journal of vol. no. 13, issue of may 5, pp. q of ... · the journal of biological chemistry q 1987...
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY Q 1987 by The American Society of Biological Chemists, Inc
Vol. 262, No. 13, Issue of May 5, pp. 6266-6279,1987 Printed in U.S.A.
Quantitative Studies of Hydroperoxide Reduction by Prostaglandin H Synthase REDUCING SUBSTRATE SPECIFICITY AND THE RELATIONSHIP OF PEROXIDASE TO CYCLOOXYGENASE ACTIVITIES*
(Received for publication, September 8, 1986)
Christine M. Markey, Abdo Alward, Paul E. Weller, and Lawrence J. Marnett$ From the Department of Chemistry, Wayne State University, Detroit, Michigan 48202
The peroxidase activity of prostaglandin H (PGH) synthase catalyzes reduction of 5-phenyl-4-pentenyl hydroperoxide to 5-phenyl-4-pentenyl alcohol with a turnover number of approximately 8000 mol of 5- phenyl-4-pentenyl hydroperoxide/mol of enzymelmin. The kinetics and products of reaction establish PGH synthase as a classical heme peroxidase with catalytic efficiency similar to horseradish peroxidase. This sug- gests that the protein of PGH synthase evolved to fa- cilitate peroxide heterolysis by the heme prosthetic group. Comparison of an extensive series of phenols, aromatic amines, 8-dicarbonyls, naturally occurring compounds, and nonsteroidal anti-inflammatory drugs indicates that considerable differences exist in their ability to act as reducing substrates. No correlation is observed between the ability of compounds to support peroxidatic hydroperoxide reduction and to inhibit cy- clooxygenase. In addition, the resolved enantiomers of MK-410 and etodolac exhibit dramatic enantiospecific differences in their ability to inhibit cyclooxygenase but are equally potent as peroxidase-reducing sub- strates. This suggests that there are significant differ- ences in the orientation of compounds at cyclooxygen- ase inhibitory sites and the peroxidase oxidation site(s). Comparison of 5-phenyl-4-pentenyl hydroper- oxide reduction by PGH synthase and horseradish per- oxidase reveals considerable differences in reducing substrate specificity.
Both the cyclooxygenase and peroxidase activities of PGH synthase inactivate in the presence of low micro- molar amounts of hydroperoxides and arachidonic acid. PGH synthase was most sensitive to arachidonic acid, which exhibited an IBO of 0.6 p~ in the absence of all protective agents. Inactivation by hydroperoxides requires peroxidase turnover and can be prevented by reducing substrates. The IBO values for inactivation by 15-hydroperoxy-5,8,11,13-eicosatetraenoic acid are 4.0 and 92 p ~ , respectively, in the absence and pres- ence of 500 p~ phenol, a moderately good reducing substrate. The ability of compounds to protect against hydroperoxide-induced inactivation correlates di- rectly with their ability to act as reducing substrates. Hydroquinone, an excellent reducing substrate, pro- tected against hydroperoxide-induced inactivation when present in less than 3-fold molar excess over
* This work was supported in part by Research Grant GM 23642 from the National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ Recipient of Faculty Research Award FRA 243 from the Ameri- can Cancer Society. To whom correspondence should be addressed 435 Chemistry, Wayne State University, Detroit, MI 48202.
hydroperoxide. The presence of a highly efficient hy- droperoxide-reducing activity appears absolutely es- sential for protection of the cyclooxygenase capacity of PGH synthase. The peroxidase activity is, therefore, a twin-edged sword, responsible for and protective against hydroperoxide-dependent inactivation of PGH synthase. As such, it may constitute an important tar- get for pharmacological modulation of eicosanoid bio- synthesis.
The first step in the biosynthesis of prostaglandins, throm- boxane, and prostacyclin is the oxygenation of arachidonic acid to the hydroperoxy endoperoxide PGG, (1, 2). This reaction is catalyzed by a heme-containing oxygenase, called cyclooxygenase, that requires no external source of electrons (3-5). The hydroperoxy group of PGG, is reduced to a hydroxy group (PGH, ’) by a peroxidase that utilizes a wide variety of compounds to provide the requisite pair of electrons (3, 6). Ohki et al. (7) discovered that the cyclooxygenase and perox- idase activities of bovine seminal vesicle copurify and are contained on a single 71-kilodalton protein. This finding was confirmed for the activities in ovine seminal vesicle, and it has been shown that monoclonal antibodies raised against the cyclooxygenase coprecipitate the peroxidase (8, 9). The puri- fied protein, in the presence of an excess of reducing substrate, converts arachidonic acid to PGH, and is therefore termed PGH synthase (Equation 1).
Both cyclooxygenase and peroxidase activities require the presence of one molecule of ferriprotoporphyrin IX/molecule of enzyme (a dimer of 71-kDa subunits) (10). The fact that heme participates in the oxidations catalyzed by the two different activities raises intriguing questions about the loca- tion of the active sites, the role of the peroxidase in cycloox- ygenase catalysis, and the reason for the presence of both enzyme activities in a single protein. One could presumably answer these questions by performing a detailed study of the reduction of hydroperoxides by the peroxidase and comparing it to turnover of the cyclooxygenase. This has been technically difficult because the natural substrate, PGG,, is difficult to prepare in high purity, and the assays for its reduction are tedious and time-consuming. Readily available hydroperox- ides such as cumene hydroperoxide and t-butyl hydroperoxide are not substrates for the peroxidase (7). We recently devel- oped a general assay for peroxidases in which one quantitates by HPLC the enzyme-catalyzed reduction of 5-phenyl-4-pen-
The abbreviations used are: PG, prostaglandin; HPLC, high pres- sure liquid chromatography; PPHP, 5-phenyl-4-pentenyl hydroper- oxide; PPA, 5-phenyl-4-pentenyl alcohol; DDC, diethyldithiocar- bamic acid HPETE, hydroperoxy-5,8,11,13-eicosatetraenoic acid.
6266

The Peroxidase of PGH Synthase
2y . 1 : ; C H D P ,: PGH Synthase PGH Synthase
6OH PGG2 (Peroxidase) 20:4 (cyclooxygenase) Heme
Llsplrln lnhlblts
tenyl hydroperoxide (PPHP) to 5-phenyl-4-pentenyl alcohol (PPA) (Equation 2) (11).
The availability of this new method has enabled us to carry out a comprehensive study of hydroperoxide reduction by the peroxidase of PGH synthase. The results reveal that 1) the peroxidase of PGH synthase reduces hydroperoxides hetero- lytically a n d is among the most active of any known heme peroxidases; 2 ) there is no correlation between the ability of a series of compounds to inhibit the cyclooxygenase a n d t o reduce the peroxidase; 3) dramatic stereochemical differences exist in the interaction of compounds with the cyclooxygenase a n d peroxidase; and 4) the peroxidase is a critical determinant of the ability of cyclooxygenase to oxygenate arachidonic acid.
MATERIALS AND METHODS
Sulindac sulfide, MK-447, 5-hydroxyindomethacin, and (+)- and (-)-MK-410 were gifts from Merck Sharp and Dohme (Rahway, NJ); (+)- and (-)-A were gifts from Sandoz Ltd. (Basel, Switzerland); BW-755C was a gift from Wellcome Laboratories (Beckenham, Eng- land); nafazatrom was a gift from Miles Pharmaceuticals (West Haven, CT); oxyphenbutazone and diclofenac were gifts from Ciba- Geigy Corp. (Summit, NJ); D- and L-etodolac were gifts from Ayerst Laboratories (Montreal, Canada); meclofenamic acid and isoxicam were gifts from Warner-Lambert Pharmaceutical Co. (Ann Arbor, MI); piroxicam was a gift from Pfizer Central Research (Groton, CT); and ibuprofen and flurbiprofen were gifts from The Upjohn Co. The following chemicals were purchased as analytical grade reagents and used without further purification: L-ascorbic acid, butylated hydrox- yanisole, butylated hydroxytoluene, diethyldithiocarbamic acid (DDC), L-epinephrine, guaiacol, reduced glutathione, indole-3-acetic acid, reduced nicotinamide adenine dinucleotide, reduced nicotina- mide adenine dinucleotide phosphate, polyoxyethylene sorbitan mon- olaurate (Tween 20), DL-thiotic acid (lipoic acid), L-tryptophan, mon- ophenylbutazone, phenylbutazone, indomethacin, acetylsalicylic acid (aspirin), n-propyl gallate, acetominophen, flufenamic acid, luminol, uric acid, and hematin (Sigma); phenol and phenylhydrazine (Mathe- son, Coleman and Bell Manufacturing Chemists); aniline, sodium dibasic phosphate, and sodium monobasic phosphate (Spectrum Chemical Manufacturing Corp.); mitomycin C (Bristol Laboratories); L-methionine (Behring Diagnostics), potassium iodide, 30% hydrogen peroxide, and hydroquinone (J. T. Baker Chemical Co.); and 1,3- diphenylisobenzofuran (Aldrich). HPLC-grade methanol was pur- chased from Fisher; octadecyl solid-phase extraction columns were purchased from J. T. Baker Chemical Co.; and arachidonic acid (>99%) was purchased from NuChek Preps.
15-HPETE was prepared by the method of Funk et al. (12); 12- HPETE was prepared by the method of Porter et al. (13); PPHP was prepared as previously described (11); and 9-hydroperoxy-10-octade- cenoic acid and 10-hydroperoxy-8-octadecenoic acid were gifts from Regine Labeque (Department of Chemistry, Wayne State University, Detroit, MI).
