i modelli matematici nei sistemi metabolici: analisi del...
TRANSCRIPT

Dottorato di Ricerca in Ingegneria dei Sistemi
XXII ciclo
Tesi di Dottorato
I modelli matematici nei sistemi metabolici:
Analisi del metabolismo del glucosio in
situazioni normali e patologiche
Relatore Prof.ssa Serenella Salinari
Candidato Ing. Simone Asnaghi
Coordinatore del Dottorato Prof. Carlo Bruni
ANNO ACCADEMICO 2008-2009


Alla mia famiglia,
testimone e partecipe dell’impegno e della fatica.
A tutti i ricercatori,
perché trovino sempre la luce che li illumini nel loro lento e difficile
peregrinare.
Agli amici,
agli amici veri, compagni di viaggio e di meravigliosi momenti di scoperta
della vita.
All’amore
all’amore paziente,
gioia passata o sorriso che aspetta dietro l’angolo.
Che sia la fonte che disseti la vostra sete di conoscenza
o, semplicemente, la felicità,
qualunque cosa voi cerchiate,
vi auguro di trovarla seguendo sempre il vostro cuore,
imprevedibile e non razionale, ma sempre sincero.

- 2 -
Indice 1. Introduzione..................................................................................................................6 1.1 La modellizzazione dei sistemi biologici ................................................................6 1.2 Contenuti della tesi ................................................................................................6
2. Nozioni di anatomia e fisiologia ....................................................................................9 2.1 Ghiandole endocrine .............................................................................................9 2.2 Il pancreas...........................................................................................................11 2.2.1 Forma, posizione e rapporti..........................................................................12 2.2.2 Dotti escretori, vasi e nervi ...........................................................................12 2.2.3 Pancreas esocrino........................................................................................13 2.2.4 Pancreas endocrino .....................................................................................14
2.3 L’insulina .............................................................................................................16 2.3.1 Sintesi e meccanismo di azione dell’insulina................................................17 2.3.2 Il recettore dell’insulina.................................................................................19 2.3.3 Diabete e insulina.........................................................................................21
2.4 Regolazione della secrezione dell’insulina ..........................................................22 2.5 Glucagone ...........................................................................................................22 2.6 Insulina, sport e diete ..........................................................................................26 2.7 Insulino-resistenza...............................................................................................27 2.8 Iperinsulinemia ....................................................................................................30
3. Modelli matematici per la rappresentazione dei pathway metabolici ..........................32 3.1 Modelli monocompartimentali e bicompartimentali ..............................................32 3.1.1 Esempio di modello monocompartimentale: assorbimento ed eliminazione 34 3.1.2 Esempio di modello monocompartimentale: infusione endovenosa continua 35 3.1.3 Esempio di modello bicompartimentale........................................................36
3.2 Alcune note sul significato del Volume di Distribuzione.......................................37 3.3 Michaelis-Menten ................................................................................................39 3.4 Modulazione negativa..........................................................................................41 3.5 Panoramica dei modelli matematici della secrezione dell’insulina indotta dal glucosio ..........................................................................................................................41 3.5.1 Modello Storage-Limited e modelli Signal-Limited: nomenclatura e definizioni comuni 43 3.5.2 Modello Bi-Compartimentale ........................................................................44 3.5.3 Modelli Signal-Limited ..................................................................................46 3.5.3.1 Delta-Signal Model ...................................................................................47 3.5.3.2 Modello eccitatore-inibitore .......................................................................48 3.5.4 Modello di Toffolo .........................................................................................50
3.6 La modellizzazione del metabolismo dell’insulina................................................54 3.7 Le grandezze fondamentali derivate dai modelli..................................................57
4. Metodi di analisi qualitativa e di analisi quantitativa dei modelli mediante reti di Petri 64 4.1 Reti di Petri ..........................................................................................................64 4.1.1 Significato di posti e transizioni, regola di scatto ..........................................65 4.1.2 Non determinismo ........................................................................................67 4.1.3 Strutture fondamentali ..................................................................................67 4.1.4 Proprietà delle PN ........................................................................................69 4.1.5 Proprietà comportamentali ...........................................................................69
4.2 Metodi di analisi...................................................................................................71 4.3 Proprietà strutturali ..............................................................................................74 4.4 Analisi matriciale..................................................................................................74

- 3 -
4.5 Utilizzo delle reti di Petri per l’analisi qualitativa dei modelli di sistemi biologici ..77 4.5.1 Il legame tra modelli qualitativi e modelli quantitativi e l’utilizzo delle reti di Petri 77 4.5.2 Il framework “modello qualitativo – modello quantitativo” .............................78 4.5.3 Modelli qualitativi basati su PN.....................................................................79 4.5.4 Analisi qualitativa del modello a rete di Petri ................................................80 4.5.4.1 T-Invarianti realizzabili e feasible T-Invariant............................................81 4.5.4.2 MCT, Maximal Common Transition set.....................................................82
4.6 Modelli quantitativi basati su PN..........................................................................82 4.7 Reti di Petri continue e ibride...............................................................................86 4.7.1 Reti di Petri discrete, continue e ibride .........................................................86 4.7.2 Reti di Petri temporizzate .............................................................................88 4.7.3 Reti di Petri ibride .........................................................................................90 4.7.4 Reti di Petri ibride autonome ........................................................................90 4.7.5 Reti di Petri ibride temporizzate....................................................................91
5. Progettazione di un software di supporto per l’analisi e la simulazione di modelli biologici..............................................................................................................................93 5.1 Le necessità che hanno portato alla progettazione del software .........................93 5.2 Le funzioni solver per la risoluzione numerica delle equazioni differenziali .........94 5.3 L’ambiente di lavoro e i moduli progettati ............................................................96 5.3.1 L’utilizzo di XML per la memorizzazione della rete.......................................98 5.3.2 L’interfaccia grafica e la memorizzazione dei flussi della rete ....................100 5.3.3 La generazione degli script Matlab.............................................................101 5.3.4 Esempio di utilizzo del programma.............................................................102
6. Modello Glu-Ins-NEFA: analisi qualitativa e quantitativa ..........................................104 6.1 Modello del pathway Glucosio-Insulina-NEFA...................................................105 6.1.1 Il pathway Glu-Ins-NEFA............................................................................105 6.1.2 Modellizzazione tramite rete di Petri del pathway.......................................107 6.1.3 Analisi qualitativa del modello PN del pathway Glu-Ins-NEFA ...................109 6.1.4 Analisi quantitativa del modello Glu-Ins-NEFA...........................................112 6.1.4.1 Alcuni miglioramenti computazionali del modello....................................116
7. Conclusioni: prossimi passi e proposte di sviluppo...................................................118 7.1 Analisi della reversibilità dal diabete a seguito di BPD ......................................118 7.1.1 Esperimento effettuato ...............................................................................118
7.2 Sviluppo del software di supporto analisi...........................................................125 7.3 Uso dell’analisi stocastica..................................................................................126
8. Appendice: script Matlab utilizzati per il pathway Glu-Ins_NEFA..............................131 9. Bibliografia................................................................................................................143

- 4 -
Indice delle figure Figura 1 - Ghiandole endocrine in un soggetto maschile...................................................10 Figura 2 – Pancreas ..........................................................................................................11 Figura 3 – A sinistra, insulina all’interno delle cellule-beta; al centro, glucagone nelle cellule alfa;.........................................................................................................................15 Figura 4 - Molecola di proinsulina: si notino le due catene polipeptidiche unite da ponti disolfuro e il C-peptide di congiunzione .............................................................................17 Figura 5 - Pre Proinsulina ..................................................................................................18 Figura 6 - Esocitosi ............................................................................................................18 Figura 7 - Esamero ............................................................................................................19 Figura 8 - Recettore dell'insulina........................................................................................20 Figura 9 - Effetti dell'assunzione di carboidrati sull'indica glicemico ..................................25 Figura 10 - Modello bicompartimentale del metabolismo del glucosio (fonte: [DaM2006]) 36 Figura 11 - Equazioni costitutive del modello bicompartimentale del glucosio (fonte: [DaM2006]) ........................................................................................................................36 Figura 12 - Modello a bicompartimentale...........................................................................45 Figura 13 - Modello Signal-Limited ....................................................................................47 Figura 14 - Equazioni del modello di Toffolo......................................................................49 Figura 15 - Delta-Signal model (a sinistra) e modello Eccitatore-Inibitore (a destra) .........50 Figura 16 - Modello bicompartimentale del C-Peptide .......................................................51 Figura 17 - Gestionde statica (a sinistra) e dinamica (a destra) del coefficiente k per la secrezione dinamica ..........................................................................................................52 Figura 18 - Modelli minimali di sensibilità all'insulina .........................................................61 Figura 19 - Modelli minimali di risposta della cellula beta ..................................................62 Figura 20: Esempio di rete di Petri e di scatto di una transizione ......................................66 Figura 21 - Esempio di sequenza in una rete di Petri ........................................................67 Figura 22 - Esempio di concorrenza in una rete di Petri ....................................................68 Figura 23 - Esempio di conflitto in una rete di Petri ...........................................................68 Figura 24 - Esempio di rete marcata contenente transizione di sincronizzazione e transizione di concorrenza .................................................................................................69 Figura 25 - Esempi delle combinazioni delle 3 proprietà di vivezza, limitatezza e reversibilità (Fonte: [Mur89]). .............................................................................................71 Figura 26 - Esempio di PN e relativi albero e grafo di copertura........................................72 Figura 27 - A HFPN realization of the circadian rhythm model due to Ueda et al (fonte: [Tan2003]) .........................................................................................................................78 Figura 28 - Framework di modellizzazione qualitativa e quantitativa di sistemi biologici secondo Gilbert e Heiner (Fonte:[Gil2006]) .......................................................................79 Figura 29 - Esempio di modello qualitativo di una reazione biochimica rappresentato da una PN (Fonte:[PoZ2005]).................................................................................................80 Figura 30 - Esempio di PN discreta ...................................................................................87 Figura 31 - Esempio di PN continua tratto dalla PN discreta di Figura 10 .........................87 Figura 32 – Esempio di PN ibrida ......................................................................................87 Figura 33 - Esempio di PN discreta temporizzata e sua evoluzione ..................................88 Figura 34 - Esempio di processo (fluido in movimento) modellato da una PN continua: si noti come cambiando gli ordini di grandezza allo stesso modo per la velocità e per i posti, il modello usato abbia lo stesso andamento dell'altro ........................................................89 Figura 35 - Influenza della parte discreta sulla parte continua...........................................90 Figura 36 - Influenza della parte continua su quella discreta .............................................91 Figura 37 - Esempio di PN ibrida temporizzata..................................................................92 Figura 38 - Pathway di esempio per l'utilizzo del programma..........................................102

- 5 -
Figura 39 - Reti di Petri associata al pathway di esempio................................................103 Figura 40 Modello della IS stimolata da glucosio e modulata dai NEFA..........................106 Figura 41 - Modello PN del pathway Glu-Ins-Nefa...........................................................108 Figura 42 - Modello PN con T-Invarianti individuati .........................................................111 Figura 43 - Andamenti dei valori misurati e simulati per due pazienti non soggetti a BPD.........................................................................................................................................115 Figura 44 – Dati del IVGTT per un paziente obeso..........................................................120 Figura 45 – Valori plasmatici di GLP1 e GIP in un paziente obeso..................................121 Figura 46 - Framework di utilizzo delle PN per l'analisi dei pathway biochimici ...............128 Figura 47 - Esempio di applicazione di SPN in un contesto IT per l'analisi di processi aziendali...........................................................................................................................129

- 6 -
1. Introduzione
1.1 La modellizzazione dei sistemi biologici
La complessità dei sistemi biologici, dalle reazioni biochimiche alle interazioni cellulari,
richiede strumenti di modellizzazione che siano utili allo scopo di progettare e interpretare,
in modo appropriato, esperimenti biologici.
L’avvento delle ultime tecniche di manipolazione genetica a livello molecolare ha reso
evidente a molti biologi che i sistemi da essi studiati sono più complessi e dinamici di
quanto preventivamente assunto.
Contemporaneamente, la quantità di dati dovuti alle ultime tecniche e progetti scientifici
(fra tutti, si pensi al progetto GENOMA) hanno inondato gli scienziati di una mole abnorme
di dati che rendono ancora più difficile, prima ancora dell’utilizzo, alla descrizione degli
stessi.
Quindi, un modello è utile per decidere quali dati sono necessari e quali possono essere
scartati nel proseguimento dell’indagine scientifica.
Questo numero enorme e crescente di dati richiede anche un approccio modellistico
integrato: le tecnologie attuali permettono di tracciare il comportamento delle strutture
biologiche più complesse su vari livelli di scala (dal livello atomico all’espressione dei
pattern genetici) e, allo stesso tempo, è possibile testare più facilmente le teorie
scientifiche di nuova concezione, ma ciò di cui gli scienziati hanno oggi più bisogno è la
possibilità di passare facilmente dall’approccio più teorico a quello più sperimentale e
viceversa.
Per essere utili a chi fa un esperimento, i modelli teorici devono essere facilmente
collegabili ai dati biologici e questo richiede dei tool matematici in grado di essere estesi
facilmente a sistemi via via più complessi e interfacciabili con sistemi di gestione e
rappresentazione dati eterogenei.
1.2 Contenuti della tesi
Lo studio della modellistica dei sistemi metabolici è di fondamentale importanza per alcune
rilevanti patologie (obesità, diabete di tipo 2), in particolare per migliorare le condizioni di

- 7 -
vita e per evitare di ricorrere a tecniche invasive per la determinazione dello stato di salute
generale dei pazienti.
A partire da modelli recentemente sviluppati dell’interazione glucosio-grassi e modelli
relativi al calcolo della sensibilità dei tessuti all’insulina, si è notato che la resistenza dei
tessuti all’insulina e le relazioni glucosio-grassi a livello della cellula pancreatica sono
strettamente connesse [Sal2007] (la normalizzazione della sensibilità all’insulina si riflette
in generale in una normalizzazione della secrezione dell’insulina (vedi [Cob2005] e
[Dam2006]).
In tale contesto sono stati analizzati i dati relativi a soggetti diabetici obesi sottoposti ad un
intervento chirurgico (Bilio Pancreatic Diversion, BPD) per la riduzione dell’obesità, per i
quali si verifica una remissione del diabete dopo l’intervento. Tali dati (concentrazione
plasmatiche di glucosio, insulina, acidi grassi e C-Peptide) fanno riferimento ad una
condizione sperimentale classica nello studio del metabolismo del glucosio quale il test di
tolleranza al glucosio, somministrato per via orale, effettuato prima e dopo l’intervento.
I risultati ottenuti hanno suggerito l’interessante ipotesi che l’esclusione del transito del
cibo attraverso il digiuno e il duodeno conseguente all’intervento inibisca la produzione di
una particolare sostanza responsabile di provocare insulino resistenza. I risultati di tale
ricerca sono stati presentati in [Sal2008].
Tale ipotesi è in corso di analisi attraverso esperimenti e l’elaborazione di modelli
sviluppati ad hoc. I modelli compartimentali sviluppati per l’analisi quantitativa permettono
la simulazione di differenti condizioni fisiologiche e/o patologiche, oltre alla determinazione
di parametri globali di interesse di possibile utilizzazione nella pratica clinica.
All’analisi quantitativa è stata associata la modellizzazione dei sistemi metabolici mediante
reti di Petri (PN). Riprendendo gli studi sui modelli biochimici sviluppati di Monika Heiner
(tra cui [PoZ2005],[Gil2006],[ Sac2006]) e altri, l’analisi qualitativa della PN associata al
pathway studiato ha permesso la validazione del modello creato, mediante l’analisi e il
matching delle proprietà del modello e della PN stessa. Si è proceduto con lo sviluppo di
un prototipo di codice Java per la modellizzazione di pathway biochimici e la creazione
automatica di codice Matlab simulabile e di file XML rappresentativi della rete stessa, utili
per l’analisi qualitativa mediante tool di analisi delle PN disponibili su Internet.

- 8 -

- 9 -
2. Nozioni di anatomia e fisiologia
Prima di entrare nel vivo della discussione sui modelli legati al metabolismo dell’insulina, è’
fondamentale introdurre alcune nozioni di anatomia e di fisiologia del corpo umano ed in
particolare dell’organo principe legato al metabolismo dell’insulina e degli zuccheri, cioè il
pancreas.
Nel seguito del capitolo, dopo una breve e generale introduzione sul ruolo delle ghiandole
endocrine, verranno introdotti alcuni concetti relativi all’anatomia del pancreas (forma,
posizione, comunicazione con gli altri organi) e relativi alla produzione dell’insulina e al
ruolo del glucagone, ormone antagonista dell’insulina.
Infine, viene spiegato il ruolo dell’insulina nella vita di tutti i giorni e alcune delle patologie
legate ad un metabolismo anormale dell’insulina.
2.1 Ghiandole endocrine
Le ghiandole del corpo umano sono di due tipi:
• a secrezione esterna, o esocrine: riversano i loro prodotti all’esterno del corpo o in
certi organi, come ad esempio le ghiandole salivari che riversano la saliva in bocca,
e le ghiandole sudoripare, che riversano il sudore all’esterno del corpo;
• a secrezione interna , o endocrine: versano i loro prodotti,gli ormoni, nel torrente
sanguigno. Le ghiandole che producono ormoni sono molto diverse tra di loro e
sono distribuite in vari punti del nostro organismo.

- 10 -
Figura 1 - Ghiandole endocrine in un soggetto maschile
Come mostra Figura 1 l’apparato endocrino raggruppa un insieme di organi ghiandolari
che, pur diversi per origine embriologica e struttura, hanno alcune caratteristiche
morfologiche comuni. Sono dette ghiandole endocrine pluricellulari (origine e strutture
diverse, attività di tipo endocrino, diversa natura chimica degli ormoni prodotti):
• Ipofisi
• Epifisi
• Tiroide
• Paratiroidi
• Pancreas (Isolotti pancreatici)
• Ghiandole surrenali
• Ghiandola interstiziale del testicolo
• Formazioni endocrine dell’ovaia (teca interna follicolare, ghiandola interstiziale,
corpo luteo).

- 11 -
C’è ne sono altre classificate come ghiandole endocrine unicellulari(origine comune dalle
creste neurali o dal neuroectoderma, attività di tipo endocrino o di tipo paracrino, natura
polipeptidica e/o amminica dal secreto) e sono classificate come:
• Ghiandole endocrine dell’apparato digerente (cosidetto sistema
gastroenteropancreatico, GEP)
• Cellule endocrine dell’apparato respiratorio e urogenitale
• Cellule C della tiroide, cellula della midollare surrenale, melanociti cutanei, ecc.
Queste ultime sono di acquisizioni recenti e tuttavia in via di elaborazione, basati
principalmente su dati di microscopia elettronica e di immunoistochimica che hanno
permesso l’identificazione di tutta una serie di elementi cellulari capace di produrre
sostanze attive di tipo ormonale, tali da poterle inquadrare in un unico sistema detto
appunto sistema endocrino diffuso.
2.2 Il pancreas
Il pancreas (Figura 2) è una voluminosa ghiandola dell’apparato digerente, produce il
succo pancreatico coinvolto nei processi digestivi, ma svolge anche funzione di ghiandola
endocrina.
Misura circa 17-20 cm in lunghezza, 4-5 cm in larghezza e 2-3 cm in spessore; il suo peso
si aggira intorno agli 80 g, è piuttosto friabile e si presenta di colorito grigiastro o roseo
quando è in condizioni di intensa attività.
Figura 2 – Pancreas
Costituito principalmente da tessuto esocrino, secerne e riversa nell’intestino tenue diversi
enzimi per la demolizione di grassi, carboidrati e proteine. Il suo secreto (il succo
pancreatico) è infatti ricco di enzimi proteolitici, glicolitici e lipolitici e presenta inoltre un

- 12 -
grado elevato di alcalinità che contribuisce a neutralizzare il pH del chimo gastrico allorché
questo giunge nel duodeno. La parte esocrina di questa ghiandola è formata da strutture
acinose.
Gli ammassi di cellule endocrine, dette isole del Langherans, invece producono ormoni
coinvolti nella regolazione della concentrazione di glucosio nel sangue (glicemia). I due
ormoni vengono riversati direttamente nel sangue, dal quale vengono trasportati alle
cellule. Essi risultano essenziali per la regolazione del metabolismo degli zuccheri.
2.2.1 Forma, posizione e rapporti
Il pancreas è uno degli organi nascosti del corpo, infatti, è localizzato nella porzione
superiore della cavità addominale, dietro allo stomaco, in posizione trasversale. Può
raggiungere, in posizione alta, il corpo della 12 vertebra toracica o, in posizione bassa
quello della 3 vertebra lombare. Il suo asse maggiore non è perfettamente trasversale, ma
risulta verso sinistra e verso l’alto. Su questo asse, il pancreas si presenta incurvato con
una convessità volta verso l’avanti e determinata dal rapporto posteriore con la colonna
vertebrale e i grossi vasi posti al davanti di quest’ultima ([Bal87]).
Nel pancreas si distinguono una testa, un corpo e una coda (Figura 2). La testa è accolta
nella concavità dell’ansa duodenale, misura 6-7 cm di altezza, 3-4 cm di larghezza e 2-3
cm di spessore; il corpo incrocia da destra a sinistra e dal basso verso l’alto i corpi della 1
e 2 vertebra lombare; la coda è l’estremità sinistra della ghiandola e presenta una forma e
un’estensione assai variabili: può essere più o meno assottigliata e appiattita, oppure
arrotondata e tozza.
Fra la testa e il corpo esiste una porzione ristretta che viene chiamata istmo, ed è
delimitata da due incisure, una superiore e una inferiore. I mezzi di fissità del pancreas
sono rappresentati, dunque, dal duodeno che ne accoglie la testa cui aderisce, dal
peritoneo parietale posteriore che ricopre anteriormente il pancreas e lo mantiene
aderente alla parete posteriore dell’addome e agli organi retrostanti, e infine dal legamento
pancreaticolienale che ne fissa la coda all’ilo della milza ([Bal87]).
2.2.2 Dotti escretori, vasi e nervi

- 13 -
Il succo pancreatico si versa nella seconda porzione del duodeno attraverso due condotti
escretori che si aprono rispettivamente a livello della papilla duodenale maggiore e di
quella minore. Essi sono il dotto pancreatico principale (di Wirsing)e il dotto pancreatico
accessorio (di Santorini). Il dotto principale decorre nello spessore del parenchima
pancreatico con una direzione corrispondente a quella dell’asse maggiore dell’organo. I
numerosi affluenti del dotto lo raggiungono formando con esso un angolo generalmente
retto.
Il dotto accessorio decorre nello spessore della parte superiore delle testa del pancreas e
prende in genere origine dal dotto principale nel punto in cui quest’ultimo, a livello
dell’istmo, forma un gomito. Dall’origine, il dotto accessorio riceve affluenti propri ma nel
complesso può essere considerato come un ramo collaterale del dotto principale.
Le arterie del pancreas provengono dall’ arteria epatica ,dall’arteria lienale e dall’arteria
mesenterica superiore . Infine, dall’ arteria splenica, in corrispondenza del tratto che
decorre sul margine superiore del pancreas, hanno origine diversi rami arteriosi che
penetrano nella ghiandola dove si dividono ciascuno in un ramo destro e uno sinistro che
si scambiano numerosi anastomosi.
Le vene sono tributarie della vena porta. Alcuni tronchi venosi si gettano nella vena
lienale, altri nelle mesenteriche e alcuni direttamente nella vena porta. I linfatici del
pancreas sono tributari di stazioni linfonodali diverse.
I nervi diretti al pancreas derivano dal plesso celiaco per mezzo di plessi secondari che
seguono le arterie proprie del pancreas e con esse penetrano nel contesto della ghiandola
(Cf. [Bal87]).
2.2.3 Pancreas esocrino
La componente esocrina, che rappresenta dall’80% all 85% dell’intero organo, è costituita
da numerose piccole ghiandole (acini), contenenti cellule epiteliali colonnari o piramidali,
orientate in modo radiale attorno alla circonferenza della ghiandola. Le cellule aciniche
sono intensamente basofile per l’abbondante contenuto di reticolo endoplasmatico rugoso,
localizzato nella parte basale della cellula. Il complesso di Golgi è ben sviluppato e
costituisce parte di un complesso secretorio orientato verso l’apice cellulare, in grado di
produrre abbondanti granuli di zimogeno rivestiti da membrana, contenenti enzimi
digestivi.

- 14 -
Quando le cellule aciniche vengono stimolate, queste vescicole contenenti zimogeno
migrano verso l’apice cellulare, qui si fondono con la membrana cellulare liberando il loro
contenuto nel lume acinare.
Il pancreas secerne 2-2,5 litri al giorno di un liquido alcalino che contiene enzimi e
proenzimi, la cui secrezione è regolata da un meccanismo complesso che coinvolge fattori
umorali e neurologici. I più importanti fra questi sono gli ormoni secretina e
colecistochinina, prodotti nel duodeno. Il primo stimola la secrezione di acqua e
bicarbonato da parte delle cellule duttali, mentre il secondo induce la liberazione di
zimogeno da parte delle cellule acinari. I grassi e il livello di acidità del materiale gastrico
stimolano in modo particolarmente attivo la produzione di secretina. La colecistochinina è
secreta dalla mucosa duodenale per lo più in risposta ad acidi e a prodotti proteici della
digestione, come amminoacidi e peptidi (Cf. [Ram2000]).
2.2.4 Pancreas endocrino
Il pancreas endocrino è formato dalle Isole del Langherans, le cui cellule producono
insulina, glucagone e somatostatina, immettendole direttamente nel sangue (eccetto
l’ultima).
Le Isole del Langherans, o isolotti pancreatici che nell’uomo sono circa un milione e
rappresentano l’1-2% del peso del pancreas, sono costituite da cordoni irregolari di cellule,
dispersi nel tessuto pancreatico acinoso. Nei cordoni le cellule sono circondate da un
ricchissimo sistema di capillari e di sinusoidi ematici. Il sangue refluo dalle Isole del
Langherans confluisce, come quello refluo dall’intestino, nella vena porta e va al fegato.
Perciò gli ormoni insulari, a differenza di quelli secreti da altre ghiandole endocrine,
incontrano, come primo organo, il fegato e su questo esplicano prevalentemente, anche
se non esclusivamente, la loro azione. Il fegato, in effetti riduce notevolmente la
concentrazione ematica degli ormoni insulari, come dimostra il loro basso livello nel
sangue sistemico rispetto a quello portale.
Il pancreas è innervato da sistemi nervosi ortosimpatico e parasimpatico e quindi anche
nelle Isole del Langherans sono presenti fibre nervose adrenergiche e colinergiche in
stretto contatto con le cellule insulari. Non è chiara, però, la loro funzione nel controllo
della secrezione ormonale.
Nei mammiferi le Isole del Langherans contengono tre tipi di cellule: α, β e δ, (dette anche
A B D ), distinguibili per la diversa colorabilità e forma dei granuli in esse contenuti.

- 15 -
Le cellule α rappresentano circa il 20% degli elementi insulari e mentre nell’uomo sono
sparse tra le cellule insulari, in altre specie (roditori) sono disposte nelle parti periferiche
degli isolotti. Hanno forma irregolare e contengono granulazioni citoplasmatiche colorabili
in rosso con la floxorina e in nero con alcuni metodi di impregnazione argentina. Questi
granuli sono solubili in acqua, insolubili in alcol e resistono all’autolisi.
Producono il glucagone (Figura 3), un ormone di natura polipeptidica, che agisce sul
ricambio degli zuccheri promovendo la glicogenolisi epatica ed elevando cosi la glicemia.
Inoltre, le cellule α sono spesso associate a cellule gangliare e a fibre nervose e
rispondono alla stimolazione simpatica e alla somministrazione di farmaci simpatico-
mimetici ([Bal87]).
Figura 3 – A sinistra, insulina all’interno delle cellule-beta; al centro, glucagone nelle cellule alfa;
a destra, somatostatina nelle cellule delta.
Le cellule β sono le più numerose, hanno forma poliedrica od ovale e contengono granuli
ben colorabili con l’ematossilina cromica e con la paraldeide fucsina, ma con i metodi
dell’argento; i granuli sono insolubili in acqua, solubili in alcool e non resistono all’autolisi.
Al microscopio elettronico le cellule β presentano un evidente apparato di Golgi,
mitocondri più grandi e numerosi delle cellule α, un evidente reticolo endoplasmatico e
ribosomi liberi; i loro granuli sono caratteristici in quanto costituiti da uno o pi´u cristalli di
forma rettangolare o poligonale, di media densità elettronica, a struttura periodica
circondati da una matrice chiara che li separa dalla membrana limitante. Si ritiene che
questi cristalli siano costituiti da un polimero dell’ormone elaborato dalle cellule. Dei
polimeri dell’ormone fa parte anche lo zinco. L’ormone in questione è l’insulina (Figura 3),
una proteina che agisce in antagonismo al glucagone abbassando la glicemia; essa
favorisce la glicogenosintesi epatica e l’utilizzazione del glucosio da parte dei muscoli. In
generale, l’ insulina aumenta la permeabilità al glucosio di tutti i tipi cellulari. In alcune
specie animali (ratto,coniglio) i granuli delle cellule β non presentano il tipico aspetto

- 16 -
cristallino e la loro massa centrale appare omogenea, rotondeggiante o reniforme
([Bal87]).
Le cellule δ sono le meno numerose circa il 5% e si trovano sparse irregolarmente tra le
altre cellule. Sono presenti nell’uomo, ma non in tutte le specie animali. Sono molto simili
alle cellule α e presentano granuli argirofili, che tuttavia si differenziano per la loro diversa
colorabilità con alcuni metodi istologici e per le caratteristiche ultrastrutturali. Producono
somatostatina (Figura 3), che agirebbe localmente regolando l’immissione in circolo
dell’insulina e del glucagone.
2.3 L’insulina
L'insulina è un ormone di natura proteica, prodotto da gruppi di cellule pancreatiche,
chiamate "cellule β delle isole del Langerhans". Fu scoperta nel 1921 dall'inglese John
James Macleod e dal canadese Frederick Grant Bating, Premio Nobel per la medicina nel
1923.
L'insulina è l'ormone anabolico per eccellenza, infatti tramite la sua azione:
• Facilita il passaggio del glucosio dal sangue alle cellule ed ha pertanto azione
ipoglicemizzante (abbassa la glicemia). Favorisce l'accumulo di glucosio sotto
forma di glicogeno (glicogenosintesi) a livello epatico ed inibisce la degradazione di
glicogeno a glucosio (glicogenolisi).
• Facilita il passaggio degli aminoacidi dal sangue alle cellule, ha funzione
anabolizzante perché stimola la sintesi proteica e inibisce la neoglucogenesi
(formazione di glucosio a partire da alcuni aminoacidi).
• Facilita il passaggio degli acidi grassi dal sangue alle cellule, stimola la sintesi di
acidi grassi a partire da glucosio e aminoacidi in eccesso ed inibisce la lipolisi
(utilizzazione degli acidi grassi a scopo energetico).
• Facilita il passaggio di potassio all'interno delle cellule.
• Stimola la proliferazione cellulare.
• Stimola l'uso del glucosio per la produzione di energia.
• Stimola la produzione endogena di colesterolo.

