genomic dna extraction and detection of bacteria ... · immobilized in polyvinyl alcohol host...
TRANSCRIPT

This thesis was elaborated and defended at the Institute of Chemical Technology Prague within the framework of the
European Erasmus Mundus Programme “Erasmus Mundus International Master of Science in Environmental Technology
and Engineering " (Course N° 2011-0172)
Erasmus Mundus Master Course: IMETE
Thesis submitted in partial fulfilment of the requirements for the joint academic degree of:
International Master of Science in Environmental Technology and Engineering
an Erasmus Mundus Master Course from Ghent University (Belgium), ICTP (Czech Republic), UNESCO-IHE (the Netherlands)
Genomic DNA extraction and detection of bacteria immobilized in polyvinyl alcohol
Host University:
Department of Water Technology and Environmental Engineering
Probyn, Rhys Edward Promoter: Co-promoter:
Prof. Ing. Jiří Wanner. DrSc. Ing. Jan Bartáček, Ph.D.
Tutor:
Hana Stryjová, MSc.
2011 - 2013

ii

iii
iii

iv
iv

v
v

Acknowledgements
VI
Very special thanks to Hana Stryjová, MSc. for going above and beyond the call of duty
in her role as my mentor and for becoming a cherished friend.
Very special thank you to Serena Fraraccio, MSc. for helping us with DGGE and
demonstrating unbelievable kindness and patience while working with us to overcome
technical issues and obtain positive results.
Thank you to Ing. Ondrej Vopicka, Ph.D. for helping us develop and execute the liquid
nitrogen treatment for Lentikat’s Biocatalysts
Thank you to Petr Kelbich, MSc. for operating the reactors and performing the chemical
analyses in Ch. 4 of this thesis and for being a great friend.
Thank you to Prof. Ing. Jiří Wanner, DrSc. for hosting me in his laboratory for this
project.
Very special thanks to ir. Maja Šimpraga, PhD. in the IMETE Coordination unit for
tirelessly working to make this program run smoothly.
Thank you the IMETE Management Board for conceiving of and implementing this
program.
Thank you to Ing. Jana Bartáčková, Ph.D. of ICTP and Ineke Melis of UNESCO-IHE for
handling all arrangements in Prague and Delft.
Thank you to Isabel Del Agua Lopez for helping me prepare for all of the toughest exams
in this masters and for being a great friend.
Very special thanks to the following people without whom I would not have gotten this
far in life: Mom and Dad, Richard F. Commenzo esq., Jonathan Todd, Meredyth Ramsay,
Thomas Brew, William Mebane, Inna V. Grishkan MD. Ph.D., Vansa Chatikavanij MSc.,
David C. Gadsby Ph.D, Stephen M. Highstein MD. Ph.D., Scott Lindell MSc., Ed Enos,
Captain Bill Klimm, William Grossman, Gene Tassinari, Alexi Shalapyonok Ph.D., and
Greg Salamida.
This project was supported by a grant of Ministry of Industry and Trade of Czech
Republic FR-TI4/254 and by Research Plan grant MSM 6046137308.

Abstract
VII
In the first part of this investigation, the ability to extract pure high quality DNA from
Lentikat’s Biocatalysts and activated sludge for downstream PCR based applications was
examined with four different commercial DNA isolation kits. DNA extractions were carried out
in triplicate using the Powersoil® DNA Isolation kit, the QIAmp® DNA Stool kit, the Chemagic
DNA Bacteria Kit, and the MasterPure™ DNA Purification Kit. All kits were found to be
compatible with all Lentikat’s Biocatalyst and activated sludge samples and isolated DNA readily
amplified via Touchdown Polymerase Chain Reaction (PCR). Subsequent denaturing gradient gel
electrophoresis (DGGE) showed insignificant extraction bias between isolation kits applied to the
same samples. The Powersoil® DNA Isolation Kit performed the best in terms of processing time
and DNA extract purity. The MasterPure™ DNA purification kit performed the best in terms of
yield, cost, waste generation, and was second best in DNA extract purity. Results also indicated
that flash freezing Lentikat’s Biocatalysts with liquid nitrogen and grinding them prior to DNA
extraction increased the DNA yield and phylogenetic richness of the isolate, thus further
investigation into enhanced lysis methods is recommended.
In the second part of this investigation the effects of a known NOB inhibitor,
hydroxylamine, on Lentikat’s Nitrifying Biocatalysts (PVA biocarriers) was examined in
laboratory scale partial nitrification reactors. Two separate dosing regimes were tested on
individual reactors. The activity of nitrifying bacteria was determined throughout the experiment
with water chemistry analyses while the presence of AOB and NOB within biocarriers was
determined with fluorescent in situ hybridization (FISH). Water chemistry analyses revealed that
daily dosing to 484 µM hydroxylamine for 10 days followed by 49 days at 121 µM achieved
nearly complete nitritation during peak dosing but was not sufficient to achieve long term
sustained NOB inhibition. On the other hand hydroxylamine dosing to 1,211 µM for 14 days
followed by 4 days at 302 µM achieved nearly complete nitritation that was sustained for nearly
78 days and greater than nitratation for 182 days. FISH analyses revealed significant populations
of NOB remained in biocarriers after 14, 54, and 116 days of the onset of severe nitratation
inhibition. This implies a greater resistance of hydroxylamine inhibited NOB immobilized in
PVA biocarriers to washout in comparison with those in RBC biofilms or aerobic granules. NOB
detected by FISH are suspected to include: a small fraction performing limited nitratation, a
significant proportion of dead biomass, an unknown fraction that switched to alternative organic
substrates for survival, and in one case an unknown fraction that may have been seeded from
anaerobic digester effluent.

Table of Contents
VIII
Assignment of Diploma Thesis………………………………………………………………………iii
Certification…………………….…………………………………………….……………………….V
Acknowledgements...……………….……………………………………….……………………….VI
Abstract……….…………………………….………………………………..……………………...VII
List of Figures……………………………………….…………………….…………………………..X
List of Tables……………………………………………………………..…………………………..XI
Abbreviations…………………………………………………………..……………………………XII
Chapter 1 Introduction 1.1 Introduction ....................................................................................................................................... 1
Chapter 2 Literature Review
2.1 The Importance of Nitrogen Removal in Wastewater Treatment ..................................................... 1
2.2 Biological Nitrogen Removal (BNR) Pathways ............................................................................... 2
2.2.1 Nitrogen Speciation ................................................................................................................... 2
2.2.2 Nitrification ................................................................................................................................ 3
2.2.2.1 Ammonia Oxidation (Nitritation) ....................................................................................... 3
2.2.2.2 Nitrite Oxidation (Nitratation) ............................................................................................ 4
2.2.3 Denitrification ............................................................................................................................ 5
2.2.4 Anaerobic Ammonia Oxidation (Anammox) ............................................................................ 6
2.3 Applications of Biological Nitrogen Removal (BNR) Pathways in Wastewater Treatment and the
Case for Partial Nitrification ................................................................................................................... 7
2.4 Lentikat Biocatalysts ......................................................................................................................... 9
2.5 Molecular Methods for Assessing Microbial Communities in WWTP .......................................... 10
2.5.1 DNA Isolation .......................................................................................................................... 11
2.5.2 Polymerase Chain Reaction (PCR) .......................................................................................... 12
2.5.3 Denaturing Gradient Gel Electrophoresis (DGGE) ................................................................. 13
2.5.4 Fluorescence in Situ Hybridization (FISH) .............................................................................. 14
Chapter 3 Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl
Alcohol Biocarriers
3.1 Objectives ....................................................................................................................................... 18
3.2 Materials and Methods .................................................................................................................... 19
3.2.1 Poly Vinyl Alcohol Pellets and Activated Sludge Samples ..................................................... 19
3.2.2 Activated Sludge Characterization ........................................................................................... 20
3.2.3 DNA Isolation .......................................................................................................................... 21
3.2.3.1 Comparison of Commercial DNA Isolation Kits .............................................................. 21
3.2.3.2 Liquid Nitrogen (LN) Enhanced Cell Lysis ...................................................................... 22
3.2.4 DNA Yield and Purity ............................................................................................................. 23
3.2.5 PCR Amplification ................................................................................................................... 24
3.2.6 DGGE ...................................................................................................................................... 26
3.3 Results ............................................................................................................................................. 28

Table of Contents
IX
3.3.1 DNA Isolation and Purity ........................................................................................................ 28
3.3.2 PCR Amplification ................................................................................................................... 32
3.3.3 DGGE ...................................................................................................................................... 35
3.4 Discussion ....................................................................................................................................... 37
3.4.1 Waste Generation, Processing Time, and Cost .................................................................... 37
3.4.2 DNA Yield ........................................................................................................................... 38
3.4.3 Purity .................................................................................................................................... 39
3.4.4 Phylogenetic Comparison of Extracts .................................................................................. 41
3.5 Conclusions ..................................................................................................................................... 43
Chapter 4 In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification
SBRs
4.1 Objectives ....................................................................................................................................... 44
4.2 Materials and Methods .................................................................................................................... 45
4.2.1 Partial Nitrification Reactor Setup and Operation ................................................................... 45
4.2.2 Chemical Analyses ................................................................................................................... 48
4.2.2.1 Ammonia Nitrogen (NH4+) ............................................................................................... 48
4.2.2.2 Nitrite Nitrogen (NO2-) ..................................................................................................... 48
4.2.2.3 Nitrate Nitrogen (NO3-) ..................................................................................................... 49
4.2.2.4 Hydroxylamine (NH2OH) ................................................................................................. 49
4.2.3 FISH ......................................................................................................................................... 49
4.2.3.1 Reagents and Probes ......................................................................................................... 50
4.2.3.2 Fixation with Paraformaldehyde (based on Amann 1995 for Gram-negative bacteria) ... 51
4.2.3.3 Hybridization .................................................................................................................... 51
4.2.3.4 Imaging ............................................................................................................................. 52
4.2.4 Live/Dead Staining .................................................................................................................. 52
4.2.5 DNA Isolation, PCR, DGGE, and Sequencing ........................................................................ 52
4.3 Results ............................................................................................................................................. 53
4.3.1 FISH Images, Inorganic Nitrogen Speciation, and Live/Dead Staining .............................. 53
4.3.2 DGGE and Sequencing ........................................................................................................ 62
4.4 Discussion ....................................................................................................................................... 63
4.4.1 Inhibition of Nitratation ....................................................................................................... 63
4.4.2 In situ detection and characterization of NOB community .................................................. 65
4.5 Conclusions ..................................................................................................................................... 68
Chapter 5 Summary of Conclusions...................................................................................................69
References...............................................................................................................................70
Appendix 1…………………………………………………………………………………...79
Appendix 2…………………………………………………………………………………...80

List of Figures
X
Chapter 2 Literature Review
Figure 1. Lentikat Biocatalyst structure from Bouskova et al. (2011) .................................................... 9
Chapter 3 Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl
Alcohol Biocarriers
Figure 2. 2% Agarose gel profiles of PCR products ............................................................................. 33
Figure 3. DGGE Profile of 16S rDNA amplicons from all samples. .................................................... 35
Figure 4. UPGMA Dendrogram and BSI for Sample N1. .................................................................... 35
Figure 5. UPGMA Dendrogram and BSI for Sample N2. .................................................................... 36
Figure 6. UPGMA Dendrogram and BSI for Sample D1. .................................................................... 36
Figure 7. UPGMA Dendrogram and BSI for Sample D2. .................................................................... 36
Figure 8. UPGMA Dendrogram and BSI for Sample L1...................................................................... 37
Figure 9. UPGMA Dendrogram and BSI for Sample P1 ...................................................................... 37
Chapter 4 In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification
SBRs
Figure 10. Image of nitrifying SBRs in laboratory in ICT Prague. ....................................................... 45
Figure 11. Hydroxylamine dosing regimes for treatment reactors........................................................ 47
Figure 12. FISH images of newly manufactured nitrification biocatalyst ............................................ 53
Figure 13. Nitrogen speciation in control reactor (Reactor C) effluent throughout operational life. ... 54
Figure 14. FISH images from control reactor (Reactor C) biocatalysts sampled on Day 30. ............... 55
Figure 15. Inorganic nitrogen speciation and hydroxylamine dosing in Reactor A. ............................ 57
Figure 16. FISH images of biocatalysts sampled from treatment Reactor A. ....................................... 58
Figure 17. Inorganic nitrogen speciation and hydroxylamine dosing in Reactor B .............................. 59
Figure 18. FISH images of biocatalysts sampled from treatment Reactor B. ....................................... 60
Figure 19. Live/Dead Images of biocarriers sampled from Reactor B. ................................................ 62
Figure 20. DGGE Profile of amplified 16S rDNA extracted from Reactor B biocarriers on Day 274. 62
Appendix 2. Omission of Q-D2-LN Justification
Figure 21. DGGE Profiles for Sample D2 and Sample D1…………………………………………...80
Figure 22. UPGMA Dendrogram of Sample D2 Including Q-D2-LN………………………………..80

List of Tables
XI
Chapter 2 Literature Review
Table 1. Typical nitrogen values for various wastewaters adapted from Zanetti et al. (2012) ............... 1
Table 2. Stoichiometric comparison of BNR pathways taken from Zanetti et al. (2012) ....................... 8
Chapter 3 Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl
Alcohol Biocarriers
Table 3 Touchdown PCR Master Mix Formula .................................................................................... 25
Table 4. Preparation of ureumformamide (UF)/6% acrylamide solutions employed in denaturing
gradient gels. ................................................................................................................................. 27
Table 5. DGGE Gel Casting Reagents .................................................................................................. 27
Table 6. Plzeň and Lochovice Activated Sludge Characteristics .......................................................... 28
Table 7. Average cost, processing time, and mass of waste generated during a 9 sample DNA
Isolation......................................................................................................................................... 28
Table 8. DNA yields (µg/g), absorbance 260 nm/280nm, and absorbance 260nm/230nm.
...................................................................................................................................................... 29
Table 9. PCR amplification of 16S rDNA isolated from Lentikat Biocatalysts® and activated sludge
samples .......................................................................................................................................... 32
Table 10. Mean BSI of all DGGE extracts excluding MB-LN-D2 ....................................................... 41
Chapter 4 In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification
SBRs
Table 11. Summary of treatments administered to Reactors A and B throughout their operations. ..... 47
Table 12. Hybridization probes employed. Table adapted from Nielsen et al. 2009. ........................... 51
Table 13. Residual hydroxylamine concentrations measured in each reactor in µM ........................... 55

Abbreviations
XII
Amo Ammonia Monooxygenase
Anammox Anaerobic Ammonia Oxidation
AOA Ammonia Oxidizing Archaea
AOB Ammonia Oxidizing Bacteria
ARDRA Amplified Ribosomal DNA Restriction Analysis
ATAD Autothermal Aerobic Sludge Digester
BABE Bioaugmentation Batch Enhanced
BNR Biological Nitrogen Removal
BSA Bovine Serum Albumin
BSI Band Sharing Index (Phylogenetic richness compared to synthetic lane)
CANON Completely Autotrophic Nitrogen Removal over Nitrite
CARD Catalyzed Reporter Deposition
CLSM Confocal Laser Scanning Microscopy
COD Chemical Oxygen Demand
DEMON De-ammonification
DNA Deoxyribonucleic Acid
dsDNA Double stranded DNA
DO Dissolved Oxygen
DGGE Denaturing Gradient Gel Electrophoresis
FISH Fluorescent in situ Hybridization
Hao Hydroxylamine Oxidoreductase
Hh Hydrazine Hydrolase
Hzo Hydrazine Oxidoreductase
IQR Inner Quartile Range
LN Liquid Nitrogen
MAR Microautoradiography
MSW Municipal Solid Waste
MSc Master of Science
Nar Nitrate Reductase
Nir Nitrite Reductase
NirK Nitrite Reductase variation K
NirS Nitrite Reductase variation S
NOB Nitrite Oxidizing Bacteria
Nor Nitric Oxide Reductase
Nos Nitrous Oxide Reductase
Nxr Nitrite Oxidoreductase
OLAND Oxygen Limited Autotrophic Nitrification-Denitrification
OTU Operational Taxonomic Unit
PCR Polymerase Chain Reaction
PFA Paraformaldehyde
PNA Peptide Nucleic Acid
PVA Poly Vinyl Alcohol
qPCR Quantitative Real Time Polymerase Chain Reaction

Abbreviations
XIII
RBC Rotating Biological Contactor
RNA Ribonucleic Acid
rRNA Ribosomal Ribonucleic Acid
SBR Sequencing Batch Reactor
scBNR Short Cut Biological Nitrogen Removal
SEM Scanning Electron Microscopy
SHARON Single Reactor System for High Ammonium Removal over Nitrite
SNAP Single Stage Nitrogen Removal Using Anammox and Partial Nitrification
TKN Total Kjeldahl Nitrogen
UF Ureum-formamide
t-RFLP Terminal-Restriction Fragment Length Polymorphism
WWTP Waste Water Treatment Plant
WW Waste Water

Ch 1. Introduction
1
1.1 Introduction
In the early days of activated sludge wastewater treatment, the primary objectives were
the removal of carbonaceous organic material and the transformation of ammonia to nitrate
(Seviour and Nielsen, 2010). Nowadays, a greater understanding of the environmental
impacts of nitrogen and phosphorus compounds as well as the identification of ecologically
sensitive areas with economic and recreational importance has led to an emphasis on their
removal in wastewater treatment plants (WWTPs). Despite the importance of phosphorus
removal, the subsequent review and research presented will focus on the applied
microbiology of nitrogen removal.
As our understanding of microbiologically mediated nitrogen removal (BNR) processes
advances, techniques continue to emerge to facilitate the proliferation of beneficial organisms
to optimize WWTP performance. While some technologies require the construction of new
facilities, many also focus on retrofitting existing activated sludge WWTPs by altering
process flows, bioaugmenting with biocarriers in existing reactors, and/or by adding new side
stream reactors. Regardless of the approach taken, the ability to characterize and monitor the
community of nitrogen removing microorganisms is essential for maintaining and optimizing
the environmental and economic performance of WWTPs as well as diagnosing the cause of
problems.
The research presented in this MSc. Thesis is meant to 1) develop an internal protocol for
total genomic DNA extraction from nitrifying and denitrifying microorganisms immobilized
in porous polyvinyl alcohol pellets (Lentikat Biocatalysts®) and activated sludge by
comparing commercial DNA isolation kits and 2) to assess the development of nitrifying
bacteria communities immobilized in Lentikat Biocatalysts® and exposed to a known nitrite
oxidizing bacteria (NOB) inhibitor, hydroxylamine, in lab scale sequencing batch reactors
(SBRs) using Fluorescence in situ Hybridization (FISH) and chemical analyses. The
following review is meant to establish the current state of knowledge regarding: biological
nitrogen removal (BNR) pathways, the organisms involved in them, the utility of biocarriers
namely Lentikat Biocatalysts®, and the molecular tools at our disposal for characterizing
microbial communities in environmental and WWTP derived samples.
Ch 2. Literature Review
1
2.1 The Importance of Nitrogen Removal in Wastewater Treatment
The importance of Nitrogen removal from wastewater prior to discharge has become
increasingly clear in light of the negative impacts caused by high concentrations of inorganic
nitrogen species on aquatic environments. Although nonpoint sources such as agricultural
runoff and atmospheric deposition are considered far more problematic, WWTP effluents are
recognized as a significant point source contributing to nitrogen enrichment of both surface
and ground waters (Smith et al., 1999). Some typical values for nitrogen content expressed in
terms of Total Kjeldahl Nitrogen (TKN) and ammonium nitrogen (NH4-N) in various waste
streams requiring treatment are given in Table 1. The three primary environmental impacts
resulting from inorganic nitrogen enrichment are: acidification of freshwater ecosystems;
eutrophication of fresh, brackish and marine ecosystems; and direct toxicity to aquatic
organisms and even humans (Smith et al., 1999; Kloep et al. 2000; Camargo and Alonso,
2006).
Table 1. Typical nitrogen values for various wastewaters adapted from Zanetti et al. (2012)
TKN (mg/L) NH4-N (mg/L)
Raw Domestic Wastewater 60-110 50-100
Swine Wastewater 996-1,520 534-1154
Anaerobic Digester Reject
Water
3,000-5,000 2,000-4,000
While nitrogen induced acidification is most often attributed to atmospheric deposition of
nitrous acid (HNO3) into freshwater ecosystems, the discharge of ionized ammonia (aka
ammonium; NH4+) into systems with low buffering capacities can also be a cause. This in
particular, is due to the release of hydrogen ions (H+) during the naturally occurring oxidation
of ammonium (Camargo and Alonso, 2006). Though worth mentioning, acidification
resulting from WWTP effluent is not as great of a concern as eutrophication or direct toxicity
to aquatic organisms.
High concentrations of ammonia, nitrite, and/or nitrate all exert direct toxic effects on
aquatic organisms (Kloep et al., 2000). Unionized ammonia, NH3, can be acutely or
chronically toxic particularly to fish and is present in significant quantities at elevated pH
(>8) and temperatures (>20oC) (Randall and Tsui, 2002). Notable effects on fish include:
immune system repression, inhibition of ATP production, reduction in blood oxygen carrying
capacity, and suppression of gill tissue function leading to asphyxiation (Camargo and
Alonso, 2006). Likewise, nitrite (NO2-) toxicity has been attributed with causing severe
electrolyte imbalances, disruption of membrane potentials, neurotransmission, muscle

Ch 2. Literature Review
2
contractions, and repression of immune function in fish and crustaceans (Camargo and
Alonso, 2006). However the most important toxic effect of nitrite and nitrate (NO3-) in fish
(and humans) is the oxidation of iron atoms in hemoglobin thus converting it to
methemoglobin and rendering it useless for oxygen distribution. Because toxicity varies by
species, the following water quality criteria for long term and short term maximum exposure
concentrations for aquatic organisms to ammonia, nitrite and nitrate are recommended by
Camargo and Alonso (2006): 0.02-0.35 mg NH3-N/L, 0.35- 3 mg NO2-N/L, and 2-5 mg
NO3-N/L.
The last and perhaps most significant impact of nitrogen enrichment in aquatic
ecosystems is eutrophication. This is defined as a state of being “well nourished” in regards
to the concentrations of growth-limiting nutrients, most often nitrogen and to an even greater
extent phosphorus (Smith et al., 1999). The result of such nutrient enrichment is the excessive
proliferation of algae and macrophytes followed by oxygen depletion as dead biomass
accumulates and decomposes (Camargo and Alonso, 2006). Additional problems induced by
eutrophication include: shifts in macroinvertebrate, vascular plant, and algae composition,
toxic algal blooms, fish kills, reduced water clarity, elevated pH in the water column, loss of
coral reef communities (marine), disruption of drinking water treatment processes, recreation
restriction, and many more (Smith et al., 1999; Camargo and Alonso, 2006). As reported by
Smith et al. (1999) the following values for total nitrogen (mg/m3) may constitute a
“eutrophic” state in lakes, streams and coastal marine systems respectively:
650-1,200; >1,500; and >400.
2.2 Biological Nitrogen Removal (BNR) Pathways
2.2.1 Nitrogen Speciation
The most common forms of nitrogen entering municipal and industrial wastewater
treatment plants are ammonium (NH4+) and ammonia (NH3), in pH dependent equilibrium
skewed towards NH4+
below pH 9, and organic nitrogen compounds including urea, proteins
and amino acids (Seviour and Nielsen, 2010). As biologically catalyzed reactions proceed, a
portion of ammonium and organic nitrogen are directly incorporated into heterotrophic
biomass however a majority is mineralized into NH3/NH4+ in a process referred to as
ammonification (Zeng et al., 2012). Further microbial processes, namely nitrification, give
rise to aqueous oxidized inorganic nitrogen species including nitrite (NO2-), and nitrate (NO3
-
) while denitrification and anammox processes further transform these into gaseous species,

Ch 2. Literature Review
3
primarily dinitrogen (N2) and to a lesser extent nitric oxide (NO) and nitrous oxide (N2O)
which are off-gassed to the atmosphere particularly in oxygen limiting conditions (Seviour
and Nielsen, 2010). It’s worth noting that in conditions of high aeration, typical of many open
air biological reactors, some ammonia (NH3) volatilization to the atmosphere will also occur.
2.2.2 Nitrification
Nitrification is an aerobic two-stage biologically catalyzed process through which
ammonia is fully oxidized to nitrate by way of nitrite as an intermediate.
NH3/NH4+ → NO2
- → NO3
-
Each stage of nitrification is primarily catalyzed by separate functional groups of
chemolithoautotrophic bacteria that obtain energy from oxidizing inorganic compounds and
carbon from CO2 fixation and in some cases the degradation of simple organic compounds.
Unfortunately, the oxidation of inorganic nitrogen species yields relatively low energy, thus
nitrifying bacteria are notoriously slow growers in comparison with most heterotrophic,
organic compound degrading, microbes present in WWTPs (Seviour and Nielsen, 2010;
Whang et al., 2009). As a result, special design and operational parameters must be taken into
consideration in order to facilitate their presence and activity. Furthermore, the stability of
nitrification in wastewater treatment, particularly in activated sludge plants, is highly
sensitive to alterations in environmental and operational parameters. Sudden or even seasonal
changes in temperature, pH, dissolved oxygen (DO), wastewater composition and
concentrations can cause disruptions in nitrification performance that may require many
weeks to recover from (Whang et al., 2009; Zeng et al., 2012). Nevertheless nitrification,
particularly ammonia oxidation, remains a critical component of our wastewater treatment
strategy in terms of nitrogen removal, as no known viable alternatives exist at this time
(Seviour and Nielsen, 2010).
2.2.2.1 Ammonia Oxidation (Nitritation)
Ammonia oxidation to nitrite is the first, and rate limiting, stage of nitrification. It is
mostly attributed to the activity of a functional group of microbes referred to as Ammonia
Oxidizing Bacteria (AOB) that employ the following metabolic reaction pathway (Seviour
and Nielsen, 2010):
NH4+ + O2 + 2H
+ + 2e
- → NH2OH
NH2OH + H2O → HNO2 + 4H
+ + 4e
-

