investigation of thermal and fire performance of novel hybrid geopolymer composites
TRANSCRIPT

J O U R N A L O F M A T E R I A L S S C I E N C E 3 9 (2 0 0 4 ) 4721 – 4726 L E T T E R S
Investigation of thermal and fire performance of novel hybrid
geopolymer composites
M. HUSSAINMonash University, School of Physics and Materials Engineering, Clayton, Vic-3168, AustraliaE-mail: [email protected]
R. J. VARLEYCSIRO, Molecular Division, Australia
Y. B. CHENG, G. P. SIMONMonash University, School of Physics and Materials Engineering, Clayton, Vic-3168, Australia
Many advanced polymer composites consist of an or-ganic phase of a highly cross-linked epoxy resin, thusthe flammability of the virgin polymer matrix limitsthe use of these materials in many applications, suchas marine platforms, automotive, and aerospace [1–3].Various methods have been proposed for forming inor-ganic protective layers on the surface of burning poly-mers, but their positive effect is generally accompaniedby certain disadvantages. Inorganic filler particles im-prove fire performance through a dilution effect andreduced heat feedback. Silica additives of high surfacearea are most efficient, as these particles accumulateon the surface instead of sinking into the polymer [4].A current area of great interest involves polymer-clay(layered silica) nanocomposites, when introduced in in-tercalated or exfoliated form, which may accumulate onthe surface of a polymer during burning to form a barrierlayer to either outgoing degradation products or incom-ing gases [5, 6]. Their effect is clearly indicated by thereduced rate of heat release of horizontal samples in thecone calorimeter. Recent work also suggests that evenvery low levels (1–4%) of nano-clay lower the peak heatrelease rate in a cone-calorimeter by some 30% [7]. Inparticular, a family of new materials called geopoly-mers (poly-silates) has emerged during the last decadeas one of the most promising replacement candidates,for plastics and even ceramics, in certain applicationssince they have certain advantageous physical proper-ties of ceramics. The term “geopolymer” was first usedby Davidovits [8, 9] to describe a family of mineralbinders closely related to artificial zeolites.
The most attractive geopolymers (polysialates) areinorganic polymers made from alumino-silicates sincethey can be synthesized at low temperatures and haveuseful properties such as high compressive strengthand are stable at temperatures up to 1300–1400 ◦C.By changing the Si/Al ratio, it is possible to producecomposites with very high fire resistant properties.Polysialates are readily synthesized from natural alumi-nosilicates such as kaolinite, a very abundant source ofalumina and silica. The formation of geopolymeric ma-terials follows the same routes as that of most zeolites.Geopolymers are formed by the co-polymerization ofalumino and silicate species, which originate from the
dissolution of silicon and aluminium-containing sourceof materials at a high pH, in the presence of solu-ble alkali metal silicates. The current commercial useof geopolymers alone, compared to plastics, is lim-ited because of the complexity of large scale process-ing, high density and problems with machining andmolding, and most importantly, their brittleness. Gen-erally geopolymers are linear poly(metasilicate), withtetra-coordinate aluminate crosslinks. The geopoly-merization involves the chemical reaction of alumino-silicate oxides (Al3+ in IV-fold coordination) withalkali polysilicates yielding polymeric Si O Albonds, the amorphous to semi-crystalline three di-mensional silico-aluminate structures are of thepoly(silate) type ( Si O Al O), the poly(silate-siloxo) type ( Si O Al O Si O ), the poly(silate-disiloxo) type ( Si O Al O Si O Si O ).
In this paper synthesized geopolymer is incorpo-rated into the cross-linked polymeric structure sys-tems by manipulating the chemical composition of thegeopolymer and hence compatibility, rather than phys-ical blending. In so doing, we will make use of theprocessability and properties of the cross-linked epoxyresin, in combination with the geopolymers to pro-duce inorganic organic hybrid materials, which haveexcellent mechanical properties such as stiffness andstrength, and in particular are more fire resistant. Thusthe first system reported involves the choice of a stan-dard, bi-functional epoxy resin, diglycidyl ether ofbisphenol A (DEGEBA) to be incorporated with thegeopolymers. The results are also compared with aphysically blended epoxy–kaolin blend to investigateany synergistic effect of producing a more homoge-neously dispersed network on the fire performance.This paper presents, to our knowledge, the first inves-tigation of the successful incorporation of a geopoly-mer system into an organic polymer system, such as anepoxy, and is advantageous to improving properties ofthe matrix.
