investigation of the pressure dependence of so3 formation./67531/metadc4403/m2/1/high... · sulfur...
TRANSCRIPT

APPROVED: Paul Marshall, Major Professor Angela Wilson, Committee Member Ruthanne Thomas, Chair of the Department of
Chemistry Sandra L. Terrell, Interim Dean of the Robert B.
Toulouse School of Graduate Studies
KINETIC STUDY OF THE PRESSURE DEPENDENCE OF SO3 FORMATION
Jacinth Naidoo, BSc (Honors)
Thesis Prepared for the Degree of
MASTER OF SCIENCE
UNIVERSITY OF NORTH TEXAS
December 2003

Naidoo, Jacinth, Investigation of the Pressure Dependence of SO3 Formation.
Master of Science (Chemistry), December 2003, 85 pp., 7 tables, 25 illustrations,
references, 120 titles.
The kinetics of the pressure dependent O + SO2 + Ar reaction have been
investigated using laser photolysis resonance fluorescence at temperatures of 289 K, 399
K, 581 K, 699 K, 842 K and 1040 K and at pressures from 30-665 torr. Falloff was
observed for the first time in the pressure dependence. Application of Lindemann theory
yielded an Arrhenius expression of k(T) = 3.3 x 10-32exp(-992/T) cm6 molecule-1 s-1 for
the low pressure limit and k(T) = 8.47 x 10-14exp(-468/T) cm3 molecule-1 s-1 for the high
pressure limit at temperatures between 289 and 842 K. The reaction is unusual as it
possesses a positive activation energy at low temperature, yet at higher temperatures the
activation energy is negative, illustrating a reaction barrier.

ii
ACKNOWLEDGEMENTS
I would sincerely like to thank my advisor Dr. Paul Marshall for so freely and
interestingly sharing his wealth of knowledge, his interest and enthusiasm about gas
phase kinetics with me.
I would also like to thank Dr. A Goumri and Dr. L.R Peebles for their mentoring and
assistance.
I am grateful for the support, encouragement and advice of my dear husband Derrick, and
my parents Elaine and Meg Govender.
I would like to acknowledge financial assistance from the educational fund of Murial and
Harold Onishi.
Finally I would like to thank the Robert. A Welch Foundation and the National Science
Foundation for their financial support of this work.

iii
TABLE OF CONTENTS
LIST OF TABLES…. ………………………………………….………… v
LIST OF ILLUSTRATIONS.......….......………………………………… vi
Chapter
1. INTRODUCTION
1.1 Coal and its combustion products ............................................. 6
1.2 Fuel desulphurization................................................................. 10
1.3 Flue gas desulphurization
1.3.1 Limestone-based method...................................... 11
1.3.2 Magnesium based method………………….…... 12
1.3.3 Ammonium sulfate based method……………… 13
1.3.4 Dry injection method…………………………… 14
1.4 Relevance of SO2 to flame combustion………………………. 15
1.5 SO3 formation………………………………………………… 17
1.6 The effect of sulfur on NOx emission………………………… 19
2. EXPERIMENTAL METHODS
2.1 Flash Photolysis Resonance Fluorescence (FP-RF) technique… 21
2.2 Kinetic experimental procedure……………………………….. 24
2.3 Materials……………………………………………………….. 26
2.4 Data analysis…………………………………………………… 27
2.5 SO2 absorption cross-section determination…………………... 29
3. RESULTS AND DISCUSSION
3.1 Results…………………………………………………………... 33
3.2 Discussion………………………………………….…………… 33

iv
3.3 Background to Lindemann theory…………………………………. 35
3.4 Third body contribution to the third order rate…………………….. 47
3.5 Comparison of rate constant with those from prior determinations. 40
3.6 Spin considerations…………………………………………….…. 43
3.7 Statistical analysis of O + SO2 + Ar reaction……………………… 45
4. CONCLUSION………………………………………………………… 48
APPENDIX A………………………………………………………….. 50
APPENDIX B…………………………………………………………. 53
REFERENCES………………………………………………………… 76

v
LIST OF TABLES
Table Page
1. Summary of rate constant for the low and high pressure limits
for the O + SO2 + Ar reaction.……………………………… 39
2. Summary of rate constant measurements for O + SO2 + Ar .
at 289 K ……………….…………………………………. 54
3. Summary of rate constant measurements for O + SO2 + Ar
at 399 K…………………………………………………… 58
4. Summary of rate constant measurements for O + SO2 + Ar
at 581 K…………………………………………………… 60
5. Summary of rate constant measurements for O + SO2 + Ar
at 699 K…………………………………………………… 63
6. Summary of rate constant measurements for O + SO2 + Ar
at 842 K…………………………………………………… 64
7. Summary of rate constant measurements for O + SO2 + Ar
at 1040 K………………………………………………… 67
8. Summary of the rate constants available for the SO2+O +Ar
reaction................................................................................ 41

vi
LIST OF ILLUSTRATIONS
Figure Page
1. Plot of estimated sulfur emission from biomass burning,
biogenic and non-biogenic sources…………………… 5
2. Plot of estimated global emission of sulfur in 1980…… 9
3. Illustration of a flash- photolysis resonance setup………. 23
4. Plot of fluorescence intensity including background of
SO2+ O + Ar at 297 torr and 1047 K……………………. 28
5.Plot of pseudo first order rate constant for the loss of O radicals
at 297 torr and 1047 K........................................................ 29
6. Beer-Lambert plot of SO2 at room temperature………… 51
7. Beer-Lambert plot of SO2 at room temperature………… 51
8. Beer-Lambert plot of SO2 at room temperature………… 52
9. Plot of temperature dependence of SO2 cross section absorption 32
10. Plot of first order rate constant vs. pressure at 289 K…… 57
11. Plot of first order rate constant vs. pressure at 399 K…… 59
12. Plot of first order rate constant vs. pressure at 582 K…… 61
13. Plot of first order rate constant vs. pressure at 699 K……. 63
14. Plot of first order rate constant vs. pressure at 841 K…….. 65
15. Plot of first order rate constant vs. pressure at 1040 K…….. 72

vii
16. Lindemann plot at 289 K…………………………………. 73
17. Lindemann plot at 399 K………………………………… 73
18. Lindemann plot at 581 K………………………………… 74
19. Lindemann plot at 699 K………………………………… 74
20. Lindemann plot at 842 K………………………………… 75
21. Lindemann plot at 1040 K………………………………. 75
22. Plot of low- pressure limit for O + SO2 + Ar vs. T……… 38
23. Arrhenius plot of extrapolated kinf for
O + SO2 + Ar recombination…………………………….. 38
24. Comparative plot of rate constant of reaction 3.1 obtained from
various experimental studies…………………………..… 42
25. A simple energy diagram for the reaction mechanism
as suggested by Davis………………………………………….. 44
26. A simple energy diagram for the reaction mechanism
as suggested by Westenberg and deHaas……..………………… 43
27. A simple energy diagram for the reaction mechanism
as suggested by Troe et al……..………………………..……… 47

1
CHAPTER 1
INTRODUCTION
Sulfur compounds such as SO2, SO3, H2S, COS, CS2, C4H4S, CH3SCH3 and CH3SH are
emitted into the atmosphere from non-biogenic, biogenic and anthropogenic sources.
Estimates of total sulfur emissions have varied widely.1-4 Identified as a major pollutant,
sulfur dioxide, SO2 is the central focus of this study. In flame chemistry there is a direct
relationship between sulfur compounds and radical reactions. SO2 is believed to be a sink
for radicals such as O, H and OH. There is also evidence to suggest that SO2 may
influence NOx chemistry in flames and flue gases.5-13
Emissions of sulfur compounds into the atmosphere have non-biogenic, biogenic and
anthropogenic sources. Vegetation, marine algae, soils, wetlands, and sulfur reducing
bacteria are the major biogenic sources of sulfur compounds released into the
atmosphere.14-16 Vegetation contains on average 0.25% dry weight sulfur.17 Sulfur
compounds may be released from living plant leaves17 and decaying leaves,18 although
sulfur emission rates from decaying leaves are 10 to 100 times higher than emissions
from living leaves of the same species.18 In addition, many fungi and bacteria are known
to release sulfur compounds during plant decomposition. H2S is emitted from some
plants.19-23 Emission rates of H2S have varied between 0.006 to 0.25 g S m-2 yr-1,22 from
several lawns and a pine forest on aerobic soil in France, to 0.24 to 2.4 g S m-2 yr-1 from

2
humid forests in the Ivory Coast, West Africa.23 Other sulfur compounds known to be
emitted from plants are dimethyl sulfide, DMS,24,25 carbonyl sulfide, COS,20 carbon
disulfide, CS2,24-29 and possibly ethyl mercaptan.28,29 H2S and DMS are the major sulfur
species emitted from crops such as corn, soybeans, oats and alfalfa.28,30,31
Another biogenic source of sulfur is wetlands. The major compounds emitted are H2S and
DMS. Emission of DMS is dependent on temperature,32 and on the bacterial species S.
alterniflora.33 Emission of H2S is estimated to be 5.3 x 10-4 to 52.6 g m-2 yr-1 and is
closely associated with tidal cycling.32,34-38 Biogenic sulfur emissions originate also from
soil. The major sulfur species emitted are H2S, OCS, CS2, DMS and DMDS. Soil surface
temperature,31 soil nitrogen content,39 soil type and moisture content, are factors
determining the flux of sulfur gases from soils, which range from 1.2 to 23.4 mg S m-2yr-1
for temperatures between 20 and 30 0C.31,40,41 Global emission of sulfur from the
terrestrial biosphere is approximated at 0.91 Tg S yr-1.4 This includes 0.86 Tg S yr-1 from
vegetation and 0.05 Tg S yr-1 from soils.
The marine biosphere is the leading source of biogenic sulfur emissions. DMS, CS2,
CH3SH and CH3SSCH3 gases are produced biologically and H2S and OCS is produced
photochemically.42 DMS, the most abundant compound emitted,42,43 was first reported in
1972 in oceanic waters,18,43 before being measured throughout the Pacific, Atlantic and
southern seas. DMS concentrations are in the range of 0.5 to 5 nmol/L in open ocean

3
surface seawater, although the concentration varies depending on the region and season.
Dimethyl -sulfoniopropionate (DMSP), a precursor to DMS, has been identified in
marine algae, P. fastigiata,44 and phytoplankton,45 and is enzymatically cleaved to yield
DMS and acrylic acid. The initial investigation of global DMS flux was based on
observations in the Atlantic and eastern tropical Pacific and a value of 32 Tg S yr-1 was
proposed.42,46,47 When seasonal variations of DMS concentration were accounted for, the
estimate of global DMS flux was revised to 16 ± 11 Tg S yr-1.48 CS2 and OCS are also
present in surface open ocean waters at concentrations of 16 ± 8 pmol/L and 10 to 100
pmol/L respectively.49-54 The fluxes of OCS and CS2 were approximated at 1.2 % of the
flux of DMS.20,50,52,53 CH3SH and CH3SSCH3 are volatile sulfur species suspected of
being present in marine sediments, and decomposing algal mats. Recent evidence
however suggests that these compounds are produced as an artifact of sampling if
plankton undergoes anaerobic decomposition.55
Geothermal emissions, such as sulfur springs and volcanoes, are non-biogenic sources of
SO2. Including emissions from lava, volcanoes are estimated to contribute 3.9 Tg of
emitted S per year.56 Erupting and non-erupting degassing volcanoes emit sulfur
compounds into the stratosphere, although erupting volcanoes account for the majority of
sulfur emitted.57 While SO2 is the major specie emitted, SO42- and H2S comprise less than
1%,58 and OCS less than 0.1% of the total sulfur emission.59,60 SO2 emissions from
volcanoes are periodic and vary with eruption activity. Remote sensing correlation

4
spectrometry was used to measure SO2 emissions from eruptions in Japan,61 Central
America,62 Hawaii 63 and Italy.64,65 The volcanic contribution to atmospheric SO2
emission was estimated at 5 Tg S yr-1 by extrapolation to cover all of the earth’s surface
and excluding the big eruptions.62
Sea spray is a more important source of atmospheric sulfur. The amount of sulfur emitted
depends on the sulfur concentration in seawater, which is roughly constant at 0.27%, and
the extent to which sulfur ions are enriched relative to Na+ and Cl- ions by fractionation
during spray formation. About 7-10 % of spray generated sulfate is deposited on land
surfaces.66,67 The total emission of sulfur from sea spray is accepted as 44 Tg S yr-1.
Classification of biomass burning as a source of atmospheric sulfur has varied in previous
studies of global sulfur emissions, from not treated,2 to a source separate from man made
sources,3,4 to a natural source of sulfur.1 Since about 95 % of biomass burning is human
initiated,68 it is considered an anthropogenic source of sulfur here. Biomass burning is a
significant source of sulfur, with SO2 the major compound emitted. An estimated 50 to 60
% of global emissions are derived from savannah fires.69 Total sulfur emissions from
biomass burning was calculated at 1.44 to 2.94 Tg Syr-1.1

5
0
20
40
60
80
100
120
biomassburning
volcanoes marine bios. terrestrialbios.
man-made
Tg
S pe
r ye
ar
Spiro et al for 1980
Bates et al for 1990
Cullis and Hirschlerfor 1980
Figure 1: Estimates of sulfur emission from biomass burning, biogenic and non-biogenic
sources and anthropogenic sources.
Large differences in estimates of sulfur emission from biomass burning, and the marine
and terrestrial biosphere may be attributed to varying models and emission factors used in
the three studies.1,2,4 Bates et al1also reported limited resolution and few specific source
types in their global estimate of sulfur emission. Differences in global sulfur emission
from biomass burning, biogenic and non-biogenic sources were also introduced in
different seasonal and latitudinal considerations.

