investigation of structural, electronic and magnetic...
TRANSCRIPT

Investigation of Structural, Electronic and Magnetic
Properties of the Half-Metallic Ferromagnetic
Materials
By
MUHAMMAD ATIF SATTAR
A DISSERTATION
Submitted in the fulfillment of the requirements
for the degree
DOCTOR OF PHILOSOPHY
IN
PHYSICS
Department of Physics
The Islamia University of Bahawalpur Pakistan
2018


In the name of ALLAH, most gracious most merciful!
The Prayer of the believers!
Praise be to ALLAH,
The Cherished and sustainer of the worlds;
Most Gracious, most Merciful;
Thee do we worship,
And think and we seek,
Show us the straight way,
The way of those one whom
Thou haste bestowed the grace,
Those whose (portion)
Is not wrath,
And who go not astray.
(Surah Fatiha)

A dissertation entitled Investigation of Structural,
Electronic and Magnetic Properties of the Half-
Metallic Ferromagnetic Materials
In partial fulfillment of the requirements
for the degree of
DOCTOR OF PHILOSOPHY
IN
PHYSICS
By
MUHAMMAD ATIF SATTAR
Submitted to
Department of Physics
The Islamia University of Bahawalpur Pakistan
2018

i
Preface
By An initial look at the title of this dissertation “Investigation of Structural,
Electronic and Magnetic Properties of the Half-Metallic Ferromagnetic Materials” may
possibly emerge two thoughts in our minds. The very first phrase is about the physical
properties like structural arrangement of the atoms, different states of electrons in the unit
cell of the compound and origin of magnetism. The second term deals with the half-metallic
(HM) ferromagnetic (FM) materials or half-metallic ferromagnets (HMFs). What are the
HMFs and what makes them so special and important in terms of research and applications.
In this study, we explore the issue like this one by one to learn how to discover brand new
HM materials as well as how we can really utilize these types of HM materials into the
spintronic applications.
More recently scientist making spintronic devices with brand new Ferro-magnetic
(FM) materials. These types of materials are referred as half-metals or HM materials. They
display multiple properties of metals and semiconductors and can be viewed as hybrids in
between metals and semiconductors. HMFs are the materials which have one spin channel
conductive, showing the metallic behavior and the other spin channel is showing a semi
conducting gap at the Fermi level (EF) or showing insulating behavior.
This unique feature encourages materials scientists to produce ideal devices for
spintronic. For the better spintronic device characteristic, the spin of the material should be
100% aligned. In HM materials, the spin of the each consisting element is totally lined up.
For instance, when the spin of the FM electrode is injected into a semiconductor (SCs), the
more alignment of the spin results in a greater injection of the spins.
HMFs are very important prospective materials regarding spintronic. These HM
materials may also be defined through their integer value associated with magnetic
moments according to the Slater–Pauling rule (SPR) which µ𝑡𝑜𝑡 = 𝑁𝑣𝑎𝑙 − 24 for the Full-
Heusler (FH) and µ𝑡𝑜𝑡 = 𝑁𝑣𝑎𝑙 − 18 for the Half-Heusler (HH) materials respectively where
µ𝑡𝑜𝑡 stands out as the total magnetization and 𝑁𝑣𝑎𝑙 is the total number of valance electrons.
It is very essential to produce HMFs, like HM with different elements which are called
hetero structures.

ii
In general, HMFs are formed by combining three elements which make the crystal
structure more complex as compared to the FM iron which contains only one element.
Therefore, In HMFs, there is benefit of 100% spin polarization (SP) but the drawback of
the very complex crystal structure.
It is a problem to determine how to prepare top quality materials with HM features
to ensure that these types of HMFs may be used to create practical spintronic applications.
In this project, not only the new HMFs will be find out but also step by step the basic
physical properties of these HMFs understood and physics behind these properties will be
thoroughly discussed and how to utilize these half-metals into practical spintronic
applications.

iii
CERTIFICATE
It is certified that the research work in this dissertation entitled
“Investigation of Structural, Electronic and Magnetic Properties of the Half-Metallic
Ferromagnetic Materials”
completed by Muhammad Atif Sattar to fulfill the partial requirements for the degree of
Doctor of Philosophy (Ph.D.) in Physics has been conducted under my supervision in the
department of Physics, The Islamia University of Bahawalpur.
Supervisor: Prof. Dr. Sheikh Aftab Ahmad
Department of Physics
The Islamia University of Bahawalpur
Chairman: Dr. Saeed Ahmad Buzdar
(Associate Professor)
Department of Physics
The Islamia University of Bahawalpur

iv
DECELRTAION
I, Muhammad Atif Sattar student of Ph.D. in the subject of physics, hereby declare and
certify that material printed in this dissertation “Investigation of Structural, Electronic
and Magnetic Properties of the Half-Metallic Ferromagnetic Materials” is my individual
research and that it has not been submitted concurrently to any other university or any
research institution for any degree or diploma in Pakistan or abroad.
MUHAMMAD ATIF SATTAR

v
FORWARDING CERTIFICATE
The research entitled “Investigation of Structural, Electronic and Magnetic Properties of
the Half-Metallic Ferromagnetic Materials” is carried out under my supervision and the
dissertation is submitted to The Islamia University of Bahawalpur, Pakistan in fulfillment
of the requirement for the degree of DOCTOR OF PHILOSOPHY (Ph.D.) in physics with
my permission.
Prof. Dr. S. A. Ahmad

vi
RIGHT OF DISSERETATION
All Rights of the dissertation are reserved to the author (researcher). No part of this
research may be reproduced or transmitted in any form or by any means, electronic or
mechanical, including photocopy, recording or any information storage and retrieval
system, without permission in writing from the researcher.
MUHAMMAD ATIF SATTAR

vii
ACKNOWLEDGEMENTS
All praises and thanks to Almighty Allah, the Beneficent and the Merciful, the
Creator of the universe, Who enabled me to complete my research work successfully. I
would like to offer my humblest thanks to His prophet Hazrat Muhammad (peace be upon
him), who is a source of guidance and knowledge for humanity.
I am especially thankful to my kind supervisor Prof. Dr. Sheikh Aftab Ahmad,
department of physics, The Islamia University of Bahawalpur, who provided me all
possible facilities in the laboratory and encouraged me to complete my research work. He
has always been very kind, friendly, easy to reach and helpful.
I am grateful to my mentor, Dr. Altaf Hussain, department of physics, The Islamia
University of Bahawalpur and my foreign supervisor Dr. Claudio Cazorla, School of
Material Science, University of New South Wales (UNSW), Australia, for their valuable,
sincere suggestions, guidance and providing me all the lab facilities to complete my thesis.
I appreciate my beloved brothers and close collaborators, Dr. Fayyaz Hussain, Dr.
Muhammad Rashid, Dr. Imran, Dr. Zafar, Dr. Shabir, Dr. Shakil, whose guidance and
co-operation really helped me in learning and solving different simulation problems
related to DFT investigation.
I have the honor to express my deep sense of indebtedness to ever affectionate my
dear friends, Muhammad Ali Abbas and PhD Scholars, Muhammad Nasir Rasul,
Muhammad Raza Hashmi, Salman Mehmood for their encouragement during my
research period and lastly to my all lab mates because they were always kind to me in every
matter of my research during PhD.
I am thankful to my friends Dr. Tariq , Dr. M. Ali and Dr. Jawad Ahmad for their
valuable guidance, keen interest and encouragement, kind and understanding spirit during
my research work at the UNSW, Australia. I express my special gratitude to all of them for
their endless support and make my stay comfortable.
Finally, I wish to express my nice feelings toward my mother for her affection and
to all family members who remembered me in their prayers and heartened me to continue
higher studies.
MUHAMMAD ATIF SATTAR

viii
DEDICATION
TO
My great parents whose prayers are always with me,
who are the sources of my success in every field of
life and whose substitutes are impossible,
innocent sister
and
Loving brothers.

ix
Abstract
Spintronics or magneto-electronics develop the focus on the fascinating class of
intermetallic Heusler alloys (HA) which is the active field of scientific research because
numerous HA appeared to be half-metallic ferromagnets (HMFs). It is very interesting by
counting the valence electrons of the HA, one can predict their half-metallic (HM)
properties. Half-Heusler (HH) alloys with 1:1:1 composition tend to be appealing prospects
pertaining to spintronic applications due to their structural resemblance towards the binary
semiconductors (SC), spin-polarization is offered 100% at the Fermi level (EF) and possess
higher values of Curie Temperature (TC). Prospective fields of applications and brand-new
properties arise continuously because one can easily control their atomic disorder and
structure interface. Devices depending on multifunctional properties i.e. the combined
magnetic and remarkable transport properties can revolutionize technological applications
as HH materials possess higher importance for the advancement of the spintronic devices.
To make use of the substantial prospective of HH materials, in this dissertation, a
detailed Density Functional Theory (DFT) investigation on the understanding of the basic
structural, electronic and magnetic properties of the HH XYZ family are explored.
Numerous newly HM HH materials like CrTiZ (Z= Si, Ge, Sn, Pb), FeVZ (Z= Si, Ge, Sn),
YMnZ (X= Si, Ge, Sn) along with YCrSb & YMnSb followed by the series of 90 HH XYZ
materials where (X = Li, Na, K, Rb, Cs & Y= V, Nb, Ta & Z = Si, Ge, Sn, S, Se, Te) are
studied by the First-principles calculations. The detailed results for the different series of
the HM HH XYZ materials are described below:
HM properties of new HMFs CrTiZ (where Z = Si, Ge, Sn, Pb) are studied by means
of the first-principles band structure calculations within the framework of DFT. From the
spin-polarized calculations using full-potential linearized augmented plane-wave (FP-
LAPW) method, we found that all these compounds are stable in the FM MgAgAs-type
crystal structure. The lattice parameters of CrTiZ compounds increase with increasing
atomic radius of X atom and ranges from 5.76 Å to 6.38 Å. The calculated electronic
structure of these compounds in MgAgAs-type structure shows that they are HM materials
with an integer magnetic moment of 4 µ𝐵. Densities of states, electronic band structure,
and origin of ferromagnetism have been discussed, and robust HM nature of these
compounds is analyzed which makes them fascinating compounds for spintronic devices.

x
DFT-based ab-initio calculations are utilized to investigate the electronic and
magnetic properties of new series of HH FeVZ (where Z=Si, Ge, Sn) compounds. The C1b-
type structure is considered for these materials in three different atomic configurations,
termed as α, β, and γ phases, to find the most stable geometric structure. The structural
properties of all three phases have been determined and the effect of spin-polarization has
been studied. Our calculated electronic properties suggest that the studied materials under
study are HMFs and stable in the α-phase. The Trans-Blaha modified Becke-Johnson (TB-
mBJ) local spin density approximation functional is employed for a better description of
the HM response of HH FeVZ materials. We have also shown that the HM nature of FeVZ
compounds is robust for a wide range of lattice constant, making these materials suitable
for spintronic applications.
Structural, electronic, and magnetic properties of newly predicted half-Heusler
YCrSb and YMnSb compounds within the ordered MgAgAs C1b-type structure are
investigated by employing first- principal calculations based on DFT. Through the
calculated total energies of three possible atomic placements, we find the most stable
structures regarding YCrSb and YMnSb materials, where Y, Cr (Mn), and Sb atoms occupy
the (0.5, 0.5, 0.5), (0.25, 0.25, 0.25), and (0, 0, 0) positions, respectively. Furthermore,
structural properties are explored for the non-magnetic (NM), FM and anti-ferromagnetic
(AFM) states and it is found that both materials prefer FM states. The electronic band
structure shows that HM HH YCrSb has a direct band gap of 0.78 eV while YMnSb has an
indirect band gap of 0.40 eV in the majority spin channel. Our findings show that YCrSb
and YMnSb materials exhibit HM characteristics at their optimized lattice constants of 6.67
Å and 6.56 Å, respectively. The half-metallicities associated with YCrSb and YMnSb are
found to be robust under large in-plane strains which make them potential contenders for
spintronic applications.
The basic structural stability, electronic, magnetic and thermoelectric properties of the
newly predicted HM YMnZ (Z=Si, Ge, Sn) alloys in HH phase are contemplated with
optimized lattice constants by ab-initio FP-LAPW method using DFT. The MgAgAs (C1b-
type) structure of these HH YMnZ materials in three different atomic arrangements (X-
type1, X- type2, X-type3) have been explored and X-type1 structure is found to be
energetically more favorable for YMnSi and YMnGe whereas YMnSn prefers the X-type2
structure. Moreover, NM, FM and AFM states computed for YMnZ HH materials favor
FM states. The presence of the energy gap in the majority spin bands and density of the

xi
states within the HH YMnZ are indications of potential HMFs. For the lattice constant
range of 6.2 Å to 7.4 Å, the total magnetic moment (µ𝑡𝑜𝑡) remains an integral value of 4.0
µ𝐵 per formula unit and obeys the modified Slater-Pauling rule (SPR). The calculations
reveal that YMnZ displays HM ferromagnetism having the µ𝑡𝑜𝑡 of 4.00 µ𝐵 which primarily
arises from the spin-polarization of d-electrons of Mn atom and partial involvement of p-
electrons of Z-atom. The half-metallicity of HH YMnZ materials might show that they are
ideal for applications in spin polarizers and spin injectors of magnetic nano devices due to
their larger EBG, which mean that they are steady at ambient conditions. The robustness
associated with half-metallicity contrary to the lattice constant is additionally ascertained
for desirable spintronics applications. Thermoelectric properties of the YMnZ materials are
additionally computed over an extensive variety of temperature and it is discovered that
YMnSi demonstrates a higher figure of merit than YMnGe and YMnSn.
Finally, in this dissertation, DFT-based systematically investigation on structural,
electronic, magnetism and vibrational stability of the unexplored 90 half-Heusler (HH)
XYZ materials where (X= Li, Na, K, Rb, Cz; Y= V, Nb, Ta & Z= Si, Ge, Sn, S, Se, Te) in
the C1b structure is carried out by using First-principles calculations. The energetically
most stable structure is determined among the three different atomic arrangement types
(T1, T2, T3) inside the C1b unit cell. The magnetic ground state; FM and AFM are also
checked for these HH XYZ materials. The electronic and magnetic properties are calculated
by using the TB-mBJ functional as it is proven to give the accurate values for the energy
band gaps. Among 90 HH XYZ materials, 28 HH XYZ materials show HM properties at
their respective stable phase (T1 or T3) with FM arrangement and obey the modified SPR.
The 5 NM SC, 2 FM SC, and 21 AFM HH XYZ materials are discovered in our findings.
Furthermore, the Curie temperature (TC) and mixing energy of the vibrational stable 28
HM HH XYZ materials are also calculated. The larger values of the energy band gap (EBG)
and HM gap (EHM) along with magnetic moments (up to 4 µ𝐵) show that these compounds
can be excellent spin-injectors for the spintronic applications.

xii
List of Publications
This dessertation is based on the following publications:
1. Sattar, M. A.; Rashid, M.; Rasool, M. N.; Mahmood, A.; Hashmi, M. R.; Ahmad,
S.; Imran, M.; Hussain, F., Half-metallic ferromagnetism in new half-Heusler
compounds: an ab initio study of CrTiX (X= Si, Ge, Sn, Pb). Journal of
Superconductivity and Novel Magnetism 2016,29 (4), 931-938.
2. Sattar, M. A.; Rashid, M.; Hashmi, M. R.; Rasool, M. N.; Mahmood, A.; Ahmad,
S., Spin-polarized calculations of structural, electronic and magnetic properties of
Half Heusler alloys FeVX (X= Si, Ge, Sn) using Ab-initio method. Materials
Science in Semiconductor Processing 2016,51, 48-54.
3. Sattar, M. A.; Rashid, M.; Hashmi, M. R.; Ahmad, S.; Imran, M.; Hussain, F.,
Theoretical investigations of half-metallic ferromagnetism in new Half–Heusler
YCrSb and YMnSb alloys using first-principle calculations. Chinese Physics B
2016,25 (10), 107402.
4. Sattar, M. A.; Rashid, M.; Hussain, F.; Imran, M.; Hashmi, M. R.; Laref, A.;
Ahmad, S., Physical properties of half-Heusler YMnZ (Z= Si, Ge, Sn) compounds
via ab-initio study. Solid State Communications 2018,278, 10-19.
5. Sattar, M. A.; Ahmad, S.A; Hussain, F.; Claudio, C.; First-principles prediction of
magnetically ordered half-metals above room temperature, Available online 11
April 2019, Journal of Materiomics.
Other Publications
6. Rasool, M. N.; Mehmood, S.; Sattar, M. A.; Khan, M. A.; Hussain, A.,
Investigation of structural, electronic and magnetic properties of 1: 1: 1: 1
stoichiometric quaternary Heusler alloys YCoCrZ (Z= Si, Ge, Ga, Al): An ab-initio
study. Journal of Magnetism and Magnetic Materials 2015,395, 97-108.
7. ur rehman Hashmi, M. R.; Zafar, M.; Shakil, M.; Sattar, A.; Ahmed, S.; Ahmad,
S., First-principles calculation of the structural, electronic, and magnetic properties
of cubic perovskite RbXF3 (X= Mn, V, Co, Fe). Chinese Physics B 2016, 25 (11),
117401.

xiii
8. Imran, M.; Hussain, F.; Rashid, M.; Ullah, H.; Sattar, A.; Iqbal, F.; Ahmad, E.,
Comparison of Electronic and Optical Properties of GaN Monolayer and Bulk
Structure: a First Principle Study. Surface Review and Letters 2016,23 (04),
1650026.
9. Behram, R. B.; Iqbal, M.; Rashid, M.; Sattar, M. A.; Mahmood, A.; Ramay, S. M.,
Ab-initio investigation of AGeO3 (A= Ca, Sr) compounds via Tran–Blaha-
modified Becke–Johnson exchange potential. Chinese Physics B 2017,26 (11),
116103.
10. Hussain, F.; Imran, M.; Sattar, M. A.; Tailoring magnetic characteristics of
phosphorene by the doping of Ce and Ti: A DFT study. Physica E: Low-
dimensional Systems and Nanostructures 2018.
11. Hussain, F.; Imran, M.; Rana, A. M.; Khalil, R. A.; Khera, E. A.; Kiran, S.; Javid,
M. A.; Sattar, M. A.; Ismail, M., An insight into the dopant selection for CeO 2-
based resistive-switching memory system: a DFT and experimental study. Applied
Nanoscience 2018, 1-13.
12. Hussain, F.; Imran, M.; Siddiqa, A.; Khalil, R. M. A.; Rana, A. M.; Sattar, M. A.;
NIAZ, N. A.; ULLAH, H.; AHMAD, N., Ab initio STUDY OF POINT DEFECTS
IN 2D GRAPHENE LAYER. Surface Review and Letters 2018, 1850142.
13. Maryam, A.; Abbas, G.; Rashid, M.; Sattar, A., Directional mechanical and
thermal properties of single-layer black phosphorus by classical molecular
dynamics. Chinese Physics B 2018,27 (1), 017401.
14. Rasul, Nasir; Anum, Asifa; Sattar, Atif; Manzoor, Alina; Hussain, Altaf., DFT
based structural, electronic and optical properties of B(1-x)InxP(x=
0.0,0.25,0.5,0.75,1.0) compounds: PBE-GGA vs mBJ-approaches. Chinese Journal
of Physics 2018, 56 2659.
15. Imran, M.; Hussain, F.; Sattar, M. A., A study of surface diffusion of ternary (Cu-
AgZr) adatoms clusters for applications in thin film formation, Surface and
Interface Analysis, Published online on 15 January 2019
16. Hussain, F.; Imran, M.; Khalil, R. A.; Rana, A. M.; Rasheed, U.; Khera, E. A.;
Mumtaz, F.; Sattar, M. A.; Javid, M. A., Effect of Cu and Al doping in ZrO2 for
RRAM device applications using GGA and GGA+U approach, submitted.
17. Hussain, F.; Imran, M.; Sattar, M. A.; Khalil, R. A.; Rana, A. M., An insight of
Mg doping in ZnO thin films: a comparative experimental and first-principle
investigations, submitted.

xiv
Table of Contents
Title Page No.
Preface………………………………………………………………………………….i
Certificate……………………………………………………………………………..iii
Declaration……………………………………………………………………………iv
Forwarding Certificate………………………………………………………………...v
Rights of Dissertation....………………………………………………………………vi
Acknowledgments……………………………………………………………………vii
Dedications………………………………………………………………………….viii
Abstract……….……………………………………………………………………....ix
List of Publications………………………………………………………………….xii
Table of Contents…………………………………………………………………….xiv
List of Tables…………………………………………………………………………xix
List of Figures……………………………………………………………………….xxii
List of Abbreviations……………………………………………………………….xxix
Chapter 1: Fundamentals……………………………………………………………1
1.1 Heusler Alloys (HA)………………………………………………………1
1.2 Types of Heusler Alloys (HA)…………………………………………….2
1.2.1 Full Heusler (FH) Alloys……………………………………...2
1.2.2 Half Heusler (HH) Alloys……………………………………..4
1.2.3 Quaternary Heusler (QH) Alloys……………………………...4
1.3 Crystal Framework of Heusler Alloys (HA)………………………………4
1.4 Structural Arrangement……………………………………………………8
1.4.1 Order-Disorder Phenomena in Half-Heusler (HH)
Materials……………………………………………………....8
1.5 Heusler Alloys (HA) and Magnetism……………………………………..9
1.5.1 Half-Metallic Ferromagnets (HMFs)…………………..........12

xv
1.5.2 The Slater-Pauling Rule (SPR)………………………………12
1.6 Applications of Heusler Alloys (HA) for Spintronic Devices…………...14
1.6.1 The Effect of Giant Magnetoresistance (GMR)
and Tunneling Magnetoresistance (TMR)…………..............16
1.6.2 Spin Polarization……………………………………..............18
1.6.3 Current-perpendicular-to-plane (cpp)
Giant-magnetoresistance (GMR)…………………………….19
1.6.4 Perpendicular Magnetic Anisotropy………………….………20
1.6.5 Spin Injection………………………………………………....22
1.6.6 Shape-Memory Compounds………………………………….22
1.6.7 Superconductors……………………………………………...23
1.6.8 Thermoelectric Compounds………………………………….24
1.6.9 Topological Insulators………………………………………..25
1.7 Heusler Moves Nano……………………………………………………..26
1.8 Dissertation Scheme……………………………………………………...26
Chapter 2: Literature Review……………………………………………………...29
2.1 Motivation for the work………………………………………………….32
Chapter 3: Basics of Density Functional Theory (DFT)………………………….34
3.1 Computational Material Science…………………………………………34
3.2 Many Particle Problem in Solids…………………………………...…….35
3.3 The Born Oppenheimer Approximation………………………………….35
3.4 Why Density Functional Theory (DFT) Needed?......................................37
3.5 Density Functional Theory (DFT)………………………………………..37
3.6 Hohenberg and Kohn (at the heart of DFT)……………………………...38
3.6.1 Theorem I…………………………………………………….38
3.6.2 Theorem II……………………………………………………38
3.7 The Kohn-Sham Scheme…………………………………………………39
3.8 Self-Consistency Scheme………………………………………………...39

xvi
3.9 Crystalline Solids and Plane-wave Density Functional Theory (DFT)….40
3.9.1 Cutoff Energy…………………………………………………..41
3.9.2 K-points………………………………………………………...42
3.10 Pseudo-Potentials……………………………………………………….42
3.11 WIEN2K………………………………………………………………...43
Chapter 4: Half-Metallic Ferromagnetism in New Half-Heusler Compounds:
an Ab-Initio Study of CrTiZ (Z = Si, Ge, Sn, Pb)……………………45
4.1 Introduction………………………………………………………………45
4.2 Crystal Structure and Computational Details…………………………….46
4.3 Results and Discussion…………………………………………………...49
4.3.1 Ground State Properties………………………………………...49
4.3.2 Electronic and Magnetic Properties……………………………52
Chapter 5: Spin-polarized Calculations of Structural, Electronic and Magnetic
Properties of Half-Heusler Alloys FeVZ (Z= Si, Ge, Sn)
Using Ab-Initio Method……………………………………………..61
5.1 Introduction………………………………………………………………61
5.2 Computational Details……………………………………………………63
5..3 Results and Discussions…………………………………………………63
5.3.1 Crystal Structure Stability……………………………………...63
5.3.2 Electronic Properties…………………………………………...65
5.3.3 Magnetic Properties…………………………………………….71
5.3.4 Half-Metallic (HM) Robustness.…….…………………………75

xvii
Chapter 6: Theoretical Investigations of Half-Metallic Ferromagnetism in
New Half-Heusler YCrSb and YMnSb Alloys Using First-Principle
Calculations…………………….……………………………………...79
6.1 Introduction………………………………………………………………79
6.2 Computational Details……………………………………………………80
6.3 Results and Discussion…………………...………………………………81
6.3.1 Structural Arrangements and Stability………..………………..81
6.3.2. Electronic Properties…………………………………………..87
6.3.3 Magnetic properties…………………………….………………93
6.3.4. Location Associated with Half-Metallicity……………………94
Chapter 7: Physical Properties of Half-Heusler (HH) YMnZ (Z = Si, Ge, Sn)
Compounds via Ab-Initio Study………………………………………96
7.1 Introduction………………………………………………………………96
7.2 Computational Insights…………………………………………………..98
7.3 Results and Discussions………………………………………………….99
7.3.1 Structural Properties……………………………………………99
7.3.2 Electronic Properties………………………………………….105
7.3.3 Magnetic Properties…………………………………………...109
7.3.4 Thermoelectric Properties…………………………………….114
Chapter 8: Structural Chemistry and Physical Properties of the Newly
Designed Half-Heusler XYZ Materials with Large Spin-Gap…….119
8.1 Introduction…………………………………………………….……….119
8.2 Computational Methods………………………………………………...121
8.3 Results and Discussion………………………………………………….122
8.3.1 Ground State Properties….…………………………………....122
8.3.2 Electronic Properties……………….…………………………128
8.3.3 Mixing Energy (Emix)………………………………………....130

xviii
8.3.4 Curie Temperature (TC)……………………………………….132
8.3.5 Band Structure and Density of States ……………...................134
8.3.6 Vibrational Properties ………………………………………...141
8.3.7 Magnetic Properties…………………………………………...143
Chapter 9 Overview of the Results……………………………………………….144
9.1 Future Directions………………………………………………………..147
References………………………………………………………………...………..148
Appendix-I……………………………...………………………………………….174

xix
List of Tables
Table No. Page No.
Table 1.1 Different atomic placement inside the C1b-type
framework. The ZnS-type sublattice is formed by the
4a and 4c Wyckoff positions whereas octahedral
holes are filled by the 4b position……………………………...7
Table 1.2 Various inequivalent Wyckoff positions and the
general formula with space group notations for the
different HA ………..………………………………………….11
Table 4.1 Different configurations of atomic arrangements in
HH structure…………………………………………………...48
Table 4.2 a (Å): lattice parameter, B (GPa): bulk modulus; ΔE
(Ry): Energy difference between FM and NM states
and EFor (eV): Formation Energy……………………………....55
Table 4.3 Total and individual magnetic moments of CrTiZ……………55
Table 5.1 Atomic arrangement of atoms X, Y and Z in α, β and
γ phases. The 4d position is empty…………………………….64
Table 5.2 Atomic optimization of the HH FeVZ alloys at the α,
β and γ phases, a(Å) is the lattice constant. Etot and
µ𝑡𝑜𝑡 are the total energy and magnetic moment per
formula unit respectively………………………………………67
Table 5.3 Total and partial magnetic moments (µB) of the HH
FeVZ (Si, Ge, Sn) compounds in the α-phase at the
equilibrium lattice constant……………………………………67
Table 5.4 Different physical properties of HH FVX (Si, Ge, Sn)
at the equilibrium lattice constant in the α-phase. VXC
is the exchange correlation potential, VBM is the
maximum value of the valance band, and CBM is the
minimum value of the conduction band, EBG is the
energy band gap, EHM is half-metallic gap. Transition
between the bands and nature of compound is also
given…………………………………………………………...70

xx
Table 6.1 The Site preferences of X, Y and Z atoms in three
atomic arrangements XI, XII and XIII in the C1b HH
structure. The 4d site is empty…………………………………82
Table 6.2 Values of optimized lattice constant aopt (Å), the bulk
modulus B (GPa), the pressure derivative of the bulk
modulus B, the total energy (Ry) of the YCrSb and
YMnSb materials………………………………………………85
Table 6.3 Calculated values of formation energy Efor (in eV) per
formula unit, spin-up band gap Eg (eV), half-metallic
gap EHM (eV), and the energy difference between FM
and NM states ΔEFM-NM (eV)
for HH YCrSb and YMnSb materials…………………………85
Table 6.4 Calculated values of total and local magnetic moment
(µB) of the individual atom and interstitial site for HH
YCrSb and YMnSb materials…………………………...…….85
Table 7.1 Inequivalent atomic arrangement inside the C1b-type
framework in which atoms placed on Wyckoff
positions 4a and 4b make a ZnS-type sublattice
whereas the octahedral holes occupied by the atoms
on 4b…………………………………………………………101
Table 7.2 Computed total energy (Ry/f.u.) at three unique
structural phases (X-type1, X-type2, X-type3) with
NM, magnetic (FM and AFM) states of the HH YMnZ
(Z=Si, Ge, Sn) materials. Also, predicted lattice
parameter a (Å), bulk modulus B (GPa) and the
formation energy Efor (Ry) of these studied materials
are given in the preferred FM state………………………....101
Table 7.3 The computed total µtot (µ/f.u.) and local magnetic
moments (µatomic/f.u.) of the half-Heusler YMnZ
(Z=Si, Ge, Sn) materials with X-type1 phase at the
two exchange-correlation potential (VXC) is given.
Band gap energy: Eg (eV) and half-metallic gap: EHM
(eV) is also described at the Electronic conductivity

xxi
(metallic, HM, or semiconducting)……………..………..…..112
Table 8.1 Equilibrium lattice parameter: a (Å), electronic
conductivity (SC and HM) represent the
semiconductor and half-metallic characteristics, µB is
equivalent to total magnetic moment, EBG& EHM are
the energy band gaps and HM gap, FM & AFM along
with EFM& EAFM shows the magnetic ground state and
energy of the FM and AFM respectively, TC & Emix
shows the Curie Temperature and mixing energy of
the each 30 interesting HH XYZ materials at their
preferred ground state (T1 or T3) with vibrationally
stability check at the γ-point………………………………….135

xxii
List of Figures
Figure No. Page No.
Fig. (1.1) Periodic table showing the large numbers associated with
HA could be created through mixture of the various
components based on the color plan…………………………………..3
Fig. (1.2) Crystal structures of (a) NaCl-type (Rock salt), (b) Zinc-
blende (c) HH & (d) FH……………………………………………….5
Fig. (1.3) Atomic placement inside the unit cell of the HH, FH, QH
and inverse Heusler alloys. In the lattice, there are four
interpenetrating f.c.c lattices for all the cases. It can also be
noted that the lattice will be b.c.c if all the atoms are same…………..7
Fig. (1.4) HA based on Mn2 with respect to the position of Y element
for each type, the inverse Heusler and the normal Heusler
framework…........................................................................................10
Fig. (1.5) Unit cell of (a) inverse Heusler framework CuHg2Ti as
well as quaternary edition LiMgPdSn……………………………….10
Fig. (1.6) (a) XYZ HH materials and TMs occupy only the octahedral
site and display magnetic moment (b) X2YZ FH materials.
The HH and FH have the one and two magnetic sublattice
respectively which means FH can be coupled in both FM
and AFM phases while the HH has only FM
and AFM phases while the HH has only FM phase…………………11
Fig. (1.7) Schematic outline for the DOS (a) metal (b) spin projected
metal (c) a ferro-magnet (d) a HM ferromagnet
(e) HM ferrimagnet…………………………………………………..13
Fig. (1.8) Various distinctive important physical properties of the
outstanding class of HA……………………………………………...15
Fig. (1.9) Outline of the fundamental spintronic gadgets………………………17

xxiii
Fig. (1.10) When conducting electrons move towards magnet, their
own spin preferentially line up within the magnet’s path.
Since the electrons experience the nanomagnet, sandwiched
in between levels associated with NM materials near to the
set alignment magnetic, the actual path associated with their
own spin is repolarized to complement from the
nanomagnet. Consequently, nanomagnet has the magnetic
moment starts to precess,
turns just like a spinning-top on its pivot……………..………………21
Fig. (1.11) Selected elements from the periodic table for the studied HH
XYZ materials in this dissertation based on the color
plan……………………………………………………………………28
Fig. (4.1) Crystal structure of HH XYZ compound with
(MgAgAs) C1b for Type 1……………………………………………48
Fig. (4.2) Volume optimization at various atomic positions (Type 1,
Type 2, Type 3) for (a) CrTiSi (b) CrTiGe (c) CrTiSn
and (d) CrTiPb…..……………………………………………………50
Fig. (4.3) Volume optimization for (a) CrTiSi (b) CrTiGe (c) CrTiSn
(d) CrTiPb for magnetic FM & NM state.……………………………51
Fig. (4.4) Electronic band structures of HH (a) CrTiSi, (b) CrTiGe, (c)
CrTiSn & (d) CrTiPb compounds for spin-up (↑)
and spin-down (↓)……………………………………………………..53
Fig. (4.5) Total and partial DOS of (a) CrTiSi, (b) CrTiGe,
(c) CrTiSn & (d) CrTiPb for spin- up (↑) and spin-down (↓)…………56
Fig. (4.6) Total and orbital resolved partial DOS of the HH (a) CrTiSi
(b) CrTiGe, (c) CrTiSn & (d) CrTiPb compounds……………………57
Fig. (4.7) Origin of semiconducting gap in the majority spin channel
in the α-phase for the CrTiGe as a prototype…………………………59
Fig. (4.8) Total Magnetic moment (µ𝑡𝑜𝑡) of the HM HH
CrTiZ (Z= Si, Ge, Sn, Pb) as a function of lattice constant…………..60
Fig. (5.1) Unit cell of cubic C1b-type structure for the HH FeVGe in
α-phase………………………………………………………………...64

xxiv
Fig. (5.2) Total energy as a function of volume for FeVGe in
different phases (α, β, γ) of atomic positions…………………………66
Fig. (5.3) Total energy as a function of volume for the HH FeVGe in
α-phase for the magnetic and NM states………...……………………66
Fig. (5.4) Spin-dependent total and partial DOS of HM FM materials
(a) FeVSi (b) FeVGe (c) FeVSn at equilibrium lattice
constant at the α-phase. Fermi level is set at zero. The top
portion (spin-up) displays the majority-spin channel and the
lower portion (spin-dn) is for the minority spin channel.
Solid and dotted lines show the DOS’s of GGA & mBJ-
GGA potential respectively ………………………………………….69
Fig. (5.5) Spin polarized band structure of the FeVSi for the α-phase
at equilibrium lattice constant. Solid and dashed lines
denote for the GGA and mBJ-GGA potential respectively…………...72
Fig. (5.6) Spin polarized band structure of the FeVGe for the α-phase
at equilibrium lattice constant. Solid and dashed lines
denote for the GGA and mBJ-GGA potential respectively…………...73
Fig. (5.7) Spin polarized band structure of the FeVGe for the α-phase
at equilibrium lattice constant. Solid and dashed lines
denote for the GGA and mBJ-GGA potential respectively…………...74
Fig. (5.8) Band structure of the HH FeVGe compound with mBJ
potential of α-phase at equilibrium lattice constant…………………..76
Fig. (5.9) Magnetic moment as a function of lattice constant of the
HH (a) FeVSi, (b) FeVGe & (c) FeVSn materials……………………77
Fig. (6.1) Conventional unit cells of YCrSb HH alloy in the MgAgAs
(C1b) structure for the three distinct XI, XII and
XIII atomic arrangements……………………………………………...82

xxv
Fig. (6.2) Variations of computed FM total energy with volume per
unit cell for the three feasible atomic arrangements XI, XII
and XIII of both HH (a) YCrSb (b) YMnSb with MgAgAs
(C1b) structure……………………………...………………………...84
Fig. (6.3) Variations of calculated total energy with volume of HH (a)
YCrSb (b) YMnSb materials in stable XI phase for NM,
FM and AFM states…………………………………………………..86
Fig. (6.4) Spin-resolved band structures of HH YCrSb (a) spin up
(b) Spin down. Fermi level is set to be zero…………………………..88
Fig. (6.5) Spin-resolved band structures of the HHYMnSb
(a) spin up (b) Spin down. Fermi level is set to zero………………….89
Fig. (6.6) Spin-polarized densities of state for the total and individual
atoms at the equilibrium lattice constant for the XI phase of
the HH (a) YCrSb (b) YMnSb materials……………………………...91
Fig. (6.7) Schematic representations of origin of semiconducting gap
in the majority spin state in the stable XI structure for the
HH YCrSb material…………………………………………………...92
Fig. (6.8) Lattice parameter dependences of the total magnetic
moment, and the spin moments of Y, Cr/Mn and Sb atoms
for the HH (a)YCrSb and (b) YMnSb, respectively…………………..95
Fig. (7.1) Conventional cubic unit cell of the HH YMnSi at the diverse
atomic arrangement X-type1, Xtype2, and X-type3……….………...100
Fig. (7.2) Total energy to be functionality connected with volume inside
three unique atomic positions X-type1, X-type2 and X-type3 for
the HH (a) YMnSi (b)YMnGe and YMnSn alloys. These curves
represent the FM state………………………………………………..103
Fig. (7.3) Total energy to be functionality connected with volume
inside ternary magnetic states (NM, FM, and AFM) for the
HH (a) YMnSi (b) YMnGe and (c) YMnSn alloys………....……….104

xxvi
Fig. (7.4) Spin-projected band structure with the HH YMnSi alloy.
Black solid lines show the GGA and red dotted lines are
for the GGA+mBJ…………………………………………………..106
Fig. (7.5) Spin-projected band structure with the HH YMnGe
compound. Black solid lines show the GGA and red dotted
lines are for the GGA+mBJ………………………………………...107
Fig. (7.6) Spin-projected band structure with the HH YMnSn
compound. Black solid lines show the GGA and red dotted
lines are for the GGA+mBJ………………………………………...108
Fig. (7.7) Total and partial DOS of the HH (a) YMnSi,
(b) YMnGe and (c) YMnSn compounds using GGA+mBJ……......110
Fig. (7.8) The computed total magnetic moment (µ𝐵) for the HH (a)
YMnSi, (b) YMnGe and YMnSn materials corresponding to
variation of lattice constant. The dashed vertical line
shows the optimized equilibrium lattice constant………………….115
Fig. (7.9) (a) Electrical conductivity, (b) thermal conductivity
(c) See beck coefficient and (d) Figure of merit as a function
of temperature …………………………………………………......117
Fig. (8.1) Conventual unit cell of the HH XYZ materials in three
different atomic arrangement types T1 [4c (1
4,
1
4,
1
4), 4d
(3
4,
3
4,
3
4), 4a(0, 0, 0)], T2 [4a(0, 0, 0), 4d (
3
4,
3
4,
3
4),
4c (1
4,
1
4,
1
4)] and T3 [4b (
1
2,
1
2,
1
2), 4d (
3
4,
3
4,
3
4), 4a(0, 0, 0)]…………..123
Fig. (8.2) Volume optimization of the HH HM NaVTe material at the
(a) three different atomic arrangement types (T1, T2 & T3)
(b) FM and AFM ground state…………………………………….125
Fig. (8.3) Summary of the 90 HH XYZ materials, at their preferred
stable ground state among three different types T1, T2 & T3
phases and magnetic ground state (NM, FM, AFM).
Illustration of the electronic properties of the each HH
materials is also presented at their preferred stable type and
magnetic ground state……………………………………………..127

xxvii
Fig. (8.4) Colors shows the width of the (a) Energy band gap (EBG) (b)
HM gap (EHM) of 90 HH XYZ materials. The species X, Y
and Z which represent the HH XYZ materials are signifying
on the three coordinates. Blue and yellow colors represent
the successively increasing values of these
energy band gaps…………………………………………...............129
Fig. (8.5) Mixing energy (eV/atom) of the 28 HM HH XYZ materials
at their respective stable state with FM configurations which
are also vibrationally stable at the gamma point. Blue shades
show the mixing energy less than 0.2 (eV/atom). Each
coordinate of the 3D plot symbolizes the X, Y, Z species of
the associated HM HH
XYZ materials………………………………………………..…….131
Fig. (8.6) The Curie temperature of the 28 HM HH XYZ materials at
their respective stable state with FM configurations which
are also vibrationally stable at the gamma point. The color
bar shows the values of the calculated TC (K) with +/- 25 K
tolerance. Each coordinate of the 3D plot symbolizes the X,
Y, Z species of the associated HM HH XYZ material
XYZ material……………………………………………………….133
Fig. (8.7) Phonon full spectrum curve for the (a) HM HH NaVSi &
(b) FM SC LiVGe ……………………………………………….....138
Fig. (8.8) Band structure of the HH FM SC LiVGe material in the
spin up and spin down channel. The horizontal dashed line
represents the Fermi level (EF) which is fixed at zero eV………….139
Fig. (8.9) Band structure of the HM HH CsVSe material in the spin up
and spin down channel. The horizontal dashed line
represents the Fermi level (EF) which is fixed at zero eV………….140
Fig. (8.10) Fig. (8.10) Spin projected total and partial density of state
(DOS) of HM HH RbVTe material at the equilibrium lattice
constant. The vertical dashed line in the middle
shows the Fermi level (EF = 0 eV)………………………………….142

xxviii
List of Abbreviations
HMFs = Half-metallic Ferromagnets
HM = Half-metallic
HA = Heusler Alloys
HH = Half-Heusler
FH = Full-Heusler
QH = Quaternary Heusler
TC = Curie temperature
TMs = Transition metals
VBM = Valance Band Maxima
VCM = Conduction Band Minima
𝑁𝑣𝑎𝑙 = Number of valance electrons
ZA = Atomic number
NM = Non-magnetic
FM = Ferromagnetic
AFM = Anti-ferromagnetic
SPR = Slater-Pauling Rule
µtot = Total magnetic moment
SP = Spin polarization
mBJ = modified Becke-Johnson approximation
TB-mBJ = Tran-Blaha modified Becke-Johnson
DOS = Density of States
EBG = Energy band gap
EHM = Half-metallic gap

xxix
EF = Fermi Energy
𝐸𝑓𝑜𝑟
= Formation Energy
𝐸𝑚𝑖𝑥 = Mixing Energy
SCs = Semiconductors
BZ = Brillouin Zone
TMR = Tunneling Magnetoresistance
GMR = Giant Magnetoresistance

1
CHAPTER 1
Fundamentals
Prior to presenting physical facets of this thesis, the introduction of half-metallic
ferromagnets (HMFs) must be presented. Up to now, half-metallic (HM) properties are already
noticed in several alloys, for instance, Heusler alloys (HA) ferromagnetic (FM) materials
(Bayar et al., 2011; Özdog and Galanakis, 2009; Sharma et al., 2010b), dilute magnetic
semiconductors (SCs) and zincblende transition metals (TMs) oxides (Soeya et al., 2002; Song
et al., 2009a; Szotek et al., 2004), pnictides and chalcogenides (Xie et al., 2003). Among
several HMFs are pointed out above, HA considered as the most inspiring possibility for
practical utilizations of spintronic materials because of high Curie temperatures (TC), high
magnetic moments, and their crystal structures equivalent with the traditional SCs. HMFs are
considered this kind of novel compounds. Consequently, it is much more useful to review the
Heusler framework along with brief history of the HA before the HMFs term must be cleared
up.
1.1 Heusler Alloys (HA)
Heusler alloys (HA) are recognized, more than a century. These are referred to after
Friedrich Heusler, a German engineer as well as a chemist, who found out in 1903 that
Cu2MnAl acts as FM, although, combination (Cu2MnAl) is made of non-FM or non-magnetic
(NM) constituents (Cu, Mn, Al). This exceptional material and its family members that right
now exist consist of huge selection more than 1000 alloys and are referred as HA.
The simultaneous advancement of computational materials modelling, and growth
methods at the nanoscale develop scientific interest on the research of many magnetic materials
such as HA. Interest in the HA has enhanced as the fact is established that their physical
properties may effectively adjusted by replacement of elements. Heusler materials undoubtedly
are a huge class of intermetallic materials representing different kinds associated with
electronic and magnet attributes (Webster and Ziebeck, 1988a). Many one of these HA are
HMFs, ferrimagnets, and antiferromagnets.

2
These materials have distinct attention because of the quite high TC range which often
surpasses 1000 k producing all of them well suited for spintronic applications (Hirohata and
Takanashi, 2014).
1.2 Types of Heusler Alloys (HA)
HA are type associated with intermetallic compounds which can be divided into four
main families: (a) the typical Full-Heusler (FH) alloys such as Co2MnSi with the chemical
composition is X2YZ in which valence associated with the X is larger than the valence
associated with the Y and Z atoms are similar, (b) the semi-Heusler also referred as HH alloys
like NiMnSb possess the chemical composition XYZ with X and Y represents the TMs,
whereas (c) the Quaternary Heusler (QH) alloys such as CoFeMnSi which have comparable
attributes like the FH alloys and lastly (d) the inverse-HA such as Cr2CoGa that also have the
chemical composition of X2YZ however right now valence associated with Z atom scale down
compared to Y and so result of modification of atomic placement in the crystal, the two Z
atoms are no longer similar (Webster and Ziebeck, 1988a).
1.2.1 Full Heusler (FH) Alloys
In earlier times, HA had been frequently recognized as an intermetallic material, even
though the framework as a possible intermetallic material is certainly more appropriate because
of their feature atomic order. Ternary FH alloys hold the basic general chemical formula X2YZ
in which X & Y = TMs whereas Z represents main group of the periodic table. An illustration
of FH alloys is presented in Fig. (1.1). Though, in certain cases, Y atom is exchanged with
alkaline earth metal or rare earth constituent. Usually, metals, that are available two times, is
set in the beginning with the formula, whilst the Z atom is put at the last, for example, Co2MnSi,
Fe2VAl (Nishino et al., 1997; Ritchie et al., 2003). These FH alloys have a stoichiometry of
2:1:1 and crystalizes in the cubic L21 structures.
Half-metallicity in the FH alloys may be blended both with all the physical appearance
regarding ferrimagnetism if the Z atoms inside the X2YZ could be the Mn and together with
ferromagnetism when Z atom will be Co. In each instance, the FH alloys have the total
magnetic spin moment in µB and follow the SPR of 𝑍𝑣𝑎𝑙 – 24 (Galanakis, 2002a).
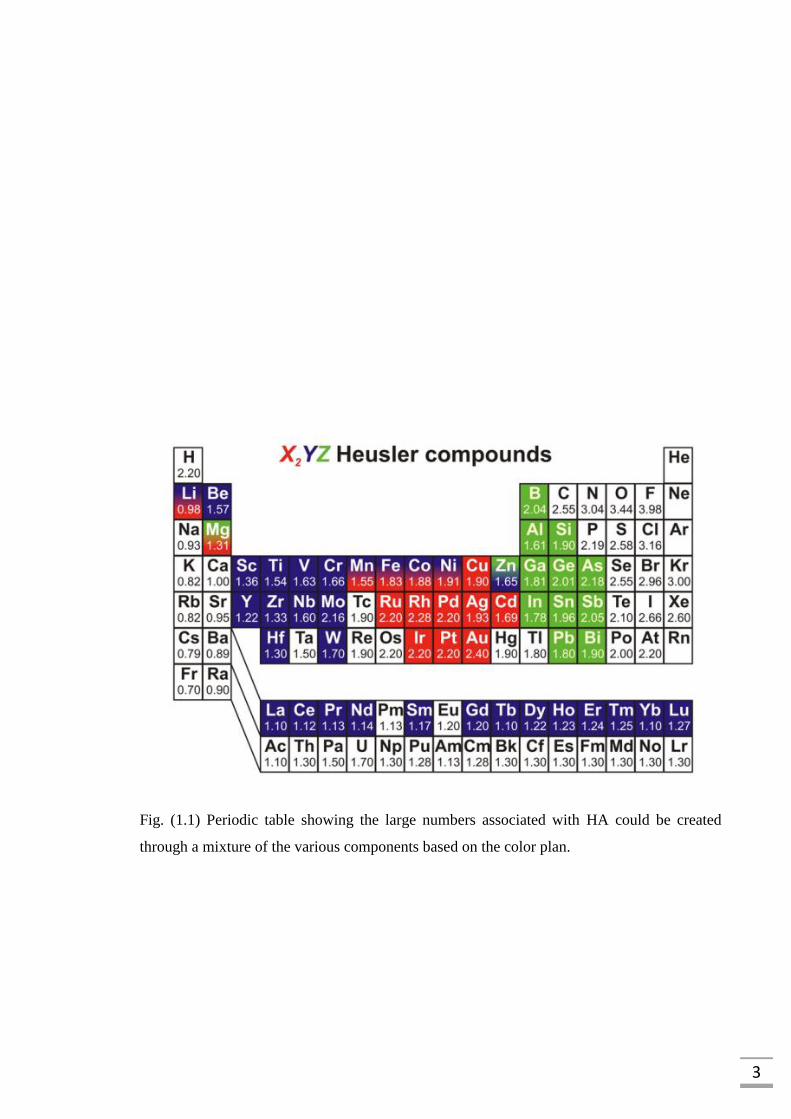
3
Fig. (1.1) Periodic table showing the large numbers associated with HA could be created
through a mixture of the various components based on the color plan.

