hogenom a phylogenomic database simon penel, pascal calvat, jean-francois dufayard, vincent daubin,...
TRANSCRIPT

HOGENOM a phylogenomic database
Simon Penel, Pascal Calvat, Jean-Francois Dufayard, Vincent Daubin, Laurent Duret ,
Manolo Gouy, Dominique Guyot, Daniel Kahn, Vincent Miele, Vincent Navratil, Guy
Perriere, Remi Planel

Several phylogenomic databasesdeveloped at LBBE/PRABI
• HOVERGEN– Verterbrate Proteins from UniProt – Clustering with SiLiX
• HOMOLENS– Proteins from Ensembl Complete Genomes– Clustering from Ensembl– Trees calculated and annoated (S,D,L) with new methods
(PhylDog,LBBE)
• HOGENOM– Proteins from all available complete genomes – (Bacteria, Eukaroyota, Archaea)– Clustering with SiLiX and post-processing with HiFiX– Trees will be annotated (S,D,L,T)

HOGENOM characteristics
• all complete genomes from the whole tree of life (not restricted to particular phylum)
• Propose « gene families » : full length homologous sequences (different of « domain families »)

Domain vs. gene families
Families of homologous protein domains (ProDom): - Evolution by domain shuffling (duplication, loss, translocation)
Protein domain family

Domain vs. gene families
Families of homologous protein domains (ProDom): - Evolution by domain shuffling (duplication, loss, translocation)Homologous Gene families (HOGENOM): - Evolution of homologous genes by speciation or by gene duplication, or horizontal transfer - Sequences are homologous over their entire length (or almost)
Homologous gene family

Orthologs and paralogs in HOGENOM
HOGENOM is centered on phylogenetic trees of gene families.
Information on orthologs and paralogs can be deduced from gene trees:
- from the annotation of gene trees (Duplication, Speciation,
Transfer)- from query tools such as tree-pattern
matching

BuildingCompare all proteins against each other (BLAST)
Cluster homologous sequences into families (SILIX + HIFIX)
Compute multiple alignments for each family
Compute phylogenetic trees for each family
Annotate phylogenetic trees (gene duplications, losses, transfers)

Compare all proteins against each other
• Iterative BLAST calculation
• Use of a non-redundant protein sequence database …
(all know proteins , about 20,000,000 non redondant sequences)
… associated with a resulting BLAST hits database(from which blast hits may be extracted)
• Cluster, grid and cloud computing

BuildingCompare all proteins against each other (BLAST)
Cluster homologous sequences into families (SILIX + HIFIX)
Compute multiple alignments for each family
Compute phylogenetic trees for each family
Annotate phylogenetic trees (gene duplications, losses, transfers)

SiLiX
BLASTP
BLOSUM62E ≤ 10-4
Protein database
Local pairwise alignments
1st step : similarity search

SiLiX
2nd step : SiLiX clustering
Use the all-against-all BLAST hits

SiLiX SiLiX : Selection of consistent
HSPs S2 S4S1S3Seq. A
Seq. B
S2S1’
∆lg1 lgHSP1 ∆lg2 ∆lg3lgHSP2
Seq. A
Seq. B

SiLiX
SiLiX : single linkage clustering
A
B
A
C
HSP ≥ 80 % lengthIdentity ≥ 35 %
C
B
ACluster A, B, C

SiLiX
SiLiX : single linkage clustering with alignment coverage constraints (Miele et al. BMC Bioinformatics 2011)
• Computing efficiency:– Ultra-fast– Memory efficient– Scalable (parallel architecture)
• Clustering quality: – At least as good as the previously published
methods

However …
• Because of over-extension of BLAST alignments, some sequences that share only partial homology may be clustered in a same family
• The risk of alignment over-extension is low, but becomes a problem for very large protein families
• Use more stringent clustering criteria ? No : optimal clustering criteria are not the same
for all families

HiFiX
• The mode and tempo of evolution is specific to each protein family
• A multiple alignment provides information about the specific pattern of evolution of a family
• => this can be used to decide whether or not a new sequence belongs to that family

HiFiX
• Step 1: rapid clustering (SiLiX) pre-families
• Step2: sub-clustering of pre-families into homogeneous protein clusters⇒ sub-families
• Step3: progressive merging of sub-families into families, with evaluation of multiple alignment quality at each step⇒ families