6
Copper extraction, and to a much lesser extent, lead and zinc extraction are
anthropogenic contributors to sulfur emissions in the smelting of non-ferrous metals.
Sulfur emissions from smelting have been on the decline and in 1976 these emissions
were estimated at 21.4 Tg SO2 (10.7 Tg S), of which 18.8 Tg SO2 (9.4 Tg S) was emitted
during the production of copper. For the year 1980, sulfur emission from the smelting of
copper, lead and zinc was estimated to be 6.8 Tg S yr-1. Countries leading sulfur
emissions from smelting of ores are Chile, Peru, Zaire and Zambia. A small contributor
to total atmospheric sulfur is the manufacture of sulfuric acid and the total annual
emission was calculated to be 1.25 Tg S in 1976.2 In the year 2000, an estimated 1 Tg S
was emitted globally from lead and zinc smelting, and sulfuric acid production.
Petroleum refinery and petroleum products are the second major source of anthropogenic
atmospheric sulfur with an average refinery in 1965 emitting 25 tons of S per 100,000
barrels of petroleum.70 In 1974, an estimated 29.15 Tg S was emitted from petroleum
products,2 while in 2000 the estimate was reduced to 23 Tg S produced from oil refining
processes.3
1.1 Coal and its combustion products
Combustion of coal and petroleum, petroleum refining and smelting of non-ferrous ores
are the main industrial sources of atmospheric sulfur. During the combustion of coal SO2
is evolved through the oxidation of sulfur resulting in flue gas concentrations of 500-
2000 ppmv. Total sulfur emission from coal was calculated to be 61.9 Tg S in 1976.2 A

7
dramatic increase is observed in the total sulfur emitted into the atmosphere over the last
hundred years. Over 1990-1999, US coal consumption increased by 16.7%, reaching
1,039 million tons in 1999.71 About 90.5% of domestic consumption in 1999 was by the
electric power sector. Accurate estimations of emissions from coal combustion require
the knowledge of the magnitude of coal consumption as well as the sulfur content of the
coal, which is highly variable between 0.2–10 % sulfur by weight.72,73
Sulfur compounds present in coal are classed into organic and inorganic sulfur containing
compounds. Almost all inorganic sulfur is pyrite sulfur. The ratio of inorganic: organic
sulfur is approximately 2:1,74 although the ratio may vary from 4:1 to 1:3.75 Most bound
sulfur was determined to be in the form of thiophenic, aromatic and aliphatic structures.75
Total yield of sulfur compounds from coal depends on the rank of the coal and the
temperature. The carbon content of a coal determines its rank. Coals with the least to the
most carbon content are: - lignite, sub-bituminous, bituminous and anthracite. Anthracite
yields about 5% of sulfur compounds while highly volatile lignite may yield a maximum
of 50% of gaseous sulfur compounds.76
The coal combustion process involves the ignition and burning of crushed and pulverized
coal in a combustion chamber. Fine particles (fly ash) are suspended in the flue gas.
Course particles settle at the bottom of the chamber and have two components: bottom
ash and boiler slag. The fourth product of coal combustion is coal ash, which is derived

8
from inorganic impurities, and either remains in the combustion zone or is carried in the
flue gas stream.
In an attempt to control sulfur emissions from coal combustion, the US government
implemented the Clean Air Acts Amendments in 1990, which took effect in two phases.
The first phase began in 1995 and limited the 110 power plants built before 1978 to 2.5
pounds of SO2 per million British thermal units (BTU) of energy generated. The second
phase took effect in 2001 and limits emissions by all power plants to 1.2 pounds of SO2
per million BTU of energy generated.
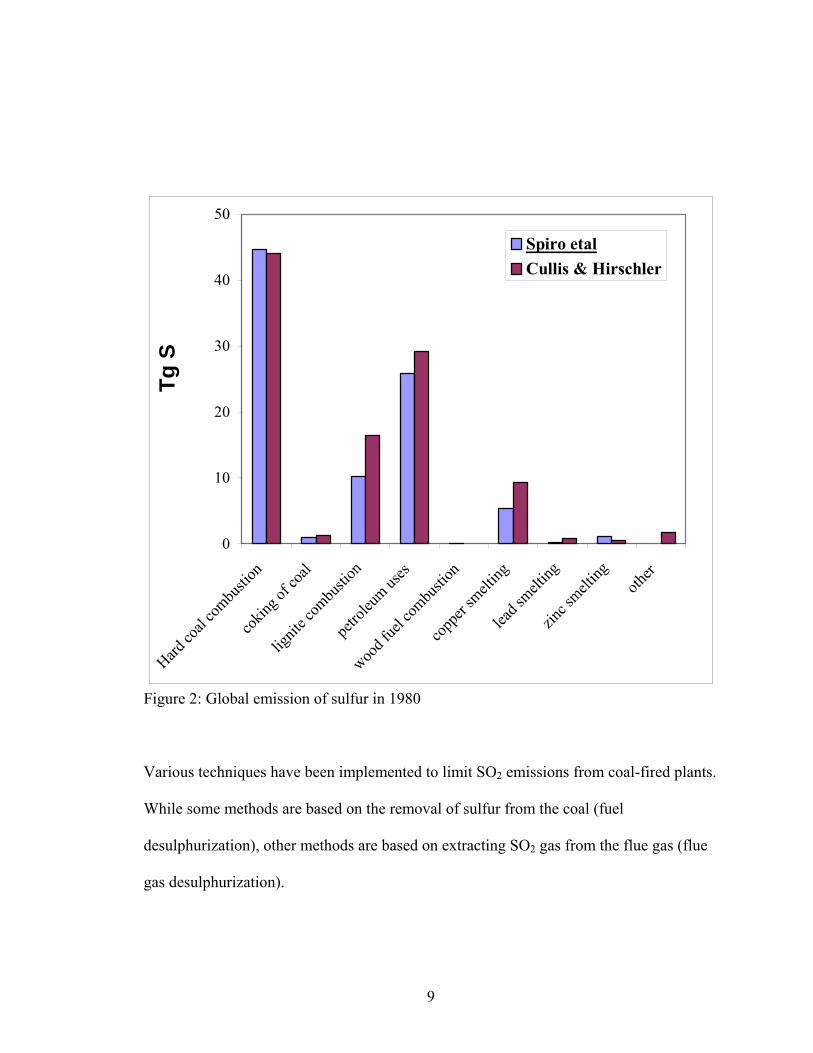
9
0
10
20
30
40
50
Hard co
al co
mbusti
on
cokin
g of c
oal
lignit
e com
busti
on
petro
leum us
es
wood f
uel c
ombu
stion
copp
er sm
elting
lead s
melting
zinc s
melting
other
Tg S
Spiro etalCullis & Hirschler
Figure 2: Global emission of sulfur in 1980
Various techniques have been implemented to limit SO2 emissions from coal-fired plants.
While some methods are based on the removal of sulfur from the coal (fuel
desulphurization), other methods are based on extracting SO2 gas from the flue gas (flue
gas desulphurization).

10
1.2 Fuel desulphurization
Sulfur is commonly removed from coal by cleaning. Traditional methods of coal cleaning
are based on the reduction of ash-forming materials. The physical coal cleaning processes
such as crushing and separation mainly removes inorganic sulfur. However, these well-
established techniques do not completely remove pyrites from coal, thereby reducing SO2
emission by less than 30%. It is hoped that the advanced yet underdeveloped physical
cleaning methods such as flotation, agglomeration and flocculation will remove more of
the inorganic SO2.24,77
1.3 Flue gas desulphurization (FGD)
The general trend in reduction of SO2 emissions from flue gas has been the switch to low
sulfur containing coal or the blending of low sulfur containing coal with high sulfur
containing coal. The plant may also be co-fired with natural gas. Alternatively flue gas
desulphurization equipment may be installed. Although 200 FGD methods have been
identified, only four of these methods are economically and technically feasible. The four
listed FGD methods are classified into two categories: wet and dry processes.
- Lime-limestone based method
- Magnesium based method
- Ammonium sulfate based method
- Dry injection method
The first three of these methods are wet processes and the fourth method is a dry process.

11
1.3.1. Lime- limestone based method
In the lime process, lime is slaked on site to form calcium hydroxide slurry, which reacts
with sulfur gases to form a calcium sulfite (CaSO3) and calcium sulfate (CaSO4) as
illustrated by the following reactions:
SO2 (aq) + Ca (OH) 2 (aq) CaSO3· ½ H2O (s) + ½ H2O (1.1)
SO2 (aq) + ½ O2 (aq)+ Ca (OH) 2 (aq)+ H2O CaSO4· 2 H2O (s) (1.2)
where (aq): slurry or solution; (s):solid and (g):gas.
In the process utilizing limestone, similar chemistry is observed, although CO2 is
generated. The process is described by the following reactions:
SO2 (aq) + CaCO3 (aq) + ½ H2O CaSO3·½ H2O (s) + CO2 (g) (1.3)
SO2 (aq) + ½ O2 + CaCO3 (aq) + 2 H2O CaSO4·2 H2O (s) + CO2 (g) (1.4)
The limestone reacts with the gaseous SO2 to form calcium sulfate (CaSO4) or gypsum
under oxidizing conditions. The formation of gypsum sometimes poses a problem with
sludge disposal, although gypsum has been used for gypsum binders, plasters, and
plasterboard manufacture and as additives in Portland cement production.78 It has been
found that sulfation in reaction 1.4 causes fouling in boilers firing high sulfur fuels.79-81
Fouling in boilers has been attributed to insufficient seed crystals in the slurry when a
supersaturated state has been reached. Almost pure deposits of CaSO4, meters in length,
have been found on the walls of the upper furnace, in the cyclone and on the super
heaters. It was thought that these solid deposits are derived from various fuel ash species

12
within the system, but detailed investigation of the solid demonstrated that fouling was
linked to an agglomeration mechanism.82 Often the lime or limestone is recirculated in a
scrubber. Although calcined limestone is useful in reacting with SO2 enabling reduction
of SO2 emissions, it is an active catalyst for CO oxidation83 and the oxidation of nitrogen
containing compounds, leading to the formation of NO and N2O.84
1.3.2. Magnesium based method
In this regenerative process, SO2 is captured by formation of magnesium sulfite. Reactive
MgO is slaked, forming Mg(OH)2 slurry, which becomes the absorber. SO2 and SO3
react with MgO forming MgSO2 and MgSO3 respectively. The process is illustrated by
the following reactions:
Mg(OH)2 + 5 H2O + SO2 MgSO3·6 H2O (1.5)
Mg(OH)2 + 2 H2O + SO2 MgSO3·3 H2O (1.6)
Mg(OH)2 + 6 H2O + SO3 MgSO4·7 H2O (1.7)
SO2 + MgSO3·6 H2O Mg(HSO3)2 + 5 H2O (1.8)
SO2 + MgSO3·3 H2O Mg(HSO3)2 + 2 H2O (1.9)
Mg(HSO3)2 + MgO + 11H2O 2 MgSO3·6 H2O (1.10)
Mg(HSO3)2 + MgO 5 H2O 2 MgSO3·3 H2O (1.11)
The aqueous sorbent slurry containing MgO, MgSO3 and MgSO4 is concentrated in a
clarifier and then fed into a continuous centrifuge. MgSO3·6 H2O, MgSO3·3 H2O and

13
MgSO4·7 H2O and unreacted MgO crystals are contained in this “wet cake”. The
supernatant is returned to the main recirculation stream. The “wet cake” is dried at a
temperature of 176-232 0C. The dry mixture is then calcined at 800-1000 0C. This
calcining regenerates MgO and releases SO2, which is used in the production of H2SO4 or
elemental sulfur. Typically an excess of 95% of sulfur gas is removed by this method
during operation at a pH of 5.5-6.5.
1.3.3. Ammonium sulfate based method
This popular European method uses ammonia as sorbent. Fly ash and other particulates
are removed from the flue gas by being passed through a spray drier and an electrostatic
precipitator. The following chemistry is observed upon entry of the flue gas into the
scrubber that contains ammonia:
SO2 + 2 NH3 + H2O (NH3) 2SO3 (aq) (1.12)
CO2 + 2 NH3 + H2O (NH3) 2CO3 (aq) (1.13)
(NH3) 2SO3 + ½O 2 (NH3) 2SO4 (aq) (1.14)
Injection of oxidized liquor, containing ammonium sulfite and smaller concentrations of
ammonium carbonate and ammonium sulfate, into a spray drier decomposes the sulfite
and carbonate fractions. Ammonium sulfate remains and is used as sulfur blending stock
in chemical fertilizer formulations. A second washing of the clean flue gas leaving the
scrubber prevents scaling in this FGD unit. This method is highly advantageous as there
is direct reaction of ammonia with SO3, leading to the formation of ammonium sulfate.

