inflammation as a therapeutic target in alzheimer’s … as a therapeutic target in alzheimer’s...
TRANSCRIPT

Inflammation as a Therapeutic Target in Alzheimer’s Disease
•“Cocktail” approach to AD therapy
Andrea Tenner, PhDSeptember 20, 2013
Countdown to 2025: Progress on Ending Alzheimer’s Disease

Hallmarks of Alzheimer’s Disease Pathology
• ß-amyloid containing plaques
• Neurofibrillartangles
• Neuronal loss
AD Control
à dementia

Catastrophic PhasefAβ, C’ activation,tangles,neuron and synapse loss
Losesindependence
Death
AD as a Progressive Disease
Oligomeric AβDiffuse plaques
InitiatingFactor
(?) PromotingFactor(s)
(?) Clinicalsymptomsappear
Preclinical Phase
ReparativeActivities (?)

Overview• NeuroInflammation is a component of Alzheimer’s Disease
• The Complement Cascade is inducer of AD Neuroinflammation
• Some components of this response can be reparative or protective.
àIdentifying and screening novelCandidate Therapeutics

Immune Response
Adaptive
AntibodiesT cellsCytotoxic T cellsCD4 T Cell-
mediated killing
Goal: Recognition and Elimination of Danger
Phagocytes/MicrogliaComplementMolecular Danger
SensorsCytokines
Innate

Inflammation• An immune response to a perceived injury or infection: – Heat (fever) – Swelling– Redness– Pain
Neuroinflammation-no heat, no swelling, no redness, no pain
**Deleterious to Neurological Function

Microglia Responses
NeuroinflammationProliferation and activation of microglia-Induction of proinflammatory cytokines à**Deleterious to Neurological Function
Phagocytosis
GoodBad
Inflammation Repair/ remodelingGood

Activation States of Microglia
• Classical Activation – stimulated by the “cytokine” IFNγ; attack function
• Alternative Activation – stimulated by IL-4 and IL-13; anti-inflammatory
• Acquired Deactivation – stimulated by TGF-ß, IL-10 and dying cells; phagocytic but immunosuppressive
Can coexist - (provides targets for therapeutics)

Complement
• A group of >30 interacting proteins in blood, extracellular fluids and on cells that are activated by injury or infection.
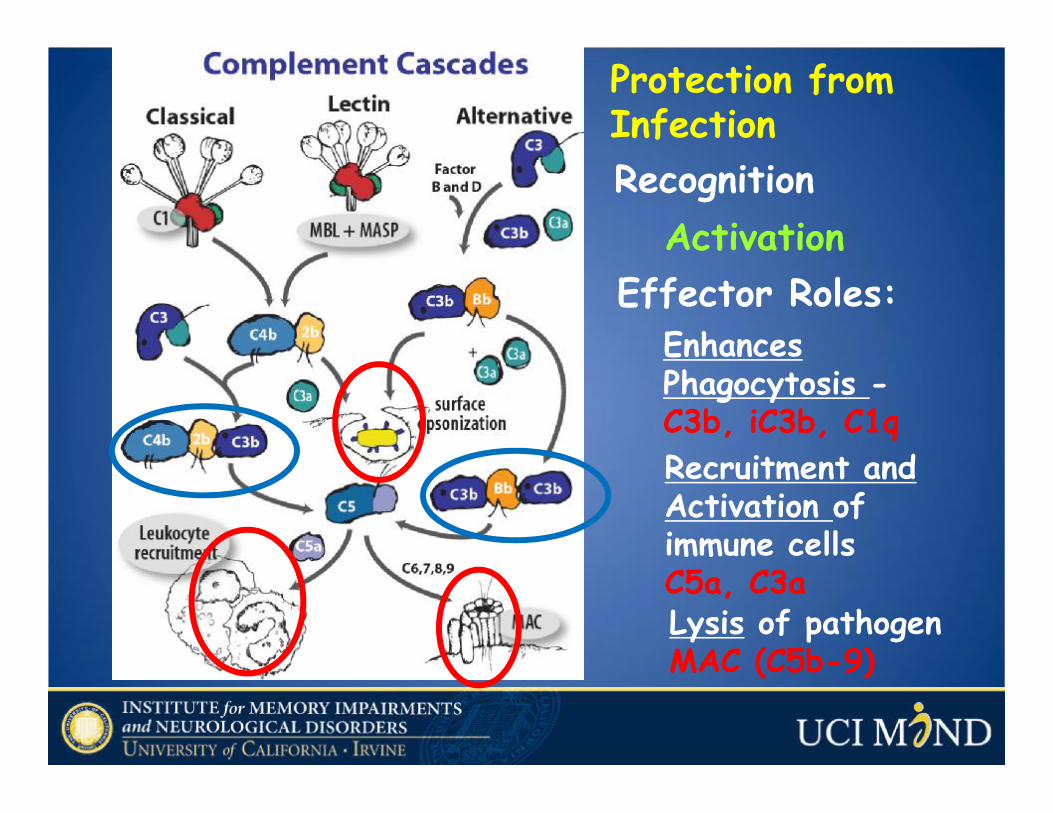
Enhances Phagocytosis -C3b, iC3b, C1q
Recognition
Recruitment and Activation of immune cellsC5a, C3aLysis of pathogen MAC (C5b-9)
Effector Roles:Activation
Protection from Infection

