introduction to polymer science - kit
TRANSCRIPT

Institute of Chemical Technology and Polymer Chemistry [email protected] http://www.itcp.kit.edu/wilhelm/
KIT – Universität des Landes Baden-Württemberg und nationales Forschungszentrum in der Helmholtz-Gemeinschaft www.kit.edu
Introduction to Polymerscience
Prof. Dr. Manfred Wilhelm
private copy 01/2019

Content
1 Introduction 1 1.1 Literature 1 1.2 Definition, materials 3 1.3 Definition, polymers 3 1.4 History and nomenclature
12
2 Polymer chemistry 13 2.1 Molecular architectures 13 2.2 Separation/classification of polymers into classes 16 2.3 Typical monomers, polymers 19 2.4 Synthesis 28 2.5 Carothers equations 30 2.6 Kinetics 31 2.7 Size distribution in linear polymers for step reaction 34 2.8 Chain growth reaction, e.g. radical 38 2.9 The ceiling temperature 42 2.10 Suspension polymerization 43 2.11 Emulsion polymerization 44 2.12 Ionic polymerization 45 2.13 Anionic polymerization 47 2.14 Kinetics and molecular weight distribution of ionic polymerization 49 2.15 Copolymers 54 2.16 Coordinative polymerization 57 2.17 Constitution, conformation and configuration isomers 58
3 Polymer physics, physical chemistry 62 3.1 The lonesome chain 62 3.1.1 End to end distance, contour length 62 3.1.2 Radius of gyration 64 3.1.3 Random-walk and Gaussian chain 65 3.1.4 Entropy-Elasticity, basic idea 68 3.1.5 Deviation from simple-statistics for end to end distance 70 3.1.6 Kuhn segment 72 3.1.7 Persistence length (“how stiff is a polymer”) 73 3.2 Polymer physics of melts 75 3.2.1 The reptation model 75 3.2.2 The amorhous state 79 3.2.3 The crystalline state 88 3.2.4 Kinetics of crystallization 100 3.2.5 How to reach 100% crystallinity in a solid polymer
102
4 Molecules and characterisation 104 4.1 Distribution of molar mass and determination of molar mass of polymers 104 4.2 Experimental determination of molecular weight and distribution 108 4.3 GPC, gel permeation chromatography 110 4.4 Ultracentrifuge 113 4.5 Light scattering of polymer solutions 116 4.6 IR-Spectroscopy 126

4.7 Mass spectroscopy 135 4.8 NMR-spectroscopy
149
5 Engineering properties 160 5.1 Mechanical properties 160 5.2 Dielectric properties 166 5.3 Processing of thermoplast; extrusion, injection molding, calendaring
169
6 Special topics 6.1 Polyelectrolytes 176 6.1.1 Definition, examples 176 6.1.2 Theory: Poisson-Boltzmann, Debeye-Hückel, Skolnick-Fixmann-Odijk 177 6.1.3 Experiments 183 6.1.4 Application: super absorbing polymers (SAP) 184 6.1.5 Application: oil production 187 6.1.6 Conclusion polyelectrolytes 189 6.1.7 Literature, polyelectrolytes 1906.2 Spatially heterogeneous systems, e.g. blends or blockcopolymers 191 6.2.1 Definition 191 6.2.2 Why does the introduction of heterogeneity make sense? 191 6.2.3 How can heterogeneity be achieved 192 6.2.4 Theory of mixing, Flory-Huggins theory 196
7 Appendix 203
Staudinger - Nobel Lecture, December 11, 1953 Ziegler - Nobel Lecture, December 12, 1963 Natta - Nobel Lecture, December 12, 1963 Flory - Nobel Lecture, December 11, 1974
De Gennes - Nobel Lecture, December 9, 1991

Material classes, development and history of polymers 1
1 Introduction
1.1 Literature:
1) History:
- Polymers, The origin and growth of a science, Herbert Morawetz, Dover Pub.
1985
2) Introduction:
- Große Moleküle, Hans-Georg Elias, Springer 1985
- Introduction to polymers, R.J. Young, CRC Press 1991
3) Chemistry:
- Grundriss der Makromolekularen Chemie, Bruno Vollmert, E. Vollmert-
Verlag 1988
- Makromolekulare Chemie, Bernd Tieke, VCH 2005
- Makromolekulare Chemiei, Lechner, Gehrke, Nordmeier, Springer 2003, incl.
CD
- I.M.G. Cowie, V. Arrighi, Polymers: Chemistry and Physics of modern
Materials, CRC Press, 2008
4) Polymer and engineering:
- Material science for Polymers for engineers, Oswald/ Menges, Hanser 1995
- Polymermechanik, Schwarzel, Springer 1990
5) Encyclopaedia, dictionary:
- Concise, Encyclopaedia of polymer science and engineering, Kroschwitz
(editor) Wiley 1990
- I-IV von H.G. Elias, Chemie + Physik + Industrie + Anwendung,Wiley VCH
6-te Edition 1999-2003
6) Physics:
- Physik kondensierter Materie, G. Strobl, Springer 2001
- The physics of polymers, 1995, G. Strobl, Springer 1996
- Polymer physics, U.W. Gedde, Chapman & Hall 1996
7) Characterisation:
- Principles of instrumental analysis, Skoog, Leavy, Saunders College
Publishing 1992
- Polymer Charakterisierung, Arndt/ Müller, Hanser 1996
- Polymer characterisation, B.J. Hunt, M.I. James, Blackie academy 1997
i Title is misleading; covers very detailed all aspects, in German.

Material classes, development and history of polymers 2
- Polymer characterisation- physical techniques, D. Campell, R.A. Pethrick, I.R.
White, Staley-Thornes 2000
- Spectroscopy of polymers, J.L. Koenig, Elsevier 1999
8) Polymer technology:
- Handbuch der technischen Polymerchemie, A. Echte, VCH 1993
9) Internet:
- http://scholar.google.com
- www.vke.de (Verband der Kunststofferzeugenden Industrie)
- www.pslc.ws/macrorg.htm
- web.umr.edu/~mwlf/

Material classes, development and history of polymers 3
1.2 Definition, materials
- Materials are synthetically or biologically built chemicals that we generally use
due to there physical, dominatingly mechanical and chemical properties,
specifically their 2 and 3 dimensional structure.
- As a consequence: material science is inherent interdisciplinary: biology,
chemistry, physics and mechanical engineering are interacting.
- Currently, not only mechanical properties (module, T-stability,) chemical
resistance), but also “functional” properties are more and more developed,
investigated and applied. For example: magnetic ( storage of data), electric
( computer, CPU), chemical (medicine) or optical functions are added
- Materials can be classified as follows
i. Metal (high, low density, conducting, semi-conducting, reactive, …)
ii. Glass (organic, inorganic), frozen liquid
iii. Ceramics (in general: inorganic, but crystalline)
iv. Polymers (organic materials)
1.3 Definition, polymers:
Polymers are high molecular weight synthetically or biologically built chemical
structures that contain at least one repeat unit that is covalently bound and repeated.
The dominant elements are C, H, O, N, Cl, F, S, P, Si,…
How much materials do we need (world wide)?
1990 => 95 10 people if one 1t per person and year,95 10 t
Steel: 9 kgt10 200year person and year
Polymer: 6 kg60 10 12 person and yeart
year
Al: 615 10 3kg person and yeart year
Note: Germany 50-70 kgperson and year
Factor 15 steel polymers, but: 3Feg7 8cm
, , 3Polymerg1cm

Material classes, development and history of polymers 4
Polymers 2002: 6 t227 10 year
Note:
There is an empirical correlation between price and production in general
-0.4price ~ production
Factor 10 in production factor 2-3 in price

Material classes, development and history of polymers 5
Source: Schwarzel 1990

Material classes, development and history of polymers 6
Use of polymers in our environment:
Nature: construction, storage, clothing, protection
Food: carbon hydrates, proteins
Packing: bags, storage tanks (PE for H2, full cell in cars)
Traffic: super plastified concrete (1-5% polymers), security glass, belts, airbag, oil
additives
Clothes: wool, cotton, gore-tex® (teflon), polyester
Cosmetics: hairspray
Hygiene: super absorber, surfactants
Medicine: dialysis, contact lenses, controlled drug release
Optics: light conducting fibres, organic LED (O-LED), NLO (non linear optics)
Electronics: Photoresists, primer for UV-etching, electrically conducting polymers
What do we want?
- mechanical => high E, G module, low compressibility, M
- low weight, low density
- low price, P
(For non functional polymers these properties are very important)
Additionally:
- inert
- non toxic
- temperature stability
- dielectric, magnetical, optical properties
- easy to form and shape !
If we assume that the importance for a specific applications scales with a scaling exponent
(e.g. , ,a b cM P )
We might generate a “figure of merit” (Kenngröße), F
; , ,a
c b
MF a b c 0
P
If price does not matter (e.g. space application, formula 1, professional sports)
a
b
MF

Material classes, development and history of polymers 7
Do not take this too literately!
Typical production (source: Nachrichten aus der Chemie 2004, p. 324):
year 2002: 6 t227 10 year , 620 10 tGermany year
polymer Production 6 t10 y growth
PE 56 (24%) 5,5%
PP 32 (14%) 9,1%
PET 32 (14%) 8%
PVC 20 (12%) 4,7%
In case we have an average price of 1.5 €kg €:PE 1 kg
Production 9 €330 10 year
Assuming a business volume of year and person€t200 in a company
. 61 65 10 people in primary production!
average production per person: yeart150
Typical factory: PE, PP, worldscale factoryabout 1000 people per factory
yeart100000 1000000
Why investigating materials “Knowledge is power” Francis Bacon (1561-1626)
Two examples:
1) Iron at times of Nebukhadnezar in bible (1125-1104 b.Chr.)

Material classes, development and history of polymers 8
Daniel 2

Material classes, development and history of polymers 9

Material classes, development and history of polymers 10
2) PE in WW II
Source: Morawetz

Material classes, development and history of polymers 11
Read this every year in “Nachrichten aus der Chemie“ or “Macromol. Chem. Phys.“ around Feb. or March

Material classes, development and history of polymers 12
1.4 History and nomenclature:
Polymer: greek: poly: many; meros: parts
Plastic: Polymer + additives
Natural rubber: Kautschuk (German) from Cahuchu = “caa” (wood) and “o-chu” (tears)
in the native South American language. The word rubber originates
from first use to erase lead pencil marks from paper by rubbing
Rubber trees first mentioned 1516
Short history:
5000 b.Chr. cotton (mexico)
3000 b.Chr. silk (china)
2000 b.Chr. bitumen (sealing of boats)
1500 a.Chr. rubber
1832-1838 F. Lüdersdorf + Charles Goodyear => vulcanisation of rubber via sulphur
1870 cellulose nitrate by Isaak Hyatt and John Hyatt films, packing, first thermoplast
1907 Bakelite, Leo Baekeland, phenol-formaldehyde resin, fist synthetic thermo set
1924 Hermann Staudinger (Freiburg, Germany) proposed polymers as linear chains built of
covalent bonds; this concept was first heavily criticized by colleagues, Nobel price 1953
1930-1940 Wallace Carothers at Du Pont worked on Polyester and polyamids (Nylon®)
1930-1950 H. Staudinger, Nobel price in 1953
1961, Ziegler Natta, Nobelprice polyolefin catalysis
1974 Nobel price for Paul Flory (chemistry) he worked on physical chemistry
1991 Nobel price for De Gennes, liquid crystals and polymers (reptation theory)
2000 A. Heeger, A.G. Mac Diarmid, H. Shirakawa,, Conducting polymers
2005 Grubbs, Chauvin, Schrock, Metathese

Polymer chemistry 13
2 Polymer chemistry
2.1 Molecular architectures
One monomer:
Linear
Comb
Branched
Network
Different topologies (conectivities)
Typical molecular weight: . . g50 000 500 000 mol ,
if e.g. made of 2CH 35 000. units 2CH units kg500 mol .
“In a plastic bag their wont be two molecules with the same architecture.”
1 5A 1 4 A 1 3A
C C C C C C
, , ,
; ; i
C
C
C
.1 5 A
1 5 A 35 000 units
5 400nm 5 4 m
. ,
, . contour length
i 101A 10 m
Polymer chemistry 14
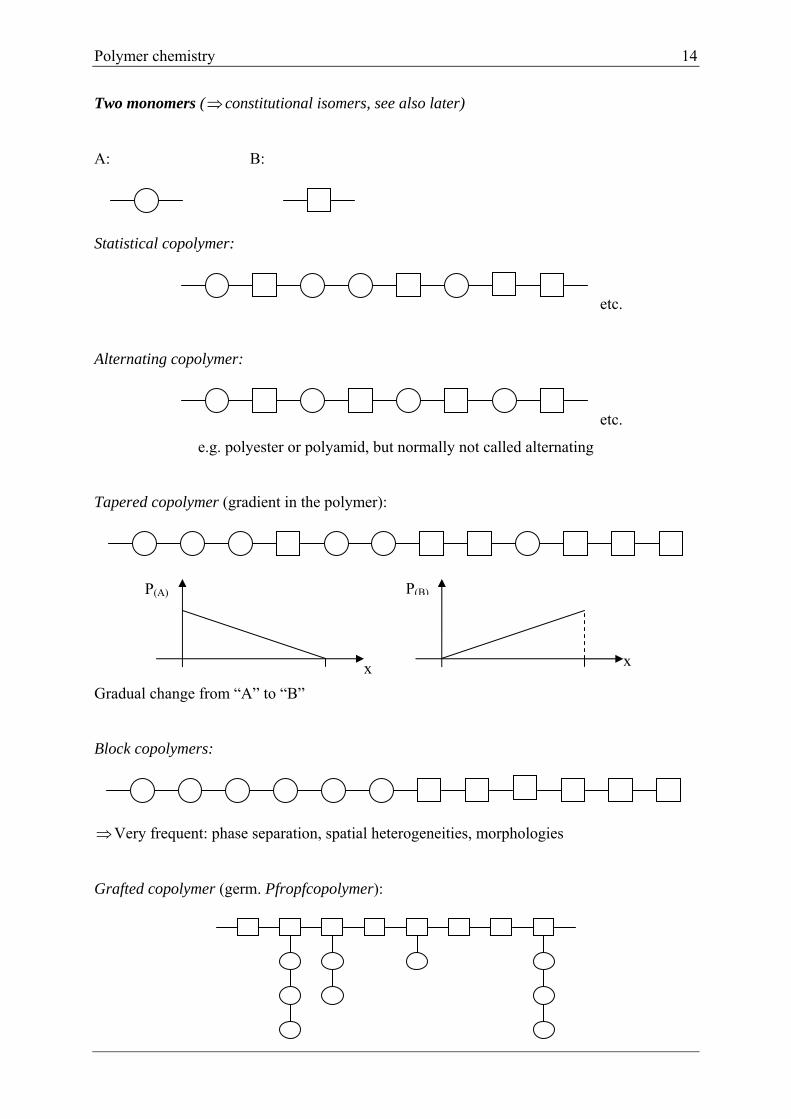
Two monomers ( constitutional isomers, see also later)
A: B:
Statistical copolymer:
etc.
Alternating copolymer:
etc.
e.g. polyester or polyamid, but normally not called alternating
Tapered copolymer (gradient in the polymer):
Gradual change from “A” to “B”
Block copolymers:
Very frequent: phase separation, spatial heterogeneities, morphologies
Grafted copolymer (germ. Pfropfcopolymer):
P(A)
x x
P(B)

Polymer chemistry 15
both topology and morphology!
Blend:
In general, blends are phase separated, typical size is
A
B B
1 10 m
+

Polymer chemistry 16
2.2 Separartion/ classification of polymers into classes
Polymers
Thermoplast - Linear or branched - Can be melted
rubber Thermoset (germ. Duroplast)
Slightly crosslinked network
Elastic properties, e.g. can stretch
Very mobile polymer => glass transition
Cannot melt Examples: PI (Polyisoprene)
PB (Polybutadiene)
Semi crystalline Amorphous, non crystalline
Less than 100% crystalline, typical: 20-50%
Mostly linear topology, low amount of branching
Stereo chemistry is regular
Examples: Isotactic PP (i-PP) HDPE (high
density PE)
Linear and branched structures
Irregular stereo chemistry
Examples: Polystyrene PMMA
(Plexiglas®)
PC
Strongly crosslinked, dense, 3D network
Rigid, intractable Degrades rather than
melt Examples : Phenol-formaldehyde resin Urea-formaldehyde
resins Epoxy resins

Polymer chemistry 17
Remark:
Crystalline material is generally more dense than amorphous material (typically 3-
10%), if semicrystalline material is exposed to light, scattering will happen if
crystalline size is in dimension of (wavelength of light, ca. 400-800 nm)
material is scattering, white not
transparent.
Please note! For Polymers crystallinity does not mean that the crystalline part is not mobile/ moving! Most
polymers are packed in Zick-Zack or helix shape. It can be, that the helix jumps up to
110000 s (e.g. PEO CH2 CH2 O ) at room temperature (poly-1-butene
ca. 11 s )! This motion is like a stochastical move of a screw in a threat1
The motion is not caused by a coherent move of the helix but rather by diffusive motion of a
defect along the threat.
Note: about number of different molecules for a statistical polymer
Assume:
20 different monomers (e.g. amino-acids in nature)
With ngM 100 mol (typical)
Polymerize to a polymer with ,ngM 100 000 mol
n = 1000, No, molecular weight distribution assumed, no polydispersity! Only
linear topology!
How many molecules are allowed?
1 German: Schraube in Gewinde
l
l l : size of crystallite

Polymer chemistry 18
3
3301000 1000 1000 3 1000 330 1000 1330
2 1010
20 2 10 2 10 10 10 10
if molecules carry information, basically infinite amount of information can
be encoded!
Please compare this to other large numbers:
e.g. number of water molecules on earth:
Earth surface: 2 6O 4 r r 6 10 m, (6000km)
Assume 1km H2O: h = 1000m
22 6 3 3 12 3 3 16 3
10O
V 4 r h 4 6 10 10 m 10 36 10 10 m 36 10 m
.31m 50 000mol since :n 2g gM H O 2 1 16 18 20mol mol
231mol 6 10 molecules
Therefore
,
32
2
m H O
16 4 23 46H O earthN 36 10 5 10 6 10 10

Polymer chemistry 19
2.3 Typical monomers, polymers
a) Ethylene
C CH
H H
H
CH2 CH2
Three different ways to show the same.
Polyethylene (PE)
C C C C CC C
H
H H
H H
H
H
H H
H
H
H
H H
H
CH2 CH2n
:n degree of polymerization
why not
CH2
2n ? not the chemical building block
Use:
Moulded objects, tubing, films, waste bags, electrical insulation, low dielectric loss
Several subtypes (no need to memorize):
- HDPE (High density PE)
- LDPE (Low density PE)
- UHMWPE (Ultra high molecular weight PE)
- LLDPE (linear low density PE)
- M-LLDPE (metalocane, linear low density)
Cheapest polymer, prize €1 kg (year 2005)
b) Propylene
C C
C
H
H
H
H H
H
CH2 CH
CH3
Polypropylene (PP)

Polymer chemistry 20
Use:
similar to PE, lighter, stiffer, very high growth rates in production over last 20 years
(+8 y% !)
c) Tetrafluorethylene
C C
F
FF
F
Polytetrafluorethylene PTFE
C C C C C C
F
F
F
F
F
F
F
F
F
F
F
F
or
C C
F
F
F
F
n
Use:
Mouldings or films, high temperature polymer useT 350 C , excellent electric
insulation, low sliding friction, expensive, tradename: Teflon®, also used for Gore-
Tex® membranes
d) Styrene
C
C C
C
CC
C
C
H
H
H
H
H
HH
H
CH2 CH
C C C C C C C C C
H
H CH3
H
H
H
CH3
H
H
H
CH3
H
H
H
CH3
H
H
H
relative orientation is called "stereochemistry", e.g. atactic (a-PP), isotactic (i-PP), syndiotactic (s-PP)=> later changes drastically e.g. melting point! hinders crystallisation (=> a-PP) => later
phenylgroup

Polymer chemistry 21
Polystyrene (PS)
CH2 CH CH2 CH CH2
or
CH2 CH
n
Question:
Use:
Cheap moulded objects, amorph, transparent, copolymerised with butadiene
to make high impact PS (HIPS), expanded with pentane to make foam
(styropur® => BASF)
Note: atactic PS is softening at ca. gT 90 C (glass-transition temperature Tg), if
stereoselective catalysts are used: ! !m
melt
T 260 C T 150 C
e) Methyl Methacrylates
(Ethyl Methacrylates, Propyl Methacrylates, Methyl Methacrylate) in analogy, less in
use
acrylic acid COOH ester
are called acrylates, e.g. ethyl acrylate 2
methacrylic acid COOH ester
are called methacrylates, e.g. ethyl methacrylate 3
2 E.g. ethyl acrylate (compare: sodium Na chlorid Cl ) COOC2H5
3 E.g. ethyl methacrylate COOC2H5
Why not? CH2 CH CH CH2
n/2 Head-head polymerisation

Polymer chemistry 22
CH2 C
CH3
COO CH2 CH3
poly methyl methacrylate (PMMA)
CH2 C
CH3
C
CH2 C
CH3
CO
O
CH3
OO
CH3
Question: What is the more rigid structure acrylates or methacrylates, which should
have higher Tg (“Brittle-point“)?
Use:
Amorphous, transparent sheets and tubing, more expensive than PS, airplane windows,
tradename Plexiglas®, Prespex®, Lucite®, Diakon®
f) Vinyl chloride
CH2 CHCl
Poly(vinyl chloride) (PVC)
CH2 C
H
Cl n
Use:
Records, waste water pipes (very inert), rain coat, bags for blood, floor, toys (=>
problem plasticicer e.g. dioctyl phthalate)
COO
COO CH2 CH
H5C2
C4H7
CH2 CH
C2H5
C4H7
g) Vinyl acetate
Dioctyl phthalate, (DOP)

Polymer chemistry 23
CH3 COOH
CH2C
OC
O
CH3
Hvinyl
acetic acid
CH2 CH2
Poly (vinyl acetate) (PVA;PVAc)
CH2 CH
OCO CH3
n
Use:
Chewing gum ( gT body temperature!), adhesive, coatings, copolymer to make
superplastified concrete
Remark: If ester is hydrolysed in PVAc the result is poly (vinyl alcohol)
CH2 CH
OHn
, exists also partly hydrolized (“Mowiol®” former Hoechst)
Hydrolysis, specifically called saponification in english (“Verseifung”): split of an
ester function via the addition of water into an alcohol and carboxylic acid, invers of
esterification, e.g.
CH3 CH2 CO
O CH2 CH3
+ OH2 CH3 C
O
OH+ CH3 CH2 OH
acetat ethyl acetic acid ethanol
exercise: draw PVAc and PMA [poly (methyl acrylate)], look for similarities and
differences
C C
OC
O
CH3
H
H H
n
C C
CO
H
H H
O
CH3
n
PVAc PMA
why not synthesize via 2 ?HOHH C C

Polymer chemistry 24
h) Acrylonitril
Polyacrylonitril (PAN)
C C C C
H
H
C
H
H
H
C
H
N Nn
or
CH2 CH
C Nn
Use: textile fiber, Orlon®, Acrylan®, superglue: polycyanacrylate
CH2 C
C
C N
O
O CH3
2-Cyanacrylacidmethylester
i) Ester
R1 C O R2
O
e.g.
Ethylene glycol:
OH CH2 CH2 OH
Terephthalic acid:
HOOC COOH
Poly (ethylen eterephthalate) (PET)
CH2 CH2 O C
O
C
O
O
n
Draw structure: PBT poly (butylene terephthalate)
Polyester in general:
R1 C O R2
O
n
Use: textile fibres (“polyester“ mostly PET), bottles for soft drinks,
COOH
acrylicacid
C N
nitril
CH2 CH
C N
Polymer chemistry 25
sympatex® membranes
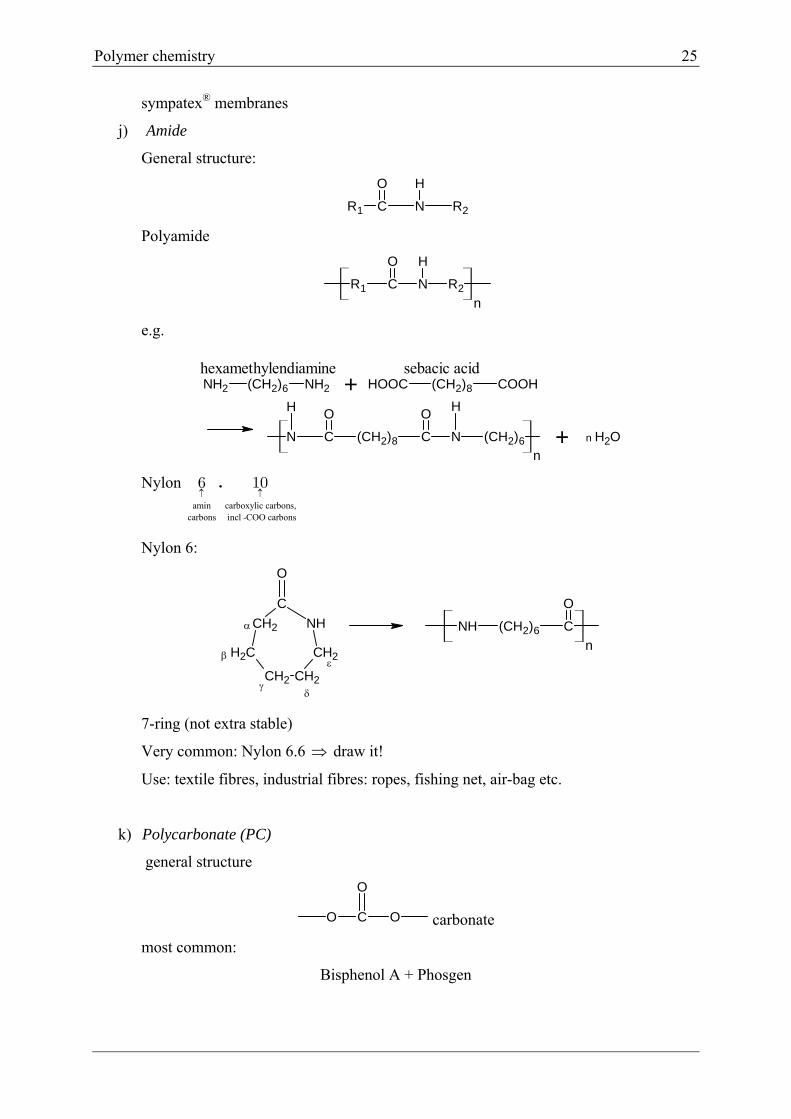
j) Amide
General structure:
R1 C N R2
O H
Polyamide
R1 C N R2
O H
n
e.g.
NH2 (CH2)6 NH2 HOOC (CH2)8 COOH
N C (CH2)8 C N (CH2)6
O OH H
n
hexamethylendiamine sebacic acid+
+ OH2n
Nylon amin carboxylic carbons,
carbons incl -COO carbons
.6 10
Nylon 6:
NH
CH2
CH2CH2
CH2
CH2
C
O
NH (CH2)6 C
O
n
7-ring (not extra stable)
Very common: Nylon 6.6 draw it!
Use: textile fibres, industrial fibres: ropes, fishing net, air-bag etc.
k) Polycarbonate (PC)
general structure
O C O
O
carbonate
most common:
Bisphenol A + Phosgen

Polymer chemistry 26
OH C
CH3
CH3
OH + C
O
Cl Cl
O C
CH3
CH3
O C
O
On
Amorphous, non crystaline, colourless, high Tg, tough
Use: like PMMA, CD’s are made out of PC.

Polymer chemistry 27
Source: Lechner 2003

Polymer chemistry 28
2.4 Synthesis
Two main types:
- step growth, polycondensation, e.g. polyester or polyamids, stable
intermediates during reaction, elimination of small molecules
- chain growth, e.g. olefines ( C C ), free radical polymerisation, no elimination of
small molecules, reactive intermediate
2.4.1 Polycondensation => step growth
Remember basic chemistry,e.g. for ester
CH3 COOH+ OH CH2 CH3 CH3 COO C2H5 OH2+
ethyl - acetat
Na+
Cl-
acetic acid ethanol
C2H5+
Na+
Cl-
chemically not correct
sodium chloride
analogy: CH3 COO-
Note:
1. water is produced => need to remove it, H , solubility, permeability!
2. products are called ester
To get a polymer we need two or more functional sites at each molecule
HOOC COOHOH
CH2CH2
OHHOOC C
O
O (CH2)2 OH OH2+ +
Simplified chemistry:
+ +
each is a bifunctional monomer => linear polymer: bifunctional monomer needed
During reaction, oligomers (2<n<50) and any polymer can react with each other, e.g.
+ +

Polymer chemistry 29
In case of +
we need equimolar mixtures!
Small access will stop the polymerisation, since all endgroups will either be hydroxy (-OH)-
groups or carboxylic (-COOH) acid!
Solution to this problem:
n-1
+n x n x
Chemically:
CH2OH COOH
n
-hydroxy-carboxylicacid
”end”
Reaction:
monomer polymer + H2O
We need to remove condensate (e.g. water, methanol,…)
Ringformation is possible
or
Note:
In early “polymer days“ people assumed that polymers where dominately ring molcules.

Polymer chemistry 30
2.5 Carothers equations
p : extend of reaction or probability to react
number of groups reactedThis can be determined easily, with spectroscopy or titration,
number of groups initially
therefore important quantity
p
N0: number of groups initially
N: number of groups at time t
00
0 0
N N Np 1 N p 1 N
N N
N: number of molecules present after a certain extend of reaction
01 number of groups intially
1 Number of groups at time t n
NX
p N
nX : number averaged degree of polymerisation
n
1X
1 p
Carothers equation
e.g. if during a polyester condensation 98% of the groups have reacted (in “normal” chemistry
one would say “quantitative”) the degree of polymerisation is nX = 50 , the product is hardly
a polymer!
Consequence: e.g. very pure chemicals, very defined and quantitative reaction with no side
reactions are needed.

Polymer chemistry 31
2.6 Kinetics
Definition:
Kinetics studies the rate of chemical reaction
e.g.
2R COOH HO R R COO R H O
d HO R d R COO Rd R COOH
dt dt dt
x : concentration of chemical x in 3mol
mor mol
l
The rate of the chemical reaction must be a function of probability ( concentration) of the
chemicals that take place in the time determine step (= elementary reaction).
Please be aware, that the elementary reaction is frequently not the same as the stoichometric
reaction equation!
Furthermore, it is often assumed that the reactivity of the reactive groups do not change as the
polymer is formed.
e.g. elemantary reaction
+2
He.g. R-COOH
These moleculesls have to meet
R-COOH+H-O-R + cat. R COO R H O cat.
.
molecules that have to meet, "educts"the "change" of concentration or rate of reaction
d R COOHk R COOH R OH cat
dt
k : rate constant for reaction
Here reaction is catalysed via H+, protons e.g. coming from R-COOH RCOO- + H+
If no other catalyst is added self-catalyzed
2d R COOHk R COOH R OH
dt
If R COOH R OH is given, same number of functional groups and
R COOH c
3dckc
dt (differential equation)
3
dckdt
c
Integration from ; 0t 0 to t t c c to c

Polymer chemistry 32
0
c t
3c 0
1dc k dt
c
0
c2
c
1c kt
2
202 2
0
1 12kt c
c c
2
2002
c1 2ktc
c
Using Carothers equations:
0 0N 1 c
N 1 p c
20 2
12ktc 1
1 p
plot
If plot gives us a linear relation self catalysed
For same reaction a catalyst is added, so [cat.] = const.
We have:
2
`[ ] [ ][ ,]
'OH COOHcat k
dck c
dt
2
dck dt
c
0
c t
2c 0
1dc k dt
c
0
c1
cc k t
00
1 1k t c
c c
time
21
1 p
e.g. via titration

Polymer chemistry 33
0 00
c c 11 k tc
c c 1 p
0
11 k tc
1 p
Plot:
If the plot gives us a linear relation the reaction is not self catalysed.
time
11
1 p

Polymer chemistry 34
2.7 Size distribution in linear polymers for step reaction
i 1i H O R COOH H O R COOH R COOH[ ]
For (i-1) ester linkages
If “p” is probability that reaction has taken place, then
1- ester bond : p
+ 2- ester bond : 2p
+ 3- ester bond : p3
+ 4- (i-1)- ester bond next to (i-2) ester bond : i 1p
Last bond has not reacted probability: (1-p)
i 1
iP p 1 p
If N polymer molecules ( monomers) are present, the number of molecules Ni with length i
is given by:
i 1i iN P N N p 1 p (A)
But N can not be measured, only N0 is known.
Carothers (see chapter 2.5):
0N N 1 p
We can say
2i 1i 0N N p 1 p (1)
There are two different and common ways to quantify the average molecular weight:
a) number average Mn
b) weight average Mw
Mn: we “ask” each polymer molecule: “what is your mass?” and built the average, number
average of polymer molecules
Mw: we “ask” each monomer: “what is the molecular weight of your polymer”
Example:
2 Polymers
n 4
n 1

Polymer chemistry 35
n
4 1M 2 5
2,
(1st momentum of the distribution)
w
4 4 4 4 1 17M 3 4
5 5,
(related to 2nd momentum of the distribution)4
w nM M , this is a general statement! (memorize!)
i
mass of molecules of length iw
total mass of all molecules , weight probability
M0: mass of monomers
i 0 i
i0 0 0
N iM i Nw
N M N
(2)
(1) in (2)
2i 1iw ip 1 p (3)
For reaction p=0,99
Knowing Ni and wi we can determine Mn and Mw.
i in
i
N MM
N
If the sample contains N-molecules iN N and i 1iN N p 1 p , see eq. (A)
4 Side comment
P(x) P x 1 , x xP x dx ; 2 2x x P x dx ; n nx x P x dx , nth moment
standart deviation 2 2 2x x ; 2
w
xM
x
100 200 300 400 500 600 700
i0w 00
1
2
3 i ww x
nx

Polymer chemistry 36
iM
i 10 i 1
n 0
Np 1 p i MM M 1 p ip
N
In mathematical textbooks we find (e.g. Bronstein)
i 1
2i 1
1ip
1 p( )
( ) if p 1
0n 0 2
1 MM M 1 p
1 p1 p
nn
0
M 1x
M 1 p
See Carothers!
iM
2i 1w i i 0
2 2 i 10
M w M iM ip 1 p
M 1 p i p
We find in math textbooks
2 i 1
3i 1
1 pi p
1 p
2
w 0 03
1 p 1 p 1 pM M M
1 p1 p
ww
0
M 1 px
M 1 p
Weight averaged degree of polymerisation
Often the ration of w
n
M
M is used to characterize the polydispersity (width of the molecular
weight distribution) sometimes called “heterogeneity index”, or PDI, polydispersity index
0w
n
MM1
M
1 p 1 p
1 p 0M1 p
Memorize: For ideal (full) step reaction w
n
Mp 1 2
M
Note:
Anionic polymerization
w
n
M1 03 1 2
M. .
Industrial samples up to
w
n
M10 20
M (starting from 1.03)

Polymer chemistry 37
In case we use functionalities f greater than f 2 we can generate branched and networks
(networks only if reactants are more than difunctional) structures. Functionality and
concentration are the main control parameters, e.g.
Monomers
Interpenetrating networks (networks which were polymerized in another network), IPN
Branched:
Only if one reactant is more than difunctional.
CH2
CH
CH2
OH
OH
OH
glycerol
f=3
OH
CH2
C CH2CH2
CH2
OHOH
OH
pentaerythritol
f=4

Polymer chemistry 38
2.8 Chain growth, e.g. radical
Addition reactions are conducted in three steps
1) Initiation via special initiator ( e.g. ester formation)
2) Propagation of reactive species ( e.g. ester formation since there every step leads to
stable molecules)
3) Termination, side reaction of reactive species
Monomers contain double bonds
C C
R1
R2
H
H
needs space reactive species "attack" from this side
Examples:
CH2 CH2
CH2 CH Cl
CHCH2
CH2 CH
O C
CH3
O
CH2 CH
C N
CH2 C
CH3
CO
OCH3
CH2 C
Cl
Cl
Ethylene:
Vinyl chloride:
Styrene:
Vinyl acetate:
Acrylnitril:
Methyl methacrylate:
Vinylidene chloride:
PE
PVC
PS
PVA
PAN
PMMA
PVDC
The three steps of the addition reaction
Initiaton via radicals
A radical is a reactive species with an unpaired (paramagnetic) electron as denoted R .
In chemistry a line e.g. C C is a symbol for two electrons C C
Initiators form radicals in a controlled way via heat or electromagnetic radiations (e.g. light).

Polymer chemistry 39
Examples:
Peroxy or azo components
C O O C
instable
C
O
O O C
Oe.g.
Benzoyl peroxide
C
O
O O C
O
CO2C
O
O +2x
conjugated system
makes radical more stable
2x
Azo:
N N
R R
N N
R
R
cis trans
Compare
N N (very stable) preformed
e.g.
CH3 C
CH3
C
N N
N
C
CH3
C
CH3
N
AIBN (2, 2 Azoisobutyronitril)
CH3 C
CH3
C
N N
N
C
CH3
C
CH3
N
T
hlight or
heat
CH3 C
CH3
CN+ N22x
very stable
peroxy:
h v

Polymer chemistry 40
Propagation:
After the initiation I I 2 I active radical
1I M IM
If M is
CH2 CR1
R1
There are two possibilities
CH2 CR1
R1
I CH2 C
R2
R1
C
R1
R2
I
H
H+I
more probable
1) size
2) radical is stabilized
1 2
not index degree of
polymerisation
I M M I M typical time 1 10ms “turn over rate“.
Again two possibilities for next step
I CH2 C
R1
R2
CH2 C
R2
R1CH2 C
R1
R2
CH2 C
R1
R2
I
CH2 C
R1
R2
C
R1
R2
CH2I
+head to tail (more probable)
head to head
Termination:
- via combination:
R1 CH2 CH
X
CH CH2 R2
X
+ R1 CH2 CH CH CH2 R2
X X
Head to head

Polymer chemistry 41
- via disproportion:
R1 CH2 CH
X
CH CH2 R2
X
+ R1 CH CH
X
+ R2 CH2 CH2
X
can act as macromonomer
The kinetic equation of the three different processes (initiation, propagation,
termination) can be analysed and lead to the following expression Literature.
nM M , the more monomers the longer the polymer
n 1
2
1M
I the less initiator, the longer the polymer
n 12
MM
I
[I] : Initiator concentration, why inverse?
[M] : monomer concentration
The distribution is
w
n
M1
M
Where rate of propagation
rate of propagation + rate of all other reactions
If prop restk k then 1 and w
n
M2
M , identical to step growth reaction
Auto acceleration
If reaction growths/ continuous, polymer gets less mobile, solution becomes more
viscous, the active centers do not meet any more rate of termination is reduced.
Monomer and initiator still the same mobility. Reaction takes place normally (heat
production exothermic!)
As consequence: catastrophic failure (e.g. explosion, called Trommsdorf-Norish
effect or gel effect)
Solve the problem:
− Dilute solutions, stop or slow down reaction chemically
− Emulsion or suspension polymerisation
Inhibitors and retarders:
Retarder: slow down reaction e.g.
NO2
for

Polymer chemistry 42
Inhibitor: Stops reaction after consumption normal rate. Used for storage and transport,
removed prior reaction (e.g. via distillation or addition of extra initiator)
2.9 The ceiling temperature TC (=> recycling, e.g. PMMA)
In case we have reaction
(1) pki i 1M M M , for all i
pk : Rate of polymerization
We can also loose a monomer
(2) dpki 1 1M M M , for all i
dpk : Rate of depolymerization
For (1) we have the kinetic equation
p i
d Mk M M
dt
For (2) we have
dp i
d Mk M
dt
If d M
0dt
dp pk k M
The constant kdp and kp are temperature dependant, [M] is assumed to be pure monomer.
These conditions define the ceiling temperatuer
CT ceiling temperature:
time
reaction Normal, h t
retarder f t a h t
inhibitor f t h t t
consumption inhibitor

Polymer chemistry 43
CT : Maximum temperature where polymer can be formed thermodynamically at dilute
solution.
Examples TC:
Methylacrylate 493 K 120 C (recycle via destillation!),
styrene 583K,
methylstyrene 334 K 61 °C
To reduce the problem of heat transfer two specific ways of radical polymerization are shown.
1) suspension polymerization
2) emulsion polymerization
2.10 Suspension polymerization
E.g. styrene, vinyl chloride, methyl methacrylate
Monomer droplet: size: !10 m 5mm
Shape: spherical pearl polymerisation
Initiator is soluble in monomer droplet
- stabilizer change surface tension and reduce coalescence, e.g. special polymers at
interface as surfactants
- reaction is “normal” and water cooled, high surface area helps to get rid of heat, no
problem if droplets become viscous as long as suspension can be pumped
- fire-fighter built in!
- in real life discontinuously acting reactors (up to 3200m )
Example:
Brita Filter®: pearl polymerized + sulfonated polystyrene (sulfonated for ion exchange) +
active carbon
H2O
Monomer droplet

Polymer chemistry 44
2.11 Emulsion polymerization
At the beginning:
- monomer droplet, 1 m ca. 103
droplets10cm
- empty micells, built of 100-1000 surfactants ca.
18 83
micells10 monomer droplet, factor 10cm
- monomer in water + micells, factor 100 more than pure water
- water soluble initiator starts reaction (e.g. K2S2O8)
22 8 4S O 2SO
The addition of monomer reduces solubility enters micelle
- In the micell, all the monomer will be polymerized and reaction continues via
diffusion of new monomer into micelle (shrinks monomer droplet). Typical size of
product 100 300nm
- Solid content can be up to 80% (multimodal distribution!)
I
I
Empty micell Micell + monomer
Micell + polymer
Called Latex
Monomer droplet with surfactant (rare)I : Initiator, water soluble : Monomer, partly water soluble
: surfactant (germ. : Tenside)
Charged head

Polymer chemistry 45
2.12 Ionic polymerization
Generally subdivided into
cationic (positive charged)
anionic (negative charged)
general scheme:
anionic:
1I M IM anions start reaction, fast
n n 1IM M IM growth, n 1
Cationic:
1I M IM cations start reaction, fast
n n 1IM M IM growth, n 1
Generally:
- type of reaction depends on initiator, monomer and solvent. E.g. solvent can stabilize
the ions and via dielectric constant, changes energy of the separated ions
- often rapid reactions, high degrees of polymerization via low temperatures
- control of stereochemistry ( isotactic, syndiotactic, 1.2 addition; 1.4 addition cis or
trans)
- reactive ends repel each other
less side reactions
very stable
“living” polymerization
Example for cationic polymerization
CH2 C
H
CH3 C+
H
H+ClO4-
ClO4-+
perchloric acid,
strong acid
ion pair
In principle three types of initiators exist:
a) Proton donating molecules, acid
b) Electron accepting molecules, “Lewis-acids” e.g. AlCl3
c) Positive charged carbons, “carbenium” salts
n mCoulomb!
IM IM

Polymer chemistry 46
Technical important system:
AlCl3 + C
CH3
CH3
CH2 C CH2
CH3
CH3
n
iso-butylene
4xC+1doublebondCH3
CCH3
R1
R2
polyisobutylene
CCH3
CH3
OH
H
CH3 CH2 CH2 OH
e.g. isopropyl alcohol (after shave)
propanol:
Rubbery material trade name: Oppanol® (BASF)

Polymer chemistry 47
2.13 Anionic polymerization
I M IM
In case we would allow any 2H O or 2O we would get
2cannot start reaction again
H O IM IM H OH
2
cannot start reaction again
O IM I M O O
very, very clean reaction conditions are needed!
Initiation scheme:
I- C C
R1
R2
H
H
to attack, we would like to have not to much electron density in the double bond to reduce Coulomb repulsion!
Typical monomers:
CHCH2styrene:
butadiene:
isoprene:
CH2
CHCH
CH2
CH2
C
CH3
CHCH2
Typical initiators:
Na, K (BuNa –Werke Leuna: Butadien + germ. for sodium
Natrium )
The product depends a lot on the solvent, example butadiene:
Counterion Solvent Cis Trans 1,2
Li hexane 0.35 0.58 0.07 unpolar solvent 1,4
Li THF 0.06 0.06 0.88 polar solvent 1,2
Na THF 0.06 0.14 0.80
1,4

Polymer chemistry 48
Butadiene:
1,4 cis
1,4 trans
1,2
n
n
n
n
n
n
For rubber tires we need: cis-1,4-polybutadiene

Polymer chemistry 49
2.14 Kinetics and molecular weight distribution of ionic polymerisation
(copied from lecture Prof. Sillescu)
Assume:
- no termination reaction
- concentration of ions is constant and equal c0
- reaction constant (rate) independent of molecular weight
C C
H
H
H
C C-
H
H
H
C
H
H
I-
I
k
2monomerinitiator
M M I living polymerisation
k2 3 M
k3 4
kn n 1
M M M c c
M M M
M M M
I
assume:
2 3 Mc c c c const, , , ... .I
12 0 3 0
00
0 2 31
... 0
... for all t
nt
I t
I ni
c c c
c c
c c c c c
II I
2I
2I
M
M M 2
2
dck c c kc
dtdc
k c c k c cdt
dck c k c
dt
M
ktI 0
k c const k
pseudo first order, solution:
c c e
.
for c3 we find:
33 2
dck c k c
dt
In general
nn n 1
dck c k c
dt in math differential equation set of Kolmogoroff

Polymer chemistry 50
Stepwise solution for this set of differential equations, including induction prove
kt22 I I 0
dck c kc c c e
dt
kt22 0
dck c k c e
dt , (I)
“Ansatz”
kt
2
to be found
c t U t e
kt kt2dc dUe U k e
dt dt in (I)
2
kt kt kt2 0
c
dUe k U e k c k c e
dt
0kt kt ktdU
e k c e edt
0
dUk c
dt
0
integrationconstant
U t k c t
In “Ansatz“
kt2 0c t k c t e
2c t 0 0, see assumptions
kt kt2 0 0c t k c t e c kt e
!
!
33 2
kt33 0
kt3
2kt
3 0
3kt
4 0
dck c kc
dtdc
k c kc kt edt
c U e Ansatz
ktc c e
2
ktc c e
3
Assumption:
n 1kt
n 0
ktc c e
n 1 !

Polymer chemistry 51
nkt
n 1 0
ktc c e
n !
, (II)
To be put into:
n 1n 1 n
dck c k c
dt
, induction prove in maths (III)
Derivative of (II):
n 1ktn 1
0 n 1
n k tdcc k e kc
dt n n 1 !
(product rule)
In (III)
n 1kt
0 n 1 n 1 n
n k tc k e k c k c k c
n n 1 !
!
n
n 1kt
0 n
c
k tk c e k c
n 1
The probability of a molecule to have a degree of polymerization “n“ is as follows:
n nn
0n
i 1see "assume"
c cw
cc
n 1ktn
n0
ktcw e
c n 1 !
Poisson distribution, special case of binomical distribution, only characterized by one
parameter (Gauß 2: , )
Average in general:
P x dx 1
x x x P x dx , first moment in math textbooks
2 2x x P x dx , second moment
n nx x P x dx , nth moment
n 1 n 1kt kt
n nn 1 n 1 n 1
kt ktP n nw n e e n
n 1 n 1! !

