implementation and characterization of protein folding on ... · implementation and...
TRANSCRIPT

ImplementationandCharacterizationof ProteinFoldingona DesktopComputationalGrid
Is CHARMM asuitablecandidatefor theUnitedDevicesMetaProcessor?
B. Uk1, M. Taufer1, T. Stricker1, G. Settanni2, A. Cavalli2
1 Departmentof ComputerScience 2 Departmentof BiochemistryETH Zurich Universityof Zurich
CH-8092Zurich,Switzerland CH-8057Zurich,Switzerlandbuk,taufer,[email protected] settanni,[email protected]
Abstract
CHARMM is a popularmoleculardynamicscodefor computationalbiology. For many CHARMM appli-cationssuchasproteinfolding, desktopgridscouldbecomeviablealternativesto clustersof PCs.In this technicalreport,we presenta prototypeanddiscussthe viability of a protein folding applicationwith CHARMM on the United DevicesMetaProcessor, a platform for widely distributedcomputing. Weidentify thealgorithmicapproachof proteinfolding asa hybrid searchalgorithmwith best-first,depth-firstandbreadth-firstcomponentsandaddresstheissuesof taskschedulingandfault tolerance.Theperformanceevaluationof our systemindicatesthat thecalculationis robustagainsttheheterogeneityof computenodesandlimited communicationcapabilitiestypically found in desktopgrids. We show thatthereis aninterestingtradeoff betweenaccuracy andtaskparallelismresultingin optimalwork-poolsizeforagivenplatformandagivensimulation.Surprisinglytheplatformheterogeneityof adesktopgrid positivelyaffectsthequality of proteinfolding simulations.Proteinfolding calculationswith CHARMM turn out to be well suitablefor desktopgrids like e.g. theUnited Devices MetaProcessor. Our software systemcan make a large amountof nearly free computecyclesavailableto computationalbiologists.
Keywords: proteinfolding, widely distributedcomputing,desktopgrids,workloadcharacterization,searchalgorithms,CHARMM, UnitedDevicesMetaProcessor.
1 Introduction
Millions of PCsare in useeverywherebut remainunder-usedmostof the time, utilizing only 18% of theirpower on averageduring normaluse(word processing,web browsing, e-mail etc.) [3, 31, 23]. At the sametime,somecomputationallyintensive problemsaretoo largefor eventodayslargestsupercomputersto solve inreasonableamountsof time. Wefind examplesof suchcomputationsin many fieldsof computationalsciences,cryptographyandmathematics.Thegoalof a wide varietyof technologieslike Globus [11, 12], Condor[2], EU DataGrid[27, 16], andGri-PhyN [10], is to transformthe Internetinto a powerful computationalplatform. Moreover, a significantin-dustrialeffort will provide standardsoftwarefor similar systemsin thenearfuture (e.g. Entropia[5], UnitedDevices[1]).
1

Many projectstry to utilize theunused computing power availableontheIntranetor theInternetfor large-scaleproblems.The possibility of harnessingthepower of idle processorsin a company or all over theworld hasbeenproven by the successof technologydemonstratorslike Seti@home for the analysisof radio telescopedata[24] or distributed.net for testingcryptographickeys againstsecurityflaws [7]. Theseprojectsinvolveso-calledembarrassinglyparallelproblems,meaningthateachparticipatingnodeperformsa lot of work thatisnotdependenton thework of othernodes.Proteinfolding researchis anareaof computationalbiology thatcouldgreatlybenefitfrom freecomputationalpower. Proteinfolding simulationsusingmoleculardynamicsrequirehugeamountsof computingpower, butunlike cryptographickey-spacesearchesor cosmicsignalanalysisthey arenotembarrassinglyparallel.Previous researchaddressesthe issueof protein folding simulationson distributed platforms,but no projectexplicitly investigatestheamountof taskparallelismavailableandtheeffect of taskingon theaccuracy of theresults.CHARMM onthedistributedcomputingplatformLegionhasbeenstudiedin [20]. Theimplementationdescribedtherespecializesin high-performance,stronglyinterconnectedclustersanda fairly smallnumberoftasks.Ourwork appliesmorebroadlyto desktopgridsbuilt from commodityPCsconnectedby a wide varietyof strongerandweakernetwork technologies.TheFolding@home projectconductedby researchersatStanfordUniversity[9, 35, 34] is basedon theTINKER moleculardynamicspackageandusesplain taskparallelismonPCsconnectedvia Internet. For a first implementationthesupercomputercodewasre-designedso that onlytrajectoriesovercomingenergy barrierswerecontinued[25]. The resultingstudydealtwith a collectionoffoldingsof a16residueβ-hairpinpeptide[35]. In amoreadvancedimplementationthesameteamremovedalldependenciesamongtrajectoriesresultingin amostscalablecodedesignedto collectasmuchsimulationtimeaspossible[34]. The latterversionof thecodewasappliedto theunfoldedstateof 3 smallproteins(rangingfrom 12 to 36residues)andnoneof theproteinsshowedclearfolding eventsin thesimulatedtime.As a startingpoint to the work describedin this technicalreport,we alsouseplain taskparallelismto suc-cessfullymigratetheCHARMM codefor a proteinfolding simulationto thewidely distributedplatform[30]of United Devices(UD): the UD MetaProcessor. However, in our studyof the viability andeffectivenessofCHARMM on theUD MetaProcessorwe focusour attentionon thewholecomplex systemincludingboththeplatformandtheapplicationparameters.In this first prototypeof theproteinfolding application,we carefullystudyissueslike theeffect of theheterogeneityof thecomputationplatformunderinvestigationandtheeffectof severalworkloadparameters,suchasthenumberof moleculardynamicsstepsperwork-unit (or work-unitsize)andthemaximumnumberof work-unitsin progressat any time (i.e. work-poolsize).As aresultof this studyonworkloadcharacteristicsandapplicationperformance,we find thattherearesignif-icant limitationsof thescalabilitydueto thelimited amountof taskparallelismgeneratedby a proteinfoldingcalculation. In particular, large work-unit sizesproviding simulationswith betterquality factors,andcalcu-lation methodssuchasPME providing higheraccuracy, both increasetheamountof work perwork-unit andthereforetheturn-aroundtimeof asingletask.On theotherhand,simulationswith ashortoverall turn-aroundtimemake thebiologistsmoreproductive in their research.In Section2, we briefly introducethe protein folding processusing CHARMM code. We also presenttheUnited DevicesMetaProcessor, a commercialplatform for widely distributing computingthat we usedasagrid platform in our investigation. We show the adaptationandthe migrationof the proteinfolding processto our platformfor widely distributedcomputingandidentify thealgorithmicapproachof proteinfolding asahybrid searchalgorithmwith best-first,depth-firstandbreadth-firstcomponents.In Section3, we presentourexperimentalresultsof the studyof critical issueslike the effect of heterogeneity, work-unit sizeandwork-pool sizeon theexecutiontime andthequality of the resultsfor theproteinfolding of a specificprotein,theSH3domain.In Section4, we concludesummarizingour resultsandpointingout somerelevant limits of taskparallelismfor proteinfolding applicationonsuchawidely distributedplatform.
2

