human hypoxanthine-guanine phosphoribosyltransferase ... · the journal of biological chemistry q...
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY Q 1989 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 264, No. 1, Issue of January 5, pp. 520-525, 1989 Printed in U.S.A.
Human Hypoxanthine-Guanine Phosphoribosyltransferase Deficiency THE MOLECULAR DEFECT IN A PATIENT WITH GOUT (HPRTASHVILLE)*
(Received for publication, April 5, 1988)
Beverly L. DavidsonS, Mohammad PashmforoushS, William N. KelleySBIl, and Thomas D. PalellaBlII From the Departments of $Biological Chemistry and Slnternal Medicine and the TRackhnm Arthritis Research Unit, University of Michigan Medical School, Ann Arbor, Michigan 48109
The genetic basis of hypoxanthine-guanine phos- phoribosyltransferase (HPRT) deficiency has been identified by nucleotide sequence analysis of HPRT cDNAs cloned from a patient with gout. A single nu- cleotide change was identified in two independent clones: an A to G transition at nucleotide 602. Confir- mation of a mutation at this site was provided by RNase mapping analysis. The predicted consequence of this transition is an aspartic acid to glycine substitution at amino acid 201. We have designated this variant
Prior to this report, enzyme activity in HPRTAshVi11. had not been detected by routine assay. Using more sensitive techniques, including an in situ gel assay for HPRT activity, we were able to demonstrate electro- phoretic, kinetic, and structural differences between HPRT~sh~ill~ and normal HPRT. Electrophoretic migra- tion of HPRTAshVille is more cathodal than normal, con- sistent with the predicted amino acid change. Addition- ally, HPRTA.hville has elevated Michaelis constants for 5-phosphoribosyl- 1-pyrophosphate and hypoxanthine. Predicted secondary structural alterations may result from the aspartic acid to glycine substitution.
HPRTAshvi1le-
Hypoxanthine-guanine phosphoribosyltransferase (HPRTl; 1MP:pyrophosphate phosphoribosyltransferase, EC 2.4.2.8) catalyzes the conversion of hypoxanthine and guanine to their respective mononucleotide forms, IMP and GMP. The catalytic mechanism occurs through an ordered bi-bi reaction in which the substrate 5-phosphoribosyl-l- pyrophosphate (PP-ribose-P) binds first (1).
In humans, complete deficiency of HPRT causes the Lesch- Nyhan syndrome which is characterized by hyperuricemia, hyperuricaciduria, and devastating neurological abnormalities (2). Partial HPRT deficiency leads to severe precocious gout and uric acid nephrolithiasis due to overproduction of uric acid (3). The mutations responsible for human HPRT defi- ciency are strikingly heterogeneous (4-6).
* This work was supported by National Institutes of Health Grant R01-DK19045. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
11 To whom correspondence should be addressed University of Michigan, Dept. of Internal Medicine, 1150 W. Medical Center Dr., 5520 Medical Science Research Bldg. I, Ann Arbor, MI 48109.
The abbreviations used are: HPRT, hypoxanthine-guanine phos- phoribosyltransferase; PP-ribose-P, 5-phosphoribosyl-1-pyrophos- phate; P.C., patient’s initials; W.E., patient’s initials; bp, base pair; CRM, immunologically cross-reactive material; PAGE, polyacryl- amide gel electrophoresis; V,,,, maximal velocity; AMV, avian mye- loblastosis virus.
The structure and organization of the HPRT gene have been determined (7) and the roles of certain 5’-regulatory regions have been defined in the mouse (8). Less is known regarding critical structure-function relationships of the pro- tein itself. Although predictions have been made regarding substrate binding sites by comparison of its primary structure to other nucleotide binding proteins whose three-dimensional structure is known (9), three-dimensional structural analysis and active site analyses have not yet been performed. The importance of certain regions within the molecule that are strongly conserved among bacterial, rodent, and mammalian phosphoribosyltransferase has also been inferred (10).