Ch 2. Literature Review
4
The first step of this reaction, the oxidation of ammonia to hydroxylamine, is catalyzed by the
enzyme ammonia monooxygenase (Amo) while the second step, the oxidation of
hydroxylamine to nitrite, is catalyzed by hydroxylamine oxidoreductase (Hao) (Simon and
Klotz, 2013; Seviour and Nielsen, 2010). Based on extensive research, AOB are credited with
carrying out a majority of ammonia oxidation in WWTPs, however it is worth noting that
recently discovered Ammonia Oxidizing Archaea (AOA) and Anaerobic Ammonia Oxidizers
(Anammox) are also thought to play a role in some nitrifying reactors and marine biofiltration
systems (Sakami et al., 2012; Kawagoshi et al., 2012). The significance of the role played by
AOA however, remains poorly characterized therefore continued discussion will focus on
AOB. Anammox will be discussed further in section 2.2.4.
Most AOB are members of the phylum Proteobacteria and more specifically the classes
Betaproteobacteria and Gammaproteobacteria and are widespread throughout terrestrial,
freshwater, and marine ecosystems (Seviour and Nielsen, 2010; Whang et al. 2009). Two
species belonging to the genus Nitrosococcus are the only known AOB from the class
Gammaproteobacteria, and are obligate halophiles widely found in brackish and marine
environments and occasionally brackish biofilters (Seviour and Nielsen, 2010; Kumar et al.,
2013). On the other hand, according to Seviour and Nielsen (2010), Betaproteobacterial
AOB belonging to the genera Nitrosomonas and Nitrosospira are far more diverse and widely
distributed across terrestrial and aquatic ecosystems. Among these, Nitrosospira are believed
to be the dominant AOB in soil ecosystems while Nitrosomonas seem to include a broader
array of physiologically diverse/tolerant species with greater presence in a various aquatic
environments, especially WWTPs. This being said, Whang et al. (2009) and Sakami et al.
(2012) report that Nitrosospira-like AOB do occasionally exist and play a role in nitrifying
WWTP reactors and marine aquaculture biofilters.
2.2.2.2 Nitrite Oxidation (Nitratation)
Nitrite oxidation to nitrate is the second and final stage in the aerobic process of
nitrification and is mostly carried out by a functional group referred to as Nitrite Oxidizing
Bacteria (NOB). Unlike AOB, NOB are not all strict chemolithoautotrophs in that some are
also known to oxidize simple organic compounds like acetate and pyruvate (Seviour and
Nielsen, 2010). Furthermore, they possess greater diversity of enzymatic machinery for the
oxidation of nitrite than AOB do for ammonia. The only well characterized nitrite oxidation
enzyme is nitrite oxidoreductase (Nxr) belonging to the genus Nitrobacter which catalyzes

Ch 2. Literature Review
5
the following metabolic reaction (Seviour and Nielsen, 2010; Vanparys et al., 2007; Simon
and Klotz, 2013):
NO2- + H2O
↔ NO3
- + 2H
+ + 2e
-
Phylogenetically speaking, NOB, include at least one member from the phylum Chloroflexi
as well as Proteobacteria genera from the classes Alphaproteobacteria (Nitrobacter),
Deltaproteobacteria (Nitrospina), and Gammaproteobacteria (Nitrococcus) as well as
Nitrospira (name of class and genus) from the phylum Nitrospirae (Vanparys et al., 2007;
Seviour and Nielsen, 2010; Sorokin et al., 2012).
Of these genera, Nitrococcus and Nitrospina, have only been isolated from marine
ecosystems while Nitrobacter and Nitrospira have been found to tolerate a wide swath of
aquatic and terrestrial environments and are the most commonly detected NOB in WWTPs
(Seviour and Nielsen, 2010; Han et al., 2012).
2.2.3 Denitrification
Denitrification is a four step anaerobic/anoxic process whereby nitrate and nitrite are
transformed to gaseous species through the following metabolic reaction pathway (Seviour
and Nielsen, 2010):
NO3- → NO2
- → NO
→ N2O
→ N2
While the end product of N2 is most desirable because of its inert nature in the atmosphere,
releases of the intermediates NO and N2O (both greenhouse gasses) can be significant
depending on the denitrifying community present and environmental conditions (Seviour and
Nielsen, 2010; Kong et al., 2013).
Bacteria carrying out denitrification are far more diverse and heterogeneous a group
than AOB or NOB. Typically, denitrifiers are facultative anaerobes that utilize nitrate and
nitrite as alternative terminal electron acceptors in their respiration when ample oxygen is not
available. The ability to do this has been found across the spectrum of prokaryotes from
organotrophs, lithotrophs, and diazotrophs, to halophiles and thermophiles (Seviour and
Nielsen, 2010). This heterogeneity makes denitrifiers a more difficult functional group to
define, however common genera reported in literature include: Bacillus, Pseudomonas,
Paracoccus, Hyphomicrobium, Azoarcus, Marinobacter, Halomonas, Methylophaga, and
many more (Yoshie et al., 2004; Song et al., 2012; Osaka et al., 2008).
The metalloenzymes essential for denitrification are Nitrate reductase (Nar), Nitrite
reductase (Nir), Nitric oxide reductase (Nor), and Nitrous oxide reductase (Nos). As is the

Ch 2. Literature Review
6
case for Amo in AOB, there are different forms of these enzymes coded for by different gene
sequences, for example the nitrite reductase variations NirS and NirK. Due to the diversity of
denitrifying organisms, molecular tools discussed in section 2.5, enable the quantification of
these gene sequences and have become the preferred method for assessing the abundance of
denitrifying bacteria in environmental samples and WWTPs (Warneke et al., 2011).
2.2.4 Anaerobic Ammonia Oxidation (Anammox)
Anaerobic ammonia oxidation is a nitritation-denitritation alternative to the classical
nitrification-denitrification BNR pathway discussed previously. As the name suggests,
anammox, is an anaerobic microbially catalyzed process whereby ammonia serves as the
electron donor, nitrite as the electron acceptor, and dinitrogen gas is the final product (Feng et
al., 2007). The process proceeds via the following reaction pathway (Feng et al., 2007; Jetten
et al., 2009; Seviour and Nielsen, 2010):
Overall Reaction: NH4+ + NO2
- N2 + H2O
Metabolic Pathway: NO2- → NO, NO + NH4
+ → N2H4
→ N2 + H2O
Anammox Stoichiometry: NH4
+ + 1.32NO2
- + 0.066HCO3
- + 0.13H
+ 1.02N2 + 0.26NO3- + 0.066CH2O0.5N0.15 + 2.03H2O
First, nitrite is reduced to nitric oxide by nitrite reductase (NirS); next hydrazine hydrolase
(Hh) reduces nitric oxide while simultaneously oxidizing ammonium to form hydrazine.
Lastly, hydrazine is oxidized by hydrazine oxidoreductase (Hzo) to produce dinitrogen gas,
water, and a very small amount of byproduct nitrate (Feng et al., 2007; Jetten et al., 2009).
Anammox bacteria are slow growing chemolithoautotrophs belonging to phylum
Planctomycetes. They are obligate anaerobes with high sensitivity to oxygen (>2 µM O2 is
fatal), they rely on CO2 fixation and bicarbonate as their carbon sources, have doubling times
between 10-20 days (lower in situ), and have relatively low biomass yields (Jetten et al.,
2009; Seviour and Nielsen, 2010). Owing to their low growth rate, they tend to thrive in
natural environments that have low substrate concentrations. On the other hand they do not
all adhere strictly to chemolithoautrophic metabolism as some are known to employ ferrous
iron and/or specific organic compounds as alternative electron donors and ferric iron,
manganese oxides, and/or nitrate as alternative electron acceptors (Jetten et al., 2009). To
date, 5 candidatus genera have been described including Kuenenia, Brocadia, Jettenia,
Anammoxoglobus, and Scalindua. The first four were all enriched from activated sludge

Ch 2. Literature Review
7
while Scalindua was found in marine sediments. Anammox is estimated to be responsible for
33-50% of global nitrogen removal from marine ecosystems (Dalsgaard et al., 2005).
2.3 Applications of Biological Nitrogen Removal (BNR) Pathways in
Wastewater Treatment and the Case for Partial Nitrification
The earliest activated sludge WWTPs focused on the removal of organic carbon
compounds and the transformation of ammonia to less toxic species like nitrite and nitrate
(Seviour and Nielsen, 2010). As discussed in section 2.1, the negative environmental impacts
of these species have since been well documented thus necessitating the implementation of
more comprehensive nitrogen removal systems. To accommodate this requirement, there has
been a massive proliferation of reactor types, multistage configurations, process control
strategies, and bioaugmentation techniques used at WWTPs. From an engineering standpoint,
there are far too many nitrogen removal approaches to go into any detail here, however from
the standpoint of BNR pathways the options are far more limited.
Nitrification-denitrification via nitrate has long been the dominant BNR pathway
employed in WWTPs around the world (Kuenan and Robertson, 1994; Zanetti et al., 2012;
Seviour and Nielsen, 2010). Its rise to dominance is due to its simplicity in regards to
providing more or less stable performance, while relying on a simple staged aerobic/anoxic
configuration (though modern systems can be far more complex), the occasionally necessary
dosing of simple organic substrates (COD) to facilitate adequate denitrification, and
prescribed (fixed) operational controls (Seviour and Nielsen, 2010; Zanetti et al., 2012).
Although effective, this strategy is quite expensive due to the high cost of aeration and the
dosing of COD when needed. This has fueled great interest in the development of potentially
cheaper alternatives aimed at employing so called “short cut” biological nitrogen removal
(scBNR) pathways particularly for use on waste waters low in COD.
There are two known scBNR pathways that both rely on partial nitrification, namely
nitritation facilitated by AOB, as their initial step. The first option is nitritation followed by
heterotrophic denitritation which cuts out the conversion of nitrite to nitrate thereby reducing
the amounts of oxygen and COD consumed (Aslan and Dahab, 2008; Zanetti et al., 2012).
The second option employs only partial nitritation followed by anammox which not only
further reduces oxygen consumption but also eliminates COD consumption (though
bicarbonate and/or CO2 are needed) (Lan et al., 2011; Zanetti et al., 2012). The O2 and COD
requirements of the three BNR pathways are summarized in Table 2.

Ch 2. Literature Review
8
Table 2. Stoichiometric comparison of BNR pathways taken from Zanetti et al. (2012)
gO2 / gN gCOD/ gN
Nitrification-Denitrification 4.57 4
Nitritation-Denitritation 3.43 2.4
Partial Nitritation-Anammox 1.72 NA
The key to achieving partial nitrification is selecting for AOB over NOB. Currently
established strategies for accomplishing this include: maintaining reactor temperatures over
25oC, maintaining free ammonia (NH3) levels above 1 mg/L, maintaining a free nitrous acid
(H-NO2-N) concentration between 0.011-0.10 mg/L, limiting oxygen concentrations to
between 0.5-1.5 mg/L (though difficult to control), and chemical inhibition with
hydroxylamine (Zanetti et al., 2012; Park and Bae, 2009; Xu et al., 2012). Several reactors
have been developed that employ one or more of these strategies including the high
temperature nitritation Single Reactor System for High Ammonium Removal over Nitrite
(SHARON), the oxygen limited AOB/anammox single reactor Completely Autotrophic
Nitrogen Removal over Nitrite (CANON) and Oxygen Limited Autotrophic Nitrification-
Denitrification (OLAND) systems, as well as the De-ammonification (DEMON), Single
Stage Nitrogen Removal Using Anammox and Partial Nitrification (SNAP), and
Bioaugmentation Batch Enhanced (BABE) systems (Kumar and Lin, 2010).
Chemical inhibition of NOB via the dosing of hydroxylamine (NH2OH) is a less
studied approach that has yet to be applied outside of laboratory settings. Kindaichi et al.
(2004) followed up on previous studies that established NH2OH dosing as a method for
stimulating AOB growth but noted inhibitory effects on NOB by examining this inhibition.
They found that the addition of 250 µM NH2OH completely inhibited the growth on NOB in
rotating biological contactor (RBC) biofilms. Xu et al. (2012) then investigated the use of
NH2OH inhibition as a strategy for establishing a partial nitrification reactor using aerobic
granules that are not compatible with the free ammonia or oxygen limitation strategies.
Though they were unsuccessful at achieving the appropriate ratios of NH4+ and NO2
- for
subsequent anammox treatment, they reported great compatibility of inhibiting NOB with
NH2OH in aerobic granules. Extrapolating upon this conclusion, it seems as though
controlled NH2OH dosing has great potential as a strategy to facilitate the start-up and
performance of partial nitrification reactors thus further research is warranted.

Ch 2. Literature Review
9
2.4 Lentikat Biocatalysts
Biocatalyst, also called biocarrier, technology is based on the encapsulation of
functional microorganisms within porous hydrogel matrices for use in biologically mediated
processes. This concept has been successfully applied for the production of bioethanol,
pharmaceutical enzymes, artificial seeds, and artificial cells in addition to medical treatments
and bioaugmentation of BNR at WWTPs (Park and Chang, 2000).
Lentikat’s Inc. is a market leading producer of biocatalysts based in Prague, Czech
Republic. Their patented biocatalysts (Lentikat Biotechnology: German Patent
# DE 198 27 552) are made from nontoxic, non-biodegradable, non-abrasive, and highly
elastic polyvinyl alcohol (PVA) (Bouskova et al., 2011; Park and Chang, 2000; Vackova et
al., 2012). Each lens shaped PVA pellet is approximately 3-4 mm in diameter and 200-
400 µm in width as shown in Figure 1. They are produced at room temperature by blowing a
mixed solution, containing the cell suspension and dissolved polymer, through a jet nozzle
into a rotating wire wheel composed of 1 µm wires. The solution is cut into appropriately
sized beads which land on a moving film where they harden with the help of a warm air
blower (Park and Chang, 2000; Bouskova et al., 2011). Following immobilization, pellets
undergo a 6 week cultivation period in the laboratory (Vackova et al., 2011).
Figure 1. Lentikat Biocatalyst structure, the porous PVA hydrogel is depicted in blue while the orange
spots represent encapsulated microorganisms (Bouskova et al., 2011)
Lentikat’s wastewater treatment biocatalysts are specially designed to facilitate
enhanced nitrogen removal. They currently produce three separate pellets, one for
nitrification that contains encapsulated Nitrosomonas europaea and Nitrobacter winogradskyi
and the other two for denitrification containing either encapsulated Paracoccus denitrificans
or Pseudomonas fluorescens (Bouskova et al., 2011). Because the production technique is
carried out at room temperature, nitrifying organisms in Lentikat’s Biocatalysts retain far
greater levels of activity compared to PVA biocatalysts produced at higher temperatures,
which most nitrifiers do not tolerate (Park and Chang, 2000).

Ch 2. Literature Review
10
Biocatalyst technologies have demonstrated a number of advantages when compared
with conventional suspended culture bioaugmentation technologies. They are easy to handle
and recover from solutions and provide enhanced volumetric nitrogen removal by facilitating
a high density of cells without the threat of washout and with reduced susceptibility of
nitrifiers to predation (Vackova et al., 2011; Ravnjak et al., 2013). Bouskova et al. (2011)
reported nitrogen removal efficiencies of 98% in well maintained systems (provided with
ample oxygen and organic substrate) treating concentrations of N-NH4+ and N-NO3
- as high
as 2500 mg/L and 4000 mg/L respectively. They also demonstrated that Lentikat’s
Biocatalysts retained high nitrogen removal efficiency when applied to separate wastewaters
containing high concentrations of inorganic salts (20 g/L NaCl and 2 g/L Na2SO4) and toxic
compounds (Aniline) at concentrations far greater than previously reported to cause
inhibition. These results correlate well those of Barber and Stuckey (1999) who found that
Nitrobacter immobilized in porous PVA beads demonstrated decreased susceptibility to
unionized Ammonia (NH3) concentrations previous reported to be inhibitory.
These data support the claims that encapsulation of biomass enhances adaptability to
harsh conditions and increases robustness towards fluctuating environmental parameters such
as chemical shocks, pH changes, and temperature shifts (Bouskova et al., 2011; Trogl et al.,
2011; Ranjak et al., 2013; Barber and Stuckey, 1999; Park and Chang, 2000). Consequently,
nitrifying bacteria immobilized within PVA seem to possess great potential to enhance the
performance of new and existing WWTPs as legislative restrictions on nitrogen emissions get
tighter.
2.5 Molecular Methods for Assessing Microbial Communities in WWTP
Molecular biological techniques for assessing microbiological communities in
environmental samples and WWTPs have proven effective at overcoming the biases and
shortfalls of the culture based approaches that previously dominated such analyses (Gilbride
et al., 2006). Some of the powerful advantages of molecular biological approaches include:
the ability to characterize the structure of complex mixed communities in situ as well as
identify representative populations of difficult to culture anaerobes, currently unculturable
species, and functional groups, such as nitrifiers and denitrifiers, based on unique DNA
sequences and/or genes coding for functional enzymatic machinery. These tools can therefore
provide engineers with a greater ability to optimize WWTP performance by facilitating

Ch 2. Literature Review
11
beneficial organisms while combating problematic ones on a continual basis (Gilbride et al.,
2006; Sanz and Kochling, 2007).
2.5.1 DNA Isolation
The isolation, or extraction, of pure genomic DNA from environmental or WWTP
samples is a critical initial step in all molecular analyses of microbial populations that require
subsequent polymerase chain reaction (PCR) DNA amplification (Mahmoudi et al., 2011).
The process of DNA isolation includes cell lysis and homogenization which typically
involves heating, detergents, and/or mechanical force followed by the stepwise removal of all
non DNA constituents and the eventual elution of DNA in a suitable storage buffer
(Whitehouse and Hottel, 2006; Mahmoudi et al., 2011). Isolating acceptable quantities of
high purity DNA from soil and activated sludge samples can be particularly challenging due
to presence of compounds that may inhibit downstream PCR such as humic acids and other
recalcitrant organic compounds and/or pollutants (Whitehouse and Hottel, 2006; Guobin et
al., 2008; Mahmoudi et al., 2011).
Nowadays, numerous commercial DNA extraction kits are available that enable the
processing of high volumes of samples with relatively lower cost and time consumption in
comparison with previous established methods (Whitehouse and Hottel, 2006; Dauphin et al,
2009). An additional advantage of these commercial kits is the incorporation and continuing
improvement of PCR inhibitor removal techniques (Whitehouse and Hottel, 2006). On the
other hand, previous investigations report differential suitability of kits in their applications to
environmental and WWTP derived samples and some degree of “extraction bias” when
different kits are applied to the same samples. This implies that microbial community
assessments obtained from subsequent analyses may not fully represent reality and therefore
adequate investigation is needed to determine the most suitable DNA extraction protocol for
each particular sample type (e.g. activated sludge or Lentikat Biocatalysts)(Gilbride et al.,
2006; Mahmoudi et al., 2011).
Despite the apparent limitations, DNA Isolation remains essential for the
characterization of microbial communities in soils and WWTPs using molecular techniques
including: Amplified Ribosomal DNA Restriction Analysis (ARDRA), Denaturing Gradient
Gel Electrophoresis (DGGE), Terminal-restriction Fragment Length Polymorphism (t-
RFLP), Multiplex PCR, Real Time PCR (qPCR), and Nucleic Acid Microarrays (Gilbride et
al., 2006; Sanz and Kochling, 2007).

Ch 2. Literature Review
12
2.5.2 Polymerase Chain Reaction (PCR)
PCR is a nucleic acid amplification technique developed in the 1980’s that has
become a cornerstone of many molecular analyses employed across nearly the entire
spectrum of biological sciences. PCR proceeds via the cyclical denaturation of DNA
molecules by heating, followed by cooling which triggers hybridization with target sequenced
primers (most often targeting genes coding for 16S rRNA in Prokaryotes and 18S rRNA in
Eukaryotes) that are assembled into complimentary strands, or copies, of the original
fragment by DNA polymerase (Mullis et al., 1986). During each subsequent cycle the DNA
fragments generated in all previous cycles also serve as templates for further amplification
thus resulting in the exponential increase in the concentration of target DNA molecules
(Mullis et al., 1986). While the input concentrations of nucleic acid may be extremely small,
the sensitivity of successful PCR enables the output of sufficient DNA concentrations for
comprehensive analyses including the assessment of subdominant species (Gilbride et al.,
2006; Postollec et al., 2011).
Since its inception numerous PCR techniques and PCR based analyses have been
developed and successfully employed to assess microbial communities in WWTP’s. Among
the most commonly used for nitrifying bacteria and archaea are quantitative real time PCR
(qPCR) and conventional PCR followed by denaturing gradient gel electrophoresis (PCR-
DGGE) (Sanz and Kochling, 2007; Gilbride et al., 2006). qPCR involves the generation of an
amplification curve by quantifying the fluorescence given off by amplicons at every cycle
throughout the amplification process. This provides an estimate of the initial concentration of
the target DNA molecule and thus its source organism’s abundance in the initial sample
(Postellec et al., 2011). This technique is frequently applied to assess the populations of total
bacteria, AOB, NOB, anammox, and bulking organisms in WWTPs (van den Akker et al,.
2010; Davery et al., 2013; Wang et al., 2010; Wang et al., 2012 ). A major advantage of
qPCR is that it does not require post PCR manipulation and therefore minimizes additional
contamination risk (Postellec et al., 2011). On the other hand, while its inherently high
sensitivity enables the identification of subdominant species with high accuracy, it also
dictates that extra controls over experimental design must be applied to eliminate errors
(Postellec et al., 2011). The present study undertaken in this MSc. thesis employs PCR-
DGGE which is described in more detail in the following section (2.5.3).
As mentioned in section 2.5.1, PCR is vulnerable to inhibition by a variety of
materials and chemical constituents associated with WWTP and environmentally derived

Ch 2. Literature Review
13
samples. Besides insufficient initial DNA concentration, inhibition is the greatest cause of
PCR failure (Alaeddini, 2012). Substances reported to cause PCR inhibition include: humic
compounds, polysaccharides in feces, heme in blood samples, proteinases and phenols used
in DNA extraction, heavy metals, certain divalent ions, nanoparticles, excessive DNA, urea,
certain microfluidic chips, and many others (Alaeddini, 2012; Kodzius et al., 2011). While
the precise mechanisms of PCR inhibition are not yet well understood, several methods for
overcoming inhibition are routinely used starting with selecting the most appropriate DNA
extraction protocol. As noted by Whitehouse and Hottel (2006), many commercial DNA
extraction kits are designed to work with particular sample types (e.g. soil or stool) by
incorporating washing buffers that target commonly associated inhibitors for removal.
Additional, techniques used to overcome PCR inhibition include: DNA dilution, addition of
amplification facilitators like bovine serum albumen (BSA), the addition of extra polymerase
enzymes, or DNA purification (Alaeddini, 2012).
2.5.3 Denaturing Gradient Gel Electrophoresis (DGGE)
DGGE is a genetic finger printing technique for determining the dominant members
of microbial communities in environmental samples and WWTP sludges with high precision
(Sanz and Kochling, 2007; Hesham et al., 2011). The principle of this technique is that PCR
amplified DNA fragments of the same length are separated in a polyacrylamide gel
containing a gradient of DNA denaturants. The basis of this denaturation is the differing
sequence of base pairs in each fragment giving rise to unique melting domains. This refers to
a section of the DNA fragment with identical melting temperature. Once one of these sections
reaches its melting temperature the helical DNA structure breaks down creating drag that
severely restricts continued migration through the gel (Muyzer and Smalla, 1998). The end
result is a series of bands in the gel, each representing an accumulation of DNA fragments
with an identical sequence of base pairs thus representing an individual “species”. DGGE
profiles can be used to make simultaneous comparisons of samples (e.g. in time series
investigation) and/or bands can be cut from the gel and the DNA therein can be sequenced to
determine the species represented (Muyzer and Smalla, 1998).
The applications of PCR-DGGE in the analyses of WWTP communities have been
rapidly increasing over the past decade. Successful applications include: characterizing the
communities carrying out essential functions such as anaerobic sludge digestion (Kim et al.,
2009) and autothermal aerobic sludge digestion (Hayes et al., 2011); analyzing the effects of

Ch 2. Literature Review
14
changing operational parameters such as temperature (Niu et al,. 2012) and aeration regimes
(Tocchi et al., 2012.) in aerobic activated sludge reactors; comparing raw sewage and aerobic
sludges from different WWTPs to assess performance differences (Liu et al., 2006); and
comparing compartments and treatment trains within a single WWTP to assess bulking
problems (Hesham et al., 2011).
Particularly relevant to this study, DGGE has also long been applied to detect
differences in DNA extraction protocols (e.g. differential cell lysis techniques) and PCR
biases (Muyzer and Smalla, 1998; Sanz and Kochling, 2007). Quigley et al. (2012) and
Mahmoudi et al. (2011) used DGGE profiles to compare DNA isolated using different
commercial DNA extraction kits on raw milk and soil samples respectively in terms of yield,
purity, and the presence of PCR inhibitors. Both studies found that significant differences
occurred between kits in terms of purity, yield (total and variability), and species composition
in the case of soil. The aim of this current study is to use DGGE as a tool to help establish an
internal DNA extraction protocol for future characterization and long term evaluation of
communities encapsulated within and fixed upon Lentikat Biocatalysts® and activated sludge
flocs.
2.5.4 Fluorescence in Situ Hybridization (FISH)
FISH is a cultivation independent technique employed to assess the phylogenetic and
spatial composition of microbial communities derived from environmental samples and
WWTP sludges. The principle behind this method is that samples are immediately fixed, then
the cells are permeabilized, nucleic acids are hybridized with fluorescently labeled
oligonucleotide probes, thoroughly washed and examined using flow cytometry or
epifluorescent or confocal laser scanning microscopy (CLSM) (Amman et al., 1995; Gilbride
et al., 2006). FISH is extremely useful in WWTP studies in that it enables the characterization
of the structure and quantity of morphologically in-tact microorganisms present in complex
communities down to the single cell level (Amman et al., 1995; Nielsen et al., 2009). Some
additional highly publicized advantages of FISH over other molecular techniques are: the
speed with which it can be carried out; the simplicity of microscopic analyses; the specificity
of RNA, DNA, and more recently PNA (peptide nucleic acid) probes to target whole domains
(e.g. Bacteria, Archaea, or Eukarya) on down to single species and sub-species; and its
relative immunity to inhibition or extraction and amplification biases that can affect PCR
based analyses (Amman et al., 1995; Nielsen et al., 2009; Okten et al., 2012; Machado et al.,