2 Protein Folding on Computational Desktop Grid
2.1 Protein Folding with CHARMM
CHARMM is a codefor simulatingthe structureof biologically relevant macromolecules(proteins,DNA,RNA) [4]. It usesclassicalmechanicalmethodsto investigatepotentialenergy surfacesderived from experi-mentaland”ab initio” quantumchemicalcalculations[18]. WeuseCHARMM for MolecularDynamics(MD)simulationsat constanttemperatureto investigatethe proteinfolding process,in which the Newton equationof motion of the system(protein+ thermalbath)is discretizeandsolved by an integrationprocedure(Verletalgorithm).Theforceon theatomsis thenegative gradientof theCHARMM potentialenergy [18].Basically, in aproteinfolding simulationweperformasearchfor trajectoriesleadingto conformationsclosetothenative (folded)conformationof a protein,startingfrom anunfoldedconformation.An entiretrajectoryforwhich a certainamountof sub-trajectoriesor work-unitshave beencompletedis characterizedby its startingconformationwith a startingquality factor, aswell asthebestcalculatedquality factorfor theentiretrajectorywith therelatedconformationandthethesetof work-unitsleadingfrom thestartingconformationto thebestconformation.In eachwork-unit, an MD simulationof a certainnumberof moleculardynamicssteps(or work-unit size)andwith constanttemperatureis performed.Theentirefolding processis guidedby themeasureof a qualityfactor, which is repeatedcalculatedfor a certainamountof snapshotsalongeachwork-unit computation.Thebestquality factorfound in a sub-trajectory(lower quality factorsarebetter!) guidesthe overall branchandboundsearchfor the bestconformation. The bestconformationwith lower quality factor is returnedat theendwhen the work-unit is completed. The quality factor usedhereis the φ-RMSD introducedin [33]. Inour implementationwe usea slightly differentdefinition[15]. Accordingto this definition,theconformationsminimizing thequality factorshouldbelongto thefolding transitionstateensemble(i.e. they lie on thetop ofthefreeenergy barrierseparatingthefoldedfrom theunfoldedpopulation).During a proteinfolding simulation,multiple work-unitsareprocessedsimultaneouslyin parallel.New work-unitsarealwaysgeneratedusingthebestresultobtainedso far asstartingconformationandassigningto thisconformationnew randomatomvelocitiesat thesametemperature.Theeffectivenessof thiskind of algorithmsin moleculardynamicssimulationshasbeendiscussedin [26].
2.2 The United Devices MetaProcessor
TheUnitedDevices(UD) MetaProcessorplatform(MP platform)[31, 32] providesanenvironmentfor runningcomputeintensive tasksdistributedover many desktop-classmachinesin a corporate-wideIntranetor over theworldwideInternet.TheMP platformis well establishedfor problemswith low communicationto computationratio and involving coarse-grainparallelismwith no dependenciesbetweenwork-units. Figure1 shows theMetaProcessorplatformarchitectureandcomponents.TheMP server is thelink betweentheMP platformandtheparticipatingagents.It is responsiblefor scheduling,aswell asfor the distribution of taskmodules,residentdataandwork-units to the agents,andfor receivingresultsreturnedby the agents. The managementserver allows accessto the internaldatastructuresvia theManagement API (Application ProgrammingInterface),allowing for instancethe submissionof work-unitsandretrieval of results.Thedatabasecontainsall relevant informationof theMP platform. Thedatabase,MPserver andManagementserver form theserver sideof theMP platform.TheUD agentis asmallprogramwhichrunsoneachparticipatingdevice(or worker). Theagentcommunicateswith theMP server to requestwork-units,residentdataandtaskmodulesasneeded,executesthetaskmoduleontheparticipatingdevice,andreturnsthetask’sresultsto theserver. Themanagementconsoleprovidesaweb-basedinterfaceto theMP platform,allowing administrative tasksto beperformed.Work-units aresubmittedto
3

Figure1: MetaProcessorplatformoverview.
theserver via theManagementAPI by a controller process which performsthegenerationof work-unitsandtheretrieval of results.
2.3 Porting Protein Folding to the UD MetaProcessor
Many uniquepropertiesof the UD MetaProcessorplatform mustbe taken into accountto successfullyporta protein folding applicationto this distributed platform. An application-specific controller processwhichgenerateswork-unitsandsubmitsthemto theMP server hasto bedesignedandimplemented.However, thiscontrollerdoesnot interactdirectly with theworkers. Workerscommunicateonly with theMP server, whichperformstheschedulingdecisions.For our protein folding application,we implementedsucha controllerprocess,taking into accountboth theissuesrelatedto theproteinfolding listedin theprevioussectionandthefeaturesof theUD MetaProcessor.At the beginning of a protein folding simulation,our controllerprocesscreatesandsubmitsa setof startingwork-unitsto the MP server (initialization phase).Thesework-unitsall have the sameproteinconformationbut adifferentrandomseednumber. Thework-unitsarealsocharacterizedby astartingbestquality factor. Thework-unit setis definedasthe work-pool. The userdecidesthedimensionof thework-pool. Thenumberofwork-unitsin thework-poolis calledthework-pool size.During thewholesimulation,besidesthesetof work-unitswhich have beencompletedandfor which resultshave alreadybeenreturned,we have thesetof work-unitswhich areeitherin processby a worker or waitinguntil they areassignedto a worker. At regular intervalsduringa socalledupdatephase,thestateof thework-pool is inspectedby thecontrollerprocess.Thecontrolprocessis responsiblefor generatingandsubmittingtothework-pooladditionalwork-unitsin substitutionto theoneswhich have reachedtheendsothat thesumofthework-unitsin processby a worker andtheonesin waiting in theserver queueis the work-pool size. The
4