4
1.2.2 Half Heusler (HH) Alloys
The materials which have a stoichiometry of 1:1:1 tend to be a ternary SC or metallic
compounds additionally identified as half-Heusler (HH) alloys. Generally, HH alloys have
general formula XYZ can end up being recognized as materials comprising the covalent as
well as an ionic component. The X and Y possess unique cations identity although Z represents
the anionic version.
In literary works, there is a wide range of variations in the nomenclature of these
compounds. Three possible arrangements are found as they sorted out as alphabetically,
randomly or depending upon on electronegativity. The most-well recognized semi-Heusler
material is NiMnSb.
1.2.3 Quaternary Heusler (QH) Alloys
Apart from the most common and inverse FH materials, one more FH relative will be
the LiMgPdSn-type kinds, also referred to as LiMgPdSb-type Heusler material. These types of
materials tend to be QH materials using the chemical composition of XX′YZ exactly, in which
X, X′, as well as Y tend to be metallic atoms and Z is equivalent to sp-element. The valence
electrons involving X′ is lesser as opposed to valence involving X element, plus valence of Y
is simply lesser as opposed to valence involving the two X along with X′. The arrangement
from the atoms across the fcc cube’s diagonal is actually X-Y-X′-Z and vigorously probably
uttermost dynamic stable phase (Alijani et al., 2011). A couple of LiMgPdSn-sort of HM
materials have been analyzed (Gökoğlu, 2012; Izadi and Nourbakhsh, 2011) and some reviews
demonstrated that one can likewise discover spin gapless SCs among them (Xu et al., 2013).
1.3 Crystal Framework of Heusler Alloys (HA)
HA mainly divided into two unique families in which one has the composition 2:1:1
and the other has 1:1:1 stoichiometry. The very first family of HA analyzed were crystallizing
within the L21 structure that includes four fcc sublattices. Later, is found that if one of the fcc
sublattice is withdrawn from the L21 framework leads to the C1b framework. HH crystal is a
variant of the ternary ordered CaF2 structure. When the ZnS-type framework is filled by the
octahedral positions then it leads to the HH structure Fig. (1.2) (Graf et al., 2011).

5
Fig. (1.2) Crystal structures of (a) NaCl-type (Rock salt), (b) Zinc-blende (c) HH & (d) FH

6
Prominent aspect of the HH structural framework (C1b type) is that it contains three
filled and one vacant fcc sublattice which are hosted by X, Y and Z elements (Webster and
Ziebeck, 1988b). The HH structure crystallizes into a cubic non-centrosymmetric lattice with
F-43m space group (No. 216). The materials with C1b structural framework tend to be referred
as half- or feasibly semi-Heusler materials or just Heusler, although L21 materials are usually
called FH materials. The atomic arrangement filled by 4a (0, 0, 0), 4b (1
2,
1
2,
1
2) along with 4c
(1
4,
1
4,
1
4 ) Wyckoff positions for the HH materials. In theory, three inequivalent atomic
placements tend to be feasible in this structural framework which is described inside Table 1.1.
Usually, the HH structure can be considered as the ZnS sublattice in which Wyckoff position
was taken by 4a and 4c whereas the octahedral site generally filled by 4b.
This depiction underlines the covalent bonding among the two contained components
that perform a significant part to illustrate the electronic properties. An attention is drawn to
consider the appropriate atomic placements in the crystal which is very important to
comprehend structural properties for HA, as well as unique treatment, needs to be used
whenever carrying out theoretical research to acquire proper outcomes.
The FH X2YZ materials crystallizes into a cubic (L21-type) structural framework and
have space group (No. 225) Fm-3m using Cu2MnAl as a model (Bradley and Rodgers, 1934;
Heusler, 1934). The Wyckoff position for the X is 8c (1
4,
1
4,
1
4 ), whilst the Y & Z situated at 4a
(0, 0, 0) & 4b (1
2,
1
2,
1
2) individually .
Like HH materials, this framework comprises of 4 interpenetrating fcc sublattices, 2
associated with that are similarly involved by X atom, whereas the Y and Z comprise of
slightest and most electropositive elements form the rock salt-type lattice. The elements in FH
are usually synchronized octahedrally due to their ionic interaction persona. Alternatively, all
tetrahedral gaps are usually positioned by the X atom. Family of HA is highlighted within Fig.
(1.3). Besides the unit cell of HH and FH materials which are explained in the previous pages,
an inverse Heusler construction can be made when X and Y TMs selected from the same period
of periodic table and arranged in a unique manner that the atomic number (ZA) of Y, is greater
than the ZA of X. If TMs are taken from other periods, the Inverse Heusler framework also
exists in the HA (Puselj and Ban, 1969).

7
Table 1.1 Different atomic placement inside the C1b-type framework. The ZnS-sort sublattice
shaped by the 4a & 4c Wyckoff positions whereas octahedral holes are filled by the 4b position.
Structural phase 4a 4b 4c
Type I X Y Z
Type II Z X Y
Type III Y Z X
Fig. (1.3) Atomic placement inside the unit cell of the HH, FH, QH and inverse HA. In the
lattice, there are four interpenetrating f.c.c lattices for all the cases. It can also be noted that the
lattice will be b.c.c if all the atoms are same.

8
For every situation, X will be a lot more electropositive as compared to the Y. As a
result, X & Z shape NaCl-type crystal and X takes an octahedral coordination. By using 4-fold
symmetry, rest of the X & Y packed to tetrahedral gaps. Still, there are 4 interpenetrating fcc
sublattices inside the unit cell of the Inverse HA, however, a basic cubic unit cell is not shaped
by X anymore.
Rather the X occupy the Wyckoff placements 4a (0, 0, 0) and 4d (3
4,
3
4,
3
4 ), even though
the Y, as well as the Z, are situated with 4b (1
2,
1
2,
1
2) and 4c (
1
4,
1
4,
1
4 ), respectively. This
framework has the prototype of CuHg2Ti along with space group F-43m (No. 216).
Additionally, it is also feasible to distinguish the ordinary HA by indicating the chemical
composition as (XY)X′Z (Graf et al., 2011). The compounds based on Mn2 are often
considered as Inverse HA in which ZA(Y) > ZA (Mn) as outlined in the Fig. (1.4).
In Fig.(1.4), a properly researched illustration is shown for the inverse Heusler
materials i.e. Mn2CoSn or (MnCo)MnSn. Regarding QH materials, there are not one, but two
distinct elements X and X′. The Wyckoff positions 4a (0, 0, 0) & 4d (3
4,
3
4,
3
4 ) are taken by the
X & X′, whereas Y and Z are placed at 4b (1
2,
1
2,
1
2) and 4c (
1
4,
1
4,
1
4 ) respectively. LiMgPdSn is
the prototype of the QH alloys. An excellent example for the inverse Heusler framework and
of the QH alloys are provided within Fig. (1.5).
1.4 Structural Arrangement
1.4.1 Order-Disorder Phenomena in Half-Heusler (HH) Materials
The order and placement of the atoms inside the crystal, are very important and greatly
influence the electronic structure, and as a result effect the physical properties of HA. On that
basis, a detailed investigation on the structural framework is important to comprehend, along
with the prediction of the physical properties, for the different compounds. Sometime, a
halfway intermixture can adjust the electronic structure. HH materials tend to be tetrahedrally
stuffed structures that strongly linked to the binary SCs. Covalent bonding interactions perform
a substantial part, as well as their own crystalline arrangement, is maintained up to the
composed temperature (Skovsen et al., 2010).

9
Therefore, the structural disorder results in an occupancy from the empty lattice site
which happens seldom within HH materials, while the FH materials (X2YZ) structural
arrangements frequently show huge extents with atomic disorder. Different types of disorder
are possible within HH structural arrangement which is described in Table 1.2.
1.5 Heusler Alloys (HA) and Magnetism
HA initially pulled enthusiasm among the scientist in 1903, when F. Heusler
discovered, how the alloy Cu2MnAl gets to be distinctly FM, although none of its constituent
components is FM on its own. Even so, the idea needed about three ages prior to the amazingly
structural composition ended up being identified which has a deal with structure fcc lattice
(Bradley and Rodgers, 1934; Heusler, 1934). However, they pale virtually throughout oblivion
in the next ages, and the only a couple of reviews upon the experimental formation of the brand
new HA had been published in the 1970s (Webster, 1971).
It had been not really before anticipation of HM ferromagnetism within MnNiSb
through de Groot et al. in 1983, then scientific curiosity came back to Heusler compounds. The
HH XYZ compounds display a single magnet sublattice considering that the atoms around the
octahedral sites bring any magnet moment, which is suggested inside Fig. (1.6).
Mostly from the experiment, magnetic XYZ HH alloys can be found just for X = Mn
and rare earth metals. However, from experiment, it is found that a little magnetic moment is
observed for the Ni along with the late TMs. Certainly, by considering some basic principles,
situation like this can be overlooked. On the other hand, the majority of HA consisting of rare
earth elements which are discovered within the literature so far tend to be semiconducting or
semi-metallic frameworks or anti-ferromagnetic (AFM) with lower Néel temperature range
[24].
Merely, a few FM HH materials are portrayed in literature, as an illustration, NdNiSb
and VCoSb (Hartjes and Jeitschko, 1995; Heyne et al., 2005). The HH materials which contain
Mn atom are usually HMF along with higher TC. For the FH (X2YZ) materials, now the two
atoms are placed at the tetrahedral sites and hence situation is entirely different as compared
to the HH (XYZ) materials which result in an interaction between X atom and development of
the subsequent additional delocalized magnetic sublattice.

10
Fig. (1.4) HA based on Mn2 with respect to the position of Y element for each type, the inverse
Heusler, and the normal Heusler framework.
Fig. (1.5) Unit cell of (a) inverse Heusler framework CuHg2Ti as well as (b) the quaternary
edition LiMgPdSn.

11
Table 1.2 Various inequivalent Wyckoff positions and the general formula with space group
notations for the different HA (Graf et al., 2011).
Fig. (1.6) (a) XYZ HH materials and TMs occupy only the octahedral site and display magnetic
moment (b) X2YZ FH materials. The HH and FH have the one and two magnetic sublattice
respectively which means FH can be coupled in both FM and AFM phases while the HH has
only FM phase.

12
This is the reason that FH (X2YZ) materials display all types of a magnetic
phenomenon like ferromagnetism, ferrimagnetism and HM ferromagnetism today.
1.5.1 Half-Metallic Ferromagnets (HMFs)
In last three decades, different magneto-optical properties of a few HA inspired the
analysis of their electronic structure that results in a surprise outcome. For example, a number
of HA demonstrate metallic together with insulating properties simultaneously, an attribute
identified as HMF (De Groot et al., 1983; Kübler et al., 1983). De Groot and his associates
created an arrangement aiming out that three distinct sorts of HMF can be recognized.
The formal illustration of the density of states (DOS) is offered in the Fig. (1.7) which
show (a) A metal along with a limited DOS at the EF and (b) spin-projected depiction for the
metal. The Fig. 1.7 (c) demonstrates the DOS associated with the FM material for both
channels, the spin-up (majority) and spin-down (minority) are moved next to one another,
prompting to a quantifiable total magnetic moment (µtot), whereas, the Fig. 1.7 (d) illustrate
the HMFs which have metallic character for one spin-channel as well as insulator or SC for
the opposite spin-channel.
Basically, 100% spin polarization (SP) in the HMFs is merely achieved, at the absolute
temperature and half-metallicity often disappears when spin-orbit interactions are considered.
Many HA that contains just 3d TMs do not display any kind of spin-orbit interactions, they are
perfect applicants for HMFs.
1.5.2 The Slater-Pauling Rule (SPR)
The µtot associated with 3d TMs evaluated on the premise of counting the normal
valence electron number (𝑁𝑣𝑎𝑙) for each element present in the compound (Pauling, 1938;
Slater, 1936). This fact was discovered by Slater and Pauling. The µtot is provided by, in
multiple of Bohr magnetrons (µB) as:
µ𝑡𝑜𝑡 = 𝑁𝑣𝑎𝑙 − 2𝑛↓
where 2𝑛↓ signifies the quantity of electrons in the spin down (minority states). The minimal
in the spin down DOS pushes the quantity of d-electrons in the minority band being around
three.
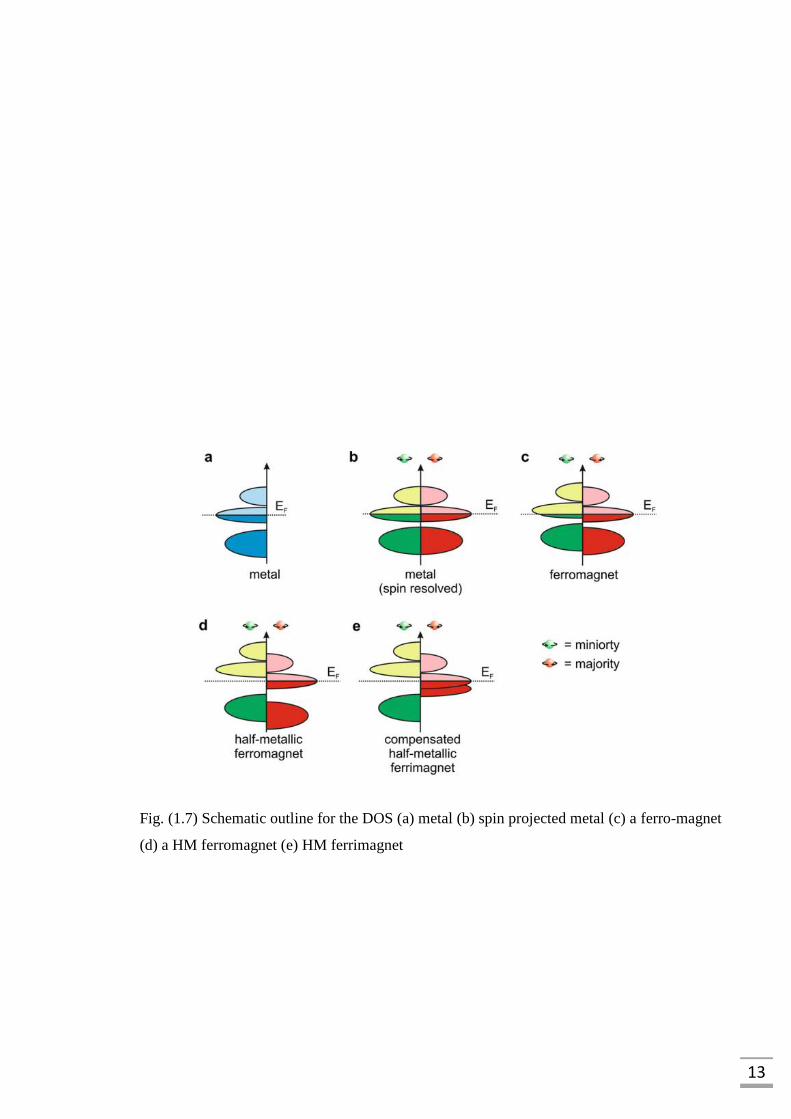
13
Fig. (1.7) Schematic outline for the DOS (a) metal (b) spin projected metal (c) a ferro-magnet
(d) a HM ferromagnet (e) HM ferrimagnet

14
By ignoring the (s & p) electrons, the µ𝑡𝑜𝑡 inside localized section of the SP-curve is
determined in accordance with
µ𝑡𝑜𝑡 = 𝑁𝑣𝑎𝑙 − 6
It means that magnet moment for every atom is simply the typical quantity of valance
electron less than six. HMFs display energy band gap (EBG) in any one of the spin-channel
(spin-up or spin-down) DOS at the EF according to their definition because of this EBG, the
quantity of filled minority states should be a whole number, which is precisely satisfied for the
case µ = 𝑁𝑣𝑎𝑙 − 6 (Kübler, 2000; Wurmehl et al., 2005). This principle can result in a non-
integer values, if normal 𝑁𝑣𝑎𝑙 is not a whole number. Therefore, it is much simpler to use the
actual 𝑁𝑣𝑎𝑙 for each formula unit.
Regarding HH XYZ materials which have three atoms for every formula per unit cell,
the SPR becomes
µ𝑋𝑌𝑍 = 𝑁𝑣𝑎𝑙 − 18
and the HM materials which have less 𝑁𝑣𝑎𝑙 than the congenital HH materials, this rule is
modified by the Damewood et al. (Damewood et al., 2015a) as,
µ𝑋𝑌𝑍 = 𝑁𝑣𝑎𝑙 − 8
In the case of FH (X2YZ) materials, four atoms are placed inside each unit cell prompting to
the equation
µ𝑋2𝑌𝑍 = 𝑁𝑣𝑎𝑙 − 24
1.6 Applications of Heusler Alloys (HA) for Spintronic Devices
Summary of the different factors associated with HA will be talked considered, in this
evaluation section. The Fig. (1.8), condenses all the essential information regarding these types
of outstanding materials, varying through SCs, more than alloys as well as magnets to
topological insulators along with lots of technical programs within spintronic, thermoelectric,
opto-electronics and much more. Numerous intriguing studies will arise within the long term
that make the most of their own multiple’s benefits.

15
Fig. (1.8) Various distinctive important physical properties of the outstanding class of HA.

16
1.6.1 The Effect of Giant Magnetoresistance (GMR) and Tunneling Magnetoresistance
(TMR)
In 1986, the breakthrough of the giant magnetoresistance (GMR) influence the FM
multilayers and sandwiching of these layers by A. Fert (Baibich et al., 1988) & P. Grünberg
(Grünberg et al., 1986), changed the arena of information technology. In 2007, these two-
scientist privileged by a Noble prize for their exceptional breakthrough in Physics.
Nowadays, we are interacting with spintronic in our daily life due to the application of
GMR effect, the sort of spin-valves that can be utilized as a part of FM disks. The spin-valve
device contains a couple of magnetic levels, sandwiched with an extremely slim NM metallic
part.
Among the magnetic levels, one layer is "trapped" with AFM compound which is not
responsive to controlled FM fields, whereas the 2nd coating is “free” from the magnetization,
meaning that it can be balanced by rotating and using little magnetic fields. In contrast of GMR
spin-valves, the magnetoresistance increases 10%, if the metallic layer is replaced by an
insulating material. This increase is due to the tunneling of electrons from the insulating
material. Such materials are called tunneling magneto resistance (TMR) or tend to be referred
as magnetic tunnel junctions (MTJs).
The symbolic representation of the GMR and TMR is illustrated in Fig. (1.9) and see
review (Moodera et al., 1999) for the additional point of interest. Amazingly, a definitive
objective of spintronic, i.e. a tunneling gadget having a GMR impact associated with thousands
of percentages, could be attained simply by couple of distinct routes: The first path leads to
build insulating layer, and the second route guides to grow brand new HMFs with 100% SP.
At the very top of Fig. (1.9), GMR multilayers tend to be revealed in which the
magnetic coupling could be modified through different width from the nonmagnetic (NM)
spacer coating. While the bottom of Fig. (1.9) illustrates the TMR gadget in which the
tunneling current comes after opposite to the film surface.

17
Fig. (1.9) Outline of the fundamental spintronic gadgets.

18
1.6.2 Spin Polarization (SP)
Earlier groundbreaking research about the issue associated with spin-dependent
tunneling had been carried out within the 1970s through G. Michael. Tedrow as well as Ur.
Meservey (Tedrow and Meservey, 1973), by Michael. Jullière (Julliere, 1975), and also Utes.
Maekawa along with Ough. Gäfvert (Maekawa and Gafvert, 1982). Two decades later,
nevertheless, the primary substantial magnetoresistance within magnetic tunnel junctions had
been noticed at room temperature through T. Utes. Moodera (Moodera et al., 1995) as well as
Capital t. Miyazaki (Miyazaki and Tezuka, 1995). Pursuing the Jullière demonstration [219],
the TMR proportion of junction is identified with the SP of the electrodes based on the equation
⧍𝑅
𝑅𝑇𝑀𝑅=
2𝑃1𝑃2
1 + 𝑃1𝑃2
in which 𝑃1 and 𝑃2 are the polarization of the primary and secondary electrodes, respectively.
Also, the “SP” is characterized by
𝑆𝑃 = 𝑑 ↑ − 𝑑 ↓
𝑑 ↑ + 𝑑 ↓
whereas 𝑑 ↑ and 𝑑 ↓ presents the densities of the spin up and spin down states at the EF. The
Julliére model gives the basic estimation for the tunneling effect.
The very first theoretical work associated with half-metallicity within MnNiSb
triggered huge investigation curiosity, striving in the usage associated with HA within MTJs.
Actually, for the HH MnNiSb bulk material, a SP of just about 100% at the EF has been noticed
by method for spin polarized positron annihilation (SSPA) (Hanssen and Mijnarends, 1986).
However, the actual preparing of these thin-films of MnNiSb ended up being not
without challenges. Subsequently, diverse growth techniques, including co-sputtering and
molecular beam epitaxy (MBE), must be applied to get ready epitaxial films. At last, the crystal
arrangement was affirmed through XRD and the existence of the magneto crystalline
anisotropy. The assembly of HA into TMR appliances prompted an extraordinary boost in the
TMR proportion in the next years.
Nonetheless, the listing of guaranteeing applicants is lengthy, and numerous diverse
supplies happen to be examined, for example Co2Fe0.5Mn0.5Si, Co2FeAl0.5Si0.5 are among the

19
QH materials and Co2FeSi, Co2MnSi, Co2MnGe are the FH alloys. Consequently, a noticeable
advancement associated with the thin film high quality resulted in a definite enhancement from
the MTJs depending on Heusler materials.
This ended up, which not just an adequate crystallinity from the thin films performs a
significant part within MTJs, yet in which additionally the outer surface roughness, as well as
the user interface morphology in which electrode and the barrier are made of HA, includes an
excellent impact on the TMR esteem. In addition, the place of EF for actual half-metallic (EHM)
gap have a great importance and essential aspect within temperature reliance from TMR
percentage.
Accordingly, a big reduction in the TMR percentage can also be related to the little
energy splitting up between the EF and CBM. This is because of the thermal variations at
ambient conditions, which tend to be two times as large as this energy splitting up. Some key
features of the spintronics devices are the following:
➢ Higher values of TC
➢ Higher spin-polarization
➢ Manipulate involving atomic dysfunction
➢ Manipulate of the interface structure
These desired requirements regarding HMFs as well as their applications in spintronic
gadgets highly signifies that HA tends to be anticipating compounds, for a huge TMR because
of coherent tunneling along their own modified electronic properties as well as magnetic
attributes.
1.6.3 Current-Perpendicular-To-Plane (cpp) Giant-Magnetoresistance (GMR)
Besides the manufacture regarding TMR gadgets, current-perpendicular-to-plane (cpp)
GMR gadgets in which electrodes are made of HA, not too long ago surfaced in an area of
spintronics. Half-metallicity is frequently damaged at the electronic state on the interfaces, so
the cpp GMR devices have a great advantage over to the TMR devices because they are
insensitive at the digital surface.

20
The primary cpp-GMR gadgets contained a couple of Co2MnSi electrodes in which
each electrode is sandwiched with 3 nm Cr spacer (Yakushiji et al., 2006). It ought to be
mentioned that the selection of the spacer layer is critical problem.
The substantial spin dissemination length as well as reduced width are likewise
essential for the spacer coating to acquire wide cpp-GMR values. Such types of factors, joined
with a little lattice confound, prompted to the determination of silver as a perfect spacer
coating. Through an application perspective, a reliable cpp-GMR results 30% with area
temperature will be completely appropriate to make high performance gadgets.
1.6.4 Perpendicular Magnetic Anisotropy
In the magnetoresistance process (GMR or TMR), which are discussed in the previous
part permits to manage the electron circulation via FM nanostructure through their FM
condition. The opposite of this method is also available. The flow of spin-projected current can
easily affect the magnetic state when it flows through the magnetized Nano-structure. Spin-
transfer torque is a highly noticeable, amongst the better encouraging innovations these days,
to fulfill the actual growing need about quicker, scaled-down as well as non-volatile consumer
electronics.
Convoluting this improvement in the direction of scaled-down gadget dimensions is
the truth that power-consumption needs tend to be growing because transistor dimensions
reduce in size towards the sub-100 nm routine as it is illustrated in the Fig. (1.10).
Exchanging the current with a spin is also conceivable because of the mutual connection
among the spin of the inbound conducting carriers and the spin of the electrons in charge of
the area magnetization. Some important features of the spin-torque devices can be portrayed
in the following:
➢ Higher values of TC
➢ Higher spin-polarization
➢ Lower magnetic-damping
➢ Lower saturation-magnetization
➢ High perpendicular-anisotropy

21
Fig. (1.10) When conducting electrons move towards the magnet, their own spin preferentially
lines up within the magnet’s path. Since the electrons experience the nanomagnet, sandwiched
in between levels associated with NM materials near to the set alignment magnetic, the actual
path associated with their own spin is repolarized to complement from the nanomagnet.
Consequently, nanomagnet has the magnetic moment starts to precess, turns just like a
spinning-top on its pivot.

22
Above are the standard layouts for the compounds regarding prospective applications
inside spin-torque appliances. The look for brand new compounds with appropriately planned
properties is a dynamic field progressing research. Particularly tetragonally twisted HA are
typically emphases when innovative magnetic layers inside spin-torque gadgets.
1.6.5 Spin Injection
In spintronic, spin-injection treatment directed into degenerate SCs, for instance, GaAs
is additionally a region of extraordinary technological intrigue (Awschalom and Samarth,
2002). Truth to be told, the scientific utilizations of spin-injection are heap, which incorporate
the control of established data conveyed by spin, low-level formatting, and read-out of spin
qubits (Loss and DiVincenzo, 1998) as well as lucid control of spin associated with the
suggested spin field effect transistor (Schliemann et al., 2003).
The injected polarization of HA is considerably beneath the estimation of 100% that
would be normal for an HM. Conceivable clarifications for this trend involve a neighborhood
atomic mix-up and little EBG (≈ 200 meV) for the minority spin, for instance inside the
Co2MnGe (Picozzi et al., 2002). As a result, Heusler materials along with bigger EBG, for
example, Co2MnSi, might be proficient injectors (Ishida et al., 1998; Schmalhorst et al., 2004).
Given that spin-injection treatment studies investigate the actual SP in the interface, a
reasonable concept does not just need to think about the electronic framework from the
interface, but additionally the actual existence associated with atomic disorder along with the
results associated with non-zero heat. Certainly, these types of elements perform an important
part within interpretation spin-injection dimensions in brand new compounds.
1.6.6 Shape-Memory Compounds
These days, the FH Ni2MnGa framework is a standout amongst the most seriously
researched compounds attributable to its shape memory conduct along with its prospective
function in actuator gadgets, through which strains are manipulated by simply use of an outer
magnetic field. From this framework, under a FM changeover at TC of 376 K, the cubic phase
transition occurs (Webster et al., 1984).

23
Moreover, stoichiometric Ni2MnGa experiences any structural stage changeover
against the higher-temperature cubic L21 arrangement into a lower-temperature martensite
stage (Webster et al., 1984). Because of the structural transition feasibility, a shape memory
impact can be seen now through this L21 framework.
Shape memory compounds were broadened in order to a significant number of
materials, as an illustration, Ni2MnAl, Co2NbSn along with Fe2MnGa (Mañosa et al., 2004;
Zhu et al., 2009) and furthermore QH materials were researched inside this kind of
circumstance (Ito et al., 2008; Kainuma et al., 2008), e.g. magnetic field prompted shape
recuperation was accounted for the compressively distorted NiCoMnIn (Kainuma et al., 2006).
Stress greater than 100 MPa could be produced with this compound by applying the magnetic
field.
1.6.7 Superconductors
The group of HA incorporate not just metallic as well as semiconducting alloys, but
additionally superconducting materials. The initial superconducting HA Pd2RESn and also
Pd2REPb (where RE = rare earth metals) have been investigated simply by Ishikawa et al. in
1982 (Ishikawa et al., 1982). So far, several brand-new superconductors inside the Heusler
composition happen to be documented, their critical temperature, on the other hand, being too
low from an applications perspective. Typically, superconductivity can be discovered
frequently with FH materials which have 𝑁𝑣𝑎𝑙 = 27.
In the course of HH materials, absolutely no superconductor is identified, so these
persist into a non-centrosymmetric compound. One of the main exemption for the HH LaPtBi,
which has a basic exchange temperature around 0.9 K (Goll et al., 2008). About this type of
semimetal having a low transporter concentration, superconductivity had not been anticipated
and it is presently talked about within the framework associated with topological insulators.
Nevertheless, a reasonable comprehension of the cause of superconductivity, magnetism, as
well as their own concurrence in HA is yet absent.

24
1.6.8 Thermoelectric Compounds
As of late, HA materials has drawn excellent technological curiosity because of their
conceivable uses in the field of thermoelectric. Some important features of the thermoelectric
compounds can be portrayed in the following:
➢ Semi-metals with adjustable EBG
➢ Semi-metals with adjustable charge carrier concentration
➢ High Seebeck coefficient
➢ Low thermal conductivity
➢ Industrial processable
➢ Availability of resources
➢ Thermoelectrically compatible
➢ Low cost materials
As we already know that HH compounds along with 𝑁𝑣𝑎𝑙 = 18, display SC attributes.
The band structure information exposed narrow bands, resulting in the higher efficient bulk
along with a big thermo power (Uher et al., 1999). An incredibly favorable position of HA can
be likelihood of doping each of the 3 involved fcc sublattices exclusively keeping in mind, the
goal to enhance the thermoelectric properties.
Probably the most appealing attributes associated with HH compounds for
thermoelectric tend to be their higher Seebeck coefficient S of about 𝑆 ≈ 300 µ𝑉𝐾−1at the
ambient temperature and also their particular large electrical conductivity approximately
ranges from 1000 to 10000 Sc𝑚−1 (Kimura et al., 2009; Schwall and Balke, 2011; Uher et al.,
1999; Xie et al., 2008). The only real disadvantage could be the higher energy conductivity,
which possibly will be up to 10𝑊𝑚−1𝐾−1.
Numerous HH materials have been researched in the previous couple of years to
enhance their thermoelectric attributes (Bhattacharya et al., 2000; Mastronardi et al., 1999;
Uher et al., 1999; Xie et al., 2008). Lately, the actual manufacturing associated with Fe2VAl
thin films, along with higher Seebeck principles as well as reduced thermal conductivity,
brought on by the actual feed framework. These films had been documented permitting their
applications, in the thin film thermoelectric products.

25
1.6.9 Topological Insulators
For the topological insulators, a direct EBG is necessary at the middle position (Γ-point
of the BZ). There are 50 materials are reported in the HA which show the band inversion like
the topological insulator (e.g. HgTe). Topological insulators have several advantageous and a
continuous effort is still in progress for the development of topological insulators.
Specifically, the HH YPtSb, YPdBi, as well as ScAuPb tend to be near the edge. In
between, trivial or topological insulators are astoundingly fascinating materials claiming the
quantum transition which were produced by altering the definite lattice parameter or even little
variation of the atomic arrangement.
Some important features of the topological insulators can be portrayed in the following:
➢ SCs having adjustable EBG
➢ EBG in the bulk
➢ EBG in the quantum well framework
➢ Direct EBG at the Γ-point
➢ Dirac cones of odd numbers
➢ Huge spin orbit-coupling
➢ Parity alteration
➢ Band reversal
➢ Addition of brand-new qualities
➢ Having multiple functions
HH materials are an exceptionally tunable as well as the versatile course of compounds.
The numerous completely unique quantum phenomena like topological superconductivity and
quantized anomalous Hall Effect which also have the experimentally acknowledgment can
exist in the HH structure. Additionally, they unlock the new investigation and gives the
instructions in the direction of multifunctional topological gadgets regarding spintronic as well
as fault-tolerant quantum computing.

26
1.7 Heusler Moves Nano
There is no uncertainty, nowadays, how the development associated with
nanotechnology has already established a massive effect on a variety of research territories.
The basis behind this situation is the truth that nanocrystals compounds display physical
attributes which are very not the same as their own bulk partners. Limited size impacts that
result into the quantum containment which have crystal in the Nano range prompt the
development of innovative HMFs marvels. These HMFs may be used inside the huge range of
appliances associated with various applications.
Specifically, magnet nanoparticles have obtained massive attention regarding
appliances in several fields, for instance, drug delivery, data-storage gadgets, biomedical
imaging and catalysis (Hyeon, 2003; Raj and Moskowitz, 1990; Schladt et al., 2010; Sun,
2006). Given that a lower particle dimension refers to some bigger surface-to-volume
percentage, also numerous uncompensated spins can be found upon scaled-down
contaminants, consequently ensuing within improved values of µ𝑡𝑜𝑡.
The ball milling method is the typical strategy to manufacture the nanoparticles out of
the bulk compounds (De Santanna et al., 2008; Hatchard et al., 2008; Peruman et al., 2010;
Zhang et al., 2003a). As an example, FM Ni2MnGa nanoparticles have been well prepared by
firstly employing the ball milling approach besides post annealing method (Wang et al., 2007).
Just lately, ternary Heusler nanoparticles had been effectively synthesized through precursors
as well as their magnetic along with structural attributes have been explored (Kodama et al.,
1997; Wang et al., 2010). The immersion magnetization with lower temperatures is comparable
to the bulk worth that signifies, how the HM properties tend to be maintained within the
nanostructured compound.
1.8 Dissertation Scheme
HA sponsor an array of unpredicted unique physical features that cannot be obtained
through the properties from the exclusive atoms within the unit cell. Although different types
of HA exist, like FH, HH, QH, Inverse FH alloys, but our focus was only on the HH materials
among different types of HA. In this dissertation, a scientific contribution has been made to
study and discover the new HM HH XYZ materials to achieve higher EHM for the spintronic
applications.

27
The design of the selected elements from the periodic table of the investigated HH XYZ
materials is illustrated in the Fig. (1.11). A plethora of exotic HM properties of the new HH
XYZ materials is studied based on the first-principle calculations in this dissertation. The
structural, electronic, magnetic, thermal and HM properties of the series of HH CrTiZ (where
Z=Si, Ge, Sn, Pb), FeVZ (where Z= Si, Ge, Sn), YCrSb & YMnSb, YMnZ (where Z= Si, Ge,
Sn) and the HH XYZ materials where (X= Li, Na, K, Rb, Cs & Y=V, Nb, Ta & Z=Si, Ge, Sn,
S, Se, Te) are thoroughly discussed in the coming chapters of this dissertation. The ground
state properties like lattice parameters, the correct ground, and magnetic state, structural
stability, their equilibrium volume, total minimized energy, bulk modules and its pressure
derivative, total and partial magnetic moments, energy band gaps and many other important
aspects of the HH materials are explored. The computational calculations are performed by
using different DFT codes like WIEN2K, VASP, BoltzTrap, Phonopy for the study of the HH
XYZ materials in this dissertation. The dissertation scheme is distributed as follows:
➢ Chapter 1 covers the fundamentals of the HA and their applications in the spintronic
devices
➢ Chapter 2 provides the brief literature survey of the Heusler materials
➢ Chapter 3 gives the basic idea about computational parameters, techniques, and
details about DFT
➢ Chapter 4 presents an ab-initio study of new series of HH CrTiZ (where Z = Si, Ge,
Sn, Pb) materials in which HM ferromagnetism is explored
➢ Chapter 5 is all about the spin-polarized calculations of structural, electronic and
magnetic properties of the HH FeVZ (where Z=Si, Ge, Sn) alloys by using ab-initio
method
➢ Chapter 6 describes the theoretical investigations of HM ferromagnetism in new
HH YCrSb and YMnSb alloys using first-principle calculations
➢ Chapter 7 depicts about the physical properties of HH YMnZ (where Z= Si, Ge,
Sn) compounds via ab-initio study
➢ Chapter 8 demonstrate the structural chemistry and physical properties of the newly
designed HH XYZ materials with large spin-gaps
➢ Chapter 9 draws the conclusion and give a summary of this thesis and suggest the
future framework for the HH materials

28
Fig. (1.11) Selected elements from the periodic table for the studied HH XYZ materials in this
dissertation based on the color plan.

29
Chapter 2
Literature Review
Magnetism is referred to as an old phenomenon which has numerous cutting-edge
surprises. The man’s fascination with magnetism dates back hundreds of years, however, their
perception of it, is recent and still inadequate. In naturally existing materials, almost all
magnetic materials contain Fe and experimentally made magnetic compounds comprises of
one or more with the FM TMs like Fe, Co or Ni (Webster, 1969). In 1903, Fritz Heusler
synthesized Cu2MnAl alloy which acts as an FM even though none of involving component
aspects can be a magnet on its own (Heusler, 1904).
Due to the potential applications of the HMF materials from the perspective of
spintronic and magneto-electronics, they are under intensive investigation from the various
research groups in the last two decades (Hirohata and Takanashi, 2014; Žutić et al., 2004).
There are numerous distinctive advantages such as ultrafast processing speed of increased data,
enhanced integration densities, much lower consumption of electric power by the manipulation
and actively controlled of the spin degrees of freedom into the electronic appliances other than
conventional SCs (Prinz, 1998; Prinz, 1999; Wolf et al., 2001).
In 1983, by using the first-principles calculations, de Groot and his coworkers (De
Groot et al., 1983) from the electronic structure for the NiMnSb showed that it is fully HMFs.
The NiMnSb is the most researched compound (Watanabe, 1976) which show 100% SP at the
(EF). This means, the band structure of the NiMnSb display metallic character at one spin
channel whereas exhibit SC behavior at the opposite channel. The existence of EBG in one of
the spin channels gives 100% SP at the EF which ultimately leads the 100% SP current. In this
way, HMFs are potential contenders for the magneto-electronic appliances.
Other than HH and FH alloys (Galanakis, 2004; Zhang et al., 2003b; Zhang et al.,
2004), HM behavior is identified in several compounds as well like diluted magnetic SCs in
which impurity in the form of Mn is added in Si or GaAs (Akai, 1998; Stroppa et al., 2003),
the manganites i.e. La0.7Sr0.3MnO3 and some CrO2 and Fe3O4 oxide materials (Soulen et al.,

30
1998), also in CaAs which is called d0 ferromagnets (Coey, 2005), the pyrites i.e.
CoS2(Shishidou et al., 2001), the pnictides i.e. CrAs and the TMs chalcogenides in the wurtzite
or zinc-blende structure (Akinaga et al., 2000; Continenza et al., 2001; Galanakis, 2002a;
Mavropoulos et al., 2004; Mizuguchi et al., 2002; Nagao et al., 2004; Pask et al., 2003; Sanyal
et al., 2003; Shirai, 2001; Xie et al., 2003; Zhang et al., 2003c; Zhao et al., 2003), the double
perovskites i.e. Sr2FeReO6 (Kato et al., 2004), and in the europium chalcogenides i.e. EuN
(Horne et al., 2004).
Even though, in the practical manner, 100 % SP is also achieved at the EF with the
ambient conditions for the thin films obtained from La0.7Sr0.3MnO3 and CrO2 (Kato et al., 2004;
Soulen et al., 1998), but due to the fairly higher TC (Webster and Ziebeck, 1988a) of the HA,
they are considered more fascinated for the technological applications such as spin-filters
(Kilian and Victora, 2000), devices like giant-magnetoresistance (GMR) (Caballero et al.,
1998; Hordequin et al., 1998) or spin-polarized tunnel junctions (Tanaka et al., 1999) and so
called spin-injectors (Datta and Das, 1990).
A range of diverse magnetic phenomenon such as localized and itinerant magnetism,
Pauli paramagnetism, helimagnetism, ferromagnetism, antiferromagnetism, or heavy-
fermionic behavior (Gilleßen and Dronskowski, 2010; Toboła et al., 2003; Webster and
Ziebeck, 1988a; Ziebeck and Neumann, 2001) occurs in this fascinating family of HA which
consists of a large number of magnetic members.
The HH HM NiMnSb material has drawn lots of experimental curiosity. The
experiments like positron-annihilation (Hanssen and Mijnarends, 1986; Hanssen et al., 1990)
and infrared absorption (Kirillova et al., 1995) had successfully grown the HM single crystals
of NiMnSb. The high-quality films of NiMnSb are also produced but they do not show HM
behavior (Bona et al., 1985; Clowes et al., 2004; Mancoff et al., 1999; Zhang et al., 2004; Zhu
et al., 2001).
In contrast of 100% SP of single crystals of NiMnSb, high quality films of this
compound have only 58% SP at the EF achieved by Soulen et al. (Soulen et al., 1998). Such
types of SP measures are coherent to magnetoresistance calculated for NiMnSb based spin-
valve framework (Caballero et al., 1999; Kabani et al., 1990), the tunnel magneto resistance
junction (Tanaka et al., 1997) along with a superconducting tunnel junction (Tanaka et al.,

31
1999). The reason for losing HM characteristics for the HH NiMnSb epitaxial thin films is due
to the surface separatism associated with Mn and Sb atoms, and that is not even close to staying
ideal (Caruso et al., 2003; Komesu et al., 2000; Ristoiu et al., 2000a; Ristoiu et al., 2000b). In
addition to experimental research, NiMnSb lured additionally substantial interest amongst
numerous theoretical studies and many first-principles calculations verified the HM behavior
(Galanakis et al., 2000; Halilov and Kulatov, 1991; Kulatov and Mazin, 1990; Wang et al.,
1994). Larson and his coworkers show that the most stable structure of NiMnSb is the C1b
framework by the exchange of atoms (Larson et al., 2000).
The work done by the Orgassa et al. demonstrated that HM character kept maintained
by producing atomic disorder of some percentage in NiMnSb material although it causes
minority spin states within the semiconducting gap as well (Orgassa et al., 1999). Several
studies done on the surface properties of the different materials showed that interfaces or
surface of the HM materials loses the half-metallicity when layered with other SCs (Galanakis,
2002b; Galanakis, 2005; Galanakis et al., 2008a; Jenkins and King, 2002; Ležaić et al., 2005).
However, some researchers Wijs and de Groot along with Debernardi et. al. suggested that for
few layers of thin films, the possibility to regain HM persona for the HH NiMnSb material
(De Wijs and De Groot, 2001; Debernardi et al., 2003).
Lastly, Kübler (Webster and Ziebeck, 1988a) theoretically determined that NiMnSb
has the very accurate value of the TC is equal to 770 K that was very close to the experimental
result. The magnetic properties of these exceptional class of HA can be tuned by doping of
sp-electron substitution (Galanakis et al., 2008b; Özdoğan et al., 2009). The first series of
synthesized material from the HA was the FH materials (Suits, 1976; Webster, 1971; Ziebeck
and Webster, 1974). Kübler et al. were the first one to analyze the stability mechanism of the
FM and AFM phases of these HA (Kübler et al., 1983).
On the other hand, the presence of half-metallicity in the FH materials theoretically
predicted by the Japanese researchers. Ishida and collaborators were the pioneers to investigate
the ab-initio structural electronic calculations of the FH Co2MnSi and Co2MnGe materials
(Fujii et al., 1990; Ishida et al., 1982; Ishida et al., 1995) whereas FH Fe2MnSi and Fe2MnGe
materials investigated by Fujii et. al. (Ishida et al., 1995).

32
But polarized neutron diffraction experiment done by Brown et al. demonstrated that
the HM behavior is destroyed by the appearance of the small DOS around EF. Kübler et al.
verified this fact when he performed ab-initio computations on the FH Co2MnAl and Co2MnSn
materials (Kübler et al., 1983).
Until now, several HMFs are synthesized experimentally based on HA like Mn2-, Fe2-
and Co2- etc (Balke et al., 2006; Hongzhi et al., 2007; Luo et al., 2008a; Özdogan et al., 2006;
Shan et al., 2009) but there is still need to explore the HM properties theoretically as well as
experimentally for the useful spintronic applications.
2.1 Motivation for the work
As a detailed introduction chapter and literature survey of the HM HA gives so much
information about the application of the HM HA. The discovering, designing and functionality
of the new HM Heusler materials could be a meaningful work for the applications in the
spintronic devices. The simple structural framework of the Heusler materials which also have
lattice parameter comparable to numerous SCs have the large values of the magnetic moment
and TC, therefore are fundamental for the spintronic applications.
Therefore, it is very essential scientific outcome that the Heusler materials display
magnetic and semiconducting properties simultaneously. To discover new HM materials
experimentally is very hard as the confirmation of 100% SP is needed for the expected HMFs.
So DFT investigated band structure calculations play an essential part in this regard. The First-
principle estimations, particularly DFT have grown to be an extremely helpful technique
regarding predicting the physical properties of the different alloys.
The problem with the HMFs is that, the discovered HM properties are often disappear
due to the very small values of band gaps in the HA when the small lattice mismatch exists
due to strains in the HA also when they coupled with other conventional SCs. Consequently,
discovering brand new HMFs along with the larger values of EBG and EHM are vital regarding
useful for the spintronic devices.

33
In this dissertation, some new series of HMF HH materials are predicted by using first-
principles calculations. The structural, electronic, magnetic and thermal properties of the HH
CrTiZ (Z=Si, Ge, Sn, Pb), FeVZ (Z= Si, Ge, Sn), YCrSb and YMnSb, YMnZ (Z= Si, Ge, Sn,
Pb) materials are explored for the first time. Our proposed HM HH materials can be very useful
for the spintronic applications due to their large HM gaps, capable of providing 100% SP at
the EF with their equilibrium lattice constants and have adequate magnetic moment ranging
from of 1 µB to 4 µB.

34
Chapter 3
Basics of Density Functional Theory (DFT)
There are numerous fields inside the material sciences as well as in engineering where
it is very crucial to logical and innovative advancement is being familiar and managing the
properties associated with matter from the number of distinct atoms and molecules. Density
functional theory (DFT) is really a remarkably prosperous method of discovering results to the
essential formula, which explains the actual quantum conduct associated with atoms as well as
substances along the Schrödinger equation, with configurations associated with useful worth.
This method has quickly developed as being a specific artwork used through a number of
physicists as well as chemists, in the leading edge associated with quantum mechanized
concept to some device, that have specialty to frequently use, by many scientists in
biochemistry, physics, material science technology, chemical substance architectural, geology,
along with other professions (Sholl and Steckel, 2011).
The layout of this chapter is separated into two sections. The initial segment is about
the presentation of the fundamentals of density DFT which includes solving many body
Schrödinger equations and furthermore discuss the DFT in crystalline solids which is our
general concern about. The second part includes how to perform DFT estimations which
includes the little overview of WIEN2K software which is applied to run the DFT calculations.
3.1 Computational Material Science
Computational materials science is a field that is not necessarily associated with
computer coding. Materials researchers perform tests on the computers. DFT calculations that
are also known as first-principles calculations or ab-initio (which means from the beginning)
calculations. Ab-initio calculations offer three main segments;
➢ Theory (e.g DFT)
➢ Numerical methods and code
➢ Computer

35
The whole theory does not depend on the empirical parameters. It is just plain and
simple Quantum mechanics which is building up the whole world. In computational material
science, the computational scientist set up the experiments and let the computer apply the
theory and get a lot of nice properties like structural, vibrational, electronic, magnetic, and
thermal properties and much more describing any material. The beautiful thing about the
computational materials science is to illustrate the physical attributes of the matter by using
some theoretical methods rooted in the fundamental equations.
3.2 Many Particle Problem in Solids
The aim of DFT calculations to seek the ground state (most stable state of the
framework) for the set of particles and a useful approach to be carried out for the solution of
the many body Schrödinger equations.
ĤΨ ({ri }, {RI}) = EΨ ({ri }, {RI})
So, in the above equation, Ψ is a wave function which portrays the considered framework and
Ĥ and E expresses to the Hamiltonian and Energy operator which are applied to the wave
function. Also, the Hamiltonian operator can be described as
Ĥ = �̂� + �̂�𝑐𝑜𝑢𝑙𝑜𝑚𝑏
in which �̂� operator taking care of kinetic energy and �̂�𝑐𝑜𝑢𝑙𝑜𝑚𝑏 is the coulomb potential
operator which includes all the coulomb’s interactions between the two charges. The above
equation describes the basic quantum mechanics in which if the single electron state is selected
then our calculations will be simple which make the life easier. There are also bunch of nuclei
a and electrodes which makes the equation sort of very complicated to solve. Therefore,
making this equation a somewhat less difficult and the first thing to be carried out is to utilize
the Born-Oppenheimer approximation.
3.3 The Born Oppenheimer Approximation
According to this estimation, which can be stated as
mnuclei >> me.

