incremental gmps for imps and qp batch certification presented by: karen s. ginsbury for: iff...
TRANSCRIPT

Incremental GMPs for IMPsand QP Batch Certification
Presented by: Karen S. Ginsbury
For: IFF
October 2009
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Course Objective
Provide an overview of regulations and expectations of regulatory authorities regarding manufacture and in particular, certification and release of clinical trials material
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

To be discussed…
• EU + US legislation / guidance for IMPs:– FDA’s GMPs for Phase I
guidance– EU Annex 13
• The role of the Quality Unit vs The Qualified Person in batch certification and / or release (Annex 16)
• Sponsor responsibilities and release to sites
• Documentation requirements – Product Specification File
• Contract manufacture and analysis of investigational products
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

To be discussed…• Preliminary Hazard Analysis /
risk assessment tool for determining GMP levels
• Pre-clinical batches: “GMP” or not
• Risks for early / late phase:– Analytical methods
qualification / verification / validation
– Cleaning verification / validation
– Facilities: dedicated / campaign basis / potent drugs /biological
– Vendors: qualification / approval program / auditing – at what stage
• Process: - when to• define target product
profile• identify Critical
Process Parameters•
identify Critical Quality Attributesstart to show reproducibilitythink about validationpackagingstability and retest / reuse/ expiry date
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

The Playing Field
FDA• Exempt phase I
product from21 CFR part 211
• Continue to oversee under STATUTORY requirements of FD&C Act
• Phase 2 and 3 still under CFR
EURequire ALL of GMP+ Annex 13+ Annex 16 (QP release)For all phases
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Product Control Strategy and Risk Management
• Developing a Target Product Profile
• Product Specification File
• Initial risk assessment
• Company Strategy for controlling material to be used to generate data for use in regulatory submissions: pre-clinical toxicology, phase 1 – 3
• Product specific control strategy
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

The Phases of Drug Development
• Pre-Clinical
• Phase 1
• Phase 2
• Phase 3
• Phase 4
• Toxicology, animals
• Safety in humans(usually first in humans)
• Small scale efficacy
• Large scale trial
• Post marketing
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Regulation of Clinical Trials Material
US FDA• Investigational New
Drug Application (IND)• May “choose” to inspect
generally based on IND review
• Very rare for FDA to inspect even in phase 3
EU EMEA• Investigational Medicinal
Product Dossier (IMPD) / Clinical Trial Application (CTA)
• Obliged to inspect under clinical trial directive(in third country this might be a QP audit rather than competent authority)
• Inspections are common
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCIPCI Pharmaceutical Consulting Israel Ltd. 9/48
Quality Assurance: Definition2003/94/EC
‘pharmaceutical quality assurance’ means the total sum of the organised arrangements made with the object of ensuring that medicinal products or investigational medicinal products are of the quality required for their intended use
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

IND Phase 1 – CMC Requirements
• Drug Substance
– Description (physical, chemical, biological)
– Manufacturer (name and address)
– Method of Preparation (brief description/ flow diagram, reagents, solvents, catalysts)
– Analytical Methods ( brief description, proposed criteria, certificates of analysis)
– Stability (brief description of study/test methods, preliminary tabular data)
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

IND Phase 1 – CMC Requirements
• Drug Product
– Components (grade (e.g. USP/ NF, ACS), novel excipients, etc.)– Quantitative composition– Manufacturer (name and address)– Method of Manufacture (narrative and/or flow diagrams, sterilization process for
sterile products)– Analytical Methods
• brief description of test methods and limits (dosage form dependent)– Stability of Drug product
• Information to assure the product’s stability during the planned clinical studies
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

IND Phase 1 – CMC Requirements
• Placebo (NOTE: EU wants placebo / comparator product released by QP)– Description– Composition and Controls– Control
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

FD&C Act
• 501(a)(2)(B) requires all drug products be manufactured in accordance with current good manufacturing practice (CGMP)
• CGMP Regulations, published in 1978 and codified in 21 CFR 210 and 211 primarily directed to commercial manufacturing of approved drugs and biologics
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

