glycogen synthase kinase 3 is activated by camp … · glycogen synthase kinase 3 is activated by...
TRANSCRIPT

1
Glycogen synthase kinase 3 is activated by cAMP and
plays an active role in the regulation of melanogenesis
Mehdi KHALED, Lionel LARRIBERE, Karine BILLE, Edith ABERDAM, Jean-Paul
ORTONNE, Robert BALLOTTI and Corine BERTOLOTTO*.
INSERM U385, Biologie et Physiopathologie de la peau, IFR 50, 28, avenue de
Valombrose, 06107 NICE Cedex 2, France.
Phone number: (33) 4 93 37 77 90
Fax number: (33) 4 93 81 14 04
Email: [email protected]
* Address request to Corine Bertolotto
Running title: Involvement of GSK3β during cAMP-induced melanogenesis
key words: cAMP, AKT, GSK3β, MITF, tyrosinase
Copyright 2002 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on July 1, 2002 as Manuscript M202939200 by guest on A
ugust 20, 2018http://w
ww
.jbc.org/D
ownloaded from

2
Abstract
In human and mouse, cAMP plays a key role in the control of pigmentation. cAMP,
through the activation of PKA, increases the expression of Microphthalmia-
associated transcription factor (MITF), which in turn stimulates tyrosinase gene
expression, to allow melanin synthesis. Beyond this simplified scheme, cAMP inhibits
the phosphatidylinositol-3-kinase (PI3K) and inhibition of PI3K, by a specific inhibitor,
stimulates melanogenesis. However, the link between the PI3K pathway and
melanogenesis remained to be elucidated. In this report, we showed that cAMP,
through a PKA-independent mechanism, led to inhibition of AKT phosphorylation and
activity. Consistent with the role of AKT in the regulation of GSK3β, cAMP decreased
the phosphorylation of GSK3 β and stimulated its activity. Further, experiments were
performed to investigate the role of GSK3β in the regulation of MITF expression and
function. We observed that GSK3β regulated neither MITF promoter activity nor the
intrinsic transcriptional activity of MITF, but synergized with MITF to activate the
tyrosinase promoter. Additionally, lithium, a GSK3β inhibitor, impaired the response
of the tyrosinase promoter to cAMP and cAMP increased the binding of MITF to the
M-box. Taking into account that GSK3β phosphorylates MITF and increases the
ability of MITF to bind its target sequence, our results indicate that activation of
GSK3β by cAMP facilitates MITF binding to the tyrosinase promoter, thereby leading
to stimulation of melanogenesis.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

3
Introduction
In mammals, epidermal melanocytes synthesize and transfer melanin pigment to
surrounding keratinocytes to allow skin and hair pigmentation. In human, melanins
play a crucial photo-protective role against the carcinogenic and deleterious effects of
ultraviolet radiation of the solar light. Hence, numerous efforts have been made to
understand the molecular mechanisms that govern pigment production.
In mice as well as in humans, it is now well established that pro-opiomelanocortic
(POMC) peptides, adrenocorticotrophic hormone (ACTH) and α-melanocyte
stimulating hormone (αMSH), play a key role in the control of pigmentation. Indeed,
any disturbance of αMSH or ACTH signalling, due to mutations of the melanocortin
type I receptor (MC1R) (1) or to a decrease of αMSH or ACTH expression, results in
an inhibition of melanin synthesis (2). Conversely, pathologic over-expression of
αMSH (3) or ACTH (4,5), or administration of αMSH analogue to human voluntaries
increase skin pigmentation (6,7). Binding of ACTH or αMSH to the Gαs-coupled
MC1R, leads to adenylate cyclase activation, elevation of intracellular cAMP and
activation of the protein kinase A (PKA) (8). The cAMP pathway plays a pivotal role in
the regulation of skin pigmentation. Indeed, patients with Mac Cune-Albright
syndrome display large hyper-pigmented areas caused by an activating mutation in
the Gαs protein that controls cAMP level (9,10). Further, mutations in the type Iα
regulatory subunit of PKA, leading to a constitutive activation of PKA, have been
described in patients with Carney syndrome characterized by spotty skin
pigmentation (11,12). Finally, pro-pigmenting effects of αMSH can be mimicked, in
vitro, by forskolin that directly binds and activates adenylate cyclase (13,14).
Taken together, these observations clearly demonstrate the meaningful role of the
cAMP pathway in the regulation of melanogenesis and skin pigmentation by
melanocortins.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4
cAMP increases melanogenesis mainly through the stimulation of tyrosinase
expression, the enzyme catalyzing the rate limiting reaction of the melanin synthesis
process (15-17). We have shown that cAMP, through activation of PKA and CREB
transcription factor, promotes an increase in the expression of Microphthalmia-
associated transcription factor (MITF) (18), a melanocyte-specific transcription factor
crucial for melanocyte development and differentiation (19,20). As a result, MITF
binds to and activates the tyrosinase promoter, leading thereby to stimulation of
melanogenesis (21-23).
In the course of these studies, we observed that cAMP regulates other signaling
pathways that are also involved in the control of melanogenesis. In melanocytes and
melanoma cells, elevation of the intracellular cAMP content results in the activation of
the Ras/ERK cascade (13). However, activation of Ras/ERK leads to an inhibition of
melanogenesis, and this pathway has been thought to be a feedback mechanism
preventing an excessive production of melanin that would be toxic for cells (24).
Indeed, ERK and RSK, which is activated by ERK, phosphorylate MITF and promote
its degradation thereby leading to an inhibition of tyrosinase expression and of
melanogenesis (25,26). Further, in B16 melanoma cells, we previously showed that
cAMP inhibits the phosphatidylinositol-3 kinase (PI3K) (27). Additionally, the PI3K
specific inhibitor, LY294002, stimulates melanogenesis. These observations suggest
that PI3K pathway might be involved in the regulation of melanin synthesis by cAMP.
While PKA/CREB and Ras/ERK pathways have been thoroughly dissected, the
involvement of the PI3K pathway in cAMP-induced melanogenesis remains to be
elucidated.
One of the key effector of PI3K is the serine/threonine kinase AKT. In response to
growth factors and hormones, AKT is activated by binding to the membrane PI3K
phospholipid products and phosphorylation on threonine 308 and serine 473 (28,29).
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5
Activated AKT phosphorylates, the glycogen synthase kinase 3β (GSK3β) on serine
9, and promotes its inactivation (30). GSK3β is a serine/threonine kinase, first
described to regulate glycogen synthase activity. More recently, GSK3β was
demonstrated to be a key mediator of vertebrate development, tumorigenesis and
cell differentiation (31,32).
Taking into account the role of cAMP in the regulation of melanogenesis, we wished
to evaluate the involvement of AKT and GSK3β in the regulation of melanocyte
differentiation by cAMP. In this report, we clearly demonstrated, in B16 melanoma
cells, that cAMP, by a PKA-independent mechanism, led to inhibition of AKT
phosphorylation and activity, resulting in a dephosphorylation and activation of
GSK3β. Further, we showed that GSK3β regulated neither MITF transcription nor
MITF intrinsic transcriptional activity. However, we found that lithium, a GSK3β
inhibitor, decreased the response to cAMP of the tyrosinase promoter. Additionally,
we showed that GSK3β synergized with MITF to stimulate the tyrosinase promoter.
Finally, short cAMP treatment, that did not up-regulate MITF expression, enhanced
the binding of MITF to the M-box sequence of the tyrosinase promoter. Together, our
results suggest that activation of GSK3β by cAMP, is involved in the regulation of
cAMP-induced melanogenesis by up-regulating the binding of MITF to the tyrosinase
promoter.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