Analysis of functionally important regions of the human HPRT molecule is logically the definition of the impact of mutations on the activity, stability, or kinetic behavior of the enzyme. The characterization of four unique structural var- iants of HPRT has shed some light on the functional signifi- cance of certain regions of the molecule (11). However, very few HPRT-deficient subjects have residual levels of HPRT sufficient for these types of studies. Alternative approaches available are: (i) characterization of mutant HPRT cDNA sequences; (ii) site-directed mutagenesis of cloned HPRT gene sequences; or (iii) chemically or physically induced mutagen- esis in cultured cells and subsequent selection of HPRT- deficient cells using established selection systems (12, 13) followed by analysis of gene sequences by established tech- niques. Our initial attempts to define functionally meaningful regions of HPRT have concentrated on the first of these approaches. Because 80% of deficient subjects express near normal quantities of HPRT mRNA from which cDNA clones may be derived, analysis of this heterogeneous group of mu- tations will identify regions of interest (5). These data would then provide a rational basis for the design of site-directed mutants. This approach has been successful in identifying mutations in three HPRT-deficient subjects (14-16).
In this report, we identify a single nucleotide substitution present in HPRT cDNA clones from P.C., a patient with severe partial HPRT deficiency. The resultant amino acid substitution, aspartic acid to glycine at residue 201, is in a region to which two other mutants have been mapped (17- 19).
EXPERIMENTAL PROCEDURES’
Cell Lines and RNA Isolation-B-lymphoblastoid cells were derived from lymphocytes isolated from P.C. and W.E. as previously described (20). Radioimmunoassay of lysates from P.C. cells reveals only 4% of normal levels of immunologically cross-reactive material (CRM) us-
’ Portions of this paper (including “Experimental Procedures,” Figs. 1, 6, and 7, and Footnote 3) are presented in miniprint at the end of this paper. Miniprint is easily read with the aid of a standard magnifying glass. Full size photocopies are included in the microfilm edition of the Journal that is available from Waverly Press.
520

Human HPRT Deficiency 521 ing a highly specific polyclonal anti-HPRT antiserum (5) . HPRT mRNA levels in P.C. are normal as assessed by Northern blot analysis (data not shown). Cell line W.E. was established from an individual with the Lesch-Nyhan syndrome. There is no demonstrable HPRT activity or immunoreactive protein in cell line W.E. (5). Cell line GM558, a B-lymphoblast cell line derived from a normal male, was obtained from the Human Cell Repository in Camden, NJ. All cell lines were propagated in RPMI supplemented with 2 mM glutamine and 5% fetal calf serum at 37 “C in a humidified environment con- taining 5% COz.
Total RNA was isolated from approximately 1 g (wet weight) of B- lymphoblasts using guanidium isothiocyanate (21). Poly(A)+ RNA was separated from total cellular RNA by oligo(dT)-cellulose chro- matography (22). The integrity of the RNA was examined by North- ern blot analysis (23). All reagents used in RNA isolation and cDNA synthesis were treated with diethylpyrocarbonate and autoclaved.
Native Polyacrylamide Gel Electrophoresis and in Situ Activity Staining-Nondenaturing polyacrylamide gel electrophoresis
P X
5’ HPRTcDNA 3’ pSP64 I P
Sal I/Pst I cut 1 pSP64
i
E P pSPH2 1- T
3’ 5‘
RNA Probe PC HPRT mRNA
Cleavage Products FIG. 2. Constructions of pSPH2 for RNase cleavage analy-
sis and predicted cleavage products of P.C. mRNA. pSP64 was cleaved with SalI (S) and PstI ( P ) and ligated to a 714-bp XhoI (X)/ PstI ( P ) fragment isolated from a 955-bp HPRT cDNA clone (26). The resultant plasmid was linearized with EcoRI, ( E ) and transcribed using SP6 RNA polymerase to give the antisense RNA probe con- taining vector sequence (solid line) and antisense HPRT RNA se- quence (striped bar). Hybridization with P.C. RNA and RNase diges- tion using conditions described under “Experimental Procedures” yield the cleavage products indicated. Open bar, SP6 promoter; stip- pled bar, HPRT DNA sequences; solid bar and solid line, vector sequences; T, TaqI site reconstructed on ligation of XhoI and SalI cohesive ends. Asterisk indicates the site of the mutation. 3‘ and 5’ refer to the 3’ end and 5’ end of the full-length clone shown at the top of the diagram.
HPRTAshville
C T A G
Normal HPRT
C T A G
Arg Gly Leu HPRTAshville AGG G @ - TG
602
Normal AGG G B l T G Arg Asp Leu
201 FIG. 3. A, nucleotide sequence of HPRThhville (pHPPC4) (left
panel) from nucleotide 595 (top) to 606 (bottom) compared to normal HPRT. An A to G transition is present a t nucleotide 602. The nucleotides listed between the panels are grouped into codons. Direc- tion of electrophoresis is from top to bottom. Thus, antisense sequence is read from a sense strand by assigning the order GTAC to the lanes. B, the predicted amino acid change in HPRThhville. The G to A mutation (indicated by boxes) in HPRTAshville predicts aspartic acid to be replaced by glycine. This substitution occurs at amino acid 201.
