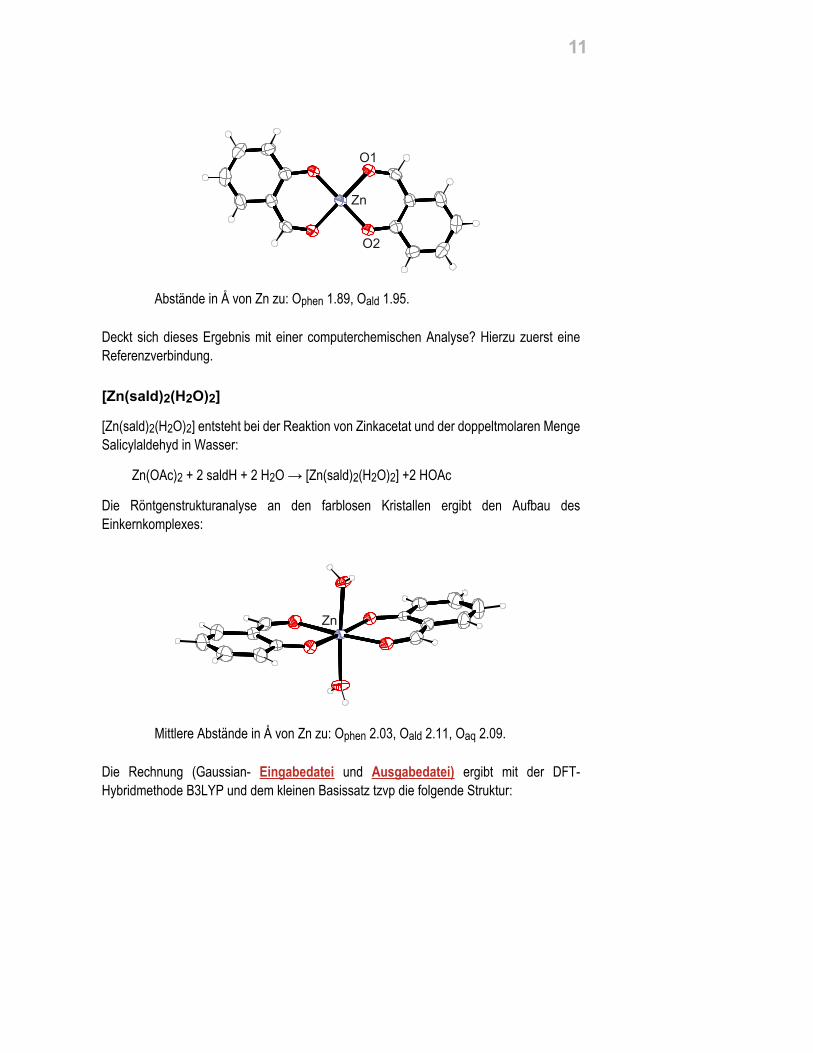
Abstände in Å von Zn zu: Ophen 1.89, Oald 1.95.
Deckt sich dieses Ergebnis mit einer computerchemischen Analyse? Hierzu zuerst eineReferenzverbindung.
[Zn(sald)2(H2O)2]
[Zn(sald)2(H2O)2] entsteht bei der Reaktion von Zinkacetat und der doppeltmolaren MengeSalicylaldehyd in Wasser:
Zn(OAc)2 + 2 saldH + 2 H2O → [Zn(sald)2(H2O)2] +2 HOAc
Die Röntgenstrukturanalyse an den farblosen Kristallen ergibt den Aufbau desEinkernkomplexes:
Mittlere Abstände in Å von Zn zu: Ophen 2.03, Oald 2.11, Oaq 2.09.
Die Rechnung (Gaussian- Eingabedatei und Ausgabedatei) ergibt mit der DFT-Hybridmethode B3LYP und dem kleinen Basissatz tzvp die folgende Struktur:
11

Abstände in Å von Zn zu: Ophen 2.00, Oald 2.12, Oaq 2.27.
Es wird deutlich, dass die Übereinstimmung der Atomabstände innerhalb der Chelatringegut ist, dass aber der Abstand zum Aqualigand in der Rechnung erheblich abweicht.Wir diskutieren den Grund hierfür, der auch für den offensichtlichen Unterschied bei derKonformation verantwortlich ist (intermolekulare Wasserstoffbrückenbindungen im Kristallgegenüber [schwachen und in der Realität meist irrelevanten] intramolekularenWasserstoffbrückenbindungen im isolierten Computer-Molekül).
[Zn(sald)2]
Die B3LYP/tzvp-Rechnung an quadratisch-planarem [Zn(sald)2] führt zu den folgendenParametern:
Abstände in Å von Zn zu: Ophen 1.92, Oald 2.08.
Für die recht großen Abweichungen zum Experiment lassen sich keine starkenintermolekularen Wechselwirkungen verantwortlich machen. Ein stärkerer Hinweis, dassetwas nicht stimmt, wird durch die routinemäßige Behandlung des Rechenergebnissesgewonnen: der Strukturoptimierung folgt stets eine sogenannte Frequenzrechung, diezeigt, ob die zu einem Extremum führende kleinste-Fehlerquadrate-Verfeinerung derStrukturoptimierung ein Energieminimum oder ein Maximum, also einen Übergangs-zustand ergeben hat. Im letzteren Fall treten Schwingungen mit negativerAnregungsenergie auf, deren Auslenkung oft schon das angestrebte Minimum erkennenlassen.
Die Minimumstruktur sieht für [Zn(sald)2] daher erwartungsgemäß nicht quadratisch-planar, sondern tetraedrisch aus:
12

Abstände in Å von Zn zu: Ophen 1.93, Oald 2.01.
Neben der Struktur ein wichtiger Unschied zwischen den beiden Formen: dieGesamtenergie. Wird die Energie des stabilen Konformers auf 0 festgesetzt, errechnetsich der quadratisch-planare Übergangszustand auf B3LYP/tzvp-Niveau zu +40 kJ mol−1.
[Cu(NH3)3]2+: „entatischer Zustand“ vs Nebenminima der Potential-hyperfläche – eine Übung
In auffallend vielen Kupfer-Enzymen gibt es ein T-förmiges CuN3-Chromophor. Beispieleaus der Vorlesung Bioanorganische Chemie sind Hämocyanin, CuZn-Superoxiddismutaseund Cytochrom-c-Oxidase. Das N-Atom wird in diesen Enzymen von Histidin-Seitenkettenzur Verfügung gestellt, die sich hinsichtlich ihrer Ligandeigenschaften nicht allzu sehr vonAmmoniak unterscheiden. Wir überzeugen uns daher in einer DFT-Rechnung, dass das[Cu(NH3)3]2+-Ion wirklich T-förmig ist und fragen nach dem Grund.
Bei diesem Beispiel wird zur Berechnung Orca verwendet. In der Eingabedatei(cu3am.inp) wird das in Orca implementierte COSMO-Modell zur Modellierung einerUmgebung mit Standardparametern genutzt, des Weiteren werden van-der-Waals-Wechselwirkungen durch Grimmes Verfahren berücksichtigt. Wegen desParamagnetismus wird „unrestricted“ gerechnet (α- und β-Spins mit unterschiedlicherEnergie). Es bedeuten: uks [unrestricted Kohn-Sham], bp [BP86-Methode], ri [technischesDetail], vdw [Grimmes van-der-Waals-Korrektur], tzvp [Basissatz], tzvp/j [Hilfsbasissatzfür ri], opt [Strukturoptimierung, Computerchemiker sagen dazu „Geometrieoptimierung“],cosmo(water) [Aufbau einer wasserartigen Umgebung]. Die nächsten vier Zeilen sorgenfür die Übernahme aller nötigen Information in die Ausgabedatei, um Orbitale ansehen zukönnen. Mit „* xyz 2 2“ beginnt die Eingabe des Moleküls: xyz sagt kartesische Koordinatenan, die erste 2 ist die Ladung, die zweite 2 die Multiplizität).
2.6 Bestimmung von Beständigkeitskonstanten
Beständigkeitskonstanten von Komplexen können durch alle Methoden bestimmt werden,die eine Aussage zur Konzentration einer Spezies zulassen. Ein Standardverfahren fürden häufigen Fall eines protonierbaren Liganden ist die Messung der H3O+-Konzentration,um die Konkurrenz eines Metallions und des Protons um den Liganden auszunutzen. Die
13

Beständigkeitskonstanten werden dann durch Anpassen von Titrationskurven gewonnen.Mit bekannten Beständigkeitskonstanten können anschließend Speziesverteilungskurvenerrechnet werden.
Die wesentlichen Definitionen sind im zweiten Kapitel der Vorlesung Koordinationschemiezusammengesellt. Wie man nun in der Praxis vorgeht, schauen wir in einer Übung an,in der es wieder um Kupfer(II)-Komplexe geht. Der Ligand ist jetzt die proteinogeneAminosäure Serin. In der Übung werden die einzelnen Schritte gezeigt, wie das in denfolgenden beiden Graphiken zusammengefasste Ergebnis erhalten wird. Als Hilfsmittelwird das Programm Hyperquad2008 verwendet. Auf eine Schwierigkeit mit der üblichenArt, Beständigkeitskonstanten protonendefizienter Spezies anzugeben, wird in einer pdf-Datei näher eingegangen.
14

Oben: Titration von 2 mmol Serin + 2 mmol H+ in 200 mL Lösungmit 0.5 M NaOH. Unten: Dieselbe Tritration nach Zusatz von 1 mmolKupfer(II)-Salz.
2.7 Literatur
[actae_retraction_2010]
IUCr: Retraction of articles.Acta Crystallogr., Sect. Sect. E, 2010, 66, e21-e22.doi: 10.1107/S1600536809054300.
15

3 Mehrkernkomplexe gezielt aufbauen,Superaustausch
Der Aspekt des Lösungsgleichgewichts bekommt in dieser Lehreinheit eine noch größereBedeutung. Wir stecken uns das Ziel, einen Mehrkernkomplex aufzubauen, und schaffendamit die Grundlage zum Aufbau interessanter Materialien, zum Beispiel molekularerMagnete. In dieser Lehreinheit geht es sehr „un-organisch“ zu. Sowohl in der OrganischenChemie als auch in der Anorganischen Molekülchemie bauen Sie Moleküle schön peu-a-peu in Einzelschritten auf, indem Sie ausnutzen, dass Ihr jeweiliges Zwischenproduktdurch hinreichend hohe Aktivierungsbarrieren darin gehindert wird, irgendetwas zu tun,was Sie nicht wollen – Sie bauen an kinetisch inerten Molekülen herum. Dieser Weg ist inder Koordinationschemie oft verschlossen. Viele wichtige Metallzentren sind bezüglich desLigandaustauschs kinetisch labil. Deren Chemie verläuft unter strikter thermodynamischerKontrolle, meist ohne dass Aktivierungsbarrieren erkennbar sind. Wir lernen eine wichtigeMethode kennen, unter dieser Randbedingung komplexe Strukturen aufzubauen: dieKontrolle durch die Stöchiometrie und den pH-Wert der Reaktionslösungen.
Konkret schauen wir folgendes Beispiel an: Wir vereinfachen Tartrat zu Tartronat (das istdas Anion der 2-Hydroxy-1,3-propandisäure, also Hydroxymalonat, wenn Sie so wollen)und setzen mindestens die doppeltmolare Menge dieses „ambidenten“ Liganden inwässrig-neutraler Lösung mit Kupfer(II)-Salzen um. Es lassen sich dann Salze wie zumBeispiel Li2[Cu(C3H2O5)2(H2O)2] · 2 H2O isolieren. Die Struktur der Diaqua-bis(tartronato-κO1,O3)-cuprat(II)-Dianionen ist nicht ungewöhnlich: über die Carboxylatfunktionen derTartronat-Liganden sind sechsgliedrige Chelatringe gebildet worden. Die Cu-O-Abständebetragen ca. 195 pm (1.95 Å). Mit 291 pm deutlich weiter entfernt sind zwei O-Atome vonAqualiganden. Das Tartronatocuprat zeigt damit die zu erwartende Jahn-Teller-Verzerrungin ungewöhnlich deutlicher Form. Die Ursache hierfür wird weiter unten behandelt.
16

Als nächstes wird nun gespart, und zwar am Liganden. Es wird nur soviel Tartronateingesetzt, dass sich ein Ligand:Metall-Verhältnis von 1:1 ergibt. Der pH-Wert muss nunmit etwas Base auf 7 eingestellt werden, um wieder eine klare Lösung zu erhalten.Wir schauen uns wieder die Struktur eines aus solchen Lösungen isolierten kristallinenProdukts an, zum Beispiel Cs3[Cu3(C3HO5)3(H2O)3] · H2O: es ist ein dreikerniger Komplexentstanden. Die Regel, dass ein verrringertes Ligandangebot zum Brückenbindungs-modus, also zur Verteilung der nun knapperen Lewis-Basizität kommt, ist ein allgemeinesPrinzip. Man denke nur an die Reaktionen von zum Beispiel AlCl3 in aprotischenLösungsmitteln: Äquimolare Mengen AlCl3 und Cl− führen zu [AlCl4]−; wird nur die halbemolare Chloridmenge zur Verfügung gestellt, entsteht das Anion [Al2Cl7]− mit einemverbrückenden Chloridoliganden gemäß [Cl3Al–Cl–AlCl3]−. Wir fragen uns, welche Regelhinter dieser Beobachtung steckt; außerdem fragen wir uns (nochmal), warum eine pH-Wert-Erhöhung bei Tartronat-Überschuss zu denselben Komplexen führt. Das heißt, wirschauen das Gleichgewicht an:
3 [Cu(C3H2O5)2]2− + 3 OH− = [Cu3(C3HO5)3]3− + 3 C3H2O52−
Nachdem die thermodynamischen Randbedingungen geklärt sind, lautet die nächsteFrage, ob besondere Eigenschaften für das dreikernige Cuprat(II) zu erwarten sind. DieStrukturdaten zeigen günstige Voraussetzungen für Superausstausch an. Die beiKupfer(II)-Komplexen sehr häufige antiferromagnetische Kopplung sollte bei dembetrachteten Tricuprat zu „Spinfrustration“ führen. die Winkel an O zeigen allerdings (wiefast immer) eine Aufhebung der C3-Symmetrie an (Cu-O-Cu-Winkel vom rechten oberenBrücken-O-Atom im Uhrzeigersinn: 130, 129, 115°). Die Spinkopplung kann über längereStrecken vermittelt werden. Ein Lehrbuchbeispiel ist Kupfer(II)-acetat, bei dem sich dieFrage stellt, ob nicht eine Kupfer-Kupfer-Bindung die bessere Erklärung für denbeobachteten S=0-Grundzustand ist.
17

Ausschnitt aus den Kristallstrukturen von Li2[Cu(C3H2O5)2(H2O)2] · 2H2O und Cs3[Cu3(C3HO5)3(H2O)3] · H2O:
Die Spins der paramagnetischen Zentralmetallatome wechselwirken in Mehrkern-komplexen über chemische Bindungen hinweg oder direkt aufgrund ihrer räumlichen Nähe.Die häufigste Wechselwirkung ist die antiferromagnetische Kopplung, bei der sich dieSpins benachbarter Zentren im Grundzustand antiparallel ausrichten. Der üblicheKopplungsweg ist der Superaustausch entlang eines Pfades CuA–O–CuB, wenn mit Kupferund μ-Oxido-Liganden formuliert wird. Das spintragende Orbital des Kupfers ist imTartronato-tricuprat das d(x2−y2)-Orbital (warum?). Wird CuA α-Spin zugeordnet, so wirdβ-Spin in den unmittelbar benachbarten Lappen eines p-Orbitals des μ-O-Atoms induziertund damit α-Spin in den abgewandten Orbitallappen. Dieser α-Spin induziert dann wiederβ-Spin in das magnetische Orbital von CuB – die beiden Metallzentren sindantiferromagnetisch gekoppelt. Das Ausmaß der Kopplung wird üblicherweise durch dieKopplungskonstante J ausgedrückt, die die Energie für die Entkopplung der Spins an denZentren A und B beschreibt. Ein negativer Wert für J entspricht einem S=0-Grundzustand,also der antiferromagnetischen Kopplung (cave: diese Regelung wird in der Literatur nichteinheitlich angewendet!). In der Chemie wird J üblicherweise in cm−1 angegeben. Beidem beschriebenen Superaustauschpfad ist der Betrag von J umso größer, je stumpferder Cu-O-Cu-Winkel ist. Solche Beziehungen werden durch die Goodenough-Kanamori-Regeln beschrieben, auf die weiter unten ausführlicher eingegangen wird. Werden zweiKupfer(II)-Zentren durch Sauerstoff-Liganden verbrückt, sollte bei Cu-O-Cu-Winkeln um110–115° antiferromagnetische Kopplung einsetzen. Bei Werten um 140° wird eine sogroße Kopplungkonstante erwartet, dass der angeregte Triplettzustand auch beiRaumtemperatur kaum populiert ist.
18

Im vorliegenden Beispiel ist ein besonders interessanter Fall nicht realisiert. Wäre dasTricuprat-Ion C3-symmetrisch, so wäre J1 = J2 = J3. Durchgehende antiferromagnetischeKopplung wäre dann nicht möglich, es läge „Spinfrustration“ vor.
Zum Schluss dieses Kapitels eine Übung zu den Goodenough-Kanamori-Regeln: WelcheSpinkopplung ist für einen zweikernigen Nickelkomplex mit verbrückendem N2-Ligandenvorherzusagen, der 2009 publiziert wurde [2ni_n2_2009]?
3.1 Literatur
[2ni_n2_2009]
S. Pfirrmann, C. Limberg, C. Herwig, R. Stößer, B. Ziemer:Ein zweikerniger Nickel(I)-Distickstoffkomplex und seine Reduktion inEinelektronenschritten.Angew. Chem. 2009, 121, 3407�3411.doi: 10.1002/ange.200805862
19

4 Supramolekulare Chemie
4.1 Helicate, Gitter, Knoten
Die zuletzt besprochenen Komplexe waren zugleich (kinetisch) labil und(thermodynamisch) stabil. Mit Komplexen, die diese Randbedingung erfüllen, wurde dieheute besonders aktuelle Teildisziplin der Supramolekularen Chemie begründet (Jean-Marie Lehn, Nobelpreis 1987). Das Grundprinzip wird besonders prägnant bei Lehns„Helicaten“ deutlich. Deren Baustein ist zum Beispiel das Komplexkation Bis(2,2'-bipyridyl)-kupfer(I), [Cu(bpy)2]+, das den zweizähnigen bpy-Chelatliganden enthält. [Cu(bpy)2]+ kannmit einem geeigneten Gegenion in methanolischer Lösung hergestellt werden. (Was darferwartet werden, wenn Wasser als Reaktionsmedium gewählt wird?) Als Komplex einesd10-Zentralmetalls mit vier Ligatoratomen ist tetraedrischer Aufbau zu erwarten (warumnicht quadratisch-planarer?). Mit diesem Bauelement werden nun mehrkernige Komplexe(mit viel größeren Metall-Metall-Abständen als bei den Tartronato-cupraten) aufgebaut,indem bpy-Moleküle durch eine flexible Brücke zusammengefügt werden. Das einfachsteBeispiel ist:
20

Drei oder vier Metallzentren lassen sich in analoger Weise in eine Helix einbauen. Einbesonders lehrreicher Fall liegt vor, wenn verschiedene Helicat-Bildner gemischt und dannmit der benötigten Mengen Kupfer(I) umgesetzt werden. Das folgende Schema zeigt dasErgebnis:
Es wäre zu erwarten, dass ein Gemisch mit zahlreichen Komplexen entsteht, was abernicht der Fall ist. Die Ursache ist die kinetische Labilität der Kupferzentren bezüglichdes Ligandaustauschs. Natürlich entstehen bei der Umsetzung von Ligandgemisch undKupfer(I)-Salz auch Komplexe, die unerwünschten Kombinationen entsprechen. EinBeispiel könnte so aussehen:
21