Kaolin (HR1-F grade) with an average particlesize of 38.20 µm was procured from CommercialMinerals, Sydney, Australia. The potassium silicatesolution was obtained from PQ Australia Pty Ltd.Potassium silicate composition was SiO2/K2O = 2.00,
0022–2461 C© 2004 Kluwer Academic Publishers 4721

SiO2 = 29.3 (wt%) and K2O = 14.5 (wt%) with den-sity 1420 kg/m3. 5M KOH solution was prepared inthe laboratory from KOH pellets from Bdh, MerckPty. Ltd. The epoxy resin used in the study wasdiglycidyl ether of bisphenol-A (DGEBA), commer-cially known as DER-331 from Dow Chemical Com-pany, Australia. The curing agent used in this experi-ment was a mixture of 3,5-diethyltoluene-2,4-diamineand 3,5-diethyltoluene-2,6-diamine (Ethacure-100) ob-tained from Albemarle Corporation, USA.
Inorganic geopolymer was synthesized by the reac-tion of kaolin, potassium silicate and potassium hy-droxide solution at room temperature. Initially, the de-sired amount of potassium silicate was mixed with 5 Mpotassium hydroxide solution and then 20 g kaolin wasadded and mixed for 5–30 min. The viscous mix wasthen added to a mixture of DGEBA epoxy resin andthe curing agent with constant stirring for 15 min. Themixture was then placed in the Teflon coated mold andwas cured at 60 ◦C for 6 h followed by post curing at180 ◦C for 2 h.
To fabricate filler dispersed composites, 20 g of purekaolin was mixed with the mixture of DGEBA epoxyresin and the curing agent for 1 h. The mix was thenplaced in a Teflon coated mold and was then curedat 80 ◦C for 6 h followed by post curing at 180 ◦Cfor 2 h. The sample was then cut, ground and pol-ished for thermal, cone calorimetry and microstructureanalysis.
The formation of geopolymers was characterized byX-ray diffraction analysis by Rigaku wide-angle go-niometer. An acceleration voltage of 40 kV and currentof 22.5 mA were applied using Ni filtered Cu Kα adi-ation. FT-IR spectra were recorded on a Perkin-ElmerFT-IR spectrometer. Wave numbers were recorded from400–4000 cm−1. The loss tangent, tan δ of cured sam-ples was determined on a rheometric scientific dy-namic mechanical thermal analyzer, DMTA IV. Thecured samples were clamped in a medium frame us-
Figure 1 DMTA spectra of (a) cured DGEBA (b) cured 20% Geopolymer-DGEBA and (c) cured 20% kaolin-DGEBA.
ing a small center clamp in the dual cantilever mode.Frequency sweep scans were performed from 80 to260 ◦C at 2 ◦C/min. Thermo gravimetric analysis wasperformed on the cured samples using a TG-92 Setaramthermal analyzer. The thermographs were obtained ata heating rate of 10 ◦C/min using 10–15 g of the pow-dered sample. The experiments were made in a staticair atmosphere. Fire performance tests including timeto ignition (TTI), rate of heat release (RHR), time toreach maximum RHR, smoke density, carbon monox-ide and carbon dioxide evolution and the sample massloss were determined by cone. The heat flux producedwas 50 kW/m2 on the specimen, which had an exposedsurface of 100 × 100 mm.