General CGMP Requirements
Quality Control Function:– established for every manufacturer of IND
products– responsibilities documented in writing and include:
• examination of components, containers, closures, in-process materials, packaging and labeling materials
• review and approval of production and testing procedures, acceptance criteria
• review of completed production records for release or rejection of each clinical batch
– is responsibility of all staff involved in production
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Multi-product Facilities
• An area or room can be used for multiple purposes and products, provided that:– only one product is produced in an area at
any given time – appropriate cleaning and change over
procedures are in place to ensure there is no carry-over of materials or products or mix-ups
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Biological and Biotechnological Products
• Additional safeguards– Some production systems may warrant
additional safeguards to protect personnel (e.g., pathogenic microorganisms, some products made from spore-forming microorganisms, live viral vaccines and gene therapy vectors)
– Equipment qualification and controls in production assure success of unit operations with safety-related functions (e.g., viral clearance, virus/ toxin attenuation, pasteurization)
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

FD
A G
uidance, July 2008
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

GMP for Phase I
A. Personnel
B. QC Function
C. Facility & Equipment
D. Component control
E. Manufacturing and Records
F. Laboratory controls
G. Packaging, Labeling, distribution
H. Record-keeping
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

EU Clinical Trials Directive2001/20/EC
• Laws, regulations relating to implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use
(12) The principles of good manufacturing practice shouldbe applied to investigational medicinal products.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

EU Clinical Trials Directive - 22001/20/EC
3. Take all appropriate measures toensure that the qualified person is responsible for ensuring:– (b) in the case of investigational medicinal
products manufactured in a third country, that each production batch has been manufactured and checked in accordance with:
• standards of good manufacturing practice• product specification file• The clinical trial authorisation (CTA)
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

EU Clinical Trials Directive – 3: Article 15Verification of compliance of investigational medicinalproducts with good clinical and manufacturing practice
• Member States shall appoint inspectors to inspect:– the trial site or sites– the manufacturing site of the investigational
medicinal product– any laboratory used for analyses in the clinical
trial– and/or the sponsor's premises.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

EU Clinical Trials Directive – 4: Article 15Verification of compliance of investigational medicinalproducts with good clinical and manufacturing practice
4. The Commission, upon receipt of a reasoned request from a Member State or on its own initiative may propose that the trial site and/or the sponsor's premises and/or the manufacturer established in a third country undergo an inspection. The inspection shall be carried out by duly qualified Community inspectors.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

The meaning of the Directive
• As of 01 May 2004 all manufacturers of IMPs including Phase 1 are open to inspection
• QPs need to demonstrate competence and have required qualifications to certify (NOT release) IMPs
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

The meaning of the Directive – 3Voluntary Inspection Findings
• Voluntary inspections ceased 01/05/04
• Findings:– Poorly developed quality systems– Inadequate facilities– Inadequate validation– Poorly defined QP responsibilities– Poor labeling / packaging practices– Poor TSE / BSE management
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