Es wird deutlich, dass durch die Fehlpassung nicht alle Metallbindungsstellen desLiganden besetzt sind. Da in ortho-Stellung zum ersten und letzten N-Atom eines jedenLiganden eine Methylgruppe verhindert, dass Polymerisation eintritt, ist dieser Auswegverschlossen, die noch fehlende Bindungsenergie zu nutzen. Unter den gegebenenVersuchsbedingungen ist jedoch eine Reparatur der Fehlpassung möglich. Das kinetischlabile System nähert sich so allmählich der Belegung aller Metallbindungsstellen, also demausgeordneten, thermodynamisch stabilen Zustand.
Dasselbe Grundprinzip, das zu Helicaten führt, eröffnet weitere aktuelle Zweige derSupramolekularen Chemie: den Aufbau von Metallatom-Gittern [supra_2004] und dieKonstruktion Metall-Organischer Netzwerke („MOFs“ von Metal Organic Frameworks)[mofs_2011]. In vielen MOFs ist dabei das Bindungsstellenmuster eines einfachenZentralmetallatoms durch eine Baugruppe hoher Stabilität ersetzt – einer sekundärenBaueinheit („SBU“ von Secondary Building Unit; siehe zum Beispiel Figures 6 und 7 in[mofs_2011]).
Die Ideen der letzten beiden Kapitel finden sich in einem besonders interessantenForschungsansatz zusammengefasst – der kontrollierten räumlichen Anordnungparamagnetischer Zentren zur Konstruktion von Quantencomputern[quantum_computing_2012]. Wir schauen uns ein Beispiel aus diesem Review an[quantum_computing_2009], bei dem ein bereits bekannter achtkerniger Komplex alsBaustein dient [quantum_computing_2003].
4.2 Literatur
[quantum_computing_2012]
G. Aromi, D. Aguila, P. Gamez, F. Luis, O. Roubeau:Design of magnetic coordination complexes for quantum computing.Chem. Soc. Rev. 2012, in press.doi: 10.1039/C1CS15115K
22

[mofs_2011]
M. O�Keeffe, O. M. Yaghi:Deconstructing the Crystal Structures of Metal��Organic Frameworks and RelatedMaterials into Their Underlying Nets.Chem. Rev. 2011, in press.doi: 10.1021/cr200205j
[quantum_computing_2009]
G. A. Timco, S. Carretta, F. Troiani, F. Tuna, R. J. Pritchard, C. A. Muryn, E. J. L. McInnes,A. Ghirri, A. Candini, P. Santini, G. Amoretti, M. Affronte, R. E. P. Winpenny:Engineering the coupling between molecular spin qubits by coordination chemistry.Nat. Nano. 2009, 4, 173-178.doi: http://www.nature.com/nnano/journal/v4/n3/suppinfo/nnano.2008.404_S1.html
[supra_2004]
M. Ruben, J. Rojo, F. J. Romero-Salguero, L. H. Uppadine, J.-M. Lehn:Metallionen-Gitterarchitekturen: funktionelle supramolekulare Metallkomplexe.Angew. Chem. 2004, 116, 3728�3747.doi: 10.1002/ange.200300636
[quantum_computing_2003]
F. K. Larsen, E. J. L. McInnes, H. E. Mkami, J. Overgaard, S. Piligkos, G. Rajaraman,E. Rentschler, A. A. Smith, G. M. Smith, V. Boote, M. Jennings, G. A. Timco, R. E. P.Winpenny:Synthesis and Characterization of Heterometallic {Cr7M} Wheels.Angew. Chem. 2003, 115, 105-109.doi: 10.1002/ange.200390002
23

5 Bindungsverhältnisse inKoordinationsverbindungen
5.1 Kristallfeldmodell und MO-Schemata
Als Vorbereitung auf speziellere Fälle soll bei einem Komplex, den Sie schon imGrundpraktikum kennengelernt haben, untersucht werden, ob eine quantitativeBehandlung der Bindungssituation Bezüge zu einfachen Modellen zeigt. Wir schauen unszu diesem Zweck das Hexaaquamangan(II)-Ion, [Mn(H2O)6]2+, näher an.
Das Th-symmetrische [Mn(H2O)6]2+-Ion.
24

Die α-Spin-Orbitale des [Mn(H2O)6]2+-Ions. Der Bezug der Rassen derPunktgruppe Th zur Obergruppe Oh ist angegeben.
Dass metall- und ligandzentrierte Orbitale energetisch nahe beieinander liegen, diskutierenwir als Folge des Ausgleichs der Orbitalstabilitäten durch Ladungsübertragung von
25

unstabilen zu stabilen Orbitalen. Ein altes Konzept, das dies für normale Bindungenversucht, ist die Idee des Elektronegativitätsausgleichs (engl. electronegativityequalisation). Wir betrachten hierzu den Ausgleich zwischen den stabileren BH3-Orbitalenund dem weniger stabilen Hydrid-Valenzorbital bei der Bildung von Tetrahydridoboranat:
Energien besetzter Orbitale in Monoboran, Hydridoboranat und Hydrid(mp2/6-31+g(d,p)].
Eine NBO-Analyse ergibt die folgenden Partialladungen:
B HBH3 +0.38 −0.13BH4− −0.54 −0.12
Anschließend ein umfangreicheres Beispiel, das Beladungen bei normaler Bindung(Hydroxido-Spezies) und koordinativer Bindung (Aqua-Komplex) zeigt. Wir machen unsan diesem Beispiel klar, dass Elektronegativitäten nicht auf koordinative Bindungenangewendet werden können:
26

Das [Zn(Me3tacn)(H2O)]2+-Ion, b3lyp/6-31+g(d,p).
Strukturparameter und Ladung nach NBO-Analyse bei [Zn(Me3tacn)X]2+/+ für X = H2O,OH−, SMe− und ohne X, Strukturoptimierung mit b3lyp/6-31+g(d,p):
X = OH2 X = OH X = SMe X = ■Zn-X/Å 2.02 1.81 2.21Zn-N/Å 2.06 2.14 2.16 2.03δ(tacn) 0.35 0.26 0.25 0.48δ(X) 0.09 −0.80 −0.57δ(Zn) 1.56 1.54 1.32 1.52e−(X→Zn) 0.09 0.20 0.43
5.2 Struktur und Spinzustand
Im letzten Beispiel lag das Zentralmetall in tetraedrischer Koordination vor (T-4 in derIUPAC-Nomenklatur). Wegen der d10-Konfiguration des Zink(II)-Zentralatoms sollten dieBindungen zu den Liganden durch das Zn(4s)- und die (Zn)4p-Orbitale vermittelt werden.
Bei unvollständig besetztem d-Niveau sind weitere Baumotive üblich. So wird für low-spin-d8-Komplexe bevorzugt quadratisch-planare Struktur (SP-4) gefunden. Welche Regelngelten für die übrigen Kombinationen aus Koordinationspolyeder und Spinzustand? Wirschauen hierzu auf den Aufsatz von Alvarez How High the Spin?.
Die bei Alvarez zusammengestellten Beschränkungen lassen sich in der Regel mit Hilfedes Kristallfeldmodells ableiten; π-Wechselwirkungen sollten dabei zusätzlich beachtetwerden. Wir sehen uns als Beispiel eine praktische Frage an: lohnen Versuche, dieSP-4-Koordination des d6-high-spin-Eisen(II)-Zentrum im seltenen Mineral Gillespit für einSpin-Crossover-Ereignis von S = 2 zu S = 0 zu nutzen?
27

Das bei Normaldruck rote Mineral Gillespit (El Rosario, Halbinsel BajaCalifornia, Mexiko, auf farblosem Sanbornit, BaSi2O5), BaFeSi4O10,kristallisiert isotyp mit Ägyptisch Blau, CaCuSi4O10. In diesemStrukturtyp ist das Übergangselement quadratisch-planar koordiniert(Foto: Michael Schwan; Inv.-Nr. 74449, GeowissenschaftlicheSammlungen der TU Bergakademie Freiberg).
Ausschnitt aus der Kristallstruktur von Ägyptisch Blau, CaCuSi4O10.Farbcode: graugrün Si, blau Cu; Ca weggelassen.
Zur Übung behandeln wir das Problem so, dass weitere Standardverfahren derKoordinationschemie sichtbar werden: Wir schauen PES-Abtastungen an (PES = PotentialEnergy Surface, Potentialhyperfläche) und wir üben uns im Umgang mit verschiedenenKoordinationssystemen.
28

Ausgangspunkt ist das Komplexkation [Fe(NH3)6]2+ aus der AC3-Vorlesung. WelcheOrbitalabfolge erwarten Sie, wenn entlang z zwei Liganden entfernt werden? Wirvergleichen mit der entsprechenden Ableitung aus einem hypothetischen, tetraedrischen[Mn(NH3)4]2+:
Grenzorbitale von [Mn(NH3)4]2+ (TPSSh/tzvp); der Beitrag der Metall-Atomorbitale zu den MOs ist angegeben.
Nächster Schritt: wir schließen π-Wechselwirkugen ein und überlegen, welches Bild sichnach dem Zufügen eines β-Spins ergibt, wenn also Eisen(II) statt Mangan(II) betrachtetwird:
29

Grenzorbitale von [MnF4]2− (TPSSh/tzvp; Gaussian 09 mitscrf=(solvent=water)-Anweisung); der Beitrag der Metall-Atomorbitalezu den MOs ist angegeben.
Das Ergebnis der entsprechenden Rechnung:
30

Grenzorbitale von [FeF4]2− (TPSSh/tzvp); der Beitrag der Metall-Atomorbitale zu den MOs ist angegeben.
Am Schluss ordnen wir die Ergebnisse anhand des Jahn-Teller-Theorems und schließenmit einem Ausblick auf Cobalt(II)
5.3 Spinkopplung vs. Metall-Metall-Bindung
Paramagnetische Metallzentren sind bevorzugte Motive, die in supramolekulare Struktureneinbaut werden. Dahinter steht die Hoffnung, magnetische Wechselwirkungen inausgedehnten, aber wohldefinierten Strukturen zu erzeugen. In dieser Lehreinheit wirdeine grundlegende Frage der Koordinationschemie beleuchtet: Führt ein räumlich engerKontakt paramagnetischer Zentren zu einer magnetischen Wechselwirkung? – zu einerBindung?
Chrom(II)-acetat: eine (schwache) Metall-Metall-Vierfachbindung
Um nebenbei koordinationschemische Grundregeln aufzugreifen, werden strukturell undchemisch nah verwandte Verbindungen betrachtet: Kupfer(II)- und Chrom(II)-acetat.Generell gilt, dass Kupfer(II)- und Chrom(II)-Verbindungen strukturell eng verwandt sind.Viele Salze sind isotyp. Ursache der Ähnlichkeit ist eine gemeinsame Besonderheit derd9- bzw. der high-spin-d4-Konfiguration. Beides sind bei oktaedrischer Koordination Jahn-
31

Teller-Ionen, deren energetisch entartete Konfigurationen sich in der Besetzung des eg-Niveaus unterscheiden. Die Aufhebung der Degeneration bei diesen direkt auf dieLiganden gerichteten Orbitale geht mit einer deutlichen Verzerrung desKoordinationsoktaeders einher, meist zu einer gestreckten quadratischen Bipyramide (Oh→ D4h).
Ein Paar besonders interessanter Strukturen stellen die beiden Acetate von Kupfer(II) undChrom(II) dar. Chrom(II)-acetat entsteht bei der Umsetzung von zum Beispiel Chrom(II)-sulfat, das bei der Umsetzung von hochreinem(?) Chrom mit hochreiner(?) Schwefelsäureentsteht, mit Natriumacetat. Aus dem blassblauen Hexaqua-chrom(II)-Ion entsteht rotesChrom(II)-acetat-Monohydrat. Bei der Strukturanalyse überrascht der recht kurze Cr-Cr-Abstand von 2.36 Å.
Werden die Aqua-Liganden entfernt, sinkt der ohnehin schon kurze Abstand drastisch.Für die Gasphase ergibt die Elektronenbeugung an [Cr2(AcO)4] 1.96 Å. In kristallinemwasserfreien Chrom(II)-acetat beträgt der Cr-Cr-Abstand 2.29 Å (können Sie die merklicheDifferenz zwischen Gasphase und Kristall erklären?).
Wird Acetat durch andere verbrückende Liganden ersetzt, so lässt sich der Metall-Metall-Abstand auf bis zu 1.828 Å verkürzen – vor der Entdeckung von Fünffachbindungendem kürzesten bekannten Abstand zweier Metallatome überhaupt. Die Vorstellung einerkovalenten Bindung zwischen den Chrom-Atomen wird vor allem durch Verbindungengestützt, bei denen eine Cr2-Einheit nicht durch Liganden überbrückt ist. So wird ineinem ebenfalls roten und diamagnetischen Dichromat [Cr2R6]2− mit R = N,N-Dimethyl-aminomethyl ein Abstand von 1.84 Å zwischen den Chromatomen gefunden[crcr_unsupported_1998].
32

Wie sollte eine solche Bindung aussehen? Werden zwei isolierte, quadratisch-planareCrO4-Fragmente betrachtet, die in z-Richtung keine weitere Liganden tragen, so befindensich die 4 Valenzelektronen in den d-Orbitalen xy, xz, yz und z2. Das bei quadratisch-planarer Koordination instabilste x2−y2-Orbital bleibt unbesetzt. Nähern sich die beiden Cr-Atome entlang z an, so kommt es zuerst zur Überlappung der z2-Orbitale, also zu einerσ-Bindung. Diese wird durch weitere Bindungen unterstützt, für deren Bildung symmetrie-geeignete Orbitale zur Verfügung stehen: zwei entartete π-Bindungen (xz ↔ xz undyz ↔ yz) sowie eine δ-Bindung aufgrund der Wechselwirkung der beiden xy-Orbitale.Die rote Farbe der Dichrom(II)-Verbindungen beruht auf einem δ→δ*-Übergang. EineBindungsordnung von 4 ist bei den Chrom-Verbindungen im großen und ganzenakzeptiert, auch wenn es Gegenargumente gibt. Man beachte, dass der gemesseneDiamagnetismus auch erklärt werden kann, wenn zum Beispiel allein von einer σ-Bindungausgegangen wird und die übrigen Spins durch Austausch-Kopplung zumS=0-Grundzustand führen.
Wie ist die Stärke der Chrom(II)-Chrom(II)-Vierfachbindung einzuschätzen? Hierzu gibtes den Bericht über den recht leichten Zerfall eines analogen Zweikernkomplexes beimeinfachen Auflösen in kaum koordinierenden Lösungsmitteln [cr_2003]. Trotz derFormulierung als Vierfachbindung ist die Wechselwirkung offensichtlich eher als schwacheinzuschätzen.
Die Vierfachbindung in Octachlorido-dirhenat(III)
Bei der ersten Metall-Metall-Vierfachbindung, die in der Literatur beschrieben ist, bestehenalle diese Zweifel nicht. Im Octachlorido-dirhenat(III), [Re2Cl8]2−, in dem die Rhenium-Atome trotz ihrer Stellung in der dritten Übergangsreihe nur 2.24 Å voneinander entferntsind (vgl. 2.75 Å in Rhenium-Metall), liegen die Chlorido-Liganden in der sterischungünstigen ekliptischen Konformation vor, in der ihr Abstand kleiner als die Summe dervan-der-Waals-Radien ist. Nur in dieser Anordnung ist eine δ-Bindung möglich. Wird in dielangwellige Absorption, dem δ→δ*-Übergang, eingestrahlt, so kommt es im angeregtenZustand zur Rotation in die gestaffelte Konformation.
Kupfer(II)-acetat: Superaustausch
Gibt es eine Analogie zwischen Chrom(II)-acetat-Monohydrat und einem entsprechendenKupfer(II)-acetat-Monohydrat? Auf den ersten Blick unbedingt. Die Strukturen weisen diegleiche Konnektivität und Symmetrie auf. Lediglich der Metall-Metall-Abstand ist bei derKupferverbindung mit 2.612 Å weniger spektakulär (vgl. 2.56 Å im Metall) – was aber auchnicht zu erwarten ist, da bestenfalls eine Einfachbindung gebildet werden kann.
33

Eine Betrachtung des Kristallfeldschemas für ein quadratisch-planares CuO4-Fragmentzeigt, dass das spintragende Orbital das x2−y2-Orbital ist. Die in Frage kommendeEinfachbindung wäre also eine δ-Bindung. Die Art der Spinkopplung entspricht dagegennicht ganz der Erwartung. Zwar ist im Grundzustand S = 0, allerdings lässt sich derGrundzustand nur bei tiefer Temperatur untersuchen; bei Raumtemperatur sind die beidenSpins merklich entkoppelt. Im Bereich teilweiser Spinkopplung zwischen ca. −200 und−100 °C beschreibt der Formalismus der antiferromagnetischen Kopplung korrekt denVerlauf der zunehmenden Entkopplung.
Die Struktur von Kupferacetat lässt die Interpretation durchaus zu, den S=0-Grundzustandals Folge antiferromagnetischer Kopplung zu verstehen. Die magnetischen x2−y2-Orbitalesind auf die O-Atome der verbrückenden Acetato-Liganden ausgerichtet und es ergibt sichein antiferromagnetischer Austauschpfad im Sinne der Goodenough-Kanamori-Regeln,der im folgenden Bild für einen der vier Acetato-Liganden formuliert ist. Die übrigendrei Liganden lassen sich auf die gleiche Weise behandeln, so dass durch dasZusammenwirkung der vier Austauschpfade die hohe Kopplungskonstante von J = −294cm−1 plausibel wird. (Die x-Richtung ist in der folgenden Abbildung senkrecht zurZeichenebene gewählt, z verläuft entlang der Cu-Cu-Achse; man beachte, dass die px-Orbitale an den O-Atomen nichtbindende Wechselwirkungen mit den x2−y2-Orbitalen derKupferatome aufweisen, dass also nicht die delokalisierte π-Bindung des Carboxylato-Liganden für die antiferromagnetische Kopplung verantwortlich ist; die üblicherweiseformulierte sp2-Hybridisierung am Carboxylat-C-Atom wurde für das Kopplungsschemaaufgehoben, ferner wurden die py- und pz-Orbitale der O-Atome in geeigneter Weiselinearkombiniert.)
34

Ist im Kupfer(II)-acetat nun eine δ-Bindung oder antiferromagnetische Spinkopplung fürden S=0-Zustand verantwortlich. Das Schema suggeriert den derzeitigen Stand derDiskussion: Werden die Orbitale in der aus quantenchemischen Rechnungen erhaltenenAusdehnung dargestellt, so ergibt sich bei dem schon recht großen Abstand derKupferzentren keine nennenswerte direkte Überlappung der beiden magnetischenOrbitale, so dass die Übertragung der Spininformation auf dem abgebildetenSuperaustauschpfad den bei weitem größeren Anteil an der beobachteten Spinkopplunghaben dürfte [cu_acetate_2010].
Eine experimentelle Elektronendichtebestimmung (Prosenc et al., unpublizierterTagungsbeitrag) modifiziert dieses Ergebnis. Hier wird ca. ein Drittel der Wechselwirkungs-energie auf eine direkte Cu-Cu-Bindung zurückgeführt.
Chrom(III)-Chrom(III)-Wechselwirkungen
Der Frage „Bindung oder antiferromagnetische Kopplung?“ kann man sich besonders gutnähern, wenn Spezies in die Diskussion eingeschlossen werden, bei denen üblicherweiseeine Bindung überhaupt nicht diskutiert wird. Ein Beispiel sind mehrkernige, oxido-verbrückte Chrom(III)-Spezies. Im folgenden soll ein Kation, in dem zwei Chrom-Atome ineinem Abstand von ca. 3 Å vorliegen, auf mögliche Austauschpfade untersucht werden,um abschließend die Frage nach einer Cr-Cr-Bindung zu klären. Aufgrund der kinetischenInertheit von Chrom(III)-Zentren gegenüber Ligandenaustauschreaktionen können hierZwischenstufen auf dem Weg [Cr(H2O)6]3+ → Cr(OH)3 isoliert werden, die bei einemZentralmetall wie Eisen oder Aluminium bislang nicht gefasst wurden. Ein Beispiel ist das[Cr2(OH)2(H2O)8]4+-Ion, für das eine Strukturanalyse vorliegt:
Die Lage der spintragenden Orbitale im Raum ergibt sich aus der Struktur und demKristallfeldmodell. Die oben erwähnten Goodenough-Kanamori(-Anderson)-Regeln eignensich zur Voraussage der zu erwartenden Spinkopplung. (1) AntiferromagnetischeKopplung ist danach auf Pfaden zu erwarten, entlang denen ein nenneswertesÜberlappungsintegral resultiert. (2) Ferromagnetische Kopplung ist dagegen zu erwarten,wenn das Überlappungsintegral 0 oder nahe 0 ist, wenn also orthogonale Orbitale inräumliche Nähe geraten. Liegen beide Pfade vor, dominiert die antiferromagnetischeKopplung.
35

