inhibition ammoniamonooxygenase nitrosomonas …jb.asm.org/content/172/9/4775.full.pdf · carbon...
TRANSCRIPT

JOURNAL OF BACTERIOLOGY, Sept. 1990, p. 4775-4782 Vol. 172, No. 90021-9193/90/094775-08$02.00/0Copyright C) 1990, American Society for Microbiology
Inhibition of Ammonia Monooxygenase in Nitrosomonas europaeaby Carbon Disulfide
MICHAEL R. HYMAN,t CHARLOTTE Y. KIM,T AND DANIEL J. ARPt*Department of Biochemistry, University of California, Riverside, California 92521
Received 5 March 1990/Accepted 7 June 1990
Carbon disulfide has long been recognized as a potent inhibitor of nitrification, and it is the likely activecomponent in several nitrification inhibitors suitable for field use. The effects of this compound on Nitrosomonaseuropaea have been investigated, and the site of action has been determined. Low concentrations of CS2 (<400,uM) produced a time-dependent inhibition of ammonia-dependent O2 uptake but did not inhibit hydrazine-oxidizing activity. CS2 also produced distinct changes in difference spectra of whole cells. These results suggestthat ammonia monooxygenase (AMO) is the site of action of CS2. Unlike the case for thiourea and acetylene,saturating concentrations of CS2 did not fully inhibit AMO, and the inhibition resulted in a low but significantrate of ammonia-dependent 02 uptake. The effects of CS2 were not competitive with respect to ammoniaconcentration, and the inhibition by CS2 did not require the turnover ofAMO to take effect. The ability of CS2-treated cells to incorporate [14C]acetylene into the 28-kilodalton polypeptide of AMO was used to demonstratethat the effects of CS2 are compatible with a mode of action which involves a reduction of the rate of turnoverof AMO without effects on the catalytic mechanism. It is proposed that CS2 may act on AMO by reversiblyreacting with a suitable nucleophilic amino acid in close proximity to the active site copper.
The major process in microbial nitrification is the oxida-tion of ammonia to nitrite, followed by the oxidation ofnitrite to nitrate. These reactions are predominantly cata-lyzed by specialized autotrophic bacteria characterized bysuch species as the ammonia-oxidizing bacterium Ni-trosomonas europaea and the nitrite-oxidizing bacteriumNitrobacter winogradskyi, respectively. Although our un-derstanding of the biochemistry and enzymology of individ-ual reactions in the nitrification process is far from complete,there is a considerable general interest in nitrification be-cause it plays an important role in controlling the balancebetween fixed and mobile forms of nitrogen in soil and watersystems. For example, in agriculture, nitrification can lead tosubstantial losses of costly ammonia-based fertilizer (andurea-based fertilizer, which hydrolyzes to ammonia) becauseboth nitrification products, nitrite and nitrate, are easilyleached from soils by water. In addition, nitrate is the majorsubstrate for denitrifying bacteria that generate gaseousproducts (N20, NO, and N2) which are lost to the atmo-sphere. These same reactions, while detrimental to agricul-ture, are regarded as positive benefits in water treatmentregimens, in which the objective is to reduce the nitrogencontent of waste waters (22).Because it is ammonia-oxidizing bacteria that initiate
nitrification as a whole, there has been considerable researchinto the inhibition of this process. Ammonia oxidation by N.europaea is a two-stage process that is now thought toinvolve the initial oxidation of ammonia to hydroxylamineby a membrane-bound enzyme known as ammonia monoox-ygenase (15) (AMO):
NH4+ + XH2 + 02- NH20H + H20 + H+ + X (1)
* Corresponding author.t Present address: Laboratory for Nitrogen Fixation Research,
Department of Botany and Plant Pathology, Oregon State Univer-sity, Corvallis, OR 97331.
t Present address: Medical School, Stanford University, PaloAlto, CA 94301.
The resulting hydroxylamine is then oxidized to nitrite, withthe release of four electrons by a complex, multiheme-containing, soluble protein, hydroxylamine oxidoreductase(11) (HAO):
NH20H + H20-> N02- + 5H+ + 4e- (2)Because hydroxylamine is the only source of reductantavailable for AMO activity during steady-state ammoniaoxidation (11, 33), AMO activity is susceptible to inhibitioneither through direct effects on the enzyme itself or throughindirect effects on HAO or other components that areinvolved in the transfer of electrons to AMO. This unusualinterrelationship is of considerable importance in determin-ing the site of action of ammonia oxidation inhibitors.One of the oldest known inhibitors of nitrification is
carbon disulfide (CS2), which was first reported by War-rington (31). Even before Warrington's work, CS2 had beenused as a soil fumigant and its effects on nitrification hadbeen recognized. However, the inhibitory effects of CS2 onnitrification at concentrations used during soil fumigationalmost certainly arise from its general bacteriocidal qualities.Powlson and Jenkinson (23) were the first to characterize thepotency of CS2 as a specific nitrification inhibitor anddemonstrated that it effectively inhibits nitrite productionfrom ammonia at concentrations as low as 0.5 jig/ml. Thisfinding indicates that ammonia-oxidizing bacteria are thesensitive agents. Powlson and Jenkinson (23) identified CS2as the active inhibitor compound released from rubberstoppers, and they concluded that CS2 was generated as abreakdown product of sulfur compounds used in the rubber-vulcanizing process. Beyond the anecdotal nature of thisdiscovery, the significance of the potent effects of CS2 onnitrification is perhaps more widespread than is generallyappreciated, since the action of many other compounds thatinhibit nitrification can also potentially be rationalized interms of the mode of action of CS2. These inhibitorycompounds include thiosulfates (18), thiocarbonates (1, 2),thiocarbamates (12, 18), xanthates (such as potassium eth-ylxanthate) (18, 29, 30), sulfur-containing amino acids such
4775
on July 9, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

4776 HYMAN ET AL.
as methionine (20), cystine and cysteine (4, 5, 17), andseveral pesticides and fungicides (18, 28), which are allcompounds known to degrade in solution or soils to releaseCS2. However, despite widespread interest in inhibitors ofnitrification and the use of CS2 as a specific nitrificationinhibitor in recent field and soil studies (1, 2, 4) neither themode nor site of action of CS2 on ammonia-oxidizing bacte-ria has been established. In this study, we have character-ized some of the effects of CS2 on whole cells of N. europaeaand have compared and contrasted these effects with thoseproduced by acetylene (16) and thiourea (20), two otherwell-characterized inhibitors of nitrification.