Horseradish peroxidase (Type IV) was purchased from Sigma and desalted on a Sephadex G-15 column (1 X 10 cm, Pharmacia P-L Biochemicals) with elution by 100 mM potassium citrate (pH 5.5).
Preparation of Purified PGH Synthase-PGH synthase was puri- fied from ram seminal vesicle microsomes as previously described (14). Enzyme preparations purified by this method had specific activ- ities in the range of 17.5-28.5 pmol of arachidonic acid oxidized per
Hem
mg of protein/min in the absence of exogenously added hematin. These preparations were 48-51% holoenzyme as measured by cycloox- ygenase activity in the presence and absence of 1 p~ hematin. The purified enzyme contained 300 p~ DDC as a protective agent against inactivation and was stored in 0.5-ml portions at -80 "C until used. Aliquots were desalted on Sephadex G-25 gel equilibrated with 20 mM sodium phosphate (pH 7.8) containing 30% glycerol using a rapid desalting procedure (15). All preparations were desalted in this man- ner unless otherwise indicated. Glycerol was added as a stabilizing agent (16) but did not serve as a protective agent against hydroper- oxide-induced inactivation at the levels present (data not shown). Preparations desalted in the presence of glycerol were stable up to 6 h at 4 "C, whereas those in the absence of glycerol were inactivated with a half-life of approximately 1 h. Protein was determined by the Bio-Rad protein assay according to the manufacturers' instructions using bovine serum albumin as standard.
Cyclooxygenase Activity Assay and Hydroperoxide-dependent Inac- tivation of PGH Synthase-The cyclooxygenase activity of PGH synthase was estimated by measuring the maximal rate of oxygen uptake in the presence of enzyme and arachidonic acid (Gilson model 5/6H oxygraph equipped with a Clarke-type electrode (Gilson Medical Electronics, Inc., Middleton, WI)). The final volume of the oxygraph chamber was 1.3 ml. The velocity was determined from a tangent to the linear portion of the oxygen uptake curve. Purified enzyme was added to 100 mM sodium phosphate buffer (pH 7.8) pre-equilibrated at 37 "C (concentrations are given in the figure legends). Hydroper- oxides, when included, were prepared as stock solutions in methanol or ethanol and added in volumes less than 30 pl. They were allowed to incubate with the enzyme for 2 min, unless otherwise indicated. Phenol (stock solution in water) was added to each assay after the 2- min incubation to a final concentration of 500 pM. Addition of phenol after hydroperoxide inactivation did not protect against hydroperox- ide-induced inactivation but enhanced the residual activity. Reactions were initiated by addition of 100 p~ arachidonic acid prepared as the sodium salt in water.
In experiments where protection from inactivation was investi- gated, reducing substrates were added in volumes less than 30 p1 from stock solutions in methanol or water. Reducing substrates were added to the enzyme in buffer and pre-equilibrated for 30 s before addition of hydroperoxide.
Cyclooxygenase Inhibition-Inhibition of cyclooxygenase was de- termined by incubating various concentrations of inhibitor with 350 nM enzyme for 1 min prior to initiation of reaction with 100 p~ arachidonic acid and measuring cyclooxygenase activity as described above. In cases where inhibition was determined in the presence of phenol, 500 p~ phenol was included in the buffer. I,, values were determined from the concentration of inhibitor that gave half-maxi- mal activity under these conditions.
Peroxidase Assay-Peroxidase activity was measured by the reduc- tion of PPHP to PPA using the chromatographic assay described elsewhere (11). PGH synthase was preincubated in 2 ml of 100 mM sodium phosphate containing 200 p M Tween 20 (pH 7.8) at 37 "C for 3 min at the concentrations given in the figures. For the inactivation experiments, 15-HPETE was added to the enzyme solution and allowed to incubate for 3 min before 500 p~ phenol was added as the reducing substrate. Three min later, the reaction was initiated by the addition of 100 p~ PPHP. Reactions were terminated after 3 min by solid-phase extraction onto C,, reverse-phase extraction columns. PPHP and PPA were eluted with 2 ml of methanol, and an internal standard, p-nitrobenzyl alcohol, was added to 100 p ~ . Samples were chromatographed isocratically on a 5-pm Zorbax Ce reverse-phase HPLC column (Du Pont) with 65% (v/v) methano1:water as the mobile phase at a flow rate of 2 ml/min. The amount of PPA produced and the unreacted PPHP were quantitated by the internal standard method previously described (11).
Substrate Efficiency Assay-The peroxidase assay was performed essentially as described above. Experiments to examine the efficiency of compounds as reducing substrates for PGH synthase contained

6268 The Peroxidase of PGH Synthase
the concentration of enzyme required to reduce 50% of PPHP (100 pM) in the presence of 200 p M phenol. The compound to be evaluated as a reducing substrate was added at 200 &I to the enzyme and allowed to incubate for 3 min. PPHP (100 p M ) was then added to initiate the reaction. Reactions were terminated and worked up as described above. The 2:l ratio of reducing substrate to hydroperoxide was chosen to reflect the stoichiometry of two electrons necessary per peroxide molecule reduced, assuming each compound is a one- electron donor. Substrate efficiency assays for horseradish peroxi- dase-catalyzed reduction of PPHP were performed in essentially the same manner except that 2.5 nM enzyme was incubated in 100 RIM potassium citrate containing 200 p M Tween 20 (pH 5.5) at 25 "C.
RESULTS
Peroxidative Reduction of PPHP by PGH Synthase-We recently developed an assay for measuring peroxidase activity that measures an enzymes's ability to reduce PPHP to the corresponding alcohol, PPA, in the presence of a reducing substrate (11). PPHP is an excellent substrate for PGH synthase. As shown in Fig. 1 (left), incubation of 100 FM PPHP with PGH synthase in the presence of 200 ~ L M phenol (a reducing substrate) gave significant reduction. In the ab- sence of phenol, PPHP was not reduced (Fig. 1, right). Thus, PGH synthase catalyzes the two-electron reduction of hydro- peroxides coupled with the oxidation of reducing substrates.
We attempted to determine steady-state kinetic parameters of PPHP reduction by PGH synthase in the presence of reducing substrates. The time course of PPHP reduction at 0 "C in the presence of 200 ~ L M BW-755C, an excellent reduc- ing substrate for PGH synthase, is shown in Fig. 2 A . Within the first 10 s of reaction, the enzyme inactivated as judged by the lack of further reduction of PPHP at longer time periods. Similar results were observed with 200 pM phenol or hydro- quinone as reducing substrate (data not shown). The absence
Phenol
*I
Ls,
0 t
4 0 6 1 0 % 1111
T i m e Cmln)
FIG. 1. High performance liquid chromatograms of PGH synthase-catalyzed reduction of PPHP. The peroxidase assay was performed as described under "Materials and Methods." Incu- bations contained 154 nM PGH synthase, 200 p~ phenol (left) or no reducing substrate (right), and 100 MM PPHP. The peak at 3.8 min
of a linear time course limited the kinetic information obtain- able. Increasing the amount of PGH synthase increased PPA production in a linear manner (Fig. 3) but did not alter the time course (Fig. 2 A ) . As shown in Fig. 2B, the time course for PPHP reduction by horseradish peroxidase in the presence of 200 FM phenol was linear for at least 8 min. A turnover number of 10,500 mol of PPA produced per mol of horseradish peroxidase/min was calculated from a Lineweaver-Burk plot using the V,,, value under saturating conditions. If a turnover number is calculated at the 10-s point of Fig. 2A for 39 nM PGH synthase, a value of 1,333 mol of PPA produced per mol of PGH synthase/lO s is obtained (-8,000/min). Due to the rapid enzyme inactivation, this value considerably underesti- mates the actual turnover number (see "Discussion"). Com- parison of time courses run at 37 "C and 0 "C showed that the inactivation process occurred to a greater extent at 37 "C than at 0 "C as judged by the higher production of PPA at 0 "C (data not shown). Thus, the inactivation process could be slowed but not prevented by decreasing the temperature.
Efficiency of Reducing Substrates for PGH Synthase-De- spite problems in determining steady-state kinetic parameters for PGH synthase, our assay was useful for evaluating the efficiency of a number of compounds as reducing substrates for the peroxidase activity of PGH synthase. The compounds evaluated included a group of phenolic compounds, several types of nonsteroidal anti-inflammatory drugs, some naturally occurring reductants, and other compounds routinely used as reducing substrates for peroxidases. The results are presented in Table I in the form of an "index of efficiency." The index value reflects the ability of the compound to act as an electron donor for the peroxidative reduction of PPHP. The index was calculated from the equation: I = [PPA]/([PPA] + [PPHP]), where [PPA] is equal to the alcohol produced during the reaction and [PPHP] is equal to the remaining hydroperoxide. High index values indicate excellent reducing substrates, whereas low index values indicate poor reducing substrates. The index was designed to rank substrates relative to phenol which was placed at an index value of 0.5 by manipulating the enzyme concentration. All potential substrates were run under identical conditions. The 3-min time point was chosen to accommodate the automated method described elsewhere (ll), although the time course information indicated that reduction was complete after 10 s. Reduction of PPHP by each of the compounds in Table I did not occur in control experiments performed in the absence of enzyme.