Ch 2. Literature Review
15
2013). Problems commonly encountered while using FISH include: autofluorescence of cells
and surrounding materials, nonspecific binding of fluorescent labeled oligonucleotides, and
low signal intensity (Amman et al., 1995; Okten et al., 2012).
While a majority of the primers used in PCR target DNA sequences coding for 16S
ribosomal RNA (rRNA) in prokaryotes and 18S rRNA for eukaryotes, the oligonucleotide
probes used in FISH target the rRNA itself (Gilg et al., 2010; Machado et al., 2013). The
highly conserved nature of these sequences makes them well suited for defining operational
taxonomic units (OTUs), or the microbiological equivalent of “species”, and is the primary
reason for their targeting by FISH probes. Another reason for targeting rRNA is that it is
present in cells in far greater quantities than DNA and thus gives off a more pronounced
signal (Nielsen et al., 2009). The establishment of extensive and periodically scrutinized
databases, such as the University of Vienna’s “probeBase” (Loy et al., 2003), has provided a
foundation for the development of a wide variety of probes now used in FISH (Lucker et al.,
2007; Nielsen et al., 2009). Refinement of probes is also continually occurring as researchers
identify regions of rRNA with highly limited access to oligonucleotide diffusion (Okten et
al., 2012) and PNA probes demonstrate greater specificity and thermal stability than some
RNA and DNA alternatives (Machado et al., 2013).
Fluorescent microscopy employed in FISH is based on the principle of exposing
hybridized specimens to short wavelength visible light to excite the fluorescent dyes,
fluorophores, which in turn leads to them emitting longer wave light which can be detected
(Seviour and Nielsen, 2010). The most common fluorophores used in FISH include:
fluorescein (FITC), tetramethylrhodamine, and indocarbocyanines (CY3, CY5, and Cy7)
(Amman et al., 1995). Recent advances in FISH image analysis are due in large part to the
use of CLSM and associated computer software that combine the ability to take three
dimensional images with automated and/or semi-automated quantification of hybridized cells
(Seviour and Nielsen, 2010).
The application of FISH in investigations of WWTP microbial communities is
extremely widespread. A majority of these studies focus on characterizing populations of
functional groups such as nitrifiers, denitrifiers, polyphosphate accumulators, glycogen
accumulators, as well problematic filamentous organisms (Nielsen et al., 2009). While
traditional FISH is still most frequently used as a standalone molecular method for analysis,
newer variations that include catalyzed reporter deposition (CARD-FISH) and

Ch 2. Literature Review
16
Microautoradiography (MAR-FISH) are becoming increasing popular (Seviour and Nielsen,
2010).
The use of FISH to assess nitrifying bacteria in WWTPs has been an extremely
common practice since the late 1990’s (Gilbride et al., 2006). More recently, FISH has
emerged the method of choice for assessing AOB vs. NOB present in partial nitrification
reactors as demonstrated by Cho et al. (2010), Zhang et al. (2012), Kong et al. (2013), Okabe
et al. (2011), and Gu et al. (2012) just to name a few.
Perhaps the most interesting trend, evident in a number of the partial nitrification
investigations mentioned above, is the evolving tendency to employ FISH in combination
with other molecular tools. FISH is increasingly being combined with scanning electron
microscopy (SEM), qPCR, and PCR-DGGE in order to provide comprehensive analyses of
microbial communities that combine the advantages of each, while attempting to mitigate and
identify their shortcomings. This trend is not confined to partial nitrification but is becoming
prevalent throughout microbial ecology with examples including: Yasin et al. (2012), who
used a combination of FISH and PCR-DGGE to characterize the hydrogen producing
communities in food waste fermenters; Cardinali-Rezende et al (2012), who combined FISH,
CARD-FISH, qPCR, and DGGE to track the evolution of the microbial community in a full
scale municipal solid waste (MSW) anaerobic digester from start up to steady state;
Fernandes et al. (2013) who used FISH to track phosphorus accumulators, nitrifiers and
sulphate reducers while using DGGE to track overall community structure in a full scale
sequencing batch reactor treating domestic wastewater for 180 days; Hayes et al. (2011) who
combined FISH, FISH-MAR, and PCR-DGGE to assess the microbial ecology of an
autothermal thermophilic aerobic sludge digester (ATAD); and Ferrero et al. (2010) who
combined PCR-DGGE and FISH to characterize the microbial populations in high altitude
soils.
Interestingly, Cardinali-Rezende et al. (2012) noted that for a majority of their
overlapping analyses, qPCR yielded lower cell enumerations than FISH by 1-4 orders of
magnitude. While this trend was not universal, they attributed the difference to potential
losses of DNA during extraction and purification. In a more complimentary combination,
Kong et al. (2013) used FISH to determine that Nitrosomonas was the dominant genus of
AOB in his lab scale partial nitrification reactor and PCR-DGGE on genes coding for
variations of Amo to determine the evolution of dominant Nitrosomonas species over time. If
nothing else, the complimentary and sometimes conflicting results presented in these articles

Ch 2. Literature Review
17
indicate that combining FISH with additional molecular tools for microbial ecology
investigations is becoming mainstream.

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
18
3.1 Objectives
The objectives of this investigation are to compare the effectiveness of four commercial DNA
isolation kits at isolating bacterial DNA from PVA biocarriers (Lentikat’s Biocatalysts) for
use in downstream PCR based applications. The primary criteria for comparison are DNA
yield, purity, waste generation, processing time, cost per sample, successful PCR
amplification, and phylogenetic richness in extracts.

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
19
3.2 Materials and Methods
3.2.1 Poly Vinyl Alcohol Pellets and Activated Sludge Samples
A total of 6 unique samples were analyzed in this project, 4 of Lentikat’s Biocatalysts
and 2 of aerobic activated sludge. Paracoccus cultures for immobilization in PVA
biocatalysts were obtained from the collection of Deutsche Sammlung von Microorganismen
and Zellkulturen GmbH while nitrifiers were enriched from activated sludge and were
cultivated at the Slovak University of Technology in Bratislava. The specific PVA employed
for immobilization was Mowiol® 28-99 (Kuraray America, Inc., Houston, USA) with a 99%
degree of saponification (hydrolysis) and relative molecular mass of 145,000 g/mol. The
preparation of lens-shaped biocatalysts (diameter, 3 - 4 mm; thickness 200–300 μm) was
described in section 2.4 and was carried out by LentiKat´s Incorporated (Prague, Czech
Republic).
Two of the biocatalyst samples, N1 and N2, were nitrification biocarriers containing
immobilized Nitrosomanas europaea and Nitrobacter winogradskyi. Sample N1 was
obtained from LentiKat’s Inc. immediately after immobilization and prior to the subsequent 6
week in lab cultivation period that all commercially distributed biocatalysts undergo. Sample
N2 was obtained following 4 months of implementation treating effluent from a municipal
root zone WWTP in Kotenčice, Czech Republic. Both samples, N1 and N2 were collected
and transported to ICT Prague on February 15, 2013
The other 2 biocatalyst samples, D1 and D2, were both denitrification biocarriers
containing immobilized Parococcus denitrificans (strain DSM 1403). Sample D1 was
obtained following 38 months of implementation performing post denitrification on effluents
from mixed sewage and industrial waste water at a private pharmaceutical production plant.
Sample D2 was obtained following 9 months of implementation performing post
denitrification of chemically treated underground water from uranium mining (DIAMO
Corp). Organic substrate augmentation at both of these of these facilities was carried out with
the controlled dosing of Brenntaplus vp1 (Brenntag N.V., Deerlijk, Belgium) carbon rich
nutrient blend. Both samples, D1 and D2, were collected and transported to ICT Prague on
February 15, 2013.

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
20
The two activated sludge samples, L1 and P1, were collected from aeration basins in
conventional suspended growth activated sludge WWTPs. Sample L1 was obtained on March
20, 2013 from the Lochovice, CZ WWTP operated by Envi-Pure Inc. (Prague, Czech
Republic). This plant treats primarily Industrial WW. Sample P1 was obtained on March 20,
2013 from the Plzeň, CZ municipal WWTP operated by Veolia Water (Paris, France). This
plant treats primarily MSW and industrial WW primarily from Beer Breweries.
Once in the laboratory, all biocatalyst samples were transferred to 50 mL Falcon
tubes, residual water was removed by pipetting, and tubes were stored at -20oC until DNA
extraction. Activated sludge samples were stored briefly in 3L aerated carboys. Prior to DNA
extraction samples were thoroughly homogenized, small amounts were then transferred to
sterile 2 mL microcentrifuge tubes, centrifuged for 1 min after which residual water was
removed by pipetting and the thickened product was used for isolation.
3.2.2 Activated Sludge Characterization
The TSS and VSS of activated sludge samples were determined following protocols
adapted from 2540 D in Standard Methods for the Examination of Water and Wastewater.
22nd
Edition (2012):
TSS
1. Filter 10 mL of thoroughly homogenized sludge under vacuum onto a pre-
weighed Pragapor #6 membrane filter (50 mm diameter, 0.4 µm thick)
(Pragochema spol. s.r.o, Prague, CZ)
2. Place filter in over to dry at 105oC for 2 hours and weigh
3. Calculate TSS with the following formula:
TSS =
TSS total suspended solids [g/L];
SF weight of filter and sludge after drying [mg];
F weight of filter [mg];
V homogenized sample volume used for analysis [mL].

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
21
VSS
1. Following determination of TSS place the filter in a pre-weighed porcelain dish
2. Add 2-3 drops of glycerin and place in an electric oven at 550oC for 2 hours, dry
in the desiccator and weigh.
3. Calculate VSS with the following formula:
VSS =
VSS … volatile suspended solids [g/L];
SFD weight of dish, filter, and residue after drying at 105 °C but prior to burning
[mg]
SBD weight of dish, filter, and residue after burning [mg];
F weight of the filter [mg];
V homogenized sample volume used for analysis [mL].
3.2.3 DNA Isolation
3.2.3.1 Comparison of Commercial DNA Isolation Kits
Bacterial DNA was isolated from all 6 samples in triplicate, using four different
commercial DNA extraction kits selected to include several nucleic acid extraction
methodologies. The manufacturer´s protocols for gram-negative bacteria were followed for
each kit and can found online (as of 3/6/2013) at the links provided in Appendix 1. Slight
modifications to some protocols were made based upon manufacturer recommended
troubleshooting following a test run to familiarize ourselves with each procedure. The
average processing time of a 9-sample run was determined for each kit beginning with the
addition of the first reagent and ending with the final elution of DNA and calculated based
upon two runs. In addition, the cost per sample was determined based upon the ratio of kit
price/preps and the mass of waste generated in each 9 sample run was recorded and averaged.
The cost of pipette tips, eppendorf tubes, standard laboratory equipment, and inexpensive
(< €200) kit specific equipment (Chemagic: Magnetic stand; PowerSoil®: Vortex adapter
tube holder) were excluded from these cost analyses. All extracted DNA was eluted in the
buffers provided by the manufacturers and subsequently stored at -20 °C until downstream
use. The kits evaluated and methodologies employed include:

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
22
The QIAamp® DNA Stool kit (50 preps) (QIAGEN Inc., Valencia, CA, USA)
Principle: Lyse bacterial cells with enzymes and heating (70oC); bind Impurities
to inhibit-ex tablets (a fluidized adsorption media); bind DNA to membrane and
wash; elute DNA in buffer.
Protocol Modifications: Reduced elution buffer from 200 μL to 100 μL
Justification: Preliminary results yielded very low concentrations of DNA in
eluate and thus unreliable Nanodrop™ results.
The PowerSoil® DNA Isolation kit (50 preps) (MoBio Laboratories Inc., Carlsbad, CA,
USA) Bead Beating and Membrane.
Principle: Lyse bacterial cells with combined mechanical and enzymatic force;
precipitate initial impurities; bind DNA to membrane and wash; elute DNA in
buffer.
Protocol Modifications: A) Increased step 5 (mechanical lysis) vortexing time
from 10 to 15 minutes, B) Reduced elution buffer from 100 μL to 50 μL
Justification: A) Optional manufacturer recommendation, B) Preliminary results
yielded very low concentrations of DNA in eluate and thus unreliable Nanodrop™
results.
The Chemagic DNA Bacteria Kit (100 preps) (PerkinElmer chemagen Technologe
GmbH, Baesweiler, Germany)
Principle: Lyse bacterial cells with enzymes and mild heating (37oC); bind DNA
to magnetic beads; washout proteins, RNA, Lipids, etc.; elute DNA in buffer.
Protocol Modifications: Used 4 μL of RNAse A (5 mg/ml) in lysis (Step 1)
Justification: Optional Manufacturer Recommendation
The MasterPure™ DNA Purification kit (200 preps) (Epicentre Biotechnologies,
Madison, WI, USA)
Principle: Lyse cells with enzymes and heating (65oC); cool and precipitate
impurities; precipitate DNA with alcohol; rinse with alcohol and elute DNA in
buffer.
Protocol Modifications: None
3.2.3.2 Liquid Nitrogen (LN) Enhanced Cell Lysis
Prior to this study, attempts to extract bacterial DNA from Lentikat’s biocatalysts
using the UltraClean® Microbial DNA Isolation Kit (MoBio laboratories, Carlsbad, CA
USA) consistently yielded very low concentrations of DNA (>1µg DNA/g sample). For this
reason, during preliminary tests to familiarize ourselves with the different DNA Isolation
protocols we performed liquid nitrogen (LN) enhanced cell lysis to determine if it would

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
23
increase DNA yields. As these tests were considered preliminary they were only carried out
in duplicate and only using sample D2. At the time, the resulting difference in yields between
LN treated biocatalysts and untreated biocatalysts were not considered significant and the
treatment was abandoned. Upon revisiting the data months later it became apparent that LN
treatment seemed to deliver higher yields and comparable purity across the board. Although
there is not enough data to draw definitive conclusions we have chosen to present it. Also,
LN treated samples extracted with the Powersoil® and QIAmp® kits were subjected to PCR-
DGGE for further phylogenetic comparison.
We developed the following protocol for LN treatment of polyvinyl alcohol
biocarriers:
Wear thick thermally protective gloves and full face masks!
1. Transfer approximately 10 g of fresh biocatalyst pellets to a 50 mL falcon tube and
remove as much water as possible using a 1 mL pipette
2. Transfer 2-3 g of dewatered pellets to a sterile smooth ceramic mortar
3. Pour enough liquid nitrogen into the mortar to cover to pellets, once it has evaporated
completely, repeat this process. After multiple repetitions (4-6) the rate of evaporation
will decrease significantly.
4. Once all liquid nitrogen has evaporated thoroughly grind the pellets with the pestle.
Note: They should be extremely brittle at this point, if they remain rubbery and resist
grinding into a wet powder repeat step 3.
5. Transfer ground up pellets to a clean 50 mL falcon tube and process immediately or
store at -20oC
3.2.4 DNA Yield and Purity
Isolated DNA was evaluated using a NanoDrop™ 1000 Spectrophotometer with the
software package ND-1000 Version 3.8 (Thermo Fisher Scientific Inc., Waltham, MA,
USA). The operating protocol was as follows:
1. Thaw frozen samples (if necessary), vortex for 3-5 seconds, and spin briefly to
remove drops from the tube walls and lid.
*Note: To calibrate the instrument follow steps 2-5 using only fresh elution buffer
corresponding to that in each sample and by selecting “Blank” instead of “Measure”.
Recalibration was performed after every 15-20 measurements.
2. Load 1.5-2 µL of sample onto the measurement pedestal

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
24
3. Close the sampling arm and initiate the measurement by selecting “Measure” on the
PC operating software
4. Upon completion of the measurement open the sample arm and clean the upper and
lower sampling pedestals with a clean lab wipe
5. If errors occur clean the upper and lower pedestals with a lab-wipe lightly moistened
with distilled water and repeat measurement.
This instrument measures isolated DNA concentration based upon the measured
absorbance at 260 nm (A260) using Beer’s Law, and has lower and upper detection limits for
dsDNA of 2 ng/μL and 3,700 ng/μL respectively. To estimate the purity of extracted nucleic
acid, the absorbencies at 280 nm (A280) and 230 nm (A230) were measured and the ratios of
averages between the A260 nm and A280 nm (A260/A280) and the A260 nm and A230 nm
(A260/A230) were calculated for each measurement. Samples with A260/A280 ratios
between 1.8 and 2.0 were presumed to be free of contamination (Dauphin et al., 2009).
Samples with mean A260/280 ratios below 1.8 were presumed to contain protein or other
contaminants, whereas samples with ratios above 2.0 were presumed to be contain RNA
contamination (Thermo Fisher Scientific, 2010; Mahmoudi et al., 2011; UMTK 322).
A260/A230 ratios are a supplementary measure of purity where values between 2-2.2 are
considered pure. Lower A260/A230 values often attributed to the presence of humic
compounds and/or residual phenol or other extraction reagents while higher values are often
attributed to errors in blanking the instrument (Mahmoudi et al., 2011; Thermo Fisher
Scientific, 2010).
Each sample of isolated DNA (6 samples>4 kits>Triplicate of each) was measured a
minimum of 5 times and the values for total DNA concentration, A260/A280, and
A260/A230 were statistically analyzed in order to characterize the products of each isolation.
3.2.5 PCR Amplification
The DNA extracts were used as a templates for Touchdown PCR amplification of the
variable V3 region of 16S rDNA (product 566 bp long + 40 bp GC clamp). The nucleotide
sequences of eubacterial-specific universal primers were as follows: 341F (5´-
CTACGGGAGGCAGCAG-3´) and 907R (5´-CCGTCAATTCMTTTGAGTTT-3´), (Schäfer
and Muyzer, 2001) to facilitate subsequent DGGE, we employed a forward primer, which
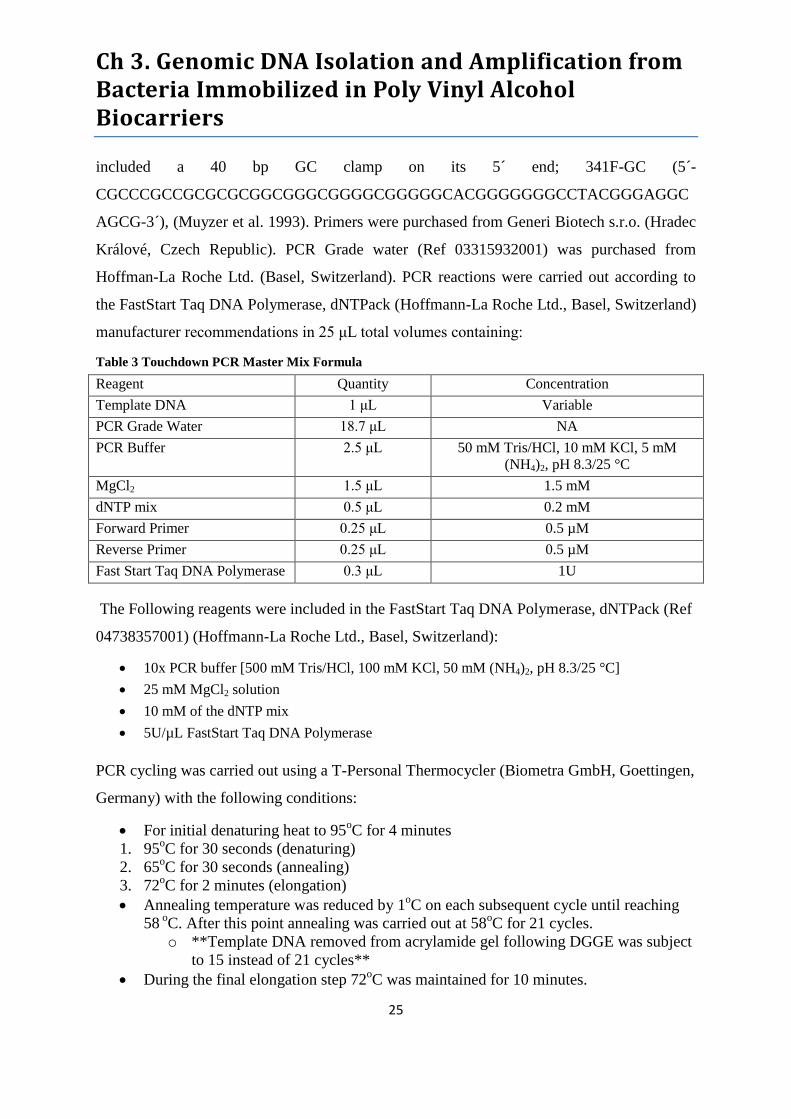
Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
25
included a 40 bp GC clamp on its 5´ end; 341F-GC (5´-
CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGGGCCTACGGGAGGC
AGCG-3´), (Muyzer et al. 1993). Primers were purchased from Generi Biotech s.r.o. (Hradec
Králové, Czech Republic). PCR Grade water (Ref 03315932001) was purchased from
Hoffman-La Roche Ltd. (Basel, Switzerland). PCR reactions were carried out according to
the FastStart Taq DNA Polymerase, dNTPack (Hoffmann-La Roche Ltd., Basel, Switzerland)
manufacturer recommendations in 25 μL total volumes containing:
Table 3 Touchdown PCR Master Mix Formula
Reagent Quantity Concentration
Template DNA 1 μL Variable
PCR Grade Water 18.7 μL NA
PCR Buffer 2.5 μL 50 mM Tris/HCl, 10 mM KCl, 5 mM
(NH4)2, pH 8.3/25 °C
MgCl2 1.5 μL 1.5 mM
dNTP mix 0.5 μL 0.2 mM
Forward Primer 0.25 μL 0.5 µM
Reverse Primer 0.25 μL 0.5 µM
Fast Start Taq DNA Polymerase 0.3 μL 1U
The Following reagents were included in the FastStart Taq DNA Polymerase, dNTPack (Ref
04738357001) (Hoffmann-La Roche Ltd., Basel, Switzerland):
10x PCR buffer [500 mM Tris/HCl, 100 mM KCl, 50 mM (NH4)2, pH 8.3/25 °C]
25 mM MgCl2 solution
10 mM of the dNTP mix
5U/µL FastStart Taq DNA Polymerase
PCR cycling was carried out using a T-Personal Thermocycler (Biometra GmbH, Goettingen,
Germany) with the following conditions:
For initial denaturing heat to 95oC for 4 minutes
1. 95oC for 30 seconds (denaturing)
2. 65oC for 30 seconds (annealing)
3. 72oC for 2 minutes (elongation)
Annealing temperature was reduced by 1oC on each subsequent cycle until reaching
58 oC. After this point annealing was carried out at 58
oC for 21 cycles.
o **Template DNA removed from acrylamide gel following DGGE was subject
to 15 instead of 21 cycles**
During the final elongation step 72oC was maintained for 10 minutes.