userdecidesthedimensionof theupdateinterval. For thegenerationof new work-units,thecontrol processsortsoutwhetherbetweenthesetof work-unitswhichhave reachedtheendsincethelastupdate,thereareanywork-unitswhichimprovethebestquality factorat thattime. In casethereis awork-unitwith improvedqualityfactorthenthenew work-unitsaregeneratedusingits conformationwhile the relatedquality factorbecomesthenew bestquality factor. In thecasethatno improvementhasbeenfound,thelastbestconformationis usedfor generatingthework-unitsto addto thework-poolwhile thebestquality factorremainsunchanged.For schedulingthework-units(tasks)we incorporatethemaster-worker settingprescribedby theUD MetaPro-cessorsoftware toolkit becauseit is well suitablefor the task parallelismresultingfrom the applicationofCHARMM to proteinfolding.Oncenew work-unitsarequeuedto theMP server in its work-pool, it is theserver which distributesthenewwork-unitsandtheonesalreadypresentin thequeueacrosstheagentsin thedistributedsystem.Theschedulingdecisionsoverwhich taskto selectnext areleft to theUnitedDevicesMetaProcessorandin thecurrentversionof thesoftwarethereis little influenceleft to theapplicationwriter.The schedulerin the MP server providesso-calledeager scheduling, which basicallymeansthat as long astheserver haswork-unitsqueuedthatarenot beingprocessedby otherworkers,it will sendout oneof thesework-unitsto aworkerrequestingwork [17]. A workeronwhichafinishedwork-unithasbeenrunningasksforandreceivesanew work-unit from theMP server. If however, all work-unitshavealreadybeensentto workers,theserver will re-sendwork-unitsfor whichno resultshave beenobtainedyet. Thiseagerschedulingprovidesasimplemethodfor fault tolerance,i.e. if aworkerhascrashed,eventuallythework-unit it wasprocessingwillgetresentto adifferentworker. This schemecanalsohave negative effects,for instancecausingwork-unitsofslow machinesto berescheduled,resultingin redundantresultsandthusapotentialwasteof computationtime.Sothereschedulingfeatureof theMP platformis in factnotdesirable.To avoid reschedulingof work-units,wetake a simpleapproachby ensuringthat thesizeof thework-pool is alwayslarger thanthenumberof workerscontributing.Sincethestartingconformationof eachnewly generatedwork-unitdependson theresultsreturnedsofar, therearedependenciesbetweenwork-units,makingthetaskgenerationandschedulingof thisapplicationabit morechallengingthantypical “embarrassinglyparallel” caseof astaticallygeneratedsearchapplication.Theproteinfolding algorithmusedis in itself fault tolerant, becausethework-unitsgeneratedfrom a certainconformationare identical except from a randomnumberseedthat is usedto assignnew atom velocities.Thereforetheresultsareonly marginally affectedby theinfrequentlossof anonlinework-unit.
2.4 Protein Folding as a Search Algorithm
The processof protein folding for a given protein is a searchthrougha treeof a large numberof possibleconfigurationof the atomsin the protein, a so called conformation,underinvestigation. Eachnodein thetreecorrespondsto a differentconfigurationof theatomsin themolecule.For eachconfigurationthereis anheuristicfunctionto guidefurtherdecisionsin thesearch.This functioncalculatesthequality factorbasedonthetotalenergy of theatomconfiguration.In theprocessof lookingfor bestconformations,weperformasearchin breadthandin depthonatreeof three-dimensionalatomconfigurations.Correspondinglythereare two kinds of changesto the three-dimensionalatomconfigurations.
2.4.1 Energy minimization as search in depth
Thefirst kind of changesaresmoothchangesanddueto themechanicalforcesbetweentheatoms.Theprocessof calculationfor thesechangesis a physicalsimulationcalledenergy minimization.A sub-trajectoryof stateswithin suchanenergy minimizationis a linearchainwithout any branchesandis encapsulatedin a work-unit
5

or a task, whichwill beourbasicquantumfor thedistribution andtheschedulingof theproteinfolding compu-tation. Sinceit is too expensive to evaluateevery configurationduringenergy minimization,thecalculationofthequality factoris limited to certainsnapshotsduringtheenergy minimization.In thecalculationsconsideredfor this report,snapshotsweretakenevery 100simulationsteps.
Figure 2: Quality factor along the sub-trajectorycomputedby awork-unit (50’000steps,asnapshotevery 100steps).Thesnapshotwith thebestqual-ity factor(minimum)is at thebeginning.
Figure 3: Quality factor along a sub-trajectorycomputedby awork-unit (50’000steps,asnapshotevery 100steps).Thesnapshotwith thebestqual-ity factor (minimum) is in the secondhalf of thesub-trajectory.
Figures2 and3 show examplesof thepaththe quality factortakesalongtwo differentsub-trajectoriesof anenergy minimizationwith thesamework-unit sizeof 50000steps.Onepropertyof this simulationis that it isnot possibleto predictif or whenan improvementof thequality factorwill occur. Specificallyin our casesasimulationconcludesaftera fixednumberof stepsbut in generaladaptive criteriabasedon thequality factorsarealsosupported.The resultof the simulationis the snapshotof configurationwith the bestquality factorobtainedwithin theentiresub-trajectory. At a higherlevel of abstraction,theprocessof energy minimizationfor asingleconformationcandidatecanbecollapsedinto asingleedgein thehigherlevel searchtree.
2.4.2 Randomized conformations as search in breadth
The searchin breadthis triggeredby a secondkind of changesto the configurationsof a moleculeunderinvestigation.Thefolding processintentionallyintroducesdisturbancesto themolecularsystemby reassigningtheatomvelocitiesof aconformationwith randomnumberswhile preservingthetemperature.Eachwork-unitgeneratedin theprocessof randomizationreceivesa differentrandomseed.Multiple work-unitsaregeneratedfrom onesingleconformationusingdifferentseeds.This resultsin thebranchesof abreadth-firstsearch.Figure4 showsapossibleconfigurationof thesearchtree.In thepicture,wehavethreeupdatephasesfor whichnew work-unitsaregeneratedandqueuedin awork-pool.In Figure4, theproteinconformation(Mol1) in thefirst updateis thebestfoundat thattimeandweassumethatanemptywork-poolis fully completedby new work-unitseachonewith adifferentrandomseed.In thesecondupdatephaseonly two work-unitshave reachedtheendwhile two othersareeitherin progressby workersorwaiting at the MP server. Betweenthe work-unitswhich reachthe end, the conformationwith bestqualityfactor(Mol2) is chosenandtwo new work-unitsaregeneratedandaddedto the server queueby the controlprocess.Note that the work-unitsnot finishedyet remainin the work-pool queue. During the third update,
6