Im folgenden Schema sind antiferromagnetische Pfade gezeigt. Die Überlappungsintegralefür die einzelnen Wechselwirkungen werden nicht so groß sein wie bei den Kupfer(II)-Beispielen. Während dort Wechselwirkungen mit lokaler σ-Symmetrie charakteristischwaren, ergeben sich bei Chrom(III) π-Bindungen:
Ferromagnetische Austauschpfade verlaufen über orthogonale Orbitale (Blick auf dieCr2(μ-OH)2-Ebene, definiert als xy-Ebene:
Beim ferromagnetischen Spinkopplungspfad fällt auf, dass die einfach besetzten Chrom-Orbitale in der passenden Symmetrie vorliegen, um eine Cr-Cr-Bindung aufzubauen.Diese Möglichkeit wird in der Literatur jedoch kaum erwogen. Die Ursache deutet aufeinen wesentlichen Unterschied zwischen Metallen der ersten Übergangsreihe in höhererOxidationsstufe und Metallen der beiden folgenden Übergangsreihen hin: Bei den 3d-Elementen reichen die d-Orbitale nicht wirksam in den Raum hinaus, so dass zum Beispielin der gezeigten Anordnung keine Überlappung wirksam wird. Aus demselben Grundwurde bereits bei Kupfer(II)-acetat einer einzelnen δ-Bindung keine Bedeutungbeigemessen. Chrom(II)-acetat mit der Cr-Cr-Vierfachbindung erscheint demnach alsbemerkenswerte Ausnahme, man beachte aber die Argumente dafür, dass die Cr-Cr-
36

Bindung nur schwach ist. Die geringe Orbitalausdehnung bei Chrom(III), wo die Positionin der ersten Übergangsreihe mit einer hohen Oxidationsstufe zusammentrifft, beeinflusstauch die Größe der magnetischen Kopplung. Die Goodenough-Kanamori-Regeln sagenaufgrund der konkurrierenden Austauschpfade voraus, dass die dann dominante antiferro-magnetische Kopplung vorherrschen sollte. Dies wird in der Tat bei einem von zweiuntersuchten Salzen gefunden, allerdings ist aufgrund der eher geringen Überlappung derSauerstofforbitale mit den kontrahierten Chrom-Orbitalen die Kopplungskonstante klein (J= −5.7 cm−1) [2cr2oh8aq_xray_1997]. In dem zweiten Salz liegt ferromagnetischeKopplung vor, wobei die Kopplungskonstante ebenfalls klein ist (J = 5 cm−1).
Eine neuere Arbeit an einem zweikernigen Chromkomplex zeigt, mit welchen Methodendie antiferromagnetische Wechselwirkung untersucht werden kann. Wir diskutieren bei[criii_criii_af_2010] die dort vorgestellte magnetische Messung und die DFT-Untersuchung.
Übung: Berechung der magnetischen Kopplungskonstante im[Cr2(OH)2(H2O)8]4+-Ion
Wir berechnen den oben qualitativ betrachteten Dichrom(III)-Komplex mit denselbenMethoden, die in [criii_criii_af_2010] angewendet wurden. Wir schauen Eingabe-und Ausgabedatei einer BS-DFT-Rechnung mit ORCA an.
Chrom(I)-Chrom(I)-Fünffachbindungen
Die Diskussion um Metall-Metall-Vielfachbindungen konzentriert sich auf dinukleareChrom(I)-Komplexe, seit 2005 ein Aryl-chrom(II)-Komplex durch Reduktion mit KC8 ineinen zweikernigen Chrom(I)-Komplex mit einer Cr-Cr-Fünffachbindung überführt wurde[cr2_2005]. Der dort gefundene Cr-Cr-Abstand von 1.83 Å wurde inzwischen inAmidinato-chrom(I)-Komplexen nochmals deutlich unterboten: 1.75 Å in [cr2_2008],1.74 Å in [cr2_2008a], 1.73 Å in [cr2_2009]. Wir diskutieren Details einerFünffachbinding, indem wir Figure 3 in [cr2_2008a] interpretieren.
5.4 Literatur
[cu_acetate_2010]
M. Kyuzou, W. Mori, J. Tanaka:Electronic structure and spectra of cupric acetate mono-hydrate revisited.Inorg. Chim. Acta 2010, 363, 930-934.doi: 10.1016/j.ica.2009.12.035
[criii_criii_af_2010]
V. V. Semenaka, O. V. Nesterova, V. N. Kokozay, V. V. Dyakonenko, R. I. Zubatyuk, O. V.Shishkin, R. Boca, J. Jezierska, A. Ozarowski:
37

CrIII-CrIII Interactions in Two Alkoxo-Bridged Heterometallic Zn2Cr2 Complexes Self-Assembled from Zinc Oxide, Reinecke��s Salt, and Diethanolamine.Inorg. Chem. 2009, 49, 5460–5471.doi: 10.1021/ic1000123
[cr_2009]
A. Noor, G. Glatz, R. Müller, M. Kaupp, S. Demeshko, R. Kempe:Metal-Metal Distances at the Limit: Cr-Cr 1.73 Å – the Importance of the Ligand andits Fine Tuning.Z. Anorg. Allg. Chem. 2009, 635, 1149–1152.doi: 10.1002/zaac.200900175
[cr_2008a]
C.-W. Hsu, J.-S. K. Yu, C.-H. Yen, G.-H. Lee, Y. Wang, Y.-C. Tsai:Quintuply-Bonded Dichromium(I) Complexes Featuring Metal��Metal Bond Lengthsof 1.74 Å.Angew. Chem. Int. Ed. 2008, 47, 9933–9936.doi: 10.1002/anie.200803859
[cr_2008]
A. Noor, F. R. Wagner, R. Kempe:Metal��Metal Distances at the Limit: A Coordination Compound with an UltrashortChromium��Chromium Bond.Angew. Chem. Int. Ed. 2008, 47, 7246–7249.doi: 10.1002/anie.200801160
[noninnocent_2007]
K. Ray, T. Petrenko, K. Wieghardt, F. Neese:Joint spectroscopic and theoretical investigations of transition metal complexesinvolving non-innocent ligands.Dalton Trans. 2007, 1552–1566.doi: 10.1039/b700096k
[cr_2005]
T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long, P. P. Power:Synthesis of a Stable Compound with Fivefold Bonding Between Two Chromium(I)Centers.Science 2005, 310, 844–847.doi: 10.1021/ja035082g
[cr_2003]
A. R. Sadique, M. J. Heeg, C. H. Winter:A Weak, Short Metal-Metal Bond in a Chromium(II) Amidinate Complex.J. Am. Chem. Soc. 2003, 125, 7774–7775.doi: 10.1126/science.1116789
38

[crcr_unsupported_1998]
F. Becke, P. Wiegeleben, T. Rüffer, C. Wagner, R. Boese, D. Bläser, D. Steinborn:A Homoleptic [(Dimethylamino)methyl]chromium Complex with an Extremely ShortCr��Cr Bond, [{Li(THF)}2Cr2(CH2NMe2)6].Organometallics 1998, 17, 475�478.doi: 10.1021/om970821l
[2cr2oh8aq_xray_1997]
A. Drljaca, D. C. R. Hockless, B. Moubaraki, K. S. Murray, L. Spiccia:A Supramolecular Approach to the Crystallization of Polynuclear Aqua Ions:Structure and Magnetism of an 18-Crown-6 Adduct of Bis(μ-hydroxo)octaaquadichromium(III) Mesitylene-2-sulfonate Trihydrate.Inorg. Chem. 1997, 36, 1988�1989.doi: 10.1021/ic9614125
39

6 Metall-Metall-Bindungen bei frühen 4d-und 5d-Elementen
Das Fazit der Lehreinheit lautet, dass die d-Orbitale bei den Elementen der zweiten unddritten Übergangsreihe viel deutlicher den Charakter von Valenzorbitalen haben als beiden 3d-Elementen. Bei den Elementen der ersten Übergangsreihe sind die d-Orbitalenäher am Atomrumpf lokalisiert und zeigen – umso mehr, wenn eine hohe Oxidationsstufevorliegt – geringe Überlappung mit den Orbitalen der Bindungspartner. Paramagnetische,eventuell antiferromagnetisch gekoppelte, seltener ferromagnetisch gekoppelteGrundzustände sind daher bei vielen Komplexen mit 3d-Elementen die Regel, während4d- und 5d-Metalle in homologen Verbindungen Metall-Metall-Bindungen aufbauen.Lanthanoide verhalten sich in dieser Hinsicht noch extremer als die Metalle der erstenÜbergangsreihe. Hier stehen die teilweise besetzten f-Orbitale für eine Orbitalüberlappungnicht zur Verfügung, nicht einmal eine merkliche Kristallfeldaufspaltung trägt zur Chemiedieser Elemente bei.
6.1 [MoV2O2(μ-O)2(H2O)6]2+
Das Prinzip findet sich zum Beispiel bei Molybdän(V). In wässrig-saurer Lösung liegteine kationische Spezies der Summenformel MoO2(H2O)3+ vor. In der Formulierung alsEinkernkomplex läge ein d1-Zentrum vor – eine Situation, die bei den 3d-Elementenwohlbekannt ist, man denke an das Hexaaqua-titan(III)-Ion [Ti(H2O)6]3+ oder an dashydratisierte Pentaaqua-oxido-vanadium(IV)-Ion („Vanadyl“-Ion) [VO(H2O)5]2+. Mit dem4d-Element Molybdän jedoch dimerisieren die hypothetischen d1-Radikale und es entstehteine Mo-Mo-Einfachbindung:
40

6.2 [RuVINCl4]−
Bei der Diskussion der Mo-Mo-Einfachbindung führt das Kristallfeldmodell zu einerplausiblen Deutung der Bindung als einer Wechselwirkung mit lokaler σ-Symmetrie. Sinddie Liganden sehr verschieden oder ist die Struktur des Komplexes ungewöhnlich, fällt esschwerer, die metallständigen Orbitale hinsichtlich ihrer energetischen Abfolge zu ordnen.Ein Beispiel ist das [RuVINCl4]−-Ion, ein d2-Komplex, der aus RuO4, HCl und Azidzugänglich ist. Die Strukturanalyse zeigt quadratisch-pyramidalen Aufbau. Der Ru-N-Abstand ist mit 1.58 Å recht kurz. Die Bindungsverhältnisse lassen sich durch eine DFT-Rechnung klären (Orca-Eingabe- und -Ausgabedatei ).
Die Rechung auf bp/def2-tzvp-Niveau (van-der-Waals-Anziehung und wässrige Umgebungberücksichtigt) zeigt unter den fünf 4d-Orbitalen das z2-Orbital (64) als das unstabilste.Hier hätten wir es mit dem Kristallfeldmodell schwer gehabt zu entscheiden, ob der sehrnah an Ru gebundene Nitrido-Ligand die z-Richtung so sehr destabilisiert, dass auch diefreie trans-Position dies nicht wieder ausgleicht. Hat man aber aus der Rechnung diesenFixpunkt, ist die Orbitalreihenfolge klar: unter z2 finden wir x2−y2 (63), dann xz und yz(61 und 62), schließlich, als HOMO, xy (60). Ganz typisch für Komplexe der 4d- und5d-Elemente: der Singulett-Zustand, hier ermöglicht durch die Ru-N-π-Wechselwirkungen.Im Bild ist deutlich der stark π-antibindende Charakter zu sehen, der die Orbitale 61und 62 fast energiegleich mit dem x2−y2-Orbital (63) macht. Es resultiert ein so großerEnergieunterschied zwischen den Orbitalen 60 und (61, 62), dass nur das stabilere unterSpinkopplung besetzt wird.
61
60
63
64
62
Das Fazit bis hierhin: dn-Zustände sind in der zweiten und dritten Übergangsreiheentweder Metall-Metall-bindend, oder sie führen zu low-spin-Konfigurationen.
41

[MoIV3(μ3-O)(μ-O)3(H2O)9]4+
Ein weiteres Beispiel für die d2-Konfiguration, nun aber Metall-Metall-Bindungenverursachend. Die Bindungsordnung zwischen den Molybdän(IV)-Atomen im dargestelltenTetrakation ist 1.
6.3 Nb3Cl8
Die Tendenz zur Bildung von Metall-Metall-Bindungen wird bei den 4d- und 5d-Elementenherangezogen, um ungewöhnliche Eigenschaften zu deuten. Leitfähigkeitsmessungenzeigen zum Beispiel, dass in Kristallen von Nb3Cl8 1 bewegliches Elektron proFormeleinheit vorliegt. In der Kristallstruktur liegen Nb3-Fragmente in Oktaederlückeneiner dichtesten Chlorid-Packung vor. Die Ladung des Nb3-Fragments ist 8+, die formaleOxidationsstufe der Metallatome ist 8/3 entsprechend einer d-Elektronenzahl von 5 − 8/3 =7/3. Werden nun jeweils 6/3 = 2 d-Elektronen für jeweils zwei Metall-Metall-Bindungen proNiob-Atom verwendet, so bleibt 1/3 Elektron pro Niob-Atom, also 3 × 1/3 = 1 Elektron proFormeleinheit übrig – passend zum Experiment:
42

6.4 3d- und 4d-Metalle im Vergleich: CrCl2 und MoCl2
Ein direkter Vergleich der d4-Zentren CrII und dem schweren Homologen MoII zeigt dieUnterschiede zwischen der ersten und zweiten Übergangsreihe in besonders instruktiverWeise. Die Diskussion um Chrom(II)-acetat hat als Regel ergeben, dass Metall-Metall-Bindungen in den Strukturen sehr auffallend sind, aber hinsichtlich der Bindungsenergienicht dominant sind. Ohne passgenau die Cr2-Einheit unterstützende Brückenligandenwird mit einer CrII-CrII-Bindung daher eher nicht zu rechnen sein.
An dieser Stelle gelingt der Bezug zur Festkörperchemie: Welcher Aufbau lässt sichaufgrund dieser Gesetzmäßigkeiten für Chrom(II)-chlorid ableiten? Welche Unterschiedesind zu erwarten, wenn anstelle von Chrom dessen schweres Homologes eingesetzt wird?Bei Molybdän(II)-chlorid treten nämlich die deutlich stärkeren MoII-MoII-Bindungen beimAufbau des Festkörpers hervor.
Zurück zur Koordinationschemie: Welcher Aufbau darf für das Reaktionsprodukt vonMoCl2 und 2 Cl− erwartet werden? Sind in ReCl3 = 1/3 Re3Cl9 Re-Re-Bindungen möglich?
6.5 Fazit
Der Schwerpunkt des Kapitels lag auf Beispielen, bei welchen die Ausbildung von Metall-Metall-Bindungen als charakteristisches Verhalten der d-Elektronen von 4d- und 5d-Elementen auftrat. Generell ist diese Möglichkeit vor allem bei den frühenÜbergangsmetallen zu finden. Verbindungen später, elektronenreicherer Metalle derzweiten und dritten Übergangsreihe sind durch low-spin-Zustände charakterisiert, die beiBedarf von Metall-Metall-Bindungen begleitet werden. Das Prinzip wurde bereits bei[RuNCl4]− sichtbar. Als weiteres Beispiel werden Aufbau und Rh-Rh-Bindungsordnung in[Rh2(H2O)2(OAc)4] diskutiert.
6.6 Übung: Die M–M-Wechselwirkung in Kupfer(II)-,Chrom(II)- und Rhodium(II)-acetat-Monohydrat inbroken-symmetry-DFT-Rechnungen
Die in den beiden letzten Kapiteln diskutierten Fragen, ob eine Metall-Metall-Wechsel-wirkung in einem binuclearen Komplex durch eine ferro- oder antiferromagnetischeSpinkopplung beschrieben werden sollte oder ob vielleicht von einer Bindung gesprochenwerden sollte, ist immer dann nicht trivial, wenn die Metallatome recht nahe nebeneinanderliegen – sie also nicht nur durch einen ausgedehnten Brückenliganden miteinanderkommunizieren können. Die besprochenen Metall(II)-acetate sind solche Beispiele. Ein
43

Brückenligand ist vorhanden und bietet Superaustauschpfade, der M–M-Abstand aber liegtim Bereich des Atomabstands im Metall selbst und könnte eine Bindung anzeigen.
Das übliche Routineverfahren, mit geringem Aufwand die Austauschwechselwirkung vonMetallatomen zu berechnen, ist eine broken-symmetry-DFT-Rechnung (BS-DFT). DasVerfahren ist einfach und „billig“ im Sinne geringer Rechnerressourcen, hat aber fürdie Lehre den Nachteil, dass der dort verwendete broken-symmetry-Zustand weniganschaulich ist. Zuerst aber der technische Teil. Wir verwenden Orca, um zuerst diejeweilige Molekülstruktur zu optimieren (lässt man in der Literatur oft auch weg und fixiertden M-M-Abstand auf den gefundenen Wert; hier wurden die drei Strukturen mit bp/tzvp +vdW + cosmo(water) verfeinert, also reines Dichtefunktional mit nicht zu teurem Baissatz,Grimmes van-der-Waals-Korrektur und Kontinuummodell mit der Dielektrizitätskonstantevon Wasser); im Detail wird optimiert: Cu im BS-Zustand, Cr auch, Rh im spingepaartenSingulettzustand.
Anschließend werden mit dem tpssh-Hybridfunktional und def2-tzvp (außerdem wie zuvorvdw und cosmo) Eingabedateien geschrieben für Cu, Cr, Rh und dabei die Atom-koordinaten aus den Optimierungen verwendet.
Bei der Rechnung wird nun dem ferromagnetisch gekoppelten Zustand (in der Literaturleider „high-spin“ genannt) ein broken-symmetry-Zustand gegenübergestellt, indem dieSpins an dem Zentrum mit der geringeren Zahl ungepaarter Elektronen herumgedrehtwerden; sind wie hier die beiden Zentren gleich, ist es egal welches. In der Orca-Eingabedatei geht das so: Die Multiplizität, die wie üblich der letzte Eintrag auf der xyz-Anweisung ist, gilt für den ferromagnetisch gekoppelten Zustand. Bei Cu steht daher eine„3“, da die beiden α-Spins dann ein Triplett bilden. Bei Chrom(II) sind die beiden Bausteinehigh-spin-d4 („high-spin“ jetzt wieder individuell), beide zusammen in ferromagnetischerKopplung also S = 2 + 2 = 4, 2S + 1 = 9, und: *xyz 0 9. Für Rhodium(II) gehen wir von zweiZentren aus, die jedes für sich low-spin-d7 sind, also 1 Spin pro Zentrum, die zusammenwie bei Kupfer(II) ein Triplett ergeben.
Anschließend wird der broken-symmetry-Zustand eingerichtet, und zwar zwischen „%scf“und „end“: flipspin 1 heißt: drehe die Spins von Atom 1 um (das ist bei Orca, bei demAufzählungen immer mit 0 beginnen, das zweite Atom in der Liste). Dann: finalms 0, lies:final Ms = 0, womit die Spinorientierungsquantenzahl im antiferromagnetisch gekoppeltenZustand angegeben wird.
%scfflipspin 1finalms 0
end
* xyz 0 3Cu ...Cu ......
Das Programm geht jetzt hin und beginnt mit der ferromagnetisch gekoppelten Anordnung.Anschließend werden die Spins umgedreht und es wird versucht, in einer unrestricted-
44