Ricklin and Lambris, Nature Biotech., 2007
Pathological Conditions Associated with Complement Activation
Induction of excessive inflammation à cell damage

Indicators of Inflammation in Alzheimer Disease - 1
•Neuritic amyloid plaques colocalize with reactive microglia (infiltrating macrophages?) and astrocytes
•complement proteins•Increased proinflammatorycytokines
• GWAS SNPs in CR1,Clusterinand Trem2 as contributing to the risk of AD.

Genetic Clues to Late Onset AD:GWAS – SNPs -2
Genome wide association studies - single nucleotide polymorphisms
• ApoE4 - lipid biology • TREM2 – phagocytosis*• CR1 – phagocytosis; regulation of
inflammation• Beclin1 – endosomal trafficking
*ingestion of pathogens and debris

Neurodegenerative Diseases
• Signals from other injured cells
• Imbalance between pro- and anti-inflammatory processesInflammation Neuronal
Dysfunction
• Accumulation of protein that is aggregated/misfolded/modified

“Inflammasome” – activated by infection and debris
Including fibrillar amyloid
• accumulation of undigested protein
• Dysfunctional phagocytosis
• improper trafficking within cells for digestion
• regulation of the inflammatory products
GWAS identified SNPs Hansson & Klareskog Nature med 17: 790-791, 2011
Aß

Clinical Studies• Epidemiological
Prospective studies NSAID use ~ decreased risk /delay onset
• Treatment Trials – without success• Prevention Trials – celecoxib; naproxen ~
no effect• The multiple effects of inflammation make
specificity of therapeutic targets critcal;• Differential properties of peripheral inflammation
vs. neuroinflammation may be key