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
26
PCR products were run on 2% agarose gel (m/v) stained with of Nancy-520 DNA Gel Stain
(Sigma-Aldrich Corporation, St. Louis, MO, USA) to evaluate successful amplification.
Results were visualized with the Major Science documentation system (Major Science,
Saratoga, CA, USA) provided with Canon G11 camera (Canon Inc., Tokyo, Japan). Samples
that failed to amplify were diluted 20x and 50x with PCR grade water and re-amplified.
3.2.6 DGGE
Bacterial PCR amplicons were run through a 6% polyacrylamide gel with a
denaturing gradient of 20% - 70% ureumformamide (80% denaturant solution contained
5.6 M urea and 32% (v/v) formamide). Visual quantification of PCR products was carried out
by comparing bands in the 2% agarose gel with the GeneRuler™ 100 bp Plus DNA Ladder
(Thermo Fisher Scientific Inc., Waltham, MA, USA). Between 10-13 μL of each sample
were mixed with 2 μL of 6x loading dye (Thermo Fisher Scientific Inc., Waltham, MA, USA)
and loaded into wells in the stacking gel. Electrophoresis was performed in a 17 L bath of 1x
Tris-acetate-EDTA buffer at 200 V and 60°C for 5 h using an INGENYphorU-2x2 DGGE
apparatus (Ingeny, Goes, The Netherlands).
The step by step manufacturer’s protocol for the INGENYphorU-2x2 DGGE
apparatus can be found online (as of 3/6/2013) at the link provided in Appendix 1. This
protocol was followed closely with only the following modifications:
Step 3.2: We applied a small amount of High-Vacuum Grease (Dow Corning®,
Midland, MI USA) to the U-shaped spacer in the glass plate sandwich to create a
better seal and prevent the gradient gel from leaking prior to polymerization.
Step 5.9: Electrophoresis was run for 5 hrs. at 200 V
Step 7.2: Stock solutions were prepared in 50 mL batches and were adapted from 9%
acrylamide in the manufacturer’s protocol to 6% acrylamide for our purposes.
The ureumformamide/acrylamide stock solutions and reagent mixtures employed in gel
casting (section 4 of the manufacturer’s protocol) are listed in Tables 3 and 4 below.
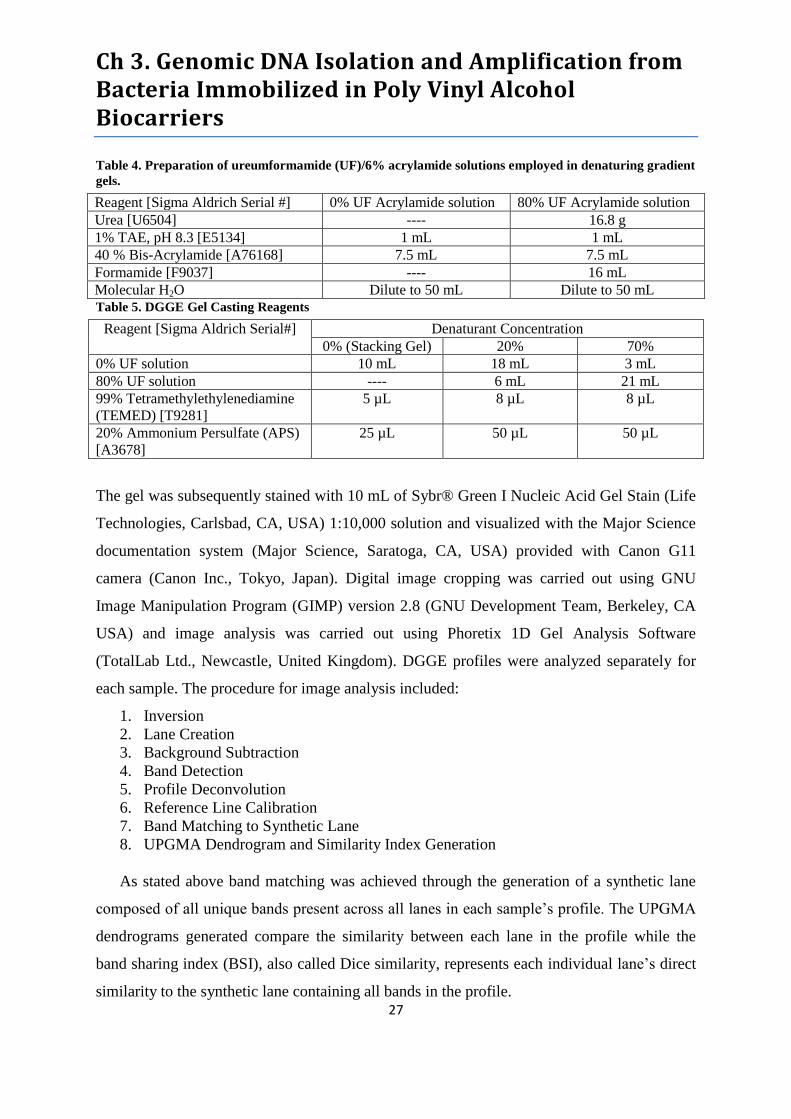
Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
27
Table 4. Preparation of ureumformamide (UF)/6% acrylamide solutions employed in denaturing gradient
gels.
Reagent [Sigma Aldrich Serial #] 0% UF Acrylamide solution 80% UF Acrylamide solution
Urea [U6504] ---- 16.8 g
1% TAE, pH 8.3 [E5134] 1 mL 1 mL
40 % Bis-Acrylamide [A76168] 7.5 mL 7.5 mL
Formamide [F9037] ---- 16 mL
Molecular H2O Dilute to 50 mL Dilute to 50 mL
Table 5. DGGE Gel Casting Reagents
Reagent [Sigma Aldrich Serial#] Denaturant Concentration
0% (Stacking Gel) 20% 70%
0% UF solution 10 mL 18 mL 3 mL
80% UF solution ---- 6 mL 21 mL
99% Tetramethylethylenediamine
(TEMED) [T9281]
5 µL 8 µL 8 µL
20% Ammonium Persulfate (APS)
[A3678]
25 µL 50 µL 50 µL
The gel was subsequently stained with 10 mL of Sybr® Green I Nucleic Acid Gel Stain (Life
Technologies, Carlsbad, CA, USA) 1:10,000 solution and visualized with the Major Science
documentation system (Major Science, Saratoga, CA, USA) provided with Canon G11
camera (Canon Inc., Tokyo, Japan). Digital image cropping was carried out using GNU
Image Manipulation Program (GIMP) version 2.8 (GNU Development Team, Berkeley, CA
USA) and image analysis was carried out using Phoretix 1D Gel Analysis Software
(TotalLab Ltd., Newcastle, United Kingdom). DGGE profiles were analyzed separately for
each sample. The procedure for image analysis included:
1. Inversion
2. Lane Creation
3. Background Subtraction
4. Band Detection
5. Profile Deconvolution
6. Reference Line Calibration
7. Band Matching to Synthetic Lane
8. UPGMA Dendrogram and Similarity Index Generation
As stated above band matching was achieved through the generation of a synthetic lane
composed of all unique bands present across all lanes in each sample’s profile. The UPGMA
dendrograms generated compare the similarity between each lane in the profile while the
band sharing index (BSI), also called Dice similarity, represents each individual lane’s direct
similarity to the synthetic lane containing all bands in the profile.
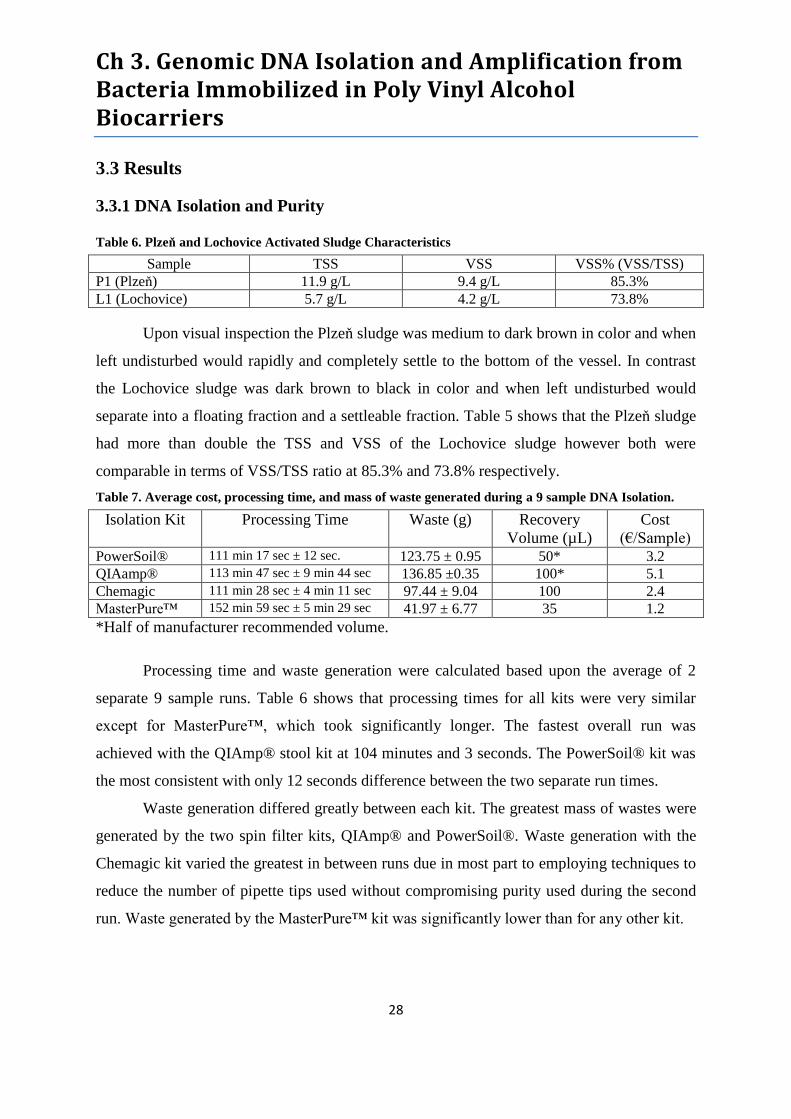
Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
28
3.3 Results
3.3.1 DNA Isolation and Purity
Table 6. Plzeň and Lochovice Activated Sludge Characteristics
Sample TSS VSS VSS% (VSS/TSS)
P1 (Plzeň) 11.9 g/L 9.4 g/L 85.3%
L1 (Lochovice) 5.7 g/L 4.2 g/L 73.8%
Upon visual inspection the Plzeň sludge was medium to dark brown in color and when
left undisturbed would rapidly and completely settle to the bottom of the vessel. In contrast
the Lochovice sludge was dark brown to black in color and when left undisturbed would
separate into a floating fraction and a settleable fraction. Table 5 shows that the Plzeň sludge
had more than double the TSS and VSS of the Lochovice sludge however both were
comparable in terms of VSS/TSS ratio at 85.3% and 73.8% respectively.
Table 7. Average cost, processing time, and mass of waste generated during a 9 sample DNA Isolation.
Isolation Kit Processing Time Waste (g) Recovery
Volume (µL)
Cost
(€/Sample) PowerSoil® 111 min 17 sec ± 12 sec. 123.75 ± 0.95 50* 3.2
QIAamp® 113 min 47 sec ± 9 min 44 sec 136.85 ±0.35 100* 5.1
Chemagic 111 min 28 sec ± 4 min 11 sec 97.44 ± 9.04 100 2.4
MasterPure™ 152 min 59 sec ± 5 min 29 sec 41.97 ± 6.77 35 1.2
*Half of manufacturer recommended volume.
Processing time and waste generation were calculated based upon the average of 2
separate 9 sample runs. Table 6 shows that processing times for all kits were very similar
except for MasterPure™, which took significantly longer. The fastest overall run was
achieved with the QIAmp® stool kit at 104 minutes and 3 seconds. The PowerSoil® kit was
the most consistent with only 12 seconds difference between the two separate run times.
Waste generation differed greatly between each kit. The greatest mass of wastes were
generated by the two spin filter kits, QIAmp® and PowerSoil®. Waste generation with the
Chemagic kit varied the greatest in between runs due in most part to employing techniques to
reduce the number of pipette tips used without compromising purity used during the second
run. Waste generated by the MasterPure™ kit was significantly lower than for any other kit.
Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
29
Table 8. DNA yields (µg/g), absorbance 260 nm/280nm, and absorbance 260nm/230nm. [
]
PowerSoil® QIAamp®
Yield
µg/g
A260/A280 A260/A230 Yield
µg/g
A260/A280 A260/A230
N1 1.79
1.60
1.86
1.95
1.84
2.00
1.51
0.84
1.58
1.29
1.23
1.61
1.73
1.65
2.12
0.93
0.83
1.06
N2 6.23
3.79
6.56
1.91
1.89
1.95
1.86
1.53
2.04
9.95
7.84
10.57
1.99
1.95
2.05
1.89
1.14
1.95
D1 3.93
2.70
4.59
1.89
1.82
1.96
1.47
1.37
1.69
3.97
3.39
4.45
1.73
1.72
1.81
1.38
1.10
1.44
D2 7.37
6.52
8.53
1.93
1.90
1.96
1.82
1.78
1.87
6.87
6.56
7.26
1.91
1.84
1.95
1.59
1.50
1.73
D2 LN*
10.47
9.87
11.20
1.98
1.96
2.02
1.65
1.59
1.70
11.84
11.19
12.02
2.03
1.98
2.04
1.30
1.22
1.30
L1 24.17
19.54
26.22
1.93
1.92
1.95
1.99
1.97
2.04
15.23
14.05
15.72
1.91
1.85
1.95
1.25
0.80
1.30
P1 17.04
13.32
17.37
1.91
1.90
1.92
1.97
1.76
1.99
30.66
30.35
38.96
2.14
2.13
2.16
2.08
1.76
2.12
Chemagic MasterPure™ Yield
µg/g
A260/A280 A260/A230 Yield
µg/g
A260/A280 A260/A230
N1 67.69
58.17
173.53
1.68
1.39
1.72
1.11
0.90
1.34
35.45
28.82
95.83
1.85
1.81
2.20
1.23
1.16
1.92
N2 160.88
156.02
165.89
1.69
1.67
1.71
1.11
0.95
1.34
298.83
138.05
311.46
2.02
1.96
2.05
1.95
1.90
1.97
D1 299.53
283.46
353.88
1.80
1.77
1.82
1.08
1.03
1.10
236.98
203.72
288.90
1.93
1.92
1.95
1.64
1.60
1.71
D2 173.40
113.86
182.97
1.42
1.42
1.44
0.74
0.73
0.80
334.51
308.32
351.17
1.83
1.81
1.84
1.44
1.40
1.47
D2
LN* 276.73
269.27
290.58
1.59
1.58
1.59
0.75
0.73
0.76
385.02
279.55
436.48
1.79
1.76
1.85
1.36
1.33
1.45
L1 179.52
154.24
563.28
1.64
1.53
1.66
1.26
1.24
1.27
512.95
404.09
527.56
1.76
1.70
1.76
1.41
1.28
1.45
P1 221.70
170.19
230.56
1.64
1.63
1.67
1.21
1.18
1.23
557.83
493.85
623.77
1.85
1.82
1.86
1.57
1.53
1.61
* Values for liquid nitrogen treated samples represent duplicate rather than triplicate samples.

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
30
DNA yields varied significantly between sample origin, extraction kits, and even in
some case triplicate isolations. Due to variability in yield and purity ratios obtained between
triplicate isolations the assumption of normal distribution could not always be made and thus
median and interquartile range were determined to best describe the data in Table 7. A
common trend was that 2/3 triplicate isolations would return very similar results while the
third would differ greatly and cause either positive of negative skew. The extent and direction
of such skews can be observed by examining the Q1 and Q3 values provided in Table 7.
DNA yield varied significantly between both isolation kits and sample origin however
many trends in the data were clearly evident and are summarized below. Note that LN
treatment treated biocarriers are considered separately from all other analyses:
Liquid Nitrogen Treatment (D2-LN vs. D2)
o Sample D2-LN Yielded higher median amounts of DNA for all isolation kits
and Q1 values greater than the Q3 values of D2 for all kits except
MasterPure™
Sample N1 yielded the lowest amount of DNA for each kit across the board
Sample D1 yielded the next lowest amount of DNA for all kits except Chemagic for
which it was the highest.
Sample L1 yielded the highest amounts of DNA for the PowerSoil® kit and the
second highest amounts for both the QIAmp® stool kit and the MasterPure™ kit
Sample P1 yielded the highest amount of DNA for the QIAmp® stool kit and the
MasterPure™ kit and the second highest for the PowerSoil® kit
Samples N2 and D2 were in the middle of the pack for median DNA yield although
sample N2 yielded lower amounts of DNA from each kit except the QIAmp® stool
kit
The QIAmp® stool kit yielded median DNA concentrations of 15.23 – 30.66 µg/g for
activated sludge samples and below 9.95 µg/g for all biocarrier samples
The PowerSoil® kit yielded median DNA concentrations of 17.04 - 24.17 µg/g for
activated sludge samples and below 7.37 µg/g for all biocarrier samples

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
31
The Chemagic kit yielded median DNA concentrations of 179.52-221.70 µg/g for
activated sludge samples and 67.69 - 299.53 µg/g for all biocarrier samples with only
1 median yield below 160.88 µg/g
The MasterPure™ kit yielded median DNA concentrations of 512.95 - 557.83 µg/g
for activated sludge samples and 35.45 - 334.51 µg/g for all biocarrier samples with
only 1 median yield below 236.98 µg/g
DNA purity was assessed based upon two ratios, the absorbance at 260nm to the
absorbance at 280 nm (A260/A280) and the absorbance at 260 nm to the absorbance at
230 nm (A260/A230). A260/A280 values between 1.8–2.0 (considered pure) were achieved
by all kits for at least 1 sample. Median A260/A230 values of 2.0-2.2 (considered pure) were
achieved only in one instance thus this measure of purity was assessed based upon proximity
to this range. Data presented in Table 7 imply that purity is more highly correlated with
particular isolation kits rather than with sample origin. Purity results are summarized below:
Liquid Nitrogen Treatment D2-LN vs. D2
o The median values of A260/A280 for sample D2-LN were 0.03-0.12 higher for
each kit except MasterPure™ which was 0.04 lower. Only the PowerSoil® kit
achieved a pure A260/A280 ratio of 1.98 while others were 0.01 (MasterPure™),
0.03 (QIAmp®), and 0.21 (Chemagic) out of the range. Interestingly, median
A260/A230 values of LN treated biocarriers were lower by 0.17 (PowerSoil®),
0.2 (MasterPure™), 0.29 (QIAmp®) and 0.01 (Chemagic). It’s worth noting that
the despite the miniscule drop in A260/A230 for the Chemagic D2-LN, the
untreated D2 extract was already well below 1.0 and represented the worst purity
measurement obtained for any other extract.
The PowerSoil® kit achieved median pure A260/A280 ratios across the board and ranked
first with the lowest average inner quartile range (IQR) of 0.092. It also and had the
highest A230/A260 ratios for samples N1, D2, and L1 and second highest for D2 and P2.
Overall, all median A260/A230 ratios exceeded 1.46, four exceeded 1.8, and two exceed
1.95, however the average IQR of 0.33 ranked third out of the four kits.
The MasterPure™ kit achieved median pure A260/A280 ratios for 4/6 samples, was
within 0.04 of the desired range for the remaining two samples (N2 and L1) and ranked

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
32
third with an average IQR of 0.11. It also achieved the highest median A260/A230 ratios
for samples N2 and D1 and the second highest values for samples N1, D2 and L1.
Overall, all A260/230 ratios exceeded 1.22 with 3 exceeding 1.5, one equaling 1.95, and
ranked second with an average IQR of 0.21.
The Chemagic Kit only achieved a median pure A260/A280 ratio for sample D2 with the
remaining values between 0.11 – 0.38 below the desired range. Despite the comparatively
poor A260/A280 purity it did rank second with an average IQR of 0.098. In terms of
A230/A260 ratios it also yielded the lowest values across the board with no value
exceeding 1.26 and two values below 1.0 despite the top ranked (smallest) IQR of 0.18.
The QIAmp® stool kit achieved median pure A260/A280 ratios for 3/6 samples with the
remainder falling 0.07 – 0.14 above or below the desired range. The average A260/A280
IQR was larger than for any of the other kits at 0.15. In terms of A260/A230, this was the
only kit to yield a median ratio in the desired 2.0-2.2 range which was 2.08 for sample P1.
Overall however, it yielded median A260/A230 values ranking third lowest out of the
four kits for 4/6 samples, ranked second once, and even had one value below 1.0. The
average A260/A230 IQR was also worse than any other kit at 0.41.
3.3.2 PCR Amplification
Table 9. PCR amplification of 16S rDNA isolated from Lentikat Biocatalysts® and activated sludge
samples
Sample PowerSoil® QIAamp® Chemagic MasterPure™
N1 + + + +
N2 + + + -/+
D1 + + + +
D2 + + -/-/+ -/-/+
D2-LN +/- +/- NA NA
L1 + + + -/+
P1 + + + -/+ Note: + Indicates successful amplification of undiluted template DNA; -/+ Indicates failed
amplification of undiluted template DNA but successful amplification at 1:20 dilution; -/-/+ Indicates
failed amplification of undiluted and 1:20 dilution but successful amplification at 1:50 dilution.

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
33
Figure 2. 2% Agarose gel profiles of PCR products
A. PCR Products optimized for downstream DGGE. B. PCR Products optimized for downstream DGGE
C. PCR products of PowerSoil® and QIAmp® with full buffer D. PCR products of QIAmp® with full buffer
K= Concentrated (½ manufacturer recommended elution buffer); Q= QIAamp®; MB= (MoBIO)
PowerSoil®; CH= Chemagic; MP= MasterPure™; L= Ladder; NC= Negative control Hy= Sample
from partial nitrification SBR (Section 4.2.5). LN = indicates treatment with liquid nitrogen prior to
DNA extraction. Samples presented /20 or /50 (e.g. D2/50) indicate dilution ratios where applied. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 A L NC K-Q
N1 K-MB-N1
CH N1
MP N1
K-Q N2
K-MB N2
CH N2
MP N2/20
K-Q D1
K-MB D1
CH D1
MP D1
B L K-Q-
D2
K-MB
D2
CH
D2/50
MP
D2/50
K-Q
L1
K-MB
L1 CH
L1
MP
L1/20
K-Q
P1
K-MB
P1 CH
P1
MP
P1/20
K-MB
Hy
C L NC MB
N1
MB
N2
MB
D1
MB
D2
MB
L1
MB
P1
MB
D2-LN
MB-D2 LN/20
Q
N1
Q
N2
Q
D1
Q
D2
D L Q
L1
Q
P1
Q-D2
LN
Q-D2
LN/20
L
PCR Amplification was performed on one of the three DNA extracts from each
sample chosen based upon NanoDrop® data to best represent the median values for yield and
purity. In the case of incongruence between yield and purity values, the extract was selected
that best represented median purity.
Table 8 and Figure 2 Images A and B indicate that all kits isolated DNA that
successfully amplified via Touchdown PCR for all samples, but that dilution was necessary in
2 3 4 5 6 7 8 9 10 11 12 13 14 2 3 4 5
2 3 4 5 6 7 8 9 10 11 12 13 14 2 3 4 5 6 7 8 9 10 11 12 13 14

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
34
some cases. Table 8 and Figure 2 Images C and D indicate that DNA extracted from all
samples using the PowerSoil® and QIAmp® stool kits successfully amplified when the
manufacturers recommended volume of elution buffer was used. Unfortunately due to an
error in programming the thermocycler, images C and D depict an amplification that was cut
short by 6 cycles. However, these results are still interesting in that these images indicate that
the concentration of PCR products obtained from the QIAmp® stool kit extracts were lower
than those extracted with the PowerSoil® kit possibly indicating some PCR interfering
contaminants.
Table 8 and Figure 2 Images C and D also show that while undiluted extracts from
sample D2 treated with LN readily amplified, 1:20 dilutions with PCR grade water did not.
Note that both were tested as insurance to avoid running PCR again in the event that the
undiluted extracts would not amplify.
As indicated above and show in Table 8, the undiluted MasterPure™ extracts from
samples N2, D2, L1, and P1 as well as the Chemagic extract for sample D2 failed to amplify
despite successful positive control and other sample amplification (not shown). Another
round of PCR was then performed on these samples using a 1:20 dilution with PCR grade
water which resulted in successful amplification for all but two samples which amplified after
a final round using 1:50 dilution ratio.

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
35
3.3.3 DGGE
Figure 3. DGGE Profile of amplified V3 region of 16S rDNA from all samples. From left to right:
Samples L1, N1, D2, Reactor B (Ch. 4), D1, P1, N2 (Order: QIAmp®, PowerSoil®, Chemagic,
Masterpure™). *Note the image of Sample L1 was taken from a separate gel.*
Figure 3 shows the DGGE profiles from all samples and kits examined. For each
sample the order of extracts in each lane from left to right is QIAmp®, Powersoil®,
Chemagic, and Masterpure™. The only deviation from this ordering is for sample D2, where
LN treated biocarrier extracts from QIAmp® and Powersoil® were added adjacent to their
untreated analogues. Note that Q-D2-LN is marked with an X. Upon close investigation it
was determined that Q-D2-LN was in fact a mistakenly amplified extract of Sample D1.
Evidence of this mistake and justification of its omission are presented in Appendix 2.
Figure 4. UPGMA Dendrogram and BSI for Sample N1.
Figure 4 shows that for sample N1, all extracts shared 90% similarity with eachother. A total
of 15 distict bands were identified across all N1 extracts. Both Masterpure™ and Chemagen
represented 93% of total phylogenetic richness (BSI) in the synthetic lane while QIAmp®
represented the least at 85%.
BSI (%)
93
89
85
93
N1 N2 D1 D2 L1 P1 Reactor B
-Ch.4
X MB
LN
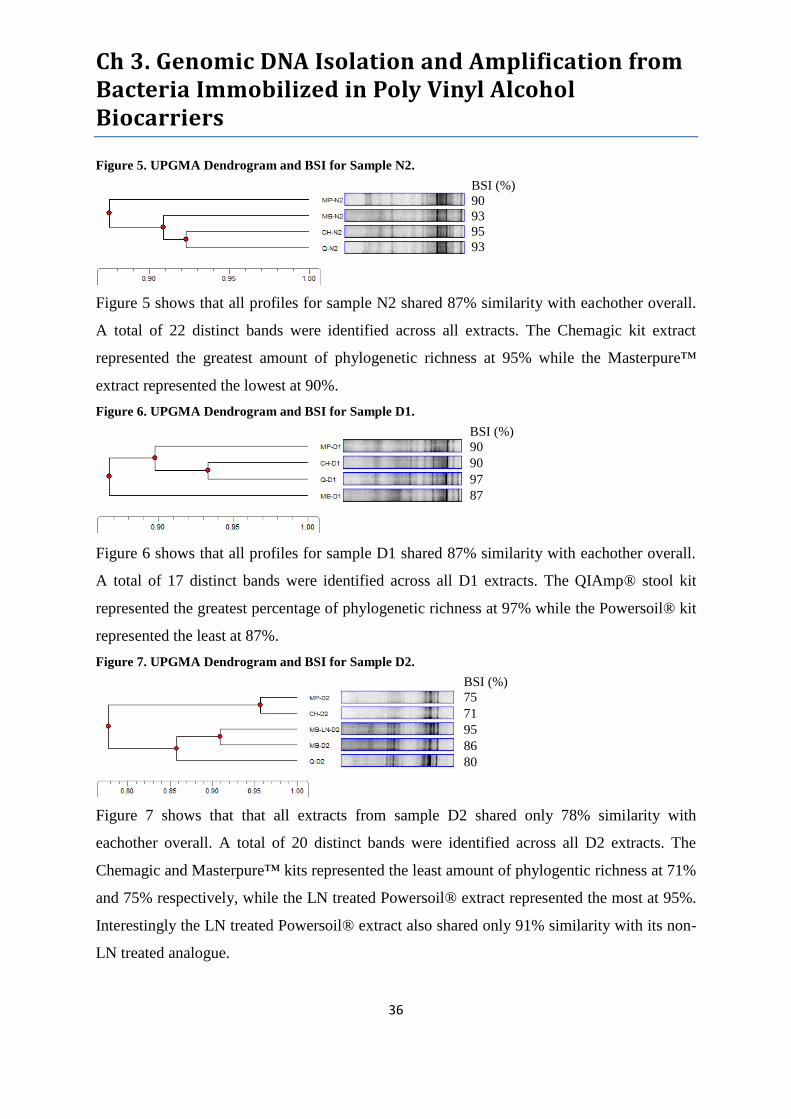
Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
36
Figure 5. UPGMA Dendrogram and BSI for Sample N2.
Figure 5 shows that all profiles for sample N2 shared 87% similarity with eachother overall.
A total of 22 distinct bands were identified across all extracts. The Chemagic kit extract
represented the greatest amount of phylogenetic richness at 95% while the Masterpure™
extract represented the lowest at 90%.
Figure 6. UPGMA Dendrogram and BSI for Sample D1.
Figure 6 shows that all profiles for sample D1 shared 87% similarity with eachother overall.
A total of 17 distinct bands were identified across all D1 extracts. The QIAmp® stool kit
represented the greatest percentage of phylogenetic richness at 97% while the Powersoil® kit
represented the least at 87%.
Figure 7. UPGMA Dendrogram and BSI for Sample D2.
Figure 7 shows that that all extracts from sample D2 shared only 78% similarity with
eachother overall. A total of 20 distinct bands were identified across all D2 extracts. The
Chemagic and Masterpure™ kits represented the least amount of phylogentic richness at 71%
and 75% respectively, while the LN treated Powersoil® extract represented the most at 95%.
Interestingly the LN treated Powersoil® extract also shared only 91% similarity with its non-
LN treated analogue.
BSI (%)
90
93
95
93
BSI (%)
90
90
97
87
BSI (%)
75
71
95
86
80