(PAGE) was used to fractionate membrane-free lysates from normal (GM558), W.E., and P.C. lymphoblasts. Lysates were prepared as described (20) and exhaustively dialyzed against 10 mM Tris, pH 7.4, 0.02% azide. Lysates from P.C. and W.E. were concentrated using a Centricon apparatus. Protein concentration was measured according to Lowry et al. (31). Protein was electrophoresed through 6% poly- acrylamide gels according to the method of Laemmli (32) except that sodium dodecyl sulfate was omitted. HPRT activity was assayed in the polyacrylamide gels as described (33), with the following modifi- cations: (i) the reaction mixture consisted of 1 mM [8-”C]hypoxan- thine (specific radioactivity, 38.5 mCi/mmol), 15 mM PP-ribose-P, and 30 mM MgC12; and (ii) the incubation time was extended to 2 h.
RNase A Mapping-A 714-bp fragment of normal HPRT cDNA spanning the mutation in P.C. was cloned into the SP64 plasmid to form the recombinant plasmid pSPH2 (Fig. 2). The fragment was cloned in an orientation such that transcription using the SP6 pro- moter results in an antisense probe. pSPH2 was linearized with EcoRI before transcription. The labeled RNA probe was hybridized with 100 pg of total RNA isolated from P.C. or GM558, transferred to message affinity (poly(U)) paper, and digested with RNase A and RNase T1 using described conditions (6). The digestion products were electro- phoresed on a 6% polyacrylamide gel containing 8 M urea.
Kinetic Analysis-Measurements of Michaelis constants (Km) for hypoxanthine and PP-ribose-P were done on membrane-free extracts of cell line P.C. Cells were harvested as described (20), and protein concentration determined according to Lowry et al. (31). The standard radioisotopic assay time was increased from 10 min to 1 h (34). HPRT activity remained linear with respect to time up to 2 h (data not shown). HPRT activity assays were done on 25 pl of cell-free extract in a total assay volume of 50 p1 containing 0.25 to 1.5 mM [8-“C] hypoxanthine (5.28 mCi/mmol) and 1 to 15 mM PP-ribose-P. The MgClz concentration was kept a t 4 mM in excess of the PP-ribose-P concentration. The K,,, values for hypoxanthine and PP-ribose-P were estimated from Lineweaver-Burke plots.
The maximal velocity ( Vmm) for HPRTbh,.iu. was calculated by measuring enzyme activity at saturating conditions and dividing by the level of HPRT immunoreactive protein (ng of CRM/mg of pro- tein).
RESULTS
Analysis of HPRTashu~ cDNA Clones-Two recombinant phages, pHPPCl and pHPPC4, were isolated and sequenced

522 Human HPRT Deficiency
P
’ 4
4
262
” 7 1 4
-452
FIG. 4. RNase cleavage products of normal HPRT and HPRTMN~ mRNA. Lane 1, normal HPRT. Lane 2, HPRThh~Ue. Cleavage of H P R T d d e mRNA hybrids yields two fragments, 452 and 262 bp, consistent with a nucleotide mismatch at position 602 in the coding sequence. Cleavage of normal HPRT yields a fully pro- tected fragment (714 bp). The conditions for hybridization and the strategy used in the RNase A mapping experiment are described under “Experimental Procedures” and in Fig. 2.
FIG. 5. Activity stain of lymphoblast HPRT after nonde- naturing PAGE. Membrane-free protein from P.C. cells (HPRThh. dl.; 2 mg) (PC) and normal cells (2.5 pg) ( N ) were electrophoresed and stained for HPRT activity as described. Migration of HPRTht,de is more cathodal than normal HPRT. The cathode is at the top of this gel.
TABLE I HPRT specific oligonucleotides: sequences and sites of priming
Oligodeoxy- nucleotide Sequence (5’ to 3’) Priming site Strand”
1 AGTGATGATGAACCAG 2 CACTGAATAGAAATAGT 251-267 -
31-47 - 3 GATATAATTGACACTGG 403-419 - 4 CCCCTGTTGACTGGTCA 337-321 + 5 AGTCCTGTCCATAATTA 138-122 +
‘ The strand to which the primer anneals is represented as either sense (+) or antisense (-).