MATERIALS AND METHODSMaterials. CS2 (spectrophotometric grade, 99+%) was
obtained from Aldrich Chemical Co., Inc. (Milwaukee,Wis.). Thiourea was obtained from EM Science (Gibbstown,N.J.). [14C]acetylene was generated from Ba14CO3 andtrapped in dimethyl sulfoxide DMSO, as described by M. R.Hyman and D. J. Arp (submitted for publication). Ba14CO3(specific activity, 57.5 mCi/mmol) was obtained from Amer-sham Corp. (Arlington Heights, Ill.). Electrophoresis mate-rials were supplied by Bio-Rad Laboratories (Richmond,Calif.).Growth and harvesting of N. europaea cells. N. europaea
was grown in 1-liter shake flask cultures as described previ-ously (14). Cells were harvested by centrifugation (20,000 xg for 20 min), and the sedimented cells were suspended in 50mM sodium phosphate buffer, pH 7.8, containing 2 mMMg2+. The cells were then sedimented again, suspended toapproximately 0.2 g (wet weight)/ml in buffer (as describedabove), and stored on ice until used (within 3 h of harvest-ing).02 uptake measurements. Rates of 02 uptake of cell
suspensions were measured by using a Clark-type oxygenelectrode (Yellow Springs Instrument Co., Yellow Springs,Ohio) mounted in an all-glass, water-jacketed reaction vessel(1.6-ml volume). All measurements were made at 30°C; 50mM sodium phosphate, pH 7.8, containing 2 mM Mg2+ wasused as the buffer in all experiments.
Anaerobic incubation of cells. Incubations of cells underanaerobic conditions were conducted in an all-glass, microreaction vessel. The vessel consisted of a borosilicate glasslower chamber (1.5 by 0.5 cm; inside diameter, 0.3 cm) witha female ground-glass neck joint into which a male capillarysection (4 by 0.8 cm; inside diameter, 1 mm) could beinserted. The volume of the complete vessel was approxi-mately 250 ,ul. Cells were added to fill the lower chamber,and the capillary section was then inserted. This procedureforced air bubbles out of the chamber; the displaced cellswere retained in the capillary section and minimized thediffusion of air into the lower chamber. All additions to themicro reaction vessel were made from anaerobic stocksolutions, using gas-tight microsyringes.
Spectroscopic studies with whole cells. Difference spectraobtained with whole cells were recorded with a BeckmanDU-7 spectrophotometer. A whole-cell suspension (1 ml of0.05 g [wet weight] of cells per ml) was added to a quartzcuvette (1-cm path length; total volume, 1.2 ml), which wassealed with a Teflon stopper. A base line was recorded andmemorized. Additions to the cell suspensions were made byremoving the stopper from the cuvette and adding substratesand inhibitors (10 Pdl) from stock solutions. The cuvette wasthen stoppered, inverted twice, and replaced in the spectro-photometer. Spectra were then recorded at the indicatedtimes.
Fractionation of whole cells and sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of membrane pro-teins. After exposure to mixtures of inhibitors and ["4C]acetylene, whole cells were sedimented by centrifugation ina microfuge (12,000 x g for 2 min) and then resuspended toapproximately 0.2 g (wet weight)/ml in 50 mM sodiumphosphate buffer, pH 7.8, containing 2 mM Mg2+. The cellswere then broken by using freezing and thawing cycles andfractionated into soluble and membrane fractions as de-scribed previously (16). After fractionation, 100 p,g of mem-brane protein was solubilized in microfuge tubes in samplebuffer containing 1% (wt/vol) sodium dodecyl sulfate, 10%(wt/vol) glycerol, 10% (vol/vol) P-mercaptoethanol, and 62.5mM Tris hydrochloride, pH 6.8. The samples were solubi-lized at 60°C for 3 min, and nonsolubilized material wassedimented by centrifugation. Electrophoresis was con-ducted at room temperature in 13.5% acrylamide slab gels.The gels were stained and prepared for fluorography asdescribed previously (16). Fluorographs were produced byusing Kodak XAR5 X-ray film and a 5-day exposure time.
Protein was determined by using the biuret assay on cellsand membrane fractions that had been solubilized in 3 MNaOH for 30 min at 65°C. Bovine serum albumin was usedas a standard. The solubility of 02 in air-saturated buffer at30°C was taken as 230 puM (15), and the solubility ofacetylene in water at 25°C was taken as 42 mM (16).
RESULTS
Since both the oxidation of ammonia to hydroxylamine(reaction 1) and hydroxylamine oxidation of nitrite (reaction2) ultimately consume 02, the simplest means by whichthese activities can be assayed and the effects of inhibitorsdiscriminated is by following 02 uptake rates in the presenceof appropriate substrates. In the absence of any inhibitor,the addition of 10 mM NH4Cl to a whole-cell suspensionresulted in a rapid rate of 02 uptake which, after an initialshort lag phase (<5 s), remained constant until in excess of95% of the remaining 02 had been consumed (Fig. 1, traceA). The addition of low concentrations of CS2 (25 to 400 ,uLM)during the period of steady-state ammonium oxidation re-sulted in a time-dependent inhibition of oxygen uptake(Traces B to F). Increases in the CS2 concentration from 25to 100 p.M increased the rate of inhibition. With higherconcentrations of CS2 (200 and 400 ,uM), the rate of inhibi-tion was the same, which indicated that the system wassaturated with respect to inhibitor concentration. Even withthe highest inhibitor concentration, a residual rate of 02uptake was observed when the time-dependent effects ofCS2 appeared to have taken full effect. The inhibition ofammonia-dependent 02 uptake activity by CS2 did notrequire the presence of NH4'. The kinetics of the inhibitioncaused during preincubation of cells with CS2 were the sameas those observed when CS2 was added during steady-staterates of ammonia oxidation (data not shown). Although CS2was added as a solution in DMSO (final concentration insample of 0.08 M), DMSO alone did not inhibit eitherammonia or hydrazine oxidation (data not shown).The site of action of CS2 was investigated in an experiment
in which the effects of acetylene, thiourea, and CS2 onammonia and hydrazine oxidation were compared. All threeinhibitors produced time-dependent inhibitions Of 02 uptakein the presence of 10 mM NH4' (Fig. 2). In all cases, theaddition of 600 p.M hydrazine (an alternative substrate forHAO) to each incubation 20 min after addition of theinhibitor led to a stimulation of the rate of02 uptake. In each