Polymer chemistry 52
2 3
2 3
2 3
2 3
21 3 4 ...
1! 2! 3!side calculation:
1 ...1! 2! 3!
2 3...
2! 3!
3 421 ...
1! 2! 3!
ktn
kt
kt
kt ktktP e
kt ktkte
kt ktkte kt
kt ktkt
kt kt kt ktnP e e kte e
nP n 1 kt kt n 1 here: n n t;
n 1n 1
n n
n 1w e x
n 1 !
Number average of molecular weight
0 0 0(1 )n nM M P M n M kt
Note:
nM can be determined via colligative properties (e.g. osmotic pressure), GPC or end-group
analysis (titration or NMR).
Weight fraction gn of molecules with a degree of polymerization Pn:
n0 nIn n n n n n
nn n
0n n n nnIn 1 n 1n 1
c Mcc M W M W M
gc Mc M W MM
c
n 0 n
n 0
2w n n n n
M M Pn nM n M
1 1P n g n M W n W
M P
22n n 0
0 w W n nn
to be proofed, next pagesecond moment diveded by mean
W M MM P M M g kt 1 kt
M kt 1
22wn n 2 22
n n
1 1 ktM M W kt 1 kt 1M M kt 1 kt 1
General statement for Mw Specific for anionc

Polymer chemistry 53
If n kt 1 1
w
n
1M 1M n
, in reality: w
n
M1 01 to 1 2
M. .
Side calculation:
Assumption; to be proofed
2 2 2 +ktnn W kt 1 kt kt 3kt 1 e( ) ( )
For
n 1kt
n
ktW e
n 1 !
n 1 0 1
kt 2 kt 2
1 2 3
1 2 3kt
1 2 3kt
32 2kt
1 2 3
kt kt kte n e 1 2
n 1 0 1
4 kt 9 kt 16 kt1
1 2 3this should be equal to:
kt kt kt1 e 1
1 2 3
3 kt 3 kt 3 kt3kt e
1 1 2
ktkt e kt
1
4 kt 9 kt 16 kt1
1 2 3
...! ! !
! ! !
...! ! !
...! !
...!
..! ! !
.
Note: in textbooks you find:
n
vvP v n e
n,
!
v k expection value, Erwartungswert
v n 1 n , mean degree of polymerization since n 1
v

Polymer chemistry 54
2.15 Copolymers:
Remember ( earlier)
1) random copolymers: A A B A B B A B ...
2) alternating copolymers: A B A B A B ...
in a strict sense a condensation of e.g. dicarboxylic acid and a diol is an alternating
copolymer, but generallyA B
n is treated as the repeat unit
3) block-copolymers A A A A B B B B ...
A and B: different monomers, or different tacticity
copolymer composition:
active end, e.g. radical or charge
4 possibilities for next step (for di block copolymers)
k
k
k
k
AA
AB
BB
BA
*
*
*
*
k11 and k22: self-propagation
k12 and k21: cross-propagation
Assumption:
Reactivity depends only on last attached monomer, not on chain length or previous sequence!
Consumption of A:
n
11 21
different M
d Ak A A k A B
dt* *
(I)
Consumption of B:
22 12
d Bk B B k B A
dt* * (II)
(I) divided by (II)

Polymer chemistry 55
11 21
12 22
1Ak k
B Bd A A1Ad B B
k kBB
** *
***
(III)
Assumed steady state:
d A
0dt
* and
d B0
dt
*
New A * are created via cross-propagation from B *
21
d Ak A B
dt
**
Destruction of A * via cross-propagation to B *
12
d Ak B A
dt
**
At steady state, both must be equal
21 12k A B k B A* *
21
12
Ak A
k B B
*
*
Substituting into (III):
2111 21
12 21
2112 22
2112
Ak Bk k
d A A k B kBAkd B B
k kkk B
1112
2221
Ak B
d A A kBd B B A kk
If we define a reactivity ratio:
111
12
222
21
kr
k
kr
k
Note: 1r and 2r can be determined and found in the literature.
Then:

Polymer chemistry 56
1
2
d A A r A B
d B B A r B
Copolymer equation!
Different cases:
A) 1 2r r 1
no preference, random distribution
ideal statistic copolymer
B) 1 2r r 0
11k and 22k are small compared to cross propagation
completely alternating copolymer (e.g. polycondensation)
C) 1r 1 and 2r 1
lot of A will be incorporated, only rarely B
The Q-e-scheme
Semi empirical method to predict reactivity ratios for a pair of monomers, specifically for
free-radical polymerization.
“Q” and ”e” are measured (and tabulated) relative to styrene, where Q 1 0. and e 0 8. .
The reactivity ratios are given by:
11 1 1 2
2
Qr e e e
Qexp
22 2 2 1
1
Qr e e e
Qexp
Only estimate of 1r and 2r !

Polymer chemistry 57
Lit.: Tieke

Polymer chemistry 58
2.16 Coordinative polymerization (insertion polymerization)
Characterized via:
1) monomer is “attached“ (coordinated) to transition metal catalyst (e.g. Cr, Hf, Ti, Zr,
…)
2) monomer is added, inserted into the still attached (to the metal) polymer chain
examples:
German, PE Italy,PP
3
- polyethyleneZiegler - Natta , Nobelprice 1963 via, TiCl (typical combination)
- polypropylene
- polybutadiene
3AlEt
The coordinative polymers have often a high degree of stereoregularity. To understand this,
we have to distinguish: constitution, configuration and conformation (next chapter).
Basic idea of coordination polymerization: a transition metal can have a “coordinative”
chemical bond ( covalent or ionic)
Ti4-
CH2 Cl
Cl Cl
Cl vacancy
CH2
polymer
+CH2=CH2
This can happen timess10 100 000. !
e.g. 1g cat => 106 g polymer
in ppm range metal impurities
Oktaeder
Ti
polymer
Cl
Cl
Cl
Cl vacancy C C
Orbitals
Monomer is preoriented attached to vacancy

Polymer chemistry 59
2.17 Constitution, conformation and configuration isomers
Constitution:
If chemicals (e.g. polymer) exihibit the same sum formula, but different covalent
connectivity (“constitution”) the chemicals are constitutional isomers
Example:
1) PE PP 2 2n nC H
CH2 CH2CH2 CH
CH3n m
2) Block-copolymers: (5 5; )
3) Head-tail, head-head polymerization
To change the constitutional isomers, we have to break bonds!
Configuration:
In case the same atoms are connected, but the two molecules can not be put “on top of each
other” a configuration isomer is defined
Examples: (left hand + right hand !, same connectivity of bones, but can not be on top of each
other!)
1)
natural rubber, elastic
cis 1,4 polyisoprene
trans 1,4 polyisoprene
Guttapercha, rigid resin

Polymer chemistry 60
2)
CH3 CCH3
CH3 CH3 CH3 CH3 CH3 CH3
H H H H H H
CH3
CH3
CH3CH3
CH3
CH3
CH3CH3H
H
H
H
H
H
CH3
CH3
CH3 CH3
CH3
CH3 CH3
CH3H H
H
H H
H
iso-tactic polypropylene (crystalline)
syndio-tactic polypropylene
a-tactic polypropylene
polypropylene (all head-tail !)
zick-zack chain in plane, CH3 always above (or: always below!)
CH3 is alternating above and below zick-zack plane
CH3 is randomly above and below zick-zack plane
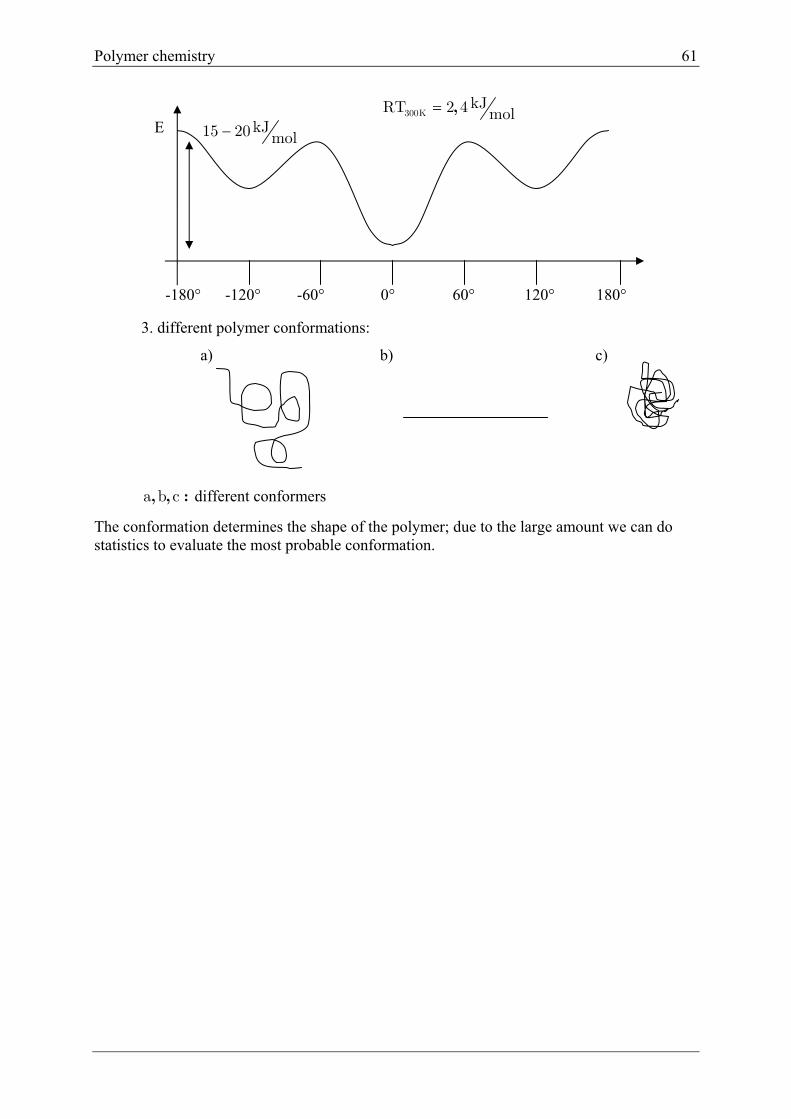
Conformation:
Single C-C bonds can be rotated with low energy (thermally). Two snap-shots of same
polymer are called conformation isomers in case they are non-identical.
e.g.
1. rotation (e.g. 1210 in 1 second) to slow down this rotation you need 1-2 Kelvin!
C
H
H
H
2.
C C
CH3CH3
H
H
HH
We expect:
Max. + min. 180° apart
360° self repeating
Every 60° Max MinMax
-CH3
(crystalline)
not crystalline, waxy
Polymer chemistry 61
3. different polymer conformations:
a b c, , : different conformers
The conformation determines the shape of the polymer; due to the large amount we can do statistics to evaluate the most probable conformation.
E
-180° -120° -60° 0° 60° 120° 180°
kJ15 20 mol
,300KkJRT 2 4 mol
b)a) c)

Polymer physics, physical chemistry 62
3 Polymer physics, physical chemistry
Topics covered:
1. – Polymers in “vacuum” or ideal solution
2. – Polymer dynamic in melt (reptation)
3. – Polymers in crystal, motion, determination of crystallinity
4. – speed of crystallization (Avrami equation)
3. 1.1 End to end distance, contour length
Assumptions:
- All lengths between atoms are the same
- All angles possible, all have same probability
- No own volume of atoms
Picture:
h 0
Since vector points with equal probability in all directions
2h ?
expectation value
N N2
i ji 1 j 1
h l l
For i j :
all same probability
2i j i jl l l l l 0cos cos
Reason:
1
2 3
N
h N+1
1l
2l Contour length:
N l , odometer along the polymer chain, total length, end to end distance if totally stretched
N
ii 1
h l

Polymer physics, physical chemistry 63
Normali-zation
P const
11 d
A .
cos cos
first moment of cos
For :i j i jI I I
; 1I
N N N
2 2i j i ij
i 1 j 1 i 1i j
h l l l l N l
2 2h N l
:2 2h h
h N l
The size h of ideal polymer growths linear with “l“ and as a square roots “N”, e.g. factor 10
in nM is only factor 3.1 in size. Typical polymers might differ factor 100 in nM and factor 10
in size, e.g nM from 410 /g mol to 610 /g mol .
-1
1
2
cos cos2
0
d
02

Polymer physics, physical chemistry 64
3.1.2 Radius of gyration (can be measured, e.g. light scattering)
Moment of inertia, see also physics books:
2 2I r dm M R
Radius of gyration of polymer chain
N
2 2i i
i 1
1R m r
M
N
2 2i i
i 1
1R m r
M
; im m for all i,
N
2 2i
i 1
1R r
N
; M N m
Without any proof (see e.g. Lechner, Gehrke p.42-43)
2 2
2 h NlR
6 6
2 h l NR Rg
6 6
The radius of gyration is factor .6 2 4 smaller than end to distance h. Both are second
moments with respect to different distribution.
i 1
il
i 1r
S ir
S
: center of gravity (“Schwerpunkt“)

Polymer physics, physical chemistry 65
3.1.3 Random-walk and Gaussian chain
Let’s assume a lattice with a 3-D grid. We
throw a dice (“Würfel”) if we get
1 move +1 in first dim
2 move -1 in first dim
3 move +1 in second dim
4 move -1 in second dim
5 move +1 in third dim
6 move -1 in third dim
The shape of this “Monte Carlo”1 simulation should tell us something about the shape of
polymers!
Poor man’s 1-D version:
At t 0 we are at x 0 we throw a coin to determine if we move left or right (50:50
probability) by l.
binominaldistribution
. ; .K N K Nn k
N Nw p q p p 0 5 q
K K
If .p q 0 5 and limN
Gaussian distribution (see maths textbooks)
;2x 2w e Nl
Where N: number of coins thrown, molecular weight
K width of distribution, standard deviation , !h Rg
length l!
Question:
In a random walk we move stochastically as a function of time, when we do the analysis of
chain conformation we do not need to consider time, so what does time stand for?
1 This type of computer simulation is really named after the gambling place!
0 1 2 3 x
.0 5 t 1
t 0 1
y
2
1
0
-1
-2
0 1 2 3 4 x
2-D picture
1
2
3
4
10

Polymer physics, physical chemistry 66
!nM , number of monomers added.
In 1-D
2
2x
21w x e
2
In 3-D:
The probabilities are the same and independent for all three directions
, ,
2 2 2
2
x y z
21w x y z w x w y w z e
2
2 2 2 2x y z r
Where 2 2Nl
Picture:
One end is fixed at origin, freely joint chain
To find chain end (“x”) in distance r (at any angle , ) we need to calculate probability to
find chain end in shell of thickness dr with volume dV
2dV 4 r dr
, ,2
2r
21w x y z e
2
2
2
r22
1w r dV e 4 r dr
2
x y
z
r

Polymer physics, physical chemistry 67
Question where is , ,w x y z 0 ? for r ! What do we know for r N l (contour
length)?
, ,w x y z w r 0 !
Logic?
100 200 300 r A
, ,w x y z e.g. if
; .4N 10 l 2 5A
100 200 300 r A
w r

Polymer physics, physical chemistry 68
3.1.4 Entropy- Elasticity, basic idea
Let’s assume:
2
2x
2w x e Gaussian statistics, polymer chain
G H T S
Boltzmann:
lnS k w (better: 1
2
lnw
S kw
)
We stretch a polymer (e.g. rubber) and see what we would expect for ,F x T
0
G H T S
ln2x 2G T S Tk e Tx
Hook !k T
Hook!
d GF T x
dx
Polymer “spring“get’s stiffer as temperature increases.
Picture:
change in temperature moves mass!
Compare combustion engine:
M M
Rubber band
1k T increase
xMass is lifted!
2k
2 1k k Hook

Polymer physics, physical chemistry 69
There must be a set up, where a temperature gradient in combination with rubber strings
must create a rotation!
Staudinger wheel:
1T 2T
Hot gas, after explosion
<
Change in temperature moves mass rotation!
Cool gas prior explosion
Rubber string
Cold, zone, soft rubber
Hot, zone rubber is rigid spring
motion
Centre of rotation below centre of mass rotation to reduce
IR-lamp
potential energy
center of mass

Polymer physics, physical chemistry 70
3.1.5 Deviation from simple- statistics for end to end distance
Remember (3.1, this chapter)
- all length between atoms are the same okay
- all angles possible, all have same probability not okay
better:
sp-orbital:
,iC C 180 0
sp2-orbitals:
C
O
Oi = 120°, = 60°
sp3-orbitals:
C i=109.5°, = 70,5°
after longer calculation (see Lechner, Gehrke p. 36-38), .P const
modification
2 2 1h N l
1
cos
cos where i180
e.g.
. .i 109 5 70 5
2 2h N l 2
,i 1 i 2
i 1l
,i i 1
il
i 2l
Since bond angle are generally fixed at 180° (sp), 120° (sp2) and 109.5° (sp3) for ,i i 1 for all ,ii
still same probability
P( )=const.
, 1 180i i

Polymer physics, physical chemistry 71
if has not same probability ( remember conformation!) the equation is further modified:
cos cos
cos cos2 2 1 1
h N l1 1
Remember: for simple statistics: all segments have no volume.
- It is trivial that this can not be correct, but what do we expect if we include chain
volume?
Simple statistics
If we include chain volume, the chain must try to avoid to overlap with itself “self
avoiding walk” this leads generally to larger sizes.
1instead of 2
35h N
- Gaussian statistics must also fail for larger elongations, since
;2xw x e w x 0 at x N l (contour length)
can’t be
Kuhn and Grün 1942:
sinus hyperbolicus
exp lnsinhk
hw h k N
N l
kl : Kuhn-segment, see next page
; :k
h
N l
-1 -1L L inverse Langevin-function
Possible, chain folds back on its own
This bond had to change direction
hh
1 2
4
3
5
6
7 8
9
1 2 3
4 5
6
7 8
9

Polymer physics, physical chemistry 72
Approximation:
2 4 6
k k k
Gauss
3 h 9 h 99 hw h k N
2 Nl 20 Nl 350 Nlln ...
3.1.6 Kuhn segment:
Remember:
2 2h N l , most simple case
cos
cos2 2 1
h N l1
For fixed , equal probable
cos cos
cos cos2 2 1 1
h N l1 1
We guess that a relation exists:
2 2sh N l
. s1 5l l 3l Typical value for sl
The unitless quantity at lim :N
slcl
Is a measure of conformative constrains
:sl apparent bond length (“s: scheinbar”)
For N we assume a Gaussian chain with different length and segment number N N
(a) 2 2sh N l
(b) *2 2kh N l ; * :N statistical segment number
( )( )
ba :
*
*
2k2
s
N l N1
N l N
*
:2
k2
s
N lk
N l
:k ratio between chemical segment number and statistically needed number

Polymer physics, physical chemistry 73
2 2k sl k l
:kl measure of stiffness
3.1.7 Persistence length (“how stiff is a polymer“)
If define a tangential unit vector t
that moves along the contour from x 0 to x N l ,
what will happen if we correlate two 1t x
and 2t x
vectors with each other?
: 1 1g 0 t x t x 1
for all 1x
: 1 1g t x t x 0
for very large, 2 1x x
We expect a strictly monotonic decaying correlation function of the shape:
plg 1 e
:pl persistence length
The angular correlation has decayed after pl to 1
e, or cos ij
1
e polymer has curved by
typically 68° (memorize: 90°)
0
1
1e
pl
1t x
N l
0
2t x

Polymer physics, physical chemistry 74
Note:
A) the persistence length can range from 1 3nm (polyolefines) to
10nm (polyparaphenylene, PPP) up to 1 2 m for rigid viruses (even with
7 8nm diameter) ”bamboo”
B) correlation functions are frequently used with time as a variable (here: space x for
persistence length)
general definition
2g t g t dt
gg t dt
Correlation time:
:c
0
g d
used: e.g. quasielastic light scattering later
C) For special cases we can approximate (see e.g. Lechner p. 40-41)
p k2 l l
:pl persistence length
:kl Kuhn length

Polymer physics, physical chemistry 75
3.2. Polymer physics of melts
3.2.1 The reptation model, De Gennes (Nobelprice 1992)
Basic idea: one dimensional stochastic process of chain along own contour, restricted by
neighbouring chain (reptate: reptile)
Simplified
Tube with diameter d, other chains are static, typical distance s for constrains of other chains
s d 30 80A
.
If we have a certain position at time t 0 , we use one-D Fick equation to describe chain
distribution probability
2
1d 2
p pD
t x
, 1-D second Fick equation
[first Fick equation:
c
D Jx
]
Solution for ,P x t in case of 1dimensional random walk:
, exp2
1d 1d
1 xP x t
4 D t 4D t
Gaussian:
2
2
x
2e
2 2r 2nDt with :n Dimensionality
Mean square displacement ( second moment) ,2 21dx x P x t dx 2 D t
Stochastic motiond
s constrain (not moving)

Polymer physics, physical chemistry 76
If we assume stochastic friction coefficient , of the polymer where this friction coefficient
is proportional to N, therefore M (molar weight)
N
: friction per monomer unit
Using the Einstein relation for 1-d diffusion
11d
kT kTD M
N
The time needed to diffuse along L, will allow a fully different conformation, so that all
memory of the other constrains is erased
;21dr 2D t t
r L contour length 1N l M
21dL 2D
2 2
31
1d
L MM
2D M
3M
The longest relaxation time in a linear, amorphous, monodisperse homopolymer is 3M .
The self-diffusion coefficient sD is given by the time to move the centre of mass by a typical
coil diameter R (3-D problem)
;2sr 2nD t n 3
2
2s 3
R MD M
6 M
2 2R Nl M
2sD M
Assuming a Maxwell model
Pa s
G Pa
G
With:
3M + chapter 5, this script
0G M
G
see rheology lecture next semester!

Polymer physics, physical chemistry 77
Molecular weight independent G for rubber plateau, given by temporary entanglements, mesh
length Me
polymer ,3M De Gennes 1971, experiment .3 4M
For low shear rates!
Non entangled polymers: 1M friction of polymer contour
Rule of thumb for flexible monomers with 2 carbons per backbone (so not true for PPP,
polyparaphenylene large persistence length)
en 100 200 monomers (200-400 backbone carbons)
- entanglement is rare event
- local correlation and parallel orientation (Pechhold, meander 1970 )
- spaghetti picture is misleading, to much free volume (to much space for “sauce”) for
melt
Me
cM logM
1
3
log 0
cM 3Me “3 fingers to hold a stick“
pivot point
log0
1 log
Slope, typically -0.8-0.9

Polymer physics, physical chemistry 78
examples
PE: g828 mol
PDMS: . kg12 3 mol
PMMA: kg10 mol
1,4PI: . kg5 4 mol
PS: kg13 mol
PIB: . kg7 3 mol
1,4-PB: . kg1 8 mol
might differ depending on literature source + definition eM
typical response for shear module as function of frequency Engineering properties (later),
chapter 5 and rheology lecture

Polymer physics, physical chemistry 79
3.2.2 The amorphous state
Or: When do polymers crystallize?
a) X is small :
CH2 CH
Xn
stereo regular semi-crystalline example: i-PP
not stereo regular amorphous example: a-PP
X is very small e.g. X: ,F OH
b) X is very large and regular: e.g.
m
n
CH
C O
O
CH2
CH3
CH2
n 11 side chains will crystallizes, how to suppress crystallisation in side chains?
How to prohibit crystallisation?
a) for random copolymers; do not crystallize or have reduced crystalline amount, if
copolymer is in the range 1-5%
b) if cooling rate is sufficient high, the freeze out of motion is faster than the
crystallisation kinetics ( see later). The amorphous parts can not rearrange to form
crystals, below the “brittle point” ( : ,gT glass transition temperature) of the
amorphous parts.
specific volume v as function of temperature
v
T mTgTPT
Problem: amorphous becomes as dense as crystalline
If .v
constT
“kinetic“ 2nd order phase transition
liquid
Liquid is 100% crystallized First order phase transition
Liquid glass

Polymer physics, physical chemistry 80
:PT if .v
constT
at pT the specific volume v of disordered frozen liquid ( glass)
would be below crystal not logic! “something” must happen before! glass transition
at gT , e.g. measured via DSC (heat flux as function of T, differential scanning calorimetry)
Typical DSC curve for semicrystalline monopolymer
Typical heating rates:
minK2 20
To low takes forever and noisy spectra
To high it takes time to crystallize
Melting enthalpy for crystals is in the range of 0.1-0.3kJg
determination of crystallinity
What influences gT ?
a) Molecule weight
If endgroups are “different” the probability to have endgroups in a certain volume n
1
M ,
therefore gT of a finite size polymer needs a correction, since gT of an infinite nM
system will not be reached
g gn
AT T
M
b) Size of “X” in
CH2 CHX
n
X: inflexible and large gT
Very small and smeared out
Diff. Heat flux
exothermic
Endoth.
gT mT
peak step
T
=> first order phase transition
step=> second order
Integral degree of crystallinity (be carefull with this analysis)

Polymer physics, physical chemistry 81
X
CH3
CH3
[ ]gT C -10 100 115 135 145
a) If “H” is exachenged by CH3
alkyl acrylates alkyl methacrylates
CH2 C
X
H
CH2 C
CH3
X
CH2 C
C O
O
CH2
CH3
H
m
n-1
C O
O
CH2
CH3
CCH2
CH3
m
n-1
Stiffer!
gT
100
0
-50
1 7
n
T 100 C
The CH3 in main chains increases rigidity,
( )2 n 1 3O CH CH acts as plasticizer for amorphous polymer. Longer side chains can crystallize, e.g. n > 11.
acrylates
methacrylates

Polymer physics, physical chemistry 82
b) Electric dipol increases gT
Empirical rule;
for semi crystalline polymers
g
m
T2
3 T
mT in °C or K? we need an energy K
Tg of blends and copolymers
Generally 2 1g gT T , the rigid polymer 2 is more “plasticized” as mobile polymer 1 is
“rigidified”.
Compare:
100
-60
30 50 mol% Cl in PE
PVC
Tg [°C]
exo 1g
T
2gT
1gT 2gT
T
Blend size > 1µm, clearly separated
Copolymer heterogen size 5nm, otherwise “homogenious“on DSC length scale

Polymer physics, physical chemistry 83
In case only one gT is detectable ( heterogeneities < 5-10nm) several equations can
approximately predict the common glass transition, most known equation is the Fox-equation
(1950)
1 2
1 2
g g g
1 w w
T T T
:iw weight fraction of polymers
Low molecular weight additions can act as plasticizer ( )gT (opposite effect exists too).
PVC is most prominent example
Most common plasticizer: DOP (dioctyl phthalate added 0-50% !)
why not n-alkyl??
C
O
O
C
O
O
CH3
CH3
CH3
CH3
Daily use example:
Extreme dry cellulose (“T-shirt”) has gT 225 C each 1% of H2O lowers gT
by more than 10°C wet ironing (steam!) afterwards T-shirt is plane and gT is increased
due to dry state. Fox-eq. does not apply since 1 polymer plus one low Mn plasticizer.
General considerations for all glass forming materials (polymers, low nM
glass formers, inorganic material)
- free volume allows motion (5-10%), T-dependant
- similarity with second order phase transition
1R 1 2R 100
1 2
1 1 1
R R R
Is R1 more influenced by
R2, or R2 more by R1?
no symmetry

Polymer physics, physical chemistry 84
- non equilibrium, time dependant (e.g. different gT for different cooling rates)
hysteresis
- kinetic theories: free volume mobility
- thermodynamic theories (Gibbs, Di Marzio 1958) lattice theory with intermolecular
contributions
- heterogeneity (1-3nm):
spatial and dynamic (time and space) solid state NMR, Prof. Spiess
WLF-equation
Dynamics as a function of temperature for polymer melts William-Landel-Ferry (WLF-
equation)
Semiempirical:
log 1 r10 T
2 r
C T Ta
C T T
:Ta shift factor
:rT reference temperature g r gT T T 100 C
T T refT
ref ref T
a
If we chose . ; .r g 1 2T T C 17 4 C 51 6K other rT (reference temperatures) are
possible, but change ,1 2C C
.
log.T
17 4 Ta
51 6K T
At T 50 C singularity Vogel-Fulcher temperature
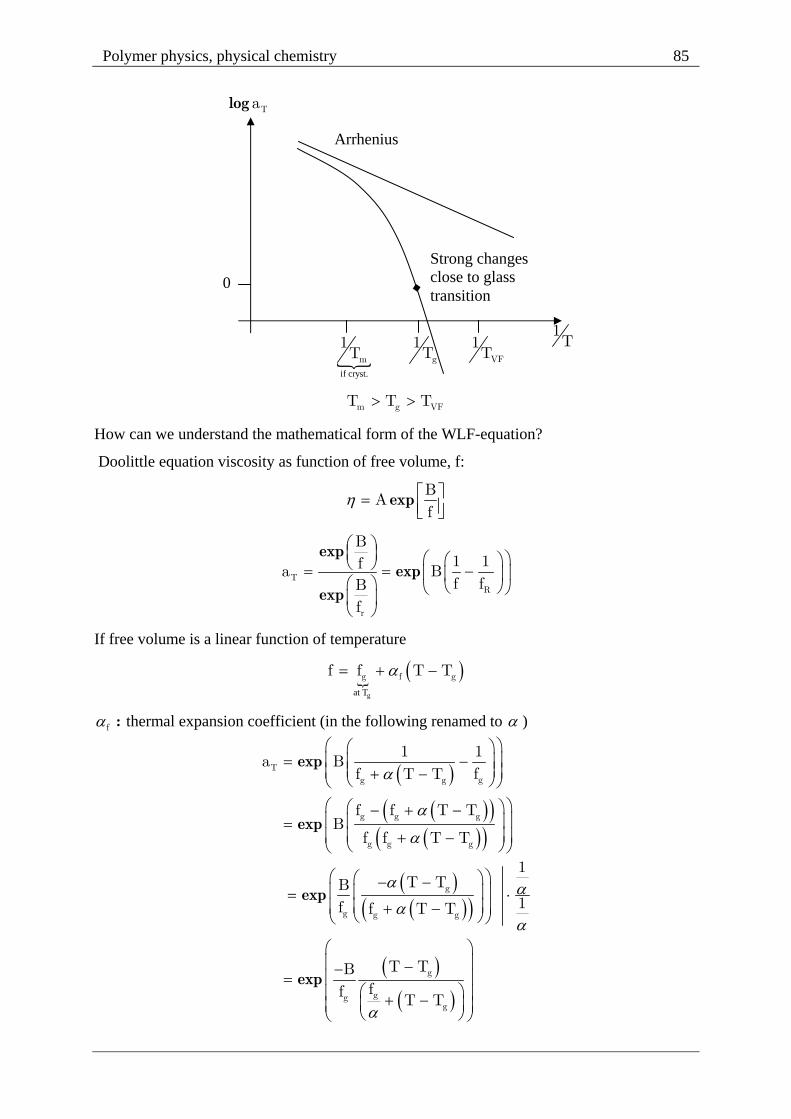
Polymer physics, physical chemistry 85
m g VFT T T
How can we understand the mathematical form of the WLF-equation?
Doolittle equation viscosity as function of free volume, f:
expB
Af
exp
exp
expT
R
r
B1 1fa Bf fB
f
If free volume is a linear function of temperature
gat T
g f gf f T T
:f thermal expansion coefficient (in the following renamed to )
exp
exp
exp
exp
Tgg g
g g g
g g g
g
g g g
g
ggg
1 1a B
ff T T
f f T TB
f f T T
1T TB
1f f T T
T TBff
T T
log Ta
0
if cryst.
m
1T g
1T VF
1T
1T
Arrhenius
Strong changes close to glass transition

Polymer physics, physical chemistry 86
.log
1
2
C
gg
Tg
g
C
B T T2 303fa
fT T
Combination of linear expansion coefficient for free volume with exponential inverse
dependence of dynamics towards free volume makes WLF-equation plausible.
shift-factor
log( )
1 ref10 T
2 ref
C T Ta
C T T
: WLF-equation
expB
f
with f: free volume
For several techniques e.g. (dielectric spectrometry, rheology, induced aging ...) the change in
temperature is used to accelerate in a known plus predictable way (WLF-eq!) molecular
processes, to reduce time of experiments or access, otherwise not easy reachable frequency
ranges. This is called time temperature superposition (TTS).
Please be aware:
If WLF-equation is used, within the temperature range investigated any first order phase
transition must be excluded.
E.g. melting, smectic- nematic (liquid crystalline phase transition), LCST, UCST (lower/
upper critical solution temperature), TODT (temperature for order-
disorder transition) and of course Tm (melting temperature)!
A B T A+B

Polymer physics, physical chemistry 87
Note on Liquid crystals:
Centre of mass
correlation
+ - - +
Director
correlation
+ - + -
state: crystalline Liquid/
amorphous
Liquid crystal Conformation
disordered crystal
(condis cryst.)
examples: NaCl Water (liquid) Biphenyl Adamantan,
Fullerene
M
director
centre of mass

Polymer physics, physical chemistry 88
3.2.3 The crystalline state
Crystals are arrangements of molecules and/or atoms in regular, repeated and three
dimensional periodic pattern with translation symmetrie.
There are seven crystalline systems
,
,
a b c
90 120
In polymer science: c-axis is polymer axis
Called:
Triclinic, monoclinic, orthorhombic, tetragonal, hexagonal, rhombohedral, cubic
They exist in 14 space filling lattices (so called “Bravis” lattices) and 230 space
groups.
Bravis e.g. cubic
,a b c 90
Polymer: crystalline mostly in “all-trans” conformation or in helices.
Example for all-trans: PE-orthorhombic
,a b c 90
Cubic (primitiv) Only one type of repeat structure
Body centred cubic(bcc)
Face centred cubic (fcc)
a
c
c c
b
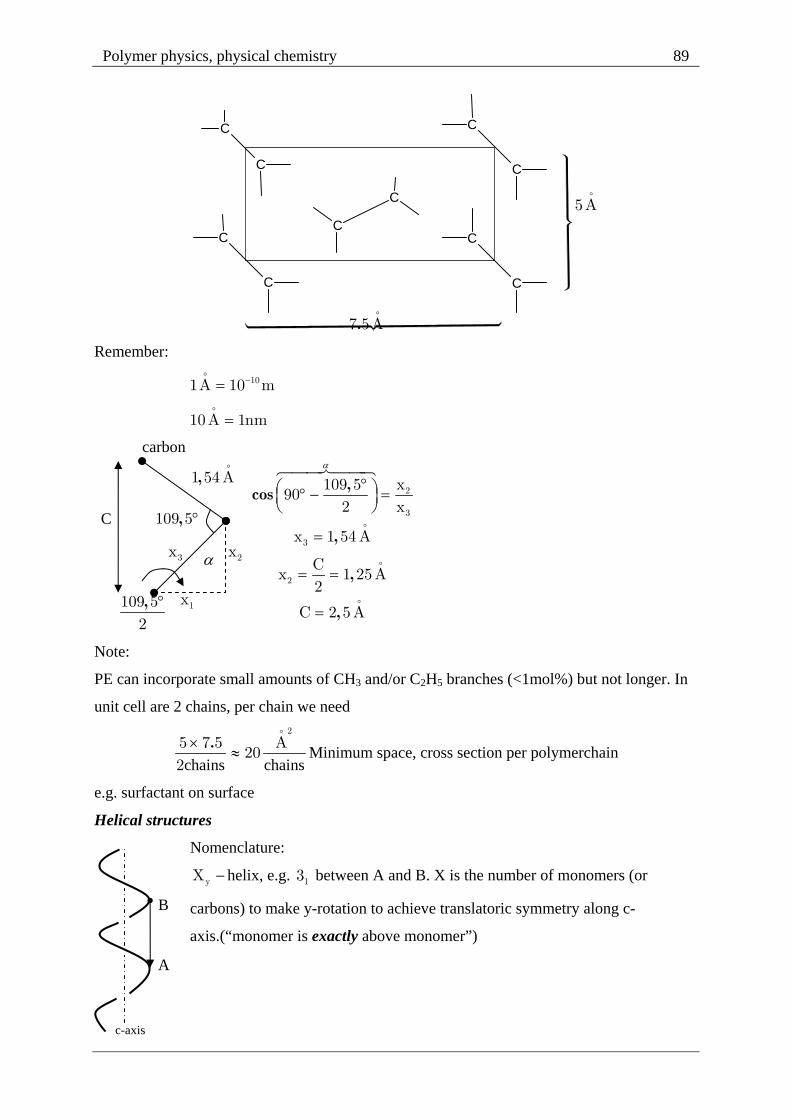
Polymer physics, physical chemistry 89
Remember:
101A 10 m
10A 1nm
Note:
PE can incorporate small amounts of CH3 and/or C2H5 branches (<1mol%) but not longer. In
unit cell are 2 chains, per chain we need
chains chains
.2
5 7 5 A20
2
Minimum space, cross section per polymerchain
e.g. surfactant on surface
Helical structures
Nomenclature:
yX helix, e.g. 13 between A and B. X is the number of monomers (or
carbons) to make y-rotation to achieve translatoric symmetry along c-
axis.(“monomer is exactly above monomer”)
.7 5A
C
C
C
C
C
C
C
C
C
C
5A
1x,109 5
2
3x
,109 5
,1 54A
carbon
C
,
cos
,
,
,
2
3
3
2
109 5 x90
2 x
x 1 54 A
Cx 1 25A
2
C 2 5A
2x
c-axis
B
A

Polymer physics, physical chemistry 90
How can polymers arrange to form crystals (lamella)?
a) folded chain crystal
Adjacent re-entry (from dilute solution)
Adjacent re-entry with loose folds
Random switchboard (Flory 1962)
Super folded crystal consistent with SAXS (small angel x-ray
scattering) and SANS
(small angel neutron scattering)
Plane folds on plane
10-100 nm

Polymer physics, physical chemistry 91
b) fringed micelle (“Fransenmizellen”)
generally not correct but historically important
In both types of arrangements (a, b) the polymer chains can participate in more than one
crystallite.
In early polymer days (1920) it was believed that a (unit) cell must have size of a molecule
(this is frequently true for low Mn material).
PE: c-axis is .2 5A
, contour length ( /610 g mol ), 10 µm (factor 40,000) 1920 polymers can not
exist as long and covalent bond molecules.
How can we determine crystalinity?
Main examples:
- X-Ray, Röntgen
- IR
- Dilatometry (density measurement)
- NMR mobility
- DSC thermodynamic properties
Either structural (x-ray, long range spatial correlation, IR: defined energy potential for
vibration), dynamic (NMR, contrast in mobility between crystal and amorphous) or
thermodynamic properties (dilatometry: density or DSC: heat flux) are used.
We can not expect 100% the same values for all techniques using the same sample! But we
assume a strong monotonic correlation between the values determined via different
techniques.
regular structure

Polymer physics, physical chemistry 92
Basic idea of x-ray (“Röntgen”)
We find sin1l
d
“extra path” for beam (2) sin12l 2d
To have constructive interference we need
sinn 2d Bragg-equation (memorize!) n , e.g. 1,2,3,…
for X-ray has dimension of inter-atomic distances and most commonly used sources
,Cu 1 54A
(copper anode)
Problem:
X-Ray source are normally monochromatic but not coherent (similar phase of
radiation) but in the derivation we used a coherent X-ray beam, since (1) and (2) were
in phase!
Explanation:
On small size ( 10 50nm ) only coherent contributions need to be considered, since
“incoherent rest” kills itself.
In case we have only first order peak (n = 1) ,Cu 1 54A
and 30 we can use the
following simplification:
grad
. sin ...!
. ,
.
3
rad
rad
1 1 54A 2d 2d3
1 54A 2d
21 54A 2d
360
(1)
(2)
d
Regular array of objects on a lattice e.g. polymer chains
Incoming beam
1l 2l

Polymer physics, physical chemistry 93
A88 2d
grad simplified Wilhelm
e.g. 2 20 .d 4 4A
!
cc
c a
AX
A A
:cA crystalline peak
:aA amorphous peak (called “amorphous halo”)
Please be aware:
Crystallinity does not exclude mobility!
e.g.
PEOjumps
POMs
PE
10000
- Small side-chains stops monomer to move
outside the crystalline area.
- PE under load deforms with time, because the
chains move to avoid load.
- Side-chains can force the chains to stay at there place.
IR: (Theory later), just basic idea
Different resonance (-H vibrations) or deformation for amorphous and crystalline areas
k
m
C H
I
0
I
0
30 10
Ac
In“ “ information about crystal
thickness! 1
D
Aa
For ideal crystalline material Real life: semi-crystalline material
Scherrer equation
Sharp peak, Bragg condition
Small branches
Crystalline area
up to
stochastic motion

Polymer physics, physical chemistry 94
Typically:
Useful bands:
1650 1500cm e.g. 11000cm
Using Lambert-Beer law of absorbance:
, ,c d0I c d I e
From cd0
dII I I e
dx
: absorption coefficient
:c concentration
:d thickness
Problem of scattering, size of crystalls light
absorbance
4000 600 V wavenumber1cm
Ia
Ic
deconvolution
n 1
per n 1000 1cm
Nk 500
m
10 m
for single bond

Polymer physics, physical chemistry 95
Dilatometry (density measurements)
Typically:
. .a
c
0 85 0 95
(at same temperature)
5-15% more free volume for amorphous material
e.g. PE
.c 3
g1 000
cm good packing of chains
.a 3
g0 853
cm
But poly(4-methyl-1pentene) c a !
1
23
45
We assume
c aV V V volume (what does this imply?!)
c am m m weight
c c a aV V V ; m
V
With a cV V V
c c a cV V V V
c c a aV V V
a c c aV V
acc
c a
V
V
Volume fraction
ac c c cc
c a
V mX
V m
mass fraction
,a c : known, : measured
Assumption:
Two phase model (crystalline and amorphous phase)
In case the density difference c a is 3kg100
m and we want to determine cX with <1%
accuracy. We need to determine relative density differences 310
. 3( 1000 / )Kg m
Technically:

Polymer physics, physical chemistry 96
: 3 410 2 10
is achieved
e.g. use of solvent gradient
T !!!
Typically:
31 110
V T K
, thermal expansion coefficient
Extension 2 phasemodel: 3 phasemodel
Why not modify “c”?
Suspended polymer particle location is
10cm
l 1m
l 1mm
Solvent density x
T control T 1Kwater
c a
c ao ano
:c crystallin :a amorphous :oa amorphous
oriented :noa amorphous not
oriented
reason

Polymer physics, physical chemistry 97
NMR (theory later)
In case we can find a temperature T, where a contrast in bulk mobility is high between
crystalline material and amorphous and the mobility (WLF) is sufficient for the amorphous
part to change the NMR resonance, position or shape (but not for the crystalline parts) we can
separate the two contributions.
Generally g mT T T (close to mT ) gT T 40K (TTS)
Main reason for line broadening (simplified picture) in solid state NMR
Fdx 12
1F E r
r
Field of (1) and (2) should be a function of r and .
Mathematically similar problem Legendre polynoms
cos23
13 1
r
If we could calculate the pre-factor, we would get (for protons!)
20 30kHz for rigid solid for 2 3r
Question: how close are protons?
C
HH H H
N
S
B
r
(1)
(2)
N
S
Monopol – monopol ~ 2F r
Dipol – monopol ~ 3F r
Dipol – dipol ~ , ~4 3F r E r

Polymer physics, physical chemistry 98
If rate of motion2 is large > line broadening mechanism line narrowing, e.g. liquid
1Hz
Therefore the extreme cases
DSC
Already described in chapter 2.2 polymer physics
Question:
When cX was determined what was assumed?
two phase model
If we improve, where is further differentiation needed?
2 Mobility effects the line shape, mostly the width, less the location
Experimental:
0
res
6res 300 900 10 Hz
dynamic contrast everything rigid, amorphous + crystalline
res
6300 900 10 Hz
Dynamic contrast, integration .cX very simple NMR machines 20 60MHz wide
line spectroscopy
.g m measT T T
Rigid material Solution NMR
20 – 30 kHz
1 Hz
No dynamic contrast, everything mobile, no crystals exist anymore
Tlow Tmed Thigh
Crystalline area, everything same, at least in average homogeneous
Amorphous area, everything same, at least in average homogeneous
two phase model
„minispec“

Polymer physics, physical chemistry 99
3 phase model with 2 amorphous and 1crystalline would be appropriate extension
(Maximum speed of crystallization halfway between gT and mT )
ordered amorphous
crystalline
C a
C a0 aa 3 phase model
disordered amorphous

Polymer physics, physical chemistry 100
3.2.4 Kinetics of crystallization (Avrami equation)
Growth of spherulites
Probability that number of spheroids “C“ be at point “P” until time t?
Poisson distribution (1837), E=E(t)
exp
!
CE EP C
C
not to be a crystal ( amorphous); C=0
(0) exp( )P E
All nuclei grow at t 0 number does not change = athermal nucleation growth rate
.r const
radius r t r t
volume occupied by crystal = probability of crystals to hit “P”
34E t rt g
3
:g concentration of nuclei
exp 3 3c
4P 0 1 V r t g
3
General Avrami equation
exp nc1 V kt
,k n they are typical constants depending on type of nucleation (athermal or thermal) and
growth mechanism, e.g. line, circular, fibrillar, sheaf3
3 Bündel, Garbe
P P
PL
t 0 start all nucleation takes place at t 0 , athermal
time
( )P t
t
E P
Averrage value of crystalline fronts passing P
0C
0C 0C

Polymer physics, physical chemistry 101
experimental determination of “n”
hint about local mechanism
Typically:
typical
1 3 n 4 6 ; n dimensionality of the problem
At early times, Taylor expansion of Avrami
ncV k t Göler equation

Polymer physics, physical chemistry 102
3.2.5 How to reach 100% crystallinity in a solid polymer
Start from melt ?
Topological constrains impossible to reach 100%
Idea:
Single crystal of monomers + thermal or ionizing radiation (1970, Prof. Wegner)
source: Young
C
C
CRC
R
C
C
CRC
R
topochemical reaction
polymer

Polymer physics, physical chemistry 103
Unusual optical, electronical and mechanical properties, e.g. anisotropic, defect free, conjugated.

Molecules and characterisation 104
4 Molecules and characterisation
4.1 Distribution of molar mass and determination of molar mass of polymers
In general we have a discrete distribution, but approximate it with a continuous distribution.
With n
0
P dn 1
The distribution does not need to be symmetric
Or have a single maximum
Number average (number of polymers number of monomers !)
1n 0 nM M P n dn (first moment)
Probability Pn
n
Probability Pn
n
Gauß
Probability Pn
n

Molecules and characterisation 105
Weight average:
weight:ng
2 20 n n 0 0 0
w
0 n 0 n
M P n dn P n M n M dn MM
M P ndn M P ndn
n
0
g ndn
M
second moment
first moment
n
0
P ndn
M
z- average: (Centrifuge average)
third moment
second moment
3 3n 0
z 02 2n 0
P n M dnM M
P n M dn
If we would know all moments, we would have the full information about the distribution
(functional analysis, maths), but most experimental techniques give “only” , , ,n w zM M M M .
average: viscosity average, measured via viscosity
an
a 1n
P n dnM
P n dn
. . ,a 1 5 1 9 close to wM
,a no necessary 1, 2, 3,…
pM ; peak molecular weight:
The most probable molecular weight is called pM
In general:
n w zM M M M
To describe the “width” of the distribution we can use the standard deviation :
mathematical definition
2 2 2x x
where 2 2x P x x dx and x P x xdx
Probability Pn
nnM wM zM
M
Width of distribution
pM

Molecules and characterisation 106
Using
2w nM M x second moment of nP
nM x first moment of nP
2 2n w n n w nM M M M M M (I)
Most commonly in polymer science the width of the distribution is described via the
polydispersity (PDI, polydispersity index):
always,w
n
M1
M
In German literature often the term “Uneinheitlichkeit” is used to describe the width of the
distribution
:U 2wn
n
M1 M
M
( )
2 2 2n n w n n w n
IU M M M M M M M
2 2n
n
U M
M U
Three examples of molecular weight distribution
:iw weight distribution (mass)
5041
gM 10 mol 405
2gM 5 10 mol
73
gM 2 10 mol
(1) 0.1 0.9 0
(2) 0 0.9 0.1
(3) 0.05 0.9 0.05
, ,n w zM M M in 5 g10 mol , U
nM wM zM U
(1) 0.85 4.51 4.99 4.3
(2) 5.54 24.5 164.18 3.4
(3) 1.47 14.5 139.48 8.9
Factor 200, even if . !2M 0 9

Molecules and characterisation 107
Small amounts of low nM , do not change much andw zM M , but nM .
Small amounts of high nM , changes drastically andw zM M .

Molecules and characterisation 108
4.2 Experimental determination of molecular weight and distribution, most
common examples
absolute:
Several of these methods will be presented.
Please note, that frequently even the full information about nP is not sufficient, if topology is
not known, or chemistry e.g. block-copolymers is not clear!
1 “Siedepunkt“
Method Determined quantity Optimum case: range gmol
Osmotic pressure nM 4 610 10
Vapour pressure osmosis nM 42 10
Cryoscopy, ebulloscopy1 nM 45 10
Ultra centrifuge , ,n w zM M M 210
Light scattering (static) wM 35 10
X-ray wM 35 10
Dynamic light scattering wM 35 10
NMR (endgroups) nM 52 10
Titration nM 45 10
Mass spectrometry , ,n w zM M M 610
Viscosity n wM M 210
GPC , ,n w zM M M 42 10
Colligative properties
relative:

Molecules and characterisation 109
Example of colligative properties: osmotic pressure
Scheme:
- similar chemical potential of solvent left and right of membrane
- additional pressure is generated since solvent concentration at the right is lower and
polymer is restricted to right side (“entropy”!)
g h
: density
:g gravity
:h height
If polymer would behave as a gas, ideal gas-law (p ) for polymer
c
p V n R T
nR T
V
c RT
Range:
3 5n
g g20 10 M 2 10mol mol
Lower limit:
Polymer is so small, can diffuse across membrane holes
Upper limit:
is to small to be detected
Solvent + polymer
solvent
h
Membrane: solvent can cross membrane. Polymer can not cross through membrane.