36
This approximation says that nuclei tend to be very large plus heavy which makes them
slow to move whereas electrons are tending to be small and that is the reason they move very
fast. This means that the actual dynamics of the nuclei and electrons can be decoupled,
therefore the duration of time for the electrons must discover their ground state for just one
placement from the nuclei is a lot quicker compared to nuclei can proceed which means the
electrons begin to see the exterior potential associated with static nuclei, therefore by
decoupling the wave function
Ψ({𝑟𝑖 }, {𝑅𝑖}) → 𝛹𝑁 ({𝑅𝐼}) + 𝛹𝑒 ({𝑟𝑖 })
In the above equation 𝛹𝑁({𝑅𝐼}) represents the nuclear wave function and 𝛹𝑒 ({𝑟𝑖 }) is
termed as electronan ic wave function. It means, now the focus will be made only on to solve
the ground state of the electrons such as static ga roup of atomic placements therefore by
decreasing the list of parameters a little into the Hamiltonian,
Ĥ𝛹(𝑟1, 𝑟2, 𝑟3, … … … , 𝑟𝑁) = 𝐸𝛹(𝑟1, 𝑟2, 𝑟3, … … … , 𝑟𝑁)
So, the more detailed description of the Schrödinger equation becomes when the multiple
electrons are interacting with multiples nuclei making the equation more complex, and now
the Hamiltonian contains the three terms consisting of electronic variables, therefore
Ĥ = −ħ2
2𝑚𝑒∑ 𝛻𝑖
2
𝑁𝑒
𝑖
+ ∑ 𝑉𝑒𝑥𝑡
𝑁𝑒
𝑖
𝑟𝑖 + ∑ ∑ 𝑈 (𝑟𝑖 , 𝑟𝑗)
𝑗 >1
𝑁𝑒
𝑖=1
here, 𝑚𝑒 stands for the mass of the electron and one of three terms define the kinetic and
potential energy and second term shows the electrons interacting with the nuclei which see
themselves as an external potential and then the last term represents the electron-electron
repulsion. Let just pause and think of how big our problem this turns out and how fast this
escalates in terms of dimensions.

37
3.4 Why Density Functional Theory (DFT) Needed?
For the real materials, just take an example of carbon dioxide which has 22 electrons.
CO2: 6 + 16 = 22 electrons
Each electron in the CO2 molecule are described by three spatial dimension coordinates that
means solving the Schrodinger equation becomes a 66-dimensional problem. Consider another
example is of Pb nanocluster in which each Pb atom consists of 82 electrons. So, if the
nanocluster has 100 atoms then the specific nanocluster will contain 8200 electrons and
therefore Schrodinger equation becomes a 24,600-dimensional problem. So, by solving the
many body Schrodinger equation is for all practical materials a bit annoying. So, that is the
reason to approach the DFT.
3.5 Density Functional Theory (DFT)
To move from a wave function to electron density, which is true observable that in
principles, can be measured and defined from the wave function that reduces from 3N
dimensions to 3 spatial dimensions. So, the electron density is of only three dimensional,
𝑛 (𝑟) = 𝛹∗(𝑟1, 𝑟2, 𝑟3, … … … , 𝑟𝑁) 𝛹 (𝑟1, 𝑟2, 𝑟3, … … … , 𝑟𝑁)
So, that seems to be sort of a hint. Let’s go in that direction. Let’s try to solve the Schrodinger
equation in terms of considering the electron density intent. Now, make another approximation,
in which spouse that the Jth electron as a point charge, which is placed in all other electrons
field, that will simplify the many-electron problem to many-one electron problem.
So, situation now becomes like this
𝛹 (𝑟1, 𝑟2, 𝑟3, … … … , 𝑟𝑁) = 𝛹1(𝑟1) ∗ 𝛹2(𝑟2) ∗ 𝛹3(𝑟3) ∗ … … … ∗ ∗ 𝛹𝑁(𝑟𝑁)
which is a Hartree Product. Still, there is necessity to know about the molecules to
calculate. So that means, the electron density in terms of the single electron wave function
can be defined as
𝑛 (𝑟) = 2 ∑ 𝛹𝑖∗
𝑖
(𝑟)𝛹𝑖(𝑟)
which is another step to approaching at the heart of DFT.

38
3.6 Hohenberg and Kohn (at the heart of DFT)
So, the heart of DFT is based on two very fundamental theorems.
3.6.1 Theorem I
The 1st theorem conditions about the ground state energy from the Schrödinger
equation, which can be a unique functional for the density of electrons.
𝐸 = 𝐸 [𝑛(𝑟)]
The electron density is all which is needed to be defined the ground state energy. So that is the
good thing and 2nd theorem is about to find out the ground state density.
3.6.2 Theorem II
To precisely minimize the energy functional means that to moving down the well until
the ground state electron density has been found.
𝐸 [𝑛(𝑟)] > 𝐸0[𝑛0(𝑟)]
Now, let’s talk about a bit of energy functional.
𝐸[{𝛹𝑖}] = 𝐸𝑘𝑛𝑜𝑤𝑛[{𝛹𝑖}] + 𝐸𝑋𝐶[{𝛹𝑖}]
In which
𝐸𝑘𝑛𝑜𝑤𝑛[{𝛹𝑖}] = −ħ
𝑚𝑒∑ ∫ 𝛹𝑖
∗𝛻2𝛹𝑖
𝑖
𝑑3𝑟 + ∫ 𝑉(𝑟)𝑛(𝑟) 𝑑3𝑟 + 𝑒2
2∬
𝑛(𝑟)𝑛(𝑟′)
𝑟 − 𝑟′𝑑3𝑟𝑑3𝑟′
+ 𝐸𝑖𝑜𝑛
and the term, 𝐸𝑋𝐶[{𝛹𝑖}] represents the Exchange-correlation (XC) functionals, which include
all the quantum mechanical terms and the not known terms needs to be approximated. Some
basic XC functionals are the following
➢ Local density approximation (LDA),
➢ Generalized gradient approximation (GGA),
➢ modified Becke-Johnson (mBJ) approximation etc.

39
The energy functional can be divided into two parts. One that is known, and the other
is unknown. So, the known part is basically all the energy terms that are already described, like
the kinetic energy and all the potential energy terms which are connected just to the coulomb’s
reaction. The exchange-correlation functional that take cares of all the quantum mechanical
interactions between the electrons and about the not known terms that something exists, but
unfortunately, the information about them is lacking.
So in all calculations of DFT, this is something needs to be approximated, so the
simplest XC functional that one will stumble upon in the DFT is called the LDA, which is only
basic approximation of local electron density and then the GGA which is also taken into
account to the gradient of the electron density and there is a lot of developments going on all
the time on improving the XC functionals.
3.7 The Kohn-Sham Scheme
The Kohn and his post doctorate Sham have discovered that how to obtain the ground
state energy of the electron density in practice. They developed the scheme by considering the
exact state of single electron wave functions which are not interacting. So, single electron wave
functions are non-interacting systems. The interactions are sort of implicitly accounted for, in
these potentials.
[−ħ2
2𝑚∇2 + 𝑉(𝑟) + 𝑉𝐻(𝑟) + 𝑉𝑋𝐶(𝑟)] 𝛹𝑖(𝑟) = ∈𝑖 (𝑟)𝛹𝑖
So, the Hamiltonian for the single electron wave functions, 𝑉(𝑟) is the external potential and
then of the Hartree potential 𝑉𝐻(𝑟) that is just one electron interacting with the electron density
and then this exchange-correlation potential 𝑉𝑋𝐶(𝑟), which must be approximated.
3.8 Self-Consistency Scheme
In the DFT estimations, an iterative method also called self-consistency field (SCF) is
performed during the calculations. The SCF cycle consists of following steps:
• The 1st step, an initial guess of atomic density𝑛(𝑟)is made depending on the placement
of atoms.

40
• In 2nd step, the Hamiltonian operator is placed on the electronic system and set of Khon-
Sham equations are solved to calculate the electron wave functions 𝛹𝑖(𝑟).
• In 3rdstep, calculation of electron density is made depending upon the single electron
wave function. i.e 𝑛(𝑟) = 2 ∑ 𝛹𝑖∗(𝑟)𝛹𝑖(𝑟)𝑖
• In the last 4th step, a comparison is made between the 1st initial guess electron density
n(r) and the obtained electron density. If the values are same, then the true ground
density is achieved and if different then the calculations start again from the 2nd step
with the new value of electron density n(r)
So, one equation for each electron to obtain a set of wave functions and it is already
discussed, how this correlates with the electron densities. After that, recalculate the electron
density and if the electron density resulted, was the same one as the initial guessed value which
has been set, in the self-consistency loop, so that means the true ground state density is
achieved. If it is not the same, then replace the old electron density and put it as new trial and
then just loop through again.
That is how minimization of the electron density is done and try to search for the lowest
ground state and electron density by loop through this (SCF cycle). Now it is super easy to find
the ionic ground state. By electron density, the forces on ions can easily be calculated,
𝐹𝐼 = − 𝑑𝐸
𝑑𝑟𝐼= − < 𝛹𝑖 |
𝜗𝐻
𝜗𝑟𝑖| 𝛹𝑖 >
and then just move along the steepest distance of the ionic forces to obtain the ionic ground
state. So, in practice, an algorithm is used. After getting to the ground state, one can go even
further and move the ions, a bit away from their equilibrium position. Then, force constants
and vibrational frequencies are obtained, and the phonon dispersion curve can be calculated
for the ions. There are lot of possibilities.
3.9 Crystalline Solids and Plane-wave Density Functional Theory (DFT)
Now, moving towards the more specific case of crystalline solids. Now, the concern is
about the plane wave DFT. The crystal is a periodic arrangement of atoms. Consider the nuclei
which have some positive charge and represent the periodic potential U(r).

41
A free electron is represented by a plane wave (𝑒𝑖𝑘𝑟), but what happens with the wave
function when the electron is considered into the crystal. The Bloch finds the Bloch waves
𝛹𝑛𝑘(𝑟), by considering the electrons in a periodic potential.
𝛹𝑛𝑘(𝑟) = 𝑒𝑖𝑘𝑟 . 𝑢𝑛𝑘(𝑟)
So, Bloch waves are the plane waves which are modulated by some random
potential 𝑢𝑛𝑘(𝑟) which is also periodic with the lattice. Basically, they are perturbed free
electrons. So, these are the plane wave DFT which sort of comes out into the discussion. Now,
some important concepts in terms of DFT calculations or plane wave DFT are discussed.
3.9.1 Cutoff Energy
The first key parameter is cutoff energy. A very central concept in the DFT is reciprocal
space. Mathematically, it is just the Fourier transformation of the real axis where all the
dimensions are turned upside down.
𝛹𝑛𝑘(𝑟) = exp(𝑖𝑘. 𝑟) 𝑢𝑛𝑘(𝑟) = exp(𝑖𝑘. 𝑟) ∑ 𝐶𝑘
𝐺
exp (𝑖𝐺. 𝑟)
Basically, everything which is large in real space turns small in the reciprocal space
and the small things in real space extend far in the reciprocal space. The periodic function can
be expanded in terms of the Fourier series. So, the Bloch functional which is the electronic
wave function in the unit cell and the 𝑢𝑛𝑘(𝑟) is periodic with the lattice. By expanding it, the
wave vector turns out to be sort of reciprocal lattice vector. The Bloch wave are represented
as a summation of plane waves which has a wave vector of G + K. The kinetic energy of each
plane wave in the sum can be given as,
𝐸 = ℎ
2𝑚[𝐾 + 𝐺]2
Thus, the sum over an infinite number of reciprocal lattice vectors is needed. The
problem numerically is of course that one cannot deal with the infinite sums. In DFT
calculations, some sort of cut off energy must be defined so each plane wave in this sum has a
kinetic energy which is given by the above expression. So, for a larger reciprocal lattice and
vector, upsurge of kinetic energy is needed.

42
During the DFT calculations, for the considered plane waves, a cutoff energy is defined
which have the higher kinetic energy then this specific cutoff. This is an important input
parameter for the DFT calculations. To choose the suitable high enough cutoff energy, a
convergence test in terms of total energy is needed to be performed.
3.9.2 K-points
Another important concept is k-point sampling. In the reciprocal space, wave vectors
are plane wave vectors. They expand in the reciprocal space and the primitive unit cell is
termed as Brillouin Zone (BZ) or first BZ.
𝐾 = 2𝜋
𝜆[
1
𝑚]
Any k-vector which extends the primitive cell and reciprocal space, or it extends the
BZ. It can be written as the sum
𝑘′ = 𝑘 + 𝐺
The k-vector is just differing by some reciprocal lattice vector G which is basically the
same wave with some sort of face shifted. It means in terms of considering the plane wave
vector, only consider the first BZ. Without K-point sampling, numerically integrations cannot
be done, so in DFT terms of integral evaluation, only integrate over the first BZ.
It is also an important part of the DFT calculations that the researcher should map and
define the BZ in terms of k-points and the number of k-points must be sufficiently large so that
true, reliable and converged values of the energies can be obtained.
3.10 Pseudo-Potentials
Another important concept is the pseudo potentials in the DFT calculations. In terms
of the physical properties like chemical bonding and other characteristics of the material which
must be taken special care of, are mainly characterized by the outer electrons or so called the
valence electrons.
So, let’s make life a bit easier for ourselves. Instead of considering all the electrons of
the system, the DFT calculations can be made simpler by considering only the valance

43
electrons and freezing the inner electrons of the system while minimizing the electron energy
which makes the calculations much easier to do. A nice thing about the currently DFT code is
that they provide the pseudopotentials. It is not like the researcher must recalculate the Pseudo-
Potentials every time. There is often a library with the pseudopotentials for the use, of each
element in the periodic system.
3.11 WIEN2K
To examine the different physical properties, to develop the modern devices from the
solid materials at the atomic level, electronic structure calculations based on DFT are
performed, to the ideal crystal at ambient condition. This enables the quantum mechanized
discourse of the physics of the different physical properties, for instance, ground state
properties, volume optimization, electric, elastic, optical, chemical bonding, the dynamic
stability of the crystal, mechanical, the transition of phases, magnetic properties and so on. To
solve the equations of DFT, several procedures are designed.
There are numerous DFT simulations codes available but WIEN2K is probably the
speediest and trustable simulation package between other computational techniques. The
WIEN2K code is embedded in the DFT, which is already discussed in the earlier sections of
this chapter. WIEN2K includes numerous impartial F90 applications, that are connected
collectively by C-shell or Perl-scripts. WIEN2K utilizes the full-potential linearized
augmented plane wave method, which is probably the most accurate approach, developed by
the Blaha et al (Blaha et al., 2001b) and is founded on the Kohn-sham formalism regarding
DFT.
These days, greater than 500 research groups apply the WIEN2K simulation code
around the world to unravel the crystal properties on the nanoscale. These WIEN2K
computations operated on any kind of Unix/Linux systems through computers, work stations,
servers, supercomputers for the different crystal systems consisting up to 100 atoms per
formula unit cell. WIEN2K is very user-friendly simulation code based on the web graphical
user interface (w2web) along with a very good graphical user interface (GUI). The accuracy,
efficiency, and performance of the WIEN2K code are maintained as high as possible which
manage into a benchmark simulation package for the solids.

44
This dissertation contains the work on the HM HH XYZ materials which are fully
performed by using the state-of-art WIEN2K simulation code. Some other DFT simulation
packages like VASP, BoltzTrap, Phonopy are also used for the study of the HH XYZ materials
in this dissertation. This dissertation provides the general summary of an exceptional types of
HH alloys which have countless functionalities, ranging from semiconductors to metals and
HMFs which have lot of opto-electronics, thermoelectric and spintronic applications.

45
Chapter 4
Half-Metallic Ferromagnetism in New Half-Heusler Compounds:
An Ab-initio Study of CrTiZ (where Z= Si, Ge, Sn, Pb)
4.1 Introduction
Half metallic ferromagnets (HMFs) have attracted many researchers in the last decades
due to their unique properties and potential applications in spintronic devices (Jimbo et al.,
1993; Julliere, 1975; Ohno, 1998). Research on spintronic is currently very vigorous
worldwide since HMFs have the capability to use both charge and spin degree of freedom in
the solid materials to achieve multifunctional electronic devices (Prinz, 1998; Wolf et al.,
2001). The perspective of encryption and decryption of data from magnetic hard disk drives
makes these materials highly desired in the computer industry (Birsan and Palade, 2013a;
Carey et al., 2007). In addition, the SP at the EF allows these materials to be incorporated in
applications which include nonvolatile magnetic random access memories (MRAM) and
magnetic sensors (De Groot et al., 1983; Li et al., 2014; Wolf et al., 2001; Žutić et al., 2004).
The earliest of theoretical works carried out by de Groot et al. on NiMnSb and PtMnSb
HH compounds (De Groot and Buschow, 1986; De Groot et al., 1983) stimulated numerous
groups for exploring half metallicity in other compounds based on zinc blende SCs (Galanakis
and Mavropoulos, 2003; Xu et al., 2003; Yao et al., 2005a; Yao et al., 2005b; Zhang et al.,
2004) , FM metallic oxides (Lv et al., 2011; Song et al., 2009a; Szotek et al., 2004), transition
metal oxides (Lewis et al., 1997), perovskite manganite (Kato et al., 2002), transition metal
chalcogenides (Xie et al., 2003), dilute magnetic SCs (Saeed et al., 2010; Zhang et al., 2008),
FH compounds (Bai et al., 2011; Kandpal et al., 2007; Luo et al., 2009; Sharma et al., 2010a;
Xu et al., 2012; Zenasni et al., 2013) and HH compounds (Casper et al., 2012; Chen et al.,
2011; Lakdja et al., 2013; Nanda and Dasgupta, 2003; Rozale et al., 2013; Umamaheswari et
al., 2014).

46
Among these materials, HM HA are most prominent owing to their probability of
achieving higher value TC (Şaşıoğlu et al., 2004). Moreover, the structure of Heusler
compounds matches considerably with zinc blende and diamond structures which prevail in a
large variety of known SCs (Lin et al., 2014).
Additionally, HM HA show conduction of electrons in one spin direction around the
EF i.e. possess metallic behavior and are insulator or SCs in opposite spin orientation (Birsan
et al., 2012). Although in recent years some HH compounds like NiVM (M=P, As, Sb, S, Se
and Te) (Zhang et al., 2004), NiCrZ (Z=Al, Ga, In, P, As, Sb, S, Se and Te) (Galanakis et al.,
2008a; Luo et al., 2008b; Van Dinh et al., 2009), XMZ (X=Fe, Co and Ni; M=Ti, V, Nb, Zr,
Cr, Mo and Mn; Z=Sb and Sn) (Nanda and Dasgupta, 2003), XYZ (X=Li, Na, K and Rb;
Y=Mg, Ca, Sr and Ba; Z=B, Al and Ga) (Umamaheswari et al., 2014), XYZ (X(Nanda and
Dasgupta, 2003), Y=V, Cr, Mn, Fe, Co, and Ni; Z=Al, Ga, In, Si, Ge, Sn, P, As, and Sb) (De
Groot et al., 1986) have been predicted which possess HMFs nature by means of first-
principles calculations but a little work has been done experimentally on these compounds due
to the difficulty in synthesizing the chemical composition of HH materials like CoMnSb and
CrMnSb.
Nonetheless, it is very meaningful to investigate structural stability, electronic,
magnetic and HM behaviors of these compounds. According to our knowledge, no theoretical
or experimental research on the half-metallicity of CrTiZ (Z= Si, Ge, Sn, Pb) has been reported
yet. Motivated by this, structural, electronic and magnetic properties of these HH CrTiZ (Z=
Si, Ge, Sn, Pb) compounds are investigated by using density functional theory (DFT)
calculations.
4.2 Crystal Structure and Computational Details
HH compounds are intermetallic ternary with a 1:1:1 stoichiometry XYZ and
crystallizes into non-centrosymmetric cubic (MgAgAs) C1b configuration (Webster and
Ziebeck, 1988b) using space group of F-43m consisting on four percolate face centered cubic
sub lattices passed through the three atoms X, Y and Z, and a vacant site.
In the unit cell of this structure, the atoms X, Y, and Z are located at the corresponding
Wyckoff positions a1= (1
2,
1
2,
1
2), a2= (
1
4,
1
4,
1
4), a3=(0, 0, 0) sites, respectively, while (
3
4,
3
4,
3
4) site is

47
empty (Umamaheswari et al., 2014). In general X and Y are an alkali metal, transition metal
or rare-earth metal and Z is the main group element.
The X atom is placed at a1 and Z at a2 forming a rock salt lattice. On the other hand,
the Y atom is located at the center of the tetrahedron molded by X and Y atoms as shown in
Fig. (4.1). Generally, there are six modes in which X, Y and Z atoms can be distributed above
the three sub lattices. The interchanging of atoms at a1 and a2 results at equivalent positions
due to symmetry, which means that X, Y and Z atoms can be prescribed at (a1, a2, a3), (a3, a1,
a2), and (a2, a3¸ a1).
Due to this, to form C1b structure, there are three possible arrangements (Type 1, Type
2, Type 3) given in Table 4.1. Some experimental studies give the evident that the HH
compounds structure relies on the disorder of the atoms. For volume optimization, we utilized
self-consistent full-potential linearized augmented plane wave (FP-LAPW) method (Kohn and
Sham, 1965), which depends on DFT instigated into the WIEN2K simulation package (Blaha
et al., 2001a). We follow the Perdew–Burke–Ernzerhof (PBE) generalized gradient
approximation (GGA) for the electronic exchange–correlation interaction (Perdew et al.,
1996a). However, the DOS and the band structure were calculated by using mBJ potential.
A mesh of 12×12×12, consisting on 72 special k points that were taken in the BZ of the
irreducible wedge for the integrations within the modified tetrahedron method and for the wave
function broadening inside the atomic sphere, orbital momentum is taken lmax = 10. For the
convergence of the Eigen-value energy, the expansion of plane waves is organized by cut-off
parameter Kmax × Rmt = 8.0 where Kmax shows the uttermost value of the reciprocal lattice
vector used in plane wave function and Rmt represents the smallest muffin tin sphere radii. The
value of Gmax is set to 12 where Gmax is the largest vector value in charge density Fourier
expansion.

48
Table 4.1 Different configurations of atomic arrangements in HH structure.
Types X Y Z
Type 1 (¼, ¼, ¼) (0, 0, 0) (½, ½, ½)
Type 2 (¼, ¼, ¼) (½, ½, ½) (0, 0, 0)
Type 3 (½, ½, ½) (¼, ¼, ¼) (0, 0, 0)
Fig. (4.1) Crystal structure of HH XYZ compound with (MgAgAs) C1b for Type 1.

49
4.3 Results and Discussion
4.3.1 Ground State Properties
We employed ab-initio calculations to search HM compounds for spintronic
applications. In the present work, we have studied the structural, electronic and magnetic
properties of unreported ternary HH compounds CrTiZ (Z= Si, Ge, Sn, Pb). Because of no
experimental lattice constant has been reported, so we employed geometrical optimize
calculations to obtain theoretical equilibrium lattice constant of DFT investigated compounds
CrTiZ (Z= Si, Ge, Sn, Pb) in HH composition, the total energy varies with the empirical
Murnaghan’s equation as a function of volume for three possible phases (Type 1, Type 2 and
Type 3) in order to find out the correct arrangement of atoms in the crystal as shown in the Fig.
(4.2).
It is noted that all compounds are dynamically most stable in Type 1. So, structural,
electronic and magnetic properties are calculated for this type only. The theoretical lattice
constant of CrTiZ (Z= Si, Ge, Sn, Pb) for Type 1 is determined by minimizing the total energy
as a function of volume in the unit cell.
Magnetic ground state properties are also determined by minimizing the total energy
as a function of volume for both magnetic and NM states. The total energy curve with
deference to comparative volume in magnetic (FM) and NM phases are shown in the Fig. (4.3)
for all the compounds. In this structural optimization process, it is found that magnetic FM
calculation has a lower minimum total energy than NM type, indicating the magnetic state is
more stable in energy than NM form.
In addition, the value of energy difference ΔE is positive which is also indicating that
the magnetic state is preferable than the NM state as clearly shown in Fig. (4.3). The same
behavior is reported in the previous work (Umamaheswari et al., 2014). So, the structural,
electronic and magnetic properties are calculated for the Type 1 only. The theoretical lattice
constant of CrTiZ (Z= Si, Ge, Sn, Pb) for Type 1 is determined by minimizing the total energy
as a function of volume in the unit cell.
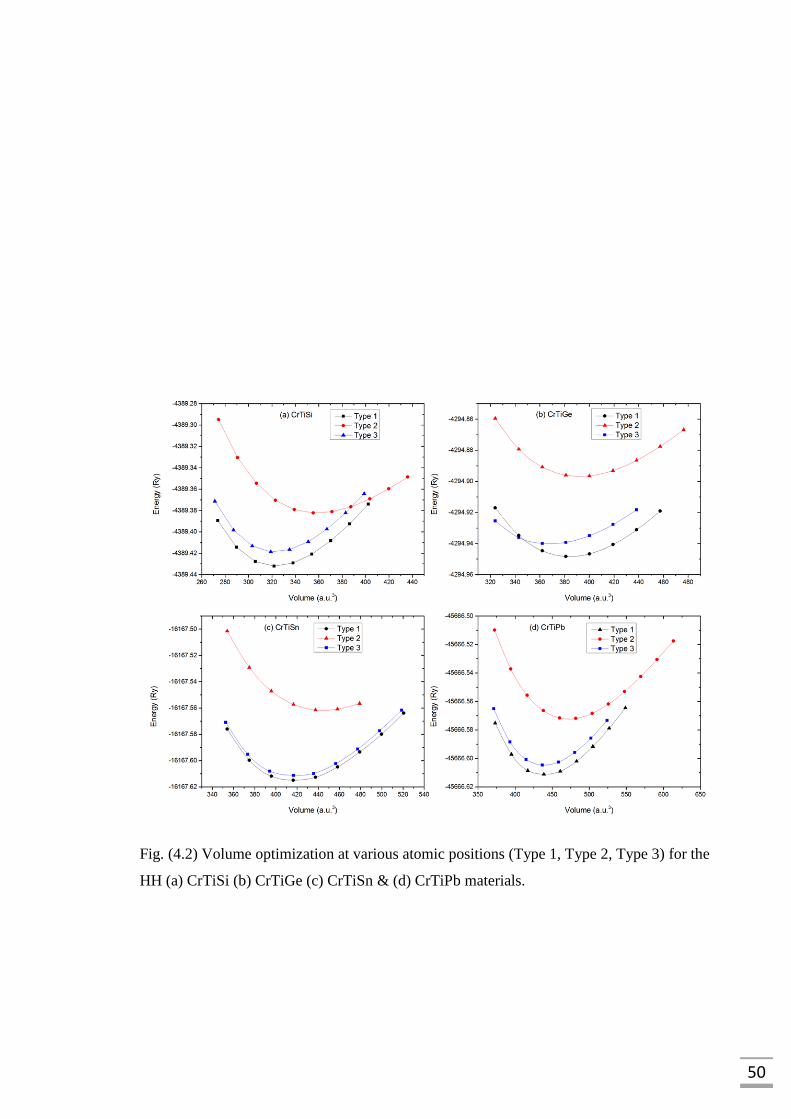
50
Fig. (4.2) Volume optimization at various atomic positions (Type 1, Type 2, Type 3) for the
HH (a) CrTiSi (b) CrTiGe (c) CrTiSn & (d) CrTiPb materials.

51
Fig. (4.3) Volume optimization for (a) CrTiSi (b) CrTiGe (c) CrTiSn (d) CrTiPb for magnetic
& NM states.

52
The predicted essential lattice constants, equilibrium volume, bulk modulus (B0), and
energy differences ΔE = ENM - EFM between the NM and FM states are given in Table 4.2.
Until now, an experimental or maybe theoretical value of CrTiZ has not been claimed to
compare with the present calculations.
The Formation energy (𝐸𝐹𝑜𝑟) of these compounds is significant to know the stability
of these putative compounds. The Formation energy is premeditated using the relation
𝐸𝐹𝑜𝑟 = 𝐸𝐶𝑟𝑇𝑖𝑍 − (𝐸𝐶𝑟 + 𝐸𝑇𝑖 + 𝐸𝑍)
where 𝐸𝐶𝑟𝑇𝑖𝑍 is the total energy of compound in a unit cell and 𝐸𝐶𝑟, 𝐸𝑇𝑖, and 𝐸𝑍 are total energy
of pure essential elements in the compound ECrTiZ and the calculated values are given in Table
4.2. These values of 𝐸𝐹𝑜𝑟 show that these compounds would not decompose as soon as they
have got been shaped.
4.3.2 Electronic and Magnetic Properties
For the prediction of the HMF and magnetic properties, electronic structure plays a
significant role. The electronic and magnetic properties of HH CrTiZ (where Z= Si, Ge, Sn,
Pb) compounds are discussed in this section. The band structures of CrTiZ (where Z= Si, Ge,
Sn, Pb) HH compounds revel that all CrTiZ (where Z=Si, Ge, Sn, Pb) compounds are HM in
nature because it is seen that minority spin (spin down) has metallic behavior at the EF which
is the indication of metallic nature.
On the other hand, there is EBG in the majority spin (spin up) showing the majority spin
is semi-conducting which clues to 100% SP near the EF as showing in Fig. (4.4). At the
optimized equilibrium volume and lattice constant, the Fig. (4.4) shows that all compounds
CrTiZ (where Z=Si, Ge, Sn & Pb) having metallic behavior for a spin up and semiconducting
for a spin down, which shows HM properties. In addition, (as given in Table 4.3) the particular
computed µtot of all the CrTiZ compounds is integral that is a usual quality connected with
HMFs (Felser et al., 2007).
The energy bands near the EF appears typically a result of the hybridization regarding
d-orbitals in the daptation of TMs. The band structure of the CrTiZ tends to be primarily
engaged using the 3d-Cr as well as 3d-Ti electrons near the EF.

53
Fig. (4.4) Electronic band structures of HH (a) CrTiSi, (b) CrTiGe, (c) CrTiSn & (d) CrTiPb
compounds for spin-up (↑) and spin-down (↓) channel.

54
It can be evidently identified that for all the materials there be found major splitting
among the majority and minority spin of the 3d-Cr and 3d-Ti states. The 3d-Cr along with 3d-
Ti are generally segregated into the t-2g state in the valance band (at low energy) and eg state
in the conduction band (at high energy).
Band structure also reveals that for the HH CrTiGe as a prototype, the upper part of the
valence band for a spin down lies at -0.17 (eV) and the lower part of conduction band for spin
up lies at 0.66 (eV) from the EF. By using these energies at the uppermost occupied band at L
point and the lowermost unoccupied band at the X point, the width of the EBG (indirect gap)
can be calculated which is 0.84 (eV) for the HH CrTiGe and EHM is 0.17 (eV) which can be
demarcated as the minimum energy requisite for an electron to flip the spin from the valance
band maxima to the EF. The majority spin EBG and as well as the EHM for other compounds is
provided within Table 4.2. The µtot of the compounds and individual magnetic moment of the
atoms are also given in Table 4.2.
We have also calculated spin-polarized total density of state (DOS) and partial density
of state (PDOS) for all the CrTiZ compounds at their equilibrium volume to understand the
origin of the ferromagnetism. Total and PDOS of CrTiZ (Z= Si, Ge, Sn, Pb) are presented in
the Fig. (4.5), which reveals that total and partial DOS of all the CrTiZ (Z=Si, Ge, Sn, Pb)
materials are HM since each alloy in the compute, the Cr and Ti sites rules portion of the plots.
For both spin up and down conditions it could be noticed from the total DOS that
uppermost part of valence bands originates from the hybridization associated with Cr and Ti
states. The spin splitting in these types of materials mostly come up from Cr-d states with a
small share coming from Ti-d states and the hybridization among Cr-d and Ti-d states.
Hence the magnetism comes up generally owing to the spin splitting of Cr-d like states.
The share regarding Z-atom is very small when compared to Cr and Ti. Because of interaction
between Cr-d like states and Ti-d like states, Because of interaction between Cr-d and Ti-d like
states, in the majority (spin-up) state, the d-t2g orbitals of Cr & Ti are fully occupied and d-eg
of Cr and Ti are partially occupied”. The hybridization is present among Cr-Ti within the 3d
orbital.

55
Table 4.2 Here, a (Å): lattice parameter, B (GPa): bulk modulus; ΔE (Ry): Energy difference
between FM and NM states and EFor (eV): Formation Energy
Table 4.3 Total magnetic moment µ𝑡𝑜𝑡 (µB) of compounds CrTiZ and local magnetic moments
(µB) of Cr, Ti, and Z atom. MI (µB) is the magnetic moment of the interstitial region, band gap
EBG (eV), and HM gap EHM (eV) in the spin up channel.
Material 𝐦𝐂𝐫
(µB)
𝐦𝐓𝐢
(µB)
𝐦𝒁
(µB)
𝐌𝐈
(µB)
µ𝐭𝐨𝐭
(µB)
𝐄𝐁𝐆
(eV)
𝐄𝐇𝐌
(eV)
CrTiSi 2.54 0.85 -0.02 0.64 3.99 0.86 0.12
CrTiGe 2.79 0.75 -0.06 0.53 4.00 0.84 0.17
CrTiSn 2.97 0.60 -0.05 0.49 4.00 0.91 0.31
CrTiPb 3.10 0.49 -0.04 0.45 4.00 0.94 0.33
Compounds a (Å) B0 (GPa) ΔE (eV) EFor (eV)
CrTiSi 5.7633 133.49 0.571 0.386
CrTiGe 5.9721 102.30 0.952 0.531
CrTiSn 6.2738 88.40 1.387 0.640
CrTiPb 6.3844 78.26 1.714 0.912

56
Fig. (4.5) Total and partial DOS of the HH (a) CrTiSi, (b) CrTiGe, (c) CrTiSn and (d) CrTiPb
materials, for spin- up (↑) and spin-down (↓).

57
Fig. (4.6) Total and orbital resolved partial DOS of the HH (a) CrTiSi, (b) CrTiGe, (c) CrTiSn
and (d) CrTiPb materials, for spin- up (↑) and spin-down (↓).

58
The reason for the absence of semiconducting gap in the majority spin is due to the
dominance of the Ti-t2g and Cr-t2g electrons round the EF for all the CrTiZ materials and the
cause of the origin of presence of semiconducting gap in the minority spin is due to the
depletion of Cr-t2g and Ti-t2g electrons which turns these occupied states into unoccupied states
and produces the semiconducting gap which is shown in Fig. (4.6) as a prototype for HH
CrTiGe alloy.
An understanding of half-metallicity along with the lattice constant is very crucial
regarding practical applications in spintronics. To analyze the sensitivity of half-metallicity,
the lattice constant must be changed in detail because by altering the lattice constant of the HM
materials could possibly have an impact on the HM nature. So, for this purpose, we focus on
the robustness of the ferromagnetism with respect to lattice distortion. The Fig. (4.7) shows
the computed µtot as a function of lattice parameter. We noted that integer value of the magnetic
moment remains unchanged up to the critical value of the lattice constant. These critical values
are 5.7 Å, 6.1 Å, and 5.9 Å for CrTiSi, CrTiGe, CrTiSn, CrTiPb, respectively.

59
Fig. (4.7) Origin of the semiconducting gap in the majority spin channel in the type T1 for the
HH CrTiGe as a prototype.

60
Fig. (4.7) The total magnetic moment (µtot) of the HM HH CrTiZ (where Z= Si, Ge, Sn, Pb) as
a function of lattice constant.

61
CHAPTER 5
Spin Polarized Calculations of the Structural, Electronic and
Magnetic Properties of New Half Heusler Alloys FeVZ (where Z =
Si, Ge, Sn) by GGA and mBJ Approaches
5.1 Introduction
In recent years, HM materials have been a focus of considerable attention from both
academic as well as industrial point of view. The interest in these materials stems from the fact
that these materials are capable of showing complete SP at the EF (De Groot and Buschow,
1986; Wolf et al., 2001; Žutić et al., 2004). In the HMFs, there is totally dissimilar behavior in
the two spin bands. In one spin orientation, these materials display a metallic character while
the other spin orientation shows a semi-conductive nature that leads to a 100% SP at EF
(Kandpal et al., 2007).
HM materials have attracted many technologists in the past decade owing to their
versatile electronic property which may lead to potential applications in the spintronic devices
such as nonvolatile magnetic random-access memories (MRAM) and magnetic sensors (De
Groot and Buschow, 1986; Wolf et al., 2001; Žutić et al., 2004), spin LED (Jonker et al., 2000),
spin FET (Schliemann et al., 2003), spin-tunneling devices (Lou et al., 2007; Miyazaki et al.,
1997).
Since the earliest of theoretical studies carried out by de Groot et al. (De Groot et al.,
1983) numerous HM compounds have been forecasted by different research groups (Xiao et
al., 2010). To-date, HM nature has been explored in dozens of materials such as dilute magnetic
SCs (Akai, 1998; Ogawa et al., 1999; Yao et al., 2005b), binary transition metal pnictides
(Jaiganesh et al., 2010), chalcogenides with zinc-blende structure (Tan et al., 2010), Full
Heusler (FH) compounds (Birsan and Palade, 2013b; Birsan et al., 2012; Birsan et al., 2013;
Fang et al., 2013; Huang et al., 2012; Kervan and Kervan, 2012; Kervan and Kervan, 2011;
Lei et al., 2011) and HH alloys (Casper et al., 2012; Chen et al., 2011; Lakdja et al., 2013;
Nanda and Dasgupta, 2003; Rozale et al., 2013; Umamaheswari et al., 2014; Yadav and
Sanyal, 2015; Zhang et al., 2003b).

62
The HH alloys have particularly attracted many researchers and material scientists to
predict the half-metallicity in various materials due to their high TC, structural resemblance to
the zinc-blende phase, magnetic behavior and other diverse properties. HH alloys provide a
chance for developing magnetic devices directly into SCs technology. Although a lot of
Heusler compounds have been theoretically anticipated to exhibit HM attributes, and recently,
many theoretical researchers have taken a keen interest in HH compounds until now to be in
our best knowledge HH FeVZ (where Z= Si, Ge, Sn) compounds have not received much
attention both theoretically and experimentally.
In the present study, the structural, electronic and magnetic properties of HH FeVZ (Z=
Si, Ge, Sn) compounds with C1b-type structure are investigated for the first time by performing
Perdew-Burke-Ernzerhof generalized gradient approximation (PBE-GGA) (Perdew et al.,
1996a) while the electronic properties are probed using both GGA and the state-of-the-art mBJ
local density approximation functional calculations (Tran and Blaha, 2009). As the GGA
functional is known for underestimation of the band gap, so mBJ functional is employed which
has previously been shown to predict correct properties of magnetic materials.
For intestacy, GGA calculations of MnAs zincblende predict to display non-HM
properties (Sanvito and Hill, 2000), because conduction band minima touch or even crosses
the EF whereas experimental studies (Ono et al., 2002; Yokoyama et al., 2005) shows that it is
a truly HM compound which can be confirmed by mBJ calculations. Recently, Abdelaziz et
al. (Lakdja et al., 2013) also compares the results related to electronic and magnetic properties
of HH XCsBa alloys (where X= C, Si & Ge) with GGA and mBJ-GGA potential and concluded
that mBJ-GGA potential gives the accurate bandgap.
Although numerous feasible approaches are available in DFT calculations of SC gaps,
for instance, exact-exchange approach (Kotani, 1995; Sharma et al., 2005; Städele et al., 1997),
different GW methods (Faleev et al., 2004; Hybertsen and Louie, 1986), hybrid functionals
(Betzinger et al., 2010) which lead to the correct interpretation of the electronic properties, but
the mBJ potential (as an orbital independent, semi-local exchange correlation potential) has
been proved to produce accurate gaps for wide band gap insulators, sp-SCs, and 3d-TMs oxide.
Consequently, to discover the actual HM properties of the materials, it is very appropriate and
essential to make use of mBJ-GGA potential which significantly enhances the electronic
properties and its simple form as well as cheaper computationally cost makes mBJ functional
ideal for studying HM materials.

63
5.2 Computational Details
The structural optimization and the electronic calculations are performed by using the
self-consistent full potential linearized augmented plane wave (FPLAPW) method. FPLAPW
is employed into the WIEN2K code (Blaha et al., 2001b) based on density functional theory
(DFT). For the exchange correlation function, we used the PBE-GGA (Perdew et al., 1996a;
Perdew et al., 1996b). In this technique, space is consisting of muffin-tin (MT) spheres which
are non-overlapping and an interstitial region between these spheres. To separate the valance
and core electrons states, cut-off energy is set to -6 Ry. The spherical harmonic functions and
Fourier series, originate from a basis function, are employed for MT spheres and interstitial
region, respectively.
The cut-off parameter for the planewave was set to 𝐾𝑚𝑎𝑥 × 𝑅𝑀𝑇 = 9, where 𝐾𝑚𝑎𝑥 is
the maximum modulus for the reciprocal lattice vector. For the self-consistency cycles, energy
was set to 105 Ry per formula unit and by using the modified tetrahedron method (Blöchl et
al., 1994) for the BZ integration, 72 specific number of k-points are taken in the irreducible
partition of BZ (2000 k-points in the full BZ). The total energy calculation is also performed
with larger k-points and found negligible differences in the total energy computed using 2000k-
points. These kinds of variables assure excellent convergence for total energy.
5.3 Results and Discussions
5.3.1 Crystal Structure Stability
Ternary HH compounds, often referred as ternary intermetallic compounds having the
chemical formula XYZ with stoichiometry 1:1:1 crystalize in the face centered cubic C1b
structure with the space group F-43m (No. 216) (Nanda and Dasgupta, 2003). These HH
compounds can be derived from the L21 structure of a FH alloy X2YZ by omitting the one X
element, where X is the transition metal element and Y may be either transition metal or a rare-
earth metal and Z is from the main group element. Three phases α, β, and γ can be found for
the XYZ HH alloys because, in the unit cell of the HH alloy, three different atomic
arrangements are possible. Crystal structure of the HH FeVGe is shown in Fig. (5.1), consisting
of three interpenetrating, face-centered-cubic sub-lattices, which are occupied by Fe, V and Z
elements.

64
Table 5.1 Atomic arrangement of atoms X, Y, and Z in α, β and γ phases. The 4d position is empty.
Phase 4a (X) 4b (Y) 4c (Z)
α (1
4,
1
4,
1
4) (0, 0, 0) (
1
2,
1
2,
1
2)
β (0, 0, 0) (1
2,
1
2,
1
2) (
1
4,
1
4,
1
4)
γ (1
2,
1
2,
1
2) (
1
4,
1
4,
1
4) (0, 0, 0)
Fig. (5.1) The unit cell of cubic C1b-type structure for the HH FeVGe in α-phase

65
Atomic arrangement of the Studied HH FeVZ materials are described in Table 5.1. To
find out the lattice constant, bulk modulus and energy deviations as a function of volume,
geometrical optimization of HH alloys having generic formula FeVZ (X= Si, Ge, Sn) C1b -
type structure have been performed by using the Murnaghan's equation of state (Murnaghan,
1944a). Total energy as a function of volume is plotted in Fig. (5.2) for three possible phases
α, β, and γ, to reveal the accurate atomic arrangement of atoms in a unit cell which is very
essential because some studies show that nature of bond existing between the neighboring
atoms strongly influences the physical properties (Casper et al., 2012; Graf et al., 2011).
Therefore, the correct position of atoms is determined by minimizing the energy as a
function of volume at their equilibrium lattice constant. The three possible phases of the HH
FeVZ (Z= Si, Ge, Sn) alloys and position of these atoms for each phase (α, β & γ) are given in
Table (5.1). The Fig. (5.2) shows that α-phase is more favorable than other β and ɤ phases
because it has the lowest minimized energy. These results show that given compounds reside
in the α-phase.
To determine the magnetic ground state at the most stable structure (α-phase), both
spins polarized (magnetic) and spin un-polarized NM calculations are also performed. The HH
FeVGe as a prototype, for the FM and NM states as a function of volume, is shown in Fig.
(5.3), which is clearly indicating that the magnetic state has the lower energy as compared to
the NM state and therefore, is more favorable. Computed structural parameters such as
equilibrium lattice constant a(Å), total energy and magnetic moment at the three different
phases (α, β, γ) for the three HH FeVZ (where Z= Si, Ge, Sn) compounds in the FM state are
listed in Table 5.2, Moreover, the partial magnetic moments for the α-phase are also arranged
for the HH FeVZ (where Z = Si, Ge, Sn) in Table 5.3.
5.3.2 Electronic Properties
To explore the electronic properties of the FeVZ (where Z= Si, Ge, Sn) at equilibrium
volume, spin polarized calculations have been studied. The electronic and magnetic properties
of the FeVZ HH alloys are discussed in this section and we will compare our GGA outcomes
with the state-of-art mBJ-GGA results. It is already known fact that mBJ-GGA computations
only effects the electronic properties of the compounds that are underestimated or even
overestimated along with GGA or LDA calculations.