The data in Table I demonstrate that a smooth gradation exists in the ability of different compounds to act as reducing substrates for PGH synthase. Thus, the PPHP reduction assay not only determines whether a compound is a peroxi- dase-reducing substrate but also ranks it relative to a series of other reducing substrates. To determine whether the series in Table I is specific for PGH synthase, a limited number of compounds were tested with horseradish peroxidase. As shown in Table 11, several compounds that are excellent reducing substrates for PGH synthase (Table I) are essentially inactive as electron donors for horseradish peroxidase. These data indicate obvious differences in the substrate specificity of the two peroxidases.
Effect of Chirality on Reducing Substrate Efficiency-Sev- era1 compounds that are good peroxidase-reducing substrates are also inhibitors of cyclooxygenase, but a cursory inspection of Table I reveals a poor correlation between the two activities. A more definitive probe with which to study the relationship between cyclooxygenase inhibitory potency and reducing sub-
annotated I.S. is the internal standard,p-nitrobenzyl alcohol. Vertical strate efficiency is based on StereoselectivitY. Three corn- numbers are associated with retention times. AU, absorbance unit. pounds that contain chiral centers, namely MK-410, A ana-

The Peroxidase of PGH Synthase
A 193
0 97
- 39
* 10
1 1
10 20 30 Time (sec)
FIG. 2. Comparison of time courses for reduction of PPHP by PGH synthase and by horseradish peroxidase. The peroxidase assay was performed as described under “Materials and Methods.” A, the concentration of PGH synthase was varied as noted. Re- actions were run in 100 mM sodium phosphate containing 200 p M Tween 20 (pH 7.8) at 0 “C and 200 pM BW-755C was used as the reducing substrate. B, the concentration of horseradish perox- idase was 2.5 nM. Reactions were run in 100 mM potassium citrate containing 200 p~ Tween 20 (pH 5.5) at 25 “C with 200 p~ phenol as the reducing substrate. In- cubations were terminated at the indi- cated time points by solid-phase extrac- tion as described. Data points are the average of three determinations.
p ”-E 30 20 10 100 [PGH Synthase] 200 300 (nM) 400
FIG. 3. Dependence of PGH synthase-catalyzed reduction of PPHP on protein Concentration. The peroxidase assay was performed as described under “Materials and Methods.” Enzyme concentration was varied as indicated and 200 p~ phenol was used as the reducing substrate. Data points represent three determinations.
logs of MK-447, and etodolac, were examined to determine if they exhibit stereoselective peroxidase-reducing substrate ac- tivity. The enantiomers of the analog of MK-447, A, exhibit differential activity in in vivo assays in their ability to act as anti-inflammatory agents (17). MK-410 isomers are differ- entially active as inhibitors of the cyclooxygenase (18). Under our assay conditions, the (+)-isomer of MK-410 inhibited oxygen uptake by 270 nM PGH synthase in the presence of 100 p~ arachidonic acid with an I,, of 21 pM. Under identical conditions, the (-)-isomer did not inhibit up to 125 pM. In the presence of 500 p~ phenol, the I,, of the (+)-isomer was 15 PM, whereas the (-)-isomer inhibited only 30% a t 300 p~ (Table 111). The two enantiomers of etodolac possess differ- ential anti-inflammatory activity in vivo and inhibitory po- tency toward the cyclooxygenase activity of an acetone powder of ram seminal vesicle microsomes (in the presence of 600 p~ phenol) (19). In vitro and in vivo, the D-isomer is more effective than the L-isomer. In cyclooxygenase assays using 92 nM purified PGH synthase in the presence of 500 p~ phenol and 100 p~ arachidonic acid, the Is, for the D-isomer was 2 p ~ , whereas the L-isomer had no effect at concentra- tions up to 308 p ~ . In contrast to their effects on cyclooxy- genase, there appears to be no enantioselectivity in the ability of any of these optically active compounds to serve as electron donors for the peroxidatic reduction of P P H P by PGH syn- thase (Table 111). In fact, the index values for the etodolac enantiomers appear to exhibit opposite stereoselectivity for their electron donor activity compared to their cyclooxygenase inhibitory activity, although the difference is small.
Inactivation of PGH Synthase by Hydroperonides-In the absence of reducing substrates, PGH synthase is rapidly in-
nM
nM
nM
nM
100 1 6269
B
Time (min)
activated by low levels of hydroperoxide (20). We have deter- mined the hydroperoxide-induced inactivation of PGH syn- thase by following both the cyclooxygenase and peroxidase activities. The inactivation of the cyclooxygenase activity in the presence of varying amounts of hydroperoxide was meas- ured as described under “Materials and Methods.” Inactiva- tion was complete within 2 min when 352 nM PGH synthase was incubated with 4 p~ 15-HPETE as shown in Fig. 4. The concentration of hydroperoxide that gave half-maximal inac- tivation of the cyclooxygenase component of 352 nM PGH synthase in 2 min is given in Table IV for several hydroper- oxides and for arachidonic acid. The values range from 1.6 to 4.6 p ~ , and it appears that the hydroperoxide is required for inactivation. PPA, the alcohol analog of PPHP, did not inactivate PGH synthase at 10 p~ (data not shown). The second method we employed measured the ability of hydro- peroxides to inactivate the peroxidase activity of PGH syn- thase. Inactivation by 15-HPETE as measured by the reduc- tion of PPHP to the corresponding alcohol, PPA, occurred within the same time period as cyclooxygenase inactivation and with a similar of 2.5 p~ (Fig. 5B).
Protection of PGH Synthase from Hydroperoxide-induced Inactivation by Reducing Substrates-Addition of a reducing substrate prior to treatment with hydroperoxide provided substantial protection of both the cyclooxygenase and perox- idase activities. Fig. 5 ( A and B ) illustrate the effect of increasing amounts of 15-HPETE on both the cyclooxygenase and peroxidase activities of PGH synthase in the absence and presence of 500 p~ phenol. In the absence of phenol, both the cyclooxygenase and peroxidase activities inactivated with I,, values of 4.0 and 2.5 pM, respectively, for 352 nM PGH synthase. In the presence of 500 pM phenol, the for inac- tivation of the cyclooxygenase activity was 92 p ~ , and there was no inactivation up to 10 p~ 15-HPETE for the peroxidase activity (Table IV). The in the presence of phenol was not determinable for the peroxidase activity for practical reasons.
Protection of PGH synthase by reducing substrates oc- curred in a dose-dependent manner (Fig. 6). The ability of a compound to protect against the hydroperoxide-induced in- activation by 10 p~ 15-HPETE correlated with the efficiency of the compound to serve as an electron donor (Fig. 6 and Table I). Hydroquinone, with an index of efficiency of 0.99, was an excellent protective agent a t 25 p~ and eventually led to stimulation of enzyme activity at 70 p ~ . Phenol, a moderate reducing substrate with an index value of 0.5, protected ap- proximately 70% of the activity at 38 pM, with essentially

6270 The Peroxidase of PGH Synthase TABLE I
Index of efficiency of reducing substrates for PGH synthase-catalyzed reduction of PPHP
The peroxidase assay was performed as described under “Materials and Methods.” Each potential reducing substrate was added to a final concentration of 200 FM from stock solutions prepared as described under “Materials and Methods.” Results are presented as the fraction of PPA produced, calculated as described in the text. Enzyme samples were not desalted prior to use and therefore contained approximately 4 FM DDC. The sample denoted “NO reducing substrate” is the control incubation containing enzyme and PPHP.
Compound Index
n-Propyl gallate Butylated hydroxyanisole Hydroquinone (-)-A Sulindac sulfide BW-755C (+)-A L-Epinephrine Acetaminophen Nafazatrom MK-447 Oxyphenbutazone Flufenamic acid Guaiacol Methylphenyl sulfide Lipoic acid L-Etodolac
Phenol Luminoi
Diclofenac D-Etodolac Indoleacetic acid Ascorbic acid Uric acid Meclofenamic acid 5-Hydroxyindomethacin Piroxicam Butylated hydroxytoluene L-Tryptophan (-)-MK-410 (+)-MK-410 Isoxicam Monophenylbutazone Phenylbutazone Mitomycin C Glutathione (reduced) L-Methionine 2-Arnino-4,5-diphenylthiazole Aniline NADPH NADH Iodide Phenylhydrazine No reducing substrate (control) Indomethacin Ibuprofen 1,3-Diphenylisobenzofuran Flurbiprofen Salicylic acid Aspirin
1.00 * 0.00 1.00 f 0.01 0.99 f 0.01 0.99 f 0.01 0.96 f 0.05 0.96 f 0.02 0.92 -+ 0.12 0.92 C 0.01 0.86 f 0.04 0.79 f 0.03 0.72 f 0.01 0.71 f 0.04 0.62 f 0.03 0.64 -C 0.05 0.62 C 0.01 0.59 f 0.07 0.55 f 0.03 0.53 C 0.02 0.50 2 0.07 0.50 f 0.03 0.46 f 0.01 0.45 C 0.03 0.43 If: 0.01 0.37 f 0.01 0.35 ? 0.03 0.34 f 0.04 0.34 f 0.00 0.27 f 0.03 0.27 f 0.00 0.27 f 0.01 0.26 f 0.01 0.25 f 0.02 0.23 ? 0.09 0.23 f 0.00 0.21 f 0.01 0.21 1- 0.09 0.21 ? 0.12 0.20 f 0.03 0.18 f 0.11 0.17 2 0.04 0.17 ? 0.02 0.15 f 0.00 0.14 f 0.03 0.13 f 0.03 0.12 f 0.00 0.12 f 0.00 0.12 f 0.09 0.12 +- 0.01 0.06 f 0.06 0.04 * 0.01 -.
complete protection occurring at 308 WM (data not shown). Tryptophan was a poor protective agent with only 40% of the activity remaining at 160 pM, reflecting the low index value of 0.27. Protection by tryptophan was only 78% at 308 pM (data not shown). Thus, the ability of a compound to protect PGH synthase from hydroperoxide-induced inactivation de- pends on its ability to support enzyme-catalyzed hydroper- oxide reduction.