Molecules and characterisation 110
4.3 GPC, gel permeation chromatography also called size exclusion
chromatography (SEC)
Basic principle:
Who comes out first?
high nM fraction, since it can not enter all the holes and interact with all surface area, low
nM fraction more trapped.
Note:
Size entropy
Compare: HPLC (high pressure liquid chromatography) and GPC
Principle Related effect
HPLC Energetic interaction Specific adsorption
GPC Entropic interaction Size exclusion
Low nM polymer
solution High nM polymer
solution
Flowing solvent through porous media, large surface area+ holes in nm range
Detectors: what would you want to detect?
0
Concentration polymer
High nM Low nM
Elution volume
transform High nM
nM
Low nM nP M

Molecules and characterisation 111
Detection Information
UV, IR Chemical constitution + concentration
Viscosity nM (via calibration), detection of pressure loss, Hagen-
Poisseuille
n (refractive index) Concentration, RI-detector (refractive index)
Light scattering , ,n wM M special set-up called MALLS (multi-angle laser
light scattering)
To correlate detected concentration with nM , frequently viscosity is used:
Newton:
F
A (shear stress)
For solution we expect:
, nc M Concentration + molecular weight
solventspecific
solvent
To “take out solvent”, no units!
Normalize to concentration
specificred c
, reduce viscosity, unit of inverse concentration
To take away polymer- polymer interaction:
redlim : ,c 0
intrinsic viscosity, unit of inverse concentration, “Staudinger index”
What can we guess for:
nM ?
Einstein (1906+1911)
solvent specific. .1 2 5 2 5
: volume fraction rigid
If molecules would collapse to spheres with same density where solvent can not enter
!nf M
sample
LASER Several detectors, simultaneous detection

Molecules and characterisation 112
If Gaussian chain would immobilize solvent:
2 2h Nl
mass
polymer
Volume of spheres. 13 1 5 3 22h N l N l N l
specific olume sphere 13 22
massV h N l N l , Einstein!
specific
c
1 12 2N M
information about shape?!12M
Kuhn-Mark-Houwink-equation, also called Staudinger-Mark-Houwink
ak M
, :k a empirical constants good solventtheta solvent
.0 5 a 1 , typically in tables for different polymer-
solvent combination:
35 mk 10 kg
:M molecular weight (monodisperse)
,h end to end vector
Immobilized sphere, volume V

Molecules and characterisation 113
4.4 Ultracentrifuge
Principle
Particles or molecules feel gravity. Shape, size, density difference and gravitational forces
determine sedimentation velocity.
To increase speed of this process ultracentrifuge
Pioneering work: T. Svedberg (from Sweden) (since 1923, Nobel price 1927)
Method to proof hypothesis of Staudinger that high molecular weight polymers exist (but:
Staudinger had no money to buy it, so he developed viscosity measurements…)
Range: 3 7 g10 10 mol
Determination: , ,n w zM M M
Disadvantage:
long measurement times (h to days), expensive equipment
Force towards molecule under rotation:
cos cos,
sin sin2
t tx r x r
t t
acceleration
2F m x m r
m is effective mass:
pp
L
Mm 1 V
N
:LN Avogadro number
: density solvent
Particles sink down, sedimentation + diffusion due to Brownian motion
Liquid mediumgravity
x

Molecules and characterisation 114
:pV specific volume 3cm g
polymer
:pM molar mass polymer
This centrifuge force is balanced by friction proportional to sedimentation velocity.
drF f
dt
:f friction coefficient
centrifugal
acceleration velocity
p2p
L
M drr 1 V f
N dt
Svedberg:
L Lp 2
p pS
drfN s fNdtM1 V r 1 V
How can we determine “f” ?!
Bk Tf
D
Einstein relation
:D diffusion constant
p
p
s RTM
D 1 V
Svedberg equation
D light scattering or via Stokes-Einstein:
kTD
6 r
Apparatus (“Beckmann”)
Typically: 510 rpm
Solvent can be with density gradient
Applications:
- particle size of polymer emulsions in water or organic solvent
610 g (earth acceleration)
optics, triggered: UV, absorption filter
sample + solvent
detectors
refractive index
rotor axis

Molecules and characterisation 115
- absolute nM for extreme high nM where GPC and light scattering is problematic (e.g.
6 g10 mol )
- molecular weight distribution for polymers that interact with GPC column
- determination of chemical heterogeneity using density gradients; seperation of
polymer mixtures and copolymers

Molecules and characterisation 116
4.5 Light scattering of polymer solutions
Two types:
- coherent, elastic scattering, frequency is not affected
- incoherent, inelastic scattering, frequency is modulated via polymer motion (Doppler
shift) (also called: inelastic, quasi-elastic, dynamic light scattering)
general set-up:
note:
for special cases, e.g. crystallisation or phase separartion the transmitted light I t is
measured, making use of the “Tyndall” effect.
Basic equations:
cos0E E t
electric field of light beam
cos0p E t
polarisation of molecule via light
22
s 2
d pI
dt scattered light (change in dipole)2,
Two types of polarisation p
a) for unpolar molecules+ polar molecules shift of centre of mass of electron cloud
relative to protons, fast process shift polarisation
b) polar molecules only: if a permanent dipole exists, it can be oriented. Orientation of
molecule results in total polarisation, can be calculated using Boltzmann distribution
Langevin equation with final result
dipole of molecule
field2
0
pp E
3kT
but this process is for light
scattering to slow! light14 1
v 10s
incident beam
,0I
frequently Laser Sample-cell polymer solution
detector
Scattered light 4
010 I
transmitted beam
c d0I I e

Molecules and characterisation 117
orientation polarisation, detected via dielectric spectroscopy
(not covered in this chapter, see chapter 5)
How to vibrate electrons?
p em 2000 m
No need to use reduced mass
e pe
e p
m mm
m m
No external force: ,0e
k
m resonance frequency 2
0 ek m
With electric field sin0E E t
sin2
2e e 0 02
d xm m x eE t
dt
:e charge
Solution:
sin02 2
0 e
eEx t t
m
In phase, even if not at resonance.
Typical resonance frequency for vibration of atoms is in IR region
( , ,1 14 1503000cm 10 Hz 10 )
Nk 500 m for single bond
We guess:
Changes into
,0
k
m with Nk 500 m and 30
em 10 kg
+ proton
- electron
All interactions are summed up, Taylor + stop after quadratic term
k
proton proton proton
electron
factor 2000 in mass

Molecules and characterisation 118
for electrons :
160 2 10
light 0 x Ray
For light scattering:
0 sin02
0 e
eEx t t
m
For X-ray:
0 sin02
e
eEx t t
m
Dipole moment is:
p t e x t
22
s 2
pI
t
light scattering: sin22 2
0s 2
0 e
e EI t
m
X-ray scattering: sin22
0s
e
e EI t
m
4sI for light scattering! Blue sky, Rayleigh scattering (1871)
sI f X-ray!
s 2e
1I
m if number of electrons is proportional to 3r (size of scattering particle)
number of electrons
2
6s
e
1I r
m
large particles scatter much more
Scattering of light is dependant an orientation of dipole and polarisation of light

Molecules and characterisation 119
non-polarized :
For a certain volume we find:
impotant term
222 22
s 0 0 02
pI 2
t three sources of scattering intensity
:0 mean
: fluctuation
Idea of Einstein 1910:
2s 0 s 0I I 2 0
Reason:
In scattering volume v we will find a lot of electric dipoles. Always a pair is found that will
cancel each other. Therefore only the quadratic fluctuation leads to scattering intensity (
Doppler effect, frequency shift)
n
y
x
z
Dipole vibrates along z-axis
sin2sI
x
y
x
y
No scattering along dipol axis
Isotropic in x-y plane
y
x
z
Dipole vibrates along y-axis
Incomming light x-y plane
Incomming light x-z plane
cos21

Molecules and characterisation 120
density fluctuation polymer concentration fluctuation,polymer + solvent 0 important contribution
22
2 2 2 2 20 0 B
B
n nn n c
c
n : refraction index
: density
thermo-dynamic
;2
2 2 2 Bs 0 B B B
TB
n cI n c c RT c
c
To compare scattering intensities we define
2
0 0
I rR
I V
0V : scattering volume
0I : incident beam intensity
Reduced scattering intensity or Rayleigh ratio R .
To get scattering intensity from polymer
solution solvent :R R R R
BB
T
cR k c RT
: Osmotic pressure
no increment, no scattering
isorefractive solvent, index match
,22 2
04
L 0 B
4 n nk
N c :k optical constant
B
B T
k c 1
R RT c
M light scattering can measure polymer weight M!
For polymers we find for osmotic pressure :
(*)
...2 2 3 3BB B
cRT A c A c
M

Molecules and characterisation 121
(*) second virial coefficient, may differ from osmotic second virial coefficient
B
BW
k c 12Ac
R M for non-monodisperse solutions, fundamental scattering equation
Holds, if ,gR20
e.g. 5 gM 10 mol
In case :gR20
Debye scattering, includes intra- and intermolecular scattering
B
2 Bw
k c 12A c
R M P
(I)
,I
PI 0
form factor, scattering factor.
Debye 1915:
sin ij
2i j ij
q r1P
N q r
. sin4
q2
(II)
:ijr distance scattering centre i from j
sin!
3
3
and 2 2ij2
1r r
2N
2 21P 1 q r
3 Guinier 1939
General, independent of shape
2 21 1
1 q rP 3
(III)
Putting (I), (II) and (III) together
if polydispersity included
sin,
22 2B
2 B2 zB w
k c 1 161 r 2A c
R c M 3 2

Molecules and characterisation 122
Analysis via Zimm-plot
Form factor P contains information about shape of molecule, e.g. sphere, stick, Gaussian
shape…
Incoherent, inelastic, quasi-elastic or dynamic light scattering
Idea:
k c
R
wM
slope 2A
2
zr
extrapolate to c 0
variation
extrapolate 0
free
parameter
21q k c
140 3 10 Hz
frequency distribution „in“
0
broadened 140
6
3 10 Hz
1 10 Hz
frequency distribution „out“, but 810
can not easy be measured in frequency space
,0 0I
velocity
particl
,I t intensity modulation and
frequency shift (Doppler-shift)

Molecules and characterisation 123
Solution to this problem:
Wiener-Khintschin theorem (1930+1934)
“The spectral density (=spectrum) is the Fourier-transform of the autocorrelation function”
i t1S g t e dt
2
i tg t S e d
What is the time autocorrelation function?
2I t I t dt
gI t dt
c typical time need to be again at mean value or cross this value I .
Note:
I is function of t due to concentration fluctuation
general form of autocorrelation functions
exp ,c
tG A 1 B
calculated by “autocorrelator”
Large particle
t
I
I
c
Small particle
t
I
I
c
slow fluctuations fast fluctuations
4 1 2 3 t
I t
tt

Molecules and characterisation 124
c should depend on?
- Scattering angle, sin ,4
q2
unit: 1
length
- Diffusion coefficient, unit: area
time
- Size of spherical particle
- Viscosity of media
Stokes- Einstein:
kTD
6 r
:r size of particle, hydrodynamic radius
: medium, e.g. H2O, 1mPas
For large D small ?c c
1
D
Autocorrelation for monodisperse spheres
At low concentration:
2c
t
Dq tg t e e
c 2
1
Dq
Generally: no single exponential function
Analysis:
distribution of size auto-intensity
diffusion distributioncorrelationfluctuationcoefficient
I t g t D r P r
The step from autocorrelation to the distribution of diffusion coefficient is not easy
(mathematically), via e.g.:
- cumulated method
- non-negative least square (NNLS)
- Contin algorithm
g t
1e
ct
g t
clog t

Molecules and characterisation 125
Experimentally conducted via two techniques:
Application of dynamic light scattering
- polymers in solution
- suspension
- emulsion
Typical:
:r 10 1000 nm
:2
12 14 mD 10 10
s
Be aware of the following relation:
10 100
r
nm
1 : 1
10 100
r
nm
1 : 1000
10 100
r
nm
1 : 1000000
number volume scattering intensity
Photo multiplier
Auto-correlator
data
Photo multiplier
Auto-correlator
data
scattered light
non-scattered light,
scatterd light
Homodyne (simpler, mostly in commercial application)
heterodyne

Molecules and characterisation 126
4.6 IR-Spectroscopy
Spectral regions
m 1cm Hz
Near 0.78-2.5 12800-4000 . .14 143 8 10 1 2 10 most
important Middle 2.5-50 4000-200 . 14 121 2 10 6 10
Far 50-1000 200-10 12 116 10 3 10
Remember:
1
1
1eV 9000 cm
kT 200 cm
wavenumber energy E=h
1h c
: how many waves per 1 cm, e.g. , . ,14000cm 2 5 m since
. 6 3 22 5 10 4 10 10 m 1cm
Important since:
- analysis + characterization
- isotope selective
- not selective towards optical isomers (chiral)
- combination, gas chromatography, GPC
- FT resulted in factor 100-1000 increase in sensitivity per time, e.g. monomolecular
layers detectable via IR
IR: vibrations between nuclei in molecules can be excited in case electrical dipole is present
Vibration frequency:
Newton:
F m a m x k x (Hook)
A B
vibration
Electric dipol
l
Q l
+ -
Raman: polarizibility, CO2!

Molecules and characterisation 127
“Ansatz”:
i tx t A e
2 i t i tm Ae kAe
2 k
m
k
m
1 k
2 m
:k force constant via chemical bond, electrons
:m nucleus mass, sensitive to isotope!
Potential: harmonic, if force is linear in x
1F k x
21V Fdx kx
2
How many degrees of freedom for vibrations if N-atoms in molecule are present?
f 3N 5 for linear molecule
e.g. H2O: f 3
these vibrations are separated in:
distance angle, torsion
- stretch + bending vibrations
A
A
A
x
vibrational levels Quantum mechanics
E x
E
A2
, ,x y z
f 3N 6
Translation or rotation of whole molecule does not lead to vibration

Molecules and characterisation 128
- in principle we should be able to detect all degrees of freedom in spectrum, via
anharmonic potential even more
but:
1) symmetry, (degeneration) reduces number of resonances (also called bands or
vibration modes)
2) often the energy differences between different resonances are small (especially for
bending)
3) resonances are forbidden (e.g. no dipole moment) very low intensity
4) resonances are outside investigated spectral region
treatment of harmonic potential in quantum mechanics leads to:
symmetric asymmetric
In plane rocking In plane scissoring
wagging twisting
bending
stretch
+ - + +
1 h k
E n2 2
Zero point energy (Heisenberg), particle cannot
stand still typically .0 01A
for vibrations
Reduced mass 1 2
1 2
m m
m m
, , , ...
n
n 0 1 2
2
;xH e H: Hermite polynoms

Molecules and characterisation 129
Selection rule:
Golden rule (Fermi), perturbation theory
Absorbance ˆ2
f 1 iH
ˆ ˆ1H E
electric field
ˆ : dipole moment
space
operator
ˆ x̂ Q charge
ˆ ˆˆ1
x I I2 ladder operator ˆ ˆˆ ˆ ˆ ˆ;I x iy I x iy
ˆ ˆ2
f iI I
, :i f set of orthogonal functions
n 1
Typical force constants
2 Nk 5 10
m single bond
3 N1 10
m double bond
. 3 N1 5 10
m triple bond
v k
1 molecule H2O should be stretched by 1A
, how many molecules H2O
are needed to generate this force simply by weight?
10 8F k x 500 10 N 5 10 N
-8
molecules
5 10 molecules
. 23218gH O 0 18N 6 10
N x
.
. .
8 23 16175 10 6 10 3 10
x 1 5 100 18 0 18
!!!
the spring is very, very rigid !
H2O
O
H
H
e.g.
Selection rule for IR but: spin-orbit coupling, heavy atom effect, Born-Oppenheimer…

Molecules and characterisation 130
Hardware:
IR-light sources:
- blackbody radiation at hot iron red
1500 2200K
(sun: 6000K visible)
- Nernst glower (rare earth oxide, electrically heated)
- Globar source (SiC)
- Mercury arc (high pressure to widen the resonance lines)
- Tunable CO2- laser ( 1900 1100cm ) high intensity, not vissible
Detectors:
- thermal ( T of 610 K detectable, V6 8 W
)
- photo conducting detectors, e.g. Hg Cd Te, PbS
IR-instruments:
1) dispersive instruments
2) FT-IR (see later)
3) Non- dispersive at fixed frequency ( atmosphere measurements)
4) Reflective instruments, e.g. in emission, for example to detect chemicals via large
distance
FT-instruments
- multiplex instruments ( information theory) more than a single information is
simultaneous transmitted via a single channel
compare telephone wire: several conversations simultaneous via one wire
prism
3
2 Not multiplex
1 1I 2I
3I
prism
3
2Generally double beam
1

Molecules and characterisation 131
FT-advantages
1) flux- or Jaquinot advantage, less optical elements (e.g. collimation, slits etc.) lead to
higher signal, factor: 10 100
2) higher precision averaging of spectra easier possible, , n:scansS n
3) multiplex or Fellgett advantage all vibrations are simultaneous excited,
factor: 10 100
FT conducted via Michelson interferometer; A.A. Michelson (first built 1891, Nobel prize
1907)
Intensity varies as a function of absorption (reduction)
detector
Semi-transperent (reflectant) mirror,
e.g. Ag ca. 100A
lamp
fixed mirror
moving mirror
1
2 0 1
2
x
sample
1
2 0 1
2
P t
No sample

Molecules and characterisation 132
Setup at “0” constructive interference, “maximum”
Interference pattern
Source: Skoog
P x t
0 x [cm]
x DW SW e.g. 14000cm
. ( )1
DW 1 25 m SW2DW
mechanically demanding
Amplitude of mirror motion x AQ spectral resolution
e.g. . 110cm 0 1cm

Molecules and characterisation 133
12-25: methylacetophenon
C
O
CH3CH3
12-26: acrolein
CH2 CH CHO + OH2
12-27: propannitril
CH3 CH2 C N

Molecules and characterisation 134
IR-spectroscopy for polymers, applications:
- chemical composition and additives
- kinetics (IR is very fast) of polymerization
- analysis of sequences in copolymers, changes of resonance
- detection of small amount of branches, e.g. in PE (dimension 1 per 1000 CH2)
- semi quantitative using Lambert-Beer cd0I I e ( :c concentration, : extinction
coefficient)
- stereo regularity (e.g. a-PP, i-PP)
- blend composition
- orientation via rotation of sample along 2I P second Legendre polynom
- detection and quantification of crystallinity
- characterisation of polymer surfaces
Note: Raman spectroscopy and SERS (surface enhanced Raman spectroscopy) is getting
more and more applications due to high intense Laser light sources.

Molecules and characterisation 135
4.7 Mass spectrometry
How to measure the weight of a molecule???
In general no spectroscopy since non coherent and non resonant
Definition of atomic mass:
1amu 1Dalton 2(unit Dalton frequently used in Biology)
12
molatoms
mol
atom
.
.
12
23
24
C12g112 6 0221 10
g1 66054 10C
Question:
Why is Dalton.3517Cl 34 9688 3
Dalton .0 0312 energy to bind nuclear particles together
2E mc
- atoms of same element can be in different isotopes
e.g. . % , . % , %1 2 3H 99 985 H 0 015 H 0
isotopesi iA A p
:A mean atomic mass
:iA mass of isotope “i”
:ip relative probability of isotope “i”
Mass spectrometer:
- measures amount and mass of charged atoms, molecules and molecular fragments
- relative precision: 5m10
m
- absolute price: €5 610 10
2 amu (atomic mass unit) 3 Explanation
protons + neutrons
protons X
, X: element abbreviation

Molecules and characterisation 136
- set up, each part in more detail afterwards
inlet system:
- ng might be enough
- different for liquid or gas phase samples coupling to gas- chromatography or TGA
(for polymers)
- samples should be in gaseous phase so that they can be ionized
Detectors:
Most ions are cations (ca ions, positive charged)
- dynode amplifier photomultiplier
- Faraday cup, continuous decrease in electric potential along electric resistance,
decreasing distance ,U
Ed
electric field
Inlet system
Ion source
Mass analyser separation
detector
Signal processor
Read out
Vacuum system
Why? mean free path length of ions
sample
5 810 10 Torr, 760 Torr 1 bar

Molecules and characterisation 137
Source: Skoog
Mass resolution
- e.g. to determine sum formula
- purine C6H5N4 .m 120 044
N
N
N
N
H
- benzamidine C7H8N2 .m 120 069
C
N
NH2
H
- acetopheone C8H8O .m 120 058
C
O
CH3
About 20 times, each factor 2-3 . !20 7 82 5 10 10
Why?!

Molecules and characterisation 138
!
Needed resolution: .0 01
80ppm120
, commercial:1 to 1ppm
Information also from isotope distribution
, , , ...12 13 14 15 I M 1C C N N
I M
: . %
: . %
: . %
: . %
: . %
: . %
1
2
14
15
28
29
12
13
35
37
16
17
H 1 0 015H
N 1 0 34N
Si 1 5 1Si
C 1 1 1C
Cl 1 32 5Cl
O 1 0 04O
:
: . % . %
: . % . %
: . % . %
: . % . %
. %
6 4 2 4
13
2
15
17
C H N O
C 6 1 1 6 6
H 4 0 015 0 06
N 2 0 37 0 74
O 4 0 04 0 16
7 56
:
: . % . %
: . % . %
. %
12 24
13
2
C H
C 12 1 1 13 2
H 24 0 015 0 36
13 56
Mass analyser
- separation of mass with respect to different acceleration (at similar force) in B-/ or E-
fields, magnetic or electric
1) acceleration in electric field
21z e V mv2 (1)
2) force in B-field (Lorentz-force)
F z e v B
with
v B
F z e v B (2)

Molecules and characterisation 139
3) Centrifugal force
cos
sin2
0
tr t r a t r
t
v
r
2 22
2
v vF m a m r m r m
r r (3)
From (2) = (3)
2vB e z v m
r
B e z rv
m
In (1)
2 2 2 2
2
B e z r1z e V m2 m
2 2 2 2B e z r m B e r1V 2 m z 2 V
Variation of , ,B r V
Molecules and characterisation 140
Source: Skoog
ca.15 cm
4-10mm
Faraday cup

Molecules and characterisation 141
Quadrupole mass filter
Monopole:
Dipole:
Quadrupole:
The electric potential at the poles contain a time dependant oscillatory and constant dc-offset.
The trajectory for the ions is different for different m
z and v (when entering the quadrupole).
The quadrupole acts simultaneous as filter for low and high masses.
TOF: time of flight
Scheme:
Time of flight:1 30 s repetition rate
Further:
- FT-transform
- Combination of B
and E-fields
Ion sources
- depending on ion source, the molecules will experience different degrees of
fragmentation hard and soft sources
common ways to produce ions:
- electron impact
- chemical ionisation
Volt3 410 10
Detector
No electric field,ions fly
All ions generated simultaneous ( .0 25 s )

Molecules and characterisation 142
- field ionisation
- field desorption
- Maldi- TOF, very important for polymers ( 6nM 10 g
mol ) matrix assisted laser
desorption of ions, + time of flight detection
Electron impact (hard)
Acceleration voltage: 70V binding energy 3 4eV
strong fragmentation
Fragmentation ( mass spectrometry books), e.g.
Norrish I:
scission
Norrish II:
scission
- CO2, H2O elimination
gas M
M
Anode
X
X

Molecules and characterisation 143
Source: Skoog
Advantage
- via fragmentation chemicals have unique decomposition
Disadvantage:
- very complex spectra
- sample must be in gas phase
-H2O 18
-COOH 45
63
Electron impact (hard)
Field ionisation
Field desorption (soft)

Molecules and characterisation 144
Chemical ionisation (soft)
Idea:
reactive, will transfer
proton with charge4
4 4 5 3
CHe
CH CH CH CH
Field ionisation (middle)
Desorption methods ( ,m 10000 amu soft)
- field desorption, sample on Tungsten anode, similar to field ionisation
- fast atom bombardment:
Maldi-TOF
10 m
Needles, ca. 1 m thick
Very high E-field at tip ionisation!
kathode, 10-20kV
Trungsten anode
Xe, Ar
Glycol + sample 1000 : 1
Matrix
Laser
ions
Sample + COOH
+ Salt (e.g. silver)
Very low amount, e.g. polymer

Molecules and characterisation 145
Further important for MS (mass spectrometry):
- combination with GC, gas chromatography
- Tandem MS, also called MS/MS, (e.g. QQQ: 3quadrupole)
First: soft ionisation, only molecular ions
Second: selection of specific mass, e.g. via quadrupole
Third: hard fragmentation via ions (e.g. He) in quadrupole
Forth: analysis of fragmentation fingerprint
Advantage: very fast (compared to GC/MS), ms versus min!
- Detection of elements via extreme hard sources (plasma) ppb.0 1 10 detection limit
- SIMS (secondary ion, mass spectrometry)
Depth resolution: 100A
Sensitivity: 152
g10m
- Laser microprobe mass spectrometry
Sensitivity: 2010 g !
Ar+, Cs+, N2+
O2+
MS
Sample t = 0
Sample t = later
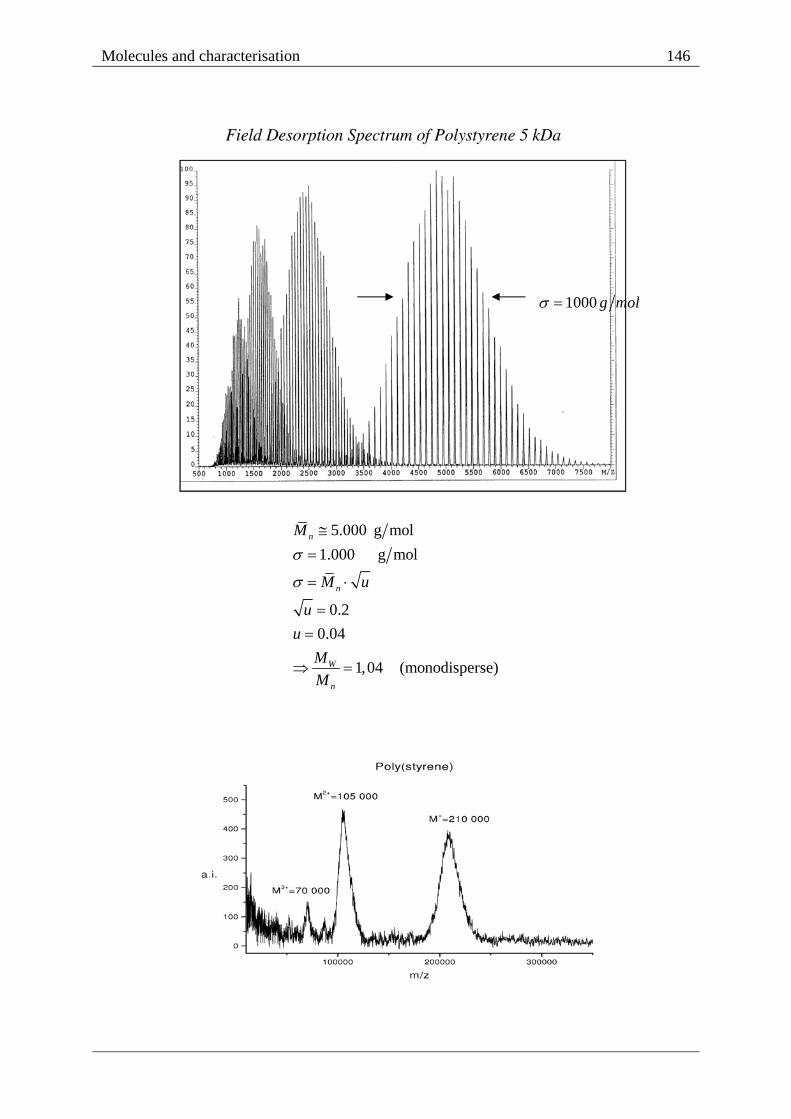
Molecules and characterisation 146
5.000 g mol
1.000 g mol
0.2
0.04
1,04 (monodisperse)
n
n
W
n
M
M u
u
u
M
M
1000 g mol

Molecules and characterisation 147
Source: J. Räder, MPI-P

Molecules and characterisation 148
MS- application to polymers (examples)
- identification of polymeric systems
- identification of additives (e.g. combination with TGA, thermo gravimetry)
- molecular weight and molecular weight distribution (but: ionisation probability is
function of Mn separation via GPC, quantification via MS) calibration of GPC
- end group analysis
- characterisation of copolymers (distribution of each component)
- oligomer characterisation

Molecules and characterisation 149
4.8 NMR- spectroscopy
-Very versatile spectroscopy of the nucleus with respect to chemical structure, dynamics and
orientation.
This spectroscopy is mostly used for solutions but can also investigate bulk material. Large
dynamic range ( 9 510 s 10 s ) and spatial range ( 12 010 m l 10 m NMR imaging).
NMR- spectroscopy is isotope (not element!) sensitive. Most important isotopes to be
investigated: 1H, 2H, 13C, 14N, 19F, 29Si, 31P…
Principle (QM):
The nuclear spin is related to a quantified, absolute angular momentum, eigenvalue of
the 2̂I operator (QM)
2̂I I I 1 ;
:I nuclear spin quantum number, e.g.: , , , ...10 12
Spin 12 : 1H, 13C, 15N, 19F, 29Si, 31P
The z-component of the angular momentum is given by
ˆ ;z II m magneticquantumnumber
: , , ...Im I I 1 I
For spin 12 only two possible states: 12 and 1
2 exist.
The total magnetic moment is given by
;zI
: gyromagnetic ratio, isotope specific
Since the energy for a classical magnetic moment is given by:
z IE B I B m B

Molecules and characterisation 150
We expect:
- energy gap linear in B-field
Transition between two states 12 and 1
2 can happen if electromagnetic wave has energy
that “fits” the difference
Larmor frequency
L
1 1E B B h B2 2
Tesla, .HMHz42 B 1 5 22T
: resonance frequency:
range: 60 900 MHz for protons
15 225 MHz for 13C
Spectroscopically used, only population difference (Boltzmann)
exp exp exp34
1 2 L
1300MHz2
N E h N 6 10 300
N kT RT
610 6 2310
8 300
exp 5 56 10 1 6 10
Very, very small!!
Picture for magnetisation (spin 12 )
QM: Spin components with respect to absolute value and z-component spin classical
magnetisation M
Transition possible! E h
1m 2
B-field
1m 2 E

Molecules and characterisation 151
Mx,y
M
Mz
If B1 is switched off after 90 , magnetisation is in xy-plane, rotating with Lamor
precision ( ) sin0 LM M t around B1-field.
Since:
cosL 0 L
d dMU M t
dt dt, we expect periodic signal for induced voltage in a coil.
The exponential decay is given, since magnetisation wants to go back to equilibrium (along z-
axis). Any vector M
can be separated into two vector components: along z-axis (B0-field)
and perpendicular z-axis. Both components have individual relaxation constants.
Along z:
T1, spin-lattice, longitudinal relaxation time
:z
T2, spin-spin, transversal relaxation time
B0
z M
y
x
y
applied
B1-field for some time so that
e.g.
or 180°1t B t 90
B t 90
t 5 20 s
T
t
Exponential envelope
Free induction decay = FID
FT
0 1T
B1
2
1
T 1 e
2T
50ms-1s

Molecules and characterisation 152
Who will relax magnetisation back to equilibrium? fluctuation of local magnetic field,
analogue light scattering or dielectric spectroscopy, spectral density I
Along z:
T1, energy is changed, spins “look” for fluctuations.
at Larmor frequency range e.g. 100MHz to make transition information about fast
motions, LI
:z
T2, energy is not changed, magnetisation “fans” out 0 I information about
slow motion
Question:
Why the hell would we do NMR if we would see only 1 peak?
chemical shift!
Simple picture:
Electrons on orbit (moving charge) will counteract applied B0-field and generate a Lenz-field
BLenz. Nucleus “feels” sum of both. Lenz-field is function of local electron density (order
number z), anisotropy ( solid state NMR), hybridisation (e.g. sp3, sp2, sp)…
We expect small changes. Since we have no “naked” 1H or 13C we detect relative
differences.
Chemical standard is TMS ( 3 4CH Si 1H: = 0 ppm, 13C: = 0 ppm, 29Si: = 0 ppm)
Isotropic chemical shift sample TMS
TMS
B0
nucleusBohr orbit
BLenz

Molecules and characterisation 153
Range:
1
13
0 10 ppm H
0 200 ppm C
Note :
Since TeslaIHMHz42 and
1
13
H
C
4
a 1ppm changes means 300 Hz at 7 Tesla for
protons and 75 Hz for 13C, current limit: 1Hz ! (
up to 910 )4 ”theoretically”
3000 resonances in 1H and 15,000 resonances in 13C can be separated 13C better resolution
power (selectivity).
The isotropic chemical shift (“chemical shift”) is very sensitive to chemical environment, up
to 4-6 bonds away influence vanishes.
Samples:
%1 10wt solution in 5-10 mm test tube (1-100 mg), typically
Temperature:
-150°C up to 200°C typically
Spectrum can be integrated to achieve relative distribution of 1H (13C more complicated), up
to 1 relative accuracy
4 Record Mösbauer: 1410 !

Molecules and characterisation 154
Source: Skoog

Molecules and characterisation 155
For solution NMR, two more interactions are important called I -coupling and NOE-effect.
I -coupling, dimension: 1-200Hz, information about spin state transferred along the covalent
bonds, via s-electrons
Idea:
C
C
OHA
HH
HB
HB
H
Proton HA “sees” proton HB having the following magnetic quantum states:
or 1:
or 2:
1:
spectra HA:
“triplet” (others: singulet, doublet, quartet…)
NOE-effect:
Spin 12 systems can simplified be viewed as stick magnets.
Depending on orientation and distance r (6
1
r ) they
interact. It is possible to use this information to detect spatial
proximity ( 5A
)
N
S
B0
r
(1)
(2)
N
S
s-electrons “dive” into nucleus along the bond
Intensity 1 2 1 Pascale triangle
I

Molecules and characterisation 156
Decoupling:
The magnetic moments of e.g. 1H affect the 13C spectra. To switch off this influence one can
irradiate at 1H while detecting 13C,
2D-NMR:
The basic idea is to correlate resonances of different isotopes (e.g. 1H. 13C) and/or different
interactions (e.g. with or without decoupling, along the bond using J-coupling, through space
using NOE) in analogy to a joint probability 1 2P . This joint probability is displayed like
a chess board. The correlation is generated via complicated pulse-sequences that switch on
and off specific interactions. 2D-NMR answers questions like:
“Proton with 1 has covalent 1H neighbours with 2 ” COSY
“Proton with 1 has spatial neighbours with 2 ” NOSY
“Carbon with 1 has covalent 13C with 2 ” inadequate (NMR abbreviation)
Currently perhaps 100-300 NMR selection methods “pulse sequences” are known!
Experimental realisation (idea), assuming 1T
2 types of resonance, A+B
Pulse generates magnet. In x-y plane
1 2 3
3
2 1
1 1t
To via pulse
Magnetisation along z
1 is correlated
1
t
Direct detection
B
BA
B
A
assumption
sequence
2 2t

Molecules and characterisation 157
Spectra:
The information for resonance A during 1t is “transported” to 2t via the amplitude at 1 .
Variation of distance between the first two pulses transfers the whole information as
amplitude modulation to 2t (Ernst, Nobel prize 1991 idea: Jeener 1971).
Applications to polymers:
- Mode of addition (head to head, head to tail etc.)
- Stereo chemistry, tacticity
- Isomers of dien polymers (1,4 or 1,2 addition, cis-, trans-,…)
- Chain branching
- End groups
- Copolymer composition
- Sequences of monomers copolymerization constant ,1 2r r
- Coupling to GPC
Solid-state NMR:
Solid state NMR allows to study bulk properties (e.g. orientation, dynamics and chemical
structure) of polymers.
Lines are generally broader due to non-averaged motion and dipolar interaction, e.g. 1H-liquid-resonance:
1-100 Hz 1H-rigid-solid-resonance:
10-20 kHz, Gaussian shape
To achieve better resolution and sensitivity several hardware and HF-techniques might be
applied:
- CP, cross polarisation, transfer of e.g. 1H magnetisation to 13C using Hartmann-Hahn
condition
2
1
B
A
projections
1 2
0
0

Molecules and characterisation 158
- DD, dipolar decoupling, high intensity irradiation to reduce 1H 13C dipolar coupling
- MAS, fast rotation of sample (e.g. 7 mm rotor, 200 mg) along “most trivial”
orientation (1,1,1) ,54 7 relative to z-axis ”magic angle”. If rotation frequency is
fast, interaction is detected only as averaged quantity. rotor
2: 3 35kHz
mechanical
motion! MAS: magic angle spinning
- Solid-state NMR can (as liquid NMR) be performed in 1D, 2D, 3D and 4D!
4D: 4 frequency axis + 1 intensity axis
Solid state NMR application on polymers:
- quantification of crystallinity
- determination of orientation distribution e.g. fibres and films
- chemical assignment of insoluble or cross linked material
- dynamics in bulk material, e.g. spatial and dynamic heterogeneity for glasses
- motion in polymer crystals (helix jumps sec110 to sec1000 )
- spatial proximity in H-bond systems, DNA, bio polymers, fuel cell membranes…

Molecules and characterisation 159
Source: Diploma thesis Wilhelm, 1992

Engineering properties 160
5 Engineering properties
Polymeric materials are mostly used (and have to be processed) due to optical, mechanical
and electrical properties.
5.1 Mechanical properties
For melts, typically three measurements
.F v
A d Newton
A) shear rate dependant viscosity
linear homo polymer
: unit of inverse time 1s
: unit of pressure time Pas
0 : plateau viscosity, relaxation time ; 30 nn M , reptation model
: bending point “knee”, if applied shear rate exceeds inverse of longest relaxation
time
n : scaling exponent, polymer is frequently shear thinning . .n 0 7 0 9
Fit of shear rate dependant viscosity via 2-6 parameter models, e.g.
0n
1
log0
log
n
Bending point 1
d
v

Engineering properties 161
x t
sample
B) step experiment
G Hook in shear
:c correlation time, 3nM , if network : no decay to t 0
Very slow processes can be seen, since t can be measured for minutes to hours,
related frequency range
2 4
c
110 to10 Hz
C) oscillatory measurements (small amplitudes, , ,0 0G G G G )
idea: apply: sin0
1x t x t
d
sin0t t
What happens for: spring, elasticity (memory, storage)
G t Hook (1634-1703)
sin0G t in phase
- perfect viscosity, dash pot, viscosity (no memory, loss)
Newton (1642-1727): ; : constant
sin
cos
0
0
t
t
0 t 0 t
0 e.g.
c
t
e
Fast step, 0
Torque (time)
t
t
t
t
;t
t
0 , amplitude

Engineering properties 162
t
t t
( )t
cos ,0t t 90° out of phase
In “real” live superposition of spring and dash pot
e.g. Maxwell or Kelvin-Voigt
in series S DP S DP
S DP S DP
Many more combinations exist!
Separation of response in “in-phase” (=elasticity) and “out of phase” (=viscosity)
contributions
phase lag
tan
sin
sin cos
0
0 0G
G
t
t G t G t
:G storage modulus, shear
:G loss modulus, shear
Typical shape of ,G G for monodisperse linear polymer melt
length scales Rg 10-50nm ; Re 2-3nm
Large frequency range available via TTS, e.g. 10 decades
1
2
pG
pG
tan
I II III
IV
1
log
logG
analog
TTS
Temp.
G Pa Glass 109
Rubber plateau 106
flow
Polymer

Engineering properties 163
Zone I:
analog Maxwell-model
2
1
G
G
G G viscosity dominated at low frequency length scale probed: Rg
Longest relaxation time: 1 , for tan 1
Zone II:
G G elasticity dominated, physical networks of entanglements, maximum relative
elacticity at tan : minimum
Entanglement molecular weight via
e
RTM
G
, typical values: 3
kg1000m
,
, . , ekgJT 450K R 8 3 M 10molK mol
!5 6G 10 10 Pa memorize, rubber plateau
Plateau length in is strong function of nM since 3M
e.g. 1 2M 10M Zone II 3 decades larger
Zone III: Transition zone towards polymer glass
Zone IV: ,9G 10 Pa length scale probed 2 3nm , polymer glass
In solids, polymer materials are tested in elongation using test bones
Elongation 0
0
l l1
l , zero for no change
Deformation ratio: 0
l
l , one for no change
For small elongations: E
In shear: G-modulus, in elongation: E-modulus, Young’s
modulus
For simple elongation the following types of response can be identified in a stress-strain
curve:
5-10nm
0l
Force

Engineering properties 164
Yield point with y , yield stress and y yield elongation
Van-der-Waals bounds are destroyed, necking happens
: :SS maximum tensile stress, tensile strength
:S elongation at break
Young’s E-modulus are also determined under oscillatory conditions (DMTA: dynamic
mechanical thermo analyser), 1 frequency, T-variation, simple apparatus
E (storage) and E (loss)
1 0
2 0
2 0
x x
y y
z z
Simplify: 0 0 0x y z 1
Volume conservation, incompressibility: 0 0 0 0 1 0 2 0 2x y z x y z
21 21
21
1
; :1
22
1
(cross section area)
For Gaussian chain
, ,2 2 2
0 0 0x y z
1 0 0 0P x y z e and , ,
2 2 2 2 2 20 0 2 0 2x y z
2P x y z e
ln 1
2
PS P Boltzmann 1896
y s
Limit of linearity
Maximum: yield point
S
rupture
soft
Brittle and hard Tough and hard
Tough and soft
F F
0x 0y 0z
xyz
Uni-axial: y=z

Engineering properties 165
0 0 0
2 2 2 2 2 2 20 0 0 0 0 0
2 2 2 20 0 0
x y z 1
2 2 2
1 1S x y z x y z
1 1x 1 y 1 z 1
1 1 2 21 1 1 3 3
1, H 0 force S
22
12 1 2 2
2, G T S W Fd
2
modulus
force 1stress
area'
2
Young s
1E
Careful: for experimental reasons, the non-stretched area is used ( 0 0z y )
For simple model (incompressible, small, isotropic) it can be shown that
3 G E , more general:
EG
2 1
; .0 0 5
: Poisson number

Engineering properties 166
5.2 Dielectric properties
Since polymers are frequently used for electrical insulation the interaction of electric fields as
a function of frequency is practically importanti. From a spectroscopic point one can learn
something about molecular motions as function of frequency and temperature.
Principle (macroscopic):
i t0U U e applied voltage
i t0I I e electric current
UE
d electric field
Dielectric displacement, D
* , . 120 0
AsD E 8 9 10
Vm
Complex dielectric function (dielectric permittivity)
* i
Polarization:
*0 0 i
1P D D 1 E p
V
ip : individual dipole molecular quantity ip Q l , charge times distance
In contrast to pi is P a macroscopic quantity.
The individual dipole moments result from:
,p fast in time large:
1. electronic polarization, shift of electronic orbits
2. atomic polarization, shift of relative position of nucleus
studied, molecular motion , :
3. dipole polarization as result of orientation of permanent dipoles under influence of
electric field
the dielectric spectrum is an analogy to quasi-elastic light scattering (see there) and use of the
Wiener-Khinchine theorem the Fourier transform of the dipole autocorrelation function
i E.g. radar technology in WWII, superior dielectric properties of polyethylene

Engineering properties 167
*i t
FT0
g t e dt
Autocorrelation function:
2
P P 0g t
P
e.g. exp
t
called Debye relaxation
For Debye relaxation:
1 -(i +
0
complex conjugated
real part
e
)*
,
tt i t ti t
0 01
t
1i t
t 0
2
22 2
2 2
2
e e dt e dt dt
1i1 1
e1 1 1i i i
1i
1 1
1
part
,
2
imaginary
i
1
For dielectric spectroscopy a Debye relaxation is described by
21
21
: relaxation strength
To describe non-(single)-exponential relaxation, the most common equation used is the
Havriliak-Negami function (1966).
HN
HN1 i
: symmetric broadening of
: asymmetric broadening of
!!HN MAX but close
The local arrangement of dipoles in macromolecules was classified by Stockmeyer (1967):

Engineering properties 168
Typical spectrum amorphous polymer: type A:
e.g. Li+ from anionic polymerisation
Experimentally: 15 decades!
Normal mode:
end-to-end vector only for type “A” 3M , strong molecular weight dependence.
Segmental:
Glass transition, cooperative, non single Debye, T-dependant, apparent activation
energy called relaxation ( , , : increasing frequency), several monomers
( 1 3x nm), main chain involved, determination of Tg.
Local:
Single exponential, Debye, Arrhenius type (Ea) -relaxation, frequently side chain
motion.
Be careful, for semi crystalline polymers: : crystalline motion, : segmental, : local
Type “A” e.g. 1,4 cis Polyisoprene
end-to-end vector! Normal mode!
Type “B” e.g. PVC
Type “C”e.g. PMMA
-3 12 log10
Conductivity 1
Normal mode segmental
local
´´ , !T
glass

Engineering properties 169
5.3 processing of thermoplast; extrusion, injection molding, calendaring
For practical applications rarely pure polymer systems are used. Most of the time polymers
are combined with other polymers to form a blend (e.g. PS + Polybutadiene called HIPS, high
impact polystyrene), plasticizer, flame retardants, antistatic agents, fillers, blowing agents,
processing agents or pigments.
Plasticizer:
Reduce crystallinity, modulus (up to 103), viscosity, easier processable, similar effect
as raising temperature (but without degradation), example: DOP (dioctyl phthalate),
low Mn polyester, chlorinated polyester,…
Flame retardants:
Flammability serious problem, parameter used, e.g. limiting oxygen index (LOI) or
critical oxygen index (COI). Recommended: LOI > 0.27 (27% O2 in N2 per volume),
e.g.
PE: 0.18; PTFE: 0.95; PC: 0.27; PEO: 0.15
Halogens (Cl, Br) or phosphorous, trap radical production during burning.
Antistatic agent:
For specific use, static charging can not be tolerated, e.g. when dust ( explosions) is
present or sensitive parts (microcomputers) are involved agents. Carbon black,
carbon nanotubes (why?) and conducting surfaces (H2O, PVA) are used.
Fillers:
Two main groups:
o to improve mechanical strength composites, e.g. glass fibres in epoxy resin,
o to reduce the amount of polymers needed (called: extender) e.g. CaCO3, SiO2,
clay, wood, etc., must be cheap < 1 €kg
Blowing agents:
Production of open or closed cellular structure, expand plastics
( 3EPkg2 1000
m ), e.g.
o thermal decomposition ,2 2N CO
o heat + low boiling liquid ( ,5 12 6 14C H C H ), e.g. styropor
o chemical reaction, e.g. 2 2 2N C O H O NH CO
o gas expansion, foams can be very heterogeneous , , !x y z

Engineering properties 170
Processing agents:
To improve processing ability, e.g. salts of fatty acids, fluorinated alkanes, mineral
oils,…
Stabilizer:
Combination of heat + oxygen affects final properties (strength, colour yellow),
antioxidants or peroxide decomposers, PVC is thermally most problematic since HCl
acts as autocatalyst, example: sterically hindered phenols
Pigments:
To colour the material
To get a homogenious distribution, mixing is needed
Two main types:
- distributive mixing, number of particles constant, shape changes (surface area)
important is viscosity ration 2
1
and surface tension between two liquids
- dispersive mixing: break up of agglomerates and/or particles (e.g. carbon black for
rubbers)
balance between shear forces and surface tension is described by the “capillary
number”
; surface ,2 2shear shear sh
F F r A rA
2 1
shear
or elongation
or
shear
or elongation
or
Breakup of droplets
(satellites, misting)
shear
or elongation
or