66
Fig. (5.2) Total energy as a function of volume for FeVGe in different phases (α, β, γ) of atomic
positions.
Fig. (5.3) Total energy as a function of volume for the HH FeVGe in α-phase for the magnetic
and NM states.
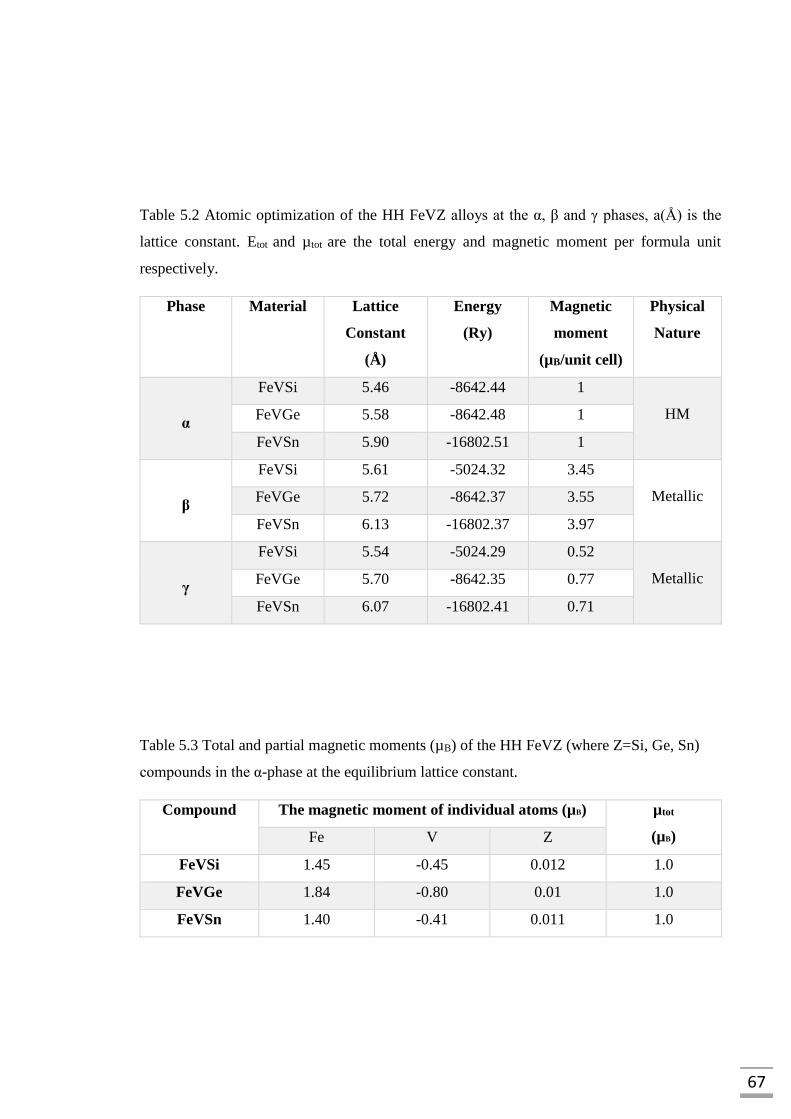
67
Table 5.2 Atomic optimization of the HH FeVZ alloys at the α, β and γ phases, a(Å) is the
lattice constant. Etot and µtot are the total energy and magnetic moment per formula unit
respectively.
Phase Material Lattice
Constant
(Å)
Energy
(Ry)
Magnetic
moment
(µB/unit cell)
Physical
Nature
α
FeVSi 5.46 -8642.44 1
HM FeVGe 5.58 -8642.48 1
FeVSn 5.90 -16802.51 1
β
FeVSi 5.61 -5024.32 3.45
Metallic FeVGe 5.72 -8642.37 3.55
FeVSn 6.13 -16802.37 3.97
γ
FeVSi 5.54 -5024.29 0.52
Metallic FeVGe 5.70 -8642.35 0.77
FeVSn 6.07 -16802.41 0.71
Table 5.3 Total and partial magnetic moments (µB) of the HH FeVZ (where Z=Si, Ge, Sn)
compounds in the α-phase at the equilibrium lattice constant.
Compound The magnetic moment of individual atoms (µB) µtot
(µB) Fe V Z
FeVSi 1.45 -0.45 0.012 1.0
FeVGe 1.84 -0.80 0.01 1.0
FeVSn 1.40 -0.41 0.011 1.0

68
The plots of the total DOS and partial DOS of the individual atoms of the HH FeVZ
(where Z= Si, Ge, Sn) compounds are only presented in α-phase at the optimized lattice
constant as shown in Fig. (5.4). We have not shown some states such as s and p states of Fe
and V because their contribution to the total DOS is very small. From Fig. (5.5), it can easily
be visualized that total DOS is mainly contributed by the 3d states of Fe and V while p state of
Ge near the EF makes the most contribution to the total DOS which is also in consistent with
previous HH transition-metal alloys [48, 49]. The d-orbital split up into two d-eg and d-t2g
orbitals. A small energy gap is found in the spin up channel due to the major contribution of
d-t2g orbitals of Fe and V atoms which have contributed more than d-eg orbitals of Fe and V.
The band d-t2g is dominated near the Fermi level (EF).
On the contrary, in the spin down channel, the p orbitals of the Z atom cross the EF,
leading to a metallic character for all three compounds. The most part of d-state of Fe in the
spin up channel is positioned around -2.0 to -1.2 eV, whereas, in the spin down channel, this
state is situated a little up around -1.8 to -0.8 eV, but d-states of V atom prevail in the energy
range from -1 to 0.3 eV. The p state of Z atom is quite symmetrical in the spin up directions
and crosses EF a little in the spin down the channel with a little share to the magnetism. It is
also revealed from the Fig. (5.5), that hybridization occurs between the 3d states of Fe and V
atoms. Among all the three atoms in a compound, the most part of the µtot is contributed by
Fe-atom.
The different values of the physical properties of the HH FeVZ (Z=Si, Ge,
Sn) such as, valence band maxima (eV), conduction band minima (eV), EBG (difference
between the valence band maxima to the conduction band minima) and EHM (which is the
minimum energy required to flip the electron spin across the EF from valence band maxima),
physical nature of the material and band transition calculated with both GGA and mBJ
potentials, are given in Table 5.4.
The spin up DOS has an EHM of 0.16, 0.21 and 0.02 eV for the HH FeVSi, FeVGe and
FeVSn materials respectively and show the semiconducting nature. So, in consequence, at the
equilibrium lattice constant for the HH FeVZ (Z = Si, Ge, Sn) alloys in α-phase, an ideal 100%
SP of the conducting electrons has been resulted because there appears a gap around EF in the
spin up state and at the same time DOS peak also crosses the EF in the spin-down state.

69
Fig. (5.4) Spin-dependent total and partial DOS of HM HH (a) FeVSi (b) FeVGe (c) FeVSn at
equilibrium lattice constant at the α-phase. EF is set at zero. The top portion (spin-up) displays
the majority-spin channel and the lower portion (spin-down) is for the minority spin channel.
Solid and dotted lines show the DOS’s of GGA & mBJ-GGA potential respectively.

70
Table 5.4 Different physical properties of HH FVX (Si, Ge, Sn) at the equilibrium lattice
constant in the α-phase. VXC is the exchange correlation potential, VBM is the maximum value
of the valance band, and CBM is the minimum value of the conduction band, EBG is the energy
band gap, EHM is a half-metallic gap. The transition between the bands and nature of compound
is also given
Material VXC VBM
(eV)
CBM
(eV)
EBG
(eV)
EHM
(eV)
Magnetic
moment
(µB)
Band
transition
Physical
State
FeVSi
GGA
0.10
0.67
0.57
-----
0.92
W X
Nearly
HM
mBJ -0.16 0.72 0.88 0.16 1.00 W X HM
FeVGe
GGA -0.05 0.79 0.84 0.04 1.00 L X HM
mBJ -0.21 0.61 0.82 0.21 1.00 T X HM
FeVSn
GGA
0.07
0.72
0.65
-----
0.98
T X
Nearly
HM
mBJ -0.23 0.60 0.83 0.23 1.00 T X HM

71
Computed results also suggest that p-state of Z-atom has the lowest part in the valance
band in both majority and minority spin states. In the locality of the EF, d-state of Fe and p-
state of Z atom point out the certain dominance showing the origin of the µtot will arise from
Fe and Z atoms. Also, the negative magnetic moment for V represents the AFM alignment of
the magnetic moment of V with Fe. There is large splitting near EF due to the Fe and Z-atoms
and splitting causes the appearance of the energy gap in the minority spin channel (in the spin-
up state). It also reveals that Fe and Z-atoms governed the energy gap in the spin up state.
Band structure calculations must be carried out very carefully because, for the
prediction of HM ferromagnetism and magnetic properties of the HH compounds, electronic
structure plays an important role. The band structures for all the magnetic compounds of FeVZ
with PBE-GGA and mBJ-GGA potentials are calculated and presented in Figs. (5.5-5.7), at
their equilibrium lattice constants in the α-phase. Left side shows the spin up (majority-spin)
state and the right side shows the spin down (minority-spin) state. It can clearly be noticed that
for all the HH FeVZ compounds, the minority spin state shows no semiconducting gap and is
of a metallic nature, whereas the majority spin state shows the semiconducting behavior.
When GGA potential is employed to calculate band structures for the FeVGe HH
compound, which shows that it is truly HM because electronic states in the minority spin
channel depicting the metallic behavior, however, in the majority spin channel, there exist an
EBG of 0.84 eV and electronic state do not cross EF. The band structures of FeVSi and FeVSn
compound calculated with PBE-GGA potential show that these compounds are nearly half
metals because in the spin-up state very little part of the valance band crosses the EF.
To improve these results obtained from PBE-GGA, mBJ-GGA potential is employed.
From PBE-GGA calculations, only HH FeVGe is an HM compound but when calculations are
made from mBJ-GGA potential, it is observed that valance bands are now shifted downwards,
and conduction bands are shifted upwards for all the FeVZ compounds. The values of both
EBG and EHM are also increased and now, all these compounds are truly HM materials.
5.3.3 Magnetic Properties
The total and partial magnetic moment µtot and magnetic moment of the individual
atoms (in the units of µB) for the HH FeVZ (where Z=Si, Ge, Sn) per formula unit cell for the
α-phase are summarized in Table 5.4.
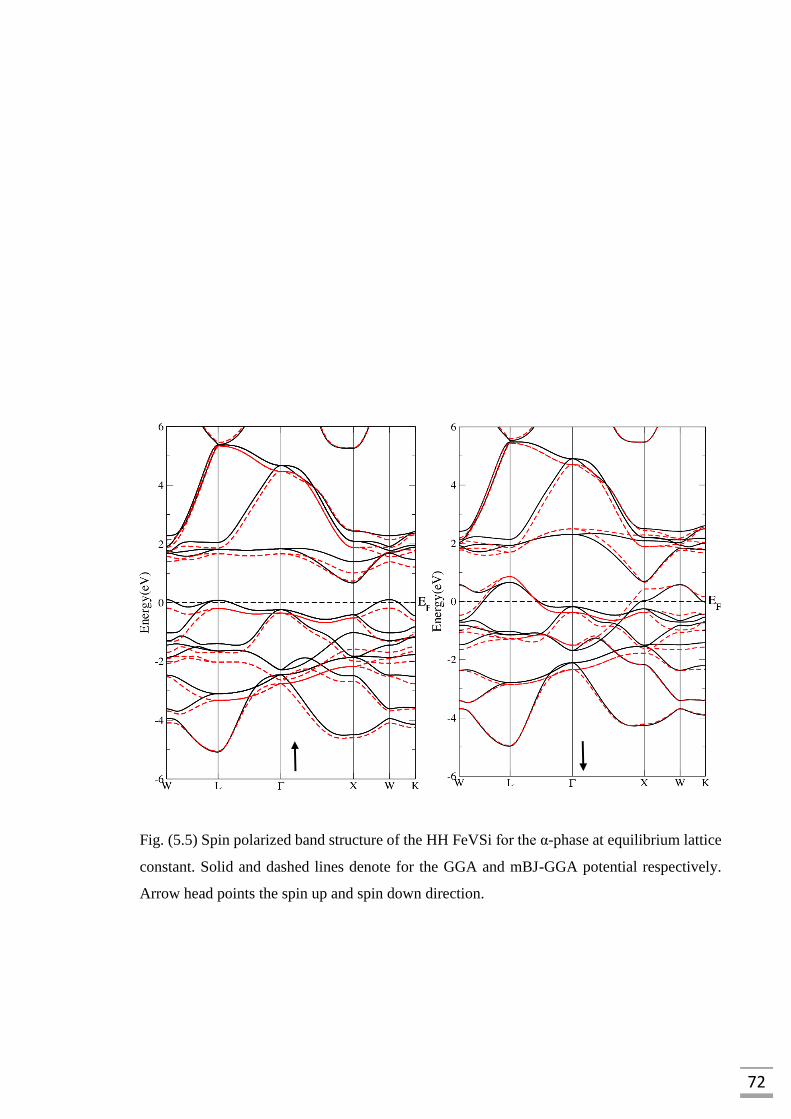
72
Fig. (5.5) Spin polarized band structure of the HH FeVSi for the α-phase at equilibrium lattice
constant. Solid and dashed lines denote for the GGA and mBJ-GGA potential respectively.
Arrow head points the spin up and spin down direction.

73
Fig. (5.6) Spin polarized band structure of the HH FeVGe for the α-phase at equilibrium lattice
constant. Solid and dashed lines denote for the GGA and mBJ-GGA potential respectively.
Arrow head points the spin up and spin down direction.

74
Fig. (5.7) Spin polarized band structure of the HH FeVSn for the α-phase at equilibrium lattice
constant. Solid and dashed lines denote for the GGA and mBJ-GGA potential respectively.
Arrow head points the spin up and spin down direction.

75
To understand the semiconducting gap, Fig. (5.8) illustrates the spin-polarized band
structure of the HH FeVGe (up-state) in the α-phase at the equilibrium lattice constant. Band
1 is of the s-state of the Ge, which has the lowest part in the valance band for both majority
and minority spin states. Bands 2-4 and 12-14 are because of the p-state of Ge whereas bands
3-6 are because of d-states of the Fe and 7-11 consists of d-states of V.
The d-orbitals further splits up into double degenerated d-eg(2) and triplet degenerated
d-t2g(3) states due to the crystal field effect. Bands 7-9 at Г are because of the d-t2g states of
the Fe atom below EF. Above EF, bands 10 and 11 located at X are due to the d-eg states of the
V atom. Bonding and antibonding states are formed because these triplet d-t2g states of Fe
interact with the sp states of V atom. It means that triply degenerated d-t2g state of Fe and
doublet degenerated the d-eg state of V in the spin up channel should be occupied as in the spin
down channel, but by the exchange interaction, electrons get depleted in these occupied states
and become unoccupied and govern the semiconducting gap.
5.3.4 Half-Metallic (HM) Robustness
The HM robustness is the transition of a material from HM to pure metallic nature.
Finally, for the useful applications of HH alloys in the spintronic, HM stability for the FeVSi,
FeVGe, and FeVSn compounds is explored over a wide range of lattice constant. As thin films
or multiple layers of HM materials are used to grow on a suitable substrate for spintronic
devices, therefore, the lattice constant of the deposited thin films may change, and this can
destroy the half-metallicity of the HH materials. Hence, it is very essential to know how far
lattice constant of the HH FeVSi, FeVGe, FeVSn materials should be varied so that they keep
their half-metallicity.
Due to this reason, the relationship between the µtot and the spin magnetic moment of
Fe, V, and Z atoms and its reliance on the lattice parameters are shown in Fig. (5.9). It can be
revealed that as lattice parameter of FeVZ (Z= Si, Ge, Sn) compounds are extended or
contracted from their theoretical equilibrium lattice constant, the hybridization between the Fe
and V atoms changes. For all the three compounds, when the lattice parameter is boosted, it
increases the magnetic moment of the Fe and decreases the magnetic moment of the V but the
µtot per unit cell changes slightly and remains approximately at 1 µB, which is an integral
multiple of Bohr magnetron.

76
Fig. (5.8) Band structure of the HH FeVGe compound with mBJ potential of α-phase at
equilibrium lattice constant. The different colors show the s, p and d orbitals of atoms.

77
Fig. (5.9) Magnetic moment as a function of lattice constant of the HH (a) FeVSi, (b) FeVGe
& (c) FeVSn materials

78
The HH FeVSi, FeVGe, and FeVSn materials retain their HM property within the
lattice constant range of 5.25Å to 5.75Å, 5.18Å to 5.8Å and 5.20Å to 5.90Å, respectively. i.e,
EF remains in the gap and these alloys keep their half-metallicity when the lattice constants of
FeVSi, FeVGe, and FeVSn are contracted and extracted up to 3.84% to 5.31%, 3.91% to 5.91%
and 3.94% to 11.8% respectively, from their theoretical equilibrium lattice constant.

79
CHAPTER 6
Theoretical Investigations of Half-Metallic Ferromagnetism in
New Half-Heusler YCrSb and YMnSb Alloys Using First-Principle
Calculations.
6.1 Introduction
To meet the essentials of the advance technological applications, the search for the best
materials in general and for spintronic applications is a challenge. Spin-polarized FM materials
are generally supposed to be the best replacements for conventional materials (Bhat et al.,
2015). The quest for brand-new materials in the field of spintronic has guided towards HA in
the last three decades due to their ability to be strong candidates for spin based electronic
materials. The important part regarding spintronic is a way to obtain spin-polarized charge
carriers.
HMFs are a type of brand new material because of their distinctive characters and tend
to be probably the most essential components designed for spintronic (Umamaheswari et al.,
2014). HMFs have attracted considerable interest within last three decades due to their unique
property of possessing a semi-conducting behavior in one spin direction with a narrow gap at
the EF producing 100% polarization at the EF and metallic behavior in other spin direction.
HMFs will be appealing materials that can result in high performance applications in
spintronics devices, as a source of spin polarized charge carriers injected, such as spin field-
effect transistor (spin-FET), spin light emitting diode (spin-LED) along with tunneling devices
(Huang et al., 2014).
HMFs with HH structure offer the great possibility of integrating magnetic devices into
SC technologies and potential applications in spintronics due to their structural resemblance to
the zinc-blende phase and relatively high value of TC. In 1983, de Groot et al.(De Groot et al.,
1983) initially predicted the HM ferromagnetism by exploring the band structure calculations
of Mn-based materials in semi-Heusler NiMnSb, which is right now more successful to be
synthesized experimentally with single crystalline nature.

80
Several studies relevant to HMFs are already expected theoretically and many of HMFs
are validated experimentally. Half-metallicity is located in Heusler compounds (Alijani et al.,
2011; Chatterjee et al., 2010; Jaiganesh et al., 2010; Ko et al., 2010; Liu et al., 2008; Luo et
al., 2011) and several other kinds of materials which include FM metallic oxides (Jedema et
al., 2001; Li et al., 2009; Soeya et al., 2002; Song et al., 2009b), nanostructures (Son et al.,
2006) binary TM pnictides (chemical compounds) as well as chalcogenides acquiring zinc-
blended and rock-salt structural arrangements (Ahmadian and Alinajimi, 2013; Dong and
Zhao, 2011; Galanakis and Mavropoulos, 2003; Gao et al., 2007; Liu, 2003).
Numerous studies have already been conducted on these types of materials and plenty
of them have become HMFs. But often an experimental synthesis of these materials at the room
temperature is difficult, as half-metallicity is lost due to very small EHM and very large
magnetic moments arises. Small EHM and large magnetic moment mean high stray field. The
EHM would frequently vanish in each of these HH materials when strain mismatch rises at the
interface with the traditional SCs.
In addition, HM materials having a large magnetic moment are not ideal for spintronic
practical applications, since the big magnetic moment indicates higher stray fields as well as
large energy deficits. This deficiency inspires us to find brand new HM alloys that have a
modest magnetic moment and larger EHM. The outcomes offered by this study might clarify
the applications of these HH materials in the arena of spintronics. Structural, electronic and
magnetic properties of HH YCrSb and YMnSb are explored in this study for the sake of their
novel applications.
6.2 Computational Details
To cope with the exchange and correlation potential, all computations are executed
within density function theory (DFT) using the generalized gradient approximation (GGA)
available as Perdew–Burke–Ernzerhof (PBE) functional (Perdew et al., 1996a). A cycle of the
self-consistent scheme is performed to find out the structural and electronic properties of
YCrSb and YMnSb HH materials by solving the Kohn–Sham equations (Sham and Kohn,
1966) through utilizing the full-potential linearized augmented plane wave method (FPLAPW)
(Andersen, 1975) as implemented within the WIEN2K simulation code (Schwarz et al., 2002).
A k-point mesh of 15×15×15 is chosen for the calculations of these HH materials each
with muffin-tin sphere radius of 2.5 a.u, for Y, Cr(Mn) and Sb atoms, respectively, and the

81
value of RMT × Kmax is set to be 9. Expansion of site-centered potentials and densities is taken
with the angular momentum up to lmax = 10. The particular BZ integration is completed from
the standard tetrahedron approach (Jepson and Anderson, 1971). For the two consecutive
computations, the actual convergence criterion in this self-consistent information about ionic
relaxations is 10-5 eV/unit cell.
6.3 Results and Discussion
6.3.1 Structural Arrangements and Stability
The HH alloy with general formula XYZ has only one magnetic sublattice, where X
and Y are the transitional metals and Z is the main group element. HH materials belong to a
family relating to traditional SCs, for instance, Si or GaAs and crystalize into the non-
centrosymmetric cubic MgAgAs-C1b structure (space group F-43m, No. 216) having 1:1:1
stoichiometry, which is ternary arranged different from the CaF2 and can be derived from the
tetrahedral ZnS type structure (Graf et al., 2011).
The Wyckoff positions of the three interpenetrating fcc lattices are 4a(0, 0, 0), 4b(𝟏
𝟐,
𝟏
𝟐,
𝟏
𝟐), and 4c(
𝟏
𝟒,
𝟏
𝟒,
𝟏
𝟒), and the 4d(
𝟑
𝟒,
𝟑
𝟒,
𝟑
𝟒) site is empty. In essence, X, Y and the Z atoms can occupy
these Wyckoff positions 4a, 4b and 4c sites, respectively. Three unique phases (XI, XII, XIII)
are possible for X, Y and Z atoms by changing these atomic positions in a unit cell, for instance
XI, XII, and XIII phases can be organized at distinct Wyckoff positions (Helmholdt et al., 1984).
Atomic layout for each phase is presented in Table 6.1. For an illustration, the crystal structures
of HH YCrSb alloy for three possible atomic arrangements are shown in Fig. (6.1).
Exploration of XYZ materials within three feasible arrangements is essential because
a few experimental types of research display that the composition associated with HH materials
rely on the atomic disorder (Aliev, 1991; Helmholdt et al., 1984; Ishida et al., 1997). The
crystalline framework of C1b-type structure associated with this kind of material can be
reviewed properly from Refs. (Gruhn, 2010; Umamaheswari et al., 2013). In our latest
information, there is no experimental nor theoretical report so far, relating to both YCrSb and
YMnSb HH materials. Murnaghan’s equation of state (Murnaghan, 1937) is utilized to find
out the lattice constants. Prior to studying electronic and magnetic properties, stabilities of the
crystal structure of YCrSb and YMnSb are checked by optimizing the total energy as a function
of volume for all the three possible states.

82
Table 6.1 The Site preferences of X, Y and Z atoms in three atomic arrangements XI, XII and
XIII in the C1b HH structure. The 4d site is empty.
Phase 4a (0,0,0) 4b(𝟏
𝟐,
𝟏
𝟐,
𝟏
𝟐) 4c (
𝟏
𝟒,
𝟏
𝟒,
𝟏
𝟒)
XI Z X Y
XII X Y Z
XIII Y Z X
Fig. (6.1) Conventional unit cells of HH YCrSb alloy in the MgAgAs (C1b) structure for the
three distinct XI, XII and XIII atomic arrangements.
XI
Structure
XII
Structure
XIII
Structure

83
Variations of total energy with volumes of HH YCrSb and YMnSb compounds for all
the three possible XI, XII and XIII phases are shown in Fig. (6.2). It is obvious that XI
structures for both the compounds obtain the least minimized total energies compared with
those of the other XII and XIII feasible structures. Consequently, it is concluded that XI
structure is the most preferred phase because of its least minimized total energy and all further
computations are performed at this specific ground state phase.
The outcomes are ascribed to the following reasons: (a) within the XI structure, Cr sorts
the nearest neighbor (NN) surrounds with both Y and Sb, although Sb has both Cr and Y as
NN pairs for the XII structure; (b) the size of Y is than that of Cr. Therefore, a strong bond is
established between Cr and Y resulting in the minimum total energy in the XI structure.
In the XIII structure, Sb is not in the NN arrangement with the Cr. The optimized lattice
constants of the HH YCrSb and YMnSb materials for stable XI phase are 6.673 Å and 6.565
Å, respectively. For these two studied compounds with the FM state, the computed lattice
constant, total energy, bulk modulus B (in unit GPa) and first order derivative of the modulus
B´ evaluated by using unit cell volume at zero pressure for the three unique phases are detailed
in Table 6.2. Yet, no experimental data for the bulk moduli nor the lattice constant of the
studied compounds are available to be compared with the theoretical results.
Spin-polarized (magnetic phase) and non-spin polarized (NM phase) calculations are
also carried out for each of YCrSb and YMnSb compounds within the stable XI structure. The
variations of total energy with volume are presented in Fig. (6.3) respectively for the NM, FM
and AFM states, which clearly indicate that FM state is energetically more favorable than NM
and AFM states. Furthermore, the total energy difference between the NM and FM phases
(ΔEFM-NM) in stable XI-structure is given the Table 6.3.
As can be seen, the values of ΔEFM-NM are negative for both the studied compounds,
implying that the FM phase of such materials is much steadier than NM phase. Therefore, the
next discussion provides the actual FM phase. To confirm that the studied materials can
possibly be synthesized experimentally, formation energy (𝐸𝑓𝑜𝑟) is also taken into
consideration for the YCrSb and YMnSb materials which can be explained by using the
following equation:
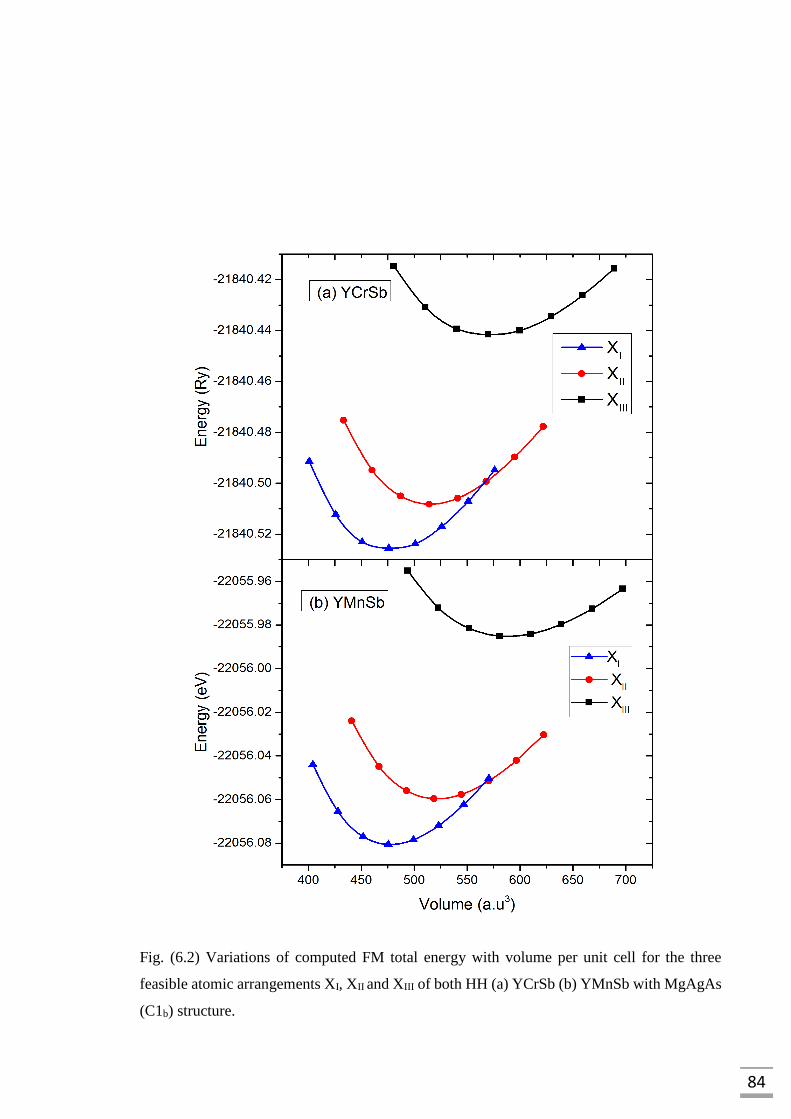
84
Fig. (6.2) Variations of computed FM total energy with volume per unit cell for the three
feasible atomic arrangements XI, XII and XIII of both HH (a) YCrSb (b) YMnSb with MgAgAs
(C1b) structure.

85
Table 6.2 Values of optimized lattice constant aopt (Å), the bulk modulus B (GPa), the pressure
derivative of the bulk modulus B, the total energy (Ry) of the HH YCrSb and YMnSb
materials.
Table 6.3 Calculated values of formation energy (𝐸𝑓𝑜𝑟) (in eV) per formula unit, spin-up
energy band gap EBG (eV), half-metallic gap EHM (eV), the energy difference between FM &
NM states ΔEFM-NM (eV) and the spin-polarization SP (%) for YCrSb and YMnSb HH
materials.
(X)4a:(Y) 4b:(Z) 4c EBG
(eV)
EHM
(eV)
ΔEFM-NM
(eV)
𝑬𝒇𝒐𝒓
(eV)
SP (%)
YCrSb 0.78 0.43 -1.68 -3.557 100
YMnSb 0.40 0.13 -1.19 -6.770 100
Table 6.4 Calculated values of total and local magnetic moment (µB) of the individual atom
and interstitial site for HH YCrSb and YMnSb materials.
Compound Y
(µB)
Cr
(µB)
Sb
(µB)
Interstitial
(µB)
µ𝐭𝐨𝐭
(µB)
YCrSb 0.17336 3.41594 -0.06046 0.47179 4
YMnSb -0.00442 2.94109 -0.02969 0.09358 3
Compound Structure aopt
(Å)
B
(GPa)
B´ Etot (Ry) Half-
metallicity
YCrSb
XI 6.673 64.6 4.71 -21840.5260 Yes
XII 6.839 57.8 4.44 -21840.5081 No
XIII 7.068 44.25 4.07 -21840.4419 No
YMnSb
XI 6.565 68.10 4.78 -22056.0804 Yes
XII 6.760 60.86 4.61 -22056.059 No
XIII 7.038 42.33 4.20 -22055.9851 No
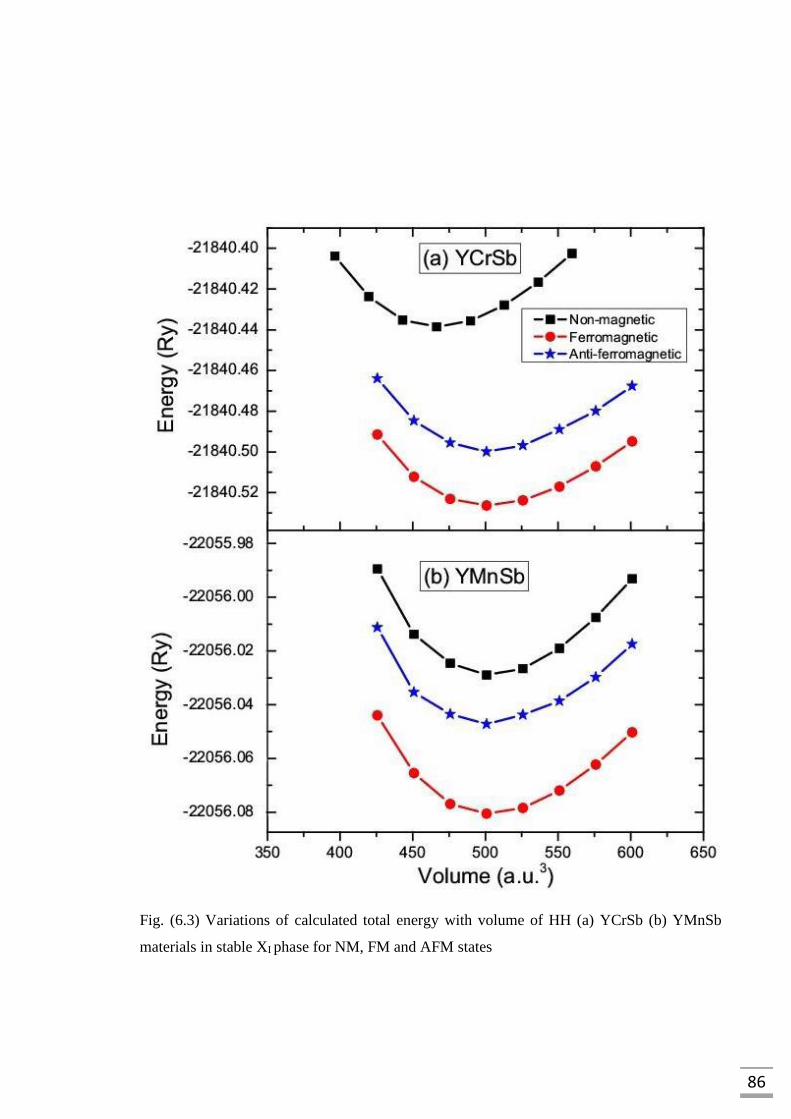
86
Fig. (6.3) Variations of calculated total energy with volume of HH (a) YCrSb (b) YMnSb
materials in stable XI phase for NM, FM and AFM states

87
𝐸𝑓𝑜𝑟 = 𝐸𝑌𝐶𝑟(𝑀𝑛)𝑆𝑏 − 𝐸𝑌 − 𝐸𝐶𝑟(𝑀𝑛) − 𝐸𝑆𝑏
where 𝐸𝑌𝐶𝑟(𝑀𝑛)𝑆𝑏 is the total energy of the YCrSb and YMnSb materials calculated by the
first-principles calculations and 𝐸𝑌, 𝐸𝐶𝑟(𝑀𝑛), 𝐸𝑆𝑏 are energies of the corresponding individual
atoms. Usually, negative formation energy signifies the stableness of the compound. Formation
energies for the YCrSb and YMnSb HH materials are listed in Table 6.3. The lower value of
formation energies for the studied compounds indicate that these materials can be synthesized
experimentally.
6.3.2 Electronic Properties
The electronic structure affects an essential part to identify the HM properties
associated with HH materials. Band structures of the HH YCrSb and YMnSb alloys are shown
in Figs. (6.4) and (6.5), respectively. The left panel demonstrates the spin-up (majority) state,
and the right panel indicates the bands for the spin-down (minority) state.
The different colors in Figs. (6.4 & 6.5) represent different physical meanings. They
show the contributions of s, p and d orbitals of Y, Mn/Cr and Sb atoms to electronic band
structure. They reveal the information about which orbital of the atoms in the alloy is
contributing more near the EF and which atom orbital is in the core state. They also represent
each eigenvalue along the k-path which we have previously selected during the SCF cycle.
Obviously, it can be noted that half-metallicity has semiconducting EBG around the EF
in the majority spin (spin-up) state Figs. (6.4 (a) & 6.5 (a)), and that the band cross the EF and
thus displays metallic nature in minority spin (spin-down) state Figs. (6.4(b) and 6.5(b)). For
both HH YCrSb and YMnSb materials in the majority spin channel, there are clear gaps
between EBG and EHM in the majority band. The EBG is the energy band gap which is the spacing
between the valance band maximum and the conduction band minimum, and the EHM is the
shortest distance twixt the most occupied valance band energy and the EF.
This parameter EHM possess specific significance for the half metallicity of the FM
material rather than EBG along the EF. The presence of non-zero flip gap (EHM) for both the
compounds in the majority spin channel indicates that they are true HMFs. The values of EBG,
EHM, and EF calculated for both HH YCrSb and YMnSb materials are presented in Table 6.3.

88
Fig. (6.4) Spin-resolved band structures of HH YCrSb (a) spin up (b) spin down. EF is set to
zero.
Spin-up Spin-dn
d0n

89
Fig. (6.5) Spin-resolved band structures of the HH YMnSb (a) spin up (b) Spin down. The
Fermi level, EF is set to zero.
Spin-up Spin-dn

90
It can also be seen that the band transition for the YCrSb is direct (Γ-Γ) while an indirect
band transition is found for the YMnSb (Γ-X) HH material. It can also be noted that bands near
the EF, are triply degenerate at Γ for the majority spin channel.
The number of states for each period of energy which is occupied by the specific energy
levels is usually explained by the DOS of the system. To examine the electronic natures of the
YCrSb and YMnSb materials, total and partial DOS within the magnetic phase designed for
spin-up and spin-down channel are also measured by utilizing the PBE-GGA. The DOS
graphics are displayed in Fig. (6.6) to analyze the EBG in the majority spin state of the studied
materials at the equilibrium lattice constant. The spin-dependent total and orbital electronic
DOS of the HH YCrSb and YMnSb are presented in Fig. (6.6).
Large spin splitting in these materials occurs from Cr/Mn (d-t2g) states with a small
contribution from Y (d-t2g) states. The contribution by the Sb atom near the EF is quite small
in comparison with by the Cr/Mn (d-t2g) and Y (d-t2g) atoms. Also, Sb atom has symmetrical
state below the EF having energy around -10 eV. A solid hybridization among the d-orbitals of
Y, Cr, and Mn atoms is found, which divides the d orbitals of these atoms into d-eg and d-t2g
states. A semiconducting gap is found for the majority-spin channel. It can be visualized that
states near the EF for both majority and minority spin states are mostly contributed due to the
Cr/Mn (d-t2g) and Y (d-t2g) atoms, which are comparable to various other TMs based on HH
materials (Huang et al., 2014; Huang et al., 2015).
To clarify the origin of the semiconducting gap, d-d hybridization nearby the EF can be
revealed inside Fig. (6.7). The Cr and Mn atoms are enclosed by Sb atoms, seeing them at NN
and Y as next neighbor. Both Cr and Mn 3d states split up into the triplet associated with d-t2g
states and a doublet associated with eg states because of the crystal field theory. In the majority
spin channel, the d-t2g and eg states of Mn and Cr ought to be occupied like in the minority spin
states but electrons are depleted in the majority spin state due to the exchange interaction.
These states turn out to be unoccupied and develop the semiconducting gap. Furthermore, the
electronic SP at the EF can be expressed by the following relation (Monir et al., 2015),
𝑆𝑃 = 𝑑 ↑ (𝐸𝐹) − 𝑑 ↓ (𝐸𝐹)
𝑑 ↑ (𝐸𝐹) + 𝑑 ↓ (𝐸𝐹)
where 𝑑 ↑ (𝐸𝐹) and 𝑑 ↓ (𝐸𝐹) are the densities of states at the EF for the majority and
minority spin states, respectively.

91
Fig. (6.6) Spin-polarized densities of state for the total and individual atoms at the equilibrium
lattice constant for the XI phase of the HH (a) YCrSb (b) YMnSb materials.
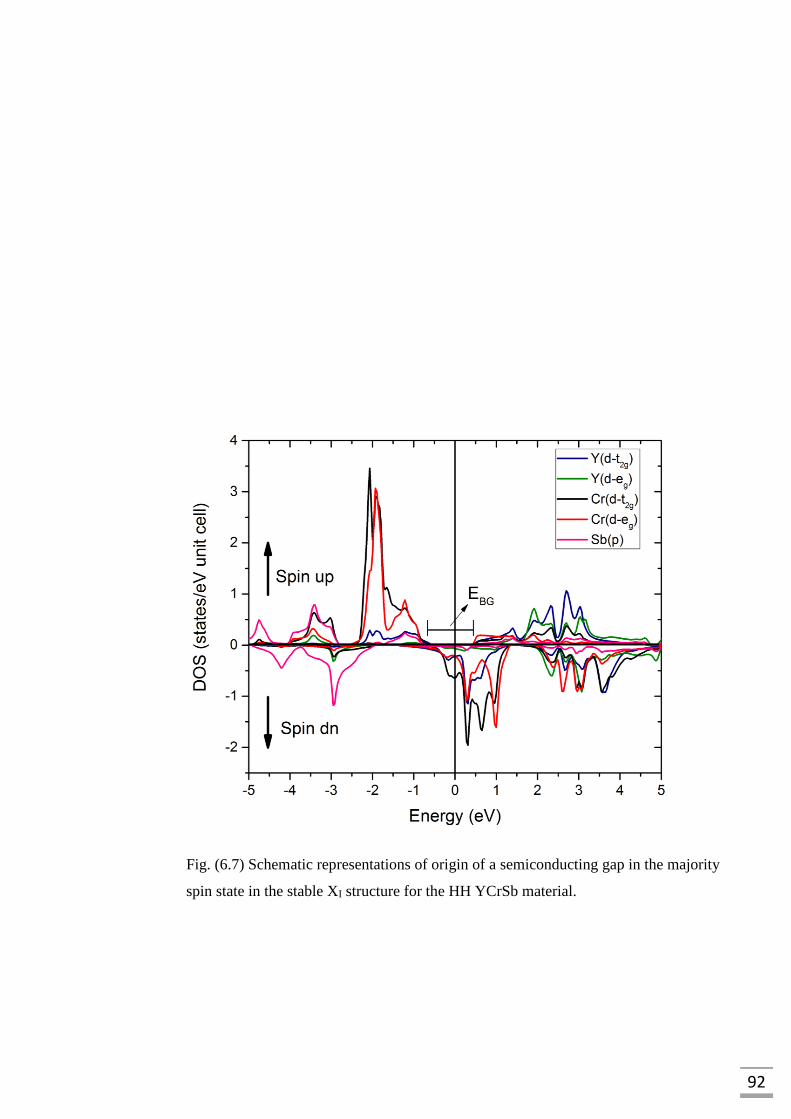
92
Fig. (6.7) Schematic representations of origin of a semiconducting gap in the majority
spin state in the stable XI structure for the HH YCrSb material.

93
The studied HH YCrSb and YMnSb materials have 100% SP (see Table 6.3), proving
that electrons at the EF are fully spin-polarized. That confirms the HM characteristics.
6.3.3 Magnetic Properties
The origin of magnetic moment for the HH alloy is described in this section. The total
and individual magnetic moment of the atoms per unit cell in the unit of multiples of Bohr
magnetron for the YCrSb and YMnSb for the XI structural phase are presented in Table 6.4.
The structures of HH materials can be decomposed right into a zinc blende (ZB) substructure
along with variants within the occupancy from the interstitial lattice sites. There are three main
mechanisms (Huang et al., 2015) which govern the magnetic properties due to HM in the ZB
structure: (a) the d-states of the Cr and Mn atoms (here it can even be Y-atom) split into triply
(t2g) and doubly (eg) due to the crystal field effect, (b) bonding and antibonding states are
formed when the sp3-type state of Sb interacts with these triplet states, (c) populations of the
electrons change in the majority and minority spin channel due to the exchange interaction.
The local magnetic moments of the crystal are formed by the remaining electrons of the Cr and
Mn atoms.
Due to the resemblance of HH alloy composition to the ZB structure, the magnetic
moment arranged in Table 6.4 can be ascribed to the Mn and Cr atoms. As the HH materials
contain only one magnetic sublattice consisting of the atoms on the octahedral sites (Graf et
al., 2011), which is also indicated in Table 6.4, the Cr and Mn atoms mainly contribute to the
µtot for the studied HH materials and occupy the octahedral sites in the stable XI structure.
It can also be understood from the electronic arrangements. Electronic configurations
for the Cr and Mn are (3d)54s and (3d)54(s)2, respectively. For pure metals, Cr and Mn,
electronic spins form the magnetic moments according to the first Hund’s rule but when these
types of TMs (Cr, Mn) are made into alloys, the availability of 3d-electrons will change
because of diverse electro-negativity. In the stable XI structure, Sb is the NN for the Cr and
Mn in the HH YCrSb and YMnSb materials because of this a small charge is transferred from
Cr and Mn to Sb by leaving lots of d-electrons at Cr and Mn atoms that govern the magnetic
moments for these HH materials respectively.

94
6.3.4 Location Associated with Half-Metallicity
In the experimental fabrication of HH alloys on a substrate to make device applications,
lattice mismatch may happen due to the influence of various factors. For this “strain
engineering” technique (Lu et al., 2001) is usually used to obtain the required electrical and
physical properties of HH alloys through developing layered alloys by lattice mismatched
substrates. Therefore, it is essential to further investigate the lattice parameter strain
engineering by exploring the robustness belonging to the HM through changing the lattice
constants of these two studied materials.
The µtot and the spin moments of the Y, Cr, Mn and Sb atoms with respect to lattice
parameter are shown in the Fig. (6.8). When the lattice parameters of HH YCrSb and YMnSb
materials are expanded, the hybridization between Y and Cr/Mn decreases, which leads to the
rise in the spin moment of Cr/Mn and decrease in Y. The EF found inside the gap and the
number of majority spin states change a little in all these lattice variations. In addition, it is
also observed that by varying the lattice constants in a wide range, there is slightly change in
the overall µtot, and the overall µtot remain 4 and 3 µB for the HH YCrSb and YMnSb materials,
respectively.
It is also discovered that for the optimized equilibrium lattice constant for the stable XI
phase, the magnetic moments have integer values for both compounds, which is viewed as the
character associated with HMF. Thus, these types of substrates may be handled as ideal
applicants for the spintronic devices. Through the computations, the HM can be found to reach
up to compression and expansion of -10.1% to 3.6% for the HH YCrSb and for -12.3% to 2.7%
for the HH YMnSb material. This verifies that magnetic properties, can be tuned for these
substrates, by expanding and compressing their lattice parameters.
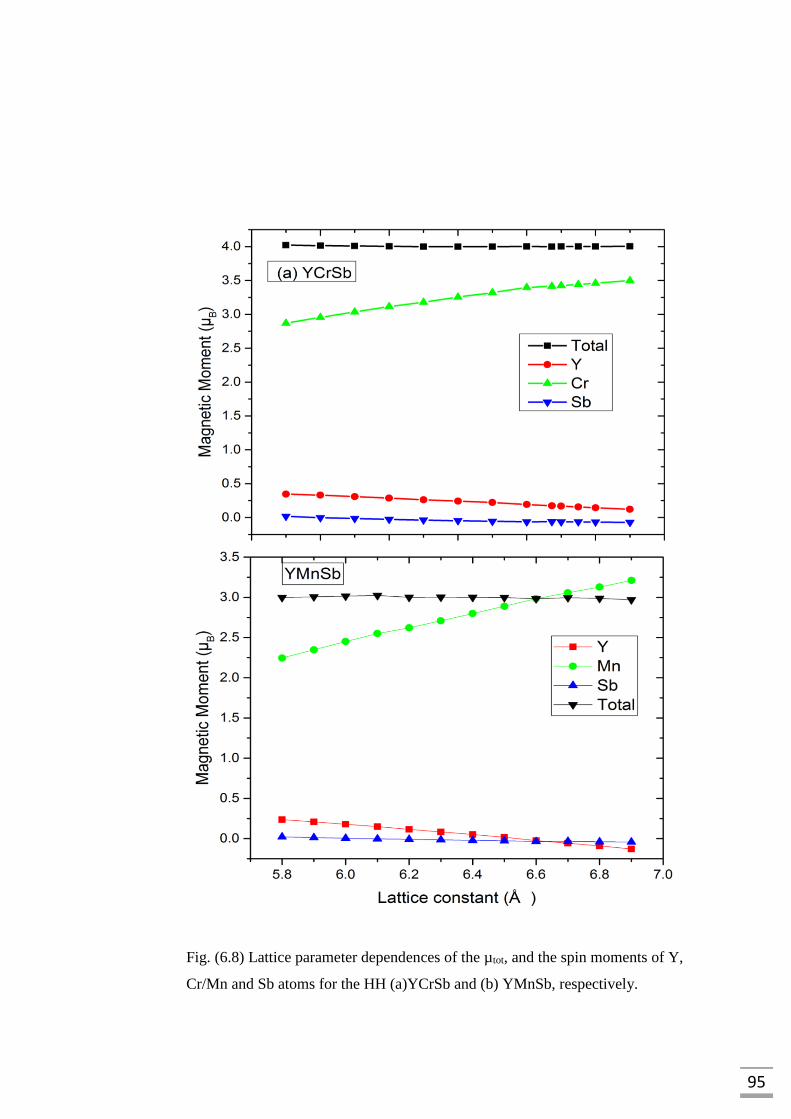
95
Fig. (6.8) Lattice parameter dependences of the µtot, and the spin moments of Y,
Cr/Mn and Sb atoms for the HH (a)YCrSb and (b) YMnSb, respectively.

96
Chapter 7
Physical Properties of Half-Heusler YMnZ (where Z = Si, Ge, Sn)
Compounds Via Ab-Initio Study
7.1 Introduction
Recently, HM magnets have drawn a plenty of attention due to their attainable uses in
spintronics and magneto-electronics (Hirohata and Takanashi, 2014; Žutić et al., 2004). Highly
spin-polarized magnetic materials had been popular to enhance the overall performance
associated with spintronics products for example spin filter as well as spin-valves (Inomata et
al., 2016) and have several advantages over conventional SC electronic devices such as
reduced energy usage, the higher processing speed of data, non-volatility as well as elevated
integration densities (Wolf et al., 2001). Numerous appealing materials, such as half-metals
(De Groot et al., 1983; Tanveer et al., 2015), SC (Tobola et al., 1998), magnetic shape memory
alloy(Ullakko et al., 1996), topological insulator (Chadov and Qi, 2010) and spingapless SC
(Ouardi et al., 2013) like properties happen to be present in HA.
Recently half-metallicity has been found by numerous research groups in the based on
zinc-blende SC compounds (Ahmad and Amin, 2013; Arif et al., 2012), dilute magnetic alloys
(Amin and Ahmad, 2009; Amin et al., 2011), HH alloys (Ahmad and Amin, 2013; Hamidani
et al., 2009; Huang et al., 2014; Tanveer et al., 2015), QH alloys (Rasool et al., 2016; Wang et
al., 2017a) and in the FH alloys not only theoretically (Deka et al., 2016; Gupta and Bhat,
2014; Hu and Zhang, 2017; Qi et al., 2015; Rauf et al., 2015; Yousuf and Gupta, 2017) but
also experimentally (Bae et al., 2012; Checca et al., 2017; Vivas et al., 2016). Heusler based
materials are more prominent because they have the probability of achieving higher value of
TC and their structure resemblance to the zinc-blende and diamond structures which prevail in
a large variety of known SC compounds.
Fritz Heusler in 1903, discovered that compound having composition Cu2MnAl acts
like a HM material. HM materials demonstrate absolutely distinct actions within two bands i.e.
semiconducting regarding electrons in one spin band showing a gap at the EF and exhibits
conventional metallic character in complete reverse spin alignment for the other band.