The glass transition temperature and dynamic-mechanical properties of DGEBA, 20% Kaolin-DGEBA and 20% Geopolymer-DGEBA has been de-termined. Fig. 1 shows the tan δ spectra from dynamicmechanical analyses of the cured of DGEBA and itscomposites respectively. Cured DGEBA showed a Tgaround 201.0 ◦C. However, on addition of kaolin the Tgof the composites decreased to 195 ◦C. The effect onthe Tg of clay addition has been widely studied by manyresearchers reporting an increase in Tg, [10, 11], whilstothers found a slight decrease or no change [12, 13].Becker [14] reported a decrease in Tg for higher func-tionality epoxy resins and suggested that the decrease inTg of clay-modified composites was due to interferenceof the clay with crosslink density, epoxy homopoly-merization and plasticization. Subsequently, addition ofgeopolymer into the DGEBA epoxy produced a lowerTg. The lower crosslink density of Kaolin modifiedDGEBA was confirmed by the greater height of thedelta relaxation observed in Fig. 1. The lower relax-ation strength and Tg of geopolymer-modified compos-ites are believed to be due to structural inhomogenetywhich were observed by SEM images.
Thermo-gravimetric analysis was performed to ex-amine the effect of geopolymer and Kaolin addition on
4722
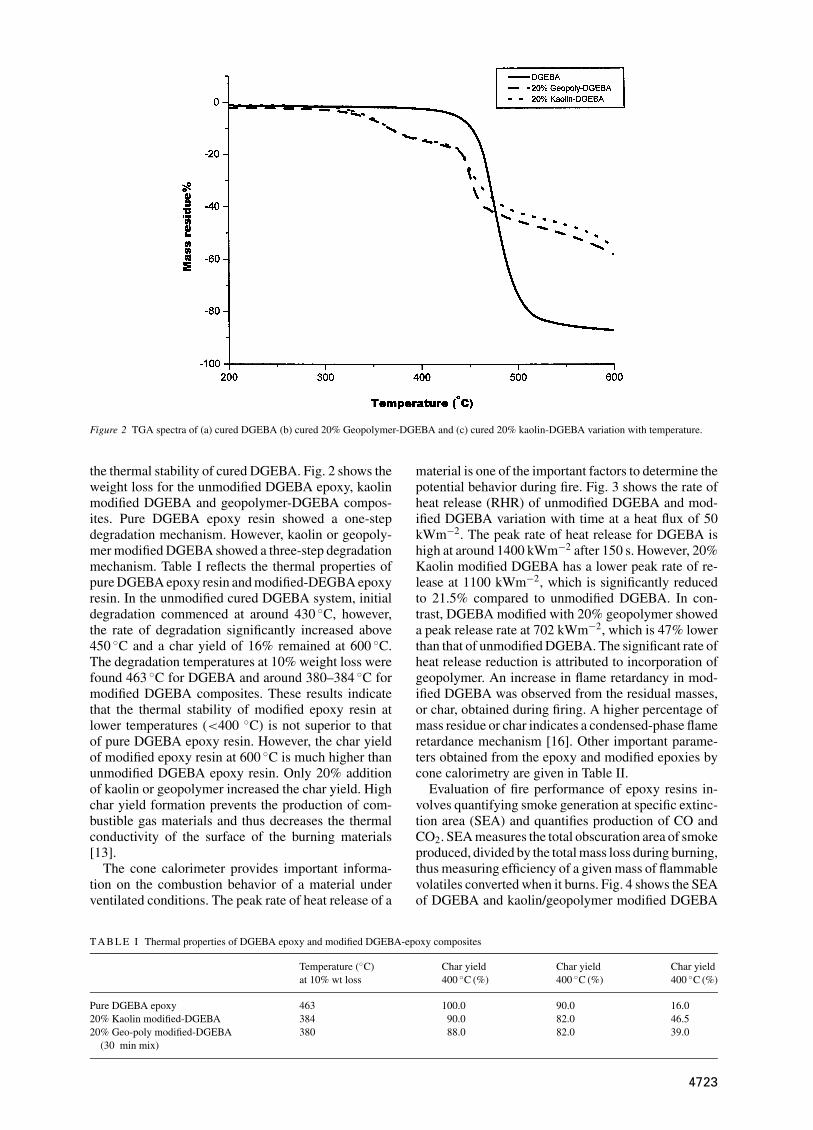
Figure 2 TGA spectra of (a) cured DGEBA (b) cured 20% Geopolymer-DGEBA and (c) cured 20% kaolin-DGEBA variation with temperature.