Aß plaques are fibrillar in AD brainwhile diffuse in nondemented brain
AD Nondemented elderly
Totalamyloid Fibrillar Aß
activates the ComplementCascade
Fibrillaramyloid
C1q
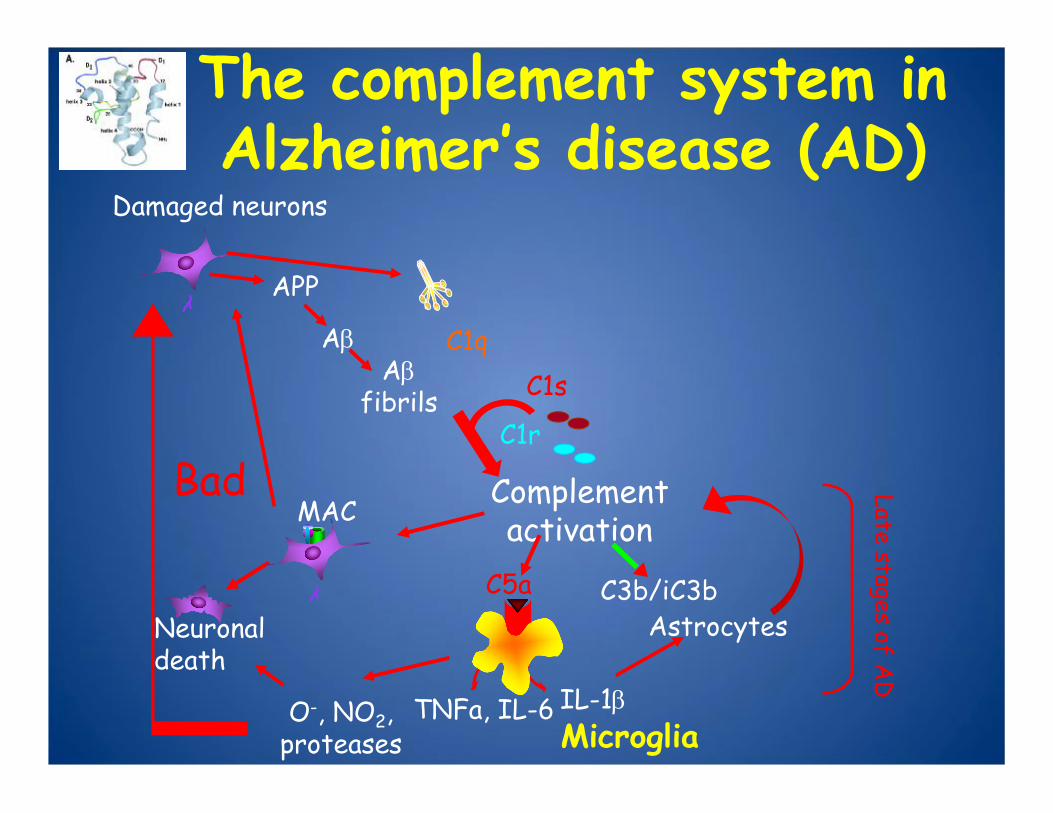
Damaged neurons
APP
C1s
C1r
AβAβ
fibrils
The complement system in Alzheimer’s disease (AD)
C1q
Complement activation
C3b/iC3b
MAC
C5aAstrocytes
Bad
O-, NO2, proteases
IL-1βTNFa, IL-6
Neuronal death
Late stages of AD
Microglia

Damaged neurons
APP
C1s
C1r
AβAβ
fibrils
Hypothesis:C5a is “Bad” inflammation in AD
C1q
Complement activationMAC
C5a C3b/iC3b
Late stages of AD
X Astrocytes
Bad
O-, NO2, proteases
IL-1βTNFa, IL-6
Neuronal death
TLR2/4
Pharmacologic and genetic evidence
C5a
Microglia

C5a Receptor antagonist: PMX205• Cyclic six amino acid
peptide based on the C-terminus of C5a.
• Interacts specifically with the C5a receptor, CD88
• Therapeutic effects in animal models of inflammatory diseases, including a rat model of neurodegeneration (Woodruff et al., 2005)
Therapeutic Target: C5a receptor – CD88
N
O
HN
OHN
O
NH
O
NHO
NHO
NH
NH
NH2
NH+
PMX205: cyclo-hydrocinnamate-[Orn-Pro-D-cyclohexylalanine-Trp-Arg]
Tg2576 – AD model12 weeks – oral12-15 months old