EU GMP Directive2003/94/EC
• Principles and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use
• “investigational medicinal product” appears 27 times
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Importer’s Responsibility2003/94/EC
• For … investigational medicinal products imported from third countries
• the importer shall ensure that:– products have been manufactured in accordance with
standards equivalent to the good manufacturing practice standards laid down by the Community
– by a manufacturer notified to the competent authorities and accepted by them for that purpose.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Manufacturer’s Responsibility - 32003/94/EC
• Appropriate technical or organisational measures shall be taken to avoid cross contamination and mix-ups.
• In the case of investigational medicinal products, particular attention shall be paid to the handling of products during and after any blinding operation.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Manufacturer’s Responsibility - 42003/94/EC
• For investigational medicinal products, the manufacturing process shall be validated in its entirety in so far as is appropriate:
• taking into account the stage of product development
• At least the critical process steps, such as sterilisation, shall be validated
• All steps in the design and development of the manufacturing process shall be fully documented.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Quality Control2003/94/EC
• For investigational medicinal products, the sponsor shall ensure that the contract laboratory complies with the content of the request referred to in Article 9(2) of Directive 2001/20/EC, as accepted by the competent authority.
• When the products are imported from third countries, analytical control shall not be mandatory.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Quality Assurance / Batch Release2003/94/EC
• During the final control of the finished product before its release..for use in clinical trials, the quality control system shall take into account, in addition to analytical results, essential information such as the production conditions, the results of in-process controls, the examination of the manufacturing documents and the conformity of the product to its specifications, including the final finished pack.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Scope of Annex 16
• This annex …gives guidance on the certification by a Qualified Person (QP) and
• batch release within the European Community (EC) or European Economic Area
• (EEA) of medicinal products holding a marketing authorisation or made for export.
• The relevant legislative requirements are contained in Article 51 of Directive 2001/83/EC or Article 55 of Directive 2001/82/EC
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• Certification of the finished product batch:the certification in a register or equivalent document by a Q.P., as defined in Article 51 of Directive 2001/83/EC and Article 55 of Directive 2001/82/EC, before a batch is released for sale or distribution.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Batch Release Certificate
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Batch Release Certificate• Investigational Medicinal Products may not be used in a
clinical trial in the EEA until completion of a two-step release procedure. The first step is the certification by the Qualified Person of the manufacturer or importer that the provisions of Article 13(a), (b) or (c) of Directive 2001/20/EC have been complied with. insofar as these provisions have been complied with, the IMPs in question shall not have to undergo any further checks if they move into another Member State provided they are accompanied by batch release certification signed by the Qualified Person.
• The GCP and GMP Inspection Services of the Member States have agreed that the content of these certificates should be in accordance with the attached format
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Batch Release Certificate
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Batch Release Certificate
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• The annex covers in particular those cases where a batch has had different stages of production or testing conducted at different locations or by different manufacturers, and where an intermediate or bulk production batch is divided into more than one finished product batch
• It also covers the release of batches which have been imported to the EC/EEA both when there is and is not a mutual recognition agreement between the Community and the third country
• The guidance may also be applied to investigational medicinal products, subject to any difference in the legal provisions and more specific guidance in Annex 13 to the Guide
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• The basic arrangements for batch release for a product are defined by its Marketing Authorisation
• Nothing in this annex should be taken as overriding those arrangements
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• Each batch of finished product must be certified by a QP within the EC/EEA before being released for sale or supply in the EC/EEA or for export
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
The purpose of controlling batch release in this way is• to ensure the batch was manufactured and checked in
accordance with:– Its marketing authorisation– the principles and guidelines of EC Good Manufacturing
Practice or the good manufacturing practice of a third country recognised as equivalent under a mutual recognition agreement and any other relevant legal requirement before it is placed on the marketand
– in the event that a defect needs to be investigated or a batch recalled, to ensure that the QP who certified the batch and the relevant records is readily identifiable
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• in an industrial situation it is usually not possible for a single QP to be closely involved with every stage of manufacture
• The Q.P. who certifies a finished product batch may need therefore to rely in part on the advice and decisions of others
• Before doing so he should ensure that this reliance is well founded, either from personal knowledge or from the confirmation by other QPs within a quality system which he has accepted
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• When some stages of manufacture occur in a third country it is still a requirement that production and testing are in accordance with the marketing authorisation, that the manufacturer is authorised according to the laws of the country concerned and that manufacture follows good manufacturing practices at least equivalent to those of the EC
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• the correct manufacture of a particular batch of product, regardless of how many sites are involved, should be the overall concern of the Q.P. who certifies the finished product batch before release
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• Different batches of a product may be manufactured or imported and released at different sites in the EC/EEA. For example a Community marketing authorisation may name batch release sites in more than one member state, and a national authorisation may also name more than one release site
• In this situation the holder of the marketing authorisation and each site authorised to release batches of the product should be able to identify the site at which any particular batch has been released and the QP responsible for certifying the batch
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16• The Q.P. who certifies a finished product batch before release
may do so based on personal knowledge of all the facilities and procedures employed, the expertise of the persons concerned and of the quality system within which they operate.
• Alternatively he may rely on the confirmation by one or more other QPs of the compliance of intermediate stages of manufacture within a quality system which he has accepted.
• This confirmation by other QPs should be documented and should identify clearly the matters which have been confirmed
• The systematic arrangements to achieve this should be defined in a written agreement in conformance with Chapter 7 (Technical Agreement / Quality Contract)
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• The agreement should include an obligation on the part of the provider of a bulk or intermediate product to notify the recipient(s) of any deviations, out-of-specification results, non-compliance with GMP, investigations, complaints or other matters which should be taken into account by the QP responsible for certifying the finished product batch
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• For assembly at different sites under a single marketing authorisation, there should be one person, normally a QP of the manufacturer of the bulk production batch, with overall responsibility for all released finished product batchesderived from one bulk production batch
• The duty of this person is to be aware of any quality problems reported on any of the finished product batches and to co-ordinate any necessary action arising from a problem with the bulk batch.
• While the batch numbers of the bulk and finished product batches are not necessarily the same, there should be a documented link between the two numbers so that an audit trail can be established
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• QC laboratories authorised under separate marketing authorisation – QP must either take responsibility or have alternative QP assigned as responsible for testing and test results and their sign off
• Same for third country manufacture (“equivalent” GMP)
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 16
• Routine duties of QP prior to batch certification:– Compliance with MA– Compliance with GMP– Manufacturing and testing processes validated– Deviations / planned changes and notification of
CA if needed– Documentation complete– Audits performed– QP’s knowledge up to date
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Retained Samples2003/94/EC
• For an investigational medicinal product, sufficient samples of each batch of bulk formulated product and of key packaging components used for each finished product batch shall be retained for at least two years after completion or formal discontinuation of the last clinical trial in which the batch was used, whichever period is the longer[Normally keep whole batch forever]
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Complaints2003/94/EC
• In the case of investigational medicinal products, the manufacturer shall, in cooperation with the sponsor, implement a system for recording and reviewing complaints together with an effective system for recalling promptly and at any time investigational medicinal products which have already entered the distribution network.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Labelling2003/94/EC
• In the case of an investigational medicinal product, labelling shall be such as to:– ensure protection of the subject and
traceability– enable identification of the product and trial– facilitate proper use of the investigational
medicinal product.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 13 – July 2003Manufacture of IMPs
• Investigational medicinal products should be produced in accordance with the principles and the detailed guidelines of Good Manufacturing Practice for Medicinal Products (The Rules Governing Medicinal Products in The European Community, Volume IV)
• [Rules to be found by entering: EudraLex Volume 4 in search engine]
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 13 cont/
• Packaging and Labeling. Self inspection or independent audits should be used to monitor these critical operations.
• Personnel - separate QA and production (FDA also require this).
• Batch release by qualified person (QP)
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 13 cont/
• Product Specification File
• A reference file containing, or referring to files containing, all the information necessary to draft the detailed written instructions on processing, packaging, quality control testing, batch release and shipping of an investigational medicinal product– constantly updated
– QP needs to be familiar with it prior to certification
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 13 cont/
44. Investigational medicinal products should remain under the control of the Sponsor until after completion of a two-step release procedure: – certification by the Qualified Person– release following fulfilment of the requirements of
Article 9 (Commencement of a clinical trial) of Directive 2001/20/EC
– The sponsor should ensure that these are consistent with the details actually considered by the Qualified Person
– Both releases should be recorded and retained in the relevant trial files held by or on behalf of the sponsor
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Annex 13 cont/
• Manufacturing and Packaging Records
• Production– Starting materials should be well defined and
documented.– Written procedures for generating
randomization codes and blinding.
• QC.
• Complaints, returns, recalls, destruction.
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