- 17 -
Il maggior stimolo per l'azione insulinica è dato da un pasto ricco di carboidrati semplici e
povero di fibre, grassi e proteine. Anche alcuni farmaci (sulfaniluree) sono in grado di
aumentarne la secrezione.
2.3.1 Sintesi e meccanismo di azione dell’insulina
La sintesi di tutte le proteine inizia nel nucleo della cellula con l'attivazione del segmento
del DNA (il gene) che specifica la composizione della proteina. Il gene è trascritto nella
catena di un'altro acido nucleico, l'RNA. A sua volta, l'RNA è trasformato in una forma nota
come RNA messaggero (m-RNA) che viene inviato nel citoplasma della cellula. Nel
citoplasma l'm-RNA si comporta come uno stampo per la produzione di proteine: dà
istruzioni ad organelli chiamati ribosomi, affinché dispongano gli aminoacidi in una
sequenza definita con precisione per costruire la catena proteica prescritta.
La proteina secreta dalle cellule beta, ossia l'insulina, è formata da due catene separate di
aminoacidi, le catene A e B unite da ponti di disolfuro. Le modalità di sintesi dell'insulina
furono chiarite dal Dr. Steiner (1960) il quale scoprì che l'insulina veniva sintetizzata come
parte di una proteina più grande (proinsulina, vedi Figura 4).
Figura 4 - Molecola di proinsulina: si notino le due catene polipeptidiche unite da ponti disolfuro e il
C-peptide di congiunzione
In realtà i ribosomi non sintetizzano ne’ insulina ne’ proinsulina, ma una molecola ancora
più grande che funge da precursore, la Pre-ProInsulina (ossia la proinsulina con una breve
sequenza aminoacidica supplementare). Successivamente la pre-proinsulina (vedi Figura

- 18 -
5) viene scissa, ad opera di alcuni enzimi prodotti dai ribosomi, prima in proinsulina e poi
slegandola dal peptide C di collegamento (quello che collega le catene A e B) in insulina.
Figura 5 - Pre Proinsulina
A questo punto l'ormone insulina è sintetizzato e immagazzinato in granuli adiacenti alla
membrana interna della cellula. In risposta ad uno stimolo appropriato, come l'aumento del
glucosio nel sangue, le membrane dei granuli si fondono con la membrana cellulare e le
catene di insulina sono libere di entrare nel circolo sanguigno: questo meccanismo viene
detto esocitosi, ed è raffigurato in Figura 6.
Figura 6 - Esocitosi
L'insulina è costituita da due catene polipeptidiche (α più piccola di 21 AA e β più grande
di 30 AA), tenute insieme da ponti disolfuro che si formano tra le cisteine 7 e 20 della
catena α e le cisteine 7 e 19 della catena β. L'insulina viene prodotta a partire dalla
proinsulina tramite taglio proteolitico di un peptide di congiunzione di 33 AA. Questo
peptide è chiamato peptide C, mentre l'enzima responsabile del taglio proteolitico è detto
endopeptidasi.
L'insulina viene rilasciata come proteina globulare a catena polipeptidica unica dai
poliribosomi; successivamente l'ormone si deposita sotto forma di granuli raggiungendo

- 19 -
una forma cristallina visibile al microscopio elettronico. All'aumentare della concentrazione,
l'insulina viene aggregata in dimeri (coppia di monomeri tenuti insieme da legami deboli) e
trimeri di dimeri o esameri (tenuti insieme da 2 ioni Zn centrali esacoordinati con le 3
tirosine dei dimeri e le tre molecole di H2O), come si può vedere in Figura 7.
Figura 7 - Esamero
Una volta riversata nel torrente circolatorio l'insulina passa, per diluizione, alla forma
dimerica e monomerica, conformazione, quest'ultima, riconosciuta dal recettore insulinico.
Alcuni ricercatori notarono che nell'insulina umana sono presenti delle regioni variabili, in
particolare la sequenza degli aminoacidi n° 28 e 29 (Pro-Lys) della catena β;
successivamente si scoprì che invertendo tali AA l'insulina passava direttamente allo stato
monometrico, saltando quello dimerico. Nacque così la "Lys Pro" o "insulina rapida", un
farmaco particolarmente utile se iniettato in prossimità di un pasto abbondante.
2.3.2 Il recettore dell’insulina
I numerosi processi cellulari regolati dall’insulina sono dipendenti dal legame con i suoi
recettori presenti nella membrana cellulare dai tessuti bersaglio, fondamentalmente
muscolo e tessuto adiposo. I recettori dell’insulina sono di natura glicoproteica, in buona
parte saldamente ancorati alla membrana plasmatica.
Il recettore per l'insulina (Figura 8) è una glicoproteina transmembrana costituita da 4
catene (2α esterne alla cellula e 2β interne alla cellula), fra loro unite da ponti di solfuro.
La molecola presenta un'emivita piuttosto breve ed è pertanto soggetta ad un rapido turn
over. Anch'essa è sintetizzata come precursore dal reticolo endoplasmatico rugoso e

- 20 -
viene poi elaborata nel Golgi. Le 2 catene α sono ricche in cisteine, le β sono ricche di AA
idrofobici, che le ancorano alla membrana cellulare, e tiroxine, rivolte verso la parte interna
al citosol.
Figura 8 - Recettore dell'insulina
Il legame insulina recettore stimola l'attività tirosin chinasica e porta al dispendio di 1 ATP
per tirosina fosforilata. Questo causa una serie di eventi a catena (attivazione delle G
proteine della fosfolipasi C) che portano alla formazione di due prodotti: il DAG che rimane
ancorato alla membrana e che interviene nella fosforilazione delle proteine, e l'IP3 che
agisce a livello citosolico permettendo il rilascio di ioni Ca++.
Quando la glicemia si alza aumenta la quantità di insulina secreta dalle cellule del
pancreas. Nelle cellule insulino-dipendenti il legame insulina recettore va ad agire su un
pool intracellulare di vescicole, liberando il trasportatore del glucosio che viene trasferito
alla membrana per fusione. Il trasportare porta il glucosio dentro la cellula, causando una
diminuzione della glicemia che a sua volta stimola la dissociazione tra l'insulina ed il suo
recettore. Questa dissociazione innesca un processo di simil-endocitosi con il quale il
carrier viene riportato all'interno delle vescicole.
La cinetica dell’associazione dell’insulina con i suoi recettori indica una cooperatività
negativa, nel senso che la capacità dei recettori liberi di legare l’insulina è tanto minore
quanti più recettori si sono gi`a legati all’ormone (Cf. [Sil2006]).
Il numero di recettori può diminuire o per diminuita sintesi, o per aumentata demolizione ,
o per internalizzazione. Questa possibilità di variazione del numero dei recettori di
membrana, quelli con i quali interagisce l’insulina, costituisce il più importante fattore di

- 21 -
controllo della sensibilità delle cellule all’insulina. La Insulino-resistenza è infatti spesso
determinata da una diminuzione del numero dei recettori di membrana. Anche uno stato di
Iperinsulinemia può indurre una riduzione del numero dei recettori cellulari.
Si tratta del fenomeno della down regulation.
2.3.3 Diabete e insulina
Il termine diabete deriva dal greco diabetes e significa passare attraverso. Uno dei segni
clinici caratteristici di tale patologia è la presenza di zucchero nelle urine, che vi giunge
attraverso il rene quando la sua concentrazione nel sangue supera un certo valore. A
questo termine è stato associato l'aggettivo mellito in quanto le urine, proprio per la
presenza di zucchero, sono dolci e, anticamente, l'assaggio costituiva l'unico modo per
diagnosticare la malattia
Il diabete mellito è una patologia cronica, caratterizzata da iperglicemia, cioè da un
aumento degli zuccheri (glucosio) presenti nel sangue. E' causata da una ridotta
secrezione di insulina o dalla combinazione di ridotta secrezione e resistenza periferica
all'azione di questo ormone.
In condizioni normali l’insulina, rilasciata dal pancreas, entra nel circolo sanguigno dove
funziona come una "chiave" necessaria per far entrare il glucosio all'interno delle cellule,
che, a seconda delle richieste metaboliche, lo utilizzeranno o lo depositeranno come
riserva. Ciò spiega come mai una carenza o un'alterata azione insulinica si accompagni ad
un aumento degli zuccheri presenti in circolo, caratteristica, questa, tipica del diabete.
Le cause delle disfunzioni delle cellule beta non sono completamente note. Le autopsie
hanno mostrato che percentuali tra il 20 e il 50%delle cellule beta erano perse dopo diversi
anni di diabete.( [War84]).
Tuttavia, ci sono evidenze sperimentali che una parziale pancreasaectomia del 65% nei
cani reduce la risposta insulinica massima, ma il resto delle cellule beta fornisce una
parziale compensazione ([War88]).
Come riportato da Porte e Kahn ([Por2001]), “the loss of ß-cell function is
disproportionately more important than the degree of ß-cell loss”. Anche In soggeti colpiti

- 22 -
da patologie tumorali del pancreas si è notato come la cellula beta mantiene attive la
trascrizione genetica e la traslazione.
Questo suggerisce ([Sem2004]) un anormale accoppiamento tra risposta insulinica e
glicemia.
2.4 Regolazione della secrezione dell’insulina
La velocità di secrezione dell’insulina da parte delle cellule β dell’isole del Langherans è
controllata, essenzialmente, dalla concentrazione di glucosio nel liquido che le bagna, e
perciò nel plasma. Il controllo è a feed-back positivo, perciò ad un aumento della glicemia
corrisponde un aumento di secrezione di insulina, e viceversa. La relazione tra
concentrazione di glucosio extracellulare e velocità di secrezione dell’insulina è
rappresentata, in vitro, da una curva di tipo sigmoidale, che mostra una concentrazione
soglia di glucosio a 90-100 mg/100ml (5,5 mM) ed una velocità massima di secrezione a
300-500 mg/100 ml. La risposta secretoria di insulina alla stimolazione da glucosio è
tipicamente bifasica: una rapida prima fase con un picco a circa 5min seguita da una lenta
ma crescente seconda fase.
Sulla base di alcuni esperimenti biochimici si è visto che le cellule β contengono due tipi di
gruppi di granuli: il primo (< 5 %) è costituito da granuli immediatamente pronti al rilascio di
insulina (RRP) e possono subire l’esocitosi senza ulteriori modificazioni dopo lo stimolo,
mentre la maggior parte dei granuli individua il secondo gruppo che per rilasciare insulina
devono essere sottoposti ad una serie di reazioni dipendenti da ATP, ADP, tempo e
temperatura. Ipoteticamente il rilascio da parte dei granuli RRP individua la prima fase
della secrezione la cui fine è dettata dall’esaurimento di tale gruppo e l’area sotto la curva
è equivalente all’insulina rilasciata da circa 40 granuli (15granuli/min *6min/2) la seconda
fase, invece, è caratteristica del secondo gruppo ( [Ror2003]).
2.5 Glucagone
L'organismo umano possiede un sistema di regolazione che consente di mantenere entro
un certo range la glicemia, ovvero la concentrazione di glucosio disciolto nel sangue.
La costanza della glicemia è necessaria per la sopravvivenza del cervello, per tre motivi:

- 23 -
• il cervello, a differenza dei muscoli, non ha la capacità di immagazzinare scorte di
glucosio.
• il glucosio ematico è praticamente l'unico carburante per il cervello.
• il cervello consuma una quantità costante di energia, a prescindere dalla sua attività
(studiando, infatti, si consumano pochissime calorie)
In assenza di glucosio, dopo pochi minuti le cellule celebrali muoiono.
Il meccanismo di regolazione della glicemia è basato sul controllo di due ormoni
antagonisti: l'insulina e il glucagone.
Nella seguente tabella sono indicati i principali effetti di questi due ormoni sul
metabolismo.
Effetti dell’insulina sul metabolismo
Effetti del glucagone sul metabolismo
Promuove l'accumulo di glicogeno
(zucchero di riserva) nel fegato e nei
muscoli
Promuove la liberazione del glicogeno dal
fegato, che viene riversato sottoforma di
glucosio nel sangue.
Deprime il consumo di grassi e proteine in
favore dei carboidrati, ovvero spinge le
cellule a bruciare carboidrati piuttosto che
proteine e grassi
Promuove il consumo di grassi e proteine a
sfavore dei carboidrati, ovvero spinge le
cellule a bruciare le proteine e i grassi
piuttosto che i carboidrati
Promuove la formazione di trigliceridi
(grassi) a partire da carboidrati e proteine
Promuove la mobilizzazione dei grassi dai
tessuti adiposi, che vengono resi disponibili
ai tessuti per essere bruciati
Promuove l'immagazzinamento di grassi nel
tessuto adiposo
Tabella 1 - Effetti dell'insulina e del glucagone sul metabolismo umano
Grazie a questo meccanismo, possiamo introdurre il glucosio (sotto forma di carboidrati)
solo poche volte al giorno, durante i pasti: a mantenere costante la sua presenza nel

- 24 -
sangue ci pensa l'asse ormonale insulina-glucagone, che utilizza come "magazzino"
per il glucosio il fegato:
• Se la glicemia scende, come durante il digiuno, il pancreas secerne glucagone che
ordina al fegato di prelevare glucosio dalle sue scorte e d'immetterlo nel sangue. Il
glucagone, inoltre, spinge le cellule all'utilizzo di grassi e proteine come fonte
energetica: in questo modo si predispone tutto l'organismo al risparmio del glucosio.
• Se invece la glicemia sale, come dopo un pasto, il pancreas secerne insulina che
comanda al fegato di prelevare il glucosio dal sangue e d'immagazzinarlo. Siccome
la capacità del fegato d'immagazzinare glucosio è piuttosto limitata (circa 70
grammi), i carboidrati in eccesso vengono convertiti in grassi e depositati nei tessuti
adiposi. L'insulina, al contrario del glucagone, spinge le cellule a utilizzare i
carboidrati come fonte energetica.
Il nostro organismo si comporta pressappoco in questo modo: quando c'è abbondanza di
glucosio si adopera per utilizzarne il più possibile, e quello in eccesso lo immagazzina
sottoforma di grassi; quando c'è carenza cerca di conservarlo il più possibile, prelevando i
grassi dalle scorte e utilizzandoli come fonte energetica.
Il meccanismo dell'insulina diventa "perverso" quando ne viene secreta troppa: in questo
caso la glicemia si abbassa troppo, il cervello va in crisi e invia all'organismo gli stimoli per
introdurre nuovo combustibile (sensazione di fame).
La quantità d'insulina secreta dal pancreas dipende dalla velocità con la quale s'innalza la
glicemia, questa velocità dipende da due fattori: l'indice glicemico e la quantità dei
carboidrati che assumiamo.
In Figura 9, in alto è riportato il caso relativo all'ingestione di carboidrati a basso indice
glicemico: la glicemia s'innalza gradualmente, viene secreta una quantità normale
d'insulina che riporta gradualmente la glicemia ai livelli precedenti l'assunzione di
carboidrati. In questo caso la fame sopraggiungerà dopo circa 3 ore.

- 25 -
Figura 9 - Effetti dell'assunzione di carboidrati sull'indica glicemico
Sempre in Figura 9 ma in basso, è riportato il caso di ingestione di carboidrati AIG (ad Alto
Indice Glicemico): a seguito della loro ingestione la glicemia subisce un brusco
innalzamento, viene secreta una quantità notevole d'insulina che causa un'altrettanto
brusca diminuzione della glicemia. In una situazione di questo tipo la fame sopraggiungerà
molto prima rispetto al caso precedente.
L'indice glicemico degli alimenti e la quantità di carboidrati non sono l'unico fattore che
influenza la quantità d'insulina che viene secreta dal pancreas, poiché esiste una diversa
reazione individuale, com'è stato dimostrato da Gerald Raven nel 1987.
Secondo i suoi studi il 25% della popolazione ha una risposta insulinica pigra. In pratica
questi fortunati individui hanno una risposta simile a quella in figura 4a anche assumendo
carboidrati ad alto indice glicemico.
Un altro 25% della popolazione ha una reazione insulinica eccessiva. Anche assumendo
carboidrati a medio indice glicemico, queste persone hanno una reazione insulinica simile
a quella di figura 4b.

- 26 -
Il restante 50% ha un comportamento che possiamo definire "normale".
Per poter mantenere l'insulina entro livelli accettabili occorre seguire queste semplici
regole:
• limitare la quantità di carboidrati giornaliera, adottando una ripartizione ottimale dei
macronutrienti (carboidrati, proteine e grassi non inferiori rispettivamente al 45%,
15% e 25%);
• assumere pasti con la corretta ripartizione dei macronutrienti (evitare i pasti a base
di soli carboidrati);
• non fare pasti troppo abbondanti;
• preferire fonti di carboidrati a bassa densità, ovvero frutta e verdura.
2.6 Insulina, sport e diete
Molte delle diete nate in questi ultimi anni si sono poste l'obiettivo di controllare la
secrezione di insulina grazie a combinazioni alimentari corrette. Un'iperproduzione di
quest'ormone conseguente al consumo di grossi quantitativi di carboidrati può infatti
portare, nel lungo periodo, a sviluppare patologie come obesità e diabete.
E’ importante notare che non è l'insulina di per sé ad essere pericolosa ma le abitudini
scorrette che ne amplificano gli effetti per cosi dire "negativi". Dunque non è importante
solo ciò che mangiamo ma anche ciò che facciamo durante la giornata. In particolare
l'organismo di uno sportivo è in grado di modulare meglio l'azione insulinica difendendosi
dai possibili effetti negativi.
Il primo punto fondamentale è che non è vero che l'insulina fa ingrassare, o meglio lo fa
solo quando si verificano contemporaneamente le seguenti condizioni:
• le scorte di glicogeno muscolare ed epatico sono sature
• si assume con la dieta un surplus di carboidrati (anche complessi) non assumendo
quantità adeguate degli altri nutrienti (grassi e proteine)
• dopo tale assunzione si svolgono attività sedentarie che impediscono l'utilizzo del
glucosio ematico in eccesso.

- 27 -
Se è vero che tali condizioni si verificano di frequente nelle persone sedentarie che si
alimentano male, è anche vero che difficilmente uno sportivo si troverà
contemporaneamente in tutte e tre le situazioni:
Un altro punto fondamentale è che l'azione dell'insulina è utile per gli sportivi: in particolare
al termine dell'attività fisica per rifornire il corpo dei carboidrati spesi durante l'esercizio
fisico. Il pasto post-allenamento deve infatti fornire il giusto apporto di zuccheri semplici in
modo da attivare un picco insulinico che ripristini le scorte di glicogeno.
Bisogna inoltre ricordare che a parità di ossigeno consumato i carboidrati hanno un
rendimento energetico superiore ai grassi. Perciò maggiori saranno le scorte di glicogeno
e migliore sarà la prestazione di un atleta impegnato in gare di durata (maratona, gran
fondo, ecc.).
Discorso a parte va fatto per body building ed attività di potenza; durante queste attività
l'organismo consuma quantità limitate di carboidrati. Ne consegue che un body builder può
trovarsi con maggiore facilità nelle tre condizioni descritte in precedenza.
Tuttavia anche gli atleti di queste discipline possono trarre vantaggio dall'azione insulinica:
infatti che l'insulina è l'ormone anabolico per eccellenza e, oltre a facilitare l'ingresso di
grassi e carboidrati all'interno della cellula, agevola anche l'ingresso delle proteine.
Ecco spiegato perché dopo l'allenamento con i pesi si consiglia di consumare carboidrati
ad alto indice glicemico (ad esempio una banana) insieme a proteine del siero. Questa
associazione causa un picco insulinico che favorisce l'ingresso degli amminoacidi nelle
cellule muscolari, dove verranno utilizzati per riparare le strutture proteiche danneggiate e
favorire l'anabolismo.
2.7 Insulino-resistenza
Si parla di Insulino-resistenza quando le cellule dell’organismo diminuiscono la
propria sensibilità all’azione dell’insulina; ne consegue che il rilascio dell’ormone, in
dosi note, produce un effetto biologico inferiore rispetto a quanto previsto.

- 28 -
Non tutte le cellule corporee necessitano di insulina per assorbire il glucosio; l’ormone è
tuttavia essenziale per il tessuto muscolare e per quello adiposo, che da soli
rappresentano circa il 60% della massa corporea.
In risposta all’Insulino-resistenza, l’organismo mette in atto un meccanismo compensatorio
basato sull'aumentato rilascio di insulina; si parla, in questi casi, di Iperinsulinemia, cioè di
elevati livelli dell’ormone nel sangue.
Se nelle fasi iniziali questa compensazione è in grado di mantenere la glicemia a livelli
normali (euglicemia), in uno stadio avanzato le cellule pancreatiche deputate alla
produzione di insulina non riescono ad adeguarne la sintesi; il risultato è un aumento della
glicemia post-prandiale. Nella fase conclamata, infine, l’ulteriore riduzione della
concentrazione plasmatica di insulina - dovuta al progressivo esaurimento delle beta-
cellule pancreatiche - determina la comparsa di iperglicemia anche a digiuno. Non
sorprende, pertanto, che l’Insulino-resistenza rappresenti spesso l’anticamera del diabete.
Brevemente, l’Insulino-resistenza determina:
• un aumento dell’idrolisi dei trigliceridi a livello del tessuto adiposo, con aumento
degli acidi grassi nel plasma;
• una diminuzione dell’uptake di glucosio a livello muscolare, con conseguente
diminuzione dei depositi di glicogeno;
• una maggiore sintesi epatica di glucosio in risposta all’aumentata concentrazione
degli acidi grassi nel sangue ed il venir meno dei processi che la inibiscono; di
riflesso si ha un innalzamento dei livelli glicemici a digiuno.
• si ritiene che l'Iperinsulinemia compensatoria renda la beta-cellula incapace di
attivare tutti quei meccanismi molecolari necessari al suo corretto funzionamento e
alla sua normale sopravvivenza. La diminuita funzionalità delle cellule pancreatiche
deputate alla sintesi di insulina apre le porte al diabete mellito di tipo II.
Il tessuto muscolare rappresenta la sede principale dell’Insulino-resistenza periferica;
tuttavia durante l’attività fisica questo tessuto perde la sua dipendenza dall’insulina ed il
glucosio riesce ad entrare nelle cellule muscolari anche in presenza di livelli insulinici
particolarmente bassi.

- 29 -
Le cause dell’Insulino-resistenza sono numerose. Dal punto di vista biologico il problema
può localizzarsi a livello pre-recettoriale, recettoriale o post-recettoriale, comprese le varie
possibili sovrapposizioni.
L’Insulino-resistenza può essere causata da fattori ormonali; è possibile, ad esempio, un
difetto qualitativo nella produzione di insulina, così come un’eccessiva sintesi di ormoni
con effetti contro-insulari. In questa classe di sostanze rientrano tutti quegli ormoni, come
l’adrenalina, il cortisolo ed il glucagone, capaci di antagonizzare l’azione dell’insulina, fino
a determinare Insulino-resistenza quando presenti in eccesso (come avviene tipicamente
nella sindrome di Cushing). Le modalità con cui questi ormoni si oppongono all’insulina
sono le più disparate: possono ad esempio agire sui recettori insulinici riducendone il
numero (è il caso del GH), oppure sulla trasduzione del segnale innestato dal legame
insulina-recettore (necessario per regolare la risposta cellulare). Quest'ultima azione
biologica consiste nella redistribuzione dei trasportatori di glucosio GLUT4* dal
compartimento intracellulare alla membrana plasmatica; tutto ciò permette di aumentare
l'approvvigionamento di glucosio. Anche l’apporto esogeno di questi ormoni (ad esempio
cortisone od ormone della crescita) può determinare Insulino-resistenza.
Possono esistere, inoltre, cause genetiche provocate da mutazioni del recettore insulinico.
Nella maggior parte dei casi, comunque, le cause dell’Insulino-resistenza non sono
chiaramente determinabili. Oltre all'immancabile componente ereditaria, nella maggior
parte dei casi l’Insulino-resistenza interessa soggetti colpiti da malattie e condizioni come
ipertensione, obesità (in particolare quella androide od addominale), gravidanza, steatosi
epatica, sindrome metabolica, uso di steroidi anabolizzanti, aterosclerosi, sindrome
dell'ovaio policistico, iperandrogenismo e dislipidemia (elevati valori di tigliceridi e
colesterolo LDL associati ad una ridotta quantità di colesterolo HDL).
Tali condizioni, associate all'immancabile componente genetica, rappresentano anche
possibili cause/conseguenze dell'Insulino-resistenza e sono importanti per la sua diagnosi;
tralasciando esami specifici, molto costosi e limitati al campo della ricerca, nella pratica
clinica si valutano le concentrazioni plasmatiche di glucosio ed insulina a digiuno. Talvolta
si utilizza anche la classica curva glicemica, che in presenza di Insulino-resistenza
presenta un andamento relativamente normale, salvo presentare poi - a distanza di varie
ore - un rapido declino della glicemia (dovuto all'Iperinsulinemia).

- 30 -
Il trattamento più efficace per l'Insulino-resistenza è dato dalla pratica di regolare attività
fisica, associata al dimagrimento e all'adozione di una dieta basata sulla moderazione
calorica e sul consumo di alimenti a basso indice glicemico. Utili anche i presidi in grado di
ridurre o rallentare l'assorbimento intestinale degli zuccheri (acarbosio ed integratori di
fibra come il glucomannano e lo psillio). Alcuni farmaci utilizzati nella cura del diabete,
come la metformina, si sono dimostrati efficaci anche nel trattamento dell'Insulino-
resistenza; tuttavia è molto importante intervenire prima di tutto sulla dieta e sul livello di
attività fisica, ricorrendo alla terapia farmacologica solo quando le modifiche dello stile di
vita non sono sufficienti.
2.8 Iperinsulinemia
Si parla di Iperinsulinemia tutte le volte che gli esami ematochimici evidenziano un
eccesso di insulina nel sangue. Questa condizione, non necessariamente patologica, è
tipica delle persone affette da diabete mellito di tipo II e - più in generale - di quelle che
hanno sviluppato una forma di resistenza all'insulina. L'Iperinsulinemia è inoltre
comunemente associata alla sindrome metabolica.
Nel corso della vita, per fattori congeniti o acquisiti, può accadere che le cellule diventino
meno sensibili all'insulina; si parla, in questi casi, di Insulino-resistenza.
Indipendentemente dalle numerose cause che possono produrre la Insulino-resistenza,
nelle fasi iniziali tale condizione determina Iperinsulinemia. Il pancreas, infatti, cerca di
compensare la ridotta sensibilità cellulare aumentando la sintesi ed il rilascio dell'ormone.
Quando tale condizione cronicizza, il superlavoro del pancreas e gli effetti negativi
dell'Iperinsulinemia stessa sulla sensibilità cellulare, provocano un declino funzionale delle
cellule adibite alla produzione di insulina e la comparsa di iperglicemia a digiuno; viene
così a cadere il meccanismo compensatorio descritto in precedenza e si può parlare a tutti
gli effetti di diabete mellito di tipo II.
Non sorprende, dunque, che l'Iperinsulinemia preceda in molti casi - anche di qualche
anno - la comparsa di diabete mellito.
Molto raramente, l'Iperinsulinemia può essere causata da un tumore che coinvolge le
cellule deputate alla produzione di insulina (insulinoma), o dalla presenza di un numero

- 31 -
eccessivo di tali cellule (nesidioblastosi). Un'Iperinsulinemia acuta (a breve termine e
transitoria) può invece essere la conseguenza di un'eccessiva assunzione di insulina o di
zuccheri.
In generale, l'Iperinsulinemia non causa segni e sintomi particolari; quando è
particolarmente accentuata può tuttavia associarsi a tremori, sudorazione, letargia,
svenimento e coma, tutti sintomi dovuti alla condizione di ipoglicemia reattiva che si viene
a creare.
Considerate le azioni endocrine dell'ormone, in presenza di Iperinsulinemia si ha anche
un'aumentata sintesi epatica di trigliceridi (ipertrigliceridemia); a livello renale, invece,
l'aumentata ritenzione di sodio favorisce la comparsa di ipertensione.
Per tutti questi motivi - e per la frequente associazione con obesità, iperandrogenismi,
steatosi epatica, dislipidemia, fumo, iperuricemia, ovaio policistico ed aterosclerosi -
l'Iperinsulinemia è considerata un importante e indipendente fattore di rischio
cardiovascolare.
Il trattamento dell'Iperinsulinemia dipende ovviamente dalle cause che l'hanno
determinata; qualora comporti una grave ipoglicemia, andrà trattata attraverso
l'assunzione di zuccheri ad alto e medio indice glicemico; nei casi più gravi si rende
necessaria un'iniezione intramuscolare di glucagone o endovenosa di glucosio.
Quando l'Iperinsulinemia è associata ad Insulino-resistenza, può essere efficacemente
trattata con la pratica di regolare esercizio fisico, dieta appropriata e perdita di peso,
eventualmente coadiuvati da integratori specifici (pectine e fibre solubili riducono
l'assorbimento intestinale di glucosio, con appiattimento della curva glicemica
postprandiale). Quando le modificazioni dietetiche e comportamentali non riescono a
riportare l'assetto glicemico a valori accettabili, si può ricorrere a speciali farmaci
ipoglicemizzanti.

- 32 -
3. Modelli matematici per la
rappresentazione dei pathway
metabolici
3.1 Modelli monocompartimentali e bicompartimentali
Il processo di eliminazione può essere rappresentato mediante il cosiddetto modello
monocompartimentale, facilmente formalizzabile in termini matematici (vedi M. Guidotti
in [Gui2008]).
Secondo questo modello, l’organismo viene considerato come costituito da un unico
compartimento aperto: si trascurano gli specifici meccanismi di trasporto attraverso la
membrana e la particolare distribuzione delle sostanze nei vari flussi e tessuti biologici.
In questo modo, i processi di assorbimento, metabolizzazione ed eliminazione sono trattati
come un semplice flusso di materia attraverso una scatola nera (black box). Tale
schematizzazione, a prima vista riduttiva, è comunque ampiamente utilizzata nelle
applicazioni per la pratica clinica.
Prendiamo come primo esempio il caso della somministrazione di una sostanza per via
endovenosa, in cui il compartimento è rappresentato dal plasma, in cui al tempo t=0
abbiamo una certa quantità q0 di sostanza e l’eliminazione della sostanza stessa tiene
conto della sua metabolizzazione.
Il modello monocompartimentale viene descritto da una semplice equazione differenziale
e
dqk q
dt= −
in cui il segno negativo implica che la velocità di l’eliminazione
dq
dt diminuisce con il
diminuire della sostanza stessa e ke è la costante di eliminazione dal compartimento.