J. BACTERIOL.
on July 9, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

CARBON DISULFIDE INHIBITION 4777
FIG. 1. Inhibition by CS2 of ammonia-dependent 02 uptake bywhole cells of N. europaea. 02 uptake was monitored with anoxygen electrode. In each experiment, 1.6 ml buffer was used andNH4Cl was added to an initial concentration of 10 mM. The reactionwas initiated by adding whole cells to a final concentration of 0.18mg/ml. After a steady rate Of 02 uptake was established, CS2 wasadded to the electrode chamber at the point indicated by the arrowto the following concentrations: (A) no addition; (B) 25 ,um; (C) 50,um; (D) 100 ,uM; (E) 200 ,uM, and (F) 400 ,M. CS2 was added froma stock solution in DMSO, and additions were equal to or less than20 ,l.
case, the rate of 02 uptake in the presence of hydrazine wasnot affected by any of the inhibitors tested. In this experi-ment, both acetylene and thiourea rapidly inactivated am-monia-dependent °2 uptake within 5 min. For both inhibi-tors, the residual 02 uptake rate was close to zero (Fig. 2,traces B and C). In contrast, the inhibition caused by CS2(200 ,uM) took longer to take full effect and resulted in a lowbut significant and constant residual rate of 02 uptake (traceB) that was slightly above 4% of the uninhibited rateobserved with 10 mM NH4+.
The residual activity observed after CS2 treatment wasfurther investigated by using alternative techniques that didnot rely on 02 uptake measurements. First, the mediumacidification associated with ammonia oxidation (see reac-tions 1 and 2) was monitored spectrophotometrically, using aweakly buffered reaction medium containing a pH indicator.Only a very slight acidification occurred when cells wereincubated in the absence of ammonium (Fig. 3, trace A). Inthe presence of 10 mM NH4', a short lag phase wasobserved, after which the medium was acidified at a constantrate until the available 02 in the reaction cuvette wasdepleted. Acidification recommenced if the cuvette wasshaken to reintroduce air into the cell suspension (trace B).Traces C to E show the effects of thiourea, acetylene, andCS2, respectively, on the time course of medium acidifica-tion during steady-state rates of ammonia oxidation. Again,both acetylene and thiourea fully inhibited ammonia-depen-dent acidification, whereas CS2-treated cells exhibited sig-nificant residual activity after CS2 inhibition had apparentlytaken full effect.The residual level of AMO activity observed after the
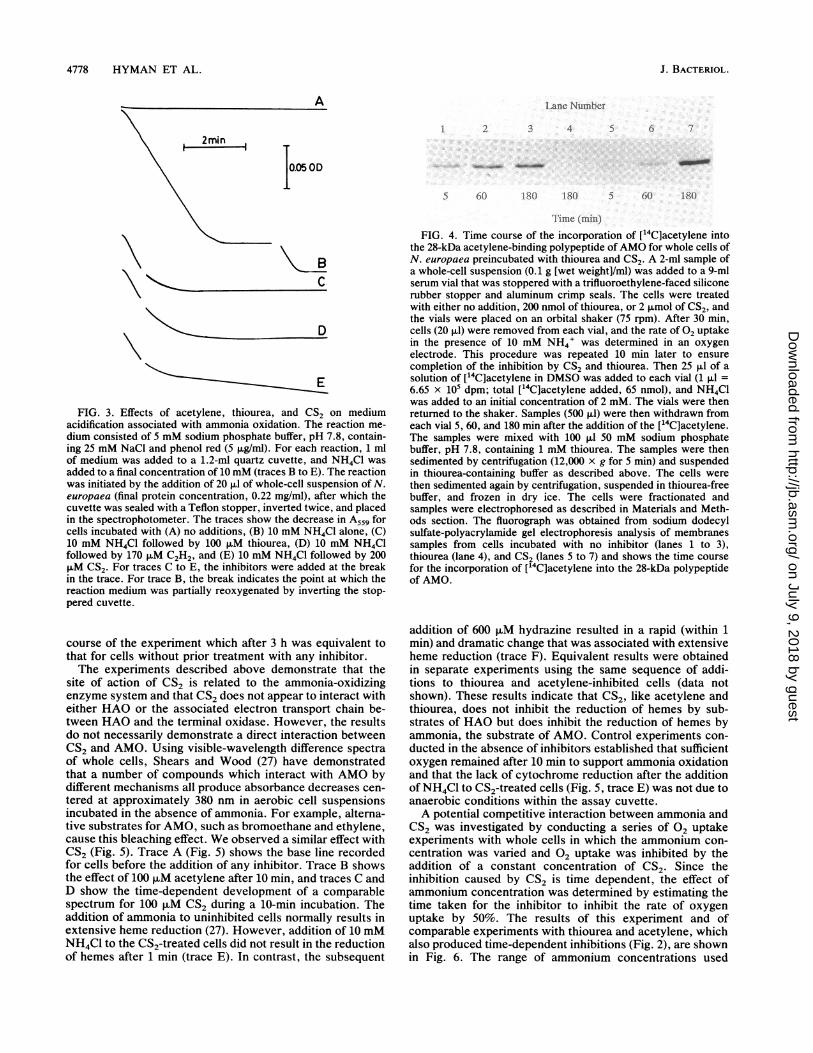
addition of CS2 was further confirmed by monitoring thesusceptibility of AMO to inactivation by acetylene aftertreatment with either thiourea or CS2. Cells were preincu-bated for 30 min with either no inhibitor, thiourea, or CS2.Residual ammonia-oxidizing activity was quantified after 30and 40 min from initial rates Of 02 uptake in the presence of10 mM NH4+ to confirm that the effects of each inhibitor hadreached completion during the preincubation period. Equalconcentrations of 14C2H2 (as a solution in DMSO) were thenadded to each incubation, and samples of cells were re-moved after 5, 60, and 180 min. The resulting fluorogram(Fig. 4) summarizes the effects of thiourea and CS2 on14C2H2 radiolabel incorporation into the 28-kilodalton (kDa)membrane-bound polypeptide that has been shown to be thesite of acetylene binding (16). For cells incubated in theabsence of either thiourea or CS2, label incorporation wasnearly complete by the first time point. For cells pretreatedwith thiourea, there was no apparent incorporation of labeleven after 3 h of exposure. For cells preincubated with CS2,there was a slow rate of label incorporation over the time
1 5 minI - ICELLS
"AN2H4
A
FIG. 2. Effects of acetylene, thiourea, and CS2 on ammonia- and hydrazine-dependent 02-uptake by whole cells of N. europaea. Theexperimental conditions were as described for Fig. 1. Once steady-state rates of ammonia-dependent 02 uptake were established, thereactions were made to 200 ,uM each of (B) CS2, (C) C2H2, and (D) thiourea. Hydrazine was added (to 600 ,uM) to reactions B, C, and D atthe point indicated by the second arrow. (E) Hydrazine (at 600 ,M) replaced NH4Cl at the initiation of the experiment.