6
Experimental procedures
Materials.
Forskolin, sodium fluoride, sodium orthovanadate, 4-(2-aminoethyl)-benzene-sulfonyl
fluoride (AEBSF), aprotinin and leupeptin were purchased from Sigma Chem.Co. The
PKA inhibitor H89-dihydrochloride and the PI3K inhibitor LY294002 were from
MERCK Eurolab. Dulbecco's modified Eagle's medium (DMEM), trypsin and
Lipofectamine reagent were from GIBCO and fetal calf serum (FCS) was from
Hyclone. C. sordelii lethal toxin (LT) was a gift from Pr. P. Boquet (Nice, France). The
peptide GRPRTSSFAEG (Crosstide) and the glycogen synthase peptide-2 (GS
peptide-2) (YRRAAVPPSPSLSRHSSPHQ-pSEDEEE) were from Euromedex.
Antibodies.
The polyclonal phospho-specific AKT (S473 and T308), GSK3β (S9), CREB (S133),
p42/44 MAPK antibodies and polyclonal antibody that recognizes AKT regardless of
its phosphorylation state were from Cell Signaling. The monoclonal GSK3β (0011-A)
and monoclonal ERK2 (D-2) antibodies were from Santa Cruz. Monoclonal anti-
hemagglutinin (HA) 12CA5 antibody was from BABCO. The phospho-glycogen
synthase antibody (Ab-1) and the phospho-specific Tau antibody (S396) were
purchased from Oncogene Research products. Horseradish peroxidase-conjugated
anti-rabbit or anti-mouse antibodies were from Dakopatts.
Cell Cultures.
B16/F10 murine melanoma cells were grown at 37°C under 5% CO2 in DMEM
supplemented with 7% FCS and penicillin/streptomycin (100 U/ml/50 µg/ml).
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

7
Western blot assays.
B16 melanoma cells were cultured in 6-well dishes with or without different effectors
for the time indicated in the figure legends. Then, cells were lysed in buffer A
containing 50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100, 10 µM leupeptin, 1
mM AEBSF, 100 U/ml aprotinin, 10 mM NaF and 1 mM Na3VO4. Samples (30 µg)
were resolved by 10% SDS-PAGE, transferred to PVDF membrane and then
exposed to the appropriate antibodies. Proteins were visualized with the ECL system
from Amersham using horseradish peroxidase-conjugated anti-rabbit or anti-mouse
secondary antibody. Western blot assays were representative of at least 3
experiments.
Expression vectors, transfection and luciferase assays.
The luciferase reporter plasmids pTyro and pMITF and the expression vector
encoding MITF were previously described (18,33). The pCDNA3 vector encoding
GSK3β was kindly provided by Dr. T.C Dale (London, UK).
B16 melanoma cells were seeded in 24 well dishes and transient transfections were
performed the following day using 2 µl of lipofectamine and 0.5 µg of total DNA
plasmid. pCMVβGal was transfected with the test plasmids to control the variability in
transfection efficiency. After 48 h, cells were harvested in 50 µl of lysis buffer and
assayed for luciferase and β-galactosidase activities. All transfections were repeated
at least three times.
Immunoprecipitation and in vitro AKT assay.
B16 cells were seeded in 6-well dishes and transient transfections were performed
the following day using 10 µl of lipofectamine and 1.5 µg of HA-tagged AKT or 1 µg
of HA-tagged AKT with 2 µg of empty pCDNA3 or vector encoding RasV12. HA-
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8
tagged AKT in the mammalian expression vector pCDNA3 was a kind gift from Dr. G.
Baier (Innsbruck, Austria) and the vector encoding RasV12 was provided by Dr. A.
Eychene (Orsay, France). Cells were treated for 1 h with 50 µM forskolin or 15 µM
LY294002 and, then were lysed in buffer A and immunoprecipitated with the anti-HA
antibody coupled to protein G-sepharose, for 3 h at 4 °C. Immunocomplexes were
washed and AKT activity was assayed with Crosstide as a substrate in a reaction
mixture containing 50 mM Tris, 10 mM MgCl2, 1 mM dithiothreitol, 5 µM ATP, 30 µM
Crosstide and 3.3 µCi of [γ-32PATP], for 30 min at 30 °C. The phosphorylation
reaction was stopped by spotting onto Whatman p81 papers and immersing them in
1% orthophosphoric acid. The papers were washed, rinsed in ethanol, air dried and
the radioactivity was determined by Cerenkov counting.
Immunoprecipitation and in vitro GSK3 assay.
B16 cells were seeded in 6-well dishes, exposed to 50 µM forskolin or 15 µM
LY294002 for the time indicated in the figure legend, lysed as previously described
for the AKT assay and, GSK3β was immunoprecipitated with an anti-GSK3β antibody
for 3 h at 4°C. The activity of GSK3β was assayed with GS peptide-2 as a substrate
in buffer containing 20 mM Tris, 10 mM MgCl2, 5 mM DTT, 20 µM glycogen synthase
peptide-2 as substrate, 10 µM ATP and 3.3 µCi of [γ-32PATP] for 10 min at 30 °C. The
following steps were as described for AKT assay.
Nuclear extracts and gel mobility shift assay.
Nuclear extracts from control cells or cells incubated with forskolin for 1 h were
prepared as previously described (34). Double-stranded synthetic M-box, 5'-
GAAAAAGTCATGTGCTTTGCAGAAGA-3' was γ32P end-labeled using T4
polynucleotide kinase. 5 µg of nuclear proteins were preincubated in a binding buffer
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