- 33 -
Risolvendo, si ha
0ek t
q q e−=
e questo risultato dimostra come l’eliminazione della sostanza segua un andamento
esponenziale: la quantità eliminata diminuisce con il diminuire della concentrazione.
Lo stesso modello monocompartimentale è utilizzabile nel caso di somministrazione orale
di una sostanza: in questo caso il compartimento è rappresentato dal tratto
gastrointestinale, in cui abbiamo una certa quantità a0 nell’istante t=0 e l’eliminazione della
sostanza coincide con l’assorbimento della sostanza da parte del plasma.
In questo caso, avremo a
dak a
dt= − , da cui 0
ak ta a e
−= .
Accanto al modello monocompartimentale, è possibile utilizzare modelli più complessi con
lo stesso criterio della black box; un esempio tipico è il modello bicompartimentale.
In questo caso ho due black box in cui la scatola “aggiuntiva” rappresenta il metabolismo
in un insieme separato di organi.
Con questo modello, ammettendo che la velocità di assorbimento sia istantanea e quindi
irrilevante, ottengo un sistema di due equazioni differenziali da risolvere in combinazione.
11 12 1 21 2
221 2 12 1
e
dqk q k q k q
dt
dqk q k q
dt
= − − + = − +
in cui, accanto alla costante di eliminazione ke, ho la costante di trasferimento k12 dal
compartimento 1 al compartimento 2 e la costante di trasferimento k21 dal compartimento
2 al compartimento 1.
Risolvendo il sistema di equazioni differenziali, ottengo la forma finita
t tq Ae Beα β− −= +

- 34 -
dove A e B sono due costanti mentre α e β sono costanti ibride che dipendono dalle
costanti cinetiche k del modello: conoscendo i valori delle costanti k e del volume di
distribuzione i valori delle costanti sono facilmente ottenibili.
Secondo il modello bicompartimentale le sostanze si ditribuiscono inizialmente in un
compartimento centrale (che comprende il plasma e gli organi altamente perfusi, quali ad
esempio fegato e rene) prima di distribuirsi nel circolo periferico (comprendente organi e
tessuti mediamente e scarsamente perfusi).
Se un farmaco raggiunge rapidamente l’equilibrio con il comportamento tissutale, allora ai
fini pratici si utilizza il modello monocompartimentale.
3.1.1 Esempio di modello monocompartimentale: assorbimento ed
eliminazione
Trattiamo il caso più generale di assorbimento di una sostanza conseguente a
somministrazione orale, a cui segue una successiva eliminazione orale della sostanza,
utilizzando un modello monocompartimentale.
Supponiamo quindi che nel compartimento (esempio: plasma) si abbia un flusso in input
ed uno in output, caratterizzati dalle costanti ka e ke rispettivamente. Dobbiamo
considerare il fatto che il flusso in output è regolato dalla costante ke relativamente alla
quantità q di sostanza che viene via via eliminata (esempio: metabolizzazione della
sostanza da parte degli organi irrorati dal sangue), mentre il flusso in input è considerato
indipendente dalla quantità q di sostanza contenuta nel compartimento (esempio:
assorbimento da parte del tratto gastrointestinale a seguito di somministrazione orale).
Avrò quindi la relazione
a e
dqk a k q
dt= −

- 35 -
È chiaro che la quantità di sostanza eliminata aumenta al diminuire della quantità di
sostanza assorbita. Sia dunque l’assorbimento caratterizzato dalla funzione 0ak t
a a e−= , si
ottiene
0ak t
a e
dqk a e k q
dt
−= −
Risolvendo questa equazione differenziale a coefficienti costanti, si ottiene
( )0e ak t k ta
a e
kq q e e
k k
− −= −−
Facciamo immediatamente alcune considerazioni analitiche sulla soluzione ottenuta. La
funzione presenta un picco, raggiunto al tempo tmax, mentre le due funzioni rappresentanti
l’assorbimento e l’eliminazione si intersecano in un punto che non coincide con tmax.
Infatti, le due equazioni di assorbimento ed eliminazione assumono lo stesso valore
quando la quantità di sostanza assorbita coincide con la quantità di sostanza
eliminata, ma ciò non implica che la velocità di assorbimento e di eliminazione
coincidano: questa ultima condizione si ottiene imponendo appunto che le due
velocità coincidano, condizione ottenuta in tmax; precedentemente a tmax si ha che la
velocità di assorbimento è maggiore di quella di eliminazione, mentre dopo tmax si ha che
la velocità di eliminazione supera quella di assorbimento.
Il punto tmax, ottenuto eguagliando le due velocità, ovvero annullando la derivata
dell’equazione sopra riportata, coincide con
max
1t ln a
a e e
k
k k k=
−
3.1.2 Esempio di modello monocompartimentale: infusione
endovenosa continua
In questo caso, molto comune in ambiente ospedaliero, ho somministrazione continua di
una sostanza per via endovenosa.
Il farmaco viene immesso continuamente, per fleboclisi, nel torrente circolatorio, per cui si
ha 0 e
dqk k q
dt= − .

- 36 -
La soluzione di questa equazione differenziale è data da ( )0 1 ek t
e
kq e
k
−= − .
3.1.3 Esempio di modello bicompartimentale
Un esempio di modello bicompartimentale è riportato in Figura 20. Questo modello
rappresenta il metabolismo del glucosio
Figura 10 - Modello bicompartimentale del metabolismo del glucosio (fonte: [DaM2006])
tratto dal modello generale di simulazione di un pasto riportato in [DaM2006].
Le equazioni che caratterizzano questo modello sono riportate in Figura 11:
Figura 11 - Equazioni costitutive del modello bicompartimentale del glucosio (fonte: [DaM2006])
in cui Gp e Gt (mg/Kg) sono rispettivamente le masse di glucosio nel plasma e nei tessuti
rapidamente irrorati, G (mg/dl) è la concentrazione di glucosio nel plasma, il suffisso b
indica il livello basale delle sostanze, EGP è la produzione endogena di glucosio
(mg/Kg/min); Ra è il “rate of appearance” nel plasma di glucosio (mg/Kg/min); Uii e Uid
rappresentano le utilizzazioni di glucosio insulino-indipendenti e insulino-dipendenti
rispettivamente (mg/Kg/min), Vg è il volume di distribuzione del glucosio (dl/Kg) e infine k1
e k2 sono rate.

- 37 -
Nello stato basale, la produzione endogena EGPb uguaglia il “rate of disappearance”, cioè
la somma dell’utilizzazione di glucosio e di estrazione renale (EGP=Ub+Eb).
Nel caso di soggetti normali (non soggetti a Diabete di tipo 2) Eb è pari a zero.
3.2 Alcune note sul significato del Volume di
Distribuzione
L’equazione dell’assorbimento-eliminazione è stata ottenuta ipotizzando una sostanza
assorbita da una black box e da questa eliminata. Tale equazione permette di calcolare
l’andamento nel tempo della quantità di sostanza presente nell’intero organismo dopo la
somministrazione di una dose orale, sottocutanea o intramuscolare.
( )0e ak t k ta
a e
kq q e e
k k
− −= −−
Se ora consideriamo che la sostanza è in equilibrio tra sangue e tessuti, è possibile
estendere la validità di detta equazione al calcolo della concentrazione ematica.
A questo scopo si introduce un fattore di correzione, detto Volume di Distribuzione, Vd,
definito come rapporto tra la quantità q di farmaco nel corpo (equivalente alla dose
somministrata) e la concentrazione c di farmaco nel plasma:
Vd = D/C0
dove D è la dose somministrata e C0 la concentrazione plasmatica al tempo zero.
Essendo le unità di misura di q e c rispettivamente mg e mg/l, ne deriva che il volume di
distribuzione si misura in litri.
Ponendo q = VdC e q0 = VdC0=D, si ha
( )0e ak t k ta
a e
kc c e e
k k
− −= −−
In condizioni di equilibrio ogni variazione delle concentrazioni plasmatiche si riflette in una
modificazione delle concentrazioni tissutali. La relazione fra concentrazione plasmatica e
tissutale è espressa appunto dal volume di distribuzione detto anche volume di
distribuzione apparente: con questo termine si vuole sottolineare che non è una entità
anatomica, ma un volume teorico che indica come il farmaco si dovrebbe distribuire per

- 38 -
avere una concentrazione uguale a quella del plasma, qualora la distribuzione fosse
monocompartimentale.
Il volume di distribuzione apparente è influenzato dalla liposolubilità e dall’entità del
legame proteico. Il suo valore corrisponde a quello del sangue solo nel caso in cui la
sostanza non forma alcun legame con le proteine del plasma o dei tessuti; in tal caso si ha
evidentemente una limitazione nelle concentrazioni tissutali del farmaco e ciò è valido
terapeuticamente solo per le sostanze che agiscono nel circolo ematico (ad esempio i
farmaci antitrombotici).
Le sostanze con alta affinità per i tessuti extravascolari possono avere un volume di
distribuzione apparente molto superiore a quello dell’acqua corporea totale: infatti per
definizione il Vd non fa alcuna distinzione fra la concentrazione nel plasma e negli organi e
tessuti.
Se ad esempio somministriamo ad un paziente una dose di 500 mg di sostanza e ottengo
una concentrazione nel plasma di 100mg/l, allora il volume di distribuzione è di
500mg/100mg l-1=5 litri, prossima al volume medio di sangue contenuto nel corpo umano
(4.5-5.5 l).
Supponiamo invece di somministrare 500 mg di sostanza ad un paziente di 70 Kg e che la
sostanza si concentri in misura pari al 10% della dose somministrata nel plasma, per il
restante 90% negli organi e tessuti.
Il volume di plasma in una persona corrisponde al 5% della massa corporea, per cui ho
0,05*70= 3,5 litri, in cui ritrovo 0,1*500=50 mg di sostanza, per cui la concentrazione
plasmatica sarà 50/3,5 = 14.2 mg/litro. Segue che il Vd è pari a 500/14.2=35 litri.
Ad un volume di distribuzione dell’ordine del volume di plasma corrisponde una sostanza
con un elevato legame con le proteine plasmatiche; nei soggetti con stati patologici che
causano una diminuzione del legame con le proteine, aumenta il volume di distribuzione.
La distribuzione è il passaggio di una sostanza dal sangue ai diversi compartimenti
tissutali dell’organismo e dipende in gran parte dalla liposolubilità, dal flusso ematico e dal
legame con le proteine plasmatiche. Nel sangue una sostanza può essere infatti libera o

- 39 -
legata alle proteine plasmatiche: solo la quota libera di un farmaco è in grado di uscire dal
letto capillare e di raggiungere gli organi bersaglio e quelli deputati al suo metabolismo ed
escrezione.
Essendo il legame tra sostanza e proteine facilmente dissociabile c’è un equilibrio
dinamico tra la quota libera e la quota legata.
Al concetto di volume di distribuzione si lega anche la clearance, cioè la capacità
dell’organismo di eliminare una sostanza: più precisamente, la clearance è la quantità di
sangue che viene “depurata” da una sostanza nell’unità di tempo.
Maggiore è il volume di distribuzione, maggiore la quantità di sostanza che viene
concentrata nei tessuti rispetto a quella presente nel plasma e quindi ne verrà depurata
una quantità minore, ho cioè una clearance minore. Ne consegue un tempo di
dimezzamento maggiore.
3.3 Michaelis-Menten
La cinetica di Michaelis-Menten è adatta a spiegare l’attività di un enzima (un catalizzatore
biologico) rispetto ad un substrato (un reagente): enzima e substrato sono in “fast
equilibrium” col loro “complesso”, che si dissocia in prodotto ed enzima libero:
E + S �� ES � E + P
L’equilibrio tra il complesso ES e le sostanze E ed S libere è regolato dalle costanti k1 nel
verso E + S �ES e k-1 nel verso E + S � ES, mentre per ES � E + P ho la costante k2.
Si assume che la reazione sia irreversibile e che il prodotto non si leghi all’enzima.
Definiamo [ ]d P
Vdt
= il reaction rate, cioè il rate di produzione del prodotto: V dipende dalla
costante k2 e dalla concentrazione [ES], che non è quasi mai misurabile, per cui deve
essere espressa mediante i valori conosciuti [E] e [S].
Si assume allora l’approssimazione “steady state”: la concentrazione di enzima legato al
substrato (e di quello “slegato”) cambia più lentamente di quella di prodotto e substrato
k-1 [ES]+ k2 [ES] = k1 [E][S]

- 40 -
da cui (essendo per l’approssimazione “steady state” k2<<k-1)
1
1 2
[ ][ ] [ ][ ][ ]
m
k E S E SES
k k k−
= ≈+
essendo 1
1
m
kk
k
−= .
Sia quindi [E0] la concentrazione totale di enzima (free enzyme [E] e bound enzyme [ES]),
da cui [E] = [E0] – [ES].
0([ ] [ ])[ ][ ]
m
E ES SES
k
−=
da cui si ricava
0[ ][ ]
1[ ]
m
EES
k
S
=+
ed essendo il rate di produzione [ ]d P
Vdt
= , segue
2 2 0 max
[ ] [ ] [ ][ ] [ ]
[ ] [ ]m m
d P S SV k ES k E V
dt S k S k= = = =
+ +
Esprimendo gli inversi della velocità di produzione V e della concentrazione di substrato S,
si ha
max
1 11[ ]
mk
V V S
= +
che può essere rappresentata come una retta di 1/V nella variabile 1/[S].
La concentrazione del substrato cambia come la reazione prende piede, per cui si utilizza
il rate di reazione iniziale V0, con [S] come concentrazione iniziale.
Se [S]>>km, allora V=k2[E0]. Se [ ] mS k� , allora V=0,5 Vmax.
Facendo diversi esperimenti con E0 costante e differenti [S], ottengo il rate della reazione.
L’equazione di Michaelis-Menten descrive il rate delle reazioni irreversibili; si basa sulla
“legge di azione di massa”, derivata dalle assunzioni di collisioni random per diffusione e
termicamente guidate.

- 41 -
3.4 Modulazione negativa
In questo caso, ciò che si cerca di modellare è l’inibizione di una sostanza A da parte di
un’altra sostanza B o, per meglio dire, il fatto che la produzione/assorbimento di una
sostanza B causi una diminuzione nella concentrazione della sostanza A.
Quello che si deve notare è che non si vuole utilizzare un modello a compartimenti: così
facendo si sarebbe rappresentato il fatto che all’aumentare della concentrazione di una
sostanza nel compartimento diminuisce la velocità di variazione nel tempo della sua
concentrazione, del tipo dG/dt = -k*G.
Si vuole invece rappresentare la modulazione negativa di una sostanza su di un’altra, per
cui si potrebbe utilizzare una formula del tipo
A = A*/B
dove A* è una costante e in cui la concentrazione della sostanza A è inversamente
proporzionale alla concentrazione della sostanza B.
3.5 Panoramica dei modelli matematici della secrezione
dell’insulina indotta dal glucosio
Con il passar degli anni, la nostra conoscenza sulla secrezione dell’insulina da parte delle
cellule β del pancreas si è notevolmente evoluta. Tutto ciò grazie a nuove tecniche
emerse, come l’uso di proteine fluorescenti che sono in grado di individuare i granuli
secretori, immagini in tempo reale che seguono il traffico dei granuli nelle cellule viventi, le
misure della capacità della cellula che permettono di esaminare le variazioni dell’aria
superficiale della cellula dovuta al processo di fusione tra granulo-membrana.
Fin dalle prime osservazioni sulla prima e seconda fase della secrezione dell’insulina in
risposta ad uno step della concentrazione di glucosio diversi sono stati gli aspetti della
risposta che solo recentemente sono stati analizzati.

- 42 -
Le varie considerazioni fatte sull’associazione stimolo-secrezione sono state ben definite:
nella depolarizzazione lo stimolo del glucosio porta ad un aumento del rapporto ATP/ADP,
alla chiusura dei canali potassio dipendenti da ATP, alla depolarizzazione della membrana
cellulare e all’affluenza del Ca2+ che stimola l’esocitosi. L’ATP e il calcio hanno dunque un
ruolo molto importante nel processo che regola la traslocazione dei granuli dall’apparato di
Golgi alla membrana cellulare, nella fusione e nel rilascio del contenuto nello spazio
extracellulare. Altri importanti aspetti del comportamento degli isolotti sono: l’eterogeneità
della sensibilità di risposta al glucosio da parte della popolazione delle β-cellule; la
presenza di spontanee e prolungate oscillazioni —nella concentrazione di segnalare
molecole— e le oscillazioni durante il rilascio di insulina , manifestate negli isolotti isolati
sia nei modelli di animali e sia di quelli di pazienti umani.
In questo capitolo presenteremo i principali modelli matematici utilizzati per descrivere la
secrezione dell’insulina stimolata dal glucosio, in particolare ci occuperemo del Modello
Bicompartimentale, dei modelli Signal-Limited e del modello di Toffolo . Non tratteremo
invece modelli più complessi del tipo di Hodgking-Huxley che descrivono il comportamento
dell’attività elettrica delle β-cellule e delle oscillazioni da parte dell’ ATP e del Ca2+( per
esempio Chay e Kaizer 1983; Bertram e Sherman, 2004, 2005). Una rappresentazione
schematica della secrezione e della cinetica dell’insulina e del C-peptide è mostrata nella
figura seguente:
Come abbiamo visto nel precedente capitolo, una parte dell’insulina rilasciata viene
degradata dal fegato mentre tutto il C-peptide arriva nel liquido extra-cellulare, per questo
alcuni modelli, da esempio quello di Toffolo, fanno riferimento all’analisi dei dati del C-
peptide rilasciato e non all’insulina.

- 43 -
3.5.1 Modello Storage-Limited e modelli Signal-Limited:
nomenclatura e definizioni comuni
Un signal-limited system è un sistema in cui il rilascio multifasico è determinato da
variazioni di valore o espressione di un segnale, avendo lo stesso obiettivo mentre un
storage-limited system è un sistema in cui il rilascio multifasico è determinato da un
segnale costante applicato a più di un obiettivo con differente sensibilità. Il primo sarà
rappresentato come modello ad un solo compartimento a differenza del secondo che sarà
a due compartimenti chiamato dunque modello bicompartimentale. Di seguito riportiamo la
nomenclatura e le definizioni comuni ai due modelli, ovvero Signal-limited e
Bicompartimentale.
Definizioni
• t: tempo; t = 0 alla prima concentrazione di glucosio diversa da zero (minima
concentrazione);
• G(t): concentrazione esterna di glucosio(mg/dl);
• S(G,t):velocità di secrezione istantanea dell’insulina;
• S1(G, t): secrezione dell’insulina per minuto(µg/min);
• P(G,t): fattore di quantità (µg);
• K: 3.3; costante esponenziale nell’espressione per X(G,0) trovata con il metodo dei
minimi quadrati. Questa costante si adatta alla relazione sigmoidale tra quantità di
insulina secreta nella prima fase e la concentrazione del glucosio ([mg/dl])k;
• C: 1.52x107; costante nel denominatore nell’espressione X(G,0) trovata con il
metodo dei minimi quadrati. Anche questa costante si adatta alla relazione
sigmoidale tra quantità di insulina secreta nella prima fase e la concentrazione del
glucosio([mg/dl])k;
• Kp: 7.0; costante trovata con il metodo dei minimi quadrati conforme alla relazione
sigmoidale tra P(G,∞)e la concentrazione di glucosio ([mg/dl])kp ;
• Cp: 2.25x1015; costante trovata con il metodo dei minimi quadrati conforme alla
relazione sigmoidale tra P(G,∞)e la concentrazione di glucosio ([mg/dl])kp ;
• α: 0.0337 min−1; variazione del fattore P(G,t).
Equazioni: dP(G, t)/dt = α (P[G,∞] − P[G, t])

- 44 -
P(G,∞) = G KP / (Cp + GKp) Input-Output input : G(t) output : S(G, t) Assunzioni comuni ai due modelli:
• ´è valido usare in ogni modello la risposta sigmoidale per le quantità di insulina
secreta nella prima fase derivante dalla somministrazione di glucosio
• l’insulina è immagazzinata in uno o più compartimenti delle β cellule del pancreas
• esiste un fattore P(G,t) che controlla la velocità di secrezione della seconda fase; è
assunto che vari di valore dopo 50/60 min (vedi [OCo80]).
Le differenze tra i due modelli e quindi l’equazioni matematiche rispettive sono presentate
nei prossimi paragrafi.
3.5.2 Modello Bi-Compartimentale
Questo modello è stato studiato e sviluppato da G. Grodsky e si basa sulla
rappresentazione delle riserve di insulina suddivise in due compartimenti come mostrato in
Figura 12. Il compartimento Stable è il più grande dei due, contiene circa il 98%
dell’insulina complessivamente immagazzinata nel pancreas, al contrario il compartimento
Labile contiene soltanto il restante 2%. I due compartimenti si differenziano per il
comportamento dinamico ovvero, nel momento in cui viene superato il livello basale di
glucosio è il compartimento Labile ad occuparsi dell’emissione dell’insulina per ristabilire
l’equilibrio. L’emissione di insulina secondo questo modello, avviene in due distinte fasi:
1. la prima d’intensità elevata e durata breve. In questa fase il compartimento Labile
perde l’insulina contenuta.
2. la seconda di durata molto maggiore, circa 50 minuti; in questa fase la frequenza di
emissione e linearmente crescente e coincide con il trasferimento progressivo
dell’insulina contenuta nel compartimento Stable verso il compartimento Labile.
Questo trasferimento è regolato dal fattore P(G,t).
In Figura 12 è riportata la schematizzazione del modello (vedi [OCo80]).

- 45 -
Figura 12 - Modello a bicompartimentale
Inizialmente il modello presentava un inconveniente per il fatto che una misura costante
del compartimento labile non era capace di rilevare che una concentrazione più alta del
glucosio provocava un aumento in ampiezza della prima fase della risposta. Per questa
ragione successivamente il modello è stato ridefinito assumendo che il compartimento
labile non era omogeneo nel senso che conteneva una popolazione di β-cellule
(denominate pacchetti) avente differente soglia di sensibilità al glucosio. L’insulina
immagazzinata nei pacchetti veniva dunque rilasciata tutta-o-niente quando si superavano
le soglie di sensibilità al glucosio.
Le equazioni del modello sono
Parametri:
• K1(G, t)= K0 + γ P(G,t)/Xs(G, 0); parametro che controlla la velocità con cui avviene
lo scambio di insulina tra i due compartimenti(min−1);
• Xs(G, 0)= XsmaxGK/(C+GK); quantità dell’ insulina nel compartimento stabile(µg);
• K0= 0.0002(min−1); velocità costante dal compartimento stabile a quello labile prima
della stimolazione del glucosio (quando P(G,t)=0);
• γ= 0.5(min−1);efficienza del fattore di quantità P(G,t);
• Xsmax= 82.5(µg); quantità iniziale totale di insulina nel compartimento stabile;
• Xmax= 1.65(µg); quantità iniziale totale di insulina nel compartimento labile;
• K2= 0.01 (min−1); velocità costante;

- 46 -
• m= 0.622(min−1); velocità costante di rilascio dell’insulina dal compartimento
piccolo.
Condizioni iniziali :
• t=0;
• K0 Xs(G, 0)= K2X(G,0);
• P(G,t)= 0;
• XS(G, t)= Xs(G, 0);
• X(G,t)= X(G,0);
• X(G,0)=Xmax GK/(C+GK); quantità iniziale di insulina disponibile nel compartimento
labile come una funzione della concentrazione di glucosio(
3.5.3 Modelli Signal-Limited
I modelli signal-limited sono stati studiati e sviluppati da Cerasi e altri colleghi. Questi
abbandonano il modello a compartimenti in favore dell’idea che il rilascio bifasico di
insulina è determinato dalla interazione dinamica tra stimolazione e inibizione da parte del
glucosio. Questi due eventi hanno una propria cinetica e dose dipendente. In questi
modelli si assume che un alto livello di glucosio rapidamente provoca un segnale che da
inizio al rilascio di insulina (initiation). Questo segnale è dunque proporzionale all’aumento
della concentrazione del glucosio dal valore basale. Oltre al segnale initiation, il glucosio
accresce la sensibilità degli isolotti, un processo denotato con potentiation, che aumenta la
velocità di rilascio di insulina provocata dall’azione iniziale del glucosio. Questo processo è
assunto essere tempo-dipendente. In fine il glucosio induce un effetto inibitorio (inhibition)
sul rilascio di insulina, riducendo cosi gli effetti di stimolazione dell’initiation e potentiation.
Se il tempo di inhibition è intermedio tra quello di initiation e di potentiation la
sovrapposizione di questi tre segnali e capace di produrre la classica risposta bifasica
delle β-cellule con i suoi diversi aspetti (vedi [Cau2003]).

- 47 -
Figura 13 - Modello Signal-Limited
Presentiamo di seguito due forme di Modelli Signal-limited ovvero il delta-signal model e il
modello eccitatore-inibitore(feedback).
3.5.3.1 Delta-Signal Model
Questo modello è il risultato di associare la fase iniziale di rilascio dell’insulina con la
velocità di cambio di un segnale. Di seguito verrà assunto che l’insulina è immagazzinata
in un solo compartimento; che la differenza di quantità di glucosio determina la velocità di
rilascio della prima fase della secrezione dell’insulina; che le differenze prodotte da una
variazione di concentrazione di glucosio vengono riequilibrate dopo cinque minuti; c’è un
componente che reagisce lentamente alla somministrazione del glucosio e che si riporta in
equilibrio soltanto sessanta minuti dopo che la concentrazione di glucosio si è stabilizzata;
la velocità con cui l’insulina è trasportata nelle cellule dove è immediatamente disponibile
è illimitata (vedi [OCo80]).
Di seguito riporteremo dettagliatamente le definizioni formali del modello in oggetto.
Equazioni del modello:

- 48 -
Parametri:
• Y(G,t)= quantità del glucosio??all’interno di alcune strutture cellulari(µg); si assume
che Y’(G)vari il suo valore in maniera istantanea;
• Y(G,∞)=Ymax GK/(C+GK);
• Ymax= 1.65µg; costante scelta con il metodo dei minimi quadrati che si adatta con K
e C rispettando la relazione sigmoidale tra Y(G,∞) e G;
• D0= 0.5(min−1); costante che determina la velocità di aumento(o decremento)di
Y(G,t) dentro alcune cellule;
• D1= 0.52(min−1); costante relativa alla differenza tra Y(G,t) e la velocità di
secrezione dell’insulina S0(G, t) ;
• D2= 1.3(µg/min)−1; costante relativa al prodotto di P(G,t)e il limite di Y(G,t) quando t
tende all’infinito.
Condizioni iniziali :
• t = 0;
• Y(G,t)= 0;
• P(G,t)= 0;
Esiste anche un modello modificato del delta-signal che differisce dall’originale solo per
l’equazione della velocità di secrezione istantanea dell’insulina che diventa:
S(G, t) = {D1[Y (G,∞) − Y (G, t)] + D2[Y (G,∞)]P(G, t) + D3[Y (G,∞) − Y (G, t)]P(G, t)}+
dove D3 = 3.5(µg/min)−1.
3.5.3.2 Modello eccitatore-inibitore
Anche per questo modello, si ipotizza che l’insulina sia contenuta in un solo
compartimento; si assume che la differenza tra eccitatore e inibitore all’interno delle cellule
β controlli il rilascio costante e determini la velocità della prima fase della secrezione
dell’insulina; che la variazione della concentrazione di glucosio esterna porta rapidamente
la quantità di eccitatore ad un nuovo valore stabile; che qualche minuto dopo un veloce

- 49 -
aumento o decremento della quantità di eccitatore, anche l’inibitore si porta lentamente
allo stesso valore; c’è un componente, dipendente soltanto dall’eccitatore, che reagisce
lentamente alla somministrazione del glucosio e che si riporta in equilibrio soltanto
sessanta minuti dopo che la concentrazione si è stabilizzata; la velocità con cui l’insulina è
trasportata nelle cellule dove è immediatamente disponibile è illimitata Cf.[OCo80].
Le equazioni del modello sono :riportate in Figura 14
Figura 14 - Equazioni del modello di Toffolo
Parametri:
• E = eccitatore;
• I = inibitore;
• E(G,t) = quantità di eccitatore alla concentrazione G e al tempo t (µg);
• I(G,t) = quantità di inibitore alla concentrazione G e al tempo t (µg);
• Emax = 1.65(µg);
• H0= 3.0 (min−1); costante che determina la velocità di aumento(o decadimento)della
quantità di eccitatore E(G,t);
• H1= 0.6 (min−1); costante che determina la velocità di aumento (o decadimento)
della quantità di inibitore I(G,t);
• H2= 0.24(µg/min)−1;
• H3= 1.0(min−1); costante relativa alla differenza tra eccitatore e inibitore;
• N = 0.95; costante che determina la minima velocità di secrezione alla fine della
fase iniziale.
Condizioni iniziali :

- 50 -
• t = 0;
• E(G,t)= 0
• I(G,t)= 0
• P(G,t)=0
In Figura 15 seguente sono riportate le rappresentazioni schematiche dei due modelli
signal-limited: la lettera A indica il Delta-Signal e la lettera B il modello Eccitatore-Inibitore.
Figura 15 - Delta-Signal model (a sinistra) e modello Eccitatore-Inibitore (a destra)
3.5.4 Modello di Toffolo
Il modello minimo di Toffolo postula che la secrezione dell’insulina in risposta ad un
aumento del glucosio è data dalla somma di due componenti, una statica e l’altra
dinamica. La componente statica o controllo statico è proporzionale alla variazione di
concentrazione del glucosio nel plasma e dipende dai parametri
1. α: ritardo dinamico tra l’aumento della concentrazione del glucosio e il rilascio
dell’insulina da parte del pancreas ;
2. β: costante di proporzionalità tra la concentrazione di glucosio e la componente
statica della risposta di secrezione.
La componente dinamica o controllo dinamico è proporzionale, invece, alla derivata della
concentrazione di glucosio e dipende solo da un parametro
Kd: costante di proporzionalità tra la velocità di aumento del glucosio e la componente
derivata della risposta di secrezione (vedi [Cau2003]).