97
Considering that just one spin band is conducting, it is possible to picture a tool in
which the digital conduction might be merely switched off and on with a permanent magnetic
field inside a spin valve or even magnetic tunnel junction, similar to the electrical field-effect
products, in which the conduction is actually switched off and on, with an entrance electrode
(Behin-Aein et al., 2010). The spin–orbit coupling will be disregarded due to its tiny share for
the magnet attributes.
The presence of energy Egin majority spin band results in a 100% SP at the EF and
therefore the completely spin-polarized current ought to be achievable within these HM HA
making the most of the actual effectiveness and performance associated with magneto-
electronic devices (De Boeck et al., 2002a; De Boeck et al., 2002b) and capable of presenting
amazing attributes such as shape-memory alloys as well as tunable topological insulators
having a higher possibility of spintronics, power systems, half-metals, multi-feroics, high
temperature ferri and ferromagnets and magneto-caloric programs(Felser et al., 2015).
Materials regarding state-of-the-art applications turn out to be progressively complicated and
nowadays electronic industry mostly depends on elemental SC Si and Ge.
Latest developments in physics, chemistry, and material science empowered the
realistic layout of the fresh usages regarding various advanced technology. FH alloys and HH
alloys which show HM properties possess a plethora of probable prospective applications
within spintronics, data storage, magnetocaloric and as permanent magnets. So far, considering
the ab-initio electronic structure calculations, numerous compounds were anticipated proposed
to be HMFs. A breakthrough discovery in the half-metallicity with most materials owned by
HH family has drawn substantial interest from the scientists (Kübler et al., 1983).
Yet, HM properties are hard to be maintained at ambient conditions except if there is a
big EHM and this gap need to be effective towards diverse lattice constants. To design the shape
memory combination, not only the reduced value of cohesive energy is required to strain
effortlessly but also the reduced value of formation energy (Efor) guarantees that these
compounds could be experimentally synthesized. As compared to the FH alloys, a very few
HM HH composites can be synthesized experimentally. Although, some ternary HH
compounds like YPtBi and YAuPb are found to be non-trivial topological insulators with zero
EBG while YAuSn, YPtSb, YNiBi, YPdSb, and YPdBi are found to be trivial band insulators
(Al-Sawai et al., 2010).

98
In one of our previous theoretical work, YMnSb and YCrSb also show HM properties
(Sattar et al., 2016a). This research is aimed to investigate the magnetic, electronic and
transport properties of HH YMnZ (Z= Si, Ge, Sn) alloys to identify at which lattice constant
range the HM character is maintained along with high magnetization. The study has predicted
some important results above and beyond 100% SP and half-metallicity in these alloys. The
present investigation also aims to evaluate theoretically the thermoelectric efficiency of HH
YMnZ alloys by calculating the Seebeck coefficient, electrical conductivity, and thermal
conductivity.
Due to increase in demand of thermal-electrical energy conversion, the thermoelectric
properties of various materials such as clathrates, filled-skutterudites, layered cobalt oxides,
and HH alloys have been studied (Mohankumar et al., 2015). Particularly, in HA, the
thermoelectric properties of many HH materials have been investigated theoretically and
experimentally (Hu and Zhang, 2017; Mikami et al., 2013; Xu et al., 2011; Yadav and Sanyal,
2015; Yousuf and Gupta, 2017). The advantage of HA is that one can play with the
thermoelectric properties by doping easily.
In the light of the above, in this chapter, structural stability, electronic, magnetic and
transport properties of the YMnZ materials are investigated based on ab-initio calculations
with the HH structure which has the general formula XYZ and crystallizes in the space group
F-43m (No. 216). We have theoretically investigated to identify at which lattice constant range
the HM character is maintained along with high magnetization. The present investigation also
aims to evaluate theoretically thermoelectric efficiency of YMnZ HH alloys by calculating the
Seebeck coefficient, electrical conductivity, and thermal conductivity. The present work is
totally original.
The remaining chapter is sorted out as follows: In section 7.2, method and
computational points of interest are discussed. In section 7.3, the crystal structure and stability
of HH alloys are portrayed and the fundamental results are elaborated.
7.2 Computational Insights
Geometrical optimization and also electronic structural computations are performed by
using the density functional theory (DFT) which has been carried out by means of self-
consistent full potential linearized augmented plane wave (FP-LAPW) method accomplished
through Wien2K simulation code (Blaha et al., 2001b).

99
The exchange-correlation outcomes have been explained with the parameterization
from the generalized gradient approximation (GGA). In order to explore the electronic and
magnetic properties with accurate Eg, the state-of-art mBJ local density approximation method
offered by Tran and Blaha (Tran and Blaha, 2009) is utilized. In present calculations, the value
of Rmt×Kmax is selected to 9 that decides the good convergence for the matrix size in which
Kmax is the plane wave cutoff energy and Rmt is equivalent to the minimum value of muffin-tin
sphere radii. For the denser BZ sampling, a k-point mesh of 21×21×21 in the first BZ (10000
k-points in full BZ) is chosen. For the convergence requirements, total energy and force have
been preserved as 10-5 eV and 10-4eV/Å, respectively.
7.3 Results and Discussions
7.3.1 Structural Properties
Both theoretical and experimental (Fecher et al., 2005; Miura et al., 2004a; Miura et
al., 2004b; Picozzi et al., 2004; Pierre et al., 1997; Wurmehl et al., 2006) research work about
Heusler compounds acknowledge that their structural arrangement has a significant effect on
their physical properties. This kind of robust relationship among the structural arrangement of
HA and their useful properties require an intensive structural depiction when their physical
attributes are usually reviewed.
The HM YMnZ alloys are characterized in the HH with a face-centered cubic (fcc) C1b
phase having space group of F-43m (Feng et al., 2014) which are directly relevant to zinc-
blende and diamond structure. The crystalline framework (C1b) of this kind of materials can
be reviewed properly in the literature (Gruhn, 2010; Sattar et al., 2016b).
Possibly, there are three unique sorts of atomic placement in the conventional cubic
unit cell X-type1: 4a(0,0,0), 4b(½, ½, ½), and 4c(¼, ¼, ¼); X-type2: 4c(¼, ¼, ¼), 4a(0,0,0),
4b(½, ½, ½), and X-type3: 4b(½, ½, ½), 4c(¼, ¼, ¼) and 4a(0,0,0) as suggested by the T. Graf
et al (Graf et al., 2011) and placed in Table 1. Just to illustrate, the conventional cubic unit cell
of the HH YMnSi material in the three different types is pointed out in Fig. (7.1). In our finest
details, there is not a single experimental or hypothetical review performed on these alloys.
Thus, we calculated the lattice constant by using Murnaghan equation of state
(Murnaghan, 1944b) within GGA with a specific goal to discover the ground state properties.
For the initial first stage, to search for stabilize structural layout along with the correct magnetic
state with an objective to determine the exact ground state of the HH YMnZ alloys.

100
Fig. (7.1) The conventional cubic unit cell of the HH YMnSi at the diverse atomic
arrangement X-type1, Xtype2, and X-type3.
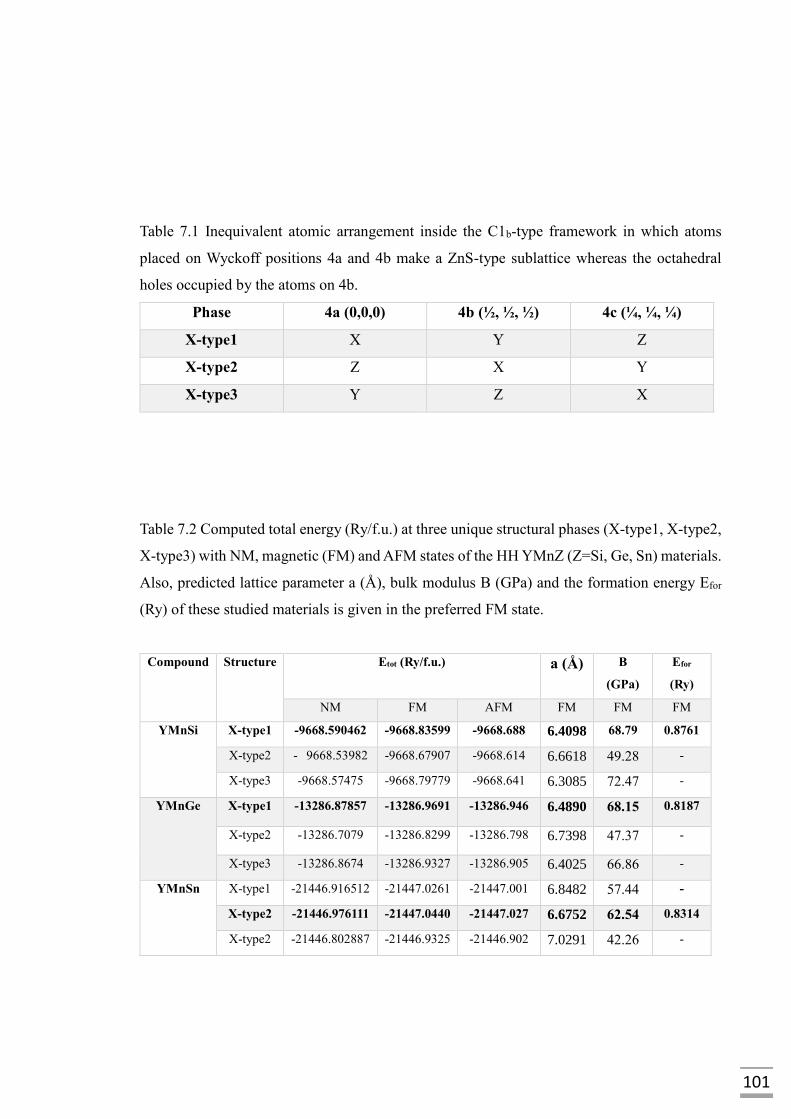
101
Table 7.1 Inequivalent atomic arrangement inside the C1b-type framework in which atoms
placed on Wyckoff positions 4a and 4b make a ZnS-type sublattice whereas the octahedral
holes occupied by the atoms on 4b.
Phase 4a (0,0,0) 4b (½, ½, ½) 4c (¼, ¼, ¼)
X-type1 X Y Z
X-type2 Z X Y
X-type3 Y Z X
Table 7.2 Computed total energy (Ry/f.u.) at three unique structural phases (X-type1, X-type2,
X-type3) with NM, magnetic (FM) and AFM states of the HH YMnZ (Z=Si, Ge, Sn) materials.
Also, predicted lattice parameter a (Å), bulk modulus B (GPa) and the formation energy Efor
(Ry) of these studied materials is given in the preferred FM state.
Compound Structure Etot (Ry/f.u.) a (Å) B
(GPa)
Efor
(Ry)
NM FM AFM FM FM FM
YMnSi X-type1 -9668.590462 -9668.83599 -9668.688 6.4098 68.79 0.8761
X-type2 - 9668.53982 -9668.67907 -9668.614 6.6618 49.28 -
X-type3 -9668.57475 -9668.79779 -9668.641 6.3085 72.47 -
YMnGe X-type1 -13286.87857 -13286.9691 -13286.946 6.4890 68.15 0.8187
X-type2 -13286.7079 -13286.8299 -13286.798 6.7398 47.37 -
X-type3 -13286.8674 -13286.9327 -13286.905 6.4025 66.86 -
YMnSn X-type1 -21446.916512 -21447.0261 -21447.001 6.8482 57.44 -
X-type2 -21446.976111 -21447.0440 -21447.027 6.6752 62.54 0.8314
X-type2 -21446.802887 -21446.9325 -21446.902 7.0291 42.26 -

102
For this, we performed minimization of energy as a function associated with a lattice
constant towards three distinctive conceivable site occupation for each NM, FM as well as
AFM structures. The acquired results are demonstrated in Figs. (7.2 & 7.3).
Computed optimized lattice constant ɑ (Å) combined with bulk modulus B (GPa) and
the total energy Etot (Ry) within their distinctive structural phases (X-type1, X-type2, and X-
type3) as well as various NM and magnetic states (FM, AFM) for the HH YMnZ materials are
listed in Table 7.2. The X-type1 structure with an FM ground state for both studied compounds
have the most reduced energy among the three conceivable structures in the respective different
magnetic configurations and is presented as the ground state structure which is clearly visible
in the Figs. (7.2 & 7.3).
As the atomic radii of Sn is greater than the Si/Ge and Z (Si, Ge and Sn atoms) share
the same group in the periodic table, the optimized lattice constant of HH YMnSi is smaller
than the HH YMnGe and YMnSn alloy, as a result, bulk modulus reduces due to the weak
hybridization among the atoms. Our results are the DFT predictions as there is no experimental
or theoretical evidence present regarding the lattice constant and bulk modulus for these
studied compounds.
To examine the stableness of these DFT investigated HH YMnZ studied compounds,
the estimation of the formation energy (EFor) is important which decides whether a compound
can be synthesized experimentally or not. It can be defined as the adjustment in energy when
the compound is shaped through its constituent components to their compound form and can
be measured as,
EForYMnZ = Etot
YMnZ - EY - EMn – EZ
where EtotYMnZ are the first-principles calculated equilibrium total energies of the HH YMnZ
compounds per formula unit. The EY, EMn, EZ are the equilibrium total energies for each atom
of the natural distinct elements Y, Mn and Z (Si, Ge, Sn) within their individual stable state,
respectively. It is observed that Y take the hcp structure, while Mn crystallizes in bcc along
with the Si, Ge and Sn prefers fcc diamond structure.
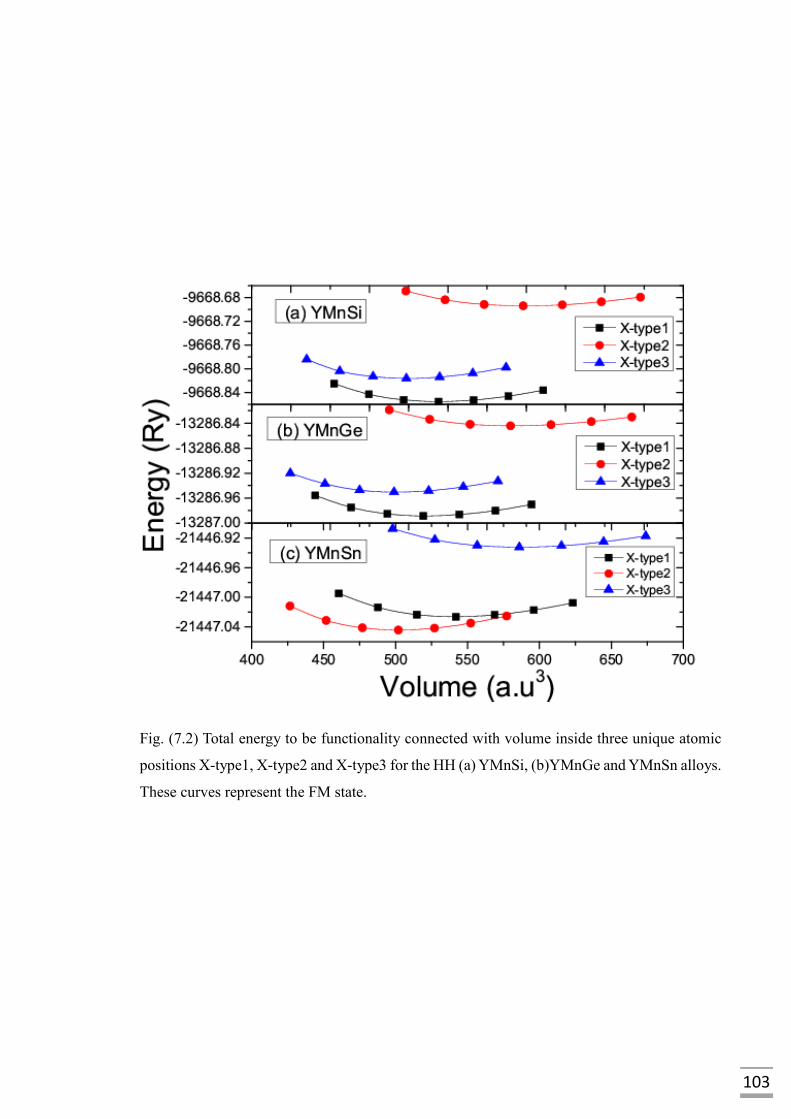
103
Fig. (7.2) Total energy to be functionality connected with volume inside three unique atomic
positions X-type1, X-type2 and X-type3 for the HH (a) YMnSi, (b)YMnGe and YMnSn alloys.
These curves represent the FM state.
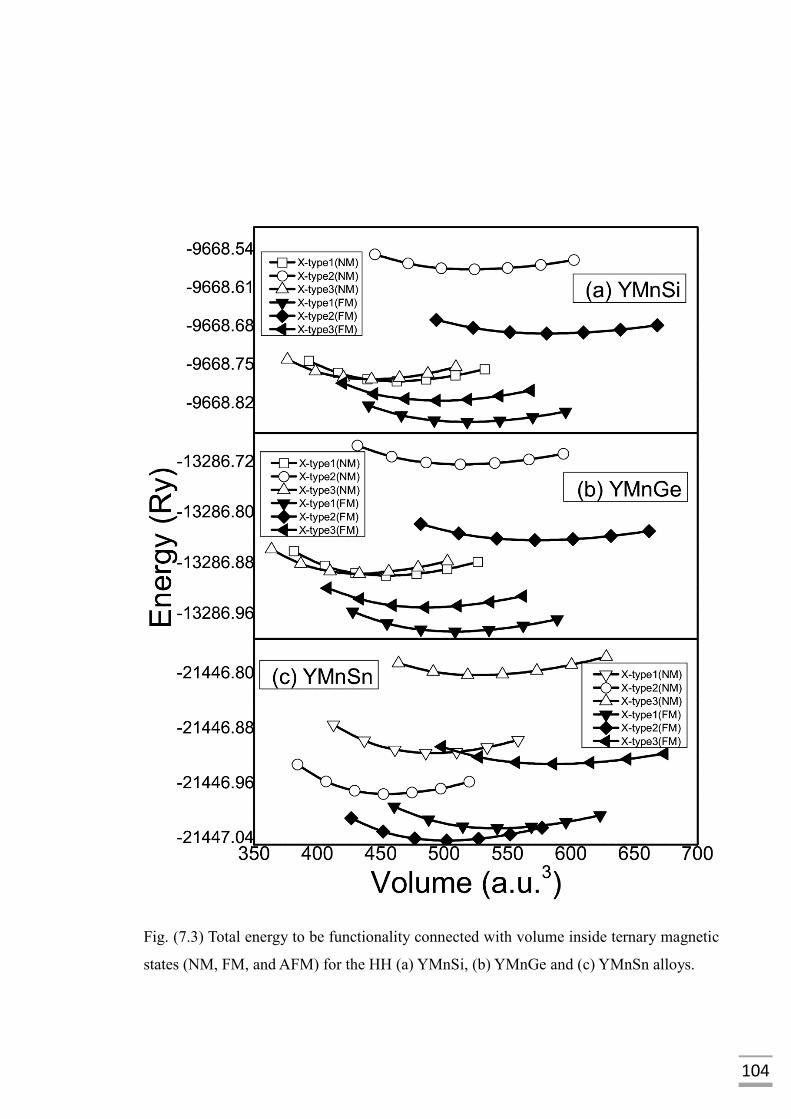
104
Fig. (7.3) Total energy to be functionality connected with volume inside ternary magnetic
states (NM, FM, and AFM) for the HH (a) YMnSi, (b) YMnGe and (c) YMnSn alloys.

105
7.3.2 Electronic Properties
In this segment, the electronic and magnetic properties for both studied HH
materials are examined through GGA and GGA-mBJ for the comparison between them.
The GGA-mBJ predicts energy EBG with favorable accuracy and work of Guo et al. (Guo
and Liu, 2011) on FM materials are in phenomenal concurrence with experimental
outcomes.
Spin-dependent band structure on the optimized relaxed lattice constant with high
symmetry guidelines regarding the first BZ in the stable X-type1 structural phase of HH
YMnSi and YMnGe alloys have been computed and exhibited in Figs. (7.4 &7.5) whereas
the band structure of the HH YMnSn is shown in Fig. (7.6) at their equilibrium lattice
constant with the most stable phase of X-type2.
For GGA computations, it is clearly visible from these plots that the physical nature
of YMnSi is of metallic and YMnGe and YMnSn are half-metal. The GGA-mBJ
estimations reveal that spin-down states (minority bands) stay metallic but in the spin-up
states (majority-bands), valence band maxima (VBM) shifts downwards to the reduced
energy area and conduction band minima (CBM) is moved upwards towards the greater
energy region comparatively to the EF and thus displaying the EBG.
To demonstrate the character associated with the electronic structure, we have
additionally plotted the computed spin-polarized total as well as the partial DOS. The Fig.
(7.7) demonstrate the computed spin-dependent total DOS and atom projected partial DOS
for the two HH YMnSi and YMnGe materials with the X-type1 structure and for the HH
YMnSn material with the X-type2 structure at the optimized lattice constants.
It can be viewed from the Fig. 7.7 (a, b & c) that the overall general features of the
DOS for the three studied materials are comparable because of the likeness of their crystal
framework and chemical ambiances. It has been observed that DOS acquired from GGA
and mBJ adopts almost the similar pattern with a difference of gap.
It can be easily visualized from the total DOS that HH YMnZ are HM materials
because EF falling in the semiconducting gap in the spin-up channel with EBG is found of
0.4214 eV, 0.5761 eV and 0.5903 eV for the HH YMnSi, YMnGe and YMnSn compounds
respectively and shows the 100% SP for both HH materials. On the other hand, there is an
intersection at the EF for the spin down channel which strongly suggests that these materials
are true HMFs.

106
Fig. (7.4) Spin-projected band structure with the HM HH YMnSi alloy. Black solid lines
show the GGA and red dotted lines are for the GGA+mBJ
Spin-up Spin-dn

107
Fig. (7.5) Spin-projected band structure with the HH YMnGe compound. Black solid lines
show the GGA and red dotted lines are for the GGA+mBJ.
Spin-up Spin-dn

108
Fig. (7.6) Spin-projected band structure with the HH YMnSn compound. Black solid lines
show the GGA and red dotted lines are for the GGA+mBJ.
Spin-up Spin-dn

109
Besides, the partial DOS of the HH YMnSi and YMnGe materials are for the most
part involved by the Mn d-states with a little share of Y d-states electrons and it can be
obviously observed that for both materials a substantial exchange splitting exists among
the spin-up and spin-down channel due to the d-states of Mn and Y atoms.
These d-states of Mn and Y atoms are separated into eg states at the low energy and
presence of t2g states declare at the high energy due to the hybridization between them. The
s-bands are not shown in Fig. 7.7 (a, b & c) because these bands are very low in energy and
thus easily can be distinguished from other bands. It is also clear from the plot that the
lower energy region of the valance band is related to the p-bands of Z atoms. For both spin-
channels, the s and p-states are almost alike. Furthermore, it is noted that the partial DOS
of Mn 3d-orbitals represents the same tendency. It is also observed that the maximum peak
of DOS around EF is mainly linked to Mn 3d-states. This affirms that the bonding state, for
the most part, exists at the greater valence transition element Mn, as the contribution of
partial DOS for Y d-orbital is quite small when compared to the Mn d-orbitals. Symmetrical
states are found for the Y atom below and above the EF. States below and above the EF for
both spin channels are because of d-states of Mn and Y atoms, as several other TMs HH
alloys studies (Huang et al., 2014; Huang et al., 2015) suggest that states around the EF for
both spin channels are mostly contributed by the d-orbitals of the TMs.
7.7.3 Magnetic Properties
For the HH YMnZ studied materials, the estimated total magnetic moment per unit
cell at their optimized lattice parameter is precisely 4.00 μB. The integer value associated
with magnetic moment shows the halfmetallicity of the HH alloys. The computed total
magnet moments atomic-resolved and share regarding interstitial locations inside the
crystal (interstitial magnetic moments) in the HH YMnZ compounds at their optimized
lattice constant with the stable structure X-type1are listed in Table 7.3.
It is observed that the maximum contribution in the µtot comes from Mn which is
the result of the immense exchange splitting among the majority as well as minority spin
states of Mn. It is furthermore pointed out that partial moments of the Y and Z(Si, Ge, Sn)
atoms are extremely little and their share for the overall magnetic moment is quite little
which can be noticed from the Table 7.3 as well.
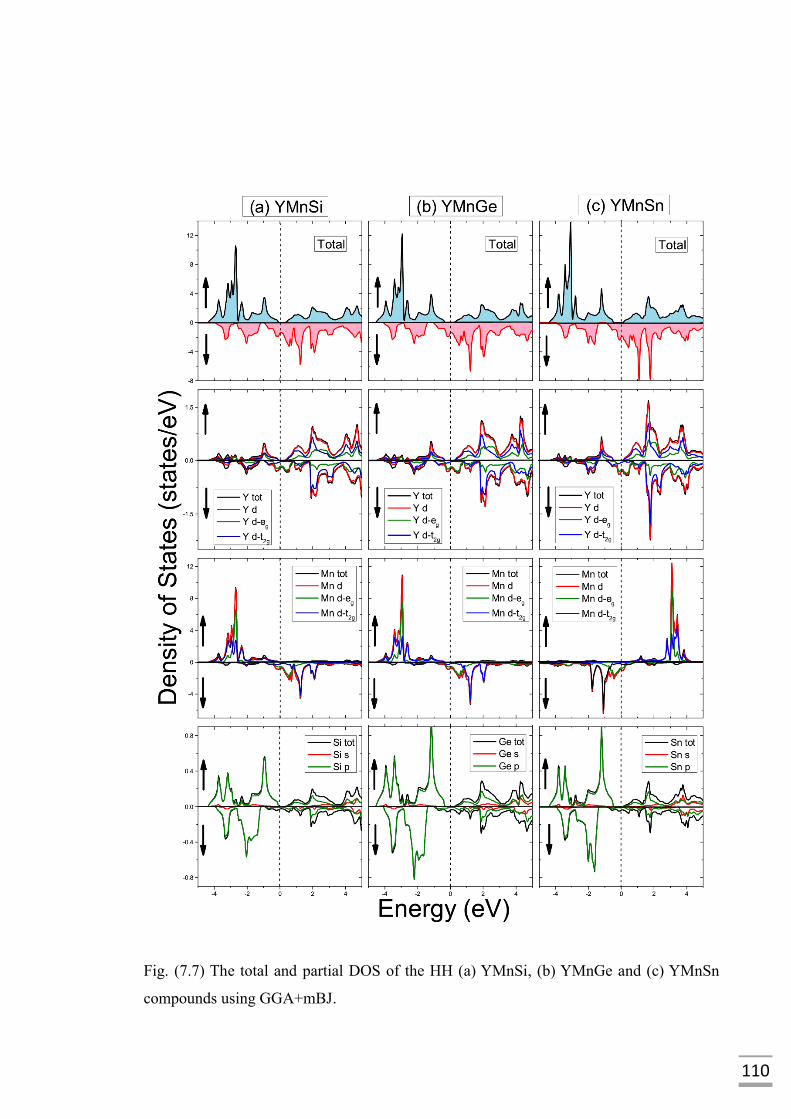
110
Fig. (7.7) The total and partial DOS of the HH (a) YMnSi, (b) YMnGe and (c) YMnSn
compounds using GGA+mBJ.

111
The expression of the SP of a magnetic material can be provided by
𝑆𝑃 = 𝑁 ↑ − 𝑁 ↓
𝑁 ↑ + 𝑁 ↓
in which 𝑁 ↑ and 𝑁 ↓ are the number of spin-up and spin down states and 𝑃𝑁 measures the
spin imbalance of valence electrons. Alternately, at the EF, the electron SP can be expressed
as
𝑆𝑃 = 𝑑 ↑ (𝐸𝐹) − 𝑑 ↓ (𝐸𝐹)
𝑑 ↑ (𝐸𝐹) + 𝑑 ↓ (𝐸𝐹)
in which 𝑑 ↑ (𝐸𝐹) and 𝑑 ↓ (𝐸𝐹) are the spin dependent DOS at the EF for the spin up and
spin down channel, respectively. In this study, the HH YMnZ materials calculated with
GGA-mBJ have 𝑆𝑃 (%) = 100% . It means that the electrons at the EF are completely
polarized, thus confirming that these two studied compounds possess HM characteristics.
The calculated values of SP (%) are also listed in Table 7.3.
The magnetic moment of Z and Y atoms are quite small and almost negligible when
compared to the magnetic moment of Mn and do not play a considerable role to the µtot of
the materials. A very minor negative interstitial magnetic moment of the interstitial region
is also discovered as suggested by the Table 7.3. It is notable that during the experimental
synthesis of the materials like the non-equilibrium melt-spun as well as ball milling process
for the spintronics appliances, the lattice parameter is broadly impacted.

112
Table 7.3 The computed µtot (µ/f.u.) and local magnetic moments (µatomic/f.u.) of the HH
YMnZ (Z=Si, Ge, Sn) materials with X-type1 phase at the two exchange-correlation
potential (VXC) are given. Band gap energy: EBG (eV) and half-metallic gap: EHM (eV) is
also described at the Electronic conductivity (metallic, HM, or semiconducting).
Material VXC EBG
(eV)
EHM
(eV)
µtot
(µB)
µY
(µB)
µMn
(µB)
µZ
(µB)
µI
(µB)
Electronic
conductivity
YMnSi GGA 0.24 ----- 3.98 -0.02 3.92 -0.02 0.10
Nearly
HM
GGA+mBJ 0.42 0.16 4.00 -0.02 3.98 -0.02 0.06 HM
YMnGe GGA 0.32 0.05 4.00 -0.07 4.06 -0.03 0.04 HM
GGA+mBJ 0.57 0.36 4.00 -0.04 4.04 -0.02 0.02 HM
YMnSn GGA 0.33 0.23 3.98 -0.06 4.01 -0.02 0.06 HM
GGA+mBJ 0.59 0.50 4.00 -0.07 4.11 -0.02 -0.1 HM

113
The lattice constant can be changed considerably because of the uncontrolled
change in the strain. So, the lattice constant deviates from its optimized equilibrium value.
Furthermore, when the multilayers of the materials are prepared by molecular beam epitaxy
(MBE) or by some different techniques, the strain due to the substrate likewise is not
avoidable. Therefore, for the future hypothetical investigation as well as the experimental
synthesis of these HH YMnZ HM materials, it is very meaningful and useful to explore the
effect of lattice constant variation on these studied materials to ensure the half-metallicity
for the extensive variety of lattice parameter.
For the studied materials YMnZ, the computed total as well as a local magnetic
moment of these elements (Y, Mn, and Z) as a function of lattice constant are displayed in
Fig. 7.8 (a, b & c). It can be noticed that the magnetic moment of Mn atoms slightly
improves by the increase of the lattice parameter, even though the magnetic moment of Y
reduces. The actual change within Mn as well as Y magnetic moments changes one another
and keeps the entire magnetic moments being an integer.
It means that spin moment of Mn and Y atoms improve together with expanding
lattice parameter continues till a µtot within the unit cell. Once the lattice constant is
broadened, the improvement associated with the partial spin magnetic moment is a
consequence of the adjustment from atomic-like character ensuing through the decline
associated with hybridization in between nearby atoms. According to the SPR, for the HH
materials, the relationship between the µtot and final amount associated with 𝑁𝑣𝑎𝑙 inside the
unit cell is
µtot = (Z - 18) µB
But for the HM HH materials such as in the present study, the amount of valence
electrons is less than 18 and EBG is in the majority-spin state instead of minority spin state.
In this manner, the above the principle is not applicable and the suitable relationship will
be adopted. The µtot is a number of uncompensated spins results in
µtot = (Nmaj - Nmin) µB = (2Nmaj - Nval) µB
In which Nmaj and Nmin are the quantities of the majority and minority spin state,
respectively (Galanakis, 2002a). For all the three studied materials, the majority-spin bands
tend to be occupied with a total of 9 electrons for each unit cell.

114
In this manner, the suitable relation for such materials is,
µtot = (18 – Nval) µB
where µtot is equivalent to twice the quantity of possessed majority electrons minus the Nval.
In the case of HH YMnZ materials, the number of valence electrons is equivalent to 14 in
which Mn atom contribute 7, whereas 3 from Y atom and 4 from Z= (Si, Ge & Sn) atom.
Therefore, the magnetic moment is exactly 4 µB per unit cell. This µtot agrees with our DFT
calculated results. It also means that the majority band takes the 9 electrons for each unit
cell whilst the minority channel consists of 5 electrons providing the magnetic moment of
4 µB for each unit cell.
To examine the reliance of conducting behavior and magnetic properties on the
optimized lattice constant, we performed calculations to find out the aggregate magnetic
moment to be a functionally connected with lattice parameter for the wide range of 6.0 Å
to 7.4 Å. The half-metallicity preserve itself for the lattice constant range of around 6.3 Å
to 7.1 Å for YMnSi whereas for the YMnGe this HM range is in between 6.2 Å to 7.0 Å
and for the HH YMnSn half-metallicity is maintained from lattice constant range of 6.4 Å
to 7.1 Å. The graph between the µtot versus lattice constant is plotted in Fig. 7.8 (a, b & c).
Varieties of valence band maxima (red circles) and conduction band minima (black
squares) are also presented in the Fig. 7.8 (a, b & c) in the spin-up channel to related with
lattice parameter. It can be spotted that HM region is almost the same for these studied
materials.
The HH YMnSi, YMnGe, and YMnSn continue to retain the HM behavior with all
the lattice constants inside the array of 6.3-7.1 Å, 6.2-7.0 Å and 6.4-7.1 Å respectively.
This kind of outcome affirms in which the lighter component has the steady half-metallicity
regarding the variety of the lattice parameter (Özdogan et al., 2006). The EBG in the spin-
up band (majority channel) is given by the difference amongst VBM to CBM. It is visible
(Fig. 7.8) that EBG increases with the increasing lattice constant in the majority spin-band.
7.3.4 Thermoelectric Properties
In the present era, where the generation of the energy mainly presides over fossil
fuels, requires efficient and easily manipulated substitutes that are eco-friendly in nature
(Yousuf and Gupta, 2017). The interest in supportable energies has started a substantial
investigation into various sorts of power change advancements during the past ages.

115
Fig. (7.8) The computed total magnetic moment (µtot) for the HH (a) YMnSi, (b) YMnGe
and YMnSn materials corresponding to the variation of lattice constant. The dashed vertical
line shows the optimized equilibrium lattice constant.

116
Thermoelectric compounds, which can straight forwardly transform waste material
heat into feasible electrical energy, have obtained a growing number of consideration for
appealing applications within power collection (Snyder and Toberer, 2008).
To affirm the current outcomes, the electronic transport properties, and the
electronic conductivity along with, thermal conductivity, Seebeck coefficient and power
factor have been computed by applying the Boltzmann transport equation (Wooten, 1972)
as applied inside the BoltzTrap code (Madsen and Singh, 2006). This is an established fact
that the relaxation time for the scattering in BoltzTrap code is treated as constant.
In the present research, we estimated the thermoelectric variables to be functionally
connected with temperature shifting from 300 K to 800 K. The averaged value of the
electrical conductivity of HH YMnZ materials is plotted as a function of the temperature
in Fig. 7.9 (a), demonstrating distinctive patterns of the present HH materials and
uncovering a robust reliance on temperature. Moreover, at 300 K, it is obvious that
electrical conductivity associated with YMnSi offers higher value (7.55×1019 (Ω.m.s)-1)
compared to the estimations of YMnGe (i.e. 7.22×1019 (Ω.m.s)-1) and (i.e. 7.0×1019
(Ω.m.s)-1) YMnSn. Past 300 K, the electrical conductivity of all the studied materials has
expanded with temperature (till 800 K). The expansion of temperature prompted to an
increment of the carrier concentration number and in addition portability of carriers, which
at long last outcome in more prominent electrical conductivity. The studied materials tend
to be p-sort HH, henceforth the registered conductivity is essentially prompted by the hole
carrier concentrations.
Fig. 7.9 (c) demonstrates the averaged value of Seebeck coefficient (S) versus
temperature fluctuating from 300 K to 800 K. We acquired a positive value of S for all the
studied materials showing the dominance of holes over electrons. The Seebeck coefficient
(S) of HH YMnZ materials are incremented to 800 K with the expansion of temperature.
Essentially, we note two imperative issues concerning the impact of these studied HH
materials on the Seebeck coefficient: Firstly, at low temperature the value of S (YMnSi) >
S (YMnGe) > S (YMnSn) because of contrast of several carrier concentration for all the
studied materials. Secondly, interchanging Si by Ge/Sn (is much like include more
electrons or lessen the holes concentrations) sometimes appears as changing the carrier
concentration that triggers an extensive anisotropy in the estimated S of YMnZ materials.
The YMnSi has the greatest value of S (at 800 K) since it consists of more hole
concentration for a little scale in comparison with the YMnGe and YMnSn.

117
Fig. (7.9) (a) Electrical conductivity, (b) thermal conductivity, (c) See beck coefficient and
(d) Figure of merit as a function of temperature.

118
Temperature reliant on averaged thermal conductivity associated with HH YMnZ
materials are expressed within Fig. 7.9 (c). Because restricted through the formalism put in
place within BoltzTraP code, we talk about right here just the electronic thermal
conductivity and disregard the part associated with lattice thermal conductivity.
Wiedemann-Franz rule clarifies in which thermal conductivity associated with metals as
well as a SC is dependent on temperature along with electrical conductivity.
Inside the regarded temperature extend, thermal conductivity associated with HH
YMnZ materials rises directly. For the most part, thermal conductivity fluctuates based on
the variance as indicated by the amount of carrier concentration, electrical conductivity as
well as mobility. Each one of these guide lines differs using the variance associated with
the temperature. At optimum temperature, k (YMnSi) > k (YMnGe) >k (YMnSn) summing
17×1014 (W/m.K.s) as well as 16×1014 (W/m.K.s) and ×1014 (W/m.K.s) respectively,
thermal conductivity merely reveals heat circulation inside the materials. By changing the
Si element with Ge and Sn, we demonstrate a capacity to reduce the actual thermal
conductivity, which is the most imperative parameter for assessment of compounds in term
of modern significance.
Fig. 7.9 (d) provides power factors based on the electrical conductivities as well as
Seebeck coefficients as an element of temperature for YMnZ materials. The power
component of HH YMnZ materials fluctuates persistently within a temperature variation
from 300 K to 800 K. By evaluating each studied material, HH YMnSi has the most
noteworthy computed value of power factor. The explanation behind this high-power factor
in the HH YMnSi material is the greater value of S as well as thermal conductivity. Like
other thermoelectric variables such as electrical and thermal conductivities and Seebeck
coefficient, the power factor also shows a similar pattern (YMnSi) > (YMnGe) > (YMnSn).
Based on these types of results, YMnSi can be probably the most noticeable material for
thermoelectric applications.

119
Chapter 8
Structural Chemistry and Physical Properties of the Newly
Designed Half-Heusler XYZ Alloys with Large Spin Gaps
8.1 Introduction
Spintronics is a versatile domain which involves physics, physical chemistry, as
well as engineering, and it is the new research region for material scientists. Various new
materials should be discovered to meet distinct requirements. To find out the HMFs and
FM SCs along with TC greater than ambient conditions is always a priority for the material
science researchers (Felser et al., 2007). Logic circuits based on the spin of electrons
possess a long hold anticipation for a spintronic application. Even so, the constrained
variety of compounds designed for different system parts offers apparent difficulties for the
layout of the spintronic appliances. Compounds regarding spintronic purposes demand
appropriate mix of magnet moments and large values of band gaps also capable of being
operative at room temperature. It is observed that HH materials are ideal prospects
pertaining to spintronic applications because their crystal structure resemblance to the zinc-
blend phase and present large values of TC also they can maintain the half-metallicity under
small lattice mismatch.
In the last three decades, the half-metallicity has been widely investigated
particularly in the HA due to its unique feature that their electronic band structure is of
metallic nature in any one of two spin channels and show EBG or insulating behavior in the
other spin channel resulting in a complete (100%) SP at the EF. This sort of substantial SP
can be enabled to improve the functionality of the spintronic devices for example spin
valve, spin diode, and spin filter (Žutić et al., 2004).
HMFs are considered most suitable electrode materials designed for injecting a
spin-polarized current into SCs (Van Roy et al., 2000), MTJs (Tanaka et al., 1999), and for
GMRs devices (Hordequin et al., 1998). The significant amount of potential HH materials,
their multifunctional properties and latest recognition that HM Heusler has a tendency to
stay HM when layered with additional HH or FH (Azadani et al., 2016) boosts the
opportunity of discovering, tailoring, and synthesizing compounds suitable for distinct
practical applications to next generation spintronics devices.

120
Generally, the remarkable class of intermetallic materials which is in intense
investigations from last decade are the Heusler materials having general formula X2YZ
with L21 (2:1:1stichometry) and HH materials with C1b framework (1:1:1 composition) --
display exactly the same vast variety of properties similar to the perovskites, such as
topological insulators (Chadov and Qi, 2010; Feng et al., 2010; Yan and de Visser, 2014),
Kondo behavior (Ślebarski et al., 2001), non-centrosymmetric superconductivity
(Winterlik et al., 2008) as well as traditional, tunable magnetic properties, non-collinear
magnetism, semiconductivity, magnetoresistance effects, Li-ion-conductivity along with
other physical properties.
The surprising results about the Heusler materials were firstly reported by Fritz
Heusler in 1903 when he discovered that Cu2MnAl has FM behavior at the room
temperature even though none of the involving component Cu, Mn, or Al displays
magnetism. HA sponsor a plethora of surprising unique properties, that cannot be
discovered through primary properties of the atoms within the crystal framework.
Today, more than 1000 compounds have been identified in this fascinating class of
Heusler family which synthesized through 40 different combinations of the elements and
still, new materials with intriguing properties are continuous discovered for numerous
technological applications such as thermoelectric SCs (Sakurada and Shutoh, 2005;
Sootsman et al., 2009) optoelectronics SCs (Kieven et al., 2010) and piezoelectric SCs (Roy
et al., 2012).
A systematic research on the structural stability of HH (C1b) family is essential to
furnish direction for potential findings. Despite the fact that, a lot of first-principles
calculations anticipated several HH materials show half-metallicity (Galanakis et al., 2006;
Graf et al., 2011), though, a detailed investigation on the structural stability, electronic and
magnetic properties of HH family is essential, because it is not apparent which of the
numerous HM HH alloys that can be predicted, usually are stable.
To explore half-metallicity in the materials, it is a more effective way to implement
first-principles computations to design HM compounds first for the spintronics applications
and then synthesize them experimentally rather use costly trial-and-error experimental
plan. Employing a priori details with the structural stableness permits experimentalists to
pay attention to favorable properties to synthesize half-metals with ease.

121
The use of acoustic phonon spectra is the easiest method to deal with the stableness
regarding anticipating HM compounds. A recent study of the HH LiCrS and LiCrSe at their
optimized lattice constant show that both compounds show half-metallicity at the bulk and
Cr-S, Cr-Se at (001) surfaces (Hussain, 2018). Another theoretical research suggests that
ternary intermetallic compounds LiMnZ (Z = N, P) using an HH framework reveal HM
character at their elongated lattice constants and their magnet moments are anticipated to
the highest up to 5µ𝐵 per f.u. (Damewood et al., 2015b).
The work of Xiaotian et. al. (Wang et al., 2017b) on the XCrZ (X = K, Rb, Cs; Z
= S, Se, Te) shows that these HH materials have a largest magnetic moment of 5 µ𝐵 and a
semiconducting gap of more than 2 eV. Lately, studies on alkali-metals chalcogenides NaX
and LiX where (X= S, Se & Te) also predicts the HM properties and show very large EBG
with magnetic moments of 1 µ𝐵 when they crystalizes in zinc-blend and wurtzite structures
(Sadouki et al., 2018a; Sadouki et al., 2018b).
Based on the previously mentioned details, it is essential to discover new HM HH
alloys with larger EBG and high magnetic moment. In this computational investigation, HM
properties are systematically investigated and explored. We addressed, the series of 90 HH
XYZ compounds where (X= Li, Na, K, Rb, Cz; Y= V, Nb, Ta & Z= Si, Ge, Sn, S, Se, Te)
by the help of First principle calculations. We have produced a database of the structural,
electronic and magnetic properties, that will help us to recognize possibly practical
electrode/spacer supplies intended for long-term spintronics applications.
The computational methods, simulation guidelines, and code utilized in this DFT
calculations are presented in section 8.2. In section 8.3, we examine the three possible
types of structural arrangement (Wang et al., 2017b) for the HH alloys for each material to
figure out the energetically most stable structural framework using the lowest minimized
energy and discover the ground magnetic state of these studied materials.
8.2 Computational Methods
The ground state properties for all the 90 HH XYZ materials are performed by using
the Perdew–Burke–Ernzerh of (PBE) scheme (Perdew et al., 1996a) which is variant of the
generalized gradient approximation (GGA) to DFT whereas the electronic and magnetic
properties of all the HH XYZ materials are performed by using the TB-mBJ local density
approximation (Tran et al., 2007) implemented in the WIEN2K simulation code.

122
The TB-mBJ functional gives the accurate values of the band gaps compared to
PBE and computationally very less expensive. We have also used the Vienna Ab-initio
Simulation Package (VASP) to determine the correct magnetic ground state (FM, AFM)
and to check the vibrational stability at the gamma point and to perform phonon full
spectrum calculations for the some interesting HH XYZ materials. Phonopy software is
also used to determine the force constants between the lattice when the atoms are slightly
distorted from their equilibrium position. The force constants help to determine the phonon
frequency and the dynamic stability of the crystal structure. Mean field approximations
(MFA) are mapped into the Heisenberg spin model. The Monte Carlo simulations are also
carried out to use the Heisenberg spin model to determine the values of TC.
8.3 Results and Discussion
8.3.1. Ground State Properties
The general formula for ternary HH materials is XYZ where X, Y are usually an
alkali or TMs and Z is the main group element. Here, in this chapter, X represents the alkali
metals (Li, Na, K, Rb & Cs) whereas Y shows the group-V elements (V, Nb, & Ta) and Z
is for the sp-elements (Si, Ge, Sn, S, Se & Te). The unit cell of the HH XYZ materials
features a face-centered cubic (fcc) structure which has a Structurbericht representation of
C1b and space group of 216 (F-43m) according to the International Tables of
Crystallography consisting on three inequivalent interpenetrating fcc sublattices filled by
X, Y and Z atoms respectively.
Physical properties of the HH compounds are greatly influenced by the atomic
placement inside the unit cell. There exist three unique possible phases of atomic
arrangements in type1 (T1), type2 (T2) & type3 (T3) by interchanging the distinct Wyckoff
positions of X, Y & Z atoms in the C1b structure (Wang et al., 2017b). The illustration of
the unit cell of the HH XYZ materials of these three types is displayed in Fig. (8.1).
Geometrical optimization has been performed to determine the most stable type
among the three different types (T1, T2 & T3) for all the HH XYZ materials before
calculating the electronic properties. The lattice constants are determined by using the PBE-
GGA potential as there are no prior reports found on our studied HH XYZ materials. The
lattice constant can be changed considerably because of the uncontrolled change in the
strain. So, the lattice constant deviates from its optimized equilibrium value.
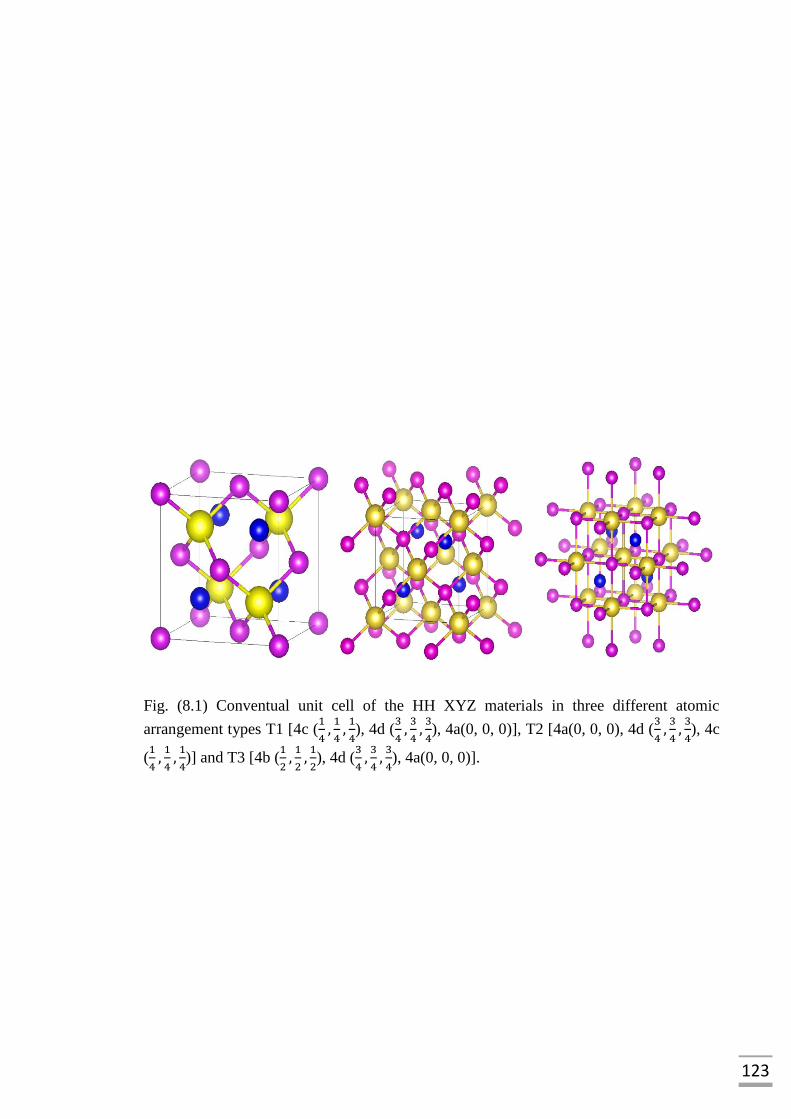
123
Fig. (8.1) Conventual unit cell of the HH XYZ materials in three different atomic
arrangement types T1 [4c (1
4,
1
4,
1
4), 4d (
3
4,
3
4,
3
4), 4a(0, 0, 0)], T2 [4a(0, 0, 0), 4d (
3
4,
3
4,
3
4), 4c
(1
4,
1
4,
1
4)] and T3 [4b (
1
2,
1
2,
1
2), 4d (
3
4,
3
4,
3
4), 4a(0, 0, 0)].