14
This is helpful in eliminating corrosion related problems in the reactor. Efficiencies in
sulfur removal with this process are reported to be about 95%.
1.3.4. Dry injection method
This method is one of the dry processes, of which there are three types: spray drying, dry
injection and simultaneous combustion of fuel sorbent mixtures. The method is based on
sulfur oxides reacting with reagent in the duct and on the surface of filter bags.
Commonly used reagents are Nahcolite and trona, which closely commercially resemble
sodium hydrogen carbonate. The reaction chamber is heated to the temperature at which
the sorbent decomposes. Nahcolite and trona decompose at 1350C and 93 0C,
respectively. Decomposition of the sorbent increases porosity, reaction surface and the
reaction rate.
Decomposition of sorbent is described by the following reactions:
2NaHCO3 NaCO3 + CO2(g) + H2O(g) (1.15)
2(Na2CO3)·NaHCO3·2H2O 3 Na2CO3 + CO2(g) + H2O(g) (1.16)
Reaction of SO2 with the sorbent is described by the following reaction:
Na2CO3 + SO2 + ½O 2 Na2 SO4 + CO2(g) (1.17)
Dry methods have an advantage over wet lime-limestone based methods, as their end
products are solid and can be treated by fly ash handling systems. This eliminates the
handling of wet sludge, however, sodium based byproducts must be properly disposed of,
to prevent the rise of potential environmental problems from the leaching of highly

15
soluble sodium based FGD byproducts. Dry methods however require a higher ratio of
sorbent to sulfur than wet methods, as gas-solid reactions proceed slower than gas-liquid
reactions.
As part of their Clean Coal Technology program, the Department of Energy instituted a
new process called the Integrated Gasification Combined Cycle (IGCC), where coal is
not burned directly but converted to gas, then combusted in a combined-cycle gas
turbine. Gasification of coal occurs in an enclosed pressurized reactor under reducing
conditions. Synthesis gas or syn gas is a mixture of CO and H2, and is produced from
gasification. The syn gas is cleaned before it is burned in air or oxygen and combustion
products are generated at high temperature and pressure. Under reducing conditions,
sulfur is present mainly as H2S and some COS. H2S is more easily removed than SO2.
Sulfur is produced in elemental form as a by-product in most units.
This method uses a combined cycle format where a combusted syn gas drives a gas
turbine. Heat exchange between hot exhaust gas and water and or team is used to
generate superheated steam, which drives a steam turbine. This has reduced SO2
emissions by 98%, and increased plant efficiency by 40%.
1.4 Relevance of SO2 to flame combustion
In a flame, radicals are generated by sequences of elementary reactions such as:
H + O2 OH + O (1.18)

16
H + O2 HO2 (1.19)
OH + H2 H2O + H (1.20)
O + H2 OH + H (1.21)
The rate of overall combustion is determined in large part by the elementary reaction
between H atoms and O2 molecules. The higher the temperature the larger the
contribution of reaction 1.18 relative to reaction 1.19.
Interest in SO2 lies in its ability to affect basic flame chemistry, as the coupling of sulfur
chemistry with radical chemistry has been evident. Recombination of radicals is
catalyzed by SO2 through the following mechanism:85,86
X + SO2 + M X SO2 + M (1.22)
Y + X SO2 XY + SO2 (1.23)
where X and Y may be O, H or OH radicals. These reactions have been found to
influence flame behavior and explosion limits.87 Three mechanisms have been identified
depending on which radical initially attacked SO2: the “H cycle”, “the O cycle” and “the
OH cycle”. This study focused on reaction 1.22 with X being O radicals. The “O-cycle”
in a lean flame is comprised of the following reaction sequence.
O + SO2 + M SO3 + M (1.24)
O + SO3 SO2 + O2 (1.25)

17
Reaction 1.24 is considered to be one of the elementary steps in aerosol formation. (Refer
to section 1.5 for more information on aerosol formation). Formation of SO3 through the
recombination of SO2 and O atoms (reaction 1.24) is spin forbidden when the ground
states of O (3P), SO2 (1A1) and SO3 (1A1') are involved. Several experimental studies of
reaction 1.24 have illustrated positive activation energies at low temperature,88 while at
higher temperatures the reaction rate decreased with temperature.89,90
Although both reactants are present in the atmosphere, reaction 1.24 is of little
consequence there. In the atmosphere the ratio of molecular oxygen to SO2 molecules is
so high that the following reaction with atomic oxygen radicals, produced from the
photolysis of NO2 or O3, is ensured.
O + O2 O3 (1.26)
Photodissociation of SO2 into SO molecules and O atoms require 565 kJ/mol,91 which is
impossible energetically for wavelengths of light greater than 218 nm. Solar radiation
reaching the lower atmosphere is of wavelength greater than 290 nm; thus only molecular
reactions involving the ground and electronically excited states of SO2 can occur at the
300-400 nm wavelength.
1.5 SO3 formation
Reaction 1.24 has been determined to be the only major homogenous source of SO3 in
flames,92 which is highly corrosive and contributes to aerosol formation. While

18
consumption of SO3 is not well characterized, competition between reaction 1.24 and
reaction 1.25 yields the net SO3 formed. The catalytic effects of surface deposits also
contribute to SO3 formation. When the vanadium content of a fuel is high, especially in
large oil fired units, SO3 formation becomes very important, as vanadium catalyzes
reaction 1.24. SO3 is not readily removed from exhaust gases by conventional flue gas
desulphurization methods. SO3 readily reacts with water to form sulfuric acid. The
reaction is so exergonic that a fine aerosol of H2SO4 is formed that passes through
scrubbers. SO3 emissions may be reduced by addition of methanol, CH3OH, or hydrogen
peroxide (H2O2), which lead to HO2 formation. Hydrogen peroxide reacts directly with
SO3 to produce the HO2 radical as illustrated by the following reactions:
SO3 + H2O2 HSO3 + HO2 (1.27)
HSO3 + M OH + SO2 + M (1.28)
Methanol reacts with hydroxyl radicals in a flame to produce the HO2 radical as
illustrated by the following reactions:
CH3OH + OH CH2OH + H2O (1.29)
CH2OH + O2 CH2O + HO2 (1.30)
HO2 formation in a combustion system is desirable as this radical is the active specie that
converts NO to NO2 and SO3 to SO2 by the following pathways:
NO + HO2 NO2 + OH (1.31)
SO3 + HO2 HSO3 + O2 (1.32)
HSO3 + M SO2 + HO + M (1.33)

19
1.6 The effect of sulfur on NOx emission
NOx is the collective term for the oxides of nitrogen NO and NO2. NOx gases are a major
contributor to acid rain and photochemical smog. NOx gases in combustion systems are
derived from nitrogen contained in combustion air and from nitrogen contained in fuel,
such as coal or heavy oil. Nitric oxide, NO, is formed when N2 reacts with O2 in air
during combustion at high temperature and during oxidation of fuel nitrogen.
N + O2 NO + O (1.34)
NO2 is produced from the further oxidation of NO:
NO + O2 NO2 + O (1.35)
In a cyclic set of reactions, NO is formed from the reactions of NO2 with O, H and OH:
NO2 + O NO + O2 (1.36)
NO2 + H NO +HO (1.37)
NO2 + OH NO + HO2 (1.38)
The interest in sulfur combustion products lies in their potential to influence NOx
chemistry in flames and exhaust gases. Sulfur can either reduce or enhance NOx
concentration in flames, depending on the conditions.5,6,8-13Fuel sulfur-nitrogen
interactions in exhaust gases are of particular interest, as they may shift the balance
between NO2 and SO2, and the less desirable NO and SO3. NO is relatively inert which
makes removal difficult. NO2 even though undesirable is efficiently removed by SO2
scrubbers, which cannot remove highly corrosive SO3.93 For efficient SOx and NOx

20
removal, conversion of NO to NO2 and SO3 to SO2 is required. In an experiment
simulating flue gas, 90% NO-to-NO2 and SO3-to-SO2 conversion was achieved by
injection of methanol into gas.94
Chief strategies utilized in NOx reduction in combustion are minimizing the excess air
supply, reducing the optimum combustion temperature, and staging of the combustion
process. A successful fuel staging method for NOx control has been reburning, which
exploits the sequence of combustion stages. About 80-90% of fuel is burned in the main
combustion zone in a fuel lean environment, forming NOx. More fuel is injected into the
secondary combustion zone at 1400- 1700 K, establishing a fuel-rich environment, where
NOx removing reactions occur. At optimum conditions, NOx emissions may be reduced
by 50-70%.
Although many kinetic investigation of reaction 1.24 have been performed,89,90,95-104 the
rate constant of the reaction has not been determined with certainty. Most kinetic
determinations have been performed at low temperatures (300-500 K). At high
temperatures (1700-2500 K) the rate of reaction 1.24 has been estimated using the reverse
dissociation rate constant for SO3. In the intermediate temperature range, no experimental
measurements are available. The mechanism of the reaction has also not been clearly
established as evidenced by discussions on the state of the reaction product, SO3.90,99,105

21
CHAPTER 2
EXPERIMENTAL METHOD
2.1 Flash (Laser) Photolysis/ Resonance Fluorescence (FP-RF) Technique
In 1967 Norrish and Porter received the Nobel Prize for the development of the flash
photolysis technique, which was designed to overcome the shortcomings of other
contemporary kinetic techniques. The basis of the technique is the pulsed photolysis of a
precursor compound with light (UV or visible), which creates a reactive specie. The light
source is either a flashlamp or a laser. The latter was used in these experiments. The pulse
of light should have a shorter duration than the reaction being studied.
The radicals generated by the flash are excited by absorption of continuous radiation, in
resonance with a higher electronic state, from the resonance lamp. Decay of radicals to
the ground state produces fluorescence radiation, which is detected as photons by the
photomultiplier tube (PMT), which is situated perpendicular to the laser and the
resonance lamp. The PMT is connected to a multichannel scaler with photon counting
electronics to interpret the fluorescence detected by the PMT. Fluorescence radiation is
monitored as a function of time. Since fluorescence is proportional to radical
concentration, the relative radical concentration as a function of time is obtained.
An excimer laser operating at 193 nm was used as a light source for flash photolysis in
these experiments. A laser has a short pulse duration, a precisely defined wavelength

22
range and a well-defined spatial profile, which makes it a good light source. Refer to
figure 3 for the flash photolysis-resonance fluorescence apparatus.
O (3P) radicals are generated by pulsed photolysis of sulfur dioxide (SO2) diluted in argon
(Ar) bath gas, by 193 nm radiation from an excimer laser (PSX-100, MCB). The
radiation passed through a suprasil quartz window transmitting light at λ > 165 nm. The
energy of the laser was varied between 0.014 – 0.20 mJ, by adjusting the number of
filters (copper mesh and steel micro fiber sheets) between the laser and the reactor. When
the energy output of the laser was too low for experimental operation, the laser was
evacuated and filled with a fresh F2, Ar, and Ne gas mixture.
The concentration of the reaction product, atomic O (3P), was monitored during the
course of the reaction by time resolved resonance fluorescence at a wavelength of 130-
131 nm (O (3s) 3S O (2p) 3P2,1,0).106 Resonance radiation was generated by a
microwave discharge lamp through which a mixture of 0.9% O2 in Ar gas at a pressure of
300 mtorr was passed. Fluorescence was detected by a solar-blind PMT (Thorn EMI,
9423 B), situated orthogonal to the resonance lamp and the laser. The fluorescence is
passed through a multichannel scaler (EG & G Ortec ACE) with photon counting
electronics. A digital delay-pulse generator (model DG535, Stanford Research Systems
Inc.) triggered the laser. The delay pulse generator also provided trigger pulses to a
computer controlled multichannel scaler.

23
Figure 3: flash-photolysis resonance fluorescence apparatus.
Kinetic measurements of O radicals were investigated as a function of temperature and
pressure. Experiments were performed at ambient temperature, 399 K, 581 K, 699 K, 842
K, and 1040 K and at pressures from 25- 660 torr. For the experiments at 399 K, 581 K,
699K, 842 K, and 1040 K, the temperature was measured before and after each
experiment by inserting a movable thermocouple into the reaction zone. The
thermocouple (Omega, type K, chrome (+) vs. alumel (-)) was corrected for radiation

24
errors, which occur from loss of heat out of the reaction zone through the windows of the
reactor.107
2.2 Kinetic Experimental Procedure
A stainless steel reactor was employed in this kinetic study. This type of reactor can
successfully be employed at temperatures up to about 1100 K. The reactor has a window
cooling system, which prevents overheating of the rubber vacuum seals, especially during
operation at higher temperatures. Acetone was used in routine cleaning of the reactor.
The reactor and the gas handling system were evacuated overnight using a mechanical
pump. In preparation for an experiment, the gas handling system was vacuumed to ≤ 4
mtorr, using a combination of a mechanical pump and a diffusion pump. The system was
evacuated to similar vacuum levels before gas mixtures were made up or diluted.
At the beginning of the project, the flow meters were calibrated using soapsuds, Ar gas
and calibrated cylinders. The actual flow rate was calculated from how long the suds took
to reach a given volume. At least five flow rates within the operation range of each flow
meter were tested and each volume tested timed several times (with a 0.1 s difference).
Flow meter readouts were always zeroed when no gas was flowing. Pressure in the
reactor was adjusted using a needle valve, which is connected to the outlet port of the
reactor. The needle valve is also connected to a stopcock, which opens to the vacuum
pump and the gas handling system. During an experiment the flows of SO2 and Ar were

25
complementary to some total volume flow rate so as to prevent fluctuations in reactor
pressure.
A steady slow flow of reactant in bath gas was allowed to flow into the reactor for at least
20 minutes to saturate the reactor walls with reactant. The effect of secondary chemistry
was investigated using different laser energies; at least a doubling of the lower energy, at
a single pressure at each temperature that the reaction was investigated at. A large
difference in the pseudo-first rate constants would indicate a large effect of secondary
reactions.
The gas residence time is the average time a sample of gas spends from entry into the
reactor until reaching the center of the reaction zone. Varying the flow rates and the
pressure varied the residence time of the gas, which is useful in detection of systematic
error, arising for example from thermal decomposition. Pulses of light at a wavelength of
193 nm from the laser passed into the reactor through a suprasil quartz window.
Fluorescence from the resonance lamp is focused into the reactor through a CaF2 window
transmitting at λ >125 nm. Radical detection in the reactor is achieved by detection of
fluorescence at 130.2 nm and is focused through a CaF2 lens before the PMT. In the
reactor, Ar sweeper gas passes around the windows to prevent adsorption onto the optics,
especially at higher temperatures.

