inhibition of the hedgehog pathway targets the tumor- associated
TRANSCRIPT

Angiogenesis, Metastasis, and the Cellular Microenvironment
Inhibition of the Hedgehog Pathway Targets the Tumor-Associated Stroma in Pancreatic Cancer
Rosa F. Hwang1, Todd T. Moore1, Maureen Mertens Hattersley3, Meghan Scarpitti2, Bin Yang3,Erik Devereaux3, Vijaya Ramachandran2, Thiruvengadam Arumugam2, Baoan Ji2, Craig D. Logsdon2,Jeffrey L. Brown3, and Robert Godin3
AbstractPurpose: The Hedgehog (Hh) pathway has emerged as an important pathway in multiple tumor types and is
thought to be dependent on a paracrine signalingmechanism. The purpose of this studywas to determine the role ofpancreatic cancer-associated fibroblasts (human pancreatic stellate cells, HPSCs) in Hh signaling. In addition, weevaluated the efficacy of a novel Hh antagonist, AZD8542, on tumor progression with an emphasis on the role ofthe stroma compartment.Experimental Design: Expression of Hh pathway members and activation of the Hh pathway were analyzed in
bothHPSCs and pancreatic cancer cells.We tested the effects of Smoothened (SMO) inhibitionwith AZD8542 ontumor growth in vivo using an orthotopic model of pancreatic cancer containing varying amounts of stroma.Results:HPSCs expressed high levels of SMO receptor and low levels ofHh ligands, whereas cancer cells showed
the converse expression pattern. HPSC proliferation was stimulated by Sonic Hedgehog with upregulation ofdownstream GLI1 mRNA. These effects were abrogated by AZD8542 treatment. In an orthotopic model ofpancreatic cancer, AZD8542 inhibited tumor growth only when HPSCs were present, implicating a paracrinesignaling mechanism dependent on stroma. Further evidence of paracrine signaling of the Hh pathway in prostateand colon cancer models is provided, demonstrating the broader applicability of our findings.Conclusion: Based on the use of our novel human-derived pancreatic cancer stellate cells, our results suggest
that Hh-targeted therapies primarily affect the tumor-associated stroma, rather than the epithelial compartment.Mol Cancer Res; 10(9); 1147–57. �2012 AACR.
IntroductionThe Hedgehog (Hh) pathway is an essential developmen-
tal pathway involved in regulating key aspects of embryo-genesis, stem cell maintenance, and tumor biology (1). Inhumans, the Hh ligands Sonic, Indian, and Desert Hedge-hog (SHH, IHH, and DHH) bind to the Patched1 protein(PTCH1) on target cells, which results in the release ofinhibition of the SMO receptor. SMO is a 7-transmembraneG-protein–coupled receptor-like protein that, when activat-ed, results in activation of GLI transcription factors andexpression of downstream targets GLI1, PTCH, BCL2,myc, and IGF2.
Aberrantly activated Hh has recently been identified inseveral malignancies, including basal cell carcinomas (2–4),medulloblastomas (5, 6), lung cancer (7–9), prostate cancer(10–12), and gastrointestinal malignancies (10, 13). Inpancreatic adenocarcinoma (PDAC), the Hh pathway isconsidered one of the "core" signaling pathways that isaltered, and the majority of these cancers show abnormalexpression of SHH, PTCH1, and SMO (14). Moreover,these factors are expressed early in preneoplastic pancreaticintraepithelial neoplasia (PanIN) lesions, whereas they areabsent in normal pancreatic tissue, suggesting that thesefactors are important for early tumorigenesis. Blockade ofHh with SMO inhibitors such as cyclopamine inhibitedinvasion and metastasis and prolonged survival in mousemodels of pancreatic cancer (15–17).Recently, intriguing evidence has emerged suggesting that
the Hh pathway is highly active in a paracrine signalingmanner in the tumor microenvironment of some pancreatictumors. When SMO was genetically ablated in the pancre-atic epithelium of PDAC-susceptible mice, development ofPDAC tumors was not affected, suggesting thatHh signalingin PDAC does not occur in an autocrine manner (18). Usinghuman–tumor xenograft models, Yauch and colleagues (19)used species-specific expression profiling to show that Hhpathway antagonist treatment resulted in downregulation of
Authors' Affiliations: Departments of 1Surgical Oncology and 2CancerBiology, The University of Texas M.D. Anderson Cancer Center, Houston,Texas; and 3AstraZeneca Pharmaceuticals, Waltham, Massachusetts
Note: Supplementary data for this article are available at Molecular CancerResearch Online (http://mcr.aacrjournals.org/).
Corresponding Author: Rosa F. Hwang, Department of Surgical Oncol-ogy, The University of Texas M.D. Anderson Cancer Center, P.O. Box301402, Unit 444, Houston TX 77230. Phone: 713-563-1873; Fax: 713-745-1462; E-mail: [email protected]
doi: 10.1158/1541-7786.MCR-12-0022
�2012 American Association for Cancer Research.
MolecularCancer
Research
www.aacrjournals.org 1147
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

Hh target genes only in the murine stromal microenviron-ment but not within the human tumor epithelial compart-ment. Similarly, expression of SMO in mesenchymal cells,but not epithelial cells, in the pancreas led to Hh pathwayactivation, further supporting a paracrine model of Hh-mediated tumorigenesis (20). Finally, treatment of a genet-ically engineered mouse model of PDAC with the Hhinhibitor IPI-926 resulted in depletion of desmoplasticstroma in pancreatic tumors (21). These observations areconsistent with a model in which tumor cells produce Hhligands that trigger signaling in the stromal microenviron-ment in a paracrine manner.Despite these initial observations, the precise role of
stromal cells in Hh signaling in pancreatic cancer is notwell understood.We have previously shown that the cancer-associated fibroblasts in PDAC (human pancreatic stellatecells, HPSCs) produce secreted factors that promote tumorprogression and metastasis in vitro and in vivo (22). In thisstudy, we analyzed the role of HPSCs from the tumor-associated stroma in Hh signaling. In addition, we evaluatedthe efficacy of a novel SMO inhibitor (AZD8542) onpancreatic tumor progression with an emphasis on the roleof theHPSCs from the stroma.We present data that stronglysuggests the primary mechanism of action of Hh signaling inPDAC occurs in a paracrine manner with ligand expressionby the cancer cells and activation of SMO on neighboringHPSCs in the stromal microenvironment.
Materials and MethodsCell cultureNIH-3T3, human embryonic palatal mesenchyme
(HEPM), C3H10T1/2, HeLa, and human colon cancerColo205 cells as well as BxPC3, Panc1, SU86.86,MiaPaca2,and Capan2 pancreatic cancer cell lines were obtained from
American Type Culture Collection (ATCC,Manassas, VA).Human pancreatic cancer MPanc96 and human pancreaticductal epithelial (HPDE) cells were obtained from DrTimothy J. Eberlein (Washington University, St. Louis,MO, USA) and Dr M. Tsao (Ontario Cancer Institute,Toronto, Ontario, Canada), respectively. L3.6pl cells wereobtained from Dr I. Fidler (23) and immortalized HPSCswere isolated as previously described (22). Primary HPSCswere established and cultured as previously described (22).Both immortalized (using hTERT and SV40T) and non-immortalized primary cells were used in these studies.NIH-3T3, HeLa, pancreatic carcinoma cells, and HPSCswere cultured in Dulbecco's minimal essential medium(DMEM) with 10% FBS (Invitrogen) and 1% L-glutamine.HEPM and C3H10T1/2 cells were cultured in Eagle'sminimal essential medium (EMEM) with Earle's BSS þ2 mmol/L L-glutamine þ 1.0 mmol/L nonessential aminoacids þ 1.5 g/L sodium bicarbonate þ 10% FBS. HPDEcells were cultured in keratinocyte serum-free media con-taining 50 mg/mL bovine pituitary extract and 0.2 ng/mLrecombinant epidermal growth factor (all from Invitrogen).Colo205 was maintained as an adherent culture in DMEMcontaining 10% FBS at 37�C in a humidified atmosphere of6% CO2. All other cells were cultured at 37�C in ahumidified atmosphere of 5% CO2.