Rechnung („uks“ = unrestricted Kohn Sham) von diesem antiferromagnetisch gekoppeltenZustand zu retten, was in einer SCF-Rechnung zu retten ist. Im Falle magnetischerKopplung kommt dann ein broken-symmetry-Zustand heraus, der stark vomErwartungswert für S(S+1)=0 abweicht (Tabelleneintrag <S2>BS). Man sagt, dass er„spinkontaminiert“ sei. Energetisch liegt er zwischen dem wahren antiferromagnetischgekoppelten und dem ferromagnetisch gekoppelten Zustand (der Hintergrund derGeschichte ist, dass der antiferromagnetisch gekoppelte Zustand im Gegensatz zumferromagnetisch gekoppelten Zustand ein Multikonfigurationszustand ist, der sich durchDFT-Methoden nicht darstellen lässt). Erstaunlicherweise ergibt sich die Kopplungs-konstante J aus einer solchen Rechnung oft ganz gut; schauen Sie sich ziemlich amEnde der Ausgabedateien den Wert für J(3) an, der als Energiedifferenz zwischenferromagnetisch gekoppeltem und broken-symmetry-Zustand dividiert durch die Differenzder <S2>-Werte für die beiden Zustände berechnet wird (der experimentelle Wert bei derKupferverbindung wird zwischen −290 und −300 cm−1 angegeben).
Sie finden alles in den out-Dateien für Cu, Cr, Rh (die out-Dateien sind sehr lang, da sie diegesamte Orbitalinfomation enthalten, falls Sie sich zum Beispiel in Gabedit Orbitalwechsel-wirkungen anschauen wollen).
Nach diesen wenig befriedigenden rechentechnischen Details können wir aber jetzt aufden Punkt kommen – und dabei viele Aussagen wiederentdecken, die wir zuvor qualitativgemacht haben. Wir schauen dazu die folgende Tabelle an, in der J das J(3) der Rechnungist. Sehr nützlich ist der Eintrag Sαβ, der ein Überlappungsintegral darstellt, das in Orcain einer Auflistung von „Corresponding Orbitals“ zu finden ist (cave: auch Orbitale zählenin Orca ab 0, beim Anschauen in Gabedit ab 1, außerdem ist das Formelzeichen „S“ jetztwirklich gut ausgelastet – Sαβ wird in der Literatur meist ohne „αβ“ hingeschrieben undist trotzdem kein Spin). Sαβ = 0 sagt uns: keine Überlappung, Sαβ = 1 heißt: ein ganznormales spingepaartes Elektronenpaar (das sind mit Abstand die meisten in der Liste),0 < Sαβ < 1: zeigt ein mehr (ca. 0) oder weniger (ca. 1) spinpolarisiertes Elektronenpaaran. Der Zahlenwert bei Cu von 0.17 ist dabei keine Überlappung von zwei allein aufKupfer liegenden Atomorbitalen, sondern es ist die Überlappung zwischen einem α-MOund einem β-MO, die sich auch über die Brückenliganden hinweg erstrecken. Zusammenmit dem negativen Vorzeichen von J sieht so eine durch Superaustausch vermittelteantiferromagnetische Spinkopplung aus.
Das Gegenstück ist der Rhodiumkomplex. Hier war Ihnen erzählt worden, dass es sichum eine normale Rh-Rh-Bindung handelt, man würde sich also eine broken-symmetry-Rechung ersparen und das ganze (restricted) als normales Singulett behandeln (so wurdedie Struktur optimiert). Macht man aber überflüssigerweise eine broken-symmetry-Rechnung, kommt ein klares Ergebnis heraus: der BS-Zustand ist nicht spinkontaminiert(<S2>BS = 0), der ferromagnetisch gekoppelte diradikalische Zustand ist extrem unstabil(siehe J in kJ mol−1), und das Überlappungsintegral ist 1.
Und der Chromkomplex, dem die Literatur die hohe Bindungsordnung 4 zuweist, dieBindung dann aber gleich wieder als sehr schwach einstuft? Schauen Sie hier vor allemauf die vier Überlappungsintegrale im Grenzorbitalbereich, die in der Tabelle in σ, π (inder Orca-Ausgabe die beiden vorletzten Einträge) und δ unterschieden sind. Man kommt
45

zu dem Schluss, dass nur die σ-Bindung nennenswert ist, aber wie bei Kupfer ist auchhier J eher bescheiden, wenn wir uns das Ergebnis auf einer Kilojoule-pro-Mol-Skalaklar machen. Ganz interessant: die δ-Wechselwirkung stellt sich beim Kupfer- und beimChromkomplex sehr ähnlich dar.
CuII (d9) CrII (d4) RhII (d7)M-M/Å (Xray) 2.612 2.362 2.386M-M/Å (DFT) 2.529 2.290 2.430J/cm−1 −309 −809 −6380J/kJ mol−1 −3.7 −9.7 −76.3<S2>BS 0.97 3.27 0.00Sαβ (σ) – 0.73 1.00Sαβ (π) – 0.29 –Sαβ (δ) 0.17 0.19 –
Zum Schluss noch einmal der Hinweis: der Rhodiumkomplex würde in einer Publikationsicher nicht eine broken-symmetry-Behandlung erfahren.
46

7 Donor-Akzeptor-Liganden undNichtunschuldige Liganden
7.1 Cyanido-Komplexe: allgemeines
High-spin-Cobalt(II) illustriert besonders anschaulich das Prinzip, dass die d-Elektronender 3d-Elemente nur begrenzt die Chemie dieser Zentralmetalle mitbestimmen, sich alsowie Valenzelektronen verhalten. Metall-Metall-Bindungen werden nicht beobachtet,darüber hinaus bedingt die ungefähr gleiche Ligandfeldstabilisierungsenergie beiverschiedenen Koordinationsfiguren, dass Kristallfeldeffekte kaum wahrnehmbar sind.
Dies alles gilt jedoch nur im high-spin-Fall. Starkfeldliganden, die eine d7-low-spin-Anordnung verursachen können, verändern dieses Bild völlig. [Co(CN)4]2− ist quadratischplanar, [Co(CN)5]3− neigt zur Dimerisierung zu [Co2(CN)10]6− unter Aufbau einer Co-Co-Bindung, ein dem [Fe(CN)6]4− entsprechendes [Co(CN)6]4− ist nicht bekannt. Vor allemdie Bildung einer Metall-Metall-σ-Bindung bei der Dimerisierung rückt das low-spin-Cobalt(II) in die Nähe seiner schweren Homologen. Völlig analog bildet Rhodium(II) eineRh-Rh-σ-Bindung im oben erwähnten [Rh2(H2O)2(OAc)4].
Der Unterschied zwischen Cyanido- und Halogenido-Liganden wird im Orbitalschemadeutlich. Halogenido-Liganden sind σ- und π-Donoren. Cyanido-Liganden sind bessereσ-Donoren, deren HOMO, das 3σ-Orbital, wirksam auf das Zentralmetall zuweist. DasHOMO−1, das 1π-Orbital, ist dagegen auf das elektronegativere N-Atom ausgerichtet, sodass dessen Überlappung mit dem Zentralmetall schwach ist (vgl. die analoge Situationbei den Grenzorbitalen des Carbonyl-Liganden). Cyanid ist also aufgrund seines Aufbausaus einem elektronegativen N- und einem weniger elektronegativen C-Atom ein Ligand,dessen σ-Donoreigenschaft verstärkt und dessen π-Donoreigenschaft geschwächt ist.
Beides trägt zur Stellung des Cyanido-Liganden in der spektrochemischen Reihe bei.Liegt ein Zentralmetall in niedriger Oxidationsstufe vor, kommt ein weiterer Aspekt hinzu,der in der Literatur jedoch kontrovers behandelt wird. Das LUMO, das 2π-Orbital, istebenfalls deutlich auf das Zentralmetall ausgerichtet. Dessen Symmetrie erlaubt eineÜberlappung mit lokaler π-Symmetrie mit geeigneten d-Orbitalen des Zentralmetalls, beieinem oktaedrischen Komplex mit den t2g-Orbitalen. Sind diese besetzt, wird eineRückbindung erhalten. Das Metall wirkt hierbei als Lewis-Base, der Ligand als Säure. In
47

der Summe zeigt sich Cyanid als starke σ-Base, als schwache π-Base und eventuell alsπ-Säure, wenn geeignete besetzte Orbitale am Zentralmetall zur Verfügung stehen. Unterwelchen Umständen eine π-Rückbindung einen merklichen Beitrag hat, ist Gegenstandder Diskussion. Aus Röntgenspektren wurde abgeleitet, dass selbst low-spin-Eisen(II) imHexacyanidoferrat(II) trotz seiner recht hohen Oxidationsstufe und seiner folglich eherstärker kontrahierten 3d-Orbitale eine „nennenswerte“ Rückbindung aufbauen[cyano_2001].
Die bei den Cyanidocobaltaten(II) gefundenen Verhältnisse zumindest lassen sichzwanglos mit der starken σ-Basizität der Cyanido-Liganden allein erklären. In einemoktaedrischen [Co(CN)6]4− wäre die Aufspaltung zwischen eg und eg* groß, eg* wäre starkantibindend. In dieses Schema sind 7 + 6 × 2 = 19 Elektronen einzufüllen, 1 Elektron würdesich also – anders als im [Fe(CN)6]4−-Ion – im stark antibindenden Zustand wiederfinden.Komplexe mit stark σ-basischen Liganden, zu denen zum Beispiel auch Hydrido- undAlkyl-Liganden zählen, beachten daher eine Obergrenze von 18 Elektronen. Im Fall des[Co(CN)4]2−-Ions führt die gleiche Betrachtung zur quadratisch-planaren Ligandanordnunganstelle der tetraedrischen. Auch ist es günstig, bei starkem Feld das stark antibindendex2−y2-Orbital nicht zu besetzen. Bei tetraedrischer Anordnung gibt es dagegen keineinzelnes unstabiles Orbital.
7.2 Cyanido-Komplexe: Rückbindung?
Die qualitativen Aussagen, vor allem die sich häufiger ändernde Einschätzung, ob eineRückbindung in Cyanidokomplexe von Metallen in positiver Oxidationsstufe nennenswertist oder nicht, sollen anhand dreier Komplex-Ionen näher betrachtet werden. Nebenbeiwenden wir eine weiter oben schon vorgestellte Publikation an, mit deren Hilfe diemöglichen Spinzustände eines Formeltyps eingegrenzt werden[orbital_deformation_2006] und gehen der Frage nach, ob das Erstaunen derAutoren von [cyano_2005] gerechtfertigt ist, das erste high-spin-Cyanidochromathergestellt zu haben. Die Formel des high-spin-Chromats ist [Cr(CN)5]3−; im Kristall liegenquadratisch-planare Konformere neben verzerrt trigonal-bipyramidalen vor. Die fünf α-Spin-d-Orbitale spalten in der folgenden Weise auf:
48

Die Aufspaltung der d-Orbitale im high-spin-Cyanido-Komplex[CrII(CN)5]3− für die beiden Konformere (bp/tzvp). Die vier stabilenOrbitale enthalten ein Elektron.
Wir diskutieren als Erstes, warum bei der d4-Konformation die Spinzustände S = 0 und 1nicht vorkommen sollen, sondern nur S = 2. Anschließend gehen wir auf einen weiterenPunkt der Publikation ein, und zwar auf die Bedeutung einer p-Orbital-Zumischung zu d-Orbitalen. Hierzu betrachten wir das quadratisch-planare Konformer:
Der high-spin-Cyanido-Komplex [CrII(CN)5]3−, HOMO (Orbital 48a)des C4v-Konformers (auf bp/tzvp-Niveau 3.2 kJ mol−1 unstabiler alsdas D3h-Konformer); isovalue = 0.04.
Nun zur Frage der Rückbindung. Das α-MO 45 sollte eine solche Bindung zeigen, siescheint aber keine besondere Bedeutung zu haben:
[CrII(CN)5]3−, Orbital 45a des D3h-Konformers; isovalue = 0.04.
49

Man könnte einwenden, dass zu einer richtigen Rückbindung Elektronenpaare gehören,die der high-spin-d4-Komplex nicht zu bieten hat. Wir vergleichen daher das analogaufgebaute low-spin-d8-Ion [NiII(CN)5]3−:
Das D3h-Konformer des [NiII(CN)5]3−-Ions, Orbital 45a (das C4v-Konformer ist auf bp/tzvp-Niveau 0.6 kJ mol−1 unstabiler; isovalue =0.04.
Auch hier zeigt sich keine nennenswerte Rückbindung. Zum Vergleich schauen wir aufdas [Cr(CO)5]2−-Ion, das alle Voraussetzungen für eine Rückbindung mit sich bringt: dieOxidationsstufe des Metalls ist sehr niedrig, der Ligand ist CO.
[Cr(CO)5]2−, Orbital 45a; isovalue = 0.04.
Tatsächlich ist es nun überhaupt kein Problem, die Delokalisation des Metall-Orbitals aufdie C-Atome der Carbonylliganden zu erkennen.
Das Fazit: bei positiver Oxidationsstufe des Metalls und Cyanid als Ligand ergibt sichdie große Kristallfeldaufspaltung aus der Kovalenz der M-C-Bindung, nicht aus einerRückbindung. Die hohe σ-Basizität unterscheidet den Cyanido-Liganden von isosterenTeilchen wie CO und NO+. Man vergleiche hierzu die Stabilitäten von HCN und HCO+.Trotz der formalen Gemeinsamkeiten unterscheidet sich die Carbonyl-Komplexchemieerheblich von der Cyanid-Metall-Chemie. Stabile Cyanido-Komplexe von Metallen mitpositiver Oxidationsstufe gibt es in großer Zahl, nicht jedoch die analogenCarbonylkomplexe. Man vergleiche [Cu(CN)4]3− und einen der unbeständigen Carbonyl-kupfer(I)-Komplexe wie [Cu(NH3)3(CO)]+ oder Cu(CO)Cl.
Besonders illustrativ ist der Vergleich zwischen [Fe(CN)6]4− und [Fe(CO)6]2+. Das Aniondes gelben Blutlaugensalzes ist seit fast 200 Jahren bekannt; es ist so stabil, dasses ungiftig ist. Das Hexa(carbonyl)eisen(II)-Ion wurde dagegen erst kürzlich von Willner
50

und Aubke in supersauren Medien hergestellt, also in Abwesenheit aller konkurrierenderLiganden. Stabile Carbonylmetall-Komplexe sind dagegen unter Bedingungen bekannt,die die höhere π-Acidität des Kohlenmonoxids ausnutzen, also mit Metallen in niedrigerOxidationsstufe, die eine hinreichend große Metallbasizität aufbringen, um die M-CO-Rückbindung zu stärken. Komplexe mit dem NO+-Ligand setzen diesen Trend fort.Beständige Nitrosyl-Komplexe vom NO+-Typ sind daher nur von elektronenreichenMetallen bekannt.
Um diese Prinzipien verständlich zu machen, werden die folgenden Eisen-Komplexionenverglichen: [Fe(CO)3NO]−, [Fe(CN)5NO]2− und [Fe(H2O)5NO]2+, dem farbgebendenKomplex des „braunen Rings“. Alle drei Nitrosyl-Eisen-Komplexe haben eineGemeinsamkeit: das Fe-N-O-Fragment ist linear. Dies legt in einem einfachen VB-Bildnahe, dass der Ligand als NO+ vorliegt, isoster mit CO und CN−. So einfach ist es abernicht, denn – Nitrosylliganden sind nicht-unschuldig.
7.3 Der nicht-unschuldige Nitrosyl-Ligand
Nicht-unschuldige Liganden, engl. non-innocent ligands, entziehen sich den üblichenIUPAC-Regeln zur Bestimmung der Oxidationsstufe eines Metalls. Wichtig wird nun diephysikalische (spektroskopische) Oxidationsstufe. Ob ein Ligand unschuldig oder nicht-unschuldig – also redox-aktiv – ist, hängt auch vom Zentralmetall, seiner Oxidationsstufeund seinem Spinzustand ab.
Einschub: Biochemisch wichtige nicht-unschuldige Liganden
Diese werden vor allem in der Vorlesung Bioanorganische Chemie behandelt. Es handeltsich um
NO, das als NO+, NO-Radikal, Singulett-NO− oder Triplett-NO− gefunden wird.Dieser Ligand wird im Folgenden ausführlich behandelt.
O2, das als Triplett- oder Singulett-O2 (Paulingsches Modell vonsauerstoffbeladenem Myoglobin/Hämoglobin), als Hyperoxido-Ligand (WeissschesMb/Hb-Modell), oder Peroxid (Hämerythrin, Hämocyanin) gebunden sein kann.
Tyrosinat, das als Phenolat-Anion oder als Phenoxy-Radikal gefunden wird(Galactose-Oxidase).
Porphyrine, deren normale por2−-Form zu einem radikalischen por•−- oxidiert seinkann (Cytochrom P450).
[Fe(CO)3NO]−
Der Standardfall des beständigen Nitrosylkomplexes mit einem fest gebundenen NO-Ligand ist im [Fe(CO)3NO]−-Ion realisiert. Die Reaktionsbedingungen für die Synthese
51

wässriger Lösungen der intensiv gelben Alkali-tricarbonyl-nitrosylferrate(1−) zeigen schon,dass das Komplexanion keine unbeständige Spezies ist. So wird eine wässrige, schwachalkalische Alkalinitrit-Lösung mit Pentacarbonyl-eisen(0) solange am Rückfluss zumSieden erhitzt, bis kein [Fe(CO)5] – eine mit Wasser nicht mischbare farblose Flüssigkeit –im Rücklauf des Kühlers mehr erkennbar ist:
[Fe(CO)5] + NO2− + Ca(OH)2 → [Fe(CO)3NO]− + CO + H2O + CaCO3
Sowohl [Fe(CO)5] als auch [Fe(CO)3NO]− sind typische Komplexe mit stark π-acidenLiganden, die der 18-e-Regel genügen.
Um die Elektronenbilanz festzustellen, wird üblicherweise (1) aus der Ladung desKomplexes und der (von der IUPAC festgesetzten) Ladung der Liganden dieOxidationsstufe des Zentralmetallatoms bestimmt, dann (2) die Zahl der (n−1)d-Elektronendes Metalls ermittelt (der ns-Zustand wird dabei als unbesetzt angenommen) und (3) diesed-Elektronenzahl und die Zahl der (laut IUPAC-Regel) von den Liganden beigetragenenElektronen addiert.
Für [Fe(CO)5] ergibt sich wegen des Neutralliganden CO 0 als Oxidationsstufe des Eisensentsprechend 8 3d-Elektronen, hinzugezählt werden 5 × 2 = 10 Elektronen, da CO als 2e-Donor zählt, so dass sich insgesamt 18 e ergeben. Diese Zählweise steht im Einklang mitden physikalischen Eigenschaften des Pentacarbonyleisens – formale und physikalische(spektroskopische) Oxidationsstufe sind gleich. Dies ist bei Nitrosylkomplexen eher dieAusnahme. Der Nitrosylligand ist als neutraler Dreielektronen-Donor definiert. Für das[Fe(CO)3NO]−-Ion ergibt die Rechnung dann −I als Oxidationsstufe des Zentralmetallsentsprechend 9 d-Elektronen. Hinzuaddiert werden 3 × 2 = 6 Elektronen von den dreiCarbonyl-Liganden und 3 Elektronen von NO. Die Summe ist wieder 18 Elektronen.
Die Behandlung von NO als Neutralligand führt jedoch zu einer Reihe von Problemen beider Deutung der physikalischen Daten. So zeigt die Strukturanalyse eine lineare Fe-N-O-Einheit, während das VSEPR-Modell eine an N gewinkelte Struktur nahelegt. WerdenAbstände (in der Abbildung in Å, die Zahlenwerte sind Mittelwerte im Natriumsalz) undWinkel hinzugenommen, so ergibt sich eher die Vorstellung von einem mit CO isosterenNO+-Ligand, der den Carbonyl-Liganden hinsichtlich der π-Acidität deutlich übertrifft:
52