HiFiX

HiFiX

HiFiX

Results of clustering
Number Sequences Number of Families – at least 2 296,920– 2:10 242,398 – 10:500 53,450 – 500:2000 1,026 – more than 2000 79
Family size distribution:
About 7,000,000 proteins clustered into 300,000 families

BuildingCompare all proteins against each other (BLAST)
Cluster homologous sequences into families (SILIX + HIFIX)
Compute multiple alignments for each family
Compute phylogenetic trees for each family
Annotate phylogenetic trees (gene duplications, losses, transfers)

Compute multiple alignments
All alignments ( ~ 300, 000) have been calculated
with ClustalΩ

BuildingCompare all proteins against each other (BLAST)
Cluster homologous sequences into families (SILIX + HIFIX)
Compute multiple alignments for each family
Compute phylogenetic trees for each family
Annotate phylogenetic trees (gene duplications, losses, transfers)

Question: what about the alternative splicing ?
Compute phylogenetic tree

In eukaryotes, due to alternative splicing , one unique gene may be
be transcripted into several transcripts
Alternative splicing

We selected all the transcripts for each gene.
Because the longest transcript is not allways the best!
Transcripts in HOGENOM6

We don’t want several proteins for a same gene in a phylogenetic tree: may be seen as a duplication
We want 1 protein per gene for statistic comparison
among organisms
Selection of a representaitive isoform in HOGENOM
Because:

Selection of a representaitive isoform : how ?

Selection of a representative isoform : how ?
Archaea and bacteria1 transcript per gene
Eukarya1 or more transcripts per gene

Selection of a representative isoform : how ?
Archaea and bacteria
Eukarya
clustering

Selection of a representative isoform : how ?
First step: when a gene has
isoforms in different families ( ), choose a family for the
gene

2
Selection of a representative isoform : how ?
We select the family with the highest number of eukaryotic genes (and not proteins)
1
2
3
3 genes
2 genes
2 genes
1
2
1

2
Selection of a representative isoform : how ?
We select the family with the highest number of eukaryotic genes (and not proteins)
1
2
3
3 genes
2 genes
2 genes
1
2
1
If the number of eukaryotic genes are identical, we select the family with the highest number of eukaryotic proteins

2
Selection of a representative isoform : how ?
We select the family with the highest number of eukaryotic genes (and not proteins)
1
2
3
3 genes
2 genes
2 genes
1
2
1
If the number of eukaryotic genes are identical, we select the family with the highest number of eukaryotic proteins
If the number of eukaryotic proteins are identical, we select the family with the highest number of proteins

2
Selection of a representative isoform : how ?
We select the family with the highest number of eukaryotic genes (and not proteins)
1
2
3
3 genes
2 genes
2 genes
1
2
1
If the number of eukaryotic genes are identical, we select the family with the highest number of eukaryotic proteins
If the number of eukaryotic proteins are identical, we select the family with the highest number of proteins
SOME FAMILIES MAY FINALLY BE EMPTY AFTER THIS
The « rejected » isoforms are called « ISOFORMEX »

2
Selection of a representative isoform : how ?
1
2
3
3 genes
2 genes
2 genes
1
2
1Second step:
when a gene has isoforms in a
family, choose a representative isoform for the
gene

2
Selection of a representative isoform : how ?
1
2
3
3 genes
2 genes
2 genes
1
2
1Second step:
when a gene has isoforms in a
family, choose a representative isoform for the
gene
?
?