9
containing 10 mM Tris pH 7.5, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 4 % glycerol,
80 µg/ml of salmon sperm DNA, 0.1 µg poly (dIdC), 10% FCS, 2 mM MgCl2 and 2
mM spermidine for 15 min on ice. Then, 30,000-50,000 cpm of 32
P labeled probe
were added to the binding reaction for 10 min at room temperature. DNA-protein
complexes were resolved by electrophoresis on a 4 % polyacrylamide gel (37.5:1
Acrylamide-Bisacrylamide) in TBE buffer (22.5 mM Tris-borate, 0.5 mM EDTA, pH 8)
for 1 h 30 at 100 V. For supershift assays, 0.3 µl of pre-immune serum or anti-MITF
antibody (33) was preincubated with nuclear extracts in the binding reaction buffer
before adding the labeled probe.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

10
Results
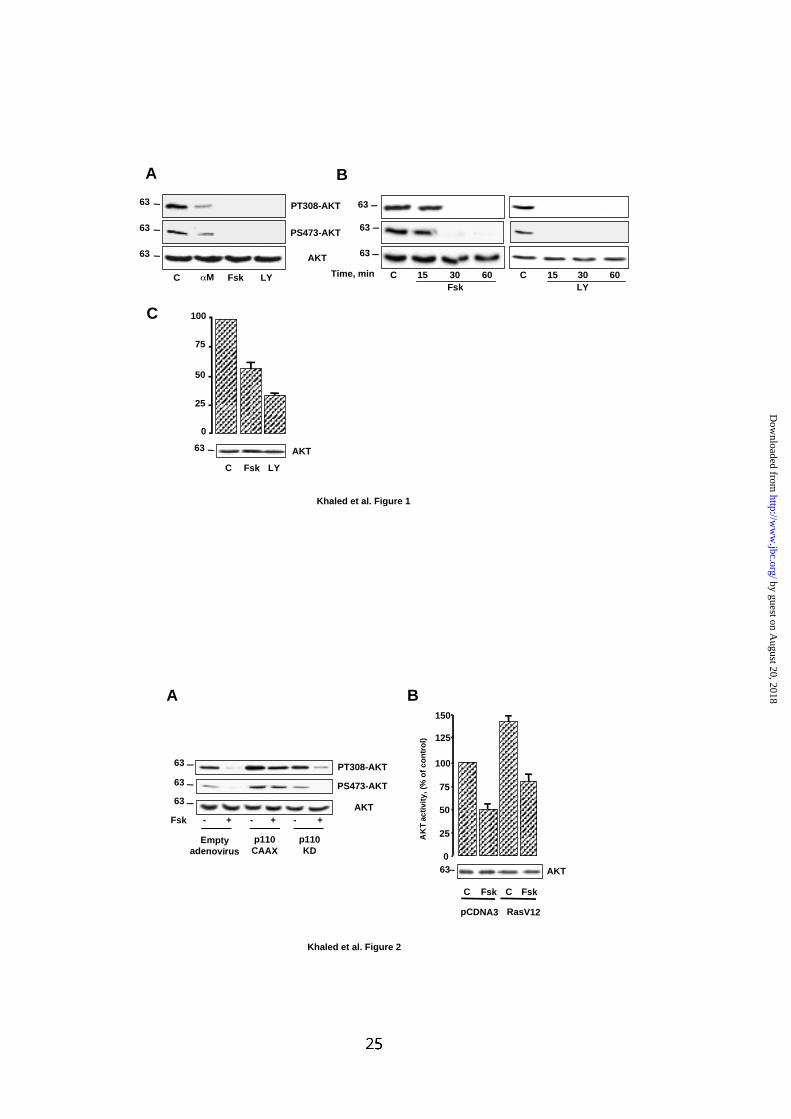
In B16 melanoma cells, cAMP leads to an inhibition of AKT phosphorylation
and activity.
First, we investigated, in B16 melanoma cells, the effects of two cAMP-elevating
agents, αMSH and forskolin on AKT. Immunoblotting of cell lysates, with phospho-
specific antibodies to AKT, revealed that αMSH, a physiologic melanocyte
differentiating agent, as well as forskolin, induced a strong inhibition of AKT
phosphorylation on both threonine 308 and serine 473 compared to control cells (Fig.
1A). As expected, LY294002, a specific pharmacological inhibitor of PI3K, also
abolished AKT phosphorylation on both residues. Increase in cAMP content led to a
complete dephosphorylation of AKT after 30 min (Fig. 1B). The effect of LY294002
was even more rapid since no phosphorylation persisted after 15 min of treatment.
Further, B16 cells, transfected with a vector encoding a HA-tagged AKT were left
untreated, exposed to forskolin or to LY294002 for 30 min. Then, HA-AKT was
immunoprecipitated to perform an in vitro kinase assay. As shown in Fig. 1C,
forskolin and LY294002 reduced the activity of AKT to about 50 and 70%
respectively indicating that the decrease in AKT phosphorylation correlated with an
inhibition of its activity. In each experiment, detection of AKT regardless of its
phosphorylation state ensured even loading of each lane (Fig. 1).
To investigate whether cAMP mediates its effect through a phosphatidylinositol-3-
kinase (PI3K)-dependent mechanism, B16 cells were infected with an adenovirus
encoding a constitutively active (p110CAAX) or a kinase-dead (p110KD) form of the
p110 sub-unit of PI3K. B16 cells, were then exposed or not to forskolin and
phosphorylation of AKT was examined. p110CAAX stimulated the phosphorylation of
AKT over the basal and markedly reduced the effect of cAMP compared to cells
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