C5aR antagonist decreases pathology in the Tg2576 AD mouse
*
n=5 n=6p<0.003
UT PMX205Fibrillar Ab
n=11 n=17p<0 .002
Microglia
44-54%
n=11 n=17p<0 .001
Astrocytes
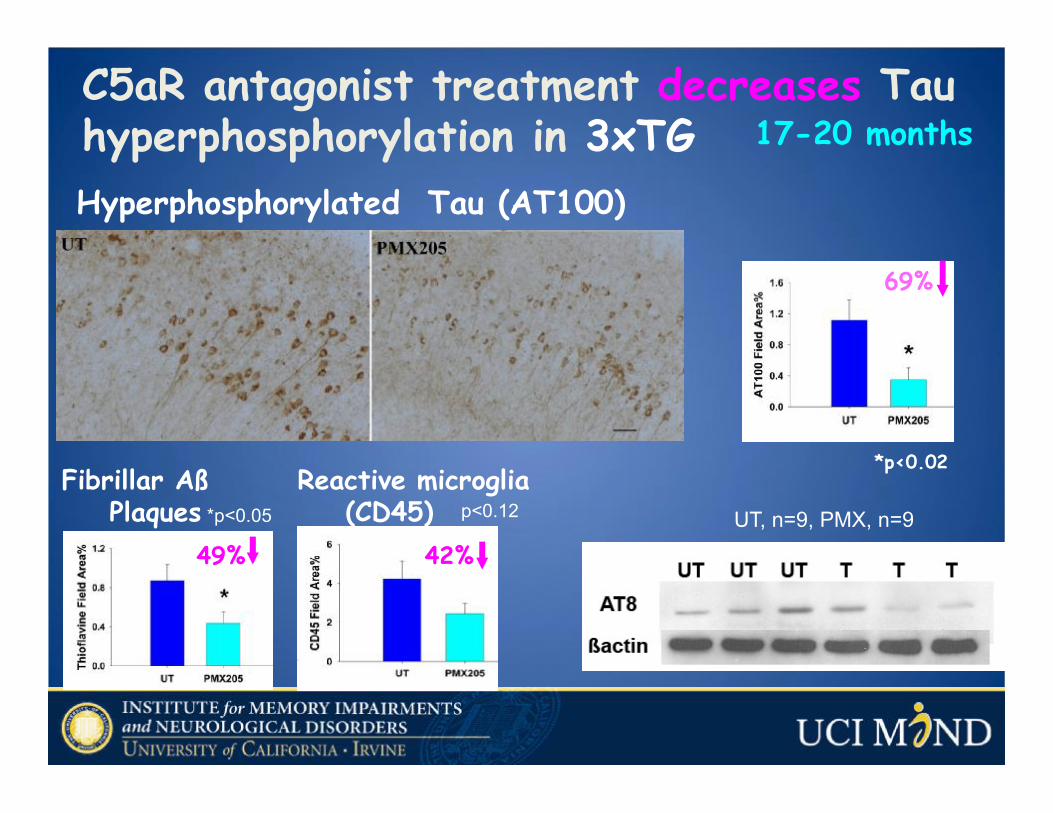
Hyperphosphorylated Tau (AT100)
C5aR antagonist treatment decreases Tau hyperphosphorylation in 3xTG 17-20 months
69%
Fibrillar Aß Plaques *p<0.05
Reactive microglia (CD45)
49% 42%
p<0.12 UT, n=9, PMX, n=9
*p<0.02

C5a – C5a RECEPTOR INTERACTION
C5a C5a Receptor (CD88)
Peter Ward, 2010

Object Location Memory
Genetic Deletion of CD88/C5aR Partially Protects Against Loss of Spatial Memory
Arctic APP AD mouse model x C5aR-/-
Tracy Cole, 2013
*10 Months
*Kruskal-Wallis ANOVA < .01

Object Location Memory
Overproduction of C5a Accelerates Loss of Spatial Memory in AD Mouse Models
Arctic APP x GFAP-C5a
Tracy Cole
**
*One way ANOVA is significant p=0.001
7 Months5 Months
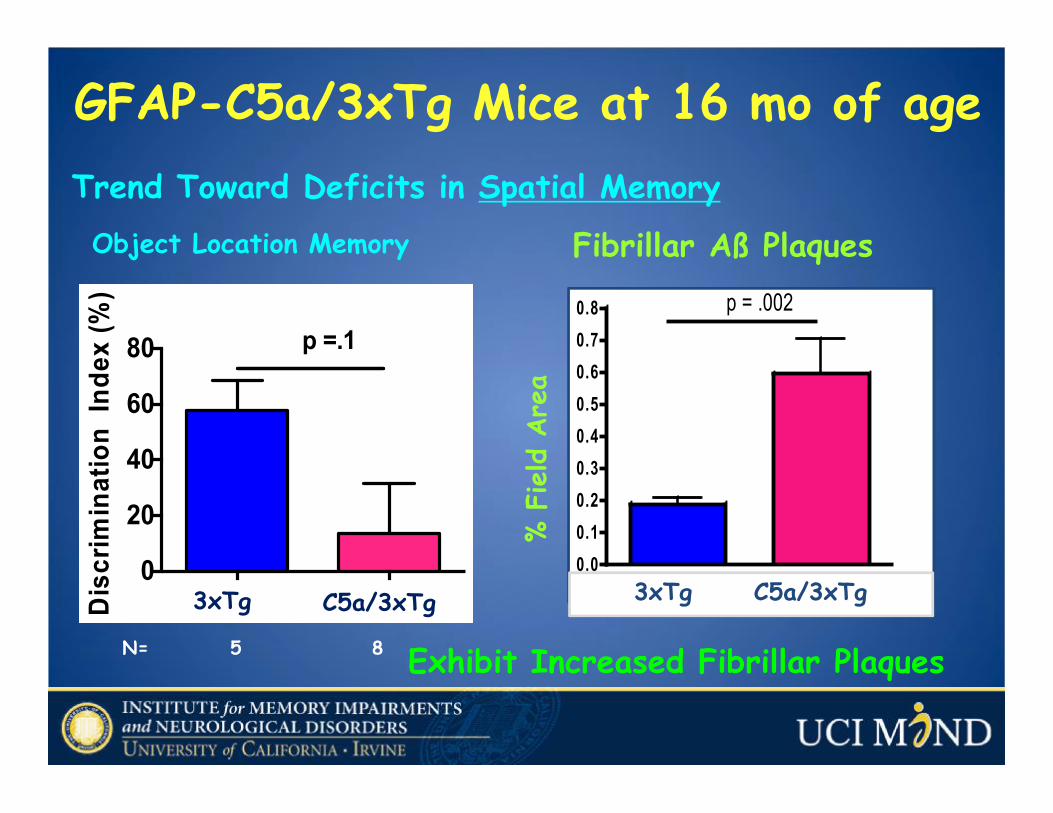
GFAP-C5a/3xTg Mice at 16 mo of age
Object Location Memory
0
20
40
60
80
Disc
rimin
atio
n In
dex
(%)
p =.1
3xTg C5a/3xTg
N= 5 8 Exhibit Increased Fibrillar Plaques
3xTg GFAP/C5a 3xTg0.00.10.20.30.40.50.60.70.8 p = .002
Fibrillar Aß Plaques
% F
ield A
rea
3xTg C5a/3xTg
Trend Toward Deficits in Spatial Memory