Engineering properties 171
:r size
surface tension surface tesion STF F rl
2sh sh
aST ST
r rC
r
capillary number
Often elongation is more effective compared to shear for mixing
In processing, mixing is achieved via static or dynamic mixing mostly via extruders.
Extruders are snail pumps (analogue Archimedes screw). They are single or double crew type.
The double screw can be intermeshing or non intermeshing and co-rotating or counter
rotating.
Depending on friction of screw and wall the extruder is solely a pump or locally rotating the
melt. For optimum mixing the shaft should have low friction (polished) and the wall should
have high friction.
Generally an extruder transports the solid, melts it and pumps the material e.g. for injection
moulding.
To intensity mixing process special sections are included along the extruder, e.g. pinmixing,
pinbarrel extruder (QSM: Quer-Strom-Mischer) pineapple mixer, cavity transfer mixer, cok-
neader, shear torpedo, Maddock-Le Roy-mixer, congestion ring, kneading block, etc. …
After exiting the extruder a dye is responsible for the shape of the continuous extrudate. This
can be hollow or filled, spherical, quadratic, rectangular shape, covering of wires etc.
Since polymer melts exhibit a shape memory the dye shape and dimension is not necessarily
the product shape and dimension (dye swell, first normal stress coefficient rheology,
Weissenberg effect).
Energy consumption of extruder is highly reduced if processing is fast since .0 8 ,
upper limit are shear instabilities called shark skin, stick-slip and melt fracture.
The polymer exiting the dye are either cooled (air, water) and cut into pieces (granulate) or
directly put into shape via injection molding.
Note:
The outer screw diameter might vary between 16 and 900 mm, the output between
0.5kgh and 50,000 !5kg t5 10 yh (PE, PP, world scale). The mechanics of the extruder
is designed to last 100,000h (12y) with only 10% failure probability, for production purpose.
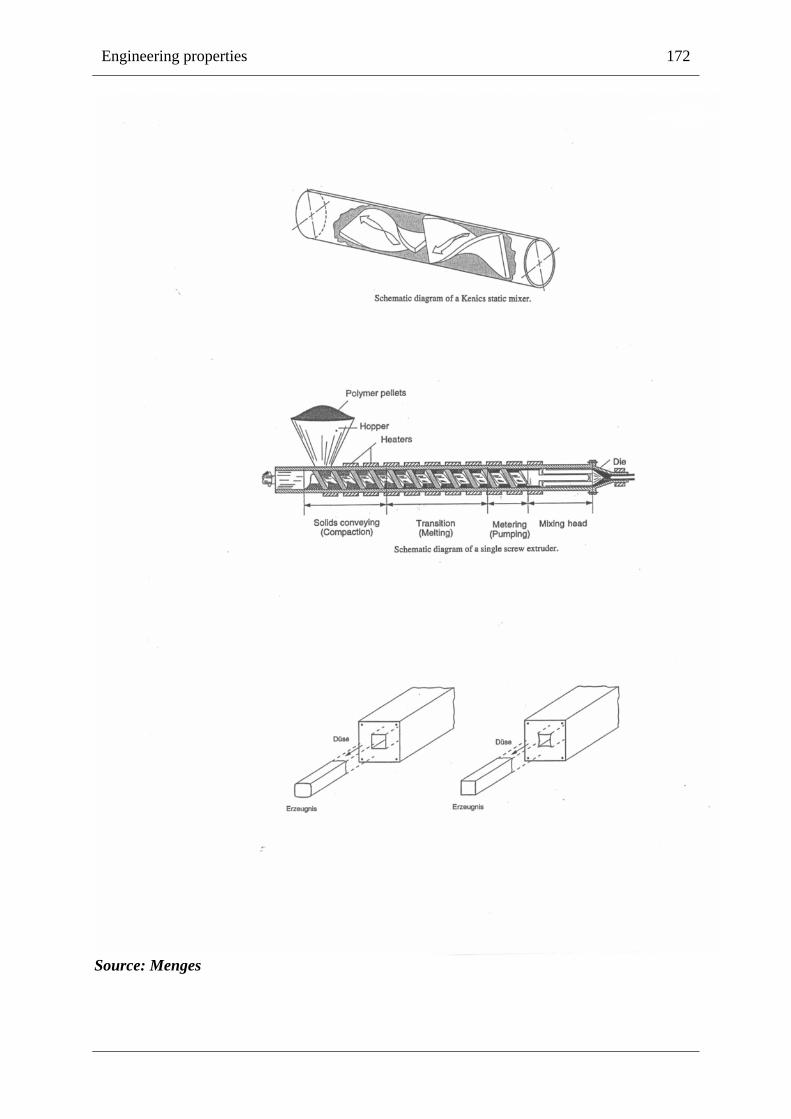
Engineering properties 172
Source: Menges
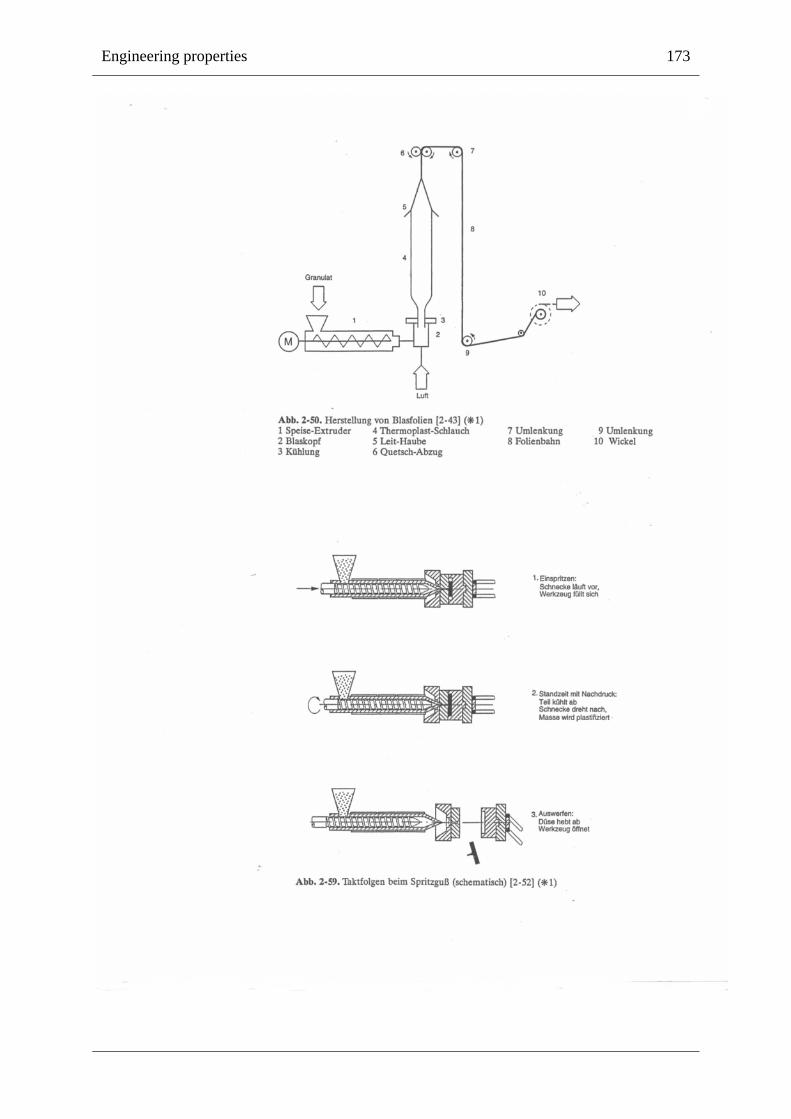
Engineering properties 173
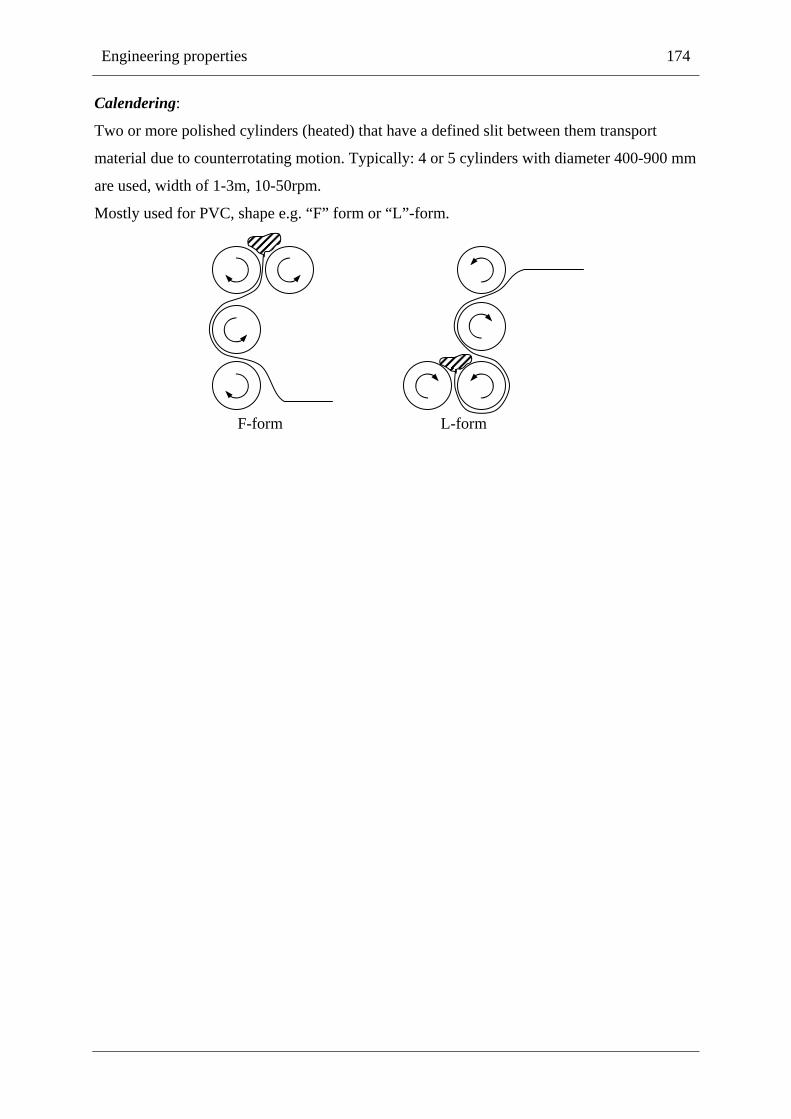
Engineering properties 174
Calendering:
Two or more polished cylinders (heated) that have a defined slit between them transport
material due to counterrotating motion. Typically: 4 or 5 cylinders with diameter 400-900 mm
are used, width of 1-3m, 10-50rpm.
Mostly used for PVC, shape e.g. “F” form or “L”-form.
F-form L-form

Engineering properties 175

Special topics 176
6.1 Polyelectrolytes
6.1.1 Definition, examples
Polyelectrolytes are covalent bound macromolecules which could carry charges (electric
monopoles). These charges are compensated via low molecular counterions.
Question: -[O-CH2-CH2]n- polyelectrolyte?
-[O-CH2]n-
Examples:
CH2 CH
NC
O
npoly(vinylpyrrolidon):
CH2 CHn
SO3-
CH2 CH
N+
n
CH2
CH2 CHn
COO-
poly(styrolsulfonacid)
poly(vinyl-pyridinium-bromide)
Br- Na+
CH2 Cn
COO-
poly(acrylacid)
poly(methacrylacid)
Na+
Na+
H2C
N+
CH3H3C
H2C
poly(diallyldimethyl-ammoniumchloride)poly(DADMAC)
CH3
Cl-
n
poly(acrylicacid)
poly(styrolsulfonicacid)
poly(methacrylicacid)

Special topics 177
6.1.2 Theory: Poisson-Boltzmann, Debye-Hückel, Skolnick-Fixman and Odijk…
simplified: polyelectrolyte = polymer + charges
+ +
+
+ + +
+ ++
+
+
- -
- ---
- -
-
- -
++
+
+ ++
+ ++
+
+
--
- -- -
- -
-
- -
Typical chemical groups:
-COO-, -CSS-, -O-SO3-, -SO3
-, -O-PO32-
-NH3+, -NRH2
+,-NR2H+, -NR3+
Subgroup of polyelektrolytes: ionene
Polyelectrolytes with quaternary ammoniumgroups in the main chain: "ionic amines", via "m"
adjustment of charge density
(CH2)m N+
R
Rn

Special topics 178
n = 24
n = 1
l n = 24 Entropy via conformational degrees of freedom
Coulomb-law:
lnr
AezyxW nl
r
2/12
2 2
2
),,(
TdSdHdG
constPTdGfdr
nl
rkcWkS
0
2
2
,;
2ln
Trdr
dSTf
20
21
4 r
QQF
r
Mean value for the maximum distance between
neighbouring ions and simple cubic lattice.
1:1 electrolyte:
mol/lin
nm1
nm94,0
m001,02
33
33
c
ccl
cNl L
e.g.: 10-3 M: 10 nm 1 M: 1 nm
++
+
+++
+ ++
+
+
--
- ---
- --
-
l

Special topics 179
Maxwell-equation (1865):
equation-isson Po
potential electric: ;
density charge : ;
0
2
0
r
r
E
E
:scoordinate spherical in :2
r
rrdr
d
02
2 11
Approximation of via Boltzmann-distribution, has the form of a screened Coulomb
potential
+ -
)()(
))(
1())(
exp((r)
0
homo
00
rkT
rQ
kT
rQ
kT
rQ
eff
equation aldifferenti-Hückel- Debyeequation, ellfirst Maxw
equation value-en Eig1 2
2
2
rrdr
d
nm 3r M,0.01c
nm 304,0
1r
:eelectrolyt 1:1for
function waveelectronic of despence radialfor polynome-Laguerre
(0,5Å) ,r radius- Bohratom,- Horbital,-1S :compare
length,- Debye:r ;/1
)exp(4
e-(r)
);exp(4
)(
D
D
B
D
2
)0(
0
c
r
rr
rr
qr
D
r
The source of the electric field are charges

Special topics 180
Bjerrum-length in water
Bjerrum-length lb:
At what length do we gain kT per molecule if we bring a charge from infinity close to another
charge? The medium is assumed to have a dielectric constant r.
Problems:
macroscopic dielectric constant r:
scalar?
polarization?
tensorial property?
as simple as possible
RT = 2,4 kJ/mol
r = 80
for distance < lb:
Manning condensation: reduction of the efficient charges density
please be aware:
Manning condensation Mannich reaction (organic chemistry)
lb
electrostatic interaction
+
lb + H O H
medium
drr
eekT
fdrW
bl
r
204
lb = 7 Å

Special topics 181
Extended theory
Skolnick-Fixman-Odijk (1977):
Additional stiffness via charges in the polymer backbone
uncharged:
charged:
d
l = l0 + le
L: contour length
l: persistenz length l = l0 + le
d: distance between charges
cs: salt concentration for large L/rD (high cs, large polymer chains),
incl. Manning condensation:
cs high pure polymer behaviour, no “charge” contribution
l0
2
2 2
2
1; large distance between charges, low charge density along chain
4
1; high charge density along polymer chain
4
b De b
S
De b
b S
l rl l d
d d c
rl l d
l c
++
+++ +
+++

Special topics 182
Slow mode (Exp.):
Dynamic light scattering on poly(vinylpyridin), g(t)= A exp (-q2Dt)
Aggregation? Cluster? Cooperativeness? Large domains?
Pearl-necklace (Sim.):
generation of local temporary aggregates along the polyelectrolyte chain
electrostatic energy hydrophobic energy
log cp/cs
1 0,1
10-9
10-7
slow mode
scm
D
/
log2
- -
-
-
-
- - -
- - -
- - -
-
-
- -
counterions
polymer with charges

Special topics 183
6.1.3 Experiments
1 g super absorber, polyelectrolyte, sap (super absorbing polymer)
1. + 150 ml dest. H2O ( beaker), approximately 2 min. stir, 2 min. wait 2. add salt (NaCl)
Remark:
- Isotonic NaCl solution; c = 9 g NaCl/l; 0,15 M, posmose = 7,5 bar; rD = 8 Å ( Bjerrum-
length)
- Ocean water: c = 30 g NaCl/l; 0,5 M; rD = 4 Å!!
- Experiments of Stanley Miller and Harald Urey, 1952 (Science 117 528 (1953), but: Loeb
1913! „Ursuppe“ made of H2O, H2, NH3, CH4 plus electric charges aminoacids; no
salts!

Special topics 184
6.1.4 Application
- baby diapers
- packing (food)
- sealant
- agriculture: but: 1 mm thickness 10.000 kg SAP/ha!! grain, sereals (e.s. rye, corn): 1 t = 100 EURO harvest: ~ 7 t/ha
Superabsorber
Swelling is influenced via physical or chemical influenced: pH, ions, temperature…
Intelligent gels
Swell up to 1000 – times of the own-weight
R=1
R=10
compare: Hobermann sphere as mechanical model

Special topics 185
Chemistry super absorbing polymers ( SAP)
afterwards: cut, dry, grind
particle size distribution: 150 - 850 µm
LD50/oral/rat: > 2g/kg!
polymer network (mesh) with carboxylate groups:
poly(acrylacid - co -acrylacidsalt)
further types:
- crosslinked poly(acrylate) or poly(acrylamide)
- cellulose- oder starck-acrylnitril-graft-copolymers
- crosslinked maleic acid anhydrid-copolymers
difunctional crosslinker:
alkylene-bisacrylamid

Special topics 186
trifunctional crosslinker: tetrafunctional crosslinker:
triallylamin tetraallyloxyethan Strukture: SAP 1. generation
optimization of water absorption
2. generation optimization of water uptake under load
3. generation combination of both advantages via cove-shell systems
small crosslink density, max swelling high amount of extractable polymer small gel strength ( G’)
high crosslink density appropriate absorption under load (AUL) high gel strength small capacity
highly crosslinked shell and weakly crosslinked cove high capacity good mechanical strength high AUL 4-generation? Including ionexchange
$$$ + Euros: SAP
assume:
8 x 109 people (average: 20 years old)
0 – 3 years kids have diapers
5 diapers per day
10 g sap per diaper, 1 kg sap ≈ 2 €

Special topics 187
6.1.5 Application: oil production
constraints:
oilreserves, definition: via drilling known, and currently useful with profit: 141 x 109 t
(1998)
consumption: 3,35 x 109 t/year factor 42
but: 1% higher yield compensates 1 - 2 years world consumption!
drill fluids:
per 1000 m increase:
P = 100 bar, T = 30 K
in 5000 m:
- 500 bar pressure
- 400 - 450 K
- high shear rates
- variable pH
- high salt concentration
approx. 1 - 3 % addition of polyelectrolytes
CH2 CH CH2n
HOOC
polyacrylamide, partly hydrolysed
CH
CONH2
m
CH2 CH CH2n
HOOC
CH
CONH2
mCH2 CH
COm
NH
C CH3H3C
CH2
SO3H
acrylamido-2-methylpropan-sulfonacid
OOHO
HO
OH-OOC
Na+O
O
HO OH
O
carboxymethylcellulose
O-OOC
Na+

Special topics 188
Scale-inhibitors
about 10 ppm, e.g.
demulgator:
+
H2C
N+
CH3H3C
H2C
Poly(diallyldimethyl-ammoiumchlorid)Poly(DADMAC)
Cl-
n
CH2 CHn
R3NOC
quart. Polyacrylamide
Mn: 104 - 107 g/mol
O CH2 CH2 OHC CH2 O
n m
O CH2 CH2 OnH
m
Ethylenoxid-Propylenoxid-Blockcopolymere, "Pluronics"
Alklyphenol-Formaldehydharze
Mn: 5 x 102 - 104 g/mol
CH3
CH2 CHn
HOOC
polyacrylacid
CH CHn
polymaleinacid
HOOCHOOC
CH2 CHn
COOH
CH2 CHm
SO3H
polyacrylat-polyvinylsulfonatcopolymer
N CH2 P
O
OH
OHH2CP
O
HO
OH CH2
PO OH
OH
amino-tri-methylenphosphonacid
ethylenoxid-propylenoxid-blockcopolymer, „pluronics“
alkylphenol-formaldehydresin
poly(diallyldimethyl-ammoniumchlorid) poly(DADMAC)
quart. polyacrylamide
polyacrylicacid

Special topics 189
6.1.6 Conclusion polyelectrolytes
- polyelektrolytes are more complex as polymers or electrolytes
- structure can partly be explained via superposition of Coulomb and entropic effects
Several reasons for complex behaviour:
- many particle interaction
- long distance electrostatic interaction
- no separation of involved time scales
- dielectric constant
- ions have volume
application polyelectrolytes:
- flocculation additives
- extraction of metal, oil
- adjustment of rheological properties
- storage of water
- reduction of energy needed for transportation (pumping-systems)
- blood plasma expander
- contolled drug release

Special topics 190
6.1.7 Literature
review:
1) Polelectrolytes in Solution; S. Förster, M. Schmidt; Advances in Polymer Science; 1995, 120, 51
2) Polyelectrolytes, Hanser Publisher München (1994); H. Dautzenberg, W. Jaeger, J. Kötz, B. Philipp, Ch. Seidel, D. Stscherbina; Advances in Chemical Physics, 1996, 94, 1; J.-L. Barrat, J.-F. Joanny; Theory of Polyelectrolyte Solutions; T. Radeva (Ed.);
3) Physical Chemistry of Polyelectrolytes; Surfactant Science Series; M. Dekker Inc.; Vol. 99; 2001
4) Enzyclopedia of Polymers Science and Engineering; Wiley 1990, 788; Concise
5) Intermolecular & Surface Forces; Academic Press; 1992; J. Israelachvili
6) Macroions in Solution and Colloidal Suspensions; VCH (1994); K.S. Schmitz
layer formation of polyelectrolytes:
1) G. Decher; Science 1997, 277, 1232
2) Ölindustrie: W. Gulden; Kein Erdöl ohne die Chemie; Chemie in unserer Zeit; 2001, 35, 82
3) J. P. Gerling, F.W. Wellmer; Wie lange gibt es noch Erdöl und Erdgas; Chemie in unserer Zeit; 2005, 39, 236
superabsorbing polymers:
1) www.gia.com/~cricher/history.htm
2) www.basf.de/de/corporate/innovationen/erklaert/baby

Special topics 191
6.2 Spatially heterogeneous systems, e.g. blends or blockcopolymers
6.2.1 Definition
In case two or more polymers are blended or covalently bound, e.g. blockcopolymers, they
can be homogeneous or heterogeneous with respect to their properties (e.g. mechanical,
chemical).
please distinguish:
homogeneous heterogeneous
- spatially dependant scalar property and
isotropic non isotropic
- orientation dependant vectorial (or tensorial) property
Question: can a homopolymer be heterogeneous?
can a homopolymer be nonisotropic?
Obviously the term homogeneous must depend on the observed length scale, e.g. everything
is heterogeneous on a length scale of 1-5 Å. Consequently this term becomes only meaningful
for length scales > 1nm.
In case of a homogeneous blend, crystallisation is often not possible and the common glass
transition temperature is influenced by both compounds (see chapter 3.2, Fox-equation).
6.2.2 Why does the introduction of heterogeneity make sense?
homogeneous polymer
crack
if force is applied
high local stress (force per area)
easy to propagate catastrophic failure
crack or craze propagates
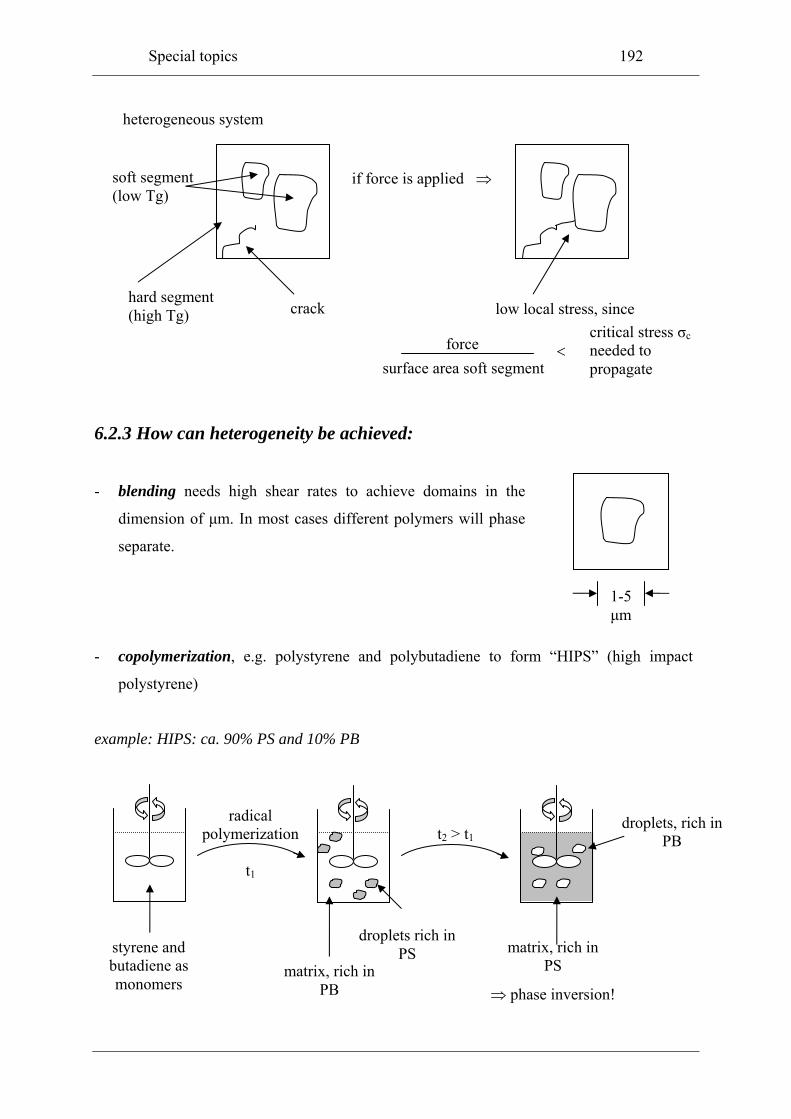
Special topics 192
6.2.3 How can heterogeneity be achieved:
- blending needs high shear rates to achieve domains in the
dimension of μm. In most cases different polymers will phase
separate.
- copolymerization, e.g. polystyrene and polybutadiene to form “HIPS” (high impact
polystyrene)
example: HIPS: ca. 90% PS and 10% PB
t2 > t1
droplets rich in PS
radical polymerization
t1
styrene and butadiene as monomers
matrix, rich in PB
matrix, rich in PS
phase inversion!
droplets, rich in PB
surface area soft segment
force
heterogeneous system
crack
if force is applied
low local stress, since
soft segment (low Tg)
hard segment (high Tg)
critical stress σc
needed to propagate
1-5 μm

Special topics 193
at the end “salami” morphology
Example: ABS – polymers
Acrylonitrile-butadiene-styrene polymer
H2C CH
C N
homopolymerH2C C
H
C N
nhead-tail + mostly isotactic
acrylonitrile
rigid: acrylonitrile + styrene
flexible: butadiene ̂ rubber part ( 1,2 and 1,4 cis + trans)
very variable due to: three component system, Mn, grafting, rubber particle size and
morphology, further monomers
synthesis: free radical polymerization of styrene and acrylonitril in
presence of polybutadiene (or PB-copolymer)
1μm
PS covered with PB
PS
additionally: grafting of PS on PB during
polymerization gives optimized
mechanical properties

Special topics 194
- block-copolymerization
[A]n - [B]m e.g. PS - PI diblock high Tg – low Tg
PS – PB – PS triblock high Tg – low Tg – high Tg
Mostly via anionic synthesis
From a mechanical point of view PB – PS – PB does not make sense, why?
Type of micro phase separation depends on volume fraction of the components, minimum
interface
Via Mn, the spacing can be adjusted typically in the range of 5 – 100 nm
RgB RgA
B
A
phase separation should be on
the order of Rg f (Mn)
sometimes gyroide
? ?
fB fA
fA › fB
columns
fA ≈ fB
lamellar
fB » fA
fB › fA
fA » fB
spheres
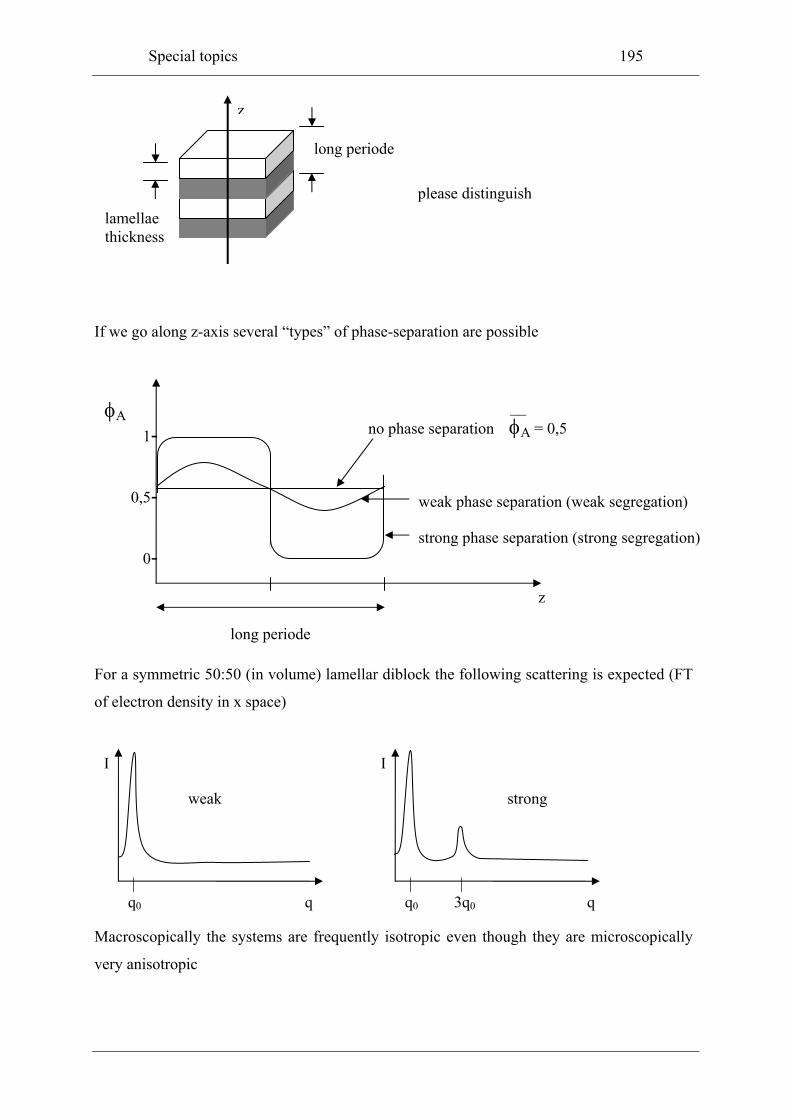
Special topics 195
If we go along z-axis several “types” of phase-separation are possible
For a symmetric 50:50 (in volume) lamellar diblock the following scattering is expected (FT
of electron density in x space)
Macroscopically the systems are frequently isotropic even though they are microscopically
very anisotropic
lamellae thickness
long periode
z
please distinguish
A 1-
0,5-
0-
long periode
weak phase separation (weak segregation)
strong phase separation (strong segregation)
__
z
I
| q0 q
I
| | q0 3q0 q
weak strong
no phase separation A = 0,5

Special topics 196
In case shear is applied there are three possibilities:
6.2.4 Theory of mixing, Flory-Huggins theory
In case we mix polymers, two types of curvature are in principal possible:
orientation
via - B-field - E-field - shear (elongation)
same long period!
| | | α β A volume fraction, A
Δ Gmix
or or
parallel perpendicular transversal
02
2
d
Gd
stable against demixing

Special topics 197
generally:
as function of temperature:
LCST: highest temp. where no phase separation takes place
UCST: lowest temp. where no phase separation takes place, sometimes kinetically hindered,
since Tg of one polymer makes structure to rigid
A volume fraction, A
Δ Gmix
stab
le
one
pha
se
met
asta
ble
met
asta
ble
unst
able
tw
o ph
ases
binodal spinodal
| | | α β A volume fraction, A
Δ Gmix
A volume fraction, A
T
LCST, lower critical solution temp.
UCST, upper critical solution temp.
binodal
mixed single phase
spinodal metastable
phase sep.
02
2
d
Gd
unstable against demixing
stab
le
one
pha
se

Special topics 198
In most cases polymer blends tend to phase separate due to conformation entropy:
Nevertheless several systems exist that do no phase separate, e.g.
- PS/PXE PXE: polyphenyleneoxide O
H3C
H3C
n
- PS/PVME PVME: poly(vinylmethylether) nCH2 CH
O CH3
- PMMA/PVF2 PVF2: polyvinylidenfluoride
H2C
F2C
n
- PMMA/PC
To understand why phase separation occurs, thermodynamic of solutions has to be
considered
- ideal solution of polymer in solvent
ΔGM = G1,2 – (G1 + G2)
G1,2: free enthalpy of solution
G1, G2: free enthalpy of solvent respective polymer
additionally we have:
ΔGM = ΔHM - TΔSM
ΔSM: always positive
ΔHM: for ideal solutions: ΔHM=0

Special topics 199
ΔSM is calculated via a lattice model (monomer in solution)
possibilities: !!
!
!!
!
2121
21
NN
N
NN
NN
using:
!!
!ln
ln
21
21
NN
NNkS
kS
M
M
using the Stirling approximation: ln ! lnN N N N
proof:
!ln N NN ln...ln3ln2ln1ln)...321ln(
1 1 1
ln ln lnNNN
x
x xdx x x x
since: ln 1
ln 1 lnd x x x
x x xdx x
!ln N Nxxx 1ln
11ln1ln0
NNN
NNNNNN ln1ln
q.e.d further:
AN
Nn 1
1 number of moles of “1”
AN
Nn 2
2 NA: Avogardo number, number of moles of ”2”
21
11 nn
nx
analog
21
22 nn
nx
, molefraction
1 2 3 4 5 6 7 8 9 here: 1 81 lattice position 2 Ο
3 5 monomers N2
4 Ο N1+N2=N
5 76 solvent molecules N1
6 Ο Ο 7 8 Ο 9

Special topics 200
!!
!ln
21
21
NN
NN !ln!ln!ln 2121 NNNN
212121 ln NNNNNN 111 ln NNN
222 ln NNN
2211 lnlnln NNNNNN
221121 lnlnln NNNNNNN
2211 lnlnlnln NNNNNN
N
NN
N
NN 2
21
1 lnln
N
N
N
N
N
N
N
NN
AAA
2211 lnln
2211 lnln xnxnN A
MS 2211 lnln xnxnR (1)
MS 1 1 2 2ln lnk N x N x (2)
for ideal case we assume 0 MH , therefore
MG M MH T S
MG 1 1 2 2( ln ln )RT n x n x
for polymers this formula is not sufficient, due to:
− polymer molecules are much larger than solvent molecules
− MH often 0
− monomers in a polymer are covalent-bounded, can not move fully free, joint
probability 1 2( | )P x x

Special topics 201
Flory-Huggins-Theory
theory for non-ideal polymer-solutions
MG M MH T S
a) : MS N1: number of solvent molecules
N2: number of polymer molecules
n: number of segments (?monomers?) in a single polymer
n·N2: volume occupied by N2 polymer chains
using (2)
MS 1 2
1 21 2 1 2
ln lnnumber
volume fraction
N nNk N N
N nN N nN
using the volume fraction i
1
11 2
N
N nN
2
21 2
nN
N nN
1 1
2 2
N
nN
note: 2 1 1 2nN N (3)
and NA we receive
MS 1 1 2 2ln lnR n n
b) :MH we have the following interaction energies
− solvent - polymer: 12
− solvent – solvent: 11
− polymer – polymer: 22
If we start to dissolve a polymer, one polymer – polymer contact, one solvent –
solvent contact is broken, but two solvent – polymer contacts are generated.
12 11 22
1
2
Please note: to have 0 (to be ideal) it is not needed to have 12 = 11 = 22 !
212 = 11 + 22 is already sufficient.
The total number of polymer – solvent contacts “p” for a rather concentrated, non-
ideal solution is
2 1p n N z
nN2: total number of segments

Special topics 202
z: co-ordination number
1: probability of having a lattice cell occupied by a solvent molecule (= volume
fraction)
using eq. (3) 2 1 1 2n N N
we receive 1 2p z N
therefore 1 2MH p z N
if we define z
kT
, knowing 1 1N k n R
1 2MH N kT
1 2MH n R T
to be put into M M MG H T S
1 1 2 2 1 2ln lnMG RT n n n (4)
The Flory-Huggins theory describes MG for non-ideal, but not for dilute solutions.
Eq. (4) becomes ideal again, if =0 and n=1 (n = number of segments in polymer)
1 1 2 2ln lnMG RT n x n x
is called Flory-Huggins parameter and is the normalized (to RT) local change in energy if a
polymer is dissolved.
If two polymers of the same degree of polymerization (n) are mixed, phase separation can
occure if n >2
0 0,5 1 A
G/R
T
0
!
·n>2
·n=2
·n<2

HE R M A N N S T A U D I N G E R
Macromolecular chemistry
Nobel Lecture, December 11, 1953
Macromolecular chemistry is the youngest branch of organic chemistry andas such has experienced the honour of the award of the Nobel Prize forChemistry. I sincerely hope that this great distinction will be the meanswhereby macromolecular chemistry will undergo further fruiful devel-opment.
Some few months after I had the opportunity of speaking in this audito-rium on the development of macromolecular chemistry into a new branchof organic chemistry at the International Congress for Pure and AppliedChemistry 1, it is today my duty to describe to you the characteristic featuresof macromolecular chemistry and demonstrate the new features which itintroduces into organic chemistry.
The macromolecular compounds include the most important substancesoccurring in nature such as proteins, enzymes, the nucleic acids, besides thepolysaccharides such as cellulose, starch and pectins, as well as rubber, andlastly the large number of new, fully synthetic plastics and artificial fibres.Macromolecular chemistry is very important both for technology and forbiology.
In common with all organic compounds, the structure of the organicmacromolecular compounds (inorganic macromolecular compounds arenot discussed in the following) involves in addition to carbon atoms, chieflyhydrogen, oxygen, and nitrogen atoms which in accordance with the lawsof Kekulé’s structural theory are bound by chief valences2. The only dif-ference between macromolecules and the small molecules of low molecularsubstances is one of structural size. If it is desired to lay down a boundarybetween macromolecular and low molecular compounds - there are ofcourse transitions linking the two groups - the substances with a molecularweight greater than ten thousand, i.e. the molecules of which consist of onethousand and more atoms, may be classified as macromolecular. Beyondroughly this size, characteristic macromolecular properties occur. So far noupper limits can be given for the size of the macromolecules. Macromolec-ular compounds with a molecular weight of several millions are known,

398 1 95 3 H .ST A UD IN GER
i.e. compounds in which one million and more atoms form the macro-molecules in the manner prescribed by Kekulé’s structural theory.
In recent decennia the field of macromolecular chemistry has been thescene of very intensive scientific and technical activity. I personally havebeen concerned3 with macromolecular chemistry since 1920 initially at theFederal Institute of Technology in Zurich. Since my move to the chemicallaboratory in Freiburg University I have devoted myself entirely to extend-ing this field which, since my retirement, has been further studied in a specialresearch institute in Freiburg.
In this work I have been assisted by a number of first-class colleagues whohave published valuable research on this field. Here I should like to mentionthe oldest of them, Signer4, now in Bern, whose support during the discus-sions on the structure of these compounds in the 1920’s was very valuableand who introduced the flow birefringence method for studying the particleshape of macromolecular substances. In addition Schulz, now in Mainz,extended the physico-chemical studies in the last two decennia, especiallythe molecular weight determination of macromolecular substancess. He alsoworked on the kinetics involved in the formation of macromolecular sub-stances 6
. Kern7, now in Mainz, studied the behaviour of polyelectrolytes.Husemann 8, Freiburg i. Br., studied polysaccharides, i.e. starch, wood poly-saccharides and, in conjunction with Schulz, the fine structure of cellulose9.Kohlschütter 10, now in Darmstadt, investigated topochemical reactionsusing polyoxymethylenes. Batzer 11 has in recent years successfully continuedCarothers’ work on polyesters. Hengstenberg18, Sauter13 and Plötzer14 car-ried out X-ray studies at different periods. Staudinger (M.)15 conductedmorphological studies of macromolecular substances, and introduced light,ultraviolet and electron microscopy - and for some time now, phase contrastmicroscopy as well - into macromolecular chemistry. In addition, over thelast 25 years in which almost 400 publications have appeared, she has collab-orated in these and a series of books. She is the originator in particular ofnew considerations in respect of the relations between macromolecularchemistry and biology16.
I cannot mention here the names of all the individual assistants and grad-uate students but today I remember with gratitude the assistance of all thosecolleagues who have participated in expanding this field.
Since, as I explained, macromolecular compounds are built up accordingto the laws of Kekulé’s structural theory in exactly the same manner as thelow-molecular organic compounds, i.e. they are genuine organic com-

M A C R O M O L E C U L A R C H E M I S T R Y 399
pounds but with particularly large molecules, the question arises whetherthere is any need at all to classify macromolecular chemistry as a new fieldof organic chemistry. It has been found, however, that owing to the sizeof the macromolecules, a whole series of new problems do actually arisehere so that in many respects macromolecular chemistry differs substantiallyfrom low molecular chemistry.
Even the classification of the macromolecular compounds is based onother criteria than in low molecular chemistry: the naturally occurringmacromolecular substances are conveniently treated separately from thefully synthetic compounds (Table 1).
Table I. Classification of macromolecular substances.
I. Substances occurring in nature1. Hydrocarbons - rubber, guttapercha, balata.2. Polysaccharides - celluloses, starches, glycogens, mannans, pectins, polyuronic
acids, chitines.3. Polynucleotides (nucleic acids).4. Proteins and enzymes.5. Lignins and tans (transition from low- to macromolecular substances).
II. Cowersion products of natural substancesVulcanized rubber, rayon, cellophane, cellulose nitrate, leather, lanital, galalith,etc.
III. Synthetic materialsPlastics (polyplastics) formed by
polymerization - buna, polystyrene, poiymethacrylic ester.polycondensation - bakelite, nylon, Perlon, Terylene.polyaddition - polyurethane.
For the low molecular compounds such a division is unnecessary and irrel-evant; the low molecular substances occurring in nature can largely bemanufactured synthetically and these synthetic compounds are indistin-guishable from the natural products. The situation is otherwise for the ma-cromolecular compounds. It has so far proved impossible to build a macro-molecular natural substance by a clear, stepwise synthesis from low molecularcompounds. Thus synthetic polyisoprene is not identical with, say, naturalrubber but has an essentially different constitution and hence other physicalproperties as well. In deriving macromolecular natural substances from veg-etable and animal material the original macromolecules are in many casesmodified to a greater or lesser extent by isolation and purification, and thus

400 1 9 5 3 H . S T A U D I N G E R
the macromolecules examined are not the same as those formed by Nature.Problems of this type prevail for instance in the production of cellulose,starch, and many proteins. Low molecular products on the contrary can beisolated from natural products without modification. It is furthermore pos-sible that Nature creates macromolecules of uniform size but positive proofof this fact has so far been found only in a few cases. In 1926 for exampleSvedberg I7 made the astounding discovery that a number of respiratoryproteins are monodisperse, i.e. that they are composed of uniformly largemacromolecules. Recently a whole series of enzymes and hormone proteinshave been crystallized so that here too the occurrence of natural productswith uniformly large macromolecules is very probable18.
Against that the technically manufactured transformation products of nat-ural macromolecular substances which can be termed semi-synthetic prod-ucts are invariably polymolecular mixtures since the original macromol-ecules are broken down to a greater or lesser extent during processing. Inall fully synthetic products, i.e. in plastics and fibres, there are also insep-arable mixtures of polymer homologues. It is thus impossible to preparecompletely uniform products by synthesis as was formerly considered nec-essary by the organic chemist to elucidate the constitution of organic com-pounds.
Macromolecular products are created in various ways. The first to deservemention is polymerization, constituting a particular chain reaction such asis unknown in low molecular chemistry19 (Formula 1). Owing to the
Formula I. Polymerization of styrene.

M A C R O M O L E C U L A R C H E M I S T R Y 401
technical importance of the polymerisates, e.g. polystyrenes, polymetha-crylic esters and polyvinyl chlorides, this reaction has been studied verythoroughly with reference to the kinetics and the influence of catalysts onthe course of the polymerization20.
In addition, macromolecular substances can be produced by polyconden-sation, a process which has long been known to technology and the onewhereby Baekeland secured the phenoplasts which are of unusual technicalimportance. This field was then further studied by Carothers21 and led tosuch technically valuable products as nylon (Formula 2).
Formula 2. Polyamides.
A further technique to produce macromolecular compounds evolved byO. Bayer is polyaddition with diisocyanates (Formula 3).
Formula 3. Polyurethanes.
In the most favourable case therefore, in the manufacture of fully syntheticproducts, uniform polymeric compounds are formed, i.e. mixtures of fil-amentary molecules of equal constitution but different length. Very recently,research conducted by Melville23 and others showed that polymerizationprocesses are frequently more complex than had formerly been assumed sothat the resultant products are mixtures of polymer isomers.
Since most natural macromolecular substances as studied are polymolec-ular mixtures in common with all synthetic macromolecular substances, a

402 1 9 5 3 H . S T A U D I N G E R
macromolecular substance must be identified by different criteria than a purelow molecular substance. Ostwald (Wi.)24, in his Analytical Chemistry, haspointed out that the task of the analytical chemist is facilitated by the factthat the agreement of just a few properties of two substances is sufficientfor regarding them as identical. This principle applies only for low molecularcompounds in which, owing to the small size of the molecules or ions,relatively large differences in the properties occur, i.e. they differ abruptlyfrom one compound to another. The situation is different for macromo-lecular compounds; a plastic such as, say, a polystyrene can be consistent withanother polystyrene in a number of essential properties and still differ incomposition. It is moreover also impossible to characterize and identifymacromolecular substances by the melting point and by the mixed melting-point test, a technique which has contributed greatly to the rapid successesin the identification of natural substances in low molecular chemistry.
Initially identifying the constitution of macromolecular compounds seem-ed to be further complicated by the fact that they do not yield normal mono-disperse solutions but that - if they dissolve at all - they mostly form poly-disperse, colloidal solutions. The procedure for elucidating the constitutionof organic compounds can again be referred to here: It consists in dissolvingthe particular compound after elementary analysis and then determining thesize and finally the intrinsic structure of the dissolved particles by furtherstudies. With completely insoluble substances occurring as a single aggregateonly25, details of the type of bond in the smaller groups in their moleculescan in many cases be disclosed by decomposition; but the molecular weightof such a substance is indeterminable. Against that, however, it may not beinferred from the insolubility of a substance alone that it has a particularly
Table 2. New classification of disperse systems by the number of atoms.
Increasing degree of dispersion.Coarse dispersions The particles are composed of more than 109
atoms, do not passthrough filter paper and can be made visible microscopically. They are poly-disperse.
Colloids The particles are composed of 103 to 109 atoms, pass through filter paper,cannot be resolved microscopically and cannot be dialysed. They are polydisperseor monodisperse.
Low molecular dispersions The particles are composed of 2 to 103 atoms, cannot beresolved microscopically, diffuse and dialyse readily. The molecules or ions ofuniform substances are identical in structure and size.

M A C R O M O L E C U L A R C H E M I S T R Y 403
high molecular weight, a fact which emerged during studies on amino-plasts26 in the laboratory at Freiburg.
When considering the various modes of dispersion of organic substancesin a liquid, it is expedient to indicate the size of the dispersed particles bythe number of atoms they contain, the convention27 adopted in Table 2.The particles of the low molecular organic compounds thus contain up toa maximum of 103 atoms and as a rule are identical with the molecules. Themolecule concept is defined here in the same way as in low molecular chem-istry: a molecule is the smallest particle in which all the atoms are linkedby chief valences2. It can readily be ascertained whether such particles con-.tain not molecules but associations or double molecules, as applies withe.g. solutions of fatty acids in benzene.
The particles of colloidal size which are composed of 103 to 109 atomscan have a much more varied structure than the particles made up of feweratoms (Table 3).
It follows from Table 3 that for one thing lyophobic colloids can occurwhich formerly roused special interest when Ostwald (Wo.)29 drew atten-tion to the "field of neglected dimensions" and pointed out that every sub-stance can be broken down into particles of colloid size by appropriate
Table 3. Classification of colloidal solutions of organic substances.

404 1953 H.STAUDINGER
dispersion. A further group of colloids are the lyophilic colloids to whichbelong on the one hand the micellar colloids and on the other the solutionsof macromolecular substances. Since macromolecules are the size of colloidparticles they can dissolve in no other way than colloidally, even if the sol-vent is changed, whereas with micellar colloids, e.g. the soaps, low molec-ular disperse solutions are also possible in certain solvents. The solutions ofmacromolecular substances may hence also be termed molecular colloids30.
Surprisingly, one group of macromolecular compounds, the linear macro-molecular substances, exhibit in many respects properties like those of typ-ical micellar colloids. In particular there is great similarity here to the soaps,the colloidal nature of which was recognized at the beginning of the centurythrough the work of Krafft31, Zsigmondy32, McBain33, and others. Sincethe solutions of linear macromolecular substances, however, differ substan-tially from the solutions of low molecular substances, it can be understoodwhy, a few decennia ago, the colloid particles in these solutions of macro-molecular substances were assumed to have a micellar structure similar tothose in aqueous soap solutions (Table 4).
Table 4. Properties of various substances.
Initially therefore, the efforts of the workers studying rubber, cellulose,starch, and proteins were aimed at determining the size of the moleculesmaking up these micells. A number of workers such as Karrer 34, Bergmann35,Pummerer 36 and Hess37 assumed them to be small molecules whereas Meyerand Mark38 held that these micells consisted of rather long chief valencechains.

M A C R O M O L E C U L A R C H E M I S T R Y 405
Notwithstanding this similarity in the behaviour of micellar colloids andlinear molecular colloids which derives from the elongated shape of thecolloid particles in both cases, there is a profound difference between thetwo groups: the particles of the micellar colloids are loose aggregates ofsmall molecules, in the case of the molecular colloids they are the macro-molecules themselves.
The proof was obtained by the conventional methods of organic chem-istry, i.e. by determination of the "macroradicals". In this context the word"radical" is used in the meaning given to it by Liebig in his studies of thebenzoyl radical. In many instances a polymeric compound can be trans-formed into derivatives of a different type without any change in the degreeof polymerization of the compound in exactly the same way as small mol-ecules can be transformed. A polymer compound can hence be transformedinto polymer analogous derivatives, the transformation proving that all thebasic molecules contained in the colloid particles of these polymeric com-pounds are linked together by chief valences, in other words the colloid par-ticles are macromolecules. This proof becomes especially clear because out ofa series ofpolymer homologues various ones can be transformed into polymeranalogous derivatives. Anexplanation can be given with reference to cellulose.
The bond of the glucose radical in cellulose was establishedby the studies of Haworth39 and Freudenberg40; furthermore Sponsler andDore4I demonstrated that the results of X-ray studies are consistent with thechain structure of cellulose. Subsequent studies then clarified the questionwhether the colloid particles in the solutions of celluloses and their deriv-atives have a macromolecular or micellar structure42.
Table 5. Comparison of the molecular weights of decomposed cellulose triacetates asdetermined by the osmotic and end-groups methods, and the Km-constants of these
compounds.