ICH Quality Vision (July 2003)Q8, Q9, Q10 – The Trilogy
“Develop a harmonized pharmaceutical quality system applicable across the life cycle of the product emphasizing an integrated approach to quality risk management and science.”
PCI Pharmaceutical Consulting Israel LtdPCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCI Pharmaceutical Consulting Israel Ltd80/60
Definitions: Target Product Profile
• A target product profile is a prospective and dynamic summary of the quality characteristics of a drug product that ideally will be achieved to ensure that the desired quality, and hence the safety and efficacy, of a drug product is realised
• The target product profile forms the basis of design for the development of the product
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..
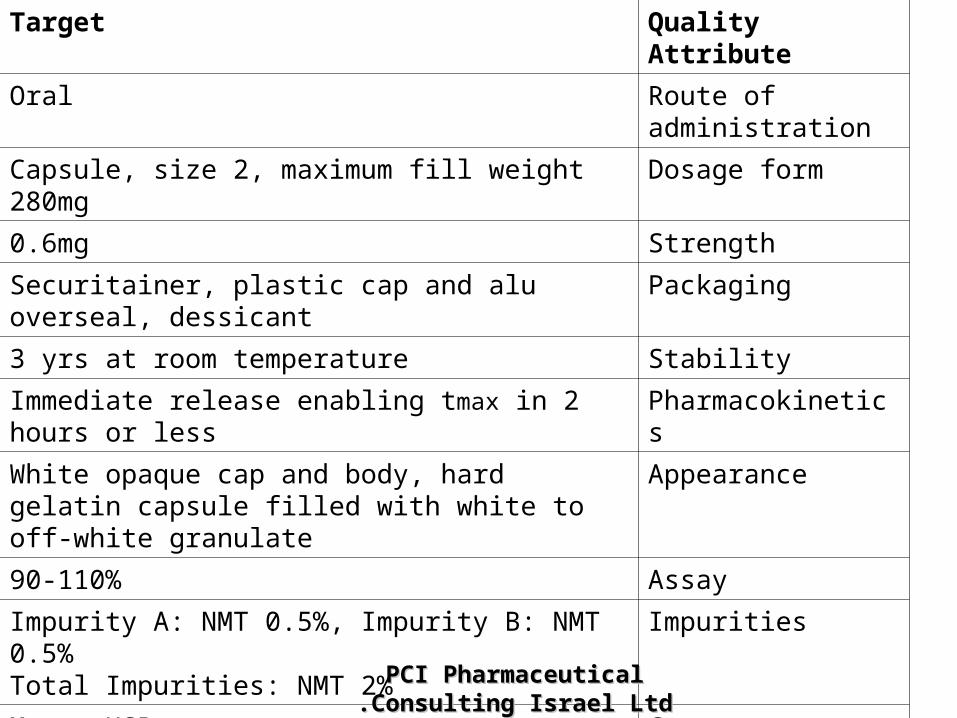
Quality AttributeTarget
Route of administration
Oral
Dosage formCapsule, size 2, maximum fill weight 280mg
Strength0.6mg
PackagingSecuritainer, plastic cap and alu overseal, dessicant
Stability3 yrs at room temperature
PharmacokineticsImmediate release enabling tmax in 2 hours or less
AppearanceWhite opaque cap and body, hard gelatin capsule filled with white to off-white granulate
Assay90-110%
ImpuritiesImpurity A: NMT 0.5%, Impurity B: NMT 0.5%Total Impurities: NMT 2%
Content UniformityMeets USP
DissolutionNLT 70% of labeled amount is dissolved in 30 min : (500 ml water; USP apparatus II {paddles}; 50 rpm)
MicrobiologyMeets USP criteria (NMT 1000cfu /g; 100cfu fungi)PCI Pharmaceutical Consulting PCI Pharmaceutical Consulting Israel LtdIsrael Ltd..