26
For each set of experiments, the pseudo-first-order rate constants for at least five different
concentrations of reactant were determined. The maximum SO2 concentration in a given
experiment was at least 1.0 x 1016 molecules/cm3. Higher SO2 concentrations were
permitted by a good signal and small uncertainty of the pseudo-first-order rate constant.
Lower and higher flows were alternated for easy detection of systematic errors.
2.3 Materials
Ar (99.9999%, Air Liquide) and N2 (industrial grade, Air Liquide) were used directly
from the cylinder. SO2 (99.98%, MG Industries) was purified by distillation from a trap
first cooled by liquid N2. The SO2 gas was then subjected to several freeze, pump and
thaw cycles using a trap cooled to about 175 K by a liquid N2 /methanol slush. Cold
methanol at 263 K was used in the distillation of impurities from SO2 at atmospheric
pressure. An SO2 gas mixture was prepared by firstly pumping on the pure SO2, frozen
by liquid N2. The liquid N2 was replaced by cold methanol at 223 K to thaw the solid SO2
to vapor slowly. SO2 gas mixtures were prepared by filling a bulb with a few torr of pure
SO2 vapor which is diluted with Ar to some total pressure at about 1000 torr. Once
pressure of the gas mixture had fallen to a few hundred torr, the gas mixture was often
diluted, depending on the dilution. O2 (99.999%, Air Liquide) was stored in a bulb and a
few percent of pure O2 was diluted with Ar and stored in a separate bulb. Gas mixtures
were prepared the day before commencement of an experiment to ensure good mixing
and proper distribution of gas molecules.

27
2.4 Data Analysis
Following generation, atomic oxygen is mainly lost in two process:
O + SO2 SO3 (2.1)
O wall loss (2.2)
-d [O]/dt = k1 [O][ SO2] + kw [O] (2.3)
Since [SO2]0 >> [O]0, then the [SO2] remains approximately constant and the rate law is
ln ([O]/[O]0) = - (kw + k1 [ SO2] )t = -kps1t (2.4)
[O] = [O]0 exp (-kps1t) (2.5)
where the pseudo-first-order rate constant, kps1, consists of two terms: kw and k1[SO2],
which is the rate of decay of [O] in reaction (2.1). The second term, kw, is the rate of
decay of O radicals in the absence of SO2, due to its diffusion out of the reaction zone
and its slow reaction with impurities in the bath gas. In many of the lowest pressure
experiments kw was large compared to a change in the pseudo-first-order rate. These
experiments were excluded from the analyses. The pseudo-first-order rate is determined
from a non-linear least squares fit to fluorescence decay of O radicals, although
background from scattered light must be considered in the analysis algorithm to account
for the observed fluorescence signal I. This is achieved by modifying equation 2.5 above:
I = A exp (-kps1t) + B (2.6)
where A and B are constants and B represents the background.( Refer to figure 4 for the
plot of fluorescence decay of O atoms with time). The second order rate constant, k1 was

28
then obtained from the gradient of a linear plot of kps1 versus SO2 as illustrated in figure
5.
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.350
200
400
600
800
1000
1200
Fluo
resc
ence
Inte
nsity
Time/s
Figure 4: Plot of fluorescence intensity including background of SO2+O+Ar at 297 torr
and 1047 K with an SO2 concentration of 7.9 x 1015 molecules cm-3

29
0 2 4 6 8 10 12 14 160
50
100
150
200
250
300
K ps1 /s
-1
[SO2]/1015 cm3 molecule-1
Figure 5: Plot of pseudo-first-order rate constant (kps1) for the loss of O radicals at 297
torr and 1047 K. Open symbol corresponds to decay in figure 4.
2.5 SO2 Absorption Cross-Section (σ) Determination
The ultraviolet absorption cross-section of SO2 is required to calculate the concentration
of SO2 photolyzed, hence the concentration of O radicals produced. Calculating O
radicals produced involves the Beer-Lambert law:
Itrans = I0 exp (-σ cl) (2.7)
Itrans and I0 represent the intensity of transmitted and incident light respectively. σ is the
molecular absorption cross coefficient with units of cm2 molecule-1, c is the concentration
of the absorbing specie in molecules cm-3 and l is the path length in cm. The cross section
may also be measured in terms of ε, which is the molar absorption coefficient with units
of cm2 mole-1.

30
Although the absorption coefficients in the vacuum ultraviolet region have been
published,108-111 fine structure around 193.3 nm leads to varying estimations of the cross
section at the laser photolysis wavelength. The absolute cross-section of SO2 at room
temperature was determined in a set of three experiments using an excimer laser source
and a flowing gas cell. Mixtures of slightly less than 1 % SO2 in Ar gas were passed
through the cell. Complete saturation of the cell was attained usually after two hours of
constant flow before attenuated light was measured with a pulse energy meter (Molectron
detector Inc, model J25LP). Saturation of the cell was determined by the consistency of
the absorbance at a constant SO2 flow. The attenuated signal was averaged by a digital
oscilloscope (Tektronix Inc., model 2440). The SO2 concentration in the cell was
changed by altering the gas pressure in the cell.
Error limits generated from the determination of absolute absorbance at room
temperature were obtained from the flowmeter corrections and the limits of the digital
temperature and pressure readouts. Statistical errors of the constrained fit Beer-Lambert
plots (figures 6-8 in Appendix A) were 2.1 %, 1.9 % and 1.7 %. With a path length of 35
cm the absorption coefficients derived from these plots are 7.40 x 10-18, 7.44 x 10-18 and
7.40 x 10-18 cm2 molecule-1. The mean absorption coefficient is (7.4 ± 0.4) x 10-18 cm2
molecule-1. Error limits of the mean absorption coefficient of SO2 were generated from
error limits of the SO2 concentration of each Beer-Lambert plot. Deviation from the best
fit Beer-Lambert plot were obtained from a best fit to the edges of the error limits of the

31
SO2 concentration, yielding closely symmetrical positive and negative deviation from the
best fit. The best fits were constrained to pass through the origin.
The absorption coefficient of SO2 has been estimated by Fockenberg and Preses,113 from
prior literature,108,109 to be 6 x 10-18 cm2 molecule-1 at room temperature At 193 nm, the
absorption cross of SO2 has been measured to be 8.24 x 10-18 cm2 molecule-1 at 300 K.112
The temperature dependence of the absorption cross section of SO2 has been investigated
at 345 and 925 K at 193 nm,113 and between 293 and 1070 K at 200 – 350 nm.114
Fockenberg and Preses reported a 40 % decrease in the SO2 absorption cross-section
between 345 and 925 K, which fixes the SO2 absorption cross section at 925 K relative to
the cross section at 345 K. SO2 absorption cross sections at 873 and 1073 K were
determined relative to the cross section at room temperature, assuming that relative
absorption cross sections remained the same for 200 and 193 nm. The temperature
dependence of the SO2 absorption cross-section can then be estimated at other
temperatures from a linear interpolation illustrated on the following page:

32
200 400 600 800 1000 1200
0.6
0.7
0.8
0.9
1.0
Rel
ativ
e Ab
sorb
ance
T, K
Figure 9: Temperature dependence of cross section absorption of SO2. The equation of
the above interpolation is: Rel. abs. = (1.16 ± 0.11)T – (5.24 x 10-4 ± 1.29 x 10-4)

33
CHAPTER 3
RESULTS AND DISCUSSION
3.1. Results
SO2 + O + (M) SO3 + (M) (3.1)
Second order rate constants for reaction 3.1 at 289 K, 399 K, 581 K, 699 K, 840 K and
1040 K were obtained under different conditions and are listed in tables 1-6 in appendix
B. The rate constants have statistical errors of 1σ. Results are independent of the laser
energy, verifying the isolation of the reaction 3.1 from any secondary chemistry.
3.2 Discussion
Previous studies of reaction 3.1 have been limited by temperature and pressure
considerations. In this study the kinetics of reaction 3.1 have been assessed at pressures
between 30 torr and pressures close to atmospheric pressure (660 torr) and at
temperatures between ambient and 1040 K.
Lifting of previous temperature and pressure restrictions allows for the study of the
pressure dependence of reaction 3.1. The reaction is found to be in the falloff region at
temperatures studied here, which is a new observation. Unlike with other temperatures of
this study, the plot of first order rate constant vs. pressure at 1040 K does not yield a y
intercept of zero, but 5 x 10-15 cm3molecule-1s-1. The y intercept reflects some overall rate
and since the y intercept increased from 0 to 5 x 10-15 cm3molecule-1s-1, there might be a

34
shift in the chemistry at this temperature. It was initially speculated that the abstraction
reaction
SO2 + O → SO + O2 (3.2)
might be a competing reaction at this temperature. If so, the rate of reaction obtained at
1040 K from a log k vs. temperature plot for reaction 3.2 is the contribution to the
observed rate of reaction 3.1
The study of the reverse rate of reaction 3.2 was carried out over a temperature range of
450-585 K at a total pressure of 20 torr by Garland.115 Incorporating data from literature
together with the measured rates, she derived the rate expression at 250-3500 K:
k(T)= 1.5 x 10-13 T1.4exp(-1868/T) cm3 s-1.
The rate of the forward and reverse reactions are related by the equilibrium constant:
Keq = kforward/kreverse (3.3)
The equilibrium constant of the forward reaction 3.2 was determined from the following
relationship:
-RT ln Keq = ∆reactionG0 (3.4)
where ∆reactionG0 is the Gibbs free energy of the forward reaction in kJ/mol and calculated
from thermodynamic tables.116 R is the universal gas constant and T is the temperature in
K. At 1040 K the derived rate of the forward reaction 3.2 is 7 x 10-16 cm3molecule-1s-1,
which is a small contribution to the y intercept of rate vs. pressure for reaction 3.1 at
1040 K. The origin of the non-zero intercept is therefore unknown.

35
To analyze the pressure dependence of the reaction 3.1, the Lindemann-Hinshelwood
theory was implemented.117,118
3.3. Background to Lindemann theory
In 1922 Frederick Lindemann suggested a reaction sequence to account for the observed
first order kinetics of spontaneous unimolecular reactions, such as isomerizations and
decompositions, instead of implied second order kinetics. The sequence for the reverse of
bond decomposition, i.e., recombination of radicals is:
A + B C* (3.5)
C* A +B (3.6)
C* + M C +M (3.7)
C* is an energized molecule of C, which has sufficient energy to isomerizes or
decompose. C* is formed by the transferal of kinetic energy of M. C* is either de-
energized to C by the transferal of energy to M, or C* can be transformed to products A
or B when it has the extra vibrational energy to disrupt the necessary bond. Reaction 3.6
is favored at lower pressure while at higher pressures reaction 3.7 is the dominant
pathway. The reaction rate is:
ν = d[C]/dt = -d[A]/dt = krec[A][B] (3.8)
d[A]/dt =-ka[A][B] + kb[C*] (3.9)
where a, b and c represent the rate constants of reactions 3.5, 3.6 and 3.7 respectively.

36
Applying the steady state approximation to C* yields:
d[C*]/dt = -kb[C*]+ ka[A][B] –kc[C*][M] = 0 (3.10)
Substitution into equation 3.10 into equation 3.9 yields a rate constant of:
krec =(-kakc[M]/(kb+ kc [M]) (3.11)
There are two limiting cases in determining the rate constant k. The first case, when kc
[M] >> kb, is favored at higher pressure and the rate constant,
k∞ = ka (3.12)
Equation (3.12) is the high-pressure limit. The high-pressure rate law is second order.
The second case, when kc [M] << kb, occurs at lower pressure, where the rate determining
step is stabilization by collision and the rate constant,
k0= kakc[M]/kb (3.13)
Equation (3.13) is the low-pressure limit and the low-pressure rate law is third order.
Application of Lindemann-Hinshelwood theory to reaction 3.1 yields the following:
SO2+O SO3* (3.14)
SO3* O + SO2 (3.15)
SO3*+ M SO3+ M (3.16)
The reaction rate for reaction 3.1 is:
ν = d[SO3]/dt= -d[O]/dt = krec[O][SO2] (3.17)
If k1, k2 and k3 represent the rate constants of reactions 3.14, 3.15 and 3.16 respectively,
then

37
d[O]/dt =-k1[O][SO2] + k2[SO3*] (3.18)
d[SO3*]/dt = k1[O][SO2] – k2 [SO3*] –k3 [SO3*][M] (3.19)
From the steady state approximation
[SO3*] = k1[O][SO2]/(k2+k3[M]) (3.20)
Substituting equation 3.20 into equation 3.18 yields:
d[O]/dt =k1[O][SO2] + k2k1[O][SO2]/(k2+k3[M]) (3.21)
d[O]/dt = [O][SO2] (-k1+ k2k1/(k2+k3[M])) (3.22)
krec = (k1k3[M]/(k2 + k3[M])) (3.23)
1/krec = 1/k1+ (k2/k3k1)(1/[M]) (3.24)
A plot of 1/krec vs. 1/[M] therefore yields k1-1 as the y intercept and (k2/k3k1) as the slope.
Therefore the rate constant k1 and the ratio of k2 to k3k1 may be determined from a plot of
1/krec vs. 1/[M].
When [M] is small:
krec,0= k1k3[M]/k2 (3.25)
and when [M] is large:
krec,∞= k1 (3.26)
See figures 15-20 in Appendix B for data fit to Lindemann-Hinshelwood theory. Rate
constants at the low and high pressure limits, calculated from the Lindemann-
Hinshelwood plots, were then plotted as a function of temperature. See figures 22 and 23
for plots of the rate constants at the low and high pressure limits. See table 1 for a
summary of these rate constants.

38
200 300 400 500 600 700 800 900
1x10-33
1x10-32
k o, c
m6 m
olec
ule-2
s-1
Tem perature, K
Figure 22: Plot of low-pressure limit for O + SO2 + Ar vs. T. The interpolated curve is a
quadratic fit of the form log k(T) = [(-6.3 ± 2.6) x 10-6]T2 + [(8.6 ± 3.0) x 10-3]T + (-35.0
± 0.8) cm6 molecule-1s-1.
1.0 1.5 2.0 2.5 3.0 3.5
2.0x10-14
4.0x10-14
6.0x10-14
1000K/ T
k inf, m
olec
ule-1
cm
3 s-1
Figure 23: Arrhenius plot of extrapolated k∞ for O + SO2 + Ar recombination. A linear fit
is shown and has the form k(T) = 8.5 x 10-14exp(-468/T) cm3 molecule-1 s-1.