Der größere mittlere N-Fe-C-Winkel gegenüber einem kleinen C-Fe-C-Winkel zeigt einehöhere Fe-N- im Vergleich zur Fe-C-Bindungsordnung an. Im Einklang hiermit sind die Fe-N/C-Abstände. Bezeichnend aber ist vor allem die deutliche Aufweitung des N-O-Abstandsim Komplex gegenüber dem Wert für eine N-O-Dreifachbindung. Die entsprechendenWerte unterscheiden sich für CO viel weniger (C-O-Abstand in freiem Kohlenmonoxid:1.128 Å). Mit der Ladung +1 für den Nitrosylligand ergibt sich für Eisen die physikalischeOxidationsstufe -II, die einer d10-Konfiguration entspricht, wie sie auch im isosterenTetracarbonyl-ferrat(−II) gefunden wird. Dieses homoleptische Ferrat entsteht bei derUmsetzung von [Fe(CO)5] mit wässriger Lauge (Hiebersche Basereaktion):
[Fe(CO)5] + 4 OH− → [Fe(CO)4]2− + 2 H2O + CO32−
Der Zweielektronen-Reduktion des Eisenzentrums steht bei beiden Reaktionen dieOxidation eines Äquivalents CO zu CO2 gegenüber, das unter den basischenReaktionsbedingungen als Carbonat anfällt. Das Natriumsalz des Tetracarbonylferrats wirdals „Collmans Reagenz“ verwendet. Die höhere π-Acidität des NO+-Liganden im Vergleichmit CO lässt sich auch anhand der C-O-Schwingungsfrequenzen nachvollziehen: 1790cm−1 bei Tetracarbonylferrat gegenüber ca. 1900 cm−1 bei Tricarbonylnitrosylferraten, beidenen der NO-Ligand mehr Metallbasizität auf sich zieht (vgl. 2143 cm−1 bei freiem CO).
[Fe(H2O)5NO]2+
Die Vorstellung, dass eine lineare M-N-O-Einheit einen CO-analogen NO+-Ligandenanzeigt, ist in Lehrbüchern verbreitet, aber falsch. Der von der Nitratprobe bekanntebraune Ring, dessen Chromophor bei der Nitritprobe im gesamten Probevolumen entsteht,wird durch den Pentaaqua-nitrosyl-eisen(II)-Komplex [Fe(H2O)5NO]2+ hervorgerufen. Fürdieses Ion liegt keine Strukturanalyse vor, es herrscht jedoch Einigkeit, dass diespektroskopischen Daten und die Ergebnisse von Rechnungen ein lineares Fe-N-O-Fragment hinreichend belegen. Der Grundzustand ist ein Quartett (S = 3/2). In älterenLehrbüchern wird der Nitrosyl-Ligand daher als NO+ eingestuft, der an ein high-spin-d7-Eisen(I)-Zentralatom gebunden ist. Die physikalische Oxidationsstufe des formalenEisen(II)-Zentrums wäre demnach +I.
Die Unterschiede der beiden Nitrosyleisen-Komplexe [Fe(CO)3NO]− und [Fe(H2O)5NO]2+
sind gravierend. Während das Ferrat(−II) thermisch belastbar ist und hohe Reaktivität nurgegenüber Oxidationsmitteln zeigt, ist das Ion des braunen Rings nur wenig stabil undzerfällt leicht wieder. Aktuelle Rechnungen zeigen, dass das Pentaaqua-nitrosyl-eisen-Ioneher als FeII-NO• oder als FeIII-NO−-Komplex zu verstehen ist [brown_ring_2004],[brown_ring_2002]. Wie aber ist dann die lineare Anbindung des Nitrosyl-Ligandenan das Eisenzentrum zu verstehen? Wäre gemäß dem VB-Bild zum Beispiel für denFeIII-NO−-Komplex nicht ein Fe-N-O-Winkel von ca. 120° zu erwarten? Man beachtejedoch die geringe Stabilität der Fe-NO-Bindung. Das VB-Bild steht für die Koordinationeines Metallzentrums an eine Singulett-NO−-Spezies. Ein high-spin-Eisenzentrum derOxidationsstufe +II oder +III führt jedoch nicht zu einem so drastischen Eingriff in dieElektronenstruktur des Liganden. Dieser ist isoelektronisch zu O2 und liegt wie dieses ineinem Triplett-Grundzustand vor. Die nun mögliche Wechselwirkung zwischen den beiden
53

π*-Orbitalen des Liganden und symmetrisch passenden Eisen-Orbitalen ist für eine vonzwei Wechselwirkungen dargestellt; die gezeigte Spinkopplung wird meist als antiferro-magnetische Kopplung verstanden:
Es wird deutlich, dass sowohl Triplett-NO− als auch, in alternativer Betrachtung, dasNO-Radikal linear an das Eisenzentrum binden. Aus der DFT-Rechnung in[brown_ring_2004] folgt ein Energieschema für die Grenzorbitale des Komplexes.Dargestellt sind diejenigen Orbitale, die im Bereich des FeNO-Fragments lokalisiert sind.Die Nummern verweisen auf Tabelle 8 in [brown_ring_2004], außerdem sind diebeteiligten Fe-Orbitale genannt; man beachte aber, dass hiermit ein Elektron nicht alleindem jeweiligen Orbital zugewiesen wird; die beiden besetzten β-Spin-Orbitale haben zumBeispiel jeweils zur Hälfte Metall- und NO-π*-Charakter. Die braune Farbe von[Fe(H2O)5NO]2+ geht auf Übergänge des Typs π→σ* zurück:
54

Noch ein Kommentar zur Nomenklatur: Zur Beschreibung von Nitrosyl-Metall-Komplexenist die von Enemark und Feltham eingeführte Angabe der Elektronenzahl des MNO-Fragments allgemein üblich. Im braunen Ring liegt zum Beispiel ein {FeNO}7-Zentrum vorentsprechend 7 Elektronen als Summe der d-Elektronen des Metalls und der Elektronenin antibindenden NO-Orbitalen. Durch diese Definition umgeht man eine Festlegung aufeinen speziellen Bindungsmodus.
[Fe(CO)3NO]− und [Fe(H2O)5NO]2+ erscheinen als zwei Extremfälle für die Bindung einesNitrosylliganden. Das Eisen(−II)-Zentrum baut eine starke Rückbindung zum NO-Ligandenauf und es entsteht ein Komplex, der nicht zur Dissoziation des Nitrosyl-Liganden neigt.Das Lewis-saure high-spin-Eisen(+II)-Zentrum dagegen ist nicht in der Lage, einenennenswerte Rückbindung aufzubauen, umgekehrt ist der Nitrosyl-Ligand kein besondersguter Donor – heraus kommt ein zersetzlicher Komplex.
[Fe(CN)5NO]2−
Die für eine starke Rückbindung nicht förderliche Oxidationsstufe +II liegt jedoch auchim lange bekannten Nitroprussid-Anion vor. Dessen Natriumsalz, Natrium-pentacyanido-nitrosyl-ferrat(III)-Dihydrat, Na2[Fe(CN)5NO] · 2 H2O, ist ein geläufiges Medikament, daszum Beispiel bei Operationen nach Bedarf infundiert wird um den Blutdruck des Patientenschnell und wirksam zu senken. Nach der Entdeckung der Hormonwirkung von NO wurdeerkannt, dass das Ferrat-Ion unter physiologischen Bedingungen NO abspaltet, worauf dieWirkung beruht. NO ist also offensichtlich auch an diesem Eisen(II)-Zentrum weniger festgebunden als es die Strukturanalyse nahelegt:
Die Strukturparameter des {FeNO}6-Zentrums im Natrium-nitroprussid zeigen wieder einlineares Fe-N-O-Fragment. Die Fe-N-Bindung ist recht kurz, die gegenüber demCarbonylferrat deutlich geschwächte Rückbindung zeigt sich jedoch am stark verkürztenN-O-Abstand. Um zu verstehen, dass die Fe-N-Bindung bei weitem nicht so schwachist wie beim Aqua-nitrosyl-Komplex, ist die low-spin-Konfiguration des Cyanidokomplexeszu beachten. Die sich andeutende Mittelstellung der Fe-N-Bindung – nicht so stabil wiebei einem Eisen(−II)-Zentrum, nicht so schwach wie bei high-spin-Eisen(II) – wird am
55

besten deutlich, wenn eine aufsehenerregende Eigenschaft des Natrium-nitroprussids indie Diskussion einbezogen wird (Hauser, 1977). Beim Bestrahlen von SNP-Kristallen (SNP= sodium nitroprusside) mit grünem Laserlicht bei tiefer Temperatur wird ein metastabilerZustand (MS2) erhalten, der einer Population von etwa der Hälfte aller Ferrat-Ionenentspricht. Beim Aufwärmen über 165 K geht MS2 in einen weiteren metastabilen ZustandMS1 über. Beide metastabile Zustände haben in der Kälte eine beliebige Lebensdauer.Der Grundzustand kann entweder durch Aufwärmen auf Raumtemperatur oder durchBestrahlen mit rotem Laserlicht wieder erreicht werden.
Die ungewöhnliche Lebensdauer deutet an, dass es sich bei den metastabilen Zuständennicht um elektronische Anregungen handelt, sondern dass eine strukturelle Veränderungstattfindet. Diese wurde 1997 erstmals durch Röntgenbeugung nachgewiesen. AktuelleRechnungen unterstützen den Befund, dass in MS2 die Nitrosylgruppe eine Art π-Donor-Bindungsmodus einnimmt, der in die Isonitrosyl-Struktur MS1 relaxiert. Im Einklang mit derArbeitshypothese, dass eine Fe-N-Rückbindung bei Eisen(II)-Zentren nicht nenneswert ist,unterscheiden sich die N-O-Abstände in den drei Formen kaum:
Eine Übersicht zu der an SNP beschriebenen photoinduzierten Bindungsisomerie (engl.photoinduced linkage isomerism) gibt [pli_review_2002]. Wir diskutieren, warum esgenau die beiden beobachteten metastabilen Isomere gibt. Hierzu machen wir uns dieverschiedenen Komponenten der Fe-NO-Wechselwirkung klar, bei MS2 zum Beispiel diefolgenden:
56

7.4 Aktuelle Entwicklungen
Der NO-Bindungsmodus und die Anregung von Bindungsisomeren hat offensichtlichbiochemische Bedeutung. Im Mittelpunkt aktueller Arbeiten stehen daher Nitrosyl-Häm-Komplexe, die im natürlichen Stickstoffkreislauf (Scheme 1 in [noh_heme_2010]) eineRolle spielen. Die Frage linearer oder gewinkelter Fe-N-O-Bindung im {FeNO}7-Fall wird in[feno7_dft_2010] umfassend behandelt.
7.5 Literatur
[no_pli_heme_2010]
N. Xu, J. Yi, G. B. Richter-Addo:Linkage Isomerization in Heme-NOx Compounds: Understanding NO, Nitrite, andHyponitrite Interactions with Iron Porphyrins.Inorg. Chem. 2010, 49, 6253–6266.doi: 10.1021/ic902423v
[no_heme_dynamics_2010]
W. R. Scheidt, A. Barabanschikov, J. W. Pavlik, N. J. Silvernail, J. T. Sage:Electronic Structure and Dynamics of Nitrosyl Porphyrins.Inorg. Chem. 2010, 49, 6240–6252.doi: 10.1021/ic100261b
[no_heme_bonding_2010]
L. E. Goodrich, F. Paulat, V. K. K. Praneeth, N. Lehnert:Electronic Structure of Heme-Nitrosyls and Its Significance for Nitric OxideReactivity, Sensing, Transport, and Toxicity in Biological Systems.Inorg. Chem. 2010, 49, 6293–6316.doi: 10.1021/ic902304a
[noh_heme_2010]
M. R. Kumar, J. M. Fukuto, K. M. Miranda, P. J. Farmer:Reactions of HNO with Heme Proteins: New Routes to HNO–Heme Complexes andInsight into Physiological Effects.Inorg. Chem. 2010, 49, 6283�6292.doi: 10.1021/ic902319d
[feno7_dft_2010]
J. Conradie, K. H. Hopmann, A. Ghosh:Understanding the Unusually Straight: A Search For MO Insights into Linear {FeNO}7
Units.
57

J. Phys. Chem. B 2010, 114, 8517–8524.doi: 10.1021/jp101847y
[orbital_deformation_2006]
S. Alvarez, J. Cirera:How High the Spin? Allowed and Forbidden Spin States in Transition-MetalChemistry.Angew. Chem. Int. Ed. 2006, 45, 3012�3020.doi: 10.1002/anie.200503492
[cyano_2005]
K. J. Nelson, I. D. Giles, W. W. Shum, A. M. Arif, J. S. Miller:The Myth of Cyanide Always Being a Strong-Field Ligand: Synthesis and StructuralCharacterization of Homoleptic S = 2 Pentacyanidochromate(II), [CrII(CN)5]3−, andNonacyanidodichromate(II), [CrII2(CN)9]5−.Angew. Chem. 2005, 117, 3189–3192.doi: 10.1002/ange.200462763
[brown_ring_2004]
H.-Y. Cheng, S. Chang, P.-Y. Tsai:On the ��Brown-Ring�� Reaction Product via Density-Functional Theory.J. Phys. Chem. A 2003, 108, 358–361.doi: 10.1021/jp031136x
[pli_review_2002]
P. Coppens, I. Novozhilova, A. Kovalevsky:Photoinduced Linkage Isomers of Transition-Metal Nitrosyl Compounds and RelatedComplexes.Chem. Rev. 2002, 102, 861–884.doi: 10.1021/cr000031c
[brown_ring_2002]
A. Wanat, T. Schneppensieper, G. Stochel, R. van Eldik, E. Bill, K. Wieghardt:Kinetics, Mechanism, and Spectroscopy of the Reversible Binding of Nitric Oxide toAquated Iron(II). An Undergraduate Text Book Reaction Revisited.Inorg. Chem. 2001, 41, 4–10.doi: 10.1021/ic010628q
[cyano_2001]
A. S. Vinogradov, A. B. Preobrajenski, A. Knop-Gericke, S. L. Molodtsov, S. A. Krasnikov,S. V. Nekipelov, R. Szargan, M. Hävecker, R. Schlögl:Observation of back-donation in 3d metal cyanide complexes through N Kabsorption spectra.J. Electron Spectr. Relat. Phenomena 2001, 114–116, 813–818.doi: 10.1016/s0368-2048(00)00272-3
58

8 Die 18-Elektronen-Regel I
Das folgende qualitative MO-Schema zeigt für den Bereich der Grenzorbitale einesoktaedrischen d6-Komplexes qualitativ den Einfluss zunehmender Ligandorbital-Stabilität.So sieht es aus, wenn man die Abfolge CN− → CO → NO+ durchläuft (man beachte, dassdas Schema für NO hypothetisch ist, so etwas wie [Ti(NO)6]4+ ist unbekannt):
Die 18-Elektronen-Regel gilt dann, wenn die xy-, xz- und yz-Orbitale (t2g unter Oh) bindendsind. Der in der Abbildung skizzierte Unterschied zwischen Cyanido- und Nitrosyl-Ligandenergibt sich bei konstanter Energie der Metallorbitale. Vor allem die in der Literaturumstrittene Charakteristik des Cyanido-Liganden – ist er im wesentlichen nur σ-Donor-oder auch π-Akzeptorligand? – wird durch eine solche Darstellung transparent. LiegenMetalle in höherer Oxidationsstufe vor, sind die Metallorbitale stabilisiert und energetischweit von den Ligand-π*-Orbitalen entfernt – eine Rückbindung wird erschwert sein. BeiErniedrigung der positiven Ladung des Metalls nimmt die Orbitalenergie zu, dieMetallorbitale bewegen sich also entlang der Energieachse auf die gleiche Weise, wiees in der Abbildung dem Gang vom Cyanido- zum Nitrosylliganden entspricht. Zumindestbei Cyanidokomplexen von Metallen in niedriger Oxidationsstufe ist demnach mit einerdeutlichen Beteiligung von Rückbindung zu rechnen. Die signifikante Zunahme vonRückbindung zeigt sich besonders deutlich in isoelektronischen Reihen. Man verfolge
59

als Maß für die zunehmende Rückbindung in der Reihe [Fe(CN)5NO]2−, [Mn(CN)5NO]3−,[Cr(CN)5NO]4− die NO-Valenzschwingungsfrequenzen von 1939 cm−1 (Fe), 1725 cm−1
(Mn) und 1515 cm−1 (Cr). Übung: Man gebe die formale und die spektroskopischeOxidationsstufe der Metallzentren an. Zum Vergleich: Die N-O-Valenzschwingungs-frequenz beträgt im [Fe(CO)3NO]−-Ion 1640 cm−1.
Den in der Abbildung gezeigten Fällen ist gemeinsam, dass HOMO und LUMOmetallständige Orbitale sind. Die elektronische Anregung mit der niedrigsten Energiewird daher ein Übergang zwischen den d-Orbitalen des Metalls sein. Die besonderenEigenschaften einiger Nitrosylkomplexe, nach Bestrahlung langlebige metastabileZustände unter Rotation des Nitrosylliganden zu entwickeln, wird so nicht plausibel.Rechnungen am Nitroprussid-Anion zeigen jedoch, dass durch die Substitution eines vonsechs Cyanido-Liganden eines Hexacyanidoferrat(II)-Ions durch NO+ ein antibindendesNO-π*-Orbital unter den eg*-Zustand eingeschoben ist (P. Boulet, M. Buchs, H. Chermette,C. Daul, F. Gilardoni, F. Rogemond, C. W. Schläpfer, J. Weber, J. Phys. Chem. A 2001,105, 8991–8998; die Orbitalenergien sind für ein Anion unerwartet niedrig, die Autorengeben aber keine Einzelheiten an, vermutlich haben sie ein isotropes positives Feldzugeschaltet):
Die optische Anregung ist als Folge der NO-π*-Lage ein MLCT-Übergang (MLCT = metalto ligand charge transfer), der die Fe-N-Bindung schwächt und so die Rotation einleitet. DieBildung dieser metastabilen Zustände ist an den Festkörper gebunden, die Rotation findetnach der Bindungsschwächung in einem begrenzten Hohlraum der Kristallstruktur statt. InLösung wäre die Abdissoziation des Liganden zu erwarten – siehe das oben Gesagte zurmedizinischen Anwendung von SNP.
Bei Komplexen, welche die 18-Elektronenregel beachten, ist die Elektronendonorfähigkeitdes Metallzentrums mit der Ausbildung von Rückbindungen oft noch nicht erschöpft.Solche Zentren weisen Metallbasizität auf, sie leiten sich oft von Fragmenten ab, dieisolobal zu Nichtmetall-Fragmenten sind und deren recht hohe Elektronegativität teilen.
60