- 51 -
La trattazione matematica fa riferimento alle misure del C-peptide presente nel plasma,
perciò facendo riferimento alle sue cinetiche il modello è rappresentato da due
compartimenti come mostra la Figura 16:
Figura 16 - Modello bicompartimentale del C-Peptide
Le equazioni del modello sono:
dove:
• CP1 (pmol/l) è la concentrazione del C-peptide nel compartimento1(circa basale);
• CP2 (pmol/l) è la concentrazione del C-peptide nel compartimento 2 (circa basale),
equivalente al C-peptide del compartimento2 diviso quello del compartimento 1 ;
• k12 e k21(min−1) sono i parametri che regolano la velocità di trasferimento tra i due
compartimenti;
• k01(min−1 ) è la perdita irreversibile dal compartimento 1;
• SR (pmol * l−1 *min−1) è la secrezione pancreatica entrante nel compartimento1
(circa basale), normalizzata al volume dalla distribuzione del compartimento 1.
Come per il modello IVGTT (vedi [Tof95]) il rapporto funzionale tra la secrezione
dell’insulina e la concentrazione di glucosio nel plasma è derivata da un modello
precedentemente proposto (vedi [Gro72], [Lic73]) basato sull’ipotesi di immagazzinamento
di insulina sottoforma di pacchetti. SR è descritto come la somma di due termini,
rispettivamente, la concentrazione del glucosio( controllo statico) e la velocità di variazione
della concentrazione del glucosio(controllo dinamico):
SR(t) = SRs(t) + SRd(t)
SRs è assunto essere uguale a Y (pmol l-1 min−1), il rifornimento di nuova insulina alle β-
cellule

- 52 -
SRs(t) = Y (t) quest’ultimo è controllato dal glucosio secondo la seguente equazione:
˙Y(t) = −αY (t) − β[G(t) − Gb] Y (0) = 0 SRd rappresenta la secrezione dell’insulina immagazzinata nelle β cellule pronta al
rilascio.
L’insulina labile non si comporta in maniera omogenea rispetto alla stimolazione del
glucosio: per un dato gradino di glucosio, solo una frazione dell’insulina labile è
mobilizzata. Si assume quindi che la quantità di insulina rilasciata sia direttamente
proporzionale all’incremento della quantità di glucosio somministrata (dG):
dQ = kd dG
Il flusso della secrezione dell’insulina, SRd, è proporzionale alla derivata del glucosio
Il parametro kd descrive il controllo dinamico del glucosio sulla secrezione dell’insulina(
variazione positiva dG dt ).
In realtà il modello descritto non si adatta a tutti i soggetti sottoposti all’esperimento (vedi
[Tof2001]), per questo è stato studiato un secondo modello che differisce dal precedente
per una più flessibile gestione del controllo dinamico (Figura 17): SRd è ancora
proporzionale alla derivata del glucosio, ma il fattore di proporzionalità varia con il glucosio
Figura 17 - Gestionde statica (a sinistra) e dinamica (a destra) del coefficiente k per la secrezione
dinamica

- 53 -
Come si evince dalla precedente equazione il controllo dinamico è massimo quando il
glucosio aumenta dal suo valore basale, decresce linearmente con la concentrazione del
glucosio e diventa nulla quando quest’ultima supera la soglia Gt; da li in poi un ulteriore
incremento di glucosio non produce effetto sulla secrezione dell’insulina.
Di fatto quest’ultimo modello (M2) per valori elevati di Gt può essere approssimato con il
modello descritto inizialmente(M1); il termine
( )1 b
t b
G t G
G G
−−
−
si approssima ad 1 ed M2 si riduce ad M1.
Entrambi i modelli permettono di ricostruire l’andamento della secrezione dell’insulina
durante la somministrazione del glucosio in base alle seguenti equazioni:
1
01 1
01 1
1: ( ) { ( )}
{ ( ) } , 0 ( )
{ ( )} ,
b
b d b
b
M ISR t SR SR t V
dG dGk CP Y t k V se e G t G
dt dt
k CP Y t V altrimenti
= + ⋅ =
= ⋅ + + ⋅ > >
= ⋅ + ⋅
1
01 1
01 1
2 : ( ) { ( )}
( ){ ( ) [1 ] } , 0 ( )
{ ( )} ,
b
bb d b
t b
b
M ISR t SR SR t V
G t G dG dGk CP Y t k V se e G t G
G G dt dt
k CP Y t V altrimenti
= + ⋅ =
−= ⋅ + + ⋅ − ⋅ ⋅ > >
−
= ⋅ + ⋅
dove SRb è la secrezione dell’insulina allo stato basale, e V1(in litri) è il volume del C-
peptide nel compartimento1.
Per concludere si definiscono tre indici di sensibilità:
1. statico: φs(min−1)=β, per entrambi i modelli.
2. dinamico: φd = X0 Gmax−Gb

- 54 -
Il parametro X0 (pmol/l) è la quantità di insulina (per unità di volume del C-peptide)
rilasciata in seguito alla massima concentrazione di glucosio ottenuta durante
l’esperimento.
Per M1:
X0 = kd(Gmax − Gb)
Per M2:
X0 = kd [1-(Gmax − Gb)/2(Gt − Gb)] (Gmax − Gb), se Gt > Gmax
X0 = kd (Gt − Gb)/2, se Gt < Gmax
3. basale: φb(min−1)=SRb/Gb , per entrambi i modelli. I tempi di risposta da parte delle β-cellule sono:
• Tdown = 1/α, tempo con cui le β-cellule rispondono ad un decremento del glucosio ;
• Tup = 1/α- φd/φs, tempo con cui le β-cellule rispondono ad un aumento del glucosio.
Il modello minimo del C-peptide è stato applicato con successo al’IVGTT per stimare la
secrezione effettuata dalla cellule β durante perturbazioni di glucosio (vedi [Tof99]).
3.6 La modellizzazione del metabolismo dell’insulina
Le anormalità nella secrezione dell’insulina sono importanti fattori che determinano il
diabete mellito e altri stati di “glucose intolerance”. Tuttavia, l’assessment della funzione
della cellula beta negli umani in condizioni fisiologiche è di difficile determinazione dato il
complesso rapporto tra le variabili fisiologiche.
Benché la funzionalità della cellula beta è comunemente associabile ai valori di insulina
plasmatici, questa misurazione è caratterizzata da un fattore di errore molto importante

- 55 -
dovuta all’estrazione epatica di insulina; d’altra parte, le misure del C-peptide e
dell’insulina consentono di avere simultaneamente un assessment della funzionalità della
cellula beta e dell’estrazione epatica.
Una difficoltà aggiuntiva è che la sola misura dell’insulina fornisce dettagli limitati, in
quanto sarebbe utile capire il funzionamento della cellula beta sotto condizioni fisiologiche,
cioè si vuole capire l’influenza della cellula beta sui livelli di grassi, zuccheri e altre
sostanze nutritive con un semplice test fisiologico che tenga conto di tutte queste variabili.
Quali sono I metodi disponibili e quali le loro limitazioni? La Tabella 2 fornisce una lista dei
protocolli più comunemente utilizzati con i loro vantaggi e limitazioni.
La conoscenza dei valor plasmatici di glucosio e insulina fornisce importati ma limitate
informazioni sulla funzionalità della cellula beta. Infatti, le concentrazioni basali riflettono
solamente dei punti singoli della complessa curva dose(glucosio)-risposta(insulina) e
quindi non possono fornire dettagli sulla risposta della cellula beta all’alzarsi o
all’abbassarsi della curva glicemica, corrispondente alle condizione fisiologiche ottenute
dopo aver mangiato.
Sono stati proposti approcci alternativi basati su protocolli orali e intra-venous.
Il Hyperinsulinemic euglycemic clamps (EHC)[DeF79] è in grado di misurare l’azione
dell’insulina data una certa concentrazione di insulina; tuttavia non fornisce un assessment
della funzionalità della cellula beta.
In aggiunta, dato che sotto certe condizioni il livello di insulina non rimane a concentrazioni
elevate per molte ore e la stessa insulina è secreta nella vena porta invece di essere
infusa nelle vene periferiche, questo metodo non è fisiologico.
Quindi i test Hyperglycemic clamps e graded glucose infusions possono essere usati per
l’assessment della cellula beta ma sono limitati dal fatto che tali test non sono fisiologici.

- 56 -
Tabella 2 - Protocolli, loro attributi e contenuto informativo
I modelli di secrezione insulinica permettono di valutare la funzionalità della cellula beta
seguendo l’iniezione intravenosa di un bolo di glucosio, ad esempio durante un IVGTT
(intravenous glucose tolerance test) o dopo l’ingestione di glucosio, come in un OGTT
(oral glucose tolerance test) o in un mixed meal (pasto misto). I protocolli del tipo oral
perturbation (OGTT, meal) sono senza dubbio quelli più fisiologici rispetto a quelli
intravenous per la presenza di nutrienti quali proteine e grassi.
L’azione dell’insulina e l’estrazione epatica di insulina possono anch’esse essere valutate
con questi metodi (IVGTT, OGTT e meal). E’ bene notare che, rispetto all’azione
dell’insulina, i modelli intravenous sono più semplici da creare rispetto a quelli orali, perché
l’input nella circolazione sanguigna (cioè la quantità di glucosio iniettato) è noto. Al
contrario, la misura dell’azione insulinica durante un protocollo orale richiedono un
assessment del rate o f appearance del glucosio ingerito: per questo motivo, questi
modelli orali sono stati utilizzati solo recentemente, grazie alle ultime tecniche di
tracciamento multiplo.
La secrezione di insulina può essere valutata immediatamente a seguito di una
perturbazione di glucosio, misurando la concentrazione di C-peptide e quindi facendo la
deconvoluzione. Questo approccio è semplice e virtualmente indipendente dal modello
([Eat80], [Pol86]); tuttavia è più difficile misurare la funzionalità della cellula beta, in quanto
ciò richiederebbe di prendere in considerazione il cambiamento di glucosio nella risposta
della cellula beta. Per far ciò, è necessario trovare un meccanismo causale che colleghi gli
andamenti temporali delle due grandezze o, come si dice più comunemente, occorre
trovare un modello della funzionalità della cellula beta.

- 57 -
Infine, si vuole rimarcare il fatto che il test di tipo clamp viene considerato “Gold Standard
Data”: ciò significa che la bontà di un test viene verificata sempre rispetto ai risultati
ottenuti dal clamp.
3.7 Le grandezze fondamentali derivate dai modelli
Per valutare l’efficienza del sistema di regolazione del glucosio è cruciale misurare la
Insulin Sensiticity (SI), cioè l’abilità dell’insulina nel controllare la produzione e
utilizzazione di glucosio.
Questo indice è utile non solo a scopi diagnostici ma anche per valutare l’efficacia di una
terapia.
In genere la Insulin Sensitivity è misurata con metodi che comportano l’uso di iniziazioni
endovena di glucosio o insulina, quali il glucose clamp technique o l’IVGTT interpretato
con il modello minimale.
Entrambi i metodi sono ampiamente usati in contesti di laboratorio e in particolare l’uso del
modello minimale è preferito in quanto richiede l’utilizzo di tecniche di calcolo semplici,
senza dover appoggiarsi troppo alle apparecchiature del laboratorio steso.
Prendiamo ad esempio il modello minimale IVGTT presentato in Figura 18 a sinistra.
Come si può vedere, il modello si basa sulle misurazioni di glucosio G(t) e di insulina I(t).
Definiamo con i(t) il livello dell’insulina scalato del suo valore basale Ib (misurato a
digiuno), per cui
i(t) = I(t) - Ib
Come si può vedere dalla figura, ho nel compartimento dell’insulina remota i’(t) un flusso in
ingresso dovuto all’insulina del plasma e un flusso in uscita dovuto alla sua utilizzazione,
per cui si ha:
2 3
'( )( ) '( )
di tk i t k i t
dt= ⋅ − ⋅

- 58 -
Contemporaneamente, ho un bilancio relativo al glucosio, che da una parte vede l’azione
da parte del fegato (NHGB, Net Hepatic Glucose Balance) dall’altra l’utilizzazione del
glucosio da parte delle cellule.
Si ha quindi:
0 5 6
1 4
( )
( ) '( ) ( )
( ) '( ) ( )
dG tNHGB U
dt
NHGB B k G t k i t G t
U k G t k i t G t
= −
= − ⋅ − ⋅ ⋅
= ⋅ + ⋅ ⋅
Per il bilancio epatico, si nota un rilascio costante B0, un uptake dipendente solo dal
glucosio e un uptake mediato dall’insulina.
Analogamente, l’utilizzazione del glucosio dipende in parte dal solo glucosio e in parte è
mediato dall’insulina.
Segue, con pochi semplici passaggi
0 1 5 4 6
( )( ) ( ) ( ) '( ) ( )
dG tB k k G t k k i t G t
dt= − + ⋅ − + ⋅ ⋅
in cui si distinguono i contributi del solo glucosio e quelli mediati dall’insulina.
Definiamo inoltre come Insulin Action
4 6 4 6
2 3
4 6
4 6 2 4 6 3
( ) '( )( ) '( ) ( ) ( )
1 ( ) '( )( ) '( )
( )
( )( ) ( ) ( ) '( )
dX t di tX t i t k k da cui k k
dt dt
dX t di tk i t k i t
k k dt dt
dX tk k k i t k k k i t
dt
= ⋅ + = + ⋅ ⇒
⇒ ⋅ = = − ⋅ ⇒+
⇒ = + ⋅ ⋅ − + ⋅ ⋅
e infine:

- 59 -
4 6 2 3 3 2
0 1 5 4 6 4 1
( ) ( )( ) ( ) ( ) ( ) ( ), (0) 0
( ) ( )( ) ( ) ( ) '( ) ( ) ( ) ( ) ( ), (0) b
dX t dX tk k k i t k X t p i t p X t x
dt dt
dG t dG tB k k G t k k i t G t p p G t X t G t G G
dt dt
= + ⋅ ⋅ − ⋅ = ⋅ − ⋅ = ⇒
= − + ⋅ − + ⋅ ⋅ = + ⋅ + ⋅ =
Si definisce Glucose Effectiveness E, la variazione della velocità di scomparsa del
glucosio rispetto alla variazione di glucosio stesso, per cui:
1
( )
( )
Gd
tE p X tdG
∂∂= = +
da cui la Insulin Sensitivity
( )I
E dX tS
i di
∂= =∂
Considerando condizioni stazionarie, si ha:
3 33 2
2 2
( )( ) ( )0 ( ) ( ) 0 ( )
p i t pdX t dX tp i t p X t X t
dt p di p
⋅= ⇒ ⋅ − ⋅ = ⇒ = ⇒ =
e quindi
3
2
I
pS
p=
Per calcolare i parametri p2 e p3, basta analizzare i campioni di glucosio e insulina
plasmatici. Infatti:
33 2
2 3 0
3 2
2 0
( ) ( )0 ( ) ( ) 0 ( )
( )
( )
I
I
pdX t X tp i t p X t X t dt
dt i t p pS
p pS i t dt
p
∞
∞
= ⇒ ⋅ − ⋅ = ⇒ =⇒ = =
=
∫
∫

- 60 -
dove l’integrale a denominatore è semplicemente l’area sotto la curva della
concentrazione di insulina, mentre l’integrale a denominatore dipende dalla
concentrazione di glucosio.
Le tecniche clamp e IVGTT sono non fisiologiche in quanto nelle condizioni di vita di ogni
giorno un individuo non è mai soggetto ad una concentrazione costantemente elevata di
insulina, non coordinata con un innalzamento di glucosio, cosa che accade appunto con i
test clamp e IVGTT. E’ quindi preferibile avere un test più fisiologico, ad esempio usare un
pasto opportunamente bilanciato: questo tipo di test è anche più adatto ad effettuare test
di massa su centinaia o addirittura migliaia di soggetti!
Per utilizzare un modello in cui il glucosio è introdotto per via orale, si deve considerare il
Rate of Appearance Ra del glucosio nel plasma, in quanto, diversamente dal caso di un
bolo di glucosio introdotto per via endovenosa e che si presenta nel plasma come un picco
di glucosio (spesso viene associato ad un delta di Dirac), nel caso orale è il tratto gastro-
intestinale che “addolcisce” la curva con cui il glucosio appare nel sangue.
* *
3 2
* *
4 1
( )( ) ( )
( ) ( )( ) ( ) ( )
dX tp i t p X t
dt
dG t Ra tp p G t X t G t
dt V
= ⋅ − ⋅ = + ⋅ + ⋅ +
dove Ra(t) è in genere una equazione lineare a tratti o, nei modelli più complessi, uno
spline model.
Si vuole qui rimarcare il fatto che durante questi test orali, avviene comunque una
infusione endovenosa di glucosio: infatti, quella che si andrebbe a misurare nel sangue
sarebbe una concentrazione di glucosio già degradata dagli effetti dell’insulina. L’infusione
di glucosio da’ una base di da degradare da parte dell’insulina: per questo motivo il
glucosio ingerito e quello per via endovenosa sono caratterizzati da due diversi traccianti.
La funzionalità della cellula beta deve essere interpretata anche rispetto alla sensibilità
all’insulina: una possibilità per far ciò è utilizzare il Disposition Index, introdotto nel 1981 da
Bergman ([Ber81]), secondo cui il prodotto tra sensibilità all’insulina e funzionalità della
cellula beta è costante; questo concetto è abbastanza accettato, in quanto in caso di una

- 61 -
tolleranza al glucosio normale, a seguito di una diminuzione della sensibilità all’insulina si
avrebbe un aumento della funzionalità della cellula beta. Invece, un aumento non
adeguato della funzionalità della cellula beta corrisponderebbe ad una intolleranza al
glucosio.
Questo metodo, introdotto per l’IVGTT, è stato adottato anche per l’OGTT e il clamp.
Certamente, è da rimarcare, relazioni più complesse di quella iperbolica sono più adatte
alla descrizione del sistema insulinico e il Disposition Index mal si adatta nel caso della
comparazione tra popolazioni diverse, oltre al fatto che non tiene conto di differenti livelli di
estrazione epatica.
Figura 18 - Modelli minimali di sensibilità all'insulina
L’indice AIR (acute insulin response) è forse quello più popolare tra quelli utilizzati; dopo
l’iniezione, il glucosio rapidamente sale da 90 a 350 mg/dl. Il livello di insulina plasmatica è
misurato nei primi 10 minuti e la sua area sotto la curva (AUC) viene calcolata.
L’AIR riflette il rilascio istantaneo di insulina dai granuli precedentemente agganciati alla
membrana della cellula beta che caratterizzano il rilascio immediato della cellula beta.
Tuttavia AIR, basandosi sulla secrezione di insulina, riflette anche l’estrazione epatica non
fornisce un assessment indipendente della secrezione insulinica.
Come discusso precedentemente una accurata misura della funzionalità della cellula beta
deve tenere in considerazione anche la sensibilità all’insulina. Il modello IVGTT consente
di effettuare un assessment dell’indice di sensibilità all’insulina che è stato validato in
numerosi studi e che moltiplicato per AIR consente di calcolare un tipo di Disposition

- 62 -
Index, così da determinare se il secrezione insulinica è appropriato per il livello di azione
insulinica.
Gli indici e i ritardi di prima e seconda fase misurati con l’AIR del C-peptide hanno due
limitazioni: prima di tutto, sono dei “compositi” di funzionalità della cellula beta e
dell’estrazione epatica dell’insulina e poi, sondano il comportamento della cellula beta in
un breve periodo di tempo e immediatamente dopo uno stimolo di glucosio marcatamente
sopra-fisiologico.
La funzionalità della cellula beta può essere misurata dalle concentrazioni plasmatiche di
C-peptide misurate durante un IVGTT standard o modificato con insulina (vedi Figura 19 a
sinistra), usando il modello minimale mostrato, basato sui primi lavori di Grodsky e colleghi
( [Gro72], [Lic75]). L’uso del C-peptide invece che delle concentrazioni di insulina
permettono di evitare il problema dell’estrazione epatica.
Figura 19 - Modelli minimali di risposta della cellula beta
La cinetica del C-peptide è rappresentata da u n modello bi-compartimentale in cui la
secrezione insulinica è caratterizzata da due componenti di prima e seconda fase, dove la
prima fase rappresenta l’esocitosi dei granuli docce (e dove viene introdotto il controllo
legato alla derivata dell’andamento del glucosio) e la seconda fase deriva dal rifornimento
di nuovi granuli prodotti proporzionalmente al alla concentrazione di glucosio e che
raggiungono il pool di granuli rilasciabili con un ritardo costante T.
Un test orale differisce da un IVGTT per diverse ragioni. Queste includono il route orale
della fornitura di glucosio, la associata secrezione di ormoni incretinici, il pattern più

- 63 -
fisiologico di rilascio di glucosio, insulina e C-peptide e la presenza di nutrienti diversi dal
glucosio quali aminoacidi e grassi, nel caso di mixed meal.
Tutti i componenti della risposta già presentati per l’IVGTT sono presenti anche nei modelli
orali (vedi Figura 19 a destra), ad eccezione del fatto che la risposta immediata è più
dolce, dovuta al fatto che ho una introduzione di glucosio più lenta.
Quindi nel modello orale ho un una componente che rappresenta la risposta dinamica
sensibile al rate di cambiamento di glucosio e una statica, comprensiva di ritardo, sensibile
alla concentrazioni di glucosio.
E’ da notare che, mentre nel IVGTT la risposta dinamica contribuisce per i primi due
minuti, nel modello OGTT e in generale in quelli orali, tale risposta contribuisce per i primi
60-90 minuti.

- 64 -
4. Metodi di analisi qualitativa e di
analisi quantitativa dei modelli
mediante reti di Petri
4.1 Reti di Petri
Nel 1962 Carl Adam Petri presentò, nella propria dissertazione di dottorato, un formalismo
utile alla modellistica ed analisi del comportamento dinamico di sistemi ad eventi discreti:
le reti di Petri.
Si trattava di uno strumento generale per la modellizzazione di sistemi concorrenti, con
particolare attenzione alla descrizione dei rapporti di causalità tra eventi.
L’utilizzo delle PN condente di superare alcuni dei limiti degli Automi a Stati Finiti (ASF).
Infatti un ASF presenta, tra le altre, queste caratteristiche:
• Modello computazionale a memoria finita (proporzionale al numero degli stati )
• Il numero di stati aumenta velocemente all'aumentare della complessità del
sistema: componendo automi che descrivono sottosistemi, l'automa globale ha N x
M stati (a meno di minimizzazioni) a cui però in pratica si ovvia gerarchizzando gli
automi:
• Natura intrinsecamente sequenziale
o lo stato corrente di un automa è singolo, concentrato
o l'automa può effettuare una sola transizione per volta
• Difficile modellare sistemi concorrenti in cui più parti evolvono indipendentemente,
con tempi propri
I motivi che rendono interessante l’utilizzo delle PN sono molteplici: innanzitutto le PN
sono state proposte come uno strumento di modellizzazione formale necessario sia per
eliminare ogni fonte di ambiguità nella rappresentazione sia per effettuare analisi e

- 65 -
verifiche sul comportamento del sistema. Inoltre, sono dotate di una rappresentazione
grafica intuitiva che ne facilita l’uso anche con strumenti dedicati assistiti da calcolatore,
ma soprattutto sono una estensione significativa degli automi: la PN infatti possono
rappresentare in modo compatto concetti generali quali sincronizzazione tra processi, il
succedersi asincrono di eventi, operazioni concorrenti, conflitti e condivisione di risorse.
Formalmente, una rete non marcata è una 5-pla N=(T,P,F,W,M0) dove P è l’insieme dei
posti, T è l’insieme delle transizioni, F è la relazione di flusso, W la funzione peso e M0 la
marcatura iniziale della PN; in particolare si ha
• P∩T=0
• P∪T≠0
• F⊆(PxT) ∪(TxP)
dove la prima condizione significa che gli insiemi dei posti e delle transizioni devono
essere disgiunti, la seconda che la rete per esistere deve avere almeno una transizione o
un posto e la terza ci indica la relazione che lega posti a transizioni e viceversa (ma non
posti a posti e transizioni a transizioni).
Quindi una rete di Petri (PN) è un particolare tipo di grafo bipartito (in cui sono cioè
presenti due tipi di nodi) pesato, con uno stato iniziale detto marcatura iniziale M0; i due
tipi di nodi sono detti posti e transizioni e sono rappresentati rispettivamente da cerchi e
barrette collegati mediante archi orientati che rappresentano gli elementi della relazione di
flusso. Gli archi sono etichettati con dei pesi (interi positivi)
Una marcatura (stato) assegna a ciascun posto un intero non negativo e se una marcatura
assegna ad un posto un valore k, allora si dice che il posto è marcato con k gettoni
(token).
4.1.1 Significato di posti e transizioni, regola di scatto
Tipicamente un posto rappresenta una condizione mentre una transizione rappresenta un
evento.

- 66 -
Una transizione ha un certo numero di posti in ingresso e uscita che rappresentano
rispettivamente pre-condizioni e post-condizioni dell’evento.
Nel caso di un posto che sia contemporaneamente in input ed in output ad una
transizione, allora si parla di self-loop: una PN priva di self-loop è detta pura.
Il comportamento di un sistema può essere rappresentato in termini stato del sistema e di
cambiamenti di stato. Per simulare il cambiamento di stato di un sistema dunque, deve
cambiare la marcatura della PN, in accordo ad una regola di transizione (o firing rule).
Seconda questa regola, prima di tutto una transizione deve essere abilitata: una
transizione è abilitata se ciascun posto di input della transizione è marcato con un numero
di tokens almeno pari a quello indicato dalla funzione peso che collega il posto alla
transizione. Quindi la transizione può o meno avvenire (a seconda che l’evento accada o
meno) e quando avviene da ciascun posto in input viene tolto un numero di tokens pari a
quelli indicati nella funzione peso che collega i posti in input alla transizione, mentre ai
posti in output alla transizione viene aggiunto un numero di tokens pari a quello indicato
nella funzione peso che collega la transizione al posto di output.
Nella Figura 20 viene riportato l’esempio di una PN che rappresenta una ben nota
reazione chimica e dove viene mostrato l’effetto dello scatto di una transizione.
Figura 20: Esempio di rete di Petri e di scatto di una transizione
Si osservi quindi che il numero totale di tokens non deve necessariamente rimanere
inalterato a seguito dello scatto di una transizione.

- 67 -
Inoltre si osservi che lo scatto di una transizione rappresenta un evento locale della rete:
infatti sia l’abilitazione che lo scatto di una transizione dipendono solo dai posti in input
collegata ad essa e lo scatto influenza solo i posti in output ad essa; lo scatto di una
transizione non può indurre effetti collaterali a nessun posto non direttamente collegato ad
essa.
4.1.2 Non determinismo
La regola di scatto definita non è sufficiente a determinare compiutamente l’evoluzione di
una rete: infatti può accadere che in una generica marcatura corrente siano abilitate più di
una transizione: Nelle PN standard, dato l’insieme S delle transizioni abilitate in una certa
marcatura M, solamente una di queste viene scelta a caso all’interno di S: si parla quindi
di “non determinismo”. Si faccia attenzione che una volta avvenuto lo scatto può aver
modificato l’insieme S e quindi è necessario analizzare ad ogni scatto la PN per decidere
quale sarà la futura transizione abilitata a scattare.
4.1.3 Strutture fondamentali
Le PN semplici (con questo aggettivo indichiamo le PN così come formulate inizialmente
da Petri) sono in grado di rappresentare in modo efficace alcuni fenomeni che avvengono
in molti sistemi da esse rappresentati e che vengono di seguito riportati.
Sequenza: due transizioni t1 e t2 si dicono in sequenza e t1 precede t2 in una data
marcatura M quando, con abilitata t1 e t2 non abilitata, lo scatto di t1 abilita t2 (Figura 21).
Figura 21 - Esempio di sequenza in una rete di Petri
Concorrenza: due transizioni t1 e t2 si dicono in concorrenza quando non condividono
alcun posto di ingresso, cioè quando lo scatto di ciascuna transizione non disabilita l’altra.
Più precisamente, quella appena data è la definizione di concorrenza strutturale, mentre si
ha concorrenza effettiva quando la marcatura M abilita entrambe le transizioni(Figura 22).

- 68 -
Figura 22 - Esempio di concorrenza in una rete di Petri
Conflitto: transizioni t1 e t2 si dicono in conflitto se e solo se hanno almeno un posto
d’ingresso in comune; più precisamente, questa è la definizione di conflitto strutturale,
mentre il conflitto effettivo si ha quando le due transizioni, nella marcatura M, sono
entrambe abilitate e i gettoni presenti nei posti di input non sono sufficienti a soddisfare i
pesi degli archi che li collegano alle transizioni (Figura 23).
Figura 23 - Esempio di conflitto in una rete di Petri
Infine, in Figura 24, è riportato un esempio di rete marcata contenente due strutture legate
al concetto di concorrenza: la transizione t4 rappresenta una transizione di
sincronizzazione mentre la transizione t1 rappresenta una transizione di concorrenza.
Tipicamente questa transizione, con il suo scatto, attiva due sequenze concorrenti, mentre
la fase di concorrenza termina con lo scatto della transizione di abilitazione.

- 69 -
Figura 24 - Esempio di rete marcata contenente transizione di sincronizzazione e transizione di
concorrenza
4.1.4 Proprietà delle PN
Dopo aver detto cosa è una PN e dopo averla utilizzata per modellizzare dei sistemi, la
questione che sorge spontanea è: cosa si può fare con questi modelli?
Una delle caratteristiche più importanti delle PN è la possibilità di analizzare le proprietà e i
problemi dei sistemi concorrenti rappresentati; con le PN è possibile studiare due tipologie
di proprietà: quelle dipendenti dalla marcatura iniziale (proprietà comportamentali) e
quelle indipendenti dalla marcatura iniziale (proprietà strutturali).
4.1.5 Proprietà comportamentali
Raggiungibilità: una marcatura Mn si dice raggiungibile dalla marcatura M0 se esiste una
sequenza di scatti che porta da M0 a Mn. Si scrive quindi M0[σ>Mn dove σ è la sequenza di
scatti t1, t2, tn che porta da M0 a Mn. Il problema della raggiungibilità consiste quindi nel
capire se Mn fa parte dell’insieme di raggiungibilità di R(M0) di M0.
Limitatezza: una PN si dice k-limitata o semplicemente limitata se il numero di tokens per
ciascun posto non eccede un numero finito k per ogni marcatura raggiungibile dalla
marcatura iniziale.
Se una rete è limitata allora non può avere un numero illimitato di marcature distinte.

- 70 -
In generale una qualsiasi rete può essere resa limitata con l’aggiunta di opportuni posti
complementari.
Vivezza: la definizione di vivezza esprime una condizione molto forte sulla possibilità che
una transizione possa scattare nell’evoluzione futura della PN. Informalmente questa
proprietà rappresenta la possibilità che una rete possa continuare ad evolvere “bene” in
qualsiasi situazione si possa trovare e che quindi nessuna sua parte si blocchi mai.
Una transizione si dice viva se, per ogni marcatura M raggiungibile dalla marcatura
iniziale M0, esiste una marcatura M* raggiungibile da M per cui la transizione t è
abilitata in M*.
Si osservi che essendo la transizione abilitata in M*, essa può scattare raggiungendo una
marcatura M^ che per definizione è raggiungibile da quella iniziale M0.
Dunque, essendo t viva, esisterà M** raggiungibile da M^ per cui t sarà ancora abilitata.
In una rete viva (cioè una rete in cui tutte le transizioni sono vive) tutte le transizioni
possono scattare infinite volte, qualunque sia la marcatura iniziale.
Una marcatura si dice viva se e solo se per ogni transizione t esiste una marcatura M*
raggiungibile da M in cui t è abilitata. Dunque da una marcatura viva posso far scattare
qualunque transizione, ma può accadere che si raggiungano marcature che non abilitano
una, alcune o tutte le transizioni. Se però tutte le marcature raggiungibili dalla marcatura
iniziale sono vive, allora la rete è viva.
In finale, una PN è viva se, raggiunta una certa marcatura, è possibile far scattare una
transizione t passando attraverso una qualunque sequenza di scatto.
Reversibilità: una PN con marcatura iniziale M0 si dice reversibile se per ogni marcatura
raggiungibile da M0, la marcatura M0 è raggiungibile da M. Questa proprietà è molto utile
quando ad esempio si chiede ad un sistema di tornare all’inizio di una data sequenza
ciclica di operazioni, una volta raggiunta la fine della stessa.
Si noti che le tre proprietà di limitatezza, vivezza e reversibilità sono indipendenti l’una
dall’altra. In Figura 25 sono rappresentate alcune PN ciascuna con una combinazione
delle 3 proprietà di vivezza, raggiungibilità e limitatezza; in questa figura, valgono le
seguenti assunzioni:
• B= boundedness
• L=Liveliness

- 71 -
• R=reversibility
• Le lettere sopralineate indicano la mancanza di una di queste proprietà.
Figura 25 - Esempi delle combinazioni delle 3 proprietà di vivezza, limitatezza e reversibilità (Fonte:
[Mur89]).
4.2 Metodi di analisi

- 72 -
Per analizzare una PN, possono essere utilizzati: il metodo dell’albero di copertura,
l’approccio matrice di incidenza - equazione di stato ( le tecniche di riduzione e
decomposizione non vengono per ora riportate).
Data una PN con una marcatura iniziale, da questa è possibile raggiungere altre
marcature a seconda della transizione che viene fatta scattare e da ciascuna marcatura
arrivare ad altre tramite ulteriori scatti di transizioni: rappresentando graficamente le
marcature come stati della rete e le transizioni come passaggi di stato, ottengo un albero
di raggiungibilità. Bisogna notare che tale rappresentazione può crescere indefinitamente
se la rete non è limitata, perciò si introduce un simbolo ω a rappresentare l’infinito. Se la
PN è limitata, l’albero si dice di raggiungibilità. In Figura 26 è rappresentata una PN con il
suo albero di copertura e relativo grafico di copertura.
Figura 26 - Esempio di PN e relativi albero e grafo di copertura
Attraverso l’albero di copertura è possibile studiare la limitatezza di una PN (non compare
il simbolo ω) mentre non è possibile studiare i problemi di vivezza e raggiungibilità. Può
accadere infatti che due PN, una viva ed una non viva, abbiano stesso grafo di copertura.
Nel caso invece della rappresentazione matriciale, quello che si vuole fare è ricondursi ad
una rappresentazione del tipo delle equazioni differenziali o algebriche tipiche di molti
sistemi.