PCI Pharmaceutical Consulting Israel Ltd80/62
Control Strategy: Definition
• A planned set of controls, derived from current product and process understanding, that assures process performance and product quality
• The controls can include parameters and attributes related to drug substance and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control (ICH Q10)
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCI Pharmaceutical Consulting Israel Ltd80/63
Definitions
Critical Quality Attribute (CQA):• A physical, chemical, biological or
microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality
Critical Process Parameter:• A process parameter whose variability has an
impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCI Pharmaceutical Consulting Israel Ltd
Control Parameters vs CQAs
Process Control Parameters– Risk assessment can be used
to identify material attributes and process parameters that can affect CQAs
– Risk assessment tools can be used to identify and rank parameters (e.g., operational, equipment, input material) with potential to have an impact on product quality based on prior knowledge and initial experimental data
– Batch production records are developed with paramter ranges designed to ensure the CQAs are achieved
Critical Quality Attributes– physical, chemical, biological,
or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality
– CQAs are generally associated with the drug substance, excipients, intermediates, and drug product
– Drug product CQAs include the properties that impart the desired quality, safety, and efficacy
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Product Specification File – Annex 13
• Should be continually updated during development to allow traceability to previous versions (audit trail)
• Includes (but not necessarily limited to):– Specifications and analytical methods for starting materials,
packaging materials– intermediate, bulk and finished product– Manufacturing methods– In-process testing and methods– Approved label copy– Relevant clinical trial protocols and randomisation codes– Relevant technical agreements with contract givers– Stability data– Storage and shipment conditions
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Product Specification File – Annex 13
• The contents will vary depending on the product and stage of development
• The information should form the basis for assessment of the suitability for certification and release of a particular batch by the Qualified Person and should therefore be accessible to him/her
• Where different manufacturing steps are carried out at different locations under the responsibility of different
• Qualified Persons, it is acceptable to maintain separate files limited to information of relevance to the activities at the respective locations
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCI Pharmaceutical Consulting Israel Ltd80/67
Risk Assessment in R&D: Control Parameters
Ishikawa (Fishbone) Diagram
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCI Pharmaceutical Consulting Israel Ltd80/68
Q8 R1: Risk Management in R&D
• A cross-functional team of experts could work together to develop an Ishikawa (fishbone) diagram that identifies all potential variables which can have an impact on the desired quality attribute
• The team could then rank the variables based on probability, severity, and detectability using failure mode effect analysis
• (FMEA) or similar tools based on prior knowledge and initial experimental data
• Design of experiments or other experimental approaches could then be used to evaluate the impact of the higher ranked variables, to gain greater understanding of the process, and to develop a proper control strategy
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCI Pharmaceutical Consulting Israel Ltd80/69
Control Strategy
• Controls should be based on product, formulation and process understanding and should include, at a minimum, control of the critical parameters and attributes
• A comprehensive pharmaceutical development approach will generate process and formulation understanding that identifies sources of variability
• Critical sources of variability that can lead to product failures should be identified, appropriately understood, managed or controlled
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCI Pharmaceutical Consulting Israel Ltd80/70
Control Strategy
• Understanding sources of variability and their impact on downstream processes or processing, intermediate products and finished product quality can provide flexibility for shifting of controls upstream and
• minimise the need for end product testing• Control of process parameters allows variability
of raw materials to be compensated for in an adaptable process to deliver consistent product quality
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCIPCI Pharmaceutical Consulting Israel Ltd.
Recommendations for complying with CGMP Requirements - FDA
• Utilize appropriate quality control (QC) standards, i.e.:– well defined procedures– adequately controlled equipment– accurate recording of all data
• Application of QC standards leads to implementation of CGMPs consistent with good scientific methodology