GLI1 reporter assays: mouse and human versionsA subset of the proprietary AstraZeneca compound col-
lection (40,000 compounds) with similarity to cyclopaminewas screened using a GLI1 luciferase reporter assay toidentify inhibitors of the Hh pathway (24).The GLI1luciferase construct consists of 8 Gli-binding sites upstreamof a luciferase reporter gene (25). The construct was trans-fected into NIH-3T3 cells along with a constitutively activeRenilla luciferase construct as a control. Stable cells wereselected and stimulated with 50% SHH-containing condi-tioned medium. Conditioned media was generated bytransfecting HEK293 cells with an SHH expression vectorand collecting media 48 to 96 hours after transfection. Cellswere treated with Hh inhibitor compounds (in dimethylsulfoxide) at varying concentrations for 24 hours and assayedfor luciferase activity using the Dual-Glo Luciferase AssaySystem (Promega) per the manufacturer's instructions.HEPM cells and C3H10T1/2 cells were treated with inhib-itor compounds for 24 hours and assayed for luciferasereporter activity using the Steady-Glo Luciferase AssaySystem (Promega) per the manufacturer's instructions.Plates were read on the Tecan Ultra microplate reader(Tecan, Mannedorf, Switzerland) at 50 ms integration timeper well.
Differentiation assayTo evaluate the efficacy of SMO inhibitor compounds, an
osteoblast differentiation assay was done using C3H10T1/2cells, which differentiate into osteoblasts after stimulationwith SHH or Wnt. In brief, C3H10T1/2 cells were platedinto 384-well plates, and media was changed to low serum(2% FBS) or conditioned media containing either SHH or
Translational RelevanceThe Hedgehog (Hh) pathway has emerged as an impor-
tant pathway in multiple tumor types including pancreaticcancer (PDAC). Recent evidence suggests that the Hhpathway functions in a paracrine fashion that is dependenton the tumor microenvironment of PDAC. In this study, wedetermined the role of carcinoma-associated fibroblastsisolated from human pancreatic cancer (pancreatic stellatecells, PSCs) in Hh signaling. The Smoothened (SMO) recep-tor was highly expressed in PSCs but was absent in pan-creatic cancer cells. Activation of the Hh pathway in PSCsresulted in increased cell proliferation that was reversed bya novel SMO inhibitor (AZD8542). In vivo, AZD8542 waseffective in reducing tumor growth in an orthotopic modelof PDAC only when stroma was present, indicating astroma-dependent paracrine signaling mechanism. Ourresults suggest that Hh antagonists primarily target thetumor-associated stroma and may be novel therapies forthe treatment of pancreatic cancer.
Hwang et al.
Mol Cancer Res; 10(9) September 2012 Molecular Cancer Research1148
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

WNT3a. Cells were then treated with inhibitor compoundsfor 72 hours, and alkaline phosphatase activity was mea-sured. Cells were lysed in 15 mL of 1� RIPA cell lysis buffer,incubated at �80�C for 30 minutes, treated with p-nitro-phenyl phosphate at 1mg/mL in diethanolamine buffer (pH9.8), incubated at 30�C overnight for color development,and read at absorbance of 405 nm.
SMO binding assayHeLa cells were transfected with pcDNA vector expres-
sing human or mouse myc-tagged SMO (GeneCopoeia;FuGENE transfection reagent, Promega).After 24 hours incubation, media was changed to low
serum (0.5% FBS). The cells were then pretreated withvarious concentrations of SMO inhibitors for 20 minutes,followed by 3 nmol/L BODIPY-labeled cyclopamine (Tor-onto Research Chemicals) and incubated at 37�C for 4hours. After incubation, the cells were fixed with 3%paraformaldehyde and 0.5% Triton X þ 1� PBS (Invitro-gen). The cells were then washed, blocked with 10% goatserum (Fisher Scientific), and incubated with myc-taggedantibody (Cell Signaling Technology) for 2 hours followedby Alexa Fluor 594 anti-rabbit antibody (Invitrogen). Cellswere stained with Hoechst dye (Invitrogen), and fluores-cence was detected using an ImageXpress system (MolecularDevices).
Proliferation assayHPSCs were seeded at 2,000 cells per well in triplicate in
96-well plates and cultured inDMEMcontaining 10%FBS.After overnight attachment, media was changed to DMEMcontaining 1% FBS, and varying concentrations (0, 1.0, 1.5,and 2 mg/mL) of recombinant SHH (rSHH; R&D Systems)were added to the wells. Cell proliferation was analyzed at 72hours using MTS reagent (Promega) added 1 hour beforetaking a spectrophotometric reading according to the man-ufacturer's instructions.
Hh stimulation of HPSCsHPSCs were seeded in 6-well plates and grown to 70%
confluence in DMEM containing 10% FBS. The media wasthen changed to serum-free DMEM overnight. rSHH orrIHH (R&D Systems) was added to the wells (2 mg/mL)along with varying concentrations (0, 10, 100, and1000 nmol/L) of the SMO inhibitor AZD8542 (AstraZe-neca). Cells grown in 1%DMEMwithout rSHH, rIHH, orAZD8542 served as controls. RNA was isolated after 24hours and expression of GLI1 was measured by quantitativereverse transcriptase (RT)-PCR (qRT-PCR) and normalizedto HPRT.
Colon cancer subcutaneous xenograft modelsAnimal protocols for the colon cancer models were
approved by the AstraZeneca R&D Boston site's Institu-tional Animal Care and Use Committee. All animal workwas done in accordance with applicable internal standardsand external local and national guidelines, regulations, andlegislation. Female Ncr nude mice aged 6 to 8 weeks were
maintained under specific-pathogen-free conditions in afacility accredited by the Association for Assessment andAccreditation of Laboratory Animal Care International.Colo205 cells were implanted subcutaneously in the right
flank (4 � 106 cells per mouse) in 0.1 mL of serum-freemedia. Tumors were allowed to grow until they reached anaverage volume of 200 mm3, and mice were randomized(N ¼ 5 per treatment group.) AZD8542 was suspended in0.5% (v/v) hydroxypropyl methyl cellulose (HPMC) insterile water and administered orally once daily (20 or40 mg/kg). Tumor and blood samples were collected inRNAlater (Ambion) and EDTA, respectively, at 1, 4, 6, 8,12, and 16 hours after dosing. In a coimplantation xenograftmodel of colon cancer, mice were injected with both HT29(0.3� 106) andMEF (1.5� 106) cells subcutaneously in theright flank (100 mL per injection) for a final tumor:stroma(T/S) ratio of 1:5. When tumors reached 70 to 100 mm3,animals were randomized to receive either SMO inhibitor(20–80 mg/kg) or vehicle (0.5% HPMC/0.1% Tween 80;N¼ 10 per group). Tumor measurements and body weightswere recorded twice weekly. Data are expressed as percentagetumor growth inhibition at day 29. For pharmacodynamicstudies, tumors were collected 8 hours after each dose inRNAlater (Qiagen) or formalin (Newcomer Supply). Bloodwas collected for pharmacokinetic analysis.Tumors were lysed using a Lysing Matrix D tube (MP
Biomedicals) with RLT buffer containing 1% b-mercarp-toethanol and homogenized with the Fast-Prep-24 Instru-ment (MP Biomedicals). RNA was isolated using theRNeasyMidi Kit Column (Qiagen) and converted to cDNAusing the High-Capacity cDNA Archive Kit (Applied Bio-systems). Quantitative RT-PCR was done using primers formouseGLI1 (Applied Biosystems) on the 7900HTTaqManreal-time PCR instrument (Applied Biosystems). Data werenormalized to housekeeping gene HPRT.
