genexpressionsanalysen bei dem biotechnologisch relevanten
TRANSCRIPT

Genexpressionsanalysen bei dem biotechnologisch relevanten Hyphenpilz
Acremonium chrysogenum:
Verwendung des autofluoreszierenden Proteins DsRed
Dissertation
zur Erlangung des Grades
eines Doktors der Naturwissenschaften
der Fakultät für Biologie und Biotechnologie
der Ruhr-Universität Bochum
angefertigt am
Lehrstuhl für Allgemeine und Molekulare Botanik
vorgelegt von
Danielle Irina Margarete Janus
aus Herne
Bochum 2007

MEINEN LIEBEN ELTERN
UND BORIS

DANKSAGUNG
Meinem akademischen Lehrer und Doktorvater Herrn Prof. Dr. U. Kück danke ich herzlich
für die Überlassung des interessanten Themas, das mir entgegengebrachte Vertrauen,
seinem steten Interesse am Fortgang der Arbeit sowie seiner fortwährenden
wissenschaftlichen Förderung. Frau Prof. Dr. N. Frankenberg-Dinkel danke ich für die Übernahme des Korreferats. Danken möchte ich auch allen MitarbeiterInnen des Lehrstuhls für Allgemeine und
Molekulare Botanik für die gute Atmosphäre und die ständige Hilfs- und Diskussions-
bereitschaft. Frau Eva Szczypka und Frau Gabriele Frenssen-Schenkel danke ich für ihre
Hilfe bei der graphischen Gestaltung dieser Arbeit. Ein ganz liebes Dankeschön geht an
Elke Adolph für das Autoklavieren von Unmengen von Gerätschaften und Medien. Kerstin
Kalkreuter und Stefanie Mertens möchte ich für ihre exzellente und z.T. auch spontane
technische Mitarbeit bei einigen Projekten dieser Arbeit danken. Ingeborg Godehardt
danke ich für die Herstellung von allgemeinen Fluoreszenzvektoren. Mein besonderer Dank geht an Frau Dr. Minou Nowrousian und Frau Dr. Birgit Hoff, die
mich mit ihren wertvollen Ratschlägen und durch das kritische Lesen dieses Manuskriptes
unterstützt haben. Birgit möchte ich weiterhin für ihre stetige Unterstützung und
Aufmunterung vor allem während der knockout-Phase danken. Bei meinen Doktorbrüdern und Doktorschwestern möchte ich mich herzlich für den Spaß
im und außerhalb des Labors bedanken. Mein besonderer Dank geht hier an meine Labor-
und Büronachbarin M. Sc. Jacqueline Dreyer, die es mehr als vier Jahre mit mir
ausgehalten hat und immer ein offenes Ohr für mich hatte. Der Firma Sandoz GmbH (Kundl, Österreich) danke ich für die Bereitstellung finanzieller
Mittel und ihrem steten Interesse am Fortgang des gesamten Forschungsprojektes.
Insbesondere möchte ich mich hier bei Herrn Dr. H. Kürnsteiner und Herrn Dr. E. Friedlin
für ihr Interesse, die interessanten Diskussionen und ihre Unterstützung bedanken. Meinen Eltern bin ich dankbar für die Unterstützung, ihre Kraft und ihre Liebe, die mich
all die Jahre durch mein Studium begleitet haben. Dem Rest meiner Familie danke ich für
ihr großes Interesse und ihre Unterstützung. Meinem Freund Boris möchte ich für seine
große Geduld und sein Vertrauen während der letzten Jahre danken.

INHALTSVERZEICHNIS
I
INHALTSVERZEICHNIS
Abkürzungsverzeichnis III
I. EINFÜHRUNG 1 1. Einleitung 1 2. Enzym-kodierende Reportergene 2 3. Autofluoreszierende Proteine 7 3.1 Das grün-fluoreszierende Protein (GFP) und seine Varianten 7 3.2 Rot-fluoreszierende Proteine 10 4. Anwendung von Reportergenen in Hyphenpilzen 13 4.1 Lokalisationsstudien 13 4.2 Sekretion 14 4.3 Promotoranalysen 15 4.4 RNAi-Analysen 16 5. Zusammenfassung 17 II. PROBLEMSTELLUNG 18 1. Entwicklung eines RNAi-Systems zur Anwendung in A. chrysogenum 19 2. Analysen des A. chrysogenum cre1-Promotors 21 III. JANUS D, HOFF B, HOFMANN E, KÜCK U (2007) 23 „An Efficient Fungal RNA-Silencing System Using the DsRed Reporter Gene“ IV. JANUS D, HORTSCHANSKY P, KÜCK U (2007) 24 „Identification of a minimal cre1 promoter sequence promoting glucose-
dependent gene expression in the β-lactam producer Acremonium chrysogenum“

INHALTSVERZEICHNIS
II
V. GESAMTDISKUSSION 25 1. Etablierung eines RNAi-Systems in A. chrysogenum 25 2. Das RNAi-System kann zur Aufklärung regulatorischer Komponenten des Sekundärmetabolismus beitragen 29
3. Die CT-reiche Region des cre1-Promotors ist verantwortlich für eine erhöhte Genexpression und interagiert mit dem Glukoserepressor CRE1 32
4. Der cre1-Promotor besitzt das Potential zur modifizierten Expression von Cephalosporin C-Biosynthesegenen 39 5. Das rot-fluoreszierende Protein (DsRed) – eine zuverlässige Alternative zur Selektion pilzlicher Transformanten 41
VI. ZUSAMMENFASSUNG 43 VII. SUMMARY 45 VIII. LITERATUR 47 IX. APPENDIX 58

ABKÜRZUNGSVERZEICHNIS
III
ABKÜRZUNGSVERZEICHNIS
Abb. Abbildung AcFKH1 forkhead-Transkriptionsfaktor 1 aus Acremonium chrysogenum AS Aminosäuren bp Basenpaar(e) cefEF Deacetoxycephalsporin C/Deacteylcephalosporin C-Synthetase-Gen cefG Acetyl-CoA:Deacetylcephalosporin C-Acetyltransferase-Gen cDNA komplementäre DNA CPCR1 Cephalosporin C Regulator 1 aus Acremonium chrysogenum CRE1/CREA Glukoserepressor DNA Desoxyribonukleinsäure DsRed rot-fluoreszierendes Protein dsRNA doppelsträngige RNA EGFP „enhanced“ grün-fluoreszierendes Protein GFP grün-fluoreszierendes Protein kb Kilobasen kDa Kilodalton lacZ β-Galaktosidase-Gen aus Escherichia coli mRNA Boten-RNA („messenger“-RNA) PCR Polymerase-Kettenreaktion („polymerase chain reaction“) PACC pH-abhängiger TRanskriptionsfaktor pcbC/ipnA Isopenicillin N-Synthase-Gen RNA Ribonukleinsäure RNAi RNA-Interferenz rRNA ribosomale RNA RT Raumtemperatur SPR Surface Plasmon Resonance uidA β-Glucuronidase-Gen aus E. coli Sowie Aminosäuren im Ein- und Drei-Buchstaben-Code. Allgemein gebräuchliche Abkürzungen sowie übliche Maßeinheiten sind nicht gesondert
aufgeführt.

I. EINFÜHRUNG
1
I. EINFÜHRUNG
1. Einleitung Hyphenpilze haben wichtige Funktionen in der industriellen Mikrobiologie. Seit
Jahrzehnten werden sie für die Produktion verschiedener Verbindungen, wie Antibiotika,
hydrolytischer Enzyme (z.B. Cellulasen, Glucoamylasen) und heterologer Proteine (z.B.
Lysozym) genutzt (Saunders et al. 1989, Gouka et al. 1997). Ferner finden sie Einsatz in
der Lebensmittelindustrie zur Produktion von z.B. Zitronensäure oder zur Entfernung von
Bitterstoffen (Mayer und Staples 2002, Schuster et al. 2002). Mit der zunehmenden
Identifizierung neuer Gene, die für die Produktion einer Vielzahl von Metaboliten
verantwortlich sind, steigt auch das Interesse an den Regulationsmechanismen der
Expression dieser Gene (Hoffmeister und Keller 2007). Dank der Entwicklung und
Optimierung verschiedener genetischer, biochemischer und molekularbiologischer
Untersuchungsmethoden konnte das Verständnis der Genfunktion und deren Regulation in
Hyphenpilzen während der letzten Jahre stark erweitert werden (Weld et al. 2006).
Reportergensysteme bestehen aus Genen, deren Produkte leicht messbare Phänotypen
hervorrufen, die sich leicht vom Hintergrund endogener Proteine abzeichnen. Für
gewöhnlich werden solche Reporter aufgrund ihrer Sensitivität, ihrer dynamischen
Spannweite, der einfachen Handhabung und ihrer Zuverlässigkeit ausgewählt (Naylor
1999). Dabei können Analysen genetisch regulierender Elemente, des Proteintransports
und die Funktion von Proteinen über einen definierten Zeitraum im Zusammenhang der
komplexen Physiologie lebender Zellen erfolgen.
Im Allgemeinen werden in vitro- und in vivo-Reportergensysteme unterschieden. Bei der
Anwendung von in vitro-Reportergenanalysen erfolgt eine Quantifizierung des
Reporterproteins aus Zell- oder Gewebelysaten oder aus dem Kulturüberstand. In vivo-
Reportergenanalysen hingegen werden mit lebenden Zellen oder Geweben durchgeführt,
sie eigenen sich weniger für quantitative Analysen und werden daher meistens zur
Lokalisation von Proteinen oder zur Ermittlung von zell- bzw. gewebespezifischen
Genexpressionen verwendet. Die Auswahl eines geeigneten Reportergensystems sollte vor
allem an die entsprechende experimentelle Situation angepasst werden, da z.B. endogene
Enzymaktivitäten die Ergebnisse verfälschen können. Weiterhin sind einige Reporter vor
allem zur Untersuchung der räumlichen Expression geeignet, während andere eher
quantitative Messungen ermöglichen. Wenn beispielsweise eine zeitliche Regulation

I. EINFÜHRUNG
2
analysiert werden soll, ist es ratsam, Reporterproteine mit einer geringen Halbwertszeit
auszuwählen, da stabile Reporter zur Akkumulation neigen. Im weiteren werden die zwei
großen Klassen der Reportergene, die Enzym-kodierenden und die autofluoreszierenden
Proteine detaillierter vorgestellt.
2. Enzym-kodierende Reportergene Reporterproteine mit enzymatischer Aktivität katalysieren chromogene oder fluorogene
Reaktionen unter Verwendung eines bestimmten Substrats, welches nach Umsetzung eine
Farb- oder Lichtreaktion hervorrufen kann. Die Enzymaktivität des Reporters wird somit
anhand des Umsatzes der Indikatorsubstanz gemessen. Es existiert eine große Vielfalt und
Diversität innerhalb der Reportersysteme, so finden z.B. die β-Galaktosidase, die
β-Glucuronidase, Glucoamylasen, Glukose-Oxidasen, die Chloramphenicol-Acetyl-
transferase, Luciferasen, uvm. einen häufigen Einsatz als Reporter in der angewandten
Biologie. Diese Systeme werden in der Regel Spezies-spezifisch eingesetzt, da je nach
Organismus und Art der Versuchsdurchführung unterschiedliche Voraussetzungen
vorherrschen. Reportergensysteme können sowohl pro-, als auch eukaryotischen Ursprungs
sein. In diesem Kapitel werden primär Enzym-kodierende Reportergene beschrieben, die
erfolgreich in Hyphenpilzen etabliert werden konnten.
Das erste jemals verwendete Reportergen ist das lacZ-Gen aus dem Darmbakterium
Escherichia coli (Bassford et al. 1978). Es kodiert für die cytoplasmatische
β-GALAKTOSIDASE, eine Glykosidase, die für die Hydrolyse verschiedener β-Galaktoside
zu Monosacchariden verwendet werden kann. In Bakterien ist dieses Gen Teil des lac-
Operons, welches beim Abbau des Disaccharids Laktose in seine monomeren Bestandteile
Galaktose und Glukose eine essentielle Rolle spielt (Beckwith 1970, Fowler und Zabin
1977). Aufgrund ihrer beachtlichen Substratvielfalt kann die β-Galaktosidase sowohl für
in vitro-, wie auch für in vivo-Analysen genutzt werden. Als Substrate können entweder
chromogene Substrate, die nach Hydrolyse durch die β-Galaktosidase eine Farbreaktion
hervorrufen, oder fluorogene Substanzen, deren Umsetzung fluorometrisch bestimmt
werden kann, verwendet werden. Ein Beispiel ist das farblose Substrat X-Gal (5-Brom-4-
chlor-3-indoxyl-β-D-galactopyranosid), das durch die β-Galaktosidase zu einen tiefblauen,
unlöslichen Farbstoff hydrolysiert wird (Miller 1972). Häufig in der Molekularbiologie