Annex 13 - Principle
• Procedures flexible to provide for changes as knowledge of the process increases, and appropriate to the stage of development of the product
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Quality System - SOPs
• There is a need for a quality system in any functional organization
• A quality system means:– Quality policy– Management commitment– CAPA system (of some description)– Monitoring, evaluation and continual
improvement activities
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Process Performance and Product Quality Monitoring Through Lifecycle
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Application of CAPA Through Lifecycle
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Change Management Through Lifecycle
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Management Review Through the Lifecycle
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCIPCI Pharmaceutical Consulting Israel Ltd.
Recommendations for complying with CGMP Requirements (FDA)
• Use available technology and resources to facilitate product development, CGMP compliance, and lessen CGMP burden, i.e.:– disposable equipment and process aids– prepackaged water for injection (WFI) and
sterilized containers– contract manufacturing and testing facilities

PCIPCI Pharmaceutical Consulting Israel Ltd.
Recommendations for complying with CGMP Requirements (FDA)
• Prevent contamination and cross-contamination:
– evaluate production environment to identify potential hazards
– ensure that substances (i.e. chemicals, adventitious agents) from previous or concurrent research or production are removed

PCIPCI Pharmaceutical Consulting Israel Ltd.
General CGMP Requirements
• Personnel: – education, experience and training, (or any
combination of the three) to perform assigned functions, e.g. training to include GMPs outlined in guidance

Batch Release – Annex 13
• The Qualified Person should be responsible for ensuring that there are systems in place that meet the requirements of this Annex and should therefore have a broad knowledge of pharmaceutical development and clinical trial processes
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Batch Release – Annex 13• IMPs should remain under control of the sponsor until
after completion of a two-step procedure: certification by the Qualified Person and release by the sponsor for use in a clinical trial following fulfillment of the requirements of Article 9 (Commencement of a clinical trial) of Directive 2001/20/EC
• Both steps should be recorded and retained in the relevant trial files held by or on behalf of the sponsor
• The Sponsor should ensure that the details set out in the clinical trial application and considered by the Qualified Person are consistent with what is finally accepted by the Competent Authorities
• Arrangements should be established to meet this requirement e.g. through a change control process for the Product Specification File and defined in a Technical Agreement between the QP and the Sponsor
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