39
Table 1: Summary of rate constants at the low and high pressure limits.
Temperature, K k0 k∞
293 8.0 x 10-34 1.8 x 10-14
399 4.6 x 10-33 2.2 x 10-14
581 6.3 x 10-33 4.0 x 10-14
699 9.2 x 10-33 3.4 x 10-14
842 7.3 x 10-33 5.5 x 10-14
3.4 Third body contribution to the third order rate
Since reaction 3.1 is slow, relatively large concentrations of SO2 were used in this kinetic
study. It is therefore necessary to evaluate the ratio of the contribution of SO2 to Ar as a
third body in the determination of the rate constant. The overall rate of reaction 3.1 is a
function of a third order rate and O, SO2 and Ar concentrations.
d[O]/dt = -kIII[O][ SO2][M] (3.27)
kIII [M] = kIII,SO2[SO2] + kIII,Ar[M] (3.28)
The third order rate constant kIII is the sum of the product of the third order rate of each
third body and its concentration. At room temperature,102
kIII,SO2 = (9.5 ± 3.0) x 10-33 cm6 molecule-2 s-1
kIII,Ar = (1.05 ± 0.21) x 10-33 cm6 molecule-2 s-1

40
SO2 is about 9 times more efficient than Ar as a third body and therefore at low total
pressures (low [Ar]) SO2 could potentially interfere with determination of kIII,Ar. Data
reflecting a third body contribution from SO2 greater than 11 % of Ar third body
contribution were eliminated from the analysis.
3.5 Comparison of rate constants with those from prior determinations
At room temperature, the value of the rate constant falls between the rate constants
quoted by Davis,103 and Atkinson and Pitts.102 The value obtained in this study lie closely
outside the statistical error limits quoted in the Atkinson paper. Unfortunately no
statistical error limits are available from the Davis paper. The rate constant quoted by
Halstead and Thrush96 is about an order of magnitude greater than the rate determined in
this study at room temperature. The rate constant of 2.8 x 10-33 cm6 molecule-1
s-1 obtained by Mulcahy et al using afterglow detection121 has been preferred over their
previous determinations of 3.9 x 10-33 cm6 molecule-1 s-1 and 6.6 x 10-32 cm6 molecule-1 s-
1 by ESR detection,99 because of the greater sensitivity of the afterglow method.121 At 399
K, the rate constants obtained in this study are about a factor of two larger than those
quoted by Atkinson and Pitts. Study of the reaction over the other temperatures, cover a
range which has not been previously explored, hence no prior kinetic data are available
for the 580 – 1040 K region.

41
Table 7: Summary the rate constants available for the SO2+O +Ar reaction including
results from this study.
Experimental Method Temperature, K k0, cm6 molecule-2 s-1 Reference
SO2 afterglow 299
2.8 x 10-33 121
SO2 afterglow 300
1.3 x 10-32 96
FP-RF 353-220
3.4 x 10-32exp(-1120/T) 103
FP-NO2 chem. 299-440
3.1 x 10-32exp(-2000/RT) 102
Shock wave 1700-2500
2.9 x 10-35exp(7870/T) 90
LP-RP 289-1040
3.3 x 10-32exp(-992/T) this study ESR, electron spin resonance spectroscopy; FP, flash photolysis; LP, laser photolysis
RF,Resonance fluorescence; FP-NO2 chem, flash photolysis NO2 chemiluminescence
All of the tabulated kinetic studies of reaction 3.1 have employed Ar as a bath gas, while
other kinetic determinations of reaction 3.1 have employed various bath gases such as N2,
He, O2, SO2 and N2O.99-103 The different collisional efficiencies of these bath gases have
resulted in rate constants varying as much as a factor of 40 at room temperature.101
Estimations of the third body relative efficiencies have also varied. Collisional
efficiencies are principally a function of molecular complexity and mass. In Davis’ study
of reaction 3.1, several bath gases including Ar were used. A conversion factor of 0.87
was adopted there in the conversion from N2 to Ar efficiency.

42
0.000 0.001 0.002 0.003 0.004 0.00510-34
1x10-33
1x10-32
k 0,
cm6 m
olec
ule-2
s-1
1/Temperature, K-1
Figure 24: Comparative plot of rate constant of reaction 3.1 obtained from various
experimental studies, including this.
(—— reference 90; - - - reference 103, ▲ reference 102 , ■ this study ,○ reference 96,
reference 99, - . - reference 89.)

43
3.6. Spin Considerations
The reaction of SO2 with oxygen atoms violates spin conservation rules when the ground
states of O (3P), SO2 (1A1) and SO3 (1A1') are concerned. Referring to Figure 24 it is
evident that at low temperature reaction 3.1 has a positive activation energy (from the
slope of the plot) which is suggestive of a barrier to reaction 3.1. At high temperature the
rate of the reaction 3.1 was determined from the reverse dissociation reaction and the
equilibrium constant and shows a negative activation energy.
Davis103 accounts for the positive temperature dependence of reaction 3.1 in a two step
mechanism, the first of which is the formation of a spin-allowed triplet SO3 molecule.
The second step involves intersystem crossing between triplet and singlet ground state
SO3. Intersystem crossing is often associated with spin-orbit coupling, which arises when
a heavy atom such as S is present. Formation of singlet SO3 violates the spin conservation
rule, and may account for the energy barrier of reaction 3.1.

44
Figure 25: A simple energy diagram for the mechanism of reaction3.1 as suggested by
Davis.103 ISC represents intersystem-crossing.
Westenberg and deHaas101 suggest that the positive temperature dependence of reaction
3.1 occurs when the positive energy of the excitation process of :
SO3(1A)→SO3*(3A) excitation energy = E3
*
is greater than the heat of enthalpy for reaction 3.1 (∆rxnH). So a positive temperature
dependence is observed when E3*>|∆rxnH|. A negative temperature dependence of
reaction 3.1 at high temperature can then be explained in terms of E1*< |∆rxnH|, where E1
*
is the energy of SO3*(1A) formation after intersystem crossing. This mechanistic theory
may be verified by quantum mechanical calculations of the triplet and singlet state
energies of SO3.

45
Figure 26: A simple energy diagram for the proposed intermediates in the mechanism of
reaction 3.1 as suggested by Westenberg and deHaas.101 ISC represents intersystem-
crossing.
3.7 Statistical Analysis of O + SO2+Ar
Troe has suggested a broadening factor, which when incorporated with the Lindemann
scheme gives a better estimation of the high pressure limit.119 In the Lindemann-
Hinshelwood model, a fall-off curve is described by:
k/k∞ = (k0/k∞)/(1 + k0/k∞) ≡ FLH(k0/k∞) (3.29)
The broadening of the falloff curve is accounted for in a collision broadening factor
k/k∞ = FLH(k0/k∞)FBF(k0/k∞) (3.30)

46
where FLH and FBF represent the Lindemann-Hinshelwood and broadening factor
functions. Detailed analysis of FBF in terms of Troe’s statistical adiabatic channel model
may be found elsewhere.120,121
Troe applied this kind of theoretical analysis to reaction 3.1.89,90 The dash-dot curve of
figure 24 is a fit of this analysis. Troe calculated the barrier of reaction 3.1 as the
difference between the threshold energy, which was best fit to experimental data, and the
heat of enthalpy for the reverse dissociation reaction 3.1. The estimated barrier at 0 K is
13.8 ± 4 kJ/mol.
The rate constant at the high-pressure limit at room temperature obtained from Troe’s
theoretical analysis89 is k∞= P x (2.16 x 10-13) cm3 molecule-1 s-1 where P represents the
triplet-singlet transition probability. Troe’s rate constant compares favorably with the rate
constant of k∞= 1.8 x 10-14 cm3 molecule-1 s-1 calculated in this study for the high pressure
limit. The implied value of P is ~ 0.1, which is consistent with Troe’s lower limit of 0.03.

47
Figure 27: A simple energy diagram for the proposed intermediates in the mechanism of
reaction 3.1 as suggested by Troe et al.90 ISC represents intersystem-crossing.

48
CHAPTER 4
CONCLUSION
The rate constant for the O + SO2 +(Ar) reaction has been measured between 289 and
1040 K by the laser photolysis resonance fluorescence technique. The reaction is spin
forbidden and slow, so large concentrations of SO2 were used in this kinetic study.
Conditions were selected to make the contribution of SO2 to the third order reaction
minor to the contribution from M = Ar. The rate obtained in this study illustrates a barrier
to the reaction because at low temperature the reaction has a positive activation energy
while at higher temperature it possesses a negative activation energy.
For the first time fall-off behavior was observed in O + SO2 recombination. The rate
expression for the O + SO2 +(Ar) reaction over the temperature range of 289 to 842 K is:
log k(T) = [(-6.3 ± 2.6) x 10-6]T2 + [(8.6 ± 3.0) x 10-3]T + (-35.0 ± 0.8) cm6 molecule-1 s-1
for the low pressure limit and k(T) = 8.5 x 10-14exp(-468/T) cm3 molecule-1 s-1 for the
high pressure limit. The kinetic study at 1040 K may be revisited, as concerns over
possible side reactions have arisen due to a non-zero first order rate at zero pressure at
this temperature.
The temperature dependence of the absorption coefficient of SO2 was estimated relative
to the absorption coefficient at room temperature, which was measured at

49
(7.4 ± 0.4) x10-18 cm2 molecule-1.

50
APPENDIX A: Spectroscopic data

51
0 . 0 5 . 0 x 1 0 1 4 1 . 0 x 1 0 1 5 1 . 5 x 1 0 1 5 2 . 0 x 1 0 1 5 2 . 5 x 1 0 1 5 3 . 0 x 1 0 1 5 3 . 5 x 1 0 1 50 . 0
0 . 1
0 . 2
0 . 3
0 . 4
0 . 5
0 . 6
0 . 7
0 . 8
Ln(I 0/I)
[ S O 2 ] m o l e c u l e s / c m 3
Figure 6: Beer-Lambert plot of SO2 at room temperature. (Temperature = 295 K,
I0 = 0.047 mJ, τres = 4.4-10.0 s, average τres = 6. 7 s, laser repetition rate =2 Hz)
0 1 x 1 0 1 5 2 x 1 0 1 5 3 x 1 0 1 50 . 0
0 . 1
0 . 2
0 . 3
0 . 4
0 . 5
0 . 6
0 . 7
0 . 8
0 . 9
[ S O 2 ] m o l e c u l e s / c m 3
Ln(I 0/I)
Figure 7: Beer-Lambert plot of SO2 at room temperature. (Temperature = 296 K,
I0 = 0.040 mJ, τres = 7.6-17.7 s, average τres = 11.2 s, laser repetition rate =10 Hz)

52
0 .0 5 .0 x 1 0 1 4 1 .0 x 1 0 1 5 1 .5 x 1 0 1 5 2 .0 x 1 0 1 5 2 .5 x 1 0 1 5 3 .0 x 1 0 1 50 .0
0 .1
0 .2
0 .3
0 .4
0 .5
0 .6
0 .7
0 .8
[S O 2 ]m o le c u le s /c m 3
Ln(I 0/I)
Figure 8: Beer-Lambert plot of SO2 at room temperature. (Temperature = 294 K,
I0 = 0.051 mJ, τres = 9.6-20.5 s, average τres = 12.7 s, laser repetition rate =10 Hz)

53
APPENDIX B: Kinetic Data
Codes:
A: datum included in analysis
B: datum excluded in analysis due to SO2 contributing more than 11 % to the third body
efficiency than Ar.
C: datum excluded from analysis due to non-linearity in the kps1 vs. [SO2] plot.
D: datum excluded from analysis due to a large uncertainty of the rate derived from the
kps1 vs. [SO2] plot.
E: conditioning of the reactor questionable; datum excluded in analysis
F: percentage ratio of [SO2] to [Ar] contribution as a third body
Int: y intercept

54
Table 2: Rate constant measurements for O + SO2 + Ar at 289 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
287 202 3.5 15.8 7.5 15.0 1.93 0.09 4.95 0.10 6.6 6.80 3.5 0.4 -5 12.0 A
287 201 3.5 85.0 30.6 81.2 1.41 0.09 4.85 0.10 6.5 6.77 3.6 0.1 3 2.1 A
287 200 3.5 34.6 9.1 33.6 0.99 0.09 4.80 0.10 6.5 6.73 3.5 0.03 1 1.0 A
286 122 4.4 13.8 5.4 13.3 1.54 0.12 4.89 0.13 10.7 4.12 2.6 0.1 -2 2.4 A
287 77 2.9 13.8 6.8 11.5 2.02 0.08 3.97 0.09 13.9 2.59 1.5 0.1 10 2.8 B
287 650 14 13.8 4.1 10.8 0.42 0.11 3.65 0.12 1.5 21.88 7.4 0.1 6 1.7 A
287 652 14 17.8 6.8 14.0 0.42 0.11 4.81 0.12 2.0 21.95 7.7 0.3 20 8.1 A
287 522 11 17.8 1.7 16.2 0.33 0.09 4.52 0.10 2.3 17.57 7.1 0.2 11 4.4 A
288 404 10 11.9 3.7 11.0 0.32 0.09 4.63 0.10 3.1 13.55 6.7 0.4 11 10.1 A
288 406 11 17.8 1.6 16.6 0.32 0.08 4.67 0.10 3.1 13.62 8.6 0.2 22 6.8 C
294 300 13 16.8 4.2 13.8 0.28 0.10 3.88 0.11 3.6 9.86 5.9 0.4 15 7.0 A
294 302 13 32.6 8.2 26.5 0.28 0.10 3.84 0.11 3.5 9.92 6.1 0.2 17 3.3 A
293 664 28 15.8 1.3 13.0 0.30 0.07 3.88 0.08 1.6 21.89 9.5 0.3 28 4.9 A