11
infected with a control adenovirus (Fig.2A). The kinase dead-mutant, had no effect on
AKT regulation by cAMP. Therefore, in B16 melanoma cells, cAMP leads to an
inhibition of AKT phosphorylation and activity that appears to be mediated through a
PI3K-dependent mechanism.
To better define the site of cAMP action, experiments were designed to evaluate
whether cAMP acted upstream of p21Ras, that is described to activate PI3K. B16
cells were co-transfected with vectors encoding a HA-tagged AKT and a
constitutively active mutant of p21Ras, RasV12. Cells were treated or not with
forskolin and HA-AKT was immunoprecipitated to perform an in vitro kinase assay.
Consistent with fig. 1C, forskolin reduced the activity of AKT to about 50% in
presence of a pCDNA3 empty plasmid (Fig. 2B). Further, co-expression of HA-AKT
with RasV12 increased the basal AKT activity that was also inhibited by forskolin to
about 50%. These results indicate that cAMP acts independently of Ras to inhibit
AKT.
In B16 cells, cAMP decreases GSK3 phosphorylation and promotes its
activation.
Next, we wanted to determine if AKT inactivation by cAMP had an impact on
downstream events in B16 melanoma cells. In this aim, we focused our attention on
the glycogen synthase kinase 3β (GSK3β), a cellular substrate of AKT.
Immunoblotting experiments of cells exposed to forskolin or to LY294002, with
phospho-specific antibody revealed a decrease of GSK3β phosphorylation on serine
9 compared to control cells (Fig. 3A). Next, we investigated the effect of this
dephosphorylation on GSK3β activity. After treatment of the cells with forskolin or
LY294002, GSK3β has been immunoprecipitated and subjected to an in vitro kinase
assay. Results presented in Fig. 3B showed that forskolin and LY294002 stimulated
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

12
GSK3β activity. Immunodetection of total GSK3β indicated that these differences
were not due to variations in the level of protein expression. To confirm the activation
of GSK3β, the effect of forskolin and LY294002 were then analyzed, in intact cells,
toward the phosphorylation of endogenous substrates of GSK3β such as the
glycogen synthase and the microtubule-associated protein, Tau. Using phospho-
specific antibodies, we observed that forskolin and LY294002 increased
phosphorylation of both glycogen synthase and Tau protein compared to cells in
basal condition (Fig. 3C). The blot was also incubated with an anti-ERK2 antibody to
control for the equal loading of the gel. In conclusion, these results clearly
demonstrate that, in B16 melanoma cells, GSK3β is activated in response to cAMP.
The effect of cAMP on AKT and GSK3 is not mediated by PKA and Ras/ERK
pathways.
Although PKA is the major cellular target of cAMP, this nucleotide appears to have
some PKA-independent actions (35-37). Thus, to define whether PKA was involved
in the regulation of AKT and GSK3β following cAMP treatment, we used H89, a cell-
permeable and selective inhibitor of PKA. Treatment of melanoma cells with H89 had
no effect on forskolin-induced inhibition of AKT phosphorylation on threonine 308 and
serine 473 (Fig. 4A). Immunoblotting also indicated that H89 did not prevent the
inhibition of GSK3β phosphorylation on serine 9 induced by forskolin (Fig. 4A). The
functionality of H89 was assessed by its ability to block forskolin-induced
phosphorylation of CREB at serine 133. Detection of ERK2 ensured that each lane
was even loaded. These results clearly indicate that, in B16 melanoma cells, PKA is
not involved in the regulation of AKT and GSK3β by cAMP.
Interestingly, we have recently reported that cAMP stimulates the Ras/ERK pathway
via a PKA independent mechanism (35). Therefore, we wished to explore the
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

13
possibility that the Ras/ERK pathway was involved in the effect of cAMP on AKT and
GSK3β. To this purpose, we took advantage of C. sordelii lethal toxin (LT), which is
an inhibitor of p21Ras, and thus allows the inhibition of the downstream cascade.
Immunoblotting with phospho-specific antibodies demonstrated that LT failed to
abolish the inhibition of AKT and GSK3β phosphorylation induced by forskolin
exposure (Fig. 4B). The functionality of LT was demonstrated by its ability to block
forskolin-induced phosphorylation and activation of ERK1/ERK2. Together, in B16
melanoma cells, cAMP regulates the AKT/GSK3β cascade via a mechanism that
involves neither PKA nor Ras/ERK signaling pathways.
Involvement of GSK3 in the effect of cAMP on the tyrosinase promoter.
As MITF, through the regulation of tyrosinase expression is a key actor in cAMP-
induced melanogenesis, we explored the possibility that GSK3β regulates MITF
expression through a stimulation of the transcriptional activity of its promoter. As
shown in Fig. 5A, GSK3β did not alter basal or cAMP-induced MITF promoter
activity demonstrating that GSK3β did not affect MITF transcription. We also
investigated the possible involvement of GSK3β in the stimulation of MITF activity.
Using a mammalian one hybrid system, we found that GSK3β did not up-regulate the
intrinsic transcriptional activity of MITF (Fig. 5B). Finally, we wished to evaluate the
potential role of GSK3β in the regulation of tyrosinase expression, the enzyme that
controls melanin synthesis. To do so, B16 cells were transfected with the tyrosinase
promoter and then exposed to forskolin in presence or in absence of lithium, an
inhibitor of GSK3β. The results presented on Fig. 6A showed that lithium reduced by
50% the response to cAMP of the tyrosinase promoter. Additionally, we showed that
GSK3β by itself did not significantly affect the activity of the tyrosinase promoter but
synergized with MITF to stimulate the tyrosinase promoter (Fig. 6B). Together, these
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

14
data suggest that GSK3β, through the control of MITF function, is involved in the
regulation of tyrosinase transcription by cAMP. Finally, gel shift assays were
performed with nuclear extracts from control cells or cells that have been exposed to
cAMP for 1 h. We observed an increased binding of nuclear proteins from cAMP-
treated cells to the M-box probe compared to control cells (Fig. 6C, lower panel).
The complexes were shifted by specific MITF antibody, indicating that cAMP
stimulated the formation of MITF/M-box complexes. Since, cAMP did not stimulate
MITF expression at 1 h (Fig. 6C, upper panel), we concluded that cAMP increased
the ability of MITF to bind its target sequence. Together, these results suggest that
GSK3β plays a key role in cAMP-induced melanogenesis by enhancing the binding of
MITF to the tyrosinase promoter.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