9 Metallcluster: Strukturen und Reaktionen
9.1 [Fe3(CO)12] (C2v) vs. [Os3(CO)12] (D3h)
Pentacarbonyleisen spaltet beim Erhitzen CO ab. Zuerst entsteht Fe2(CO)9, dannFe3(CO)12, das gezielter durch Oxidation von Tetracarbonylferrat(2−) erhalten werdenkann. Strukturanalyse und IR-Spektren zeigen für den Zweikernkomplex im Kristall und inLösung eine D3h-symmetrische Struktur mit 3 verbrückenden Carbonylgruppen, also einHexacarbonyl-tris(μ-carbonyl)-dieisen(0). (Wir schauen uns in einer Übung an, wie mandie Zahl der zu erwartenden C-O-Valenzschwingungen für eine bestimmten Molekülgestaltermittelt).
Der Aufbau von Fe3(CO)12 war längere Zeit umstritten. Die erste Strukturanalyse warnicht eindeutig, und eine auf verbrückende CO-Gruppen hinweisende IR-Bande ist nebeneiner Hauptabsorption bei 2050 cm−1 und weiteren Banden im Bereich terminalerCarbonylliganden nur schwach bei 1870 cm−1 zu erkennen. Die seit 1993 vorliegendekorrekte Strukturanalyse wird durch neuere DFT-Rechnungen bestätigt: Im Grundzustandist die Struktur nicht D3h-symmetrisch ohne verbrückende Carbonyl-Liganden, sondernzwei der drei Eisenatome sind von zwei Carbonylgruppen überbrückt. Die Symmetriedes Moleküls ist C2v, es entsteht formal durch Ersatz einer der drei μ-CO-Liganden inFe2(CO)9 durch ein Fe(CO)4-Fragment, das nur terminale CO-Liganden aufweist. DieD3h-symmetrische Struktur ist ca. 20–30 kJ mol−1 weniger stabil. Sie hat in Lösungwahrscheinlich Bedeutung. Darüberhinaus fluktuiert die C2v-Struktur in Lösung (1 Signalim 13C-NMR-Spektrun) und wahrscheinlich auch im Feststoff, indem die Brückenligandenständig ihren Platz wechseln. Man beachte, dass die IR-Spektroskopie als „schnelle“Methode terminale und verbrückende Liganden zeitaufgelöst zeigt, während dielangsamere NMR-Spektroskopie den äquilibrierten Zustand wiedergibt.
Das homologe Os3(CO)12 liegt dagegen in eben dieser D3h-symmetrischen Form ohneverbrückende CO-Liganden vor. Die DFT-Rechnung weist hier Isomere mit μ-CO-Ligandenals unstabil aus. Ursache ist die stärkere Kontraktion der 3d-Orbitale der Eisenzentren imVergleich zu den 5d-Orbitalen des Osmiums. So lässt sich zeigen, dass die Bindung zuμ-CO-Gruppen die Metall-Metall-Bindung schwächen. Während der Energieverlust durcheine schwächere Metall-Metall-Bindung bei den aufgrund der geringeren Orbital-überlappung ohnehin schwächeren Eisen-Eisen-Bindungen durch den Aufbau von mehr
61

Fe-C-Kontakten überkompensiert wird, ist dies bei Osmium umgekehrt. Wie schon in derKoordinationschemie höherer Oxidationsstufen ergibt sich der Unterschied zwischen ersterÜbergangsreihe und den schweren Homologen aus der stärkeren Lokalisierung der d-Elektronen am Atomrumpf im Fall eines 3d-Elementes [cluster_1999].
9.2 Bindungsmöglichkeiten von CO-Liganden
Die Bindung von Carbonylliganden als terminale oder verbrückende Liganden ändert inder Regel nichts an der Elektronenbilanz. Ist ein verbrückender Carbonylligand nur überdas C-Atom an zwei oder drei Metallatome gebunden, steht für die Hinbindung wie beimterminalen Bindungsmodus das freie Elektronenpaar am Kohlenstoffatom zur Verfügung.CO ist ein 2-Elektronen-Donor. Die üblichen Bindungsmodi verbrückender CO-Ligandensind im folgenden Schema zusammengestellt.
Die rechts gezeichneten Fälle des linear halbverbrückenden Modus, bei dem auch M= M' sein kann, sind selten. Sie haben besondere elektronische Verhältnisse zurVoraussetzung. Ein Beispiel für den η2, μ3-6e-Fall liegt im dreikernigen Komplex[Nb3(cp)3(CO)7] vor [nb3cp3co7_1981]. Hier ist M = M' = Nb(cp)(CO)2. Der die Bildungeines Clusters verursachende Ligandmangel wurde durch CO-Abspaltung aus demeinkernigen Carbonylkomplex [Nb(cp)(CO)4] durch Bestrahlen mit UV-Licht herbeigeführt.Ein cyclischer [{Nb(cp)(CO)n}m]-Cluster mit zwei Niob-Niob-Einfachbindungen proMetallatom darf für n = 3 erwartet werden (siehe unten). Ein dreikerniger Cluster (m =3) hätte dann die Zusammensetzung [Nb3(cp)3(CO)9] – was wohl zu einer sterischenÜberladung führen würde. Der gefundene Cluster enthält zwei Carbonyl-Liganden wenigerund trotzdem vermittelt die Struktur den Eindruck eines beachtlichen „Gedränges“: jedesNiob-Atom ist von zwei terminalen Carbonyl-Liganden koordiniert; der Cyclopentadienyl-Ligand ist wie üblich η5 gebunden. Man beachte, dass im Bild an jedem Cyclopentadienyl-
62

Liganden der Übersichtlichkeit halber 5 H-Atome nicht eingezeichnet sind, die abernatürlich auch noch Platz brauchen.
Die weiter unten zusammengefassten Elektronenabzählregeln zeigen, dass dem Clusterwegen des Verzichts auf zwei CO-Liganden vier Elektronen zur Erfüllung der18-Elektronenregel fehlen – wenn alle CO-Liganden als 2-Elektronendonoren gezähltwerden. Dass die 18-Elektronen-Regel einen hohen Stellenwert hat, wird bei einemgenaueren Blick auf den μ3-CO-Liganden deutlich, der eine Reihe von Besonderheitenaufweist. Üblicherweise ist das Sauerstoffatom eines verbrückenden Carbonyl-Ligandenvon den Metallatomen weggeneigt (siehe oben im Schema). Aus dem im folgenden Bilddargestellten Ausschnitt aus der Struktur des Niobclusters geht jedoch hervor, dass dasFe-C-O-Fragment nicht nur entsprechend der Einstufung “linear semibridging” mit einemWinkel von 169° am C-Atom nur wenig geknickt ist, sondern das vielmehr die geringeAbwinklung darauf zurückzuführen ist, dass das O-Atom sich zwei Nb-Atomen zuneigt.Der Nb-O-Nb-Winkel von 86° (Nb-C-Nb 85°) liegt nahe am rechten Winkel – so wie manes für zwei Donorbindungen erwarten darf, die von den senkrecht aufeinander stehendenbindenden π-Orbitalen zwischen C und O ausgehen. Die C-O-Bindung wird durch dieseneuen Bindungen zu zwei Niob-Atomen merklich geschwächt. Der C-O-Abstand ist mit1.30 Å gegenüber freiem CO (1.13 Å) deutlich aufgeweitet; man vergleiche den obenangegebenen Wert von 1.15 Å im [Fe(CO)3NO]−-Ion, 1.20 Å einer C=O-Doppelbindungund 1.43 Å einer C–O-Einfachbindung. Die deutlich herabgesetzte Bindungsordnung führtzu einem für Carbonylkomplexe ungewöhnlich niedrigen Wert für die Energie der C-O-Streckschwingung von nur 1330 cm−1. Vor allem dieser letzte Wert passt zu derInterpretation, dass der verbrückende Carbonylligand als 6-Elektronen-Donor fungiert.Es ist jedoch zu beachten, dass eine 18-Elektronenbilanz auch durch 1 Nb=Nb-Doppelbindung erreicht würde. Die im Bild angegebenen Nb-Nb-Abstände zeigen, dass
63

die Metall-Metall-Abstände tatsächlich nicht alle gleich groß sind. Da keine neuerenRechnungen an diesem Cluster vorliegen, bleibt hier Deutungsspielraum.
9.3 Carbidocluster
Dass die Aktivierung eines Carbonyl-Liganden bis zum C-O-Bindungsbruch führen kann,zeigen die beiden folgenden Beispiele: Wird Fe(CO)5 erhitzt, so entstehen unter CO-Abspaltung mehrkernige Carbonylmetall-Komplexe wie Fe2(CO)9 oder Fe3(CO)12. Wirdweiter erhitzt, so werden bei ca. 200 °C Carbidocluster erhalten, in denen neben intaktenCarbonyl-Liganden Carbido-Liganden, C4−, als Spaltprodukte auftreten. So lässt sich ein[Fe5C(CO)15] isolieren. Auch Cluster dieser Größe gehorchen der 18-Elektronen-Regel.Da der Umgang mit Oxidationsstufen hier sehr sperrig wäre, werden die Clusterbausteineals elektroneutral angesehen. Die bei Fe und C eingesetzten Elektronenzahlen sind alsodie der neutralen Atome. CO zählt wie üblich als 2-Elektronen-Donor. Man beachte, dassbei dieser summarischen Behandlung 2 Elektronen pro Metall-Metall-Bindung zu zählensind, da erst im letzten Schritt durch die Zahl der Metallatome dividiert wird:
[Fe5C(CO)15]5 Fe 5 × 8 = 40
15 CO 15 × 2 = 301 C 1 × 4 = 4
8 Fe–Fe 8 × 2 = 16Σ = 90
pro Fe : 5 = 18
9.4 Fischer-Tropsch-Synthese
Weitere Beispiele für die CO-Aktivierung sind alle katalytischen Reaktionen, bei denenKohlenmonoxid – meist zusammen mit Wasserstoff als „Synthesegas“ – als Reaktandeingesetzt wird. Zu den Reaktionen, bei denen die C-O-Bindung vollständig gebrochen
64

wird, gehört die Fischer-Tropsch-Reaktion (FT-Synthese), die mit Cobaltkatalysatoren derfolgenden Gleichung folgt:
CO + 2 H2 → (-CH2-)n/n + H2O
In allen neueren Varianten werden Eisenkatalysatoren eingesetzt, die außerdem dasWassergasgleichgewicht (CO + H2O = CO2 + H2) katalysieren. Die Gleichung für die FT-Synthese lautet dann:
2 CO + H2 → (-CH2-)n/n + CO2
Die FT-Synthese ist eine heterogen katalysierte Polymerisation von C1-Bausteinen,die sich auf einer Metalloberfläche (Eisen, Cobalt, Ruthenium) primär zu 1-Alkenenzusammenfinden. Die Kettenlänge ist steuerbar. Typischerweise werden Kohlen-wasserstoffgemische angestrebt, die als Treibstoffe eingesetzt werden können. Die FT-Synthese gehört neben der Kohlehydrierung zu den Verfahren, mit deren Hilfe Erdöl durchKohle ersetzt werden kann.
In der Literatur werden einige mechanistische Vorschläge gemacht. In einemSchlüsselexperiment konnte Brady zeigen, dass der Carbid-Methylen-Mechanismuswohl der realistischste ist. Dieser formuliert die C-O-Bindungsspaltung und diePolymerisation als zwei völlig voneinander getrennte Schritte mit sauerstofffreien C1-Bausteinen bei Kettenstart und Kettenfortpflanzung. Das folgende Schema zeigtschematisch die Einzelschritte: Adsorbiertes CO wird zu oberflächengebundenem Carbidund Oxid. Das Oxid reagiert mit adsorbiertem Wasserstoff zu Wasser und verlässt dieOberfläche. Carbid reagiert ebenfalls mit Wasserstoff, es bildet sich oberflächen-gebundenes Methylidin (CH) und Methylen (CH2), außerdem wird Wasserstoff als Hydrido-Ligand an die Metalloberfläche gebunden. Der Kettenstart ist die Insertion einerMethylengruppe in eine Metall-Hydrid-Bindung, es entsteht ein Methyl-Ligand. DasKettenwachstum stellt eine Folge von Methylen-Insertionen in Metall-Alkyl-Bindungendar, durch welche die Alkylreste verlängert werden. Der Kettenabbruch schließlich ist eineβ-Wasserstoff-Eliminierung, bei der das oberflächengebundene Hydrid wiederhergestelltwird und ein 1-Alken entsteht (im technischen Prozess werden Folgeprodukte anstelledieses reaktiven Primärproduktes erhalten). Schematisch:
In Bradys Schlüsselexperiment wurden oberflächengebundene Methylen-Liganden durchUmsetzung einer sauberen Metalloberfläche mit Diazomethan erzeugt. Als Metalle
65

wurden FT-Katalysatoren und Kupfer eingesetzt. Beim Erhitzen aller Methylen-belegtenOberflächen entstand Ethen durch Dimerisierung der Methylengruppen. In Anwesenheitvon Wasserstoff jedoch bildeten die FT-Katalysatoren 1-Alkene – nur mit Kupfer, dasnicht zur Bildung von Hydrid neigt (Kupfer ist kein Hydrierkatalysator!), blieb es auch beiAnwesenheit von Wasserstoff bei der Ethen-Bildung.
Der erste Teilschritte der FT-Synthese, die Spaltung von CO sowie die Bildung vonWasser und CHn-Spezies, ist auf molekularer Ebene nachgestellt worden (1981). DieVersuchsreihe wird in Lehrbüchern häufig zitiert, da es nur sehr wenige Beispiele gibt,bei denen so viele Einzelschritte einer Heterogenkatalyse an einem Metallclusternachgebildet wurden. Ausgangspunkt ist ein Carbonylferrat, das sich neben anderen beider Umsetzung höherkerniger Eisencarbonyle mit Base bildet, und zwar [Fe4(CO)13]2−. Indiesem vierkernigen Cluster sind jeweils drei terminale CO-Liganden an jedem Eisenatomgebunden. Der dreizehnte Carbonyl-Ligand liegt im η1,μ3-Bindungsmodus vor, indem dasC-Atom eine Dreiecksfläche des tetraedrischen Clusters überbrückt. Das Ferrat befolgt die18-Elektronen-Regel:
[Fe4(CO)13]2−
4 Fe 4 × 8 = 3213 CO 13 × 2 = 26[ ]2− = 2
6 Fe–Fe 6 × 2 = 12Σ = 72
pro Fe : 4 = 18
Im folgenden Schema ist die Reaktionssequenz dargestellt, die nach stufenweiserProtonierung mit starker Säure (Fluor- oder Trifluormethansulfonsäure) abläuft und die beitiefer Temperatur in Einzelschritte aufgelöst werden konnte:
Im ersten Schritt lagert sich ein Proton an den Cluster an. Dabei hat es die Wahl zwischenden weich-basischen Metallzentren und den hart-basischen O-Atomen der Carbonyl-Liganden. Unter den CO-Gruppen wiederum sollte das O-Atom des verbrückendenLiganden die höchste Basizität aufweisen, da durch Rückbindungen am meistenLadungsdichte auf diese Carbonylgruppe übertragen sein sollte. Das Experiment zeigt,dass sich das Proton für die weich-basische Position entscheidet und sich an zweiMetallatome anlagert, die es als formaler μ2-Hydrido-Ligand verbrückt.
Die sich nun anschließende strukturelle Veränderung im Cluster spiegelt die bislangbetrachteten Gesetzmäßigkeiten wider: Die 18-Elektronen-Regel wird durch dieProtonierung zuerst einmal nicht berührt. Trotzdem kommt es zu einer strukturellenVeränderung, indem eine der sechs Metall-Metall-Bindungen im Produkt aufgehoben ist(im Schema ist diese im Edukt rot gezeichnet).
Eine mögliche Interpretation ergibt sich aus der vergleichenden Betrachtung derBindungsverhältnisse in den homologen Carbonylen [Fe3(CO)12] und [Os3(CO)12]. Es war
66

gezeigt worden, das bei einem Element der ersten Übergangsreihe die d-Orbitale wenigausladend sind. Die Überlappung mit einem Brücken-CO-Ligand ist daher oft günstigerals die direkte Metall-Metall-Bindung. Die Protonierung des Ferrats(2−) vermindert denanionischen Charakter des Clusters und führt zur Kontraktion der Metallorbitale. AlsFolge wird eine der geschwächten Eisen-Eisen-Bindungen aufgegeben. Hierdurch fehlendem Cluster 2 Elektronen zur 18-Elektronen-Bilanz. Diese werden nun durch den μ3-Carbonyl-Liganden ausgeglichen, indem sich dieser einem seiner drei Bindungspartnerzuneigt, so dass der oben beschriebene η2-Mehrelektronen-Bindungsmoduseingenommen wird (in neuerer Nomenklatur wird aus einem CO-κC-Ligand ein CO-κC,O-Ligand).
Die zweite Protonierung erfolgt nun am O-Atom des verbrückenden Carbonyl-Liganden,der nun hinsichtlich Basenstärke und -härte der geeignete Bindungspartner für das Protondarstellt. Das dritte und vierte Proton erzeugt die Abgangsgruppe H3O+, welche sich vomCluster löst; dieser Schritt gelingt nur unter reduzierenden Bedingungen. Hierbei wird nichtnur die Ladungsdichte erhöht – also Basizität „nachgeladen“ –, sondern der entstehendeCarbidocluster genügt erst nach Aufnahme zweier Elektronen der 18-Elektronen-Regel(man beachte, dass eine plausible Elektronenzahl für den η2,μ4-COH-Ligand im Eduktnicht ohne weiteres angegeben werden kann, dass aber der entstehende Carbido-Clusterwieder eindeutig abgezählt werden kann: 4 × 8 e für 4 Fe + 12 × 2 e von 12 CO + 1 e vonH + 1 e Anionladung + 4 e von C + 5 × 2 e aus 5 Fe–Fe-Bindungen = 72 e; dividiert durch4 Fe ergibt 18 e pro Fe).
Mit dem letzten Schritt ist die CO-Aktivierung abgeschlossen, die C-O-Bindung istvollständig aufgehoben, es ist ein Carbido-Ligand entstanden. Das nächste Protonerzeugt aus diesem einen Methylidin-Liganden. Die letzten beiden Protonen führenschließlich zur Destabilisierung und zum Zerfall des Clusters. Aus dem Methylidin-Ligandist zum Schluss Methan geworden. (Im folgenden Schema steht das Symbol Fe im Clusterfür ein Fe(CO)3-Fragment.)
67

9.5 Wie zählt man Elektronen?
Bei den beiden letzten Beispielen wurde eine vereinfachte Methode verwendet, um zuprüfen, ob die 18-Elektronen-Regel erfüllt ist. Die Vereinfachung bestand darin, dassauf die Zuweisung formaler Oxidationstufen verzichtet wurde. Das Verfahren ist überalldort üblich, wo die Oxidationsstufe des Zentralmetalls keine besondere Bedeutung hatwie in weiten Bereichen der Organometallchemie. Solange nur die Frage nach derGesamtelektronenbilanz eines Metallkomplexes interessiert, ist es belanglos, welchesZählverfahren angewendet wird. Wichtig ist nur, die Regeln der verschiedenen Verfahrennicht zu vermischen. Zur Illustration werden die beiden oben benutzten Rechenmethodenan einigen Beispielen gegenübergestellt.
Zum Vergleich noch einmal die beiden Rezepte:
Mit formalen Oxidationsstufen: (1) Jedem Ligand wird seine von der IUPAC festgelegteLadung zugewiesen; (2) die Oxidationsstufe des Metalls wird bestimmt; (3) dieElektronenzahlen von Zentralmetall und Liganden werden addiert. In diese Rechnunggehen geläufige Neutralmoleküle wie Amine, Phosphane, Wasser, CO, Alkene, etc. als 2e-Donoren ein; mehrzähnige Liganden werden sinngemäß behandelt. NO ist ein neutraler3e-Donor. Anionen wie Hydrid, Halogenid, Chalkogenid, Amid, Phosphanid, Alkyl, Alkylen,Alkyliden, etc. tragen 2 Elektronen je Zentralmetall bei, maximal so viel wie sie freieElektronenpaare in geeigneter räumlicher Ausrichtung aufweisen; π-Hinbindungen werdenüblicherweise nicht mitgezählt. Ein Oxido-Ligand ist daher 2e-Donor im terminalenBindungsmodus, 4e-Donor als μ2-Ligand und 6e-Donor als μ3-Ligand; der Hydrido-Ligandist 2e-Donor in allen Bindungsmodi. Ein η5-Cyclopentadienyl-Ligand ist ein anionischer 6e-Donor, η6-Benzol ist ein neutraler 6e-Donor.
68