Mol_1
Mol_2
Mol_3
80
94
100
r1r2
98r3 r4
r6
95
r5
93
r7
r8
r10
Work−pool:
for protein foldingtime
upda
teup
date
upda
teMol_1,r1 Mol_1, r2 Mol_1,r3 Mol_1,r4
Mol_3,r7 Mol_3, r8 Mol_3,r9 Mol_3,r10
Mol_1,r3 Mol_1, r4 Mol_2,r5 Mol_2,r6
96
r9
Figure4: Exampleof a pieceof a searchtreefor a proteinfolding simulationin which for eachupdatephasenew work-unitsaregeneratedfrom improvementsof thequality factorobservedandqueuedin awork-pool.
all the four work-poolsarefinished. This time the bestconformationfound (Mol3) is usedto addfour newwork-unitsto thework-pool.
Mol_3
80Mol_3,r7 Mol_3, r8 Mol_3,r9 Mol_3,r10
Work−pool:
up
da
teu
pd
ate
88 84
r7r8
r9 r10 Mol_3,r9 Mol_3,r10
time
for protein folding (a)
Mol_3
80Mol_3,r7 Mol_3, r8 Mol_3,r9 Mol_3,r10
Mol_3
80
Work−pool:
up
da
teu
pd
ate
r9 r10
time
for protein folding
r11 r12
Mol_3,r9 Mol_3,r10 Mol_3,r11 Mol_3,r12
(b)
Figure5: Exampleof a pieceof a searchtreefor a proteinfolding simulationin which at theendof anupdatephaseno improvementof thequality factorcanbeobservedfor thework-unitscompleted.
Figure5.ashows the casein which no improvementof thequality factorcanbe observed for the work-unitscompletedbetweentwo updatephases.In thiscase,thetwo new conformationsarediscardedandMol3, whichis still the conformationwith the bestquality factor, is usedagainas the startingconformationof the twonewly generatedwork-units(Figure5.b). If thissituationoccursrepetitively, theproteinfolding simulationhasreacheda local minimum,andnew strategiesareneededto escapethisminimum.
7

2.4.3 Parameters affecting the search process
Thebranchingfactorof thesearchin breadthis adaptive anddependson thework-pool sizeandthenumberof taskcompletedin themeantime, while thesearchin depthdeterminesprimarily thewaiting time andturn-aroundtime of thework-unitsin thequeues.Thewaiting time dependson theratio of thework-poolsizeover thenumberof workersthatareavailableona particularplatform. Theturn-aroundtime of thesinglework-unitsis dueto thework-unit sizeaswell astheCPUclock ratesandthenetwork technologiesof thenodeson theheterogenousplatform.
3 Characterization of Protein Folding on UD MetaProcessor
3.1 The Biological Structure Folded
As anexampleof workloadfor our studywe chosethesrc-SH3domain. It is a 56 residueproteinconsistingof two anti-parallelβ-sheetspacked to form a singlehydrophobiccore(seeFigure6). The folding character-isticsof src-SH3domain(andstructurallyhomologousproteins)have beenthoroughlystudiedboth from theexperimentalandthetheoreticalpointof view [6, 13, 19, 22], andadetailedpictureof thefolding nucleus[21]andthetransitionstateensemble[22] is now available. TheSH3domainrepresentsa sortof testingtablefortheoriesor algorithmsdealingwith proteinfolding. Its shortlengthandtheabundanceof comparablestudiesmake this proteintheoptimal target for our computations.Thesizeof this proteindoesnot allow to simulateits behavior with an explicit treatmentfor water; neverthelessthe implicit solvation modelusedhere,that isbasedon thesolventaccessiblesurfaceareaof theatoms(SASA) [8] successfullydescribedseveralaspectsofthedynamicpropertiesof thisprotein[13, 14].
Figure6: Thesrc-SH3domainis a56 residueproteinconsistingof two anti-parallelβ-sheetspackedto form asinglehydrophobiccore.
3.2 Platform and Application Parameters
We usedifferenttestbedconfigurationswith machinesat ETH Zurich andat theprivateresidencesof severalresearchersfor studyingthe effect that platform and applicationparametershave on quality and total turn-aroundtime of theproteinfolding of theSH3domain.
8