the thermal stability of cured DGEBA. Fig. 2 shows theweight loss for the unmodified DGEBA epoxy, kaolinmodified DGEBA and geopolymer-DGEBA compos-ites. Pure DGEBA epoxy resin showed a one-stepdegradation mechanism. However, kaolin or geopoly-mer modified DGEBA showed a three-step degradationmechanism. Table I reflects the thermal properties ofpure DGEBA epoxy resin and modified-DEGBA epoxyresin. In the unmodified cured DGEBA system, initialdegradation commenced at around 430 ◦C, however,the rate of degradation significantly increased above450 ◦C and a char yield of 16% remained at 600 ◦C.The degradation temperatures at 10% weight loss werefound 463 ◦C for DGEBA and around 380–384 ◦C formodified DGEBA composites. These results indicatethat the thermal stability of modified epoxy resin atlower temperatures (<400 ◦C) is not superior to thatof pure DGEBA epoxy resin. However, the char yieldof modified epoxy resin at 600 ◦C is much higher thanunmodified DGEBA epoxy resin. Only 20% additionof kaolin or geopolymer increased the char yield. Highchar yield formation prevents the production of com-bustible gas materials and thus decreases the thermalconductivity of the surface of the burning materials[13].
The cone calorimeter provides important informa-tion on the combustion behavior of a material underventilated conditions. The peak rate of heat release of a
T ABL E I Thermal properties of DGEBA epoxy and modified DGEBA-epoxy composites
Temperature (◦C) Char yield Char yield Char yieldat 10% wt loss 400 ◦C (%) 400 ◦C (%) 400 ◦C (%)
Pure DGEBA epoxy 463 100.0 90.0 16.020% Kaolin modified-DGEBA 384 90.0 82.0 46.520% Geo-poly modified-DGEBA 380 88.0 82.0 39.0
(30 min mix)
material is one of the important factors to determine thepotential behavior during fire. Fig. 3 shows the rate ofheat release (RHR) of unmodified DGEBA and mod-ified DGEBA variation with time at a heat flux of 50kWm−2. The peak rate of heat release for DGEBA ishigh at around 1400 kWm−2 after 150 s. However, 20%Kaolin modified DGEBA has a lower peak rate of re-lease at 1100 kWm−2, which is significantly reducedto 21.5% compared to unmodified DGEBA. In con-trast, DGEBA modified with 20% geopolymer showeda peak release rate at 702 kWm−2, which is 47% lowerthan that of unmodified DGEBA. The significant rate ofheat release reduction is attributed to incorporation ofgeopolymer. An increase in flame retardancy in mod-ified DGEBA was observed from the residual masses,or char, obtained during firing. A higher percentage ofmass residue or char indicates a condensed-phase flameretardance mechanism [16]. Other important parame-ters obtained from the epoxy and modified epoxies bycone calorimetry are given in Table II.