Mechanism of PMX205 Protection? • Blocking a synergistic effect of C5a-C5aR
and plaque fAß – TLRs that induces more robust inflammatory response of microglia and/or infiltrating macrophages?
• Is this in the CNS or in periphery?
•Is PMX205 signaling in neurons? Astrocytes? And/or Endothelial cells?

Summary #1• Amyloid deposits in AD brain activatethe complement pathway
• Inflammatory markers increase in part as a result of complement activation in mouse models of AD brain
• Targeted Complement inhibitors may be one approach to prevent and/or slow the progression of cognitive loss in AD

Damaged neurons
C1s
C1r
APP
Aβ
Aβfibrils
The complement system in Alzheimer’s disease (AD)
C1q
Benoit et al., 2013
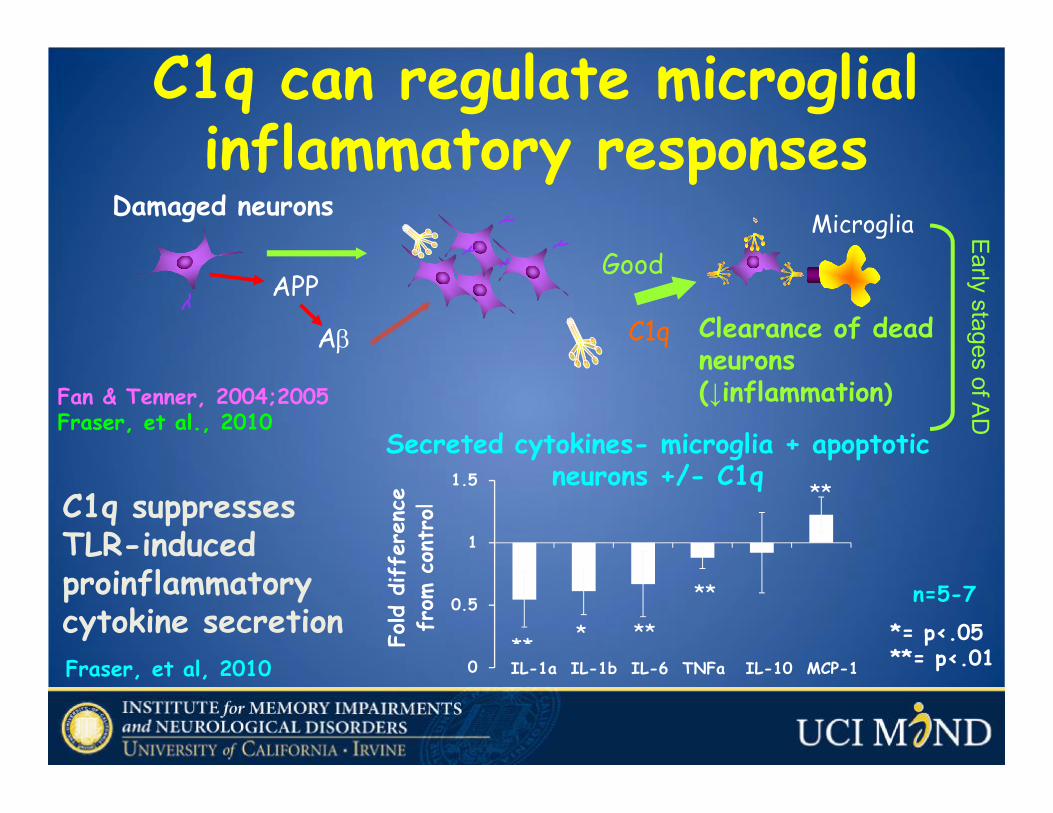
Damaged neurons
APP
Aβ
Microglia
Clearance of dead neurons (↓inflammation)