I. EINFÜHRUNG
3
verwendete Substrate der β-Galaktosidase werden in Tabelle 1 aufgeführt. Ein Nachteil des
Einsatzes der E. coli β-Galaktosidase ist die Tatsache, dass die meisten Pilze eine hohe
endogene β-Galaktosidaseaktivität aufweisen, die zu verfälschten Ergebnissen führen
kann. In den 70er Jahren wurde allerdings festgestellt, dass die Expression des bgdA-Gens,
welches für die Aspergillus nidulans β-Galaktosidase kodiert, in Gegenwart von Glukose,
Glycerol oder Saccharose so stark reprimiert wird, dass keine Umsetzung des Substrates
erfolgt (Fantes und Roberts 1973, Fekete et al. 2002). Durch die Verwendung
reprimierender Zucker kann somit die endogene Enzymaktivität inhibiert und die
bakterielle β-Galaktosidase als Reporter in Hyphenpilzen genutzt werden.
Nach der erfolgreichen Etablierung der bakteriellen β-GLUCURONIDASE (GUS) als
Reporter in Bakterien, den Nematoden Caenorhabditis elegans und höheren Pflanzen,
konnte in den späten 80er Jahren das uidA-Gen aus E. coli in Hyphenpilzen, wie
A. nidulans, Aspergillus niger und Fulvia fulva (syn. Cladosporium fulvum), exprimiert
werden (Jefferson et al. 1986, Jefferson et al. 1987a, Jefferson et al. 1987b, Roberts et al.
1989). Dieses Protein katalysiert die Hydrolyse von β-Glucuroniden zu D-Glucuronat und
einem Alkohol (Jefferson et al. 1986). Aufgrund der großen Substratdiversität wird ein
vielseitiger Einsatz des uidA-Gens als Reporter ermöglicht. So hydrolysiert die
β-Glucuronidase unterschiedlichste Substrate wie z.B. X-Gluc (5-Bromo-4-chloro-3-
indolyl-β-D-glucuronid), bei denen farbige oder fluoreszierende Reaktionsprodukte
entstehen, die sowohl spektrofluorometrische in vitro Aktivitätsmessungen, als auch
in vivo-Anfärbungen von beispielsweise Hyphen gewährleisten (siehe Tabelle 1). Die
Verwendung der β-Glucuronidase als Reporterprotein bietet gegenüber der
β-Galaktosidase den Vorteil, dass in Hyphenpilzen keine endogenen GUS-Enzym-
aktivitäten existieren (Roberts et al. 1989).
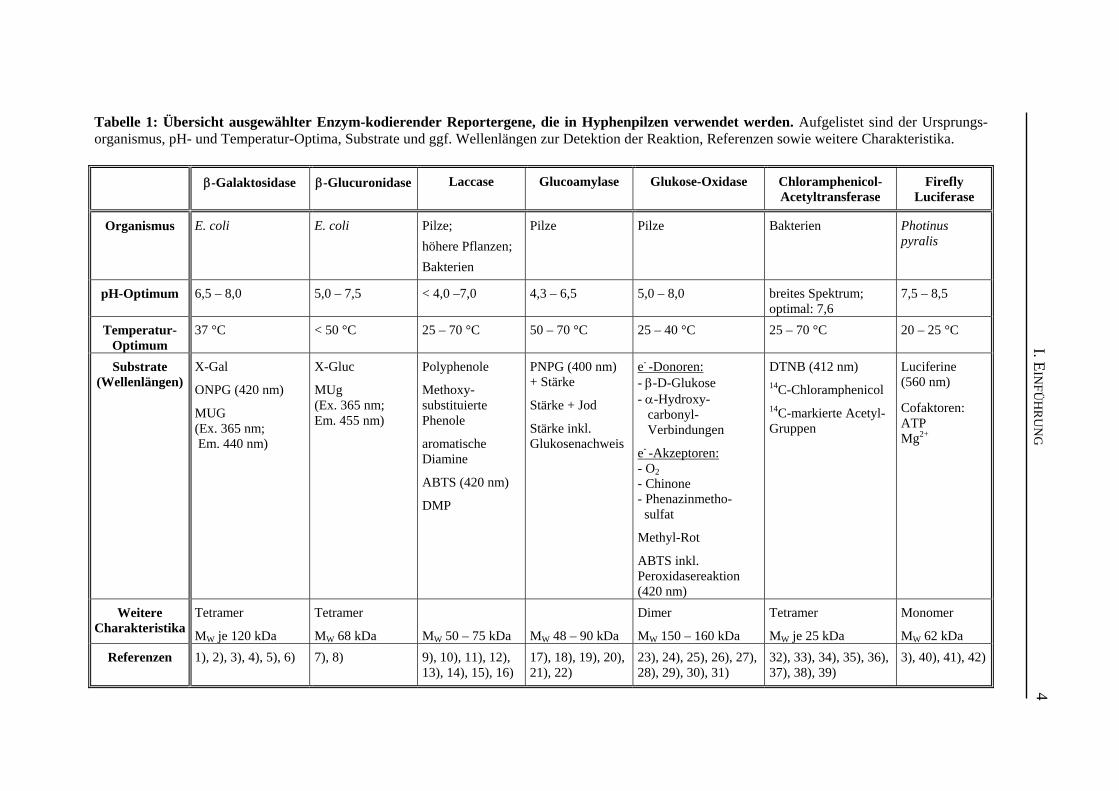
Tabelle 1: Übersicht ausgewählter Enzym-kodierender Reportergene, die in Hyphenpilzen verwendet werden. Aufgelistet sind der Ursprungs-organismus, pH- und Temperatur-Optima, Substrate und ggf. Wellenlängen zur Detektion der Reaktion, Referenzen sowie weitere Charakteristika.
β-Galaktosidase
β-Glucuronidase
Laccase
Glucoamylase
Glukose-Oxidase
Chloramphenicol-Acetyltransferase
Firefly
Luciferase
Organismus E. coli
E. coli
Pilze; höhere Pflanzen; Bakterien
Pilze
Pilze
Bakterien
Photinus pyralis
pH-Optimum
6,5 – 8,0
5,0 – 7,5
< 4,0 –7,0
4,3 – 6,5
5,0 – 8,0
breites Spektrum; optimal: 7,6
7,5 – 8,5
Temperatur-
Optimum
37 °C
< 50 °C
25 – 70 °C
50 – 70 °C
25 – 40 °C
25 – 70 °C
20 – 25 °C
Substrate
(Wellenlängen)
X-Gal ONPG (420 nm) MUG (Ex. 365 nm; Em. 440 nm)
X-Gluc MUg (Ex. 365 nm; Em. 455 nm)
Polyphenole Methoxy-substituierte Phenole aromatische Diamine ABTS (420 nm) DMP
PNPG (400 nm) + Stärke Stärke + Jod Stärke inkl. Glukosenachweis
e- -Donoren: - β-D-Glukose - α-Hydroxy- carbonyl- Verbindungen e- -Akzeptoren: - O2 - Chinone - Phenazinmetho- sulfat Methyl-Rot ABTS inkl. Peroxidasereaktion (420 nm)
DTNB (412 nm) 14C-Chloramphenicol 14C-markierte Acetyl-Gruppen
Luciferine (560 nm) Cofaktoren: ATP Mg2+
Weitere
Charakteristika
Tetramer MW je 120 kDa
Tetramer MW 68 kDa
MW 50 – 75 kDa
MW 48 – 90 kDa
Dimer MW 150 – 160 kDa
Tetramer MW je 25 kDa
Monomer MW 62 kDa
Referenzen 1), 2), 3), 4), 5), 6)
7), 8)
9), 10), 11), 12), 13), 14), 15), 16)
17), 18), 19), 20), 21), 22)
23), 24), 25), 26), 27), 28), 29), 30), 31)
32), 33), 34), 35), 36), 37), 38), 39)
3), 40), 41), 42)
I. EIN
FÜH
RU
NG
4

I. EINFÜHRUNG
5
Referenzen: 1) (Bassford et al. 1978); 2) (Fowler und Zabin 1977); 3) (Bronstein et al. 1994); 4) (Miller 1972); 5) (Lederberg 1950); 6) (Roth 1969); 7) (Jefferson et al. 1986); 8) (Jefferson et al. 1987a); 9) (Baldrian 2006); 10) (Bollag und Leonowicz 1984); 11) (Lalitha Kumari und Sirsi 1972); 12) (Kiiskinen et al. 2002); 13) (Wang und Ng 2006); 14) (Mander et al. 2006); 15) (Minussi et al. 2002); 16) (Givaudan et al. 1993); 17) (Norouzian et al. 2006); 18) (Holm 1986); 19) (Talabardon und Yang 2005); 20) (Scorpione et al. 1993); 21) (Gordon et al. 2000); 22) (Sauer et al. 2000); 23) (Leskovac et al. 2005); 24) (Chan und Bruice 1977); 25) (Markwell et al. 1989); 26) (Hodgkins et al. 1993); 27) (Leiter et al. 2004); 28) (Simpson et al. 2007); 29) (Bhatti et al. 2006); 30) (Geisen 1995); 31) (Karmali et al. 2004); 32) (Shaw und Leslie 1991); 33) (Thibault et al. 1980); 34) (Kleanthous und Shaw 1984); 35) (Shaw 1967); 36) (Sleigh 1986); 37) (Seed und Sheen 1988); 38) (Davis et al. 1992); 39) (Turner et al. 1992); 40) (Greer und Szalay 2002); 41) (Shimomura und Johnson 1975); 42) (de Wet et al. 1987) Abkürzungen : ABTS: 2,2’-Azino-di-(3-ethylbenzthiazolin-sulfonat); DMP: 2,6-Dimethoxyphenol; DTNB: 5,5’-Dithiobis(2-nitrobenzoesäure); Em.: Emission; Ex.: Extinktion; MUG: 4-Methylumbelliferyl-β-D-galactopyranosid; MUg: 4-Methylumbelliferyl-β-D-glucuronid; ONPG: o-Nitrophenyl-β-D-galacto-pyranosid; PNPG: p-Nitrophenyl-α-D-glycopyranosid; X-Gal: 5-Brom-4-chlor-3-indoxyl-β-D-galacto-pyranosid; X-Gluc: 5-Brom-4-chlor-3-indolyl-β-D-glucuronid
Im folgenden werden einige in Hyphenpilzen verwendete Reportersysteme, wie
beispielsweise die Laccase und die Glukose-Oxidase vorgestellt, die selbst einen pilzlichen
Ursprung haben und somit in einer Vielzahl unterschiedlicher Arten identifiziert werden
können.
Die von den Reportergenen kodierten Enzyme sind zumeist auch von hohem industriellen Nutzen, sie werden beispielsweise in der Lebensmittelindustrie zur Entfernung von Bitterstoffen, Sauerstoff oder Glukose aus verschiedenen Nahrungsmitteln (z.B. Wein, Kakao, Fruchtsäfte, getrocknete Eier) und zur Saccharifizierung von Stärke verwendet (Nunberg et al. 1984, Geisen 1995, Mayer und Staples 2002, Norouzian et al. 2006). Weitere Einsatzgebiete sind biologische „Sanierungsprozesse“, bei denen Industrieabwässer oder Herbizide, Pestizide und Sprengstoffe im Erdreich entgiftet werden. Zudem finden sie Anwendung in in vivo-Analysen des Blutzuckerspiegels von z.B. Diabetes-Patienten (Rhemrev-Boom et al. 2002, Rodriguez Couto und Toca Herrera 2006).
Ein erst im Jahr 2006 etabliertes pilzliches Reportersystem stellt der Einsatz von
LACCASEN dar, deren Wirkungsweise schon seit einigen Jahrzehnten untersucht wird
(Baldrian 2006). Die Isolierung des Laccase-Gens erfolgte aus dem Cellulose-abbauenden
Ascomyceten Stachybotrys chartarum, und seine Nützlichkeit als Reporter wurde mit Hilfe
verschiedener induzierbarer Promotoren aus A. nidulans, A. niger und Trichoderma reesei
bereits verifiziert (Mander et al. 2006). Laccasen gehören zu der Gruppe der
Polyphenoloxidasen und katalysieren die Reduktion von Sauerstoff zu Wasser begleitet
von der Oxidation eines Substrates, wie z.B. verschiedener Polyphenole, Methoxy-
substituierter Phenole, aromatischer Diamine und einer Reihe anderer Verbindungen
(Baldrian 2006). Der Nachweis der Laccase-Aktivität kann in vitro durch Farbreaktionen
mit dem Substrat ABTS (2,2’-Anzino-di-(3-ethylbenzthiazolin-sulfonat) erfolgen, bei