Orthotopic model of pancreatic cancerAll pancreatic cancer animal experiments were reviewed
and approved by the M.D. Anderson Institutional AnimalCare andUseCommittee. An orthotopic nudemousemodelof pancreatic cancer using BxPC3 pancreatic tumor cellslabeled with firefly luciferase (BxPC3-FL) has previouslybeen described by this lab (22). All mice were divided intogroups with varying tumor:stroma (T/S) ratios with intra-pancreatic injections of: (a) 1 � 106 BxPC3-FL cells alone(T/S 1:0), (b) 1� 106 BxPC3-FL cells and 1� 106 HPSCs(T/S 1:1), or (c) 1 � 106 BxPC3-FL cells and 3 � 106
HPSCs (T/S 1:3) suspended in 50 mL of HBSS (Sigma-Aldrich). Groups were further subdivided into those treatedwith AZD8542 and those receiving vehicle only. Treatmentwith 80mg/kg AZD8542 suspended in 200 mL of vehicle—0.5% (hydroxypropyl) methyl cellulose (Sigma-Aldrich)with 0.1% Tween 80 (Fisher Scientific)—or with vehiclealone was administered daily by oral gavage starting on day 4after tumor implantation. Bioluminescence imaging wasdone twice weekly to assess the luciferase signal from BxPC3cells using the IVIS imaging system (Caliper Life Sciences;ref. 22). Mice were euthanized, and tumors were harvested,
Hedgehog Inhibition Targets Pancreatic Cancer Stroma
www.aacrjournals.org Mol Cancer Res; 10(9) September 2012 1149
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

measured, and either snap-frozen or fixed in formalin forfurther processing. The same experiment was done usingMPanc96-FLcancer cells.
ImmunohistochemistryBriefly, for Ki67 staining, formalin-fixed paraffin embed-
ded slides were deparaffinized in xylene, rehydrated, andblocked with 3% H2O2. Antigen retrieval was done bysteaming in antigen unmasking solution (Vector Lab). Slideswere blocked with 4% fish gelatin and incubated overnightwith rabbit anti-Ki67 antibody (1:300; Fisher) at 4�C andwashed with PBS. After incubation with biotinylated goatanti-rabbit IgG (Vector Lab) for 1 hour at room tempera-ture, slides were washed and incubated for 30 minutes withVECTASTAIN� ABC Reagent (Vector Lab). A positivereaction was detected by exposure to stable 3,30-diamino-benzidine (Trevigen) for 10 to 20 minutes. Slides werecounterstained with Gill's hematoxylin. The Aperio Scan-Scope CS Slide Scanner (Aperio Technologies) was used todigitally scan the slides with a 20� objective and images wereanalyzed using Aperio Imagescope software for nuclearstaining with Ki67.For CD31 staining, frozen slides were fixed in cold
acetone and endogenous peroxidase was blocked with 3%H2O2. Slides were blocked with 5% normal horse serum þ1%normal goat serum in PBS, incubatedwith rat antimouseCD31 antibody (1:50; BD PharMingen) at room temper-ature for 2 hours and washed with PBS. After incubationwith peroxidase-AffiniPure goat anti-rat IgG (1:200; Jack-son) for 1 hour at room temperature, a positive reaction wasdetected by exposure to stable 3,30-diaminobenzidine (Tre-vigen) for 5 minutes. Nuclei were counterstained with Gill'shematoxylin.
Quantitative RT-PCRRNA was isolated from homogenized snap-frozen tumor
tissue and cultured cell lines using TRIzol (Invitrogen)according to the manufacturer's instructions. cDNA wasmade from RNA using the QuantiTect Reverse Transcrip-tion System kit (Qiagen) according to the manufacturer'sinstructions. qPCR for SMO, SHH, PTCH1, GLI1, andHPRT (Applied Biosystems) was done using TaqMan geneexpression assays (Applied Biosystems) according to themanufacturer's specifications. Quantification was accom-plished using the standard curve method. qPCR was alsodone for GLI1 in HPSCs labeled with green fluorescentprotein (HPSC-GFP) with or without coculture withBxPC3 cells for 96 hours.
Western blottingCell lysates were prepared from HPSC and pancreatic
cancer cells and protein concentrations were measured byBioRad reagent. Protein (40 mg) was loaded onto 10% SDS-PAGE gels and Western blotting was done using a rabbitprimary antibody against SHH or IHH (Cell Signaling) at a1:1000 dilution and goat anti-rabbit secondary antibody(LiCor) at a 1:10,000 dilution. Blots were reprobed for
b-Actin (1:1000 dilution; Abcam), which served as loadingcontrol.
Statistical analysisExperiments were done in triplicate and representative
data are shown. Statistical analysis was done usingGraphPadPrism software (GraphPad). Comparisons were made usingthe 2-tailed Student t test, and significant difference wasdefined as P < 0.05. Data are shown as mean � SEM.
ResultsIdentification of potent hedgehog pathway inhibitorsScreening of the AstraZeneca compound library using a
GLI1 luciferase reporter assay identified several compoundswith an IC50 of <10 nmol/L (Supplementary Fig. S1A) andno activity against the control Renilla vector. To showspecificity of the compounds for theHh pathway, we utilizedan osteoblast differentiation assay stimulated by 2 differentligands known to act at different points in the differentiationprocess. After stimulation with conditioned media contain-ing either SHH or Wnt3A, mouse C3H10T1/2 cells wereanalyzed for secreted alkaline phosphatase as an indicator ofosteoblast differentiation. Several compounds were identi-fied to have an IC50 of <20 nmol/L with SHH stimulation(Supplementary Fig. S1), but none were shown to inhibitosteoblast differentiation when stimulated withWnt3a (datanot shown), indicating the specificity of the compounds forthe Hh pathway. None of the compounds identified wereable to inhibit proliferation in a panel of 60 tumor cell lines,consistent with previous reports (19, 26). The most prom-ising clinical candidate was AZD8542 (Fig. 1A).
AZD8542 binds SMO and inhibits Hh pathwaysignalingTo determine whether the inhibitors acted specifically on
SMO,we used a cyclopamine displacement assay. HeLa cellsengineered to express either human or mouse SMO werepretreated with SMO inhibitor compounds, followed bycyclopamine labeled with BODIPY. To validate the assay,unlabeled KAAD-cyclopamine was used as a competitor forBODIPY-labeled cyclopamine; it showed a similar affinityfor bothmouse and human SMO(2 nMvs. 0.8 nM, data notshown). Fluorescent images of cells treated with AZD8542are shown in Fig. 1B. In this assay, AZD8542 shows aKd of 5nmol/L for mouse SMO and a Kd of 20 nmol/L for humanSMO, indicating that the compound has a similar range ofaffinity for both mouse and human SMO (Fig. 1C).Furthermore, we tested the ability of AZD8542 to not
only bind SMO but also block downstream GLI1 signalingin both mouse and human cells. C3H10T1/2 (mouse) andHEPM (human) cells expressing GLI1-responsive luciferasereporter genes were stimulated with SHH conditionedmedia and treated with AZD8542. AZD8542 was able toeffectively inhibit GLI1 activity with an IC50 of 2.9 nmol/Lin the mouse cell line and an IC50 of 0.02 nmol/L in thehuman cell line, indicating increased potency against thehuman reporter line (Supplementary Fig. S1B). These data
Hwang et al.