TABLE I1 Kinetic properties of HPRTA,I,,,~~o
Michaelis constants Maximal
Hypoxanthine PP-ribose-P
” units Normal* 3.5 k 2.4 (3) 16 -C 7.0 (3) 44 (3)
Defined as the maximum enzymatic activity divided by the total
*From Ref. 5. Mean f S. D. with number of determinations in
HPRThh* 595 7,200, 12,500’ 43.2, 38.7‘
quantity of HPRT enzyme as measured by radioimmunoassay (5).
parentheses. Values from two separate determinations.
using the strategy depicted in Fig. 1. Normal HPRT cDNA contains 590 bp of 3 -untranslated sequence up to but not including the poly(A) tail, 657 bp of coding sequence, and from 100 to 170 bp of 5”untranslated sequence (8, 26, 38). pHPPCl was 1380 bp and included 130 bp of 5’-untranslated sequences. pHPPC4 was 1250 bp, containing sequences up to but not including the A of the ATG start codon. Both clones were sequenced in their entirety in both orientations as de- scribed under “Experimental Procedures.”
HPRTbhde differed from normal HPRT by a single nu- cleotide in both clones: an A to G transition at nucleotide position 602 (Fig. 3A). This mutation alters a GAT codon to GGT, predicting the substitution of glycine for aspartic acid 201 (Fig. 3B). No other differences amongpHPPC1, pHPPC4, and normal were noted.
RNase A Mapping Analysis-A 714-bp antisense RNA probe was generated by transcription of pSPH2 by PS6 RNA polymerase (Fig. 2). The probe, spanning 77% of the coding sequence, was cleaved into fragments of 452 and 262 bp upon digestion with RNase A and RNase T1 when hybridized to HPRTbhae RNA (Fig. 4, lune 2) but not when hybridized to normal (Fig. 4, lune 1 ). These are expected products for a mutation at bp 602 in HPRTbhviue cDNA sequence. A mis- match in this context (AGGGUUCCCA) should allow suffi- cient melting for RNase cleavage 30 to 50% of the time (39). This is consistent with the results obtained.
In addition to the 714-bp probe used, a 232-bp probe span- ning the remaining 49 codons and the 5”untranslated se- quences was hybridized to HPRTbhae RNA. RNase digestion of this hybrid yielded only the fully protected 232-bp band (data not shown).
In Situ Activity Staining-Two mg and 2.5 pg of total protein from cell lines P.C. and GM558, respectively, were fractionated through polyacrylamide gels and assayed in situ as described under “Experimental Procedures.’’ The autora- diograph of this assay demonstrated that HPRThha. not only retains residual HPRT activity, but also migrates more cathodal than does normal HPRT (Fig. 5). A more cathodal migration is consistent with the replacement of an aspartic acid in normal HPRT with a glycine in HPRThhville.
Activity per mg of protein of HPRTbhville was determined by counting the radioactive spots in a scintillation counter.

Human HPRT Deficiency 523
TABLE I11 COOH-terminal mutations in HPRT
Variant Codon change Amino acid change K,,, hypoxanthine K,,, PP-ribose-P
HPRTAshville GAT -+ GGT Asp2"' + Gly 170-foldT 600-foldf H P R T K ~ , , ~ ~ ' NDb AsplS4 + Asn 100-fold? 500-fold? NBR4' GAT + AAT Asp2"' + Asn 2-foldf 24-foldT
a From Ref. 7. ND, not determined. The amino acid substitution in this variant was determined by amino acid sequencing.
e From Refs. 18 and 19.
Membrane-free extracts of P.C. cells retain approximately 0.045% of HPRT activity compared to GM558 lysates assayed under these conditions. This level of activity is not detectable in the standard HPRT assay.
To confirm that the electrophoretic difference between HPRTAshville and normal HPRT is a reflection of the amino acid substitution and not the amount of protein loaded, we used an HPRT- cell line, W.E. (See "Experimental Proce- dures"). Aliquots of W.E. cell lysate containing 500 pg of protein were added to 2.5 pg of GM558 cell lysate. The pooled lysates were then fractionated by polyacrylamide gel electro- phoresis and assayed in situ as before. This concentration did not alter the electrophoretic mobility of HPRT (data not shown), indicating that the difference observed between HPRTA,hviI1. and normal HPRT are authentic.