I. EINFÜHRUNG
6
denen anschließend die Absorption bei 420 nm bestimmt wird. Ferner kann es in vivo in
sogenannten Plattentest mit dem Substrat ABTS verwendet werden. Die Expression der
Laccase-Gene ist mit bestimmten Entwicklungsstadien verknüpft oder wird durch
phenolische Substrate induziert, so dass keine oder kaum endogene Enzymaktivitäten zur
Verfälschung von Testergebnissen beitragen (Übersicht bei Mayer und Staples 2002,
Baldrian 2006). Weitere Analysen werden in den nächsten Jahren zeigen, ob Laccasen sich
zu geeigneten Reportergensystemen etablieren können.
Das letzte System, das vorgestellt werden soll, ist die GLUKOSE-OXIDASE. Die Aktivität
dieses Glycoproteins konnte in vielen Pilzen, wie verschiedener Aspergillus- und
Penicillium-Spezies identifiziert werden (Markwell et al. 1989). Dieses Enzym katalysiert
die Oxidation von Glukose zu Glucono-δ-lacton. Hierbei wird das Enzym-gebundene FAD
zu FADH2 reduziert, welches folgend mit molekularem Sauerstoff reagiert, so dass H2O2
freigesetzt werden kann. Das Glucono-δ-lacton wird anschließend entweder spontan oder
enzymatisch zur Gluconsäure umgesetzt. Zur Bestimmung der Enzym-Aktivität wird zum
einen Methyl-Rot als pH-Indikator der Gluconsäure und zum anderen das entstehende
H2O2 in einer anschließenden Peroxidasereaktion verwendet (Geisen 1995). Die Substrate
der Glukose-Oxidase können in zwei Gruppen eingeteilt werden. Zum einen gibt es die
Substrate, die als Elektronen-Donor (z.B. Glukose) während der reduktiven Reaktion
fungieren, zum anderen gibt es Elektronen-Akzeptoren (z.B. O2) der oxidativen Reaktion
(Übersicht bei Leskovac et al. 2005). Einige Beispiele der in Reportergenanalysen
verwendeten Elektronen-Donoren und -Akzeptoren sind in Tabelle 1 aufgeführt. Ein
Nachteil in der Verwendung ist die Anwesenheit endogener Glukose-Oxidasen, deren
Aktivität in vergleichenden Analysen des Wildtyp-Stammes aus dem eigentlichen
Versuchsergebnis herausgerechnet werden muss. Ein weiterer Nachteil in der Anwendung
ist dadurch gegeben, dass es sich hierbei um einen gekoppelten Aktivitätsnachweis
handelt. Ferner müssen die Zellen bei Plattentests in Gegenwart von Glukose, einem
chromogenen Substrat und der Peroxidase inkubiert werden. Die Etablierung weiterer, aber seltener genutzter, Reportergensysteme, wie z.B. die
Chloramphenicol-Acetyltransferasen, die Luciferasen und die Glucoamylasen erfolgte
ebenfalls in verschiedenen Hyphenpilzen. Einige charakteristische Merkmale dieser
Reporterproteine sind in der Tabelle 1 dargestellt.

I. EINFÜHRUNG
7
3. Autofluoreszierende Proteine In den letzten Jahrzehnten konnte die Etablierung und verstärkte Nutzung von
fluoreszierenden Proteinen als Reporter zum grundlegenden Verständnis vieler
biologischer Prozesse beitragen. Autofluoreszierende Proteine sind die am weitesten
analysierten und genutzten Reportersysteme in der Biochemie und Zellbiologie. Sie finden
ihren Einsatz als Marker für Genexpressionsanalysen und zur Markierung von Proteinen
zur Analyse der Lokalisation, des Transportes, der Dynamik und der Interaktion mit
anderen Proteinen in lebenden Zellen. Die Anwendung von fluoreszierenden Proteinen in
der Molekularbiologie bietet viele verschiedene Vorteile. So kann der Nachweis des
Reportergens mittels Fluoreszenzmikroskopie ohne eine Zerstörung der Zellen erfolgen.
Des weiteren entstehen keine Probleme mit der Zellpermeabilität während der Aufnahme
von Substraten, da autofluoreszierende Proteine generell keine Substrate oder andere
Kofaktoren benötigen (Lorang et al. 2001). Mittlerweile gibt es fluoreszierende Proteine,
die das gesamte Farbspektrum abdecken, so dass eine Vielzahl unterschiedlich
fluoreszierender Proteine innerhalb einer Zelle angewendet werden können, um
beispielsweise verschiedene Organellen zu markieren oder die Lokalisation
unterschiedlicher Faktoren zeitgleich innerhalb einer Zelle zu bestimmen. Im folgenden
werden das grün-fluoreszierende Proteine und seine Varianten, sowie rot-fluoreszierende
Proteine detailliert vorgestellt. 3.1 Das grün-fluoreszierende Protein (GFP) und seine Varianten
Bereits 1955 wurde beschrieben, dass die Qualle Aequorea eine grün-fluoreszierende
Substanz im Ring an der Peripherie der Umbrella besitzt (Davenport und Nicol 1955).
Aber erst Anfang der 90er Jahre gelang es einer Forschergruppe, die cDNA des gfp-Gens,
welches für das grün-fluoreszierende Protein kodiert, aus der Qualle Aequorea victoria zu
isolieren (Prasher et al. 1992). 1994 gelang es Chalfie und Kollegen mit Hilfe der isolierten
cDNA sowohl in pro-, als auch in eukaryotischen Organismen (E. coli und Caenorhabditis
elegans) grüne Fluoreszenz zu erzeugen (Chalfie et al. 1994). Zeitgleich gelang einer
weiteren Forschergruppe ebenfalls die Expression des gfp-Gens in E. coli, sowie der
eindeutige mikroskopische Nachweis der grünen Fluoreszenz (Inouye und Tsuji 1994).
Das grün-fluoreszierende Protein besteht aus 238 Aminosäuren und hat ein
Molekulargewicht von 26,8 kDa (Prasher et al. 1992). Es setzt sich aus 11 β-Strängen
zusammen, die ein sogenanntes β-Barrel ausbilden in dessen Mitte sich eine α-Helix

I. EINFÜHRUNG
8
befindet. Im Zentrum dieser α-Helix ist der Chromophor kovalent gebunden. Dieser
besteht aus einem p-Hydroxybenzylidenimidazolinon und wird durch Modifikationen und
Oxidation der Aminosäurereste 65 bis 67 (Ser65-Tyr66-Gly67) ausgebildet (Prasher et al.
1992, Cody et al. 1993, Yang et al. 1996a). Wird dieses Protein mit einer Wellenlänge von
395 bzw. 475 nm angeregt, so konnte gezeigt werden, dass es grünes Licht mit einer
Wellenlänge von 508 nm emittiert, das mikroskopisch erfasst, aber auch
spektrofluorometrisch quantitativ gemessen werden kann (Übersicht bei Tsien 1998).
Eine optimale Faltung des Wildtyp-GFPs erfolgt bei niedrigen Temperaturen, wie sie den
Lebensbedingungen der Qualle entsprechen. Erfolgt die Reifung des Proteins bei diesen
niedrigen Temperaturen, so ist es stabil und fluoresziert auch bei Temperaturen von 65 °C
(z.B. Lim et al. 1995). Die Entwicklung von mutierten Varianten führte aber in den letzten
Jahren zur Verbesserung der Protein-Reifung bei höheren Temperaturen, der reduzierten
Aggregation bei höheren Konzentrationen, zur Verbesserung des Diffusionsvermögens
innerhalb der Zellen und zu reduziertem Ausbleichen (Übersicht bei Tsien 1998).
Mutationen im Bereich des Chromophors, wie beispielsweise S65T, resultieren weiterhin
in einer verbesserten Leuchtkraft des modifizierten Proteins und veränderten Emissions-
und Extinktionsspektren. Die Maxima verschieben sich auf 488 und 511 nm, der
zellschädigende Absorptionspeak im UV-Bereich bei 395 nm entfällt. Weiterhin werden
durch diese Mutation eine schnellere Oxidation des Fluorophors und ein weniger starkes
Ausbleichen gewährleistet. Die Faltung bei Temperaturen bis Raumtemperatur erfolgt
ähnlich effizient wie beim Wildtyp-GFP unter optimalen Bedingungen, allerdings können
Missfaltungen und nicht-fluoreszierende Aggregate bei höheren Temperaturen nicht
ausgeschlossen werden (Heim et al. 1995). Insgesamt haben diese Modifikationen dazu
geführt, dass das GFP ein optimales, vielfach anwendbares Reportersystem darstellt. Ein
weiterer großer Vorteil dieses Systems ist die Existenz von Farbvarianten des GFPs, die in
den letzten Jahren ebenfalls durch das Einbringen weiterer Mutationen erzeugt werden
konnten. Die stärksten Wellenlängenverschiebungen wurden durch den Austausch des
Thr203 gegen aromatische Aminosäuren (His, Trp, Phe, Tyr) erzeugt und bewirken eine
Verlagerung der Emissions- und Extinktionswellenlänge um bis zu 20 nm (Ormö et al.
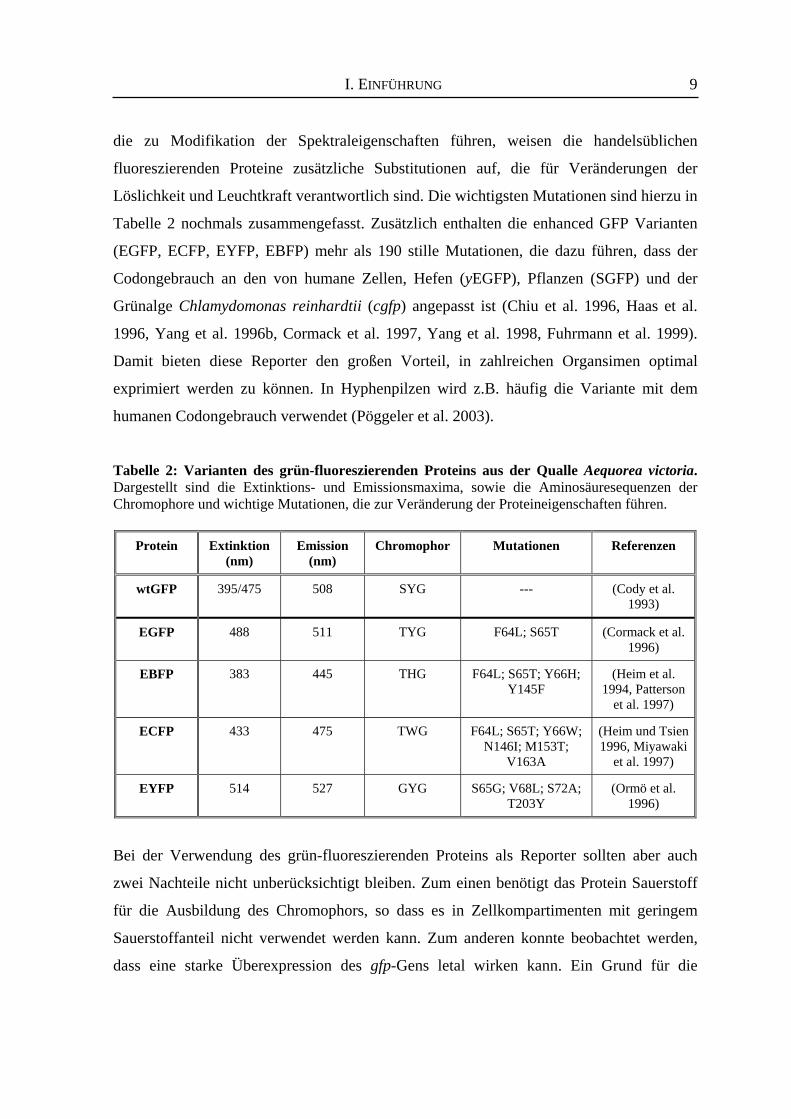
1996, siehe Tabelle 2). Somit decken die unterschiedlichen Farbvarianten (blau, cyan, gelb,
grün) ein Extinktionsspektrum von 383 bis 514 nm und ein Emissionsspektrum von 445
bis 527 nm ab (siehe Tabelle 2). Neben den hier aufgeführten Aminosäuresubstitutionen,
I. EINFÜHRUNG
9
die zu Modifikation der Spektraleigenschaften führen, weisen die handelsüblichen
fluoreszierenden Proteine zusätzliche Substitutionen auf, die für Veränderungen der
Löslichkeit und Leuchtkraft verantwortlich sind. Die wichtigsten Mutationen sind hierzu in
Tabelle 2 nochmals zusammengefasst. Zusätzlich enthalten die enhanced GFP Varianten
(EGFP, ECFP, EYFP, EBFP) mehr als 190 stille Mutationen, die dazu führen, dass der
Codongebrauch an den von humane Zellen, Hefen (yEGFP), Pflanzen (SGFP) und der
Grünalge Chlamydomonas reinhardtii (cgfp) angepasst ist (Chiu et al. 1996, Haas et al.
1996, Yang et al. 1996b, Cormack et al. 1997, Yang et al. 1998, Fuhrmann et al. 1999).
Damit bieten diese Reporter den großen Vorteil, in zahlreichen Organsimen optimal
exprimiert werden zu können. In Hyphenpilzen wird z.B. häufig die Variante mit dem
humanen Codongebrauch verwendet (Pöggeler et al. 2003).
Tabelle 2: Varianten des grün-fluoreszierenden Proteins aus der Qualle Aequorea victoria. Dargestellt sind die Extinktions- und Emissionsmaxima, sowie die Aminosäuresequenzen der Chromophore und wichtige Mutationen, die zur Veränderung der Proteineigenschaften führen.
Protein
Extinktion
(nm)
Emission
(nm)
Chromophor
Mutationen
Referenzen
wtGFP
395/475
508
SYG
---
(Cody et al.
1993) EGFP
488
511
TYG
F64L; S65T
(Cormack et al.
1996) EBFP
383
445
THG
F64L; S65T; Y66H;
Y145F
(Heim et al.
1994, Patterson et al. 1997)
ECFP
433
475
TWG
F64L; S65T; Y66W;
N146I; M153T; V163A
(Heim und Tsien 1996, Miyawaki
et al. 1997)
EYFP
514
527
GYG
S65G; V68L; S72A;
T203Y
(Ormö et al.
1996)
Bei der Verwendung des grün-fluoreszierenden Proteins als Reporter sollten aber auch
zwei Nachteile nicht unberücksichtigt bleiben. Zum einen benötigt das Protein Sauerstoff
für die Ausbildung des Chromophors, so dass es in Zellkompartimenten mit geringem
Sauerstoffanteil nicht verwendet werden kann. Zum anderen konnte beobachtet werden,
dass eine starke Überexpression des gfp-Gens letal wirken kann. Ein Grund für die