124
So structural parameters of these materials are derived by optimizing the energy
extracted from their respective optimized volume around their equilibrium lattice constant and
then their data points are fitted to Murnaghan’s equation of state (Murnaghan, 1944b). Fifty-
six of the studied HH XYZ materials have the lowest values of energies at T1 phase and thirty-
two energetically preferred to T3 phase as a stable ground state. Atomic arrangement of T2
phase is found to be a ground state for only two HH LiNbSn and LiTaSn materials out of the
90 HH XYZ materials.
We further calculated the correct magnetic ground state of the studied HH XYZ
materials by volume optimization at their respective ground phase (T1, T2 or T3). For the
illustration purpose, only HM HH NaVTe is selected to describe the variation of volume versus
total energy curve at the three possible T1, T2 & T3 phases along with magnetic ground state
(FM & AFM) in the Fig. (8.2) out of the 90 DFT investigated HH XYZ materials.
It can be clearly depicted from the Fig. (8.2) that most stable phase of the HH NaVTe
is a T1 phase with FM configuration. For the complete details about the lattice parameters,
electronic behavior, values of magnetic moments, energies of the FM, AFM states, EBG and
EHM at their preferred type and magnetic ground state of all the 90 HH XYZ materials at the
three possible phases (T1, T2 & T3) are provided as supplementary information in the
Appendix-I at the end. The number of materials which preferred to structural stability among
three unique different atomic positions in the C1b unit cell of the HH structure, with the correct
magnetic ground state along with the electronic properties of all the 90 DFT investigated HH
XYZ materials are presented in Fig. (8.3). From this figure, one can easily see that how many
numbers of materials have the stable ground phase (T1, T2 or T3) with correct magnetic (NM,
FM or AFM) configurations and their electronic properties for all the 90 DFT investigated HH
XYZ materials. After structural stability, we determined the magnetic ground phase (NM, FM
or AFM) at their preferred stable ground phases (T1, T2 or T3). We identified 14 HH materials
as a NM material, 55 HH are energetically favorable to the FM and 21 materials prefers to the
AFM ground state. Furthermore, the electronic properties of the 90 HH XYZ materials are also
calculated, out of which, 44 materials are found to be metallic in nature (NM metallic = 9, FM
Metallic = 14 & AFM metallic = 21). Also, 7 HH materials found to be SCs, in which 2
materials are magnetic SCs and 5 are NM SCs.

125
Fig. (8.2) Volume optimization of the HH HM NaVTe material at the (a) three different atomic
arrangement types (T1, T2 & T3) (b) FM and AFM ground state.

126
Additionally, half-metallicity was found in total 39 HH XYZ materials. The electronic
properties are discussed here are calculated in the most stable phase of the HH XYZ materials
with their correct magnetic ground state. Out of 90 HH XYZ materials, 21 HH materials
comparatively have the lowest energy at the AFM state rather than FM state confirms that their
magnetic ground state is AFM.
If we generalize, the series of HH XYZ materials which energetically preferred T1 as
a stable ground phase are the HH XYZ where (X= Li, Na, K & Y=V, Nb, Ta & Z= Si, Ge, Sn,
S, Se, Te) and the series of HH XYZ (X= K, Rb & Y= V, Nb & Z= S, Se) along with the series
of HH LiYZ where (Y=Nb, Ta & Z =Si, Ge, S, Se, Te) followed by the series of RbTaZ where
(Z=, Si, Ge, S, Se). On the other hand, the series of the materials which prefer T3 phase as a
most stable ground state are CsYZ where (Y= V, Nb, Ta & Z= Si, Ge, Sn, S, Se, Te) and XYZ
where (X= K, Rb, & Y=V, Nb, & Z = Si, Ge, Sn, Te) along with RbTaZ where (Z=S, Te).
Only the NM HH LiNbSn and LiTaSn materials which show the metallic character have the
lowest energy in the T2-phase.
It can be noted that the alkali metals with smaller atomic radius (e.g. Li, Na, K) tend to
prefer in the T1 stable phase while when the size of the atomic radius increases of each element
(Rb, Cs) or coupled with TMs in the XYZ material then they energetically prefer to be sable
in the T3 phase. The well-known empirical procedures also verify the energetical stable phases
of the HH XYZ materials that the most electronegative elements like (Si, Ge, Sn, S, Se & Te)
and the most electro positive elements (alkali metals Li, Na, K, Rb, Cs) forming the NaCl like
sublattice whereas the elements which have intermediate electronegativity e.g. (V, Nb & Ta)
prefer to occupy the tetrahedral sites (Zeier et al., 2016).
Among 90 HH XYZ materials, seven materials are also found to be a SC. The HH
LiVSi and LiVGe are the magnetic SC at their stable phase T1 with FM configuration. Both
FM SC materials show a semiconducting gap at both the spin channels (up & down) with a
magnetic moment of 2 µ𝐵. Reaming five CsNbS, CsNbSe, and series of three HH materials
CsTaX (where X=S, Se & Te) are NM SC with µtot of 0 µ𝐵.

127
Fig. (8.3) Summary of the 90 HH XYZ materials, at their preferred stable ground state among
three different types T1, T2 & T3 phases and magnetic ground state (NM, FM, AFM).
Illustration of the electronic properties of the each HH materials is also presented at their
preferred stable type and magnetic ground state.

128
8.3.2 Electronic Properties
In this section, the electronic structure and magnetic properties, are discussed, which
are very important and core of the present work. These properties are performed for all the 90
HH XYZ materials on their respective most stable and meta stable phases (T1, T2 or T3) with
their correct magnetic configurations (NM, FM or AFM) at their equilibrium lattice constants.
The complete information about their equilibrium lattice constant, electronic properties
at all the T1, T2 and T3 phases, µ𝑡𝑜𝑡, the EBG (eV), the EHM (eV), the info about their vibrational
stability at the gamma point and the values of NM, FM and AFM energies of all the 90 HH
XYZ materials at their respective stable and meta-stable ground state and magnetic ground
state can be found in Appendix-I along with the supporting information.
The energy difference between the valance band maxima (VBM) and conduction band
minima (VBM) forms the EBG whereas the EHM is termed as the minimum energy required by
the electrons to flip the spin gap. Our DFT investigated HM HH XYZ materials show very
large values of the EBG and EHM. In Fig. (8.4), the values of the energy (a) band gap EBG (eV)
and (b) half-metallic gap EHM (eV) of all the 90 HH XYZ materials are presented only on their
respective energetically most stable ground phase in the 3D plot.
As suggested by the general formula of HH XYZ materials, alkali-metals (X= Li, Na,
K, Rb, Cs) are assigned on the X-direction, the group-V elements (Y = Si, Ge, Sn, S, Se, &
Te) on the Y-axis and the main group elements (Z = Si, Ge, Sn, S, Se, & Te) on the Z-direction.
The color bar indicates the width of the EBG and EHM. As out of 90 HH XYZ materials, 46 HH
materials show the metallic nature, thus they have the zero values of EBG and EHM energies
which is indicated by the dark blue color. The values of the EBG and EHM increases from blue
to yellow successively.
Furthermore, the vibrational stability at the gamma point of the crystal structure for all
the 90 HH XYZ materials are also explored thoroughly. Among these, 39 are HM HH XYZ
materials. only 28 HM HH XYZ materials have the vibrational stability at the gamma point. It
means that their phonon frequencies are positive and show optic modes at the gamma point.
Remaining 11 HM HH XYZ materials show the acoustic modes and have the imaginary
(negative) phonon frequency greater than f/i = 1 THz.

129
Fig. (8.4) Colors show the width of the (a) EBG (b) EHM of 90 HH XYZ materials. The species
X, Y, and Z which represent the HH XYZ materials which are signifying on the three
coordinates. Blue and yellow colors represent the successively increasing values of these
energy band gaps.
(b) (a)

130
If the crystal has a very large values of imaginary (negative) frequency then it means
that it is vibrationally unstable and may be in a structural phase transition phase (meta stable
state). So, these 11 HM HH XYZ materials which are vibrationally unstable at the gamma
point are discarded for the further discussion and our focus will be only on the 28 vibrationally
stable HM HH XYZ materials with FM phase for the further analysis.
8.3.3 Mixing Energy
To determine either our DFT calculated HM HH XYZ materials can be experimentally
synthesized with the C1b structural framework, the mixing energy of the 28 vibrationally stable
HM HH XYZ materials is calculated with their respective stable phases (T1 or T3) with FM
ground state at their equilibrium lattice constants. The mixing energy of these 28 vibrationally
stable HM HH XYZ materials is calculated by the following formula,
𝐸𝑚𝑖𝑥 = 𝐸𝑋𝑌𝑍𝑓𝑐𝑐
− ( 𝐸𝑋𝑓𝑐𝑐
+ 𝐸𝑌𝑓𝑐𝑐
+ 𝐸𝑍𝑓𝑐𝑐
)
where 𝐸𝑋𝑌𝑍𝑓𝑐𝑐
is the total energy (eV/atom) of the HH XYZ materials with C1b crystal structure
and 𝐸𝑋𝑓𝑐𝑐
, 𝐸𝑌𝑓𝑐𝑐
and 𝐸𝑍𝑓𝑐𝑐
are the energies of the each X, Y or Z species of the HH XYZ materials
in the fcc structure. The mixing energies of these 28 vibrationally stable HM HH XYZ
materials are displayed in a 3D plot of the Fig. (8.5).
Now, we examine the variation in mixing energies of our selected vibrationally stable
28 HM HH XYZ materials at their respective stable phases (T1 or T3) with FM ground state.
It is obvious from the blue circles that HM HH XYZ materials with X = (Li, Na, Rb, Cs), Y
=V and Z = (Si, Ge, Sn, S, Se) tend to have lower values of the mixing energies which is a
good indicator that these 28 vibrationally stable HM HH XYZ materials can be synthesized
experimentally and may be more stable than its constituting elements at the zero temperature.
Only HM HH NaVTe along with magnetic SCs LiVSi & LiVGe have the negative
values of Emix (eV/atom). The list of mixing energy of the 28 vibrationally stable HM HH XYZ
materials with FM ground state along with 2 FM SCs is also presented in Table 8.1. The trend
can be easily seen from the Table 8.1 that the values of the mixing energy for the 28
vibrationally stable HM HH XYZ materials increases when the atomic size of the Y element
increases where (Y= V, Nb, Ta).

131
Fig. (8.5) Mixing energy (eV/atom) of the 28 vibrationally stable HM HH XYZ materials at
their respective stable state with FM configurations. Blue shades show the mixing energy less
than 0.2 (eV/atom). Each coordinate of the 3D plot symbolizes the X, Y & Z species of the
associated 28 HM HH XYZ materials.

132
The understanding of the mixing energy offers essential significance for the prospective
applications associated with HH materials. It is expected that the 28 HM HH XYZ materials
with FM phase which have lower values of the mixing energy (blue circles in Fig. 8.5)
especially the HM HH LiVSn, NaVZ (Z = Si, Ge & Te) with their stable phase T1, whereas
HM HH RbVSe, CsVZ (Z= Sn, S) materials with T3 phase have a greater prospect from the
spintronic application point of view because their mixing energy is around -0.1 to 0.2
(eV/atom). Thus, they have the greater chance to be stable materials and can be synthesized
experimentally when decomposed into respective constituent elements at zero the temperature.
8.3.4 Curie Temperature (TC)
For the practical spintronic applications, TC is an additional essential requirement to
use the FM HH XYZ materials as spin injectors. The TC of the interesting 28 HM HH XYZ
materials is also calculated by using the mean field approximation (MFA) and mapping the
total energy of FM and AFM states of the HH XYZ materials into a Heisenberg model.
The construction of Heisenberg spin model is made from the expression (Hu et al., 2018),
𝐸𝑠𝑝𝑖𝑛 = 𝐸0 +1
2 ∑ 𝐽𝑖𝑗
𝑖𝑗𝑆𝑖𝑆𝑗,
in which 𝐸0 is the reference energy, the magnetic moment of the ith atom is interpreted by 𝑆𝑖
and 𝐽𝑖𝑗 represents the values of the exchange constant obtained from the DFT calculations at
absolute temperature. The magnetic exchange interactions are considered for the nearest
neighbors (𝑛𝑛 = 8) inside the unit cell. The exchange coupling 𝐽𝑖𝑗 can be obtained as follows,
𝐸0 = 1
2 (𝐸𝐹𝑀 + 𝐸𝐴𝐹𝑀)
𝐽𝑖𝑗 = 1
8 |𝑆|2 [𝐸𝐹𝑀 − 𝐸𝐴𝐹𝑀]
where 𝐸𝐹𝑀 and 𝐸𝐴𝐹𝑀 are the energy of the crystal assuming the unit cell has the ideal FM and
AFM spin configurations.

133
Fig. (8.6) The Curie temperature (TC) of the 28 HM HH XYZ materials at their respective
stable state with FM configurations which are also vibrationally stable at the gamma point. The
color bar shows the values of the calculated TC (K) with +/- 25 K tolerance. Each coordinate
of the 3D plot symbolizes the X, Y, Z species of the associated HM HH XYZ material.

134
The values of the TC for the 28 vibrationally stable HM HH XYZ materials along
with 2 FM SCs are presented in the Table 8.1 and their 3D plot is illustrated in the Fig.
(8.6). From the Table 8.1, our selected 28 vibrationally stable HM HH XYZ materials have
the lower values of TC, at the range of room temperature. The quite low values of the TC
ranging from the 100 K to 300 K as it is indicated by the color plan of the Fig. (8.6). Dark
blue circles show the value of TC = 100 K. Most of our explored HM HH alloys are in the
range of 150 K to 250 K (light blue to green). Only HM HH RbNbS, RbNbZ (Z = Si, Sn,
Te) and CsNbTe have the maximum value of TC = 300 K (yellow circles).
For the practical spintronic applications, HMFs are needed which have negative
(low) values of the mixing energy and high values of the TC. Now, by looking at the Table
8.1, the materials which have the high values of TC also have higher values of the mixing
energy (greater than 0.2 eV/atom) and the HM HH XYZ materials which have lower values
of mixing energy have the TC below or around room temperature.
For instance, HM HH NaVTe although have the negative mixing energy of -0.123
eV/atom but its Tc temperature is just 100 K. The HM HH XYZ materials in which
transition metal Y= Nb, Te have the greater values of the TC but their mixing energy is
high, only the HH XYZ materials with Y=V have the lower values of mixing energy, which
indicates that HH XYZ materials with Y= V have the greater chances to be experimentally
synthesized.
Out of 28 vibrationally stable (at gamma point) HM HH XYZ materials, only HM
HH NaVSi, RbVSe and CsVTe along with FM SCs LiVSi and LiVGe materials have the
moderate and acceptable values for both mixing energy as well as TC. Remaining HM HH
XYZ materials are discarded for the further discussion in this chapter because they have
the mixing energy greater than 0.2 (eV/atom) and our focus will be only on HM HH NaVSi,
RbVSe and CsVTe along with FM SCs LiVSi and LiVGe materials.
8.3.5 Band Structure and Density of States
In our DFT investigated 90 HH XYZ materials, seven materials are found to be a
SCs. Among them, HH LiVSi and LiVGe are the magnetic SCs with the magnetic moment
of 2 µB with their stable phase of T1 at their optimized lattice constant. The NM SCs
includes the series of CsNbZ (Z= S, Se) and CsTaZ (Z= S, Se, Te) in the HH C1b
composition with their optimized lattice constant which are all energetically preferred to
T3 as a stable phase.

135
Table 8.1 Equilibrium lattice parameter: a (Å), electronic conductivity (SC and HM)
represent the semiconductor and HM characteristics, µ𝑡𝑜𝑡 is equivalent to total magnetic
moment, EBG & EHM are the energy band gap and HM gap, FM & AFM along with EFM &
EAFM shows the magnetic ground state and energy of the FM and AFM states respectively,
TC & Emix show the Curie Temperature and mixing energy of the each 30 interesting HH
XYZ materials at their preferred ground state (T1 or T3) with vibrationally stability
checked at the γ-point.
XYZ Preferred
Types
a
(Å)
Electronic
Behavior µ𝑡𝑜𝑡
(µB)
EBG
(eV)
EHM
(eV)
Mag-
netic
State
Stabl
e
EFM
(eV)
EAFM
(eV)
TC
(K)
+/-
25K
𝐸𝑚𝑖𝑥
(eV/
atom)
LiVSi T1 5.85 SC 2.00 1.43 ---- FM Yes -63.25
-62.72
200 -0.11
LiVGe T1 5.93 SC 2.00 1.06 ---- FM Yes -60.23
-59.61
250 -0.11
LiVSn T1 6.37 HM 2.00 0.89 0.47 FM Yes -54.94
-54.66
150 0.05
NaVSi T1 6.25 HM 2.00 1.11 0.52 FM Yes -55.56
-55.15 200 0.21
NaVGe T1 6.32 HM 2.00 1.16 0.50 FM Yes -53.12
-52.80
150 0.21
NaVTe T1 6.84 HM 4.00 4.18 0.54 FM Yes -52.55
-52.38
100 -0.12
KVSi T3 6.55 HM 2.00 1.49 0.21 FM Yes -48.80
-48.26
250 0.56
KVGe T3 6.64 HM 2.00 1.59 0.15 FM Yes -46.74
-46.41
150 0.53
RbVTe T3 7.27 HM 4.00 3.73 0.83 FM Yes -48.01
-47.35
250 0.14
CsVSn T3 7.32 Nearly
HM
1.99 0.66 0.15 FM Yes -42.91
-42.58
150 0.78

136
Table 8.1 (continued)
XYZ Preferred
Types
a
(Å)
Electronic
Behavior µ𝑡𝑜𝑡 (µB)
EBG
(eV)
EHM
(eV)
Mag-
netic
State
Stable EFM
(eV)
EAFM
(eV)
TC
(K)
+/-
25K
𝐸𝑚𝑖𝑥
(eV/
atom)
CsVS T3 6.93 HM 4.00 3.92 0.68 FM Yes -49.97
-49.46
200 0.012
CsVSe T3 7.13 HM 4.00 3.66 0.66 FM Yes -48.62
-48.04
250 0.111
KNbSi T1 6.79 HM 2.00 1.13 0.54 FM Yes -52.38
-52.03
150 0.595
KNbGe T1 6.86 HM 2.00 1.15 0.46 FM Yes -50.14
-49.79
150 0.604
KNbSn T1 7.25 HM 2.00 1.10 0.34 FM Yes -47.53
-47.24
150 0.685
RbNbSi T3 6.77 HM 2.00 1.24 0.57 FM Yes -49.37
-48.68
300 0.831
RbNbSn T3 7.19 HM 2.00 1.19 0.33 FM Yes -45.83
-45.14
300 0.828
RbNbTe T3 7.41 HM 4.00 3.82 0.75 FM Yes -47.59
-46.90
300 0.402
CsNbSi T3 6.87 HM 2.00 0.47 0.11 FM Yes -49.23
-48.64
250 0.907
CsNbGe T3 6.96 HM 2.00 0.62 0.14 FM Yes -47.12
-46.52
250 0.915
CsNbSn T3 7.28 HM 2.00 0.69 0.16 FM Yes -45.72
-45.09
250 0.94
CsNbTe T3 7.55 HM 4.00 3.23 0.39 FM Yes -47.16
-46.45
300 0.49
NaTaSn T1 6.71 HM 2.00 1.18 0.17 FM Yes -56.88
-56.68
100 0.55
KTaSi T1 6.70 HM 2.00 1.34 0.50 FM Yes -56.60
-56.42
100 0.74
KTaGe T1 6.80 HM 2.00 1.41 0.69 FM Yes -54.08
-53.82
150 0.77
KTaSn T1 7.17 HM 2.00 1.31 0.54 FM Yes -51.29
-50.91
200 0.88
RbTaSi T1 6.89 HM 2.00 1.36 0.66 FM Yes -53.13
-52.72
200 0.92
RbTaGe T1 6.99 HM 2.00 1.39 0.55 FM Yes -50.82
-50.32
200 0.97
RbTaSn T3 7.16 HM 2.00 1.05 0.35 FM Yes -49.09
-48.56
250 1.03
RbTaTe T3 7.37 HM 4.00 3.70 0.24 FM Yes -48.91
-48.37
250 0.75

137
For an illustration purpose, only the electronic band structure of the FM SC LiVGe
along with HM HH CsVSe and DOS of HM HH RbVTe are selected among all the 28
vibrationally stable HM HH XYZ materials to illustrate the electronic properties. Their
band structure and DOS are presented in the Figs. (8.7-8.9).
For the FM SC HH LiVGe material, Fig. (8.7), one can see the semiconducting gap
in both spin channels. In the majority (spin-up) channel and minority (spin-down) channel,
FM SC HH LiVGe material have the semiconducting gap of 0.63 eV and 1.65 eV
respectively. In the spin-up channel, band gap is direct while in the spin down channel, the
indirect band gap is found.
In the Fig. (8.8), the band structure of the HM HH CsVSe (as an example) is shown
to illustrate the HM properties of the 28 found HM HH XYZ materials. In the spin up
channel (majority bands), metallic characteristics are found while on the opposite spin
channel (minority bands), there is a large indirect EBG of 3.7 eV found at the equilibrium
lattice constant which leads to the 100% SP at the EF. The VBM found to be situated at Γ
and the conduction band minima CBM is situated at the X of the wave vectors (k-points) of
the BZ.
The value of the spin-flip gap or EHM is EHM = 0.7 eV which can be defined as the
minimum energy required to flip the gap for the HM HH CsVSe material. The values of
these EBG and EHM can be clearly seen in the Fig. (8.8) for the HM HH CsVSe alloy. The
EHM has much greater importance in the HM properties around the EF rather than EBG. The
non-zero value of the EHM in the spin down channel is the clear indication that the HH
CsVSe is true HM at the EF.
The density of state (DOS) explains the different energy levels occupied by the
electrons for the specific number of states at each energy level. For the analyses of the
electronic nature of the 90 HH XYZ material, only DOS of the HM HH RbVTe is selected
to explain the HM properties at its equilibrium lattice constant. The Fig. (8.9) presents the
total and partial DOS of the individual atoms which are present in the HM HH RbVTe
material. The DOS calculations for this material are performed at the optimized lattice
constant calculated by the DFT with the stable T3 phase and FM configurations. Some state
of elements like s & p of Rb and V are not offered in the DOS plot because they do not add
any contribution to the total DOS of the RbVTe.

138
Fig. (8.7) Band structure of the HH SC LiVGe material with FM ground phase in the spin
up and spin down channel. The horizontal dashed line represents the EF which is fixed at
zero eV.

139
Fig. (8.8) Band structure of the HM HH CsVSe material in the spin up and spin down the
channel. The horizontal dashed line represents the EF which is fixed at zero eV.

140
Fig. (8.9) Spin projected total and partial density of state (DOS) of HM HH RbVTe material
at the equilibrium lattice constant. The vertical dashed line in the middle shows the Fermi
level and fixed at EF = 0 eV.

141
On the contrary, in the spin up channel the p orbitals of Te cross the EF along with
the d-t2g orbitals of Rb and V, leading to a metallic character for all three compounds. The
p-state of Z atom is quite symmetrical in the spin up directions and crosses EF a little in the
spin down channel with a little share to the magnetism. It is also revealed from the Fig.
(8.9), that hybridization occurs between the 3d states of Rb and V atoms.
8.3.6 Vibrational Properties
Vibrational properties perform a significant part within most of the physical
properties associated with solids, like the electric conductivities, superconducting
temperature, Debye temperature, thermal properties as well as stableness of the structure.
After performing the volume optimization and extracting the most stable ground
structure of the each HH XYZ material, we further tested their dynamic stability of the
structure by two-step phonon calculations. First, we calculated the vibrational properties of
our DFT investigated 90 HH XYZ materials at their preferred stable phase at the gamma
point. If it behaves well at the gamma point and has the interesting properties, then we
carried out the phonon calculations for the selected HH XYZ materials. Out of 90 HH XYZ
materials, 63 HH XYZ materials found to be vibrationally stable at the gamma point
whereas 27 HH XYZ materials show imaginary frequencies greater than 1 THz.
Furthermore, among 27 unstable HH XYZ materials, 10 materials are unstable at
the FM phase which are KVZ (Z = S, Se, Te), RbVZ (Z = Si, Ge, S, Se), CsVZ (Z= Si, Ge,
Te) and while remaining 17 HH XYZ materials like LiVS, NaVZ (Z= S, Se), KVSn,
RbVSn, NaNbZ (Z= Se, Te), KNbZ (Z=S, Se, Te), RbNbZ (Z= S, Se), KTaZ (Z= S, Se,
Te) and RbTaZ (Z = S, Se) have the AFM ground state. It can be noted that the majority
(17 of 21) of the HH XYZ materials which have AFM as a ground state are vibrationally
unstable at the gamma point.
To ensure the structural stability of the FM HM HH NaVSi and FM SC HH LiVSi
and LiVGe materials other than gamma point of the first Brillion zone, we have calculated
the phonon full spectrum calculations as well. These two FM SC materials vibrationally
well behaved for all the k-points within the BZ at their equilibrium lattice constant. For the
illustration purpose, only the result of the phonon spectrum of SC HH LiVGe material is
shown in the Fig. (8.10) because both have the similar phonon dispersion curve due to the
resemblance of the crystal structure.

142
Fig. (8.10) Phonon full spectrum curve for the (a) HM HH NaVSi and (b) FM SC LiVGe

143
The three acoustical and six optical branches of vibrational modes at nay q-point
can be seen in the dispersion curve as our DFT calculated HH XYZ materials contains three
atoms in the primitive unit cell. As there are no imaginary frequencies are found and all
frequencies are positive at the symmetry path of the Brillion zone which confirms the
dynamical stability of the SC HH LiVGe materials.
8.3.7 Magnetic Properties
There is a unique feature related to the HM system that the µtot of the system should
be an integer and integral multiple of Bohr magnetron. The SPR for the conventional HH
XYZ materials which have three atoms in the primitive unit cell, is given by
µ𝑡𝑜𝑡 = (𝑍𝑣𝑎𝑙 − 18)µ𝐵
where µ𝑡𝑜𝑡 is the total magnetic moment of the HH XYZ material and 𝑍𝑣𝑎𝑙 represents the
total number of valance electrons inside the unit cell. In our investigated HH XYZ system,
the values for the 𝑍𝑣𝑎𝑙 is smaller than the usual TMs related to HH XYZ materials.
A researcher, Damewood et al. (Damewood et al., 2015a) has modified the SPR for
the systems which have the smaller values of 𝑍𝑣𝑎𝑙
µ𝑡𝑜𝑡 = (𝑍𝑣𝑎𝑙 − 8)µ𝐵
The value of 𝑍𝑣𝑎𝑙 is 10 and 12 for the series of HH HM XYZ (Z= Si, Ge, Sn) and
XYZ (Z= S, Se & Te) respectively. Our DFT investigated 39 HM HH XYZ (Z= Si, Ge, Sn)
and the series of XYZ (Z= S, Se & Te) materials have also a µ𝑡𝑜𝑡 of 2 µB and 4 µB
respectively and obeys the modified SPR.
The µ𝑡𝑜𝑡 of the selected 28 vibrationally stable HH XYZ materials along with the
two FM SCs are shown in the Table 8.1. Our all DFT investigated 28 vibrationally stable
HM HH XYZ materials show the 100% SP at the EF at their preferred ground stable phase
with the FM configurations.

144
CHAPTER 9
Overview of the Results
This dissertation provides an extensive overview of an exceptional class of HH
materials which have countless functionalities, ranging from SCs to metals and HMFs with
plenty of technological applications in spintronics, thermoelectric, and opto-electronics.
The structural, electronic, and HM properties of the DFT investigated HH
compounds CrTiZ (Z = Si, Ge, Sn, Pb) investigated by using the first-principles DFT
calculations. Spin-polarized ferromagnetism explored in three different atomic
arrangements (Type 1, Type 2, Type 3) and halfmetallicity has been found in all these
compounds at their optimized lattice constants in Type 1 with EHM ranging from 0.12 to
0.33 eV. As other HH materials which are based on TMs, the hybridization between d-d
orbitals of Cr and Ti atoms causes the semiconducting gap in the majority-spin state for all
the CrTiZ (Z= Si, Ge, Sn, and Pb) materials in the Type 1. Furthermore, the outcome
suggests that the half-metallicity is availably robust contrary to the lattice distortion which
gives the possibility to grow these compounds on various substrates and especially the HH
CrTiSn and CrTiPb materials are suitable for spintronics applications because of their
larger EHM. Due to a large integral magnetic moment (4 µB/unit cell) of all these HH
materials, they might perform as electrodes to insert spin-electrons into II-VI magnetic SCs.
A detailed investigation of new series of potentially HH FeVZ (where Z= Si, Ge,
Sn) compounds performed by employing GGA mBJ exchange-correlation energy
functional of density functional theory. Our GGA results indicate that FeVGe is HM, while
FeVSi, FeVSn are metallic. On the other hand, the mBJ calculations reveal that FeVSi,
FeVGe, and FeVSn compounds are excellent HM having integer magnetic moment of 1 µB
and EHM of 0.16 eV, 0.21 eV, and 0.23 eV respectively. We found that the ferromagnetism
arises from Fe-d states along with a small contribution of p-states of Z-atoms. The band-
gap appearing in the spin up channel is due to strong d-d hybridization of TMs (Fe and V).
The HH FeVSi, FeVGe, and FeVSn retain their HM nature when their lattice parameters
are changed in the range of 3.84–5.31%, 3.91–5.91% and 3.94–11.8% respectively and

145
therefore, they could be considered as a potential candidate for the useful spintronic
applications.
The structural, electronic, and magnetic properties of HH YCrSb and YMnSb
materials with three different atomic arrangements (XI, XII, and XIII) are investigated by
using FP–LAPW method. The calculated formation energies confirm that these compounds
are chemically stable. Results reveal that both YCrSb and YMnSb are true HMFs having
integral magnetic moments of 4 µB and 3 µB, respectively. The µ𝑡𝑜𝑡 of the studied materials
are mainly contributed by the Cr and Mn atoms respectively. The d–d hybridization
between the transition-metal elements Y and Cr/Mn sorts the semiconducting gap inside
the majority spin as some other transition-metal centered HH materials. The HM gaps are
0.43 eV and 0.13 eV are found for the HH YCrSb and YMnSb compounds, respectively.
The electronic structure and DOS calculations indicate that both HH YCrSb and YMnSb
materials show semiconducting nature in majority spin channel and in contrast, conducting
trend observed in minority spin channel. It is found that HH YCrSb and YMnSb preserve
their half-metallicity for lattice constant ranges of 5.77 Å 6.85 Å and 5.92 Å–6.81 Å,
respectively and retain 100% SP at the EF.
The structural, electronic and magnetic properties of the HH YMnZ (Z = Si, Ge,
Sn) alloys are explored in three different atomic arrangements (X-type1, X-type2, and X-
type3) by using GGA and GGA+mBJ exchange-correlation energy potential. The HH
YMnSi and YMnGe compounds are energetically more favorable in the X-type1 phase
while HH YMnSn prefers the X-type2 structure. Furthermore, it is also noted that these
materials prefer FM state than the NM and AFM state. The computed GGA results show
that HH YMnSi is metallic whereas HH YMnGe and YMnSn materials show the HM
properties. In contrast, the GGA+mBJ estimation indicates that all three studied materials
are truly HM with total integer magnetic moment of 4 μB. It is made clear that GGA+mBJ
provides much better spin magnetic moments and band gaps as compared to those
computed with GGA. Furthermore, the half-metallicity is witnessed to be robust based on
the extended array of lattice parameter. Consequently, HH YMnZ materials are anticipating
alloys for the upcoming spintronics appliances. The transport properties of the YMnZ
materials have been curtained utilizing BoltzTrap simulation code. The computed figure of
merit computed an extensive variety of temperature demonstrates that HH YMnSi exhibits
preferred thermoelectric conduct over HH YMnGe and YMnSn materials.

146
Finally, half-metallicity explored for the new series of 90 hypothetical HH XYZ
materials (where X=Li, Na, K, Rb, Cs & Y= V, Nb, Ta and Z= Si, Ge, Sn, S, Se Te) by
using first-principles calculations. The T2 ordering never is most favorable in the HH XYZ
materials except for two HH LiNbSn and LiTaSn materials; the debate is in between T1
and T3 phases. All the HH XYZ materials energetically prefer either T1 or T3 phase as a
ground state. The examined 90 HH XYZ materials, 15 HH materials are found to be NM,
54 HH are energetically favorable to the FM and 21 materials prefer to the AFM ground
state. These 21 AFM HH XYZ materials are metallic in nature and do not possess HM
properties while remaining HH materials are stable at the FM state. Out of 90 HH materials,
39 materials are found to be HM at their equilibrium lattice constant with the most stable
state with FM configurations while remaining materials show HM properties in their meta
stable phase. Out of 39 HM HH XYZ materials, 28 materials are vibrationally stable at the
gamma point while remaining 11 HM HH materials show the imaginary phonon frequency
greater than 1THz. The HM HH NaVTe has the lowest negative mixing energy while others
have lower values of mixing energy, while HM HH RbVSe and CsVTe materials have the
greater values of the Curie temperature. The reason for the gap is due to the 3d orbital
splitting of the TMs (V, Nb & Ta) according to the crystal field theory with p-d
hybridization of the main group elements (Si, Ge, Sn, S, Se, Te). The phonon dispersion
curve for the RbVSe and CsVTe show the soft modes while FM SC LiVSi, LiVGe and
NaVSi do not have any soft modes indicating the dynamical stability of these materials.
The lower values of mixing energy and higher value of the Curie temperature along with
dynamical stability, make the two FM SCs LiVSi and LiVGe along with HM HH NaVSi
materials potential contenders for the spintronic applications.

147
9.1 Future Directions
Numerous intriguing theoretical as well as experimental studies will arise in near
future, which will exploit the plenty of benefits of the HM properties for the HH XYZ
materials that are presented in this dissertation.
➢ This dissertation contains the work on the structural, electronic and
magnetic properties for the different newly designed HH CrTiZ (where
Z=Si, Ge, Sn, Pb), FeVZ (where Z= Si, Ge, Sn), YCrSb & YMnSb, YMnZ
(Z= Si, Ge, Sn) and the 90 HH XYZ alloys (where X= Li, Na, K, Rb, Cs &
Y=V, Nb, Ta & Z=Si, Ge, Sn, S, Se, Te) which are presented for the first
time. So, there is a still scope for the more work for these mentioned HH
XYZ materials.
➢ Several other physical properties like optical, mechanical, thermoelectric,
elastic, Curie temperature should be determined for the presented HM HH
XYZ alloys in this dissertation. The most importantly their dynamical
stability should be checked from the phonon dispersion curve.
➢ So far, no experimental research is performed for all the HH XYZ materials
which are presented in this dissertation, therefore, our DFT investigated HM
HH XYZ materials provide a useful information that these HM HH XYZ
materials can be synthesize experimentally and are potential contenders for
the spintronic applications.

148
References
Ahmad, I and Amin, B. (2013). Robust Half-Metallicity in Ga 1-Xmnxp and Ga 1-
Xmnxas. Computational Materials Science, 6855-60.
Ahmadian, F and Alinajimi, R. (2013). First-Principles Study of Half-Metallic
Properties for the Heusler Alloys Sc 2 Crz (Z= C, Si, Ge, Sn). Computational
materials science, 79345-351.
Akai, H. (1998). Ferromagnetism and Its Stability in the Diluted Magnetic
Semiconductor (in, Mn) As. Physical Review Letters, 81(14): 3002.
Akinaga, H, Manago, T and Shirai, M. (2000). Material Design of Half-Metallic Zinc-
Blende Cras and the Synthesis by Molecular-Beam Epitaxy. Japanese Journal of
Applied Physics, 39(11B): L1118.
Al-Sawai, W, Lin, H, Markiewicz, R, Wray, L, Xia, Y, Xu, S-Y, Hasan, M and
Bansil, A. (2010). Topological Electronic Structure in Half-Heusler Topological
Insulators. Physical Review B, 82(12): 125208.
Aliev, F. (1991). Gap at Fermi Level in Some New D-and F-Electron Intermetallic
Compounds. Physica B: Condensed Matter, 171(1): 199-205.
Alijani, V, Winterlik, J, Fecher, GH, Naghavi, SS and Felser, C. (2011). Quaternary
Half-Metallic Heusler Ferromagnets for Spintronics Applications. Physical
Review B, 83(18): 184428.
Amin, B and Ahmad, I. (2009). Theoretical Investigation of Half Metallicity in
Fe/Co/Ni Doped Znse Material Systems. Journal of Applied Physics, 106(9):
093710.
Amin, B, Arif, S, Ahmad, I, Maqbool, M, Ahmad, R, Goumri-Said, S and Prisbrey,
K. (2011). Cr-Doped Iii–V Nitrides: Potential Candidates for Spintronics. Journal
of electronic materials, 40(6): 1428-1436.
Andersen, OK. (1975). Linear Methods in Band Theory. Physical Review B, 12(8):
3060.
Arif, S, Ahmad, I and Amin, B. (2012). Theoretical Investigation of Half‐Metallicity in
Co/Ni Substituted Aln. International Journal of Quantum Chemistry, 112(3):
882-888.
Awschalom, DD and Samarth, N. (2002). Optical Manipulation, Transport and Storage
of Spin Coherence in Semiconductors. In Semiconductor Spintronics and
Quantum Computation, pp. 147-193: Springer.

149
Azadani, JG, Munira, K, Romero, J, Ma, J, Sivakumar, C, Ghosh, AW and Butler,
WH. (2016). Anisotropy in Layered Half-Metallic Heusler Alloy Superlattices.
Journal of Applied Physics, 119(4): 043904.
Bae, T, Lee, S, Hong, J, Lee, DR and Roh, J. (2012). Effects of Magnetic Seed-Layers
on the Structural and Magnetic Properties of Co 2 Mnsi Heusler Alloy. Materials
Chemistry and Physics, 136(2): 577-581.
Bai, J, Raulot, J, Zhang, Y, Esling, C, Zhao, X and Zuo, L. (2011). Crystallographic,
Magnetic, and Electronic Structures of Ferromagnetic Shape Memory Alloys Ni 2
Xga (X= Mn, Fe, Co) from First-Principles Calculations. Journal of Applied
Physics, 109(1): 014908.
Baibich, MN, Broto, JM, Fert, A, Van Dau, FN, Petroff, F, Etienne, P, Creuzet, G,
Friederich, A and Chazelas, J. (1988). Giant Magnetoresistance of (001)
Fe/(001) Cr Magnetic Superlattices. Physical Review Letters, 61(21): 2472.
Balke, B, Fecher, GH, Kandpal, HC, Felser, C, Kobayashi, K, Ikenaga, E, Kim, J-J
and Ueda, S. (2006). Properties of the Quaternary Half-Metal-Type Heusler
Alloy Co 2 Mn 1− X Fe X Si. Physical Review B, 74(10): 104405.
Bayar, E, Kervan, N and Kervan, S. (2011). Half-Metallic Ferrimagnetism in the Ti 2
Coal Heusler Compound. Journal of Magnetism and Magnetic Materials,
323(23): 2945-2948.
Behin-Aein, B, Datta, D, Salahuddin, S and Datta, S. (2010). Proposal for an All-Spin
Logic Device with Built-in Memory. Nature nanotechnology, 5(4): 266-270.
Betzinger, M, Friedrich, C and Blügel, S. (2010). Hybrid Functionals within the All-
Electron Flapw Method: Implementation and Applications of Pbe0. Physical
Review B, 81(19): 195117.
Bhat, IH, Yousuf, S, Bhat, TM and Gupta, DC. (2015). Investigation of Electronic
Structure, Magnetic and Transport Properties of Half-Metallic Mn 2 Cusi and Mn
2 Znsi Heusler Alloys. Journal of Magnetism and Magnetic Materials, 39581-
88.
Bhattacharya, S, Pope, A, Littleton IV, R, Tritt, TM, Ponnambalam, V, Xia, Y and
Poon, S. (2000). Effect of Sb Doping on the Thermoelectric Properties of Ti-
Based Half-Heusler Compounds, Tinisn 1− X Sb X. Applied Physics Letters,
77(16): 2476-2478.
Birsan, A and Palade, P. (2013a). Band Structure Calculations of Ti2fesn: A New Half-
Metallic Compound. Intermetallics, 3686-89.

150
Birsan, A and Palade, P. (2013b). Band Structure Calculations of Ti 2 Fesn: A New
Half-Metallic Compound. Intermetallics, 3686-89.
Birsan, A, Palade, P and Kuncser, V. (2012). Half-Metallic State and Magnetic
Properties Versus the Lattice Constant in Ti2cosn Heusler Compound: An Ab
Initio Study. Solid state communications, 152(24): 2147-2150.
Birsan, A, Palade, P and Kuncser, V. (2013). Prediction of Half Metallic Properties in
Ti2cosi Heusler Alloy Based on Density Functional Theory. Journal of
Magnetism and Magnetic Materials, 331109-112.
Blaha, P, Schwarz, K, Madsen, G, Kvasnicka, D and Luitz, J. (2001a). Vienna
University of Technology. Vienna, Austria.
Blaha, P, Schwarz, K, Madsen, G, Kvasnicka, D and Luitz, J. (2001b). Wien2k. An
augmented plane wave+ local orbitals program for calculating crystal
properties.
Blöchl, PE, Jepsen, O and Andersen, OK. (1994). Improved Tetrahedron Method for
Brillouin-Zone Integrations. Physical Review B, 49(23): 16223.
Bona, G, Meier, F, Taborelli, M, Bucher, E and Schmidt, P. (1985). Spin Polarized
Photoemission from Nimnsb. Solid state communications, 56(4): 391-394.
Bradley, A and Rodgers, J. (1934). The Crystal Structure of the Heusler Alloys.
Proceedings of the Royal Society of London. Series A, Containing Papers of a
Mathematical and Physical Character, 144(852): 340-359.
Caballero, J, Park, Y, Childress, J, Bass, J, Chiang, W-C, Reilly, A, Pratt Jr, W and
Petroff, F. (1998). Magnetoresistance of Nimnsb-Based Multilayers and Spin
Valves. Journal of Vacuum Science & Technology A: Vacuum, Surfaces, and
Films, 16(3): 1801-1805.
Caballero, J, Reilly, A, Hao, Y, Bass, J, Pratt Jr, W, Petroff, F and Childress, J.
(1999). Effect of Deposition Parameters on the Cpp-Gmr of Nimnsb-Based Spin-
Valve Structures. Journal of magnetism and magnetic materials, 19855-57.
Carey, MJ, Childress, JR and Maat, S. (2007). Magnetoresistive Sensor Based on Spin
Accumulation Effect with Free Layer Stabilized by in-Stack Orthogonal Magnetic
Coupling: Google Patents.
Caruso, A, Borca, C, Ristoiu, D, Nozieres, J and Dowben, P. (2003). A Comparison of
Surface Segregation for Two Semi-Heusler Alloys: Ticosb and Nimnsb. Surface
science, 525(1-3): L109-L115.

151
Casper, F, Graf, T, Chadov, S, Balke, B and Felser, C. (2012). Half-Heusler
Compounds: Novel Materials for Energy and Spintronic Applications.
Semiconductor Science and Technology, 27(6): 063001.
Chadov, S and Qi, X. (2010). S. Chadov, X. Qi, J. Kübler, Gh Fecher, C. Felser, and Sc
Zhang, Nat. Mater. 9, 541 (2010). Nat. Mater., 9541.
Chatterjee, S, Singh, V, Deb, A, Giri, S, De, S, Dasgupta, I and Majumdar, S. (2010).
Magnetic Properties of Ni2+ Xmn1-Xin Heusler Alloys: Theory and Experiment.
Journal of Magnetism and Magnetic Materials, 322(1): 102-107.
Checca, N, Caraballo-Vivas, R, Torrão, R, Rossi, A and Reis, M. (2017). Phase
Composition and Growth Mechanisms of Half-Metal Heusler Alloy Produced by
Pulsed Laser Deposition: From Core-Shell Nanoparticles to Amorphous
Randomic Clusters. Materials Chemistry and Physics, 196103-108.
Chen, J, Gao, G, Yao, K and Song, M. (2011). Half-Metallic Ferromagnetism in the
Half-Heusler Compounds Gekca and Snkca from First-Principles Calculations.
Journal of Alloys and Compounds, 509(42): 10172-10178.
Clowes, S, Miyoshi, Y, Bugoslavsky, Y, Branford, W, Grigorescu, C, Manea, S,
Monnereau, O and Cohen, L. (2004). Spin Polarization of the Transport Current
at the Free Surface of Bulk Nimnsb. Physical Review B, 69(21): 214425.
Coey, J. (2005). D0 Ferromagnetism. Solid State Sciences, 7(6): 660-667.
Continenza, A, Picozzi, S, Geng, W and Freeman, A. (2001). Coordination and
Chemical Effects on the Structural, Electronic, and Magnetic Properties in Mn
Pnictides. Physical Review B, 64(8): 085204.
Damewood, L, Busemeyer, B, Shaughnessy, M, Fong, C, Yang, L and Felser, C.
(2015a). Stabilizing and Increasing the Magnetic Moment of Half-Metals: The
Role of Li in Half-Heusler Limn Z (Z= N, P, Si). Physical Review B, 91(6):
064409.
Damewood, L, Busemeyer, B, Shaughnessy, M, Fong, CY, Yang, LH and Felser, C.
(2015b). Stabilizing and Increasing the Magnetic Moment of Half-Metals: The
Role of Li in Half-Heusler Limn Z (Z= N, P, Si). Physical Review B, 91(6):
064409.
Datta, S and Das, B. (1990). Electronic Analog of the Electro‐Optic Modulator. Applied
Physics Letters, 56(7): 665-667.