First of all, we investigatethe impactof the heterogeneityon the quality of the protein folding results. Inparticular, we look at thekind of CPUclock ratesandnetwork technologyasa representative of theplatformparametersof theheterogenouscomputationalgrid.We also study the effect of aspectsdirectly relatedto the application(i.e. applicationparameters)like thework-unit sizeandwork-poolsizeon thequalityandturn-aroundtimeof theproteinfolding.
3.3 Effect of Heterogeneity on the Quality Parameter
3.3.1 Testbed for the study of heterogeneity effect
Two long-runningtestsusingmachinesavailablearoundtheclockonafirst testbedareconductedoveraperiodof 20 days.Themachinesrangein CPUclock ratefrom 350MHz to 1.6GHz, providing a wide performancespectrum.Also, a widely varyingrangeof network connectivity technologiesarein use,from machinesin thesameLAN segmentasthe server (100 Mbit/s switchedEthernet)to machinesat homeconnectedvia ADSL(128kbit/s to 512kbit/s).Most of the machinesinvolved areusedduring the courseof the testsfor normalday-to-daytasks. Becausethe UD agentspawns the taskmoduleprocessesat low priority, only otherwiseunusedprocessorcyclesareconsumedby thetests.Thefollowing applicationparametershave beenchosenfor thesetests:
� 100’000simulationstepsperwork-unit,
� platformsize:45 machines,
� awork-poolof 50 work-units,
� polling frequency: onceevery 5 minutes.
Thetwo testsaresubjectto a numberof unforeseendisruptions(e.g.network failures,failureof machines,in-cludingthemachinerunningthecontrollerprocess,server’sdiskfull), whichcausethetestto stalloccasionally,exhibiting many propertieswhichusuallycharacterizerealwidely distributedgrid platforms.
3.3.2 Experimental results of heterogeneity effect
An importantquestionin aheterogeneousenvironmentlike theonewearestudying,is whetherslow machinesareableto provide enoughcontributionsto theoverall computationto justify their participation.In our case,work-unitsaregeneratedbasedon the resultsof previous work-units. The time at which a result is returnedis thereforea parameterwhich cannot be ignored. It is possiblethat,during the processingtime of a work-unit on onemachine,the stateof the entiresystemhasprogressedso that the resultof the work-unit of theconsideredmachineis obsoleteand thereforenot usedfor the generationof new work-unitsconfigurations.If slow machinesreturna disproportionatelylarge amountof obsoleteresults,their contribution to the entirecalculationwill belessvaluable.Thelong-runningtestsshow thatresultsreturnedby all machines,fastandslow, areacceptedasgoodconfigu-rations.Figures7 and8 show theCPUclock ratefor eachresultwhichwasacceptedin thelong-runningtests.Most resultsarefrom machineswith CPU clock ratesbetween910 MHz and1010MHz. This is dueto thefact thatmany suchmachinesareused,andthusthetotal numberof work-unitscompletedby thesemachineswaslarge. Nevertheless,slower machinesmake contributionsduringall phasesof thesimulation.NotethatinFigure8, no machinefasterthan1010MHz participatedin thecalculation.
9

Figure 7: CPU clock rate of nodesreturningac-ceptedresults,for thefirst long-runningtest.
Figure 8: CPU clock rate of nodesreturningac-ceptedresults,for thesecondlong-runningtest.
3.4 Effect of Work-Unit Size on the Execution Time and the Quality Parameter
3.4.1 Testbed for the study of work-unit size effect
An importantparameterof our simulationsis thework-unit size. To studytheeffect of the work-unit size onthe quality factorandthe resourceusage,a numberof short-runningtestsof 12 hoursareconductedon twotestbedconfigurationswith differentnumbersof machinesconnectedby 100 Mbit/s switchedEthernet.Thefirst testbedcomprises:
� 12 machines400MHz PentiumIII (Katmai)
� 12 machines500MHz PentiumIII (Katmai)
� 12 machines600MHz PentiumIII (Katmai)
� 12 machines933MHz PentiumIII (Coppermine)
� 12 machines1000MHz PentiumIII (Coppermine)
Thesecondtestbedusesasubsetof theabove listedmachinesandcomprises:
� 6 machines400MHz PentiumIII (Katmai)
� 6 machines500MHz PentiumIII (Katmai)
� 6 machines600MHz PentiumIII (Katmai)
� 6 machines933MHz PentiumIII (Coppermine)
� 6 machines1000MHz PentiumIII (Coppermine)
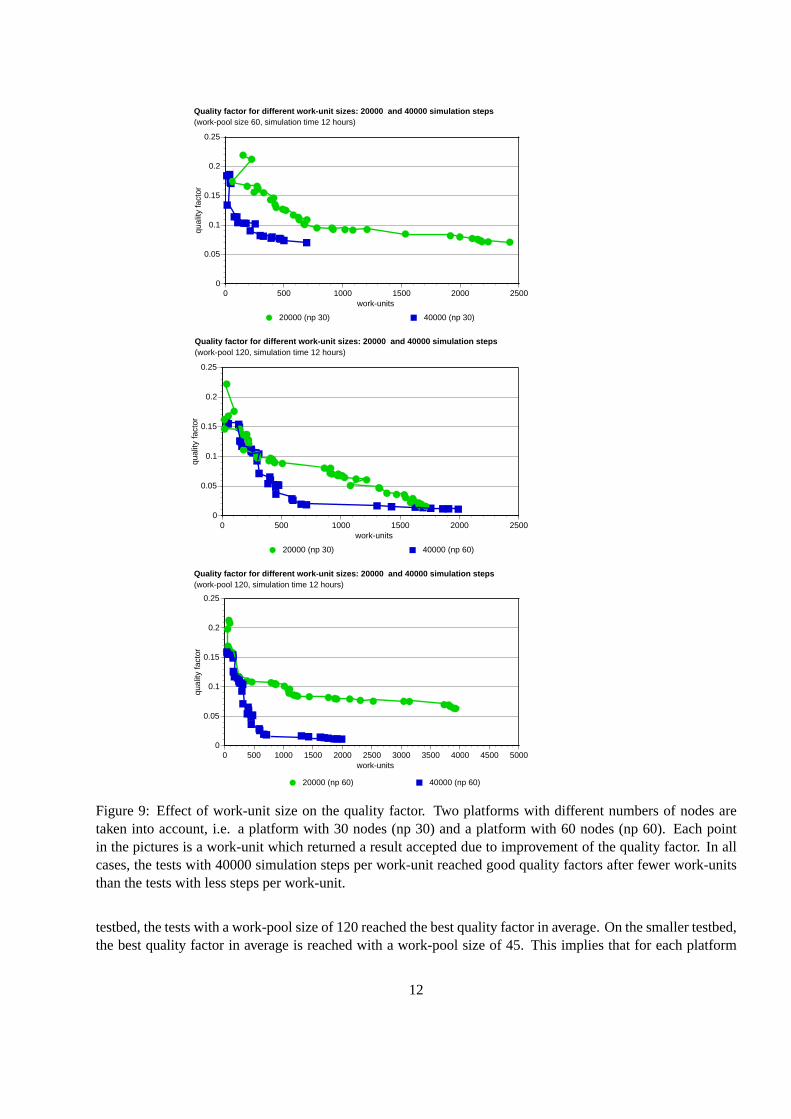
In thesetests,two differentwork-unit sizesareconsideredandtheir effect on theturn-aroundtime aswell asthequality factoris investigated.Thework-unit sizesare:20000simulationstepsand40000simulationstepsperwork-unit.
10