15
Discussion
In the course of investigating the molecular mechanisms involved in the regulation of
melanocyte differentiation, we have previously observed, in B16 cells, an inhibition of
the phosphatidylinositol-3 kinase (PI3K) during cAMP-induced melanogenesis (27).
Since the inhibition of PI3K stimulates melanogenesis, we wished to dissect, in the
present report, the molecular events that connected the PI3K pathway to melanin
synthesis. We first focused our attention on AKT, a well-characterized kinase that
functions downstream of PI3K. Depending on the cell type, cAMP has been reported
to inhibit or stimulate AKT (38,39). Hence, we verified the effect of cAMP on AKT in
B16 melanoma cells. Our results showed that cAMP potently inhibited the
phosphorylation of AKT at threonine 308 and serine 473 and led to an inhibition of
AKT activity. Additionally, cAMP effect on AKT was abolished in presence of a
constitutively active mutant of PI3K, demonstrating that cAMP promotes an inhibition
of AKT through a PI3K-dependent mechanism. However, over-expression of a
constitutively active mutant of p21Ras did not block the inhibition of AKT evoked by
cAMP, indicating that the inhibition of AKT by cAMP is mediated through a p21Ras
independent pathway. It should be noted that we have previously reported an
activation of p21Ras by cAMP in melanocytes and melanoma cells (35). Thus, in the
conditions in which we observed an inhibition of AKT, p21Ras is activated
demonstrating that the activation of p21Ras cannot prevent the inhibition of AKT.
AKT phosphorylates GSK3β at serine 9, leading to GSK3β inactivation. Therefore,
the inhibition of AKT by cAMP should lead to the activation of GSK3β. However, it
has been also reported that PKA directly phosphorylates GSK3β at serine 9 and
inhibits its activity (39,40). In B16 cells, cAMP decreased the phosphorylation of
GSK3β at serine 9, and led to the stimulation of its kinase activity. Consistent with
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

16
the effect of AKT on GSK3β and the effect of cAMP on AKT, we demonstrate for the
first time that cAMP promotes an activation of GSK3β.
As PKA is the major intracellular target of cAMP, it was tempting to propose that
effect of cAMP on AKT and GSK3β involved the PKA. However, a pharmacological
inhibitor of PKA did not impair the effect of cAMP on AKT and GSK3β. As mentioned
above, in B16 cells, cAMP also stimulates the Ras/ERK pathway via a PKA
independent mechanism (35). Thus, we hypothesized that the activation of the
Ras/ERK pathway could mediate the effect of cAMP on AKT and GSK3β. However,
inhibition of p21Ras, and consequentely of ERK, by C. sordelii lethal toxin, did not
alter the effect of cAMP on AKT and GSK3β. Hence, cAMP leads to an inhibition of
AKT and a stimulation of GSK3β through PKA and p21Ras/ERK independent
pathways. Recently, it has been also reported that cAMP inhibits AKT through a
mechanism that does not involved PKA (41).
GSK3β has been widely implicated in cell homeostasis by its ability to phosphorylate
a broad range of substrates including the glycogen synthase, the microtubule-
associated protein Tau and β-catenin (42). Interestingly, cAMP-induced activation of
GSK3β, stimulated phosphorylation of the microtubule-associated protein, Tau (Fig.
2C). Considering the role of Tau in the organization of microtubule and actin
cytosqueleton (43,44), as well as in the regulation of organelles traffic (45), our
results point out to a potential role of GSK3β and Tau in the regulation of cAMP-
induced dendritogenesis and melanosome transport, two key parameters of
melanocyte differentiation.
Further, phosphorylation of β-catenin by GSK3β promotes its degradation and
prevents its translocation to the nucleus, where β-catenin acts in concert with the
lymphoid enhancer factor-1/T-cell factor (LEF-1/TCF) family of transcription factors to
transactivate target genes. The promoter of MITF contains a LEF-1 binding site and
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

17
β-catenin stimulates the transcriptional activity of the MITF promoter (46). Thus, the
activation of GSK3β following cAMP exposure should lead to an inhibition of the
MITF promoter activity due to β-catenin degradation. However, in B16 cells, elevation
of the cAMP content did not induce the phosphorylation of β-catenin and did not
affect the activity of a TOPFlash reporter plasmid bearing LEF-1 binding sites (data
not shown). Cotransfection of MITF promoter reporter construct with GSK3β confirms
that GSK3β did not regulate the transcriptional activity of the MITF promoter (Fig.
5A).
Recently, Takeda et al., have demonstrated that GSK3β phosphorylates MITF on
serine 298 thereby enhancing its binding to the tyrosinase promoter (47).
Interestingly, mutation of the MITF serine 298 has been identified in patients with
Waardenburg syndrome type II which are characterized by pigmentary disorders,
emphasing the physiopathologic importance to understand the role of GSK3β in the
control of pigmentation. Therefore, since GSK3β stimulates neither the MITF
promoter activity nor the intrinsic transcriptional activity of MITF, we hypothesized
that cAMP, through GSK3β activation, could increase the ability of MITF to bind its
target sequence. Consistent with the work from Takeda et al., we demonstrated that
GSK3β stimulated the action of MITF on the tyrosinase promoter. Additionally,
lithium, a GSK3β inhibitor, decreased cAMP-induced stimulation of the tyrosinase
promoter. Finally, we demonstrated that cAMP increases the binding of MITF to the
M-box sequence. To demonstrate the key role of the serine 298 phosphorylation, we
studied the effect of a MITF mutant, MITF S298A. This mutant cannot be
phosphorylated by GSK3β and thus the effect of this mutant should not be increased
by GSK3β or by cAMP. However, in agreement with the previous observation of
Takeda et al., this mutant is devoid of any transcriptional activity (data not shown).
Further, we constructed two other mutants in which the serine 298 was replaced
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