152
De Boeck, J, Van Roy, W, Das, J, Motsnyi, V, Liu, Z, Lagae, L, Boeve, H, Dessein, K
and Borghs, G. (2002a). Technology and Materials Issues in Semiconductor-
Based Magnetoelectronics. Semiconductor science and technology, 17(4): 342.
De Boeck, J, Van Roy, W, Motsnyi, V, Liu, Z, Dessein, K and Borghs, G. (2002b).
Hybrid Epitaxial Structures for Spintronics. Thin Solid Films, 412(1): 3-13.
De Groot, R and Buschow, K. (1986). Recent Developments in Half-Metallic
Magnetism. Journal of Magnetism and Magnetic Materials, 541377-1380.
De Groot, R, Mueller, F, Van Engen, P and Buschow, K. (1983). New Class of
Materials: Half-Metallic Ferromagnets. Physical Review Letters, 50(25): 2024.
De Groot, R, Van der Kraan, A and Buschow, K. (1986). Femnsb: A Half-Metallic
Ferrimagnet. Journal of magnetism and magnetic materials, 61(3): 330-336.
De Santanna, Y, De Melo, M, Santos, I, Coelho, A, Gama, S and Cótica, L. (2008).
Structural, Microstructural and Magnetocaloric Investigations in High-Energy
Ball Milled Powders. Solid State Communications, 148(7): 289-292.
De Wijs, G and De Groot, R. (2001). Towards 100% Spin-Polarized Charge-Injection:
The Half-Metallic Nimnsb/Cds Interface. Physical Review B, 64(2): 020402.
Debernardi, A, Peressi, M and Baldereschi, A. (2003). Structural and Electronic
Properties of Nimnsb Heusler Compound and Its Interface with Gaas. Materials
Science and Engineering: C, 23(6-8): 743-746.
Deka, B, Kundu, A, Ghosh, S and Srinivasan, A. (2016). Effect of Electron-Electron
Correlation and Site Disorder on the Magnetic Moment and Half-Metallicity of
Co 2 Fega 1− X Si X Alloys. Materials Chemistry and Physics, 177564-569.
Dong, S and Zhao, H. (2011). First-Principles Studies on Magnetic Properties of
Rocksalt Structure Mc (M= Ca, Sr, and Ba) under Pressure. Applied physics
letters, 98(18): 182501.
Faleev, SV, van Schilfgaarde, M and Kotani, T. (2004). All-Electron Self-Consistent G
W Approximation: Application to Si, Mno, and Nio. Physical Review Letters,
93(12): 126406.
Fang, Q-L, Zhang, J-M, Xu, K-W and Ji, V. (2013). Electronic Structure and
Magnetism of Ti 2 Fesi: A First-Principles Study. Journal of Magnetism and
Magnetic Materials, 345171-175.
Fecher, GH, Kandpal, HC, Wurmehl, S, Morais, J, Lin, H-J, Elmers, H-J,
Schönhense, G and Felser, C. (2005). Design of Magnetic Materials: The

153
Electronic Structure of the Ordered, Doped Heusler Compound Co2cr1− Xfexal.
Journal of Physics: Condensed Matter, 17(46): 7237.
Felser, C, Fecher, GH and Balke, B. (2007). Spintronics: A Challenge for Materials
Science and Solid‐State Chemistry. Angewandte Chemie International Edition,
46(5): 668-699.
Felser, C, Wollmann, L, Chadov, S, Fecher, GH and Parkin, SS. (2015). Basics and
Prospective of Magnetic Heusler Compounds. APL materials, 3(4): 041518.
Feng, L, Liu, E, Zhang, W, Wang, W and Wu, G. (2014). First-Principles Investigation
of Half-Metallic Ferromagnetism of Half-Heusler Compounds Xyz. Journal of
Magnetism and Magnetic Materials, 35192-97.
Feng, W, Xiao, D, Zhang, Y and Yao, Y. (2010). Half-Heusler Topological Insulators:
A First-Principles Study with the Tran-Blaha Modified Becke-Johnson Density
Functional. Physical Review B, 82(23): 235121.
Fujii, S, Sugimura, S and Asano, S. (1990). Hyperfine Fields and Electronic Structures
of the Heusler Alloys Co2mnx (X= Al, Ga, Si, Ge, Sn). Journal of Physics:
Condensed Matter, 2(43): 8583.
Galanakis, I. (2002a). Surface Half-Metallicity of Cras in the Zinc-Blende Structure.
Physical Review B, 66(1): 012406.
Galanakis, I. (2002b). Surface Properties of the Half-and Full-Heusler Alloys. Journal
of Physics: Condensed Matter, 14(25): 6329.
Galanakis, I. (2004). Appearance of Half-Metallicity in the Quaternary Heusler Alloys.
Journal of Physics: Condensed Matter, 16(18): 3089.
Galanakis, I. (2005). Electronic and Magnetic Properties of the (1 1 1) Surfaces of
Nimnsb. Journal of magnetism and magnetic materials, 288411-417.
Galanakis, I and Mavropoulos, P. (2003). Zinc-Blende Compounds of Transition
Elements with N, P, as, Sb, S, Se, and Te as Half-Metallic Systems. Physical
Review B, 67(10): 104417.
Galanakis, I, Mavropoulos, P and Dederichs, PH. (2006). Electronic Structure and
Slater–Pauling Behaviour in Half-Metallic Heusler Alloys Calculated from First
Principles. Journal of Physics D: Applied Physics, 39(5): 765.
Galanakis, I, Ostanin, S, Alouani, M, Dreyssé, H and Wills, J. (2000). Ab Initio
Ground State and L 2, 3 X-Ray Magnetic Circular Dichroism of Mn-Based
Heusler Alloys. Physical Review B, 61(6): 4093.

154
Galanakis, I, Özdoğan, K and Şaşıoğlu, E. (2008a). Ab Initio Electronic and Magnetic
Properties of Half-Metallic Nicrsi and Nimnsi Heusler Alloys: The Role of
Defects and Interfaces. Journal of Applied Physics, 104(8): 083916.
Galanakis, I, Şaşıoğlu, E and Özdoğan, K. (2008b). Magnetic Phase Transition in Half-
Metallic Comnsb and Nimnsb Semi-Heusler Alloys Upon Cu Doping: First-
Principles Calculations. Physical Review B, 77(21): 214417.
Gao, G, Yao, K, Şaşıoğlu, E, Sandratskii, L, Liu, Z and Jiang, J. (2007). Half-
Metallic Ferromagnetism in Zinc-Blende Cac, Src, and Bac from First Principles.
Physical Review B, 75(17): 174442.
Gilleßen, M and Dronskowski, R. (2010). A Combinatorial Study of Inverse Heusler
Alloys by First‐Principles Computational Methods. Journal of computational
chemistry, 31(3): 612-619.
Gökoğlu, G. (2012). Ab Initio Electronic Structure of Nicocrga Half-Metallic Quaternary
Heusler Compound. Solid State Sciences, 14(9): 1273-1276.
Goll, G, Marz, M, Hamann, A, Tomanic, T, Grube, K, Yoshino, T and Takabatake,
T. (2008). Thermodynamic and Transport Properties of the Non-Centrosymmetric
Superconductor Labipt. Physica B: Condensed Matter, 403(5): 1065-1067.
Graf, T, Felser, C and Parkin, SS. (2011). Simple Rules for the Understanding of
Heusler Compounds. Progress in solid state chemistry, 39(1): 1-50.
Gruhn, T. (2010). Comparative Ab Initio Study of Half-Heusler Compounds for
Optoelectronic Applications. Physical Review B, 82(12): 125210.
Grünberg, P, Schreiber, R, Pang, Y, Brodsky, M and Sowers, H. (1986). Layered
Magnetic Structures: Evidence for Antiferromagnetic Coupling of Fe Layers
across Cr Interlayers. Physical Review Letters, 57(19): 2442.
Guo, S-D and Liu, B-G. (2011). Improved Half-Metallic Ferromagnetism of Transition-
Metal Pnictides and Chalcogenides Calculated with a Modified Becke-Johnson
Exchange Potential. EPL (Europhysics Letters), 93(4): 47006.
Gupta, DC and Bhat, IH. (2014). Full-Potential Study of Fe 2 Niz (Z= Al, Si, Ga, Ge).
Materials Chemistry and Physics, 146(3): 303-312.
Halilov, S and Kulatov, E. (1991). Electron and Magneto-Optical Properties of Half-
Metallic Ferromagnets and Uranium Monochalcogenide. Journal of Physics:
Condensed Matter, 3(33): 6363.

155
Hamidani, A, Bennecer, B and Boutarfa, B. (2009). Structural and Elastic Properties of
the Half-Heusler Compounds Irmnz (Z= Al, Sn and Sb). Materials Chemistry and
Physics, 114(2): 732-735.
Hanssen, K and Mijnarends, P. (1986). Positron-Annihilation Study of the Half-
Metallic Ferromagnet Nimnsb: Theory. Physical Review B, 34(8): 5009.
Hanssen, K, Mijnarends, P, Rabou, L and Buschow, K. (1990). Positron-Annihilation
Study of the Half-Metallic Ferromagnet Nimnsb: Experiment. Physical Review B,
42(3): 1533.
Hartjes, K and Jeitschko, W. (1995). Crystal Structures and Magnetic Properties of the
Lanthanoid Nickel Antimonides Lnnisb (Ln= La Nd, Sm, Gd Tm, Lu).
Journal of alloys and compounds, 226(1): 81-86.
Hatchard, T, Thorne, J, Farrell, S and Dunlap, R. (2008). Production of Ni100− X−
Ymnxgay Magnetic Shape Memory Alloys by Mechanical Alloying. Journal of
Physics: Condensed Matter, 20(44): 445205.
Helmholdt, R, De Groot, R, Mueller, F, Van Engen, P and Buschow, K. (1984).
Magnetic and Crystallographic Properties of Several C1 B Type Heusler
Compounds. Journal of Magnetism and Magnetic Materials, 43(3): 249-255.
Heusler, F. (1904). Über Manganbronze Und Über Die Synthese Magnetisierbarer
Legierungen Aus Unmagnetischen Metallen. Angewandte Chemie, 17(9): 260-
264.
Heusler, O. (1934). Kristallstruktur Und Ferromagnetismus Der Mangan‐Aluminium‐
Kupferlegierungen. Annalen der Physik, 411(2): 155-201.
Heyne, L, Igarashi, T, Kanomata, T, Neumann, K, Ouladdiaf, B and Ziebeck, K.
(2005). Atomic and Magnetic Order in the Weak Ferromagnet Covsb: Is It a Half-
Metallic Ferromagnet? Journal of Physics: Condensed Matter, 17(33): 4991.
Hirohata, A and Takanashi, K. (2014). Future Perspectives for Spintronic Devices.
Journal of Physics D: Applied Physics, 47(19): 193001.
Hongzhi, L, Zhiyong, Z, Li, M, Shifeng, X, Heyan, L, Jingping, Q, Yangxian, L and
Guangheng, W. (2007). Electronic Structure and Magnetic Properties of Fe2ysi
(Y= Cr, Mn, Fe, Co, Ni) Heusler Alloys: A Theoretical and Experimental Study.
Journal of Physics D: Applied Physics, 40(22): 7121.
Hordequin, C, Nozieres, J and Pierre, J. (1998). Half Metallic Nimnsb-Based Spin-
Valve Structures. Journal of magnetism and magnetic materials, 183(1-2): 225-
231.

156
Horne, M, Strange, P, Temmerman, W, Szotek, Z, Svane, A and Winter, H. (2004).
The Electronic Structure of Europium Chalcogenides and Pnictides. Journal of
Physics: Condensed Matter, 16(28): 5061.
Hu, S, Cazorla, C, Xiang, F, Ma, H, Wang, J, Wang, J, Wang, X, Ulrich, C, Chen, L
and Seidel, J. (2018). Strain Control of Giant Magnetic Anisotropy in Metallic
Perovskite Srcoo3-Δ Thin Films. ACS applied materials & interfaces.
Hu, Y and Zhang, J-M. (2017). Thermodynamic Stability, Magnetism and Half-
Metallicity of Various (100) Surfaces of Heusler Alloy Ti 2 Fesn. Materials
Chemistry and Physics, 192253-259.
Huang, H, Luo, S and Yao, K. (2012). First-Principles Study of Half-Metallic Properties
of the Heusler Alloy Ti 2 Coge. Journal of Magnetism and Magnetic Materials,
324(16): 2560-2564.
Huang, W, Wang, X, Chen, X, Lu, W, Damewood, L and Fong, C. (2014). Structural
and Electronic Properties of Half-Heusler Alloy Pdmnbi Calculated from First
Principles. Materials Chemistry and Physics, 148(1): 32-38.
Huang, W, Wang, X, Chen, X, Lu, W, Damewood, L and Fong, C. (2015). Structural
and Electronic Properties of Half-Heusler Alloys Ptxbi (with X= Mn, Fe, Co and
Ni) Calculated from First Principles. Journal of Magnetism and Magnetic
Materials, 377252-258.
Hussain, MK. (2018). Investigations of the Electronic and Magnetic Properties of Newly
(001) Surface Licrs and Licrse Half-Heusler Compounds. Applied Physics A,
124(4): 343.
Hybertsen, MS and Louie, SG. (1986). Electron Correlation in Semiconductors and
Insulators: Band Gaps and Quasiparticle Energies. Physical Review B, 34(8):
5390.
Hyeon, T. (2003). Chemical Synthesis of Magnetic Nanoparticles. Chemical
Communications(8): 927-934.
Inomata, K, Ikeda, N, Tezuka, N, Goto, R, Sugimoto, S, Wojcik, M and Jedryka, E.
(2016). Highly Spin-Polarized Materials and Devices for Spintronics∗. Science
and Technology of Advanced Materials.
Ishida, S, Akazawa, S, Kubo, Y and Ishida, J. (1982). Band Theory of Co2mnsn,
Co2tisn and Co2tial. Journal of Physics F: Metal Physics, 12(6): 1111.

157
Ishida, S, Fujii, S, Kashiwagi, S and Asano, S. (1995). Search for Half-Metallic
Compounds in Co2mnz (Z= Iiib, Ivb, Vb Element). Journal of the Physical
Society of Japan, 64(6): 2152-2157.
Ishida, S, Masaki, T, Fujii, S and Asano, S. (1997). Theoretical Predicts of Half-
Metallic Compounds with the C1 B Structure. Physica B: Condensed Matter,
239(1): 163-166.
Ishida, S, Masaki, T, Fujii, S and Asano, S. (1998). Theoretical Search for Half-
Metalliic Films of Co2mnz (Z Si, Ge). Physica B: Condensed Matter, 245(1):
1-8.
Ishikawa, M, Jorda, J and Junod, A. (1982). Superconductivity in D-and F-Band
Metals. Kernforschungszentrum Karlsruhe, Germany.
Ito, K, Ito, W, Umetsu, RY, Nagasako, M, Kainuma, R, Fujita, A, Oikawa, K and
Ishida, K. (2008). Martensitic Transformation in Nicomnsn Metamagnetic Shape
Memory Alloy Powders. Materials transactions, 49(8): 1915-1918.
Izadi, S and Nourbakhsh, Z. (2011). Structural, Electronic and Magnetic Properties of
Nanolayer Andábulk of Mnco2si and Mnfecosi Compounds. Journal of
superconductivity and novel magnetism, 24(1-2): 825-831.
Jaiganesh, G, Eithiraj, R and Kalpana, G. (2010). Theoretical Study of Electronic,
Magnetic and Structural Properties of Mo and W Based Group V (N, P, as, Sb and
Bi) Compounds. Computational materials science, 49(1): 112-120.
Jedema, FJ, Filip, A and Van Wees, B. (2001). Electrical Spin Injection and
Accumulation at Room Temperature in an All-Metal Mesoscopic Spin Valve.
Nature, 410(6826): 345-348.
Jenkins, S and King, D. (2002). Formation of Interface States on a Half-Metal Surface.
Surface science, 501(3): L185-L190.
Jepson, O and Anderson, O. (1971). The Electronic Structure of Hcp Ytterbium. Solid
State Communications, 9(20): 1763-1767.
Jimbo, M, Kanda, T, Goto, S, Tsunashima, S and Uchiyama, S. (1993). Giant
Magnetoresistance in Soft Magnetic Nifeco/Cu Multilayers with Various Buffer
Layers. Journal of magnetism and magnetic materials, 126(1-3): 422-424.
Jonker, B, Park, Y, Bennett, B, Cheong, H, Kioseoglou, G and Petrou, A. (2000).
Robust Electrical Spin Injection into a Semiconductor Heterostructure. Physical
Review B, 62(12): 8180.

158
Julliere, M. (1975). Tunneling between Ferromagnetic Films. Physics Letters A, 54(3):
225-226.
Kabani, R, Terada, M, Roshko, A and Moodera, J. (1990). Magnetic Properties of
Nimnsb Films. Journal of Applied Physics, 67(9): 4898-4900.
Kainuma, R, Imano, Y, Ito, W, Sutou, Y, Morito, H, Okamoto, S, Kitakami, O,
Oikawa, K, Fujita, A and Kanomata, T. (2006). Magnetic-Field-Induced Shape
Recovery by Reverse Phase Transformation. Nature, 439(7079): 957-960.
Kainuma, R, Ito, W, Umetsu, R, Oikawa, K and Ishida, K. (2008). Magnetic Field-
Induced Reverse Transformation in B 2-Type Nicomnal Shape Memory Alloys.
Applied Physics Letters, 93(9): 091906.
Kandpal, HC, Fecher, GH and Felser, C. (2007). Calculated Electronic and Magnetic
Properties of the Half-Metallic, Transition Metal Based Heusler Compounds.
Journal of Physics D: Applied Physics, 40(6): 1507.
Kato, H, Okuda, T, Okimoto, Y, Tomioka, Y, Oikawa, K, Kamiyama, T and
Tokura, Y. (2002). Metal-Insulator Transition of Ferromagnetic Ordered Double
Perovskites:(Sr 1− Y Ca Y) 2 Fereo 6. Physical Review B, 65(14): 144404.
Kato, H, Okuda, T, Okimoto, Y, Tomioka, Y, Oikawa, K, Kamiyama, T and
Tokura, Y. (2004). Structural and Electronic Properties of the Ordered Double
Perovskites a 2 M Reo 6 (a= Sr, Ca; M= Mg, Sc, Cr, Mn, Fe, Co, Ni, Zn).
Physical Review B, 69(18): 184412.
Kervan, N and Kervan, S. (2012). A First-Principle Study of Half-Metallic
Ferrimagnetism in the Ti 2 Coga Heusler Compound. Journal of Magnetism and
Magnetic Materials, 324(4): 645-648.
Kervan, S and Kervan, N. (2011). Spintronic Properties of the Ti 2 Cob Heusler
Compound by Density Functional Theory. Solid State Communications, 151(17):
1162-1164.
Kieven, D, Klenk, R, Naghavi, S, Felser, C and Gruhn, T. (2010). I-Ii-V Half-Heusler
Compounds for Optoelectronics: Ab Initio Calculations. Physical Review B,
81(7): 075208.
Kilian, K and Victora, R. (2000). Electronic Structure of Ni 2 Mnin for Use in Spin
Injection. Journal of Applied Physics, 87(9): 7064-7066.
Kimura, Y, Ueno, H and Mishima, Y. (2009). Thermoelectric Properties of
Directionally Solidified Half-Heusler (M0. 5a, M0. 5b) Nisn (Ma, Mb= Hf, Zr, Ti)
Alloys. Journal of electronic materials, 38(7): 934-939.

159
Kirillova, M, Makhnev, A, Shreder, E, Dyakina, V and Gorina, N. (1995). Interband
Optical Absorption and Plasma Effects in Half‐Metallic Xmny Ferromagnets.
physica status solidi (b), 187(1): 231-240.
Ko, V, Han, G and Feng, Y. (2010). Electronic Band Structure Matching for Half-and
Full-Heusler Alloys. Journal of Magnetism and Magnetic Materials, 322(20):
2989-2993.
Kodama, RH, Makhlouf, SA and Berkowitz, AE. (1997). Finite Size Effects in
Antiferromagnetic Nio Nanoparticles. Physical Review Letters, 79(7): 1393.
Kohn, W and Sham, LJ. (1965). Self-Consistent Equations Including Exchange and
Correlation Effects. Physical review, 140(4A): A1133.
Komesu, T, Borca, C, Jeong, H-K, Dowben, PA, Ristoiu, D, Nozières, J, Stadler, S
and Idzerda, Y. (2000). The Polarization of Sb Overlayers on Nimnsb (100).
Physics Letters A, 273(4): 245-251.
Kotani, T. (1995). Exact Exchange Potential Band-Structure Calculations by the Linear
Muffin-Tin Orbital–Atomic-Sphere Approximation Method for Si, Ge, C, and
Mno. Physical Review Letters, 74(15): 2989.
Kübler, J. (2000). Theory of Itinerant Electron Magnetism: Oxford University Press.
Kübler, J, William, A and Sommers, C. (1983). Formation and Coupling of Magnetic
Moments in Heusler Alloys. Physical Review B, 28(4): 1745.
Kulatov, E and Mazin, I. (1990). Extended Stoner Factor Calculations for the Half-
Metallic Ferromagnets Nimnsb and Cro2. Journal of Physics: Condensed Matter,
2(2): 343.
Lakdja, A, Rozale, H, Chahed, A and Benhelal, O. (2013). Ferromagnetism in the
Half-Heusler Xcsba Compounds from First-Principles Calculations (X= C, Si, and
Ge). Journal of alloys and compounds, 5648-12.
Larson, P, Mahanti, S and Kanatzidis, M. (2000). Structural Stability of Ni-Containing
Half-Heusler Compounds. Physical Review B, 62(19): 12754.
Lei, F, Tang, C, Wang, S and He, W. (2011). Half-Metallic Full-Heusler Compound Ti
2 Nial: A First-Principles Study. Journal of Alloys and Compounds, 509(17):
5187-5189.
Lewis, SP, Allen, PB and Sasaki, T. (1997). Band Structure and Transport Properties of
Cro 2. Physical Review B, 55(16): 10253.

160
Ležaić, M, Galanakis, I, Bihlmayer, G and Blügel, S. (2005). Structural and Magnetic
Properties of the (001) and (111) Surfaces of the Half-Metal Nimnsb. Journal of
Physics: Condensed Matter, 17(21): 3121.
Li, J, Li, Y, Dai, X and Xu, X. (2009). Band Structure Calculations for Heusler Phase
Co 2 Ybi and Half-Heusler Phase Coybi (Y= Mn, Cr). Journal of Magnetism and
Magnetic Materials, 321(5): 365-372.
Li, L, Yu, Y, Ye, GJ, Ge, Q, Ou, X, Wu, H, Feng, D, Chen, XH and Zhang, Y. (2014).
Black Phosphorus Field-Effect Transistors. Nature nanotechnology, 9(5): 372.
Lin, S-Y, Yang, X-B and Zhao, Y-J. (2014). First-Principle Prediction of Robust Half-
Metallic Te-Based Half-Heusler Alloys. Journal of magnetism and magnetic
materials, 350119-123.
Liu, B-G. (2003). Robust Half-Metallic Ferromagnetism in Zinc-Blende Crsb. Physical
Review B, 67(17): 172411.
Liu, G, Dai, X, Liu, H, Chen, J, Li, Y, Xiao, G and Wu, G. (2008). Mn 2 Co Z (Z= Al,
Ga, in, Si, Ge, Sn, Sb) Compounds: Structural, Electronic, and Magnetic
Properties. Physical Review B, 77(1): 014424.
Loss, D and DiVincenzo, DP. (1998). Quantum Computation with Quantum Dots.
Physical Review A, 57(1): 120.
Lou, X, Adelmann, C, Crooker, SA, Garlid, ES, Zhang, J, Reddy, KM, Flexner, SD,
Palmstrøm, CJ and Crowell, PA. (2007). Electrical Detection of Spin Transport
in Lateral Ferromagnet–Semiconductor Devices. Nature Physics, 3(3): 197-202.
Lu, H, Schaff, WJ, Hwang, J, Wu, H, Koley, G and Eastman, LF. (2001). Effect of an
Aln Buffer Layer on the Epitaxial Growth of Inn by Molecular-Beam Epitaxy.
Applied physics letters, 79(10): 1489-1491.
Luo, H, Liu, G, Meng, F, Wang, L, Liu, E, Wu, G, Zhu, X and Jiang, C. (2011).
Slater–Pauling Behavior and Half-Metallicity in Heusler Alloys Mn 2 Cuz (Z= Ge
and Sb). Computational materials science, 50(11): 3119-3122.
Luo, H, Meng, F, Feng, Z, Li, Y, Zhu, W, Wu, G, Zhu, X, Jiang, C and Xu, H.
(2009). Ferromagnetism in the Mn-Based Heusler Alloy Mn 2 Nisb. Journal of
Applied Physics, 105(10): 103903.
Luo, H, Zhang, H, Zhu, Z, Ma, L, Xu, S, Wu, G, Zhu, X, Jiang, C and Xu, H.
(2008a). Half-Metallic Properties for the Mn 2 Fe Z (Z= Al, Ga, Si, Ge, Sb)
Heusler Alloys: A First-Principles Study. Journal of Applied Physics, 103(8):
083908.

161
Luo, H, Zhu, Z, Liu, G, Xu, S, Wu, G, Liu, H, Qu, J and Li, Y. (2008b). Ab-Initio
Investigation of Electronic Properties and Magnetism of Half-Heusler Alloys
Xcral (X= Fe, Co, Ni) and Nicrz (Z= Al, Ga, in). Physica B: Condensed Matter,
403(1): 200-206.
Lv, S, Li, H, Han, D, Wu, Z, Liu, X and Meng, J. (2011). A Better Ferrimagnetic Half-
Metal Lucu3mn4o12: Predicted from First-Principles Investigation. Journal of
magnetism and magnetic materials, 323(5): 416-421.
Madsen, GK and Singh, DJ. (2006). Boltztrap. A Code for Calculating Band-Structure
Dependent Quantities. Computer Physics Communications, 175(1): 67-71.
Maekawa, S and Gafvert, U. (1982). Electron Tunneling between Ferromagnetic Films.
IEEE Transactions on magnetics, 18(2): 707-708.
Mancoff, F, Clemens, B, Singley, E and Basov, D. (1999). Infrared Probe of the
Electronic Structure and Carrier Scattering in Nimnsb Thin Films. Physical
Review B, 60(18): R12565.
Mañosa, L, Planes, A, Acet, M, Duman, E and Wassermann, E. (2004). Magnetic
Shape Memory in Ni–Mn–Ga and Ni–Mn–Al. Journal of Magnetism and
Magnetic Materials, 2722090-2092.
Mastronardi, K, Young, D, Wang, C-C, Khalifah, P, Cava, R and Ramirez, A.
(1999). Antimonides with the Half-Heusler Structure: New Thermoelectric
Materials. Applied Physics Letters, 74(10): 1415-1417.
Mavropoulos, P, Galanakis, I and Dederichs, P. (2004). Multilayers of Zinc-Blende
Half-Metals with Semiconductors. Journal of Physics: Condensed Matter,
16(24): 4261.
Mikami, M, Ozaki, K, Takazawa, H, Yamamoto, A, Terazawa, Y and Takeuchi, T.
(2013). Effect of Ti Substitution on Thermoelectric Properties of W-Doped
Heusler Fe^ Sub 2^ Val Alloy. Journal of electronic materials, 42(7): 1801.
Mikhailov, GM and Rai, D. (2018). Electronic, Elastic and X-Ray Spectroscopic
Properties of Direct and Inverse Full Heusler Compounds: Dft+ U Study. arXiv
preprint arXiv:1805.02618.
Miura, Y, Nagao, K and Shirai, M. (2004a). Atomic Disorder Effects on Half-
Metallicity of the Full-Heusler Alloys Co 2 (Cr 1− X Fe X) Al: A First-Principles
Study. Physical Review B, 69(14): 144413.
Miura, Y, Shirai, M and Nagao, K. (2004b). First-Principles Study on Half-Metallicity
of Disordered Co2 (Cr1-Xfex) Al. Journal of applied physics, 957225-7227.

162
Miyazaki, T, Kumagai, S and Yaoi, T. (1997). Spin Tunneling in Ni-Fe/Al2o3/Co
Junction Devices. Journal of applied physics, 813753-3757.
Miyazaki, T and Tezuka, N. (1995). Giant Magnetic Tunneling Effect in Fe/Al 2 O 3/Fe
Junction. Journal of Magnetism and Magnetic Materials, 139(3): L231-L234.
Mizuguchi, M, Akinaga, H, Manago, T, Ono, K, Oshima, M, Shirai, M, Yuri, M,
Lin, H, Hsieh, H and Chen, C. (2002). Epitaxial Growth of Zinc-Blende
Cras/Gaas Multilayer. Journal of Applied Physics, 91(10): 7917-7919.
Mohankumar, R, Ramasubramanian, S, Rajagopalan, M, Raja, MM, Kamat, S and
Kumar, J. (2015). Effect of Fe Substitution on the Electronic Structure, Magnetic
and Thermoelectric Properties of Co 2 Fesi Full Heusler Alloy: A First Principle
Study. Computational Materials Science, 10934-40.
Monir, MEA, Khenata, R, Baltache, H, Murtaza, G, Abu-Jafar, M, Bouhemadou, A,
Omran, SB and Rached, D. (2015). Study of Structural, Electronic and Magnetic
Properties of Cofein and Co 2 Fein Heusler Alloys. Journal of Magnetism and
Magnetic Materials, 394404-409.
Moodera, JS, Kinder, LR, Wong, TM and Meservey, R. (1995). Large
Magnetoresistance at Room Temperature in Ferromagnetic Thin Film Tunnel
Junctions. Physical Review Letters, 74(16): 3273.
Moodera, JS, Nassar, J and Mathon, G. (1999). Spin-Tunneling in Ferromagnetic
Junctions. Annual Review of Materials Science, 29(1): 381-432.
Murnaghan, F. (1944a). The Compressibility of Media under Extreme Pressures.
Proceedings of the national academy of sciences of the United States of
America, 30(9): 244.
Murnaghan, F. (1944b). The Compressibility of Media under Extreme Pressures.
Proceedings of the National Academy of Sciences, 30(9): 244-247.
Murnaghan, FD. (1937). Finite Deformations of an Elastic Solid. American Journal of
Mathematics235-260.
Nagao, K, Shirai, M and Miura, Y. (2004). Ab Initio Calculations of Zinc-Blende
Cras/Gaas Superlattices. Journal of Applied Physics, 95(11): 6518-6520.
Nanda, B and Dasgupta, I. (2003). Electronic Structure and Magnetism in Half-Heusler
Compounds. Journal of Physics: Condensed Matter, 15(43): 7307.
Nishino, Y, Kato, M, Asano, S, Soda, K, Hayasaki, M and Mizutani, U. (1997).
Semiconductorlike Behavior of Electrical Resistivity in Heusler-Type Fe 2 Val
Compound. Physical Review Letters, 79(10): 1909.

163
Ogawa, T, Shirai, M, Suzuki, N and Kitagawa, I. (1999). First-Principles Calculations
of Electronic Structures of Diluted Magnetic Semiconductors (Ga, Mn) As.
Journal of Magnetism and Magnetic Materials, 196428-429.
Ohno, H. (1998). Making Nonmagnetic Semiconductors Ferromagnetic. science,
281(5379): 951-956.
Ono, K, Okabayashi, J, Mizuguchi, M, Oshima, M, Fujimori, A and Akinaga, H.
(2002). Fabrication, Magnetic Properties, and Electronic Structures of Nanoscale
Zinc-Blende Mnas Dots. Journal of applied physics, 91(10): 8088-8092.
Orgassa, D, Fujiwara, H, Schulthess, T and Butler, W. (1999). First-Principles
Calculation of the Effect of Atomic Disorder on the Electronic Structure of the
Half-Metallic Ferromagnet Nimnsb. Physical Review B, 60(19): 13237.
Ouardi, S, Fecher, GH, Felser, C and Kübler, J. (2013). Realization of Spin Gapless
Semiconductors: The Heusler Compound Mn 2 Coal. Physical Review Letters,
110(10): 100401.
Özdog, K and Galanakis, I. (2009). First-Principles Electronic and Magnetic Properties
of the Half-Metallic Antiferromagnet Cr2mnsb. Journal of Magnetism and
Magnetic Materials, 321(15): L34-L36.
Özdogan, K, Galanakis, I, Şaşioglu, E and Aktaş, B. (2006). Search for Half-Metallic
Ferrimagnetism in V-Based Heusler Alloys Mn2vz (Z= Al, Ga, in, Si, Ge, Sn).
Journal of Physics: Condensed Matter, 18(10): 2905.
Özdoğan, K, Şaşıoğlu, E and Galanakis, I. (2009). Tuning the Magnetic Properties of
Half-Metallic Semi-Heusler Alloys by Sp-Electron Substitution: The Case of
Aumnsn1− Xsbx Quaternary Alloys. Journal of Physics D: Applied Physics,
42(8): 085003.
Pask, J, Yang, L, Fong, C, Pickett, W and Dag, S. (2003). Six Low-Strain Zinc-Blende
Half Metals: An Ab Initio Investigation. Physical Review B, 67(22): 224420.
Pauling, L. (1938). The Nature of the Interatomic Forces in Metals. Physical Review,
54(11): 899.
Perdew, JP, Burke, K and Ernzerhof, M. (1996a). Generalized Gradient
Approximation Made Simple. Physical Review Letters, 77(18): 3865.
Perdew, JP, Burke, K and Wang, Y. (1996b). Generalized Gradient Approximation for
the Exchange-Correlation Hole of a Many-Electron System. Physical Review B,
54(23): 16533.

164
Peruman, KV, Mahendran, M, Seenithurai, S, Chokkalingam, R, Singh, R and
Chandrasekaran, V. (2010). Internal Stress Dependent Structural Transition in
Ferromagnetic Ni–Mn–Ga Nanoparticles Prepared by Ball Milling. Journal of
Physics and Chemistry of Solids, 71(11): 1540-1544.
Picozzi, S, Continenza, A and Freeman, A. (2002). Co 2 Mn X (X= Si, Ge, Sn) Heusler
Compounds: An Ab Initio Study of Their Structural, Electronic, and Magnetic
Properties at Zero and Elevated Pressure. Physical Review B, 66(9): 094421.
Picozzi, S, Continenza, A and Freeman, A. (2004). Role of Structural Defects on the
Half-Metallic Character of Co 2 Mnge and Co 2 Mnsi Heusler Alloys. Physical
Review B, 69(9): 094423.
Pierre, J, Skolozdra, R, Tobola, J, Kaprzyk, S, Hordequin, C, Kouacou, M, Karla, I,
Currat, R and Lelievre-Berna, E. (1997). Properties on Request in Semi-
Heusler Phases. Journal of alloys and compounds, 262101-107.
Prinz, GA. (1998). Magnetoelectronics. science, 282(5394): 1660-1663.
Prinz, GA. (1999). Magnetoelectronics Applications. Journal of magnetism and
magnetic materials, 200(1-3): 57-68.
Puselj, M and Ban, Z. (1969). Crystal Structure of Ticuhg2. CROATICA CHEMICA
ACTA, 41(2): 79-&.
Qi, S, Shen, J and Zhang, C-H. (2015). First-Principles Study on the Structural,
Electronic and Magnetic Properties of the Ti 2 Vz (Z= Si, Ge, Sn) Full-Heusler
Compounds. Materials Chemistry and Physics, 164177-182.
Raj, K and Moskowitz, R. (1990). Commercial Applications of Ferrofluids. Journal of
Magnetism and Magnetic Materials, 85(1-3): 233-245.
Rasool, MN, Hussain, A, Javed, A, Khan, MA and Iqbal, F. (2016). Structural
Stability, Electronic and Magnetic Behaviour of Spin-Polarized Ycovz (Z= Si,
Ge) and Ycotiz (Z= Si, Ge) Heusler Alloys. Materials Chemistry and Physics,
183524-533.
Rauf, S, Arif, S, Haneef, M and Amin, B. (2015). The First Principle Study of Magnetic
Properties of Mn 2 Wsn, Fe 2 Ysn (Y= Ti, V), Co 2 Ysn (Y= Ti, Zr, Hf, V, Mn)
and Ni 2 Ysn (Y= Ti, Zr, Hf, V, Mn) Heusler Alloys. Journal of Physics and
Chemistry of Solids, 76153-169.
Ristoiu, D, Nozieres, J, Borca, C, Borca, B and Dowben, PA. (2000a). Manganese
Surface Segregation in Nimnsb. Applied Physics Letters, 76(17): 2349-2351.

165
Ristoiu, D, Nozieres, J, Borca, C, Komesu, T, Jeong, H-K and Dowben, PA. (2000b).
The Surface Composition and Spin Polarization of Nimnsb Epitaxial Thin Films.
EPL (Europhysics Letters), 49(5): 624.
Ritchie, L, Xiao, G, Ji, Y, Chen, T, Chien, C, Zhang, M, Chen, J, Liu, Z, Wu, G and
Zhang, X. (2003). Magnetic, Structural, and Transport Properties of the Heusler
Alloys Co 2 Mnsi and Nimnsb. Physical Review B, 68(10): 104430.
Roy, A, Bennett, JW, Rabe, KM and Vanderbilt, D. (2012). Half-Heusler
Semiconductors as Piezoelectrics. Physical Review Letters, 109(3): 037602.
Rozale, H, Amar, A, Lakdja, A, Moukadem, A and Chahed, A. (2013). Half-
Metallicity in the Half-Heusler Rbsrc, Rbsrsi and Rbsrge Compounds. Journal of
magnetism and magnetic materials, 33683-87.
Sadouki, H, Belkadi, A, Zaoui, Y, Amari, S, Obodo, K, Beldi, L and Bouhafs, B.
(2018a). Prediction of Half-Metallicity in the Nas, Nase and Nate Alkali-Metal
Chalcogenides Using First Principles. International Journal of Computational
Materials Science and Engineering1850015.
Sadouki, H, Belkadi, A, Zaoui, Y, Beldi, L, Bouhafs, B, Méçabih, S and Abbar, B.
(2018b). Ab-Initio Prediction of Half-Metallicity in Lithium Chalcogenides
Compounds Lix (X= S, Se and Te) in Zinc-Blende and Wurtzite Structures.
Computational Condensed Matter, 16.
Saeed, Y, Nazir, S, Shaukat, A and Reshak, AH. (2010). Ab-Initio Calculations of Co-
Based Diluted Magnetic Semiconductors Cd1− Xcoxx (X= S, Se, Te). Journal of
magnetism and magnetic materials, 322(20): 3214-3222.
Sakurada, S and Shutoh, N. (2005). Effect of Ti Substitution on the Thermoelectric
Properties of (Zr, Hf) Nisn Half-Heusler Compounds. Applied Physics Letters,
86(8): 082105.
Sanvito, S and Hill, NA. (2000). Ground State of Half-Metallic Zinc-Blende Mnas.
Physical Review B, 62(23): 15553.
Sanyal, B, Bergqvist, L and Eriksson, O. (2003). Ferromagnetic Materials in the Zinc-
Blende Structure. Physical Review B, 68(5): 054417.
Şaşıoğlu, E, Sandratskii, L and Bruno, P. (2004). First-Principles Calculation of the
Intersublattice Exchange Interactions and Curie Temperatures of the Full Heusler
Alloys Ni 2 Mn X (X= Ga, in, Sn, Sb). Physical Review B, 70(2): 024427.
Sattar, MA, Rashid, M, Hashmi, MR, Ahmad, S, Imran, M and Hussain, F. (2016a).
Theoretical Investigations of Half-Metallic Ferromagnetism in New Half–Heusler

166
Ycrsb and Ymnsb Alloys Using First-Principle Calculations. Chinese Physics B,
25(10): 107402.
Sattar, MA, Rashid, M, Hashmi, MR, Rasool, MN, Mahmood, A and Ahmad, S.
(2016b). Spin-Polarized Calculations of Structural, Electronic and Magnetic
Properties of Half Heusler Alloys FeVZ (Z= Si, Ge, Sn) Using Ab-Initio Method.
Materials Science in Semiconductor Processing, 5148-54.
Schladt, TD, Schneider, K, Shukoor, MI, Natalio, F, Bauer, H, Tahir, MN, Weber, S,
Schreiber, LM, Schröder, HC and Müller, WE. (2010). Highly Soluble
Multifunctional Mno Nanoparticles for Simultaneous Optical and Mri Imaging
and Cancer Treatment Using Photodynamic Therapy. Journal of Materials
Chemistry, 20(38): 8297-8304.
Schliemann, J, Egues, JC and Loss, D. (2003). Nonballistic Spin-Field-Effect
Transistor. Physical Review Letters, 90(14): 146801.
Schmalhorst, J, Kämmerer, S, Sacher, M, Reiss, G, Hütten, A and Scholl, A. (2004).
Interface Structure and Magnetism of Magnetic Tunnel Junctions with a Co 2 Mn
Si Electrode. Physical Review B, 70(2): 024426.
Schwall, M and Balke, B. (2011). Niobium Substitution in Zr 0.5 Hf 0.5 Nisn Based
Heusler Compounds for High Power Factors. Applied Physics Letters, 98(4):
042106.
Schwarz, K, Blaha, P and Madsen, G. (2002). Electronic Structure Calculations of
Solids Using the Wien2k Package for Material Sciences. Computer Physics
Communications, 147(1): 71-76.
Sham, L and Kohn, W. (1966). One-Particle Properties of an Inhomogeneous
Interacting Electron Gas. Physical Review, 145(2): 561.
Shan, R, Sukegawa, H, Wang, W, Kodzuka, M, Furubayashi, T, Ohkubo, T, Mitani,
S, Inomata, K and Hono, K. (2009). Demonstration of Half-Metallicity in
Fermi-Level-Tuned Heusler Alloy Co 2 Feal 0.5 Si 0.5 at Room Temperature.
Physical Review Letters, 102(24): 246601.
Sharma, S, Dewhurst, J and Ambrosch-Draxl, C. (2005). All-Electron Exact Exchange
Treatment of Semiconductors: Effect of Core-Valence Interaction on Band-Gap
and D-Band Position. Physical Review Letters, 95(13): 136402.
Sharma, V, Solanki, A and Kashyap, A. (2010a). Electronic, Magnetic and Transport
Properties of Co2tiz (Z= Si, Ge and Sn): A First-Principle Study. Journal of
magnetism and magnetic materials, 322(19): 2922-2928.

167
Sharma, V, Solanki, A and Kashyap, A. (2010b). Electronic, Magnetic and Transport
Properties of Co 2 Tiz (Z= Si, Ge and Sn): A First-Principle Study. Journal of
Magnetism and Magnetic Materials, 322(19): 2922-2928.
Shirai, M. (2001). Electronic and Magnetic Properties of 3d Transition-Metal-Doped
Gaas. Physica E: Low-dimensional Systems and Nanostructures, 10(1-3): 143-
147.
Shishidou, T, Freeman, A and Asahi, R. (2001). Effect of Gga on the Half-Metallicity
of the Itinerant Ferromagnet Cos 2. Physical Review B, 64(18): 180401.
Sholl, D and Steckel, JA. (2011). Density Functional Theory: A Practical Introduction:
John Wiley & Sons.
Skovsen, I, Bjerg, L, Christensen, M, Nishibori, E, Balke, B, Felser, C and Iversen,
BB. (2010). Multi-Temperature Synchrotron Pxrd and Physical Properties Study
of Half-Heusler Ticosb. Dalton Transactions, 39(42): 10154-10159.
Slater, JC. (1936). The Ferromagnetism of Nickel. Physical Review, 49(7): 537.
Ślebarski, A, Maple, M, Wrona, A and Winiarska, A. (2001). Kondo-Type Behavior
in Fe 2− X M X Tisn (M= Co, Ni). Physical Review B, 63(21): 214416.
Snyder, GJ and Toberer, ES. (2008). Complex Thermoelectric Materials. Nature
materials, 7(2): 105-114.
Soeya, S, Hayakawa, J, Takahashi, H, Ito, K, Yamamoto, C, Kida, A, Asano, H and
Matsui, M. (2002). Development of Half-Metallic Ultrathin Fe3o4 Films for
Spin-Transport Devices. Applied physics letters, 80(5): 823-825.
Son, Y-W, Cohen, ML and Louie, SG. (2006). Half-Metallic Graphene Nanoribbons.
Nature, 444(7117): 347-349.
Song, W, Wang, J and Wu, Z. (2009a). Half Metallic Properties of Sr 2 Cuoso 6.
Chemical Physics Letters, 482(4): 246-248.
Song, W, Zhao, E, Meng, J and Wu, Z. (2009b). Near Compensated Half-Metal in
Sr2nioso6. Journal of Chemical Physics, 130(11): 114707.
Sootsman, JR, Chung, DY and Kanatzidis, MG. (2009). New and Old Concepts in
Thermoelectric Materials. Angewandte Chemie International Edition, 48(46):
8616-8639.
Soulen, R, Byers, J, Osofsky, M, Nadgorny, B, Ambrose, T, Cheng, S, Broussard,
PR, Tanaka, C, Nowak, J and Moodera, J. (1998). Measuring the Spin
Polarization of a Metal with a Superconducting Point Contact. science, 282(5386):
85-88.

168
Städele, M, Majewski, J, Vogl, P and Görling, A. (1997). Exact Kohn-Sham Exchange
Potential in Semiconductors. Physical Review Letters, 79(11): 2089.
Stroppa, A, Picozzi, S, Continenza, A and Freeman, A. (2003). Electronic Structure
and Ferromagnetism of Mn-Doped Group-Iv Semiconductors. Physical Review B,
68(15): 155203.
Suits, JC. (1976). New Magnetic Compounds with Heusler and Heusler-Related
Structures. Physical Review B, 14(9): 4131.
Sun, S. (2006). Recent Advances in Chemical Synthesis, Self‐Assembly, and
Applications of Fept Nanoparticles. Advanced Materials, 18(4): 393-403.
Szotek, Z, Temmerman, W, Svane, A, Petit, L, Stocks, G and Winter, H. (2004).
Half-Metallic Transition Metal Oxides. Journal of magnetism and magnetic
materials, 2721816-1817.
Tan, J-J, Ji, G-F, Chen, X-R and Gou, Q-Q. (2010). Condensed Matter: Structural,
Mechanical, and Thermal Properties: Phase Transition and Phonon Spectrum of
Zinc-Blende Structure Znx (X= S, Se, Te). Communications in Theoretical
Physics, 531160-1166.
Tanaka, C, Nowak, J and Moodera, J. (1997). Magnetoresistance in Ferromagnet-
Insulator-Ferromagnet Tunnel Junctions with Half-Metallic Ferromagnet Nimnsb
Compound. Journal of Applied Physics, 81(8): 5515-5517.
Tanaka, CT, Nowak, J and Moodera, JS. (1999). Spin-Polarized Tunneling in a Half-
Metallic Ferromagnet. Journal of Applied Physics, 86(11): 6239-6242.
Tanveer, W, Faridi, M, Noor, N, Mahmood, A and Amin, B. (2015). First-Principles
Investigation of Structural, Elastic, Electronic and Magnetic Properties of Be 0.75
Co 0.25 Y (Y S, Se and Te) Compounds. Current Applied Physics, 15(11): 1324-
1331.
Tedrow, P and Meservey, R. (1973). Spin Polarization of Electrons Tunneling from
Films of Fe, Co, Ni, and Gd. Physical Review B, 7(1): 318.
Toboła, J, Kaprzyk, S and Pecheur, P. (2003). Theoretical Search for Magnetic Half‐
Heusler Semiconductors. physica status solidi (b), 236(2): 531-535.
Tobola, J, Pierre, J, Kaprzyk, S, Skolozdra, R and Kouacou, M. (1998). Crossover
from Semiconductor to Magnetic Metal in Semi-Heusler Phases as a Function of
Valence Electron Concentration. Journal of Physics: Condensed Matter, 10(5):
1013.