VOL. 172, 1990
on July 9, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
4778 HYMAN ET AL.
Lane Number
l 2 3 4 5 6 7
- _ - __o
0168I 180 5 60 180
B
C.
D
E
FIG. 3. Effects of acetylene, thiourea, and CS2 on mediumacidification associated with ammonia oxidation. The reaction me-dium consisted of 5 mM sodium phosphate buffer, pH 7.8, contain-ing 25 mM NaCl and phenol red (5 p,g/ml). For each reaction, 1 mlof medium was added to a 1.2-ml quartz cuvette, and NH4Cl wasadded to a final concentration of 10 mM (traces B to E). The reactionwas initiated by the addition of 20 ,ul of whole-cell suspension of N.europaea (final protein concentration, 0.22 mg/ml), after which thecuvette was sealed with a Teflon stopper, inverted twice, and placedin the spectrophotometer. The traces show the decrease in A,.9 forcells incubated with (A) no additions, (B) 10 mM NH4Cl alone, (C)10 mM NH4Cl followed by 100 p.M thiourea, (D) 10 mM NH4Clfollowed by 170 p.M C2H2, and (E) 10 mM NH4Cl followed by 200p.M CS2. For traces C to E, the inhibitors were added at the breakin the trace. For trace B, the break indicates the point at which thereaction medium was partially reoxygenated by inverting the stop-pered cuvette.
course of the experiment which after 3 h was equivalent tothat for cells without prior treatment with any inhibitor.The experiments described above demonstrate that the
site of action of CS2 is related to the ammonia-oxidizingenzyme system and that CS2 does not appear to interact witheither HAO or the associated electron transport chain be-tween HAO and the terminal oxidase. However, the resultsdo not necessarily demonstrate a direct interaction betweenCS2 and AMO. Using visible-wavelength difference spectraof whole cells, Shears and Wood (27) have demonstratedthat a number of compounds which interact with AMO bydifferent mechanisms all produce absorbance decreases cen-tered at approximately 380 nm in aerobic cell suspensionsincubated in the absence of ammonia. For example, alterna-tive substrates for AMO, such as bromoethane and ethylene,cause this bleaching effect. We observed a similar effect withCS2 (Fig. 5). Trace A (Fig. 5) shows the base line recordedfor cells before the addition of any inhibitor. Trace B showsthe effect of 100 pM acetylene after 10 min, and traces C andD show the time-dependent development of a comparablespectrum for 100 FM CS2 during a 10-min incubation. Theaddition of ammonia to uninhibited cells normally results inextensive heme reduction (27). However, addition of 10 mMNH4C1 to the CS2-treated cells did not result in the reductionof hemes after 1 min (trace E). In contrast, the subsequent
Time (min)FIG. 4. Time course of the incorporation of [14C]acetylene into
the 28-kDa acetylene-binding polypeptide ofAMO for whole cells ofN. europaea preincubated with thiourea and CS2. A 2-ml sample ofa whole-cell suspension (0.1 g [wet weight]/ml) was added to a 9-mlserum vial that was stoppered with a trifluoroethylene-faced siliconerubber stopper and aluminum crimp seals. The cells were treatedwith either no addition, 200 nmol of thiourea, or 2 ,umol of CS2, andthe vials were placed on an orbital shaker (75 rpm). After 30 min,cells (20 ,ul) were removed from each vial, and the rate of 02 uptakein the presence of 10 mM NH4' was determined in an oxygenelectrode. This procedure was repeated 10 min later to ensurecompletion of the inhibition by CS2 and thiourea. Then 25 p.l of asolution of [14C]acetylene in DMSO was added to each vial (1 ,ul =6.65 x 105 dpm; total [14C]acetylene added, 65 nmol), and NH4Clwas added to an initial concentration of 2 mM. The vials were thenreturned to the shaker. Samples (500 p.l) were then withdrawn fromeach vial 5, 60, and 180 min after the addition of the [14C]acetylene.The samples were mixed with 100 p.J 50 mM sodium phosphatebuffer, pH 7.8, containing 1 mM thiourea. The samples were thensedimented by centrifugation (12,000 x g for 5 min) and suspendedin thiourea-containing buffer as described above. The cells werethen sedimented again by centrifugation, suspended in thiourea-freebuffer, and frozen in dry ice. The cells were fractionated andsamples were electrophoresed as described in Materials and Meth-ods section. The fluorograph was obtained from sodium dodecylsulfate-polyacrylamide gel electrophoresis analysis of membranessamples from cells incubated with no inhibitor (lanes 1 to 3),thiourea (lane 4), and CS2 (lanes 5 to 7) and shows the time coursefor the incorporation of [14C]acetylene into the 28-kDa polypeptideof AMO.