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
37
Figure 8. UPGMA Dendrogram and BSI for Sample L1
Figure 8 shows that all extracts from sample L1 shared 84% similarity overall. A total of 25
distinct bands were identified across all L1 extracts. The QIAmp® and Masterpure™ extracts
represented 98% and 96% of total phylogentic richness respectively while the Powersoil®
extract represented the least at 81%.
Figure 9. UPGMA Dendrogram and BSI for Sample P1
Figure 9 shows that all extracts from sample P1 shared 93% similarity overall. A total of 21
distinct bands were identified across all extracts. The Masterpure™ kit extract represented the
greatest percentage of total phylogenetic richness at 98% while the Powersoil® kit
represented the least at 89%.
3.4 Discussion
3.4.1 Waste Generation, Processing Time, and Cost
The differences in waste generation between the kits can be attributed primarily to the
difference in techniques employed for DNA isolation. The Powersoil® and QIAmp® kits
were the most similar. The greatest contributing factor to QIAmp® producing the most waste
overall was the use of a combined total of 8x 1.5-2 mL microcentrifuge tubes and 2 mL spin
collection tubes as well as all the pipette tips necessary to make all transfers. Furthermore the
“InhibitEx” tablets add extra weight as well. In contrast the Powersoil® kit uses only 4x 2mL
microcentrifuge tubes plus 1x bead beating tube, however the dense plastic construction of
the bead beating tubes plus the beads themselves add significant mass to the waste
generation. While the Chemagic kit required only 2x microcentrifuge tubes its relatively high
BSI (%)
96
98
86
81
BSI (%)
98
95
89
95

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
38
mass of waste generated was due to weight of the magnetic beads and washing reagents
required but more importantly the very high number of pipette tips required. Lastly, the very
best kit in terms of waste generation was Masterpure™. This kit was quite efficient in terms
of pipette tip usage, volume of reagents used, and required only 2x microcentrifuge tubes.
The processing times for the Powersoil®, QIAmp®, and Chemagic kits were all very
similar. On the other hand, the Masterpure™ kit took significantly longer for a 9 sample run
due to 50 minutes of incubation time throughout the protocol with periodic pauses for
vortexing and 20 additional minutes of refrigerated centrifugation.
In terms of cost, the QIAmp® stool kit was the most expensive at €5.1 per sample
followed by the Powersoil® kit at €3.2 per sample. It is worth noting that the Powersoil® kit
was the only kit tested that included all necessary bead beating, microcentrifuge, and
collection tubes in this price. On the other hand, the Chemagic kit cost €2.4 per sample
excluding microcentrifuge tubes and RNAse. The Masterpure™ kit was by far the least
expensive at a cost of only €1.2 per sample however it cannot be purchased in quantities
fewer than 200 preps unlike Powersoil® (50 prep minimum), QIAmp® (50 preps), and
Chemagic (100 preps). Furthermore, in considering costs it’s important to take into account
the greater processing time required by the Masterpure™ kit. While the fixed cost per sample
is the lowest the additional costs in terms of salary of the technician performing the
extractions may significantly reduce this benefit especially when only running a small
number of samples.
3.4.2 DNA Yield
Our results indicate that for touchdown PCR and DGGE applications all kits yielded
sufficient quantities of DNA from all samples tested when we followed the manufacturer
protocols. Before discussing and comparing the extraction kits it’s worth noting a few trends
evident between the individual samples. As expected sample N1, yielded the lowest amount
of DNA across the board, yet all extracts amplified successfully. Also as expected, both
activated sludge samples, which contain biomass to total mass ratios higher than biocarriers,
yielded the greatest amounts of DNA for each kit except Chemagen. Lastly, the data implies
that LN treatment of biocarriers prior to DNA isolation was effective at increasing the
quantity of DNA yielded. This is a logical finding as the LN treatment thoroughly reduced

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
39
the biocarriers to a homogenous mass with characteristics similar to those activated sludge.
This serves to effectively eliminate any barriers that may have impeded lysis enzymes from
contacting the biomass most deeply embedding in the biocarrier material. Furthermore, it is
consistent with the findings of Alain et al. (2011) who obtained greater DNA yields from
deep sea sediments by integrating a similar cryogenic grinding step into their lysis procedure.
In regards to the extraction kits, we did reduce the amount of elution buffer for the
QIAmp® and Powersoil® extracts in order to obtain more reliable NanoDrop® readings,
however samples diluted to the manufacturer recommended volumes amplified in touchdown
PCR without impediment. In fact, the only real trouble we encountered was that a number of
Masterpure™ extracts had too high DNA concentrations for successful amplification without
dilution. Based upon the purity ratios obtained for nearly all of these samples it seems likely
that these failed amplifications were due to excessive DNA concentrations, however the
failure of the 1:20 dilution from MP-D2 most likely indicated the presence of PCR inhibitors.
Similarly, the undiluted Chemagen extract from sample D2 also failed to amplify and
although it may have been related to excessive DNA concentration, poor purity or a
combination of both factors seems very likely. Fortunately, in both of these cases the DNA
concentration was sufficient to withstand 1:50 dilution and still amplify, while the inhibitor’s
influence was overcome.
The Powersoil® and QIAmp® stool kits both yielded similarly low quantities of DNA
from all biocarrier samples and slightly higher amounts for activated sludge samples. Despite
DNA yields below 2 µg/g of sample N1, both kits produced extracts that amplified well.
While this was an encouraging finding it raised the question of whether or not such low
yields of DNA contain enough nucleic acid diversity to be representative of the total
phylogenetic diversity present in this sample. To answer this question we moved forward
with DGGE analysis which is discussed in a later section.
Based upon these results, the Masterpure™ kit appears to be the best suited to isolate
DNA for downstream applications that require very high concentrations.
3.4.3 Purity
Overall the Powersoil® kit performed the best in terms of DNA extract purity for both
A260/A280 and A260/A230 ratios. Not only did Powersoil® extracts meet the desired

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
40
A260/A280 purity standards for all samples but they also demonstrated the lowest variability
compared to all other kits. In terms of A260/A230 purity standards, Powersoil® extracts were
again among the best nearly across the board however they show greater variability in IQR
than Chemagic or Masterpure™ extracts in this measure.
The Masterpure™ kit performed nearly as well as the Powersoil® kit both in terms of
A260/A280 and A260/A230 ratios. While the IQR of A260/A280 demonstrated greater
variability than Powersoil® the opposite was true for A260/A230 IQR. Despite purity ratios
greater that many samples that did amplify, the Masterpure™ sample D2 extract failed to
amplify at 1:20 dilution, which would almost certainly rule out excessive template DNA as a
cause. Given that sample D2 was implemented in the remediation of groundwater
contaminated with sulfuric acid from uranium mining operations, it seems most likely that
multivalent cations such as heavy metals may be the culprit. Sample D2 was also extremely
dark in color and “dirty” in appearance so humic compounds shouldn’t be ruled out entirely
either. In any event, this case should be considered as a cautionary reminder that while the
purity ratios yielded on Nanodrop® may be positive indicators, they don’t guarantee that
concentrations of all impurities are below inhibitory levels.
The QIAmp® stool kit performed fairly well despite ranking 3rd
in terms of both
A260/A280 and A260/A230 ratios. What was troubling, however was that the QIAmp®
extracts demonstrated the greatest degree of variability of all kits tested. IQR values for
A260/A230 demonstrated a particularly high degree of inconsistency, which may or may not
be the reason for the difference in the reduced cycle PCR product concentrations compared to
the Powersoil® extracts show in Figure 2 images C and D.
The Chemagic kit performed the worst by far in terms of both measures of purity.
While the A260/A280 ratios indicated the presence of impurities for all but one sample they
were not so low as to cause major concern that the DNA would not be usable for touchdown
PCR amplification. On the other hand, the A260/A230 purity ratios were dismal across the
board but particularly for biocarrier samples. The extract purity values for sample D2,
discussed earlier, were the worst of any sample measured in this project.
Overall, each of the kits tested were able to successfully extract DNA of acceptable
yield and purity for downstream applications, in this case PCR-DGGE. The most consistently

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
41
Table 10. Mean BSI of all DGGE extracts
excluding MB-LN-D2
QIAmp® Stool Kit 91.3% σ = 7%
Powersoil® 87.5% σ = 4%
Chemagic 88.3% σ = 9%
Masterpure™ 90.3% σ = 8%
troubling purity results were obtained with the Chemagic kit, however a simple and obvious
modification to the protocol may logically improve these results. The manufacturer’s protocol
for this kit is designed for the extraction of DNA from laboratory bacterial cultures and thus
is performed entirely in a single microcentrifuge tube until elution. Logically, the assumption
is that any culture medium would easily be washed out and removed with other impurities
once the cells are lysed and DNA is bound to the magnetic beads, thus there is no pressing
necessity to use extra microcentrifuge tubes. In our case, when applying the Chemagic kit to
extract DNA from biocarriers, soils (data not included in analyses), and to a lesser extent
activated sludge, the sample residual remains in the microcentrifuge tube until the very last
step. It seems logical that briefly centrifuging the sample after the initial enzymatic lysis step
and transferring the lysate supernatant into a clean microcentrifuge tube could drastically
reduce the potential for contamination by humic acids, other complex organic compounds,
extraction reagents and/or washing buffers. It seems likely that the particularly poor purity of
Chemagic extracts may be due to impurities adsorbing to or infiltrating the pores of
biocarriers and being unintentionally carried through to subsequent steps in the isolation
process. This may explain the low A260/A230 ratios which are often attributed to phenol or
other extraction reagents remaining in the eluted DNA. This scenario is also supported by
fact that A260/A230 ratios were better for activated sludge samples, which would more easily
be removed along with reagents in washing steps than biocarriers that remain until the final
stage.
Based upon these results, it seems that all kits tested can be applied to Lentikat’s
Biocatalysts in order to extract DNA of sufficient purity for use in downstream Touchdown
PCR applications. Furthermore, these data indicate that the Powersoil® kit is the best suited
for isolating DNA for use in applications that require very high purity.
3.4.4 Phylogenetic Comparison of Extracts
The results from our DGGE analyses imply that
all kits performed comparably well in terms of
phylogenetic richness and similarity. Despite some
variability between the BSI values of different samples
Table 9 shows that overall the mean BSI values for all

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
42
kits were not significantly different. Furthermore, the similarity in banding patterns between
kits ranged from 78-93% with a mean of 86.5% which is highly encouraging considering the
results of Mahmoudi et al. (2011) who reported similarity of only 2-10% in a similar study
examining soil samples. Even the result of 78% overall similarity yielded for sample D2 was
deflated by the superior performance of the LN treated biocarrier extract. Overall, these
results demonstrate a far lower degree of extraction bias than had been expected.
When considered in the context of our results for the LN treatment, it seems that
suggesting superiority of one DNA isolation kit over another in terms of phylogenetic
comparison would be misguided. While the QIAmp® stool kit did demonstrate the greatest
consistency of high BSI values, the results for MB-LN-D2 suggest that obtaining the most
comprehensive phylogenetic richness from each sample may best be achieved through
optimizing cell lysis rather than kit selection. While DGGE does not provide comprehensive
phylogenetic richness as much as it is known for characterizing dominant community
members, the differences in the intensity of identical bands between extracts apparent in
Figure 3 suggests that dominant community members may be absent or less pronounced in
some extracts. This raises the question of whether or not any DNA extraction provides a true
representation of phylogenetic dominance within a community or just susceptibility to the
cell lysis techniques employed in DNA isolation. While this is an extremely bold conclusion
to draw from such highly limited data it is supported by the findings of Alain et al. (2011)
mentioned previously and Jiang et al. (2011) who found that integrating lysis techniques
drastically reduced DNA extraction biases from mangrove sediments. Interestingly, they also
noted that more intense lysis lead to a greater release of humic compounds and thus
decreased extract purity. A similar trend was apparent in both purity ratios for LN treatments
with all kits except Chemagic, which had already yielded the lowest purity of any sample D2
extract. The mistake that prevented Q-LN-D2 from being included in these analyses is even
more disappointing in light of the data yielded by MB-LN-D2.
Based upon the results of DGGE phylogenetic comparisons, no DNA isolation kit
performed significantly better or worse than any other. On the other hand, some extraction
biases were discovered in these analyses and though limited, the data for LN treatment and

Ch 3. Genomic DNA Isolation and Amplification from Bacteria Immobilized in Poly Vinyl Alcohol Biocarriers
43
available literature suggest that these biases may be minimized through continued evaluations
of enhanced lysis techniques.
3.5 Conclusions
All commercial DNA Isolation kits tested were compatible with Lentikat’s Biocatalysts
and yielded DNA of sufficient quantities and purities for downstream PCR-DGGE analyses.
The processing times for each kit were highly comparable except for the Masterpure™ DNA
purification kit which took significantly longer. On the other hand, the Masterpure™ DNA
purification kit produced less than half the mass of waste and cost only half as much per
sample than its next closest competitor which was the Chemagic DNA Bacteria kit in both
instances.
The Masterpure™ DNA purification kit and Chemagic DNA Bacteria kit both yielded the
highest median quantities of DNA in µg/g for all samples across the board. In terms of
isolated DNA purity, the Powersoil® DNA Isolation kit outperformed all others across the
board with the Masterpure™ DNA Purification kit ranking second.
All kits demonstrated a high degree of similarity and thus low degree of extraction bias in
DGGE phylogenetic comparisons. The QIAmp® DNA Stool kit performed the best in terms
of phylogenetic richness, however no other kit was significantly worse. Though the data is
limited, LN enhanced lysis of PVA biocarriers showed promise in mitigating extraction
biases and increasing the phylogenetic richness of isolated DNA. Given the high
phylogenetic similarity between all DNA isolation kit extracts, the potential of enhanced lysis
techniques for optimizing community characterization, DNA purity, and to a lesser extent
yield should be considered most heavily in establishing laboratory protocol for working with
Lentikat’s Biocatalysts. Therefore, the Powersoil® DNA isolation kit is recommended as the
best option from those examined for applications that require the highest degree of purity as
well as further investigations of enhanced cell lysis techniques. This holds true especially
when high DNA yield is not essential and sample mass is abundant. For applications where
higher DNA yield is required or sample mass is limited, the Masterpure™ DNA Purification
kit is recommended.

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
44
4.1 Objectives
The objectives of this investigation are to observe and characterize the effects of
hydroxylamine on NOB immobilized in Lentikat’s Nitrifying Biocatalysts in laboratory scale
partial nitrification SBRs. This is accomplished through a combination of water chemistry
analyses and fluorescence in situ hybridization and supplemented through PCR-DGGE and
16S rDNA sequencing.
Please note that the operation of these reactors and chemical analyses were performed
by Petr Kelbich, MSc. and Iva Johanidesová, MSc. and were the subject Mrs. Johanidesová’s
MSc. thesis titled "Evaluation of the possibility of maintaining partial nitrification using
immobilized microbial cultures” (Johanidesová, 2013).

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
45
4.2 Materials and Methods
4.2.1 Partial Nitrification Reactor Setup and Operation
Three SBRs, Labeled A, B and C, were set
up in 1.5L internal volume glass bioreactors
with external water circulation chambers in
the laboratory at ICT Prague on July 24,
2012 (Day 1). All three reactors were
situated on top of magnetic stir plates with
constant stirring, continuously aerated, and
linked in series to a Julabo F250 water bath
(Julabo GmbH., Seelbach, Germany) that
maintained constant temperature at 30oC
through continuous circulation. Each reactor
was loaded with 100 g (66.7 g/L) of wet
LentiKat’s nitrifying biocatalysts obtained following manufacture and subsequent cultivation
or “Grow out” on July 12, 2012.
During the startup phase (day 1-14), each reactor was drained and re-filled with 1L of
synthetic WW containing 50 mg N-NH4/L, 2.3 g/L KH2PO4, 2.9 g/L K2HPO4, and 0.5 g/L
NaHCO3 five days per week. Once steady state nitrification was achieved (day 15), the
concentration of N-NH4 was increased to 300 mg/L (600 mg/L prior to weekends) and
operation was again continued until steady state nitrification was reached (day 43). At this
point, Reactors A and B were subject to separate treatment regimes by dosing hydroxylamine
hydrochloride (NH2OH·HCl) obtained from Sigma-Aldrich Spol. s.r.o (Prague, Czech
Republic), while the operation of reactor C was maintained as a control until it suffered a
catastrophic mechanical failure on day 112. The treatments for Reactors A and B are detailed
in the following paragraphs and summarized in Table 10.
Beginning on day 43, Reactor A was dosed to a concentration of 0.5 mg NH2OH/L
once daily. Dosing was automated with a peristaltic pump that delivered the appropriate
quantity of hydroxylamine over a 15 minute time period. On day 73 the dose of
hydroxylamine was increased to 8 mg/L per day which was delivered in 1 mg aliquots over a
30 minute time period every 3 hours. This dosing was automated using a storage solution
Figure 10. Nitrifying SBRs in laboratory in ICT Prague.
Note: This image is of a subsequent experiment that
employed the same physical apparatus minus 1 reactor.
Photo Credit: Petr Kelbich

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
46
with a concentration of 200 mg NH2OH/L, the peristaltic pump, and a Kanlux Cyber TM-6
digital timer. On day 87, the thermostat unit critically failed and could not be immediately
replaced so the remainder of the investigation was carried out with the reactors operating at
room temperature (20-25oC). On day 112 the dose of hydroxylamine was increased to 16 mg
NH2OH/L per day delivered in 2 mg doses, every three hours, over a 15 minute time period.
On day 121, the dose of hydroxylamine was reduced to 4 mg/L per day delivered in 2 mg
doses, every 12 hours, over a 15 time period. The experiment was concluded on day 169 and
Reactor was shut down.
Beginning on day 43, Reactor B was dosed to a concentration of 5 mg NH2OH/L once
daily. Dosing was automated with the same apparatus and delivery time used for Reactor A.
On day 73, the dose of hydroxylamine was increased to 40 mg NH2OH/L per day which was
delivered in 5 mg aliquots over a 30 minute time period every 3 hours. This dosing was
automated using a storage solution with a concentration of 40g NH2OH/L, the peristaltic
pump, and a Kanlux Cyber TM-6 digital timer. On day 87, the thermostat unit critically failed
and could not be immediately replaced so the remainder of the investigation was carried out
with the reactor operating at room temperature (20-25oC). The dose of hydroxylamine was
also reduced on this date to 10 mg/L delivered in 1.25 mg aliquots every 3 hours over a 30
minute time period. On day 91, hydroxylamine dosing was ceased and ammonia nitrogen
concentration in the synthetic WW influent was reduced to 50 mg/L. The concentration of
ammonia in the reactor influent was increased to 75 mg/L on day 97, to 150 mg/L on day 99,
and back to 300 mg/L on day 107. While this experiment was concluded on day 169, Reactor
B remains operational as of June 3, 2013 as part of a separate experiment.
Beginning on day 174, the synthetic WW used over the previous 6 months was
replaced with the liquid fraction of post anaerobically digested sludge from Prague Municipal
WWTP (Prague, Czech Republic). This digestate was diluted in order to achieve an influent
N-NH4+
concentration of approximately 300 mg/L (600 mg/L prior to weekends). Operation
was again held steady until it was decided to reduce the weekend dose of N-NH4+ to 300 g/L
on day 284.

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
47
Table 11. Summary of treatments administered to Reactors A and B throughout their operations.
Day # Reactor A Reactor B
1 Startup 30oC.
1L/d [50 mg N-NH4/L, 2.3 g/L
KH2PO4, 2.9 g/L K2HPO4, and 0.5
g/L NaHCO3]
Startup 30oC.
1L/d [50 mg N-NH4/L, 2.3 g/L
KH2PO4, 2.9 g/L K2HPO4, and 0.5
g/L NaHCO3]
14 N-NH4 increased to 300 mg/L N-NH4 increased to 300 mg/L
43 NH2OH dosing 0.5 mg*d
1 dose delivered over 15 min period
NH2OH dosing 5 mg*d
1 dose delivered over 15 min period
73 NH2OH dosing 8 mg*d
1 dose every 3 hrs.; 30 min delivery
NH2OH dosing 40 mg*d
1 dose every 3 hrs.; 30 min delivery
87 Temp from 30oC to Room Temp Temp from 30
oC to Room Temp
NH2OH dosing 10 mg*d
1 dose every 3 hrs.; 30 min delivery
91 NH2OH dosing Terminated
N-NH4 decreased to 50 mg/L
97 N-NH4 increased to 75 mg/L
99 N-NH4 increased to 150 mg/L
107 N-NH4 increased to 300 mg/L
111 NH2OH dosing 16 mg*d
1 dose every 3 hrs.; 15 min delivery
121 NH2OH dosing 4 mg*d
1 dose every 12 hrs.; 15 min
delivery
169 Experiment concluded and reactor
disassembled
Experiment concluded however
reactor operation maintained
174 Influent switched from Synthetic
WW to liquid digester effluent
diluted to ~300 mg/L N-NH4+
285 Weekend dose of N-NH4+ reduced
from 600 mg/L to 300 mg/L
Figure 11. Hydroxylamine dosing regimes for treatment reactors.

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
48
4.2.2 Chemical Analyses
The concentrations of inorganic nitrogen species in reactor effluents were analyzed on a
regular basis. Parameters assessed included NH4+, NO2
-, NO3
-, and NH2OH and were
determined using the following protocols:
4.2.2.1 Ammonia Nitrogen (NH4+)
This protocol was adapted from Standard Methods in the Examination of Water and
Wastewater 4500-NH3 (APHA, 1992).
Nessler Reagent Seignett Salt Solution
25 g HgI2
17.5 g KI
40 g NaOH (in 125 mL dH2O)
dH2O Fill to 250 mL
2.5 g KNaC4H4O6
50 mL dH2O
1. Load 5 mL of sample into a test tube
2. Add 100 µL of Seignett salt solution and 100 µL
3. Seal the test tube and mix thoroughly.
4. After 10 minutes measure the absorbance at 425 nm with a photoLab® 6100 VIS
spectrophotometer (WTW GmbH, Weilheim, DE)
5. Calculate the concentration of Ammonia using a calibration curve.
4.2.2.2 Nitrite Nitrogen (NO2-)
This protocol was adapted from Standard Methods in the Examination of Water and
Wastewater 4500-NO2- (APHA, 2012).
SANED Reagent
10 g Sulfanilamide
0.5 g N-(1-napthyl)-1,2-ethyendiamine-dichloride
25 mL H3PO4 [CONC]
dH2O to 250 mL
1. Load 5 mL of sample into a test tube
2. Add 125 µL of SANED reagent and 1.1 mL of dH2O
3. Seal the tube and mix thoroughly
4. After 20 minutes measure the absorbance at 540 nm with a photoLab® 6100 VIS
spectrophotometer (WTW GmbH, Weilheim, DE)
5. Calculate the concentration of Ammonia using a calibration curve.