I. EINFÜHRUNG
10
Schädigung der Zellen besteht darin, dass während der Reifung die Freigabe von H2O2 in
einer 1:1 Stöchiometrie mit dem reifen GFP erfolgt (Übersicht bei Tsien 1998). Der erste Hyphenpilz, in dem das grün-fluoreszierende Protein als Reporter eingesetzt
wurde, ist Ustilago maydis. Es diente hier der Analyse von Interaktionen zwischen dem
Pflanzen-Wirt und U. maydis als Pathogen (Spellig et al. 1996). Kurze Zeit später wurde
für A. nidulans ebenfalls der Einsatz des GFPs als Marker für Kerntransporte beschrieben
(Suelmann et al. 1997). In der Zwischenzeit fanden das GFP und seine Varianten
Anwendung in viele Analysen in Hyphenpilzen, so konnten in A. nidulans beispielsweise
das endoplasmatische Retikulum und Mitochondrien markiert, sowie die Dynamik
cytoplasmatischer Mikrotubuli verdeutlicht werden (Fernández-Ábalos et al. 1998,
Suelmann und Fischer 2000, Enke et al. 2007). Eine große Vielzahl weiterer GFP-
ähnlicher Proteine wurde aus unterschiedlichen marinen Organismen isoliert und sind zum
Teil auch kommerziell erhältlich (siehe z.B. Verkhusha und Lukyanov 2004, Shaner et al.
2005). Die Etablierung dieser fluoreszierenden Proteine in Hyphenpilzen steht jedoch noch
aus. 3.2 Rot-fluoreszierende Proteine
Die Veränderung der Aminosäuresequenz des grün-fluoreszierenden Proteins aus
A. victoria führte nur mit mäßigem Erfolg zur Generierung einer rot-fluoreszierenden
Mutante (Lippincott-Schwartz und Patterson 2003, Czymmek et al. 2004). Im Jahre 1999
konnte aber ein rot-fluoreszierendes Protein aus einer marinen Koralle der Gattung
Discosoma isoliert werden, nach der es die Bezeichnung DsRed (drFP583) erhielt. Obwohl
es nur eine geringe Sequenzhomologie zu dem GFP aufweist, besitzt es eine fast identische
räumliche Struktur und einen nahe verwandten Chromophor. Anstelle des Ser65 des GFPs
enthält das aus 225 Aminosäuren bestehende DsRed-Protein die Aminosäure Glutamin.
Durch eine zusätzliche Oxidationsreaktion am Glutamin werden die benachbarten
Aminosäuren bis zum Phe64 in das Elektronensystem mit einbezogen, wodurch es zu
Absorptions- und Emissionsspektren von 558 und 583 nm kommt (Matz et al. 1999, siehe
Tabelle 3).
Die Verwendung des Wildtyp-DsRed-Proteins als Reporter hat den signifikanten Nachteil,
dass es eine geringe Fluoreszenz aufweist und eine relativ lange Reifungszeit benötigt.
Aufgrund der starken Tendenz zur Aggregation kann es zur Toxizität während der
Expression führen (Baird et al. 2000, Jakobs et al. 2000). Die obligate Tetramerbildung

I. EINFÜHRUNG
11
kann des weiteren zu unerwünschten Veränderungen in der Lokalisation und Funktion von
DsRed-Fusionsproteinen führen (Lauf et al. 2001). Auch hier konnten verschiedene
DsRed-Varianten mittels zufälliger und gerichteter Mutagenese erzeugt werden. Diese
weisen eine schnellere Reifung des Chromophors, z.T. stärkere, hellere Fluoreszenz und
eine bessere Löslichkeit im Vergleich zum Wildtyp-DsRed auf (Bevis und Glick 2002).
Zwei kommerziell erhältliche Varianten, die fünf bis neun Aminosäuresubstitutionen
aufweisen, werden durch die Proteine DsRed2 und DsRed-Express vertreten (BD
Biosciences Clonetech). Das Problem der Tetramerbildung konnte mit Hilfe dieser
Mutationen allerdings nicht gelöst werden (Terskikh et al. 2002, Yanushevich et al. 2002).
Die Bildung eines monomeren ror-fluoreszierenden Proteins erfolgte über die
Zwischenstufe eines DsRed-Dimers, sowie der Substitution von insgesamt 33
Aminosäuren. Das hierbei entstandene Protein wurde mRFP1 genannt und zählt zu der
ersten Generation monomerer rot-fluoreszierender Proteine (Campbell et al. 2002, Shu et
al. 2006). Obwohl dieses Protein eine Reduktion des Extinktionskoeffizienten, der
Quantenausbeute und der Fotostabilität aufweist, reift es etwa zehnmal schneller als das
Wildtyp-DsRed, so dass Proteinaggregationen und die Zelltoxizität bei der Verwendung
als Reporter stark reduziert werden können. Durch die verschiedenen Substitutionen
erfolgte eine Verschiebung der Extinktions- und Emissionsmaxima um ca. 25 nm auf 584
bzw. 607 nm (Campbell et al. 2002). Weitere Optimierungen der bis dato existierenden rot-
fluoreszierenden Proteine hatten vor allem den Schwerpunkt, Monomere zu erzeugen und
negative Eigenschaften des mRFP zu eliminieren. Die resultierenden Varianten zeigten,
neben den erwünschten Verbesserungen in der Quantenausbeute, der Extinktions-
koeffizienten, der Fotostabilität und der Toleranz N- und C-terminaler Fusionen, mehrere
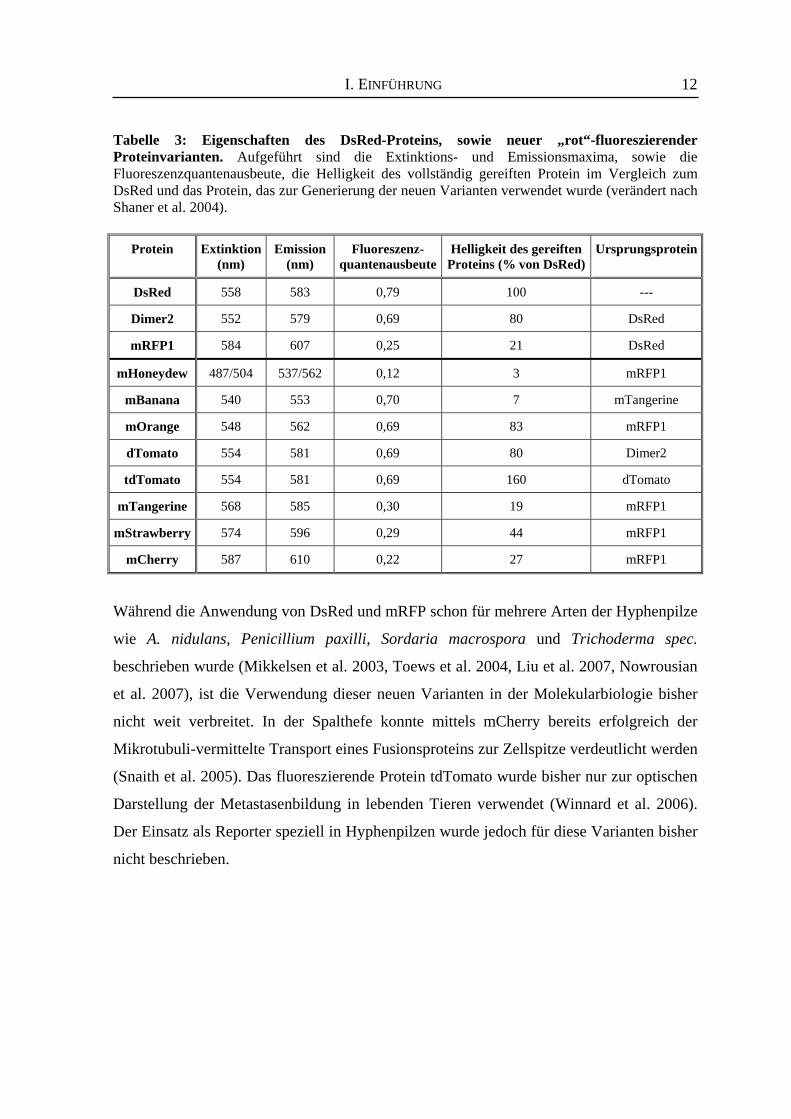
unterschiedliche Fluoreszenzfarben (siehe Tabelle 3, Shaner et al. 2004). Sie gehören zu
der zweiten Generation monomerer rot-fluoreszierender Proteine und werden als Gruppe
unter der Bezeichnung mFruits zusammengefasst. Diese neue Generation umfasst einen
bemerkenswerten Bereich an Extinktions- und Emissionsmaxima (Ex: 540-590 nm &
Em: 550-650 nm) (Shaner et al. 2004, Shu et al. 2006), so dass für jede Anwendung ein
geeignetes spezifisches autofluoreszierendes Protein zur Verfügung steht.
I. EINFÜHRUNG
12
Tabelle 3: Eigenschaften des DsRed-Proteins, sowie neuer „rot“-fluoreszierender Proteinvarianten. Aufgeführt sind die Extinktions- und Emissionsmaxima, sowie die Fluoreszenzquantenausbeute, die Helligkeit des vollständig gereiften Protein im Vergleich zum DsRed und das Protein, das zur Generierung der neuen Varianten verwendet wurde (verändert nach Shaner et al. 2004).
Protein
Extinktion
(nm)
Emission
(nm)
Fluoreszenz-
quantenausbeute
Helligkeit des gereiften
Proteins (% von DsRed)
Ursprungsprotein
DsRed
558
583
0,79
100
---
Dimer2
552
579
0,69
80
DsRed
mRFP1
584
607
0,25
21
DsRed
mHoneydew
487/504
537/562
0,12
3
mRFP1
mBanana
540
553
0,70
7
mTangerine
mOrange
548
562
0,69
83
mRFP1
dTomato
554
581
0,69
80
Dimer2
tdTomato
554
581
0,69
160
dTomato
mTangerine
568
585
0,30
19
mRFP1
mStrawberry
574
596
0,29
44
mRFP1
mCherry
587
610
0,22
27
mRFP1
Während die Anwendung von DsRed und mRFP schon für mehrere Arten der Hyphenpilze
wie A. nidulans, Penicillium paxilli, Sordaria macrospora und Trichoderma spec.
beschrieben wurde (Mikkelsen et al. 2003, Toews et al. 2004, Liu et al. 2007, Nowrousian
et al. 2007), ist die Verwendung dieser neuen Varianten in der Molekularbiologie bisher
nicht weit verbreitet. In der Spalthefe konnte mittels mCherry bereits erfolgreich der
Mikrotubuli-vermittelte Transport eines Fusionsproteins zur Zellspitze verdeutlicht werden
(Snaith et al. 2005). Das fluoreszierende Protein tdTomato wurde bisher nur zur optischen
Darstellung der Metastasenbildung in lebenden Tieren verwendet (Winnard et al. 2006).
Der Einsatz als Reporter speziell in Hyphenpilzen wurde jedoch für diese Varianten bisher
nicht beschrieben.