- 73 -
Chiamiamo matrice Pre la matrice in cui ogni elemento pre(i,j) rappresenta il peso
dell’arco che connette il posto i alla transizione j, mentre con Post indichiamo la matrice in
cui l’elemento post(i,j) rappresenta il peso dell’arco dalla transizione j al posto i.
Quindi, la matrice di incidenza C è definita come
C = Post – Pre
Dunque, se Pre(i,,j) e Post(i,,j) rappresentano rispettivamente il numero di tokens aggiunti
e tolti al posto j quando scatta la transizione i, l’elemento C(i,,j) rappresenta il numero di
tokens “cambiati” nel posto j a seguito dello scatto della transizione i.
Si osservi quindi che, nel caso di un self-loop, avendo Post(i,j)=Pre(i,,j), si avrà C(i,j)=0,
per cui la rappresentazione matriciale perderebbe di efficacia nel caso di PN non pure: per
questo motivo si considerano solo PN pure, confidando nel fatto che è sempre possibile
portarsi da una PN non pura ad una PN pura.
Quindi la marcatura Mk raggiunta a partire dalla marcatura Mk-1 attraverso lo scatto di una
transizione i è data da
Mk = Mk-1 + Cuk
essendo uk un vettore in cui ho tutti zeri ad eccezione di un 1 alla i-esima posizione.
Consideriamo allora una marcatura Md raggiungibile a partire da M0 attraverso la
successione di d scatti, per cui
Md = M0 + C Σuk
dove la sommatoria si estende per k = 1, …d, per cui posso riscrivere
∆Μ = Md - M0 = Cx
dove x = Σuk.

- 74 -
4.3 Proprietà strutturali
Le proprietà strutturali sono quelle proprietà che non dipendono dalla marcatura iniziale
della rete, nel senso che sono indipendenti dalla marcatura iniziale M0 ovvero sono legate
all’esistenza di una certa sequenza di scatti da una marcatura iniziale.
In questa sede alcune di queste proprietà vengono semplicemente accennate, senza
dimostrazioni e ulteriori approfondimenti.
Vivezza strutturale: una PN è strutturalmente viva se esiste una marcatura iniziale viva.
Controllabilità: una PN si dice completamente controllabile se ogni marcatura è
raggiungibile da ogni marcatura. Data una PN con m posti, se essa è completamente
controllabile, allora il rango della matrice di incidenza è pari ad m.
Limitatezza strutturale: una PN è strutturalmente limitata se è limitata per ogni marcatura
iniziale M0 finita.
Per altre proprietà, quali consistenza, ripetitività, conservatività, si rimanda al testo
[Mur89].
4.4 Analisi matriciale
La definizione matriciale di una PN consente di individuare alcune proprietà che non
mutano a seconda della marcatura raggiungibile: i cosiddette “Invarianti”.
Esistono due tipi di invarianti:
- P-invarianti, che riguardano proprietà dei posti e più in generale delle marcature;
- T-invarianti, che riguardano proprietà delle transizioni e più in generale delle sequenze di
scatti.
P-invarianti: si definisce strettamente conservativa una PN in cui il numero totale dei
token si mantiene costante in tutte le marcature raggiungibili, ossia:
∑∑∈∈
=∈∀PpPp
pMpMMTPRM )( )(' ),/(' .

- 75 -
Si può pensare di definire una proprietà con vincoli un po’ meno stringenti rispetto a quelli
imposti dalla stretta conservatività. Si può provare a costruire una quantità che non vari
durante l’evoluzione della rete, mediante l’attribuzione di pesi ai posti. Si definisce allora
come P-invariante la somma pesata data dal prodotto del numero dei token che in una
qualsiasi marcatura raggiungibile si trovano in ogni posto, per il peso che è associato al
posto.
Definita una funzione di peso H:P→ Z, una PN con marcatura M si dice conservativa
rispetto ad H se e solo se
∑∑∈∈
=∈∀PpPp
pMpHpMpHMTPRM )()( )(')( ),/(' .
Definendo il vettore h tale che hi=H(p(i)), un P-invariante si rappresenta così:
P-invariante = h m.
Il vettore h si può ricavare dalla soluzione di un sistema lineare omogeneo.
L’equazione fondamentale delle reti di Petri afferma che:
m’ = m + C s.
Moltiplicando da sinistra ambo i membri per il vettore h si ottiene:
h m’ = h (m + C s).
Poiché, per definizione, un P-invariante è una quantità che non cambia per ogni marcatura
raggiungibile, si ha:
),/(' ' MTPRmmhmh ∈∀= .
E dunque:
h C s = 0.
Questa relazione deve essere verificata per qualunque sequenza di scatti, cioè quale che
sia s. Allora la proprietà che caratterizza i P-invarianti è:
h C = 0

- 76 -
Come anticipato, si tratta di un sistema lineare e omogeneo, da risolvere rispetto ad h,
scartando la soluzione banale h = 0 (ogni PN è conservativa rispetto al vettore nullo!).
Si può dimostrare che in generale in questo modo non si riescono a trovare tutti i P-
invarianti; tuttavia nel caso di reti vive tutti e soli i vettori h adatti alla costruzione dei
P-invarianti si ottengono dalla soluzione di quel sistema.
Si noti che non necessariamente una PN ammette almeno un P-invariante.
Una PN si dice ricoperta da P-invarianti se per ogni posto esiste almeno un P-invariante la
cui componente in corrispondenza di tale posto sia positiva.
E’ stato dimostrato che una rete ricoperta da P-invarianti è necessariamente limitata.
T-invarianti: i T-invarianti individuano sequenze di scatti cicliche, che possono avere
luogo ripetutamente, riportando la rete esattamente nella marcatura di partenza.
Si consideri l’equazione fondamentale delle reti di Petri:
m’ = m + C s.
Imponendo che al termine della sequenza di scatti individuata dal vettore s si riproduca la
stessa marcatura di partenza si ottiene:
m = m + C s.
Cioè:
C s = 0.
Tutte e sole le (eventuali) soluzioni non banali di questo sistema lineare omogeneo sono i
T-invarianti della rete di Petri.
Sifoni e Trappole: i sifoni e trappole sono strutture fondamentali per l’analisi di vivezza e
non-bloccaggio di una rete.
Un sifone rappresenta un insieme di posti che complessivamente tende a perdere gettoni
durante l’evoluzione della rete e che, una volta persi tutti i gettoni, non è più in grado di
riacquistarne.
Le trappole sono duali ai sifoni: una trappola rappresenta un insieme di posti che
complessivamente tende ad acquistare gettoni durante l’evoluzione di una rete e che, una

- 77 -
volta preso almeno un gettone, non è più in grado di smarcare contemporaneamente tutti i
suoi posti.
Si riportano di seguito alcuni risultati importanti:
• Se S è una trappola marcata in una marcatura M allora rimane marcata in ogni
marcatura M’ raggiungibile da M;
• Se ogni sifone contiene una trappola marcata in una marcatura M, allora esiste in
[M> una marcatura morta.
4.5 Utilizzo delle reti di Petri per l’analisi qualitativa dei
modelli di sistemi biologici
4.5.1 Il legame tra modelli qualitativi e modelli quantitativi e l’utilizzo
delle reti di Petri
In questa sezione affrontiamo il problema dell’ottimizzazione relativamente ad una rete di
Petri utilizzata per rappresentare un fenomeno biologico.
Sono stati fatti numerosissimi studi sull’utilizzo delle PN per rappresentare fenomeni
biologici quali quelli legati al metabolismo cellulare.
Gli studi di Reddy ([Red93]), Hofestaedt ([Hof94]) e Genrich ([Gen2001]), hanno
dimostrato le possibilità di modellizzazione diretta dei pathway metabolici, sia a basso che
ad alto livello. La struttura delle PN riflette bene la struttura delle reazioni biochimiche e la
matrice di incidenza delle PN è la matrice stechiometrica delle reazioni coinvolte nei vari
fenomeni rappresentati.
Tramite le PN è possibile individuare i cosiddetti “modi elementari”, ovvero il set minimo di
reazioni che operano a regime, così come discusso da Schuster et all. in [Sch2000].
Spesso gli studi relativi a singoli fenomeni hanno portato a definire delle PN “di comodo”
adatte al caso singolo ma di facile estensione ad un gruppo notevole di fenomeni: ad

- 78 -
esempio, in Figura 27, è possibile osservare la rappresentazione di un meccanismo di
regolazione genetica dovuto al lavoro di Matsuno et all (vedi [Tan2003]).
Figura 27 - A HFPN realization of the circadian rhythm model due to Ueda et al (fonte: [Tan2003])
4.5.2 Il framework “modello qualitativo – modello quantitativo”
M. Heiner ha lavorato in collaborazione con I. Koch, L. Popova-Zeugmann e altri per
colmare il gap esistente tra i modelli qualitativi e i modelli quantitativi, sfruttando la teoria
delle PN. Nei suoi studi l’obiettivo è stato quello di far precedere al modello quantitativo di
un modello biologico, ad esempio un modello ODE, un modello qualitativo che
permettesse:
• Una stima dei parametri legati al modello quantitativo
• Una validazione del modello utilizzato
• Una predizione del comportamento qualitativo
secondo il framework riportato in Figura 28 1.
1 A series of ten ODEs are realized in this network, where Perm (CCn, PTn) represents the concentration of per mRNA (dCLK-CYC complex in the nucleus, PER-TIM complex in the nucleus) and C1 = 0 nM/h, S1 = 1.4

- 79 -
Figura 28 - Framework di modellizzazione qualitativa e quantitativa di sistemi biologici secondo Gilbert e Heiner (Fonte:[Gil2006])
I modelli qualitativi considerano le reti biochimiche allo stato di equilibrio, in cui cioè i
parametri cinetici sono considerati costanti e si basano su una descrizione grafica
teoretica della topologia del sistema o della struttura che è definita, nel caso di modelli
stechiometrici, dalle equazioni stechiometriche conosciute.
La maggior parte delle pubblicazioni sviluppa invece modelli quantitativi data la volontà di
predire il comportamento dinamico del sistema e si basano su misurazioni delle
concentrazioni, dei rate di reazione o delle costanti di reazione.
4.5.3 Modelli qualitativi basati su PN
Nel proseguimento del paragrafo, a meno che sia esplicitamente indicato, consideriamo il
solo comportamento nello stato di equilibrio.
Le reti metaboliche (dette anche metabolical pathways) consistono di numerose reazione
enzimatiche che trasformano delle sostanze in alcuni prodotti, passando per vari composti
intermedi.
nM/h, A1 = 0.45 nM/h, B1 = 0, L1 = 0.3 nM/h, D0 = 0.012 nM/h, D1=0.94 nM/h, R1 = 1.02 nM/h. (fonte [Tan2003])

- 80 -
Nelle reti metaboliche le reazioni chimiche dei metaboliti sono di solito conosciute dalle
equazioni stechiometriche mentre spesso sono le concentrazioni dei metaboliti o altri
parametri quantitativi ad essere ignoti: per questo motivo si inizia con modelli qualitativi.
Per derivare una rappresentazione tramite PN di una reazione chimica, sotto l’ipotesi di
stato di equilibrio, si assegna a ciascun composto biochimico un posto; le relazioni tra
composti biochimici sono stabilite da reazioni chimiche che sono rappresentate da
transizioni (rappresentano eventi biochimici ‘atomici’). Le molteplicità degli archi
rappresentano i valori stechiometrici delle equazioni stechiometriche.
Determino così una PN place-bordered che posso facilmente trasformare in una PN
transition-bordered, in cui le transizioni agli estremi rappresentano la produzione delle
specie reagenti e il consumo dei composti prodotti.
Questi passaggi sono visibili in Figura 29.
Figura 29 - Esempio di modello qualitativo di una reazione biochimica rappresentato da una PN
(Fonte:[PoZ2005])
Le transizioni agli estremi lavorano indipendentemente, perciò non sono fatte assunzioni
sulle relazioni quantitative input/input, input/output e output/output.
Quindi il comportamento atteso della PN rispecchia tutte le sequenze parzialmente
ordinate di reazioni chimiche dai componenti in input ai componenti in output nel rispetto
delle relazioni stechiometriche date. Per essere precisi, il comportamento della PN
qualitativa (cioè time-less) consiste in tutti i possibili comportamenti sotto qualsiasi
condizioni di temporizzazione.
Si noti che le transizioni senza pre-posti possono scattare infinitamente, per cui sono vive
e tutti i post-posti relativi sono non limitati.
4.5.4 Analisi qualitativa del modello a rete di Petri

- 81 -
Con riferimento al processo di analisi qualitativa riportato in [Sac2006], si procederà con le
seguenti fasi:
• individuazione degli invarianti e dei multi-insiemi della rete di Petri: P-
Invarianti, T-Invarianti, feasible T-Invariant, MCT;
• attribuzione di significato biologico agli invarianti e agli MCT;
• analisi di modelli ‘lack’ (privi di alcune sottoreti, rappresentate ad esempio da
alcuni T-Invarianti).
4.5.4.1 T-Invarianti realizzabili e feasible T-Invariant
Prima di tutto bisogna notare che il concetto di T-Invariante non considera la marcatura
antecedente al suo scatto: gli invarianti sono infatti caratterizzazioni ‘strutturali’, cioè legati
alla struttura della rete ed indipendenti dalla marcatura.
Nel contesto biologico che stiamo studiando ciò significa che dobbiamo garantire che il T-
Invariante abbia senso per la rete, data la presenza di opportune condizioni al contorno.
Si può notare che la condizione che potrebbe rendere il T-Invariante non realizzabile è
l’assenza di una marcatura sufficiente, ipotesi che scartiamo a priori (i posti saranno
riempiti opportunamente).
Invece, data una marcatura iniziale, la presenza di ‘read arcs’ rende l’individuazione dei T-
Invarianti incompleta: infatti un read arc non ‘pesa’ sulla matrice di incidenza per cui il T-
Invariante individuato non ne è influenzato.
Può quindi accadere che un T-Invariante non sia feasible in quanto, nella marcatura
iniziale, non può scattare: per renderlo feasible è necessario accoppiarlo ad altri T-
Invarianti che rendano possibile lo scatto delle transizioni relative ai read arcs.
La differenza tra un T-Invariante realizzabile e uno feasible è la seguente: un T-Invariante
è realizzabile se può scattare nella marcatura in cui si trova la rete, dunque nel caso della
marcatura iniziale questa dovrà essere opportunamente definita affinché ogni T-Invariante
minimale sia anche realizzabile; invece un feasible T-Invariant è tale se è minimale (e
quindi realizzabile) oppure è una combinazione lineare di T-Invarianti minimali creata

- 82 -
affinché il T-Invariante non feasible possa scattare in quanto la marcatura che lo rende
eseguibile è stata creata grazie ai T-Invarianti aggiunti.
Dunque, relativamente al primo step (individuazione invarianti), si deve procedere prima
con l’individuazione dei P-Invarianti e l’analisi delle loro plausibilità: ad esempio, in un
signalling pathway potremmo considerare una specie biochimica con ruolo di segnalatore
per cui questa non può essere prodotta o consumata ma può solo cambiare di stato,
dunque deve esserci un unico token da inserire in uno dei due posti che indicano lo stato
di quella specie (in genere si decide per lo stato inattivo).
Quindi si passa a ‘validare il signal flow’: dati i T-Invarianti, si rendono questi tutti
realizzabili a partire dalle condizioni iniziali; quindi se sarà necessario, oltre ai T-Invarianti
minimi si costruiranno dei T-Invarianti feasible.
La marcatura iniziale da considerare è quella rilevata nello studio dei P-Invarianti.
4.5.4.2 MCT, Maximal Common Transition set
Infine un altro multi insieme interessante è quello dei MCT, Maximal Common Transition
sets.
Dati due T-invarianti ed una transizione ti che fa parte di entrambi, fanno parte di un MCT
sia la transizione ti che tutte le transizioni tj presenti in tutti i T-Invarianti di cui ti fa parte.
Questi raggruppamenti rappresentano una relazione di equivalenza, in quanto una
transizione che fa parte di un MCT non può far parte di un altro MCT.
Gli MCT possono rappresentare una decomposizione di reti biochimiche in piccole
sottoreti, che possono essere lette come unità funzionali.
4.6 Modelli quantitativi basati su PN
L’analisi quantitativa passa attraverso l’analisi statica e dinamica della PN che si basa
sull’uso degli Invarianti e dei grafi di copertura, che qui introduciamo.
La trasformazione dal modello qualitativo al modello quantitativo avviene sfruttando una
proprietà strutturale delle PN, i T-Invarianti.

- 83 -
I T-Invarianti sono multi-insiemi di transizioni con un costo totale nullo sul marking; se tale
multi-insieme scatta, in un ordine appropriato, viene riprodotto un dato marking.
Quindi, nel contesto delle PN utilizzate per rappresentare reti biochimiche, i T-Invarianti
rappresentano multi-insiemi di reazioni chimiche capaci di riprodurre una distribuzione di
componenti chimici, cioè uno stato di equilibrio: ciò è possibile nel limite di una situazione
iniziale della distribuzione di token che permette lo scatto di tutte le transizioni implicate
(cioè se si ha un numero di token sufficiente).
Se ogni transizione di una rete appartiene ad un T-Invariante, allora la rete si dice coperta
da T-Invarianti.
Per descrivere tutti i comportamenti di un dato sistema ciclico, è ovviamente di grande
aiuto avere tutti i comportamenti ciclici basilari, i cosiddetti T-Invarianti minimi: si tratta di
multi-insiemi minimi di transizioni con la proprietà di riprodurre uno stato.
Riconosciamo due tipologie di T-Invarianti: in primis le coppie di transizioni che modellano
una reazione reversibile, detti trivial T-Invariant; quindi, tra i T-Invarianti non trivial, i
cosiddetti T-Invarianti di I/O, in quanto includono le transizioni agli estremi.
Nella rete di Figura 29, considerando la marcatura iniziale con tutti i posti vuoti, per tornare
alla stessa marcatura tutta vuota esiste un solo T-Invariante minimo, definito dal vettore di
Parikh (1,2,1,1,3), che definisce il numero di volte che ciascuna transizione deve scattare
affinché si torni allo stato iniziale tutto vuoto (facendo avvenire gli scatti secondo un ordine
opportuno). Questo T-Invariante è di I/O e copre la rete.
Data l’assunzione di stato di equilibrio, i valori del vettore di Parikh del T-Invariante minimo
corrispondono ai rate di scatto relativi delle transizioni coinvolte per mantenere, durante
sequenze di scatto continue, il dato stato stabile.
Riferendosi all’esempio suddetto, lo stato di equilibrio si mantiene se prod_B scatta due
volte e con_D tre volte rispetto alle altre transizioni, per ogni unità di tempo.
Usando un modello a transizioni temporali, i rate di scatto relativi possono essere simulati
aggiustando i tempi di transizione in modo appropriato. Per avere interi, devo normalizzare
all’unità di tempo data dal minimo comune multiplo di tutti i valori del T-Invariante minimo.
Siccome i T-Invarianti minimi possono sovrapporsi, è necessario soddisfare la condizione
di stato di equilibrio di tutti i processi, per cui i rate di scatto sono dati dal vettore somma di
tutti i T-Invarianti minimi non trivial, ridotti dal massimo comune divisore di tutti i valori del
vettore e scegliere per l’unità temporale un valore calcolato come sopra.

- 84 -
Bisogna notare che il calcolo dei T-Invarianti non comporta la costruzione dello spazio di
stato, per cui non ho il ben noto problema della ‘esplosione degli stati’ .
Riassumendo quindi il significato dei T-Invarianti, si può dire che essi rappresentano un
insieme di transizioni che:
• hanno effetto nullo sulla marcatura;
• riproducono una marcatura (cioè uno stato del sistema);
e sono interpretabili in due modi:
• rate relativi agli scatti delle transizioni: le transizioni che avvengono
permanentemente e correntemente, stiamo cioè descrivendo il
comportamento nello stato di equilibrio;
• sequenza di transizioni parzialmente ordinata: flussi di sostanze o di
“segnali”.
Strutturalmente un T-Invariante rappresenta una sottorete, composta dal suo supporto (le
sue transizioni) i pre-posti e i post-posti, più gli archi tra tutti questi nodi.
Una rete si dice quindi CTI (Covered by T-Invariants) se ogni transizione appartiene ad
almeno un T-Invariante e si ha inoltre la condizione necessaria
Boundedness e Liveliness � CTI
Analogamente, per i P-Invarianti (multi-insiemi di posti) si definiscono i P-Invarianti minimi
(non ho un supporto più piccolo) e la rete si dice CPI (Covered by P-Invariants) se ogni
posto appartiene ad un P-Invariante.
Lo scatto di una transizione non ha alcun effetto sulla somma pesata dei token
appartenenti al P-Invariante (per ogni transizione l’effetto degli archi che aggiungono token
ai posti del P-Invariante è pari all’effetto degli archi che tolgono token dai posti del P-
Invariante).
Quindi, al concetto di P-Invariante si legano le seguenti caratteristiche:

- 85 -
• preservazione dei token/composti; • un posto che appartiene ad un P-Invariante è limitato.
Un P-Invariante definisce una sottorete, costituita dai posti del P-Invariante e dalle pre- e
post-transizioni e dagli archi che congiungono i posti a queste transizioni.
L’insieme pre- del supporto è uguale all’insieme post- del supporto: il P-Invariante è self-
contained e ciclico.
Si ha la condizione sufficiente
CPI � Boundedness
A questo punto, l’utilizzo degli Invarianti consente di validare il modello: se infatti tutti i
comportamenti che ci si aspetta dal fenomeno biologico sono riconducibili ai T- e P-
Invarianti e se, viceversa, tutti gli Invarianti sono riconducibili a specifici qualità
comportamentali del fenomeno biologico e in generale si mantengono tutte le proprietà
strutturali e comportamentali del fenomeno, allora il modello si può dire validato.
All’analisi statica (fatta con gli invarianti) si può aggiungere l’analisi dinamica fatta con il
grafo di raggiungibilità: tramite questo metodo di analisi è possibile infatti ricavare
numerosi dettagli relativamente alle caratteristiche di raggiungibilità, limitatezza e vivezza
della rete.
Ora si può partire con l’analisi quantitativa della rete: la prima considerazione da fare è
che il modello quantitativo corrisponde al modello qualitativo a cui si aggiungono dei
parametri quantitativi, conosciuti o stimati.
Tipici parametri quantitativi delle bionetworks sono le concentrazioni dei composti (che
sono numeri appartenenti all’insieme dei reali), i rate di reazione e i flussi (dipendenti dalle
concentrazioni).
Ho quindi necessità di utilizzare reti dotate di nodi continui, cioè Continuous PN.
Quindi, sostanzialmente, il processo che porta dall’analisi qualitativa a quelle quantitativa
comporta:
Mantenere la struttura individuata nell’analisi qualitativa;

- 86 -
• Aggiungere i rate ad ogni transizione: i pre-posti appaiono come parametri
(dipendenza dallo stato della rete) e le funzioni dei rate definiscono il flusso
continuo
• Costruzione del marking minimo:
o a ciascun P-Invariante viene assegnato almeno un token (1 inteso come
numero reale), che rappresenta la concentrazione (continua) corrente: un P-
Invariante è infatti un deadlock strutturale e una trappola;
o tutti i T-Invarianti devo essere resi realizzabili, per rendere la rete viva;
• ottengo un marking minimo: minimizzo quindi lo spazio di stato.
Una Continuous PN definisce quindi un sistema di ODE, modellizzabile sulla PN iniziale
(non temporizzata).
4.7 Reti di Petri continue e ibride
Le reti di Petri sono per definizione dei sistemi discreti, tuttavia sono state definite le reti di
Petri continue e quelle discrete.
Queste ultime due tipologie di reti sono utilizzate per modellare sistemi continui e discreti,
prendendo o meno in considerazione il tempo.
4.7.1 Reti di Petri discrete, continue e ibride
Consideriamo nel seguito reti autonome, cioè non consideriamo la variabile tempo. Un
modello autonomo consente di effettuare valutazioni di tipo qualitativo del processo
modellato.
Come noto, in una rete di Petri discreta (vedi esempio in Figura 30), ho una marcatura
iniziale che descrive il numero di tokens presenti in ciascun posto che compone la rete
stessa. Una transizione di questa rete è abilitata se tutti i posti in input alla transizione
contengono un numero di tokens maggiore o uguale al peso indicato sull’arco
congiungente posto e transizione.
Nell’insieme di transizioni abilitate, ne scatta solo una in modo non deterministico.

- 87 -
Ad ogni transizione che scatta si ha un cambiamento nella marcatura della rete: il vettore
della marcatura è in questo caso costituito da variabili discrete (ogni variabile può
assumere un valore dell’insieme dei naturali, compreso lo zero).
Figura 30 - Esempio di PN discreta
Nel caso di reti di Petri continue (vedi Figura 31), la marcatura (a partire dalla marcatura
iniziale e durante la sua evoluzione) è costituita da un vettore di variabili continue non
negative. Una transizione è quindi abilitata finché i posti in input contengono un numero
reale strettamente maggiore di zero e lo scatto di una transizione da luogo ad una nuova
marcatura costituita sempre da variabili reali: lo scatto di una transizione toglie e aggiunge
un valore reale non necessariamente intero.
Figura 31 - Esempio di PN continua tratto dalla PN discreta di Figura 10
Una rete di Petri ibrida (vedi Figura 32) è costituita da posti e transizioni di tipo continuo e
discreto. Mentre una transizione discreta può essere preceduta solamente da posti discreti
ed ovviamente al suo scatto toglie e aggiunge un numero intero di tokens, nel caso di una
transizione continua posso avere in input posti continui e discreti.
Figura 32 – Esempio di PN ibrida

- 88 -
E’ dunque necessario che i posti, siano essi continui o discreti, abbiano un numero
sufficiente di tokens (maggiore o uguale al peso dell’arco che li congiunge alla transizione
continua). Lo scatto della transizione continua toglie un numero intero di tokens dai posti
discreti in input e un numero continuo di tokens dai posti continuo in input.
Il vettore delle marcature è perciò costituito da variabili continue e discrete.
4.7.2 Reti di Petri temporizzate
La variabile temporale, in questo tipo di reti, può essere associata ai posti o alle
transizioni. Scegliamo di associare il tempo alle transizioni (ma è possibile passare da un
tipo di modello all’altro senza difficoltà).
Nel caso di una rete di Petri discreta temporizzata, nei posti in input ad una transizione,
posso avere tokens riservati e tokens non riservati.
Per abilitare una transizione vengono considerati solamente i tokens non riservati. Allo
scatto della transizione, vengono tolti i tokens riservati dai posti di input e aggiunti dei
tokens non riservati nei posti in output.
Ad ogni transizione è associato un valore corrispondente ad un intervallo temporale: una
volta che un numero di tokens è stato riservato per una transizione, rimangono riservati
fino allo scatto della transizione stessa, che avviene quando viene raggiunto il valore
dell’intervallo temporale associato (vedi Figura 33): questo comportamento corrisponde
alla modellazione del tempo in cui avviene una azione, rappresentata dalla transizione, per
cui vengono riservate delle risorse).
Figura 33 - Esempio di PN discreta temporizzata e sua evoluzione
Bisogna notare che di per sé, una rete di Petri discreta temporizzata è un modello ibrido,
in quanto il vettore di stato è costituito dalle variabili discrete dei tokens presenti in ogni

- 89 -
posto e dalla variabile continua che rappresenta il tempo rimanente di riservazione dei
tokens.
Sia ora una rete di Petri continua temporizzata: in questo caso ad ogni transizione è
associata una velocità di scatto (Figura 34).
Bisogna ora distinguere tra transizioni fortemente abilitate (strongly enabled) e debolmente
abilitate (weakly enabled): il primo caso si ha quando ogni posto in input ad una
transizione non è vuoto, mentre il secondo caso si ha quando i posti in input che sono
vuoti vengono riempiti dallo scatto di altre transizioni.
Dunque, nel caso di transizioni fortemente abilitate la velocità di scatto è effettivamente
quella associata alla transizione, mentre nel caso di una transizione debolmente abilitata si
ha che la transizione assume la velocità minore tra quelle delle transizioni in questione: se
ad esempio un posto vuoto è seguito da una transizione a velocità v = 0.003 ma il posto
viene riempito da una transizione a velocità v = 0.002, allora la transizione che segue il
posto scatterà con una velocità pari a v = 0.002; ne consegue che la velocità associata ad
una transizione è la velocità massima.
Figura 34 - Esempio di processo (fluido in movimento) modellato da una PN continua: si noti come cambiando gli ordini di grandezza allo stesso modo per la velocità e per i posti, il modello usato
abbia lo stesso andamento dell'altro
Questo tipo di modello è una rete di Petri continua a velocità costante, CCPN (Constant
Continuous Petri Net).
Altri due modelli di reti di Petri continue temporizzate sono le VCPN (in cui le velocità
dipendono dalla marcatura della rete) e le ACPN (che sono una approssimazione delle
VCPN in cui marcatura e velocità cambiano linearmente).

- 90 -
4.7.3 Reti di Petri ibride
4.7.4 Reti di Petri ibride autonome
Una rete di Petri ibrida è costituita da nodi (posti e transizioni) di tipo continuo e discreto.
Bisogna quindi analizzare l’influenza della parte discreta su quella continua e viceversa.
Vediamo prima l’influenza della parte discreta su quella continua (Figura 35).
Supponiamo un posto discreto che sia in input sia ad una transizione discreta che ad una
transizione continua e che dalla transizione continua ci sia una arco in input allo stesso
posto discreto: in tal modo, lo scatto della transizione continua toglie e inserisce lo stesso
valore (stesso peso unitario sugli archi) al posto discreto, che quindi avrà sempre lo stesso
valore intero.
Allo scatto della transizione discreta, il token viene rimosso e quindi la transizione continua
non è più abilitata.
Figura 35 - Influenza della parte discreta sulla parte continua
Vediamo ora l’influenza della parte continua su quella discreta (vedi Figura 36).
Supponiamo ci sia una transizione discreta con in ingresso un posto continuo ed uno
continuo: se il posto continuo raggiunge il valore indicato nell’arco che lo congiunge alla
transizione discreta, la transizione scatta togliendo il token alla transizione discreta.

- 91 -
Figura 36 - Influenza della parte continua su quella discreta
Nel caso particolare in cui una transizione discreta abbia in input un posto continuo e in
output un posto discreto, allora ho la conversione di una marcatura continua in una
marcatura discreta.
Se invece in input ad una transizione discreta ho un posto discreto ed in output un posto
continuo, sull’arco che congiunge il posto discreto alla transizione alla transizione ho un
valore intero e sull’arco che congiunge la transizione al posto continuo ho un valore reale.
Lo scatto della transizione toglie quindi un numero intero di tokens dal posto discreto e
aggiunge un valore reale al posto continuo: trasformo quindi una marcatura discreta in una
marcatura continua.
E’ fondamentale notare un particolare che diversifica i posti in input o di output di una
transizione continua rispetto ad una transizione discreta: infatti, mentre in quest’ultimo
caso un posto (sia esso continuo che discreto) può essere solamente di input, solamente
di output o entrambi, nel caso di una transizione continua un posto discreto deve essere
contemporaneamente di input e di output per quella transizione, con lo stesso peso sugli
archi.
4.7.5 Reti di Petri ibride temporizzate
In questo caso, nella parte discreta ho delle transizioni a cui posso associare ritardi
deterministici o stocastici, mentre alle transizioni continue associo delle velocità (vedi
Figura 37).