Evaluation of fire performance of epoxy resins in-volves quantifying smoke generation at specific extinc-tion area (SEA) and quantifies production of CO andCO2. SEA measures the total obscuration area of smokeproduced, divided by the total mass loss during burning,thus measuring efficiency of a given mass of flammablevolatiles converted when it burns. Fig. 4 shows the SEAof DGEBA and kaolin/geopolymer modified DGEBA
4723
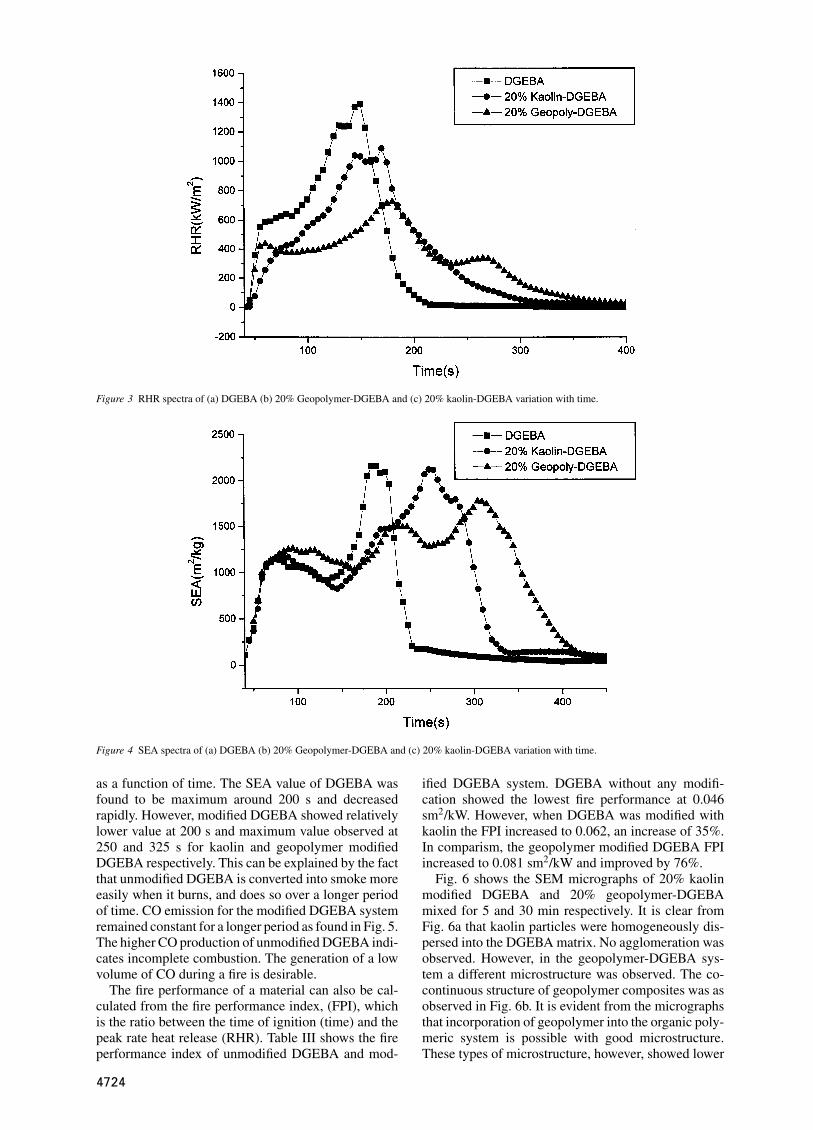
Figure 3 RHR spectra of (a) DGEBA (b) 20% Geopolymer-DGEBA and (c) 20% kaolin-DGEBA variation with time.
Figure 4 SEA spectra of (a) DGEBA (b) 20% Geopolymer-DGEBA and (c) 20% kaolin-DGEBA variation with time.
as a function of time. The SEA value of DGEBA wasfound to be maximum around 200 s and decreasedrapidly. However, modified DGEBA showed relativelylower value at 200 s and maximum value observed at250 and 325 s for kaolin and geopolymer modifiedDGEBA respectively. This can be explained by the factthat unmodified DGEBA is converted into smoke moreeasily when it burns, and does so over a longer periodof time. CO emission for the modified DGEBA systemremained constant for a longer period as found in Fig. 5.The higher CO production of unmodified DGEBA indi-cates incomplete combustion. The generation of a lowvolume of CO during a fire is desirable.
The fire performance of a material can also be cal-culated from the fire performance index, (FPI), whichis the ratio between the time of ignition (time) and thepeak rate heat release (RHR). Table III shows the fireperformance index of unmodified DGEBA and mod-
ified DGEBA system. DGEBA without any modifi-cation showed the lowest fire performance at 0.046sm2/kW. However, when DGEBA was modified withkaolin the FPI increased to 0.062, an increase of 35%.In comparism, the geopolymer modified DGEBA FPIincreased to 0.081 sm2/kW and improved by 76%.