I. EINFÜHRUNG
13
4. Anwendung von Reportergenen in Hyphenpilzen Nach der Entwicklung geeigneter Transformationssysteme für Hyphenpilze folgte die
Etablierung von Genfusionssystemen zur gezielten Analyse der Genexpression (van
Gorcom et al. 1986). Eine Vielzahl unterschiedlicher Reportersysteme konnte somit in den
letzten Jahrzehnten wirkungsvoll in Hyphenpilzen etabliert werden und zum Verständnis
der pilzlichen Biologie und der Pilz-Wirt-Interaktion beitragen. In den nächsten
Teilkapiteln werden vier Bereiche vorgestellt, die erfolgreich mit Hilfe von verschieden-
artigen Reportersystemen analysiert werden konnten. 4.1 Lokalisationsstudien
Mit Hilfe von Reporterproteinen können in Lokalisationsstudien sowohl einzelne Proteine,
als auch Proteinkomplexe und ihr Transport innerhalb einer pilzlichen Zelle beobachtet
werden. Des weiteren werden in diesem Forschungsgebiet Reportergensysteme verwendet,
um das Wachstum phytopathogener Pilze innerhalb der Gewebe ihres Wirtes sichtbar zu
machen. So kann die Expression der β-Glucuronidase und die daraus resultierende Färbung
verwendet werden, um das Wachstum phytopathogener Pilze in pflanzlichen Geweben zu
beobachten (Roberts et al. 1989). Die Expression von gfp unter einem konstitutiven
Promotor führt zur cytoplasmatischen Lokalisation des Proteins in allen pilzlichen
Strukturen (z.B. Hyphen, Sporen, Appressorien, usw.) ohne ersichtliche Veränderung des
Wachstums oder der Pathogenität. Dieses ermöglicht, den Pilz in Pflanzen sichtbar zu
machen, seine Verteilung zu beobachten und die Biomasse zu bestimmen (Übersicht bei
Lorang et al. 2001). Das GFP ist aber auch ein optimaler Reporter, um dynamische
Prozesse in pilzlichen Zellen aufzuklären. In A. nidulans konnte die Markierung von
Zellkernen, beispielsweise mit Hilfe eines Fusionsprotein aus GFP und der Kern-
lokalisationsdomäne des StuA Transkriptionsfaktors, der an der Regulation der
Konidiophorentwicklung beteiligt ist, zur Echtzeitanalyse von Kernwanderung und Mitose
verwendet werden. Die erzielten Ergebnisse trugen erstmals zum Verständnis des
Verhaltens spezifischer Kerne in verschiedenen Entwicklungsstadien bei (Suelmann et al.
1997, Fernández-Ábalos et al. 1998). Des weiteren wurden zwei unterschiedlich markierte
Pilzstämme von N. crassa verwendet, um die Bildung von Hyphenanastomosen zu
verfolgen. Hierzu wurde zum einen ein Stamm erzeugt, der ein GFP-markiertes,
cytoplasmatisches Protein synthetisiert und zum anderen ein Stamm, der das DsRed-
Polypeptid kernlokalisiert aufweist. Nach erfolgreicher Fusion der Hyphen beider Stämme

I. EINFÜHRUNG
14
konnte sowohl die GFP- als auch die DsRed-Fluoreszenz innerhalb einer Hyphe
beobachtet werden (Dementhon et al. 2006).
Ferner können das GFP und seine Varianten auch als Reporter für Protein-Protein-
Interaktionsstudien verwendet werden. Kürzlich wurde für Hyphenpilze ein System zur
in vivo-Analyse von Protein-Protein-Interaktionen etabliert (Hoff und Kück 2005). Es
handelt sich hierbei um die „bimolecular fluorescence complementation“ (BiFC)-Methode,
die auf der Bildung eines funktionellen fluoreszierenden Komplexes beruht, der sich erst
dann ausbildet, wenn zwei putative Interaktionspartner, die jeweils mit einer Hälfte des
YFP fusioniert sind, in räumliche Nähe gelangen (Hu et al. 2002). Während der
Etablierung dieses Systems konnte die nukleäre Interaktion zweier Transkriptionsfaktoren
(AcFKH1 und CPCR1) des β-Laktamproduzenten Acremonium chrysogenum bestätigt
werden (Hoff und Kück 2005).
Anhand dieser Beispiele wird deutlich, dass für die Untersuchung von Protein-
lokalisationen in Hyphenpilzen zumeist autofluoreszierende Proteine als Reporter
eingesetzt werden, da diese ein hohes Potential für unterschiedliche Anwendungen
besitzen. 4.2 Sekretion
Sekundärmetabolite (z.B. Antibiotika, Mykotoxine und andere Botenstoffe) werden von
einer Vielzahl unterschiedlicher Organismen, insbesondere auch Hyphenpilzen, gebildet.
Sie sind weder essentiell für das Wachstum, noch für die Vermehrung des Produzenten.
Die meisten Sekundärmetabolite dienen eher als Kommunikationssignale mit anderen
Lebewesen, wie Pflanzen, Tieren und Mikroorganismen, die den gleichen Lebensraum
besiedeln (Martín et al. 2005). Der Sekretionsweg dieser sekundären Stoffwechselprodukte
stellt allerdings einen limitierenden Faktor bei der industriellen Produktion dar, so dass die
Aufklärung der Systeme, die an der Sekretion von Antibiotika und anderen industriell
genutzten Enzymen beteiligt sind, von großem wirtschaftlichem Nutzen ist. In A. niger
wurde, unter Verwendung eines chimären Fusionsprotein bestehend aus einer
sezernierbaren Glucoamylase und GFP (GLA::GFP), die in vivo-Analyse von
Sekretionsprozessen in wachsenden Hyphen durchgeführt. Hierbei konnte zum einen
verdeutlicht werden, dass diese Fusion einen nützlichen Reporter für die direkte
Visualisierung des Proteintransports und der Sekretion darstellt. Zum anderen erfolgte
nach UV-Mutagenese des GLA::GFP-Stammes die Isolierung von Mutanten des

I. EINFÜHRUNG
15
allgemeinen Sekretionsweges. Es konnte beobachtet werden, dass diese Transformanten
das Fusionsprotein intrazellulär akkumulierten (Gordon et al. 2000). Die β-Glucuronidase
wird in Pilzen ebenfalls als Reporter von Sekretionsprozessen verwendet. Mit Hilfe von
Fusionsproteinen bestehend aus der β-Glucuronidase und verschiedener Xylanase-Gene
aus Penicillium funiculosum konnte deren Nutzen als Carrier-Proteine für die Etablierung
von P. funiculosum als Wirt für die Produktion heterologer Proteine beurteilt werden
(Alcocer et al. 2003).
Sekretionsprozesse spielen in Pilzen auch eine wichtige Rolle bei der Erkennung des
entsprechenden Kreuzungs-Partners. Hierzu werden Pheromone ins umgebende Medium
sezerniert. Erfolgt die Bindung des Pheromons an den korrespondierenden Rezeptor, löst
dieses eine charakteristische Antwort aus, die bewirkt, dass sich die Zellen neu orientieren
und zum möglichen Kreuzungs-Partner wachsen und miteinander fusionieren (Casselton
2002). Das S. macrospora ppg1-Gen kodiert für ein Pheromonvorläuferprotein, das ein
N-terminales Sekretionssignal besitzt. Zur Bestimmung der Sekretionsfunktion dieses
Signalpeptids wurden Fusionen der entsprechenden Aminosäuresequenz mit dem GFP
erstellt. Western Blot-Analysen des Kulturüberstandes verdeutlichten, dass dieses Signal-
peptid ausreichend und funktionell ist, um GFP über den Sekretionsweg ins Medium zu
sezernieren (Mayrhofer und Pöggeler 2005). Diese ausgewählten Beispiele zeigen, dass der
Einsatz unterschiedlichster Reportergene zur Aufklärung der Sekretionswege in
Hyphenpilzen beitragen kann. 4.3 Promotoranalysen
Cis-wirkende Elemente können funktionell analysiert werden, indem sukzessive
Deletionen, wechselnde Sequenzorientierungen oder gezielte Mutationen in Promoter-
regionen erzeugt werden. Weiterhin ermöglichen Reportergene die Analyse der
Induzierbarkeit und der Stärke eines Promoters. Hierzu steht das Reportergen unter
Kontrolle des zu analysierenden Promotors oder modifizierten Promotorfragmenten. Eine
anschließende quantitative Analyse des Genproduktes gibt Aufschluss über die Regulation
der entsprechenden Region. Bereits zu Beginn der 90er Jahre wurde zu diesem Zweck
schon ein Vektor konstruiert, der zwei prokaryotische Reportergene (lacZ & uidA) in
unterschiedlicher Orientierung enthielt, und somit eine simultane Analyse ermöglicht (Punt
et al. 1991).

I. EINFÜHRUNG
16
Die Gene der Cephalosporin C- und Penicillin-Biosynthese sind im Genom der
entsprechenden Hyphenpilze A. chrysogenum und P. chrysogenum in Clustern angeordnet,
wodurch eine koordinierte Regulation gewährleistet sein könnte. Die Biosynthesegene
werden von bidirektionalen Promotoren kontrolliert und weisen Konsensussequenzen für
verschiedene Transkriptionsfaktoren auf. Die Analyse dieser Promotorbereiche führte zur
Identifizierung essentieller, regulatorischer cis- und trans-wirkender Faktoren (Übersicht
bei Brakhage et al. 2004, Schmitt et al. 2004a). Erst vor kurzem konnte beispielsweise der
Einfluss des globalen Regulators Velvet A aus A. nidulans auf die Expression der
Penicillin-Biosynthesegene acvA und ipnA dank Fusionen mit den Reportergenen uidA und
lacZ bestimmt werden (Spröte und Brakhage 2007).
Des weiteren wurden viele pilzliche Promotoren mit Hilfe von Reportergenen auf ihre
Induzierbarkeit bzw. Reprimierbarkeit analysiert. So zeigten beispielsweise Analysen mit
dem Reporterprotein β-Glucuronidase, dass der Promotor des cfp-Gens, das für ein
cytoplasmatisches Filamentprotein in N. crassa kodiert, in Gegenwart von Glukose und
Saccharose eine Hochregulation der Reportergenexpression vermittelt und in Gegenwart
von Ethanol eine Repression der Genexpression erfährt (Temporini et al. 2004). Ferner
konnte mit Hilfe der β-Glucuronidase eine ausgeweitete Analyse der Promotorsequenz der
Endoxylanase (xylP) und die detaillierte Charakterisierung der Induktion der xylP-
Expression in P. chrysogenum erfolgen (Zadra et al. 2000). 4.4 RNAi-Analysen
Mit der Entdeckung des Phänomens der RNA Interferenz (RNAi) bei C. elegans konnte
gezeigt werden, dass die Integration doppelsträngiger RNA (dsRNA) das
Herunterregulieren Sequenz-spezifischer Gene bewirkt (Fire et al. 1998, Agrawal et al.
2003). Die Etablierung eines solchen RNAi-Systems bietet für eine Vielzahl
unterschiedlicher Hyphenpilze nun die Möglichkeit Genfunktionen durch Herunter-
regulation der Expression zu analysieren. Die Vorteile liegen vor allem in der schnellen
Umsetzung dieses Systems, ferner können nun auch Gene analysiert werden, deren
vollständiger Verlust zur Letalität führen (Übersicht bei Nakayashiki 2005). Damit ein
erfolgreicher RNAi-Versuch schnell und einfach nachgewiesen werden kann, haben
Forschergruppen Vektorsysteme entwickelt, bei denen ein Reportergen simultan mit dem
endogenen Zielgen herunterreguliert wird. Häufig wurde hierfür das gfp-Gen verwendet.
Das Ausbleiben der Fluoreszenz während der Fluoreszenzmikroskopie oder der