Dependence of Inactivation on a Functional Peroxidme- The heme prosthetic group was shown to be required for the inactivation process. Experiments involving inactivation of
TABLE I1 Comparison of index of efficiency for reducing substrates for
h.orseradish peroxidase and PGH synthase The peroxidase assay was performed as described under “Materials
and Methods.” The index value reported represents the fraction of PPA produced and was calculated as described in the text. Incuba- tions with PGH synthase were performed in 100 mM sodium phos- phate containing 200 PM Tween 20 (pH 7.8) a t 37 “C, whereas those with horseradish peroxidase were in 100 mM potassium citrate con- taining 200 ” Tween 20 (pH 5.5) at 25 “C. Reducing substrates for both enzymes were mesent at 200 UM.
Substrate
- BW-755C Sulindac sulfide Epinephrine Nafazatrom Lipoic acid Indoleacetic acid Aniline Iodide No reducing substrate
Index
0.96 & 0.02 0.96 IC 0.05 0.92 & 0.01 0.79 & 0.03 0.59 & 0.07 0.45 & 0.03 0.18 * 0.11 0.15 2 0.00 0.13 * 0.03
0.98 f 0.01 0.17 f 0.03 0.39 k 0.01 0.34 rt 0.01 0.06 f 0.01 0.14 f 0.01 0.97 f 0.01 0.97 f 0.01 0.04 k 0.01
~ ~~
TABLE I11 Comparison of cyclooxygenase Is0 values and peroxidase index values
for several chiral nonsteroidal anti-inflammatory compounds The cyclooxygenase assay was performed as described under “Ma-
terials and Methods.” Inhibitors were added at varying concentrations to 350 nM enzyme and allowed to incubate 1 min before 100 p~ arachidonic acid was added. Phenol (500 PM) was included in these assays. The peroxidase assay was performed as described under “Materials and Methods.” Reducing substrates were added to 200 p~ from stock solutions prepared as described under “Materials and Methods.” The index value represents the fraction of PPA produced and was calculated as described in the text.
Compound I W
/1M
- ”
Index
(-)-A (+)-A
0.99 f 0.01
L-Etodolac >300 0.92 f 0.12 0.55 * 0.03
D-Etodolac 2 0.46 k 0.01 0.27 k 0.01
15 0.26 * 0.01 (-)-MK-410 >300 (+)-MK-410
6 0 120 180 240 300
TIME (sec)
FIG. 4. Time course for inactivation of cyclooxygenase ac- tivity of PGH synthase by 15-HPETE. The cyclooxygenase ac- tivity was measured as described under “Materials and Methods.” PGH synthase (352 nM) was incubated with 4 PM 15-HPETE for the indicated times, and 500 p~ phenol was then added. Arachidonic acid (100 FM) was added after 30 s to initiate oxygen uptake. Data points are the average of three determinations.
the cyclooxygenase activity of PGH synthase and the depend- ence of the inactivation process on the presence of heme were performed with a 37% holoenzyme preparation and are sum- marized in Table V. Incubations were performed with enzyme

The Peroxidase of PGH Synthase 6271
TABLE IV Inactivation of cyclooxygenase activity of PGH synthase by several
hydroperoxides Inactivation of the cyclooxygenase activity of PGH synthase was
measured as described under “Materials and Methods.” PGH syn- thase (352 nM) was incubated with varying amounts of hydroperox- ides for 2 min. In experiments labeled “+DDC,” the enzyme was not desalted and therefore contained approximately 4 FM DDC. In exper- iments labeled “+Phenol,” 500 PM phenol was added prior to the hydroperoxide and allowed to incubate for 1 min. The reported 150
values were calculated based on controls assayed in the absence of hydroperoxide. Values are the average of three determinations. ~ _ _ _ ~ _ _ ~
I W
+DDC +Phenol -DDC
IrM P M PM 15-HPETE 4.0 92.5 2.75 12-HPETE 1.8 9-00H-18:l” 2.5 10-00H-18:l 1.8
Hydroperoxide _ _ _ _ ” ~ - ~ ~ _ _ _
~
PPHP 2.3
Arachidonic acid 1.6 8.7 0.6 Hz’& 1.8 0.3
_____”_
9-00H-181, 9-hydroperoxy-10-octadecenoic acid; 10-00H-181, 10-hydroperoxy-8-octadecenoic acid.
reconstituted with 1 FM hemat,in (“fully reconstituted”) or with enzyme containing only the native heme (“nonreconsti- tuted”). Inactivation of the nonreconstituted preparation with 1 FM arachidonic acid compared to nonreconstituted enzyme without arachidonic acid was 90% (Table V, Experiments 1 and 3). Addition of 1 ~ L M hematin to the nonreconstituted
A 1 4 0
120 - 0
C L .- 0 0 100
c
80 0
a c $ 60 r x 0
4 0 0”
20
-Phenol
1 I I I I 4 8 12 16 20
1
[15-HPETE] (NM)
enzyme after inactivation with arachidonic acid showed 60% of the activity of the fully reconstituted enzyme remained (Table V, Experiments 2 and 4). This reconstitutable activity approximates the portion of PGH synthase that was present as apoenzyme at the time of the arachidonic acid-induced inactivation. Reconstitution of the enzyme with 1 PM hematin prior to the addition of arachidonic acid gave 86% inactivation compared to fully reconstituted enzyme (Table V, Experi- ments 2 and 5 ) . Thus, the apoenzyme, which is devoid of peroxidase activity (4), is not subject to arachidonic acid- induced inactivation, even in the presence of holoenzyme.
The requirement for the functional peroxidase for reducing substrate-dependent protection against hydroperoxide-in- duced inactivation was also apparent from experiments in which the I,, for 15-HPETE was determined for several enzyme concentrations. As illustrated in Fig. 7, the 1,” in- creased with increasing PGH synthase concentration. Thus, the presence of efficient peroxide-reducing capacity, sup- ported by a reducing substrate and the functional peroxidase of PGH synthase, afforded protection of the enzyme from inactivation.
Arachidonic Acid-dependent Inactivation of PGH Syn- thase-The hydroperoxide that regulates PGH synthase ac- tivity is PGG2, derived from arachidonic acid (21). In experi- ments in which arachidonic acid was used as the inactivating agent in place of a hydroperoxide, time-dependent loss of cyclooxygenase activity was again observed (Fig. 8 and Table IV). Varying amounts of arachidonate were added and allowed to react for 2 min before 100 u~ arachidonate was added to
B
t- 60 1-
2 4 6 a 10
[15-HPETE] (pM)
FIG. 5. Inactivation of cyclooxygenase and peroxidase activities of PGH synthase by 15-HPETE. A , the cyclooxygenase activity was measured as described under “Materials and Methods.” Varying concentrations of 15-HPETE were added to 352 nM enzyme, and the mixture was allowed to incubate for 2 min before 500 FM phenol was added. After 30 s, 100 PM arachidonic acid was added to initiate the reaction. When phenol was used as a protector (+Phenol), it was added 30 s prior to addition of 15-HPETE. The results are presented as percent of control to normalize values. The I,, for 15-HPETE-induced inactivation of the cyclooxygenase activity in the absence of phenol was 4 PM. 23, the peroxidase activity of the enzyme was measured as described under “Materials and Methods.” Varying concentrations of 15-HPETE were added to 352 nM enzyme and allowed to incubate for 3 min before 500 FM phenol was added. After 3 min, 100 ,uM PPHP was added to initiate the reaction. Reactions were terminated after 3 min as described. PPHP and PPA were recovered and analyzed by HPLC as described. When phenol was used as a protector (+Phenol), it was added 3 min prior to the addition of 15-HPETE. Results are presented as the concentration of PPA produced. The 150 for 15-HPETE-induced inactivation of the peroxidase activity in the absence of phenol was 2.5 p M . Data points represent the result of three determinations.