Mol Cancer Res; 10(9) September 2012 Molecular Cancer Research1150
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

establish that AZD8542 is an ideal candidate to evaluateeffects on theHh pathway inmodels containing both humanand mouse components.
Expression of the Hh pathway in pancreatic cancer andstromal cellsA panel of cells including pancreatic cancer, normal
pancreatic duct, and HPSCs from PDAC were analyzed forexpression of genes involved in the Hh pathway (Fig. 2).SMO was expressed at high levels in HPSCs and at lowerlevels in all other cancer cells tested. The expression level in afew cancer cells (SU86.86, MiaPaca2, and Capan2) wasmoderate (Fig. 2A). In contrast, the SHH ligand was absentin HPSCs but was expressed in several cancer cell lines(Panc1,MPanc96, Capan2; Fig. 2B and Supplementary Fig.S2A). Similarly, IHH was expressed in cancer cells BxPC3and MPanc96 but not HPSCs (Supplementary Fig. S2A).The downstream targets PTCH1 and GLI1 were expressedin both cancer and stellate cells to varying degrees (Fig. 2Cand D). To confirm the generalizability of these results toother HPSC cell lines, we analyzed the expression of SMO,SHH, PTCH1, and GLI1 in primary HPSCs (nonimmor-talized) obtained from 3 unique patients and found thatexpression levels of these genes were similar to those of theHPSC cell line used in these studies (data not shown).
SHH-mediated stimulation of HPSCs is inhibited byAZD8542Others have shown that activation of the Hh pathway
by rSHH does not affect endogenous GLI1 messengerlevels in tumor epithelial cells (Bxpc3 and CFPAC-1),
regardless of SMO expression in these cells (19). However,it has been suggested that Hh signaling acts in a paracrinefashion because SMO inhibition in the mouse stroma ofxenograft tumor models is required for growth inhibition(19).We determined the effects of Hh pathway activation inHPSCs by evaluating cell proliferation,migration, andGLI1expression. Treatment of HPSCs with rSHH (1.5 and2.0 mg/mL) resulted in increased cell proliferation with apeak effect of 173.1% of control (P < 0.005; Fig. 3A).Accordingly, downstream GLI1 expression also increasedwith rSHH stimulation (5.7-fold vs. control, P <0.05; Fig. 3B). Inhibition of SMO by AZD8542 effectivelyabrogated the rSHH-mediated induction of GLI1 at 100 to1000 nmol/L (0.25- to 0.5-fold vs. control, P < 0.05).Similar results were obtained in a human prostate stromalcell line (Supplementary Fig. S1B). Although rSHH stim-ulated HPSC activity, it had no effect on pancreatic cancercell (Bxpc3 and Panc1) proliferation or migration (data notshown). HPSCs also responded to IHH treatment withincreased cell proliferation and increased levels of GLI1mRNA (Supplementary Fig. S2).
AZD8542 inhibits the Hh pathway in an in vivo coloncancer modelAlthough AZD8542 showed potent inhibition of Hh in
vitro, we wanted to test the ability of the compound to affectthe Hh pathway in vivo. Initially, we used a Colo205xenograft model, which expresses SHH ligand in the tumorcells but no IHH ligand (Supplementary Fig. S4A). Usingspecies-specific primers, we observed strong inhibition ofGLI1 only in the mouse stromal compartment but not the
Figure 1. Identification of a small-molecule SMO inhibitor. A, structureof lead compound AZD8542. B,treatment of HeLa cells expressingSMO with AZD8542 (0.1 and 1000nmol/L) was able to compete awayBODIPY-labeled cyclopamine in adose-dependent fashion. C, in theBODIPY-labeled cyclopaminecompetition assay, affinity ofAZD8542 for mouse and humanSMO showed similar binding Kd
values for both species (mouse,5 nmol/L; human, 20 nmol/L).
0 nmol/L 0.1 nmol/L 1,000 nmol/L
A
B
N
O
ONN
N
AZD8542
CHumanMouse
EC50: 0.020 µmol/L
AZ8542 (log µmol/L)
−4 −2 0 2 −4 −2 0 2
AZ8542 (log µmol/L)
EC50: 0.005 µmol/L
100
80
60
40
20
0
−20
% In
hib
itio
n
100
80
60
40
20
0
−20
% In
hib
itio
n
Hedgehog Inhibition Targets Pancreatic Cancer Stroma
www.aacrjournals.org Mol Cancer Res; 10(9) September 2012 1151
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

human epithelial compartment (Supplementary Fig. S4B).GLI1 inhibition was time dependent, with maximum inhi-bition at 8 hours after dosing and recovery by 16 hours.Plasma levels of AZD8542 were significant (60 mg/mL) at 1hour after dosing, with depletion by 8 hours (SupplementaryFig. S4C). Interestingly, this time frame corresponds tothe time required for maximumGLI1 inhibition, indicatinga 6-hour time delay between SMO inhibition and the effecton GLI1 transcription. These data show that AZD8542 isorally available and able to inhibit the Hh pathway in thestromal component of the Colo205 xenograft model.Although AZD8542 was able to inhibit Hh signaling in
the Colo205model, we observed no effect on tumor growth.Thus, we used a coimplant model with a different coloncancer cell line, HT29, known to express SHH combinedwith mouse embryonic fibroblasts (MEFs)—cells known tobe highly responsive to the Hh pathway (19). Animals
treated with 80 mg/kg AZD8542 showed significant tumorgrowth inhibition of 79% compared to vehicle controls (P <0.001; Supplementary Fig. S5A). The effect on tumorgrowth was dose dependent, with 52% and 46% tumorinhibition at 40 and 20 mg/kg, respectively (P < 0.001 and0.005; Supplementary Fig. S5A). The compound was welltolerated, with no significant changes in body weight (notshown). Accordingly, AZD8542 treatment resulted in adose-dependent decrease in mouse GLI1 and PTCH1expression in tumors, with nearly complete abrogation ofexpression at the highest dose (Supplementary Figs. 4B and5B). Plasma levels of AZD8542 increased with eachhigher dose (Supplementary Fig. 5C), peaking at 30 mg/mL� 12.6 with the most effective dose for tumor inhibition(80mg/kg). To further investigate the effect of AZD8542 onthe stromal compartment, we analyzed tumors for alphasmooth muscle actin (aSMA) expression. Tumors treated
BA1.0
0.8
0.6
0.4
0.2
0.0
HPSC
L3.6
pl
Panc1
BxPC3
MPan
c96
MiaPac
a2
Cap
an2
HPD
E
SU86
.86
HPSC
L3.6
pl
Panc1
BxPC3
MPan
c96
MiaPac
a2
Cap
an2
HPD
E
SU86
.86
HPSC
L3.6
pl
Panc1
BxPC3
MPan
c96
MiaPac
a2
Cap
an2
HPD
E
SU86
.86
HPSC
L3.6
pl
Panc1
BxPC3
MPan
c96
MiaPac
a2
Cap
an2
HPD
E
SU86
.86
2.5
2.0
1.5
1.0
0.5
0.0
No
rma
lize
d S
MO
exp
ressio
nN
orm
aliz
ed
PT
CH
1 e
xp
ressio
n
No
rma
lize
d G
LI1
exp
ressio
n
2.0
1.5
1.0
0.5
0.0
5
4
3
2
1
0
No
rma
lize
d S
HH
exp
ressio
n
C D
Figure 2. Hedgehog pathway geneexpression. RNA from variouspancreas cell lines was analyzedfor expression of SMO (A), SHH (B),PTCH1 (C), and GLI1 (D) by qPCR.Expression was normalized to thatof the HPRT gene.
01.
01.
52.