18
either by an aspartate or glutamate. The positive charge of these amino acids was
expected to mimic the charge brought by the phosphate group. Unexpectedly, these
two mutants were also unable to transactivate the tyrosinase promoter (data not
shown).
Together, these results show that, in parallel to the PKA/CREB pathway which
stimulates MITF expression, cAMP, through the activation of GSK3β, increases the
ability of MITF to bind to the tyrosinase promoter. These two pathways cooperate,
allowing cAMP elevating agents to efficiently stimulate tyrosinase transcription and
thereby melanogenesis. In conclusion, elevation of intracellular cAMP leads to the
activation of a complex network of signaling pathways that converge to MITF to
control melanin synthesis and melanocyte differentiation (Fig. 7). Our findings,
demonstrating that GSK3β plays an active role in cAMP-induced melanogenesis and
in melanocyte differentiation, bring information of paramount importance on the
molecular mechanisms that control melanin synthesis and skin pigmentation.
Acknowledgments
We are grateful to S. Tartare-Deckert and G. Ponzio for critical reading of the
manuscript. This work was supported by INSERM, The Ligue Nationale contre le
Cancer and the Association pour la Recherche sur le Cancer grant 5808.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

19
References
1. Valverde, P., Healy, E., Jackson, I., Rees, J. L., and Thody, A. J. (1995) Nat Genet 11, 328-
330
2. Krude, H., Biebermann, H., Luck, W., Horn, R., Brabant, G., and Gruters, A. (1998) Nat Genet
19, 155-157
3. Pears, J. S., Jung, R. T., Bartlett, W., Browning, M. C., Kenicer, K., and Thody, A. J. (1992) Br
J Dermatol 126, 286-289.
4. Lamerson, C. L., and Nordlund, J. J. (1998) Nordlund, JJ, Boissy, RE, Hearing, VJ, King, RA,
Ortonne, JP, Oxford University Press, Oxford 120, 1695-1708
5. Sowers, J. R., and Lippman, H. R. (1985) Cutis 36, 351-352, 354
6. Levine, N., Sheftel, S. N., Eytan, T., Dorr, R. T., Hadley, M. E., Weinrach, J. C., Ertl, G. A.,
Toth, K., McGee, D. L., and Hruby, V. J. (1991) Jama 266, 2730-2736
7. Lerner, A. B., and S., M. J. (1961) Nature 189, 176-179
8. Lalli, E., and Sassone-Corsi, P. (1994) J Biol Chem 269, 17359-17362
9. Schwindinger, W. F., Francomano, C. A., and Levine, M. A. (1992) Proc Natl Acad Sci U S A
89, 5152-5156
10. Weinstein, L. S., Shenker, A., Gejman, P. V., Merino, M. J., Friedman, E., and Spiegel, A. M.
(1991) N Engl J Med 325, 1688-1695
11. Casey, M., Vaughan, C. J., He, J., Hatcher, C. J., Winter, J. M., Weremowicz, S.,
Montgomery, K., Kucherlapati, R., Morton, C. C., and Basson, C. T. (2000) J Clin Invest 106,
R31-38
12. Kirschner, L. S., Carney, J. A., Pack, S. D., Taymans, S. E., Giatzakis, C., Cho, Y. S., Cho-
Chung, Y. S., and Stratakis, C. A. (2000) Nat Genet 26, 89-92.
13. Englaro, W., Rezzonico, R., Durand-Clement, M., Lallemand, D., Ortonne, J. P., and Ballotti,
R. (1995) J Biol Chem 270, 24315-24320.
14. Hunt, G., Todd, C., Cresswell, J. E., and Thody, A. J. (1994) J Cell Sci 107 ( Pt 1), 205-211
15. Hearing, V. J., Jr. (1987) Methods Enzymol 142, 154-165
16. Korner, A., and Pawelek, J. (1982) Science 217, 1163-1165
17. Prota, G. (1988) Prog Clin Biol Res 256, 101-124
18. Bertolotto, C., Abbe, P., Hemesath, T. J., Bille, K., Fisher, D. E., Ortonne, J. P., and Ballotti, R.
(1998) J Cell Biol 142, 827-835.
19. Hodgkinson, C. A., Moore, K. J., Nakayama, A., Steingrimsson, E., Copeland, N. G., Jenkins,
N. A., and Arnheiter, H. (1993) Cell 74, 395-404.
20. Steingrimsson, E., Moore, K. J., Lamoreux, M. L., Ferre-D'Amare, A. R., Burley, S. K., Zimring,
D. C., Skow, L. C., Hodgkinson, C. A., Arnheiter, H., Nakayama, N. G., Copeland, N. G., and
Jenkins, N. A. (1994) Nat Genet 8, 256-263
21. Goding, C. R., and Fisher, D. E. (1997) Cell Growth Differ 8, 935-940
22. Sato, S., Roberts, K., Gambino, G., Cook, A., Kouzarides, T., and Goding, C. R. (1997)
Oncogene 14, 3083-3092
23. Yasumoto, K., Yokoyama, K., Takahashi, K., Tomita, Y., and Shibahara, S. (1997) J Biol
Chem 272, 503-509
24. Englaro, W., Bertolotto, C., Busca, R., Brunet, A., Pages, G., Ortonne, J. P., and Ballotti, R.
(1998) J Biol Chem 273, 9966-9970.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