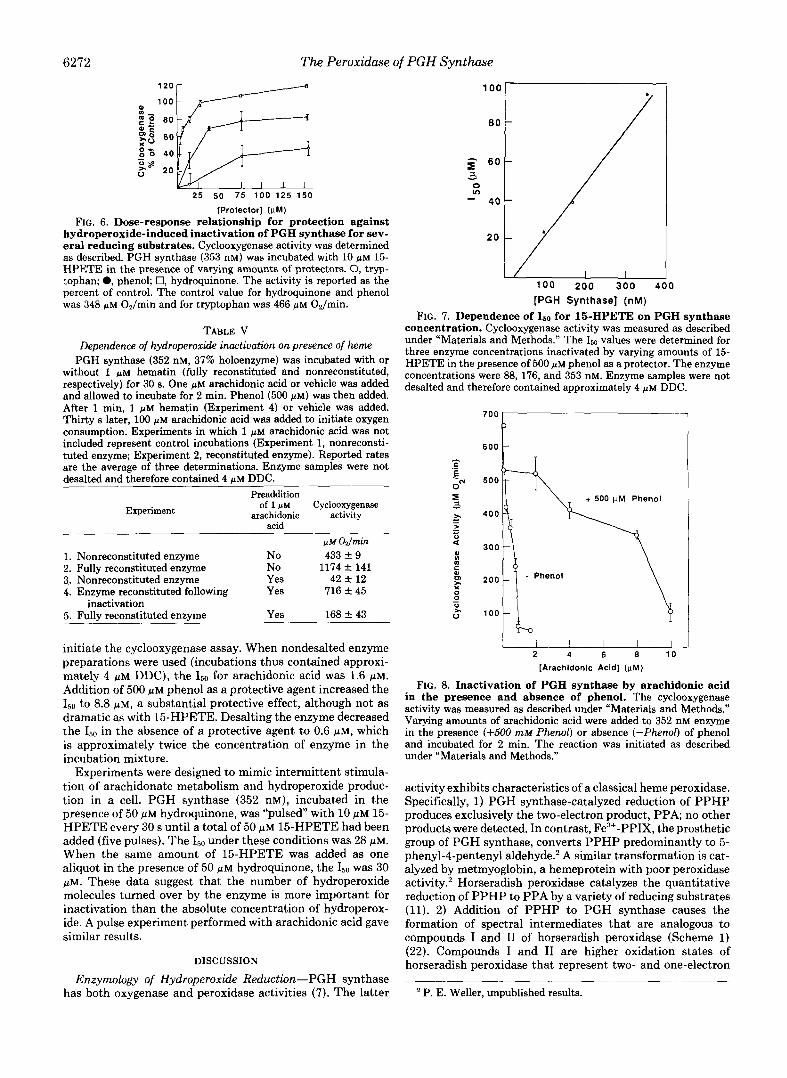
6272 The Peroxidase of PGH Synthase ::w 20
2 5 5 0 7 5 1 0 0 125 150
[Protector] (wM) FIG. 6. Dose-response relationship for protection against
hydroperoxide-induced inactivation of PGH synthase for sev- eral reducing substrates. Cyclooxygenase activity was determined as described. PGH synthase (353 nM) was incubated with 10 p~ 15- HPETE in the presence of varying amounts of protectors. 0, tryp- tophan; 0, phenol; 0, hydroquinone. The activity is reported as the percent of control. The control value for hydroquinone and phenol was 348 pM Oz/min and for tryptophan was 466 p M OJrnin.
TABLE V Dependence of hydroperoxide inactivation on presence of heme
PGH synthase (352 nM, 37% holoenzyme) was incubated with or without 1 NM hematin (fully reconstituted and nonreconstituted, respectively) for 30 s. One p~ arachidonic acid or vehicle was added and allowed to incubate for 2 min. Phenol (500 p ~ ) was then added. After 1 min, 1 pM hematin (Experiment 4) or vehicle was added. Thirty s later, 100 p~ arachidonic acid was added to initiate oxygen consumption. Experiments in which 1 p~ arachidonic acid was not included represent control incubations (Experiment 1, nonreconsti- tuted enzyme; Experiment 2, reconstituted enzyme). Reported rates are the average of three determinations. Enzyme samples were not desalted and therefore contained 4 UM DDC.
Preaddition
arachidonic activity Experiment of 1 p~ Cyclooxygenase
acid
p~ 02jmin 1. Nonreconstituted enzyme No 433 f 9 2. Fully reconstituted enzyme No 1174 f 141 3. Nonreconstituted enzyme Yes 42 f 12 4. Enzyme reconstituted following Yes 716 f 45
5. Fully reconstituted enzyme Yes 168 f 43 inactivation
initiate the cyclooxygenase assay. When nondesalted enzyme preparations were used (incubations thus contained approxi- mately 4 WM DDC), the 150 for arachidonic acid was 1.6 WM. Addition of 500 p~ phenol as a protective agent increased the 150 to 8.8 p ~ , a substantial protective effect, although not as dramatic as with 15-HPETE. Desalting the enzyme decreased the Ib0 in the absence of a protective agent to 0.6 p ~ , which is approximately twice the concentration of enzyme in the incubation mixture.
Experiments were designed to mimic intermittent stimula- tion of arachidonate metabolism and hydroperoxide produc- tion in a cell. PGH synthase (352 nM), incubated in the presence of 50 pM hydroquinone, was ‘‘pulsed with 10 ~ L M 15- HPETE every 30 s until a total of 50 WM 15-HPETE had been added (five pulses). The Is0 under these conditions was 28 p ~ . When the same amount of 15-HPETE was added as one aliquot in the presence of 50 p~ hydroquinone, the Is0 was 30 p ~ . These data suggest that the number of hydroperoxide molecules turned over by the enzyme is more important for inactivation than the absolute concentration of hydroperox- ide. A pulse experiment performed with arachidonic acid gave similar results.
DISCUSSION
Enzymology of Hydroperoxide Reduction-PGH synthase has both oxygenase and peroxidase activities (7). The latter
0 [PGH Synthase] (nM)
FIG. 7. Dependence of 160 for 15-HPETE on PGH synthase concentration. Cyclooxygenase activity was measured as described under “Materials and Methods.” The 150 values were determined for three enzyme concentrations inactivated by varying amounts of 15- HPETE in the presence of 500 p~ phenol as a protector. The enzyme concentrations were 88, 176, and 353 nM. Enzyme samples were not desalted and therefore contained approximately 4 p~ DDC.
700
600
5 0 0
4 0 0
300
200
1 0 0
I I I I I 2 4 6 a IO
[Arachidonic Acid] (@M)
FIG. 8. Inactivation of PGH synthase by arachidonic acid in the presence and absence of phenol. The cyclooxygenase activity was measured as described under “Materials and Methods.” Varying amounts of arachidonic acid were added to 352 nM enzyme in the presence (+500 mM Phenol) or absence (-Phenol) of phenol and incubated for 2 min. The reaction was initiated as described under “Materials and Methods.”
activity exhibits characteristics of a classical heme peroxidase. Specifically, 1) PGH synthase-catalyzed reduction of PPHP produces exclusively the two-electron product, PPA; no other products were detected. In contrast, Fe3+-PPIX, the prosthetic group of PGH synthase, converts PPHP predominantly to 5- phenyl-4-pentenyl aldehyde.’ A similar transformation is cat- alyzed by metmyoglobin, a hemeprotein with poor peroxidase activity.’ Horseradish peroxidase catalyzes the quantitative reduction of PPHP to PPA by a variety of reducing substrates (11). 2) Addition of PPHP to PGH synthase causes the formation of spectral intermediates that are analogous to compounds I and I1 of horseradish peroxidase (Scheme 1) (22). Compounds I and I1 are higher oxidation states of horseradish peroxidase that represent two- and one-electron
P. E. Weller, unpublished results. -

The Peroxidase of PGH Synthase 6273
OXYGENTFUNSFER / / AryR ELECTRCNTR4NSFER
R 1
SCHEME 1. Peroxidase catalytic cycles.
oxidized intermediates in the catalytic reduction of peroxide (23-26). The detection of a two-electron oxidized form of the enzyme is consistent with two-electron reduction of the hy- droperoxide (27). 3) The turnover numbers of PGH synthase- catalyzed reduction of PPHP are comparable to those exhib- ited by horseradish peroxidase and lactoperoxidase, two well- characterized heme peroxidases.* These turnover numbers are 10-100-fold higher than the turnover numbers exhibited by hematin and metmyoglobin for conversion of PPHP to the aldehyde derivative.' Thus, the products of reaction, spectral intermediates, and kinetics of PGH synthase are typical of heme peroxidases and distinct from Fe3+-PPIX alone or he- meproteins without classic peroxidase activity. These data show that the peroxidase activity of PGH synthase is not merely an adventitious one due to the presence of the heme prosthetic group, as once suggested (7).
The feature that distinguishes classical heme peroxidases from other hemeproteins is their ability to efficiently cleave peroxide bonds heterolytically (28). This requires the presence of amino acid residues distal to the heme that facilitate heterolysis. In the case of cytochrome c peroxidase, the distal amino acids appear to histidine, which acts as a general base- acid catalyst, and arginine, which stabilizes the incipient hydroxide or alkoxide ion (Scheme 2) (29). The fifth ligand to the heme group of PGH synthase is probably histidine, but the identities of the sixth ligand and the other residues at the active site are unknown (22). Considering its properties as a highly efficient peroxidase, it appears certain that the protein component of PGH synthase evolved to facilitate peroxide heterolysis, so it must contain residues that are functionally equivalent to the histidine and arginine residues of cyto- chrome c peroxidase.
PGH synthase undergoes rapid inactivation during enzy- matic turnover that is related to the peroxidase activity of the enzyme (30). The extent of inactivation is the same whether hydroperoxides are added in small aliquots or all at once, which implies that the number of hydroperoxide molecules turned over by the peroxidase is more important than the total hydroperoxide concentration. In solutions containing mixtures of holo- and apoenzyme, only the holoenzyme under- goes hydroperoxide-dependent inactivation. This underscores the importance of the heme prosthetic group to inactivation and indicates that the oxidant responsible for inactivation is not released from the enzyme.
Inactivation limits turnover of the enzyme and makes ac- curate determination of kinetic parameters difficult (53). Un- der our experimental conditions, inactivation of the peroxi- dase activity was complete within 10 s. PGH synthase is much more sensitive to peroxide-induced inactivation than horse- radish peroxidase (Fig. 2). This suggests critical differences in the protein residues at the active sites of the two proteins. We estimated a turnover number for PGH synthase-catalyzed reduction of PPHP of 8,000 mol of PPA/mol of protein/min determined from the 10-s point of the reaction with BW-755C as the reducing substrate. This value considerably underesti- mates the maximal turnover number because inactivation is complete in 10 s, and measurement at shorter time periods was not possible under the experimental conditions. Never- theless, the estimated turnover number for PGH synthase- catalyzed reduction of PPHP is comparable to that of horse- radish peroxidase and lactoperoxidase, which exhibit turnover numbers of 10,500 and 2,000 mol of PPA produced per mol of enzyme/min, respectively (11). If one compares the turnover number for the peroxidase activity with that of the cyclooxy-
-OH
P-NH, T
SCHEME 2. Mechanism of compound I formation.