- 92 -
Figura 37 - Esempio di PN ibrida temporizzata
Le transizioni discrete hanno priorità su quelle continue e non appena una transizione
discreta viene abilitata, solo questa è in grado di scattare e lo farà nel tempo dettato dal
ritardo associato alla transizione.
Se però il posto in input a tale transizione discreta è anche in input ad una transizione
continua, quest’ultima sarà comunque abilitata nel tempo che intercorre prima dello scatto
della transizione discreta: la transizione continua scatterà con la velocità ad essa
associata.

- 93 -
5. Progettazione di un software di
supporto per l’analisi e la simulazione
di modelli biologici
5.1 Le necessità che hanno portato alla progettazione
del software
La ricerca scientifica nel campo biologico ha trovato nell’informatica un valido alleato nelle
varie fasi del suo lavoro e nel tempo si sono accumulati moltissimi strumenti software per il
supporto alla ricerca nel campo della biologia.
I primi utilizzi dei computer in questo campo sono stati fatti per la memorizzazione di
grossi quantitativi di dati contenenti svariate tipologie di informazioni: dai risultati di
esperimenti specifici alle campagne di analisi effettuate su popolazioni più o meno ampie.
I supercomputer sono stati poi utilizzati per permettere l’elaborazione di questi dati in
tempi ragionevoli: il progetto GENOMA è l’esempio principe, in quanto ha permesso di
mappare in pochi mesi l’intero corredo genetico umano.
Le tecniche della biologia moderna sono passate da una fase ovviamente più artigianale e
soggetta ai gusti personali ad una forma più razionalizzata, fatta di test standard e
protocolli da seguire necessariamente e scrupolosamente, per alcune ragioni tra cui:
- accettazione da parte della comunità scientifica internazionale;
- velocità e precisione del lavoro;
- verifica da parte delle commissioni etiche.
Il proliferare degli strumenti software e dell’utilizzo dei calcolatori al contrario non è stato
accompagnato da un processo di razionalizzazione simile: spesso ogni istituto di ricerca è
dotato del suo software oppure scienziati con buone capacità di programmazione creano i
loro codici di programmazione in linguaggi spesso tra loro non compatibili ma soprattutto

- 94 -
difficilmente rimodellabili. È una norma il fatto che numerosi ricercatori, una volta creata
una struttura per il proprio codice, difficilmente la abbandonano, preferendo utilizzare il
tempo per adattare un esperimento al proprio strumento informatico piuttosto che
estendere o modificare il codice per elaborare il nuovo esperimento.
Questo fattore riduce notevolmente le possibilità di collaborazione tra ricercatori e
aumenta i tempi di studio per la necessità di comprendere il codice stesso.
È quindi fondamentale concentrare gli sforzi verso strumenti informatici che permettano di
creare un unico ambiente di lavoro in cui integrare i diversi strumenti di analisi e calcolo
già disponibili e comunicare dati, risultati e modelli attraverso l’uso di linguaggi
standardizzati.
Il software che viene presentato è un tentativo verso questa direzione e permette di
integrare strumenti di calcolo affidabili e di facile utilizzo quali Matlab con una interfaccia
grafica in grado di creare modelli grafici dei pathway biochimici e di rappresentare
attraverso metalinguaggi standard i modelli stessi.
5.2 Le funzioni solver per la risoluzione numerica delle
equazioni differenziali
Matlab è uno strumento software che mette a disposizione diversi tools matematici per
l’analisi e la simulazione di un vasto scenario di fenomeni.
Il suo utilizzo va dalla semplice analisi matematica di equazioni alla simulazione di sistemi
industriali, passando per l’analisi statica di dati, lo studio di fenomeni economici e,
ovviamente, la possibilità di analizzare e simulare fenomeni legati alla chimica e alla
biologia.
In particolare, Matlab mette a disposizione delle funzioni, dette ‘solver’, per la risoluzione
di equazioni differenziali.
Fermo restando che è possibile sfruttare soluzioni analitiche (ad esempio per integrazione
diretta di una funzione e con metodi di sostituzione) e metodi numerici (quali quelli di
Eulero e di Runge-Kutta), Matlab dispone di funzioni ‘solver’ che implementano i metodi di
Runge-Kutta con una ampiezza variabile del passo di programma.

- 95 -
Di seguito una tabella con una lista di alcuni solver disponibili in Matlab
Funzione Descrizione
ode23 Risolutore di ordine basso (combinazione di
metodi di Runge-Kutta del secondo e terzo
ordine) per equazioni non complesse
ode45 Risolutore di ordine medio (combinazione di
metodi di Runge-Kutta del quarto e quinto
ordine) per equazioni non complesse
ode113 Risolutore di ordine variabile per equazioni
non complesse
ode23s Risolutore di ordine basso per equazioni
complesse
ode23t Risolutore basato sul metodo dei trapezi
per equazioni moderatamente complesse
ode23tb Risolutore di ordine basso per equazioni
complesse
ode15s Risolutore di ordine variabile per equazioni
complesse
La sintassi di una funzione solver per risolvere equazioni differenziali di primo grado del
tipo y’ = f( t, y) è la seguente (prendiamo come esempio la funzione solver ode23):
[t, y] = ode23(‘ydot’, tspan, y0)
dove ydot è il nome del file di funzione i cui input sono t ed y, il cui output è il vettore
colonna dy/dt (cioè f( t, y) ). tspan è un vettore che contiene i valori iniziale e finale della
variabile indipendente. y0 è il valore di y nell’istante iniziale.
Nel caso di equazioni differenziali di ordine superiore al primo, Matlab permette di risolvere
il sistema di equazioni mediante le stesse funzioni solver, utilizzando come input una
notazione matriciale.

- 96 -
Infine si noti che può essere necessario introdurre delle funzioni forzanti: per far ciò,
bisogna programmarle all’interno del file di funzione derivata (utilizzando delle strutture if-
elseif-else).
5.3 L’ambiente di lavoro e i moduli progettati
Nel campo della modellizzazione matematica, software e computer ci permettono di
progettare modelli e validarli: la progettazione passa dal semplice disegno di un modello o
dall’equazione che lo rappresenta, scritti su un pezzo di carta, al disegno su computer
utilizzando dei software dotati di semplici GUI (Graphical User Interface) che permettono
di creare letteralmente in pochi minuti un modello grafico di quanto abbiamo in mente.
Ovviamente, poiché le necessità del ricercatore non si fermano alla mera progettazione
grafica, questi software hanno almeno una di due funzioni aggiuntive a quella della GUI:
1. la possibilità di rappresentare il modello mediante un apposito linguaggio
descrittivo;
2. la simulazione del modello mediante motori di calcolo dedicati.
La prima funzione permette di descrivere, tramite un linguaggio dedicato, il modello che ho
disegnato graficamente e per questo ha i seguenti vantaggi:
- l’uso di linguaggi “universali” o riconosciuti dalla comunità scientifica permette un
passaggio più rapido della conoscenza tra scienziati e/o gruppi di ricerca diversi;
- questi linguaggi descrittivi hanno una sintassi e una semantica simili a quella
umana, per cui possono essere letti anche senza passare nuovamente al modello
grafico;
- i file grafici in genere hanno una occupazione di memoria maggiore
- i file grafici non permettono una immediata elaborazione, ad esempio da parte di
motori matematici, senza il passaggio ad una versione “intermedia” come può
essere appunto un linguaggio descrittivo.
La seconda funzione permette di effettuare alcune elaborazioni basate sugli strumenti
matematici che decidiamo di utilizzare per l’analisi del modello.
Questi strumenti matematici e, di conseguenza i motori matematici utilizzati, possono
basarsi su:

- 97 -
- modelli discreti, continui o ibridi;
- modelli stocastici o deterministici;
- modelli per l’analisi qualitativa o quantitativa.
Il già citato Matlab permette, se non tutte, molte di queste operazioni, in quanto contiene
numerose funzioni o addirittura interi tool pre-costruiti.
Tuttavia, il livello richiesto per la customizzazione degli script può essere troppo
complicato, soprattutto quando abbiamo a disposizione software dotati di motori
matematici specifici.
Ad esempio, come visto precedentemente, l’utilizzo delle ODE in Matlab si basa su una
struttura standard che richiede pochi successivi ritocchi degli script prima
dell’elaborazione.
Al contrario, alcuni modelli di analisi discreta sono così versatili come altri: è il caso
dell’analisi mediante reti di Petri discrete. E’ il caso di PNTool per Matlab, che permette
l’analisi quantitativa e qualitativa delle reti di Petri, ma che presenta alcuni svantaggi
impossibilità di utilizzare GUI, necessità di sfruttare Matlab per ogni funzione, utilizzo di
linguaggi descrittivi non standard.
Le necessità che portano alla realizzazione del nostro software sono quelle di
progettazione, interfacciamento, validazione e simulazione dei modelli del metabolismo.
La progettazione di un modello richiede la presenza di una GUI, che permetta da una
parte la costruzione del modello mediante moduli grafici comuni (compartimenti, flussi,
moduli di controllo e altro ancora) dall’altra permetta di richiamare alcune funzioni, quali la
creazione dei file descrittivi o dei file da dare in input ai software di calcolo, simulazione o
analisi.
Per interfacciamento si intende la possibilità di:
- esportare il modello in formati non grafici quali i linguaggi descrittivi riconosciuti dai
software con cui ci vuole interfacciare;
- ricreare il modello grafico a partire da un file in linguaggio descrittivo;
- la possibilità di riutilizzare i risultati ottenuti da uno dei software con cui ci interfaccia
per elaborarli (e quindi interfacciandosi) con altri software.

- 98 -
La validazione e la simulazione del modello sfruttano la macrofunzionalità di
interfacciamento: infatti la validazione di un modello si basa sui risultati ottenuti dagli altri
software di analisi qualitativa e quantitativa, così simulazione si basa su specifici software.
5.3.1 L’utilizzo di XML per la memorizzazione della rete
I linguaggi descrittivi (detti anche metalinguaggi) che dobbiamo gestire si basano su XML
(eXtensible Markup Language) e sono il CSML(Cell Simulation Markup Language) e il
PNML (Petri Net Markup Language).
XML significa Linguaggio a Marcatori Estensibile: i linguaggi a marcatori consentono di
descrivere con precisione qualunque tipo di informazione, sia essa gerarchica, lineare,
relazionale o binaria. XML organizza l’informazione in una struttura gerarchica che è
possibile scorrere e navigare con semplicità. Uno dei linguaggi a marcatori più noti è
HTML (HyperText Markup Language) con cui è possibile descrivere e realizzare pagine
web.
La caratteristica più importante di XML è il fatto che è estensibile: infatti XML non definisce
una propria collezione di marcatori (detti anche TAG) ma definisce le regole sintattiche con
il quale è possibile generare dei marcatori personalizzati e i loro eventuali attributi.
XML è quindi un metalinguaggio, cioè uno strumento per realizzare altri linguaggi,
semplicemente creando nuovi tag.
Allo stesso modo è possibile creare delle regole semantiche di relazione tra marcatori.
XML inoltre è in formato testo e non binario, permettendo una alta portabilità tra
piattaforme diverse; inoltre non è legato ad una specifica azienda ma al W3C, un
consorzio di aziende e istituti di ricerca che si preoccupa del mantenimento e dello
sviluppo di XML.
PNML è quindi una estensione di XML, tramite appositi tag per la rappresentazione delle
caratteristiche di una rete di Petri in termini di posti, transizioni e archi e delle regole che

- 99 -
esistono tra questi oggetti (ad esempio: un posto non può essere preceduto o seguito da
uno o più posti ma solo da un numero qualsiasi di transizioni).
Allo stesso modo, CSML è un linguaggio con cui si descrive una cellula in termini dei flussi
che la contraddistinguono e delle relazioni tra questi flussi.
Nella prima fase di sviluppo del programma, è stato deciso di utilizzare una semplice
customizzazione di XML che permette di rappresentare i suoi elementi e i collegamenti fra
di essi.
Successivamente bisognerà sviluppare un controllo semantico della rete che preceda ogni
altra operazione relativa alla stessa rete.
La scelta della libreria che implementa il parserizzatore (lettore) ed il writer di XML è
caduta su jdom: jdom è una particolare implementazione della libreria DOM, una delle due
librerie (l’altra è SAX) che permettono di gestire i file XML in Java.
La caratteristica di jdom è il fatto che a seguito della lettura del file XML memorizza tutta la
gerarchia, per cui ogni volta che si richiede ad esempio di trovare i nodi figli di un nodo
non è necessario effettuare per l’ennesima volta la lettura del file, come avviene invece
per SAX. Poiché le reti rappresentate non sono costituite da un numero elevato di
informazioni come può invece avvenire per altri utilizzi di XML, l’occupazione di memoria
non sarà eccesiva.
Quello che segue è l’esempio di un file XML generato dalla rete in Figura 39, attribuendo il
valore 4 a K e la marcatura iniziale (1, 1, 0), da cui si possono desumere le regole
sintattiche applicate:
<?xml version="1.0" encoding="UTF-8"?>
<root>
<P1 Tokens="2.0">
<OUT Peso="1.0" Velocità ="4.0" flusso="multiplo">
<P1 Peso="1.0" />
<P2 Peso="1.0" />
</OUT></P1>
<P2 Tokens="0.0">

- 100 -
<OUT Peso="1.0" Velocità ="4.0" flusso="multiplo">
<P1 Peso="1.0" />
<P2 Peso="1.0" />
</OUT>
</P2>
<P3 Tokens="0.0">
<IN Peso="1.0" Velocità ="4.0" flusso="multiplo">
<P1 Peso="1.0" />
<P2 Peso="1.0" />
</IN>
</P3>
</root>
5.3.2 L’interfaccia grafica e la memorizzazione dei flussi della rete
L’interfaccia grafica creata è stata progettata in modo da essere semplice,
immediatamente comprensibile e leggera dal punto di vista computazionale.
L’interfaccia è infatti dotata di un’area grafica in cui inserire posti, transizioni e archi.
L’inserimento di ognuno di questi pezzi della rete si effettua cliccando uno dei bottoni
corrispondenti al componente scelto; apparirà quindi una finestra pop-up che richiederà i
dati relativi al componente, quali:
- la marcatura iniziale, nel caso di un posto;
- la velocità, nel caso di una transizione;
- il verso, il peso e i nodi collegati nel caso di un arco.
E’ da notare che è possibile creare contemporaneamente più reti: questo è
particolarmente utile quando si vuole copiare tutta o parte di una rete e
modificarla/estendere.
Infine sono presenti dei menù a tendina con cui è possibile salvare file in XML e
richiamare file già salvati e creare gli script Matlab.

- 101 -
Bisogna qui ricordare che una rete di Petri è un grafo bipartito, cioè un grafo costituito da
archi e nodi e questi ultimi sono divisi in due insiemi, posti e transizioni.
La scelta dei moduli da utilizzarsi per creare e gestire le reti di Petri è stata quindi
indirizzata su un codice già disponibile in rete, noto come JGraph ([JGraph2009]).
JGraph è una libreria open-source contenente codice Java basato su Swing, alla base
delle moderne interfacce grafiche Java.
Questa libreria contiene numerose funzioni che hanno come primo scopo la creazione di
grafi di qualunque tipo, purché basati sui concetti di nodo e di arco. A queste funzioni di
base, JGraph aggiunge alcune funzioni avanzate legate all’editing e all’analisi dei grafi.
Gli eventi di creazione dei nodi e degli archi portano quindi alla definizione grafica di una
rete e alla memorizzazione di una struttura jgraph in cui sono appunto rappresentati i nodi
e i collegamenti degli stessi tramite archi.
La generazione di XML e di script Matlab passa attraverso una nuova rappresentazione
della struttura del grafico, ottenuta memorizzando in apposite strutture informative dette
array, le informazioni relative ai nodi e archi, ciascuno con le proprie caratteristiche (pesi,
velocità, marcatura) e ai collegamenti tra questi elementi.
Questi array sono alla base di tutte le successive elaborazioni.
Infatti, l’XML che viene creato memorizza la gerarchia di collegamenti collegati al singolo
posto, per cui è stata creata una apposita classe che memorizza l’informazione relativa ad
un singolo flusso che caratterizza un singolo posto ed una classe distinta che memorizza
la collezione di flussi associata ad un singolo posto.
Attraverso queste informazioni viene prima creato l’XML contenente come nodi figli i
singoli posti e per ciascuno di questi sono memorizzati i flussi che lo caratterizzano;
successivamente vengono estratte le informazioni dei flussi per ogni nodo e viene creato il
file Matlab.
5.3.3 La generazione degli script Matlab
In questa versione ci si è concentrati molto sulla creazione dei file XML rappresentativi
della rete e sulla generazione degli script Matlab.
Gli script Matlab sono dei file in formato testo per cui si è deciso di trattarli utilizzando le
funzioni di trattamento testo presenti in Java.

- 102 -
La generazione degli script parte dall’analisi del modello grafico introdotto, passa
attraverso la generazione del file XML e crea i due file Matlab per la definizione del
sistema di ODE e per il suo calcolo.
Bisogna ricordare che nel sistema di ODE, ogni riga rappresenta i flussi attinenti un
compartimento che nella rete di Petri è rappresentato da un posto. L’algoritmo quindi
deve, a partire da ogni posto, ricostruire i flussi in input e in output dello stesso.
Per i flussi in output dovrà valutare indipendentemente le transizioni in output, calcolando il
prodotto della velocità della transizione per il peso dell’arco che le collega al posto.
Per i flussi in input il discorso è più complicato: singole transizioni non precedute da posti
vanno calcolate così come per le transizioni solitarie in output. Invece, nel caso in cui le
transizioni in input siano precedute da posti, allora la transizione porta con se il prodotto
dei posti, ciascuno moltiplicato per il peso dell’arco che le collega alla transizione e di
nuovo moltiplicato per il peso dell’arco che collega la transizione al posto analizzato.
Come detto precedentemente, dopo aver editato un grafico di una rete, si genera il file
XML contenente i singoli flussi per ogni singolo nodo: questo XML viene poi letto (parse
rizzato) da un codice che riconosce i singoli tag e ne ricava tutti i flussi per ogni nodo in
modo da derivarne le equazioni differenziali caratteristiche.
5.3.4 Esempio di utilizzo del programma
Consideriamo il seguente pathway:
Figura 38 - Pathway di esempio per l'utilizzo del programma

- 103 -
che si traduce nella seguente rete di Petri:
Figura 39 - Reti di Petri associata al pathway di esempio
Questi sono i file di Matlab script risultanti:
ode.m
[t,x]=ode45('pr',[0,7],[1.0,1.0,0.0])
plot(t,x(1))
plot(t,x(2))
plot(t,x(3))
pr.m
function xdot=foo5(t,x)
xdot(1)=-5.0*x(1)*1.0*x(2)*1.0;
xdot(2)=-5.0*x(1)*1.0*x(2)*1.0;
xdot(3)=+1.0*5.0*1.0*x(1)*1.0*x(2);
xdot=[xdot(1);xdot(2);xdot(3)];

- 104 -
6. Modello Glu-Ins-NEFA: analisi
qualitativa e quantitativa
In questo capitolo viene analizzato il modello Glu-Ins-NEFA inizialmente proposto in
[Sal2007]. Questo modello aggiunge una modulazione della secretion rate statica dovuta
agli acidi grassi o NEFA.
L’analisi qualitativa è stata effettuata mediante reti di Petri: è stato prima creato un modello
PN del pathway che è poi stato analizzato mediante le tecniche riportate nei capitoli 4 e 5.
L’importanza di questa modellizzazione è duplice in quanto, da una parte,i modelli di
pathway biochimici finora proposti in letteratura sono relativi a contesti legati a reti
genetiche o di produzione di proteine e invece in questo caso si è lavorato sulla
segnalazione biochimica che porta alla produzione di insulina a partire da glucosio e
NEFA; dall’altra parte, la particolarità del modello e soprattutto delle soluzioni utilizzate
possono essere un valido esempio da applicare ad altri pathway metabolici.
A seguire, viene presentata una analisi quantitativa del modello effettuata con Matlab:
rispetto a quanto presentato in [Sal2007], i test sono stati fatti nell’arco dei 180 minuti
invece che nelle 24 ore. L’analisi effettuata consente di validare il modello per soggetti
obesi che presentano diabete di tipo 2, non soggetti a operazioni chirurgiche come la
BPD.
Come verrà spiegato nel capitolo 7, in cui si analizzano i risultati dei modelli per pazienti
pre e post BPD, esiste qualche fattore di origine intestinale che modifica il modello
proposto nel caso dei pazienti BPD e per questo, nel lavoro futuro, si provvederà ad
estendere il modello in modo da testare l’influenza di questi fattori aggiuntivi.

- 105 -
6.1 Modello del pathway Glucosio-Insulina-NEFA
6.1.1 Il pathway Glu-Ins-NEFA
In vari modelli sviluppati negli ultimi anni (vedi [Mar2001], [Mar2002], [Tof2001]), la IS
(Insulin Secretion) delle cellule beta stimolata dal glucosio, è legata sia ad una
componente statica dovuta alla concentrazione di glucosio G nel plasma sia ad una
componente dinamica dovuta all’innalzamento della concentrazione plasmatica di glucosio
(si tratta cioè di una componente che non è legata alla concentrazione ma alla derivata nel
tempo della concentrazione dG/dt, per valori positivi di questa derivata).
E’ stato riconosciuta inoltre l’influenza di alcune ‘incretine’ (sostanze che agiscono come
fattore di potenziamento) sia per la componente statica che per quella dinamica, ad
esempio il GLP-1 (glucagon-like peptide-1) e, con minore influenza, il GIP (glucose-
dependent insulinotropic Polypeptide), potenti stimolanti della IS sia in vivo che in vitro.
Nessuno di questi modelli gestisce però il fatto che il LC-CoA citosolico agisce da ‘fattore
di accoppiamento’ per la IS e che sia il glucosio che i NEFA agiscono sulla IS
mediante il LC-CoA.
Gli acidi grassi (FA) agiscono come ‘effettori’ nella IS: il glucosio aumenta il LC-CoA
(cytosolic long-chain acyl-CoA) attraverso l’aumento di malonyl-CoA, che fa da ‘starter’
della sintesi di FA e inibisce la β-ossidazione mitocondriale, realizzando uno shift dagli FA
all’ossidazione del glucosio.
Yaney e Corkey hanno recentemente rivisto il ruolo degli FA nel controllo della IS (vedi
[Yan2003]), ed hanno sottolineato che:
1. una sorgente di FA esogena o endogena è necessaria per supportare la normale
IS;
2. un rapido incremento degli FA potenzia la IS stimolata da glucosio tramite
l’aumento della concentrazione di acyl-CoA grasso o di lipidi complessi che
agiscono indirettamente modulando enzimi chiave, come la proteino chinasi-C, o
direttamente tramite la modulazione del meccanismo esocitotico.

- 106 -
Il modello della IS stimolata da glucosio e modulata dai NEFA è rappresentato in Figura
40.
Figura 40 Modello della IS stimolata da glucosio e modulata dai NEFA
In questo modello, Fp denota la concentrazione dei NEFA nel plasma mentre F denota la
concentrazione dei NEFA dentro la cellula beta. Il modello più semplice che relaziona F e
Fp è una cinetica del primo ordine:
dF/dt = k1Fp - k2F
secondo cui le variazioni di NEFA interne alla cellula beta sono una versione smussata
della concentrazione del NEFA nel plasma.
Il termine k2F rappresenta il rate di formazione del LC-CoA nel cytosol dal NEFA della
cellula beta.
Il LC-CoA ha due possibili destini: il trasporto nel mitocondrio e l’utilizzazione nel
exocytotic machinery nel cytosol.

- 107 -
Assumendo la cinetica del “binding” di LC-CoA al CPT1 (carnitine palmitoyltransferase 1)
abbastanza veloce da potersi considerare in equilibrio nel tempo, possiamo scrivere
k2F = kint RC/(Km+C)+ k3C
dove kint è il rate costante di internalizzazione nei mitocondri, RC/(Km+C) la concentrazione
di LC-CoA legato a CPT1 (essendo Km la costante di Michaelis-Menthen) mentre k3 è la
costante di utilizzazione citosolica del LC-CoA.
Secondo il modello presentato, possiamo esprimere la componente statica S del rate della
IS come
S= αGC
dove α è una costante di sensibilizzazione.
Come dimostrato in [Sal2007], abbiamo per S la seguente espressione
S= (p1/2) ( -(p2+p3/G-F)+ sqrt [ (p2+p3/G-F)^2+ 4p2F] ) G
dove con “sqrt” si indica l’operazione di radice quadrata. Con i parametri pi da stimare;
aggiungendo il termine dinamico, si ha:
ISR =S+kd dG/dt, per dG/dt>0
ISR =S, per dG/dt<0
6.1.2 Modellizzazione tramite rete di Petri del pathway
La modellizzazione tramite rete di Petri del pathway oggetto dell’analisi deve considerare
diversi aspetti, legati al fatto che bisogna ‘tradurre’ il pathway di segnalazione biochimico
in componenti logici di una rete di Petri.
In Figura 41 è possibile vedere il modello PN ideato per modellizzare il pathway Glucosio-
Insulina-NEFA.

- 108 -
Figura 41 - Modello PN del pathway Glu-Ins-Nefa
Come si può notare da subito, la parte del modello di Figura 40 che descrive il processo
metabolico che porta ‘direttamente’ alla secrezione di insulina da parte della cellula beta
stimolata dalla presenza di glucosio è stato praticamente riportato tale e quale nella parte
sinistra di Figura 41 del modello a PN del pathway.
Diversamente, la parte del modello che fa riferimento alla modulazione indiretta del NEFA,
da parte del glucosio, relativamente alla secrezione di insulina, è stata soggetta a
particolari “rimaneggiamenti”.
Il LC-CoA infatti contribuisce come detto alla ISR e per questo motivo c’è un arco che
porta direttamente dal LC-CoA ad una transizione marcata ISR (sui dettagli di quest’ultima
transizione si tornerà più avanti).
La difficoltà maggiore è stata però nel riportare il contributo negativo della catena
Glucosio-Piruvato-Citrato-MalCoA; infatti questa catena diminuisce il numero (la
concentrazione) di CPT1 disponibili a legarsi con il LC-CoA diminuendo il rate di

- 109 -
internalizzazione del LC-CoA nel mitocondrio (transizione T11-kint). Si è voluto evitare di
utilizzare a tale scopo un arco inibitore: questo avrebbe reso non applicabile l’utilizzo delle
tecniche di analisi con invarianti delle reti di Petri.
Si è quindi scelto di modellare questo aspetto caratterizzando il posto CPT1 con due ‘read
arcs’: la presenza di MalCoA derivante dal glucosio ha un effetto ‘stealing’ sul CPT1, cioè
ruba scatti alla transizione T13 che caratterizza il binding del LC-CoA con i siti CPT1.
Si sottolinea il carattere puramente qualitativo di questa fase di analisi: infatti il concetto di
scatto di una transizione è puramente aleatorio, cioè nell’insieme delle transizioni abilitate
a scattare, quella che scatta viene scelta in maniera puramente casuale per cui nulla
vieterebbe un numero di scatti indefiniti della transizione T8 piuttosto che della T13;
l’analisi tramite PN continua invece sorpassa questo ‘problema’, in quanto gli scatti
avvengono continuamente su tutte le transizioni abilitate.
Nel pieno rispetto del modello biologico, si può notare la presenza di un ‘ciclo’tra i posti
LC-CoA e LC-CoA/CPT1, ciclo che modellizza l’equilibrio tra il LC-CoA libero e quello
legato al CPT1.
Relativamente alla ISR, è importante notare che non ho un ‘AND logico’ tra questa
sezione del pathway e quella diretta: ho un processo ‘diretto’ (funzione del solo glucosio) e
uno di modulazione da parte del NEFA alla stimolazione di glucosio (funzione del glucosio
e del NEFA).
I due processi contribuiscono entrambi alla ISR, per questo motivo si è deciso di legare la
parte di ISR del NEFA ad una transizione che ha in input anche i tokens relativi al Ca2+:
questo ha senso proprio perché questo processo non è indipendente da G.
6.1.3 Analisi qualitativa del modello PN del pathway Glu-Ins-NEFA
Relativamente al modello individuato, si individua un unico P-Invariante costituito dal posto
CPT1; sarà necessario mettere, nella marcatura iniziale, un token nel posto CTP1 per
rendere il modello operativo.
Ho i seguenti T-Invarianti:

- 110 -
T-invariante Transizioni
T1 t1, t2, t3, t4, t5 T2 t1, t2, t3, t4, t9, t10, t15 T3 t9, t10, t11, t13 T4 t1, t6, t7, t8 T5 t9, t10, t14 T6 t12, t13
Per quanto riguarda gli MCT, si individuano:
MCT-1: (t2, t3, t4);
MCT-2: (t9, t10);
MCT-3: (t6, t7, t8)
e poi le restanti transizioni a costituire MCT formate da singole transizioni.
In Figura 42 è possibile vedere il modello PN con i T-Invarianti individuati.
I primi due T-Invarianti rappresentano rispettivamente la ISR stimolata direttamente dal
glucosio e quella dovuta alla modulazione del glucosio da parte del LC-CoA prodotto dal
NEFA.
T5 rappresenta il pathway dell’utilizzazione citosolica del LC-CoA, mentre T3 rappresenta
il processo di internalizzazione del LC-CoA nei mitocondri.
T4 rappresenta il pathway con cui il glucosio causa la diminuzione di CPT1 disponibile al
binding con il LC-CoA mentre T6 rappresenta l’equilibrio tra il LC-CoA libero e quello
legato al CPT1.
Il MCT (t2, t3, t4) indica la sezione del pathway che da luogo al ISR o per stimolo diretto del
glucosio o modulato dal LC-CoA mentre il MCT (t9, t10) indica il processo che porta il NEFA
plasmatico ad essere internalizzato nella cellula beta e a produrre LC-CoA libero,
utilizzabile poi per tutti i sottoprocessi rappresentati dai vari T-Invarianti correlati al posto
LC-CoA.

- 111 -
Figura 42 - Modello PN con T-Invarianti individuati
Si nota che nella marcatura iniziale (un token nel posto CPT1) il T-Invariante T6 non è
feasible e nemmeno realizzabile: inserire un token nel posto LC-CoA lo renderebbe
realizzabile e feasible; per renderlo feasible è necessario portare un token nel posto P8 o
nel P10. Per portare un token in P10 c’è bisogno del T-Invariante T5, mentre non posso
portare un token in P8 senza averlo prima portato in P10: dunque ho un nuovo T-
Invariante, realizzabile e feasible (non minimale) ottenuto integrando T6 con il T-Invariante
T5.
Questa analisi e l’attribuzione di significato biologico ai diversi invarianti e MCT porta
anche ad alcune considerazioni sui modelli ‘lack’: se consideriamo infatti il solo invariante
T1, significa che sto considerando la sola presenza di stimolazione diretta da glucosio,
mentre non è possibile isolare il solo contributo del NEFA in quanto questo modula la
secrezione di insulina causata dal glucosio.