55
Table 1 continued: Rate constant measurements for O + SO2 + Ar at 289 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
293 664 28 33.6 2.8 27.6 0.30 0.07 3.88 0.08 1.6 21.89 9.8 0.4 45 7.6 A
294 452 19 27.7 1.4 22.3 0.18 0.05 3.77 0.06 2.3 14.85 8.2 0.4 24 7.5 C
292 651 12 15.8 2.6 16.9 0.61 0.15 5.83 0.16 2.4 21.54 7.8 0.3 37 7.9 A
292 651 12 31.6 5.3 33.9 0.61 0.15 5.83 0.16 2.4 21.54 7.9 0.5 52 16.8 C
292 548 12 11.9 1.9 12.2 0.60 0.15 5.46 0.15 2.7 18.13 7.5 0.1 15 3.0 A
292 548 12 19.8 3.2 20.4 0.60 0.15 5.46 0.15 2.7 18.13 7.4 0.1 26 4.0 A
293 375 9.6 11.9 1.8 12.4 0.56 0.12 5.57 0.13 4.1 12.36 6.1 0.0 10 1.0 A
293 650 17 11.9 1.8 12.2 0.56 0.10 5.44 0.11 2.3 21.43 8.7 0.1 20 2.6 A
293 650 17 19.8 3.0 20.3 0.56 0.10 5.44 0.11 2.3 21.43 8.8 0.2 29 5.9 A
294 450 15 18.6 3.2 19.1 0.64 0.09 5.44 0.10 3.3 14.79 7.3 0.1 17 1.8 A
293 300 9.6 11.9 0.7 8.3 0.23 0.06 3.14 0.06 2.9 9.89 5.9 0.2 9 3.1 A
293 300 9.6 25.7 1.7 18.0 0.23 0.06 3.14 0.06 2.9 9.89 6.6 0.2 13 2.9 A
296 607 15.3 32.1 6.6 35.8 0.76 0.09 4.64 0.09 2.1 19.81 8.8 0.3 41 6.4 A

56
Table 1 continued: Rate constant measurements for O + SO2 + Ar at 289 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
296 507 15.8 32.1 6.9 40.9 0.81 0.09 8.02 0.11 4.4 16.55 7.9 0.3 33 7.5 A
296 204 12.8 29.6 5.3 24.4 0.66 0.07 3.89 0.10 5.3 6.66 4.9 0.1 18 0.6 A
296 31 3.78 29.6 3.3 16.2 0.40 0.03 2.31 0.09 20.7 1.01 1.8 0.2 31 1.2 B
297 657 13.8 54.2 7.8 73.3 0.53 0.14 9.20 0.16 3.9 21.37 8.9 0.7 82 25.2 A
297 657 13.7 17.8 2.6 24.1 0.53 0.14 9.18 0.16 3.9 21.37 8.6 0.3 33 11.4 A
297 30 1.86 17.8 1.4 9.2 0.29 0.02 2.14 0.06 19.8 0.98 1.4 0.1 40 0.8 B
297 248 7.73 16.2 8.7 19.8 1.19 0.08 5.20 0.10 5.8 8.07 5.3 0.03 18 0.7 A

57
0 100 200 300 400 500 600 7000
1
2
3
4
5
6
7
8
9
10
11
Rat
e C
onst
ant,
10-1
5 cm
3 mol
ecul
e-1 s
-1
P re s s u re , to rr
Figure 10: Plot of first order rate constant vs. pressure at 289 K

58
Table 2: Rate constant measurements for O + SO2 + Ar at 399 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
398 663 20 65.2 3.3 18.3 0.19 0.05 1.13 0.05 0.6 16.09 16.3 0.4 19 2.7 A
399 528 19 65.2 2.4 19.2 0.14 0.05 1.19 0.05 0.8 12.78 16.5 0.2 12 1.7 A
399 300 9.2 88.9 2.5 31.2 0.10 0.02 1.45 0.03 1.8 7.26 12.8 0.1 9 0.8 A
399 100 6 88.9 1.5 30.7 0.06 0.02 1.42 0.04 5.3 2.42 6.4 0.2 14 1.0 A
399 75 5.6 53.4 1.1 18.8 0.08 0.02 1.46 0.04 7.3 1.82 5.1 0.2 18 0.9 A
396 30 2.9 53.4 1.6 18.0 0.11 0.03 1.38 0.06 17.1 0.73 2.3 0.2 44 1.0 B
395 200 7.5 53.4 3.0 18.9 0.21 0.09 1.46 0.09 2.7 4.89 8.5 0.1 15 1.3 A

59
0 100 200 300 400 500 600 7000
2
4
6
8
10
12
14
16
18
Rat
e C
onst
ant,
10-1
5 cm
3 mol
ecul
e-1 s
-1
P re s s u re , to rr
Figure 11: Plot of first order rate constant vs. pressure at 399 K

60
Table 4: Rate constant measurements for O + SO2 + Ar at 581 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
581 30 1.9 52.0 1.1 15.7 0.09 0.04 1.36 0.05 24.7 0.50 3.8 0.3 87 1.3 B
581 225 5.7 118.6 3.6 32.4 0.13 0.06 1.22 0.06 3.0 3.74 13.6 0.2 14 1.6 A
581 101 3.2 118.6 2.0 32.4 0.07 0.03 1.05 0.04 5.7 1.68 8.0 0.1 29 0.6 A
582 654 14 126.0 3.6 30.6 0.18 0.03 1.07 0.03 0.9 10.86 25.6 0.7 6 4.2 A
582 654 14 46.4 1.3 11.3 0.11 0.03 1.07 0.03 0.9 10.86 26.1 0.5 4 3.1 A
581 520 12 126.0 3.0 30.3 0.10 0.03 1.07 0.03 1.1 8.65 23.7 0.4 4 2.3 A
580 363 12 62.2 1.9 15.1 0.13 0.03 1.07 0.03 1.6 6.05 18.8 0.4 6 2.1 A

61
0 100 200 300 400 500 600 7000
2
4
6
8
10
12
14
16
18
20
22
24
26
28
Rat
e C
onst
ant,
10-1
5 cm
3 mol
ecul
e-1 s
-1
P re s s u re , to rr
Figure 12: Plot of first order rate constant vs. pressure at 581 K

62
Table 5: Rate constant measurements for O + SO2 + Ar at 699 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
696 255 8.9 133.4 3.4 29.4 0.11 0.02 1.04 0.03 2.7 3.54 15.90 0.50 3 2.4 A
696 255 9.1 51.38 1.3 9.0 0.11 0.02 0.82 0.03 2.1 3.54 14.60 0.90 7 3.4 A
696 75 3.9 133.4 3.3 19.5 0.11 0.01 0.68 0.02 5.9 1.04 6.00 0.20 41 0.7 A
692 655 8.8 128.5 5.1 32.8 0.18 0.07 1.22 0.08 1.2 9.14 25.80 1.00 8 7.1 A
700 518 9.2 128.5 4.9 30.8 0.17 0.08 1.15 0.08 1.5 7.15 24.50 0.90 -1 6.5 A
699 601 8 98.8 3.6 23.3 0.16 0.07 1.12 0.07 1.2 8.31 25.10 0.50 14 3.1 A
699 401 6.1 98.8 3.1 23.9 0.14 0.05 1.15 0.05 1.9 5.54 20.10 0.10 10 1.0 A
699 175 4.7 98.8 2.9 22.7 0.13 0.04 1.09 0.04 4.1 2.42 11.80 0.10 15 0.9 A
699 30 1.6 98.8 1.2 18.8 0.05 0.01 0.89 0.04 19.4 0.41 2.30 0.30 108 0.9 B

63
0 1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 00
5
1 0
1 5
2 0
2 5
3 0
Rat
e C
onst
ant,
10-1
5 cm
3 mol
ecul
e-1 s
-1
P re s s u re , to rr
Figure 13: Plot of first order rate constant vs. pressure at 699 K

64
Table 6: Rate constant measurements for O + SO2 + Ar at 842 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
844 403 4 60 1.4 11.7 0.12 0.03 1.01 0.03 2.0 4.61 19.3 0.7 21 3.7 A
844 253 7.5 69.2 1.4 9.7 0.10 0.05 0.71 0.05 2.2 2.90 15.4 0.7 13 2.9 A
843 50 1.5 33.6 0.2 5.4 0.05 0.01 0.83 0.02 13.1 0.57 3.8 0.4 94 0.9 B
841 561 5 25 0.9 5.2 0.18 0.05 1.09 0.05 1.5 6.44 24.3 1.2 15 8.7 A
842 125 3.7 25 0.6 5.2 0.12 0.04 1.08 0.04 6.8 1.43 8.2 0.3 35 1.8 A
842 200 6 109 3.5 20.7 0.16 0.06 0.98 0.06 3.9 2.29 13.4 0.7 14 3.7 A
841 25 1.1 109 2.3 12.0 0.11 0.01 0.56 0.02 17.6 0.29 2.7 0.9 185 2.6 B
843 660 7.4 91 2.1 18.3 0.11 0.04 1.04 0.04 1.2 7.56 28 0.7 14 4.0 A
842 25 1.1 61 0.8 9.2 0.07 0.01 0.78 0.03 24.6 0.29 3.1 1.3 188 3.3 B

65
0 1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 00
5
1 0
1 5
2 0
2 5
3 0
Rat
e C
onst
ant,
10-1
5 cm
3 mol
ecul
e-1 s
-1
P re s s u re , to rr
Figure 14: Plot of first order rate constant vs. pressure at 842 K

66
Table 7: Rate constant measurements for O + SO2 + Ar at 1040 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
1031 200 1.4 123.5 1.1 4.6 0.052 0.002 0.22 0.00 1.0 1.87 31.6 0.9 106 1.1 E
1032 200 1.3 133.4 1.5 6.1 0.066 0.002 0.26 0.00 1.3 1.87 30.4 2.2 98 3.7 D
1044 200 2 133.4 1.7 8.9 0.073 0.004 0.39 0.01 1.9 1.85 10.9 1.2 108 2.4 D
1045 200 1.1 123.5 0.2 3.6 0.011 0.002 0.17 0.00 0.8 1.85 9.9 1.4 92 1.6 D
1045 101 0.8 123.5 0.6 5.7 0.026 0.003 0.27 0.00 2.6 0.93 8.1 4.2 134 7.1 D
1031 102 0.8 31.6 0.1 0.8 0.020 0.001 0.15 0.00 1.4 0.96 10.2 1.1 127 1.0 D
1046 198 2.3 59.3 1.5 6.9 0.220 0.015 0.82 0.02 4.1 1.83 9.1 0.9 117 3.6 D
1046 198 2.4 197.6 4.9 27.3 0.220 0.015 0.82 0.02 4.1 1.83 9.1 0.3 115 1.5 A
1043 201 1.2 123.5 1.5 17.7 0.071 0.008 0.85 0.01 4.1 1.86 9.3 0.2 91 0.7 A
1043 201 1.2 31.6 0.4 4.5 0.071 0.008 0.85 0.01 4.1 1.86 9.8 0.2 89 0.7 C
1046 99 1.2 128.5 2.7 20.4 0.044 0.034 0.95 0.04 9.4 0.91 6.3 0.8 130 3.0 C
1047 499 3 59.3 1.5 17.9 0.140 0.085 1.88 0.09 3.7 4.60 14.9 0.4 96 3.1 C
1047 50 0.6 98.8 1.5 7.7 0.088 0.017 0.46 0.02 9.0 0.46 3.8 0.5 182 1.2 C