406 1 9 5 3 H . S T A U D I N G E R
In a polymer homologous series of decomposed low polymeric celluloseacetates, the molecular weight determined by the end groups agrees withthat determined by the osmotic method, proof that unbranched filamentarymolecules are present. The viscosity number of these compounds isproportional to the degree of polymerization as shown in Table 5.
The end-group molecular weight of higher polymer cellulose acetates cannot be determined. The relations between the degree of polymerizationdetermined by the osmotic method and the viscosity number are thesame, however, as in the low molecular compounds appearing in Table 5,testifying that these high polymer cellulose acetates are dissolved in the ma-cromolecular form and their chains are unbranched (Table 6).
Table 6. Determination of Km- constants of higher polymericcellulose triacetates in m- cresol.
With care, these cellulose acetates can be saponified to polymer analogouscelluloses, so proving that both the cellulose acetates and the celluloses arepresent in solution in the macromolecular state43 (Table 7).
Finally, with care being exercised, polymer homologous celluloses can benitrated to polymer analogous cellulose nitrates with a mixture of nitric acidand phosphoric acid, so demonstrating the macromolecular structure of thecellulose nitrates44 (Table 8).
The studies conducted at the Freiburg laboratory have frequently been con-cerned with determining macromolecular structure by polymer analogoustransformations of this type. By that means it has been proved for furtherpolysaccharides, i.e. starch’s, glycogen46, mannan47, and for a number ofplastics that the colloid particles in their solutions are identical with the ma-
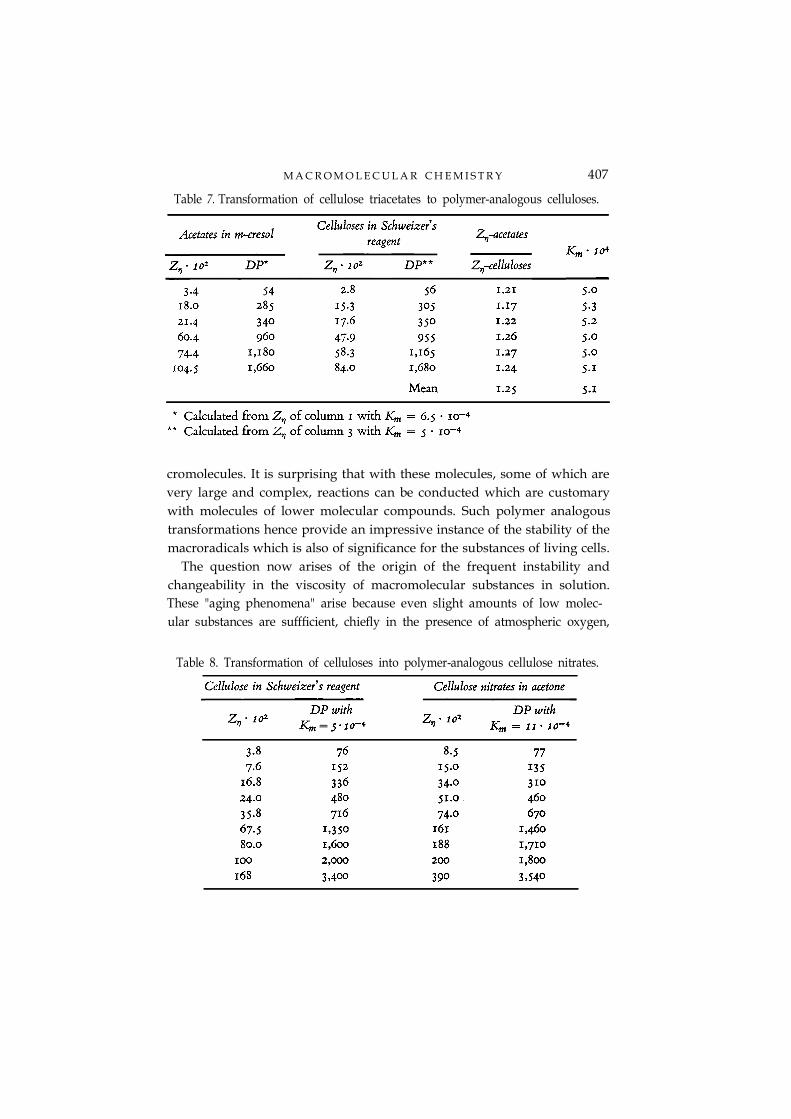
M A C R O M O L E C U L A R C H E M I S T R Y 407
Table 7. Transformation of cellulose triacetates to polymer-analogous celluloses.
cromolecules. It is surprising that with these molecules, some of which arevery large and complex, reactions can be conducted which are customarywith molecules of lower molecular compounds. Such polymer analogoustransformations hence provide an impressive instance of the stability of themacroradicals which is also of significance for the substances of living cells.
The question now arises of the origin of the frequent instability andchangeability in the viscosity of macromolecular substances in solution.These "aging phenomena" arise because even slight amounts of low molec-ular substances are suffficient, chiefly in the presence of atmospheric oxygen,
Table 8. Transformation of celluloses into polymer-analogous cellulose nitrates.

408 1 9 5 3 H . S T A U D I N G E R
to decompose the linear macromolecules, leading to a profound change inthe solution viscosity. This is shown in Table 9.
Table g.
Initially, as was the case for instance with rubber, these aging phenomenawere particularly obscure because under certain conditions oxygen decom-poses the filamentary molecules but can also link them together, this be-coming apparent in an increase of viscosity and, should the linking of thefilamentary molecules proceed further, it causes the soluble rubber to changeinto insoluble rubber. For this reason even polymer analogous transforma-tions involving macromolecular substances are mostly to be conducted onlyunder special experimental conditions since atmospheric oxygen for in-stance must be completely excluded. The tests are also complicated by thefact that the transformation products cannot be purified since all substancesare polymolecular and reprecipitation together with purification would leadto a change in the composition of the polymolecular mixture. These com-plications do not exist where low molecular substances are involved.
With the most important group of macromolecular compounds, the pro-teins and nucleic acids, scarcely any polymer analogous transformations haveso far been performed; here too it is difficult to produce polymer homol-ogous series. The size and structure of the macromolecules have to be deter-mined by different methods from those applied with the polysaccharides,rubber, and the plastics. The molecule concept as formulated for homopolarorganic compounds is not applicable here without limitation since besideschief valences, very powerful secondary valences also participate in the struc-ture of the particles.
The proof for the macromolecular structure of colloid particles is offundamental importance since it signifies that these colloids cannot be con-sidered in terms of the colloid doctrine as variable associations of small mol-ecules but rather as macromolecules which must be treated by the methods oforganic chemistry in common with molecules of low molecular substances48.

M A C R O M O L E C U L A R C H E M I S T R Y 409
There are still further profound differences between macromolecular andlow molecular compounds: these are based primarily on the fact that theshape of the macromolecules affects the physical and chemical properties ofthe substances considerably more strongly than is the case with the low mo-lecular compounds. Whereas, for example, normal nonane with elongatedmolecules, and tetraethylmethane with spherical molecules, both hydro-carbons having the composition C9H20, scarcely differ in terms of their prop-erties, a glycogen in which 5,000 glucose radicals are linked to form asphere has properties differing fundamentally from those of a cellulose inwhich the same number of glucose radicals are arranged in a long chain.This state of affairs led to the macromolecular substances being classified intotwo large groups, spheromacromolecular substances, i.e. substances withspherical molecules, and linear macromolecular substances with filamentarymolecules. These two classes are, of course, bridged by transitions: a largenumber of natural and synthetic substances have heavily branched macro-molecules, e.g. starch4 5. Table 10 indicates the significant bearing of theshape on the physical properties of natural macromolecular substances49.
Table 10. Spherical and filamentary polysaccharides and proteins.

410 1953 H . S T A U D I N G E R
In appearance, solubility, and in their further behaviour, macromolecularsubstances with spherocolloid molecules scarcely differ from low molecularsubstances; only the determination of the molecular weight proves thatmacromolecules are involved. The striking properties of macromolecularsubstances which, as listed in Table 4, caused a different type of micellar struc-ture to be assigned to these compounds, occur only with the linear macro-molecules; these include the natural and fully synthetic fibres, the rubber-elastic substances and many of the most important plastics. Linear macro-molecular substances with long filamentary molecules behave in a manneralien to low molecular substances: inclusion and swelling phenomena, highsolution viscosity, abnormal flow behaviour, etc. Since the constitution ofthese substances is known, their striking behaviour can be ascribed to thelength of the filamentary molecules. A comparison of various examples ofa polymer homologous series shows that as the filamentary molecules be-come longer, their properties change so profoundly that the earlier conceptswhich led to the conclusion that the units at the beginning and at the endhad a completely dissimilar structure (Table 11) can be appreciated.
Table II. Relation between physical properties and mean degree of polymerization(DP) of polymer homologous celluloses.

M A C R O M O L E C U L A R C H E M I S T R Y 411
It was therefore particularly difficult to study these linear macromolecularsubstances. Even to determine the molecular weight, for example, it was notpossible to apply the customary physical techniques such as the osmotic,diffusion, or Svedberg ultracentrifuge methods until the abnormal behaviourof the solutions of these substances could be clarified50.
The viscosity behaviour of the linear macromolecular substances is partic-ularly striking. Whereas Einstein’s law applies to the spheromacromolecularsubstances such as ovalbumin or glycogen, in the case of solutions of sub-stances with filamentary molecules, the viscosity of solutions of equal con-centration increases with the chain length, and with some of these substances,especially polysaccharides, there is a proportionality between the viscositynumber Zh, and the chain length n or the degree of polymerization P of fil-amentary molecules. This viscosity number is the specific viscosity of asolution, i.e. the increase in viscosity caused by 1 g in one litre, or else itslimit value. Since viscosity measurements are simple to perform, this methodhas been extensively adopted in technology to determine e.g. the degree ofpolymerization of celluloses and cellulose derivatives, as this parameter hasa considerable bearing on the fibre properties as shown in Table 11. Withother linear macromolecular substances such as e.g. the polyesters11 andmany polyvinyl derivatives, the relation between viscosity number Zh anddegree of polymerization is not as simple as with the polysaccharides, al-though there is a functional relation corresponding to an equation formulatedby Kuhn51 (Table 12).
Table 12. Viscosity relations.
The reasons. are as follows: the filamentary molecules of a linear macro-molecular substance are usually elongated in the solid crystalline state. Thelength of the elementary units and hence the length of the filamentary mol-

412 1 9 5 3 H . S T A U D I N G E R
ecules can be determined by X-ray examination, e.g. in the case of cellulose.On dissolution, owing to vibrations and intramolecular forces of attractionbetween the individual groups of the filamentary molecule, the latter be-comes convolute to a greater or lesser extent depending on the temperatureand also on the type of solvent. Thus the filamentary molecules in goodsolvents are more elongated than in poor solvents; in the former the viscos-ity number of a substance is higher than in a poor solvent. Kuhn 52 statesthat the entanglement of a filamentary molecule can be so pronounced ina poor solvent that the molecule assumes an almost spherical configurationsuch as has been experimentally determined with the polyisobutylenes53. Afilamentary molecule would adopt that configuration in the gaseous statealthough in practice this state is unattainable, since the boiling point of allmacromolecular substances lies far beyond the point at which they decom-pose.
Besides these reversible changes in shape which are governed by the na-ture of the filamentary molecule and the type of the solvent, chemical actionalso brings about irreversible changes in shape which permanently affect thephysical properties of a linear-macromolecular substance. Thus it is commonknowledge that owing to its long, filamentary molecules, rubber yieldshighly viscous solutions with swelling. Against this, the solutions of chlo-rinated rubber, which is used as a paint, are of relatively low viscosity andtherefore it was originally assumed that in the transformation from rubberto chlorinated rubber the long filamentary molecules were decomposed.Such is not the case as the degree of polymerization of chlorinated rubberis approximately the same as that of rubber54. Thus, when rubber is con-verted into chlorinated rubber, no decomposition takes place but rather astrong, chief valence cyclization which induces a permanent convolution ofthe filamentary molecules (Table 13).
Table 13. Polymer-analogous transformation of rubber and balata tochlorination products.

M A C R O M O L E C U L A R C H E M I S T R Y 413
Fig. I. Swelling of a piece of polystyrene with divinyl bonds. Before swelling (left).After swelling in benzene (right).
It is - as already mentioned - characteristic of the chemistry of macromo-lecular materials that the smallest amounts of substances are capable ofmodifying profoundly their physical properties. Hence a 0.0025% additionof divinyl benzene to styrene is sufficient during polymerization to linkthe chains of the polystyrene by divinyl benzene bonds55. Soluble poly-styrene with its unlimited swelling capacity is thus transformed into a va-riety with limited swelling capacity which absorbs solvents by solvationand so swells without altering its shape (Fig. I) and without being able todissolve.
The replacement of a hydroxyl group in low molecular compounds bya methoxyl group alters the physical and, above all, the chemical behaviourof the compound considerably. The same applies also to macromolecularsubstances except that the percentage proportion of a methoxyl end groupin the macromolecule can be so small that it readily escapes detection.Auerbach and Barschall56 described two polyoxymethylenes: one is sol-uble in sodium hydroxide, the other insoluble, yet they are identical inappearance. The product soluble in sodium hydroxide is a polyoxymethyl-ene dihydrate, the insoluble product a polyoxymethylene dimethylether57
(Formula 4).The slight percentage methoxyl end-group content thus blocks the de-
composition of the chains by sodium hydroxide. This reagent can only at-

414 1 9 5 3 H . S T A U D I N G E R
tack hydroxyl groups and so dissolve polyo-xymethylene dihydrate. In bio-logical processes as well, a slight percentage change in a macromoleculecan bring about profound changes in the chemical and physiological behav-iour of the macromolecular substance.
Possibly the most important distinction between low molecular and ma-cromolecular compounds is that the latter can exhibit properties whichcannot be predicted even by a thorough study of the low molecular sub-stances. This may be illustrated by comparing the organic molecules withbuildings in which the bricks must be joined together systematically. Witha few bricks it is impossible to erect a great variety of buildings; nevertheless,provided that 10,000 or 100,000 bricks are available it is quite possible toconstruct the most diverse buildings, viz. houses, halls, etc., the special con-struction of which cannot simply be predicted from the buildings com-prising few bricks.
One such new type of behaviour of macromolecular substances are, forexample, the swelling phenomena of the linear macromolecular substancescaused by the solvation of the long filamentary molecules on the additionof solvent without immediately being able to go into solution. These swel-ling phenomena are complicated, as has been described, in that linking ofthe filamentary molecules induces a substance with unlimited swelling capac-ity to change into a substance of restricted swelling capacity55, this also beinga very potent factor with proteins. A further instance here are inclusionphenomena. Inclusion is an indirect swelling of linear macromolecular sub-stances with liquids that are not solvents. They have mainly been studiedin the case of cellulose, between the filamentary molecules of which inertliquids such as benzene, carbon tetrachloride, and others can be embedded.It is noteworthy that these liquids cannot be altogether eliminated from thelinear macromolecular substance even under a high vacuum; they are heldmechanically between the filamentary molecules of the substances. As a

M A C R O M O L E C U L A R C H E M I S T R Y 415
Table 14. Amount of solvent included in mercerized cotton (DP 1,600) after dryingfor two days under high vacuum at 100°C.
result of this inclusion, however, the reactivity of cellulose is appreciablyraised58 (Table 14).
A particularly significant phenomenon with linear macromolecular sub-stances is a characteristic state of dissolution which is impossible with sphero-macromolecular and low molecular substances: the state in which the fil-amentary molecules are completely solvated, i.e. essentially dissolved, butowing to their large bulk, their range of action, they have no free mobility.The term "gel solution" has been proposed for this characteristic state ofdissolution which is intermediate between the normal state of dissolutionand that of swelling 5 9. The solutions of cellulose, cellulose derivatives andplastics as used technically are gel solutions.
It will be the task of macromolecular chemistry to examine further thesenew, characteristic properties which are governed by the size and configura-tion of the macromolecules since it will thus be possible to gain fresh insightinto biological processes as well.
The existence of macromolecules and the steadily deepening knowledgeof their properties have revealed the nature of the building units which theliving cell requires to create matter 1 6. The existence of macromoleculesmakes possible the vastly wide variation of the substances required; thus,for example, the very number of the isomers in protein molecules is prac-tically infinite. Assuming a protein of molecular weight 100,000 and com-

416 1953 H.STAUDINGER
posed of 20 different amino acids, the number of its isomers is 10 1270. Thesize of this number becomes clear when compared with the number of mol-ecules of water present in the seas of the earth - a mere 10 46 (Table 15).
Table 15.Number of isomers of a protein having a molecular weight 105; the protein moleculecomposed of 50 molecules of each of 20 different amino acids.
Number of isomers: 101270.
For comparison the number of molecules of water in the seas of the earth:Volume of the seas is about 1.3 x 1024 cm3; 18g water contain 6 x 1023 molecules.
The seas thus contain about 4 x 1046 molecules.
It is thus possible by isomerism alone to create an infinite number of sub-stances. This number is increased still further by variation in the configura-tion of the macromolecules while they are being built, or else by theinfluence of their environment. So, each living organism can create its ownnucleic acids, protein molecules, etc. This conclusion is necessary since everyorganism, and every human being too, differs chemically from another.
Associated with this boundless profusion of matter in the macromolecularsphere, in contrast, is a remarkable stability of the macromolecules, a stabil-ity which is governed by their structure as organic compounds in compliancewith the laws of Kekulé’s structural theory. That alone is capable of satisfyingthe "inconceivably strict demands on the integrity of the germ plasm"which are applied to the normal process of development of an organism 60.This stability of the macromolecules, associated with their reactivity, sup-plies the living substance with the necessary basis for so specific a process asthat of heredity.
The known facts of macromolecular chemistry show further that anindividual macromolecule is still not "living", however large it is and how-ever complex its structure. On the contrary, the term is relevant to a certainamount of substance comprising numerous macromolecules with the con-stituent small molecules combined together in strictly prescribed order, an"atomos" of living matter16 which is indivisible without losing its "living-ness". Living nature supplies the answer to the question how large such an"atomos" has to be in the form of the various germ cells, for nothing canbe removed from such a cell, whether a spore or a fertilized ovum, otherwiseno normal organism can be formed from it. One of the smallest units capableof se&propagation is, for example, a bacterial spore. The following estimate

M A C R O M O L E C U L A R C H E M I S T R Y 417
Table 16. A bacterial spore of 0.124p diameter weighs 10-15g (sp.gr. = I) and afterremoval of 50% water content comprises 5·107 atoms.
may afford an idea of what numbers of macromolecules and molecules arethe necessary minimum for "livingness" here (Table 16).In contrast to former opinions one of the smallest living units thus requiresa quantity of substance.
In this way macromolecular chemistry appears today to fit between lowmolecular organic chemistry and cytology. It is the connecting link betweenthem, growing systematically out of low molecular chemistry but, with theincomparably larger wealth of its chemical scope, forming living matter. Inaddition, over and above the quantitative laws of pure chemistry, macro-molecular chemistry makes use of a number of qualitative correlations:those of shape and of the associated configurational scope, up to the levelof the "atomos" of living substance, on which the game of Life ensues.
In the light of this new knowledge of macromolecular chemistry, thewonder of Life in its chemical aspect is revealed in the astounding abundanceand masterly macromolecular architecture of living matter.
1. H. Staudinger, Chem. Ztg., 77 (1953) 679.2. H. Staudinger, Die Chemie der organischen hochmolekularen Stoffe im Sinne der
Kekuléschen Strukturlehre, (Vortrag auf der Versammlung Deutscher Natur-forscher und in Dusseldorf), Ber. Deut. Chem. Ges., 59 (1926) 3019.
3. H. Staudinger, Ber. Deut. Chem. Ges., 53 (1920) 1073.H. Staudinger and J. Fritschi, Helv. Chem. Acta, 5 (1922) 785.
4. R. Signer, Z. Physik. Chem., A 150 (1930) 257, and subsequent papers.5. G. V. Schulz, Z. Physik. Chem., B 30 (1935) 379; B 32 (1936) 27; A 176( 1936) 317,
and subsequent papers.6. Cf. G. V. Schulz in Röhrs-Staudinger-Vieweg, Fortschritte der Chemie, Physik und
Technik der makromolekularen Stoffe, Vol. 1, Verlag Lehmann, München, 1939, p. 28.

418 1 9 5 3 H . S T A U D I N G E R
7. W. Kern, Z. Physik. Chem., A 181 (1938) 249, 283; A 184 (1939) 197, 302, andsubsequent papers.
8. E. Husemann, J. Prakt. Chem., 155 (1940) 13,241, and subsequent papers.9. G. V. Schulz and E. Husemann, Z. Physik. Chem., B 52 (1942) 23, and subsequent
papers.10. H. W. Kohlschütter, Liebigs Ann. Chem., 482 (1930) 75; 484 (1930) 155, and
subsequent papers.11. H. Batzer, Makromol. Chem., 5 (1950) 5, and subsequent papers.12. J. Hengstenberg, Arm. Physik, 84 (1927) 245 ; H. Staudinger, H. Johner and R.
Signer, G. Mie, and J. Hengstenberg, Z. Physik. Chem., 126 (1927) 425, and sub-sequent papers.
13. E. Sauter, Z. Physik. Chem., B 18 (1932) 417, and subsequent papers.14. E. Plötze and H. Person, Z. Physik. Chem., B 45 (1940) 193.15. M. Staudinger, Chem. Ztg., 67 (1943) 316, and other papers.16. M. Staudinger, Makromolekulare Chemie und Biologie, Verlag Wepf & Co. Basel,
1947.17. The Svedberg, Z. Physik. Chem., 121 (1926) 65; The Svedberg and K. 0.
Pedersen, Die Ultrazentrifuge, Verlag Steinkopff, Dresden, 1940.18. J. H. Northrop, Crystalline Enzymes, New York, 1939.19. H. Staudinger and W. Frost, Ber. Dart. Chem. Ges., 68 ( 1935) 2351; G. V. Schulz
and E. Husemann, Z. Physik. Chem., B 34 (1936) 187, and subsequent papers.20. L. Küchler, Polymerisationskinetik, Springer Verlag, Berlin, 1951.21. W. H. Carothers, Collected Papers, New York, 1940.22. O. Bayer et al., Angew. Chem., 62 (1950) 57.23. J. C. Bevington, G. M. Guzman, and H. W. Melville, Nature, 170 (1952) 1026.24. Wi. Ostwald, Die wissenschaftichen Grundlagen der analytischen Chemie, 2nd ed.,
Leipzig, 1897, p. 3.25. H. Staudinger et al., Liebigs Ann. Chem., 474 (1929) 168.26. H. Staudinger and K. Wagner, Makromol. Chem., 11 (1953) 79.27. H. Staudinger, Organische Kolloidchemie, Verlag Vieweg, Brunswick, 1950, 3rd
ed.; French edition, Dunod, Paris, 1953.28. H. Staudinger, Zur Nomenklatur auf dem Gebiete der Makromoleküle, Makromol.
Chem., 9 (1953) 221.29. Wo. Ostwald, Die Welt der vernachlässigten Dimensionen, 1st ed., Verlag Steinkopff,
Dresden, 1914.30. H. Staudinger, Ber. Deut. Chem. Ges., 62 (1929) 2893.31. F. Krafft and A. Stem, Ber. Deut. Chem. Ges., 27 (1894) 1747 et seq.32. R. Zsigmondy and W. Bachman, Kolloid-Z., II (1912) 145.33. J. W. McBain, Advan. Colloid Chem., 5 (1944) 102.34. P. Karrer, Helv. Chem. Acta, 3 (1920) 620; P. Karrer et al., Helv. Chem. Acta, 4
(1921) 185, 263 ; P. Karrer, Einführung in die Chemie der polymeren Koklenkydrate,Leipzig, 1925, pp. 4 and 8.
35. M. Bergmann, Angew. Chem., 38 (1925) 1141; Naturwiss., 14 (1926) 1224; Ber.Dart. Chem. Ges., 59 (1926) 2973.
36. R. Pummererer, Ber. Deut. Chem. Ges., 60 (1927) 2167.

MACROMOLECULAR CHEMISTRY 419
37. K. Hess, Chemie der Cellulose, Leipzig, 1928, p. 590,38. K. H. Meyer and H. Mark, Ber. Deut. Chem. Ges., 61 (1928) 593, 1939.39. W. N. Haworth, The Constitution of Sugars, London, 1929.40. K. Freudenberg, Tannin, Cellulose, Lignin, Berlin, 1933.41. 0. L. Sponsler and W. H. Dore, Colloid Symposium Monograph, 126 (1926) 174.42. H. Staudinger, Svensk Kem. Tidskr., 49 (1937) 3 ; Suomen Kemistilehti, A 24 (1951)
III.43. H. Staudinger and G. Daumiller, Liebigs Ann. Chem., 529 (1937) 219.44. H. Staudinger and R. Mohr, Ber. Deut. Chem. Ges., 70 (1937) 2302.45. H. Staudinger and E. Husemann, Liebigs Ann. Chem., 527 (1937) 195.46. H. Staudinger and E. Husemann, Liebigs Ann. Chem., 530 (1937) I.47. E. Husemann, J. Prakt. Chem., 155 (1940) 241.48. H. Staudinger, Die hochmolekularen organischen Verbindungen, Kautschuk und Cel-
lulose, Springer Verlag, Berlin, 1932.49. H. Staudinger, Ber. Deut. Chem. Ges., 68 (1935) 1682.50. H. A. Stuart, Die Physik der Hochpolymeren, Vol. 2: Dar Makromolekül in Lösungen,
Springer Verlag, Berlin, 1953.51. W. Kuhn, Angew. Chem., 49 (1936) 858; W. Kuhn and H. Kuhn, Helv. Chem.
Acta, 26(1943) 1394;28 (1945) 1533.52. W. Kuhn, Helv. Chem. Acta, 32 (1949) 735.53. H. Staudinger and H. Hellfritz, Makromol. Chem., 7 (1952) 274.54. H. Staudinger and Hj. Staudinger, J. Prakt. Chem., 162 (1943) 148.55. H. Staudinger and W. Heuer, Ber. Deut. Chem. Ges., 67 (1934) 1164.56. F. Auerback and H. Barschall, Arb. Kaiserl. Gesundh. Amt, 27 (1907) 183.57. H. Staudinger and M. Lüthy, Helv. Chem. Acta, 8 (1925) 65 ; H. Staudinger, R.
Signer et al., Liebigs Ann. Chem., 474 (1929) 145.58. H. Staudinger and W. Döhle, J. Prakt. Chem., 161 (1942) 219; H. Staudinger, K.
H. In Den Sir-ken and M. Staudinger, Makromol. Chem., 9 (1953) 148.59. H. Staudinger, Z. Physik. Chem., A 153 (1931) 391.60. E. Hadorn, VersammIung der Gesellschaft deutscher Naturforscher und Springer
Verlag, 1953, p. 39.

K A R L Z I E G L E R
Consequences and development of an invention*
Nobel Lecture, December 12, 1963
The awarding of the Nobel Prize for Chemistry for the year 1963 is related tothe precipitous expansion of macromolecular chemistry and its industrial ap-plications, which began precisely ten years ago at my Max-Planck-Institutefor Coal Research, in Mülheim/Ruhr. The suddenness with which this began,and the rapidity with which it was propagated are comparable to an explosion.The energy carriers in this case were the ingenuity, activity, creative imagina-tion and bold concepts of the many unnamed chemists, designers and entre-preneurs in the world who have fashioned great industries from our humblebeginnings.
If today I stand with my colleague Natta, who has been particularly effectivein promoting this explosive wave, in the limelight of distinction, and do wishto manifest, with this address, my appreciation for the honor bestowed uponme, I must begin by thanking these many anonymous persons. They, too,deserve this distinction.
The extent of this "explosion" may be illustrated by two charts1, in whichthe location of newly-established plants is indicated. The places marked byblack circles refer to the production of high molecular weight materials, thecrosses to new production facilities which, though concerned with low mo-lecular weight materials, nevertheless also have some connection with the ad-dress I am delivering today (Figs. 1 and 2).
The new development had its inception near the end of 1953, when I, to-gether with Holzkamp, Breil and Martina, observed-during only a few daysof an almost dramatic course of events-that ethylene gas will polymerize veryrapidly with certain catalysts that are extremely easy to prepare, at 100, 20and 5 atmospheres and, finally, even at normal pressure, to a high molecularweight plastic.
I would like to first describe our normal-pressure polymerization experi-ment, which actually takes about an hour but which has been condensed inthe films to a few minutes (not shown here).
*This translation of Prof. Ziegler’s Nobel Lecture is reproduced with some modifica-tions, by permission of the publishers, from Rubber Chem. Technol., 38 (1965) xxii.

C O N S E Q U E N C E S A N D D E V E L O P M E N T O F A N I N V E N T I O N 7
Fig. 1. Location of industrial applications of the Mülheim processes in Europe (as of 1963).On the figure: � "High molecular weight materials; x , Aluminium alkyls and low mo-
lecular weight materials; Under construction or planned.
Fig. 2. Location of industrial applications of the Mülheim processes in the world (as of1963). Symbols as in Fig. 1. Numbers indicate the number of factories.

8 1 9 6 3 K A R L Z I E G L E R
The catalyst is prep ared simply by simult aneous ly pouring, with exclus ion
of air , two l iqu id mater ials into about two l i ters of a gasol ine-l ike hydro-
carbon, after which ethylene is introduced, while stirring. The gas is absorbed
quickly; . within an hour one can easi ly introduce 300-400 l iters of ethylene
into the two liters of liquid. At the same time, a solid substance precipitates,
in such a way that after approximately one hour the material becomes doughy
and can scarcely be stirred any more. If the brown catalyst is then destroyed,
by the addition of some alcohol and by the introduction of air, the precipitate
becomes scow- white and can be filtered off. In its final state it will accumulate,
in amounts of 300-500g, as a dry, white powder.
The results of this experiment greatly surprised us, and, later on, many
others, since up to that time ethylene had been considered extremely difficult
to polymerize. The "polythene" of the Imperial Chemical Industries, a prod-
uct which had been known for some seventeen years, was being prepared
under pressures of 1000-2000 atmospheres, and at a temperature of 200ºC.
Our experiment thus destroyed a dogma. It led, in addition, to a polyethylene
which differed quite markedly from the high-pressure product. Low-pres-
sure polyethylene not only has a better resistance to elevated temperatures
and a higher density, but is also more rigid. This is easily demonstrated by
holding in one hand two similar objects made of the two materials, and press-
Fig. 3. Comparison between the rigidity of two beakers, one of low-pressure, one ofhigh-pressure polyethylene.

C O N S E Q U E N C E S A N D D E V E L O P M E N T O F A N I N V E N T I O N 9
ing them together (Fig. 3). Low-pressure polyethylene can be drawn without
difficulty to form fibers or ribbons of high tensi le strength . This cannot be
done at all with high-pressure polyethylene, or at best only an indication of
drawing is obtained. We establ ished these facts immediately after our dis-
covery, with test specimens which were still quite primitive1.
The differences can be attributed to the fact that in our process molecules of
ethylene are joined together l inearly, without interruption, whereas in the
high-pressure process chain growth is disturbed, so that a strongly branched
molecule results (Fig. 4).
Fig. 4. High-pressure polyethylene, structural principle.
The low-pressure process found immediate acceptance in industry. By 1955,
200 metric tons of this new type of plastic had been produced; in 1958 i t
w as 1 70 0 0 t ons , a nd i n 19 6 2 some 12 0 00 0 t on s . The muc h hi gh e r f ig ure s
occasionally cited for this and other plastics have resulted from the confusion
of available, but unused, capacit ies with actual production. The increase in
dimensions can be indicated by comparison of our first test specimens, pre-
pared ten years ago with rather primitive means, with containers that are
twenty cubic meters in capacity, the largest now being made from polyethyl-
ene. A subsequent figure shows the l ightness of the mater ial, since a very
large container can easily be carried by only a few men.
The catalyst employed in the experiment described was prepared by mixing
aluminium tr iethyl, or d iethyl aluminium chloride, with t i tanium tetrachlo-
ride. However, this is only one example, taken from the countless series of
"organometallic mixed catalysts". Most generally they will form, as we found,
whenever standard organometal lic compounds, preferably those of alumin-
i um, but a lso ma ny of o t h er me t al s , a re broug ht i nt o con t ac t w it h c om-
pounds of certain heavy metals. Those of ti tanium, zirconium, vanadium,
chromium, molybdenum, cobalt and the like are especial ly effective. S ince

10 1 9 6 3 K A R L Z I E G L E R
there are many different metal a lkyls and many different heavy metal com-
pounds, and since, furthermore, components can be mixed together in varying
proportions, and by different methods, and because all this can have an effect,
often a truly decisive effect, on the nature of the catalytic activity, it is easy to
understand why this field has grown to practically limitless proportions.
In place of the metal alkyls, one can also use metal hydrides, or the metals
themselves, whereas metal alkyls probably will still form during the catalyzed
processes.
Our catalysts then became known, at the turn of the year 1953/4, to our
friends in industry and to their foreign col leagues, in Frankfurt , the Ruhr,
Manchester, and-last but not least-Milan. Shortly thereafter this knowledge
jumped over to the U. S. A. as well, and ultimately our findings became avail-
able to all. The consequences have been characterized, elsewhere, by the state-
ment that revelation of the Mülheim catalysts had the same effect as the starting
gun of a race in which the laboratories of the interested industries had been
e nt e re d4. However, representatives of purely scientific chemistry also partici-
pated.
Because of the magnitude of the new field, arrival at further stages, or the
order of such arrivals, was necessarily dependent upon contingencies. Indeed,
many important observations were made within short spaces of t ime, inde-
pendently of one another, and at different places. Let me illustrate this with
two examples: It was pure chance that in November of 1953 the first of the
catalysts in which our invention was clearly recognizable happened to be a
relatively weak-acting combination of an aluminium alkyl with a zirconium
compound, by which ethylene could be polymerized only under a few atmo-
spheres pressure, and with which propylene, already tested the day after our
cri tical experiment with ethylene, would not polymerize at a ll . Then, for a
number o f weeks, we were absorbed in experimenting with normal-pressure
polymerizations of ethylene by means of titanium- containing catalysts. Early
in 1954 we recognized the possibility ofcopolymerizing ethylene and propyl-
ene, after which we succeeded, at Mülheim, in polymerizing propylene with
more effective catalysts, but - and this we did not know at the time - a short
while after my colleague Natta of Milan had already observed this. In a first
substantiation of his observation, and in an act of fairness, Natta had referred
to the catalyst used as a "Ziegler catalyst", and that is how this expression found
its way into the literature 5. It is surely understandable that I myself prefer to
speak of them as "Mülheim catalysts".
The second example: Near the close of 1955 work was being done in many

C O N S E Q U E N C E S A N D D E V E L O P M E N T O F A N I N V E N T I O N 11
places on the polymerization of butadiene with our catalysts. But no one had
observed that in addition to the desired high polymers, a very interesting
trimer of butadiene, namely 1,5,9-cyclododecatriene, was being produced.
Günther Wilke, of my insti tute, became aware of th is, and showed how one
can guide the reaction entirely in this new direction. While endeavouring to
explain the formation mechanism of cyclododecatriene, Wilke discovered a
way to redirect this reaction at will, either toward a dimerization to an eight-
membered ring, or - by a co-reaction with ethylene - toward a co-oligomeri-
z at i on t o a t e n -me mbe re d r i ng 6 . The result was that the Mülheim catalysts
also achieved importance for polycondensation plastics such as Nylons 8, 10
and 12, into which the ring compounds can be transformed.
These cycl izations constituted the th ird surprising development afforded
the scienti fic community by the organometal lic mixed catalysts, i f I assign
number one to the new polyethylene process. I saved the second surprise for
Fig. 5. Portions of the chains of (a) polyethylene, (b) atactic, (c) isotactic and (d) syndio-tactic polypropylene. The methyls in the polypropylene are striped, and are actually
much larger than shown.

12 1 9 6 3 K A R L Z I E G L E R
later, and I must go into that now. From the middle of 1954 on, it began to be
obvious that the Mülheim catalysts were capable of polymerizing in a struc-
turally specific, as well as a stereo-specific manner. This realization is an es-
sential contribution of my col league Natta. He had often pondered over the
mechanism of the polymerization, and very successfully strove to "train" the
catalysts in such a way that they would possess extremely high specific ity .
Without wishing to anticipate Natta 7 in any way, I nevertheless feel obliged,
for the sake of completeness, to explain briefly what this is all about.
The chain of l inear polyethylene in the model, at an enlargement of fi fty
mil lion, has approximately the following shape: (Fig. 5a). If a substituted
ethylene, for example propylene, is polymerized, only the two doubly-bound
carbon atoms of the olefin molecule will participate in the chain formation.
The substituents, as side chains, will remain on the outside. If they combine in
a purely random fashion the resultant product will show an entirely arbitrary
distribution of the substi tuents along the two sides. Previously i t had been
believed that only those polymers could be formed which Natta - so far as I
know at the suggestion of his wife - later called "atactic" (Fig. 5b). In stereo-
specific polymerizations, polymers with highly regular structure are pro-
duced, with all the substituents on one side - isotactic (Fig. 5c) according to
Natta - or with the substituents in a regular right-left sequence - syndiotactic
(Fig. 5d) according to Natta. Both these terms were again inspired by Mrs.
Natta. The particularly favorable properties of the products correspond to the
regularity of the structure.
Analogous phenomena were encountered when our catalysts were used for
polymerization of butadiene. In this instance, either only one of the two double
bonds present can take part in the polymerization process. The result is a con-
figuration comparable to that of polypropylene and containing, instead of
methyl groups, only the unsaturated residues of ethylene, the so-called vinyl
g r o u p s C2H 3, in which case it can still be isotactic, atactic, or syndiotactic.
This is a so-called 1 ,2-polymer of butadiene (Fig . 6 , upper). Or, a l l four of
the C-atoms can enter into the long chain of the polymer, in 1 ,4-polymeri-
zations, so that in the middle of each individual C4 structure unit a new double
bond is formed, which was not present previously at that particular site (Fig. 6,
l ow e r) .
In addition, because of the double bonds, and from their aspect, the va-
lences of the two adjacent carbon atoms point ei ther both toward one side,
or to opposite sides. The first is the cis configuration, and the other is the trans
( F i g . 7 ) .

C O N S E Q U E N C E S A N D D E V E L O P M E N T O F A N I N V E N T I O N 1 3
Fig.6. "1,2-"(upper) and "1,4-"(lower) polymerizations of butadiene. Hydrogenatomsare not shown.
Natural rubber is a cis-1 ,4-polybutadiene, in which, are very disable bond,
the hydrogen atom has been replaced by a methyl group. Another important
natural substance, guttapercha, corresponds to the trans-1,4-polymer( Fig. 7).
The difficulty with al l earl ier attempts to synthesize rubber or rubberl ike
materials was that it was not possible to steer the polymerization of the basic
materials - butadiene, isoprene - uniformly into the one or the other configu-
ration. For this reason synthetic products contained a chaotic array of 1,4-cis,
1,4-trans and 1,2 structural units, even in the individual molecules. Although
they resembled the natural product to some extent, none of them ever cor-
responded to it completely.
With the aid of the easily prepared Mülheim catalysts it is now possible to
synthesize all these types uniformly, as desired, in a structure-specific or
stereospecific manner. For example, 1,2-polybutadiene is formed by using a
Fig. 7. Structural principle of natural rubber (cis) and guttapercha (tram). White circles:methyl groups. Hydrogen atoms are not shown.

14 1 9 6 3 K A R L Z I E G L E R
catalyst made from ti tanium acid ester and 3 aluminium triethyl . With the
c at al ys t s obt a ine d f rom T iCl 4 + 0 .5 A l (C2H 5) 2 2C1 , trans- 1 , 4 - p o l y b u t a d i e n e
c a n b e p r o d u c e d , a n d w i t h t h o s e d e r i v e d f r o m 1 T i l4 + 1 A l ( C2H 5) 3 o r
1 CoCl2 + 1 Al(C2 H 5 ) 2 C l , cis-1,4-polybutadiene will be formed, Finally, I
would like to add that an increase of the Al : Ti ratio in the catalyst, to 5 : 1,
will lead to cyclododecatriene.
A group from the B. F. Goodrich Research Center in the U. S. A. first made
these observations with cis-1,4-polyisoprene, the synthetic "natural rubber",
a few week; after their company had learned about the essential features of our
cataiysts 8. Actually, this represented only the final, closing stages of a So-year
effort to synthesize "genuine" rubber. Corresponding polymers of buta-
diene itself were then intensely studied, in a number of places, and cis-1 ,4 -
polybutadiene is today considered to be of great technological importance.
I will close this short survey with a discussion of recent developments per-
taining to the rubber-like copolymers of ethylene and propylene, particularly
those obtained with vanadium-containing organometall ic mixed catalysts,
and to the so-cal led terpolymers, into whose molecules certain diolefins -
(dicyclopentadiene, or, again as discovered by Natta and coworkers 9, our
cyclooctadiene-1,5) - have been incorporated.
Large quantit ies of a l l these new synthetic materials, discovered in con-
n e c t i o n w i t h l o w - p r e s s u r e p o l y e t h y l e n e , a r e a l r e a d y b e i n g p r o d u c e d
throughout the world, and production is sure to continue rising at a sub-
stantial rate.
With this I have shown, in broad outline, what has resulted in the course of
ten years from our early experiments with organometallic mixed catalysts. In
order to make the sequence of events which led to such a fruitful invention
more understandable, I shall have to go back exactly forty years. Shortly after
my graduation, having been a student of Karl von Auwers at the University
of Marburg/Lahn, I began my independent scientific work with experiments
for testing the theory of so-called free radicals. I incidentally found, in 1923,
a new method for the formation of organic compounds of the alkali metals
potassium and sodium 1 0, which brought my attention to the metal alkyls as
an interesting, highly diversified field, that has continued to fascinate me, over
and over again, up to the present. The new catalysts grew out of this, as a side
sprout, in 1953. Permit me now to pursue the unbroken chain of causal rela-
t ionships that l inks Then and Now by using special block schemes (Fig . 8 ,
1, F i g s . 9 - 1 2 ) .
A few years later, in 1927, Bähr and I1 1 - 1 3 m a d e t h e d i s c o v e r y - i m p o r t a n t

C O N S E Q U E N C E S A N D D E V E L O P M E N T O F A N I N V E N T I O N 15
for t he fur t he r d e ve l op me nt - that a lkali a lkyls can be added with ease to
butadiene or styrene, at room temperature (Fig. 8,2). Repetition of the process
leads first to oligomers, in a "stepwise organometallic synthesis", and finally
to polymers and high polymer reaction products (Fig. 8,4).
This first contact of mine with "macromolecular chemistry" later gave impe-
tus to many investigations by third parties, and recently butyl lithium has also
been suggested for industrial polymerizations of isoprene. At first, however,
another, indirect result of our work was of more importance. Secondary ob-
servations suggested the conclusion that metallic lithium should be amenable
to a reaction analogous to the one by which Grignard compounds are formed
from ma gne s i um:
Fig. 8. Preliminary work (Marburg/Lahn, Heidelberg, Halle/Saale) 1931-1939. Firstresults in Mülheim/Ruhr.

16 1 9 6 3 K A R L Z I E G L E R
W i t h C o l o n i u s1 4, I was able to confirm this in 1930, and that is how the
organoli th ium compounds became easily accessible (F ig . 8 ,3).
In Mülheim/Ruhr, where I have been working since 1943, Gellert and I
succeeded in transferring the technique of a "stepwise organometallic syn-
thesis" from butadiene to ethylene 1 5. In this instance the reaction leads from
lithium alkyl directly to the higher straight-chain l ithium alkyls, and hence
also to alcohols, carboxylic acids, and the like (Fig. 8,5).
growth which a chain can undergo, since, for ethylene addition, the tempera-
tures required are such that the l ith ium alkyls wil l readily decompose to
lithium hydride + olefin. This certainly seemed to justify the following con-
clusion: If, in such decompositions, it is a question of a reversible reaction, as
we had reason to believe, then l i thium hydride and l ithium alkyls should,
under proper conditions, function as catalysts for the polymerization, or
rather, oligomerization, of ethylene to the higher -olefins (Fig. 8, 6).
We did find such a reaction in principle, but it was so complicated by sec-
ondary and subsequent reactions that we could do nothing with it. Then when
I had already decided to give up these efforts, my coworker, H. G. Gellert ,
c onduc t ed one more exp e r i me nt -an d t he la s t , he wa s con vi nc e d-w it h t he
just recently discovered lithium aluminium hydride. This led immediately to
the desired higher (F ig . 8, 7). As the decisive turning point, this
resulted in the realization that the alkali metal was not the crucial issue at all,
and that everything we already knew about the lithium alkyls, and all that we
had anticipated besides, with respect to the chemistry of the olefinic hydro-
carbons, could be achieved with a great deal more ease through use of organo-
a lumi ni um c omp oun ds16. That is:
(1) There are genuine equil ibria aluminium alkyle aluminium hydride +
olefin lying, as a rule, entirely to the left, so that, in reverse of the situation
with lithium, it is possible to synthesize the aluminium alkyls from hydride +
olefin (Fig. 8, 8,9 ).
(2) In the case of aluminium, too - and this came as a real surprise - at mod-
erately high temperatures a stepwise organometallic synthesis, or as we now

C O N S E Q U E N C E S A N D D E V E L O P M E N T O F A N I N V E N T I O N 17
call it, a "growth" or propagation reaction takes place, leading to the higher
a lumi ni um a l kyl s ; t h us , a s ynt he s i s o f t he h ig he r s t rai g ht -ch ai n p r i ma ry
monofunctional a l iphatic compounds, par ticularly the fatty alcohols (Fig. 9 ,
10,11), became possible.
( 3 ) Furthermore, we have, from about 150º on, a catalytic ol igomerization
of the ethylene to higher - olefins (Fig. 9, 21).
Here the organometallic synthesis appears as the partial reaction of a com-
pletely understood, homogeneous intermediate reaction catalysis: After a
certain number of addition steps the intermediate product decomposes to a
hydride and an olefin whereupon, after the addition of ethylene to the hydride,
the cycle is repeated.
Such a reaction was encountered in its most primitive form with propylene,
for which the homogeneous catalys is leads, without supplementary chain
gr owt h , a l most e xc lu s iv e l y t o a w e l l -de f i ne d di me r17 (Fig.9 , 1 9 ) :
Recently this reaction has achieved significance for high molecular weight
chemistry as well , since the cracking of isohexene, fol lowing the shift ing of
t he doubl e bond, p roduce s i sop ren e, in add it ion t o me th ane 18 (F i g .9 , 20 ) .

18 1 9 6 3 K A R L Z I E G L E R
The trans it ion of a l l these reactions into industr ial applications was final ly
accomplished by the so-called "direct synthesis" of aluminium alkyls from
a lumi ni um , hy droge n a nd o le f i ns , d i sc ove re d b y us a t ap p roxi mat e l y t he
same time as the new polyethylene process. Aluminium hydride, from which
t he a l umin ium t r i al ky ls ar e qu i t e e asy t o ob t ai n t hr ough t he ad di t ion o f
olefins, cannot be prepared directly from the metal and hydrogen. However,
in already-prepared aluminium trialkyls, aluminium will dissociate with hy-
drogen to dialkyl aluminium hydrides which , with ethylene, wil l give 1.5
times the original amount of aluminium triethyl,
so that any amount of aluminium trialkyl can be prepared without difficulty 1 9
(Fig.10, 12,14) .
In the charts shown at the beginning of this address, the location of the in -
dustries engaged in the production of aluminium alkyls and their low molec-
ular weight applications were included. This area o f the industrial develop-
ment init iated by Mülheim is l ikewise in a state of continuous evolution ,
though it has been less rapid than that of the high molecular weight phase.