C1q
Good
Early stages of ADC1q can regulate microglialinflammatory responses
Fan & Tenner, 2004;2005Fraser, et al., 2010
C1q suppressesTLR-induced proinflammatory cytokine secretionFraser, et al, 2010
*= p<.05**= p<.01
n=5-7
0
0.5
1
1.5
** * **
**
**
IL-1a IL-1b IL-6 TNFa IL-10 MCP-1
Fold d
iffe
renc
e fr
om c
ontr
olSecreted cytokines- microglia + apoptotic
neurons +/- C1q
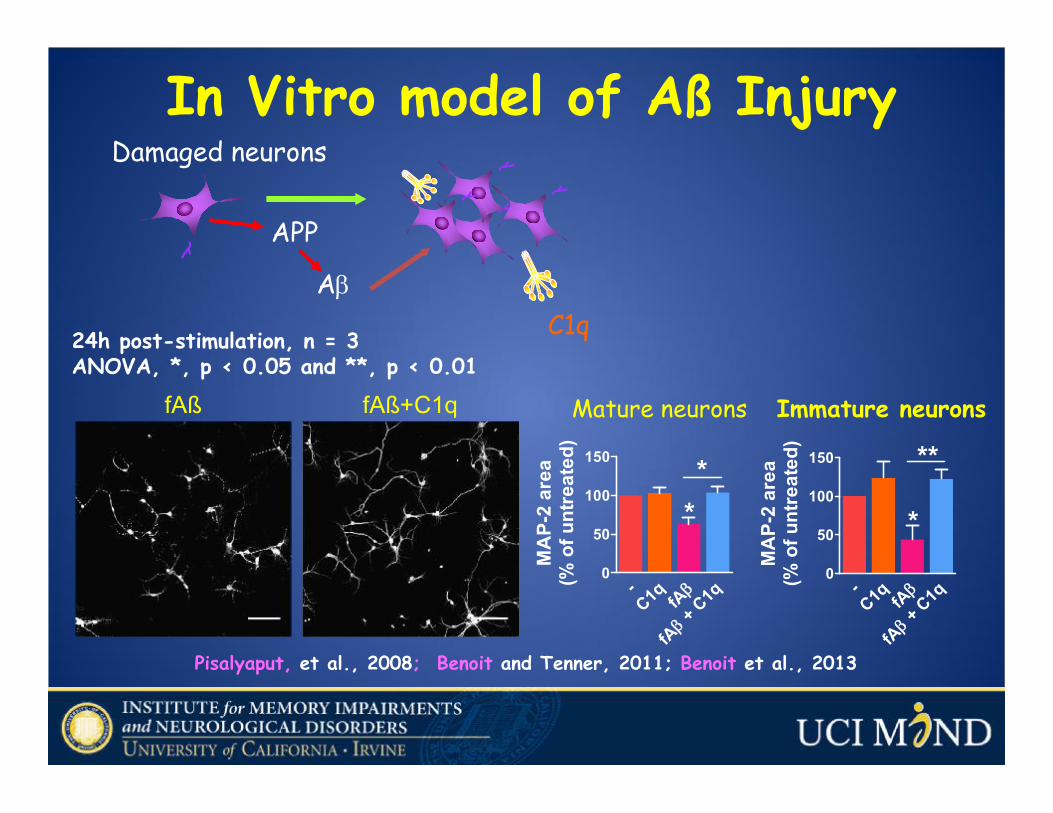
Damaged neurons
APP
Aβ
C1q
In Vitro model of Aß Injury
Pisalyaput, et al., 2008; Benoit and Tenner, 2011; Benoit et al., 2013
fAß fAß+C1q
-C1q β
fA +
C1q
βfA
0
50
100
150
*
**
MA
P-2
area
(% o
f unt
reat
ed)
Immature neurons
-C1q β
fA +
C1q
βfA
0
50
100
150
**
MA
P-2
area
(% o
f unt
reat
ed)
Mature neurons
24h post-stimulation, n = 3ANOVA, *, p < 0.05 and **, p < 0.01