I. EINFÜHRUNG
17
Fluoreszenzspektroskopie dient hierbei als Hinweis für einen erfolgreichen RNAi-Ansatz,
so konnten z.B. parallel Gene für das GFP und die Trihydroxynaphthalen-Reduktase in
Venturia inaequalis herunterreguliert werden, um nützliche Marker für Silencing-Prozesse
zu etablieren (Fitzgerald et al. 2004, de Jong et al. 2006). Im Rahmen dieser Arbeit konnte
ebenfalls ein fluoreszierendes Protein, das DsRed, als Reporter in RNAi-Analysen
verwendet werden. A. chrysogenum-Stämme, die das DsRed-Gen exprimieren, zeigen auf
Festmedien einen roten Phänotyp. Während der Etablierung des RNAi-Systems konnte
verdeutlichet werden, dass dieser Reporter zusätzlich mit einem endogenen Gen der
Cephalosporin C-Biosynthese simultan herunterreguliert werden konnte (Janus et al.
2007). Somit vereinfacht die Verwendung von Reportergenen, die z.B. einen optischen
Phänotyp hervorrufen, das Screening von Transformanten, die einen RNAi-Effekt
aufweisen.
5. Zusammenfassung Die Fortschritte in der Reportergentechnologie während der letzten Jahre führten zu
verbesserten und z.T. vereinfachten Analysemethoden unterschiedlicher Prozesse in Pro-
und Eukaryoten. So konnten für verschiedene Enzym-kodierende Reportergensysteme
unterschiedliche Substrate identifiziert werden, die einfachere, schnellere und z.T.
ungefährlichere Analysen, z.B. durch den Ersatz radioaktiver Substrate, ermöglichen. Die
Identifizierung und Modifikation fluoreszierender Proteine führte zu einer breiten Palette
spezifisch anwendbarer Reporter und ließ sie zu essentiellen „Werkzeugen“ der
molekularen Biologie werden.
Stetige Verbesserungen in der darstellenden Technologie und die Weiterentwicklung
multipler fluoreszierender Proteine, wie auch der Fluoreszenz- und Lumineszenz-
analysemethoden für Lebendzellanalysen werden zu einer drastischen Förderung der
Aufklärung molekularer und zellulärer Prozesse in den nächsten Jahren führen. Das
vielseitige Spektrum der anwendbaren Reportergensysteme kann z.B. in industriellen
Stammoptimierungsprozessen eingesetzt werden und dort zu erhöhter Produktivität von
Sekundärmetaboliten der Produktionsstämme führen.

II. PROBLEMSTELLUNG
18
II. PROBLEMSTELLUNG β-Laktam-Antibiotika stellen etwa 65 % des weltweiten Antibiotikamarktes dar und
repräsentieren ein Weltmarktvolumen von mehreren Milliarden US$. Somit gehören sie zu
einer der erfolgreichsten Wirkstoffgruppen mit antibakterieller Potenz (Elander 2003).
Aufgrund seiner Wirkung auf sowohl gram-positive als auch gram-negative Bakterien
findet das β-Laktam-Antibiotikum Cephalosporin C eine vielfache Anwendung in der
klinischen Medizin. Der industriell genutzte Hauptproduzent dieses Antibiotikums ist der
imperfekte Hyphenpilz Acremonium chrysogenum (syn. Cephalosporium acremonium),
der 1948 von Guiseppi Brotzu isoliert wurde (Brotzu 1948).
Die Biosynthese des Cephalosporin C verläuft ausgehend von den drei Aminosäuren
L-α-Aminoadipinsäure, L-Cystein und L-Valin (siehe Anhang Abb. A1), die zunächst zu
einem linearen Tripeptid verknüpft werden. Anschließend findet eine oxidative
Zyklisierung zum Isopenicillin N statt, dem ersten antibiotisch aktiven Intermediat.
Während dieser Reaktion entsteht das für β-Laktam-Antibiotika spezifische Ringsystem.
Weitere Syntheseschritte führen über mehrere Zwischenprodukte und der abschließenden
Addition einer Acetyl-Gruppe zum Endprodukt Cephalosporin C (Übersicht bei Schmitt et
al. 2004a). Die entsprechenden Biosynthesegene und ihre Genprodukte sind in Abbildung
A1 des Anhangs dargestellt. Sogenannte Hochleistungsstämme des Hyphenpilzes A. chrysogenum werden zur Zeit
mittels konventioneller Mutagenese und anschließenden, aufwendigen Selektionsverfahren
erzeugt (Elander 1999, Elander 2003). Sie zeigen eine etwa 100-fache Erhöhung des
Cephalosporin C-Titers verglichen zum Wildtyp, weisen jedoch auch hohe genetische
Instabilitäten auf. Die steigende Anzahl resistenter, pathogener Mikroorganismen erfordert
jedoch die stete Forschung und Entwicklung neuer, verbesserter β-Laktam-Antibiotika-
generationen (Elander 2003). Angriffspunkte dieser Verbesserungen waren in den letzten
Jahren vor allem die Biosynthesegene. Vergleichende Analysen von Wildtyp- und
Produzentenstämmen aus Stammoptimierungsverfahren haben verdeutlicht, dass eine
gesteigerte Antibiotikaproduktion voraussichtlich nicht durch Mutationen der Biosynthese-
gene selbst, sondern durch Veränderungen der Regulatoren und den an ihnen beteiligten
Signalkaskaden verursacht wird. Sequenzanalysen von Promotorregionen der Biosynthese-
gene verifizierten diese Hypothese, da keinerlei Sequenzunterschiede in diesen Bereichen

II. PROBLEMSTELLUNG
19
ermittelt werden konnten (Übersicht bei Brakhage und Caruso 2004). Zudem ist bekannt,
dass das Cephalosporin C nicht konstitutiv gebildet wird, sondern einer Regulation in
Abhängigkeit von Nahrungsquellen und dem pH-Wert der Umgebung unterliegt. Die
Expression der Biosynthesegene wird hierbei hauptsächlich auf der Ebene der
Transkription reguliert, so dass Transkriptionsfaktoren eine entscheidende Rolle zufällt.
Erste spezifische Transkriptionsfaktoren und globale Regulatoren (CPCR1, AcFKH1,
PACC, CRE1, VeA) konnten bereits isoliert werden, die direkt oder indirekt auf die
Cephalosporin C-Biosynthese wirken und somit einen deutlichen Einfluss auf die
Syntheseleistung nehmen (Jekosch und Kück 2000a, Schmitt und Kück 2000, Schmitt et
al. 2001, Schmitt et al. 2004b, Dreyer et al. 2007). In den letzten Jahren wurden unterschiedliche Resistenzmarker, Reportergene und
Methoden entwickelt, um die Grundlagenforschung an filamentös wachsenden Pilzen
voranzutreiben. Mit Hilfe der steigenden Anzahl neuer molekularbiologischer Techniken
ist es nun möglich, die genetische Regulation der Antibiotikasynthese zu analysieren sowie
wichtige Faktoren zu isolieren und zu manipulieren. Die kontrollierte Expression und
Manipulation spezifischer Gene stellt somit ein wichtiges Hilfsmittel für weitere
Stammoptimierungsstrategien dar.
Ziel dieser Arbeit war die Etablierung neuer Methoden basierend auf dem rot-
fluoreszierenden Protein DsRed zur Vereinfachung der Grundlagenforschung von
Acremonium chrysogenum. Zwei unterschiedliche Ansätze sollten durchgeführt werden, in
denen die Verwendung des DsRed-Gens als Reporter erfolgte.
1. Entwicklung eines RNAi-Systems zur Anwendung in A. chrysogenum Verschiedene RNA-Interferenz (RNAi)-Systeme wurden in den letzten Jahren entwickelt,
um die Genexpression von Zielgenen herunterzuregulieren (Nakayashiki 2005). Die
Reduktion der Expression wird durch Integration von dsRNA bzw. Vektoren, die eine
Ausbildung von Haarnadelstrukturen der RNA hervorrufen, erreicht.
Ein Ziel dieser Arbeit bestand darin, die Methode der RNA-Interferenz in A. chrysogenum
zu etablieren, um die Analyse von Genfunktionen in diesem industriell relevanten
Hyphenpilz zu vereinfachen und zu beschleunigen. Hierzu sollte zunächst ein Stamm
generiert werden, der das DsRed-Gen konstitutiv exprimiert. In vorangegangenen Arbeiten
konnte beobachtet werden, dass die Expression des DsRed-Gens in A. chrysogenum zur

II. PROBLEMSTELLUNG
20
sichtbaren Rotfärbung des Myzels führt, selbst wenn der Pilz auf Festmedium wächst
(Hoff persönl. Mitteilung). Dieses Phänomen wurde ebenfalls in Penicillium paxilli
nachgewiesen und es wird vermutet, dass eine Rotfärbung nur bei Pilzen mit hoher
Myzeldichte erfolgt (Mikkelsen et al. 2003). Damit stellt das DsRed-Protein einen sehr
guten Reporter in dem Hyphenpilz A. chrysogenum dar.
Ein solcher Stamm sollte als Rezipient für die Transformation eines RNAi-Konstrukts
dienen, bei dem das DsRed-Gen in „sense“ und inverser Orientierung inseriert und durch
ein Intron getrennt vorliegt. Nach erfolgreicher Transformation sollten die Transformanten
sowohl auf RNA-Ebene, als auch auf Proteinebene zur Quantifizierung der DsRed-Menge
im Gesamtproteinextrakt analysiert werden.
Die Konstruktion des RNAi-induzierenden Vektors basierte auf dem Plasmid pSilent-1,
das bereits erfolgreich in den Hyphenpilzen Magnaporthe oryzae und Colletotrichum
lagenarium zur stabilen Abnahme der Genexpression von Zielgenen verwendet werden
konnte (Nakayashiki et al. 2005). Der Vektor pSilent-1 besitzt eine transkriptionelle
Einheit zur Ausbildung von Haarnadelstrukturen, hierzu zählen der Promotor und
Terminator des trpC-Gens aus A. nidulans, das Cutinase-Intron 2 aus M. oryzae, das von
multiplen Klonierungsstellen flankiert wird und der Integration von Zielgenfragmenten in
inverser Orientierung dient sowie je eine Resistenzkassette zur Selektion pilzlicher bzw.
bakterieller Transformanten (siehe Abb. 1, Nakayashiki et al. 2005).
Aufbauend auf dieser Grundlage wurde ein weiterer RNAi-induzierender Vektor
hergestellt, der sowohl das DsRed-Gen, wie auch ein endogenes A. chrysogenum
spezifisches Gen in „sense“ und inverser Orientierung trägt. Als endogenes Gen wurde
exemplarisch das Cephalosporin C-Biosynthesegen pcbC, welches für die Isopenicillin N-
Synthase kodiert (siehe Anhang Abb. A1), mittels Induktion des RNAi-Mechanismus
herunterreguliert. In vorausgegangenen Analysen führte die Inaktivierung des pcbC-Gens
zur klaren Reduzierung des Cephalosporin C-Titers (Walz und Kück 1993). Eine mögliche
reduzierte DsRed- und pcbC-Expression innerhalb der erhaltenen Transformanten sollte
anschließend auf RNA- und Protein-Ebene analysiert werden. Zudem sollte der
Cephalosporin C-Titer dieser Stämme ermittelt werden (Stauffer et al. 1966). Mit den hier
dargestellten Analysen sollte verdeutlicht werden, dass RNAi-Analysen zur Untersuchung
der Cephalosporin C-Biosynthese beitragen können.