Selection of a representative isoform : how ?
We use the alignment
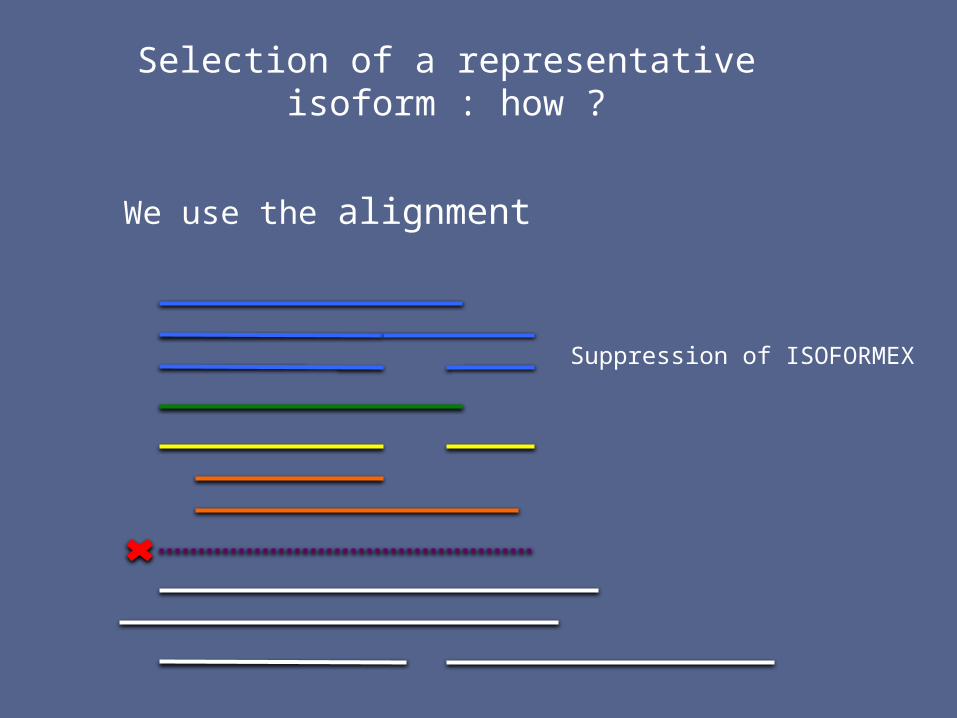
Selection of a representative isoform : how ?
Suppression of ISOFORMEX
We use the alignment

Selection of a representative isoform : how ?
Selection positions with < 50% gap
We use the alignment

Selection of a representative isoform : how ?
For each isoform of a given gene, for each position, we count for 1 each time the residue is identical to the residue in at least one of the isoforms of all other eukaryotic genes.
The isoform with the highest total is selected, the other isoforms being tagged as ISOFORMIN
1 2
2 2

Selection of a representative isoform : how ?
For each isoform of a given gene, for each position, we count for 1 each time the residue is identical to the residue in at least one of the isoforms of all other eukaryotic genes.
The isoform with the highest total is selected, the other isoforms being tagged as ISOFORMIN
1 2
2 2
1

Selection of a representative isoform : how ?
For each isoform of a given gene, for each position, we count for 1 each time the residue is identical to the residue in at least one of the isoforms of all other eukaryotic genes.
The isoform with the highest total is selected, the other isoforms being tagged as ISOFORMIN
1 2
2 2 2 2
1

Tree calculation

Tree calculation
isforminisformin
isformex
ab
c
d
efg
isformin

Tree calculation
isforminisformin
isformex
ab
c
d
efg
isformin

Tree calculation
isforminisformin
isformex
ab
c
d
efg
a
bc
d
e fg
Phyml, FastTree Gblocks

BuildingCompare all proteins against each other (BLAST)
Cluster homologous sequences into families (SILIX + HIFIX)
Compute multiple alignments for each family
Compute phylogenetic trees for each family
Annotate phylogenetic trees (gene duplications, losses, transfers)

Annotate phylogenetic trees
• Several methods are currently developed in the ANCESTROM project
– Speciation, Duplication and Loss– Speciation, Duplication, Transfert and
Loss
– See Vincent Daubin talk tomorow

Querying the database
• ACNUC server (client server application, R pacakge, python package, C API, bio++ API)

Querying the database
• Web interface on PRABI

Querying the database
• Web interface on PRABI

Querying the database
• Web interface on PRABI

Querying the database
Homologous families detected with HMM (D. Guyot)

Querying the database
• New tools ! (R. Planel, J.F. Dufayard)

Querying the databaseDisplaying the gene tree and the the syntheny context of the gene

Querying the databaseDisplaying the gene tree and the the syntheny context of the gene

Querying the databaseSearch for orthologous vertrebrate genes between mouse and man

Querying the databaseSearch for orthologous vertrebrate genes between mouse and man

Thank you for your attention
Ancestrome: Integrative phylogenetic approaches for reconstructing ancestral "-omes"