Die bei diesem Verfahren erhaltenen Oxidationsstufen sind formal und stimmen vor allembei radikalischen Liganden oft nicht mit der spektroskopischen Oxidationsstufe überein.Die Bestimung der formalen Oxidationsstufe kann hier in die Irre führen. Dicarbonyl-dinitrosyl-eisen, [Fe(CO)2(NO)2], wäre demnach ein Eisen(0)-Komplex, dieElektronenkonfiguration wäre d8. Man könnte bei dieser Konfiguration und wegen derStarkfeldliganden auf die Idee kommen, der Komplex sei quadratisch-planar, er ist jedochverzerrt tetraedrisch, wie es die spektroskopische Oxidationsstufe auch nahelegt. Umdiese zu ermitteln, muss der elektronische Zustand des Liganden bekannt sein.[Fe(CO)2(NO)2] enthält nach Struktur- und spektroskopischen Daten den 2e-Donor NO+.Die spektroskopische Oxidationsstufe des Eisens ist daher −II, es ergibt sich also eingeschlossenschaliger d10-Komplex. Dass die Idee vom quadratisch-planaren Komplexnicht gut ist, zeigt sich auch bei formaler Zählweise bei der Elektronenbilanz, dieunabhängig von der Verteilung zwischen Metall und Ligand ist. Mit der formalenOxidationsstufe ergibt sich als Summe der Beiträge von Zentralmetall, den Carbonyl- undden Nitrosyl-Liganden 8 + 2 × 2 + 2 × 3 = 18. Quadratisch-planare d8-Komplexe wie[Ni(CN)4]2− weisen jedoch 16 Elektronen auf!
Die bei Clustern und in der Organometallchemie übliche Zählweise ist schneller (wennman die etwas seltsamen Regeln verinnerlicht hat), ergibt jedoch nicht die Oxidationsstufedes Zentralmetalls – die hier jedoch meist auch wenig aussagekräftig ist (Cluster) oder nurwenig interessiert (Organometallchemie). Alle Bausteine werden als neutral angenommen.Wasserstoff, Halogenatome, Alkylreste und andere einbindige funktionelle Gruppentragen nun 1 Elektron bei. Hinzu kommen jeweils 2 Elektronen für jede zusätzlichekoordinative Bindung. Im μ2-Modus sind Halogene daher 3e-Donoren (1 normale +1 koordinative Bindung). Ein η5-Cyclopentadienyl-Ligand ist ein neutraler 5e-Donor (1normale Bindung + 2 koordinative Bindungen), η6-Benzol ist ein neutraler 6e-Donor (0normale + 3 koordinative Bindungen). Ein terminales O-Atom trägt 2 Elektronen bei (1normale Doppelbindung + 0 koordinative Bindungen), ein μ2-Oxido-Ligand in der Regel 2(2 normale Bindungen + 0 koordinative Bindung), ein μ3-Oxido-Ligand 4 (2 normale + 1koordinative Bindungen). Da Neutralmoleküle keine freie Valenzen für normale Bindungenaufweisen, sondern nur koordinative Bindungen ausbilden (siehe oben bei Benzol), werdensie genauso behandelt wie bei der ersten Zählmethode.
Beispiele für diese Zählweise sind bei den oben genannten Clustern gegeben, bei deneneine die Oxidationsstufen einschließende Rechnung sehr umständlich wäre. BeideZählweisen führen zu derselben Elektronenbilanz, wie das Beispiel des Tris(μ-bromido)-hexacarbonyl-dimanganat(1−)-Ions, [(CO)3Mn(μ-Br)3Mn(CO)3]−, zeigt. Bei einerRechnung mit Oxidationsstufen ergibt sich aus der Gesamtladung und der Ladung dermono-anionischen Bromido-Liganden +I als formale Oxidationsstufe des Metalls:
69
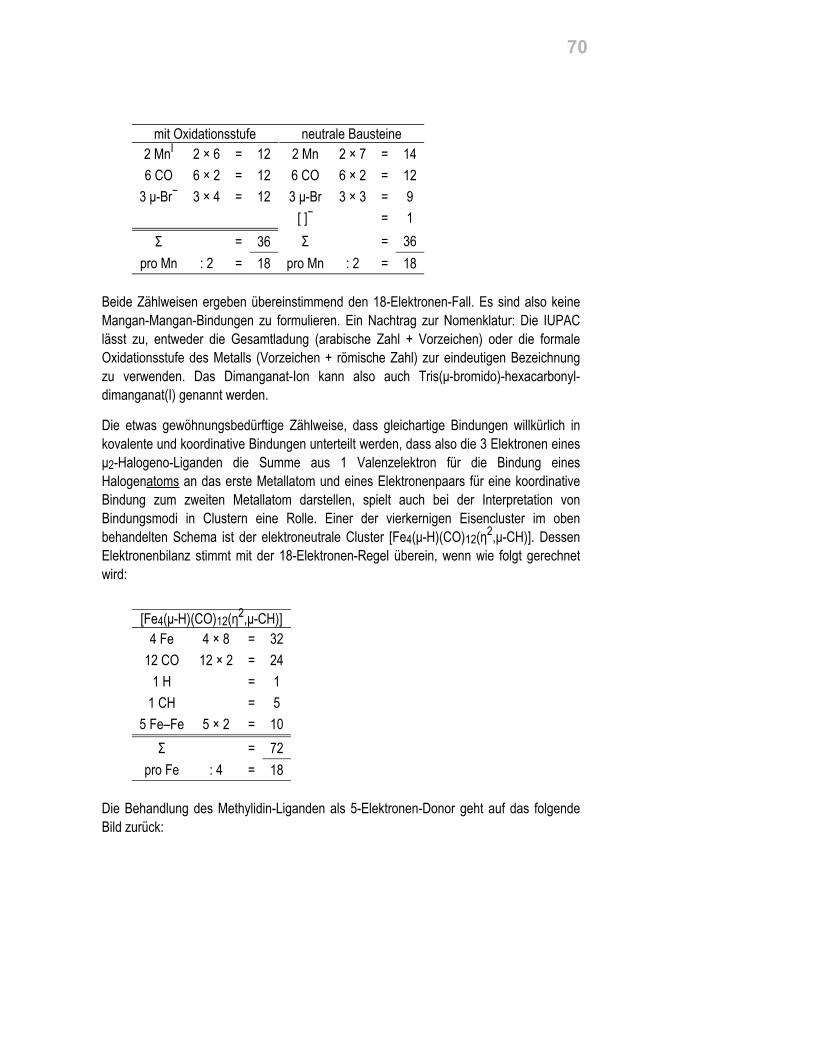
mit Oxidationsstufe neutrale Bausteine2 MnI 2 × 6 = 12 2 Mn 2 × 7 = 146 CO 6 × 2 = 12 6 CO 6 × 2 = 12
3 μ-Br− 3 × 4 = 12 3 μ-Br 3 × 3 = 9[ ]− = 1
Σ = 36 Σ = 36pro Mn : 2 = 18 pro Mn : 2 = 18
Beide Zählweisen ergeben übereinstimmend den 18-Elektronen-Fall. Es sind also keineMangan-Mangan-Bindungen zu formulieren. Ein Nachtrag zur Nomenklatur: Die IUPAClässt zu, entweder die Gesamtladung (arabische Zahl + Vorzeichen) oder die formaleOxidationsstufe des Metalls (Vorzeichen + römische Zahl) zur eindeutigen Bezeichnungzu verwenden. Das Dimanganat-Ion kann also auch Tris(μ-bromido)-hexacarbonyl-dimanganat(I) genannt werden.
Die etwas gewöhnungsbedürftige Zählweise, dass gleichartige Bindungen willkürlich inkovalente und koordinative Bindungen unterteilt werden, dass also die 3 Elektronen einesμ2-Halogeno-Liganden die Summe aus 1 Valenzelektron für die Bindung einesHalogenatoms an das erste Metallatom und eines Elektronenpaars für eine koordinativeBindung zum zweiten Metallatom darstellen, spielt auch bei der Interpretation vonBindungsmodi in Clustern eine Rolle. Einer der vierkernigen Eisencluster im obenbehandelten Schema ist der elektroneutrale Cluster [Fe4(μ-H)(CO)12(η2,μ-CH)]. DessenElektronenbilanz stimmt mit der 18-Elektronen-Regel überein, wenn wie folgt gerechnetwird:
[Fe4(μ-H)(CO)12(η2,μ-CH)]4 Fe 4 × 8 = 32
12 CO 12 × 2 = 241 H = 1
1 CH = 55 Fe–Fe 5 × 2 = 10
Σ = 72pro Fe : 4 = 18
Die Behandlung des Methylidin-Liganden als 5-Elektronen-Donor geht auf das folgendeBild zurück:
70

Würde das CH-Fragment nur mit seinen drei Valenzelelektronen zum Clusteraufbaubeitragen, wäre die 18-Elektronen-Regel nicht erfüllt. Die Bedeutung der Regel zeigt sichnun an der Orientierung des CH-Liganden. Das H-Atom, das im geläufigen 3-Elektronen-Bindungsmodus des CH-Liganden von allen Metallatomen entfernt ist, neigt sich einemFe-Atom zu. Hierdurch gehen die beiden Elektronen der C–H-Bindung eine koordinativeBindung zu diesem Fe-Atom ein, der CH-Ligand wird zum 3+2=5e-Donor und der Clusterwird elektronenpräzise im Sinne der Regel. Die hier dargestellte Wechselwirkung einesC–H-Elektronenpaares mit einem Metallatom wird agostische Bindung genannt.
Das Beispiel unterstreicht, dass in der Clusterchemie die Verbindungsstriche zwischenAtomen nicht notwendigerweise 2e,2z-Bindungen darstellen. Auch wenn unterBerücksichtigung von Elektronegativitäten und Oxidationsstufen der Methylidin-Bausteinrealistischer als CH3−-Ligand betrachtet wird, stehen für die vier eingezeichneten Fe–C-Bindungen nur drei Elektronenpaare zur Verfügung.
9.6 18-Elektronen-Regel vs. Wadesche Regeln
Die 18-Elektronenregel (die Isolobalbeziehung, die auf dieselbe Wurzel zurückgeht, führtzum gleichen Ergebnis) ist in der Regel gut erfüllt, solange weniger als ungefähr sechsMetallatome in einem Cluster enthalten sind. Oktaedrische Metallcluster folgen diesenRegeln oft nicht. Das Ligand:Metall-Verhältnis ist hier so klein, dass es sich umElektronenmangelverbindungen handelt, die den Regeln für den Aufbau andererElektronenmangelverbindungen wie den Boranen folgen. Diese Wadeschen Regelnverstehen einen Cluster als Derivat eines Deltaeders, dies ist ein Polyeder, der nurvon Dreiecksflächen begrenzt ist. Die trigonale Bipyramide, Oktaeder und Ikosaeder sindDeltaeder, Würfel nicht. Ein Cluster, dessen Gerüstatome ein Deltaeder bilden, ist eincloso-Cluster. Dieser tritt bei Nebengruppenmetallverbindungen auf, wenn bei nÜbergangsmetallatomen 14 n + 2 Elektronen eingebracht werden. Wie bei den WadeschenRegeln Elektronen zu zählen sind, zeigt ein Beispiel. Unter den vielen Osmiumcarbonylenund Carbonylosmaten finden sich das trigonal-bipyramidale [Os5(CO)16] und dasoktaedrische [Os6(CO)18]2−. Die Elektronenzahl ergibt sich als Summe der tatsächlichvorhandenen Elektronen, das heißt, Metall-Metall-Bindungen werden nun nicht mehrexplizit gezählt, da sie in den Regeln berücksichtigt sind. Das fünfkernige Carbonyl weistdaher 5 × 8 + 16 × 2 = 72 Elektronen auf. Mit n = 5 ergibt sich: 14 n + 2 = 14 × 5 + 2= 72 Elektronen. Für den Cluster wird also der closo-Typ erwartet, was auch zutrifft. Dercloso-Fall sollte bei einem sechskernigen Cluster bei 14 n + 2 = 14 × 6 + 2 = 86 Elektronen
71

auftreten. Man vergleiche mit dem Hexaosmat: 6 × 8 + 18 × 2 + 2 (die Anionladung) = 86Elektronen.
Das oktaedrische Anion lässt sich mit der 18-Elektronenregel nicht zutreffend beschreiben.Zu der Summe von 86 Elektronen würden noch 12 × 2 = 24 Elektronen hinzugezählt, umdie 12 Metall-Metall-Bindungen zu berücksichtigen. Als Summe ergibt sich 110 Elektronen;dividiert durch 6 wird 18 Rest 2 erhalten, der Cluster wäre nicht elektronenpräzise. Wieoben angemerkt, tritt bei dem nicht-oktaedrischen [Os5(CO)16] noch keine Schwierigkeitauf. Zu den 72 Elektronen der oben angestellten Rechnung wären 9 × 2 = 18 Elektronenfür 9 Metall-Metall-Bindungen zu addieren. Es ergeben sich dann für das einzelne Os-Atom90 : 5 = 18 Elektronen.
Sind mehr Elektronen vorhanden als es dem closo-Fall entspricht, entstehen offenereStrukturen, die sich von Deltaedern durch Weglassen einer oder mehrerer Ecken ableiten.Fehlt 1 Ecke, liegt eine nido-Struktur vor, 2 fehlende Ecken entsprechen einer arachno-Struktur. Die zugehörigen Elektronenzahlen sind 14 n + 4 und 14 n + 6. Kleinere Clusterfolgen oft sowohl der 18-Elektronenregel als auch den Wadeschen Regeln. Ein Beispielsind die oben behandelten Eisencluster [Fe5C(CO)15] und [Fe4(CO)13]2−, für die schongezeigt wurde, dass sie der 18-Elektronenregel genügen.
Der Fe5-Cluster weist 5 × 8 + 15 × 2 + 4 = 74 Elektronen auf. Mit n = 5 entspricht diesdem 14n+4-Fall, es ist also eine nido-Struktur zu erwarten. Bei n = 5 wäre also nach demDeltaeder mit n = 6 zu fragen, dem dann 1 Ecke fehlen sollte. Dies trifft zu: das Fe5-Gerüst hat die Struktur eines Oktaeders mit einer fehlenden Ecke. Analog stellen die 4 ×8 + 13 × 2 + 2 = 60 Elektronen des Tetraferrats die 14n+4-Bilanz des nido-Falls für n = 4dar. Entsprechend fehlt dem einfachsten Deltaeder, der trigonalen Bipyramide, 1 Ecke,wodurch sie zum Tetraeder wird.
Eine arachno-Struktur ist bei [Fe4(μ-H)C(CO)12]− zu erwarten. Dieser der18-Elektroneregel entsprechende Cluster enthält 4 × 8 + 12 × 2 + 1 (H) + 4 (C) +1 (Ladung)= 62 Elektronen. Mit n = 4 ist dies der 14n+6-Fall. Vom Deltaeder mit n = 6 wären also 2Ecken abzutrennen. Auch dies trifft zu. Die Struktur des Fe4-Gerüsts des Carbido-Clusterswird erhalten, wenn von einem Oktader 2 benachbarte Ecken entfernt werden.
9.7 Literatur
[cluster_1999]
E. Hunstock, C. Mealli, M. J. Calhorda, J. Reinhold:Molecular Structures of M2(CO)9 and M3(CO)12 (M = Fe, Ru, Os): New TheoreticalInsights.Inorg. Chem. 1999, 38, 5053–5060.doi: 10.1021/ic9905289
72

[nb3cp3co7_1981]
W. A. Herrmann, H. Biersack, M. L. Ziegler, K. Weidenhammer, R. Siegel, D. Rehder:Carbon monoxide – a six-electron ligand? Synthesis and structural characterizationof the unusual carbonylniobium cluster heptacarbonyltris(η5-cyclopentadienyl)triniobium.J. Am. Chem. Soc. 1981, 103, 1692�1699.doi: 10.1021/ja00397a018
73

10 Die 18-Elektronen-Regel II
10.1 Die 18-Elektronen-Regel in computer-chemischen Rechnungen: Ni(CO)4 und Fe2(CO)9
So etabliert und nützlich die 18-Elektronen-Regel als Richtschnur in derKoordinationschemie und der metallorganischen Chemie auch ist, so unsicher sindmanche konkreten Aussagen zur Bindungssituation eines Komplexes. Einer derkritischsten Punkte betrifft die Natur der postulierten Bindungen zwischen 3d-Metallen.In den DFT-Rechnungen, die weiter oben zur Analyse der CO-Valenzschwingungen vonFe2(CO)9 vorgenommen wurden, findet man eine Fe-Fe-Bindung nicht ohne Weiteres.Da wir bisher Ausagen zur Elektronenstruktur immer nur aus der MO-Behandlung einesoktadrischen Komplexes abgeleitet hatten, soll in diesem Abschnitt der 18-Elektronenfallbei einem tetraedrischen Carbonylkomplex analysiert werden, und zwar bei Tetracarbonyl-nickel(0). Anschließend sollen die dort gewonnenen Verallgemeinerungen genutzt werden,um in einer DFT-Rechnung nach der Eisen-Eisen-Bindung zu suchen, die durch dieAbzählregeln für Fe2(CO)9 gefordert wird.
Ni(CO)4
Nickel trägt 28 Elektronen zu Tetracarbonylnickel bei, die vier Carbonylliganden insgesamt56 Elektronen. Eine All-Elektronen-Rechnung am diamagnetischen Carbonylkomplexergibt also 42 doppelt besetzte Orbitale. Davon sind 17 Elektronenpaare stabileRumpfelektronenpaare (Ni: 1s, 2s, 3 × 2p, 3s, 3 × 3p; außerdem 8 × 1s der Leichtatome),des Weiteren darf man die aus der bindenden und antibindenden Wechselwirkung der2s-Orbitale von C und O resultierenden Orbitale 1σ und 2σ des freien CO als so stabilansehen, dass diese 8 MOs im Schema wohl folgen werden. Wir betrachten daher dieOrbitale 26–42. Um sie zuzuordnen, sollte man die Gestalt der CO-Grenzorbitale vorAugen haben, dem aus C(2pz) und O(2pz) entstandenen 3σ sowie den beiden bindendenπ-Orbitalen 1π und 2π.
Die Zuordnung der 17 Orbitale (wir verwenden GaussView und die formatierteCheckpointdatei einer b3lyp/tzvp-Rechnung, die Sie mit dieser Eingabedatei selbstnachvollziehen können oder deren Protokolldatei Sie ansehen können) ist übersichtlich,
74

auch wegen der hohen Symmetrie (Td). In der folgenden Tabelle markiert n den Beitragzur 18-Elektronenbilanz:
MO Sym. Zuordnung n40, 41, 42 t2 Ni(xy,xz,yz) π-Rückbindungen 638, 39 e Ni(z2, x2−y2) lone pairs 435, 36, 37 t2 CO(3σ) + Ni(4p) 632, 33, 34 t1 CO(π) 029, 30, 31 t2 CO(π) 027, 28 e CO(π) 026 a1 CO(3σ) + Ni(4s) 2
Es lassen sich einige Regeln erkennen: Bei der Anwendung der 18-Elektronenregelwerden mögliche π-Donor-Bindungen nicht mitgezählt. Die entsprechenden Orbitale27–34 (e + t2 + t1) haben auch nahezu reinen Ligandcharakter. Die übrigen Orbitaleschließen die Nickel-Valenzorbitale (3d, 4s, 4p) ein: entsprechend den vier Donor-Elektronenpaaren der vier CO-Liganden treten die vier MOs 26 und 35–37 auf (a1 und t2).Daneben gibt es die 3d-Elektronenpaare an Nickel (e, t2), die als einsame Elektronenpaaregesehen werden können, die ihre Elektronendichte im Sinne von Rückbindungen aberauch in CO(π*)-Orbitale delokalisieren können.
In einer VB-Betrachtung würde man ein Nickelatom mit 5 freien Elektronenpaarenzeichnen, sowie 4 koordinative Bindungen, die von den CO-Liganden ausgehen und4 sp3-Hybridorbitale am Zentralatom füllen. Hinzu kämen nun etliche mesomereGrenzstrukturen, um die Rückbindungen zu berücksichtigen.
Fe2(CO)9
Nun zum heikleren Fall Nonacarbonyldieisen(0). Die 18-Elektronenformulierung führt hierzu einer Fe-Fe-Einfachbindung (2 × 8 e− + 8 × 2 e− = 34 e− entsprechend 17 e−
pro Eisenatom; wie bei einem 7-e−-Chloratom wird also Dimerisierung unter Ausbildungeiner Einfachbindung erwartet). Eine DFT-Rechnung auf demselben Niveau wie zuvor(Eingabedatei, Protokolldatei) führt zu 89 doppelt besetzten Orbitalen. Werden die 36Rumpfelektronenpaare abgezogen, verbleiben 53 Valenzelektronenpaare. Da dasTetracarbonylnickel-Beispiel gezeigt hatte, dass alle CO-Orbitale unterhalb desGrenzorbitalbereichs lagen, subtrahieren wir noch einmal 5 × 9 = 45 Orbitale. Es verbleibenjetzt 8 Grenzorbitale. Bevor diese analysiert werden, hier noch die Gegenprobe:
Die 18-Elektronenregel betrachtet die 2 × 9 = 18 Valenzorbitale (2 × 4s, 6 × 4p, 10× 3d) der beiden Eisenatome. Da 2 × 17 e− zur Verfügung stehen, sind im Singulett-Molekül Fe2(CO)9 17 besetzte Valenzorbitale zu berücksichtigen. Zu diesen zählen auchdie 9 Orbitale mit Metall-s- und -p-Charakter, die durch die 9 Donorbindungen der 9 CO-Liganden gefüllt werden. Im Grenzorbitalbereich bleiben 17 − 9 = 8 Orbitale – die wir nunendlich anschauen wollen.
75