67
Table 7 continued: Rate constant measurements for O + SO2 + Ar at 1040 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
1048 300 3.6 98.8 2.3 17.9 0.130 0.100 1.15 0.10 3.8 2.77 11.9 0.5 117 2.7 A
1047 601 3.1 128.5 9.2 29.5 0.250 0.069 1.41 0.07 2.3 5.55 18.3 0.4 82 3.3 A
1048 25 0.3 128.5 0.5 18.4 0.074 0.007 1.12 0.03 44.0 0.23 3.1 0.7 307 2.9 B
1047 351 3.6 108.5 1.6 19.6 0.082 0.064 1.09 0.08 3.0 3.24 12.6 0.4 105 2.4 A
1046 351 1.8 98.8 2.5 29.3 0.150 0.040 1.86 0.04 5.2 3.24 13.4 1.3 86 10.9 A
1048 650 3.6 187.7 2.8 28.7 0.087 0.037 0.91 0.04 1.4 5.99 21.8 0.6 69 2.8 A
1049 550 4 187.7 2.9 28.7 0.088 0.037 0.89 0.04 1.6 5.06 20.0 0.3 64 1.6 A
1049 425 4.4 187.7 2.7 34.8 0.082 0.041 1.12 0.04 2.6 3.91 17.2 0.3 72 1.8 A
1049 251 3.1 138.3 2.4 22.2 0.099 0.029 0.96 0.03 3.8 2.31 12.1 1.1 79 3.7 A
1048 151 2.8 98.8 1.4 12.2 0.080 0.026 0.73 0.03 4.7 1.39 9.3 1.8 96 4.7 A
1047 75 1.8 88.9 1.5 9.8 0.099 0.017 0.65 0.02 8.5 0.69 8.0 1.1 123 2.8 C
1047 30 0.55 88.9 0.4 5.2 0.028 0.006 0.34 0.01 11.1 0.28 3.1 1.0 242 2.1 B
1047 30 0.56 79.1 0.3 4.7 0.025 0.008 0.35 0.01 11.4 0.28 3.2 2.0 247 3.5 B
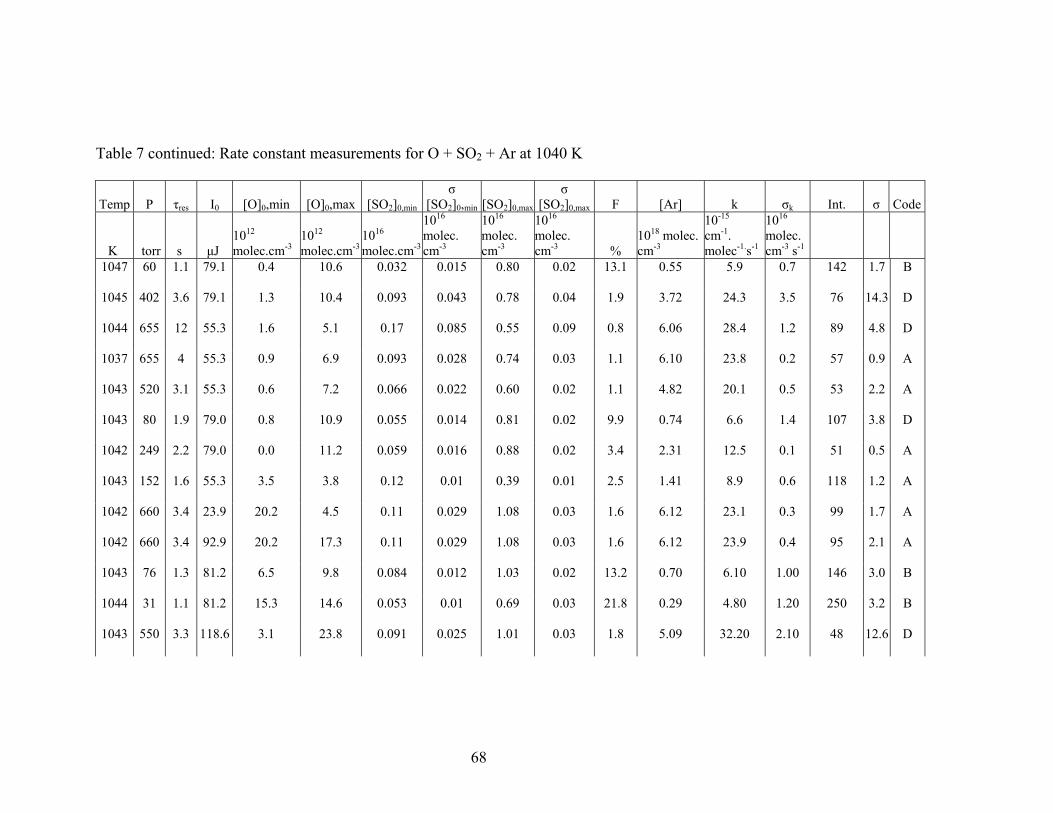
68
Table 7 continued: Rate constant measurements for O + SO2 + Ar at 1040 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
1047 60 1.1 79.1 0.4 10.6 0.032 0.015 0.80 0.02 13.1 0.55 5.9 0.7 142 1.7 B
1045 402 3.6 79.1 1.3 10.4 0.093 0.043 0.78 0.04 1.9 3.72 24.3 3.5 76 14.3 D
1044 655 12 55.3 1.6 5.1 0.17 0.085 0.55 0.09 0.8 6.06 28.4 1.2 89 4.8 D
1037 655 4 55.3 0.9 6.9 0.093 0.028 0.74 0.03 1.1 6.10 23.8 0.2 57 0.9 A
1043 520 3.1 55.3 0.6 7.2 0.066 0.022 0.60 0.02 1.1 4.82 20.1 0.5 53 2.2 A
1043 80 1.9 79.0 0.8 10.9 0.055 0.014 0.81 0.02 9.9 0.74 6.6 1.4 107 3.8 D
1042 249 2.2 79.0 0.0 11.2 0.059 0.016 0.88 0.02 3.4 2.31 12.5 0.1 51 0.5 A
1043 152 1.6 55.3 3.5 3.8 0.12 0.01 0.39 0.01 2.5 1.41 8.9 0.6 118 1.2 A
1042 660 3.4 23.9 20.2 4.5 0.11 0.029 1.08 0.03 1.6 6.12 23.1 0.3 99 1.7 A
1042 660 3.4 92.9 20.2 17.3 0.11 0.029 1.08 0.03 1.6 6.12 23.9 0.4 95 2.1 A
1043 76 1.3 81.2 6.5 9.8 0.084 0.012 1.03 0.02 13.2 0.70 6.10 1.00 146 3.0 B
1044 31 1.1 81.2 15.3 14.6 0.053 0.01 0.69 0.03 21.8 0.29 4.80 1.20 250 3.2 B
1043 550 3.3 118.6 3.1 23.8 0.091 0.025 1.01 0.03 1.8 5.09 32.20 2.10 48 12.6 D

69
Table 7 continued: Rate constant measurements for O + SO2 + Ar at 1040 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
1045 450 3.6 118.6 16.3 20.9 0.15 0.027 1.15 0.03 2.5 4.16 21.00 0.80 68 5.5 C/D
1049 433 2.58 62.0 2.0 13.3 0.19 0.021 1.31 0.02 3.0 3.99 41.6 1.5 166 9.8 D/E
1048 656 3.9 62.0 1.8 17.9 0.17 0.031 1.80 0.03 2.7 6.05 39.6 1.1 131 7.4 D/E
1050 200 1.54 62.0 0.5 14.6 0.045 0.012 1.45 0.02 7.1 1.84 26.5 1.3 110 5.4 D
1049 646 4.75 48.0 1.1 11.3 0.13 0.015 1.44 0.02 2.2 5.95 33.1 5.0 116 20.7 D
1050 330 4.65 64.0 1.4 13.2 0.13 0.017 1.25 0.02 3.7 3.04 31.1 1.6 94 7.0 D
1049 649 3.85 74.0 1.4 24.1 0.11 0.038 2.06 0.04 3.1 5.98 23.1 0.6 90 4.3 A
1049 313 3.7 68.0 1.5 16.0 0.13 0.036 1.45 0.04 4.6 2.88 17.9 1.4 105 9.2 C
1048 104 1.82 71.0 0.9 13.0 0.075 0.01 1.11 0.02 10.5 0.96 20.3 1.3 110 4.1 D
1048 428 6.1 61.0 1.0 9.4 0.092 0.025 0.93 0.04 2.1 3.95 17.0 0.9 114 3.7 A
1048 203 5.3 49.0 1.0 5.8 0.12 0.033 0.70 0.04 3.4 1.87 13.3 0.9 124 2.9 A
1048 210 3.75 70.0 1.1 14.3 0.089 0.041 1.24 0.05 5.8 1.94 17.7 0.8 90 4.3 E
1047 208 1.23 70.0 2.9 25.9 0.24 0.017 2.38 0.02 11.2 1.92 11.0 0.6 78 4.2 A

70
Table 7 continued: Rate constant measurements for O + SO2 + Ar at 1040 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
1047 109 3.83 98.8 1.9 14.8 0.11 0.042 0.90 0.05 8.1 1.01 14.4 2.9 116 12.0 E
1047 111 1.39 98.8 0.9 24.4 0.051 0.014 1.52 0.02 13.4 1.02 7.0 0.6 102 3.1 B
1046 52 1.2 98.8 2.0 19.1 0.12 0.013 1.18 0.03 22.2 0.48 7.4 0.8 159 3.2 B
1047 30 0.35 73.0 0.4 8.6 0.035 0.004 0.69 0.02 22.6 0.28 9.4 2.4 261 9.9 B
1046 32 1.11 67.0 1.3 9.6 0.11 0.013 0.86 0.03 26.3 0.30 4.0 3.3 249 10.3 B
1046 47 0.82 67.0 2.9 12.7 0.084 0.01 1.16 0.03 24.2 0.43 7.3 2.8 183 10.7 B
1047 301 3.6 63.2 1.6 12.8 0.15 0.04 1.23 0.04 4.0 2.78 13.0 0.8 92 4.6 C
1047 297 1.76 63.2 2.0 14.7 0.18 0.02 1.43 0.02 7.1 1.82 12.3 0.1 72 0.8 A
1047 547 3.9 56.1 1.9 9.4 0.08 0.022 1.01 0.02 1.8 5.05 40.2 6.8 56 29.5 C
1047 547 3.9 125.0 4.3 36.1 0.08 0.022 1.80 0.03 3.2 5.05 26.1 2.1 77 12.3 C
1047 102 1.43 56.1 0.8 9.6 0.08 0.0083 1.04 0.02 10.0 0.94 8.0 0.6 105 2.1 A
1046 551 3.91 70.2 2.1 23.5 0.17 0.047 2.12 0.05 3.8 5.09 22.1 2.6 113 18.0 C/E
1046 551 3.91 148.2 4.3 49.6 0.17 0.047 2.12 0.05 3.8 5.09 24.3 2.1 84 18.2 C

71
Table 7 continued: Rate constant measurements for O + SO2 + Ar at 1040 K
Temp P τres I0 [O]0,min [O]0,max [SO2]0,min
σ [SO2]0,min [SO2]0,max
σ [SO2]0,max F [Ar] k σk Int. σ Code
K torr s µJ 1012 molec.cm-3
1012 molec.cm-3
1016 molec.cm-3
1016 molec. cm-3
1016 molec. cm-3
1016 molec. cm-3 %
1018 molec.cm-3
10-15
cm-1. molec-1.s-1
1016 molec. cm-3 s-1
1046 33 0.77 71.9 1.2 11.7 0.10 0.0112 0.98 0.03 29.1 0.30 7.2 0.9 241 2.8 B

72
0 1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 00
2
4
6
8
1 0
1 2
1 4
1 6
1 8
2 0
2 2
2 4
2 6
Rat
e C
onst
ant,
10-1
5 cm
3 mol
ecul
e-1 s
-1
P r e s s u r e , t o r r
Figure 15: Plot of first order rate constant vs. pressure at 1040 K

73
0.0 5.0x10-20 1.0x10-19 1.5x10-19 2.0x10-19 2.5x10-19 3.0x10-190.0
5.0x1013
1.0x1014
1.5x1014
2.0x1014
2.5x1014
3.0x1014
3.5x1014
4.0x1014
1/R
ate
cons
tant
, cm
-3 m
olec
ule
s
1/[Ar]
Figure 16: Lindemann plot at 289 K
0.0 2.0x10-19 4.0x10-19 6.0x10-19 8.0x10-190.0
2.0x1013
4.0x1013
6.0x1013
8.0x1013
1.0x1014
1.2x1014
1.4x1014
1.6x1014
1.8x1014
2.0x1014
1/R
ate
cons
tant
, cm
-3 m
olec
ule
s
1/[Ar]
Figure 17: Lindemann plot at 399 K

74
0.0 1.0x10-192.0x10-193.0x10-194.0x10-195.0x10-196.0x10-197.0x10-190.0
2.0x1013
4.0x1013
6.0x1013
8.0x1013
1.0x1014
1.2x1014
1.4x1014
1/R
ate
cons
tant
, cm
-3 m
olec
ule
s
1/[Ar]
Figure 18: Lindemann plot at 580 K
0.00 2.50x10-19 5.00x10-19 7.50x10-19 1.00x10-18 1.25x10-180.0
2.0x1013
4.0x1013
6.0x1013
8.0x1013
1.0x1014
1.2x1014
1.4x1014
1.6x1014
1.8x1014
1/R
ate
cons
tant
, cm
-3 m
olec
ule
s
1 /[Ar]
Figure 19: Lindemann plot at 699 K

75
0.0 2.0x10-19 4.0x10-19 6.0x10-19 8.0x10-190.0
2.0x1013
4.0x1013
6.0x1013
8.0x1013
1.0x1014
1.2x1014
1/R
ate
cons
tant
, cm
-3 m
olec
ule
s
1 /[Ar]
Figure 20: Lindemann plot at 841 K
0.0 5.0x10-19 1.0x10-18 1.5x10-180.0
2.0x1013
4.0x1013
6.0x1013
8.0x1013
1.0x1014
1.2x1014
1.4x1014
1.6x1014
1/R
ate
cons
tant
, cm
-3 m
olec
ule
s
1/[Ar]
Figure 21: Lindemann plot at 1040 K

76
REFERENCES
(1) Bates, T. S.; Lamb, B. K.; Guenther, A.; Dignon, J.; Stoiber, R. E. Journal
of Atmospheric Chemistry 1992, 14, 315.
(2) Cullis, C. F.; Hirschler, M. M. Atmospheric Environment 1980, 14, 1263.
(3) Moller, D. Atmospheric Environment 1984, 18, 19.
(4) Spiro, P. A.; Jacob, D. J.; Logan, J. A. Journal of Geophysical Research
1992, 97, 6023.
(5) Chen, A. T.; Malte, P. C.; Thornton, M. M. 20th Symposium
(International) on Combustion 1984, 769.
(6) Corley, T. L.; Wendt, J. O. L. Combustion and Flame 1984, 58, 141.
(7) Nimmo, W.; Hampartsoumian, E.; Hughes, K. J.; Tomlin, A. S.
Proceedings of the Combustion Institute, 1998.
(8) Nimmo, W.; Hampartsoumian, E.; Gibbs, B. M. Fuel 2001, 80, 887.
(9) Pfefferle, L. D.; Churchill, S. W. Industrial Engineering and Chemistry
Research 1989, 28, 1004.
(10) Tseregounis, S. I.; Smith, O. I. Combustion and Science Technology 1983,
30, 231.
(11) Wendt, J. O. L. Combustion and Flame 1975, 25, 355.
(12) Wendt, J. O. L.; Morcomb, J. T.; Corley, T. L. 17th Symposium
(International) on Combustion., 1979.