II. PROBLEMSTELLUNG
21
Abb. 1: Schematische Darstellung des ca. 6,9 kbgroßen Vektors pSilent-1 (Nakayashiki et al.2005). Ampr: Ampicillin-Resistenzgen; Hygr:Hygromycin-Resistenzgen; IT: Intron 2 desCutinasegens aus M. oryzae; PtrpC: A. nidulanstrpC-Promotor; TtrpC: A. nidulans trpC-Terminator
2. Analysen des A. chrysogenum cre1-Promotors Das DsRed-Reportergen bietet ferner die Möglichkeit zur detaillierten Funktionsanalyse
von Genen und regulatorischen Elementen. Ein interessanter Ansatzpunkt ist hier der
Glukose-Repressor CRE1 aus A. chrysogenum, der nachweislich für die Repression
verschiedener Cephalosporin C-Biosynthesegene verantwortlich ist (Jekosch und Kück
2000b). Gleichzeitig mit der Reduktion der Transkription von Biosynthesegenen erfolgt
die Zunahme des cre1-Transkriptes, so dass für cre1 eine positive Autoregulation
postuliert wurde (Jekosch und Kück 2000a). In silico-Analysen unter Verwendung der
CRE-Konsensussequenz haben gezeigt, dass mehrere putative Bindestellen für das CRE1-
Protein in den Promotoren der Cephalosporin C-Biosynthesegene, sowie der cre1-
Promotorregion identifiziert werden konnten (Übersicht bei Schmitt et al. 2004a). In ersten
vorausgegangenen Arbeiten wurde die cre1-Promotorregion mit Hilfe von verschiedenen
Verkürzungskonstrukten analysiert. Als Reporter der cre1-vermittelten Genexpression
wurde das cefEF-Gen, welches für die Deacetoxycephalosporin C/Deacetylcephalosporin
C-Synthetase kodiert, in Northern Blot-Hybridisierungen und Western Blot-Analysen
verwendet. Die erzielten Ergebnisse ließen allerdings keinen Rückschluss auf wichtige
Bereiche bzw. essentielle CRE1-Bindestellen zu (Ogrodowczyk 2002). Der zweite Teil dieser Arbeit befasste sich somit mit der Analyse des A. chrysogenum
cre1-Promotors. Hierbei ging es vor allem darum, die für die Glukoseregulation
essentiellen Bereiche zu identifizieren und einen möglichst kleinen Core-Promotor zu
definieren. Verschiedene Deletionsderivate des cre1-Promotors wurden angefertigt, bei
denen verschiedene putative CRE1-Bindestellen und andere mögliche regulatorische

II. PROBLEMSTELLUNG
22
Bereiche entfernt wurden. Diese Derivate sollten in vivo mit Hilfe des DsRed-Reportergens
auf Transkript- und Proteinebene analysiert werden.
Zur weiteren Charakterisierung putativer CRE1-Bindestellen im cre1-Promotor sollten
Protein-DNA-Interaktionsstudien durchgeführt werden. Hierzu erfolgte zunächst die
Herstellung eines rekombinanten GST::CRE1 Proteins, das anschließend aus E. coli
gereinigt und mit verschiedenen radioaktiv-markierten DNA-Fragmenten des cre1-
Promotors in Gelretentionsanalysen verwendet werden sollte. Zur Bestätigung der
Spezifität dieser Interaktionen wurden ferner Surface Plasmon Resonance-Analysen
durchgeführt. Diese Methode ermöglicht die Bestimmung der Echtzeit-Kinetik
molekularer Interaktionen (Myszka 2000). Zusammenfassend war das Ziel dieser Arbeit, neue Methoden zu entwickeln, die zur
vereinfachten Analyse der Cephalosporin C-Biosynthese und deren Regulatoren in
Acremonium chrysogenum beitragen. Ferner sollten Bereiche des cre1-Promotors, die für
die Glukoseregulation des cre1-Gens verantwortlich sind, ermittelt werden und folglich ein
Core-Promotor definiert werden. Ein besseres Verständnis der Regulation der β-Laktam-
Biosynthese kann zur weiteren Optimierung der Produktionsstämme beitragen.

III. JANUS ET AL. 2007A
23
An Efficient Fungal RNAi-Silencing System Using
the DsRed Reporter Gene
Janus D, Hoff B, Hofmann E, Kück U (2007)
Applied and Environmental Microbiology 73: 962-970

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Feb. 2007, p. 962–970 Vol. 73, No. 30099-2240/07/$08.00�0 doi:10.1128/AEM.02127-06Copyright © 2007, American Society for Microbiology. All Rights Reserved.
An Efficient Fungal RNA-Silencing System Usingthe DsRed Reporter Gene�
Danielle Janus,1 Birgit Hoff,1 Eckhard Hofmann,2 and Ulrich Kuck1*Lehrstuhl fur Allgemeine und Molekulare Botanik, Ruhr-Universitat, Universitatsstr. 150, D-44780 Bochum, Germany,1 and
Lehrstuhl fur Biophysik, Ruhr-Universitat, D-44780 Bochum, Germany2
Received 8 September 2006/Accepted 20 November 2006
In filamentous fungi, RNA silencing is an attractive alternative to disruption experiments for the functionalanalysis of genes. We adapted the gene encoding the autofluorescent DsRed protein as a reporter to monitorthe silencing process in fungal transformants. Using the cephalosporin C producer Acremonium chrysogenum,strains showing a high level of expression of the DsRed gene were constructed, resulting in red fungal colonies.Transfer of a hairpin-expressing vector carrying fragments of the DsRed gene allowed efficient silencing ofDsRed expression. Monitoring of this process by Northern hybridization, real-time PCR quantification, andspectrofluorometric measurement of the DsRed protein confirmed that downregulation of gene expression canbe observed at different expression levels. The usefulness of the DsRed silencing system was demonstrated byinvestigating cosilencing of DsRed together with pcbC, encoding the isopenicillin N synthase, an enzymeinvolved in cephalosporin C biosynthesis. Downregulation of pcbC can be detected easily by a bioassaymeasuring the antibiotic activity of individual strains. In addition, the presence of the isopenicillin N synthasewas investigated by Western blot hybridization. All transformants having a colorless phenotype showedsimultaneous downregulation of the pcbC gene, albeit at different levels. The RNA-silencing system presentedhere should be a powerful genetic tool for strain improvement and genome-wide analysis of this biotechno-logically important filamentous fungus.
The study of gene functions in filamentous fungi has ad-vanced substantially in recent years, and diverse genetic ap-proaches have been used to disrupt target genes by homolo-gous recombination (46). However, in most filamentous fungi,substitution of the target genes by homologous recombinationoccurs at very low frequencies due to ectopic integration of thetransforming DNA. In wild-type strains of Aspergillus nidulans,Neurospora crassa, and Sordaria macrospora, for example, ho-mologous recombination has been observed at frequencies ofabout 0.1% to 5% (4, 6, 30). One traditionally used approachis generation of multiple knockout strains; however, this ap-proach is both time-consuming and labor-intensive. It alsorequires multiple resistance markers for selecting individualtransformants showing homologous recombination (9). There-fore, discovery and sustained development of the RNA inter-ference (RNAi) system are a reliable alternative for studyinggene functions.
An advantage of the RNAi mechanism is its locus indepen-dence due to its mediation by a mobile trans-acting signal inthe cytoplasm. Consequently, this mechanism can be used infungi having multinuclear hyphae or a low targeting efficiency,and even heterokaryotic fungal strains can exhibit downregu-lation of target genes (8, 25). Generation of a knockout strainoften requires a considerable length (�1 kb) of homologoussequences, whereas at least 132 bp of sequence homology issufficient for double-stranded (dsRNA)-induced silencing of atarget gene (5, 46). Typically, approximately 500 bp of coding
sequence is used for RNAi approaches (19). This is advanta-geous when the information about genomic sequences is re-stricted. Beyond this, it is possible to silence different genes orgene families simultaneously (1, 9, 19).
Another advantage of knockdown strains is the possibility ofcontrolling expression of the silencing construct. Inducible pro-moters, for example, can turn on gene expression at specificstages during development (10, 24). Even genes showing low-level expression can be downregulated efficiently with RNAiconstructs expressing hairpin dsRNA (10). Furthermore, theRNAi system is useful when genes that are indispensable forfungal growth are investigated. In such cases, downregulationof gene expression close to zero results in survival of transfor-mants.
Diverse RNAi systems have been developed for a broadrange of filamentous fungi (25). Similar to the genomes ofother eukaryotes, the genomes of filamentous fungi encodeconserved components, such as RNA-dependent RNA poly-merases that are known to be involved in the RNAi process. Infungi, downregulation of gene expression is variable in individ-ual transformants, and gene functions can be studied only withpreselected strains. To overcome these difficulties, several in-vestigators have used vector systems in which a reporter geneis simultaneously downregulated together with an endogenoustarget gene. Such RNAi systems use either strain-specificmarker genes that can be identified phenotypically (10, 19) or,alternatively, the generally applicable gfp reporter gene, encod-ing the green fluorescent protein (GFP) (8, 9). In the lattercase, fluorescence microscopy identifies nonfluorescent strainsand thus silenced transformants (8, 9, 14).
In this investigation, we used an alternative reporter systemthat is even simpler to identify silenced transformants. As the
* Corresponding author. Mailing address: Lehrstuhl fur Allgemeineund Molekulare Botanik, Ruhr-Universitat, Universitatsstr. 150,D-44780 Bochum, Germany. Phone: 49-234-32 26212. Fax: 49-234-3214184. E-mail: [email protected].
� Published ahead of print on 1 December 2006.
962

fungal organism, we used the major cephalosporin C producerAcremonium chrysogenum, which lacks an efficient host system,to study gene functions by homologous recombination. Thisbiotechnologically relevant fungus propagates strictly asexuallyand therefore can be modified genetically only by moleculargenetic techniques. Over the past few years, we and otherworkers have developed different selection and reporter genesystems to efficiently manipulate this important fungus (17, 18,45). These systems include, for example, use of the gfp gene asa reporter of gene expression (31). Recently, an alternativeapplication became feasible because of the discovery of the redfluorescent protein (DsRed) in Discosoma sp., which has anemission spectrum in the far-red zone (20). So far, the DsRedprotein has been used as a highly effective marker in only a fewfilamentous fungi, including A. nidulans, Penicillium paxilli, andTrichoderma species (22, 43).
In this study, the DsRed gene was used to monitor geneexpression in the cephalosporin C producer A. chrysogenum.Remarkably, colonies of strains carrying the DsRed reportergene on solid media are red and thus can serve as recipients tomonitor the RNA-silencing process. Consequently, a change inthe color of the mycelium is an indicator of successful down-regulation of gene expression. In addition, to quantify theRNA-silencing efficiencies, we developed a new spectrofluoro-metric approach to measure DsRed protein concentrations.We demonstrated the applicability of this RNA-silencing sys-tem by investigating cosilencing of the DsRed reporter genetogether with a gene involved in secondary metabolism.
MATERIALS AND METHODS
Construction of the RNAi vectors. Escherichia coli K-12 strain XL1-Blue(Stratagene) was used for general plasmid construction and maintenance (2).Plasmid pSilent-1 was generated previously as a vector for gene silencing inMagnaporthe oryzae and Colletotrichum lagenarium (26). For our approach, thehygromycin B resistance marker gene had to be replaced by the nat1 geneconferring nourseothricin resistance (16). Plasmid pNAT1 carrying the nat1 genewas hydrolyzed with SacI and NotI. The resulting 1,453-bp fragment was used forligation with the SacI-NotI-restricted fragment of pSilent-1, and the resultingrecombinant plasmid was designated pS-NAT1. In the next step, two 430-bpDsRed fragments, DsRedsense and DsRedanti, were amplified using primersDsRed-1 and DsRed-2 and primers DsRed-3 and DsRed-4, respectively, andplasmid pRHN1 as the template. pRHN1 carries the full-size DsRed gene(DsRed-Express; BD Biosciences Clontech, Heidelberg, Germany) under con-trol of the gpd promoter and trpC terminator of A. nidulans (I. Godehardt and U.Kuck, unpublished data). Then the DsRed fragments were inserted in inverseorientation into XhoI and StuI-ApaI restriction sites of plasmid pS-NAT1, gen-erating plasmid pREDi.
In the case of plasmid pPCBCi, two inversely oriented pcbC fragments werePCR amplified with primers pcbC-1 and pcbC-2 and primers pcbC-3 and pcbC-4using genomic DNA from A. chrysogenum A3/2 as the template. The PCRfragments were ligated into the vector pDrive (QIAGEN, Hilden, Germany) andsubsequently isolated using SnaBI-HindIII (pcbCanti) and BglII-StuI (pcbCsense)restriction sites. These fragments were ligated into plasmid pREDi using thecorresponding restriction sites. The sequences of all oligonucleotides and therecombinant plasmids are shown in Tables 1 and 2, respectively.
Fungal strains and culture conditions. Transformation of A. chrysogenumstrain ATCC 14553 and producer strain A3/2 was performed using conventionaltransformation procedures (32, 45). All strains used in this study are listed inTable 3, and their genotypes were verified by Southern blot analysis (data notshown) using gene-specific probes for hybridization of genomic DNA. To gen-erate strains for RNA silencing of the DsRed or pcbC gene, ATCC:DsRed orA3/2:DsRed was transformed with plasmid pREDi or pPCBCi. The resulting
TABLE 1. Sequences of oligonucleotides used in this work to generate PCR amplicons for constructionof RNAi-inducing vectors and for real-time PCR
Oligonucleotide Sequence (5�–3�)a Specificity
DsRed-1 CTCGAGATGGCCTCCTCCGAGGACGTCATCAAGG DsRed (positions 1 to 28) � XhoIDsRed-2 CTCGAGCCCAGCCCATAGTCTTCTTCTGC DsRed (positions 430 to 408) � XhoIDsRed-3 GGGCCCATGGCCTCCTCCGAGGACGTCATCAAGG DsRed (positions 1 to 28) � ApaIDsRed-4 AGGCCTCCCAGCCCATAGTCTTCTTCTGC DsRed (positions 430 to 408) � StuIpcbC-1 AAGCTTATGGGTTCCGTTCCAGTTCC pcbC (positions 1 to 20) � HindIIIb
pcbC-2 TACGTATGAAGAAGTCCTCGTCGCGAC pcbC (positions 520 to 500) � SnaBIb
pcbC-3 AGATCTATGGGTTCCGTTCCAGTTCC pcbC (positions 1 to 20) � BglIIb
pcbC-4 AGGCCTTGAAGAAGTCCTCGTCGCGAC pcbC (positions 520 to 500) � StuIb
DsRed_sense GATCCACAAGGCCCTGAAGC DsRed (positions 480 to 499)DsRed_anti GCTCCACGATGGTGTAGTCC DsRed (positions 637 to 618)SSU rRNA_for ATCCAAGGAAGGCAGCAGGC Small-subunit rRNA (N. crassa)SSU rRNA_rev TGGAGCTGGAATTACCGCG Small-subunit rRNA (N. crassa)
a Restriction sites are underlined.b The nucleotide positions are the positions in the accession no. M33522 sequence.
TABLE 2. Plasmids used for RNAi analysis
Plasmid Characteristics Reference
pRHN1 gpd promoter, DsRed gene (amino acids 1 to 226) and trpCterminator of A. nidulans
I. Godehardt and U. Kuck,unpublished data
pNAT1 gpd promoter of A. nidulans and nat1 gene of Streptomyces noursei I. Godehardt, D. Janus, andU. Kuck, unpublished data
pSilent-1 trpC promoter and terminator of A. nidulans; hph resistance gene andcutinase intron 2 of M. oryzae
26
pS-NAT1 gpd promoter of A. nidulans and nat1 gene in pSilent-1 This studypREDi DsRed fragments (amino acids 1 to 143) in sense and antisense
orientations in pS-NAT1This study
pPCBCi pcbC fragments (amino acids 1 to 173) in sense and antisenseorientations in pREDi
This study
VOL. 73, 2007 FUNGAL RNA-SILENCING SYSTEM USING DsRed REPORTER GENE 963