3.4.2 Experimental results of work-unit size effect
Figure9 shows theresultsof thequality factorfor the two differentwork-unit sizechosen(20000and40000simulationsteps). The testswith 40000stepsreachgood quality factorsafter fewer processedwork-units.Althoughtheothertestswith 20000stepsalsoreachedsimilarquality factorsafterlargeramountsof processedwork-units, in two of the threecasespresentedin Figure9, the testwith 40000stepsstill obtainedthe bestquality factorat theend.The amountof datacommunicatedfrom theserver to theworker perwork-unit for the applicationchosenisaround15 kilobytes. Theamountof datareturnedby theworker to theserver dependson whetherthework-unit simulationactuallygeneratesanimprovementof thequality factor. If this is thecase,theworker sendstheentiresub-trajectoryto theserver. Figure10 shows thedatacommunicatedin kilobytesfor differentwork-unitsizesfor this case.In caseof no improvementof thequality factorby a work-unit computation,which is themostcommoncase,theamountof datareturnedto theMP server is drasticallyreduced.The applicationperformanceis limited by the CPU clock rate. As the work-unit size increases,we haveobservedanunexpectednon-linearriseof thetotalfloatingpointoperationsperwork-unit. Figure11shows theamountof floatingpointoperationsfor differentwork-unitsizesandtherelatednon-linearbehavior (thedashedline shows thelinearbehavior). Thedatarelatedto theresourceusagewasmeasuredby meansof a frameworkfor monitoringperformanceon limited wideareatestbeddevelopedby ourgroup[29, 28].
3.5 Effect of Work-Pool Size on the Execution Time and the Quality Factor
3.5.1 Testbed for the study of work-pool size effect
Differentvaluesfor thework-poolsizeondifferentplatformsizesarechosento investigatethework-pooleffecton thequality factor. For theinvestigationof theeffect of thework-pool size, a numberof short-runningtestsof 12 hoursareconductedon two differenttestbedconfigurationseachonecharacterizedby a differentsubsetof work-poolsizes.Both the testbedconfigurationsareconnectedby 100Mbit/s switchedEthernetwhile thework-poolsizeis constant(10000simulationssteps).Thefist testbedcomprisesthefollowing machines:
� 12 machines500MHz PentiumIII (Katmai)
� 12 machines600MHz PentiumIII (Katmai)
� 24 machines933MHz PentiumIII (Coppermine)
Thework-poolsizesconsideredfor thefirst testbedare:70,120,200.Thesecondtestbedhasthefollowing structure:
� 12 machines400MHz PentiumIII (Katmai)
� 12 machines1000MHz PentiumIII (Coppermine)
Thedifferentwork-poolsizesconsideredfor thesecondtestbedare:30,45,60.
3.5.2 Experimental results of work-pool size effect
Figure 12 shows the minimum, averageand maximumquality factorsobtainedusing threedifferent levelsof the work-pool size for the short-runningtestson the larger testbedunder investigation. The work-unitsizeis maintainedconstant(10000simulationstepsperwork-unit). Figure13 shows thesamequality factors(minimum,averageandmaximum)for thesmallertestbed.Again thework-unit sizeis 10000.For the larger
11
0
0.05
0.1
0.15
0.2
0.25
0 500 1000 1500 2000 2500
qual
ity fa
ctor
work-units
40000 (np 30)20000 (np 30)
Quality factor for different work-unit sizes: 20000 and 40000 simulation steps(work-pool size 60, simulation time 12 hours)
0
0.05
0.1
0.15
0.2
0.25
0 500 1000 1500 2000 2500
qual
ity fa
ctor
work-units
40000 (np 60)20000 (np 30)
Quality factor for different work-unit sizes: 20000 and 40000 simulation steps(work-pool 120, simulation time 12 hours)
0
0.05
0.1
0.15
0.2
0.25
0 500 1000 1500 2000 2500 3000 3500 4000 4500 5000
qual
ity fa
ctor
work-units
20000 (np 60) 40000 (np 60)
Quality factor for different work-unit sizes: 20000 and 40000 simulation steps(work-pool 120, simulation time 12 hours)
Figure9: Effect of work-unit sizeon the quality factor. Two platformswith differentnumbersof nodesaretaken into account,i.e. a platform with 30 nodes(np 30) anda platform with 60 nodes(np 60). Eachpointin thepicturesis a work-unit which returneda resultaccepteddueto improvementof thequality factor. In allcases,thetestswith 40000simulationstepsperwork-unit reachedgoodquality factorsafterfewer work-unitsthanthetestswith lessstepsperwork-unit.
testbed,thetestswith awork-poolsizeof 120reachedthebestquality factorin average.Onthesmallertestbed,thebestquality factorin averageis reachedwith a work-pool sizeof 45. This implies that for eachplatform
12