- 112 -
E’ però possibile isolare il contributo negativo del MalCoA (T-Invariante T4), la cui assenza
rende possibile il solo scatto della t13 e non della t8: dunque non diminuirebbero i CPT1
disponibili per il binding con LC-CoA.
Si noti inoltre come gli MCT, comprensivi dei posti PRE e POST e degli archi che ne fanno
parte, sono “unità funzionali”: in questo esempio rappresentano le catene di
trasformazione di alcune sostanze (MCT-3 da Piruvato a MalCoA), l’internalizzazione di
altre (MCT-2 dal NEFA Plasmatico a quello intracellulare) e la segnalazione intracellulare
(MCT-1 dalla formazione di Piruvato alla defosforilizzazione dell’ATP).
6.1.4 Analisi quantitativa del modello Glu-Ins-NEFA
Mettere qui i risultati delle simulazioni fatte con MATLAB
Il modello specificato al paragrafo 7.3.2 è stato simulato in Matlab, con una serie di script
necessari a:
- definire le equazioni precedentemente introdotte;
- supportare la gestione dei dati a disposizione, sia dal punto di vista della
conversione dell’unità di misura che della scelta dei soggetti da analizzare tra quelli
a disposizione;
- definire il metodo di ottimizzazione scelto, con tutti i suoi parametri e la funzione da
ottimizzare.
Dati i valori di C-peptide misurato (Cm) e di C-peptide calcolato (C), la funzione che si è
scelto di minimizzare è stata quella che definisce il rapporto tra l’energia dell’errore e
l’energia del C-peptide misurato, per ogni campione, cioè
( )22
i i
ii
Cm C
Cm
−∑
e il risultato può essere visto come la percentuale di errore dell’energia dell’errore rispetto
all’energia dei valori misurati.

- 113 -
Matlab mette a disposizione varie funzioni di minimizzazione, fra queste è stata scelta la
“fmincon”, definendo il massimo numero di iterazioni pari a 3000; l’ottimizzazione della
funzione è stata fatta rispetto ai parametri pi,p2,p3 della equazione della secretion rate
statica.
%opzioni per la minimizzazione
options = optimset('Algorithm', 'active-set', 'LargeScale', 'on', 'hessian', 'off',
'LevenbergMarquardt', 'on','MaxFunEvals',3000,'MaxIter',3000);
% 'MaxIter',N,'MaxFunEvals',N,'DiffMinChange',1e-3,'DiffMaxChange',1e-7
% ,'TolFun',1e-8
%funzione per la minimizzazione dell'energia dell'errore
[x,fval]=fmincon('CalcCPep',x0,[ ],[ ],[ ],[ ],l,u,[], options);
I valori di C-peptide sono stati calcolati dalla coppia di equazioni del modello bi-
compartimentale, in cui la SR è somma della SR statica e di quella dinamica.
%valor iniziali
y0=[0;0];
% utilizzo della ode23 sul sistema di due equazioni per il C-Peptide
[t,y]=ode23('CPepN',t,y0);
% viene presa solo la prima colonna, valore di CP1
a=y(:,1)/V;
%calcolo errore tra C-Pep simulato e C-Pep misurato:
%E' L'INDICE CALCOLATO DALLA FUNZIONE
Cm=cpep;%valore misurato
a=[a(1) a(30) a(60) a(90) a(120) a(150) a(180)]; %valore calcolato
%differenza
g=Cm-a;
%indice da minimizzare
f=(g*g')/(Cm*Cm');

- 114 -
La SR statica tiene conto dei valori di glucosio e dei NEFA plasmatici misurati e contiene i
valori dei parametri pi,p2,p3 usati per l’ottimizzazione:
for i=1:181
SRsN(i)=
(p1/2)*( -(p2+ p3/G(i) - FA(i)) + sqrt( ( p2+(p3/G(i))-FA(i) )^2 + 4*p2*FA(i)) ) * G(i);
end
La SR dinamica sfrutta la derivata dell’andamento del glucosio ed è proporzionale a tale
derivata nei punti i cui essa è positiva, altrimenti risulta nulla; nello script corrispondente
troviamo quindi il codice
for i=1:d
if gliced(i)>0
%SRd(i)=KG(i)*gliced(i);
SRd(i)=Kd*gliced(i);
else
SRd(i)=0;
end
end
Passiamo a vedere i risultati della simulazione.
Analizziamo due pazienti che non hanno ancora subito l’intervento di BPD.
I risultati che si ottengono sono i seguenti:

- 115 -
Figura 43 - Andamenti dei valori misurati e simulati per due pazienti non soggetti a BPD
Nei grafici di Figura 43 sono stati riportati gli andamenti dei valori misurati (in blu), il C-
peptide calcolato (in rosso, nello stesso grafico del C-peptide misurato) e l’energia
dell’errore; il risultato dell’ottimizzazione da luogo ai corrispondenti valori per i parametri pi:
Paziente p1 p2 p3
1 0.3182 0.9117 7.9994
2 0.1758 0.2919 10.3153

- 116 -
I valori di Kd assegnati sono stati 5.5 e 3.
Come si può notare, sia l’energia dell’errore (inferiore allo 0.3 %) che gli andamenti dei
valori di C-peptide, provano la bontà di questo modello.
6.1.4.1 Alcuni miglioramenti computazionali del modello
Una più attenta analisi degli script Matlab ha evidenziato il fatto che era possibile
apportare delle modifiche degli script stessi per migliorarli dal punto di vista dei tempi di
calcolo.
Prima di tutto si è notato che era possibile eliminare l’equazione
dF/dt = k1Fp - k2F
relativa al passaggio degli acidi grassi dal plasma sanguigno alla cellula: infatti la costante
k1 è molto piccola, ad indicare un passaggio praticamente “istantaneo” dei NEFA.
Questa modifica permette di eliminare, negli script, il richiamo di una ODE, così da
diminuire notevolmente i tempi di calcolo senza andare a discapito della precisione nei
calcoli.
Quindi, nella equazione della secretion rate statica
S= (p1/2) ( -(p2+p3/G-F)+ sqrt [ (p2+p3/G-F)^2+ 4p2F] ) G
sono stati riportati i valori desunti dai livelli plasmatici dei NEFA.
Una ulteriore modifica degli script è stata nella secretion rate SR utilizzata nel modello
bicompartimentale del C-peptide, modello utilizzato per confrontare i valori di C-peptide da
esso calcolati con i rispettivi livelli plasmatici.
Il modello bi-compartimentale del C-peptide è infatti caratterizzato dalle seguenti due
equazioni:

- 117 -
dCP1 /dt = -(k01+k21)*CP1+k12*CP2+SR;
dCP2 /dt = k21*CP1-k12*CP2;
dove il valore di C-peptide considerato è il CP1, mentre la SR deriva dalla secretion rate
calcolata con le equazioni suddette (somma della SR statica e della SR dinamica) con un
passaggio ulteriore per un modello monocompartimentale ad indicare un ritardo. Questo
passaggio è stato saltato a favore dell’introduzione di un ritardo statico: benché meno
elegante dal punto di vista matematico e della ottimizzazione, questo metodo consente di
avere una idea più immediata del ritardo effettivo nella secrezione dell’insulina, ottenuto
per tentativi e con un semplice confronto grafico tra il C-peptide misurato e quello
calcolato.

- 118 -
7. Conclusioni: prossimi passi e
proposte di sviluppo
7.1 Analisi della reversibilità dal diabete a seguito di
BPD
7.1.1 Esperimento effettuato
Nell’esperimento, la Insulin Sensitivity periferica e la funzionalità della cellula beta dopo
IVGTT e OGTT e con una analisi del modello minimale è stato effettuato per 9 obesi
diabetici di tipo 2 prima e dopo aver subito un intervento di BPD (Bilio Pancreatic
Diversion) e confrontata con 6 soggetti normali attraverso il EHC. Sono stati misurati i
valori di GLP1 (glucagon-like peptide-1) e GIP (Glucose-dependent insulinotropic
polypeptide).
Soggetti analizzati: sono stati testate 9 obesi morbidi (body mass index (BMI) = 51.7+/-
8.1 kg/m2 ) con diabete di tipo 2 (secondo la American Diabetes Association.
Glycosylated, haemoglobin [HbA(1c)] tra il 7.5 e il 9.5%) e sei volontari sani corrispondenti
per sesso ed età (BMI = 24.6 +/- 1.3 kg/m2 ). I
Protocollo di studio: all’inizio del procedimento, tutti i soggetti erano a dieta con la
seguente composizione media 60 % carboidrati, 30 % grassi e 10% proteine; questo
regime dietetico è stato mantenuto per una settimana prima dello studio. In tutti i soggetti
sono stati effettuati i test OGTT, IVGTT e l’euglycemic Hyperinsulinemic clamp (EHC), in
maniera casuale, 1 mese prima e 1 mese dopo l’operazione.
Tutti i pazienti hanno ricevuto lo stesso regime dietetico dopo l’operazione, a seguito del
quale erano a dieta libera.
OGTT: a seguito di una notte a digiuno, un OGTT standard da 75 grammi è stato
effettuato per ciascun paziente e su ciascun volontario, entro il mese precedente

- 119 -
l’operazione; sono stati effettuati campionamenti del sangue nei minuti 0, 30, 60, 90, 120,
150, 180 e 240.
IVGTT: un IVGTT è stato effettuato entro il mese precedente dell’operazione e entro il
mese seguente l’operazione. Alle 8-9 del mattino, dopo 12 ore di digiuno, un catetere
intra-venoso è stato posizionato in una vena ante-cubitale un bolo intravenoso di 0.33
grammi di glucosio per kilogrammo, al 50 %, è stata iniettata nella vena ante-cubitale
contro laterale. I campioni di sangue sono stati presi nei minuti –15, –5, 2, 4, 6, 8, 10, 12,
14, 16, 18, 20, 25, 30, 40, 50, 60, 70, 80, 100, 120, 140, 160, 180, 220, e 240,
relativamente all’inizio dell’iniezione.
EHC: la Insulin Sensitivity periferica è stata valutata dal EHC entro 4 settimane prima
dell’operazione.
La sensibilità insulinica e l’uptake di glucosio mediato dall’insulina sono stati calcolati
durante un’iniezione costante di glucosio (6 pmol per litro per kilogrammo). Il
mantenimento della glicemia è stato effettuato mediante l’infusione di insulina e la
determinazione di glucosio ogni 5 minuti.
BPD: questa procedura chirurgica di tipo “malabsorptive” consiste in una restrizione
gastrica di circa il 60%. Il volume residuo di stomaco è di circa 300 ml.
L’operazione reduce l’intestino tenue a 250 cm, I cui 50 cm finali (canale comune)
rappresentano il sito dove il cibo ingerito e il succo bilio-pancreatico si mischiano.
Modelli matematici: I modelli minimali OGTT e IVGTT (10) sono stati usati per calcolare la
insulin sensitivity (SI).
Gli indici di sensibilità della cellula beta al glucosio per IVGTT (prima e seconda fase del
sensibilità della cellula beta) e per OGTT (sensibilità dinamica e statica della cellula beta)
e la sensibilità totale sono state calcolate mediante il modello minimale del C-peptide
proposto da Toffolo (11) e Breda (12).
I parametri del modello sono stati stimati con una minimizzazione ai minimi quadrati.
Risultati
Una piccola ma significativa perdita di peso (da 153.1 +/- 34.2 a 143.5 +/- 32.8 kg,) è stata
osservata un mese dopo il BPD.

- 120 -
La Tabella 3 riporta l’AUC incrementale di glucosio, insulina e C-peptide nei soggetti obesi
prima e dopo il BPD, così come per i soggetti di controllo, sia per OGTT che IVGTT.
Gli AUC incrementale di glucosio e insulina erano significativamente ridotti dopo il BPD sia
per OGTT che IVGTT e i valori post BPD non differivano dai valori trovati nei controlli.
L’AUC incrementale di C-peptide era significativamente ridotta dopo il BPD nell’OGTT,
mentre non differiva significativamente dagli AUC di C-peptide nei soggetti diabetici prima
del BPD e nei controlli.
Le stime degli indici calcolati con i modelli orale e intra-venous sono riportate in Tabella 4.
La prima fase della secrezione di insulina erano pienamente normalizzata dopo BPD,
come mostrato in Tabella 4 dall’incremento marcato di φ1. La Figura 44 mostra, in un
paziente, il recupero della prima fase di ISR nel IVGTT. L’indice di sensibilità dinamica
mostra una tendenza a crescere dopo il BPD.
Figura 44 – Dati del IVGTT per un paziente obeso.
(Top: glucose (left) and insulin (right) plasma concentrations before (dashed line) and after (solid
line) BPD. Bottom: ISR (left) and C-peptide data points with the fitting curves superimposed (right). Pre-BPD data: dashed lines and circles; post-BPD data: solid lines and crosses)
Prima del BPD, l’indice di sensibilità dinamica determinato dal OGTT era
significativamente più piccolo di quello trovato con IVGTT o euglycemic clamp. Tuttavia,
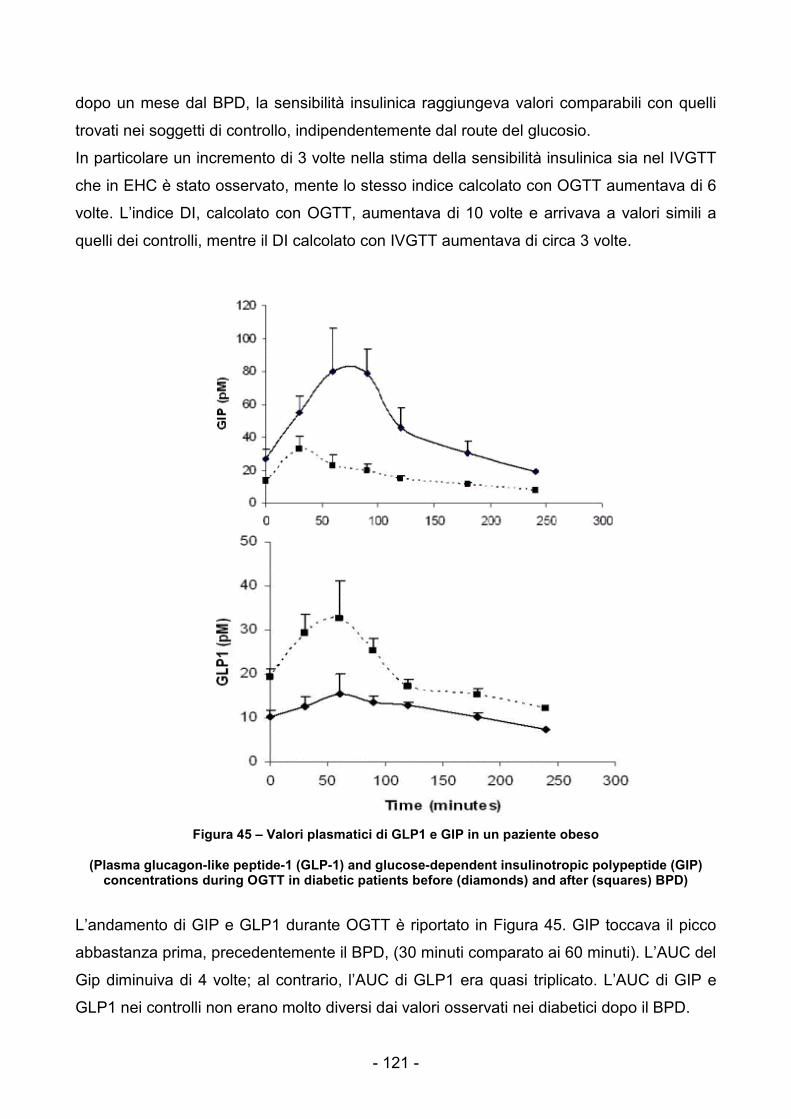
- 121 -
dopo un mese dal BPD, la sensibilità insulinica raggiungeva valori comparabili con quelli
trovati nei soggetti di controllo, indipendentemente dal route del glucosio.
In particolare un incremento di 3 volte nella stima della sensibilità insulinica sia nel IVGTT
che in EHC è stato osservato, mente lo stesso indice calcolato con OGTT aumentava di 6
volte. L’indice DI, calcolato con OGTT, aumentava di 10 volte e arrivava a valori simili a
quelli dei controlli, mentre il DI calcolato con IVGTT aumentava di circa 3 volte.
Figura 45 – Valori plasmatici di GLP1 e GIP in un paziente obeso
(Plasma glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) concentrations during OGTT in diabetic patients before (diamonds) and after (squares) BPD)
L’andamento di GIP e GLP1 durante OGTT è riportato in Figura 45. GIP toccava il picco
abbastanza prima, precedentemente il BPD, (30 minuti comparato ai 60 minuti). L’AUC del
Gip diminuiva di 4 volte; al contrario, l’AUC di GLP1 era quasi triplicato. L’AUC di GIP e
GLP1 nei controlli non erano molto diversi dai valori osservati nei diabetici dopo il BPD.

- 122 -
Discussione
I principali punti a cui si è arrivati con questo studio sono stati:
- la prima fase di secrezione insulinica è ristabilita dopo un mese dal BPD;
- la sensibilità delle cellule beta era pienamente normalizzata;
- Il DI era normalizzato grazie alla normalizzazione della sensibilità insulinica e i la
conseguente riduzione di necessità si secrezione di insulina;
- l’aumento di sensibilità dell’insulina stimato con OGTT era maggiore di quello
stimato con IVGTT.
L’associazione della disfunzione della cellula beta con l’Insulino-resistenza rappresenta il
difetto patofisiologico che maggiormente responsabile dello sviluppo di diabete di tipo 2.
La funzione delle cellule beta nel diabete di tipo 2 è caratterizzata dal progressivo declino,
dalla riduzione netta alla scomparsa della prima fase di secrezione insulinica dovuta al
glucosio, all’anormalità della seconda fase. La risposta insulinica iniziale scompare, anche
nei primi stadi quando i livelli di glucosio a digiuno sono leggermente superiori alla media.
Questo difetto è importante perché la prima fase di risposta insulinica sembra avere
l’impatto maggiore sull’escursione postprandiale del glucosio, determinando l’iperglicemia
post-pasto
E’ interessante notare come nello studio la prima fase di secrezione insulinica è reversibile
dopo il BPD, quando Il peso corporeo si reduce solo del 6%.
Briatore et al. (20) hanno recentemente riportato che l’AIR dopo IVGTT era
significativamente aumentata a seguito del BPD, negli obesi morbidi con diabete di tipo 2.
Tuttavia, basandosi sulle concentrazioni di insulina, l’AIR non da direttamente la prima
fase, perché riflette anche l’estrazione epatica.
Basandosi sugli studi sui topi, Henquin et al. (vedi [Hen08]) hanno dimostrato come la
risposta di prima fase, assente in vivo, era stata ristabilita in vitro (rispetto alle cellule beta
degli stessi animali).
Sulla stessa base, nello studio noi suggeriamo che ci sia un fattore che inibisce la
secrezione di insulina, fattore prodotto nell’intestino piccolo e la cui produzione può essere
ridotta dal bypass avvenuto con il BPD.
Le analisi del GLP1 dimostrano che l’infusione di GLP1 nei diabetici di tipo 2 è in grado di
aumentare l’AIR dopo IVGTT (da 197 ± 97 a 1,141 ± 409 pM min), ma rimane comunque 7

- 123 -
volte più bassa rispetto ai soggetti normali: ciò suggerisce la presenza di fattori intestinali
sconosciuti che regolano la normalizzazione della secrezione insulinica.
Questo fattore potrebbe anche determinare l’Insulino-resistenza. Infatti, la sensibilità
insulinica era normalizzata dopo BPD quando una piccola ma significativa diminuzione di
peso era raggiunta. A supporto di questa ipotesi, si è riscontrato che l’uptake di glucosio
mediato dall’insulina era significativamente più alto dopo il BPD quando il carico di
glucosio era amministrato oralmente invece che per via intra-venosa.
Dalla Man et al. (vedi [DaM05]) hanno dimostrato che l’azione insulinica sull’assunzione di
glucosio stimata dal modello orale era quasi identica a quella stimata con EHC,
suggerendo quindi che l’assunzione di glucosio modellizzata con il modello OTT era ben
descritta.
Dunque, almeno nei controlli, il modello OGTT della cinetica del glucosio fornisce stime
equivalenti dell’azione insulinica rispetto al EHC.
Questa osservazione rinforza quanto trovato, cioè che dopo il BPD la sensibilità insulinica
aumenta molto di più dopo un test orale rispetto ad un test intravenous. Inoltre la Insulino-
resistenza prima dell’operazione era maggiore dopo una assunzione orale che in quella
intra-venous.
Dunque, I cambiamenti anatomici indotti dall’operazione portano alla completa inversione
della sensibilità insulinica a seconda del route di assunzione del glucosio,
OGTT
∆AUCglucose x10-3 (mM⋅ min) ∆AUCinsulin x10
-4 (pM⋅ min) ∆AUCC-peptide x10-2 (nM⋅ min)
Pre BPD Post BPD Pre BPD Post BPD Pre BPD Post BPD
0.74±±±±0.08
0.22±±±±0.04
3.83±±±±0.99
1.01±±±±0.28
2.48±±±±0.35
1.10±±±±0.34
P<0.02
P<0.02
P<0.02
0.16±±±±0.03
1.86±±±±0.40
1.71±±±±0.36
Diabetic subjects Controls
NS
NS
NS
IVGTT
∆AUCglucose x10-3 (mM⋅ min) ∆AUCinsulin x10
-4 (pM⋅ min) ∆AUCC-peptide x10-2 (nM⋅ min)
Pre BPD Post BPD Pre BPD Post BPD Pre BPD Post BPD
0.63±±±±0.08
0.51±±±±0.06
3.66±±±±0.65
1.90±±±±0.27
1.63±±±±0.28
1.54±±±±0.50
P<0.05
P<0.02
NS
0.31±±±±0.02
0.50±±±±0.06
0.85±±±±0.16
Diabetic subjects Controls
NS
NS
NS
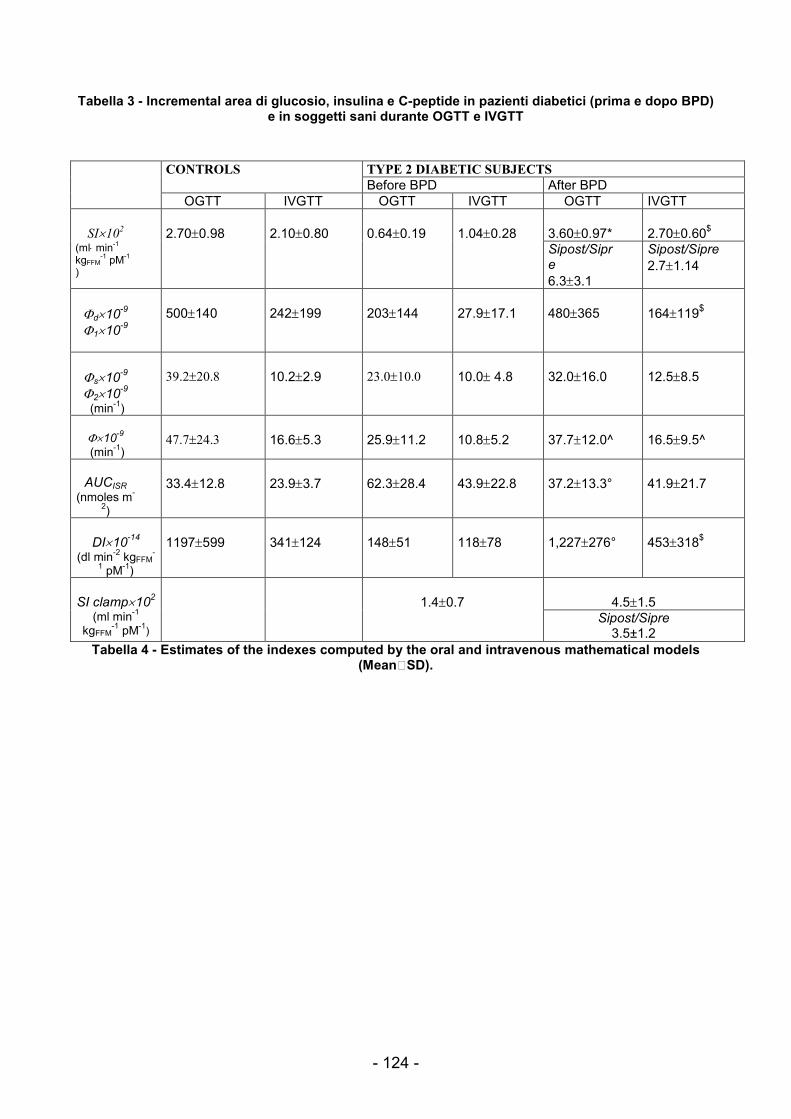
- 124 -
Tabella 3 - Incremental area di glucosio, insulina e C-peptide in pazienti diabetici (prima e dopo BPD) e in soggetti sani durante OGTT e IVGTT
TYPE 2 DIABETIC SUBJECTS CONTROLS
Before BPD After BPD
OGTT IVGTT OGTT IVGTT OGTT IVGTT 3.60±0.97*
2.70±0.60$
SI×102 (ml⋅ min-1 kgFFM
-1 pM-1 )
2.70±0.98
2.10±0.80
0.64±0.19
1.04±0.28
Sipost/Sipre 6.3±3.1
Sipost/Sipre 2.7±1.14
Φd×10
-9
Φ1×10-9
500±140
242±199
203±144
27.9±17.1
480±365
164±119$
Φs×10
-9 Φ2×10
-9
(min-1)
39.2±20.8 10.2±2.9
23.0±10.0 10.0± 4.8
32.0±16.0
12.5±8.5
Φ×10-9 (min-1)
47.7±24.3 16.6±5.3
25.9±11.2
10.8±5.2
37.7±12.0^
16.5±9.5^
AUCISR
(nmoles m-
2)
33.4±12.8
23.9±3.7
62.3±28.4
43.9±22.8
37.2±13.3°
41.9±21.7
DI×10-14
(dl min-2 kgFFM-
1 pM-1)
1197±599
341±124
148±51
118±78
1,227±276°
453±318$
4.5±1.5
SI clamp×102
(ml min-1 kgFFM
-1 pM-1)
1.4±0.7
Sipost/Sipre 3.5±1.2
Tabella 4 - Estimates of the indexes computed by the oral and intravenous mathematical models (MeanSD).

- 125 -
7.2 Sviluppo del software di supporto analisi
Nel capitolo 6 è stato presentato il progetto di un software di supporto analisi dei pathway
metabolici, sono state chiarite le necessità, il contesto di utilizzo e le sue caratteristiche più
importanti.
È importante sottolineare che questo programma è in una fase di continuo divenire, sia
perché se ne vogliono migliorare le caratteristiche sia per introdurne di nuove.
Relativamente a quest’ultima direzione, si vogliono riportare alcune delle azioni che si
vogliono fare per sviluppare il software.
Prima di tutto, la caratteristica di interfacciamento del software deve essere fatta tendere
verso standard internazionali: attualmente il programma gestisce dei file di
rappresentazione della rete basati su una estensione proprietaria di XML. Saranno quindi
riformulati opportunamente i moduli di scrittura e lettura XML in modo da supportare i
formati PNML e CSML.
Per far ciò, oltre alla sintassi di base, dovranno essere introdotte le regole grammaticali di
questi linguaggi XML-like.
Un altro obiettivo futuro è permettere la traduzione dei file che rappresentano i pathway
metabolici da un formato all’altro; gli obiettivi specifici sono:
introdurre una funzione che permetta di tradurre un formato XML all’altro;
introdurre una funzione di traduzione da un formato XML ad uno script Matlab e una
funzione che faccia l’opposto;
Quest’ultimo punto permetterebbe anche di creare lo script Matlab senza ogni volta creare
da zero la rete: il software infatti attualmente, a seguito dell’introduzione grafica della rete,
crea sia gli XML che gli script Matlab, ma non permette di aprire l’XML, editarlo e/o
trasformarlo in uno script Matlab.
Allo stesso modo, attualmente da un file XML non posso visualizzare il corrispondente
grafico: sarà introdotta una apposita funzione.

- 126 -
Infine, un importantissimo obiettivo di breve termine è la gestione di basi di dati: è infatti
importante strutturare sia l’informazione posseduta e data come input al programma così
come l’informazione che si rende disponibile in output, attraverso l’utilizzo di linguaggi e
prodotti standard, quali ad esempio SQL.
L’obiettivo di medio termine è invece quello di introdurre il supporto per la
caratterizzazione stocastica dell’analisi dei pathway biochimici.
A tal proposito si veda l’utilizzo di reti di Petri Stocastiche descritto nel paragrafo
successivo e l’interfacciamento con gli opportuni tool di calcolo: è possibile supportare le
funzioni e i tool forniti con Matlab ovvero utilizzare altri tool, specifici per le reti di Petri
Stocastiche o di generale utilizzo negli ambienti della ricerca biologica (ad esempio
StocSim).
Infine, l’obiettivo di lungo termine è la progettazione di un ambiente adatto alla gestione di
framework di lavoro: un ambiente software che permetta cioè di gestire i gruppi di lavoro,
le tempistiche e le versioni dei vari esperimenti così come delle simulazioni e di tutti i tool
necessari.
7.3 Uso dell’analisi stocastica
Come è stato visto nel capitolo 4, le reti di Petri sono un ottimo strumento di analisi dei
processi rappresentabili in uno spazio di stato discreto.
Nel capitolo 6 è stato visto come le reti di Petri permettono di rappresentare i pathway
biochimici che rappresentano il metabolismo umano e come le stesse tecniche di analisi
delle reti permettono di effettuare una analisi qualitativa del pathway.
Allo stesso tempo, il capitolo 6 ha introdotto il framework di analisi qualitativa - quantitativa
dei pathway (vedi Figura 46), facendo intendere come la stessa struttura rappresentata da
una rete poteva essere utilizzata come base sia per l’analisi e validazione del modello dal
punto di vista qualitativo, sia per l’analisi quantitativa mediante un sistema di ODE.
Una delle congiunzioni tra il mondo qualitativo - discreto e quello quantitativo-continuo
si basava sui concetti di invarianti che trasformavano una rete di processi biochimici in una
rete di segnalazione biochimica e davano una serie di ipotesi sulle condizioni iniziali del
sistema di ODE.

- 127 -
Nello stesso framework però si va ad inserire un altro tassello, che è quello dell’analisi
stocastica.
Bisogna infatti rimarcare il fatto che la modellizzazione matematica dei processi metabolici
e in generale dei processi biologici sia caratterizzata, dal punto di vista della ricerca, da
una dicotomia dei metodi di analisi: infatti, alla visione “analisi qualitativa - discreta vs
analisi quantitativa - continua”, si aggiunge la contrapposizione tra metodi deterministici
e metodi stocastici.
I modelli deterministici hanno infatti alcune limitazioni, dovute al fatto che la
rappresentazione dei pathway metabolici biochimici si basa su alcune ipotesi di fondo.
Certamente si osserva che, ragionando su scale molecolari delle reazioni biochimiche, si
vedrebbero le singole molecole interagire casualmente tra di loro: se le energie
possedute dalle singole molecole sono abbastanza elevate, allora queste potrebbero
reagire.
Per convertire questo sistema probabilistico, con la sua granularità molecolare, in un
modello continuo e deterministico, bisogna appunto fare una serie di assunzioni, che
vengono di seguito riportate:
- Per convertire una distribuzione spaziale di molecole in una singola variabile
continua rappresentante una concentrazione, bisogna assumere che il sistema
abbia volume infinito. Quindi, al diminuire del volume, il modello diviene meno
accurato e questo sicuramente accade al livello delle singole cellule dove ho volumi
minimi e dove il numero di molecole o di qualunque altro fattore di segnalazione
biochimica è nell’ordine delle decine o centinaia di unità.
- Le fluttuazioni nei sistemi fisici in cui i pathway avvengono vengono considerate
irrilevanti e quindi ignorate. Tuttavia, se tali fluttuazioni avvengono vicino a punti di
equilibrio instabile, le fluttuazioni stocastiche possono essere amplificate a livelli
macroscopici.
- La rappresentazione delle specie chimiche come variabili di concentrazione
continue ha dietro di se l’assunzione che il sistema sia assolutamente uniforme. Ciò
significa anche che assumendo costanti i rate di reazione, si ipotizza implicitamente
che ogni molecola è associabile ad una “molecola rappresentativa” il cui
comportamento è predicibile con assoluta certezza. In realtà i sistemi biochimici
presentatno spesso eterogeneità spaziale dovuta a compartimentazione,
associazione di proteine o altre specie chimiche, diffusione.