6274 The Peroxidase of PGH Synthase
TABLE VI Comparison of turnover numbers for cyclooxygenase and peroxidase
activities of PGH synthase In Part I, cyclooxygenase activity was measured as described under
“Materials and Methods” with the following changes. For Experiment 3, the enzyme was incubated in the presence of 8 p~ 15-HPETE for 2 min before the addition of 500 p~ phenol; for Experiment 4, the enzyme was incubated with 10 p~ hydroquinone and 8 p~ 5-HPETE simultaneously for 2 min. In Part 11, peroxidase activity was measured as described under “Materials and Methods” with 200 p~ BW-755 C as the reducing substrate. Enzyme samples were not desalted and therefore contained approximately 4 p~ DDC. PHs, PGH synthase.
Experiment Specific activity Total turnover
mollmol PHSlmin mol productlmol P H s I. Cyclooxygenase
1. Native enzyme 578 140 2. Native enzyme + 1542 186
3. Hydroperoxide- 60 22
4. Hydroperoxide- 1081 175
500 pM phenol
treated
treated + 10 p~ hydroquinone
11. Peroxidase 5. Native enzyme -8000 1333 _ _ _ ~
genase activity, it can be seen that both the rate and total capacity for turnover are higher for the peroxidase. This implies that the peroxide-removing capacity of PGH synthase (1,333 mol of PPHP reduced per mol of enzyme) exceeds its peroxide-generating capacity (140 mol of arachidonate oxy- genated per mol of enzyme; Table VI). In addition, the rate of peroxide turnover is approximately 14 times that of ara- chidonate turnover in the presence of peroxidase-reducing substrates (8,000 mol of PPHP/mol of enzyme/min and 578 mol of arachidonate/mol of enzyme/min, respectively).
Peroxidatic Reduction and Reducing Substrates-Reducing substrates support peroxidase-catalyzed reduction of hydro- peroxide. Enhancement of peroxidase turnover by a reducing substrate usually results from stimulation of reduction of compound I1 to resting enzyme, the rate-limiting step in the peroxidase catalytic cycle (27). In the case of PGH synthase, reducing substrates also protect the enzyme from oxidative inactivation (Fig. 6). The longer the enzyme remains active, the more catalytic cycles it can complete, thereby increasing peroxide reduction. Reduction of peroxidase higher oxidation states and protection of enzyme may represent the same event because in the absence of a reducing substrate the higher oxidation states oxidize the heme group and the protein. The index of efficiency presented in Table I illustrates the differ- ences of a number of compounds to support PGH synthase- catalyzed reduction of PPHP. All reducing substrates were tested at 200 PM to facilitate comparisons between com- pounds. Increasing the concentration of most reducing sub- strates enhanced PPHP reduction, although this is not true of very poor substrates. Therefore, doubling the concentration of a compound with an index of 0.4 produces approximately the same amount of PPA as a compound with an index value of 0.8. The compounds in Table I can be separated into groups for the purpose of discussion (structures are provided in the Miniprint S e ~ t i o n ) . ~
Portions of this paper (including Figs. 9-17 and the structures for the compounds listed in Table I) are presented in miniprint at the end of this paper. Miniprint is easily read with the aid of a standard magnifying glass. Full size photocopies are available from the Journal of Biological Chemistry, 9650 Rockville Pike, Bethesda, MD 20814. Request Document No. 86 “3067, cite the authors, and include a check or money order for $9.20 per set of photocopies. Full size photocopies are also included in the microfilm edition of the Journal that is available from Waverly Press.
~~
The phenolic type compounds include, in order of decreas- ing efficiency, n-propyl gallate, butylated hydroxyanisole, hy- droquinone, the (+)- and (---A-analogs of MK-447, L-epi- nephrine, MK-447, guaiacol, phenol, butylated hydroxytolu- ene, and salicylic acid. The structure-activity relationships of a large group of phenolic compounds has been studied exten- sively with respect to their ability to inhibit the cyclooxygen- ase activity of PGH synthase (31). Our results for the reducing substrate efficiency are in accord with the trends reported for cyclooxygenase inhibition; electron-donating substituents on the phenolic ring enhance reducing substrate efficiency. Elec- tron-donating substituents also increase the ease of oxidation of aromatic molecules, and peroxidase-reducing substrate ac- tivity is usually directly related to ease of oxidation (32). The correlation between ease of oxidation and reducing substrate ability is, therefore, not surprising. In contrast, substituents that sterically hinder the hydroxyl group, as in the t-butyl substituents of butylated hydroxytoluene, decrease the ability of the compound to serve as a reducing substrate. Salicylic acid exhibits no activity as an electron donor, which may be due in part to the stability of the hydroxyl group due to hydrogen bonding with the 0-carboxyl group.
The physiological reducing substrate for PGH synthase’s peroxidase is not known. Naturally occurring compounds tested in our assay included L-epinephrine, which was an excellent substrate, lipoic acid, indoleacetic acid, ascorbic acid, and uric acid, which were good to moderate substrates, and L-tryptophan, glutathione, L-methionine, NADPH, and NADH, which were poor substrates. L-Tryptophan and NADH have been added to preparations of PGH synthase at various stages of purification as protective agents, and tryp- tophan has been used as a reducing substrate in PGH synthase peroxidase assays at millimolar concentrations (33,34). Tryp- tophan is a poor reducing substrate in our assay, which explains why large concentrations must be employed to afford protection or to support peroxidase catalysis. Deletion of the a-amino group of tryptophan produces indoleacetic acid, which is a good reducing substrate. This suggests the impor- tance of binding interactions or partition coefficients for reduction of the peroxidase.
It has been shown that NADH is oxidized by PGH synthase incorporated into liposomes in the presence of hydrogen per- oxide, flufenamic acid, cytochrome bs, and cytochrome b, reductase (35). NADH is essentially inactive as a reducing substrate under our assay conditions, and its oxidation by liposome-entrapped PGH synthase appears to require the presence of flufenamate, a good reducing substrate. It seems likely that flufenamate is oxidized to a radical cation by PGH synthase that then oxidizes NADH. The importance of such a mechanism for NADH oxidation in cells is difficult to estimate.
We tested DDC, the additive used as a protective agent in our purification procedure. DDC reduces PPHP in the ab- sence of PGH synthase at pH 8.0. This may enhance its protective ability toward the enzyme because it does not require peroxidase turnover (data not shown). DDC is a good reducing substrate for horseradish peroxidase at pH 5.5 (11).
Gluthathione has been proposed to be the physiologically active reducing substrate (36). Experiments to support this proposal required large concentrations of glutathione relative to hydroperoxide (approximately a 400-fold excess). Under our experimental conditions, glutathione is a weak reducing substrate. There are naturally occurring compounds with better reducing substrate efficiency that may be more reason- able candidates for the endogenous co-substrate for PGH synthase peroxidase than glutathione. For example, uric acid

The Peroxidase of PGH Synthase 6275
has been isolated from the cytosolic fraction of bovine seminal vesicle microsomes (37) and is moderately active in the PPHP assay. The serum concentration of uric acid is approximately 600 p M in humans (38).
A number of nonsteroidal anti-inflammatory drugs were evaluated for their ability to support PGH synthase-catalyzed P P H P reduction. There was no correlation between their ability to serve as electron donors and their ability to inhibit the cyclooxygenase activity of PGH synthase. Sulindac sul- fide, BW-755C, and the A analogs of MK-447 are excellent reducing substrates. Sulindac sulfide is an excellent inhibitor of the cyclooxygenase (39). BW-755C is an “antioxidant’” type cyclooxygenase inhibitor and shows a characteristic bi- phasic effect on cyclooxygenase activity (40). At low concen- trations, BW-755C stimulates the cyclooxygenase activity, whereas at high concentrations, it is an inhibitor (40). BW- 755C is a potent reducing substrate. Nonsteroidal anti-inflam- matory agents with an amine functionality, in order of de- creasing efficiency, were flufenamic acid, etodolac, diclofenac, and meclofenamic acid. Flufenamic acid contains an 0-alkyl substituent and is the most potent of the fenamates (41). The presence of two chloro substituents on diclofenac reduces the relative efficiency; and finally, addition of an 0-alkyl substit- uent on the diclofenac structure to form meclofenamic acid further decreases the potency. Aniline, the simplest aromatic amine tested, was essentially inactive as a reducing substrate for PGH synthase. Compounds with a pyrazoline or pyrazo- lone ring were also tested. These include BW-755C, nafaza- trom, oxyphenbutazone, monophenylbutazone, and phenyl- butazone. Nafazatrom and BW-755C have been shown to be potent reducing substrates for the peroxidase activity of PGH synthase (14), and the present data confirm previous obser- vations. Of the phenylbutazone analogs, only oxyphenbuta- zone shows high activity as a reducing substrate. This is due to the presence of the phenolic hydroxyl group. Phenylbuta- zone and monophenylbutazone were poorer reducing sub- strates than oxyphenbutazone. Piroxicam and isoxicam, which contain /I-dicarbonyl groups, were weak reducing sub- strates, with piroxicam being the more potent of the two. Several nonsteroidal anti-inflammatory agents including in- domethacin, flurbiprofen, ibuprofen, and aspirin were inactive as reducing substrates.