addition of 600 ,uM hydrazine resulted in a rapid (within 1min) and dramatic change that was associated with extensiveheme reduction (trace F). Equivalent results were obtainedin separate experiments using the same sequence of addi-tions to thiourea and acetylene-inhibited cells (data notshown). These results indicate that CS2, like acetylene andthiourea, does not inhibit the reduction of hemes by sub-strates of HAO but does inhibit the reduction of hemes byammonia, the substrate of AMO. Control experiments con-ducted in the absence of inhibitors established that sufficientoxygen remained after 10 min to support ammonia oxidationand that the lack of cytochrome reduction after the additionof NH4Cl to CS2-treated cells (Fig. 5, trace E) was not due toanaerobic conditions within the assay cuvette.A potential competitive interaction between ammonia and
CS2 was investigated by conducting a series of 02 uptakeexperiments with whole cells in which the ammonium con-centration was varied and 02 uptake was inhibited by theaddition of a constant concentration of CS2. Since theinhibition caused by CS2 is time dependent, the effect ofammonium concentration was determined by estimating thetime taken for the inhibitor to inhibit the rate of oxygenuptake by 50%. The results of this experiment and ofcomparable experiments with thiourea and acetylene, whichalso produced time-dependent inhibitions (Fig. 2), are shownin Fig. 6. The range of ammonium concentrations used
Io.0 OD
J. BACTERIOL.
on July 9, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

CARBON DISULFIDE INHIBITION 4779
1.0
A 0.8
ca. cm0.6
0._:5E04
..O0.2
FIG. 5. Effects of acetylene and CS2 on the difference spectra ofwhole cells of N. europaea. Difference spectra of whole cells wereobtained as described in Materials and Methods. Shown are thespectra obtained from whole cells after incubation with (A) noadditions, (B) 200 ,uM C2H2 for 10 min, (C) 200 p.M CS2 for 5 min,and (D) 200 ,uM CS2 for 10 min. (E) Reaction D, 1 min after additionof NH4Cl (10 mM); (F) reaction E, 1 min after addition of 600 p.Mhydrazine.
covers concentrations both above and below the apparentKinm for NH4 , as determined from initial rates of oxygenuptake (Fig. 6A). Over the range of ammonium concentra-tions tested, there' was essentially no change in the t1j2 forinhibition caused by either thiourea or CS2, implying thattheir effects are independent of ammonium (Fig. 6B). Incontrast, the t1l2 for inactivation by acetylene was progres-sively increased with successive increases in ammoniumconcentration.Beyond competition with respect to ammonia, two addi-
tional properties of AMO-specific inhibitors have been usedto distinguish between various modes of action, namely, therequirement for 02 and the reversibility' of the inhibition.When whole cells were incubated with CS2 and acetyleneunder anaerobic conditions, these compounds caused re-spective overall losses of 8 and 21% of the ammonium-dependent 02 uptake activity when the cells were assayedunder aerobic conditions after an 80-fold dilution in theoxygen electrode chamber (Table 1). In contrast, treatmentwith thiourea under anaerobic conditions resulted in a loss of90% of the preinhibited activity. None of the inhibitorstested inhibited the oxidation of hydrazine. The effects ofCS2 were also found to be partially reversible, and up to 35%of the uninhibited ammonia-oxidizing activity was recoveredby washing CS2-treated cells in buffer containing NH4'(Table 2). Lower recoveries of activity were generally ob-
I-
.-
Li-
. n
0
w
I-4
IF
F
20'
15
10
5F
A - -I
,. ~~~~~~*_~
0
0
0
000 --0
**-* '.. .- .
0.25 0.5 1.0 2.5 5.0
[NH*] (mM)10 25 50
FIG. 6. Effects of ammonium concentration on the half-time ofinhibition of ammonia oxidation by acetylene, thiourea, and CS2. Aseries of oxygen electrode incubations was run in which whole cells(0.18 mg of total protein per nl) were assayed for rates of ammonia-dependent 02 uptake in the presence of a range of NH4Cl concen-trations between 0.25 and 50 mM. When a steady-state rate Of 02uptake had been established for each concentration of NH4Cl,inhibitor (to 200 p.M) was added. The time required for the rate Of 02uptake to decline to 50% of the preinhibited rate was then deter-mined. (A) Effect of initial NH4Cl concentration on the preinhibitedrate Of 02 uptake; (B) effect of NH4CI concentration on the half-lifeof ammonia-dependent 02 uptake activity of cells inhibited with 200,uM C2H2 (0), 200 ,uM thiourea (0), and 200 ,uM CS2 (A).
served with thiourea-treated cells, and the inclusion ofcopper in the washing medium resulted in a slight increase inthe level of recovery compared with other treatments. Incontrast, no recovery of activity was observed with anytreatment for acetylene-treated cells. It was also observedthat ammonia-oxidizing activity increased with prolongedincubation of CS2-treated cells in the oxygen electrodechamber. A 6% recovery of residual ammonia-oxidizingactivity was observed 1 min after addition of cells andNH4Cl to the electrode chamber. However, the activityincreased to 24% at 5 min and 36% at 10 min. The same levelof recovery was observed if CS2-treated cells were added tothe electrode chamber in the absence of ammonium and thenammonia oxidation was initiated 10 min later by the additionof 10 mM NH4C1. The levels of recovery reported in Table 2
VOL. 172, 1990
on July 9, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

4780 HYMAN ET AL.
TABLE 1. Effects of incubation of whole cells of N. europaeawith acetylene, thiourea, and CS2 under anaerobic conditionsa
Treatment % Inhibition of ammonia- % Inhibition of N2H4-oxidizing activity oxidizing activity
No addition 3.8 0150 ,uM C2H2 8.1 3.3150 ,uM thiourea 90.3 0150 P.M CS2 20.9 6.7
a A whole-cell suspension (0.25 ml; 12.5 mg of protein per ml) was added tothe anaerobic incubation vial described in Materials and Methods. NH4Cl wasthen added to 1 mM to allow the cells to consume remaining 02 in the vialduring a 5-min preincubation. In separate experiments, each inhibitor (to 150p.M) was then added, and the cells were incubated for a further 15 min. Asample (20 ,u) of each cell suspension was removed and added to an oxygenelectrode chamber containing 10 mM NH4Cl. The residual rate of ammonia-dependent 02 uptake was determined from steady-state rates. Thiourea (100p.M) was then added to the electrode chamber, and after complete inhibitionof 02 uptake, hydrazine (to 600 FM) was added to the reaction. The residualhydrazine-oxidizing activity was determined from steady-state rates of 02uptake.
represent the activities of cells measured 5 min after additionto the electrode chamber.