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
49
4.2.2.3 Nitrate Nitrogen (NO3-)
This protocol was derived from ISO 7890-1:1986 (ISO, 1986).
Amide-Sulfuric Acid Acid Mixture DMP
0.8 g amide-sulfuric acid
dH2O to 100 mL 2 mL concentrated H2SO4
2 mL concentrated H3PO4
0.24 g 2,6-dimethylphenol
200 mL glacial acetic acid
1. Load 0.5 mL of sample into a test tube
2. Add 50 µL of amide-sulfuric acid solution, 3.5 mL acid mixture, and 500 µL DMP
solution
3. Seal the tube and mix thoroughly
4. After 10 minutes measure the absorbance at 360 nm with a photoLab® 6100 VIS
spectrophotometer (WTW GmbH, Weilheim, DE)
4.2.2.4 Hydroxylamine (NH2OH)
This Protocol was adapted from Frear and Burrell (1955).
8-Quinolinol Solution
o 1 g 8-Quinolinol
o 100 mL 99% Ethanol
10-4
M NADH Dehydrogenase
0.025 mM NH2OH
1M Sodium Carbonate 0.001 M Manganese Chloride
12% Trichloroacetic Acid 0.05 M PBS pH 6.8
1. Add 1 mL of Sample to a test tube
2. Add 1 mL of PBS, 800 µL of dH2O, and 200 µL of trichloroacetic acid
3. Add 1 mL of 8-quinolinol and mix gently
4. Add 1 mL of 1 M sodium carbonate solution and shake vigorously
5. Seal the tube and place in boiling water bath for 1 minute.
6. After cooling for 15 minutes measure the absorbance at 705 nm photoLab® 6100
VIS spectrophotometer (WTW GmbH, Weilheim, DE)
4.2.3 FISH
This internal protocol has been adapted from Amman (1995) in order to hybridize
nitrifying microorganisms immobilized within Lenticat biocatalysts. Hybridization probes
were purchased from Generi Biotech (Hradec Králové, Czech Republic) and were pre labeled
with fluorophores listed in Table 11. The stock probes arrived in varying concentrations and
were diluted with sterile distilled water to 50mM working solutions prior to hybridization.
To establish a baseline for comparison, biocatalysts were sampled from all reactors and
subsequently fixed for hybridization on day 30 of the experiment. At this stage all three
reactors were exhibiting similar performance in regards to inorganic nitrogen speciation.
Treatments were commenced on day 43 and Reactors A and B were then sampled again on
day 130. Reactor B was sampled again on day 203. Unfortunately Reactor C suffered a
critical vessel failure on day 111 before a second round of biocatalysts could be sampled and

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
50
fixed. All biocatalysts were fixed with paraformaldehyde (see section 4.2.3.2), stored at -
20oC, and hybridized in February 2013. For supplemental comparison, nitrifying biocatalyst
samples N1 and N2 (See Section 3.2.1) we also hybridized and imaged.
4.2.3.1 Reagents and Probes
1 x Phosphate-Buffer-Saline
(PBS)
3 x Phosphate-Buffer-Saline
(PBS)
4% Paraformaldehyde (PFA)-
PBS Solution
8 g NaCL
0.2 g KCl
1.44 g Na2HPO4
0.2 g NaH2PO4
1000 mL dH2O
pH 7
24 g NaCL
0.6 g KCl
g Na2HPO4
0.6 g NaH2PO4
pH 7
66 mL ddH2O ( 60oC)
4 g PFA
2-3 drops Conc. NaOH
34 mL 3 x PBS
pH 7-7.4 (HCl)
Tris-HCl Buffer (1 M) NaCl Stock (5 M) SDS-solution (10%)
15.8 g Tris/HCl
100 mL dH2O
pH 8
29.2 g NaCl
100 mL dH2O
10 g SDS
100 mL H2Obidest
TE Buffer EDTA (0.5 M) Ethanol
1.6 g (10 mM) Tris/HCl
0.37 g (1 mM) EDTA
(Na2)-EDTAxH2O
1000 mL dH2O
pH 7.2/ pH 8
18.6 g EDTA
100 mL dH2O
50%
80%
96%
100%
Hybridization Buffer (35% Formamide) Hybridization Buffer (40% Formamide)
360 mL NaCl (5M)
40 µL Tris/HCl buffer
800 µL Formamide
800 µL dH2O
2 µL SDS (10%)
360 mL NaCl (5M)
40 µL Tris/HCl buffer
900 µL Formamide
700 µL dH2O
2 µL SDS (10%)
Washing Buffer (35% Formamide) Washing Buffer (40% Formamide)
1000 µL Tris/HCl buffer
460 µL NaCl (5 M)
500 µL EDTA (0.5 M)
50 µL SDS (10%)
Dilute to 50 mL with dH2O
1000 µL Tris/HCl buffer
700 µL NaCl (5 M)
500 µL EDTA (0.5 M)
50 µL SDS (10%)
Dilute to 50 mL with dH2O

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
51
Table 12. Hybridization probes employed. Table adapted from Nielsen et al. 2009.
Probes Target Formamide % Florophore Reference
NSO 1225 Betaproteobacterial
AOB
35 FITC Mobarry et al. 1996
NSO 190 Betaproteobacterial
AOB
35 FITC Mobarry et al. 1996
NTSPA 712 Phylum
Nitrospirae
35 Cy3 Daims et al. 2001
cNTSPA 712
(Competitor)
NTSPA 712 non
target organisms
35 None Daims et al. 2001
NTSPA 662 Genus Nitrospira 35 Cy3 Daims et al. 2001
cNTSPA 662
(Competitor)
NTSPA 662 non
target organisms
35 None Daims et al. 2001
NIT 3 Genus Nitrobacter 40 Cy3 Wagner et al. 1996
cNIT 3
(Competitor)
NIT 3 non target
organisms
40 none Wagner et al. 1996
4.2.3.2 Fixation with Paraformaldehyde
1. Collect samples of 2-10 pellets from each reactor and place in separate 2 mL eppendorf
tubes
2. Add 1.5 mL of 4% PFA-PBS solution, then add tap water until final volume reaches 2 mL
3. Mix by shaking, then refrigerate at 4oC for 4 hrs.
4. Remove and discard supernatant in PFA waste container.
5. Add 2 mL of 1xPBS washing buffer and invert several times to mix.
6. Repeat washing step 2 additional times.
7. Add 500 µL of 1xPBS and 500 µL of 100% Ethanol (not denatured)
8. Vortex to mix and store in the freezer at -20oC
4.2.3.3 Hybridization
1. Transfer a minimum of 2 pellets in a sterile 2 mL eppendorf tube
2. Dehydrate samples through successive ethanol rinses. First add enough 50% Ethanol to
cover the pellets (300 µL) and allow them to rest for 3 minutes before removing and
discarding the supernatant.
3. Repeat step 2 using 80% Ethanol
4. Repeat step 3 using 96-99% Ethanol
5. Remove the supernatant and incubate the sample tubes at 46oC for 20 minutes (or until
dry)
6. Add 50 µL of Hybridization buffer (Probe specific formula: See FISH Reagent list above)
7. Add Fluorescent probe and equal volume of complementary competitor probe (if needed)
and tap gently to mix.
a. 6.25 µL for Cy 3 labeled Probes
b. 8 µL for FITC labeled Probes
8. Incubate at 46oC for 3 hours.
9. Remove from incubator and discard the supernatant into PFA waste.
10. Add 2 mL of Washing Buffer and place in hot water bath at 48oC for 20 minutes

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
52
11. Remove from hot water bath and discard the supernatant.
12. Add 2 mL of cold Distilled H2O, shake gently and then discard the supernatant
13. Add 200-300 µL of TE Buffer (enough to cover the pellets in liquid)
14. Store refrigerated at 4oC for no more than 2 weeks.
4.2.3.4 Imaging
Fluorescence detection was carried out with an Olympus BX51-RFAA microscope,
images were captured with an Olympus MX10 CCD Camera using the fluorescence imaging
software CELLF all products of Olympus Corporation (Tokyo, Japan). Image manipulations
including: automatic white balance, manual brightness and contrast adjustment, and auto
color sharpening were carried out using GNU Image Manipulation Program (GIMP)
version 2.8 (GNU Development Team, Berkeley, CA USA).
4.2.4 Live/Dead Staining
Live/dead staining was carried out on biocatalysts sampled from Reactor B on day
224 with the LIVE/DEAD® BacLight™ Bacterial Viability kit, for microscopy and
quantitative assays (L7012, Lot 950626) (Life Technologies Corporation, Carlsbad, CA)
using the following protocol:
1. Lay Fresh PVA pellet on a sterile glass slide.
2. Apply 3 µL of a 1:1 mixture of 3.34 mM SYTO 9 dye and 20 mM Propium Iodide
(included in kit) to the pellet.
3. Incubate in the dark at room temperature for 15 minutes
4. Observe using fluorescent microscope (see section 4.2.3.4 for microscope
specifications)
4.2.5 DNA Isolation, PCR, DGGE, and Sequencing
DNA Isolation was carried out on biocatalysts sampled from Reactor B on day 274
with the PowerSoil® DNA Isolation kit (MoBio Laboratories Inc., Carlsbad, CA, USA) using
the manufacturer recommended protocol modified as detailed in Section 3.2.2 of this report.
Touchdown PCR was carried out using the protocol detailed in Section 3.2.4 prior to DGGE.
DGGE was carried out with the protocol detailed in Section 3.2.5. Bands were cut from the
acrylamide gel, soaked in PCR grade water (Hoffmann-La Roche Ltd., Basel, Swiss) for 36
hours, and subsequently re-amplified using Touchdown PCR. During this final round of PCR
amplification, the protocol detailed in section 3.2.4 was shortened by 6 cycles and an
analogue forward primer without GC-Clamp was employed. These samples were then sent to
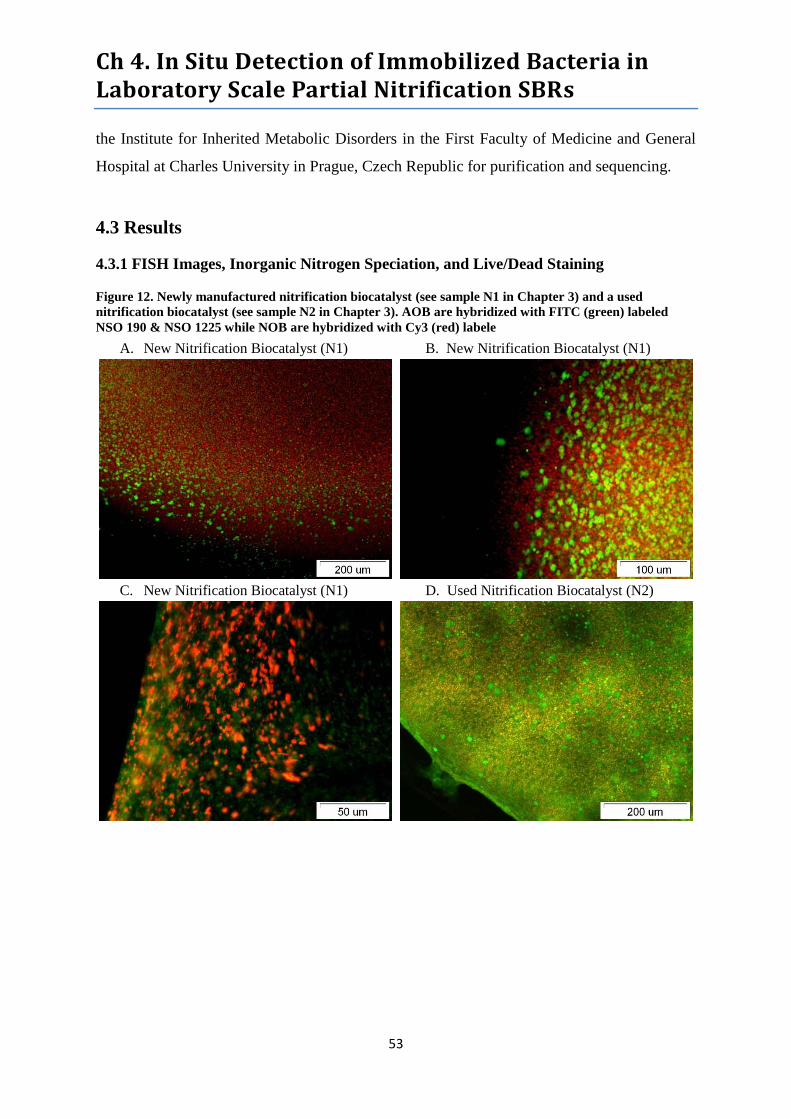
Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
53
the Institute for Inherited Metabolic Disorders in the First Faculty of Medicine and General
Hospital at Charles University in Prague, Czech Republic for purification and sequencing.
4.3 Results
4.3.1 FISH Images, Inorganic Nitrogen Speciation, and Live/Dead Staining
Figure 12. Newly manufactured nitrification biocatalyst (see sample N1 in Chapter 3) and a used
nitrification biocatalyst (see sample N2 in Chapter 3). AOB are hybridized with FITC (green) labeled
NSO 190 & NSO 1225 while NOB are hybridized with Cy3 (red) labele
A. New Nitrification Biocatalyst (N1) B. New Nitrification Biocatalyst (N1)
C. New Nitrification Biocatalyst (N1) D. Used Nitrification Biocatalyst (N2)

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
54
E. Used Nitrification Biocatalyst (N2) F. Used Nitrification Biocatalyst (N2): Cy3 only
Figure 12 Images A-F depict both new and used nitrification biocarriers hybridized
with AOB and NOB targeted probes. These images clearly show vibrant coloration consistent
with bacteria containing high concentrations of rRNA thus implying a high level of activity.
Nearly all of these images demonstrate that the distribution of AOB and NOB in Lentikats
Biocatalysts is not uniform but highly variable between individual pellets. Regardless, they
also demonstrate that each biocarrier contains substantial populations of both functional
groups. Images E and F both depict the same area of the same pellet. Image E, shows both
AOB and NOB while Image F shows only NOB. These images demonstrate the high
percentage of the biocarriers hydrogel matrix that is saturated with bacterial biomass while
also validating our hybridization technique. A close examination of both images reveals that
the bright green clusters of AOB in image E are merely blank spaces in image F, however
this is not always clear because of the 3 dimensional nature of the biocarriers.
Figure 13. Nitrogen speciation in control reactor effluent throughout operational life.
Throughout its operational life, Reactor C demonstrated complete nitrification of
ammonium to nitrate thus indicating the activity of both AOB and NOB. These results are

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
55
corroborated by Figure 14 Images A and B which clearly show that members of both
functional groups were present in biocarriers on day 30 of the experiment. In fact, the spike in
nitrite concentration to 45.07 mg/L on day 15 represents the only data point for this reactor
where the nitrite concentration exceeded 0.13 mg/L. Unfortunately, the next data point was
taken on day 49, so it is unclear exactly when the concentration of nitrite returned below
0.1 mg/L although it is likely to have occurred prior to sampling on day 30.
Table 12 below shows the residual concentrations of NH2OH in each reactor. Because
the protocol for measuring NH2OH was not discovered until this experiment was under way
there are only four data points from which to establish a baseline molar concentration of
hydroxylamine under control conditions in Reactor C. While this is too few data points to
draw definitive conclusions from, it is worth noting that the concentration never exceeded
6.05 µM NH2OH.
Figure 14. FISH images from control reactor (Reactor C) biocatalysts sampled on Day 30 of the
experiment. AOB are hybridized with FITC (green) labeled NSO 190 & NSO 1225 while NOB are
hybridized with Cy3 (red) labeled NIT3 and NTSPA 662 & NTSPA 712.
A. Day 30 B. Day 30
Table 13. Residual hydroxylamine concentrations measured in each reactor in µM
Day # Reactor A Reactor B Reactor C
87 18.16 2,664.24 6.05
91 15.13 605.51 6.05
93 51.47 3.03 3.94
107 4.54 3.33 4.54
112 13.02 3.02 NA
114 58.74 3.63 NA
122 363.31 6.05 NA
128 86.29 5.75 NA
132 4.48 5.44 NA

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
56
Figure 15 (below) shows the inorganic nitrogen speciation and NH2OH dosing in
Reactor A throughout its operational life. Note a similar spike in nitrite concentration
corresponding with the initial increase of influent ammonia to 300 mg/L on day 14.
Unfortunately, it is again unclear when this concentration returned to baseline levels of below
0.13 mg/L but it is presumed to be prior to sampling of biocarriers for hybridization on
day 30. It is also evident that the thermostat failure on day 87 corresponded with a massive
spike in ammonia to 624 mg/L and crash in nitrate production down to 10.8 mg/L. We
believe that the complete inhibition of both AOB and NOB evident at this time was due
mostly to temperature shock, but could also be a result of the combination of NH2OH dosing
and the high weekend dose (600 mg/L) of influent ammonia as well. The residual
concentration of NH2OH of 18.16 µM is above the expected baseline concentration however
it is still well below the 250 µM concentration reported by Kindaichi et al (2004) to inhibit
NOB in suspended cultures. Regardless, nitrification did recover significantly over the
following 12 days before the dose of NH2OH was increased to 16 mg/L*d on day 99, after
which nitrate concentrations fell steadily reaching their minimum levels of 5.3 mg/L on
day 128. Interestingly, this comes following the day 122 measurement in which the highest
concentration of residual NH2OH of 363.06 µM was measured in Reactor A. Perhaps most
interesting is that the dose of NH2OH was reduced on day 120, just prior to nitrate levels
bottoming out. As residual NH2OH then decreased back towards “background levels” of
below 10 µM despite continued dosing, nitrate production began a steady increase doubling
approximately every 20 days through the end the reactors operation, reaching 22.8 mg/L on
day 169.
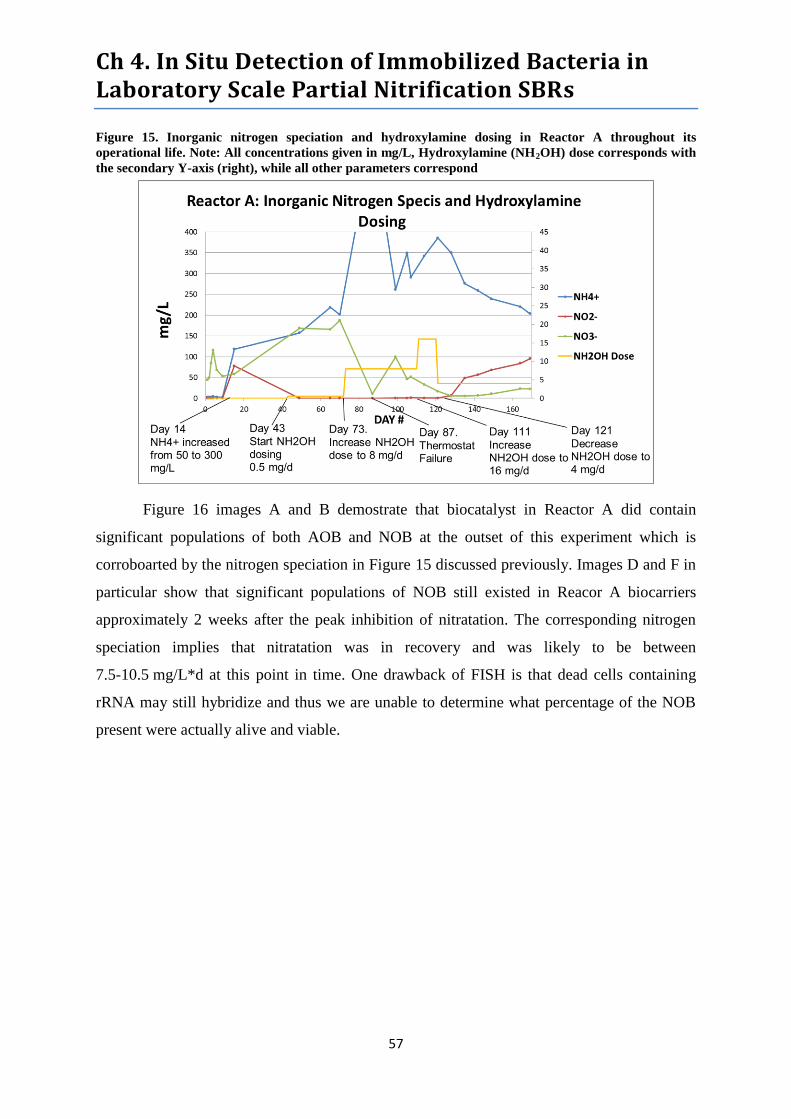
Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
57
Figure 15. Inorganic nitrogen speciation and hydroxylamine dosing in Reactor A throughout its
operational life. Note: All concentrations given in mg/L, Hydroxylamine (NH2OH) dose corresponds with
the secondary Y-axis (right), while all other parameters correspond
Figure 16 images A and B demostrate that biocatalyst in Reactor A did contain
significant populations of both AOB and NOB at the outset of this experiment which is
corroboarted by the nitrogen speciation in Figure 15 discussed previously. Images D and F in
particular show that significant populations of NOB still existed in Reacor A biocarriers
approximately 2 weeks after the peak inhibition of nitratation. The corresponding nitrogen
speciation implies that nitratation was in recovery and was likely to be between
7.5-10.5 mg/L*d at this point in time. One drawback of FISH is that dead cells containing
rRNA may still hybridize and thus we are unable to determine what percentage of the NOB
present were actually alive and viable.
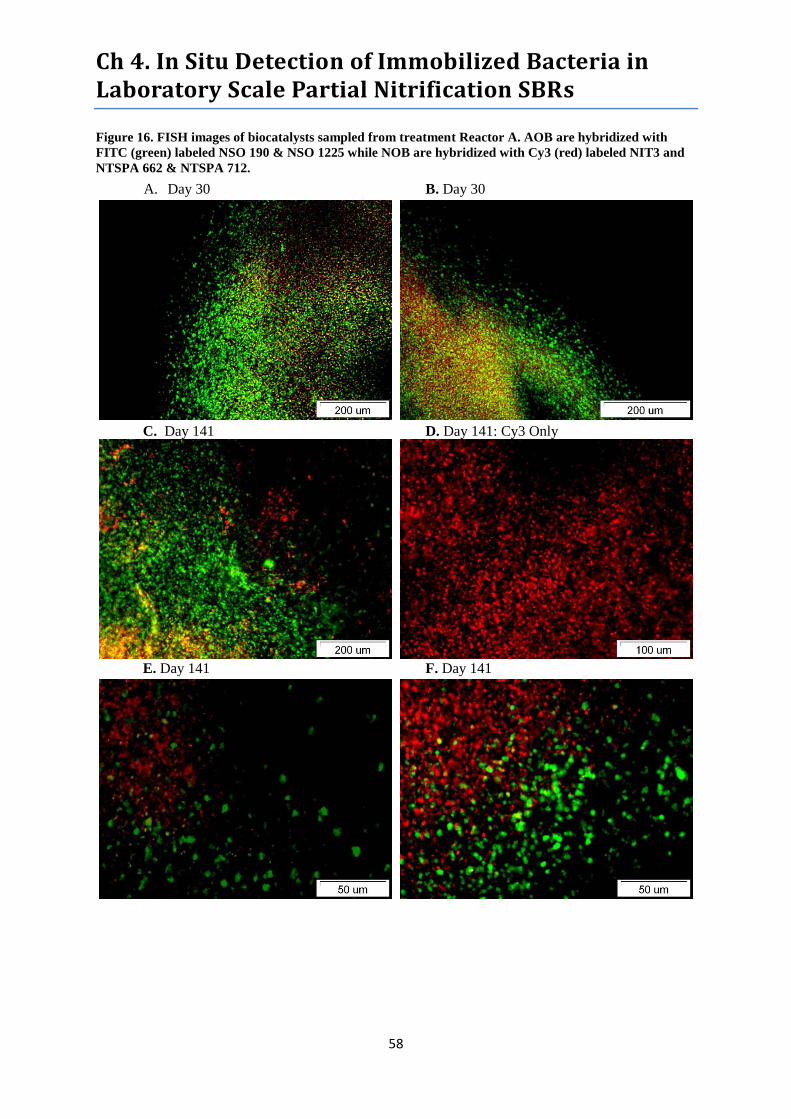
Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
58
Figure 16. FISH images of biocatalysts sampled from treatment Reactor A. AOB are hybridized with
FITC (green) labeled NSO 190 & NSO 1225 while NOB are hybridized with Cy3 (red) labeled NIT3 and
NTSPA 662 & NTSPA 712.
A. Day 30 B. Day 30
C. Day 141 D. Day 141: Cy3 Only
E. Day 141 F. Day 141

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
59
Figure 17. Inorganic nitrogen speciation and hydroxylamine dosing in Reactor B throughout its
operational life. Note: All concentrations given in mg/L, Hydroxylamine (NH2OH) dose corresponds with
the secondary Y-axis (right), while all other parameters correspond
Figure 17 shows that Reactor B exhibited nearly identical nitrogen speciation to
Reactors A and C prior to NH2OH dosing. The presence of NOB in Reactor B biocarriers is
also confirmed by Figure 18 Images A and B. Similar to Reactor A, Reactor B showed
inhibition of both AOB and NOB corresponding with the thermostat failure on day 87,
however in Reactor B this inhibition was more comprehensive and furthermore it coincided
with the highest dosing levels and highest residual concentrations of NH2OH. The amount of
residual NH2OH measured in Reactor B on day 87 was 88 mg/L or 2,664.24 µM which is just
over 10 times the 250 µM reported by Kindaichi et al. (2004) to inhibit NOB and 1.3 times
the 2000 µM they reported to inhibit AOB. Hydroxylamine dosing was then reduced by ¾
and subsequently completely ceased 4 days later. Interestingly, day 87 represented the lowest
measured level of nitrate in the reactor of 4.01 mg/L and these levels did not exceed 8.2 mg/L
until day 164, after which they hovered between 1-28 mg/L until commencing a steady
increase on day 274. Overall the data shows that nitrite levels exceeded nitrate levels in
Reactor B for as many as 182 days.
Figure 18 Images C-F show that despite a high degree of inhibition over the previous
54 days, NOB remained present in fairly large numbers in Reactor B biocarriers. These
images also show that the conditions inside of the reactor caused a high degree of mechanical
stress and breakdown of the PVA biocarriers, which is supported to a greater extent by
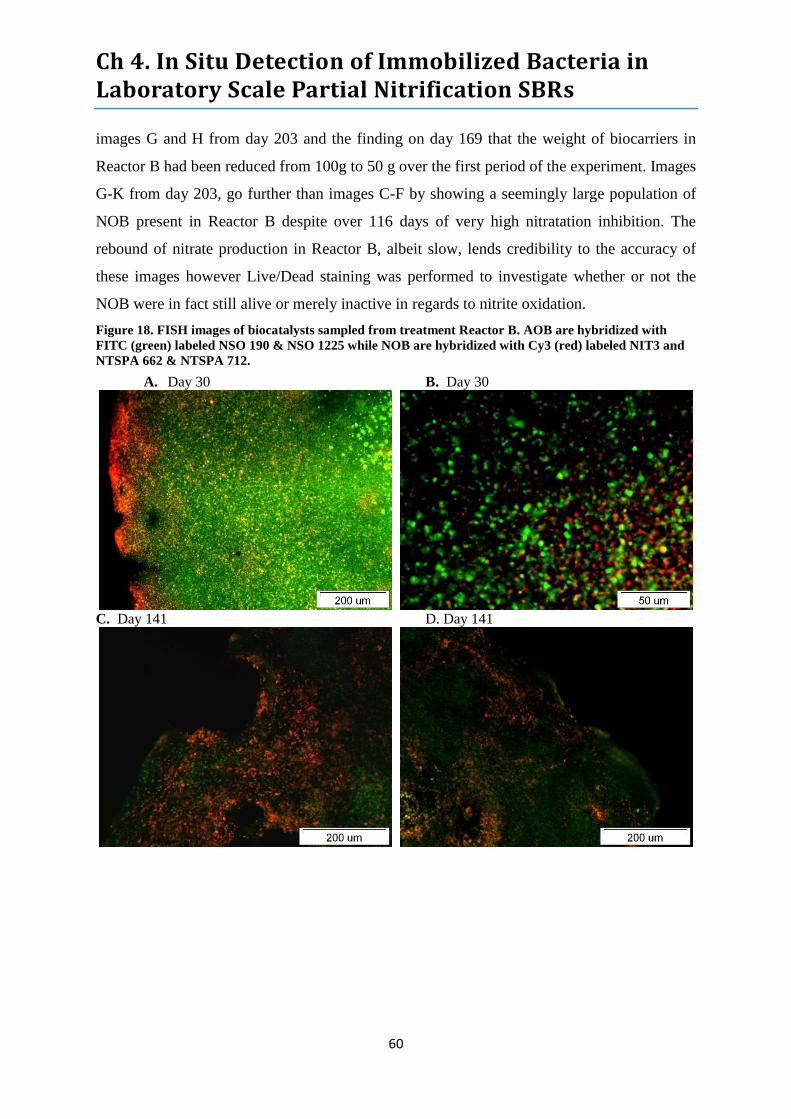
Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
60
images G and H from day 203 and the finding on day 169 that the weight of biocarriers in
Reactor B had been reduced from 100g to 50 g over the first period of the experiment. Images
G-K from day 203, go further than images C-F by showing a seemingly large population of
NOB present in Reactor B despite over 116 days of very high nitratation inhibition. The
rebound of nitrate production in Reactor B, albeit slow, lends credibility to the accuracy of
these images however Live/Dead staining was performed to investigate whether or not the
NOB were in fact still alive or merely inactive in regards to nitrite oxidation.
Figure 18. FISH images of biocatalysts sampled from treatment Reactor B. AOB are hybridized with
FITC (green) labeled NSO 190 & NSO 1225 while NOB are hybridized with Cy3 (red) labeled NIT3 and
NTSPA 662 & NTSPA 712.
A. Day 30 B. Day 30
C. Day 141 D. Day 141
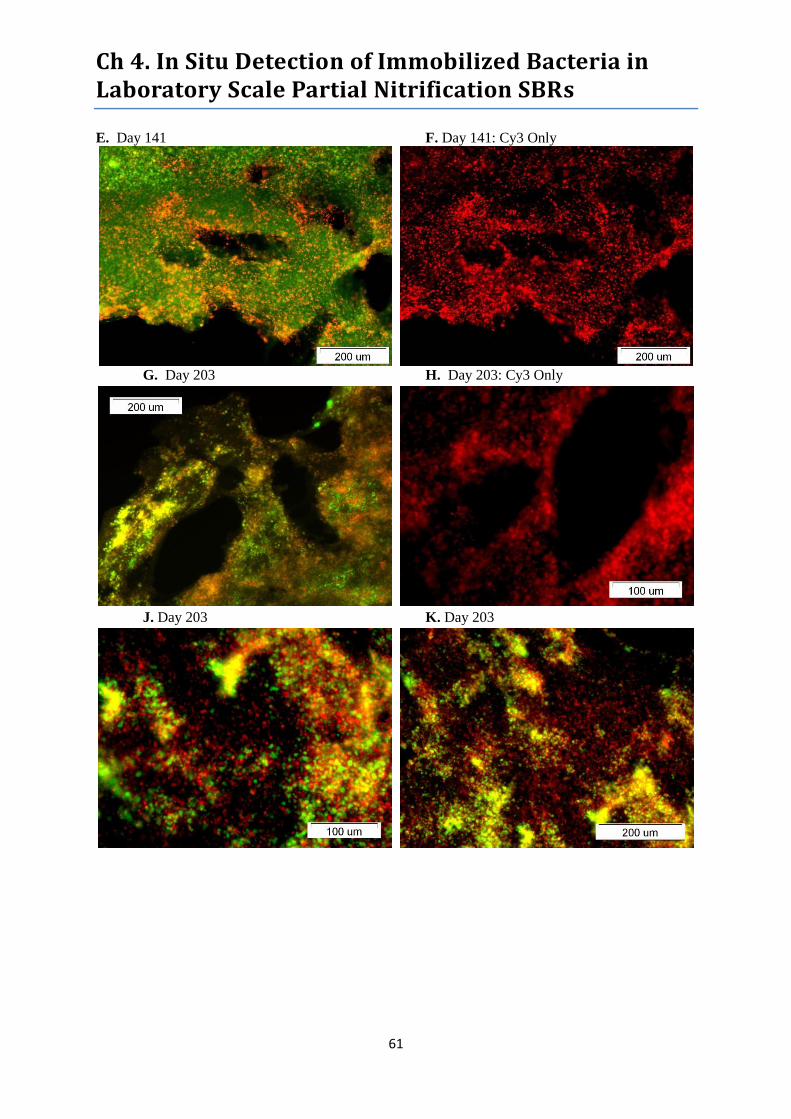
Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
61
E. Day 141 F. Day 141: Cy3 Only
G. Day 203 H. Day 203: Cy3 Only
J. Day 203 K. Day 203