77
(13) Wendt, J. O. L.; Wottan, E. C.; Corley, T. L. Combustion and Flame 1983,
49, 261.
(14) Rasmussen, R. A. Tellus 1974, 26, 254.
(15) Hitchcock, D. R. Enviromnetal Biogeochemistry; Nriagu, J. O., Ed.; Ann
Arbor Science, 1976; Vol. 1; pp 351.
(16) Hallberg, R. O.; Bagandr, L. E.; Engvall, A.-G. Enviromental
Biogeochemistry; Nriagu, J. O., Ed.; Ann Arbor Science: Ann Arbor,
1976; Vol. 1; pp 351.
(17) Maynard, D. G.; Stewart, J. W. B.; Bettamy, J. R. Biogeochemistry 1986,
1, 97.
(18) Lovelock, J. E.; Maggs, R. J.; Rasmussen, R. A. Nature 1972, 237, 452.
(19) Wilson, L. G.; Bressan, R. A.; Filner, P. Plant Physiology 1978, 61, 184.
(20) Lamb, B.; Westberg, H.; Allwine, G.; Bamesberger, L.; Guenther, A.
Journal of Atmospheric Chemistry 1987, 5, 469.
(21) Rennenberg, H. Biogenic sulfur in the environment. In ACS symposium
series 393; Saltzman, E. S., Cooper, W. J., Eds. Washington, D.C, 1988.
(22) Delmas, R.; Baudet, J.; Servant, J.; Baziard, Y. Journal of Geophysical
Research 1980, 85.
(23) Delmas, R.; Servant, J. Tellus 1983, 35B, 110.
(24) Aneja, V. P.; Overton, J. H., Jr; Cupitt, L. T.; Durham, J. L.; Wilson, W.
E. Nature 1979, 282, 493.

78
(25) Adams, D. F.; Farwell, S. O. “Biogenic Emissions in the SURE and
Extended SURE regions,” Electric Power Research Institute, 1980.
(26) Baily, S. D.; Bazinet, M. L.; Driscoll, J. L.; McCarthy, A. J. Journal of
Food Science 1961, 26, 163.
(27) Westberg, H.; Lamb, B. “Environmental Impact on Natural Emissions,”
Air Pollution Control Association, 1984.
(28) Haines, B.; Black, M.; Fail, F. J.; McHargue, L. A.; Howell, G. Effects of
acidic deposition on forests, wetlands, and agricultural ecosystems.;
Hutichson, T. C., Meema, K. M., Eds.; Springer-Verlag, 1988; pp 599.
(29) Haines, B.; Black, M.; Bayer, C. “Biogenic sulfur in the environment”;
ACS Symposium Series 393, 1988, Washington, D.D.
(30) MacTaggart, D. L.; Adams, D. F.; Farwell, S. O. Journal of Atmospheric
Chemistry 1987, 5, 417.
(31) Goldan, P. D.; Kuster, W. C.; Albritton, D. L.; Fehsenfeld, F. C. Journal
of Atmospheric Chemistry 1987, 5, 439.
(32) de Mello, W. Z.; Cooper, D. J.; Cooper, W. J.; Saltzman, E. S.; Zika, R.
G.; Savoie, D. L.; Prospero, J. M. Atmospheric Environment 1987, 21,
987.
(33) Dacey, J. W. H.; King, G. M.; Wakeham, S. G. Nature 1987, 330, 643645.
(34) Carroll, M. A.; Heidt, L. E.; Cicerone, R. J.; Prinn, R. G. Journal of
Atmospheric Chemistry 1986, 4, 375.

79
(35) Cooper, D. J.; de Mello, W. Z.; Cooper, W. J.; Zika, R. G.; Saltzman, E.
S.; Prospero, J. M.; Savoie, D. L. Atmospheric Environment 1987, 21, 7.
(36) Cooper, W. J.; Cooper, D. J.; Saltzman, E. S.; de Mello, W. Z.; Savoie, D.
L.; Zika, R. G.; Prospero, J. M. Atmospheric Environment 1987, 21, 1491.
(37) Jorgensen, B. B.; Okholm-Hansen, B. Atmospheric Environment 1985, 19,
1737.
(38) Steudler, P. A.; Peterson, B. J. Atmospheric Environment 1985, 19, 1411.
(39) Melillo, J. M.; Steudler, P. A. Journal of Atmospheric Chemistry 1989, 9,
411.
(40) Andreae, M. O.; Andreae, T. W. Journal of Geophysical Research 1988,
93, 1487.
(41) Staubes, R.; Georgii, H. W.; Ockelmann, G. Tellus 1989, 41B, 305.
(42) Andreae, M. O. The ocean as a source of atmospheric sulfur compounds.
In The role of air-sea exchange on geochemical cycling; Baut-Menard, P.,
Ed.; Dordrect: Reidel, 1986; pp 331.
(43) Nguyen, B. C.; Bonsang, B.; Gaudry, A. Journal of Geophysical Research
1983, 88, 903.
(44) Haas, P. Biochemistry Journal 1935, 29, 1297.
(45) Ackman, R. G.; Tocher, C. S.; McLachlan, J. Journal of the Fisheries
Research Board of Canada 1966, 23, 357.
(46) Andreae, M. O.; Raemdonck, H. Science 1983, 221, 744.

80
(47) Galloway, J. N. The Biogeochemical cycling of sulfur and nitrogen in the
remote atmosphere; Reidel: Dordrecht, 1985.
(48) Bates, T. S.; Cline, J. D.; Gammon, R. H.; Kelly-Hanson, S. R. Journal of
Geophysical Research 1987, 92, 2930.
(49) Ferek, R. J.; Andreae, M. O. Geophysical Research Letters. 1983, 10, 393.
(50) Ferek, R. J.; Andreae, M. O. Nature 1984, 307, 148.
(51) Turner, S. M.; Liss, P. S. Journal of Atmospheric Chemistry 1985, 2, 223.
(52) Johnsson, J. E.; Harrison, H. Journal of Geophysical Research 1986, 91,
77.
(53) Rasmussen, R. A.; Khalil, M. A. K.; Hoyt, S. D. Atmospheric
Environment 1982, 16, 1591.
(54) Matrai, P. A. Marine Chemistry 1989, 26, 227.
(55) Bates, T. S. unpublished data as cited in reference in 1.
(56) Cadle, R. D. Journal of Geophysical Research 1975, 80, 1650.
(57) Pinto, J. P.; Turco, R. P.; Toon, O. B. Journal of Geophysical Research
1989, 94, 165.
(58) Stoiber, R. E.; Williams, S. N.; Heubert, B. Journal of Volcanology and
Geothermal Research 1987, 33, 1.
(59) Khalil, M. A. K.; Rasmussen, R. A. Atmospheric Environment 1984, 18,
1805.

81
(60) Belviso, S.; Nguyen, B. C.; Allard, P. Geophysical Research Letters.
1986, 13, 133.
(61) Okita, T.; Shimozuro, D. Bulletin of the Volcanological Society of Japan
1975, 19, 151.
(62) Stoiber, R. E.; Jepson, A. Science 1973, 182, 577.
(63) Stoiber, R. E.; Malone, G., B. Transactions of the American Geophysical
Union 1975, 56, 461.
(64) Haulet, R.; Zettwoog, P.; Sabroux, J. C. Nature 1977, 268, 715.
(65) Zettwoog, P.; Haulet, R. Atmospheric Environment 1978, 12, 795.
(66) Johansson, T. B.; van Grieken, R. E.; Winchester, J. W. Journal of
Recherches Atmospheriques. 1974, 8, 761.
(67) Garrels, R. M.; Mackenzie, F. T. Evolution of sedimentary rocks; Norton:
New York, 1971.
(68) Hileman, B. “Biomass burning: Environment hurt more than thought.” In
Chemical and Engineering News, 1990; pp 4.
(69) Dignon, J.; Penner, J. E. 1990.
(70) Robinson, E.; Robbins, R. C. In Air pollution control; Strauss, W., Ed.;
John Wiley: New York, 1972; Vol. part 2; pp 1.
(71) Coal Mining Yearbook, 2001 to2002.
(72) Deurbrook, A. W. “Sulfur reduction potentials of coals of the United
States,” US bureau of Mines, 1972.

82
(73) Vernon, J. L.; Jones, T. IEA Coal Research, 1993.
(74) Gluskoter, H. J.; Simon, J. A., III “State Geological survey”, 1968.
(75) Attar, A.; Corcoran, W. H. Industrial Engineering and Chemistry Product
Research Development 1977, 16, 168.
(76) Garcia-Labiano, F.; Hampartsoumian, E.; Williams, A. Fuel 1995, 74,
1072.
(77) Couch, G. R. IEA Coal Research, 1991.
(78) Galos, K. A.; Smakowski, T. S.; Szlugaj, J. Applied Energy 2003, 75, 257.
(79) Anthony, E. J.; Iribarne, A. P.; Iribarne, J. V. Energy Resources
Technology.
(80) Anthony, E. J.; Talbot, R. E.; Jia, L. Energy and Fuels 2000, 56, 75.
(81) Anthony, E. J.; Iribarne, A. P.; Iribarne, J. V.; Talbot, R. E.; Jia, L. Fuel
2001, 80, 1009.
(82) Anthony, E. J.; Talbot, R. E.; Jia, L.; Granatstein, D. L. Energy and Fuels
2000, 14, 1021.
(83) Dam-Johansen, K.; Amand, L.-E.; Leckner, B. Fuel 1993, 72, 565.
(84) Johnsson, J. E. Fuel 1994, 73, 1398.
(85) Smith, O. I.; Wang, S.-N.; Tseregounis, S. I.; Westbrook, C. K.
Combustion Science and Technology 1983, 30, 241.
(86) Zachariah, M. F.; Smith, O. I. Combustion and Flame 1987, 69, 125.
(87) Cullis, C. F.; Mulcahy, M. F. R. Combustion and Flame 1972, 18, 225.

83
(88) Atkinson, R.; Baulch, D.; Cox, R. A.; Hampson, R. F.; Kerr, J. A.; Troe, J.
Journal of Physical Chemistry Reference Data 1992, 21, 1125.
(89) Troe, J. Annual Review of Physical Chemistry 1978, 29, 223.
(90) Astholz, D. C.; Glanzer, K.; Troe, J. Journal of Chemical Physics 1979,
70, 2409.
(91) Bufalini, M. Environmental Science and Technology 1971, 5, 685.
(92) Johnsson, J. E.; Glarborg, P. NATO Science Series. Series C:
Mathematical and Physical Sciences 2000, 263.
(93) Zemansky, V. M.; Lyon, R. K.; Evans, A. B.; Pont, J. N.; Seeker, W. R.;
Schmidt, C. E. “SO2 control”; EPRI/EPE/DOE, 1993, Boston.
(94) Lyon, R. K. EPRI/EPA. Joint symposium on stationary combustion NOx
control, 1989, San Francisco.
(95) Kaufman, F. Proceedings of the Royal Society. Series A 1958, A247, 123.
(96) Halstead, C. J.; Thrush, B. A. Proceedings of the Royal Society. Series A
1966, A295, 363.
(97) Allen, E. R.; Cadle, R. D. Photochemistry and Photobiology 1965, 4, 979.
(98) Jaffe, S.; Klein, F. S. Transactions of the Faraday Society. 1966, 62, 2150.
(99) Mulcahy, M. F. R.; Steven, J. R.; Ward, J. C. Journal of Physical
Chemistry 1967, 71, 2124.
(100) Atkinson, R.; Pitts, J. N., Jr. Chemical Physics Letters 1974, 27, 467.

84
(101) Westenberg, A. A.; deHaas, N. Journal of Chemical Physics 1975, 63,
5411.
(102) Atkinson, R.; Pitts, J. N., Jr. International Journal of Chemical Kinetics
1978, 10, 1081.
(103) Davis, D. D. Canadian Journal of Chemistry 1974, 52, 1405.
(104) Fenimore, C. P.; Jones, G. W. Journal of Physical Chemistry 1965, 69,
3593.
(105) Webster, P.; Walsh, A. D. 10th Symposium (Int) on Combustion, 1965,
Pittsburgh.
(106) Stiganov, A. R.; Sventitskii, S. N. Tables of spectral lines of neutral and
ionized atoms.; Plenum Data Corporation: New York, 1968.
(107) Shi, Y.; Marshall, P. Journal of Physical Chemistry 1991, 95, 1654.
(108) Golomb, D.; Watanabe, K.; Marmo, F. F. Journal of Chemical Physics
1962, 36.
(109) Manatt, S. L.; Lane, A. L. Journal of Quantitative Spectroscopic and
Radiative Transfer 1993, 50, 267.
(110) Phillips, L. F. Journal of Physical Chemistry 1981, 85, 3994.
(111) Wu, C. Y. R.; Judge, D. L. Geophysical Research Letters. 1981, 8, 769.
(112) Prahlad, V.; Vijay, K. Journal of Quantitative Spectroscopic and
Radiative Transfer 1997, 57, 719.

85
(113) Fockenberg, C.; Preses, J. M. Journal of Physical Chemistry A 2002, 106,
2924.
(114) Vattulainen, J.; Wallenius, L.; Stenberg, J.; Hernberg, R.; Linna, V.
Applied Spectroscopy 1997, 51, 1311.
(115) Garland, N. L. Chemical Physics Letters 1998, 290, 385.
(116) Chase, M., W, Jr. Journal of Physical and Chemistry Reference Data
1998, Monograph No. 9.
(117) Atkins, P. Physical Chemistry, Sixth Edition.; W.H Freeman and
Company, 1998.
(118) Levine, I. N. Physical Chemistry, Fourth Edition.; McGraw-Hill Inc.,
1995.
(119) Troe, J. Journal of Physical Chemistry 1979, 83, 114.
(120) Troe, J. Journal of Physical Chemistry 1979, 83, 114.
(121) Quack, M.; Troe, J. Gas Kinetics and Energy Transfer 1977, 2, 175.
(121) Mulcahy, M.F.R., Steven, J.R., Ward, J.C., and Williams, D.J. 10th
Symposium (International) on Combustion, 1968