C1q increases specific protein expression in Aß-injured neurons
LRP1B (LDL receptor-related protein 1B)Øbinds Aß and APP Ø retains APP at the
cell surface →↓amyloid-βproduction
GPR6 (G protein-coupled receptor 6)Ø increases neurite
growth by increasing intracellular levels of cAMP
MB1

Slide 32
MB1 although I like this slide, it can be removed if you need time Marie, 10/4/2012

Does the C1q-neuroprotective response to Aß require LRP1B and GPR6? Yes!
N = 3, 5 fields per condition
2-way ANOVA test **, p< 0.01 and ***, p < 0.001. -
scr siRNA
LRP1B siRNA
GPR6 siRNA0
20
40
60
80
100
120
***
***
***
***
*** *****
**
UntreatedfAβfAβ+C1q
MA
P-2
area
(% o
f unt
reat
ed c
ontr
ol)
10 nM scrambled, LRP1B or GPR6 siRNA
24h
Inhibition of LRP1B and GPR6 expression after siRNA transfection+/- C1q (10 nM)
fAβ (5 µM)
24h
Neuronal integrity

•C1q promotes neuron survival directly
•Enhancement of clearance of neuronal blebs and apoptotic debris
•Suppression of inflammatory cytokines
Summary: C1q - Good Cop

Perspective: possible therapeutics
Damaged neurons
IL-1b
C1sC1r
APP
AbAb
fibrils
Complement activationMAC
C5a C3b/iC3b
TNF, IL-6
Astrocytes
O-, NO2, proteases
Bad
Neuronal death
Late stages of AD
Microglia
Clearance of dead neurons (↓inflammation)
C1q
Enhance neuronal survival
Good
Early stages of AD
Prevent damage
Promote repair
C5aR antagonist
Fonseca, Ager et al., J Immunol, 2009
Target downstream effectors of C1q

• promotion of complement neuroprotectiveactivity (C1q and C3)
+• Targeted inhibition of detrimental
complement events (C5aR) à approach to slow the progression of neurodegenerative or developmental disorders
Conclusion:Cocktail Approach to Therapy

Potential Targets for Therapies in AD
Secretase Inhibitors Enhance ClearanceBlock oligomer/fibril formation
Block Complement Activation Block Inflammation
Pathogenic EventGenetics/Injury
Deposition of Aß1-42
Activation of glia
Neuronal damage
Target Therapy
Prevent neurotoxicity
Enhance neuronal function
>>
>

Basic Science-DiscoveryMolecules Pathology
DiseaseDrug DiscoveryTherapeuticsClinical TrialsBenefit to patients
Pathways

Team Tenner at UCI

Acknowledgements Tenner LabMarie BenoitSophie ChuElizabeth ClarkeTracy ColeMarisa FonsecaMichael HernandezNatalia TjokroAltea RocchiRahasson AgerDeb FraserKarntipa PisalyaputMinhan DinhFrancisca Benavente
T. Woodruff, Australia
S. Taylor Australia
R. Wetsel, UTS. Barnum, UABK. Hsiao-Ashe
U.MinnY. Kimura, U.PennNIHAlzheimer’s Association
LaFerla, Cotman, Cribbs, Glabe, Wood, Poulos, Chamberlin
Pooja SelvanAmy TranSamantha TranOsvaldo VasquezAnthony ChenSamantha HardinSam TonthatDan TonthatAlice BerciAndrew PakTachaporn S.Lindsey Weiner
UCI DNA and Microarray Facility