Kinetic Analyses-The apparent K, values for hypoxan- thine and PP-ribose-P for HPRTAshville in lymphoblast ex- tracts are significantly elevated (Table 11). The K, for PP- ribose-P was estimated to be 7.2 and 12.5 mM in two separate experiments, an increase of approximately 600-fold over those values we have reported previously for normal lymphoblast extracts (5). The K, for hypoxanthine in HPRTA,h,ill, was elevated nearly 17O-fold, from 3.5 p~ (5) to 595 p ~ . The apparent v,,, for HPRTAshville remained unchanged.
DISCUSSION
We have identified the mutation in HPRTAshville. An aspar- tic acid to glycine substitution at codon 201 is the predicted result of an A to G transition at nucleotide 602. This nucleo- tide substitution was identified by the bidirectional sequenc- ing of two independent clones, pHPPCl and pHPPC4. RNase mapping of HPRTAehville mRNA confirmed the presence and position of the mutation.
Prior to this report HPRT activity in lymphoblast extracts from P.C. had not been detected using a standard radioiso- topic assay (5). However, by using a sensitive in situ gel assay, extending the incubation time of the assay, and elevating substrate concentrations, we were able to detect a low level of residual HPRT activity with altered electrophoretic mobility in concentrated membrane-free extracts from HPRTA.h,ill, lymphoblasts. The cathodal migration of HPRTAehville is con- sistent with the net loss of a positive charge from the predicted aspartic acid to glycine substitution.
This substitution is within a region near the COOH-ter- minus, which may be important for the maintenance of nor- mal HPRT activity. The importance of this region for normal catalytic and kinetic behavior has been inferred from two other mutants: HPRTKin,mn, a human variant with an aspartic acid to asparagine substitution at 194, and NBR4, a mouse neuroblastoma cell line with an aspartic acid to asparagine substitution at 201 (Table 111) (17-19). Therefore, the possi- bility of a kinetic defect in HPRTAshville was investigated. Kinetically, HPRT.t,,hvi11, is very similar to HPRTKinston, in which the primary kinetic abnormality apparently relates to PP-ribose-P binding. The decreased capability to bind PP-
ribose-P may indirectly effect hypoxanthine binding, since the enzyme proceeds through an ordered bi-bi reaction mech- anism with PP-ribose-P binding first.
Conformational alterations resulting from this amino acid substitution were investigated using Chou-Fasman secondary structure prediction techniques (35, 36). The predicted struc- ture of normal HPRT in the region from amino acids 180 to 217 consists of @-turn/@-turn/@-sheetlrandom coil/@-sheet/ a-helix (Fig. 6A). The aspartic acid to glycine substitution in HPRTA.h,ill, predicts a different structure: a @-sheet replaces much of the predicted random coil (Fig. 6B). Aspartic acid has been implicated as a strong disrupter of @-structure (35). Glycine also disrupts @-structure, but to a lesser degree than aspartic acid. Whether or not the change in the K,,, values for hypoxanthine and PP-ribose-P are directly or indirectly re- lated to these predicted structural perturbations remains spec- ulative.
Structural changes may also occur by disruption of the normal hydrogen-bonding network responsible for maintain- ing tertiary structure (40,41). This can occur by changing the hydrophilicity (or hydrophobicity) of regions within a protein and thereby affect solvent accessibility, hydrogen bonding with water, and interresidue hydrogen bonding. An altered hydrophilicity profile for HPRTA,h,i~l, is shown in Fig. 7, with the hydrophilicity near amino acid 101 diminished and that preceding 198 increased. Although this analysis is indirect, in the absence of knowledge regarding the tertiary structure of the enzyme such predictions provide a basis to suggest the molecular basis for the apparent instability of HPRTA,h,ill,.
In conclusion, the identification of the mutation in HPRTAshville has enhanced our understanding of the impor- tance of certain residues near the COOH terminus of HPRT. Further refinement of this region, through site-directed mu- tagenesis, will answer many questions regarding the roles of these residues in catalysis and structural stability.
Acknowledgments-We wish to express our gratitude to My Nyu- gen for expert technical assistance and to Steve Kelley for help in the secondary structure analysis. Also, the expert secretarial assist- ance of Ardith Listeman is greatly appreciated.