Figure10: Sizeof thedatacommunicatedfor dif-ferentwork-unit sizeswhena sub-trajectoryis ac-ceptedbecauseit providesan improvementof thequality factor.
Figure11: Amountof floatingpoint operationsfordifferentwork-unit sizesandtherelatednon-linearbehavior (the dashedline shows the linear behav-ior).
70 120 2000
0.02
0.04
0.06
0.08
0.1
0.12
qual
ity fa
ctor
work-pool size
Quality factor (min, avr, max) for different work-pool sizes: 70, 120, 200(work-unit 10000, simulation time 12 hours, number of nodes (np) 48)
Figure12: Minimum, averageandmaximumqual-ity factorfor differentwork-poolsizes(i.e.70,120,200)on a testbedwith 48machines.
30 45 600
0.02
0.04
0.06
0.08
0.1
0.12qu
ality
fact
or
work-pool size
Quality factor (min, avr, max) for different work-pool sizes: 30, 45, 60(work-unit 10000, simulation time 12 hours, number of nodes (np) 24)
Figure13: Minimum, averageandmaximumqual-ity factorfor differentwork-poolsizes(i.e. 30,45,60)on a testbedwith 24machines.
sizethereis anoptimalwork-poolsizewhich leadsto thebestresults(i.e. quality factor).To keepall workersbusythework-poolsizeshouldbelargerthanthenumberof participatingworkers.Therefore,thedependencybetweenthequality factorandthework-poolsizeinherentlylimits theamountof task-parallelismavailableintheapplicationandthepotentialfor speed-upin awidely distributedcomputation.Overly largework-poolsizescancausea slow convergenceof thequality functionandcanthereforeresultinlongerturn-aroundtime asdisplayedin Figure14. Thefigurereportsthequality factorof repeatedtestson thesmallertestbed.For theseveral tests,thework-poolsizeis 10000simulationsteps.On theotherhand,we canalsoseecomparingthe threepicturesthat asthe work-pool sizeincreases,the contribution to the simulationprovided by the slower machines(400 MHz) becomesmoreandmorerelevant. Moreover, larger work-poolsizeshelpto avoid local minimumandfacilitatetheresolutionof simulationsstuckin a localminimum.
13

0 500 1000 1500 2000 2500 3000 3500 4000 4500 50000
0.05
0.1
0.15
0.2
0.25
Quality factor over work−units with work−pool size = 30
qual
ity fa
ctor
work−units
O 400MHzX 1GHz
(work−unit size 10000, number of processors 24, simulation time 12 hours)
0 500 1000 1500 2000 2500 3000 3500 4000 4500 50000
0.05
0.1
0.15
0.2
0.25
Quality factor over work−units with work−pool size = 45
qual
ity fa
ctor
work−units
O 400MHzX 1GHz
(work−unit size 10000, number of processors 24, simulation time 12 hours)
0 500 1000 1500 2000 2500 3000 3500 4000 4500 50000
0.05
0.1
0.15
0.2
0.25
Quality factor over work−units with work−pool size = 60
qual
ity fa
ctor
work−units
O 400MHzX 1GHz
(work−unit size 10000, number of processors 24, simulation time 12 hours)
Figure14: Effect of work-poolsizeon thequality factorandthekind of machinesproviding acceptedwork-units(i.e. CPUclockrate).Theseveraltestslastabout12hoursandrunson24nodes(12nodeswith 400MHzCPUclock rateand12 nodeswith 1 GHz CPUclock rate).
4 Conclusion
In this report,wemigrateaproteinfolding simulationusingCHARMM from PCclustersto theUnitedDevicesMetaProcessor- a commercialplatformfor widely distributedcomputingon Intra- andInternet.We alsolook
14

at theperformancecharacterizationof themigratedapplicationon theheterogenousplatform.The performanceevaluationwith a 56 residueprotein(the src-SH3 domain)shows that thereareinterestingtrade-offs in work-unit size and work-pool size which even affect the quality of the resultsin the foldingsimulation.Largerwork-unitsizesprovidebetterquality factorswith asmallernumberof work-units,but resultin longerturn-aroundtimesandlessparallelism. We prove that thequality of the proteinfolding simulationis sensitive to the platform configuration,i.e. the numberof workersandthe clock ratesof the workers. Inparticular, theproperchoiceof work-poolsizefor a givenplatformsize(numberof workers)canimprove thefinal quality factorby 20%-30%with thesametotal amountof computationperformed.We demonstratethatwith larger work-pool sizesthe active contribution of slower machinesto the entirefolding processbecomemoresignificantandthecomputationbecomeslesssusceptibleto local minima. But choosingwork-poolsthataretoo largewill adverselyaffect theconvergenceof thequality factor.Theexperiencewith ourresearchprototypeindicatesthattheapplicationof proteinfolding is robustagainsttheheterogeneousenvironmentsandthelimited communicationcapabilitiesof desktopgrids. In fact,all computenodesinvolveddoprovidecontributionsto theprogressof theproteinfolding simulation,despitetheirdifferentCPUclockratesandslowernetwork interconnections.Basedonourperformancestudypresentedin thispaper,we canconcludethat the platform heterogeneitypositively affectsthe quality of proteinfolding simulations,becauseof its positive influenceon taskgenerationsandscheduling.The differencesin clock ratesandnet-work speedsseemto contributeadditionalrandomnessto thesearchfor anoptimalconformationby simulatedannealingandhelpto getbetterresults.In short,we cansaythatproteinfolding calculationswith CHARMM arewell suitablecandidatesfor widelydistributed computationon a desktopcomputationalgrid. The suggestedsoftwaresystemcanmake a largeamountof nearlyfreecomputecyclesavailableto computationalbiologistsin needof a largeamountof addi-tional computepower for proteinfolding calculationsfor their research.
References
[1] D. Anderson et al. United Devices - Building the worlds largest computer, one computer at a time.http://www.ud.com.
[2] J.Basney andM. Livny. Deploying aHigh ThroughputComputingCluster.In RajkumarBuyyaEditor, editor, HighPerformance Cluster Computing, volume1, chapter5. PrenticeHall PTR,1999.
[3] W. J.Bolosky, J.R. Douceur, D. Ely, andM. Theimer. Feasibilityof a ServerlessDistributedFile SystemDeployedon an Existing Setof DesktopPCs. In Proc. of the International Conference on Measurement and Modelling ofComputer Systems (SIGMETRICS), SantaClara,California,Jun2000.
[4] B. R.Brooks,R.E.Bruccoleri,B. D. Olafson,D. J.States,S.Swaminathan,andM. Karplus.CHARMM: A programfor macromolecularenergy, minimization,anddynamicscalculations.J. Comput. Chem., 4:187–217,1983.
[5] A. Chienet al. Entropia- High PerformanceInternetComputing.http://www.entropia.com.
[6] C. Clementi,H. Nymeyer, andJ.N. Onuchic. TopologicalandEnergeticFactors:WhatDeterminestheStructuralDetailsof the TransitionStateEnsembleand“en-route” Intermediatesfor ProteinFolding? An InvestigationforSmallGlobular Proteins.J. Mol. Biol., 298:937,2000.
[7] distributed.netProjectPage.http://distributed.net/.
[8] P. Ferrara,J. Apostolakis,andCaflischA. Evaluationof a fast implicit solvent model for MolecularDynamicsSimulations.Proteins, 46(1):24–33,2002.
[9] [email protected]://folding.stanford.edu.
[10] I. Fosteret al. Grid PhysicsNetwork. http://www.griphyn.org.
15