C O N S E Q U E N C E S A N D D E V E L O P M E N T O F A N I N V E N T I O N 19
Fig. 10. Course of the Mülheim Experiments, Part II.
Up to the year 1952 we had frequently conducted "growth" reactions based
on aluminium tr iethyl , and thought we were thoroughly acquainted with
such reactions. But when, together with E. Holzkamp 20, I attempted to apply
this type of reaction to aluminium tripropyl the formation of chains, to our
great surprise, did not materialize at all. On the contrary, we obtained propyl-
ene-from the propyl aluminium - in addition to aluminium triethyl and
butylene. Even starting from aluminium triethyl our reaction now yielded
nothing but butylene and the unchanged aluminium compound.
The explanation was obvious: A catalyst in trace amounts must have gotten
into this series of experiments, leading to an uncommonly rapid acceleration
of the displacement reactions:
a n d
mediately after the first propagation step, as butylene. It is now general ly

2 0 1 9 6 3 K A R L Z I E G L E R
known that we detected a tiny trace of metallic nickel as the disturbing ele-
m e n t 2 (Fig.9 , 22). Thus our attention was again directed to the problem to
polymerize also ethylene just as, years ago, we had been able to do with
butadiene and styrene, to produce a genuine macromolecule with the aid of
metal alkyls, in this case aluminium alkyls in particular.
Our growth reaction must lead to a genuine polyethylene, if we succeeded
in adding about 1000 ethylene units to the aluminium triethyl. For this, with
our reaction, only about 100-200 atmospheres pressure, instead o f the 1000-
2000 atmospheres used heretofore, would be required. Nevertheless, in prop-
erly performed experiments we had obtained only waxy products, because
the chain at the aluminium was prematurely spl it off -apparently by a dis-
placement reaction-as an olefin, with there-formation of ethyl at the alumin -
i u m , a n o c c u r r e n c e k n o w n t o c h e m i s t s w o r k i n g i n t h e h i g h m o l e c u l a r
weight field as a "chain transfer reaction":
To the ex tent that catalyst traces , as we now might well suspect , had been
involved here also in effecting the displacement, there existed the prospect
that completely "aseptic" procedures could eventually lead to a true poly-
ethylene. In order to provide the essentials for the "asepsis", we began, in the
middle o f 1953, to systematical ly investigate subs tances which have effects
somewhat similar to those of nickel. We found, instead, the polymerization-
promoting organometallic mixed catalysts, and, in particular, we achieved a
low-pressure polymerization of ethylene, and with this I have again arrived
at my starting point (Fig. 9, 23).Fig. II , which fol lows, once again shows, in schematic form, al l that has
resulted from the discovery of organometal l ic mixed catalysts. In this con-
nection, isoprene is doubly concerned in our work: because of the aforemen-
tioned synthesis, and for polymerization purposes.
Finally, I would like to present the following scheme (Fig. 12), in order to
show the entire development. The areas in two types of hatching indicate the
important trans it ions (f rom Li to Al , from the Al-alkyls to the mixed cata-
lysts), and also a third transition to an electrochemical side branch, which I
cannot go into at this time ( cf: Fig. 10, 1 5 18). Th e i mp ort ant c onse que nc e s
of the discovery of organometal lic mixed catalysts, evident even to the lay-
man, have led many to regard me, nowadays, as a "macromolecular chemist",
and, in fact, even as a plastics expert. I have intentionally set my work in this
field within a much broader framework, to show you that I am a "macro-

C O N S E Q U E N C E S A N D D E V E L O P M E N T O F A N I N V E N T I O N 2 1
Fig. II. Course of the Mülheim Experiments, Part III.
Fig.12. The Mülheim experiments (1948-1963), overall aspect. Numbers are as inFi gs .8 - I I .

1 2 1 9 6 3 K A R L Z I E G L E R
molecular chemist" only peripherally, and that I am not at all a plastics expert.
Rather, I have always looked upon myself as a pure chemist. Perhaps that is
also why the impact of the invention has been so enduring. The new knowl-
edge has, after all, not come from macromolecular chemistry. It is the metal
alkyls that have insinuated themselves into the chemistry of macromolecules
to effectively fertilize this field. Typical of the course I have followed from
those early beginnings of forty years ago until today, is the fact that I have
never started with anything like a formally presented problem. The whole
effort developed quite spontaneously , from a beginning which was actual ly
irrational in nature, through an unbroken causal series of observations, inter-
pretations of findings, rechecking of the interpretations by new experiments,
new observations, etc. My method resembled a meandering through a new
land, during which interesting prospects kept opening up, during which one
could frequently view part of the road to be traveled, but such that one never
quite knew where this trip was actually leading. For decades I never had the
slightest notion that successful technological and industrial applications were
also to be encountered during the journey.
Twice this path seemed seriously blocked. The first t ime was before the
transit ion from l ithium hydride to l ithium aluminium hydr ide and from the
lithium to the aluminium compounds had been accomplished. The second was
when our growth reaction suddenly, in a truly mysterious way, refused to go
any more. In both cases, a capitulation in the face of these difficulties would
undoubtedly have broken the red thread of continuity, which can now be
followed clearly.
But a much more formidable impediment might have presented i tself In
order to illustrate this, I must elaborate on the paradox that the critical con-
cluding stages of the investigations I have reported took place in an institute
for "coal research".
When I was called to the Institut für Kohlenforschung in 1943, I was dis-
turbed by the objectives implied in i ts name: I was afraid I would have to
switch over to the consideration of assigned problems in applied chemistry.
Since ethylene was available in the Ruhr from coke manufacture, the search
for a new polyethylene process, for example, could certainly have represented
such a problem. Today I know for certain , however, and I suspected at the
time, that any attempt to strive for a set goal at the very beginning, in Mül-
heim, would have completely dried up the springs of my creative activity. As
a ma t t e r o f fa c t : G iv i ng up m y p re oc cupa t i on wi t h orga no me t al l i c c om-
pounds in favor of the other, "bread-and-butter" problems of coal chemis-

C O N S E Q U E N C E S A N D D E V E L O P M E N T O F A N I N V E N T I O N 2 3
try - many of my colleagues were of the opinion, at the time, that this would
be the natural consequence of my removal to Mülheim - would have cut the
leads which I already held invisibly in my hand, and which were to lead me
safely to the results that proved of such importance also for the Ruhr industry.
As a condition o f my transfer to Mülheim I st ipulated that I was to have
complete freedom of action in the entire field of the chemistry of carbon
compounds, without regard to whether any direct relation to coal research
was or was not recognizable. The acquiescence of my stipulation was in accord
w i t h t h e p r i n c i p l e s o f t h e t h e n K a i s e r - W i l h e l m , a n d n o w M a x - P l a n c k
Society, of which my institute is a part. As far as the German coal mining in-
dustry which supported my insti tute is concerned, th is was an act of great
foresight on their part which did in fact provide the conditions for everything
that occurred, particularly the present circumstance that my ins ti tute, and
I with it, have now received this very great distinction.
The institute, however, - what is it, in its distinctive spritual and intellec-
tual substance, other than the totality of its active people. I began this address
with an expression of grati tude to the many people in the world whom I
know only sl ightly , or not at a ll , and who have developed great industries
from our beginnings. I will end the address by expressing my heartfelt thanks
to the many, very well known members of my insti tute who have stood by
me faithfully throughout all these years, and who share with me the prize for
which I have been singled out.
1. Figs. I-II are taken from K. Ziegler, Arbeitsgemeinsch. Forch. des Landes Nordrhein -Westfalen, 128 (1964) 33.
2. K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 67 (1955) 541.3. The experiment corresponding to ref. I, Fig. I was shown as a movie in Stockholm.4. Hercules Chemist, 46 (1963) 7.5. K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 67 (1955) 426.6. G. Wilke, Angew. Chem., 75 (1963) 10; Angew. Chem., Intern. Ed., 2 (1663) 105.7. G. Natta, following lecture; also Angew. Chem., 76 (1964) 553.8. S. E. Honer et al., Ind. Eng. Chem., 48 (1956) 754. 9. G. Natta et al., Chimie Ind. (Milan), 45 (1963) 651.
10. K. Ziegler and F. Thielmann, Ber., 56 (1923) 1740.11. K. Ziegler and K. Bähr, Ber., 61 (1928) 253.12. K. Ziegler and H. Kleiner, Ann. Chem., 473 (1929) 57.13. K. Ziegler, F. Dersch and H. Wollthan, Ann. Chem., 511 (1934) 13.14. K. Ziegler and H. Colonius, Ann. Chem., 479 (1930) 135.15. K. Ziegler and H.-G. Gellert, Ann. Chem., 567 (1950) 195.

2 4 1 9 6 3 K A R L Z I E G L E R
16. K. Ziegler, Angew. Chem., 68 (1956) 721, 724.17. K. Ziegler, Angew. Chem., 64 (1952) 323, 326.18. V.F. Anhom et al., Chem. Eng. Prog., 57 (1964) 43.19. K. Ziegler et al., Ann. Chem., 629 (1960) 1.20. K. Ziegler et al., Ann. Chem., 629 (1960) 121, 135.

G I U L I O N A T T A
From the stereospecific polymerization to the
asymmetr ic autocata lytic synthesis
of macromolecules
Nobel Lecture, December 12, 1963
Introduction
Macromolecular chemistry is a relatively young science. Though natural and
sy nt he t ic m ac romo le c u l ar subs t a nc es ha d l ong b ee n kn own , i t w as onl y
between 1920 and 1930 that Hermann Staudinger placed our knowledge of
the chemical structure of several macromolecular substances on a scientif ic
ba s i s1. In the wake of Staudinger’s discoveries and hypotheses, macromolecu-
lar chemistry has made considerable progress.
Very many synthetic macromolecular substances were prepared both by
polymerization and by polycondensation; methods were found for the regu-
lation of the value and distribution of molecular weights; attempts were made
to clari fy the relationships exist ing among structure, chemical regularity ,
molecular weight, and physical and technological proper ties of the macro-
molecular substances. It was far more difficult to obtain synthetic macromole-
cules having a regular structure from both the chemical and steric points of
v i e w .
An early result in this field, which aroused a certain interest in relation to
elastomers, was the preparation of a polybutadiene having a very high content
of trans-1,4 monomeric units, in the presence of heterogeneous catalysts 2.
A wider development of this field was made possible by the recent discovery
of stereospecific polymerization. This led to the synthesis of sterically regular
polymers as well as to that of new classes of crystalline polymers.
Before referring to the stereospecific po lymerizations and to their subse-
quent developments, I wish to make a short report on the particular conditions
that enabled my School to rapidly achieve conclusive results on the genesis and
structure of new classes of macromolecules. I also wish to describe the main
stages of the synthesis and characterization of the first stereoregular polymers
of - olefins.

2 8 1 9 6 3 G I U L I O N A T T A
The achievement of these results has also been helped by the research I did
in 1924 when I was a trainee student under the guidance of Professor Bruni. At
that time I began to apply X-ray study of the structures of crystals to the reso -
lution of chemical and structural problems 3.
At first, investigations were mainly directed to the study of low-molecular-
weight inorganic substances and of isomorphism phenomena; but, after I had
the luck to meet Professor Staudinger in Freiburg in 1932, I was attracted by
the study of linear high polymers and tried to determine their lattice structures.
To this end I also employed the electron-diffraction methods which I had
learned from Dr. Seemann in Freiburg and which appeared particularly suit-
able for the examination of thin-oriented films 4. I applied both X-ray and
electron-diffraction methods also to the study of the structure of the hetero-
geneous catalysts used for certain important organic industrial syntheses, and
thus had the possibility of studying in the laboratory the processes for the syn-
thesis of methanols and the higher alcohols 6, and also of following their in-
dustrial development in Italy and abroad.
In view of the experience I had acquired in the field of chemical industry,
certain I tal ian Government and industrial bodies entrusted me in 1938 with
the task of instituting research and development studies on the production of
synthetic rubber in Italy.
Thus the first industrial production of butadiene-styrene copolymers was
realized in Italy at the Ferrara plants, where a purely physical process of frac-
tionated absorption was applied for the first time to the separation of buta-
diene from 1-butene 7.
At that time I also began to be interested in the possible chemical applica-
tions of petroleum derivatives, and particularly in the use of olefins and di-
olefins as raw materials for chemical syntheses such as oxosynthesis 8 a n d
polymerization9 .
The knowledge acquired in the field of the polymerizations of olefins en-
abled me to appreciate the singularity of the methods for the dimerization of
a-olefins that Karl Ziegler described 10 in a lecture delivered in Frankfurt in
1952; I was struck by the fact that in the presence of organometallic catalysts it
was possible to obtain only one dimer from each a-olefin, while I knew that
the ordinary, cationic catalysts previously used yielded complex mixtures of
isomers with different structures.
At this time I also became acquainted with Ziegler’s results on the produc-
tion of strictly linear ethylene oligomers, obtained in the presence of homo-
geneous catalysts. My interest was aroused, and in order to understand better

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N 0 F M A C R O M O L E C U L E S 2 9
the reaction mechanism 11, concerning which very little was known, I started
the kinetic study of such polymerizations. In the meantime Ziegler discovered
the process for the low-pressure polymerization of ethylene 1 2. I then decided
to focus attention on the polymerization of monomers other than ethylene; in
particular I studied the a -olefins, which were readily available at low cost in
the petroleum industry.
At the beginning of 1954 we succeded in polymerizing propylene, other
a-o l ef i ns , a nd s t y ren e; t hus we o bt ai ne d p ol yme rs hav i ng ve ry di f fe re nt
p rop e r t ie s f rom t hose sh own by t he p re vi ous l y know n p oly me rs o bt ai ne d
from t hese monomers 1 3. I soon observed that the first crude polymers ofa-
olefins and of styrene, initia lly obtained in the presence of certain Ziegler
catalysts (TiCl 4 + aluminium alkyls), were not homogeneous, but consisted
of a mixture of different products, some amorphous and non-crystal lizable,
others more or less crystal l ine or crystal lizable. Accordingly , I studied the
separation of the different types of polymer by solvent extraction and the
structures of the single separated products. Even if the more soluble polymers
were amorphous and had a molecular weight lower than that of the crystal-
l ine, but far less soluble, polymers deriving from the same crude product, I
observed that some little-soluble crystalline fractions had a molecular weight
only a li t tle higher than that of other amorphous fractions. Therefore, con-
vinced of the well-known saying natura non facit saltus, I did not attribute crys-
tallinity to a higher molecular weight, but to a different steric structure of the
macromolecules present in the different fractions 1 4.
In fact all vinyl polymers may be regarded as built from monomeric units
containing a tertiary carbon atom. Thus in a polymer of finite length, such a
carbon atom can be considered asymmetr ic , and hence two types of mono-
meric units may exist , which are enantiomorphous 1 3, 15 .
Since all the polymers of vinyl hydrocarbons previously known, even those
re c ogni z ed a s ha v in g a he ad- t o - t a i l e n ch ai nme nt l i ke po ly s ty re ne , we re
amorphous, we examined the possibi l ity that the crys tal linity we observed
was due to a chemical ly regular (head-to-tai l) structure, accompanied by
regular succession of steric configurations of the single monomeric units. In-
deed, X-ray analysis permitted us to determine the lattice constants of crys-
talline polypropylene16 and polystyrene 1 7. The identity period along the chain
axis in the fiber spectra was of about 6.5 Å and might be attributed to a chain
segment containing three monomeric units 1 8. This led us to exclude the idea
that the crystall inity was due to a regular alternation of monomeric units
having opposite steric configuration. Thus it could be foreseen, as was in fact

3 0 1 9 6 3 G I U L I O N A T T A
later proved by more accurate calculations of the structure factors, that the
polymeric chains consisted of regular successions of monomeric units , a ll
having the same steric configuration 1 4.
In the-subsequent study of the butadiene polymers, prepared by us in the
presence of organometallic catalysts (for example, catalysts containing chro-
m i u m1 9) that have 1,2-enchainment, two different types of crystall ine poly-
mers were isolated and purified.
The X-ray and electron-diffraction analyses of these products enabled us to
establish that the structure o f one of them is analogous to the structures of
o l e f i ns20-that is, characterized by the repeti t ion of monomeric units
having the same configuration. We also established that the other crystalline
product is characterized by a succession of monomeric units, which are chemi-
cal ly equivalent but have alternately opposite steric configuration 2 1, as con-
firmed by a thorough X-ray analysis of the structure. In order to distinguish
these different structures I proposed the adoption of terms coined from the
a
b
Fig. I. Models of chains of head-to-tail vinyl polymers supposed arbitrarily stretched ona plane, having, respectively, isotactic (a), syndiotactic (b), and atactic (c) successions of
the monomeric units.

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 3 1
ancient Greek, and these are now general ly used 2 2; that is, isotactic 14 a nd s y n -
d i o t a c t i2 1.
Fig. I shows the first device we used for an easy distinction of the different
types of stereoisomerism of vinyl polymers; the main chains have been sup-
posed arbitrarily stretched on a plane.
By accurate examination of the structure of isotactic polymers on fiber
spectra, we could establish that all crystalline isotactic polymers have a helical
s t ruc tur e, a nal og ous t o t ha t foun d by Pa uli ng a nd Core y2 3 f o r a - k e r a t i n
(Fig. 2); in fact only the helix allows a regular repetition of the monomeric
units containing asymmetric carbon atoms, as was foreseen by Bunn 2 4.
Fig.2. Model of chain of according to Pauling and Corey.
Soon after the first polymerizations of a -olefins we real ized the importance
and vastness of the fields that were opened to research, from both the theoret-
ical and the practical points of view.
Our efforts were then directed to three main fields of research: (1) To in-
vestigate the structures of the new polymers in order-to establish the relation-
ships existing between chemical structure, configuration , and conformation
of the macromolecules in the crystalline state. (2) To find the conditions that
allowed the synthesis of olefinic polyhydrocarbons having a determined type
of steric structure, with high yields and high degree of steric regularity 2 5, as
well as to study the reaction mechanism, and regulation of the molecular
weight. (3) To attempt the synthesis, possibly in the presence of nonorgano-
metallic catalysts, of stereoregular polymers corresponding to other classes of
monomers having a chemical nature different from that of a-olefins.
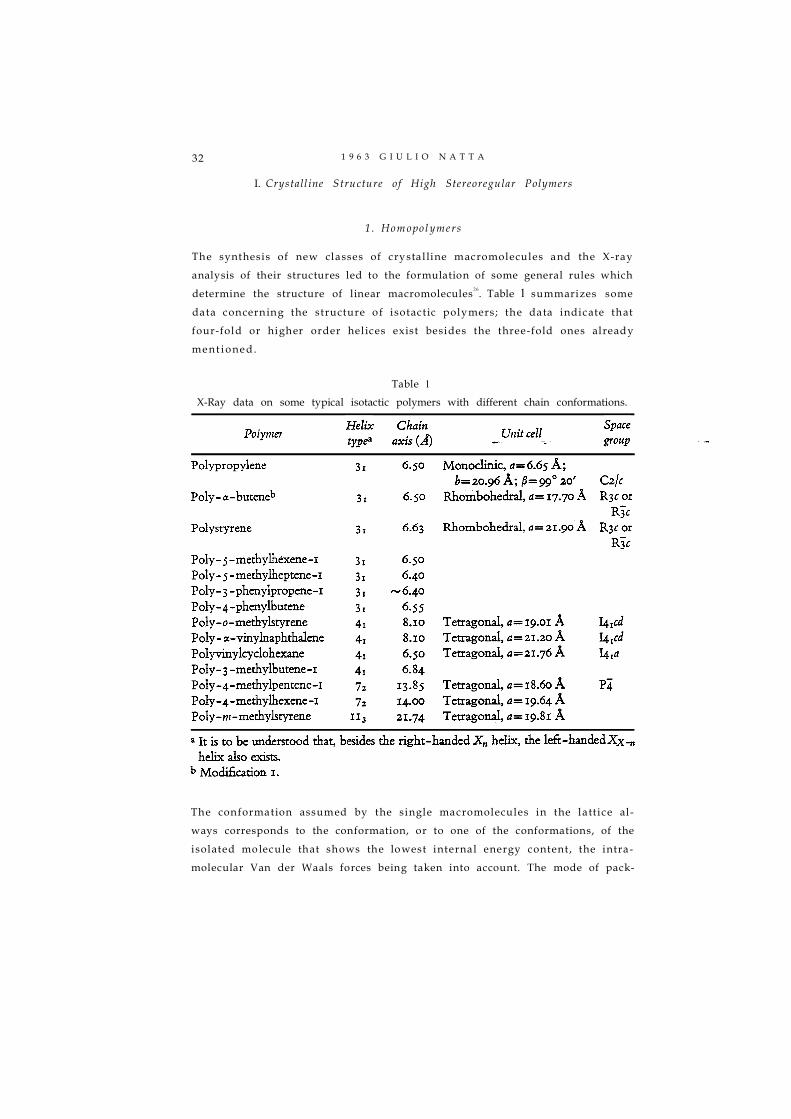
32 1 9 6 3 G I U L I O N A T T A
I. Crystall ine Structure of High Stereoregular Polymers
1 . Hom opo l ymers
The synthesis of new classes of crystal line macromolecules and the X-ray
analysis of their structures led to the formulation of some general rules which
determine the structure of linear macromolecules 2 6. Table 1 summarizes some
data concerning the structure o f isotactic polymers; the data indicate that
four-fold or higher order hel ices exist besides the three-fold ones already
me nt i one d.
Table 1
X-Ray data on some typical isotactic polymers with different chain conformations.
The conformation assumed by the single macromolecules in the latt ice al-
ways corresponds to the conformation, or to one of the conformations, of the
isolated molecule that shows the lowest internal energy content, the intra-
molecular Van der Waals forces being taken into account. The mode of pack-
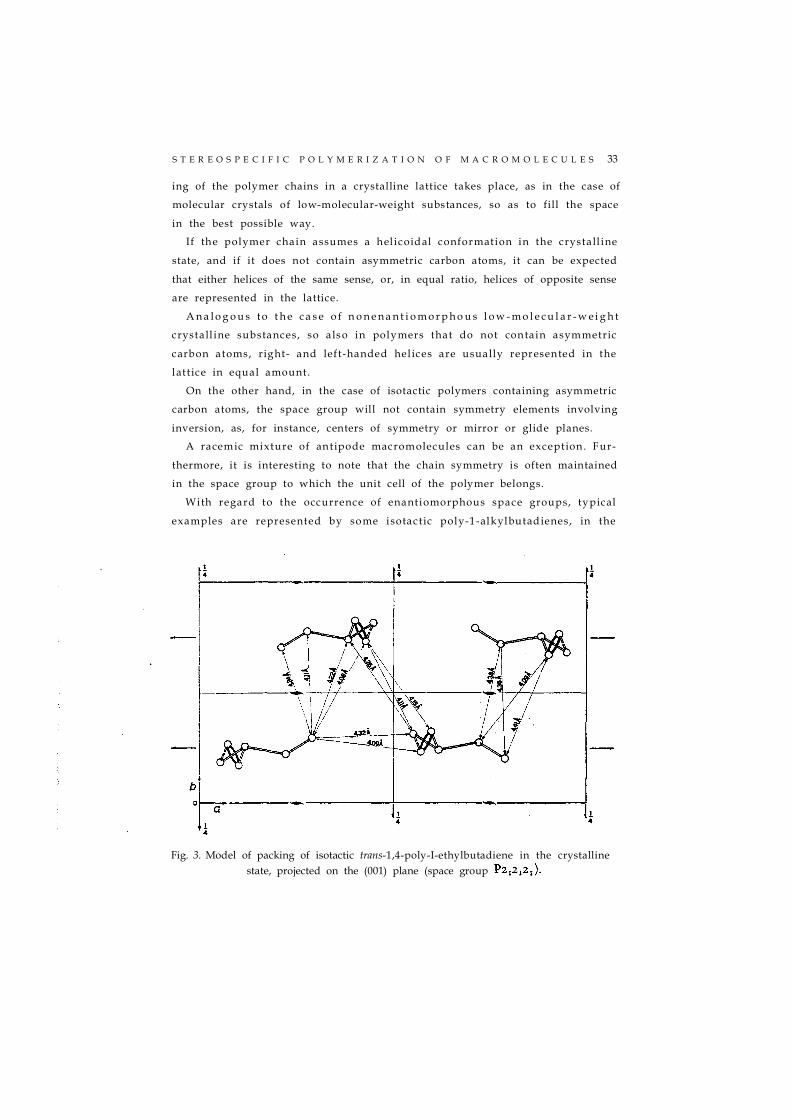
S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 33
ing of the polymer chains in a crystalline lattice takes place, as in the case of
molecular crystals of low-molecular-weight substances, so as to fill the space
in the best possible way.
If the polymer chain assumes a hel icoidal conformation in the crystall ine
state, and if it does not contain asymmetric carbon atoms, it can be expected
that either helices of the same sense, or, in equal ratio, helices of opposite sense
are represented in the lattice.
A n a l o g o u s t o t h e c a s e o f n o n e n a n t i o m o r p h o u s l o w - m o l e c u l a r - w e i g h t
crystall ine substances, so also in polymers that do not contain asymmetric
carbon atoms, right- and left-handed hel ices are usually represented in the
latt ice in equal amount.
On the other hand, in the case of isotactic polymers containing asymmetric
carbon atoms, the space group will not contain symmetry elements involving
inversion, as, for instance, centers of symmetry or mirror or glide planes.
A racemic mixture of antipode macromolecules can be an exception. Fur-
thermore, it is interesting to note that the chain symmetry is often maintained
in the space group to which the unit cell of the polymer belongs.
With regard to the occurrence of enantiomorphous space groups, typical
examples are represented by some isotactic poly-1-alkylbutadienes, in the
Fig. 3. Model of packing of isotactic trans-1,4-poly-I-ethylbutadiene in the crystallinestate, projected on the (001) plane (space group

3 4 1 9 6 3 G I U L I O N A T T A
crys tal l ine latt ice of which macromolecules with hel ices o f exclusively one
sense, right or left, exist for each crystal 27 (Fig. 3). Also in the case of isotactic
poly-tert.-butylacrylate, the helices in the lattice seem to be all of the same
s e n s e2 8.
If the chain symmetry is maintained in the crystal lattice, the possible occur-
rence of different space groups is considerably restricted. Where equal amounts
of enantiomorphous macromolecules are contained in the lattice, we must dis-
tinguish two cases concerning the relative orientation of side groups of enan-
tiomorphous macromolecules facing one another, which can be either iso-
clined or anticlined.
In t he f i r s t c ase , p oss i b l e sym me tr y op e rat ors fo r th e c ov e r i ng o f n ea r
macromolecules are either a mirror plane or a glide plane, parallel to the chain
axis.
It is, however, known that good packing is generally obtained more easily
with a glide plane than with a mirror plane, especially in the case of bodies
having periodical recesses and prominences, as in the case of spiralized polymer
chains.
In t he c ase o f a th re e - fo l d he l i x , e a c h r i gh t -ha nde d he l i x w i l l b e sur -
rounded, because of the existence of the glide plane, by three isoclined left-
handed helices, and vice versa; the space group will be R3c (Fig.4). This lattice
i s shown, for exa mp le , b y i so ta ct i c p o l ys t yre ne 2 9, by polybute ne 3 0, by 1,2-
p o l y b u t a d i e n e 3 1, and by poly- o - fl uoros tyrene 3 2; on the other hand it is not
shown by isotactic polypropylene, because i t would give rise to an insuffi -
c iently compact latt ice, i f Van der Waals contact distances , between carbon
atoms of near chains, must be maintained3 around 4.2 Å.
In the second case previously considered, in which the relative orientation
of the side groups of enantiomorphous macromolecules facing one another is
anticlined, the only symmetry operator relating neighboring macromolecules
is a symmetry center.
And again, i f the hel ix is threefold , each r ight-handed hel ix wil l be sur-
rounded, by the action of three symmetry centers at 120°C, by three left-
handed helices, and vice versa; the macromolecules are oriented so as to mini-
mize the length of the unit cell axes perpendicular to the three-fold axis, with
the best possible Van der Waals distances: the space group, which probably is
the one presented, for instance, by polyvinylmethyl ether 33 and by poly-n -
b u t y l v i n y l e t h e r3 4, will be R3 (F i g . 5) .

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 3 5

3 6 1 9 6 3 G I U L I O N A T T A
2. Copolymers
The "random" introduction of different monomeric units in a crystalline
polymer by copolymerization generally causes a decrease in crystallinity and
melting point when their content is lower than 20 to 25 percent, but at higher
content values the copolymer is generally amorphous.
As we shall remark in the section dealing with the stereoregular polymers of
hydrocarbon monomers containing an internal double bond, i t is sometimes
possible to obtain chemically and sterically regular alternating copolymers of
these monomers with ethylene, which are also crystalline. This is the case, for
instance, for the al ternating ethylene-cis-2-bute ne 3 5, ethylene-cyclopen-
t e n e3 6, and et hyl e ne -c yc lohe p te ne 37 c o p o l y m e r s .
In these cases, reaction conditions were used in which one of the monomers
is unable to homopolymerize, but can copolymerize to al ternating polymers
in the presence of a large excess of the first monomer. Moreover, in the case of
other nonhydrocarbon monomers, crystal line alternating copolymers have
be e n ob t ai ne d38 from two different monomers that are both very reactive in
the presence of stereospecific catalysts (for example, in the copolymerization
of dimethylketene with higher aldehydes 3 9), when the values of the relative
copolymerization rates are much higher than those o f homopolymerization .
In the cases mentioned above, the repeating structural unit has the structure of
a polyester obtained by treating a dimethylketene molecule with one mole-
cule of the carbonyl monomer considered.
Our researches also enabled us to find particular crystal line copolymers,
though with a "random" distribution, when the different monomeric units in
the polymeric chain showed considerable analogies both in chemical nature
and size.
This phenomenon was defined by us as isomorphism of monomeric units, even
if, in contrast to the isomorphism phenomena of low-molecular-weight sub-
stances, the crystals do not consist of physical mixtures of isomorphous mole-
cules, but of macromolecules in which monomeric units of different type can
substitute each with the other. In this case, copolymers show physical prop-
erties (density, melting temperature, and so on) which vary continuously with
the composition, and which are intermediate between those of the pure homo-
polymers. This phenomenon was observed in the copolymerization of styrene
w it h mo nof l uo ros t yre ne s4 0 and also in the copolymerization of butadiene
with 1 ,3-pentadiene to trans-1 ,4 p o ly me r4 1.
Crystalline copolymers of a completely different type are obtained by suc-

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 3 7
cessive polymerization of different monomers in the presence of catalysts able
to homopolymerize both of them. These are linear copolymers constituted by
successive blocks, each consisting of a chemically and sterically regular suc-
cession of units of the same type.
In some of these cases X-ray analysis reveals both the crystallinities corre-
sponding to the single homopolymers 4 2.
The importance of the stereospecific polymerization - from the standpoint of
both theory and practical applications - is due to the fact that in most cases
(even if not always) the stereoregularity of linear polymers determines crys-
tallinity. When the glass transition temperature and the melting temperature
are very different, the physical and especially the mechanical properties are
very different from those of the corresponding stereoirregular polymers. Due
to such properties, these materials have very interesting practical applications,
either as plastics and textiles when the melting point is high or as elastomers
when the melting point does not considerably exceed the temperature of use.
The knowledge acquired in these last 10 years in the field of the stereospeci-
ficity of the polymerization processes shows that stereoregular and, in partic-
ular, isotactic polymers can be obtained in the presence of suitable catalysts
acting through an ionic (both anionic and cationic) coordinated mechanism;
however, they cannot general ly be obtained by processes characterized by
radical mechanism.
The catalysts having a higher degree of stereospecificity are characterized
by the presence of metal atoms able to coordinate the monomer molecules in
a s t age i mme di a te l y pre c e di ng t ha t o f i nse r t io n o f t he mo nome r i c un it
between the end of the growing chain and the catalyst 4 3- 45 .
In fact , a stereospecific action is shown either by the catalysts containing
metal atoms, the coordinating properties of which are due to their charge and
t o t h ei r sma ll i on ic r ad ius al umi ni um, b er yl l ium , l i t h i um)4 4, or by com-
pounds of the transit ion metals 4 6, 47 .
S o m e a u t h o r s4 8 w er e l e d t o b el i e ve t ha t t he s t e r ic s t ruc t ure o f t he l as t
monomeric unit , or units, of the growing chain played an important ro le in
the steric regulation o f the polymerization processes. However, the low de-
gree of stereospecificity observed in the radical processes shows that this factor
alone cannot exert a determining action. In any case stereoregularity in these

38 1 9 6 3 G I U L I O N A T T A
last processes is of the syndiotactic type and may be attributed also to thermo-
dynamic factors, according to the strong increase in stereospecificity with de-
crease in temperature.
The first highly stereoregular isotactic polymers were obtained in the pres-
ence of heterogeneous catalysts; however, it soon became clear that the heter-
ogeneity of the catalytic system is an essential factor for the polymerization of
aliphatic olefins to isotactic polymers, but not for the polymerization of other
types of monomers. In fact it was found that aliphatic aldehydes and certain
monomers containing two electron-donor functional groups able to be co-
ordinated (for example, con jugated diolefins, vinyl ethers, a lkenyl ethers,
acrylic monomers, styrenes that are substituted differently in the benzene ring,
vinyl pyridine, and so on) can be polymerized in the stereospecific way also
in the presence of soluble catalysts.
It must be borne in mind that, even if the most typical highly stereospecific
catalysts for the polymerization of a-olefins contain organometal l ic com-
pounds, some classes of monomers (for example, vinyl ethers) can be poly-
merized to isotactic polymers in the presence of cationic catalysts without
the presence of organometall ic compounds 4 9.
The stereospecificity of the polymerization processes not only depends on
the catalytic system but is a property of each monomer-catalyst system. This
is particularly evident in the case of the polymerization of some conjugated
homologs of diolefins, in which the variation of the monomer changes both
the degree of stereospecificity of the process and, in some cases, the type of
stereoregularity of the polymer obtained 5 0.
Therefore, in order to attain a general view of the present state of the stereo-
specific polymerization , i t is helpful to examine the most important results
obtained in each class of monomers.
This is the most studied branch of stereospecific polymerization. As already
mentioned, isotactic polymers of a-olefins have been obtained so far only
with the use of heterogeneous catalysts.
High stereospecifici ty is observed only when one employs organometal l ic
catalysts containing a par ticular crystall ine substrate, such as that deriving
from the violet a, (ref. 51), and (ref. 52) modifications of TiCl 3, having a
l a y e r l a t t i c e4 2 ,5 3 ,5 4 . The use o f t he m o d i f i c a t i o n o f T i C l3 ( r e f . 5 5 ) , w h i c h
does not correspond to layer lattices, or of other heterogeneous catalysts (for

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 3 9
example, catalysts containing a substrate formed by metal oxides) which also
yield linear polymers of ethylene, leads to the formation of catalysts having
litt le stereospecific i ty in the polymerization of a-olefins 5 3, 56 .
The study of the catalysts prepared from organometal lic compounds con-
taining aromatic groups 56 or labeled carbon enabled us to determine the ionic
c oordi nat e d me cha ni sm of such p oly me r iz at i on a nd th e numbe r o f ac t iv e
centers on the surface of the heterogeneous catalysts 57.
Chemical and kinetic studies led to the conclusion that the stereospecif ic
polymerization of propylene is a polyaddition reaction (stepwise addition),in
which each monomeric unit, on its addition, is inserted on the bond between
an electropositive metal and the electronegative terminal carbon atom of the
growing polymeric chain. This study revealed also that some organometallic
catalysts, which contain only titanium as metal atoms, could be stereospecific
(ref. 58). The first reaction step corresponds to a coordination of the monomer
molecule to the transit ion metal belonging to the active center 4 3, 45.
The reaction chain general ly does not show a kinetic termination59 , the
length of the single macromolecules being determined by the rate of the pro-
cesses of chain transfer either with the monomer60 or with the alkyls of the
organometal lic compounds present 61 ; these transfer processes allow, after the
formation of a macromolecule, the s tart of another macromolecule on the
same active center 5 6, 62 .
The single-rate cons tants o f the different concurrent processes of chain
growth and termination have been determined for some typical catalysts 63.
Later on, the study of homogeneous catalysts based on vanadium compounds
and on alkyl aluminium monochloride permitted us to synthesize crystalline
polypropylenes with a nonisotactic structure. The detai led development of
this study led to the preparation of catalysts, obtained by treating hydro-
carbon-soluble vanadium compounds (acetylacetonates or vanadium tetra-
c hl or id e) wi t h di al ky l a l um in ium m onoc hl or id e. The se c at a ly s ts y i el d , at
low temperature, more or less crystalline polymers, free, however, from iso-
tactic crystal lizable macromolecules 64.
X-Ray analysis, applied to the fiber spectra, permitted us to establish that
this is a syndiotactic polymer; i ts latt ice structure has an identity period of
7 .4 Å , c orre sp ondi ng to four mo nome r i c uni t s6 5. Th e c omp a r is on be t we e n
isotactic and syndiotactic polypropelene structures is shown in Fig. 6.
The same type of homogeneous catalyst , which at low temperature homo-
polymerizes propylene to syndiotactic polymer, was used at higher tempera-
tures (for example, 0°C) for the production of copolymers having a random

4 0 1 9 6 3 G I U L I O N A T T A
Fig. 6. Comparison between the side and end views of the chain structure of isotactic (a)and syndiotactic (b) polypropylenes (stable modifications) in the crystalline state.
distribution of propylene with et hyl ene6 6. These polymers, which are linear,
are completely amorphous when the ethylene content decreases below 75
p e rc en t . Th e y ha v e a ve ry f l ex ib le c ha in , d ue to th e f re que n t CH 2- C H2
bon ds , wh il e t he rel a t iv e ly sm al l numb er o f CH- CH3 groups i s e noug h t o
hinder crystal lization of the polymethylenic chain segments. These copoly-
mers can be easily vulcanized through the use of peroxides; on the other hand
the terpolymers, which contain not only ethylene and propylene but also
small amounts (from 2 to 3 percent, by weight) of monomeric units, origi-
nated from the random copolymerizations of suitable dio lefins 67 (or of cyclic
compounds, such as cyclooctadiene, which can be prepared easily by dimeri-
zation of butadiene, fol lowing the method proposed by Wilke), can be vul-
canized easily by the conventional methods used for the vulcanization of low-

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 41
unsaturation rubber. They yield elastomers that are very interesting also from
the practical po int of view, because they can be obtained from low-priced
materials and also because of their physical properties and resistance to aging.
Polymers of 1-methyl-2-deuteroethylene. The study on the polymerization of
differently deuterated propylenes, undertaken by us in order to arrive at more
certain and univocal attributions of certain bands to the infrared spectrum of
isotactic polypropylene, led us to the discovery of new interesting types of
stereoisomerism in polymers of 1-methyl- 2-deutero-ethylene, and general-
ly in the case of polymers of 1,2-disubstituted ethylenes 68.
In fact, propylenes deuterated in the methylenic group can lead to monomer
units having different steric structure depending on the relative orientation of
t he CH3 and D substi tuents. Starting from these deuterated monomers show-
i ng p he nome na of ge ome t r i c isom er i sm, t wo t yp e s o f p o l y me rs w er e ob-
a
b
Fig.7. Models of the chains of head-to-tail ditactic polymers supposed arbitrarilystretched on a plane, having, respectively, threo-diisotactic (a), erythro-diisotactic (b),
and disyndiotactic (c) succession of the monomeric units.

4 2 1 9 6 3 G I U L I O N A T T A
tained. They exhibited the same X-ray spectra but different infrared spectra@.
This means that such polymers possess the same helix structure as normal
isotactic polypropylene, but that the relative orientation of D and CH 3 groups
can lead to a new type of stereoisomerism. In general, starting from a mono-
me r o f t he CH A=C HB t yp e, t hre e t yp e s o f s te re ore gula r i some rs c an be
expected (see Fig. 7).
The type of stereoisomer obtainable by stereoregular polymerization de-
pends on the mode of presentation and type of opening of the double bond of
each monomer molecule on entering the growing chain (Fig . 8).
Fig. 8. Scheme of presentation and opening of the double bond ofmonomeric units whenentering the growing chain.
Subsequently, d iisotactic polymers were obtained with the aid of cationic
catalysts, starting from monomers of the CHA = CHB type, wherein A desig-
nates an OR group and B, chlor ine 70 or an alkyl group 71 (Fig. 7).
Stereoregular homopolymers of hydrocarbons having an internal double bond. First
of all, I wish to report on the results we have obtained in the polymerization of
cyclobutene, which is of particular interest as it yields several crystalline poly-
mers having different chemical or steric structure, depending on the catalyst
u s e d72 (Fig. 9 ) .The different stereoregular polymers we have obtained and a number of
their proper ties are shown in Table 2 , from which i t may be seen that the
polymerization can take place by opening of the double bond to form cyclic
monomer units containing two si tes of optical type stereoisomerism, so that
crystalline polymers are of ditactic type.
In view of the fact that under suitable conditions it is possible to obtain two
crystalline polymers containing enchained rings that show different physical

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 4 3
Fig.9. Types of polymerization of cyclobutene: 1, cyclobutylenamer; 2, cis-1,4-poly-butadiene; 3, trans-1,4-polybutadiene.
properties, we have ascribed the differences in their properties to the different
steric structure and have attributed an erythro-diisotactic structure to one of
them and an erythro -disyndiotactic structure to the other 73 (Fig. 10).
In the presence of other catalysts the ring opens to form unsaturated mono-
mer units, which may show isomerism of geometric type. In th is case, too,

4 4 1 9 6 3 G I U L I O N A T T A
two different products are obtained (depending on the catalyst used), the
properties of which correspond to those, respectively , of cis-1,4 - and trans-
1 , 4 - p o l y b u t a d i e n e 72 (Fig. 9).
Fig. 10. Schematic drawing of the structures of erythro-diisotactic (a) and erythro-disyn-diotactic (b)cyclobutylenamer.
Ditactic polymers are also obtained from certain monomers containing in-
ternal unsaturation, which are unable to homopolymerize but, as mentioned
above, can copolymerize with ethylene, yield ing crystal line, al ternating co-
polymers of erythro-diisotactic structure. Among these monomers are cis-2-
b u t e n e3 5, c y c l o p e n t e n e3 6, a n d c y c l o h e p t e n e3 7; trans-2- b u t e n e a n d c y c l o -
hexene behave in a different way and do not give crystalline copolymers.
Unlike the ditactic polymers of deuterated propylene, the ditactic polymers
obtained by alternate copolymerization can exist in two disyndiotactic forms.
It is to be noted that the copolymerization of cis-2-butene is stereospecific
only in the presence of heterogeneous catalysts of the type used in polymeriz-
i ng a-o l e f i ns t o i so t a c t ic p o l yme rs , w hi l e t h e c op ol ym e r i za t io n o f c y cl o -
pentene and cycloheptene is also stereospecific when homogeneous catalysts
are used. We have recently74 proposed an interpretation of these facts based
essentially on steric criteria.
Stereoisomerism phenomena in the field of diolefins, and in particular of con-
jugated diolefins, are more complex than phenomena occurring in the case of
monoolefin ic monomers. In fact , besides the stereoisomerism phenomena ob-

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 4 5
served in these last (isomerism due to asymmetric carbon atoms), isomerism
phenomena of geometric type may also be present, depending on the cis- o r
trans-configuration of the residual double bonds present in the monomeric
units.
Butadienepolymers. The simplest conjugated diolefin, 1,3-butadiene, can in
fact y ield two types of polymers, according to whether the polymerization
takes place by opening of the vinyl bond (to form 1,2-enchained polymers)
or by opening of both conjugated double bonds (to form 1,4-enchained poly-
m e r s )
In the first case, the same stereoisomerism phenomena observed in other vinyl
polymers (for example, isotactic , syndiotactic , and atactic polymers) can be
e xp e c t e d.
In the second case, each monomeric unit still contains a double bond in the
2-3 posit ion , which can assume cis- or trans-configuration. Thus, four types
of stereoregular polymers could be foreseen "a priori" and precisely: trans-
1 , 4 - , cis -1,4-, isotactic-1 ,2-, and syndiotactic-1 ,2-polybutadienes. All four
these stereoisomers were prepared at my Insti tute with the aid of different
s t e re o sp e c if ic c a t a ly s t s7 5, 76 with a high degree of ster ic purity (up to above
98 percent), as shown by infrared analysis”.
X-Ray examination had made i t possib le for us not only to establish the
steric structure of the different polymers but also to determine the confor-
mation of the chains in the crystals and, for three of them, also adetailed lattice
s t ruc t ure a r 2 1, 78 . F ig . 11 shows the conformations of the chains of the various
stereoisomers, while in Table 3 a number of physical characterist ics of the
single polymers are reported.
As mentioned above, stereoregularity in the field of butadiene polymers is
not necessarily connected with the use of heterogeneous catalysts, and, in fact,
a ll four regular stereoisomers can be obtained with the aid of homogeneous
catalysts.
In the case of cis-1,4-polybutadiene, the highest steric purity is obtained by
the use of homogeneous catalysts 7 6. Of the four polybutadiene stereoisomers,

4 6 1 9 6 3 G I U L I O N A T T A
Fig. 11. Side and end views of the chain conformations of the four stereoisomers of poly-butadiene: (a) trans-1,4; (b) cis-1,4; (c) syndiotactic-1,2; (d) isotactic-1.2.
a trans-1,4-Polybutadiene exists in two crystalline modifications: one (mod. I) is stablebelow 75ºC, the other (mod. II) is stable between about 75ºC and the melting point ofthe polymer.

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 4 7
the cis-1 ,4 stereoisomer is o f particular interest also from a practical view-
point. Its preparation and properties have been investigated by a large number
o f w o r k e r s7 9.
Isoprene polymers. The two polyisoprene geometrical isomers were already
known in nature: natural rubber (cis-1 ,4 p o ly me r) a nd g ut t a-p e rc ha a nd
balata (trans-1,4 polymers). Both were obtained by synthesis through stereo-
specific polymerization .
The cis-1,4 polymer was obtained for the first time in the United States by
Goodr i c h’s w orke rs8 0, while the trans-1 ,4 p o l y me r wa s p re p are d b y us81 a t
the beginning of 1955.
The other stereoisomers, having 1,2- or 3 ,4-enchainment, have not been
prepared as yet in such a degree of steric purity as to yield crystalline products.
In fact, the only known polymer having 3,4-enchainment, obtained in the
presence of the same catalysts yield ing syndiotactic 1 ,2-polybutadiene, is
a morp hous .
1,3-Pentudiene polymers. Unlike butadiene polymers, the stereoregular poly-
mers of 1,3 -pentadiene obtained so far contain at least one asymmetric carbon
atom in the monomer unit. Furthermore, for some of them it is possib le to
expect geometric isomers, due to the presence of internal double bonds which
may have cis- or trans-configuration, so that all the polymers will show two
centers of ster ic isomerism. And in fact polymers having 3,4-enchainment,
containing two asymmetric carbon atoms, show two si tes of optical isomer-
ism; all the others exhibit one site of optical isomerism and one of geometric
isomer ism (1 ,2 and 1,4 units).
On the assumption that only polymers showing stereoregulari ty in both
possible sites (ditactic polymers) will be crystalline, 11 crystal l ine pentadiene
polymers can be expected:
(1) Pol yme rs ha v in g 3 , 4-e nc ha i nme nt (Fi g . 1 2a ) : (i) erythro-d i i so ta ct ic
p oly me r , (ii) threo-di isotactic polymer , (iii) Sy ndi ot a ct i c p o ly me r .
(2) Pol yme rs ha v in g 1 , 2-e nc ha i nme nt (Fi g . 1 2 b) : ( i v, v ) I so t ac t i c p o ly -
mers containing, respectively, one cis- or trans-double bond in the side-chain,
(vi,vi i) Sy ndi ot a ct i c p o l yme rs c ont ai ni ng , re sp e ct i v el y , on e cis- o r t r a n s -
double bond in the side-chain.
(3) Polymers having 1 ,4-enchainment (Fig. 12c): (viii,ix) cis-1 , 4- i so t a ct ic
and syndiotactic polymers, respectively , (x,xi) trans-1,4-isotactic and syn-
diotactic polymers, respectively .
Of these stereoisomers the only three so far known were prepared in my
Ins ti t u t e : trans -1 , 4- i so t a ct ic 8 2, cis-1 , 4- i so t a ct ic 8 3, and cis-1 ,4 - syndi ota ct ic

4 8 1 9 6 3 G I U L I O N A T T A
Fig. 13. Side and end views of the macromolecule of isotactic trans-poly(I-methylbuta-1,3-diene) (that is, trans-1,4-polypentadiene) in the crystalline state.

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 4 9
Table 4
p o l y m e r 8 4. In Table 4a number of physical properties character ist ic of these
isomers are reported; Figs. 13 and 14 show the conformation of the chains in
the crystals.
As could be expected, the best elastic properties in vulcanized polymers are
observed for cis-1,4-polymers, owing to their melting point, which is slightly
below the melting point of natural rubber. - - --
Fig. 14. Side and end views of the macromolecule of isotactic cis-1,4-polypentadiene(a) and syndiotactic cis-1,4-polypentadiene (b).