169
Tran, F and Blaha, P. (2009). Accurate Band Gaps of Semiconductors and Insulators
with a Semilocal Exchange-Correlation Potential. Physical Review Letters,
102(22): 226401.
Tran, F, Blaha, P and Schwarz, K. (2007). Band Gap Calculations with Becke–Johnson
Exchange Potential. Journal of Physics: Condensed Matter, 19(19): 196208.
Uher, C, Yang, J, Hu, S, Morelli, D and Meisner, G. (1999). Transport Properties of
Pure and Doped Mnisn (M= Zr, Hf). Physical Review B, 59(13): 8615.
Ullakko, K, Huang, J, Kantner, C, O’handley, R and Kokorin, V. (1996). Large
Magnetic‐Field‐Induced Strains in Ni2mnga Single Crystals. Applied Physics
Letters, 69(13): 1966-1968.
Umamaheswari, R, Yogeswari, M and Kalpana, G. (2013). Electronic Properties and
Structural Phase Transition in a 4 [M 4 O 4](a= Li, Na, K and Rb; M= Ag and
Cu): A First Principles Study. Solid State Communications, 15562-68.
Umamaheswari, R, Yogeswari, M and Kalpana, G. (2014). Ab-Initio Investigation of
Half-Metallic Ferromagnetism in Half-Heusler Compounds Xyz (X= Li, Na, K
and Rb; Y= Mg, Ca, Sr and Ba; Z= B, Al and Ga). Journal of magnetism and
magnetic materials, 350167-173.
Van Dinh, A, Sato, K and Katayama-Yoshida, H. (2009). Structural and Magnetic
Properties of Room Temperature Ferromagnets Nicrz. Journal of Computational
and Theoretical Nanoscience, 6(12): 2589-2596.
Van Roy, W, De Boeck, J, Brijs, B and Borghs, G. (2000). Epitaxial Nimnsb Films on
Gaas (001). Applied Physics Letters, 77(25): 4190-4192.
Vivas, RC, Pedro, S, Cruz, C, Tedesco, J, Coelho, A, Carvalho, AMG, Rocco, D and
Reis, M. (2016). Experimental Evidences of Enhanced Magnetocaloric Properties
at Room Temperature and Half-Metallicity on Fe 2 Mnsi-Based Heusler Alloys.
Materials Chemistry and Physics, 17423-27.
Wang, C, Guo, Y, Casper, F, Balke, B, Fecher, G, Felser, C and Hwu, Y. (2010). Size
Correlated Long and Short Range Order of Ternary Co 2 Fega Heusler
Nanoparticles. Applied Physics Letters, 97(10): 103106.
Wang, H, Wang, B, Li, Q, Zhu, Z, Wang, R and Woo, C. (2007). First-Principles
Study of the Cubic Perovskites Bi M O 3 (M= Al, Ga, in, and Sc). Physical
Review B, 75(24): 245209.

170
Wang, X, Antropov, V and Harmon, B. (1994). First Principles Study of Magneto-
Optical Properties of Half Metallic Heusler Alloys: Nimnsb and Ptmnsb. IEEE
Transactions on Magnetics, 30(6): 4458-4460.
Wang, X, Cheng, Z, Guo, R, Wang, J, Rozale, H, Wang, L, Yu, Z and Liu, G.
(2017a). First-Principles Study of New Quaternary Heusler Compounds without
3d Transition Metal Elements: Zrrhhfz (Z= Al, Ga, in). Materials Chemistry and
Physics, 19399-108.
Wang, X, Cheng, Z and Liu, G. (2017b). Largest Magnetic Moments in the Half-
Heusler Alloys Xcrz (X= Li, K, Rb, Cs; Z= S, Se, Te): A First-Principles Study.
Materials, 10(9): 1078.
Watanabe, K. (1976). K. Watanabe, Trans. Jpn. Inst. Met. 17, 220 (1976). Trans. Jpn.
Inst. Met., 17220.
Webster, P. (1971). Magnetic and Chemical Order in Heusler Alloys Containing Cobalt
and Manganese. Journal of Physics and Chemistry of Solids, 32(6): 1221-1231.
Webster, P and Ziebeck, K. (1988a). Alloys and Compounds of D-Elements with Main
Group Elements. Part, 275-184.
Webster, P and Ziebeck, K. (1988b). Landolt-Börnstein—Group Iii Condensed Matter
Vol 19c: Heidelberg: Springer) p.
Webster, P, Ziebeck, K, Town, S and Peak, M. (1984). Magnetic Order and Phase
Transformation in Ni2mnga. Philosophical Magazine B, 49(3): 295-310.
Webster, PJ. (1969). Heusler Alloys. Contemporary Physics, 10(6): 559-577.
Winterlik, J, Fecher, GH, Felser, C, Jourdan, M, Grube, K, Hardy, F, von
Löhneysen, H, Holman, K and Cava, R. (2008). Ni-Based Superconductor:
Heusler Compound Zrni 2 Ga. Physical Review B, 78(18): 184506.
Wolf, S, Awschalom, D, Buhrman, R, Daughton, J, Von Molnar, S, Roukes, M,
Chtchelkanova, AY and Treger, D. (2001). Spintronics: A Spin-Based
Electronics Vision for the Future. science, 294(5546): 1488-1495.
Wooten, F. (1972). Optical Properties of Solids.
Wurmehl, S, Fecher, GH, Kandpal, HC, Ksenofontov, V, Felser, C, Lin, H-J and
Morais, J. (2005). Geometric, Electronic, and Magnetic Structure of Co 2 Fesi:
Curie Temperature and Magnetic Moment Measurements and Calculations.
Physical Review B, 72(18): 184434.
Wurmehl, S, Fecher, GH, Kroth, K, Kronast, F, Dürr, HA, Takeda, Y, Saitoh, Y,
Kobayashi, K, Lin, H-J and Schönhense, G. (2006). Electronic Structure and

171
Spectroscopy of the Quaternary Heusler Alloy Co2cr1− Xfexal. Journal of
Physics D: Applied Physics, 39(5): 803.
Xiao, D, Yao, Y, Feng, W, Wen, J, Zhu, W, Chen, X, Stocks, GM and Zhang, Z.
(2010). Half-Heusler Compounds as a New Class of Three-Dimensional
Topological Insulators. arXiv preprint arXiv:1008.0057.
Xie, W-H, Xu, Y-Q, Liu, B-G and Pettifor, D. (2003). Half-Metallic Ferromagnetism
and Structural Stability of Zincblende Phases of the Transition-Metal
Chalcogenides. Physical Review Letters, 91(3): 037204.
Xie, W, Jin, Q and Tang, X. (2008). The Preparation and Thermoelectric Properties of
Ti 0.5 Zr 0.25 Hf 0.25 Co 1− X Ni X Sb Half-Heusler Compounds. Journal of
applied physics, 103(4): 043711.
Xu, B, Wang, Y, Zhao, W and Yan, Y. (2011). Thermoelectric Properties of Heusler-
Type Compound Fe2v1− Xnbxal. Journal of Applied Physics, 110(1): 013530.
Xu, G, Liu, E, Du, Y, Li, G, Liu, G, Wang, W and Wu, G. (2013). A New Spin
Gapless Semiconductors Family: Quaternary Heusler Compounds. EPL
(Europhysics Letters), 102(1): 17007.
Xu, N, Raulot, J-M, Li, Z, Bai, J, Zhang, Y, Zhao, X, Zuo, L and Esling, C. (2012).
Oscillation of the Magnetic Moment in Modulated Martensites in Ni2mnga
Studied by Ab Initio Calculations. Applied Physics Letters, 100(8): 084106.
Xu, Y-Q, Liu, B-G and Pettifor, D. (2003). Half-Metallic Ferromagnetism of Mnbi in
Zincblende Phase. Physica B: Condensed Matter, 3291117-1118.
Yadav, MK and Sanyal, B. (2015). First Principles Study of Thermoelectric Properties
of Li-Based Half-Heusler Alloys. Journal of alloys and compounds, 622388-393.
Yakushiji, K, Saito, K, Mitani, S, Takanashi, K, Takahashi, Y and Hono, K. (2006).
Current-Perpendicular-to-Plane Magnetoresistance in Epitaxial Co 2 Mn Si∕ Cr∕
Co 2 Mn Si Trilayers. Applied Physics Letters, 88(22): 222504.
Yan, B and de Visser, A. (2014). Half-Heusler Topological Insulators. MRS Bulletin,
39(10): 859-866.
Yao, K, Gao, G, Liu, Z and Zhu, L. (2005a). Half-Metallic Ferromagnetism of Zinc-
Blende Crs and Crp: A First-Principles Pseudopotential Study. Solid state
communications, 133(5): 301-304.
Yao, K, Gao, G, Liu, Z, Zhu, L and Li, Y. (2005b). Half-Metallic Ferromagnetic
Semiconductors of V-and Cr-Doped Cdte Studied from First-Principles
Pseudopotential Calculations. Physica B: Condensed Matter, 366(1): 62-66.

172
Yokoyama, M, Yamaguchi, H, Ogawa, T and Tanaka, M. (2005). Zinc-Blende-Type
Mnas Nanoclusters Embedded in Gaas. Journal of applied physics, 97(10):
10D317.
Yousuf, S and Gupta, DC. (2017). Investigation of Electronic, Magnetic and
Thermoelectric Properties of Zr 2 Niz (Z= Al, Ga) Ferromagnets. Materials
Chemistry and Physics, 19233-40.
Zeier, WG, Schmitt, J, Hautier, G, Aydemir, U, Gibbs, ZM, Felser, C and Snyder,
GJ. (2016). Engineering Half-Heusler Thermoelectric Materials Using Zintl
Chemistry. Nature Reviews Materials, 1(6): 16032.
Zenasni, H, Faraoun, H and Esling, C. (2013). First-Principle Prediction of Half-
Metallic Ferrimagnetism in Mn-Based Full-Heusler Alloys with Highly Ordered
Structure. Journal of magnetism and magnetic materials, 333162-168.
Zhang, L, Brück, E, Tegus, O, Buschow, K and De Boer, F. (2003a). The
Crystallization of Amorphous Fe 2 Mnge Powder Prepared by Ball Milling.
Journal of alloys and compounds, 352(1): 99-102.
Zhang, M, Dai, X, Hu, H, Liu, G, Cui, Y, Liu, Z, Chen, J, Wang, J and Wu, G.
(2003b). Search for New Half-Metallic Ferromagnets in Semi-Heusler Alloys
Nicrm (M= P, as, Sb, S, Se and Te). Journal of Physics: Condensed Matter,
15(46): 7891.
Zhang, M, Hu, H, Liu, G, Cui, Y, Liu, Z, Wang, J, Wu, G, Zhang, X, Yan, L and
Liu, H. (2003c). Half-Metallic Ferromagnetism in Zinc-Blende Crbi and the
Stability of the Half-Metallicity of Zinc-Blende Crm (M= P, as, Sb, Bi). Journal
of Physics: Condensed Matter, 15(29): 5017.
Zhang, M, Liu, Z-H, Hu, H-N, Liu, G-D, Cui, Y-T, Wu, G-H, Brück, E, de Boer, FR
and Li, Y-X. (2004). Half-Metallic Ferromagnetism in Hypothetical Semi-
Heusler Alloys Niv M (M= P, as, Sb, S, Se, and Te). Journal of Applied Physics,
95(11): 7219-7221.
Zhang, Y, Liu, W and Niu, H. (2008). Half-Metallic Ferromagnetism in Cr-Doped
Alp—Density Functional Calculations. Solid state communications, 145(11-12):
590-593.
Zhao, J, Matsukura, F, Takamura, K, Chiba, D, Ohno, Y, Ohtani, K and Ohno, H.
(2003). Zincblende Crsb/Gaas Multilayer Structures with Room-Temperature
Ferromagnetism. Materials Science in Semiconductor Processing, 6(5-6): 507-
509.

173
Zhu, W, Liu, E, Feng, L, Tang, X, Chen, J, Wu, G, Liu, H, Meng, F and Luo, H.
(2009). Magnetic-Field-Induced Transformation in Femnga Alloys. Applied
Physics Letters, 95(22): 222512.
Zhu, W, Sinkovic, B, Vescovo, E, Tanaka, C and Moodera, J. (2001). Spin-Resolved
Density of States at the Surface of Nimnsb. Physical Review B, 64(6): 060403.
Ziebeck, K and Neumann, K. (2001). Magnetic Properties of Metals. Landolt-B€
ornstein, New Series, Group III, 3264-414.
Ziebeck, K and Webster, P. (1974). A Neutron Diffraction and Magnetization Study of
Heusler Alloys Containing Co and Zr, Hf, V or Nb. Journal of Physics and
Chemistry of Solids, 35(1): 1-7.
Žutić, I, Fabian, J and Sarma, SD. (2004). Spintronics: Fundamentals and Applications.
Reviews of modern physics, 76(2): 323.

174
Appendix-I
The lattice constant, electronic properties, magnetic moment, EBG and EHM represents
energy band gap, HM gap of all the 90 HH XYZ materials at the three types. Magnetic
ground state (NM, FM, AFM) and their energy EFM & EAFM, vibrational stability at the γ-
point (f/i shows the imaginary phonon frequency) with preferred type are also illustrated
for all the DFT investigated 90 HH XYZ materials.
LiVZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Mag-
netic
State
Stable
(at
γ-
point)
EFM
(eV)
EAFM
(eV)
LiVSi T1 5.85 SC 2.00 1.43 ---- T1 FM Yes -63.25
-62.72
T2 5.58 Metallic 1.99 ---- ----
T3 5.79 HM 2.00 0.94 0.38
LiVGe T1 5.93 SC 2.00 1.06 ---- T1 FM Yes -60.22
-59.61
T2 5.68 Metallic 2.03 ---- ----
T3 5.97 HM 2.00 1.10 0.47
LiVSn T1 6.37 HM 2.00 0.89 0.47 T1 FM Yes -54.94
-54.65
T2 6.05 Metallic 2.16 ---- ----
T3 6.38 HM 2.00 0.77 0.36
LiVS T1 5.55 Metallic 1.47 ---- ---- T1 AFM f/i
1.72
THz
-65.29
-65.57
T2 5.71 Metallic 3.99 ---- ----
T3 5.87 HM 4.00 5.30 0.49
LiVSe T1 5.94 Metallic 2.64 ---- ---- T1 AFM Yes -60.73
-61.11
T2 5.95 Nearly
HM
3.99 4.05 0.07
T3 6.14 HM 4.00 4.66 0.51
LiVTe T1 6.46 Metallic 3.82 ---- ---- T1 AFM Yes -56.8
-56.95
T2 6.26 Metallic 3.99 ---- ----
T3 6.47 HM 4.00 3.58 0.78
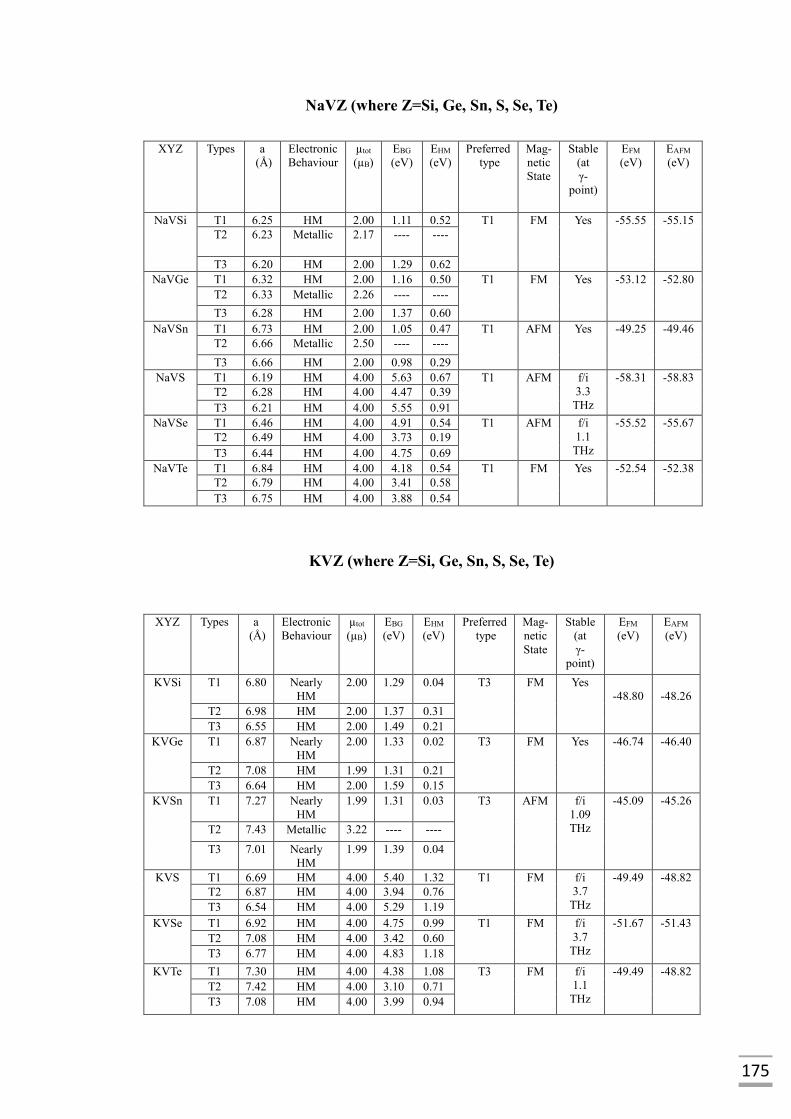
175
NaVZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Mag-
netic
State
Stable
(at
γ-
point)
EFM
(eV)
EAFM
(eV)
NaVSi T1 6.25 HM 2.00 1.11 0.52 T1 FM Yes -55.55
-55.15
T2 6.23 Metallic 2.17 ---- ----
T3 6.20 HM 2.00 1.29 0.62
NaVGe T1 6.32 HM 2.00 1.16 0.50 T1 FM Yes -53.12
-52.80
T2 6.33 Metallic 2.26 ---- ----
T3 6.28 HM 2.00 1.37 0.60
NaVSn T1 6.73 HM 2.00 1.05 0.47 T1 AFM Yes
-49.25
-49.46
T2 6.66 Metallic 2.50 ---- ----
T3 6.66 HM 2.00 0.98 0.29
NaVS T1 6.19 HM 4.00 5.63 0.67 T1 AFM f/i
3.3
THz
-58.31
-58.83
T2 6.28 HM 4.00 4.47 0.39
T3 6.21 HM 4.00 5.55 0.91
NaVSe T1 6.46 HM 4.00 4.91 0.54 T1 AFM f/i
1.1
THz
-55.52
-55.67
T2 6.49 HM 4.00 3.73 0.19
T3 6.44 HM 4.00 4.75 0.69
NaVTe T1 6.84 HM 4.00 4.18 0.54 T1 FM Yes -52.54
-52.38
T2 6.79 HM 4.00 3.41 0.58
T3 6.75 HM 4.00 3.88 0.54
KVZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Mag-
netic
State
Stable
(at
γ-
point)
EFM
(eV)
EAFM
(eV)
KVSi T1 6.80 Nearly
HM
2.00 1.29 0.04 T3 FM Yes
-48.80
-48.26
T2 6.98 HM 2.00 1.37 0.31
T3 6.55 HM 2.00 1.49 0.21
KVGe T1 6.87 Nearly
HM
2.00 1.33 0.02 T3 FM Yes -46.74
-46.40
T2 7.08 HM 1.99 1.31 0.21
T3 6.64 HM 2.00 1.59 0.15
KVSn T1 7.27 Nearly
HM
1.99 1.31 0.03 T3 AFM f/i
1.09
THz
-45.09
-45.26
T2 7.43 Metallic 3.22 ---- ----
T3 7.01 Nearly
HM
1.99
1.39 0.04
KVS T1 6.69 HM 4.00 5.40 1.32 T1 FM f/i
3.7
THz
-49.49
-48.82
T2 6.87 HM 4.00 3.94 0.76
T3 6.54 HM 4.00 5.29 1.19
KVSe T1 6.92 HM 4.00 4.75 0.99 T1 FM f/i
3.7
THz
-51.67
-51.43
T2 7.08 HM 4.00 3.42 0.60
T3 6.77 HM 4.00 4.83 1.18
KVTe T1 7.30 HM 4.00 4.38 1.08 T3 FM f/i
1.1
THz
-49.49
-48.82
T2 7.42 HM 4.00 3.10 0.71
T3 7.08 HM 4.00 3.99 0.94

176
RbVZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Mag-
netic
State
Stable
(at
γ-
point)
EFM
(eV)
EAFM
(eV)
RbVSi T1 7.04 Nearly
HM
2.00 1.38 0.01 T3 FM
f/i
1.9
THz
-46.69
-46.18
T2 7.27 HM 1.99 1.25 0.23
T3 6.72 Nearly
HM
2.00 1.16 0.004
RbVGe T1 7.12 Nearly
HM
2.00 1.36 0.03 T3 FM
f/i
1.7
THz
-44.78
-44.46
T2 7.37 HM 1.99 1.20 0.18
T3 6.80 Nearly
HM
2.00 1.25 0.04
RbVSn T1 7.51 Nearly
HM
2.00 1.33 0.06 T3 AFM
f/i
1.75
THz
-43.47
-43.52
T2 7.71 Metallic 3.84 ---- ----
T3 7.18 Nearly
HM
1.99 1.13 0.20
RbVS T1 6.95 HM 4.00 4.80 1.19 T1 FM f/i
4.04
THz
-51.29
-51.09
T2 7.18 HM 4.00 3.41 0.78
T3 6.74 HM 4.00 4.81 1.05
RbVSe T1 7.18 HM 4.00 4.26 0.91 T1 FM f/i
3.1
THz
-49.44
-49.16
T2 7.39 HM 4.00 2.96 0.65
T3 6.95 HM 4.00 4.43 1.04
RbVTe T1 7.56 HM 4.00 3.97 0.96 T3 FM Yes -48.01
-47.34
T2 7.74 HM 4.00 2.73 0.73
T3 7.27 HM 4.00 3.73 0.83
CsVZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Mag-
netic
State
Stable
(at
γ-
point)
EFM
(eV)
EAFM
(eV)
CsVSi T1 7.25 Nearly
HM
1.99 1.32 0.12 T3 FM
f/i
1.6
THz
-45.99
-45.44
T2 7.37 Metallic 2.25 ---- ----
T3 6.85 Nearly
HM
2.00 0.58 0.10
CsVGe T1 7.37
Nearly
HM
2.00 1.31 0.14 T3 FM f/i
1.2
THz
-44.18
-43.76
T2 7.49 HM 1.99 1.31 0.21
T3 6.94 HM 2.00 1.59 0.15
CsVSn T1 7.76
Nearly
HM
2.00 1.30 0.18 T3 FM Yes -42.90
-42.57
T2 7.89 HM 1.99 1.31 0.21
T3 7.32 Nearly
HM
1.99 0.66 0.15
CsVS T1 7.25 HM 4.00 4.16 1.13 T3 FM Yes -49.97
-49.46
T2 7.44 HM 4.00 3.21 0.97
T3 6.93 HM 4.00 3.92 0.68
CsVSe T1 7.50 HM 4.00 3.69 0.87 T3 FM Yes -48.62
-48.03
T2 7.69 HM 4.00 2.76 0.86
T3 7.13 HM 4.00 3.66 0.66
CsVTe T1 7.89 HM 4.00 3.54 0.94 T3 FM f/i
2.1
THz
-47.30
-46.69
T2 6.26 HM 4.00 2.44 0.92
T3 7.45 HM 4.00 3.18 0.54

177
LiNbZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Mag-
netic
State
Stable
(at
γ-
point)
EFM
(eV)
EAFM
(eV)
LiNbSi T1 5.92 Metallic 0.00 ---- ---- T1 NM Yes -68.78
-68.78
T2 5.62 Metallic 0.00 ---- ----
T3 6.01 Metallic 0.00 ---- ----
LiNbGe T1 6.01 Metallic 0.01 ---- ---- T1
NM
Yes
-64.79 -64.79
T2 5.72 Metallic 0.00 ---- ----
T3 6.11 Metallic 0.00 ---- ----
LiNbSn T1 6.44 Metallic 1.48 ---- ---- T2 NM Yes -58.29 -58.28
T2 6.06 Metallic 0.02 ---- ----
T3 6.51 Metallic 0.39 ---- ----
LiNbS T1 5.92 Metallic 1.15 ---- ---- T1 FM Yes -68.21
-68.16
T2 5.54 Metallic 1.73 ---- ----
T3 5.82 Metallic 1.15 ---- ----
LiNbSe T1 6.02 Metallic 1.27 ---- ---- T1 FM Yes -63.31
-63.16
T2 5.98 Metallic 2.71 ---- ----
T3 6.13 Metallic 1.36 ---- ----
LiNbTe T1 6.44 Metallic 1.36 ---- ---- T1 FM Yes -58.98
-58.79
T2 6.27 Metallic 2.97 ---- ----
T3 6.50 Metallic 1.48 ---- ----
NaNbZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Mag-
netic
State
Stable
(at
γ-
point)
EFM
(eV)
EAFM
(eV)
NaNbSi T1 6.25 Metallic 0.96 ---- ---- T1 AFM Yes -60.27
-60.31
T2 6.24 Metallic 1.88 ---- ----
T3 6.29 Metallic 1.30 ---- ----
NaNbGe T1 6.33 Metallic 1.51 ---- ---- T1 FM Yes -57.19
-57.08
T2 6.33 Metallic 1.99 ---- ----
T3 6.38 HM 2.00 1.03 0.13
NaNbSn T1 6.81 HM 2.00 1.05 0.36 T1 NM Yes -52.96
-52.96
T2 6.63 Metallic 2.03 ---- ----
T3 6.80 HM 2.00 0.84 0.28
NaNbS T1 6.07 Metallic 1.52 ---- ---- T1 FM Yes -60.52
-60.48
T2 6.44 HM 4.00 4.38 0.62
T3 6.10 Metallic 1.42 ---- ----
NaNbSe T1 6.47 Metallic 1.54 ---- ---- T1 AFM f/i
1.43
THz
-56.87
-56.94
T2 6.62 HM 4.00 3.89 0.59
T3 6.50 Metallic 2.60 ---- ----
NaNbTe T1 6.71 Metallic 1.51 ---- ---- T1 AFM f/i
0.9
THz
-53.49
-53.69
T2 6.88 HM 4.00 3.11 0.32
T3 6.79 Metallic 2.65 ---- ----

178
KNbZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Magnetic
State
Stable
(at
γ-
point)
EFM
(eV)
EAFM
(eV)
KNbSi T1 6.79 HM 2.00 1.13 0.54 T1 FM Yes -52.38
-52.03
T2 6.84 HM 2.00 0.57 0.19
T3 6.64 HM 2.00 1.39 0.65
KNbGe T1 6.86 HM 2.00 1.15 0.46 T1 FM Yes -50.14
-49.78
T2 6.95 HM 2.00 0.74 0.08
T3 6.72 HM 2.00 1.49 0.60
KNbSn T1 7.25 HM 2.00 1.10 0.34 T1 FM Yes -47.53
-47.24
T2 7.27 Metallic 2.22 ---- ----
T3 7.07 HM 2.00 1.32 0.42
KNbS T1 6.55 Metallic 1.85 ---- ---- T1 AFM f/i
3.4
THz
-53.72
-54.44
T2 6.94 HM 4.00 4.23 1.24
T3 6.78 HM 4.00 5.18 1.24
KNbSe T1 7.01 HM 4.00 4.98 1.22 T1 AFM f/i
2.3
THz
-51.77
-52.12
T2 7.13 HM 4.00 3.75 1.04
T3 6.97 HM 4.00 4.81 1.13
KNbTe T1 7.36 HM 4.00 4.37 1.05 T1 AFM f/i
0.5
THz
-49.63
-49.72
T2 7.42 HM 4.00 3.47 1.09
T3 7.26 HM 4.00 4.46 0.87

179
RbNbZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Magnetic
State
Stable
(at
γ-
point)
EFM
(eV)
EAFM
(eV)
RbNbSi T1 6.97 HM 2.00 1.18 0.28 T3 FM Yes -49.37
-48.68
T2 7.05 Nearly
HM 2.00 0.46 0.01
T3 6.77 HM 2.00 1.24 0.57
RbNbGe T1 7.07 HM 2.00 1.19 0.19 T3 FM Yes -47.19
-46.50
T2 7.18 HM 2.00 1.33 0.51
T3 6.85 Metallic 2.15 ---- ----
RbNbSn T1 7.45 HM 2.00 1.12 0.11 T3 FM Yes -45.83
-45.14
T2 7.54 Metallic 2.15 ---- ----
T3 7.19 HM 2.00 1.19 0.33
RbNbS T1 7.00 HM 4.00 5.08 1.46 T1 AFM f/i
3.5
THz
-51.31
-51.59
T2 7.22 HM 4.00 3.79 1.30
T3 6.93 HM 4.00 4.74 0.99
RbNbSe T1 7.23 HM 4.00 4.71 1.39 T1 AFM f/i
2.9
THz
-49.61
-49.62
T2 7.41 HM 4.00 3.38 1.11
T3 7.06 HM 4.00 4.37 0.85
RbNbTe T1 7.60 HM 4.00 4.24 1.27 T3 FM Yes -47.59
-46.89
T2 7.71 HM 4.00 3.11 1.11
T3 7.41 HM 4.00 3.82 0.75
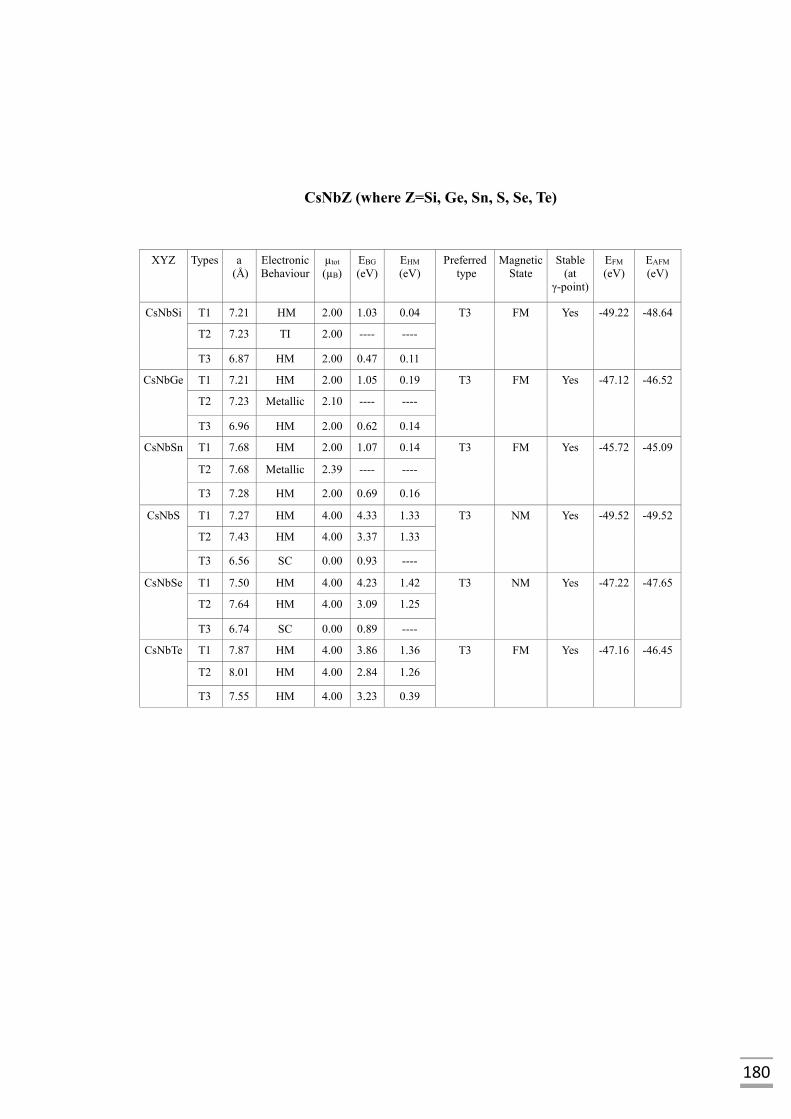
180
CsNbZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Magnetic
State
Stable
(at
γ-point)
EFM
(eV)
EAFM
(eV)
CsNbSi T1 7.21 HM 2.00 1.03 0.04 T3 FM Yes -49.22
-48.64
T2 7.23 TI 2.00 ---- ----
T3 6.87 HM 2.00 0.47 0.11
CsNbGe T1 7.21 HM 2.00 1.05 0.19 T3 FM Yes -47.12
-46.52
T2 7.23 Metallic 2.10 ---- ----
T3 6.96 HM 2.00 0.62 0.14
CsNbSn T1 7.68 HM 2.00 1.07 0.14 T3 FM Yes -45.72
-45.09
T2 7.68 Metallic 2.39 ---- ----
T3 7.28 HM 2.00 0.69 0.16
CsNbS T1 7.27 HM 4.00 4.33 1.33 T3 NM Yes -49.52
-49.52
T2 7.43 HM 4.00 3.37 1.33
T3 6.56 SC 0.00 0.93 ----
CsNbSe T1 7.50 HM 4.00 4.23 1.42 T3 NM Yes -47.22
-47.65
T2 7.64 HM 4.00 3.09 1.25
T3 6.74 SC 0.00 0.89 ----
CsNbTe T1 7.87 HM 4.00 3.86 1.36 T3 FM Yes -47.16
-46.45
T2 8.01 HM 4.00 2.84 1.26
T3 7.55 HM 4.00 3.23 0.39
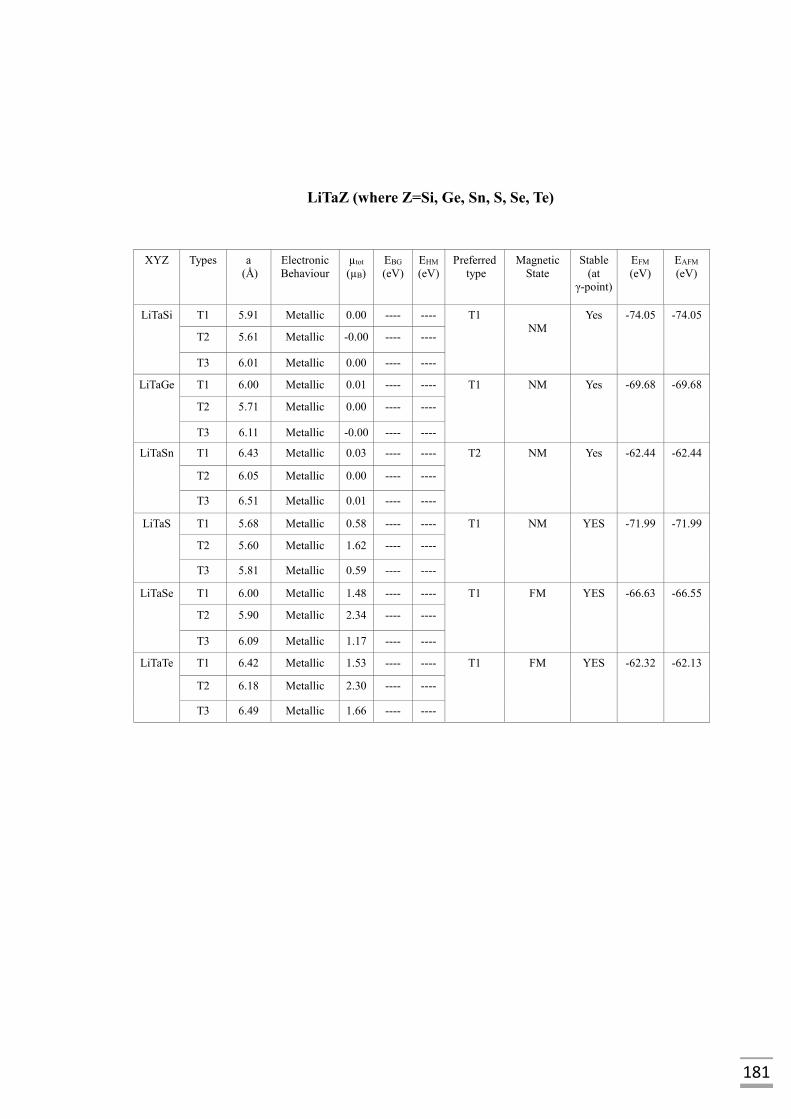
181
LiTaZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Magnetic
State
Stable
(at
γ-point)
EFM
(eV)
EAFM
(eV)
LiTaSi T1 5.91 Metallic 0.00 ---- ---- T1
NM
Yes -74.05
-74.05
T2 5.61 Metallic -0.00 ---- ----
T3 6.01 Metallic 0.00 ---- ----
LiTaGe T1 6.00 Metallic 0.01 ---- ---- T1 NM Yes -69.68
-69.68
T2 5.71 Metallic 0.00 ---- ----
T3 6.11 Metallic -0.00 ---- ----
LiTaSn T1 6.43 Metallic 0.03 ---- ---- T2 NM Yes -62.44
-62.44
T2 6.05 Metallic 0.00 ---- ----
T3 6.51 Metallic 0.01 ---- ----
LiTaS T1 5.68 Metallic 0.58 ---- ---- T1 NM YES -71.99
-71.99
T2 5.60 Metallic 1.62 ---- ----
T3 5.81 Metallic 0.59 ---- ----
LiTaSe T1 6.00 Metallic 1.48 ---- ---- T1 FM YES -66.63
-66.55
T2 5.90 Metallic 2.34 ---- ----
T3 6.09 Metallic 1.17 ---- ----
LiTaTe T1 6.42 Metallic 1.53 ---- ---- T1 FM YES -62.32
-62.13
T2 6.18 Metallic 2.30 ---- ----
T3 6.49 Metallic 1.66 ---- ----

182
NaTaZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Magnetic
State
Stable
(at
γ-point)
EFM
(eV) EAFM
(eV)
NaTaSi T1 6.22 Metallic -0.02 ---- ---- T1 NM Yes -65.34
-65.34
T2 6.16 Metallic 0.20 ---- ----
T3 6.30 Metallic -0.04 ---- ----
NaTaGe T1 6.30 Metallic -0.00 ---- ---- T1 NM Yes -57.52
-57.52
T2 6.35 Metallic 1.86 ---- ----
T3 6.40 Metallic 1.15 ---- ----
NaTaSn T1 6.71 HM 2.00 1.18 0.17 T1 FM Yes -56.87
-56.68
T2 6.55 Metallic 0.40 ---- ----
T3 6.77 Metallic 1.93 ---- ----
NaTaS T1 6.05 Metallic 1.86 ---- ---- T1 FM Yes -63.46
-63.41
T2 6.48 Metallic 3.99 ---- ----
T3 6.11 Metallic 1.72 ---- ----
NaTaSe T1 6.32 Metallic 1.90 ---- ---- T1 FM Yes -59.83
-59.70
T2 6.63 Metallic 3.99 ---- ----
T3 6.46 Metallic 2.00 ---- ----
NaTaTe T1 6.69 Metallic 1.71 ---- ---- T1 FM Yes -56.64
-56.48
T2 6.84 HM 4.00 ---- ----
T3 6.74 Metallic 2.02 ---- ----
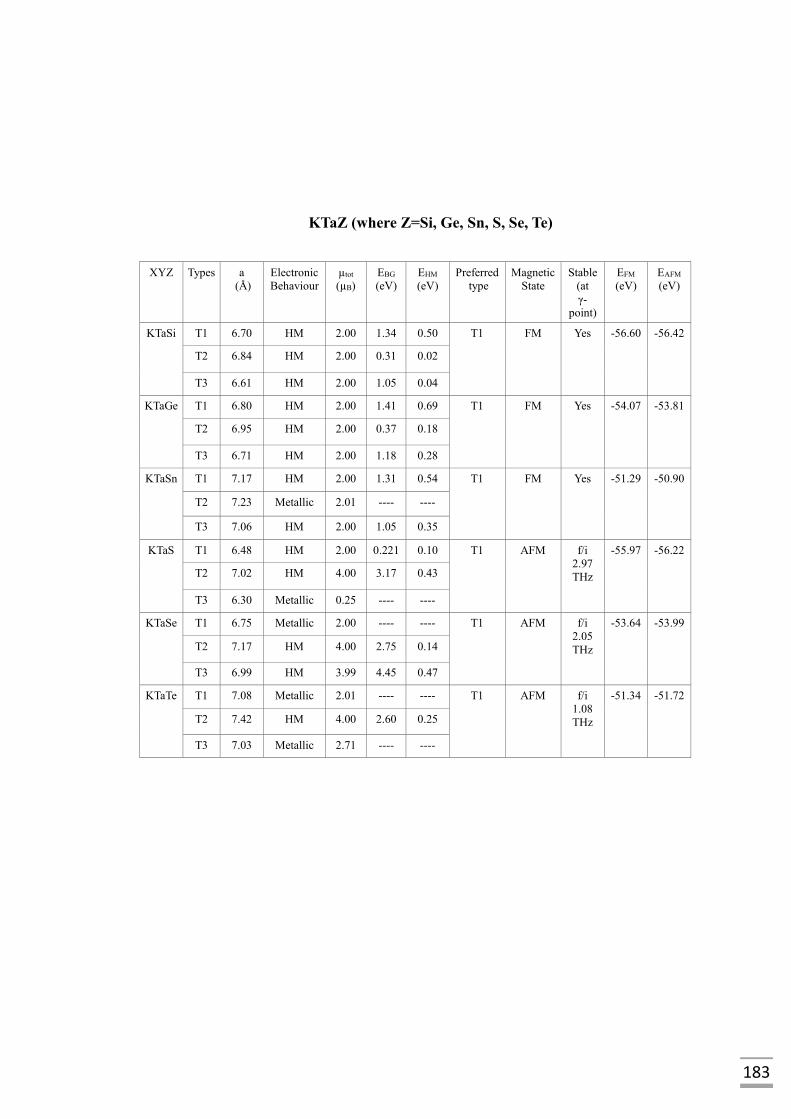
183
KTaZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Magnetic
State
Stable
(at
γ-
point)
EFM
(eV) EAFM
(eV)
KTaSi T1 6.70 HM 2.00 1.34 0.50 T1 FM Yes -56.60
-56.42
T2 6.84 HM 2.00 0.31 0.02
T3 6.61 HM 2.00 1.05 0.04
KTaGe T1 6.80 HM 2.00 1.41 0.69 T1 FM Yes -54.07
-53.81
T2 6.95 HM 2.00 0.37 0.18
T3 6.71 HM 2.00 1.18 0.28
KTaSn T1 7.17 HM 2.00 1.31 0.54 T1 FM Yes -51.29
-50.90
T2 7.23 Metallic 2.01 ---- ----
T3 7.06 HM 2.00 1.05 0.35
KTaS T1 6.48 HM 2.00 0.221 0.10 T1 AFM f/i
2.97
THz
-55.97
-56.22
T2 7.02 HM 4.00 3.17 0.43
T3 6.30 Metallic 0.25 ---- ----
KTaSe T1 6.75 Metallic 2.00 ---- ---- T1 AFM f/i
2.05
THz
-53.64
-53.99
T2 7.17 HM 4.00 2.75 0.14
T3 6.99 HM 3.99 4.45 0.47
KTaTe T1 7.08 Metallic 2.01 ---- ---- T1 AFM f/i
1.08
THz
-51.34
-51.72
T2 7.42 HM 4.00 2.60 0.25
T3 7.03 Metallic 2.71 ---- ----
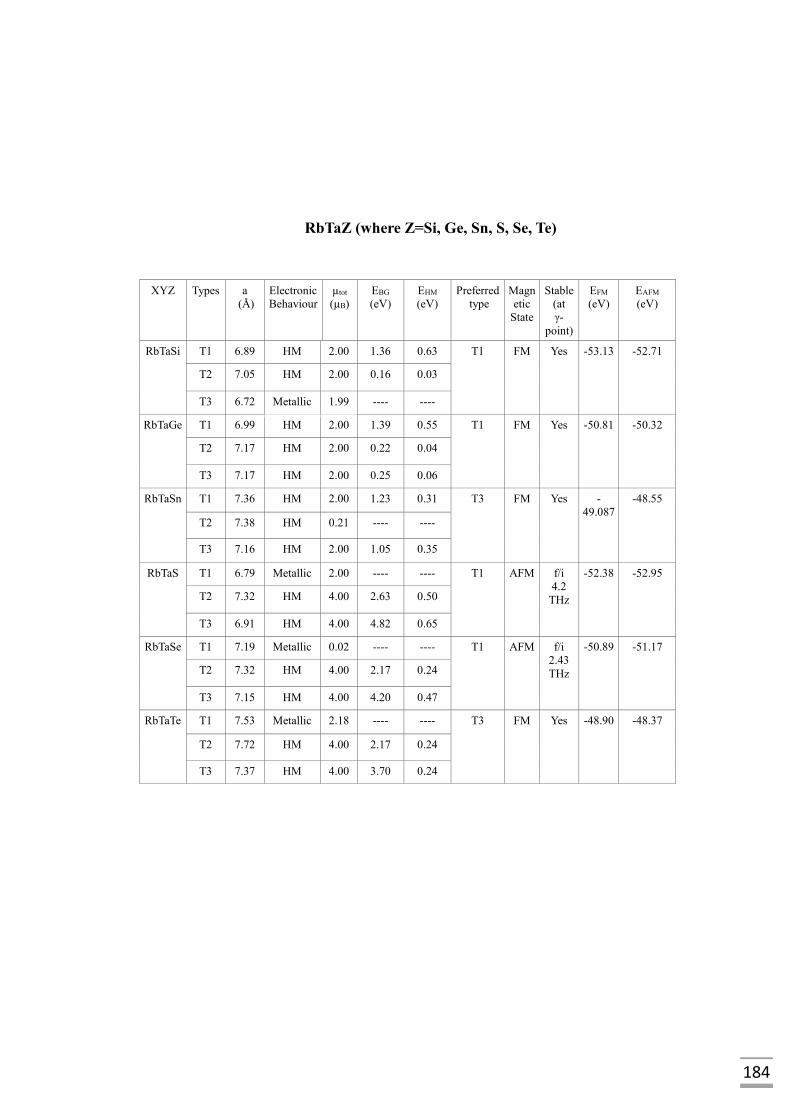
184
RbTaZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Magn
etic
State
Stable
(at
γ-
point)
EFM
(eV) EAFM
(eV)
RbTaSi T1 6.89 HM 2.00 1.36 0.63 T1 FM Yes -53.13
-52.71
T2 7.05 HM 2.00 0.16 0.03
T3 6.72 Metallic 1.99 ---- ----
RbTaGe T1 6.99 HM 2.00 1.39 0.55 T1 FM Yes -50.81
-50.32
T2 7.17 HM 2.00 0.22 0.04
T3 7.17 HM 2.00 0.25 0.06
RbTaSn T1 7.36 HM 2.00 1.23 0.31 T3 FM Yes -
49.087
-48.55
T2 7.38 HM 0.21 ---- ----
T3 7.16 HM 2.00 1.05 0.35
RbTaS T1 6.79 Metallic 2.00 ---- ---- T1 AFM f/i
4.2
THz
-52.38
-52.95
T2 7.32 HM 4.00 2.63 0.50
T3 6.91 HM 4.00 4.82 0.65
RbTaSe T1 7.19 Metallic 0.02 ---- ---- T1 AFM f/i
2.43
THz
-50.89
-51.17
T2 7.32 HM 4.00 2.17 0.24
T3 7.15 HM 4.00 4.20 0.47
RbTaTe T1 7.53 Metallic 2.18 ---- ---- T3 FM Yes -48.90
-48.37
T2 7.72 HM 4.00 2.17 0.24
T3 7.37 HM 4.00 3.70 0.24

185
CsTaZ (where Z=Si, Ge, Sn, S, Se, Te)
XYZ Types a
(Å)
Electronic
Behaviour
µtot
(µB)
EBG
(eV)
EHM
(eV)
Preferred
type
Magnetic
State
Stable
(at
γ-
point)
EFM
(eV) EAFM
(eV)
CsTaSi T1 7.01 HM 2.00 0.91 0.32 T3 FM Yes -53.19
-52.87
T2 7.05 Metallic 1.99 ---- ----
T3 6.82 Metallic 1.96 ---- ----
CsTaGe T1 7.15 HM 2.00 1.14 0.24 T3 FM Yes -50.84
-50.46
T2 7.20 Metallic 2.00 ---- ----
T3 6.91 Metallic 1.98 ---- ----
CsTaSn T1 7.55 Nearly HM 2.00 1.17 0.01 T3 FM Yes -49.37
-48.93
T2 7.55 Metallic 2.06 ---- ----
T3 7.23 Metallic 1.99 ---- ----
CsTaS T1 7.26 HM 4.00 3.82 0.64 T3 NM Yes -52.05
-52.05
T2 7.19 HM 4.00 2.85 0.82
T3 6.51 SC 0 0.53 ----
CsTaSe T1 7.48 Metallic 3.91 ---- ---- T3 NM Yes -52.02
-52.02
T2 7.70 HM 3.99 2.07 ----
T3 6.68 SC 0 0.53 ---
CsTaTe T1 7.81 Metallic -2.52 ---- ---- T3 NM Yes -49.01
-49.01
T2 8.03 HM 4.0 1.83 0.42
T3 6.98 SC 0 0.44 ----