0
10%
FBS
0
50
100
150
200 **
*
% o
f co
ntr
ol
rSHH, 2 µg/mL
rSHH, µg/mL AZD8542, nmol/L
– ++++
1,0001001000
0
2
4
6
8
** *
No
rmalize
d G
LI1
ex
pre
ssio
n
A BFigure 3. Stimulation of HPSCs byrSHH induces proliferation andGLI1 expression. A, rSHH (mg/mL)was added to HPSCs grown in 1%DMEM. A, proliferation wasmeasured by MTS assay at 72hours. �, P � 0.05, ��, P � 0.005vs. 0 mg/mL rSHH control. B,HPSCs were treated with rSHH(2 mg/mL) with or withoutAZD8542. GLI1 expression wasmeasured by qPCR andnormalized toHPRT. �,P�0.05 vs.2 mg/mL rSHH, 0 nmol/L AZD8542sample.
Hwang et al.
Mol Cancer Res; 10(9) September 2012 Molecular Cancer Research1152
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022
with AZD8542 expressed markedly less aSMA than did thecontrol group (Supplementary Fig. 5D), suggesting thatAZD8542 acts primarily on the tumor-associated stromarather than the epithelial compartment in this model.
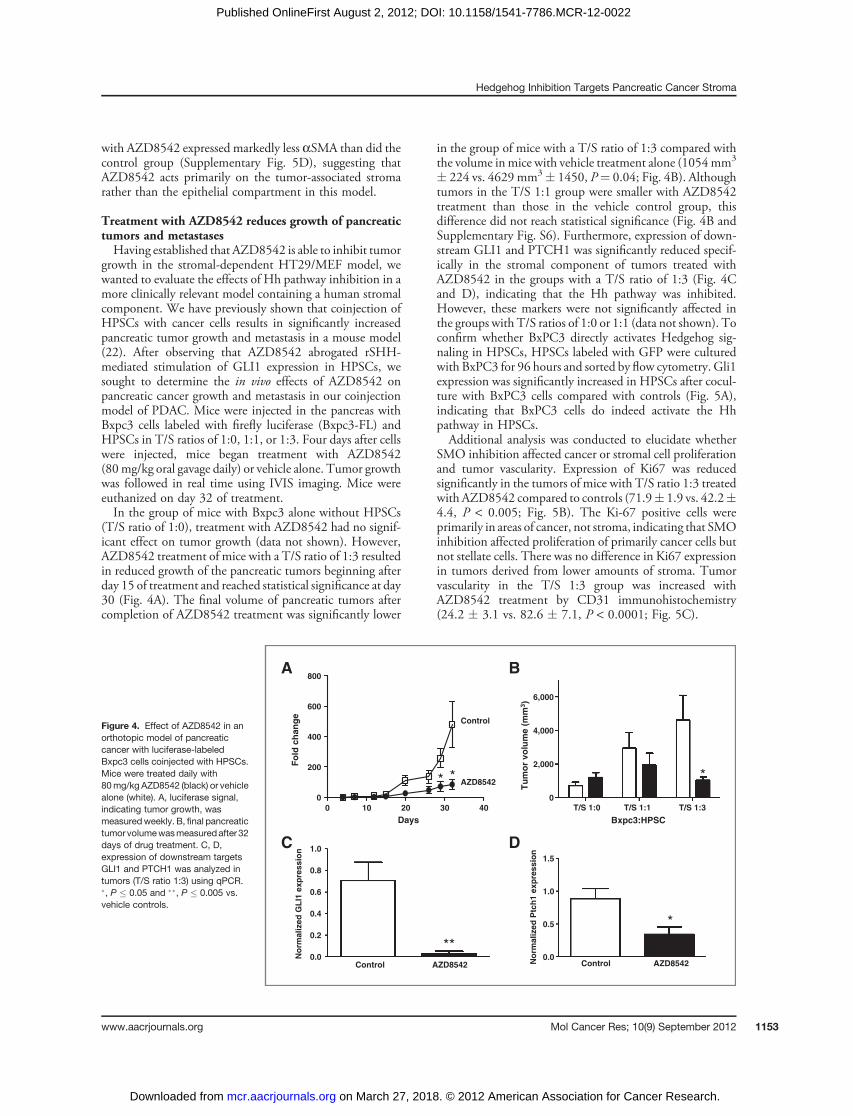
Treatment with AZD8542 reduces growth of pancreatictumors and metastasesHaving established that AZD8542 is able to inhibit tumor
growth in the stromal-dependent HT29/MEF model, wewanted to evaluate the effects of Hh pathway inhibition in amore clinically relevant model containing a human stromalcomponent. We have previously shown that coinjection ofHPSCs with cancer cells results in significantly increasedpancreatic tumor growth and metastasis in a mouse model(22). After observing that AZD8542 abrogated rSHH-mediated stimulation of GLI1 expression in HPSCs, wesought to determine the in vivo effects of AZD8542 onpancreatic cancer growth and metastasis in our coinjectionmodel of PDAC. Mice were injected in the pancreas withBxpc3 cells labeled with firefly luciferase (Bxpc3-FL) andHPSCs in T/S ratios of 1:0, 1:1, or 1:3. Four days after cellswere injected, mice began treatment with AZD8542(80mg/kg oral gavage daily) or vehicle alone. Tumor growthwas followed in real time using IVIS imaging. Mice wereeuthanized on day 32 of treatment.In the group of mice with Bxpc3 alone without HPSCs
(T/S ratio of 1:0), treatment with AZD8542 had no signif-icant effect on tumor growth (data not shown). However,AZD8542 treatment of mice with a T/S ratio of 1:3 resultedin reduced growth of the pancreatic tumors beginning afterday 15 of treatment and reached statistical significance at day30 (Fig. 4A). The final volume of pancreatic tumors aftercompletion of AZD8542 treatment was significantly lower
in the group of mice with a T/S ratio of 1:3 compared withthe volume inmice with vehicle treatment alone (1054mm3
� 224 vs. 4629 mm3� 1450, P¼ 0.04; Fig. 4B). Althoughtumors in the T/S 1:1 group were smaller with AZD8542treatment than those in the vehicle control group, thisdifference did not reach statistical significance (Fig. 4B andSupplementary Fig. S6). Furthermore, expression of down-stream GLI1 and PTCH1 was significantly reduced specif-ically in the stromal component of tumors treated withAZD8542 in the groups with a T/S ratio of 1:3 (Fig. 4Cand D), indicating that the Hh pathway was inhibited.However, these markers were not significantly affected inthe groups with T/S ratios of 1:0 or 1:1 (data not shown). Toconfirm whether BxPC3 directly activates Hedgehog sig-naling in HPSCs, HPSCs labeled with GFP were culturedwith BxPC3 for 96 hours and sorted by flow cytometry. Gli1expression was significantly increased in HPSCs after cocul-ture with BxPC3 cells compared with controls (Fig. 5A),indicating that BxPC3 cells do indeed activate the Hhpathway in HPSCs.Additional analysis was conducted to elucidate whether
SMO inhibition affected cancer or stromal cell proliferationand tumor vascularity. Expression of Ki67 was reducedsignificantly in the tumors of mice with T/S ratio 1:3 treatedwith AZD8542 compared to controls (71.9� 1.9 vs. 42.2�4.4, P < 0.005; Fig. 5B). The Ki-67 positive cells wereprimarily in areas of cancer, not stroma, indicating that SMOinhibition affected proliferation of primarily cancer cells butnot stellate cells. There was no difference in Ki67 expressionin tumors derived from lower amounts of stroma. Tumorvascularity in the T/S 1:3 group was increased withAZD8542 treatment by CD31 immunohistochemistry(24.2 � 3.1 vs. 82.6 � 7.1, P < 0.0001; Fig. 5C).