5 0 1 9 6 3 G I U L I O N A T T A
Unlike the polymerization of unsatured hydrocarbons, and particularly -
olefins, the polymerization of monomers containing functional groups, in the
p re se nc e o f c a t al ys t s ba se d on orga nom et a l l i c c omp ound s , ha s n ot be e n
investigated until recently. This is due to the fact that the functional groups
contained in such monomers can react with organometallic catalysts through
reactions that are well known in the field of classical organic chemistry, such
as Grignard reactions, Michael’s reaction, or splitting of an ether bond.
Initially it was feared that these reactions might involve both deactivation of
the catalytic agent and total or partial alteration of the said monomers.
In 1956 we demonstrated for the first time in the case of acrylonitrile 85 a n d
its homologs that , by suitably selecting the transit ion metal compounds and
organometal lic compounds forming the catalytic complex, i t is possib le to
br i ng a bout s te re osp e c if i c , ani oni c c oordi na t ed p ol yme r i z at i on o f t he se
monomers while impeding or delaying the above-mentioned side reactions
between monomer and catalyst .
Therefore, i t has been demonstrated that stereospecific polymerization of
nonh ydroc arb on monom ers ca n al so be c a rr ie d out w i th t he use o f p u re
organometallic compounds other than those of the Ziegler type, or even with
the aid of catalytic compounds that do not contain metal-to -carbon bonds.
The research work on these monomers has taken two separate but parallel
paths; that is , on the one hand it was directed to s tereospecific cationic co-
ordinated polymerization and, on the other, to stereospecific anionic poly-
merization (see Tables 5 and 6).
The cationic coordinated polymerizations carried out by us in the presence
of catalysts of the type of Lewis acids (based on organometallic compounds or
Friedel-Craft catalysts) were chiefly directed to the following classes of mo-
nom ers : v i nyl a l ky l e t he rs8 6, 87 , a l ke ny l al k yl e t he rs7 0, a l koxy-s tyre nes 8 8, vi-
nyl carba zole 89, and - chlorovinyl ethers 7 1.
The polymerization of isobutyl vinyl ethers to crystalline polymers had al-
ready been carried out by Schildknecht 49 in 1949. As a result of our further
research work i t was possib le to attr ibute their crys tal lin ity to an isotactic
s truc ture 8 6.
Stereospecific anionic coordinated polymerization, which is in general car-
ried out in the presence of basic-type catalysts (organometallic or metal amid-
ic compounds, a lcoholates ) was chiefly investigated in connection with the
fo l low in g c la sses o f monome rs : h ig he r homol ogs o f a cry lon it r i le 9 0, v i nyl -

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 5 1
Table 5
Unlike the a-olefin polymerization, which requires the presence of a cata-
lyst containing a crystalline substrate in order that it may proceed in a stereo-
specific isotactic manner, the po lymerization of nonhydrocarbon monomers
containing functional groups or atoms haying free electron pairs (such as, for
example, ethereal, carbonylic , or carboxylic oxygen; aminic , amidic, or ni-
trilic nitrogen) can proceed in a stereospecific way also in the absence of a solid
substrate - that is, in a homogeneous phase. Here the stereospecificity - which
in this case is also connected with a constant orientation and constant mode of
p re se nt at ion , on p oly me r iz i ng, o f t he m onome r uni t s w it h re spe c t t o t he
growing chain and to the catalytic agent - is due to the coordination of an

52 1 9 6 3 G I U L I O N A T T A
electron pair in the monomer with the metal of the catalytic agent by means
of a dative bond 4 7, 95. As the olefinic double bond too is necessarily bound to
t h e a c t i v e c e n t e r , s u c h m o n o m e r s a p p e a r t o b e d o u b l y l i n k e d t o t h e
complex formed by the catalytic agent and the terminal group of the growing
chain. A predetermined steric orientation is thus made possible.
Likewise, both the diolefins containing two olefin groups bound to the
c at a l ys t c om p le x an d c e r t ai n aro ma ti c a-o l e f i ns , w he re i n t he s e con d a n -
choring point is provided by the aromatic group -linked to a catalyst con-
taining a highly electropositive atom with a very small radius (lithium) 96, can
be polymerized stereospecifically even in the homogeneous phase.
The coordination of the monomer with the catalytic agent, which is the
indispensable step preceding any stereospecific po lymerization both in the
homogeneous and in the heterogeneous phase, has been particularly well
esemplified by the stereospecific polymerization of 2-vinylpyridine in the
presence of organometal lic compounds of magnesium 97.
In fact, the presence of Lewis bases in the polymerization of this monomer
exerts a determining influence on its behavior in the polymerizations. Com-
pounds having a h igher degree of basici ty than vinylpyr idine i tself (for ex-
ample, pyridine) form stable coordination compounds with the catalyst, thus
impeding the coordination of the monomer; in this way, not only does the
catalytic activity appear very much reduced, but also the stereospecific i ty
disappears and the polymer obtained is atactic . Compounds having a lower
degree of basicity than the monomer (aliphatic ethers) compete with the mo-
nomer only in so far as the association with the catalyst is concerned. Accord-
ingly this does not result in the disappearance of the catalyst reactivity, but
only in its reduction along with the degree of stereospecificity of the reaction.
The asymmetric synthesis of optically active high polymers, starting from
monomers showing no centers of optical-type asymmetry, constituted a par-
ticular, more advanced case of isotactic stereospecific polymerization.
In fact, whereas in the normal stereospecific polymerization to isotactic poly-
mers a succession of monomer units with a given configuration takes place in
each single macromolecule so that enantiomorphous macromolecules in equal
amounts are present in crude polymers, in the case ofasymmetric synthesis one

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 5 3
of the two enantiomorphous isomers of the monomer unit is contained in
hi ghe r a mount s .
It should be noted that isotactic high polymers or of other sim-
ple vinyl monomers cannot show detectable optical activity, since an ideal
isotactic polymer of infinite length does not contain asymmetric carbon atoms,
and in isotactic polymers having finite length 98 the optical activity, due to a
difference in the terminal groups, can be detected only in oligomers and de-
creases with increase in the molecular weight. This is due to the fact that the
asymmetry of each asymmetric carbon atom is to be ascribed not to the chemi-
cal d ifference o f contiguous groups l inked to the said carbon atom but to a
difference in length of the chain segments linked to it 99.
In fact, in the case of optically active polymers were ob-
tained by polymerization only from monomers having an asymmetric carbon
a t o m1 0 0 .
On the basis of our investigations it has been possible to obtain optically
active polymers from monomers containing no centers of optical asymmetry
only when, during the polymerization, monomer units are incorporated so
as to develop new asymmetric centers. The asymmetry of the new centers
arises from a difference in the chemical constitution of the groups contiguous
t o t he ca rbo n at oms t he m se lv e s1 0 1- 10 4 .
Such a result was obtained by means of stereospecific polymerization pro-
cesses, operating under conditions that allow asymmetric induction to favor
the formation of one of the two enantiomorphous structures of the monomer
unit.
The methods that have led us to the asymmetric synthesis of polymers of
substituted diolefins and of certain heterocyclic, unsaturated compounds are
of two types.
(1) The first is the use of normal stereospecific catalysts wherein at least one
group bound to the organometal l ic compound used in the catalyst prepara-
tion, which will be the terminal group of the macromolecules, is optically ac-
t i v e10 1. In this case the asymmetric induction is probably due to the particular
c onf ig urat io n o f the t e rmin al group of the g rowi ng c hai n bound to t he
catalyst .
(2) A second method is based on the use of conventional stereospecific cata-
lysts prepared without using optically active alkyls, provided they are com-
plexed with optical ly active Lewis bases, such as b-phenylalanine 10 2, or with
the use of an optical ly active transition metal compound 104 (Table 7).
In the first case, as the polymerization proceeds, the optical activity de-

5 4 1 9 6 3 G I U L I O N A T T A
creases, as could be expected in view of the fact that any accidental inversion
of configuration exerts an action not confined to one monomer unit only, but
tending to extend to subsequent units.
In the second case, on the other hand, the induction is due to the asymmetry
of the optically active counterion 1 0 5, which maintains its steric structure also
in the case where the asymmetric polymerization gives low optical yields.
These results can be extended to the interpretation of stereospecific catalysis
of vinyl monomers. They suggest that a higher stereospecificity can be expec-
ted when using catalysts, the active centers of which are per se asymmetric,
t ha n w he n sy mme t r i c c at a ly s t s a re use d, i n w hi c h t he s t e re osp e c i f i c i t y
de r i v es f r om asy mme t r i c i nduc t io n broug ht abo ut by t he con f ig urat i on
assumed by the last polymerized unit.
Even before the discovery of the asymmetric synthesis of high polymers, we
attributed 106 the stereospecificity of certain heterogeneous catalysts, prepared
by reaction of solid titanium halides, to the fact that the active centers contain
surface atoms of a transition metal having coordination number 6. In fact it is
known that, in such a case, when at least two of the coordinated groups show
a different chemical nature with respect to the others, enantiomorphous struc-
tures of the surface complexes can exist.
The high stereospecificity of such catalysts is probably due to the fact that
the initial complex maintains its asymmetry even when linked to the growing
chain.
A n i n te re s t i ng a sp e c t o f t h e as ymm et r i c p o l y me r i za t io n o f be nz ofura n

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A R O C M O L E C U L E S 5 5
consists in an autocatalytic effect observed in the first reaction period. In fact
it was noticed that the optical activity of the polymers increases as the poly-
me r i z at i on p roc e e ds107 (T ab le 8 ) .
To clari fy this phenomenon further, polymerization runs have been per-
formed in the presence of optical ly active po lybenzofuran previously ob-
tained.
Although the sign of the optical activity always corresponds to that of the
- phenylalanine complexed with the counter ion, nevertheless the presence
of preformed polymer, obtained in the same polymerization or added to the
catalytic system at the beginning of the polymerization, causes an increase in
the optical activity of the polymer newly formed.
Such an observation may have an interest that goes beyond the interpreta-
tion of stereospecific polymerization; in fact it can suggest suitable patterns
characteristic of certain biological processes in which the formation of asym-
metric molecules or groups of a given type is connected with the preexistence
of optically active macromolecules.

5 6 1 9 6 3 G I U L I O N A T T A
1. H. Staudinger, Die hochmolekularen organischen Verbidugen, Springer, Berlin,1932.
2. A. A-Morton, E.E.Magat and R.L. Letsinger, Soc., 69 (1947) 950.3. G.R.Levi and G.Natta, Atti Accad.Nazl.Lincei, Rend ., [6] 2 (1925) 1; G.Natta,
ibid., [6] 2(1925)495;G.NattaandA.Rejna, ibid., [6] 4(1926)48;G.Natta,AbovoCimento, 3(1926)114; G.Natta and E. Casazza, Atti Accad. Nazl.Lincei, Rend., [6}5(1927) 803; G.Natta , ibid., [6] 5 (1927) 1003; G.Natta, Gazz.Chim.Ital ., 58(1928) 344; G.Natta and L.Passerini, ibid., 58 (1928) 472; G.Natta and M.Strada,ibid., 58 (1928) 419; G.Natta, ibid., 58 (1928)619,870; G.Nattaand M.Strada, AttiAccad. Nazi. Lincei , Rend., [6}7 (1928) 1024; G. Natta and L. Passerini, Gazz. Chirn.Ital., 58 (1928) 597,59 (1929) 280; G. Natta and L.Passerini, Atti Accad. Nazi. Lincei,Rend., [6] 9 (1929) 557; G.Natta and L.Passerini, Gazz.Chim.Ital., 59 (1929) 129;G.Bruni and G.Natta, Rec. Trav.Chim., 48 (1929) 860; G.Natta and L.Passerini,Gazz. Chim. Ital., 59 (1929) 620; G.Natta, Atti III Congr. Nazl.Chim.Pura e Appl.,Firenze, 1929, p. 347; G.Natta and L.Passerini, ibid., p.365; G.Natta, Atti Accad.Nazl.Lincei, Rend., [6]11(1930) 679; G.Natta and A.Nasini, Nature, 125 (1930)457; G.Natta, ibid., 126 (1930)97,127(1931)129,235.
4. G. Natta, M. Baccaredda and R.Rigamonti, Gazz. Chim. Ital., 65(193 5) 182 ; G.Natta, M.Baccaredda and R.Rigamonti, Monatsh.Chem., 66 (1935) 64; G.Natta,M.Baccaredda and R.Rigamonti, Sitzunger Akad.Wiss.(Wien), 14 (1935) 196;G.Natta and M.Baccaredda, Atti Accad.Nazl.Lincei, Rend., [6] 23 (1936) 444;G.Natta and R.Rigamonti, ibid., [6] 24 (1936) 381.
5. G.Natta, Giom.Chim.Ind.ed Appl., 12 (1930) 13; G.Natta, Österr.Chemiker-Ztg.,40 (1937) 162; G.Natta, P.Pino, G.Mazzanti and I.Pasquon, Chim. Ind. (Milan),35 (1953 1705.
6. G.Natta and M.Strada, Giorn.Chim.Ind.ed Appl., 12 (1930) 169, 13 (1931) 317;G.Natta and R.Rigamonti, ibid., 14 (1932) 217.
7. G.Natta, Chim. Ina. (Milan) , 24 (1942) 43; G.Natta and G.F.Mattei, ibid., 24(1942) 271; G.Natta and G.Negri, Dechenra Monograph., 21(1952) 258.
8. G.Natta, P.Pino and R.Ercoli, J. Am.Chem. Soc., 74 (1952) 4496.9. G.Natta and E.Mantica, Gazz.Chim.Ital., 81(1951)164.
10. K.Ziegler, Angew.Chem., 64(1952)323.11. G.Natta, P.Pino and M.Farina, Ric. Sci. Suppl ., 25 (1955) 120.12. K.Ziegler, E.Holzkamp, H.Breil and H. Martin, Anger. Chem., 67 (1955) 541.13. G.Natta, P.Pino and G.Mazzanti, Brit.Pat., 810,023; U.S.Pat. 3,112,300 and
3,112,301 (Italian priority, 8 June 1954).14. G.Natta, Atti.Accad.Nazl.Lincei,Mem., [8]4(1955)61; G.Natta, J.Polymer Sci.,16
(1955) 143 ; G.Natta, P.Pino, P.Corradti, F.Danusso, E.Mantica, G.Mazzantiand G.Moraglio,J. Am.Chem. Soc., 77 (1955) 1708; G.Natta, P.Pino and G.Maz-zanti, Chim.Ind. (Milan), 37 (1955) 927.
15. G. Natta, P.Pino and G.Mazzanti, Gazz.Chim. Ital., 87 (1957) 528.16. G.Natta and P. Corradini, Atti AccadNazl.Lincei, Mem., [8]4(1955)73.17. G.Natta and P. Corradini, Atti Accad. Nazl.Lincei, Rend., [8]18(1955)19.18. G.Natta and R. Rigamonti, Atti Accad.Nazl.Lincei, Rend., [6]24(1936)381.

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 5 7
19. G. Natta, L.Porri, G. Zanini and L. Fiore, Chim. Ind. (Milan), 41(1959) 526.20. G. Natta, L.Porri, P. Corradini and D. Morero, Atti Accad. Nazl. Lincei, Rend., [8]
20 (1956) 560.21. G.Natta and P.Corradini, Atti Accad.Nazl.Lincei, Rend., [8] 19 (1955)229;G.Nat-
ta and P.Corradini, J.Polymer.Sci., 20(1956)251; G.Natta and L.Porri, Belgian Pat.549,544
22. M.L. Huggins, G.Natta, V.Desreux and H.Mark, J.Polymer Sci., 56(1962) 153.23. L.PauIing and R.B.Corey, Proc.Natl.Acad.Sci. (U.S.), 37(1951) 205.24. C. W.Bunn, Proc.Roy.Soc. (London), Ser.A, 180 (1942) 67.25. G.Natta, P.Pino and G.Mazzanti, Italian Pat. 526,101, Brit.Pat. 828,791; G.Natta,
P.Corradini, I. W.Bassi and L.Porri, Atti Accad.Nazl.Lincei, Rend., [8] 24 (1958)121.
26. G.Natta and P.Corradini, Nuovo Cimento, Suppl., [10] 15 (1960) 9; P.Corradini,AttiAccad.Nazl.Lincei,Rend., [8]28(1960)632.
27. G.Perego and I. W.Bassi, Makromol.Chem., 61(1963)198.28. I.W.Bassi, personal communication.29. G. Natta, P. Corradini and I.W.Bassi, Nuovo Cimento, Suppl. [10] 15(1960)68.30. G.Natta, P.Corradini and I. W.Bassi, Nuovo Cimento, Suppl., [10] 15(1960) 52.31. G.Natta, P.Corradini and I.W.Bassi, Atti Accad. Nazl.Lincei, Rend., [8]23(1957)
363.32. G.Natta, P.Corradini and I.W.Bassi, Nuovo Cimento, Suppl., [10] 15 (1960) 83.33. I.W.Bassi, AttiAccad.Nazl.Lincei, Rend., [8]29 (1960) 193.34. G.Dall’Asta and I.W.Bassi, Chim.Ind.(Milan), 43(1961)999.35. G. Natta, G.Dall' Asta, G. Mazzand, I.Pasquon, A.Valvassori and A.Zambelli, J.
Am.Chem.Soc., 83(1961)3343; G.Natta, G.DaII’Asta, G.Mazzauti and F.Ciam-pelli, Kolloid-Z., 182 (1962) 50; P.Corradini and P.Ganis, Makromol.Chem., 62(1963)97.
36. G.Natta, G.Dall’Asta, G.Mazzauti, I.Pasquon, A.Valvassori and A.Zarnbelli,Makromol.Chem., 54(1962)95.
37. G.Natta, G.DaII’Asta and G.Mazzauti, Chim.Ind. (Milan), 44(1962)1212.38. G.Natta, G.Ma.zzanti, G.F.Pregaglia and M.Binagbi, ].Am.Chem. Soc., 82(1960)
5511.39. G.Natta, G.Mazzanti, G.F.Pregaglia and G.Pozzi, J.Polymer Sci., 58(1962)1201.40. D.Sianesi, G.Pajaro and F.Danusso, Chim.Ind. (Milan), 41(1959)1176; G.Natta,
Makromol.Chem., 35(1960)93.41. G.Natta, L.Porri, A.Carbonaro and G.LugIi, Makromol.Chem., 53(1962)52.42. G. Natta,J.Polymer Sri., 34 (1959) 531; G. Natta and I.Pasquon, Advan.Catalysis, 9
(1 9 5 9) I .43. G.Natta, Angew.Chem., 68(1956)393; Chim.Ind. (Milan), 38 (1956) 751.44. G.Natta, Ric.Sci., Suppl., 28 (1958)1.45. G.Natta, F.Danusso and D.Sianesi, Makromol.Chem., 30(1959)238; F.Danusso,
Chim.lnd. (Milan), 44(1962) 611.46. G.Natta, Experientia, Suppl., 7(1957)21; Materie Plastiche, 21(1958)3.47. G. Natta and G.Mazzanti, Tetrahedron, 8(1960)86.

58 1 9 6 3 G I U L I O N A T T A
48. D. J.Cram and K.R.Kopecky, J.Am.Chem. Soc., 81 (1959) 2748; D. J.Cram andD.R.Wilson, ibid., 85 (1963) 1249; M.Szwarc, Chem.Ind. (London), (1958) 1589;G.E.Ham, J. Polymer Sci., 40(1959)569,46(1960)475.
49. C.E.Schildknecht, S. T. Gross, H.R.Davidson, J.M.Lambert and A. O.Zoss, Ind.Eng.Chem., 40(1948)2104.
50. G.Natta, L.Porri, A.Carbonaro and G.Stoppa, Makrornol.Chern., 77(1964)114;G. Natta, L. Porri and A. Carbonaro, ibid., 77 (1964) 126.
51. G.Natta, P. Corradini and G.Auegra, Atti Accad. Nazl.Lincei, Rend., [8]26(1959)155.
52. G.Natta, P.Corradini and G.Allegra, J.Polymer Sci., 51 (1961) 399; G.Allegra,Nuovo Cimento, [10]23(1962)502.
53. G.Natta, Actes II Congr.Intern.de Catalyse, Paris, 1960, 1961, p.39; Chim.Ind.(Milan), 42(1960)1207.
54. G. Natta, I. Pasquon, A. Zambelli and G. Gatti, J.Polymer Sri., 51(1961)387.5 5. G. Natta, P. Corradini, I. W. Bassi and L.Porri, Atti Accad. Nazl. Lincei, Rend., [8]
24(1958)121.56. G.Natta, P.Pino, E.Mantica, F.Danusso, G.Mazzanti and M.Peraldo, Chim. Ind.
(Milan), 38(1956)124..57. G.Natta, G.Pajaro, LPasquon and V. Stellacci, Atti Accad. Nazl. Lincei, Rend., [8]
24(1958)479.58. G.Natta, P.Pino, G.Mazzanti and R.Lanzo, Chim.Ind. (Milan), 39 (1957) 1032.59. G.Natta, LPasquon and E.Giachetti, Angew.Chem., 69(1957)213.60. G.Natta, I.Pasquon, E. Giachetti and F. Scalari, Chim. Ind. (Milan), 40(1958)103.61. G.Natta, LPasquon and E.Giachetti, Chim.Ind. (Milan), 40 (1958) 97; G.Natta,
LPasquon, E. Giichetti and G.Pajaro, ibid., 40 (1958) 267.62. G. Natta and I. Pasquon, Advan. Catalysis, 11(1959)1.63. G. Natta and I.Pasquon, Volume CorsoEstivo Chimica Macronrolecole, Varenna, 1961,
C.N.R., Rome, 1963, p.75.64. G.Natta, LPasquon and A.Zambelli, J. Am.Chem.Soc., 84(1962)1488.65. G.Natta, LPasquon, P. Corradini, M. Peraldo, M. Pegoraro and A. Zambelli, Arti
Accad.Nazl.Lincei, Rend., [8]28(1960)539.66. G.Natta, G. Mazzanti, A. Valvassori, G. Sartori and D. Fiumani, J.Polymer Sci., 51
(1961) 411.67. G. Natta, Rubber Plastics Age, 38(1957)495 ; G. Natta and G. Crespi, Rubber Age
(N. Y.), 87(1960) 459; G.Natta, G. Crespi and M.Bruzzone, Kautschuk Gumni, 14(1961) 54WT; G. Natta, G. Crespi, E. di Giulio, G.Ballini and M.Bruzzone, RubberPlastics Age, 42 (1961) 53 ; G. Natta, G. Crespi, G. Mazzanti, A. Valvassori, G. Sar-tori and P. Scaglione, Rubber Age (N. Y.), 89 (1961) 636; G.Natta, G. Crespi andG.Mazzanti, Proc. Rubber Technol.Conf, 4th, London, 1962; G. Crespi and E.diGiuho, Rev. Gen. Caoutchouc, 40 (1963) 99 ; G.Natta, G. Crespi, G.Mazzanti, A.Valvassori and G. Sartori, Chim. Ind. (Milan), 45(1963)651.
68. G.Natta, M.Farina and M.Peraldo, Atti Accad.Nazl.Lincei, Rend., [8]25(1958)
424.69. M.Peraldo and M.Farina, Chim. Ind. (Milan), 42(1960)1349.

S T E R E O S P E C I F I C P O L Y M E R I Z A T I O N O F M A C R O M O L E C U L E S 5 9
70. G.Natta, M.Peraldo, M.Farina and G.Bressan, Makromol.Chem., 55(1962)139.71. G.Natta, M.Farina, M.Peraldo, P. Corradini, G.Bressan and P. Ganis, Atti Accad.
Nazl.Lincei, Rend., [8]28(1960)442.72. G.Dall’Asta, G.Mazzanti, G.Natta and L.Porri, Makromol.Chem., 56(1962)224.73. G. Natta, G.Dall’Asta, G. Mazzanti and G. Motroni, Makromol.Chem., 69(1963)
163.74. G.Dall'Asta and G.Mazzanti, Makromol.Chem., 61(1963)178.75. G. Natta, L.Porri, P. Corradini and D.Morero, Chim.Ind. (Milan), 40 (1958) 362;
G.Natta,L.PorriandA.Mazzei, ibid.,41 (1959) 116; G.Natta,L.Porri and A.Car-bonaro, Atti Accad. Nazl.Lincei, Rend., [8]31(1961)I89; G. Natta, L. Porri andL.Fiore, Gazz.Chim. Ital., 89(1959) 761; G.Natta, L.Porri, G.Zanini and L.Fiore,Chim. Ind. (Milan), 41 (1959) 526; G.Natta, L.Porri, G.Zanini and A.Palvarini,ibid.,41(1959)1163.
76. G.Natta, L.Porri, A.Mazzei and D.Morero, Chim. Ind. (Milan), 41 (1959) 398;G.Natta , Rubber Plastics Age, 38(1957)495; G.Natta , Chim.Ind. (Milan), 3 9(1957)653; G.Natta, Rev. Gen.Caoutchouc, 40(1963)785.
77. D.Morero, A. Santambrogio, L.Porri and F. Ciampelli, Chim. Ind f (Milan), 4 1
(1959)758.78. G.Natta, P.Corradini and L.Porri, Atti Accad.Nazl.Lincei, Rend., [8] 20 (1956)
728; G.Natta and P.Corradini, Angew.Chem., 68 (1956) 615; G.Natta, P.Corra-dini and I. W. Bassi, Atti Accad. Nazl. Lincei, Rend., [8]23(1957)363; G. Natta andP.Corradini, Nuovo Cimento, Suppl., [10]15(1960)122.
79. See, for example, Belgian Pat. 551, 851 (1956), Phillips Company U.S.; BelgianPat.573.680 (1958) and 575, 507 (1959), Montecatini, Milan, Italy; see G.Natta,G. Crespi, G. Guzzetta, S. Leghissa and F. Sabbioni, Rubber Plastics Age, 42(1961)402; G.Crespi and U.Flisi, Makromol.Chem., 60(1963)191.
80. S.E.Home et al., Ind.Eng.Chem., 48(1956)784.81. G.Natta, L.Porri and G.Mazzanti, Belgian Pat., 545,952(Italian priority, March
1955).82. G. Natta, L. Porri, P. Corradini, G. Zanini and F. Ciampelli, Atti Accad. Nazl.Lincei,
Rend., [8]29(1960)257; J.Polymer Sci., 51(1961) 463.83. G.Natta, L.Porri, G. Stoppa, G.Allegra and F. Ciampelli, J.Polymer Sci., IB (1963)
6 784. G. Natta, L. Porri, A. Carbonaro, F. Ciampelli and G. Allegra, Makromol.Chem., 51
(1962)229.85. G.Natta and G.Dall'Asta, Italian Pat., 570,434(1956).86. G.Natta, I. W.Bassi and P.Corradini, Makromol.Chem., 18-19(1955)455.87. G. Natta, G. Dall’Asta, G. Mazzanti, U. Giannini and S. Cesca, Angew.Chem., 71
(1959) 205.88. G.Natta, G.Dall’Asta, G.Marzanti and A. CasaIe, Makromol.Chem., 58(1962)217.89. G. Natta, G. Mazzanti, G.Dall’Asta and A. Cassale, Italian Pat., 652,763(1960).90. G.Natta, G.Mazzanti and G.Dall’Asta, Italian Pat., 643, 282 (1960) ; G. Natta,
G.Dall‘Asta and G.Mazzanti, Italian Put. 648,564(1961).91. G.Natta, G. Mazzanti, G.Dall’Asta and P.Longi, Makromol.Chem., 37(1960)160.

6 0 1 9 6 3 G I U L I O N A T T A
92. G. Natta, M. Farina, P. Corradini, M.Peraldo, M.Donati and P. Ganis, Chim.Ind.(Milan), 42(1960) 1360.
93. G.Natta, G.Mazzanti, P.Longi and F.Bernardini, Chim.Ind. (Milan), 42(1960)4 5 7 .
94. G.Natta, G.Mazzanti and P.Corradini, Atti Accad. Nazl.Lincei, Rend., [8] 28(1960) 8; G.Natta, P.Corradini and I.W.Bassi, ibid., [8] 28(1960)284; G.Natta,G.Mazzanti, P.Corradini and I.W.Bassi, Makromol.Chem., 37(1960)156; G.Nat-ta, P.Corradiniand I.W.Bassi, J.Polymer Sci., 51(1961)505.
95. I.W.Bassi, G.Dall’Asta, U.Campigli and E.Strepparola, Makromol.Chem. , 6 0(1963)202.
96. D.Braun, W.Betz and W.Kern, Makromol.Chem., 28(1958) 66.97. G.Natta, G.Mazzanti, P.Longi, G.Dall’Asta and F.Bernardini, J.Polynrer Sci., 51
(1961)487.98. Actually an isotactic chain of finite length does include asymmetric carbon atoms,
but each one is neutralized by another at an equal distance from the center of themain chain:
Hence the fully isotactic polypropylene is a particular case of a meso-configurationand must be optically inactive.
99. G.Natta, P.Pino and G.Mazzanti, Gazz.Chim. Ital., 87(1957) 528.100. P.Pino, G.P.Lorenzi and L.Lardicci, Chim.Ind. (Milan), 43(1960) 711; P.Pino
and G.P.Lorenzi, Soc., 82 (1960) 4745 ; W. J.Baileg and E.T.Yates,J.Org.Chem., 25(1960)1800.
101. G.Natta,M.Farina,M.Pera.ldo andM.Donati, Chim.Ind.(Milan), 42(1960)1363;G.Natta, M.Farina and M.Donati, Makromol.Chem., 43(1961)251.
102. G.Natta, M.Farina, M.Peraldo and G.Bressan, Chim. Ind. (Milan), 43(1961)161;Makromol. Chem., 43(1961)68.
103. G.Natta, L.Poni, A.Carbonaro and G.Lugli, Chim.Ind. (Milan), 43(1961)529.104. G.Natta, L.Porri and S.Valante, Makromol.Chem., 67(1963)225.105. M.Farina and G.Bressan, Makromol.Chem., 61(1963)79.106. G.Natta, Ric. Sci., Suppl., 28 (1958) 1.107. G.Natta, G.Bresan and M.Farina, Atti Accad. Nazl.Lincei, Rend., [8]34(1963)475;
M.Farina, G.Natta and G.Bressan, Symp. Macromolecelar Chemistry I.U.P.A.C.,Paris, 1963; J.Polymer Sci., C4,(1964) 141.

SPATIAL CONFIGURATION OF MACROMOLE-CULAR CHAINSNobel Lecture, December 11, 1974
by
PAUL J. FLORY
Department of Chemistry
Stanford University, Stanford, California
The science of macromolecules has developed from primitive beginnings to aflourishing field of investigative activities within the comparatively brief span ofsome forty years. A wealth of knowledge has been acquired and new pointsof view have illumined various branches of the subject. These advances arethe fruits of efforts of many dedicated investigators working in laboratoriesspread around the world. In a very real sense, I am before you on this occasionas their representative.
In these circumstances, the presentation of a lecture of a scope commensuratewith the supreme honor the Royal Swedish Academy of Sciences has bestowedin granting me the Nobel Prize for Chemistry is an insuperable task. Ratherthan attempt to cover the field comprehensively in keeping with the generouscitation by the Royal Academy of Sciences, I have chosen to dwell on a singletheme. This theme is central to the growth of ideas and concepts concerningmacromolecules and their properties. Implemented by methods that haveemerged in recent years, researches along lines I shall attempt to highlightin this lecture give promise of far-reaching advances in our understanding ofmacromolecular substances - materials that are invaluable to mankind.
These polymeric substances are distinguished at the molecular level fromother materials by the concatenation of atoms or groups to form chains, oftenof great length. That chemical structures of this design should occur is implicitin the multivalency manifested by certain atoms, notably carbon, silicon,oxygen, nitrogen, sulfur and phosphorus, and in the capacity of these atoms toenter into sequential combinations. The concept of a chain molecule consistingof atoms covalently linked is as old as modern chemistry. It dates from theorigins of the graphic formula introduced by Couper in 1858 and advancedby Kekult, Loschmidt and others shortly thereafter. Nothing in chemicaltheory, either then apparent or later revealed, sets a limit on the number ofatoms that may be thus joined together. The rules of chemical valency, evenin their most primitive form, anticipate the occurrence of macromolecularstructures.
The importance of macromolecular substances, or polymers, is matched bytheir ubiquity. Examples too numerous to mention abound in biologicalsystems. They comprise the structural materials of both plants and animals.Macromolecules elaborated through processes of evolution perform intricateregulatory and reproductive functions in living cells. Synthetic polymers in

P.J. Flory 157
great variety are familiar in articles of commerce. The prevailing structuralmotif is the linear chain of serially connected atoms, groups or structuralunits. Departures from strict linearity may sometimes occur through the agencyof occasional branched units that impart a ramified pattern to the over-allstructure. Linearity is predominant in most macromolecular substances,however.
It is noteworthy that the chemical bonds in macromolecules differ in nodiscernible respect from those in “monomeric” compounds of low molecularweight. The same rules of valency apply; the lengths of the bonds, e.g., C-C,C-H, C-O, etc., are the same as the corresponding bonds in monomericmolecules within limits of experimental measurement. This seemingly trivialobservation has two important implications: first, the chemistry of macro-molecules is coextensive with that of low molecular substances; second, thechemical basis for the special properties of polymers that equip them for somany applications and functions, both in nature and in the artifacts of man, isnot therefore to be sought in peculiarities of chemical bonding but rather intheir macromolecular constitution, specifically, in the attributes of longmolecular chains.
Comprehension of the spatial relationships between the atoms of a moleculeis a universal prerequisite for bridging the connection between the graphicformula and the properties of the substance so constituted. Structural chemistryhas provided a wealth of information on bond lengths and bond angles. Bymeans of this information the graphic formula, primarily a topological device,has been superseded by the structural formula and by the space model thataffords a quantitative representation of the molecule in three dimensions.The stage was thus set for the consideration of rotations about chemical bonds,i.e., for conformational analysis of conventional organic compounds, especiallycyclic ones. A proper account of bond rotations obviously is essential for adefinitive analysis of the spatial geometry of a molecule whose structurepermits such rotations.
The configuration of a linear macromolecule in space involves circum-stances of much greater complexity. A portion of such a molecule is shownschematically in Figure 1. Consecutive bonds comprising the chain skeletonare joined at angles q fixed within narrow limits. Rotations may occur aboutthese skeletal bonds. Each such rotation is subject, however, to a potentialdetermined by the character of the bond itself and by hindrances imposed bysteric interactions between pendant atoms and groups. The number andvariety of configurations (or conformations in the language of organic chemistry)that may be generated by execution of rotations about each of the skeletalbonds of a long chain, comprising thousands of bonds in a typical polymer, isprodigious beyond comprehension. When the macromolecule is free ofconstraints, e.g., when in dilutesolution, all of these configurations are accessible.Analysis of the manner in which such a molecule may arrange itself in spacefinds close analogies elsewhere in science, e.g., in the familiar problem ofrandom walk, in diffusion, in the mathematical treatment of systems in onedimension, and in the behavior of real gases.

ïëè Chemistry 1974
Ú·¹ò ïò λ°®»»²¬¿¬·±² ±º ¬¸» µ»´»¬¿´ ¾±²¼ ±º ¿ »½¬·±² ±º ¿ ½¸¿·² ³±´»½«´» ¸±©·²¹«°°´»³»²¬ ¯ ±º ¾±²¼ ¿²¹´»ô ¿²¼ ¬±®·±²¿´ ®±¬¿¬·±² º±® ¾±²¼ ·ô · õ ïô »¬½ò
Inquiry into the spatial configuration of these long-chain molecules, fascinat-ing in itself, derives compelling motivation from its close relevancy to theproperties imparted by such molecules to the materials comprising them.Indeed, most of the properties that distinguish polymers from other substancesare intimately related to the spatial configurations of their molecules, theseconfigurations being available in profusion as noted. The phenomenon ofrubber-like elasticity, the hydrodynamic and thermodynamic properties ofpolymer solutions, and various optical properties are but a few that reflect thespatial character of the random macromolecule. The subject is the nexusbetween chemical constitution and physical and chemical properties of poly-meric substances, both biological and synthetic.
The importance of gaining a grasp of the spatial character of polymericchains became evident immediately upon the establishment, ca. 1930, of thehypothesis that they are covalently linked molecules rather than aggregatesof smaller molecules, an achievement due in large measure to the compellingevidence adduced and forcefully presented by H. Staudinger, Nobelist for1953. In 1932 K. H. Meyer1 adumbrated the theory of rubber-like elasticityby calling attention to the capacity of randomly coiled polymer chains toaccommodate large deformations owing to the variety of configurations acces-sible to them.
W. Kuhn 2 and E. Guth and H. Mark3 made the first attempts at mathe-matical description of the spatial configurations of random chains. The com-plexities of bond geometry and of bond rotations, poorly understood at thetime, were circumvented by taking refuge in the analogy to unrestrictedrandom flights, the theory of which had been fully developed by Lord Rayleigh.The skeletal bonds of the molecular chain were thus likened to the steps in arandom walk in three dimensions, the steps being uncorrelated one to another.Restrictions imposed by bond angles and hindrances to rotation were dismissedon the grounds that they should not affect the form of the results.
For a random flight chain consisting of n bonds each of length l, the mean-square of the distance r between the ends of the chain is given by the familiarrelation
The angle brackets denote the average taken over all configurations. Kuhn4

P. J Flory 159
argued that the consequences of fixed bond angles and hindrances to rotationcould be accommodated by letting several bonds of the chain molecule berepresented by one longer “equivalent” bond, or step, of the random flight.This would require n to be diminished and l to be increased in Eq. 1. Equiv-alently, one may preserve the identification of n and l with the actual molec-ular quantities and replace Eq. (1) with
(2)where C is a constant for polymers of a given homologous series, i.e., forpolymers differing in length but composed of identical monomeric units. Theproportionality between <r2> and chain length expressed in Eq. (2) may beshown to hold for any random chain of finite flexibility, regardless of thestructure, provided that the chain is of sufficient length and that it is unper-turbed by external forces or by »ºº»½¬ ¼«» ¬± »¨½´«¼»¼ ª±´«³» ø½ºò »¯ò÷ò
The result expressed in Eq. (2) is of the utmost importance. Closely asso-ciated with it is the assertion that the density distribution W(r) of values ofthe end-to-end vector r must be Gaussian for chains of sufficient length,irrespective of their chemical structure, provided only that the structure admitsof some degree of flexibility. Hence, for large n the distribution of values of r isdetermined by the single parameter (r2) that defines the Gaussian distribution.
Much of polymer theory has been propounded on the basis of the Kuhn“equivalent” random flight chain, with adjustment of n and ´ô or of C, asrequired to match experimental determination of (r2) or of other configura-tion-dependent quantities. The validity of this model therefore invites criticalexamination. Its ·²¬®·²·½ ¿®¬·º·½·¿´·¬§ is its foremost deficiency. Actual bondlengths, bond angles and rotational hindrances cannot be incorporated in thismodel. Hence, contact is broken at the outset with the features of chemicalconstitution that distinguish macromolecular chains of one kind from those ofanother. The model is therefore incapable of accounting for the vast differencesin properties exhibited by the great variety of polymeric substances.
The random flight chain is patently unsuited for the treatment of constitutiveproperties that are configuration-dependent, e.g., dipole moments, opticalpolarizabilities and dichroism. Inasmuch as the contribution to one of theseproperties from a structural unit of the chain is a vector or tensor, it cannot bereferenced to an equivalent bond that is a mere line. Moreover, the equivalentbond cannot be embedded unambiguously in the real structure.
Methods have recently been devised for treating macromolecular chainsin a realistic manner. They take full account of the structural geometry of thegiven chain and, in excellent approximation, of the potentials affecting bondrotations as well. Before discussing these method, however, I must direct yourattention to another aspect of the subject. I refer to the notorious effect ofvolume exclusion in a polymer chain.
At the hazard of seeming trite, I should begin by pointing out that the chainmolecule is forbidden to adopt a configuration in which two of its parts, orsegments, occupy the same space. The fact is indisputable; its consequencesare less obvious. It will be apparent, however, that volume exclusion vitiates

ïêð Chemistry 1974
Ú·¹ò îò ̸» »ºº»½¬ ±º »¨½´«¼»¼ ª±´«³»ò ̸» ½±²º·¹«®¿¬·±² ±² ¬¸» ´»º¬ ®»°®»»²¬ ¬¸»®¿²¼±³ ½±·´ ·² ¿¾»²½» ±º ª±´«³» »¨½´«·±²ô ¬¸» ½¸¿·² ¾»·²¹ »¯«·ª¿´»²¬ ¬± ¿ ´·²» ·²°¿½»ò ײ ¬¸» µ»¬½¸ ±² ¬¸» ®·¹¸¬ô ¬¸» «²·¬ ±º ¬¸» ½¸¿·² ±½½«°§ º·²·¬» ¼±³¿·² º®±³©¸·½¸ ±¬¸»® «²·¬ ¿®» »¨½´«¼»¼ô ©·¬¸ ¬¸» ®»«´¬ ¬¸¿¬ ¬¸» ¿ª»®¿¹» ·¦» ±º ¬¸» ½±²º·¹«®¿ó¬·±² · ·²½®»¿»¼ò
the analogy between the trajectory of a particle executing a random flight andthe molecular chain, a material body. The particle may cross its own path atwill, but self intersections of the polymer chain are forbidden.
The effect of excluded volume must be dealt with regardless of the modelchosen for representation of the chain. In practice, elimination of the effect ofvolume exclusion is a prerequisite to the analysis of experimental results, as Iwill explain in more detail later.
The closely related problems of random flights with disallowance of selfintersections and of volume exclusion within long-chain molecules have attractedthe attention of many theorists. A variety of mathematical techniques havebeen applied to the treatment of these problems, and a profusion of theorieshave been put forward, some with a high order of sophistication. Extensivenumerical computations of random walks on lattices of various sorts also havebeen carried out. Convergence of results obtained by the many investigatorscaptivated by the subject over the past quarter century seems at last to bediscernible. I shall confine myself to a brief sketch of an early, comparativelysimple approach to the solution of this problem.5 The results it yields contrastwith its simplicity.
Returning to the analogy of the trajectory traced by a particle undergoinga sequence of finite displacements, we consider only those trajectories that arefree of intersections as being acceptable for the chain molecule. Directions ofsuccessive steps may or may not be correlated, i.e., restrictions on bondangles and rotational hindrances may or may not be operative; this is im-material with respect to the matter immediately at hand. Obviously, theset of eligible configurations will occupy a larger domain, on the average, thanthose having one or more self intersections. Hence, volume exclusion mustcause <r2> to increase. The associated expansion of the spatial configuration isillustrated in Fig. 2. Other configuration-dependent quantities may be affectedas well.
This much is readily evident. Assessment of the magnitude of the perturba-tion of the configuration and its dependence on chain length require a morepenetrating examination.
The problem has two interrelated parts: (i) the mutual exclusion of thespace occupied by segments comprising the chain tends to disperse them over a

P. J. Flory ïêï
larger volume, and (ii) the concomitant alteration of the chain configurationopposes expansion of the chain. Volume exclusion (i) is commonplace. It isprevalent in conventional dilute solutions and in real gases, molecules of whichmutually exclude one another. In the polymer chain the same rules of exclusionapply, but treatment of the problem is complicated by its association with (ii).
Pursuing the analogies to dilute solutions and gases, we adopt a “smootheddensity” or “mean field” model. The segments of the chain, x in number, areconsidered to pervade a volume V, the connections between them beingignored insofar as part (i) is concerned. The segment need not be definedexplicitly; it may be identified with a repeating unit or some other approxi-mately isometric portion of the chain. In any case, x will be proportional to thenumber n of bonds; in general x ¹ n, however. For simplicity, we may considerthe segment density to be uniform throughout the volume V; that is, = x/Vwithin V and = 0 outside of V. This volume should be proportional to( r2 ) 3 / 2, where <r2> is the mean-square separation of the ends of the chainaveraged over those configurations ²±¬ ¼·¿´´±©»¼ ¾§ »¨½´«¼»¼ ª±´«³» ·²¬»®¿½¬·±²òAccordingly, we let
where A is a numerical factor expected to be of the order of magnitude ofunity.
It is necessary to digress at this point for the purpose of drawing a distinctionbetween <r2> for the chain perturbed by the effects of excluded volume and<r2>0 for the unperturbed chain in the absence of such effects. If a denotes thefactor by which a linear dimension of the configuration is altered, then
(4)
Equation (2), having been derived without regard for excluded volumeinteractions, should be replaced by
(2')
where C reaches a constant value with increase in n for any series of finitelyflexible chains.
The smoothed density within the domain of a linear macromolecule havinga molecular weight of 100,000 or greater (i.e., n > 1000) is low, only on theorder of one percent or less of the space being occupied by chain segments.For a random dispersion of the segments over the volume V, encounters inwhich segments overlap are rare in the sense that few of them are thus in-volved. However, the expectation that such a dispersion is entirely free ofoverlaps between any pair of segments is very small for a long chain. Theattrition of configurations due to excluded volume is therefore severe.
In light of the low segment density, it suffices to consider only binaryencounters. Hence, if b is the volume excluded by a segment, the probabilitythat an arbitrary distribution of their centers within the volume V is freeof conflicts between any pair of segments is


P. J. Flory 163
sums are executed over all configurations of the chain. The squared radius ofgyration s2, i.e., the mean-square of the distances of the segments from theircenter of gravity, is preferable to r2 as a parameter with which to characterizethe spatial distribution.’ Treatments carried out with these refinementsaffirm the essential validity of the result expressed by Eq. (12) or (12’). Theyshow conclusively 7,8 that the form of the result should hold in the limit oflarge values of i.e., for large excluded volume and/or high chain length,and hence for > > 1. In this limit, /z = 1.67 according to H. Fujitaand T. Norisuye. 8 For a < ~ 1.4, however, this ratio decreases, reaching avalue of 1.276 at = 1 .8,9
The general utility of the foregoing result derived from the most elementaryconsiderations is thus substantiated by elaboration and refinement of theanalysis, the quantitative inaccuracy of Eqs. (12) and (12’) in the range1.0 < a 1.4 notwithstanding. The relationship between a and the parameterz prescribed by these equations, especially as refined by Fujita and Norisuye,8
appears to be well supported by experiment.10,11
The principal conclusions to be drawn from the foregoing results are thefollowing: the expansion of the configuration due to volume exclusion increaseswith chain length without limit for b > 0; for very large values of relativeto it should increase as the l/10 power of the chain length. Thesustained increase of the perturbation with chain length reflects the fact thatinteractions between segments that are remote in sequence along the chainare dominant in affecting the dimensions of the chain. It is on this accountthat the excluded volume effect is often referred to as a long-range inter-a c t i o n .9-12
The problem has been treated by a variety of other procedures.9-12 Notableamongst these treatments is the self-consistent field theory of S. F. Edwards.12
The asymptotic dependence of a on the one-tenth power of the chain length,and hence the dependence of <r2> on n 6/ 5 for large values of the parameter z,has been confirmed.12
The dilute solution is the milieu chosen for most physicochemical ex-periments conducted for the purpose of characterizing polymers. The effectof excluded volume is reflected in the properties of the polymer molecule thusdetermined, and must be taken into account if the measurements are to beproperly interpreted, The viscosity of a dilute polymer solution is illustrative.Its usefulness for the characterization of polymers gained recognition largelythrough the work of Staudinger and his collaborators.
Results are usually expressed as the intrinsic viscosity defined as theratio of the increase in the relative viscosity by the polymeric solute toits concentration c in the limit of infinite dilution. That is,
the concentration c being expressed in weight per unit volume. The incrementin viscosity due to a polymer molecule. is proportional to its hydrodynamicvolume, which in turn should be proportional to <r2>3/2 for a typical polymer

164 Chemistry 1974
chain. Hence, should be proportional to the product of <r2> 3/2 a n dthe number density of solute molecules given by c/M where M is the molecularweight. It follows that
molecular weight, provided of course that the molecular weight, and hencethe chain length, is sufficiently large.
If the excluded volume effect could be ignored, the intrinsic viscosity shouldvary proportionally to M 1/ 2. Since, however, a increases with M, a strongerdependence on M generally is observed. Often the dependence of onmolecular weight can be represented in satisfactory approximation by theempirical relation
Measurement of light scattering as a function of angle, a method introducedby the late P. Debye, affords a convenient means for determining the mean-square radius of gyration. Small-angle scattering of x-rays (and lately ofneutrons) offers an alternative for securing the same information. From theradius of gyration one may obtain the parameter <r2> upon which attentionis focused here. The results are affected, of course, by the perturbation dueto excluded volume. Inasmuch as the perturbation is dependent on the solventand temperature, the results directly obtained by these methods are notintrinsically characteristic of the macromolecule. Values obtained for <r 2>from the intrinsic viscosity by use of Eq. (13), or by other methods, must alsobe construed to be jointly dependent on the macromolecule and its environ-ment.
If the factor a were known, the necessary correction could be introducedreadily to obtain the more substantive quantities, such as <r2> 0 and <s2> 0
that characterize the macromolecule itself and are generally quite independentof the solvent. Evaluation of a according to Eq. (11) and (12) would requirethe excluded volume b. This parameter depends on the solvent in a manner