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
62
Figure 19. Live/Dead Images of biocarriers sampled from Reactor B on Day 224, approximately 137 days
after inhibition of nitritation. Live bacteria appear green in color while dead bacteria appear red/orange.
A. Day 224 B. Day 224
Figure 19 Images A and B from day 224 indicate that it is indeed
possible that a significant portion of the bacteria within the biocarriers
were dead. Unfortunately with neither the mechanism of hydroxylamine
inhibition of nitratation or the ability to target only dead NOB it is not
possible to identify the population of dead bacteria. Still the fact remains
that nitratation in Reactor B had been severely restricted for over
137 days prior to Live/Dead staining and thus NOB would somehow
have to sustain their maintenance energy in order to survive this long a
period. Thus, it seems plausible that a majority of the dead bacteria in
these images are NOB although it cannot be stated with certainty.
4.3.2 DGGE and Sequencing
Figure 20 shows the DGGE profile of amplified 16S rDNA
extracted from Reactor B on day 274. The numbers in the image
correspond with the labels given to each band that was successfully
removed from the gel, reamplified, and sent for sequiencing. The results
from sequencing were obtained on May 29, 2013 and were inconclusive
due to very high background signals most likely caused by excessive
template DNA concentration due to noteable evaporation during PCR.
Alternative possibilites that should not be ruled out include
contamination, non-specific binding of primers, and UV damage.
Samples will be purified and resent for sequencing (if possible) and we
hope to present the results at the defense of this thesis on June 20, 2013.
Figure 20. DGGE
Profile of amplified
16S rDNA extracted
from Reactor B
biocarriers on Day
274.

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
63
4.4 Discussion
4.4.1 Inhibition of Nitratation
The mechanism through which hydroxylamine inhibits nitratation has not been well
characterized using modern techniques. Xu et al. (2012) cite a study by Deturk et al. (1958)
stating that hydroxylamine prevents the induction of the enzyme Nxr and affects protein
synthesis, however there do not seem to be any more recent follow up studies that definitively
prove or disprove this theory. Regardless, Xu et al. (2012) cites Hao et al. (1994) and Van der
Star et al. (2008) in stating that the inhibition of NOB by hydroxylamine is irreversible. This
is a critical assumption in assessing the performance of the partial nitrification reactors in this
experiment.
Kindaichi et al (2004) established that maintaining a hydroxylamine concentration of
250 µM in an RBC reactor fed with synthetic WW was sufficient to completely inhibit
nitratation. Xu et al. (2012) employed a slightly higher concentration of 300 µM NH2OH
dosed every 2 days to completely inhibit nitratation in an aerobic granule SBR. During their
experiment, Xu et al. (2012) ceased hydroxylamine dosing after complete nitritation had been
sustained and after 16 days nitratation began to exceed nitritation. This was attributed to the
presence and subsequent proliferation of NOB deeply embedded within aerobic granules
(confirmed by FISH) and thus shielded from exposure to and/or the effects of hydroxylamine
as well as washout. FISH images indicate that this population was extremely small which
would explain why the amount of nitrate produced throughout hydroxylamine dosing was
insignificant. The existence of such microenvironments free from sufficient hydroxylamine
concentrations to inhibit NOB is feasible considering that AOB consume hydroxylamine,
thus a reduced concentration deep within aerobic granules or suspended flocs is logical and
supported by the findings of Han et al. (2012).
Excluding the reactor crash on day 87, the inhibition of NOB in Reactor A peaked
after 37 days of hydroxylamine dosing at 242 µM followed by 10 days at 484 µM. After this
period the daily doses were dropped to 121 µM which is reported Hao et al. (1994) to be
sufficient to sustain established nitritation over nitratation in submerged aerobic filters.
Interestingly, Xu et al. (2012) performed batch tests which indicated that 150 µM was not
sufficient to suppress nitratation in an aerobic granule SBR. While nitrate production in
Reactor A did steadily increase (doubling every 20 days) through the end of the experiment,
nitrite production followed a seemingly proportional increase while ammonium decreased

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
64
over that same period. This may simply be a result of the reactor’s biota recovering from the
nearly continuous shocks evident in Figure 16 that occurred between days 60-125. These data
may therefore indicate a scenario similar to that described by Xu et al. (2012) whereby NOB
more deeply embedded in biocarriers were shielded from the peak hydroxylamine doses. As
conditions stabilized in the reactor, the may have been able to acclimate and continue
nitratation activity. It could also imply that the subsequent decrease in the daily dose to
121 µM was insufficient to prevent unaffected NOB from starting to grow outwards hence
the steady increase in nitratation.
Unfortunately, it seems unlikely that Reactor A had reached stable state equilibrium
with regards to nitritation performance at the conclusion of the experiment on day 169, thus it
is not appropriate to conclude whether or not sustained nitritation/nitratation was achieved.
Regardless, on the final day of operation, nitrate production was 23% as high as nitrite
production while Xu et al. (2012) reported < 1% in their reactors. If this did represent a stable
state without proliferation of NOB it could mean that NOB immobilized in Lentikat’s
Biocatalysts were in fact more resilient towards hydroxylamine inhibition than those in
aerobic granules or RBC biofilms. FISH images presented in Figure 16 show that NOB were
present in comparable quantities before and 14 days after peak hydroxylamine induced
inhibition.
The peak of nitratation inhibition in Reactor B occurred following 23 days of
hydroxylamine dosing at 1,211 µM. On the day of the thermostat failure and subsequent
reactor crash, the residual hydroxylamine concentration in Reactor B was high enough to
inhibit even AOB activity. Interestingly, nitritation recovered and reached more or less stable
performance 34 days later, which it sustained for an additional 48 days until the conclusion of
the synthetic WW portion of experiment on day 169. Despite the fact that hydroxylamine
dosing was ceased on day 91, nitratation remained severely inhibited over the final 78 days
preceding the switch from synthetic WW to digestate.. Following steady sate nitritation,
nitratation jumped as high as 19% of nitritation for one measurement but spent most of the
time from day 121-169 around 5% and ended on day 169 at 4.7%. Even after Reactor B was
switched to diluted digester effluent, nitratation did not exceed nitritation until a total of 182
days had passed since peak inhibition began.
The diffusion of hydroxylamine into the biocarriers may logically be a critical factor
in the difference between nitratation inhibition observed both Reactors A and B. The higher

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
65
concentration of hydroxylamine reached in Reactor B should have been accompanied with
higher concentrations diffusing deeper within biocarriers. It is possible that the only NOB
spared from exposure to inhibitory concentrations of hydroxylamine were so deeply
embedded in the biocarrier’s rigid hydrogel matrix and crowded by other organisms that
despite having ample substrate, the lack of space to reproduce and expand prevented them
from proliferating soon after dosing was ceased. With inhibitory concentrations of
hydroxylamine not diffusing as far into biocarriers in Reactor A, less deeply embedded NOB
as well as a greater proportion of the population overall may have avoided the full effects of
inhibition, which might explain the more steady recovery of nitratation observed.
The results do not clearly demonstrate threshold hydroxylamine concentrations
needed to achieve long term stable nitritation over nitratation using Lentikat’s Nitrifying
Biocatalysts. For this reason a cost analysis of large scale hydroxylamine dosed SBR’s for
partial nitrification would not be appropriate. The results do however show that the short term
high doses in Reactor B were far more effective at achieving a greater degree of long term
NOB inhibition than the lower peak dose combined with chronic low doses in Reactor A.
4.4.2 In situ detection and characterization of NOB community
The results of fluorescence in situ hybridization from both Reactors A and B indicate
that despite successful inhibition of nitratation, NOB washout was not significant. This is
most strongly demonstrated by Figure 18 Images C-K which show that significant
populations of NOB were present in Reactor B biocarriers both 54 and 116 days after peak
inhibition. Given the extremely low level of nitratation over this period, these finding imply
that one or more of the following scenarios was likely occurring: 1) the signal represented
non-specific binding of oligonucleotide probes, 2) the signal represented dead NOB cells
containing rRNA and thus remained susceptible to hybridization, 3) NOB were in fact present
and active and had switched to alternative metabolic pathways for survival 4) the signal
represented NOB that were alive and continued to oxidize nitrite to some degree. In the
absence of results from rDNA sequencing, it is impossible to determine exactly which species
of NOB were present following hydroxylamine treatments. Furthermore the DGGE results in
Figure 3 indicate that even freshly manufactured biocarriers contain more than the mere two
nitrifying organisms advertised, thus this discussion will consider the possibility that a
broader array of NOB genera were potentially present.

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
66
The non-specific binding of the oligonucleotide hybridization probes seems unlikely.
According to Nielsen et al (2009), the probes employed in this study are among the most
thoroughly tested and reliable available for use in in situ identification of NOB. The
hybridization of dead NOB cells on the other hand is likely occurring to at least some extent
particularly in Figure 18 Images C-F. At this stage in the reactors operation, it was still being
fed with synthetic wastewater, thus there should not have been any seeding of heterotrophic
organisms that might degrade dead cell material. Following the switch from synthetic WW to
anaerobic digester effluent, it is more likely that such organisms would be present in the
reactor to remove dead biomass, however the structure of the biocarriers may still have
impeded heteretrophic organisms from accessing and clearing out all dead NOB. While the
Live/Dead stains shown in Figure 19 were performed following the switch in reactor feed,
they indicate that a potentially significant portion of biomass present was dead. While it’s
unclear if this dead biomass is on the surface of the biocarrier, within the biocarrier, or even
how deep within the biocarrier it may be, it is possible, although not confirmable, that a
significant proportion are NOB. Given the fact that nitratation was not 100% inhibited
however, this potential scenario alone cannot account for all NOB present.
The results of chemical analyses clearly indicate that some NOB remained actively
performing nitratation in both reactors following peak inhibition. Based upon the extremely
low production of nitrate in Reactor B during this period however, it’s likely that only very
deeply embedded NOB continued nitratation, thus it’s possible that only a fraction if any of
the NOB shown in FISH images fit this description. An interesting alternative is that some
proportion of NOB inhibited by hydroxylamine were able to shift to an alternative substrate
for survival. The arguments in favor of this scenario include the well documented metabolic
flexibility of Nitrobacter which are known to metabolize simple organic compounds like
pyruvate or reduce nitrite to nitric oxide under anaerobic conditions (Deni et al., 2004; Ahlers
et al., 1990). Given that the reactor was maintained under constant aeration it is unlikely that
anaerobic nitrite reduction was occurring. On the other hand, the ability of Nitrobacter to
metabolize simple organic compounds may have been significant to their survival and may
explain spikes in activity just prior to, and especially in the months following the switch from
synthetic WW to digester effluent.
Deni et al. (2004) note that Nitrobacter populations were more active in soils
containing organic metabolites from diesel fuel degraded by other organisms compared to

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
67
populations in uncontaminated soils, implying that they survive better under mixotrophic
conditions. While Xu et al. (2012) found that most NOB were washed out of aerobic sludge
granules following hydroxylamine inhibition, it is possible that the rigid matrix of biocarriers
and lack of heterotrophic competition allowed them to maintain a significant presence despite
conditions in which they would normally be out competed due to retarded growth. A major
problem with this hypothesis is that the COD of synthetic WW was insignificant and thus the
only source of organic substrate should have come from dead biomass within the reactor.
Given the numerous shocks experience by Reactor B it is likely that dead biomass was
available, however the theoretical lack of heterotrophs needed to metabolize complex
organics into the simple compounds usable by Nitrobacter casts doubt on this theory.
Another detractor from this theory is that if the NOB present were alive but starving due to
substrate limitation a decrease in rRNA content would be expected and would result in a drop
in FISH signal strength while cells that were suddenly killed by shock could retain higher
levels of rRNA if not lysed (Hawkins et al., 2011). Unfortunately, it is not clear from our
FISH analyses whether or not such a drop in signal strength occurred.
The plausibility of this alternative substrate scenario increases substantially following
the switch from synthetic WW to anaerobic digestate. The first reason for this is that the
digestate theoretically contained a higher COD concentration and a diverse array of
microorganisms not present in the synthetic WW, although there is no data to show this. In
addition to the boost in simple organic compounds directly useable by Nitrobacter, the
addition of heterotrophic organisms to the reactor likely increased the degradation rate of
dead biomass already present in/on biocarriers. This could not only free up space for NOB
expansion but could also carve channels that link deeply embedded NOB performing
nitratation to such newly cleared spaces opening the door for proliferation. The 5 independent
and unsustained spikes in nitrate concentration in Reactor B between days 150-250 could be
explained by spikes in the availability of simple COD compounds to the limited population of
Nitrobacter performing nitratation and in turn those NOB surviving solely on COD. The
eventual sustained increase in nitratation following day 254 could be indicative of either
seeding of fresh NOB from digestate or the proliferation of endemic NOB freed from the
depths of biocarriers by biologically created channels, the mechanical breakdown of the
biocarriers themselves, or a combination of both. It’s difficult to conclude which is more
likely considering that the presence of NOB in digestate is rare but occasionally happens

Ch 4. In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial Nitrification SBRs
68
(Regueiro et al., 2012) and mechanical breakdown of biocarriers should have manifested
itself sooner. If neither is the case, the most likely alternative would be that the effects of
hydroxylamine are in fact reversible over time, possibly following reproduction which most
NOB subjected to hydroxylamine inhibition likely don’t get the opportunity to undergo prior
to washout. In essence, this may be the first study in which a long term investigation of the
effects of hydroxylamine on affected NOB has been possible because they do not appear to
washout from biocarriers unlike RBC biofilms and aerobic granules.
4.5 Chapter 4 Conclusions
These results demonstrate that up to 180 days of significant nitritation inhibition is
achievable after treating Lentikat’s Nitrifying Biocatalysts with only short term highly
concentrated doses of hydroxylamine. On the other hand, even when subjected to lower doses
comparable to those used in studies by Kindaichi et al (2004) and Xu et al (2012), nitratation
recovery by immobilized NOB proceeded more slowly than in RBC biofilms or aerobic
granules respectively.
These results also indicate that despite a high degree of hydroxylamine induced
nitratation inhibition, immobilized NOB were not significantly washed out of Lentikat’s
Nitrifying Biocatalysts. This finding may present a unique opportunity to investigate the long
term effects of hydroxylamine on NOB as well as characterizing the mechanisms through
which inhibition and potential nitratation recovery occur.
NOB detected by FISH within biocatalysts following inhibition are believed to
include: a small fraction performing limited nitratation, a potentially significant proportion of
dead biomass, an unknown fraction that switched to alternative organic substrates for survival,
and in one reactor an unknown fraction that may have been seeded from anaerobic digester
effluent.
Finally, the eventual recovery of nitratation observed in Reactor A was likely due to
biota acclimating to system shocks and less likely, but potentially, the proliferation of
endemic NOB unaffected by peak hydroxylamine doses. The recovery of nitratation in
Reactor B is most likely due to seeding of NOB from digester effluent, the proliferation of
endemic NOB unaffected by peak hydroxylamine inhibition, and/or the reversal of
hydroxylamine effects over time.

Ch 5. Summary of Conclusions
69
5.1 Genomic DNA Isolation and Amplification from Bacteria Immobilized in
Poly Vinyl Alcohol Biocarriers
All commercial DNA Isolation kits tested were compatible with Lentikat’s Biocatalysts
and yielded DNA of sufficient quantities and purities for downstream PCR-DGGE analyses.
The processing times for each kit were highly comparable except for the Masterpure™ DNA
purification kit which took significantly longer. On the other hand, the Masterpure™ DNA
purification kit produced less than half the mass of waste and cost only half as much per
sample as its next closest competitor which was the Chemagic DNA Bacteria kit in both
instances.
The Masterpure™ DNA purification kit and Chemagic DNA Bacteria kit both yielded the
highest median quantities of DNA in µg/g for all samples across the board. In terms of
isolated DNA purity, the Powersoil® DNA Isolation kit outperformed all others across the
board with the Masterpure™ DNA Purification kit ranking second.
All kits demonstrated a high degree of similarity and thus low degree of extraction bias in
DGGE phylogenetic comparisons. The QIAmp® DNA Stool kit performed the best in terms
of phylogenetic richness, however no other kit was significantly worse. Though the data is
limited, LN enhanced lysis of PVA biocarriers showed promise in mitigating extraction
biases and increasing the phylogenetic richness of isolated DNA. Given the high
phylogenetic similarity between all DNA isolation kit extracts, the potential of enhanced lysis
techniques for optimizing community characterization, DNA purity, and to a lesser extent
yield should be considered most heavily in establishing laboratory protocol for working with
Lentikat’s Biocatalysts. Therefore, the Powersoil® DNA isolation kit is recommended as the
best option from those examined for applications that require the highest degree of purity as
well as further investigations of enhanced cell lysis techniques. This holds true especially
when high DNA yield is not essential and sample mass is abundant. For applications where
higher DNA yield is required or sample mass is limited, the Masterpure™ DNA Purification
kit is recommended.
5.2 In Situ Detection of Immobilized Bacteria in Laboratory Scale Partial
Nitrification SBRs
The investigation of laboratory partial nitrification SBRs demonstrated that up to 180
days of significant nitritation inhibition is achievable after treating Lentikat’s Nitrifying
Biocatalysts with only short term highly concentrated doses of hydroxylamine. On the other
hand, even when subjected to lower chronic doses comparable to those used in studies by

Ch 5. Summary of Conclusions
70
Kindaichi et al (2004) and Xu et al (2012), nitratation recovery by immobilized NOB
proceeded more slowly than in RBC biofilms or aerobic granules respectively.
Results also indicate that despite a high degree of hydroxylamine induced nitratation
inhibition, immobilized NOB were not significantly washed out of Lentikat’s Nitrifying
Biocatalysts. This finding may present a unique opportunity to investigate the long term
effects of hydroxylamine on NOB as well as characterizing the mechanisms through which
inhibition and potential nitratation recovery occur.
NOB detected by FISH within biocatalysts following inhibition are believed to include: a
small fraction performing limited nitratation, a potentially significant proportion of dead
biomass, an unknown fraction that switched to alternative organic substrates for survival, and
in one reactor an unknown fraction that may have been seeded from anaerobic digester
effluent.
Finally, the eventual recovery of nitratation observed in Reactor A was likely due to biota
acclimating to system shocks and less likely, but potentially, the proliferation of endemic
NOB unaffected by peak hydroxylamine doses. The recovery of nitratation in Reactor B is
most likely due to seeding of NOB from digester effluent, the proliferation of endemic NOB
unaffected by peak hydroxylamine inhibition, and/or the reversal of hydroxylamine effects
over time.

References
71
1. Ahlers, B., König, W. and Bock, E. (1990), Nitrite reductase activity in Nitrobacter vulgaris.
FEMS Microbiology Letters, 67: 121–126. doi: 10.1111/j.1574-6968.1990.tb13847.x
2. Alaeddini, R. (2012). Forensic implications of PCR inhibition--A review. Forensic Science
International. Genetics, 6(3), 297–305. doi:10.1016/j.fsigen.2011.08.006
3. American Public Health Association (APHA), (1992). Standard Methods of Water and
Wastewater: 18th Edition. American Public Health Association, American Water Works
Association, & Water Environment Federation. Washington D.C., USA
4. American Public Health Association (APHA), (2012). Standard Methods of Water and
Wastewater: 22nd
Edition. American Public Health Association, American Water Works
Association, & Water Environment Federation. Washington D.C., USA
5. Alain, K., Callac, N., Ciobanu, M.-C., Reynaud, Y., Duthoit, F., & Jebbar, M. (2011). DNA
extractions from deep subseafloor sediments: novel cryogenic-mill-based procedure and
comparison to existing protocols. Journal of Microbiological Methods, 87(3), 355–62.
doi:10.1016/j.mimet.2011.09.015
6. Amann, R. I., Ludwig, W., & Schleifer, K. H. (1995a). Phylogenetic identification and in situ
detection of individual microbial cells without cultivation. Microbiological Reviews, 59(1), 143–
69.
7. Amann, R. I. (1995b). In situ identification of micro-organisms by whole cell hybridization with
rRNA taargeted nucleic acid probes. Molecular Microbial Ecology Manual. 3.3.6, 1-15, 1995
8. Aslan, S., & Dahab, M. (2008). Nitritation and denitritation of ammonium-rich wastewater using
fluidized-bed biofilm reactors. Journal of Hazardous Materials, 156(1-3), 56–63.
doi:10.1016/j.jhazmat.2007.11.112
9. Barber, W. & Stuckey, D. (2000). Nitrogen removal in a modified anaerobic baffled reactor
(ABR): 2, nitrification. Water Research, 34(9), 2423–2432. doi:10.1016/S0043-1354(99)00426-1
10. Boušková, A., Mrákota, J., Stloukal, R., Trögl, J., Pilařová, V., Křiklavová, L., & Lederer, T.
(2011). Three examples of nitrogen removal from industrial wastewater using Lentikats
Biotechnology. Desalination, 280(1-3), 191–196. doi:10.1016/j.desal.2011.07.001
11. Camargo, J. a, & Alonso, A. (2006). Ecological and toxicological effects of inorganic nitrogen
pollution in aquatic ecosystems: A global assessment. Environment International, 32(6), 831–49.
doi:10.1016/j.envint.2006.05.002
12. Cardinali-Rezende, J., Colturato, L. F. D. B., Colturato, T. D. B., Chartone-Souza, E.,
Nascimento, A. M. a, & Sanz, J. L. (2012). Prokaryotic diversity and dynamics in a full-scale
municipal solid waste anaerobic reactor from start-up to steady-state conditions. Bioresource
Technology, 119, 373–83. doi:10.1016/j.biortech.2012.05.136
13. Cho, S., Fujii, N., Lee, T., & Okabe, S. (2011). Development of a simultaneous partial
nitrification and anaerobic ammonia oxidation process in a single reactor. Bioresource
Technology, 102(2), 652–9. doi:10.1016/j.biortech.2010.08.031
14. Dalsgaard, T., Thamdrup, B., & Canfield, D. E. (2005). Anaerobic ammonium oxidation
(anammox) in the marine environment. Research in Microbiology, 156(4), 457–64.
doi:10.1016/j.resmic.2005.01.011