Figure 4. Effect of AZD8542 in anorthotopic model of pancreaticcancer with luciferase-labeledBxpc3 cells coinjected with HPSCs.Mice were treated daily with80mg/kg AZD8542 (black) or vehiclealone (white). A, luciferase signal,indicating tumor growth, wasmeasuredweekly. B, final pancreatictumor volumewasmeasuredafter 32days of drug treatment. C, D,expression of downstream targetsGLI1 and PTCH1 was analyzed intumors (T/S ratio 1:3) using qPCR.�, P � 0.05 and ��, P � 0.005 vs.vehicle controls.
0 10 20 30 40
0
200
400
600
800
1.0
0.8
0.6
0.4
0.2
0.0
6,000
4,000
2,000
0
**
Days
No
rmalized
GL
I1 e
xp
ressio
n
1.5
1.0
0.5
0.0
No
rmalized
Ptc
h1 e
xp
ressio
n
T/S 1:0 T/S 1:1
Bxpc3:HPSC
T/S 1:3
Fo
ld c
han
ge
Tu
mo
r vo
lum
e (
mm
3)
A B
C D
AZD8542
Control
Control Control
**
*
*
AZD8542 AZD8542
Hedgehog Inhibition Targets Pancreatic Cancer Stroma
www.aacrjournals.org Mol Cancer Res; 10(9) September 2012 1153
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

Mice in the T/S 1:3 group also developed fewer livermetastases with AZD8542 treatment comparedwith controlmice (Fig. 6A) with lower luciferase signal (1.86 � 105 vs.95.9� 105 photons, P¼ 0.06), although this difference didnot reach statistical significance. Metastatic lesions wereconfirmed histologically (Fig. 6B). Careful examination byIVIS imaging and gross examination did not reveal a sig-nificant volume of other metastatic lesions (e.g., lung, retro-peritoneum) other than the liver. Similar to our observationswith primary pancreatic tumors, AZD8542 treatment hadno effect on liver metastases in the groups with T/S ratios of1:0 or 1:1 (data not shown). Serial IVIS imaging suggeststhat the effect of AZD8542 was to inhibit tumor seeding atsites of distant metastasis, rather than to inhibit the expan-sion of metastatic lesions (Supplementary Fig. S7). Unlikemice in the control group, mice in the drug treatment groupdid not develop a signal outside the pancreas.
DiscussionIn this study, our results suggest that Hh signaling occurs
in a paracrine fashion that is dependent on activation ofSMO on neighboring pancreatic stellate cells in the tumor
microenvironment of pancreatic cancer. Furthermore, wedescribe the development of a novel Hh antagonist thatpotently binds SMOand affects the tumor-associated stromain mouse models of cancer, resulting in inhibition of tumorgrowth and metastasis.Expression of Hh pathway components revealed that
SMO and GLI1 were primarily observed in stromal-derivedHPSCs, whereas the ligands SHH and IHH were limited topancreatic tumor cells, suggesting a paracrine signalingmechanism. In support of this, treatment of HPSCs withSHH and IHH activated the Hh pathway, as measured byupregulation of GLI1 mRNA, and stimulated the prolifer-ation of HPSCs. This effect was blocked by the SMOinhibitor AZD8542. No effect of SHH stimulation or SMOinhibition was observed on pancreatic cancer cell activity.Although previous reports have suggested a possible auto-
crine role for Hh signaling, the concentration of Hh antag-onist required to downregulate the Hh pathway in cancercells was significantly higher (1.9–6mmol/L; refs. 15 and 19)than the effective dose in our studies (10–100 nmol/L). Ourdata correlate with observations by Yauch and colleagues(19), who found that the IC50 of Hh antagonist to inhibit
**
A
B
C
AZD8542Control
AZD8542
T/S 1:0
No coculture Bxpc3
coculture
T/S 1:1 T/S 1:3
Control
*
80
60
40
20
0
1.5
1.0
0.5
0.0Norm
aliz
ed G
LI1
expre
ssio
n%
po
sitiv
e c
ells
**
AZD8542Control
100
80
60
40
20
0
CD
31
+ v
esse
ls/f
ield
Figure 5. Hedgehog pathwaysignaling in PDAC in vitro and invivo. A, quantitative PCR for Gli1expression. HPSC-GFP cells werecultured with or without Bxpc3 for96 hours and sorted by flowcytometry; ��, P < 0.0001.Immunohistochemistry for Ki67 (B)and CD31 (C) was done on mousepancreatic tumors with tumor/stroma ratio 1:3 treated withAZD8542 or control. Totalmagnification 100�. Data areshown as mean � SEM;�, P < 0.005 and ��, P < 0.0001.
B
AZD8542Control
Liver0
1
2
350
75
100
125
150
Ph
oto
ns,
10
5
A
*
AZD8542Control
Figure 6. Effect of AZD8542 on livermetastases in an orthotopicmodelof pancreatic cancer withluciferase-labeled Bxpc3 cellscoinjected with HPSCs. Mice weretreated daily with 80 mg/kgAZD8542 or vehicle alone. A, livermetastases were measured byluciferase signal. B, histology wasconfirmed by H&E staining in micefrom the T/S 1:3 group (totalmagnification 200�); �. P ¼ 0.06.
Hwang et al.
Mol Cancer Res; 10(9) September 2012 Molecular Cancer Research1154
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

growth of a mesenchymal cell line was 400 times lower thanthat required for themost sensitive cancer cell line (5 nmol/Lvs. 1.9 mmol/L). High concentrations of Hh antagonistresulted in repression of unrelated transcriptional reporters,suggesting that previously observed effects in cancer cellsmay be because of off-target effect (19).Using in vivo models of colon cancer and pancreatic
cancer, we show that AZD8542 inhibits the Hh pathwayspecifically in the stromal compartments of each model. Inthe Colo205 colon xenograft model, species-specific RT-PCR showed strong inhibition ofGLI1 in themouse stromalcompartment but no downregulation in the human epithe-lial compartment. No effect on tumor growth inhibition wasobserved. However, in both the HT29/MEF and BxPC3/HPSC orthotopic pancreatic cancer models, AZD8542significantly reduced tumor growth. Interestingly, growthinhibition was observed only when tumors contained bothstromal cells and cancer cells but not cancer cells alone.Moreover, downstream Hh signaling in both tumor modelswas inhibited by AZD8542. Although the BxPC3 cells usedin the orthotopic pancreatic cancer model shown in Fig. 4 donot express appreciable levels of SHH, they do express IHHthat has similar effects on HPSCs as does SHH (Supple-mentary Fig. S2). Thus, IHH is most likely the ligandparticipating in Hh signaling in the BxPC3 orthotopicmodel. In addition, we have conducted the same experimentusing MPanc96, which expresses high levels of SHH, withsimilar effects of AZD8542 on tumor growth inhibition(Supplementary Fig. S3). Taken together, these resultsindicate that SMO inhibition blocks Hh signaling in thestromal compartment, resulting in an overall reduction intumor growth and metastasis.Our data support several recent reports showing that Hh
ligands are expressed by epithelial cells, which in turnactivate Hh signaling in the adjacent stroma with the overalleffect of promoting tumor growth (18–20). Activation of theHh pathway by expression of mutant SMO (SMOM2)specifically in pancreatic ductal epithelial cells driven byPDX promoter did not initiate tumorigenesis. In thesegenetically engineered mice, Hh signaling was active inmesenchymal cells but not neoplastic pancreatic epithelialcells (20). Furthermore, species-specific qRT-PCR analysisof primary human tumor xenografts showed correlationbetween SHH and IHH expression in the tumor andactivation ofGLI1 in the stromal compartment (19). Finally,analysis of microdissected samples of human PDAC showedthat the levels of Hh ligand (SHH and IHH) were signif-icantly higher in the tumor epithelial cells than in stromalcells, whereas GLI1 was much higher in tumor stroma (20).To our knowledge, this study is the first to investigate Hh
signaling using primary HPSCs derived from PDAC. Theprecise mechanism by which Hh signaling in HPSCs pro-motes pancreatic tumor growth and metastasis is unknown.We have shown that conditioned media from activatedHPSCs stimulates pancreatic tumorigenesis in vitro and invivo. It is likely that Hh ligand secreted from neighboringcancer cells stimulatesHPSCs to produce soluble factors thatthen feedback on cancer cells to promote their activity.