Fig. 6 shows the SEM micrographs of 20% kaolinmodified DGEBA and 20% geopolymer-DGEBAmixed for 5 and 30 min respectively. It is clear fromFig. 6a that kaolin particles were homogeneously dis-persed into the DGEBA matrix. No agglomeration wasobserved. However, in the geopolymer-DGEBA sys-tem a different microstructure was observed. The co-continuous structure of geopolymer composites was asobserved in Fig. 6b. It is evident from the micrographsthat incorporation of geopolymer into the organic poly-meric system is possible with good microstructure.These types of microstructure, however, showed lower
4724
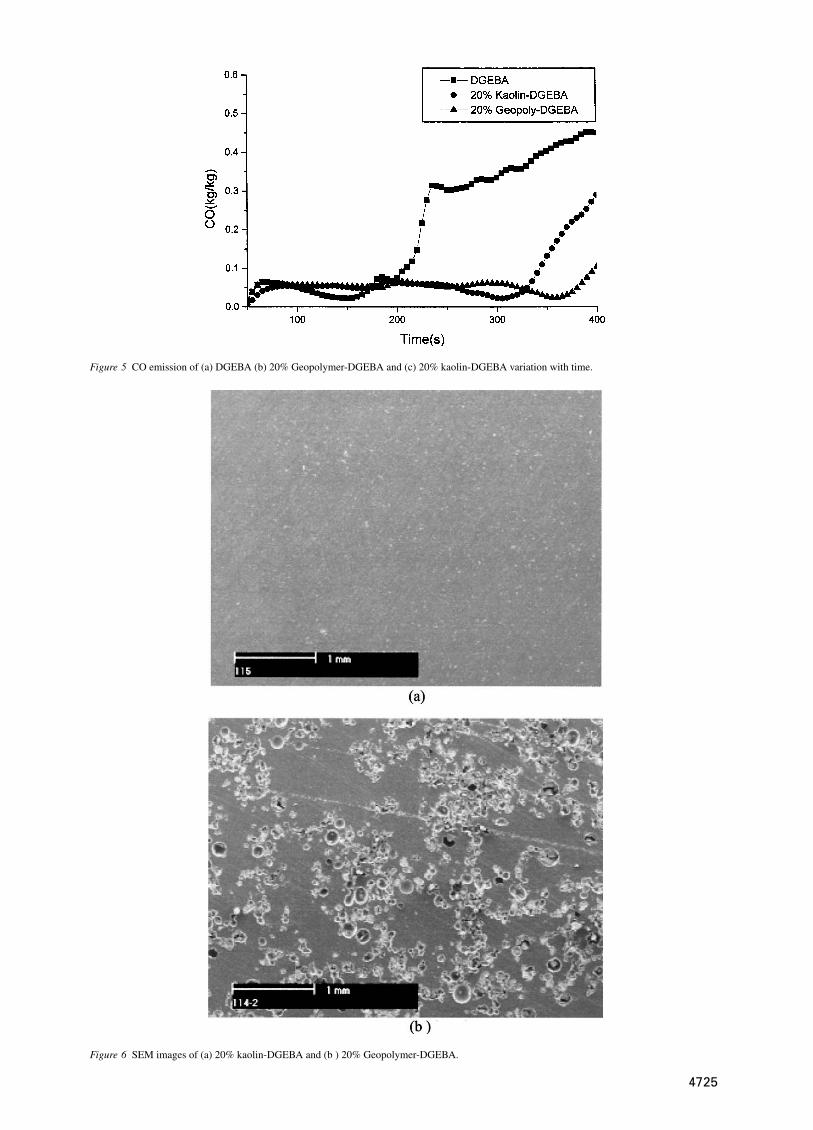
Figure 5 CO emission of (a) DGEBA (b) 20% Geopolymer-DGEBA and (c) 20% kaolin-DGEBA variation with time.
Figure 6 SEM images of (a) 20% kaolin-DGEBA and (b ) 20% Geopolymer-DGEBA.