REFERENCES
1. Henderson, J. F., Brox, L. W . , Kelley, W . N., Rosenbloom, F. M., and Seegmiller, J. E. (1968) J. Biol. Chem. 243, 2514-2522
2. Seegmiller, J. E., Rosenbloom, F. M., and Kelley, W . N. (1967) Science 155,1682-1684
3. Kelley, W . N., Rosenbloom, F. M., Henderson, J. F., and Seeg- miller, J. E. (1967) Proc. Natl. Acad. Sci. U. S. A. 67, 1735- 1739
4. Yang, T. P., Patel, P. I., Chinault, A. C., Stout, J . T., Jackson, L. G., Hildebrand, B. M., and Caskey, C. T . (1984) Nature 310,
5. Wilson, J. M., Stout, J . T., Palella, T. D., Davidson, B. L., Kelley,
6. Gibbs, R. A,, and Caskey, C. T . (1987) Science 236, 303-305 7. Patel, P. I., Framson, P. E., Caskey, C. T., and Chinault, A. C.
412-414
W . N., and Caskey, C. T. (1986) J. Clin. Inuest. 77, 188-195
(1986) Mol. Cell. Biol. 6, 393-403

524 Human HPRT Deficiency
8. Melton, D. W., McEwan, C., McKie, A. B., and Reid, A. M. (1986) 25. Gubler, U., and Hoffman, B. J. (1983) Gene (Amst . ) 25,263-269 Cell 44,319-328 26. Brennand, J., Konecki, D. S., and Caskey, C. T. (1983) J. Bid.
9. Argos, P., Hanei, M., Wilson, J. M., and Kelley, W. N. (1983) J. Chem. 258,9593-9596 Biol. Chem. 258, 6450-6457 27. Feinberg, A. P., and Vogelstein, B. (1984) Anal. Biochem. 137,
10. Hershey, H. V., and Taylor, M. W. (1986) Gene (Amst.) 43,287- 266-267
11. Wilson, J. M., Young, A. B., and Kelley, W. N. (1983) N . Engl. 29. Sanger, F., Nicklen, s., and Coulson, A. R. (1977) Proc. Natl.
12. Rubin, C. S., Dancis, J., Lily, C. Y., Nowinski, R. C., and Balis, 30. Bonthron, D. T., Markham, A. F., Ginsburg, D., and Orkin, S. H.
13. Szybalska, E. H., and Szybalski, W. (1962) Proc. Natl. Acad. Sci. 31. LOWrY, 0. H., Rosebrough, N. J.9 Farr, A. L., and Randall, R. J.
14. Davidson, B. L., Palella, T. D., and Kelley, W. N. (1988) Gene 32. Laemmlip u. K. (lg70) Nature 2279 680-685
15. Davidson, B. L., Pashmforoush, M., Kelley, W. N., and Palella, Genet. 18, 1-19 34. Holden, J. A,, and Kelley, W. N. (1978) J. Biol. Chem. 253,
16. Fujimori, S., Hidaka, Y., Davidson, B. L., Palella, T. D., and 4459-4463 35. Chou, P. Y., and Fasman, G. D. (1978) ARRU. Rev. Biochem. 47, 17. Wilson, J. M., and Kelley, W. N. (1983) J. C h . Invest. 71,1331- 36. wolf, H., Mohow, s,, Motz, M., Jameson, B., Hermann, G,, and
18. Melton, D. W., Konecki, D. S., Brennand, J., and Caskey, C. T. 37. Hopp, T. p., and Woods, K. R. (1981) proc, Natl, Acad. sei,
19. Melton, D. W. (1981) Somat. Cell Genet. 7,331-344 20. Wilson, J. M., Baugher, B. W., Mattes, P. M., Daddona, P. E.,
38. Jolly, D. J., Okayama, H., Berg, P., Esty, A. C., Filpula, D., Bohlen, P., Johnson, G. G., Shively, J. E., Hunkapillar, T., and
21. Chirgwin, J. M., Przybyla, A. E., MacDonald, R. J., and Rutter, Friedmann, T. (1983) Proc. Natl. Acad. Sci. U. S. A. 80, 477- 481
39. Myers, R. M., Larin, Z., and Maniatis, T. (1985) Science 230, 22. Aviv, H., and Leder, P. (1972) Proc. Natl. Acad. Sci. U. S. A. 69, 1242-1246
23. Fuscoe, J . C., Fenwick, R. G., Jr., Ledbetter, D. H., and Caskey, Nature 277,667-669
24. Okayama, H., and Berg, P. (1982) Mol. Cell. Biol. 2, 161-170 K., and Matthews, B. W. (1987) J. Mol. Biol. 197,315-329
293 28. Messing, J. (1983) Methods Enzymol. 101, 20-78
J. Med. 309,900-910 Acad. Sci. U. S. A. 74, 5463-5467
M. E. (1971) Proc. Natl. Acad. Sci. U. S. A. 68, 1461-1464 (1985) J. Clin. Znuest. 76,894-897
U. S. A. 48,2026-2034 (1951) J. Biol. Chem. 193,265-275
(Amst.) 68,85-91 33. Zannis, V. I., Gudas, L. J., and Martin, D. W., Jr. (1980) Biochem.