P. J. Flory 165
that eludes prediction. Fairly extensive experimental measurements arerequired for its estimation, or for otherwise making correction for the ex-pansion a.
All these difficulties are circumvented if measurements on the polymersolution are conducted under conditions such that the effects of excludedvolume are suppressed. The resistance of atoms to superposition cannot, ofcourse, be set aside. But the consequences thereof can be neutralized. We haveonly to recall that the effects of excluded volume in a gas comprising realmolecules of finite size are exactly compensated by intermolecular attractionsat the Boyle temperature (up to moderately high gas densities). At thistemperature the real gas masquerades as an ideal one.
For the macromolecule in solution, realization of the analogous conditionrequires a relatively poor solvent in which the polymer segments prefer self-contacts over contacts with the solvent. The incidence of self-contacts maythen be adjusted by manipulating the temperature and/or the solvent com-position until the required balance is established. Carrying the analogy toa real gas a step further, we require the excluded volume integral for the inter-action between a pair of segments to vanish; that is, we require that b=0.This is the necessary and sufficient condition. 5,6,13
As already noted, estimation of the value of b is difficult; the predictionof conditions under which b shall precisely vanish would be even moreprecarious. However, the “Theta point,” so-called, at which this conditionis met is readily identified with high accuracy by any of several experimentalprocedures. An excluded volume of zero connotes a second virial coefficientof zero, and hence conformance of the osmotic pressure to the celebrated lawof J. H. van’t Hoff. The Theta point may be located directly from osmoticpressure determinations, from light scattering measured as a function ofconcentration, or from determination of the precipitation point as a functionof molecular weight.6,13
The efficacy of this procedure, validated a number of years ago with thecollaboration of T. G. Fox, W. R. Krigbaum, and others, 13,17,18 is illustratedin Figs. 3 and 4 by the lower plots of data representing intrinsic viscositiesmeasured under ideal, or Theta conditions.6 The slopes of the lines drawnthrough the lower sets of points are exactly 1/2, as required by Eq. (13’) whenb = 0 and hence a = 1. The excellent agreement here illustrated has beenabundantly confirmed for linear macromolecules of the widest variety, rangingfrom polyisobutylene and polyethylene to polyribonucleotides.19 At the Thetapoint the mean-square chain vector <r2> 0 and the mean-square radius ofgyration <s2>0 invariably are found to be proportional to chain length.
A highly effective strategy for characterization of macromolecules emergesfrom these findings. By conducting experiments at the Theta point, thedisconcerting (albeit interesting!) effects of excluded volume on experimentallymeasured quantities may be eliminated. Parameters (e.g., <r2> 0 and <s2> 0)are thus obtained that are characteristic of the molecular chain. They arefound to be virtually independent of the nature of the “Theta solvent” selected.Having eliminated the effects of long range interactions, one may turn

ïêê Chemistry 1974
Ú·¹ò íò ײ¬®·²·½ ª·½±·¬·» ±º °±´§¬§®»²» º®¿½¬·±² °´±¬¬»¼ ¿¹¿·²¬ ¬¸»·® ³±´»½«´¿®©»·¹¸¬ ±² ´±¹¿®·¬¸³·½ ½¿´» ·² ¿½½±®¼¿²½» ©·¬¸ Û¯ò øïì÷ò ̸» «°°»® »¬ ±º ¼¿¬¿ ©¿¼»¬»®³·²»¼ ·² ¾»²¦»²»ô ¿ ¹±±¼ ±´ª»²¬ º±® ¬¸· °±´§³»®ò ̸» ´±©»® »¬ ±º ¼¿¬¿ ©¿¼»¬»®³·²»¼ ·² ½§½´±¸»¨¿²» ¿¬ ¬¸» ̸»¬¿ °±·²¬ò ̸» ´±°» ±º ¬¸» ´·²» ¿®» a ã ðòéë ¿²¼ðòëðô ®»°»½¬·ª»´§ò Ú®±³ ¬¸» ®»«´¬ ±º ß´¬¿®»ô ɧ³¿² ¿²¼ ß´´»²òïì
attention to the role of short range features: structural geometry, bondrotation potentials, and steric interactions between near-neighboring groups.It is here that the influences of chemical architecture are laid bare. If themarked differences in properties that distinguish the great variety of polymericsubstances, both natural and synthetic, are to be rationally understood infundamental, molecular terms, this must be the focus of future research.
Rigorous theoretical methods have recently become available for dealingrealistically with short-range features peculiar to a given structure. Most ofthe remainder of this lecture is devoted to a brief overview of these methods.Although the field is comparatively new and its exploration has only begun,space will not permit a digest of the results already obtained.
The broad objective of the methods to which we now turn attention is totreat the structure and conformations accessible to the chain molecule in sucha manner as will enable one to calculate configuration-dependent quantitiesand to average them over all conformations, or spatial configurations, of theunperturbed chain. The properties under consideration are constitutive; they

P. J. Flory 167
ïòð
ðòè
× × × × × I I
ð
Ú·¹ò ìò ײ¬®·²·½ ª·½±·¬·» ±º º®¿½¬·±² ±º °±´§ø³»¬¸§´ ³»¬¸¿½®§´¿¬»÷ ¿½½±®¼·²¹ ¬± ݸ·²¿·¿²¼ Í¿³«»´ïë °´±¬¬»¼ ¿ ·² Ú·¹ò íò ̸» «°°»® »¬ ±º °±·²¬ ©¿ ³»¿«®»¼ ·² ³»¬¸§´»¬¸§´ µ»¬±²»ô ¿ ¹±±¼ ±´ª»²¬ò ̸» ´±©»® »¬ ©¿ ¼»¬»®³·²»¼ ·² ¿ ³·¨¬«®» ±º ³»¬¸§´ethyl ketone and isopropanol ¿¬ ¬¸» ̸»¬¿ °±·²¬ò Í´±°» ¿®» ¿ ã ðòéç ¿²¼ ðòëðô ®»°»½ó¬·ª»´§ò
represent sums of contributions from the individual units, or chemicalgroupings, comprising the chain. In addition to <r2>0 and <s2>0, they include:mean-square dipole moments; the optical anisotropies underlying strainbirefringence, depolarized light scattering and electric birefringence; di-chroism; and the higher moments, both scalar and tensor, of the chainvector r.Classical statistical mechanics provides the basis for evaluating the con-figurational averages of these quantities. Since bond lengths and bond anglesordinarily may be regarded as fixed, the bond rotations ¶ are the variablesover which averaging must be carried out. The procedure rests on the rota-¬·±²¿´ ·±³»®·½ ¬¿¬» ½¸»³»ô the foundations for which were set forth in largemeasure by M. V. Volkenstein20 and his colleagues21 in Leningrad in the late1950’s and early 1960’s. It is best explained by examples.
Consider rotation about an internal bond of an n-alkane chain. As is nowwell established,22,23 the three staggered conformations, trans(t), gauche-plus(g+) and its mirror image, gauche-minus(g-), are of lower energy than

ïêè Chemistry 1974
Ú·¹ò ëò Ì©± ±º ¬¸» ¬¿¹¹»®»¼ ½±²º±®³¿¬·±² º±® ²ó¾«¬¿²»æ ¬®¿² ±² ¬¸» ´»º¬ ¿²¼ ¹¿«½¸»ó³·²« ±² ¬¸» ®·¹¸¬ò
the eclipsed forms. The t and g- conformations of n-butane are shown in Fig. 5.The energies of the eclipsed conformations separating t from g+ and t fromg- are about 3.5 kcal. mol-1 above the energy of the trans conformation. Hence,in good approximation, it is justified to consider each bond to occur in one ofthree ®±¬¿¬·±²¿´ ·±³»®·½ ¬¿¬» centered near (but not necessarily precisely at)the energy minima associated with the three staggered conformations. 20-24
The gauche minima lie at an energy of about 500 cal. mol-1 above trans.Each of the former is therefore disfavored compared to the latter by a “sta-tistical weight” factor we choose to call s » exp( -Eg/RT), where Eg is about500 cal. mol -1; thus, (s » 0.5 at T = 400 K.
A complication arises from the fact that the potentials affecting bond rotationsusually are neighbor dependent; i.e., the potential affecting depends on therotations and Bond rotations cannot, therefore, be treated indepen-dently .20,21,24,25 The source of this interdependence in the case of an n-alkanechain is illustrated in Fig. 6 showing a pair of consecutive bonds in three oftheir nine conformations. In the conformations tt, tg+, g+t, tg- and g-t, thetwo methylene groups pendant to this pair of bonds are well separated. Forgauche rotations g+g+ and g-g- of the same hand (Fig. 6b), these groups areproximate but not appreciably overlapped. Semi-empirical calculations21,24,26,
27 show the intramolecular energy for these two equivalent conformations to bevery nearly equal to the sum (cu. 1000 cal. mol-1) for two well-separated gauchebonds; i.e., the interdependence of the pair of rotations is negligible. In theremaining conformations, g+g - and g-g + , the steric overlap is severe (Fig. 6c).It may be alleviated somewhat by compromising rotations, but the excessenergy associated therewith is nevertheless about 2.0 kcal. mol-1. Hence, astatistical weight factor w » exp (-2000/RT) is required for each suchpair. 24,26,28 Inspection of models in detail shows that interactions dependentupon rotations about three, four of five consecutive bonds are disallowed byinterferences of shorter range and hence may be ignored.24 It suffices there-fore to consider first neighbors only.
The occurrence of interactions that depend on pairs of skeletal bonds is therule in chain molecules. In some of them, notably in vinyl polymers, suchinteractions may affect most of the conformations. Hence, interdependenceof rotations usually plays a major role in determining the spatial configuration

H
P. J Flory 169
H H H H
H
of the chain. The rotational isometric state approximation, whereby thecontinuous variation of each j is replaced by discrete states, provides the key tomat hematica l solut ion of t he p ro blem po sed by rot at io na l in terde-p e n d e n c e .20 ,21 ,24 ,25
It is necessary therefore to consider the bonds pairwise consecutively, andto formulate a set of statistical weights for bond i that take account of thestate of bond i-l. These statistical weights are conveniently presented in theform of an array, or matrix, as follows:
where the rows are indexed in the order t , g +, g- to the state of bondi- 1, and the columns are indexed to the state of bond i in the same order.According to the analysis of the alkane chain conformations presented brieflyabove, U i takes the form24,26,28

170 Chemistry 1974
A conformation of the chain is specified in the rotational isometric stateapproximation by stipulation of the states for all internal bonds 2 to n-linclusive; e.g., by g+ttg -g -, etc. Owing to the three-fold symmetry of theterminal methyl groups of the alkane chain, rotations about the terminalbonds are inconsequential and hence are ignored. The statistical weight forthe specified conformation of the chain is obtained by selecting the appropriatefactor for each bond from the array (15) according to the state of this bond andof its predecessor, and taking the product of such factors for all bonds 2 to
n - 1. In the example above this product is , etc. It willbe obvious that the first superscripted index in one of the factors u must repeatthe second index of its predecessor since these indices refer to the same bond.
The configuration partition function, representing the sum of all suchfactors, one for each conformation of the chain as represented by the scheme ofrotational isomeric states, is
where the subscripts are serial indexes. Each ui must be assigned as specifiedabove. The sum, which extends over all ordered combinations of rotationalstates, may be generated identically as the product of the arrays Ui treated asmatrices. That is, according to the rules of matrix multiplication
where U1 = row (1, 0, 0) and Un = column (1, 1, 1). Matrix multiplicationgenerates products precisely of the character to which attention is directed atthe close of the preceding paragraph. Serial multiplication of the statisticalweight matrices generates this product for each and every conformation of thechain, and Eq. (18) with the operators U1 and Un appended gives their sum.
The foregoing procedure for evaluation of Z is a minor variant of the methodof H. A. Kramers and G. H. Wannier 29 for treating a hypothetical one-dimensional ferromagnet or lattice. A number of interesting characteristics ofthe chain molecule can be deduced from the partition function by applicationof familiar techniques of statistical mechanics. I shall resist the temptation toelaborate these beyond mentioning two properties of the molecule that maybe derived directly from the partition function, namely, the incidences of thevarious rotational states and combinations thereof, and the equilibrium con-stants between isomeric structures of the chain in the presence of catalystseffectuating their inter-conversion. Vinyl polymers having the structuredepicted in Fig. 7 with R’ ¹ R afford examples wherein the study of equilibria

¾»¬©»»² various diastereomeric forms arising from the local chirality ofindividual skeletal bonds has been especially fruitful.30
Consider the evaluation of a configuration-dependent property for a givenconfiguration, or conformation, of the chain. Since the configuration is seldom“given”, the problem as stated is artificial. Its solution, however, is a necessaryprecursor to the ultimate goal, which is to obtain the average of the propertyover all configurations. A property or characteristic of the chain that will servefor illustration is the end-to-end vector r. Suppose we wish to express thisª»½¬±® ©·¬¸ ®»º»®»²½» ¬± ¬¸» º·®¬ ¬©± ¾±²¼ ±º ¬¸» ½¸¿·²ò For ¼»º·²·¬»²»ô ´»¬ ¿
Cartesian coordinate system be affixed to these two bonds with its X,-axis alongthe º·®¬ ¾±²¼ ¿²¼ ·¬ Çï󿨷 ·² ¬¸» °´¿²» ±º ¾±²¼ ï ¿²¼ îô ¿ ¸±©² ·² Ú·¹ò èò
·² ¬¸· ®»º»®»²½» º®¿³»ò
In order to facilitate the task of transforming every bond vector to thereference frame affiliated with the first bond, it is helpful to define a referenceframe for each skeletal bond of the chain. For example, one may place theaxis Xi along bond i, the Yi-axis in the plane of bonds i - l and i, and choosethe Zi-axis to complete a right-handed Cartesian system. Let Ti symbolize thetransformation that, by premultiplication, converts the representation of avector in reference frame i+l to its representation in the preceding referenceframe i. Then bond i referred to the initial reference frame is given by
where li is presented in reference frame i. The required sum is just
(19)
This sum of products can be generated according to a simple algorithm.We first define a “generator” matrix ß · as follows31,32

ïéî Chemistry 1974
Ú·¹ò èò Í°»½·º·½¿¬·±² ±º ¬¸» ½±±®¼·²¿¬» ¿¨» ¿ºº·¨»¼ ¬± »¿½¸ ±º ¬¸»¬¸» ½¸¿·²æ ÈïÇ ï º±® ¾±²¼ ï ¿²¼ ÈîÇ î º±® ¾±²¼ îò
º·®¬ ¬©± ¾±²¼ ±º
together with the two terminal matrices
(21)
(22)
In these equations Ti is the matrix representation of the transformation speci-fied above and 0 is the null matrix of order 1 x 3. The desired vector r isgenerated identically by taking the serial product of the A’s; i.e.,
as may easily be verified from the elementary rules of matrix multiplication.Each generator matrix Ai depends on the length of bond i and, through T i,on both the angle between bonds i and i+l and on the angle of rotation
about bond i (see Fig. 1).In order to obtain the average of r over all configurations of the chain, it is
necessary to evaluate the sum over all products of the kind given in Eq. (23)with each of them multiplied by the appropriate statistical weight for thespecified configuration of the chain; see Eq. (17). That is,

P. J Flory 173
T h e n31
The matrix a i comprises the elements of Ui (see Eq. (15)) joined with the Amatrix for the rotational state of bond i as prescribed by the column index. Itwill be apparent that serial multiplication of the a i according to Eq. (28)generates the statistical weight factor u2u3. . .un-1 for every configuration of thechain in the same way that these factors are generated by serial multiplicationof the statistical weight matrices U i in Eq. (18). Simultaneously, Eq. (28)generates the product of A’s (see Eq. (23)) that produces the vector ® for eachconfiguration thus weighted. The resulting products of statistical weights andof A’s are precisely the terms required by Eq. (24). The terminal factors inEq. (28) yield their sum.
With greater mathematical concision31,32
If each bond vector li is expressed in its own reference frame i, then
(34)

ïéì Chemistry 1974
ð îð 4 0 6 0 8 0 ïðð
ÒËÓÞÛÎ ÑÚ ÞÑÒÜÍô ²Ú·¹ò çò ݸ¿®¿½¬»®·¬·½ ®¿¬·± ä®îâ °´±¬¬»¼ ¿¹¿·²¬ ¬¸» ²«³¾»® ±º ¾±²¼ ² ·² ¬¸»½¸¿·² º±® °±´§³»¬¸§´»²»ô ¿²¼ º±® ·±¬¿½¬·½ ¿²¼ §²¼·±¬¿½¬·½ °±´§ ø³»¬¸§´ ³»¬¿½®§´¿¬»÷ �òÚ®±³ ¬¸» ½¿´½«´¿¬·±² ±º ß¾»ô Ö»®²·¹¿² ¿²¼ Ú´±®§îê ¿²¼ ±º DZ±²òíì
That is,
(35)
where G1 has the form of the first row, and Gn that of final column of Eq. (34).Evaluation of <r2> 0 proceeds exactly as set forth above for <r>0.
32,33
The foregoing method enjoys great versatility. The chain may be of anyspecified length and structure. If it comprises a variety of skeletal bonds andrepeat units, the factors entering into the serial products have merely to befashioned to introduce the characteristics of the bond represented by each ofthe successive factors. The mathematical methods are exact; the procedure isfree of approximations beyond that involved in adoption of the rotationalisometric state scheme. With judicious choice of rotational states, the errorhere involved is generally within the limits of accuracy of basic information onbond rotations, nonbonded interactions, etc.
Other molecular properties that may be computed by straightforwardadaptation of these methods24,32 include the higher scalar moments <r4>0, <r6>0,etc; the moment tensors formed from r; the radius ofgyration <s2>0 = (n+ 1)-2
the optical polarizability and its invariants that govern thei j
optical anistropy as manifested in depolarized light scattering, in strain bire-

P. J. Flory 175
fringence and in electric birefringence; x-ray scattering at small angles; andNMR chemical shifts.
For illustration, characteristic ratios <r2>0/nl2 are plotted in Fig. 9 againstthe numbers n of bonds for n-alkanes and for isotactic and syndiotactic poly-(methyl methacrylate), or PMMA. Isotactic PMMA is represented by theformula in Fig. 7 with R = COOCH, and R’ = CH, and with all dyads ofthe meso form, i.e., with R occurring consistently above (or below) the axis ofthe chain. In the syndiotactic stereoisomer, the substituents R and R’ alternatefrom one side to the other, all dyads being racemic.
For the alkane and the isotactic PMMA chains the characteristic ratiosincrease monotonically with chain length, approaching asymptotic values forn » 100 bonds. This behavior is typical. For syndiotactic PMMA, however,the characteristic ratio passes through a maximum at intermediate values of n,according to these computations by D. Y. Yoon.34 This behavior can be traced34
to the inequality of the skeletal bond angles in PMMA in conjunction withthe preference for tt conformations in the syndiotactic chain.35 The maximumexhibited in Fig. 9 for this polymer is thus a direct consequence of its constitu-tion. This peculiarity manifests itself in the small angle scattering of x-raysand neutrons by predominantly syndiotactic PMMA of high molecularweight. 36 Scattering intensities are enhanced at angles corresponding, roughly,to distances approximating <r2> 0
1/ 2 at the maximum in Fig. 9. This enhance-ment, heretofore considered anomalous, is in fact a direct consequence of thestructure and configuration of syndiotactic PMMA.
It is thus apparent that subtle features of the chemical architecture ofpolymeric chains are manifested in their molecular properties. Treatment interms of the artificial models much in use at present may therefore be quitemisleading.
The analysis of the spatial configurations of macromolecular chains presentedabove is addressed primarily to an isolated molecule as it exists, for example,in a dilute solution. On theoretical grounds, the results obtained should beequally applicable to the molecules as they occur in an amorphous polymer,even in total absence of a diluent. This assertion follows unambiguously fromthe statistical thermodynamics of mixing of polymer chains,5,6,37 includingtheir mixtures with low molecular diluents. It has evoked much skepticism,however, and opinions to the contrary have been widespread. These opposingviews stem primarily from qualitative arguments to the effect that difficultiesinherent in the packing of long chains of consecutively connected segments tospace-filling density can only be resolved either by alignment of the chainsin bundle arrays, or by segregation of individual molecules in the form ofcompact globules. In either circumstance, the chain configuration would bealtered drastically.
Whereas dense packing of polymer chains may appear to be a distressingtask, a thorough examination of the problem leads to the firm conclusion thatmacromolecular chains whose structures offer sufficient flexibility are capableof meeting the challenge without departure or deviation from their intrinsicproclivities. In brief, the number of configurations the chains may assume is

176 Chemistry 1974
«ºº·½·»²¬´§ ¹®»¿¬ ¬± ¹«¿®¿²¬»» ²«³»®±« ½±³¾·²¿¬·±² ±º ¿®®¿²¹»³»²¬ ·²
which the condition of mutual exclusion of space is met throughout the sys-tem as a whole. Moreover, the task of packing chain molecules is not madeeasier by partial ordering of the chains or by segregating them.6,37 Any state oforganization short of complete abandonment of disorder in favor of creationof a crystalline phase offers no advantage, in a statistical-thermodynamic sense.
Theoretical arguments aside, experimental evidence is compelling in showingthe chains to occur in random configurations in amorphous polymers, andfurther that these configurations correspond quantitatively with those of theunperturbed state discussed above. 38 The evidence comes from a variety ofsources: from investigations on rubber elasticity, chemical cyclization equi-libria, thermodynamics of solutions, and, most recently, from neutron scatter-ing studies on protonated polymers in deuterated hosts (or vice versa).39 Theinvestigations last mentioned go further. They confirm the prediction made¬©»²¬§óº·ª» years ago that the excluded volume perturbation should beannulled in the bulk amorphous state. 5 The excluded volume effect is thereforean aberration of the dilute solution, which, unfortunately, is the mediumpreferred for physicochemical characterization of macromolecules.
Knowledge gained through investigations, theoretical and experimental,on the spatial configuration and associated properties of random macro-molecular chains acquires added significance and importance from its direct,quantitative applicability to the amorphous state. In a somewhat less quanti-tative sense, this knowledge applies to the intercrystalline regions of semi-crystalline polymers as well. It is the special properties of polymeric materialsin amorphous phases that render them uniquely suited to many of the functionsthey perform both in biological systems and in technological applications.These properties are intimately related to the nature of the spatial configura-tions of the constituent molecules.
Investigation of the conformations and spatial configurations of macro-molecular chains is motivated therefore by considerations that go much beyond·¬ ¿°°»¿´ ¿ ¿ ¬·³«´¿¬·²¹ ·²¬»´´»½¬«¿´ »¨»®½·»ò ß½¯«··¬·±² ±º ¿ ¬¸±®±«¹¸
«²¼»®¬¿²¼·²¹ ±º ¬¸» «¾¶»½¬ ³«¬ ¾» ®»¹¿®¼»¼ ¿ ·²¼·°»²¿¾´» ¬± ¬¸» ½±³ó
°®»¸»²·±² ±º ®¿¬·±²¿´ ½±²²»½¬·±² ¾»¬©»»² ½¸»³·½¿´ ½±²¬·¬«¬·±² ¿²¼ ¬¸±»
°®±°»®¬·» ¬¸¿¬ ®»²¼»® °±´§³»® »»²¬·¿´ ¬± ´·ª·²¹ ±®¹¿²·³ ¿²¼ ¬± ¬¸» ²»»¼
of man.
REFERENCES
ïò Ó»§»®ô Õò Øòô ª±² Í«·½¸ô Ùòô ¿²¼ Ê¿´µ-ô Ûòô Õ±´´±·¼óÆô ëçô îðè øïçíî÷òîò Õ«¸²ô Éòô Õ±´´±·¼óÆô êèô î øïçíì÷òíò Ù«¬¸ô Ûòô ¿²¼ Ó¿®µô Øòô Ó±²¿¬½¸òô êëô çí øïçíì÷òìò Õ«¸²ô Éòô Õ±´´±·¼óÆòô éêô îëè øïçíê÷å èéô í øïçíç÷òëò Ú´±®§ô Ðò Öòô Öò ݸ»³ò и§òô ïéô íðí øïçìç÷òêò Ú´±®§ô Ðò Öòô Principles of Polymer Chemistry, ݱ®²»´´ ˲·ª»®·¬§ Ю»ô ׬¸¿½¿ô
ÒòÇòô ïçëíòéò Ú´±®§ô Ðò Öòô ¿²¼ Ú·µô Íòô Öò ݸ»³ò и§òô ììô îîìí øïçêê÷òèò Ú«¶·¬¿ô Øòô ¿²¼ Ò±®·«§»ô Ìòô Öò ݸ»³ò и§òô ëîô ïïë øïçéï÷òçò Ú·¨³¿²ô Óòô Öò ݸ»³ò и§òô îíô ïêëê øïçëë÷ò

P. J. Flory ïéé
ïðò Ç¿³¿µ¿©¿ô Øòô Modern Theory of Polymer Solutions, Ø¿®°»® ¿²¼ α©ô Ò»© DZ®µôïçéïò
ïïò Ç¿³¿µ¿©¿ô Øòô Ы®» ¿²¼ ß°°´ò ݸ»³òô 31, ïéç øïçéî÷òïîò Û¼©¿®¼ô Íò Úòô Ю±½ò и§ò ͱ½òô øÔ±²¼±²÷ô èëô êïí øïçêë÷òïíò Ú±¨ô Ìò Ùòô Ö®òô ¿²¼ Ú´±®§ô Ðò Öòô Öò и§ò ¿²¼ ݱ´´ò ݸ»³òô ëíô ïçé øïçìç÷ò Ú´±®§ô
P. J., ¿²¼ Ú±¨ô Ìò Ùòô Ö®òô Öò б´§³»® ͽ·òô ëô éìë øïçëð÷ å Öò ß³»®ò ݸ»³ò ͱ½òôéíô ïçðì øïçëï÷ò
ïìò ß´¬¿®»ô Ìòô ɧ³¿²ô Üò Ðò ¿²¼ ß´´»²ô Êò Îòô Öò б´§³»® ͽ·òô ßô îô ìëíí øïçêì÷òïëò ݸ·²¿·ô Íò Òòô ¿²¼ Í¿³«»´ô Îò Öòô Öò б´§³»® ͽ·òô ïçô ìêí øïçëê÷òïêò Ó¿²¼»´µ»®²ô Ôòô ¿²¼ Ú´±®§ô Ðò Öòô Öò ݸ»³ò и§òô îðô îïî øïçëî÷ô Ó¿²¼»´µ»®²ô Ôòô
Õ®·¹¾¿«³ô Éò Îò ¿²¼ Ú´±®§ô Ðò Öòô ·¾·¼òô îðô ïíçî øïçëî÷òïéò Ú±¨ô Ìò Ùòô Ö®òô ¿²¼ Ú´±®§ô Ðò Öòô Öò ß³»®ò ݸ»³ò ͱ½òô éíô ïçðçô ïçïë øïçëï÷òïèò Õ®·¹¾¿«³ô Éò Îòô Ó¿²¼»´µ»®²ô Ôòô ¿²¼ Ú´±®§ô Ðò Öòô Öò б´§³»® ͽ·òô çô íèï øïçëî÷ò
Õ®·¹¾¿«³ô Éò Îò ¿²¼ Ú´±®§ô Ðò Öòô ·¾·¼ô ïïô íé øïçëí÷ôïçò Û·»²¾»®¹ô Øòô ¿²¼ Ú»´»²º»´¼ô Ùòô Öò Ó±´ò Þ·±´òô íðô ïé øïçêé÷ò ײ²»®ô Ôò Üòô ¿²¼
Ú»´»²º»´¼ô Ùòô ·¾·¼òô ëðô íéí øïçéð÷òîðò ʱ´µ»²¬»·²ô Óò Êòô Configurational Statistics of Polymeric Chains, ¬®¿²´¿¬»¼ º®±³
¬¸» Ϋ·¿² »¼òô ïçëçô ¾§ Íò Òò ¿²¼ Óò Öò Ì·³¿¸»ººô ײ¬»®½·»²½»ô Ò»© DZ®µôïçêíò
îïò Þ·®¸¬»·²ô Ìò Óò ¿²¼ Ь·¬§²ô Ñò Þòô Conformations of Macromolecules, ¬®¿²´¿¬»¼º®±³ ¬¸» Ϋ·¿² »¼òô ïçêìô ¾§ Íò Òò ¿²¼ Óò Öò Ì·³¿¸»ººô ײ¬»®½·»²½»ô Ò»© DZ®µôïçêêò
îîò 符»®ô Õò Íòô Ü·½«·±² Ú¿®¿¼¿§ ͱ½òô ïðô êê øïçëï÷òîíò Ó·¦«¸·³¿ô Íòô ͬ®«½¬«®» ±º Ó±´»½«´» ¿²¼ ײ¬»®²¿´ ન¬·±²ô ß½¿¼»³·½ Ю»ô
Ò»© DZ®µô ïçëìòîìò Ú´±®§ô Ðò Öòô Statistical Mechanics of Chain Molecules, ײ¬»®½·»²½» Ы¾´·¸»®ô
Ò»© DZ®µô ïçêçòîëò Ù±¬´·¾ô Ç«ò Ç¿òô Ƹò Ú·¦ Ì»µ¸²ô îçô ëîí øïçëç÷ò Þ·®¸¬»·²ô Ìò Óòô ¿²¼ Ь·¬§²ô
Ñò Þòô ·¾·¼òô îçô ïðìè øïçëç÷ò Ô·¬±²ô Íòô Öò ݸ»³ò и§òô íðô çêì øïçëç÷ò Ò¿¹¿·ôÕòô ·¾·¼òô íïô ïïêç øïçëç÷ô ر»ª»ô Ýò ßò Öòô ·¾·¼òô íîô èèè øïçêð÷ò
îêò ß¾»ô ßòô Ö»®²·¹¿²ô Îò Ôòô ¿²¼ Ú´±®§ô Ðò Öòô Öò ß ³»®ò ݸ»³ò ͱ½òô èèô êí ï ø ïçêê÷òîéò ͽ±¬¬ô Îò ßòô ¿²¼ ͽ¸»®¿¹¿ô Øò ßòô Öò ݸ»³ò и§òô ììô íðëì øïçêê÷òîèò ر»ª»ô Ýò ßò Öòô Öò ݸ»³ò и§òô íëô ïîêê øïçêï÷òîçò Õ®¿³»®ô Øò ßòô ¿²¼ É¿²²·»®ô Ùò Øòô и§ò λªòô êðô îëî øïçìï÷òíðò É·´´·¿³ô ßò Üòô ¿²¼ Ú´±®§ô Ðò Öòô Öò ß³»®ò ݸ»³ò ͱ½òô çïô íïïïô íïïè øïçêç÷ò
Ú´±®§ô Ðò Öòô ¿²¼ з½µ´»ô Ýò Öòô Öò ݸ»³ò ͱ½òô Ú¿®¿¼¿§ Ì®¿²ò ××ô êçô êíî ø ïçéí÷òÍ«¬»®ô Ëò Éòô Ы½½·ô Íòô ¿²¼ з²±ô Ðòô Öò ß³»®ò ݸ»³ò ͱ½òô çé ïðïè øïçéë÷ò
íïò Ú´±®§ô Ðò Öòô Ю±½ò Ò¿¬ò ß½¿¼ò ͽ·òô éðô ïèïç øïçéí÷òíîò Ú´±®§ô Ðò Öòô Ó¿½®±³±´»½«´»ô éô íèï øïçéì÷òííò Ú´±®§ô Ðò Öòô ¿²¼ ß¾»ô Çòô Öò ݸ»³ò и§ò ëìô ïíëï øïçéï÷òíìò DZ±²ô Üò Çòô «²°«¾´·¸»¼ ®»«´¬ô Ô¿¾±®¿¬±®§ ±º Ó¿½®±³±´»½«´¿® ݸ»³·¬®§ô ͬ¿²ó
º±®¼ ˲·ª»®·¬§òíëò Í«²¼¿®¿®¿¶¿²ô Ðò Îòô ¿²¼ Ú´±®§ô Ðò Öòô Öò ß³»®ò ݸ»³ò ͱ½òô çêô ëðîë øïçéì÷òíêò Õ·®¬»ô Îò Ùòô ¿²¼ Õ®¿¬µ§ô Ñòô Æò и§·µô ݸ»³ò Ò»«» Ú±´¹»ô íïô íêí øïçêî÷ò
Õ·®¬»ô Îò Ùòô Ó¿µ®±³±´ò ݸ»³òô ïðïô çï øïçêé÷ò Õ·®¬»ô Îò Ùòô Õ®«»ô Éò ßòô ¿²¼×¾»´ô Õòô б´§³»®ô ïêô ïîð øïçéë÷ò
íéò Ú´±®§ô Ðò Öòô Ю±½ò α§¿´ ͱ½òô ßô îíìô êð øïçëê÷ò Ú´±®§ô Ðò Öòô Öò б´§³ò ͽ·òô ìçôïðë øïçêï÷ò
íèò Ú´±®§ô Ðò Öòô Ы®» ú ß°°´ò ݸ»³òô Ó¿½®±³±´»½«´¿® ݸ»³òô èô ´óïë øïçéî÷òíçò Õ·®¬»ô Îò Ùòô Õ®«»ô Éò ßòô ¿²¼ ͽ¸»´¬»²ô Öòô Ó¿µ®±³±´ò ݸ»³òô ïêîô îçç øïçéî÷ò
Þ»²±·¬ô Øòô Ü»½µ»®ô Üòô Ø·¹¹·²ô Öò Íòô з½±¬ô Ýòô ݱ¬¬±²ô Öò Ðòô Ú¿®²±«¨ô Þòô Ö¿²ó²·²µô Ùòô ¿²¼ Ѿ»®ô Îòô Ò¿¬«®»ô и§·½¿´ ͽ·»²½»ô îìëô ïí øïçéí÷ò Þ¿´´¿®¼ô Üò ÙòØòô É·¹²¿´´ô Ùò Üòô ¿²¼ ͽ¸»´¬»²ô Öòô Û«®ò б´§³»® Öòô çô çêë øïçéí÷å ·¾·¼ô îðôèêï øïçéì÷ò Ú·½¸»®ô Ûò Éòô Ô»·»®ô Ùòô ¿²¼ ×¾»´ô Õò б´§³»® Ô»¬¬»®ô ïíô íçøïçéë÷ò

ÍÑÚÌ ÓßÌÌÛÎ
Nobel Lecture, December 9, 1991
¾§
P IE RRE-GILLES DE GEN NE S
College de France, Paris, France
What do we mean by soft matter? Americans prefer to call it “complexfluids”. This is a rather ugly name, which tends to discourage the youngstudents. But it does indeed bring in two of the major features:
×÷ ݱ³°´»¨·¬§ò We may, in a certain primitive sense, say that modernbiology has proceeded from studies on simple model systems (bacterias) tocomplex multicellular organisms (plants, invertebrates, vertebrates...). Simi-larly, from the explosion of atomic physics in the first half of this century,one of the outgrowths is soft matter, based on polymers, surfactants, liquidcrystals, and also on colloidal grains.
î÷ Ú´»¨·¾·´·¬§ò I like to explain this through one early polymer experiment,which has been initiated by the Indians of the Amazon basin: they collectedthe sap from the hevea tree, put it on their foot, let it “dry” for a short time.And, behold, they have a ¾±±¬ò From a microscopic point of view, the startingpoint is a set of independent, flexible polymer chains. The oxygen from theair builds in a few bridges between the chains, and this brings in a spectacu-lar change: we shift from a liquid to a network structure which can resisttension - what we now call a ®«¾¾»® (in French: caoutchouc, a directtranscription of the Indian word). What is striking in this experiment, is thefact that a very mild chemical action has induced a drastic change inmechanical properties: a typical feature of soft matter.
Of course, with some other polymer systems, we tend to build more rigidstructures. An important example is an enzyme. This is a long sequence ofaminoacids, which folds up into a compact globule. A few of these amino-acids play a critical role: they build up the “active site” which is built toperform a specific form of catalysis (or recognition). An interesting ques-tion, raised long ago by Jacques Monod, is the following: we have a choiceof twenty aminoacids at each point in the sequence, and we want to build areceptor site where the active units are positioned in space in some strictway. We cannot just put in these active units, because, if linked directly,they would not realise the correct orientations and positions. So, in betweentwo active units, we need a “spacer”, a sequence of aminoacids which hasenough variability to allow a good relative positioning of the active sites atboth ends of the spacer. Monod’s question was; what is the minimum lengthof spacers?
It turns out that the answer is rather sharply defined(l). The magicnumber is around 13-14. Below 14 units, you will not usually succeed ingetting the desired conformation. Above 14, you will have many sequences

Pierre Gilles de Gennes 9
which can make it. The argument is primitive; it takes into account excludedvolume effects, but it does not recognise another need for a stable enzyme- namely that the interior should be built preferably with hydrophobicunits, while the outer surface must be hydrophilic. My guess is that thiscannot change the magic number by much more than one unit. Indeed,when we look at the spacer sizes in a simple globular protein like myosin, wesee that they are not far from the magic number.
Let me return now to flexible polymers in solution, and sketch some oftheir strange mechanical properties. One beautiful example is the fourroller experiment set up by Andrew Keller and his coworkers(2). Here, adilute solution of coils is subjected to a purely longitudinal shear. If the exittrajectory is well chosen (in the symmetry plane of the exit channel), themolecules are stressed over long times. What is found is that, if the shearrate exceeds a certain threshold value an abrupt transition takes place,and the medium becomes birefringent. This is what I had called a “coil-stretch transition”(3). When the shear begins to open the coil, it offersmore grip to the flow, and opens even more... leading to a sharp transition.Here, we see another fascinating aspect of soft matter - the amazingcoupling between mechanics and conformations. Indeed, Keller showedthat rather soon (at shear rates > the chains break), and they do so verynear to their midpoint - a spectacular result.
Another interesting feature of dilute coils is their ability to reduce thelosses in turbulent flows. This is currently called the Toms effect. But inactual fact it was found, even before Toms, by Karol Mysels(4). He is heretoday, to my great pleasure. Together with M. Tabor, we tried to work out ascaling model of coils in a turbulent cascade(5), but our friends in mechan-ics think that it is not realistic, the future will tell what the correct answer is.
I have talked a lot about polymers. It would be logical to do the same withcolloids, or - as I like to call it - “ultra divided matter”. But since I justgave another talk with this title at the Nobel symposium in Göteborg, I willomit the subject, in spite of its enormous practical importance.
Let me rather switch to surfactants, molecules with two parts: a polar headwhich likes water, and an aliphatic tail which hates water. Benjamin Franklinperformed a beautiful experiment using surfactants; on a pond at ClaphamCommon, he poured a small amount of oleic acid, a natural surfactantwhich tends to form a dense film at the water-air interface. He measuredthe volume required to cover all the pond. Knowing the area, he then knewthe height of the film, something like three nanometers in our current units.This was to my knowledge the first measurement of the size of molecules. Inour days, when we are spoilt with exceedingly complex toys, such as nuclearreactors or synchrotron sources, I particularly like to describe experimentsof this Franklin style to my students.
Surfactants allow us to protect a water surface, and to generate thesebeautiful soap bubbles, which are the delight of our children. Most of ourunderstanding of these soap bubbles is due to a remarkable team, Mysels,Shinoda and Frankel, who wrote the book on this subject(6). Unfortunately,

10 Physics 1991
this book is now very hard to find, I very much hope that it will be reprinted.Long ago Françoise Brochard, Jean-François Lennon and I(7) became
interested in some bilayer systems, where we have two sheets of surfactant,each pointing towards the neighbouring water. A related (although morecomplex) system of this type is a red blood cell. For many years it had beenknown that, when observed under phase contrast, these cells flicker. - It wassometimes believed that this flicker reflected an instability of a living systemunder non-equilibrium conditions. Ultimately, the thing is simpler. Theessential property of insoluble bilayers is that they optimise their area atfixed surfactant number. Thus, the energy is stationary with respect to area:the surface tension vanishes. This means that the fluctuations in shape ofthese deflated cells, or “vesicles”, are huge: the flicker is just an example ofBrownian motion for a very flexible object. What Jean-François had donewas to measure space time correlations for the flicker. Françoise thenshowed that they could be understood from a model containing no surfacetensions, but only curvature energies plus viscous forces - another goodexample of soft matter.
This was, in fact, one of the starting points for many studies on surfactantbilayers, pioneered by W. Helfrich and, on a more formal side, on randomsurfaces especially with D. Nelson. One of the great successes in this fieldhas been the invention of the “sponge phase” of microemulsions(8,9). But,more generally, it is amusing to learn from these people that there is someoverlap in thought between the highbrow string theories and the descrip-tions of soaps!
Let me now move to another corner in our garden - liquid crystals.Here, I must pay tribute first to two great pioneers:
i) Georges Friedel, who was the first to understand exactly what is a liquidcrystal, and what are the main types; ii) Charles Frank, who (after some earlywork of Oseen) constructed the elastic theory of nematics, and describedalso a number of their topological defects (“disclinations”).
I will talk here only about the smectics. Observing certain defects (“focalconics”) in smectics, Friedel was able to prove that their structure must be aset of liquid, equidistant, deformable layers(l0). By observations at the onehundred micron scale, he was thus able to infer the correct stucture at theten Å scale - an amazing achievement.
Smectics bring me naturally to another important feature of complexfluids - namely that, in our days, it is sometimes possible ¬± ½®»¿¬» ²»© º±®³ ±º³¿¬¬»®ò The sponge phase quoted above was an example. Another strikingcase was the invention of ferroelectric smectics by R.B. Meyer, in Orsay,circa 1975. He thought about a certain molecular arrangement, with chiralmolecules, which should automatically generate a phase (the “C* phase”)carrying a non-zero electric dipole. Within a few months, our local chemistshad produced the right molecule, and the first liquid ferroelectric wasborn!(ll). In our days, these materials may become very important fordisplay purposes, they commute l0 3 times faster than the nematics in ourwrist-watches.

Pierre-Gilles de Gennes 11
Another case of far smaller importance, but amusing, is the �º»®®±³»½¬·½�constructed by M. Veyssié and P. Fabre. The starting point is a water basedferrofluid; a suspension of very fine magnetic particles. (Ferrofluids wereinvented long ago by R. Rosensweig, and have many amazing properties).Here, what is done, is to prepare a
. . .“club sandwich”
A system like this, subjected to a magnetic field H, is happierwhen H is parallel to the layers. It is then interesting to observe thesandwich, with a polarizing microscope, in the frustrated situation where is normal to the layers. At very small H, nothing is seen. But beyond acertain weak threshold H c, figures like flowers grow in the field(l2). Weunderstand this as a two step process a) just above threshold there is achemical undulation instability b) later, focal conics appear, with a basic sizeimposed by the original undulation, but also with smaller conics (which arerequired to fill space correctly). This “club sandwich” is ultimately detectingrather weak magnetic fields 30 gauss).
Let me quote still another new animal: the Janus ¹®¿·²ô first made by C.Casagrande and M. Veyssié. The god Janus had two faces. The grains havetwo sides: one apolar, and the other polar. Thus, they have certain featuresin common with surfactants. But there is an interesting difference if weconsider the films which they make, for instance at a water air interface. Adense film of a conventional surfactant is quite impermeable. On the otherhand, a dense film of Janus grains always has some interstices between thegrains, and allows for chemical exchange between the two sides; “the skincan breathe”. This may possibly be of some practical interest.
The first technique used to make the Janus grains was based on sphericalparticles, half embedded in a plastic and silanated on the accessible side( 13).This produces only microquantities of material. But a group at Gold-schmidt(l4) research invented a much more clever pathway. The startingpoint is a collection of ¸±´´±© glass particles, which are available commercial-ly. There the outer surface is hydrophobized, and finally the particles arecrushed. The resulting platelets have one side hydrophilic and one sidehydrophobic. They are irregular, but they can be produced in tons.
I would like now to spend a few minutes thinking about the style of softmatter research. One first, major, feature, is the possibility of very simpleexp eriments - in the spirit of Benjamin Franklin. Let me quote twoexamples. The first concerns the ©»¬¬·²¹ ±º º·¾»®ò Usually a fiber, after beingdipped in a liquid, shows a string of droplets, and thus, for some time,people thought that most common fibers were non-wettable. F. Brochardanalysed theoretically the equilibria on curved surfaces, and suggested thatin many cases we should have a wetting film on the fiber, in between thedroplets. J.M. di Meglio and D. Queré established the existence, and thethickness, of the film, in a very elegant way(l5). They created a pair ofneighbouring droplets, one small and one large, and showed that the smallone emptied slowly into the big one (as capillarity wants it to go). Measuringthe speed of the process, they could go back to the thickness of the film

12 Physics 1991
which lies on the fiber and connects the two droplets: the Poiseuille flowrates in the film are very sensitive to thickness.
Another elegant experiment in wetting concerns the ½±´´»½¬·ª» ³±¼» ±º ¿½±²¬¿½¬ ´·²»å the edge of a drop standing on a solid. If one distorts the line bysome external means, it returns to its equilibrium shape with a relaxationrate dependent upon the wavelength of the distortion, which we wanted tostudy. But how could we distort the line? I thought of very complex tricks,using electric fields from an evaporated metal comb, or other, even worse,procedures. But Thierry Ondarcuhu came up with a simple method.
1) He first prepared the unperturbed contact line L by putting a largedroplet on a solid.
2) He then dipped a fiber in the same liquid, pulled it out, and obtained,from the Rayleigh instability, a very periodic string of drops.
3) He laid the fiber on the solid, parallel to L, and generated a line ofdroplets on the solid.
4) He pushed the line L (by tilting the solid), up to the moment where Ltouched the droplets; then coalescence took place, and he had a single, wavyline on which he could measure relaxation rates(16).
I have emphasized experiments more than theory. Of course we needsome theory when thinking of soft matter. And in fact some amusingtheoretical analogies sometimes show up between soft matter and otherfields. One major example is due to S.F. Edwards(l7). Edwards showed abeautiful correspondence between the conformations of a flexible chainand the trajectories of a non relativistic particle; the statistical weight of thechain corresponding to the propagator of the particle. In the presence ofexternal potentials, both systems are ruled by exactly the same Schrödingerequation! This observation has been the key to all later developments inpolymer statistics.
Another amusing analogy relates the smectics A to superconductors. Itwas discovered simultaneously by the late W. McMillan (a great scientist,who we all miss) and by us. Later, it has been exploited artistically by T.Lubensky and his colleagues(l8). Here again, we see a new form of matterbeing invented. We knew that type II superconductors let in the magneticfield in the form of quantized vortices. The analog here is a smectic A insidewhich we add chiral solutes, which play the role of the field. In somefavorable cases, as predicted in 1988 by Lubensky, this may generate asmectic phase drilled by screw dislocations - the so called A* phase. Thiswas discovered experimentally only one year later by Pindak and cowork-ers(19), a beautiful feat.
Let me now end up this sentimental journey into soft matter, with a briefmention of my companions. Some were met during the way, like JeanJacques, a great inventor of liquid crystals, or Karol Mysels, the undisputedmaster of surfactant science. Some others were with me all along the way;Henri Benoit and Sam Edwards, who taught me polymer science; Jacquesdes Cloizeaux and Gerard Jannink, who have produced a deep theoreticalbook on this subject. Finally, an inner core of fellow travelers, over all forms

Pierre-Gilles de Gennes 13
of land and sea: Phil Pincus, Shlomo Alexander, Etienne Guyon, MadeleineVeyssié; and last but not least, Françoise Brochard - sans laquelle leschases ne seraient que ce qu’elles sont.
The final lines are not mine: they come from an experiment on softmatter, after Boudin, which is shown on the following figure.

14 Physics 1991
An English translation might run like this:
“Have fun on sea and landUnhappy it is to become famousRiches, honors, false glitters of this worldAll is but soap bubbles”
No conclusion could be more appropriate today.
REFERENCES:
1. P G de Gennes, in Pvor. 2nd Conf. “Physique théorique et Biologie”. EditionsCNRS 1969 (15 Quai A. France 75007 Paris, France).
2. J.A. Odell, A. Keller, in Polymer-flow Interactions (ed. I. Rabin), AIP, New York1985 ; Av. Keller, J. Odell, Coll. Polym. Sci., 263, 181 (1985).
3. P.G. de Gennes, J. Chem. Phys., 60, 5030 (1974).4. For a historical review see K. Mysels, Chem. Eng. Prog., Symposium series, 67,
45, (1971).5. M. Tabor, P.G. de Gennes, Europhys. Lett., 2, 519 (1986) P.G. de Gennes,
Physica, 140 A, 9 (1986).6. K. Mysels, K. Shinoda, S. Frankel, Soap Films , Pergamon, London (1959).7. F. Brochard, J.F. Lennon, J. Physique (Paris), 36, 1035 (1976).8. G. Porte, J. Marignan, P. Bassereau, R. May, J. Physique (Paris), 49, 511 (1988).9. D. Roux, M.E. Cates, Proceedings of the 4th Nishinomya-Yukawa Symposium,
Springer (to be published).10. G. Friedel, Annales de Physique, 18, 273 (1922).11. R.B. Meyer, L. Liebert, L. Strzelecki, P. Keller, J. Physique L. 69 (1975).12. P. Fabre, C. Casagrande, M. Veyssié, V. Cabuil, R. Massart, “Ferrosmectics : A
new Magnetic and Mesomorphic Phase”, Phys. Rev. Lett., 64, 539 (1990).13. C. Casagrande, M. Veyssié, C.R. Acad. Sci. (Paris), 306 II, 1423 (1988).
C. Casagrande, P. Fabre, M. Veyssié, E. Raphael, Europhys. Lett., 9, 251(1989).
14. B. Grüining, U. Holtschmidt, G. Koerner, G. Rössmy US Patent no 4, 715, 986(Dec 1987).
15. J.M. di Meglio, CR. Acad. Sci. (Paris), 303 II, 437 (1986).16. T. Ondarcuhu, M. Veyssié, Nature, 352, 418 (1991).17. S.F. Edwards, Proc. Phys. Sot. (London), 85, 613 (1965).18. S.R. Renn, T. Lubensky, Phys. Rev., A 38, 2132 (1988).19. J.W. Goodby, M.A. Waugh, S.M. Stein, E. Chin, R. Pindak, J.S. Patel, J. Am.
Chem. Soc., 111, 8119 (1989).