[11] I. FosterandC. Kesselman.Globus: A MetacomputingInfrastructureToolkit. Intl J. Supercomputer Applications,11(2):115–128,1997.
[12] I. Foster, C. Kesselman,andS.Tuecke. TheAnatomyof theGrid: EnablingScalableVirtual Organizations.Intl J.Supercomputer Applications, 15(3),2001.
[13] J.GsponerandA. Caflisch.Roleof Native TopologyInvestigatedby Multiple Unfolding Simulationsof Four SH3Domains.J. Mol. Biol., 309:285–298,2001.
[14] J GsponerandA. Caflisch. Roleof Native TopologyInvestigatedby Multiple Unfolding Simulationsof four SH3Domains.J. Mol. Biol., 309(1):285–98,2001.
[15] J. GsponerandA. Caflisch. MolecularDynamicsSimulationsof ProteinFolding from the TransitionState. ProcNatl Acad Sci U S A., 99(10):6719–24,May 2002.
[16] W. Hoschek.A Databasefor DynamicDistributedContentandits Applicationfor ServiceandResourceDiscovery.In Proc. of Int. IEEE Symposium on Parallel and Distributed Computing (ISPDC 2002), Iasi,Romania,Jul 2002.
[17] M. Karaul. Metacomputing and Resource Allocation on the World Wide Web. PhD thesis,New York University,May 1998.
[18] A.D. MacKerellJr. andetal. All-atom empiricalpotentialfor molecularmodelinganddynamicsstudiesof proteins.J. Phys. Chem. B, 102:3586–3616,1998.
[19] J.C. MartinezandL. Serrano.TheFoldingTransitionStatebetweenSH3Domainsis ConformationallyRestrictedandEvolutionarilyConserved. Nat. Struct. Biol., 6:1010,1999.
[20] A. Natrajan,M. Crowley, N. Wilkins-Diehr, M. Humphrey, A. Fox, A. Grimshaw, andC. Brooks.StudyingProteinFolding on theGrid: ExperiencesusingCHARMM on NPACI ResourcesunderLegion. In Proc. of the 10th IEEESymposium on High-Performance Distributed Computing (HPDC-10), SanFrancisco,California,Aug 2001.
[21] J.G.B.Northey, A.A. Di Nardo,andA.R. Davidson. HydrophobicCorePackingin theSH3DomainFolding Tran-sitionState.Nature Structural Biology, 9(2):126–130,Feb2002.
[22] D. S. Riddle, V. P. Grantcharova, J. V. Santiago,E. Alm, I. Ruczinski,and D. Baker. Experimentand TheoryHighlight Roleof NativeStateTopologyin SH3Folding. Nat. Struct. Biol., 6:1016,1999.
[23] KyungDongRyu. Exploiting Idle Cycles in Networks of Workstations. PhDthesis,Universityof Maryland,2001.
[24] SETI@Home:Searchfor ExtraterrestrialIntelligenceat Home.http://setiathome.ssl.berkeley.edu/.
[25] M.R. ShirtsandV.S.Pande.MathematicalAnalysisof CoupledParallelSimulations.Phys Rev Lett., 86(22):4983–7,May 2001.
[26] M.R. ShirtsandV.S.Pande.MathematicalAnalysisof CoupledParallelSimulations.Phys Rev Lett, 86(22):4983–7,May 2001.
[27] H. Stockinger, A. Samar, B. Allcock, I. Foster, K. Holtman,andB. Tierney. File andObjectReplicationin DataGrids. In Proc. of the 10th IEEE Symposium on High Performance and Distributed Computing (HPDC-10), SanFrancisco,California,Aug 2001.
[28] M. Taufer, T. Stricker, andR. Weber. InvertingMiddlewareFramework: a Framework for PerformanceAnalysisof DistributedOLAP Benchmarkson Clustersof PCsby Filtering andAbstractingLow Level ResourceUsage.TechnicalReport367,Instituteof ComputerSystems,ETH Zurich,Switzerland,Mar 2002.
[29] M. Taufer, T. Stricker, andR. Weber. ScalabilityandResourceUsageof anOLAP Benchmarkon Clustersof PCs.In Proc. of SPAA 2002, Fourteenth ACM Symposium on Parallel Algorithms and Architectures, Winnipeg,Manitoba,Canada,Aug 2002.
[30] B. Uk. Migrationof theMolecularDynamicsApplicationCHARMM to theWidely DistributedComputingPlatformof UnitedDevices.DiplomaThesis,ETH Zurich,Switzerland,2002.
[31] United Devices, Inc. Edge Distributed Computing with the MetaProcessor Platform, 2001.http://www.ud.com/products/documentation/.
16

[32] UnitedDevices,Inc. MetaProcessorPlatform,Version2.1ApplicationDeveloper’sGuide,2001.
[33] M. Vendruscolo,C.M. Paci, E.andDobson,andM. Karplus. ThreeKey Residuesform a Critical ContactNetworkin a ProteinFoldingTransitionState.Nature, 409(6820):641–5,Feb2001.
[34] B. Zagrovic, C.D.Snow, S.Khaliq, M.R. Shirts,andV.S.Pande.Native-likeStructurein theUnfoldedEnsambleofSmallProteins.J. Mol. Biol., 323:153–164,2002.
[35] B. Zagrovic, E.Sorin,andV. Pande.BetaHairpinFoldingSimulationsin AtomisticDetailUsinganImplicit SolventModel. Journal of Molecular Biology, 2001.
17