T. D. (1988) Gene (Amst.) 63, 331-336
Kelley, W. N. (1988) Hum. Genet. 79,39-43
1335
(1984) Proc. Natl. Acad. Sci. U. S. A. 81, 2147-2151
251-276
Fortsch, B. (1988) Comput. Appl. Biosci. 4, 187-191
U. S. A. 78, 3824-3828
and Kelley, W. N. (1982) J. Clin. Znuest. 69, 706-715
W. J. (1979) Biochemistry 18, 5294-5299
1408-1412 40. Gmtter, M. G., Hawkes, R. B., and Matthews, B. W. (1979)
C. T. (1983) Mol. Cell. Bid . 3, 1086-1096 41. Gmtter, M. G., Gray, T. M., Weaver, L. H., Alber, T., Wilson,
Supplemental Material to "Human hypoxanthine guanine phoQpholiboEylt=8n.f~=~~~ deficiency:
the molecular defect in a patient with gout (HPRTAShville)
Beverly L. Dayidson, Mohammad Pashmforoush, william N. Kelley, and Thomas D. Palella
EXPERIMENTAL PROCEDURES
d(T)12-17. The first and Second strands were synthesized according to =DNA synthesis. Ten mlcrograrns of p0ly(A)'RNA were primed urth oligo
Okayama and Berg as modified by GUbler and Hoffman (24.25). The double stranded DNA was blunt ended and lia-ated to ECO RI linkers. Excess linkers were removed by ECO RI digestion fallowed by column chromatography ~ 5 1 n q a 1 5 cm column of Sephsrose CL-48. The cDNA was then ligated into iqt11 u s l q conditions recommended by the manufacturer.
S c r e e n i n q . o n i n o . and seauencing. Approximately 175,000 recombinant phages were screened with as Msp I/Taq I fragment of human HPRT =DNA representing portions Of exons 1 and 3 and all Of exon 2 ( 2 6 1 . Thls 160 nucleotide fragment was isolated from a 955 bp clone Of normal HPRT CDNA, kindly provided to us by Dr. C. Thomas Carkey of Baylor university (26). The probe was labeled vlrh [ ~ - ~ * p ] d c T P by the method of Feinberg and Vaqelsteln (27). Four positive recombinants were pfaque purified. Phages were isolated from 10 m 1 Of lysate Culture using Licl and digested with Eco RI. Inserts isolated from two of the four clones were subcloned into Ml3mplB and Hllmpl9 for single stranded phage isolation (28) and dideoxynucleatlde sequencing (291.
2Francis Callina, University o f Michigan, personal communication.
according to established procedures (29). The Ml3 universal primer and HPRT Dideaxynucleotide sequencing vas done using [o-35S]dATP incorporation
specific synthetic oligonucleotide primers were Used to determine the
orientations. This is a modification of a method described by Bonthron (301 sequence of the coding region and 5' untranslated reqlon In both
Which used sequence specific primers for Only one strand Of sequence. In this report we modified this method by using Sequence specific primers for both Strands. The regions, Sequences, and strands to which the prlmers anneal are listed in Table I.
The sequencing strategy le shown in Figure 1. For determination Of 3 ' sequences in both Orientafion5, HPRTRg CDNA clones were digested Ylth ECO RI and Bal I and subcloned. A11 cto ies were sequenced entirely ~n bath directions (sense and antlsensel.
E 0 E V I
11111111111 '1 l,,ll..l,,,l I
4" ~. ~
3 +r%- c
Figure 1. The sequencing strategy used to determine the entire mutant cDNR sequences in both ocientations. Both HPRT specific OligOnucleotlde primers
used for sequencing. The smaller &aLL lB)-EEear (El fragment was subcloned (arrows 1 through 5 ) and the Mll universal primer (unlabeled arrows) were
far determination of 3' sequences. Solid Ilne, 5' and 3 ' untranrlafed sequences; stippled bar. HPRT coding sequence: vertical hatch narks, vector sequence: arrowhead indicates site of mutation. The arrows represent primlng sites only and not the extent Of sequence read. The sequence and priming sites for the HPRT specific primers are given in Table I.