Wir laden die formatierte Checkpointdatei einer b3lyp/tzvp-Rechnung in derPunktgruppe D3h wieder mit GaussView:
MO Sym. Zuordnung88, 89 e′′ Fe(x2−y2, z2), Fe-μ-CO(π*)-Rückbindungen87 a2′′ Antibindung zu 84!85, 86 e′′ Antibindung zu 82, 83!84 a1′ Fe(xy,xz,yz), die Fe-Fe-Bindung82, 83 e′ Fe(xy,xz,yz), Fe-Fe-bindend
Es zeigt sich, dass es zu jeder Fe-Fe-bindenden Wechselwirkung eine antibindendeWechselwirkung gibt. In der Literatur wird dementsprechend seit Jahrzehnten um die Fe-Fe-Bindung gestritten. In der neuesten verfügbaren Arbeit [fe2co9_bonding_2007]wird eine schwache Bindung gefunden und zwar so: man betrachte vor allem Bindung (MO84) und Antibindung (MO 87) genauer. Die Antibindung weist an den Brückencarbonyl-Liganden höhere Koeffizienten auf, sie ist in eine Wechselwirkung zu CO(π*)-Orbitalendelokalisiert. Durch die Beteiligung an der Rückbindung schwächt sie die Fe-Fe-Bindungweniger.
Im VB-Formalismus entspricht ein doppelt besetztes Molekülorbitalpaar jeweils einemfreien Elektronenpaar an jedem Atom. Man würde also die MOs 84 und 87 an das Fe-Atompaar lokalisieren, indem man jedem Fe-Atom ein freies Elektronenpaar zuweist.Hinzu kommen nun mesomere Grenzformen, in denen eine Fe-Fe-Bindung formuliert wirdund das zweite Elektronenpaar für eine Rückbindung zu einem Brücken-CO-Ligandenverwendet wird. Die 18-Elektronenregel gilt dabei nur für die Fe-Fe-gebundenen Formen.
Das hier geschilderte Problem zeigt die Grenzen einer strikten Isolobalbehandlung. Estritt gehäuft auf, wenn in Mehrkernkomplexen von 3d-Zentralmetallen Donor/Akzeptor-Brückenliganden vorkommen.
10.2 16e-d8-Komplexe, Vaskas Komplex
16e-d8-Komplexe bilden neben großen Clustern eine wichtige Ausnahme von der 18e-Regel. Im Gegensatz zum stark antibindenden x2−y2-Orbital, das unbesetzt bleibt, istdas z2-Orbital durch das Fehlen der beiden Liganden, mit denen im Oktaeder eineantibindende Wechselwirkung vorläge, nicht destabilisiert. Durch die Besetzung des z2-Orbitals mit zwei Elektronen enthält ein quadratisch-planarer d8-Komplex zweimetallständige Valenzelektronen mehr als ein oktaedrischer Komplex. Das gefüllte, inden Raum hinausragende z2-Orbital gibt dem Metallzentrum Basizität, außerdem ist esAngriffspunkt für Oxidationsreaktionen. Solche Reaktionen sind in großem Umfang mit„Vaskas Komplex“, trans-Bis(triphenylphosphan)-carbonyl-chlorido-iridium(I),trans-[Ir(PPh3)2(CO)Cl)], untersucht worden. Die Metallbasizität zeigt sich zum Beispiel bei
76

der Reaktion mit NO+BF4−, bei dem ein Lewis-Säure/Base-Addukt entsteht, in dem dasIridium-Zentrum als Lewis-Base eintritt:
Man beachte, dass bei üblicher Zählweise das bindende Elektronenpaar zwischen Ir undN dem elektronegativeren Stickstoff zuzuweisen wäre, dass also ein NO−-Ligand vorliegt –NO+ ist oxidativ addiert worden. Die Anbindung ist nicht linear wie in dem besonderen Falldes [Fe(H2O)NO]2+-Ions, bei dem ein Triplett-NO− diskutiert wurde. Die hier vorliegendegewinkelte Ir-N-O-Gruppe (124° im Tetrafluoridoborat des abgebildeten Ions; in dessenKristallstruktur liegt das Anion in räumlicher Nähe zum Komplexkation vor, so wie diesim Schema angedeutet ist) entspricht vielmehr dem häufiger beobachteten Fall einesSingulett-NO−-Liganden, der durch eine Lewis-Formel der Art
angemessen beschrieben wird. Ist das Gegenion des Elektrophils ein besserer Ligand alsdas Tetrafluoridoborat-Ion, so ergänzt dieses die Koordinationsspäre des Iridiums. Dies giltauch für die Abgangsgruppe bei der Reaktion von Vaskas Komplex mit polaren Substratenwie den Halogenwasserstoffen oder Iodmethan (im folgenden Schema rechts):
77

Die mit HX oder CH3I ablaufende oxidative Addition an ein polares Substrat wird durcheinen nucleophilen Angriff des basischen Metallzentrums auf den positiv polarisiertenMolekülteil des Substrats eingeleitet. Bei Iodmethan entspricht dies der bekanntenSN2-Reaktion mit einem Nucleophil, bei der Iodid als Abgangsgruppe fungiert. Bei derUmsetzung mit Vaskas Komplex füllt das Iodid-Ion die Koordinationslücke am formalenIridium(III)-Zentrum auf, und zwar in trans-Position zum eingetretenen Elektrophil (daskinetische trans-Produkt lagert sich bisweilen zum cis-Produkt um, wenn dieses stabilerist). Man beachte die Ursache der 2-Elektronen-Oxidation: das Elektrophil bindet in derRegel über ein Atom an das Metallzentrum, das elektronegativer als das Zentralmetallist. Das vom basischen Metallzentrum eingebrachte Elektronenpaar wird im entstehendenLewis-Säure-Base-Addukt dem elektronegativeren Ligand zugerechnet – das Metall hatwie im Fall der NO+-Addition formal 2 Elektronen abgegeben.
Auf der linken Seite des Schemas sind zwei Reaktionen angeführt, die ebenfalls einenwichtigen Elementarschritt der Organometallchemie darstellen, die oxidative Addition anein unpolares Substrat. Diese Reaktion wird oft einfacher als cis-Addition bezeichnet. DieAddition verläuft hier prinzipiell verschieden. Das unpolare Substrat nähert sich hier mitseiner Element-Element-Bindung dem Metallzentrum. Anschließend bilden sich in einerkonzertierten Reaktion die beiden neuen Metall-Element-Bindungen. 2 Elektronen derneuen Bindungen stammen vom bindenden Elektronenpaar des Substrats, 2 Elektronenstammen vom Metall. Auch hier handelt es sich um eine 2-Elektronen-Oxidation, da alle4 bindenden Elektronen der beiden Metall-Element-Bindungen nach der Reaktion denelektronegativeren Nichtmetall-Atomen zugewiesen werden. Die Umkehrreaktion der cis-Addition ist die reduktive Eliminierung.
78

10.3 Wird [Fe(CO)4]2− H2 addieren? – Wird [Fe(CO)4]H2 addieren?
Die cis-Addition verläuft nicht nur mechanistisch anders als die Addition eines polarenSubstrats, auch die elektronischen Voraussetzungen unterscheiden sich. 18-Elektronen-Komplexe sind zur cis-Addition nicht in der Lage – sie hätten nach der Reaktion 20Elektronen. Der elektrophile Teil polarer Substrate kann jedoch oxidativ addiert werden,auch ohne dass anschließend eine Abgangsgruppe koordiniert. Beispiele wurden schonbehandelt. So ist neben der NO+-Anlagerung die Protonierung von Carbonylmetallateneine oxidative Addition ohne Veränderung der gesamten Elektronenbilanz. Man überzeugesich anhand der Reaktionen
[Fe(CO)4]2− + H+ = [HFe(CO)4]− und
[HFe(CO)4]− + H+ = [H2Fe(CO)4]
Die in Katalysecyclen wichtige cis-Addition ist demnach nur zu erwarten, wenn eine umzwei Einheiten höhere Oxidationsstufe zur Verfügung steht und zugleich in der oxidiertenForm zwei Koordinationsstellen frei sind. Diese Bedingungen werden vor allem vonquadratisch-planaren d8-Komplexen erfüllt.
Es ist nicht trivial, deren Auftreten abzuschätzen. d8-Zentren, die ein 4d- oder 5d-Metall,und neben Donor/Acceptor- auch noch Halogenido-Liganden enthalten, sind zuverlässigquadratisch-planar. Neben diesen Lehrbuchbeispielen gibt es vor allem bei den inKatalysecyclen wichtigen reaktiven Zwischenstufen einige Unsicherheit.
Ein Beispiel ist [HCo(CO)3], das im Katalysecyclus der Oxo-Synthese auftritt und das durchCO-Abspaltung aus dem 18-e-Komplex [HCo(CO)4] entsteht. Die Struktur von [HCo(CO)3]ist in der Literatur umstritten. In der jüngsten Arbeit ergibt sich als stabilste Form einplanares Molekül im Singulett-Zustand [hcoco3_2002]. Dessen C2v-Symmetrie ergibtsich aus einer quadratisch-planaren Anordnung, bei der die zum H-Atom cis-ständigenCO-Gruppen etwas vom trans-CO weg und damit zum H-Atom hingebogen sind. Die hiergezeigten Regeln (CoI als d8-Zentrum, 4 Liganden, 16e insgesamt → quadratisch-planareAnordnung) führen also zum richtigen Ergebnis – falls nicht bei weiteren Rechnungenwieder alles anders wird.
Dieses Ergebnis ist jedoch eher unerwartet. So ist das verwandte, bei der Photolyse von[Fe(CO)5] entstehende [Fe(CO)4] weder dem experimentellen Befund noch der Rechnungnach quadratisch-planar [16e1]. Die Struktur des Singulett-Zustands, dem dieseGeometrie zugetraut werden kann, ist vielmehr eine trigonale Bipyramide, der einer derdrei äquatorialen Liganden fehlt. Überdies ist das Singulett nicht der Grundzustandsondern ein Triplett mit stark verzerrter tetraedrischer Geometrie – ganz im Sinne desIsolobalkonzepts, bei dem Sie 16e-Metallcarbonyle als zweibindinge Fragmentekennenlernen. Für den Fortgang von Reaktionen sind solche Ergebnisse von Bedeutung.Für ein [Fe(CO)4] im Singulettzustand ist die oxidative Addition von H2 zum bekannten
79

[H2Fe(CO)4] ohne weiteres zu erwarten, für ein Triplett-[Fe(CO)4] ist die Reaktion jedochspinverboten [feco4_2003].
10.4 Literatur
[fe2co9_bonding_2007]
J. Reinhold, O. Kluge, C. Mealli:Integration of Electron Density and Molecular Orbital Techniques to RevealQuestionable Bonds: The Test Case of the Direct Fe–Fe Bond in Fe2(CO)9.Inorg. Chem. 2007, 46, 7142�7147.doi: 10.1021/ic700390v
[feco4_2003]
J. N. Harvey, R. Poli:Computational study of the spin-forbidden H2 oxidative addition to 16-electron Fe(0)complexes.Dalton Trans. 2003, 4100�4106.doi: 10.1039/B302916F
[hcoco3_2002]
C.-F. Huo, Y.-W. Li, G.-S. Wu, M. Beller, H. Jiao:Structures and Energies of [Co(CO)n]m (m = 0, 1+, 1−) and HCo(CO)n: DensityFunctional Studies.J. Phys. Chem. A 2002, 106, 12161�12169.doi: 10.1021/jp0270710
80

11 CO und N2: Isosterie in derKoordinationschemie
Es ist schon verrückt: N2 ist viel schwerer zu reduzieren als das isostere CO,trotzdem befasst sich die Koordinationschemie seit langem und immer intensivermit der Reduktion von gebundenem N2 – während die offensichtliche Ladungs-übertragung von Metallen in niedriger Oxidationsstufe auf einen Carbonylligandendurch eine Rückbindung kaum als Reduktion angesehen wird. Da in den letztenJahren viele neue N2-Komplexe mit dem Zentralmetall Eisen beschrieben wurden,schauen wir dort und beim homologen Ruthenium einmal genauer hin.
11.1 Elektronenstruktur von CO und N2
Ein Blick auf den Grenzorbitalbereich gibt eine Idee, warum N2 kein berauschender Ligandist:
81

E/eV
−10
−30
−40
−20
HOMO
C O
1σ
2σ
3σ1π
2π
N N
LUMO
Valenzorbitale von CO und N2 (mp2/cc-pvtz, isovalue 0.02).
11.2 CO und N2 als Liganden
CO ist ein neutraler 2-Elektronen-Donor. Eine Rückbindung wird im Rahmen der IUPAC-Regeln nicht im Sinne einer 2-Elektronen-Reduktion des CO-Liganden interpretiert. DieseBetrachtungsweise ist mit der Behandlung von zum Beispiel Wasserstoff-Liganden nicht
82

ganz konsistent. Bindet die Metallbase [Co(CO)4]− mit einem an Cobalt zentriertenElektronenpaar ein Proton, so wird das Metallzentrum formal um 2 Elektronen oxidiert, dadas Produkt als Hydrido-cobalt(I)-Komplex betrachtet wird – H ist elektronegativer als Co,so dass die beiden Bindungselektronen dem Wasserstoff zugerechnet werden. Anlagerungoder Entfernung eines CO-Liganden ist dagegen für die formale Oxidationsstufe ohneWirkung, obwohl eine ähnliche Argumentation die Rückbindung dem CO-Ligandenzuweisen könnte, der dadurch zum CO2− würde.
Bei N2-Liganden hat es sich eingebürgert, dessen Beladung eingehender zu hinterfragen(wohl deswegen, dass N2-Koordination oft durch dessen Nitrogenase-analoge Reduktionmotiviert war). Einen experimenteller Zugang erlauben Diagramme, in denen derAtomabstand in einem zweiatomigen Liganden E2 gegen die E-E-Valenzfrequenzabgetragen wird (der resultierende Verlauf wird als Badgers Regel bezeichnet [Badger'srule]). Figure 4 in [dinitrogen_2010] zeigt ein solches Diagramm, in das alsStützpunkte neben N2 auch N2H2 und N2H4 eingetragen sind. (Wir diskutieren denUnterschied zu Figure 3 in derselben Publikation.)
Ruthenium-N2-Komplexe
Das Chlorid des Pentaammin-distickstoff-ruthenim(II)-Kations war der erste isolierte N2-Komplex (1965). Ein ähnlicher Aufbau wird bei [RuIIF(tmc)(N2)]+ (tmc =1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) in[dinitrogen_ru_2010]gefunden, dessen Röntgenstrukturanlyse (Figure 1) einen N-N-Abstand von 1.144(8) Å ergibt sowie eine N-N-Valenzschwingung bei 2064 cm−1 im IR-Spektrum. Die Zahlenwerte sind typisch für einen Distickstoff-Neutralliganden.
11.3 Eisen-N2-Komplexe
Ob N2-Bindung adäquat mit oder ohne N2-Reduktion beschrieben werden sollte, hängtvon der Reduktionskraft des Zentralmetalls ab und zeigt daher die erwarteten Trendsim Periodensystem (Figure 9 in [dinitrogen_2010]) und der Oxidationsstufe desZentralmetalls.
Weitere N2-κN-Komplexe sind mit Eisen(II) bekannt. Erwartungsgemäß sind weder derN-N-Abstand noch die N2-Valenzfrequenz nennenswert anders als bei freiem N2, wiedies Tabellen mit Eisen(II)- und Eisen(0)-Komplexen in [dinitrogen_review_2010]
zeigen.
Dementsprechend ziehen derzeit Eisenkomplexe Aufmerksamkeit auf sich, in denen derDinitrogen-Ligand aktiviert zu sein scheint (Table 1 in[dinitrogen_fe_activation_2010]) und Figure 6 in [dinitrogen_2010].
83

11.4 Literatur
[dinitrogen_fe_activation_2010]
Y. Lee, N. P. Mankad and J. C. Peters:Triggering N2 uptake via redox-induced expulsion of coordinated NH3 and N2silylation at trigonal bipyramidal iron.Nat. Chem. 2010, 2, 558-565.doi: 10.1038/nchem.660
[dinitrogen_ru_2010]
T. Kizaki, T. Abe, T. Matsumoto and S. Ogo:A pH-stable Ruthenium(II)-based Sensing System for Dissolved Dinitrogen.Chem. Lett. 2010, 39, 128-129.doi: 10.1246/cl.2010.128
[dinitrogen_review_2010]
J. L. Crossland and D. R. Tyler:Iron��dinitrogen coordination chemistry: Dinitrogen activation and reactivity.Coord. Chem. Rev. 2010, 254, 1883�1894.doi: 10.1016/j.ccr.2010.01.005
[dinitrogen_2010]
P. L. Holland:Metal-dioxygen and metal-dinitrogen complexes: where are the electrons?Dalton Trans. 2010, 39, 5415�5425.doi: 10.1039/C001397H
[dinitrogen_activation2_2010]
N. Hazari:Homogeneous iron complexes for the conversion of dinitrogen into ammonia andhydrazine.Chem. Soc. Rev. 2010, 39, 4044�4056.doi: 10.1039/B919680N
84

12 Allgemeine Literatur
Aktuelle Literatur zum Gebiet Koordinationschemie ist derzeit rar. Es wird daherempfohlen, eines der aktuellen allgemeinen Lehrbücher der Anorganischen Chemie zunutzen. Hier einige neuere Bücher in umgekehrter Reihenfolge ihres Erscheinungsjahrs:
P. Atkins, T. Overton, J. Rourke, M. Weller, F. Armstrong: Inorganic Chemistry, OxfordUniversity Press, 2009,, ISBN: 978-0199236176.
C. E. Housecroft, A. G. Sharpe: Inorganic Chemistry, Pearson, 2008, ISBN:978-0-13-175553-6.
N. Wiberg: Lehrbuch der Anorganische Chemie, de Gruyter, 2007, ISBN:978-3110177701.
E. Riedel, R. Alsfasser, C. Janiak, T. M. Klapötke: Moderne Anorganische Chemie, deGruyter, 2007, ISBN: 978-3110190601.
85

13 Anhang: Zahl ungepaarter parallelerSpins (n), μeff = {n(n+2)}1/2 und χT = μeff2/8
n μeff χT0 0.00 0.0001 1.73 0.3752 2.83 1.0003 3.87 1.8754 4.90 3.0005 5.92 4.3756 6.93 6.0007 7.94 7.8758 8.94 10.0009 9.95 12.375
10 10.95 15.000
86