Expression profiling of tumor xenografts treated with Hhantagonist showed repression of the Wnt pathway andinsulin-like growth factor receptor in themouse stroma (19).Inhibition of tumor growth in the PDAC/HPSC coin-
jection model with AZD8542 was associated with decreasedcancer cell proliferation and increased vascularity. We didnot see an obvious difference in stellate cell proliferation orstroma formation in these tumors and yet our data indicatethat SMO inhibition with AZD8542 promotes the activityof stellate cells, not pancreatic cancer cells. It is plausible thatSMO inhibition may have act directly onHPSCs to alter thenature of their secreted factors, which in turn indirectly actson cancer cells in a paracrine manner to result in overalltumor inhibition. Although our observation of increasedangiogenesis with SMO inhibition correlates with studies byOlive and colleagues using a genetically engineered mousemodel of PDAC (21), a recent report byChen and colleaguesdemonstrated a proangiogenic role of Hh signaling (27).Treatment of a colorectal cancer xenograft model withanother SMO inhibitor resulted in decreased vascularity(27).These observations seem to be contradictory and thus,additional studies to elucidate the effects of Hh signaling onpancreatic stellate cells and angiogenesis are currently inprogress.Another potential mechanism by which Hh signaling in
the stroma might regulate both tumor growth and chemo-sensitivity is through the promotion of cancer stem cell-likeproperties. Several investigators have showed the role of Hhsignaling in regulating cancer stem cell renewal in breastcancer, gliomas, and leukemia (28–31). Cells that undergoepithelial–mesenchymal transition (EMT) have many prop-erties of cancer stem cells, such as self-renewal and resistanceto toxic injuries, including chemoradiation. Although pan-creatic stellate cells have not clearly been shown to participatein cancer stem cell renewal, they do promote EMT changesin pancreatic cancer cells (32). Thus,Hh signaling inHPSCsmight also affect stem cell properties in PDAC with anoverall effect of increased tumorigenesis as well as resistanceto chemotherapy.In PDAC and other solid malignancies, the tumor-asso-
ciated stroma has been implicated as a physical barrier to thedelivery of chemotherapy (33). The effects of Hh blockadeon chemoresistance were not studied in this article but havebeen described previously by others. Using a geneticallyengineered mouse model of PDAC that develops extensivestroma, Olive and colleagues (21) showed improved efficacyof gemcitabine when combined with an Hh antagonist.Tumors in this model are poorly perfused, which hampersthe delivery and efficacy of gemcitabine treatment.However,treatment with an Hh antagonist depleted tumor-associatedstroma and improved tumor vascularity, increased intratu-moral concentration of gemcitabine, and stabilized disease(21). In support of those findings, we have shown in ourHT29/MEF model a reduction of aSMA staining upontreatment with AZD8542. Not only does stroma act as aphysical obstacle to delivery of chemotherapy, but it alsoinhibits its efficacy on amolecular level. We have shown thatconditioned media from HPSCs protects cancer cells from
Hedgehog Inhibition Targets Pancreatic Cancer Stroma
www.aacrjournals.org Mol Cancer Res; 10(9) September 2012 1155
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

apoptosis induced by chemotherapy or radiation (22),although the precise mechanisms are unclear. Hh signalinghas been shown to induce chemoresistance as well as resis-tance to ionizing radiation (34–36). Therefore, it is possiblethat activation of Hh signaling in HPSCs may also inhibitneighboring cancer cell responsiveness to chemotherapeuticagents or radiation.A range of clinical trials targeting the Hh pathway has
emerged recently. A Phase II study of the SMO inhibitorvismodegib (Genentech) showed efficacy in objectiveresponse rate in patients with basal cell carcinoma, resultingin FDA approval in early 2012. However, a recent Phase IItrial by Infinity Pharmaceuticals to evaluate the SMOinhibitor saridegib (IPI-926-03) in combination with gem-citabine in patients with metastatic pancreatic cancer washalted after preliminary analysis showed a difference insurvival favoring the placebo plus gemcitabine arm. Thereasons for failure are unknown since the full data analysisfrom this trial has not been published. As showed by our dataand others, the efficacy of Hh antagonists in PDAC seems tobe dependent on the stromal compartment; whether stromais a significant component and driving force in metastaticpancreatic cancer is unknown. Perhaps the key to successwith Hh inhibitors may be the ability to better select patientpopulations that have tumors dependent on the stroma andthe Hh pathway for growth and maintenance.In summary, our results are direct evidence of Hh signal-
ing in pancreatic stellate cells in the tumor-associated stromaof pancreatic cancer. Activation of theHh pathway in PDACoccurs in a paracrine fashion, and disruption of the pathwaywith a novel SMO antagonist, AZD8542, reduced pancre-atic tumor growth and metastasis specifically by inhibitingthe stromal compartment. Thus, preclinical cancer models
that lack stroma are not adequate tools for testing Hh-targeted therapies.
Disclosure of Potential Conflicts of InterestM.M.Hattersley, M. Scarpitti, B. Yang, E. Devereaux, J. L. Brown, and R. Godin
are employees of AstraZeneca. No potential conflicts of interest were disclosed by theother authors.
Authors' ContributionsConception and design: R. F. Hwang, T. T. Moore, V. Ramachandran, B. Ji, C. D.Logsdon, J. L. Brown, R. GodinDevelopment of methodology: R. F. Hwang, T. T. Moore, M. M. Hattersley, B.Yang, E. Devereaux, R. GodinAcquisition of data (provided animals, acquired and managed patients, providedfacilities, etc.): R. F. Hwang, M. M. Hattersley, M. Scarpitti, B. Yang, E. Devereaux,V. Ramachandran, B. Ji, R. GodinAnalysis and interpretation of data (e.g., statistical analysis, biostatistics, compu-tational analysis): R. F. Hwang, T. T. Moore, M. M. Hattersley, M. Scarpitti, E.Devereaux, C. D. Logsdon, J. L. Brown, R. GodinWriting, review, and/or revision of themanuscript:R. F.Hwang,M.M.Hattersley,C. D. Logsdon, J. L. Brown, R. GodinAdministrative, technical, or material support (i.e., reporting or organizing data,constructing databases): R. F. Hwang, T. T. Moore, V. Ramachandran, R. GodinStudy supervision: R. F. Hwang, M. M. Hattersley, J. L. Brown, R. GodinTeaching techniques to authors: T. Arumugam
AcknowledgmentsThe authors thank James Janetka, Mukta Bagul, Troy Patterson, Chris Pien, Alex
Hird, Les Dakin, Qibin Su, Alex Cao, Patrick Brassil, Dan Russell, and Yan Liu fortheir contributions and support to the project team.
Grant SupportThis work is supported by the National Institutes of Health through grant
1K08CA138912-01A1 to R. F. Hwang and through MD Anderson's Cancer CenterSupport Grant, 5P30CA016672.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be herebymarked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
Received January 24, 2012; revised June 7, 2012; accepted July 13, 2012;published OnlineFirst August 2, 2012.
References1. Ingham PW, McMahon AP. Hedgehog signaling in animal develop-
ment: paradigms and principles. Genes Dev 2001;15:3059–87.2. Green J, Leigh IM, Poulsom R, Quinn AG. Basal cell carcinoma
development is associated with induction of the expression of thetranscription factor Gli-1. Br J Dermatol 1998;139:911–5.
3. Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, et al. Activatingsmoothened mutations in sporadic basal-cell carcinoma. Nature1998;391:90–2.
4. Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH Jr, Scott MP. Basalcell carcinomas in mice overexpressing sonic hedgehog. Science1997;276:817–21.
5. Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG,Watkins DN, et al. Medulloblastoma growth inhibition by hedgehogpathway blockade. Science 2002;297:1559–61.
6. Rubin JB, Rowitch DH.Medulloblastoma: a problemof developmentalbiology. Cancer Cell 2002;2:7–8.
7. Yuan Z, Goetz JA, Singh S, Ogden SK, Petty WJ, Black CC, et al.Frequent requirement of hedgehog signaling in non-small cell lungcarcinoma. Oncogene 2007;26:1046–55.
8. Chi S, HuangS, Li C, ZhangX,HeN,BhutaniMS, et al. Activation of thehedgehog pathway in a subset of lung cancers. Cancer Lett 2006;244:53–60.
9. Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, BaylinSB. Hedgehog signalling within airway epithelial progenitors and insmall-cell lung cancer. Nature 2003;422:313–7.
10. Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A,et al. Hedgehog signalling in prostate regeneration, neoplasia andmetastasis. Nature 2004;431:707–12.
11. Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM,Barrett A, et al. Inhibition of prostate cancer proliferation by interfer-ence with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad SciU S A 2004;101:12561–6.
12. Fan L, Pepicelli CV, Dibble CC,CatbaganW, Zarycki JL, LaciakR, et al.Hedgehog signaling promotes prostate xenograft tumor growth.Endocrinology 2004;145:3961–70.
13. Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, GerstenblithMR, Briggs K, et al. Widespread requirement for Hedgehog ligandstimulation in growth of digestive tract tumours. Nature 2003;425:846–51.
14. Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ,Lauwers GY, et al. alHedgehog is an early and late mediator ofpancreatic cancer tumorigenesis. Nature 2003;425:851–6.
15. Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M,et al. Blockade of Hedgehog signaling inhibits pancreatic cancerinvasion and metastases: A new paradigm for combination therapyin solid cancers. Cancer Res 2007;67:2187–96.
16. Feldmann G, Habbe N, Dhara S, Bisht S, Alvarez H, Fendrich V,et al. Hedgehog inhibition prolongs survival in a geneticallyengineered mouse model of pancreatic cancer. Gut 2008;57:1420–30.
Hwang et al.
Mol Cancer Res; 10(9) September 2012 Molecular Cancer Research1156
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

17. Hidalgo M, Maitra A. The hedgehog pathway and pancreatic cancer.N Engl J Med 2009;361:2094–6.
18. Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fern�andez-Zapico ME, et al. GLI1 is regulated through Smoothened-independentmechanisms in neoplastic pancreatic ducts and mediates PDAC cellsurvival and transformation. Genes Dev 2009;23:24–36.
19. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. Aparacrine requirement for hedgehog signalling in cancer. Nature2008;455:406–10.
20. Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ,et al. Hedgehog signaling is restricted to the stromal compartmentduring pancreatic carcinogenesis. Proc Natl Acad Sci 2009;106:4254–9.
21. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D,Honess D, et al. Inhibition of Hedgehog signaling enhances deliveryof chemotherapy in a mouse model of pancreatic cancer. Science2009;324:1457–61.
22. HwangRF,Moore T, ArumugamT,Ramachandran V, AmosKD,RiveraA, et al. Cancer-associated stromal fibroblasts promote pancreatictumor progression. Cancer Res 2008;68:918–926.
23. Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selectionand characterization of metastatic variants from human pancreaticadenocarcinoma by using orthotopic implantation in nude mice. Neo-plasia 1999;1:50–62.
24. Yang B, Hird AW, Russell DJ, Fauber BP, Dakin LA, Zheng X, et al.Discovery of novel hedgehog antagonists from cell-based screening:isosteric modification of p38 bisamides as potent inhibitors of SMO.Bioorg Med Chem Lett 2012;22:4907–11.
25. Sasaki H, Hui C, Nakafuku M, Kondoh H. A binding site for Gliproteins is essential for HNF-3beta floor plate enhancer activity intransgenics and can respond to Shh in vitro. Development 1997;124:1313–22.
26. Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, et al.Shh pathway activity is down-regulated in cultured medulloblastoma
cells: implications for preclinical studies. Cancer Res 2006;66:4215–22.
27. ChenW, Tang T, Eastham-Anderson J, Dunlap D, Alicke B, Nannini M,et al. Canonical hedgehog signaling augments tumor angiogenesis byinduction of VEGF-A in stromal perivascular cells. Proc Natl Acad SciU S A 2011;108:9589–94.
28. Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, et al.Hedgehog signaling and Bmi-1 regulate self-renewal of normal andmalignant humanmammary stem cells. Cancer Res 2006;66:6063–71.
29. Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, et al.Cyclopamine-mediated hedgehog pathway inhibition depletesstem-like cancer cells in glioblastoma. Stem Cells 2007;25:2524–33.
30. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A.HEDGEHOG-GLI1 signaling regulates human glioma growth, cancerstem cell self-renewal, and tumorigenicity. Curr Biol 2007;17:165–72.
31. Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, et al.Expansion of Bcr-Abl-positive leukemic stem cells is dependent onHedgehog pathway activation. Cancer Cell 2008;14:238–49.
32. Kikuta K, Masamune A, Watanabe T, Ariga H, Itoh H, Hamada S, et al.Pancreatic stellate cells promote epithelial-mesenchymal transition inpancreatic cancer cells. Biochem Biophys Res Commun 2010;403:380–4.
33. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and thesolid tumor microenvironment. J Natl Cancer Inst 2007;99:1441–54.
34. Sims-Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic Hedgehog pro-motes multiple drug resistance by regulation of drug transport. Onco-gene 2007;26:5674–9.
35. Queiroz KC, Ruela-de-Sousa RR, Fuhler GM, AbersonHL, Ferreira CV,Peppelenbosch MP, et al. Hedgehog signaling maintains chemore-sistance in myeloid leukemic cells. Oncogene;29:6314–22.
36. Chen YJ, Lin CP, Hsu ML, Shieh HR, Chao NK, Chao KS. Sonichedgehog signaling protects human hepatocellular carcinoma cellsagainst ionizing radiation in an autocrine manner. Int J Radiat OncolBiol Phys;80:851–9.
Hedgehog Inhibition Targets Pancreatic Cancer Stroma
www.aacrjournals.org Mol Cancer Res; 10(9) September 2012 1157
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022

2012;10:1147-1157. Published OnlineFirst August 2, 2012.Mol Cancer Res Rosa F. Hwang, Todd T. Moore, Maureen Mertens Hattersley, et al. Stroma in Pancreatic CancerInhibition of the Hedgehog Pathway Targets the Tumor-Associated
Updated version
10.1158/1541-7786.MCR-12-0022doi:
Access the most recent version of this article at:
Material
Supplementary
http://mcr.aacrjournals.org/content/suppl/2012/08/02/1541-7786.MCR-12-0022.DC1
Access the most recent supplemental material at:
Cited articles
http://mcr.aacrjournals.org/content/10/9/1147.full#ref-list-1
This article cites 34 articles, 14 of which you can access for free at:
Citing articles
http://mcr.aacrjournals.org/content/10/9/1147.full#related-urls
This article has been cited by 8 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mcr.aacrjournals.org/content/10/9/1147To request permission to re-use all or part of this article, use this link
on March 27, 2018. © 2012 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst August 2, 2012; DOI: 10.1158/1541-7786.MCR-12-0022