- 128 -
Con un modello stocastico, le concentrazioni sono rappresentate da variabili stocastiche
discrete, di solito il numero delle molecole di una particolare specie chimica, per cui non si
assume più il volume infinito. La localizzazione spaziale e le fluttuazioni possono essere
rappresentate nel modello stocastico e inoltre, nel caso di grandi volumi, il comportamento
medio presentato dal modello stocastico tende a quello del modello deterministico.
Come per una rete di Petri qualitativa, una rete di Petri stocastica ha un numero discreto di
tokens. Ma al contrario del caso time-free, alla transizione si associa una rate di scatto o
tempo di attesa, rappresentato da una variabile stocastica. Quindi, le reazioni avvengono
in un certo tempo che però non è certo, bensì è la realizzazione di una variabile
stocastica.
Dunque il sistema è descritto dallo stesso spazio di stato del modello deterministico
discreto e dalla stessa struttura topologica del modello qualitativo: di conseguenza, le
analisi (e le proprietà) di tipo strutturale sono ancora valide.
Figura 46 - Framework di utilizzo delle PN per l'analisi dei pathway biochimici
Anche per le SPN (Stochastic Petri Nets) una transizione per scattare deve essere
abilitata, ma lo scatto avviene dopo un tempo assegnato in modo aleatorio, con
distribuzione esponenziale.
La semantica di una SPN è associabile ad una catena di Markov tempo continua (CTMC)
isomorfa al grafo di raggiungibilità della PN associate.
Le applicazioni delle SPN sono le più varie, tra queste una recente ed interessante si può
trovare in [Asn2009] in cui, come si può vedere dalla Figura 47, è stato analizzato un

- 129 -
processo aziendale in un contesto IT mediante modellizzazione con reti di Petri e analisi
sia della rete che della CTMC associata.
Figura 47 - Esempio di applicazione di SPN in un contesto IT per l'analisi di processi aziendali
Volendo associare un significato biologico alla SPN, bisogna specificare il significato e tipo
di funzioni probabilistiche associate.
I tokens sono associabili a livelli di concentrazione e si assume che ci sia un valore
massimo di concentrazione e che il numero di differenti livelli sia N+1.
Quindi, i livelli astratti 0,…N rappresentano gli intervalli di concentrazione
0, (0,M/N), (M/N, 2*M/N), …( (N-1)*M/N, N*M/N ).
Questo numero finito di livelli sono quindi delle classi di equivalenza per un numero
infinito di stati continui. La funzione stocastica di livello associata alla SPN biochimica è
legata alla legge di azione di massa delle reti biochimiche, dove i tokens corrispondono ai
livelli di concentrazione e per cui si ha:
dove kt è il rate costante di transizione della specifica transizione ed N è il numero
associate al livello più alto.
Le regole di trasformazione tra rate stocastici e deterministici sono ben note (ad esempio
vedi [Wil06]).

- 130 -
In pratica I rate cinetici sono presi dalla letteratura o determinati da esperimenti.
In aggiunta a questo, l’analisi probabilistica dei comportamenti nei transienti e a regime è
spesso disponibile in lacune banche dati ed è effettuata in maniera analitica oppure in
maniera simulativa.
L’approccio analitico consiste nell’analisi esatta del comportamento e delle proprietà della
CTMC associata alla SPN ed è facilmente fattibile per CTMC finite e quindi nel caso d PN
associate limitate.
Al contrario l’analisi ottenuta a seguito dell’approccio simulativo funziona bene anche per
sistemi a spazio di stato infinito e per sistemi con dinamiche complessi come quelli
rappresentabili mediante semi-processi di Markov o processi di Markov generalizzati.
In conclusione, si vuole qui rimarcare il fatto che l’analisi stocastica permette di
quantificare le probabilità con cui le proprietà qualitative di un sistema si verificano.

- 131 -
8. Appendice: script Matlab utilizzati
per il pathway Glu-Ins_NEFA
CPepmin.m % Funzione per la ricerca dei coefficienti necessari a minimizzare la % differenza (quadratica) tra il CPep individuato dal modello matematico e % il CPep Misurato; viene presa in considerazione anche la misura del NEFA % non considerata fino alla versione quattro % versione 6.2 - 12 ottobre 2009 - Simone Asnaghi function [X,F]=CPepmin clear all global time time1 glice glice1 cpep NEFA Fp coeff a Gmax Gb d Kd time_ala contiamo k01 k12 k21 V sog;% gl; inizio=clock % memorizza data e ora iniziali time = [0 30 60 90 120 150 180]; %Coefficiente Kd per la SR dinamica din=[ 5.5; %4; 3; %6; %3; %3; 3; %3; 0.35; %3; 0.35; %0.35; ]; %Ritardo nella SR in minuti delay= [ 40; %40; 40; %40; %10;

- 132 -
%10; 25; %25; 1; %10; 40; %40; ]; %glicemia in mg/dl glu= [ 112 207 255 233 179 154 129; %, donna, diabete SI, PRE %110 124 138 147 146 138 129; % POST 135 252 252 227 182 158.5 135; %, donna, diabete SI %111 126 136 136 127 125 122; %POST %92 151 164 123 57 58 59; %, donna, diabete SI, PRE %95 107 108 105 96 100 102; % POST 130 196 238 252 196 143 126; %, donna, (NO), PRE %115 137 134 127 117 108 105; % POST 162 228 303 318 292 239 192; %, donna, diabete SI, PRE %144 157 174 177 180 174 168; % POST 99 202 185 184 125 100 92; %, uomo, diabete SI, valori CPEP presi dal CPEP dopo operazione %92 105 111 112 104 103 101; %POST ]; % cpeptide in ng/ml cp= [ 2 4.7 7.7 11.6 11.9 9.8 7.5; % Pre %2.9 3.3 4.4 5.3 4.9 4.8 4.6; % Post 1.9 3.2 3.9 4.8 4.4 4 3.5; % PRE %1.5 2 2.5 2.5 2.7 2.75 2.8; % POST %2.4 5.8 8 8.1 7.2 7.25 7.3; % PRE %2.6 3.7 4.2 4 3.6 3.3 3.8; % POST 4.4 5.8 8.4 10.6 11.8 9.9 8.9; % PRE %4.3 5 5.5 5.3 5.4 5 4.6; % POST 5 5.8 9.1 11.7 12.6 11.8 10.7; % PRE %2.8 2.9 3.5 4.3 4.5 4.65 4.8; % POST 2.8 3.1 3.8 3.9 4.1 3.6 4; % PRE %2.8 3.1 3.8 3.9 4.1 3.6 4; % POST ]; % NEFA in mmol/l NF= [ 1.06 1.23 0.93 0.67 0.47 0.47 0.48; % PRE %1.34 1.37 1.20 0.97 0.89 0.82 0.85; % POST 0.97 1.05 0.67 0.58 0.52 0.49 0.45; % PRE %0.81 0.88 0.51 0.52 0.48 0.44 0.40; % POST %1.37 2.24 3.22 2.20 1.12 0.96 0.80; % PRE %3.04 3.69 2.87 3.29 4.06 3.12 3.02; % POST 1.03 0.98 1.24 1.01 1.03 1.09 1.03; % PRE

- 133 -
%0.78 0.72 0.58 0.40 0.27 0.30 0.31; % POST 0.90 1.58 1.54 1.66 1.77 1.99 2.00; % PRE %1.05 0.94 0.54 0.42 0.31 0.35 0.39; % POST 0.93 0.77 0.45 0.35 0.27 0.25 0.23; % PRE %1.13 0.98 0.64 0.53 0.47 0.42 0.37; % POST ]; GLP1= [ %19 21 15 8 14 11 8; % (4) PRE %23 31 39 30 20 18 15; % (4) POST %7 11 18 12 10 9 8; % (7) PRE %23 32 42 31 18 17.5 17; % (7) POST %5 8 15 15 14 8 2; % PRE %24 35 33 29 21 16 11; % POST ]; % insulina in mcUI/ml %in= [ ]; %Conversione delle unità di misura glu=glu*0.05551; % glucosio in mmol/l (inizialmente in mg/dl) cp=(cp*0.0331); %conversione in nmol/l (inizialmente in ng/ml) %in=in*6/1000; % conversione a mmol/l (inizialmente in mcUI/ml) diabete = [ 0; %0; 0; %0; %0; %0; 1; %0; 1; %1; 1; %1; ]; % presenza diabete: 0=NO, 1=SI sex = [ 1; %1; 1; %1; %1; %1; 1; %1; 1; %1; 0;

- 134 -
%0; ]; % sesso soggetto: 0=uomo, 1=donna age = [ 58; %58; 48; %48; %35; %35; 53; %53; 39; %39; 29; %29; ]; weight = [ 140.3; % PRE %135.8; % POST 165; % PRE %141; % POST %145; % PRE %139; % POST 180; % PRE %169.7 % POST 138.4; % PRE %133.3; % POST 129; % PRE %125; % POST ]; height = [ 165; %165; 175; %175; %171; %171; 185; %185; 160; %160; 160; %160; ]; % valori iniziali % valori pi0 per la parte statica, utilizzati in SRsN

- 135 -
p10=0.5; %valore di p1 di partenza p20=0.8; %valore di p2 di partenza p30=8; %valore di p3 di partenza % valore iniziale p40, rappresenta la 'percentuale' di Gmax presa % per ottenere la soglia Gt, considerando sempre Gt<Gmax %p40=0.6; % valore iniziale p50, rappresenta il coefficiente 'k' da utilizzare % per l'equazione differenziale del NEFA nella SRsN %p50=0.002; x0=[p10 p20 p30]% p40 ] % p50 ] %definizioni limiti l1=0.03; % alla versione 4.6 era 0.008, abbassato per prova dalla 4.7 u1=1; l2=0.03; u2=1.2; l3=6; u3=15; %l4=0.4; %u4=0.8; %l5=0.001; %u5=0.004; l=[l1 l2 l3]% l4];% l5 ]; u=[u1 u2 u3]% u4];% u5 ]; % definizione numero iterazioni ('evaluation') per la minimizzazione %N=1000; % Calcolo tempi di interpolazione dime=size(time); dt=dime(2); fine=time(dt); for j=1:fine time_ala(j)=j; end time1=[0 time_ala]; contiamo = 0; %contatore c=5; %numero dei pazienti su cui si fa simulazione for i=1:c %conta_b = 0; % variabile di conteggio 'interna' utile per visualizzare andamento script sog=delay(i); % è il ritardo della risposta SR

- 136 -
%coefficienti age_=age(i); % dato età soggetto per calcolo coefficienti weight_=weight(i); % dato peso soggetto per calcolo coefficienti height_=height(i); % dato altezza soggetto per calcolo coefficienti inorm_=diabete(i); % dato sesso soggeto per calcolo coefficienti isex_=sex(i); %dato presenza diabete per calcolo coefficienti %Definizione coefficienti kij [k01,k12,k21,V] = coeffCPep(age_, weight_, height_, isex_, inorm_); % il valore di glicemia del paziente i è la i-esima riga glice=glu(i,:); % il valore di GLP1 del paziente i è la i-esima riga %gil=GLP1(i,:); % il valore di cpep del paziente i è la i-esima riga cpa=cp(i,:); %calcola il minimo (basale) del cpep cmin=c(1); d_c=size(cp); dt=d_c(2); for r=1:dt if cpa(r)<cmin cmin=cpa(r); end end %sottrae il valore basale al cpep cpep=cp(i,:)-cmin; % il valore di NEFA del paziente i è la i-esima riga NEFA=NF(i,:); %+0.08*gil; non consideriamo il GLP1 qui % interpolazione glucosio e NEFA e GLP1 glice1=interp1(time,glice,time1,'linear'); Fp=interp1(time,NEFA,time1,'linear'); %gl=interp1(time,gil,time1,'linear'); % Calcolo Gmax e Gb per il glucosio dim=size(glice1); d=dim(2); %Gmac Gmax=glice1(1); for ilsd=1:d if glice1(ilsd)>Gmax Gmax=glice1(ilsd); end end %G basale Gb=glice(1); for bp=1:d

- 137 -
if glice1(bp)<Gb Gb=glice1(bp); end end %Il Kd è variabile ed è preso dalla colonna "din" che si trova %all'inizio di questo script Kd=din(i); %dati antropometrici da cui si ricavano i coefficienti Kij del CPep coeff(1)=age(i); % dato età soggetto per calcolo coefficienti coeff(2)=weight(i); % dato peso soggetto per calcolo coefficienti coeff(3)=height(i); % dato altezza soggetto per calcolo coefficienti coeff(4)=diabete(i); % dato sesso soggeto per calcolo coefficienti coeff(5)=sex(i); %dato presenza diabete per calcolo coefficienti %opzioni per la minimizzazione options=optimset('Algorithm', 'active-set','LargeScale','on','hessian','off','LevenbergMarquardt','on','MaxFunEvals',3000,'MaxIter',3000); % 'MaxIter',N,'MaxFunEvals',N,'DiffMinChange',1e-3,'DiffMaxChange',1e-7 % ,'TolFun',1e-8 %funzione per la minimizzazione dell'energia dell'errore [x,fval]=fmincon('CalcCPep',x0,[],[],[],[],l,u,[],options); %registrazione risultati: in X ho i valori di p1, p2, p3 (coefficienti della risposta statica) che %minimizzano l'indice della funzione CalcCPep; in F ho la funzione indice minimizzata X(i,1)=x(1); X(i,2)=x(2); X(i,3)=x(3); %X(i,4)=x(4); %X(i,5)=x(5); F(i)=fval; % grafici risultati: sono i grafici che riportano le differenze, tra i % valori campionati e quelli previsti, del C-Pep ed un grafo che % riporta le energie degli errori di tutti i soggetti %GRAFICO CPEP CAMPIONATO IN BLU E CPEP SIMULATO IN ROSSO subplot(4,c,i); plot(time,a,'red',time,cpep,'blue'); grid ylabel('CPep') hold on %GRAFICO GLUCOSIO subplot(4,c,c+i); plot(time1,glice1); grid ylabel('Glucose') hold on

- 138 -
%GRAFICO NEFA subplot(4,c,2*c+i); plot(time1,Fp); grid ylabel('NEFA') hold on %GRAFICO GLP1 %subplot(5,c,3*c+i); %plot(time1,gl); %grid %ylabel('GLP1') %hold on %GRAFICO ENERGIA ERRORE val=1; subplot(4,c,3*c+i); bar(val,F(i)); grid ylabel('Energy Error') hold on end inizio; % mostra data e ora iniziali fine = clock % mostra data e ora finali
CalcCpep.m % Calcolo andamento C-Peptide e indice di confronto % In questa versione si considera contributo del NEFA; % versione 6.2 - 12 Ottobre 2009 - Simone Asnaghi function f=CalcCPep(x); global p1 p2 p3 cpep a time_ala contiamo V;% p4 p5; contiamo = contiamo + 1; %definizione dei valori iniziali dei coefficienti p1=x(1); p2=x(2); p3=x(3); %base dei tempi su cui si calcola la ode t=time_ala; %valori iniziali %y0=[cpep(1); cpep(1)/1.13];

- 139 -
y0=[0;0]; % utilizzo della ode23 sul sistema di due equazioni per il C-Peptide [t,y]=ode23('CPepN',t,y0); % viene presa solo la prima colonna, valore di CP1 a=y(:,1)/V; %calcolo errore tra C-Pep simulato e C-Pep misurato: %E' L'INDICE CALCOLATO DALLA FUNZIONE Cm=cpep;%valore misurato a=[a(1) a(30) a(60) a(90) a(120) a(150) a(180)]; %valore calcolato %differenza g=Cm-a; %indice da minimizzare f=(g*g')/(Cm*Cm');
CPepN.m % Definizione del sistema di equazioni differenziali del C-Peptide % Consideriamo contributo NEFA sostituendolo % "totalmente" al SRs utilizzato fino alla versione 4 % versione 6.2 - 12 Ottobre 2009 - Simone Asnaghi function ydot=CPepN(t,y) global k01 k12 k21 sog %ydot=zeros(2,1); %Definizione variabili di stato CP1=y(1); CP2=y(2); % Calcolo SRd tramite funzione SRd srd=SRd; for l=1:180 ss(l)=srd(l+1); end srd=ss'; % Calcolo SRs tramite funzione SRsN srs=SRsN; for w=1:180 sf(w)=srs(w+1); end srs=sf';

- 140 -
for i=1:180 if i<sog S(i)=0; else S(i)=srs(i+1-sog,1); end end S=S'; %totale SR sr=srd+S; %sr=srs+srd; %Definizione SR % funzione forzante if t>=179 SR=sr(180); else SR=sr(round(t+1)); end %Definizione sistema ydot(1)=-(k01+k21)*CP1+k12*CP2+SR; ydot(2)=k21*CP1-k12*CP2; ydot=[ydot(1);ydot(2)];
coeffCPep.m % Calcolo coefficienti CPeptide % versione 6.2 - 12 Ottobre 2009 - Simone Asnaghi function [k1,k2,k3,V] = coeffCPep(age, weight, height, isex, inorm) bsa=0.20247*(height^0.725d0)*(weight^0.425d0); if inorm <=0 ashort=-log(2.)/4.95; fraction=0.76; else ashort=-log(2.)/4.55; fraction=0.78; end blong=-log(2.)/(0.14*age+29.2); if isex<=0 V=1.11*bsa+2.04; else V=1.92*bsa+0.64; end

- 141 -
k2=-fraction*blong-(1.-fraction)*ashort; k3=ashort*blong/k2; k1=-ashort-blong-k2-k3;
SRs.m % Contributo della Secretion Rate Statica considerando NEFA % versione 6.2 - 12 Ottobre 2009 - Simone Asnaghi function s=SRsN global glice1 p1 p2 p3 Fp; % richiamo dei campioni del glucosio e dei NEFA precedentemente interpolati G=glice1; FA = Fp; %Calcolo della SR statica for i=1:181 SRsN(i)=(p1/2)*( -(p2+ p3/G(i) - FA(i)) + sqrt( ( p2+(p3/G(i))-FA(i) )^2 + 4*p2*FA(i)) ) * G(i); end s=SRsN;
SRd.m % Calcolo della SR dinamica SRd % basato sui campionamenti di glucosio % versione 6.2 - 12 Ottobre 2009 - Simone Asnaghi function srd=SRd global Gmax Kd glice1 d ;%KG Gb %PER ORA NON CONSIDERO IL COEFFICIENTE KG VARIABILE, PRENDO UN Kd FISSO % Definizione Gt, supponend Gt sempre minore o uguale a Gmax %Gt=Gmax; %per ora considero un Kd fisso, per cui questo ciclo è inutile % Calcolo KG %for i=1:d % if glice1(i)<Gt % if glice1(i)>Gb

- 142 -
% KG(i)=Kd*(Gt-glice1(i))/(Gt-Gb); % else KG(i)=0; % end % else KG(i)=0; % end %end % Calcolo differenze (derivata) gliced=diff(glice1); gliced=[0 gliced ]; % Calcolo SRd for i=1:d if gliced(i)>0 %SRd(i)=KG(i)*gliced(i); SRd(i)=Kd*gliced(i); else SRd(i)=0; end end srd=SRd;

- 143 -
9. Bibliografia
[Bal87] Balboni G.C., Bastianini A., Brizzi E. “Anatomia Umana”, vol 2 . edizioni-Ermes,
1987.
[Ram2000] Ramzi S. Cotran, Vinay Kumar, Tucker Collins. “Le Basi Patologiche Delle
Malattie”, PIccin, 2000
[Sil2006] Siliprandi, Tettamanti, “Biochimica medica”. Piccin, 2006.
[Ror2003] P.Rorsman, E.Renstrom, “Insulin granule dynamics in pancreatic beta
cells”, Diabetologia, 2003, 46:1029-1045.
[OCo80] O’Connor MD, Landahl H, Grodsky GM., “Comparison of storage and signal-
limited models of pancreatic insulin secretion”, The American Journal of Physiology,
1980
[Raf2002] Rafael Nesher, Erol Cerasi. “Section 2: Biphasic Insulin Release: Pools and
Signal Modulation. Modeling Phasic Insulin Release. Immediate and Time-
Dependent Effects of Glucose”,. Diabetes 51(suppl.1):S53-S59, 2002.
[Cau2003] Andrea Caumo, Livio Luzi., “First-phase insulin secretion: does it exist in
real life? Considerations on shape and function”, The American Phisiological Society ,
2003.
[War84] Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D Jr: “Diminished B. Cell
secretory capacity in patients with noninsulin-dependent diabetes mellitus”, J Clin
Invest 74:1318 –1328,1984
[War88] Ward WK, Wallum BJ, Beard JC, Taborsky GJ Jr, Porte D Jr., “Reduction of
glycemic potentiation. Sensitive indicator of beta-cell loss in partially
pancreatectomized dogs”, Diabetes, 37:723-729, 1988.
[Por2001] Porte D Jr, Kahn SE., “beta-cell dysfunction and failure in type 2 diabetes:
potential mechanisms”, Diabetes, 50:S160-3, 2001.
[Sem2004] Sempoux C, Guiot Y, Dubois D, Moulin P, Rahier J., “Human type 2 diabetes:
morphological evidence for abnormal beta-cell function”, Diabetes, 50:S172-S177,
2001.
[Ror2004] P.Rorsman, E.Renstrom, “Insulin granule dynamics in pancreatic beta
cells.” Diabetologia(2003)46:1029-1045, 2004.

- 144 -
[Tof2001] Gianna Toffolo, Elena Breda, Melissa K. Cavaghan, David A. Ehrmann, Kenneth
S. Polonsky, Claudio Cobelli, “Quantitative indexes of b-cell function during graded up
& down glucose infusion from C-peptide minimal models.”, The American
Phisiological Society , 2001
[Lic73] Licko V., “Threshold secretory mechanism: a model of derivative element in
biological control.”, Bull Math Biol 35:51-58, 1973.
[Gro72] G. M. Grodsky., “A Threshold Distribution Hypothesis for Packet Storage of
Insulin and Its Mathematical Modeling.”, The Journal of Clinical Investigation, 1972.
[Tof95] Toffolo G.,De Grandi F, Cobelli C., “Estimation of β-cell sensitivity from IVGTT
C-peptide data. Knowledge of the kinetics avoids errors in modelling the secretion.”,
Diabetes 44: 845-854, 1995.
[Tof99] Toffolo G., Cefalu W.and Cobelli C., “B-Cell function during insulin modified
IVGTT successfully assessed by the C-peptide minimal model.”, Metabolism 48:1162-
1166, 1999.
[Mur89] Tadao Murata, “Petri Nets: Properties, Analysis and Applications”, Proceeding
of IEEE, Vol.77, No.4, April 1989
[Fer2001] Luca Ferrarini, “Automazione Industriale: Controllo Logico con Reti di
Petri”, Pitagora Editrice Bologna, 2001
[Dem2006] Isabel Demongodin, Nick T. Koussoulas, “Differential Petri Net Models for
Industrial Automation and Supervisory Control”, IEEE Transaction on Systems, Man
and Cybernetics – Part C: Applications and Reviews, Vol.36, No.4, July 2006
[Zde2003] Zdenek Hanzalek, “Continuous Petri Nets and Polytopes”, IEEE, 2003
[Haa2002] Peter J. Haas, “Stochastic Petri Nets, Modelling, Stability, Simulation”,
Springer, 2002
[Red93] Reddy, V. N., Mavrovouniotis, M. L. and Liebman, M. N. (1993). “Petri Net
Representation in Metabolic Pathways“ In Proc. First Intern. Conf. on Intelligent
Systems for Molecular Biology, Hunter, L. et al, AAAI Press, Menlo Park, pp. 328-336
[Hof94] Hofestaedt, R. (1994). “A Petri Net Application of Metabolic Processes.
Journal of System Analysis, modelling and Simulation“ 16, 113-122.
[Gen2001] Genrich, H., Kueffner, R. and Voss, K. (2001). “Executable Petri Net Models
for the Analysis of Metabolic Pathways” Int. J. STTT 3, 394-404.
[Sch2000] Schuster, S., Fell, D. A. and Dandekar, T. (2000a). “A general definition of
metabolic pathways useful for systematic organization and analysis of complex
metabolic networks” Nature Biotechnol. 18, 2000, 326-332.

- 145 -
[Tan2003] Yukiko Tanaka, Hitoshi Aoshima, Atsushi Doi, Hiroshi Matsuno , Mika Matsui
and Satoru Miyano “Biopathways representation and simulation on hybrid functional
Petri net”, In Silico Biology 3, 0032 – 2003.
[Pet81] J.L. Peterson, “Petri Net Theory and the modelling of systems”, Englewood
Cliffs, NJ. Prentice Hall, 1981.
[Dav97] R.David, “Modeling of Hybrid Systems using Continuous and Hybrid Petri
Nets”, IEEE, 1997
[PoZ2005] L.Popova-Zeugmann, M.Heiner, I.Koch “Time Petri Nets for modelling and
Analysis of Biochemical Network”, Fundamenta Informaticae 67 (2005) 149-162
[Gil2006] D.Gilbert, M.Heiner, “From Petri Nets to Differential Equations An Integrative
Approach for Biochemical Network Analysis”, 2006
[Gui2008] M.Guidotti, “Farmacocinetica”, http://www.galenotech.org
[DaM2007] C. Dalla Man, R. A. Rizza, and C. Cobelli, “Meal Simulation Model of the
Glucose-Insulin System”, IEEE Transaction on Biomedical Engineering, vol. 54, No. 10,
October 2007
[Mar2001] Mari A., Camastra S., Toschi E., Giancaterini A., Gastaldelli A., Mingrone G.,
Ferrannini E. “A model for glucose control of insulin secretion in 24 h of free living”,
Diabetes 50, Suppl 1: S164–S168, 2001.
[Mar2002] Mari A., Schimtz O., Gastaldelli A., Oestergaard T., Nyholm B., Ferrannini E.,
“Meal and oral glucose tests for assessment of ββββ-cell function: modeling analysis in
normal subjects”. Am J Physiol Endocrinol Metab 283: E1159–E1166, 2002.
[Tof2001] G.Toffolo, E. Breda, M.K. Cavaghan, D.A. Ehrmann, K.S. Polonsky, C. Cobelli,
“Quantitative indexes of ββββ-cell function during graded up&down glucose infusion
from C-peptide minimal models”, Am J Physiol Endocrinal Metab 280: E2–E10, 2001.
[Yan2003] G.C. Yaney, B.E. Corkey, “Fatty acid metabolism and insulin secretion in
pancreatic beta cells”, Diabetologia 46: 1297–1312, 2003.
[Sac2006] A.Sackmann, M.Heiner, I.Koch, “Application of Petri Net based techniques
to signal transduction pathways“, BMC Bioinformatics, November 2006.
[Sal2007] S.Salinari, A.Bertuzzi, M.Manco, G.Mingrone, “NEFA-glucose comodulation
model of b-cell insulin secretion in 24-h multiple meal test”, Am J Physiol Endocrinal
Metab 292:1890-1898, 2007, First published Mar 6, 2007.
[Pal2005] William J. Palm III, “Introduction to Matlab 7 for engineer”, MCGraw-Hill,
2005.

- 146 -
[Asn2008] S. Asnaghi, “Reti di Petri e reti biochimiche: utilizzo delle PN per la
modellizzazione del metabolismo dell’insulina”, Rapporto del Dipartimento di
Informatica e Sistemistica, 2008.
[Sal2008] S. Salinari, A. Bertuzzi, S. Asnaghi, C. Guidone, M. Manco, G. Mingrone, “First
phase insulin secretion restoration and differential response to glucose load
depending on the route of administration in type 2 diabetic subjects after bariatric
surgery”, Diabetes Care, 2008.
[Cob2005] Cobelli et ali, “Insulin Sensitivity by Oral Glucose minimal models:
validation against clamp”, 2005.
[DaM2006] C. Dalla Man, M. Camilleri, C. Cobelli, “A System Model of Oral Glucose
Absorption: Validation on Gold Standard Data”, December 2006.
[DeF79] DeFronzo RA, Tobin JD, Andres R., “Glucose clamp technique: a method for
quantifying insulin secretion and resistance.”, Am J Physiol Endocrinol Metab
Gastrointest Physiol 237: E214–E223, 1979.
[Eat80] Eaton RP, Allen RC, Schade DS, Erickson KM, Standefer J., “Prehepatic insulin
production in man: kinetic analysis using peripheral connecting peptide behavior.”,
J Clin Endocrinol Metab 51: 520–528, 1980.
[Pol86] Polonsky KS, Given BD, Pugh W, Licinio-Paixao J, Thompson JE, Karrison T,
Rubenstein AH., “Calculation of the systemic delivery rate of insulin in normal man.”,
J Clin Endocrinol Metab 63: 113–118, 1986.
[Ber81] Bergman RN, Phillips LS, Cobelli C., “Physiologic evaluation of factors
controlling glucose tolerance in man: measurement of insulin sensitivity and -cell
sensitivity from the response to intravenous glucose.”, J Clin Invest 68: 1456–1467,
1981.
[Gro72] Grodsky GM., “A threshold distribution hypothesis for packet storage of
insulin and its mathematical modeling.”, J Clin Invest 51: 2047–2059, 1972.
[Lic75] Licko V, Silvers A., “Open-loop glucose-insulin control with threshold
secretory mechanism: analysis of intravenous glucose tolerance tests in man.” Math
Biosci 27: 319–332, 1975.
[Hen2008] Henquin JC, Nenquin M, Szollosi A, Kubosaki A, Notkins AL., “Insulin
secretion in islets from mice with a double knockout for the dense core vesicle
proteins islet antigen-2 (IA-2) and IA-2beta”, J Endocrinol, 196:573-581, 2008

- 147 -
[DaM2005] Dalla Man C, Yarasheski KE, Caumo A, Robertson H, Toffolo G, Polonsky KS,
Cobelli C., “Insulin sensitivity by oral glucose minimal models: validation against
clamp”, Am J Physiol Endocrinol Metab, 289:E954-E959, 2005
[Asn2009] Simone Asnaghi, Chiara Milani, “Modellizzazione di alcune classi di processi
IT mediante Reti di Petri Stocastiche”, Notiziario Tecnico Telecom Italia, anno18,
Numero 3, 2009
[Hei2008] Monika Heiner, David Gilbert, Robin Donaldson, “Petri Nets for Systems and
Synthetic Biology”, Springer-Verlag Berlin Heidelberg 2008
[JGraph2009] David Benson, “JGraph and JGraph Layout Pro User Manual“, 2009