20
25. Hemesath, T. J., Price, E. R., Takemoto, C., Badalian, T., and Fisher, D. E. (1998) Nature
391, 298-301.
26. Wu, M., Hemesath, T. J., Takemoto, C. M., Horstmann, M. A., Wells, A. G., Price, E. R.,
Fisher, D. Z., and Fisher, D. E. (2000) Genes Dev 14, 301-312.
27. Busca, R., Bertolotto, C., Ortonne, J. P., and Ballotti, R. (1996) J Biol Chem 271, 31824-
31830.
28. Bellacosa, A., Testa, J. R., Staal, S. P., and Tsichlis, P. N. (1991) Science 254, 274-277
29. Jones, P. F., Jakubowicz, T., Pitossi, F. J., Maurer, F., and Hemmings, B. A. (1991) Proc Natl
Acad Sci U S A 88, 4171-4175
30. Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995) Nature
378, 785-789.
31. Kim, L., and Kimmel, A. R. (2000) Curr Opin Genet Dev 10, 508-514
32. Plyte, S. E., Hughes, K., Nikolakaki, E., Pulverer, B. J., and Woodgett, J. R. (1992) Biochim
Biophys Acta 1114, 147-162
33. Bertolotto, C., Bille, K., Ortonne, J. P., and Ballotti, R. (1996) J Cell Biol 134, 747-755.
34. Bertolotto, C., Bille, K., Ortonne, J. P., and Ballotti, R. (1998) Oncogene 16, 1665-1670.
35. Busca, R., Abbe, P., Mantoux, F., Aberdam, E., Peyssonnaux, C., Eychene, A., Ortonne, J. P.,
and Ballotti, R. (2000) Embo J 19, 2900-2910.
36. Gray, P. C., Scott, J. D., and Catterall, W. A. (1998) Curr Opin Neurobiol 8, 330-334
37. Matthews, G. (1991) Trends Pharmacol Sci 12, 245-247
38. Filippa, N., Sable, C. L., Filloux, C., Hemmings, B., and Van Obberghen, E. (1999) Mol Cell
Biol 19, 4989-5000.
39. Li, M., Wang, X., Meintzer, M. K., Laessig, T., Birnbaum, M. J., and Heidenreich, K. A. (2000)
Mol Cell Biol 20, 9356-9363.
40. Fang, X., Yu, S. X., Lu, Y., Bast, R. C., Jr., Woodgett, J. R., and Mills, G. B. (2000) Proc Natl
Acad Sci U S A 97, 11960-11965.
41. Wang, L., Liu, F., and Adamo, M. L. (2001) J Biol Chem 30, 30
42. Frame, S., and Cohen, P. (2001) Biochem J 359, 1-16
43. Garcia, M. L., and Cleveland, D. W. (2001) Curr Opin Cell Biol 13, 41-48
44. Gundersen, G. G., and Cook, T. A. (1999) Curr Opin Cell Biol 11, 81-94
45. Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002) J Cell Biol 156,
1051-1063
46. Takeda, K., Yasumoto, K., Takada, R., Takada, S., Watanabe, K., Udono, T., Saito, H.,
Takahashi, K., and Shibahara, S. (2000) J Biol Chem 275, 14013-14016
47. Takeda, K., Takemoto, C., Kobayashi, I., Watanabe, A., Nobukuni, Y., Fisher, D. E., and
Tachibana, M. (2000) Hum Mol Genet 9, 125-132
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

21
Figure legends
FIG.1: cAMP inhibits the activity of AKT.
(A) B16 melanoma cells were left untreated (C) or were incubated with αMSH 1 µM
(αM), forskolin 50 µM (Fsk) or LY294002 15 µM (LY) for 30 min. Cell lysates was
analyzed by western blot with phospho-specific antibodies to AKT (PT308-AKT and
PS473-AKT). (B) Extracts from control B16 cells or cells exposed to forskolin 50 µM
or LY294002 15 µM for the indicated time were submitted to western blot
experiments as described in (A). (C) B16 cells were transiently transfected with an
HA-tagged AKT and then incubated or not with forskolin 50 µM or LY294002 15 µM
for 1 h. Then, AKT was immunoprecipitated and its kinase activity was evaluated
against Crosstide. Data are expressed as percentage of AKT activity in control cell
and are means ± S.E of 3 experiments performed in triplicate. (A-C) Lower panels
show western blot probed with antibody to AKT to control protein loading. Molecular
masses, indicated on the left of western blots, are expressed in Kilodaltons.
FIG.2: cAMP inhibits AKT through a PI3K-dependent, p21Ras-independent
pathway.
(A) B16 cells were infected with an adenovirus encoding either a constitutively active
(p110CAAX) or a kinase-dead (p110KD) form of the p110 subunit of PI3K. Then,
cells were exposed for 1 h to forskolin 50 µM and AKT phosphorylation was analyzed
as described in A. (B) B16 cells were transfected with an HA-tagged AKT in presence
of an empty pCDNA3 plasmid or a vector encoding RasV12 and then were incubated
or not with forskolin 50 µM for 1 h. The activity of AKT was next tested in vitro against
crosstide. Data are expressed as percentage of AKT activity in control cell and are
means ± S.E of 3 experiments performed in triplicate. Detection of total AKT ensured
that each lane was even loading.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

22
FIG. 3: cAMP activates GSK3 .
(A) B16 melanoma cells were treated as described in Fig. 1B and immunoblotting of
cell lysates was done with phospho-specific GSK3β antibody (PS9-GSK3β).
Detection of total GSK3β was used to ensure that each lane was even loaded. (B)
Endogenous GSK3β, from control B16 cells or cells exposed to forskolin 50 µM or
LY294002 15 µM for the indicated time, was immunoprecipitated and its activity was
measured with the GS peptide-2 as substrate. The results are expressed as
percentage of GSK3β activity in control cells and are means ± S.E of 3 experiments
performed in triplicate. The level of GSK3β in immunoprecipitate is shown below the
graph. (C) Cells were incubated for 1 h with forskolin 50 µM or LY294002 15 µM and
then submitted to immunoblotting using a phospho specific glycogen synthase (pGS)
antibody or an antibody that recognizes Tau phosphorylated on S396 (P-TAU).
Detection of ERK2 ensured even loading of each lane. Molecular masses, indicated
on the left of western blots, are expressed in Kilodaltons.
FIG. 4: PKA and Ras/ERK are not involved in the regulation of AKT and GSK3
by cAMP.
B16 cells were left untreated or (A) were incubated with H89 dihydrochloride 5 µM for
30 min or (B) with C. sordelii lethal toxin (LT) 70 ng/ml for 4 h, before adding forskolin
50 µM for 1 h. Western blots were next performed with phospho specific antibodies
described in figure 1 and 3. P-CREB antibody recognizes specifically CREB when
phosphorylated on serine 133. P-ERK antibody recognizes ERK1 and ERK2 when
phosphorylated and activated. Detection of ERK2 showed that each lane was loaded
with equal amounts of protein. Molecular masses, indicated on the left of western
blots, are expressed in Kilodaltons.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