DISCUSSIONIn the past, two main experimental approaches have been
used to determine the site of action of nitrification inhibitors.Since AMO has not been purified beyond a cell-free state,the combined use of these two approaches provides the mostunambiguous evidence presently available for a direct chem-ical interaction between an inhibitor and AMO. In 1973,Hooper and Terry (12) identified several groups of com-pounds that specifically inhibited ammonia oxidation, byconsidering the differential effects of these compounds onnitrite production from ammonia and hydroxylamine. More
TABLE 2. Effects of washing treatments on the reversibility ofinhibitions caused by acetylene, thiourea, and CS2"
% of activity recovereda
Unwashed Buffer Buffer plus Buffer plusInhibitor cnrlb loe 100 ~LM 1 mmcontro alone' cu2+d NH4+d
a b a b a b a b
No addition 100 100 98 104 98 104 88 104Acetylene 2 104 2 83 2 88 3 83Thiourea 6 100 16 92 24 84 17 96CS2 22 113 22 92 29 96 35 100
a A whole-cell suspension of N. europaea (2 ml, 0.1 g [wet weight]/ml) wasincubated in 9-ml serum vials sealed with trifluoroethylene-faced siliconerubber stoppers and aluminum crimp seals. In separate experiments, acety-lene (2 ,umol), thiourea (0.1 Lmol), and CS2 (2 I&mol) were added to the vials,and the reaction mixtures were shaken for 20 min on an orbital shaker (75rpm). Samples (20 p.l)kwere then removed from each incubation and assayedfor residual ammonia- and hydrazine-oxidizing activities in an oxygen elec-trode, as described for Table 1. A 1.5-ml sample of the remaining material ineach incubation was then removed, and 0.5 ml was added to each of threemicrofuge tubes. The cells were washed by three cycles of centrifugation andresuspension in various media and finally suspended to a final volume of 0.5ml in 50 mM sodium phosphate buffer, pH 7.8. Each sample was then assayedfor residual ammonia- and hydrazine-oxidizing activity as described for Table1. The reported activities were taken 5 min after the addition of cells to thechamber.
b Column a, ammonia-oxidizing activity; column b, hydrazine-oxidizingactivity.
c Buffer in all cases was 50 mM sodium phosphate, pH 7.8, containing 2 mMMg2+.
d Copper was added to buffer as CuS04; NH4+ was added as NH4Cl.
recently, Shears and Wood (27) have demonstrated thatseveral classes of AMO-specific inhibitory compounds thatinhibit ammonia oxidation through different mechanisms allproduce spectral changes in whole-cell suspensions that arecompatible with alterations in the oxidation state of thecopper, which is thought to be involved in the catalytic cycleofAMO. These compounds include (i) alternative substratesfor AMO (e.g., bromoethane and ethylene) (15, 33), (ii) ametal-binding inhibitor (allylthiourea) (12, 20), and (iii) acet-ylene, which acts as a suicide substrate for the enzyme (16).In this study we have presented both comparable kineticdata, using 02 uptake measurements (Fig. 2) and spectro-scopic evidence (Fig. 5), which suggest that CS2 is anothercompound which interacts with AMO. What is less clear isthe mode of action of CS2 on this enzyme. Various possiblemodes of action are discussed below.
Considerable interest exists in the mammalian toxicologyof CS2 since this compound is used in numerous industrialprocesses and represents an industrial hazard because of itshigh volatility and toxicity (6, 21). Studies on the effects ofCS2 in mammalian systems have concentrated on cy-tochrome P-450 monooxygenase systems which, like AMOfrom N. europaea, are typically nonspecific enzymes. Largedecreases in the levels of microsomal P-450 are reported tooccur after treatment of test animals with CS2 (9, 10, 13, 21),and the use of 14CS2 and C35S2 (9, 26) has shown that themetabolism of CS2 by microsomal fractions involves thecytochrome P-450-catalyzed oxidation of CS2 to carbonylsulfide (COS) and covalent binding of 35S to the enzyme (26).In the case of AMO, the results obtained in this studysuggest that enzymatic activation of CS2 is not a likely modeof action for this compound. Despite the apparent require-ment for oxygenated conditions, which are required forAMO activity (Table 1), we did not observe a competitiveinteraction between CS2 and ammonia (Fig. 6), as would beexpected for competing oxidizable substrates. In contrast,both an oxygen requirement (Table 1) and a competitiveinteraction with ammonia (Fig. 6) were observed for acety-lene, and inactivation by this inhibitor is known to requirethe catalytic activity of AMO (16). We also observed thatpretreatment of whole cells with CS2 only delayed and didnot prevent the complete incorporation of [14C]acetyleneinto the 28-kD acetylene-binding component of AMO (Fig.4). If the effects of CS2 involved inactivation ofAMO arisingfrom the products of CS2 oxidation, one expected effect ofpretreatment of cells with CS2 would have been a loWer levelof [14C]acetylene incorporation compared with that ob-served for cells treated with [14C]acetylene alone. This effectwas not observed (Fig. 4).An alternative and more likely mode of action for CS2
inhibition of AMO arises from consideration of the chemicalproperties of this compound. The chemistry of CS2 isdominated by its ability to reversibly react with nucleophilesto produce metal-complexing compounds (24). For example,CS2 will react with many compounds, including amino acids,which contain suitable amino, mercapto, and hydroxygroups to form dithiocarbamates, trithiocarbamates, andxanthogenates, respectively. These compounds act as che-lators of metal ions and have sufficiently high affinities forboth copper and zinc (24) that the reaction of copper saltswith dithiocarbamates generated from CS2 is the basis ofmany analytical procedures for CS2 (24). The metal-chelat-ing activity of compounds formed from the reaction of CS2with amino acids is also argued to be a significant part of thetoxic effect of CS2 on test animals and can account for theinhibitory effect of CS2 on cytochrome oxidase, monamine