The chiral anti-inflammatory agents tested for reducing substrate efficiency did not display enantiomeric specificity. Two oxidations occur on PGH synthase; one is the oxidation of arachidonic acid, and the other is the oxidation of the reducing substrate. Previous studies with the A analogs of MK-447 showed that the (+)-enantiomer exhibited anti-in- flammatory activity, whereas the (-)-isomer was inactive (17). Our results show that there is no difference in the ability of the enantiomers to act as reducing substrates for the peroxidase activity of PGH synthase. Etodolac is a cyclooxy- genase inhibitor (19) and a peroxidase-reducing substrate. It exhibits marked stereoselectivity as a cyclooxygenase inhibi- tor but no stereoselectivity as a peroxidase-reducing substrate. This indicates that the orientation of etodolac at the site where it inhibits arachidonate oxidation is different than its orientation at the site where it is oxidized by the peroxidase. Therefore, the anti-inflammatory activity of these compounds does not correspond to their ability to support PGH synthase- catalyzed peroxide reduction.
A number of compounds that are co-oxidized during ara- chidonic acid oxygenation did not exhibit activity as reducing substrates. These include diphenylisobenzofuran, benzo[a] pyrene, and 7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene. Their inability to support catalytic hydroperoxide reduction sug-
gests that they are not oxidized by higher oxidation states of the peroxidase. In fact, detailed mechanistic studies indicate that diphenylisobenzofuran is oxidized by a free radical chain reaction and that 7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene is oxidized by peroxyl radicals (42, 43). The fact that they have very low index values illustrates the utility of the PPHP assay for identification of compounds that are or are not oxidized by peroxidase higher oxidation states.
Finally, we compared several of the reducing substrates examined for PGH synthase with their activity for horserad- ish peroxidase-catalyzed reduction of PPHP. Some com- pounds that were excellent substrates for PGH synthase, notably the anti-inflammatory agents, were inactive as elec- tron donors for horseradish peroxidase. In addition, aniline and iodide, both excellent substrates for horseradish peroxi- dase, were inactive as substrates for PGH synthase. These data suggest that there is indeed specificity of the active site for the electron donor. Nonsteroidal anti-inflammatory drugs tend to be large, hydrophobic molecules, analogous to the substrate arachidonic acid. Aniline and iodide, small efficient substrates for horseradish peroxidase, may be poor substrates for PGH synthase because they are less hydrophobic or less easily oxidized than efficient substrates.
Function of Peroxidase of PGH Synthase-The 15,, values for hydroperoxide inactivation displayed in Table IV show the exquisite sensitivity of PGH synthase to hydroperoxides. It has been reported that both intact platelet and platelet homogenate cyclooxygenase are irreversibly inhibited by low levels of fatty acid hydroperoxides (44,45). Some evidence for isomeric specificity was reported (44). The small differences in 150 values for the hydroperoxides listed in Table IV may not be significant and reiterate the importance of the hydro- peroxide group for inactivation. The higher sensitivity of the enzyme to arachidonic acid compared to hydroperoxides may be due to the fact that arachidonate is converted to the hydroperoxide, PGG,, at the active site. In addition, during its conversion to PGG,, arachidonate is converted to free radicals that may inactivate PGH synthase (46). This may account for the fact that peroxidase-reducing substrates are less efficient at protecting against arachidonate-induced in- activation than against hydroperoxide-induced inactivation. If inactivation occurs every time PGG, is made, each molecule of PGH synthase would only catalyze a single turnover. In the apparent absence of all protective agents, the concentra- tion of arachidonic acid required for inactivation approaches a 3-4-fold molar excess of the enzyme concentration. (The enzyme preparations used were on the order of 50% holoen- zyme, indicating an active enzyme concentration of 175 nM.) The fact that multiple enzyme turnovers occur may indicate that traces of reductant contaminate the enzyme preparation or that the efficiency of inactivation by a single molecule of PGGz is less than 100%. In addition, results of rapid-scan spectrophotometric experiments suggest the possibility that arachidonic acid acts as an electron donor in the cyclooxygen- ase-peroxidase turnover of PGH synthase (22). This may account in part for the greater-than-stoichiometric quantities of hydroperoxide required for inactivation.
Significant protection from inactivation was achieved as a result of hydroperoxide reduction catalyzed by the peroxidase activity of PGH synthase in the presence of good reducing substrates. Comparison of the protection by hydroquinone, phenol, and tryptophan makes it clear that protection is directly related to peroxidase-reducing substrate activity. In the case of hydroquinone, an excellent reducing substrate, less than a 3-fold excess of protector over hydroperoxide is necessary to afford complete protection. The serum concen-

6276 The Peroxidase
tration of hydroperoxide is estimated to be approximately 0.5 FM (47). If comparable concentrations are present in cells, relatively low cellular concentrations of reducing substrates may be necessary to ensure maxima1 conversion of arachido- nate to PGH,. For example, nafazatrom achieves a 1-2 PM plasma concentration following an oral dose of 10 mg/kg (48). This is a dose that is antithrombotic and antimetastatic and prevents exhaustion of the capacity of vascular endothelium to biosynthesize prostacyclin (49-51). The major limitation on endothelial prostacyclin generation is self-catalyzed inac- tivation of PGH synthase (52). Nafazatrom is an excellent reducing substrate, and its plasma concentration of a phar- macologically active dose is two to four times higher than the plasma hydroperoxide concentration. Thus, nafazatrom’s pharmacological profile may result from its ability to protect PGH synthase from autoinactivation.
Overall, it seems that the peroxidase activity of PGH syn- thase serves as a twin-edged sword, responsible for inactiva- tion but necessary for protection. This paradox can be re- solved by the fact that the heme prosthetic group, which is responsible for the peroxidase activity, is required for the cyclooxygenase reaction. The hydroperoxide product of cy- clooxygenase action must be removed from the active site, and this may be achieved by reduction of the hydroperoxide functionality. The peroxide-reducing activity of simple heme proteins is so low that, without the peroxidase activity, it is unlikely that the cyclooxygenase could catalyze more than a few turnovers before complete and irreversible inactivation occurred. Viewed in this context, it seems obvious that if PGH synthase had not evolved an efficient hydroperoxide-reducing activity, it would have been unable to provide sufficient en- doperoxide intermediates for cellular prostaglandin, throm- boxane, and prostacyclin biosynthesis. Manipulation of this activity may, therefore, constitute an important new strategy for the design of pharmacologically active compounds.
REFERENCES 1. Hamberg, M., Svensson, J., Wakabayashi, T., and Samuelsson, B. (1974)
2. Nugteren, D. H., and Hazelhof, E. (1973) Biochim. Biophys. Acta 326 , Proc. Natl. Acad. Sci. U. S. A. 71,345-349
3. Miyamoto, T., Ogino, N., Yamamoto, S., and Hayaishi, 0. (1976) J. Bioi.
4. Van der Ouderaa, F.,J., Buytenhek, M., Nugteren, D. H., and Van Dorp,
5. Hemler, M., Lands, W. E. M., and Smith, W. L. (1976) J. Biol. Chem. 2 5 1 ,
6. Ogino, N., Ohki, S., Yamamoto, S., and Hayaishi, 0. (1978) J. Biol. Chem.
7. Ohki, S., Ogino, N., Yamamoto, S., and Hayaishi, 0. (1979) J. Bid. Chem.
8. Roth, G. J., Siok, C. J., and Ozols, J. (1980) J. Biol. Chem. 255,1301-1304 9. Pagels, W. R., Sachs, R. J., Marnett, L. J., Dewitt, D. L., Day, J. S., and
10. Kulmacz, R. J., and Lands, W. E. M. (1984) J. B~oi. Chem. 2 5 9 , 6358-
448-461
Chem. 251,2629-2636
D. A. (1977) Biochlm. Biophys. Acta 487,315-331
5575-5579
253,5061-5068
254,829-836
Smith, W. L. (1983) J . Biol. Chem. 258,6517-6523
6363
of PGH Synthase 11.
12. 13.
14.
15. 16. 17.
18.
19.
20.
21.
22.
23. 24. 25. 26. 27. 28.
29. 30.
31.
33. 32.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46. 47.
Weller, P. E., Markey, C. M., and Marnett, L. J. (1985) Arch. Biochem.
Funk, M. O., Isaac, R., and Porter, N. A. (1976) Lipids 11, 113-117 Porter, N. A., Lehman, L. S., Weber, B. A., and Smith, K. J. (1981) J. Am.
Marnett, L. J., Siedlik, P. H., Ochs, R. C., Pagels, W. R., Das M. Honn, Chem. Soc. 103,6447-6455
K. V., Warnock, R. H., Tainer, B. E., and Eling, T. E. \19&) Mol. Pharmncol. 26,328-335
Biophys. 243,633-643
Neal, M. W., and Florini, J. R. (1973) Anal. Biochem. 5 5 , 328-330 Titus, B. G., and Lands, W. E. M. (1982) Methods Enzymol. 86,69-72 Payne, T . G., Dewald, B., Siegl, H., Guhler, H. U., Ott, H., and Baggiolini,
M. (1982) Nature 2 9 6 , 160-162 Shen, T: Y., Ham, E. A., Cirillo, V. J., and Zanetti, M. (1974) in Prosta-
glundrn Synthase Inhattors (Robinson, H. J., and Vane, J. R., eds) pp.