References
72
15. Dauphin, L. a, Moser, B. D., & Bowen, M. D. (2009). Evaluation of five commercial nucleic acid
extraction kits for their ability to inactivate Bacillus anthracis spores and comparison of DNA
yields from spores and spiked environmental samples. Journal of Microbiological Methods,
76(1), 30–7. doi:10.1016/j.mimet.2008.09.004
16. Daverey, A., Su, S.-H., Huang, Y.-T., Chen, S.-S., Sung, S., & Lin, J.-G. (2013). Partial
nitrification and anammox process: A method for high strength optoelectronic industrial
wastewater treatment. Water Research, 47(9), 2929–37. doi:10.1016/j.watres.2013.01.028
17. Deni, J., & Penninckx, M. J. (2004). Influence of long-term diesel fuel pollution on nitrite-
oxidising activity and population size of nitrobacter spp in soil. Microbiological Research,
159(4), 323–9. doi:10.1016/j.micres.2004.06.004
18. Feng, Y.-J., Tseng, S.-K., Hsia, T.-H., Ho, C.-M., & Chou, W.-P. (2007). Partial nitrification of
ammonium-rich wastewater as pretreatment for anaerobic ammonium oxidation (Anammox)
using membrane aeration bioreactor. Journal of Bioscience and Bioengineering, 104(3), 182–7.
doi:10.1263/jbb.104.182
19. Fernandes, H., Jungles, M. K., Hoffmann, H., Antonio, R. V, & Costa, R. H. R. (2013). Full-scale
sequencing batch reactor (SBR) for domestic wastewater: performance and diversity of microbial
communities. Bioresource Technology, 132, 262–8. doi:10.1016/j.biortech.2013.01.027
20. Ferrero, M. a., Menoyo, E., Lugo, M. a., Negritto, M. a., Farías, M. E., Anton, A. M., & Siñeriz,
F. (2010). Molecular characterization and in situ detection of bacterial communities associated
with rhizosphere soil of high altitude native Poaceae from the Andean Puna region. Journal of
Arid Environments, 74(10), 1177–1185. doi:10.1016/j.jaridenv.2010.04.008
21. Frear, D.S and Burrell, R.C., (1955). Specttrophotometric Method for Determining
Hydroxylamine Reductase Activity in Higher Plants. Analytical Chemistry. Vol 27, No. 10, 1664-
1665. October 1955.
22. Gilbride, K. a, Lee, D.-Y., & Beaudette, L. a. (2006). Molecular techniques in wastewater:
Understanding microbial communities, detecting pathogens, and real-time process control.
Journal of Microbiological Methods, 66(1), 1–20. doi:10.1016/j.mimet.2006.02.016
23. Gilg, I. C., Amaral-Zettler, L. a, Countway, P. D., Moorthi, S., Schnetzer, A., & Caron, D. a.
(2010). Phylogenetic affiliations of mesopelagic acantharia and acantharian-like environmental
18S rRNA genes off the southern California coast. Protist, 161(2), 197–211.
doi:10.1016/j.protis.2009.09.002
24. Gu, S., Wang, S., Yang, Q., Yang, P., & Peng, Y. (2012). Start up partial nitrification at low
temperature with a real-time control strategy based on blower frequency and pH. Bioresource
Technology, 112, 34–41. doi:10.1016/j.biortech.2011.12.028
25. Han, Y., Liu, J., Guo, X., & Li, L. (2012). Micro-environment characteristics and microbial
communities in activated sludge flocs of different particle size. Bioresource Technology, 124(3),
252–8. doi:10.1016/j.biortech.2012.08.008
26. Hawkins, S., Robinson, K., Layton, A., & Sayler, G. (2012). Molecular indicators of Nitrobacter
spp. population and growth activity during an induced inhibition event in a bench scale
nitrification reactor. Water Research, 46(6), 1793–802. doi:10.1016/j.watres.2011.12.053

References
73
27. Hayes, D., Izzard, L., & Seviour, R. (2011). Microbial ecology of autothermal thermophilic
aerobic digester (ATAD) systems for treating waste activated sludge. Systematic and Applied
Microbiology, 34(2), 127–38. doi:10.1016/j.syapm.2010.11.017
28. Hesham, A. E.-L., Qi, R., & Yang, M. (2011). Comparison of bacterial community structures in
two systems of a sewage treatment plant using PCR-DGGE analysis. Journal of Environmental
Sciences, 23(12), 2049–2054. doi:10.1016/S1001-0742(10)60647-X
29. ISO 7890-1. Water Quality--Determination of nitrate -- Part 1: 2,6-Dimethylphenol
spectrometric method. 1986
30. Johanidesova, I. (2013). Evaluation of the possibility of maintaining partial nitrification using
immobilized microbial cultures. Unpublished Master of Science Thesis. VSCHT Department of
Water Technology and Environmental Engineering. Available in VSCHT Library.
31. Jetten, M. S. M., Niftrik, L. Van, Strous, M., Kartal, B., Keltjens, J. T., & Op den Camp, H. J. M.
(2009). Biochemistry and molecular biology of anammox bacteria. Critical Reviews in
Biochemistry and Molecular Biology, 44(2-3), 65–84. doi:10.1080/10409230902722783
32. Jiang, Y.-X., Wu, J.-G., Yu, K.-Q., Ai, C.-X., Zou, F., & Zhou, H.-W. (2011). Integrated lysis
procedures reduce extraction biases of microbial DNA from mangrove sediment. Journal of
Bioscience and Bioengineering, 111(2), 153–7. doi:10.1016/j.jbiosc.2010.10.006
33. Kawagoshi, Y., Fujisaki, K., Tomoshige, Y., Yamashiro, K., Wei, Q., & Qiao, Y. (2012).
Temperature effect on nitrogen removal performance and bacterial community in culture of
marine anammox bacteria derived from sea-based waste disposal site. Journal of Bioscience and
Bioengineering, 113(4), 515–20. doi:10.1016/j.jbiosc.2011.11.024
34. Kindaichi, T., Okabe, S., Satoh, H., Watanabe, Y. (2004). Effects of hydroxylamine on microbial
community structure and function of autotrophic nitrifying biofilms determined by in situ
hybridization and the use of microelectrodes. Water Science and Technology. Vol. 49:11-12 pg.
61-8
35. Kloep, F., Ske, È., Neu, T. R., & Ro, I. (2000). Performance and Microbial Structure of a
Nitrifying Fluidized-Bed Reactor. Water Resources 34(1), 311–319.
36. Kodzius, R., Xiao, K., Wu, J., Yi, X., Gong, X., Foulds, I. G., & Wen, W. (2012). Inhibitory
effect of common microfluidic materials on PCR outcome. Sensors and Actuators B: Chemical,
161(1), 349–358. doi:10.1016/j.snb.2011.10.044
37. Kong, Q., Liang, S., Zhang, J., Xie, H., Miao, M., & Tian, L. (2013). N(2)O emission in a partial
nitrification system: dynamic emission characteristics and the ammonium-oxidizing bacteria
community. BioresourceTtechnology, 127, 400–6. doi:10.1016/j.biortech.2012.10.011
38. Kuenen, J. G., & Robertson, L. a. (1994). Combined nitrification-denitrification processes. FEMS
Microbiology Reviews, 15(2-3), 109–117. doi:10.1111/j.1574-6976.1994.tb00129.x
39. Kumar, M., & Lin, J.-G. (2010). Co-existence of anammox and denitrification for simultaneous
nitrogen and carbon removal--Strategies and issues. Journal of Hazardous Materials, 178(1-3), 1–
9. doi:10.1016/j.jhazmat.2010.01.077
40. Lan, C.-J., Kumar, M., Wang, C.-C., & Lin, J.-G. (2011). Development of simultaneous partial
nitrification, anammox and denitrification (SNAD) process in a sequential batch reactor.
Bioresource Technology, 102(9), 5514–9. doi:10.1016/j.biortech.2010.11.024

References
74
41. Liu, Xin-chun, Yu, Z., Min, Y., Zhen-yu, W., & Wen-zhou, L. V. (2007). Analysis of bacterial
community structures in two sewage treatment plants with different sludge properties and
treatment performance by nested PCR-DGGE method. Journal of Environmental Science (China)
19, 60–66.
42. Loy, A. Horn, M., & Wagner, M. (2003). ProbeBase:an online resource for rRNA-targeted
oligonucleotide probes. Nucleic Acids Research, 31(1), 319–321. doi:10.1093/nar/gkg015
43. Lücker, S., Steger, D., Kjeldsen, K. U., MacGregor, B. J., Wagner, M., & Loy, A. (2007).
Improved 16S rRNA-targeted probe set for analysis of sulfate-reducing bacteria by fluorescence
in situ hybridization. Journal of Microbiological Methods, 69(3), 523–8.
doi:10.1016/j.mimet.2007.02.009
44. Lusk, T. S., Strain, E., & Kase, J. a. (2013). Comparison of six commercial DNA extraction kits
for detection of Brucella neotomae in Mexican and Central American-style cheese and other milk
products. Food Microbiology, 34(1), 100–5. doi:10.1016/j.fm.2012.11.007
45. Machado, A., Almeida, C., Carvalho, A., Boyen, F., Haesebrouck, F., Rodrigues, L., Cerca, N., et
al. (2013). Fluorescence in situ hybridization method using a peptide nucleic acid probe for
identification of Lactobacillus spp. in milk samples. International Journal of Food Microbiology,
162(1), 64–70. doi:10.1016/j.ijfoodmicro.2012.09.024
46. Mahmoudi, N., Slater, G. F., & Fulthorpe, R. R. (2011). Comparison of commercial DNA
extraction kits for isolation and purification of bacterial and eukaryotic DNA from PAH-
contaminated soils. Canadian Journal of Microbiology. 628, 623–628. doi:10.1139/W11-049
47. Mohd Yasin, N. H., Rahman, N. A., Man, H. C., Mohd Yusoff, M. Z., & Hassan, M. A. (2011).
Microbial characterization of hydrogen-producing bacteria in fermented food waste at different
pH values. International Journal of Hydrogen Energy, 36(16), 9571–9580.
doi:10.1016/j.ijhydene.2011.05.048
48. Mullis, K., Faloona, F., Scharf, S., Saiki, R., Horn, G., Erlich, H. (1986). Specific enzymatic
amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symposia on
Quantitative Biology. Volume: 51 Pt. 1, Issue 1, Pages 263-273
49. Muyzer, G., De Waal, E. C., & Uitterlinden, a G. (1993). Profiling of complex microbial
populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-
amplified genes coding for 16S rRNA. Applied and Environmental Microbiology, 59(3), 695–
700. Retrieved from
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=202176&tool=pmcentrez&rendertype
=abstract
50. Muyzer, G., & Smalla, K. (1998). Application of denaturing gradient gel electrophoresis (DGGE)
and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van
Leeuwenhoek, 73(1), 127–41. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9602286
51. Nielsen, P. H., Daims, H. & Lemmer, H. (2009). FISH Handbook for Biological Wastewater
Treatment, London, UK: IWA Publishing.
52. Niu, C., Geng, J., Ren, H., Ding, L., & Xu, K. (2012). The cold adaptability of microorganisms
with different carbon source in activated sludge treating synthetical wastewater. Bioresource
Technology, 123, 66–71. doi:10.1016/j.biortech.2012.06.110

References
75
53. Okabe, S., Oshiki, M., Takahashi, Y., & Satoh, H. (2011). Development of long-term stable
partial nitrification and subsequent anammox process. Bioresource Technology, 102(13), 6801–7.
doi:10.1016/j.biortech.2011.04.011
54. Okten, H. E., Yilmaz, L. S., & Noguera, D. R. (2012). Exploring the in situ accessibility of small
subunit ribosomal RNA of members of the domains Bacteria and Eukarya to oligonucleotide
probes. Systematic and Applied Microbiology, 35(8), 485–95. doi:10.1016/j.syapm.2011.11.001
55. Osaka, T., Shirotani, K., Yoshie, S., & Tsuneda, S. (2008). Effects of carbon source on
denitrification efficiency and microbial community structure in a saline wastewater treatment
process. Water Research, 42(14), 3709–18. doi:10.1016/j.watres.2008.06.007
56. Park, J. K., & Chang, H. N. (2000). Microencapsulation of microbial cells. Biotechnology
Advances, 18(4), 303–19. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/14538106
57. Park, S., & Bae, W. (2009). Modeling kinetics of ammonium oxidation and nitrite oxidation under
simultaneous inhibition by free ammonia and free nitrous acid. Process Biochemistry, 44(6), 631–
640. doi:10.1016/j.procbio.2009.02.002
58. Postollec, F., Falentin, H., Pavan, S., Combrisson, J., & Sohier, D. (2011). Recent advances in
quantitative PCR (qPCR) applications in food microbiology. Food Microbiology, 28(5), 848–61.
doi:10.1016/j.fm.2011.02.008
59. Quigley, L., O’Sullivan, O., Beresford, T. P., Paul Ross, R., Fitzgerald, G. F., & Cotter, P. D.
(2012). A comparison of methods used to extract bacterial DNA from raw milk and raw milk
cheese. Journal of Applied Microbiology, 113(1), 96–105. doi:10.1111/j.1365-2672.2012.05294.x
60. Randall, D. J., & Tsui, T. K. N. (2002). Ammonia toxicity in fish. Marine Pollution Bulletin,
45(1-12), 17–23. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12398363
61. Ravnjak, M., Vrtovšek, J., & Pintar, A. (2013). Denitrification of drinking water in a two-stage
membrane bioreactor by using immobilized biomass. Bioresource Technology, 128, 804–8.
doi:10.1016/j.biortech.2012.10.055
62. Regueiro, L., Veiga, P., Figueroa, M., Alonso-Gutierrez, J., Stams, A. J. M., Lema, J. M., &
Carballa, M. (2012). Relationship between microbial activity and microbial community structure
in six full-scale anaerobic digesters. Microbiological Research, 167(10), 581–9.
doi:10.1016/j.micres.2012.06.002
63. Rejish Kumar, V. J., Sukumaran, V., Achuthan, C., Joseph, V., Philip, R., & Bright Singh, I. S.
(2013). Molecular characterization of the nitrifying bacterial consortia employed for the activation
of bioreactors used in brackish and marine aquaculture systems. International Biodeterioration &
Biodegradation, 78, 74–81. doi:10.1016/j.ibiod.2013.01.002
64. Sakami, T., Andoh, T., Morita, T., & Yamamoto, Y. (2012). Phylogenetic diversity of ammonia-
oxidizing archaea and bacteria in biofilters of recirculating aquaculture systems. Marine
Genomics, 7, 27–31. doi:10.1016/j.margen.2012.04.006
65. Seviour, R. & Nielsen, P. H. (2010). Microbial Ecology of Activated Sludge, London, UK: IWA
Publishing.
66. Sanz, J. L., & Köchling, T. (2007). Molecular biology techniques used in wastewater treatment:
An overview. Process Biochemistry, 42(2), 119–133. doi:10.1016/j.procbio.2006.10.003

References
76
67. Schafer, H. and Muyzer, G. (2001). Denaturing gradient gel electrophorsis in marine microbial
ecology. Methods in Microbiology, Joh Paul (ed). Vol: 30, 425-468. Academic Press, London.
68. Shan, G., Jin, W., Lam, E. K. H., & Xing, X. (2008). Purification of total DNA extracted from
activated sludge. Journal of Environmental Sciences (China), 20(1), 80–7. Retrieved from
http://www.ncbi.nlm.nih.gov/pubmed/18572527
69. Simon, J., & Klotz, M. G. (2013). Diversity and evolution of bioenergetic systems involved in
microbial nitrogen compound transformations. Biochimica et Biophysica Acta, 1827(2), 114–35.
doi:10.1016/j.bbabio.2012.07.005
70. Smith, V. H., Tilman, G. D., & Nekola, J. C. (1999). Eutrophication: impacts of excess nutrient
inputs on freshwater, marine, and terrestrial ecosystems. Environmental Pollution (Barking,
Essex : 1987), 100(1-3), 179–96. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15093117
71. Song, K., Kang, H., Zhang, L., & Mitsch, W. J. (2012). Seasonal and spatial variations of
denitrification and denitrifying bacterial community structure in created riverine wetlands.
Ecological Engineering, 38(1), 130–134. doi:10.1016/j.ecoleng.2011.09.008
72. Sorokin, D. Y., Lücker, S., Vejmelkova, D., Kostrikina, N. a, Kleerebezem, R., Rijpstra, W. I. C.,
Damsté, J. S. S., et al. (2012). Nitrification expanded: discovery, physiology and genomics of a
nitrite-oxidizing bacterium from the phylum Chloroflexi. The ISME Journal, 6(12), 2245–56.
doi:10.1038/ismej.2012.70
73. Tocchi, C., Federici, E., Fidati, L., Manzi, R., Vincigurerra, V., & Petruccioli, M. (2012). Aerobic
treatment of dairy wastewater in an industrial three-reactor plant: effect of aeration regime on
performances and on protozoan and bacterial communities. Water Research, 46(10), 3334–44.
doi:10.1016/j.watres.2012.03.032
74. Trögl, J., Pilařová, V., Boušková, A., Mrákota, J., & Stloukal, R. (2011). Application of Lentikats
Biotechnology for removal of nitrates from ion-exchange brines: Implications for adaptation of
encapsulated denitrifiers. African Journal of Biotechnology, 10(79), 18304–18310.
doi:10.5897/AJB11.2302
75. Vacková, Lenka, Srb, M., Stloukal, R., & Wanner, J. (2011). Comparison of denitrification at low
temperature using encapsulated Paracoccus denitrificans, Pseudomonas fluorescens and mixed
culture. Bioresource Technology, 102(7), 4661–6. doi:10.1016/j.biortech.2011.01.024
76. Vacková, Lenka, Stloukal, R., & Wanner, J. (2012). Determination of low concentration of
Paracoccus denitrificans encapsulated in polyvinyl alcohol LentiKat’s pellets. Applied
Microbiology and Biotechnology, 94(5), 1359–64. doi:10.1007/s00253-012-4073-5
77. Van den Akker, B., Beard, H., Kaeding, U., Giglio, S., & Short, M. D. (2010). Exploring the
relationship between viscous bulking and ammonia-oxidiser abundance in activated sludge: A
comparison of conventional and IFAS systems. Water Research, 44(9), 2919–29.
doi:10.1016/j.watres.2010.02.016
78. Vanparys, B., Spieck, E., Heylen, K., Wittebolle, L., Geets, J., Boon, N., & De Vos, P. (2007).
The phylogeny of the genus Nitrobacter based on comparative rep-PCR, 16S rRNA and nitrite
oxidoreductase gene sequence analysis. Systematic and Applied Microbiology, 30(4), 297–308.
doi:10.1016/j.syapm.2006.11.006

References
77
79. Wagner, M., Rath, G., Amann, R., Koops, H.-P., & Schleifer, K.-H. (1995). In situ Identification
of Ammonia-oxidizing Bacteria. Systematic and Applied Microbiology, 18(2), 251–264.
doi:10.1016/S0723-2020(11)80396-6
80. Wang, Q. S., Sun, D.-B., Hao, W.-P., Li, Y.-Z., Mei, X.-R., & Zhang, Y.-Q. (2012). Human
activities and nitrogen in waters. Acta Ecologica Sinica, 32(4), 174–179.
doi:10.1016/j.chnaes.2012.04.010
81. Wang, X., Wen, X., Criddle, C., Wells, G., Zhang, J., & Zhao, Y. (2010). Community analysis of
ammonia-oxidizing bacteria in activated sludge of eight wastewater treatment systems. Journal of
Environmental Sciences, 22(4), 627–634. doi:10.1016/S1001-0742(09)60155-8
82. Wang, X., Zhang, Y., Wen, X., Xia, Y., Hu, M., & Ding, K. (2012). Real-time PCR quantification
of the population dynamics of ammonia-oxidizing bacteria in a pilot-scale wastewater treatment
plant. Biochemical Engineering Journal, 66, 61–65. doi:10.1016/j.bej.2012.04.015
83. Warneke, S., Schipper, L. a, Matiasek, M. G., Scow, K. M., Cameron, S., Bruesewitz, D. a, &
McDonald, I. R. (2011). Nitrate removal, communities of denitrifiers and adverse effects in
different carbon substrates for use in denitrification beds. Water Research, 45(17), 5463–75.
doi:10.1016/j.watres.2011.08.007
84. Whang, L.-M., Chien, I.-C., Yuan, S.-L., & Wu, Y.-J. (2009). Nitrifying community structures
and nitrification performance of full-scale municipal and swine wastewater treatment plants.
Chemosphere, 75(2), 234–42. doi:10.1016/j.chemosphere.2008.11.059
85. Whitehouse, C. a, & Hottel, H. E. (2007). Comparison of five commercial DNA extraction kits
for the recovery of Francisella tularensis DNA from spiked soil samples. Molecular and Cellular
Probes, 21(2), 92–6. doi:10.1016/j.mcp.2006.08.003
86. Xu, G., Xu, X., Yang, F., Liu, S., & Gao, Y. (2012). Partial nitrification adjusted by
hydroxylamine in aerobic granules under high DO and ambient temperature and subsequent
Anammox for low C/N wastewater treatment. Chemical Engineering Journal, 213, 338–345.
doi:10.1016/j.cej.2012.10.014
87. Yilmaz, G., Lemaire, R., Keller, J., & Yuan, Z. (2007). Effectiveness of an alternating aerobic,
anoxic/anaerobic strategy for maintaining biomass activity of BNR sludge during long-term
starvation. Water Research, 41(12), 2590–8. doi:10.1016/j.watres.2007.02.011
88. Yoshie, S., Noda, N., Tsuneda, S., Hirata, A., & Inamori, Y. (2004). Design of 16S rRNA-
targeted oligonucleotide probes and microbial community analysis in the denitrification process
of a saline industrial wastewater treatment system. FEMS Microbiology Letters, 235(1), 183–9.
doi:10.1016/j.femsle.2004.04.033
89. Zanetti, L., Frison, N., Nota, E., Tomizioli, M., Bolzonella, D., & Fatone, F. (2012). Progress in
real-time control applied to biological nitrogen removal from wastewater. A short-review.
Desalination, 286, 1–7. doi:10.1016/j.desal.2011.11.056
90. Zeng, Y., De Guardia, A., Ziebal, C., De Macedo, F. J., & Dabert, P. (2012). Nitrification and
microbiological evolution during aerobic treatment of municipal solid wastes. Bioresource
Technology, 110, 144–52. doi:10.1016/j.biortech.2012.01.135
91. Zhang, Liang, Zhang, S., Gan, Y., & Peng, Y. (2012). Bio-augmentation to rapid realize partial
nitrification of real sewage. Chemosphere, 88(9), 1097–102.
doi:10.1016/j.chemosphere.2012.05.025

References
78
92. Zhang, Li-sheng, Wu, W., & Wang, J. (2007). Immobilization of activated sludge using improved
polyvinyl alcohol (PVA) gel. Journal of Environmental Sciences (China), 19(11), 1293–7.
Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/18232221

Appendix 1. Links to Manufacturer Protocols Online
79
Manufacturers protocols used in this report can be found at the following links as of June 3, 2013:
1. QIAmp® DNA Stool Kit
(http://www.qiagen.com/Products/Catalog/Sample-Technologies/DNA-Sample-
Technologies/Genomic-DNA/QIAamp-DNA-Stool-Mini-Kit#resources)
2. PowerSoil® DNA Isolation Kit
(http://www.mobio.com/images/custom/file/protocol/12888.pdf)
3. Chemagic DNA Bacteria Kit
(http://www.chemagen.com/fileadmin/downloads/chemagic_DNA_Bacteria_Kit.pdf)
4. MasterPure™ DNA Purification Kit
(http://www.epibio.com/docs/default-source/protocols/masterpure-dna-purification-
kit.pdf?sfvrsn=4)
5. INGENYphorU-2x2 DGGE Apparatus
(http://www.ingeny.com/Manuals_files/INGENYphorU%20manual.pdf)
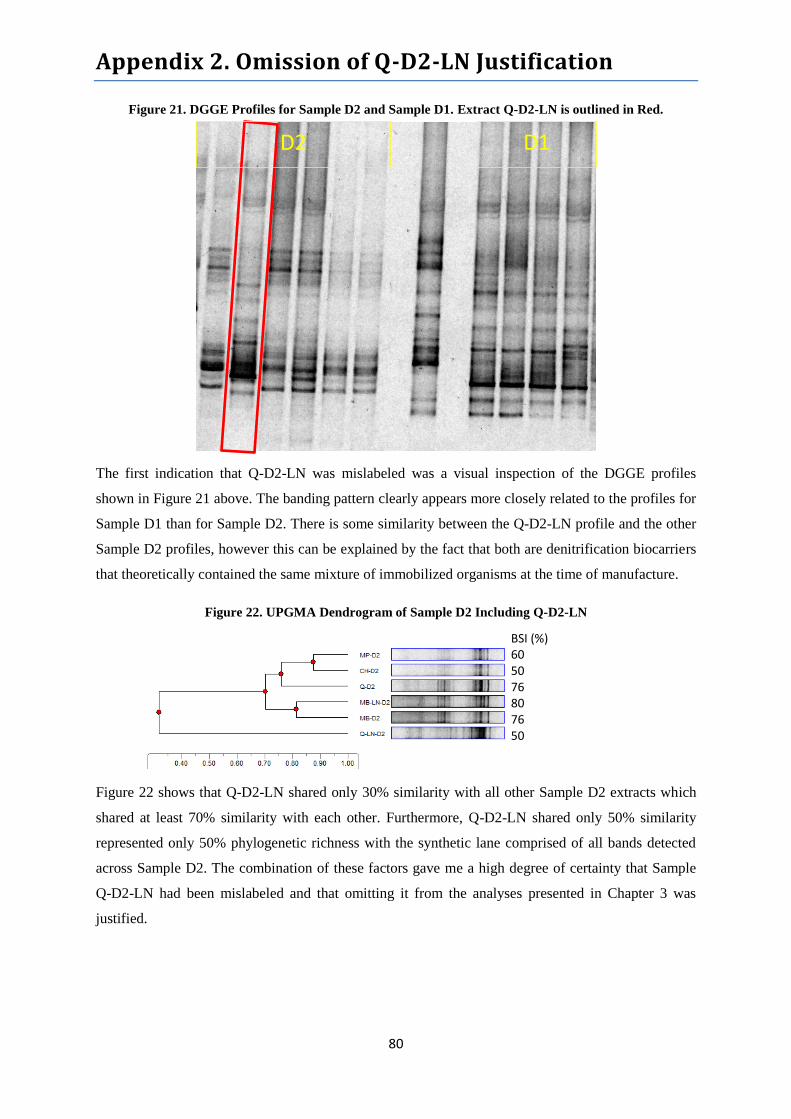
Appendix 2. Omission of Q-D2-LN Justification
80
Figure 21. DGGE Profiles for Sample D2 and Sample D1. Extract Q-D2-LN is outlined in Red.
The first indication that Q-D2-LN was mislabeled was a visual inspection of the DGGE profiles
shown in Figure 21 above. The banding pattern clearly appears more closely related to the profiles for
Sample D1 than for Sample D2. There is some similarity between the Q-D2-LN profile and the other
Sample D2 profiles, however this can be explained by the fact that both are denitrification biocarriers
that theoretically contained the same mixture of immobilized organisms at the time of manufacture.
Figure 22. UPGMA Dendrogram of Sample D2 Including Q-D2-LN
Figure 22 shows that Q-D2-LN shared only 30% similarity with all other Sample D2 extracts which
shared at least 70% similarity with each other. Furthermore, Q-D2-LN shared only 50% similarity
represented only 50% phylogenetic richness with the synthetic lane comprised of all bands detected
across Sample D2. The combination of these factors gave me a high degree of certainty that Sample
Q-D2-LN had been mislabeled and that omitting it from the analyses presented in Chapter 3 was
justified.
BSI (%) 60 50 76 80 76 50
D2 D1