23
FIG. 5: GSK3 regulates neither MITF transcription nor the intrinsic MITF
transcriptional activity.
(A) B16 cells were transfected with 0.3 µg of the luciferase reporter plasmid pMITF
plus 0.1 µg of empty pCDNA3 or 0.1 µg of pCDNA3 encoding GSK3β. Cells were left
untreated or were exposed to forskolin 50 µM for 48 h. (B) B16 cells were transfected
with 0.3 µg of a Gal4 luciferase reporter plasmid and 0.05 µg of plasmid encoding the
Gal4 DNA binding domain (Gal4) or the Gal4 DNA binding domain fused to MITF
(Gal4-MITF). When indicated, 0.1 µg of GSK3β expression plasmid was added to
the reaction. The total DNA was maintained constant at 0.5 µg per condition by
addition of empty pCDNA3 vector.
Luciferase activity was measured and normalized to β-galactosidase activity. Results
are expressed as fold stimulation of the basal luciferase activity from each
experiment. Data are means ± standard error of three experiments performed in
triplicate.
FIG. 6: GSK3 mediates the effect of cAMP on the tyrosinase promoter.
(A) B16 cells were transfected with 0.3 µg of pTyro. After transfection, cells were
incubated with forskolin 50 µM and/or lithium chloride (LiCl) 15 mM for 48 h.
(B) B16 cells were transfected with 0.3 µg of the luciferase reporter plasmid pTyro
and 0.05 µg of pCDNA3 encoding MITF and/or 0.1 µg of pCDNA3 encoding GSK3β.
The total DNA was maintained constant at 0.5 µg per point by addition of empty
pCDNA3 vector. Luciferase activity was measured and normalized to β-galactosidase
activity. Results are expressed as fold stimulation of the basal luciferase activity from
each experiment. Data are means ± standard error of three experiments performed in
triplicate. (C) Gel shift assay was performed using the 32P labeled M-box and B16
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

24
nuclear extracts from control cells (C) or cells treated for 1 h with forskolin (Fsk).
Binding reactions were performed in presence of 0.3 µl of pre-immune serum (PI) or
with 0.3 µl of a specific anti-MITF serum (I). Western blot experiment in the lower
panel shows the detection of total MITF in each condition.
FIG. 7: Model of signaling pathways involved in cAMP-induced melanogenesis.
Elevation of intracellular cAMP content leads to PKA activation and stimulation of
MITF transcription resulting in stimulation of tyrosinase expression. cAMP,
independently of PKA, also activates the Ras, B-Raf, MEKK, ERK and Rsk-1
cascade. Phosphorylation of MITF on serine 73 and serine 409 by ERK and Rsk-1
respectively promotes its degradation constituing a retrocontrol mechanism that
prevents an excessive production of melanin synthesis. Finally, cAMP, via a PKA-
independent mechanism, inhibits PI3K and AKT and promotes an activation of
GSK3β. GSK3β, by phosphorylating MITF on serine 298, increases its binding to the
M-box of the tyrosinase promoter leading to stimulation of tyrosinase expression.
Taken into account the role of Tau in cytosqueleton organization, we hypothesized a
role of Tau in dendritogenesis.
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from
C 15 30 60Fsk
Time, min
63
63
63
C 15 30 60LY
C M Fsk LY
63
63
AKT
PS473-AKT
63
PT308-AKT
B
AKT
C LYFsk
0
25
50
75
100
63
A
C
Khaled et al. Figure 1
Emptyadenovirus
Fsk - - - +++
p110CAAX
p110KD
63
63
AKT
PS473-AKT
63
PT308-AKT
A
Khaled et al. Figure 2
0
25
50
75
100
125
150
C Fsk C Fsk
pCDNA3 RasV12
AKT63
AK
T a
ctiv
ity,
(%
of
con
tro
l)
B
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

A
0
100
200
300
400
C Fsk30 LY60Fsk60
GS
K3
act
ivit
y, (
% o
f co
ntr
ol)
B
GSK346
p-TAU60
p-GS80
C Fsk LY
C
ERK236
46
C 15 30 60LY
Time, min
46
PS9-GSK3
GSK3
46
46
C 15 30 60
Fsk
Time, min
Khaled et al. Figure 3
A
C Fsk H89 Fsk+H89
36
49
46
63
63
Khaled et al. Figure 4
B
C LT Fsk Fsk+LT
63 PT308-AKT
PS473-AKT63
P-ERK36
36 ERK2
46 PS9-GSK3
PT308-AKT
PS473-AKT
P-CREB
ERK2
PS9-GSK3
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

A B
pMITF Gal4 Luc
Gal4-MITF
Gal40
10
40
50
Rel
ativ
e L
uci
fera
se A
ctiv
ity,
Fo
ld s
tim
ula
tio
n o
ver
bas
al
20
0
1
2
4
5
Rel
ativ
e L
uci
fera
se A
ctiv
ity,
Fo
ld s
tim
ula
tio
n o
ver
bas
al
3
Fsk - + - +
30
Khaled et al. Figure 5
BA
pTyro
0
10
20
30
40
50
60
70
80
pCD
NA 3
MIT
F
GSK
3
MIT
F+G
SK3
pTyro
Rel
ativ
e L
uci
fera
se A
ctiv
ity,
Fo
ld s
tim
ula
tio
n o
ver
bas
al
Cont Fsk LiCl Fsk+LiCl
0
2
4
6
8
10
12
14
C Fsk
PI I PI I
MITF
MITF
C
MITF
Khaled et al. Figure 6
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

cAMP
MC1R
MSH
PKA
MITFCREB
MITF Promoter
+
MITF
Tyrosinase
S73 S409P
Degradation
Ras
B-Raf
ERK
P
Rsk-1
MEKK
PI3K
AKT
GSK3
PS298
Tau
Dendrite outgrowth and melanosome transport
?
Khaled et al. Figure 7
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Robert Ballotti and Corine BertolottoMehdi Khaled, Lionel Larribere, Karine Bille, Edith Aberdam, Jean Paul Ortonne,
regulation of melanogenesisGlycogen synthase kinase 3b is activated by cAMP and plays an active role in the
published online July 1, 2002J. Biol. Chem.
10.1074/jbc.M202939200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on August 20, 2018
http://ww
w.jbc.org/
Dow
nloaded from