4725

T ABL E I I Cone calorimetric data measured with an irradiance of50 kWm−2
20% Kaolin- 20% Geo-Fire properties DGEBA DGEBA poly-DGEBA
Time to ignition (s) 65 69 60Peak RHR (kW/m2) 1396 1100 735Av. HRR (kW/m2) 399.8 426 364.1Av. HRR at 180 s 501 562.5 471Time to max HRR(s) 155 170 185Av. effective heat of 21.2 25.3 24.7
combustion (MJ/kg)Av. CO yield (kg/kg) 0.0454 0.06 0.059Av. CO2 yield (kg/kg) 1.48 1.99 1.955Total heat evolved (MJ/kg) 89.65 115.24 95.68Mass loss (%) 82.8 73.9 74.7
T ABL E I I I Fire performance index of unmodified DGEBA and mod-ified DGEBA system
RHR Time to FPIDGEBA system (kW/m2) ignition (sm2/kW)
DGEBA only 1396 65 0.04620% kaolin-DGEBA 1100 69 0.06220% geopolymer-DGEBA 735 60 0.081
relaxation behavior and Tg but exhibited higher thermalstability and higher fire performance capability. Otherproperties of materials, mainly the mechanical prop-erties before and after firing tests of the composites,should be investigated further.
References1. R . A . D E M A R C O , “Composites Applications at Sea, Fire Related
Issues,” in Proc. 36th Int. SAMPE Symposium, 15–18 April 1991,p. 1928.
2. W. T . H A T H A W A Y , “Fire Safety in Mass Transit Vehicle Ma-terials,” in Proc. 36th Int. SAMPE symposium, 15–18 April 1991,p. 1900.
3. R . G . H I L L , T . I . E K L U N D and C. P . S A R K O S , “AircraftInterior Panel Test Criteria Derived from Full-Scale Fire Tests,”Reported DOT/FAA/CT 85/23(1985), USA.
4. T . K A S H I W A G I , J . G I L M A N, K. B U T L E R, R . H A R R I S ,J . S H I E L D S, A. A S A N O and M. L E W I N (eds.), in “RecentAdvances in Flame Retardancy of Polymeric Materials” (Stamford,2000) Vol. 9.
5. W E I X I E , Z O N G M I N G G A O, K U N L E I L I U , W E I-P I N G
P A N, R I C H A R D V A I A, D O U G H U N T E R and A N A N T
S I N G , Thermochim. Acta 367 (2001) 339.6. J U N-C H A O H U A N G, Z I -K A N G Z H U, J I E Y I N, X U E-
F E N G Q I A N and Y A N G-Y A N G S U N , Polymer 42 (2001)873.
7. J . G I L M A N, T . K A S H I W A G I , S . L O M A K I N, E .G I A N N E L I S , E . M A N I A S , J . L I C H T E N H A N and P .J O N E S (eds.), in “Fire Retardancy of Polymers: The Use ofIntumescence” (The Royel Chemical Society, Cambridge, 1998)p. 203.
8. J . D A V I D O V I T S , Geopolymer 88, 1st Eur. Conf. Soft Mineralogy(1988) Vol. 1, p. 25.
9. Idem., Concrete Int. 16 (1994) 53.10. J . M. B R O W N, D. C U R L I S S and R. A. V A I A , Chem. Mater.
12 (2000) 3376.11. P . M U S T O, G. R A G O S T A, P . R U S S O and L . M A S C I A ,
Macromol. Chem. Phys. 202 (2001) 3445.12. J . M A S S A M and T. J . P I N N A V A I A , “Clay Nanolayer Re-
inforcement of a Glassy Epoxy polymer,” Mater. Res. Soc. Symp.Proceeding 1998, p. 36.
13. A . L E E and J . D . L I C H T E N H A N , J. Appl. Polym. Sci. 73(10)(1999) 1993.
14. O . B E C K E R, R . J . V A R L E Y and G. P . S I M O N , Polymer43 (2001) 4365.
15. E . M. P E A R C E and R. L I E P I N E S , Environmental HealthPerspect 11 (1975) 69.
16. J . R . E B D O N, B. J . H U N T, P . J O S E P H, C . S . K O N K E L,D. P R I C E and K. P Y R A H , Poly Degrad Stab 70 (2000)425.
Received 8 Julyand accepted 24 November 2003
4726