Demerson, C. A,, Humber, L. G., Abraham, N. A,, Schilling, G., Martel, R. 19-31, Raven Press, New York
Marnett, L. J., Wlodawer, P., and Samuelsson, B. (1975) J. Biol. Chem. R., and Pace-Asciak. C. (1983) J. Med. Chem. 26,1778-1780
Hemler, M. E., Graff, G., and Lands, W. E. M. (1978) Ewehem. Biophys.
Lambeir, A:M Markey, C. M., Dunford, H. B., and Marnett, L. J. (1985)
Chance, B. (1943) J. Biol. Chem. 151,553-577 George, P. (1953) J. Biol. Chem. 201,413-426
George, P. (1953) Biochem. J. 55,220-230 George, P. (1953) Biochem. J. 54,267-276
Dunford, H. B., and Stillman, J. S. (1976) Coord. Chem. Heu. 19,187-251 Marnett, L. J., Weller, P., and Battista, J. R. (1986) in Cytochrome P-450,
Structure, Mechanism, and Biochemistry (Ortiz de Mbntellano, P., ed)
Poulos, T. L., and Kraut, J. (1980) J . Bud. Chem. 255,8199-8205 pp. 29-76, Plenum Publlshing Corp., New York
Egan, R. W., Paxton, J., and Kuehi, F. A,, Jr . (1976) J. Blol. Chem. 251 ,
Dewhirst, F. E. (1980) Prostaglandins 20,209-222 Job, D., and Dunford, H. B. (1976) Eur. J. Blochem. 66,607-614 Mizuno, K., Yamamoto, S., and Lands, W. E. M. (1982) Prostaglandins 2 3 ,
250,8510-8517
Res. Commun. 8 5 , 1325-1331
J. Biol. Che;. 260,14894-14896
7329-7335
743-757 Roth, Gl J., Machuga, E. T., and Strittmatter, P. (1981) J. Biol. Chem.
Strittmatter, P., Machuga, E. T.. and Roth, G. J. (1982) J. Biol. Chern.
Eling, T. E., Curtis, J. F., Harman, L. S., and Mason, R. P. (1986) J. Biol.
Ogino, N., Yamamoto, S., Hayaishi, O., and Tokuyama, T. (1979) Biochem.
Davies, K. J. A,, Sevanian, A., Muakkassah-Kelly, S. F., and Hochstein, P.
Egan, R. W., Gale, P. H., VandenHeuvel, W. J. A., Baptista, E. M., and
Marnett, L. J., Siedlik, P. H., and Fung, L. W. M. (1982) J. Biol. Chem.
Cushman, D. W., and Cheung, H. S. (1976) Biochim. Biophys. Acta 424,
Marnett, L. J., Bienkowski, M. J., and Pagels, W. R. (1979) J. Biol. Chem.
Dix, T. A,, Fontana, R. Panthani, A,, and Marnett, L. J. (1985) J. B i d . Chem. 260,5358-5365
Sie el, M. I., McConnell, .R. T., Abrahams, S. L., Porter, N. A,, and tuatrecasas, P. (1979) Bzochem. Bm hys Res Commun. 89,1273-1280
Hashimoto Y., Naito, C. Teramoto, f., Kato,.H., Kinoshita, M., Kawa- mura, d., Hayashi, d., and Oka, H. (1985) Blochem. Biophys. Res.
Hemler, M. E., and Lands, W. E. M. (1980) J. Biol. Chem. 255 6253-6261 Conmun. 130,781-785
Warso, M. A., and Lands, W. E. M. (1984) Clin. Physiol. Biochm. 2 , 70-
256,10018-10022
257,11883-11886
Chem. 261,5023-5028
Biophys. Res. Commun. 87, 184-191
(1986) Biochem. J. 235 , 747-754
Kuehl, F. A., Jr. (1980) J. Blol. Chem. 255,323-326
257,6957-6964
449-459
254,5077-5082
77 48. Phiipp, E., Ritter, W., and Patzchke, K. (1983) Thromb. Res. IV, (Suppl.)
199 49.
50.
51.
52.
53.
Sei&, F., Busse, W. D., Meng, K., Hoffmeister, F., Moller, E., and
Honn, K. V. (1982) in Interaction of Platelets and Tumor Cells (Jamieson,
Deckmg, ,$, Van Houte, E. Verstraete, M., and Vermylen, J. (1983)
Kent, R. S., Diedrich, S. L., and Whorton, A. R. (1983) J . Clin. Inuest. 7 2 ,
Kulmacz, R. J. (1986) Arch. Biochem. Biophys. 2 4 9 , 273-285
Horstman, H. (1979) Arzneim. Forsch. 29,54-59
G. A., ed) p 295-331, Alan R. Liss, Inc., New York
Bioc m Pharmacol. 3 2 , 27k7-2762
455-465

The Peroxidase of PGH Synthase 6277
SUPPLEMENTARY MATERIALTO
PROSTAGLANDIN H SYNTHASE REDUCING SUBSTRATE SPECIFICITY AND QUANTITATIVE STUDIES OF HYDROPEROXIDE REDUCTION BY
THE RELATIONSHIPOF PERDXIDASETO CYCLOOXYGENASE ACnVlTlES
Christine M. Markey, AM" Alward. Paul E. Weller, and hwrence 1. Marnett
1 I
0 0 2 0 4.0 6.0 8.0 100 115 HPETEI (pM)
Figure9. inocliwrtion of PGH synthvse b!, 15-HPETE, rn thspwmceondobsmce of DDC.
PGH synthase (350 nM) was incubated in the presence of varying amounts of 15-HPETE for 2 mi". Phcnul (500 pM) was then added and after 30 s 1W pM arachldomc acld war added t o initiate the reaction. Exoermentr labeled "-DDC" were desalted as described m methods. Those labeled "+dDC" were not desal;ed a " i therefore comalned ap roxmately 4 uM DDC. All omts represent the mean of tri k a l e trials. The 100% v~pues for the "+DDC" and "-DL," experlmentr were 971 ph!O,/min and 947 pM
0- 0 0 2.0 4.0 6.0
IIWXIH I8 11
Figure 13. Inarliwlion o/ PCH synthase by IO~hydropnory-8-oclodrrllenoic orrd
PGH synthase I550 nM) was incubated with varying amounts of 10-00H-181 for 2 min. Arachidonic a n d was then added to initiate the reactmn. Phenol was not added after the hydroperoxide inactivation. Enz me samples were not desalted and therefore rantamed approximately 4 pM DDC. The I&% value was 293 @I O,/min
02/min, respectively.
PGH s nthare (550 nM) was incubated with varying amount^ of PPHP for 2 min. Arachidonic acid (100 pM1 was then added to mitiate the reaction. Phenol was not added after the hydroperoxide inactivation. Enzyme sampler were not desalted and therefore contained approximately 4 pM UDC. The 100% value was 183 pM OJmin.
o u 1.0 2 0 3.0 4 0
Experiments were performed ~ssentially as described in Figure 9 except phenol war not added alter hydroy,roxide inactivation. The 100% value was 267 pM 0 Imm. Enzyme s.amplrr were not e d t e d and therefore contained approxmately 4 pM D k
PCH synthase (350 "MI was incubated with var ing mncentrations of hydrogen peroxide for 2 mi". Phenol (500 pM) was added and after s 100 pM arachidonic acid was added to
Those labeled "+DDC" were not desalted and therefore contained a proxmmely 4 pM initiate the reaction. Experiments labeled " - D W were desalted as descrikd in Methods.
DDC Ail p o m r represent the mean of t r ipkale trials The 100% &es for the ' + D W and "-DDC' experiments were 853 pM 02/min and 588 pM O2lmin, respectively
PCH synthase (550 nM) was incubated wlth varying concentrations of 9-00H 181 for 2 mi". Arachidonic acld was then added to inttiate the reaction. Phenol was not added after thc hydroperoxide Inactivation Enzyme samples were not desalted and therefore contamed approximately 4 pM DDC The 100% value was 303 pM OJmm

6278 The Peroxidase of PGH Synthase
Hf-s $' O % f O . D 5
O.% i 0.02
O R h + O M
m-a, Nafazawom 0.19 taw
o . n i o . 0 1
I w+om
M K U 7 0 s f o . m
"A- analog of MK-147
L-E-hnna
o.ntow
0.57 t 0.01
Q 0.86 f 0.04 055 f 0.W I.) 0.46f0.01 (+I
L
DiLlOfenaC 0.50 i 0.03
MK-U7 0.n + 0.01
a: Medof-mir acid 035 *om 0.61 f 0.05
050 i 0.w 6 0.34 t 0.04
0.wio.m
Wkylic add MK-110 0.27 i 0.01 r-7 0.26fO.01 e+") 6 0.25 f 0.m

6279 The Peroxidase of PGH Synthase phvsioloelrallv Re Ib~mt CnmmunQ: l n d n
0.23to.w
0.23 t 0.w
0.45 t o.m o.12fo.W
012io.w
.P 0.43 f 0.01
0121001
o.r/tow
0.21 f 0.09
0.21 t o 1 2
0.17tO.M
Ol7f0.02
L-Methonine
NADPH
NADH
ELodolar
MK.410 0.27f001 C-7 0.26 f 001 Cr-1
:"l
a:; Salicylic a i d 0.06 t 0.06
ax O.MiO.01
6 0.18iO.11
0.15 f 0.w Iodide
6 UHNHl
0.14t0.00