J. BACTERIOL.
on July 9, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

CARBON DISULFIDE INHIBITION 4781
oxidase, and tyrosine oxidase (Cu containing) and on lactatedehydrogenase, carbonic anhydrase, and alcohol dehydro-genase (Zn requiring) (6). In the case of AMO, the formationof chelating species arising from the reversible reaction ofCS2 with a suitably positioned amino acid close to anactive-site copper would account not only for the timedependency and specificity of the reaction but also for thespectroscopic effects, the lack of competition between am-monia and CS2, and the apparent lack of a requirement forthe turnover of AMO during the inhibition process.Compounds that contain C=S bonds represent an impor-
tant class of potent inhibitors of ammonia oxidation by N.europaea. These compounds include CS2, thiourea, allyl-thiourea, thiosemicarbazide, thioacetamide, potassium eth-ylxanthate, and diethyldithiocarbamate. It has been arguedthat the inhibitor mechanism is largely dictated by functionalgroups (3, 25); therefore, the presence of a C=S bond in all ofthe compounds listed above tnight be taken to indicate that aproposed mechanism for one compound is generally appli-cable to all compounds of this group. In fact, the similaritybetween the inhibitory effects of these compounds on am-monia oxidation and purified copper-containing enzymesrepresents a significant proportion of the available evidencefor a role for copper in ammonia oxidation. However, it isclear from our results that the effects of CS2 are significantlydifferent than those that we observed in parallel experimentswith thiourea. This finding suggests that the mechanisms ofthese two compounds are different and that the relationshipbetween functional group and inhibitor mechanism is morecomplex than it first appeared. The significance of thedifferences between these two inhibitors are consideredmore thoroughly below.The effects of CS2 are superficially distinguished from
those of thiourea in that CS2 produces a relatively slow,time-dependent inhibition which results in a residual activitygreater than the endogenous rate of AMO-independent res-piration. In the case of partial inhibitions, the analysis of 02uptake rates cannot clearly distinguish the contributionsmade by either ammonia oxidation or hydroxylamine oxida-tion. The more significant difference between the effects ofthese inhibitors is therefore the residual activity of AMO inCS2-treated cells. This activity is most clearly demonstratedby the ability of CS2-treated cells to incorporate [14C]acetylene to the same level as do non-CS2-treated cells (Fig.4), albeit at a slower rate. This result indicates two points.First, prolonged incubation with CS2 beyond that character-ized in our short-term 02 uptake experiments (Fig. 1 and 2)does not lead to a complete inhibition of AMO. Second,since a complete inhibition does not occur, the effects of CS2appear to be equally applied to AMO, and the incompleteinactivation by CS2 does not arise from mixed populations ofinhibited and uninhibited enzyme. In contrast to the fullyinhibitory effects of thiourea, the mode of action of CS2probably therefore involves a process in which the turnoverof AMO is dramatically slowed but not halted. Potentially,incomplete inhibition can arise from allosteric inhibition orthrough a diversion of the catalytic reaction to a slower, lessefficient pathway (8). In contrast to CS2, thiourea inhibits theactivity of AMO completely. Wood et al. (32) and othershave argued that a likely mode of action for thiourea-basedcompounds involves their ability to tautomerize and formreactive thiols. The differences in the modes of action ofthiourea and CS2 may therefore be determined by the abilityof compounds such as dithiocarbamates to act as bidentateligands (and thus true chelators), whereas thiourea andrelated compounds can serve only as monodentate ligands
ARNH RN
C=S <-..........--* iSH-
NH2 NH
Bs
R-NH2 + CS2 = R-NHC
SH
FIG. 7. Differences in the modes of action of thiourea and CS2.
(24, 32). These differences are shown in diagramatic form inFig. 7.On the basis of this interpretation, it would appear that
AMO inhibitors which contain C=S bonds are all metal-complexing agents that can be further subdivided into com-pounds which act as true chelating agents (CS2 and com-pounds that are capable of releasing CS2) and simple metal-binding compounds (thiourea and other alkyl-substitutedthiourea-based compounds). From a structural point ofview, these groups can also be distinguished on the basis ofterminally located C=S groups (CS2, diethyldithiocarbamate,and potassium ethylxanthate) and internal C=S bonds (thio-urea, allylthiourea, thiosemicarbazide, and thioacetamide).
Principal assumptions behind much of the more recentresearch into ammonia oxidation has been that AMO is acopper-containing enzyme (3, 20, 27, 33) and that the modeof action of compounds such as thiourea involves a copper-selective metal-binding reaction (12, 20). These assumptionsare also reflected in this discussion. It is important to notethat one of the main lines of evidence supporting a role forcopper in AMO comes from studies which show metal-binding agents such as thiourea and allythiourea are potentinhibitors of ammonia oxidation, and it is argued that thesecompounds are copper selective (20). It is interesting to notethat outside the field of nitrification, thiourea and alkylderivatives are actually regarded as extremely nonspecificligands that can form complexes with more than 70 ionicspecies (24). More recently, thiourea has been shown to actas a potent scavenger of both hydroxyl and superoxideradicals in biological systems (7, 19); monooxygenase-cata-lyzed reactions may involve these reactive intermediates.Perhaps the inhibition of AMO by CS2 (and related com-pounds) provides more convincing evidence for a role forcopper in AMO than is provided by the inhibition bythiourea and its derivatives. A purified form of AMO and theestablishment of a role for copper in the catalytic cycle of theenzyme would undoubtedly help in the clarification of thesepossibilities.
ACKNOWLEDGMENTThis research was supported by the U.S. Department of Agricul-
ture grant 88-37120-3956.
LITERATURE CITED1. Ashworth, J., G. G. Briggs, A. A. Evans, and J. Matula. 1977.
Inhibition of nitrification by Nitrapyrin, carbon disulphide, andtrithiocarbonate. J. Sci. Food Agric. 28:673-683.
2. Ashworth, J., A. Penny, F. V. Widdowson, and G. G. Briggs.1980. The effects of injecting nitrapyrin ('N-serve'), carbondisulfide or trithiocarbonates with aqueous ammonia, on yieldand %N of grass. J. Sci. Food Agric. 31:229-237.