Batch Release – Annex 13• batch records, including control reports, in-process test reports and release reports
demonstrating compliance with the product specification file, the order, protocol and randomisation code
• include all deviations or planned changes, and any consequent additional checks or tests, and should be completed and endorsed by the staff authorised to do so according to the quality system
• production conditions;• the validation status of facilities, processes and methods;• examination of finished packs;• where relevant, the results of any analyses or tests performed after importation;• stability reports;• the source and verification of conditions of storage and shipment;• audit reports concerning the quality system of the manufacturer;• Documents certifying that the manufacturer is authorised to manufacture investigational
medicinal products or comparators for export by the appropriate authorities in the country of export;
• where relevant, regulatory requirements for marketing authorisation, GMP standards applicable and any official verification of GMP compliance;
• all other factors of which the QP is aware that are relevant to the quality of the batch
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..

PCIPCI Pharmaceutical Consulting Israel Ltd.
General CGMP Requirements (FDA)
• Facilities:– adequate work areas for intended tasks– water of appropriate source and quality– adequate air handling to prevent
contamination and cross-contamination

PCIPCI Pharmaceutical Consulting Israel Ltd.
General CGMP Requirements (FDA)
• Equipment:– in proper working condition, maintained
calibrated, cleaned and sanitized at appropriate intervals
– appropriate for intended function– should not contaminate or be reactive,
additive or absorptive with product– identified and documented in production
records

PCIPCI Pharmaceutical Consulting Israel Ltd.
General CGMP Requirements (cont’d.)
• Control of Components:– written procedures describing handling and
control of components– written, specified attributes or acceptance
criteria– COA or other documentation for components
to ensure conformance with specified attributes
– records of receipt, quantity, supplier’s name, lot number, expiration date

PCIPCI Pharmaceutical Consulting Israel Ltd.
General CGMP Requirements (FDA)
• Laboratory Controls:– tests conducted using established written
procedures under controlled conditions– scientifically sound analytical procedures– properly calibrated and maintained analytical
lab equipment– initiate stability study to support use of
product in clinical investigation

PCIPCI Pharmaceutical Consulting Israel Ltd.
General CGMP Requirements (FDA)
• Container Closure and Labeling:– Package to protect product from alteration,
contamination and damage during storage, handling and shipping
– Controlled labeling to preclude mix-ups
• Distribution: – Describes the transport of the IND product
from the point of production to the patient/subject for consumption

PCIPCI Pharmaceutical Consulting Israel Ltd.
General CGMP Requirements (FDA)
• Recordkeeping– Retain records related to quality and operation of
production processes including:• Equipment maintenance and calibration• Production records and related analytical test
records• Distribution records• All quality control functions• Component records
– Retain records for 2 years after approval of marketing application or until 2 years after shipment and delivery of the product is discontinued and FDA is notified

PCIPCI Pharmaceutical Consulting Israel Ltd.
Multi-product Facilities
• An area or room can be used for multiple purposes and products, provided that:– only one product is produced in an area at
any given time – appropriate cleaning and change over
procedures are in place to ensure there is no carry-over of materials or products or mix-ups

PCIPCI Pharmaceutical Consulting Israel Ltd.
Sterile/Aseptically Processed Products
• Investigational products intended to be sterile require specific precautions, for example:– personnel should be properly trained in aseptic
techniques – aseptic manipulation should be conducted in a
Class 100 environment e.g. laminar flow hood – controls should be in place to assure appropriate
air quality of the aseptic environment

Contractors
• Don’t forget that a lot of work is outsourced
• Quality Agreements / Technical contracts are essential – with checklists
• Batch release should always be based on a checklist to ensure relevant documentation is reviewed
• Procedures in Quality Agreement re: recalls, complaints and especially for comparator and placebo
PCIPCI Pharmaceutical Consulting Israel LtdPharmaceutical Consulting Israel Ltd..