Fasman secondary structure predictions for regions surrounding the mutatlan Secondary structure oredictions and hVdroDhiliCitv orofiks. The Chau-
in HPRTA hviil were done using the seg~ence analysis Software package from the GeneTLCS &puter Group. University Of Wisconsin Biotechnology Center
using the PEPTIDESTRUCTURE and PLOTSTRUCTURE programs. The hydrophiliclty (35.36). Secondary Stmctural preferences were determined from sequence data
profiles were plotted frvm data generated by the hydrophilicity analysls program in the Protein Idenfificatlan Resource (PIR) of Natlanal Blanet Resource Foundation, supported by the Division of Research Resources of the National Institutes of Health. The hydrophilicity indices of Hopp and Woads
and hydrophobicity moments superimposed on the two-dimensional plots (37) are used in this program, with a window size of six. The hydrophilicity
generated by PLOTSTRUCTURE were also calculated using the Hopp and Woods algorithm.
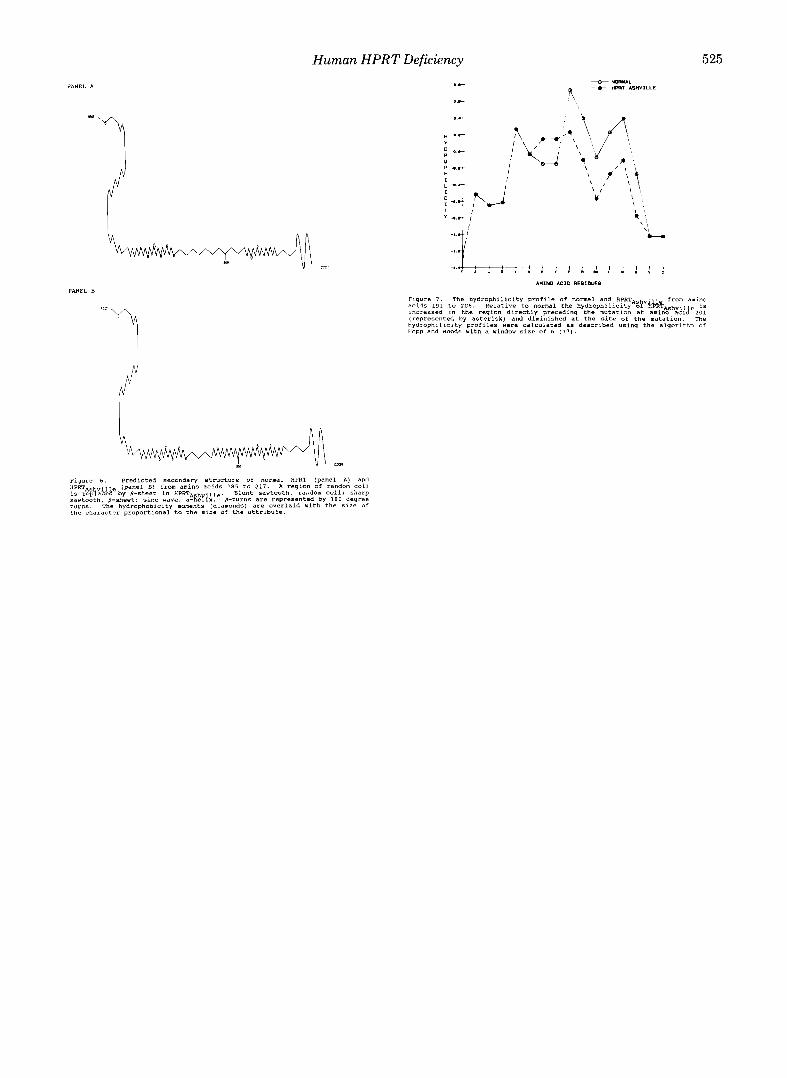
Human HPRT Deficiency 525
PANEL A
I 0 ..e- l
I I I
0 P ..r
! ! ! ! ! : ! ! ? ? : AMINO A C I D RESIDUES
PANEL B Figure 7. The hydrophilicity profile Of normal and HPRTns from amino acids 191 to 2 0 6 . Relative to normal the hydrophilicity o? kb&A,hvii e is Increased in the reqion directly precedinq the mutation at amino ac~c! 201
/:1
"i (represented by asterisk) and diminished at the site of the mutation. The hydrophlllclty profiles were calculated as deacrlbed usim the alaorithm of Happ and Woods with d window size of 6 (37).
the character 'pra6ortionai to the si;. of the.attrlbute