3. Bedard, C., and R. Knowles. 1989. Physiology, biochemistry,and specific inhibitors of CH4, NH4', and CO oxidation bymethanotrophs and nitrifiers. Microbiol. Rev. 53:68-84.
4. Bremner, J. M., and L. G. Bundy. 1974. Inhibition of nitrifica-tion in soils by volatile sulfur compounds. Soil Biol. Biochem.6:161-165.
5. Bremner, J. M., and C. G. Steele. 1978. Role of microrganisms
VOL. 172, 1990
on July 9, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

4782 HYMAN ET AL.
in the atmospheric sulfur cycle. Adv. Microbiol. Ecol. 2:155-201.
6. Brieger, H. 1967. Carbon disulphide in the living organism.Retention, biotransformation and pathophysiologic effects, p.27-31. In H. Brieger and J. Teisinger (ed.), Toxicology ofcarbon disulphide. Excerpta Medica Foundation, Amsterdam.
7. Cederbaum, A. I., E. Dicker, and G. Cohen. 1978. Effect ofhydroxyl radical scavengers on microsomal oxidation of alco-hols and on associated microsomal reactions. Biochemistry.17:3058-3064.
8. Cleland, W. W. 1970. Steady state kinetics, p. 1-65. In P. D.Boyer (ed.), The enzymes student edition. Academic Press,Inc., New York.
9. Dalvi, R. R., R. E. Poore, and R. A. Neal. 1974. Studies of themetabolism of carbon disulfide by rat liver microsomes. LifeSci. 14:1785-1796.
10. De Matteis, F. 1974. Covalent binding of sulfur to microsomesand loss of cytochrome P-450 during the oxidative desulfurationof several chemicals. Mol. Pharmacol. 10:849-854.
11. Hooper, A. B. 1984. Ammonia oxidation and energy transduc-tion in the nitrifying bacteria, p. 133-167. In W. R. Strohl and0. H. Touvinen (ed.), Microbial chemoautotrophy. Ohio StateUniversity Press, Columbus.
12. Hooper, A. B., and K. R. Terry. 1973. Specific inhibitors ofammonia oxidation in Nitrosomonas. J. Bacteriol. 115:480485.
13. Hunter, A. L., and R. A. Neal. 1975. Inhibition of hepatic mixed-function oxidase activity in vitro and in vivo by various thiono-sulfur containing compounds. Biochem. Pharmacol. 24:2199-2205.
14. Hyman, M. R., I. B. Murton, and D. J. Arp. 1988. Interaction ofammonia monooxygenase from Nitrosomonas europaea withalkanes, alkenes, and alkynes. Appl. Environ. Microbiol. 54:3187-3190.
15. Hyman, M. R., and P. M. Wood. 1983. Methane oxidation byNitrosomonas europaea. Biochem. J. 212:31-37.
16. Hyman, M. R., and P. M. Wood. 1985. Suicidal inactivation andlabelling of ammonia monooxygenase by acetylene. Biochem. J.227:719-725.
17. Jensen, H. L., and H. Sorensen. 1952. The influence of someorganic sulphur compounds and enzyme inhibitors on Ni-trosomonas europaea. Acta Agric. Scand. 2:295-304.
18. Keeney, D. R. 1986. Inhibition of nitrification in soils, p. 99-126.
In J. I. Prosser (ed.), Nitrification. IRL Press, Oxford.19. Kelner, M. J., R. Bagnell, and K. J. Welch. 1990. Thioureas
react with superoxide radicals to yield a sulflhydryl compound.J. Biol. Chem. 265:1306-1311.
20. Lees, H. 1952. The biochemistry of the nitrifying organisms. 1.The ammonia-oxidizing system of Nitrosomonas. Biochem. J.52:134-139.
21. Obreska, M. J., P. Kentish, and D. V. Parke. 1980. The effectsof carbon disulphide on rate liver microsomal mixed-functionaloxidases in vivo and in vitro. Biochem. J. 188:107-112.
22. Painter, H. A. 1986. Nitrification in the treatment of sewage andwaste-waters, p. 185-211. In J. I. Prosser (ed.), Nitrification.IRL Press, Oxford.
23. Powlson, D. S., and D. S. Jenkinson. 1971. Inhibition of nitrifi-cation by carbon disulphide from rubber bungs. Soil Biol.Biochem. 3:267-269.
24. Reid, E. E. 1963. Organic chemistry of bivalent sulfur, vol. 4 and5. Chemical Publishing Co., Inc., New York.
25. Sahrawat, K. L., and D. R. Keeney. 1985. Perspectives forresearch on development on nitrification inhibitors. Commun.Soil Sci. Plant Anal. 16:517-524.
26. Savolainen, H., J. Jarvisalo, and H. Vainiio. 1977. Specificbinding of CS2 metabolites to microsomal proteins in rat liver.Acta Pharmacol. Toxicol. 41:94-96.
27. Shears, J. H., and P. M. Wood. 1985. Spectroscopic evidencefor a photosensitive oxygenated state of ammonia mono-oxyge-nase. Biochem. J. 226:499-507.
28. Sisler, H. D., and N. N. Ragsdale. 1983. Fungicides, p. 17-47. InD. Pimentel (ed.), CRC handbook of pest management inagriculture, vol. III. CRC Press, Inc., Boca Raton, Fla.
29. Underhill, S. E., and J. I. Prosser. 1987. Inhibition and stimu-lation of nitrification by potassium ethyl xanthate. J. Gen.Microbiol. 133:3237-3245.
30. Underhill, S. E., and J. I. Prosser. 1987. Surface attachment ofnitrifying bacteria and their inhibition by potassium ethyl xan-thate. Microbiol. Ecol. 14:129-139.
31. Warrington, R. 1878. On nitrification. J. Chem. Soc. 33:44-51.32. Wood, L. B., B. J. E. Hurley, and P. J. Matthews. 1981. Some
observations on the biochemistry and inhibition of nitrification.Water Res. 15:543-551.
33. Wood, P. M. 1986. Nitrification as an energy source, p. 39-62.In J. I. Prosser (ed.), Nitrification. IRL Press, Oxford.
J. BACTERIOL.
on July 9, 2018 by guesthttp://jb.asm
.org/D
ownloaded from