transformants were selected on media containing hygromycin B and nourseo-thricin at concentrations described previously (17, 32, 45).
Liquid cultures of A. chrysogenum strains were grown at 27°C and 180 rpm for3 days in CCM medium as described previously (23). Cultivation of strains forRNA and protein isolation was started with a 5% inoculum from a 2.5-day-oldliquid culture.
Preparation and analysis of RNA. RNA was isolated as described by Jekoschand Kuck (12), and the integrity of all RNAs was verified by agarose gel elec-trophoresis and Northern blot analyses (35). The blots were hybridized with32P-radiolabeled DNA probes as described in the Results.
Quantitative real-time PCR. Quantitative real-time PCR was performed asdescribed previously (29), with the following modifications: reverse transcriptionwas performed with 400 U Superscript II (Invitrogen) and deoxynucleosidetriphosphates at a concentration of 0.33 mM, and real-time PCR was carried outwith a DNA Engine Opticon 2 (MJ Research).
Protein purification and Western blot analysis. Protein purification was car-ried out as described previously (36), except that the mycelium was resuspendedin 2 ml of buffer A (0.1 M MOPS [morpholinepropanesulfonic acid] [pH 7.5], 0.2M KCl, 10 mM MgCl2, 1 mM EDTA, 10 mM dithiothreitol, 4.2 mM phenyl-methylsulfonyl fluoride, 40% [wt/vol] glycerol). Ten micrograms of each proteinextract was separated by sodium dodecyl sulfate-polyacrylamide gel electro-phoresis and blotted onto a nitrocellulose membrane (44). Western blotting andimmunodetection of the pcbC gene product were performed by using previouslydescribed methods (13).
Preparation of A. chrysogenum crude mycelial extract. To isolate A. chrysoge-num crude extracts, mycelium was first filtered and then resuspended in 40 ml ofbuffer A (see above). The mycelium was disrupted with a standard householdblender (Siemens, Munich, Germany) for 3 min on ice. Spectrofluorometricmeasurement was performed with 20 �l of each crude extract that was diluted in2 ml of storage buffer (BD Biosciences) (10 mM EDTA, 10% glycerol in phos-phate-buffered saline [20 mM NaH2PO4�H2O, 80 mM Na2HPO4�2H2O, 100 mMNaCl]). The remaining portion was vacuum filtered and dried for 24 to 48 h at65°C to determine the dry weight of the mycelium.
Spectrofluorometric measurement. A calibration curve was produced usingpurified DsRed protein (BD Biosciences) as a prelude to determining DsRedlevels in protein or crude mycelial extracts. The fluorescence of the DsRedprotein at 576 nm was measured by using an excitation wavelength of 554 nm anda JASCO FP-6500 spectrofluorometer (JASCO, Tokyo, Japan). Both the emis-sion and excitation bandwidths were set at 5 nm. The protein or mycelial extractmeasurements were obtained using a 1:100 or 1:1,000 dilution in storage buffer.All the results described below are means of three independent measurements.The amount of DsRed protein was expressed in ng DsRed/mg mycelium or ngDsRed/�g total protein.
Quantification of cephalosporin C synthesis. Cephalosporin C synthesis wasquantified with a bioassay as described previously (45). Alcaligenes faecalis wasused as a gram-negative indicator bacterium for antibiotic production. Fungaltransformants were grown for 3 days in liquid culture, and each supernatant wasapplied to wells in the test medium seeded with A. faecalis. Test plates wereincubated for at least 12 h at 37°C. Simultaneously, the dry weight of themycelium was estimated to calculate the ratio of the halo size to the mycelialbiomass. On all plates, the halo of the A3/2:DsRed recipient was used as acontrol, and its size was defined as 100%. Accordingly, the areas of the haloswere determined and grouped into four classes: large (100 to 70% of the control
strain A3/2:DsRed halo size), medium (70 to 30%), small (30 to �0%), and nohalos.
RESULTS
Use of the DsRed gene to monitor RNA silencing. A.chrysogenum exhibits a high frequency of nonhomologous re-combination, and therefore, gene substitution to generateknockout strains is inefficient (11, 36, 45). As an alternative, wedeveloped a reliable RNA-silencing system using the DsRedgene from Discosoma sp. as a reporter. For our studies, two A.chrysogenum strains with different properties were used. ATCC14553 is closely related to the wild-type strain and gives trans-formation frequencies of about 20 transformants per �g DNA(45). The cephalosporin C titer of this strain is relatively lowcompared to that of the producer strain A3/2, and antibioticproduction can be measured with conventional bioassays onlyfor the latter strain (32). In contrast, transformation of A3/2resulted in a very low number of transformants (45), and there-fore, our initial experiments to establish the RNAi system wereperformed with ATCC 14553.
As a first step to develop an RNAi system, we generated atransgenic recipient A. chrysogenum strain expressing theDsRed gene. After transformation of ATCC 14553 withpRHN1 (Table 2), about 70% of the strains obtained were pinkor red on solid medium containing hygromycin B. To avoidheterokaryon formation, a single red transformant was se-lected and used for preparation of protoplasts. After regener-ation of protoplasts, a single red colony was propagated onsolid media. This strain had a stable phenotype even after atleast five passages on solid medium and was used as a recipient(ATCC:DsRed) in further RNAi experiments. In a comparableexperiment, strain A3/2:DsRed was generated using A3/2 as therecipient.
To silence the DsRed gene, plasmid pREDi was constructedas shown in Fig. 1. pSilent-1 carrying cutinase intron 2 from M.oryzae was used as the basic vector. This plasmid was reportedpreviously to induce strong and relatively stable silencing infungal transformants like M. oryzae and C. lagenarium (26).pSilent-1 also contained unique cloning sites for integration oftwo approximately 430-bp inversely oriented DsRed fragments.As an alternative selectable marker, the nourseothricin resis-tance gene from plasmid pNAT1 (Table 2) was inserted as
TABLE 3. Recipient and transgenic strains used in this study
Strain Characteristic(s) Source or reference
ATCC 14553 Wild-type strain American Type CultureCollection
A3/2 Producer strain 32ATCC:DsRed gpd(p)::DsRed::trpC(t), trpC(p)::hph This studyA3/2:DsRed gpd(p)::DsRed::trpC(t), trpC(p)::hph This studyATCC:DsRedi T8, T20, T25, T43, T52,
T53, T54, T123, T131, T144, T145,T179, and T180
gpd(p)::DsRed::trpC(t), trpC(p)::hph,trpC(p)::DsReds::DsRedas::trpC(t), gpd(p)::nat1
This study
A3/2:DsRedi T27 gpd(p)::DsRed::trpC(t), trpC(p)::hph,trpC(p)::DsReds::DsRedas::trpC(t); gpd(p)::nat1
This study
A3/2:pcbCi T4, T8, T9, and T10 gpd(p)::DsRed::trpC(t), trpC(p)::hph,trpC(p)::DsReds::pcbCas::pcbCs::DsRedas::trpC(t), gpd(p)::nat1
This study
964 JANUS ET AL. APPL. ENVIRON. MICROBIOL.

described in Materials and Methods. Plasmid pREDi itself isnot sufficient to generate a functional DsRed protein.
To establish the RNAi system in A. chrysogenum, the recip-ient strains and vectors described above were tested in threeindependent experiments. Transformation of pREDi intoATCC:DsRed resulted in generation of 478 nourseothricin-and hygromycin B-resistant transformants. These transfor-mants exhibited at least three different types of coloring. Asshown in Fig. 2 and summarized in Table 4, we found thatabout 20% of the colonies were dark red, while about 40% ofthe colonies were pink. More than 30% of the transformantsshowed no coloring at all and thus were most severely silenced.
To exclude the possibility that the recipient strain, or alter-natively the transformation procedure itself, was responsiblefor the phenotype observed, we conducted two different con-trol experiments. In the first experiment, the DsRed-expressingproducer strain A3/2:DsRed was transformed with plasmidpREDi. As shown in Table 4, the ratio of the three classes ofcolored transformants was very similar to the ratio obtainedwith strain ATCC:DsRed. In the second experiment, plasmidpS-NAT1, which was almost identical to pREDi but lacked thetwo 430-bp DsRed fragments, was transformed into ATCC:
DsRed and A3/2:DsRed. In these cases, all 50 and 9 transfor-mants, respectively, had a red phenotype.
In conclusion, the DsRed reporter gene provides a powerfultool for identification of silenced fungal transformants. Thesetransformants can be identified easily by their pink or colorlessphenotype compared to the red recipient.
Characterization of DsRed-silenced transformants. For fur-ther analysis, Northern blot hybridization, quantitative real-time PCR, and spectrofluorometric assays were used to char-acterize the fungal transformants mentioned above. Werandomly selected two red colonies, four pink colonies, andfive colorless colonies and examined these colonies togetherwith the wild-type strain (ATCC 14553) and the ATCC:DsRedrecipient as controls. After RNA isolation, Northern blot hy-bridization was conducted with the DsRed and gpd genes asradiolabeled probes. To detect only the full-length DsRed tran-script and not RNAs derived from the pREDi vector, a 240-bpfragment was used to detect the 3� end of DsRed. This genefragment was present in pRHN1 but not in pREDi. With theexception of the ATCC 14553 strain, all strains carried plasmidpRHN1, and thus the DsRed transcript could be detected inNorthern hybridization blots (Fig. 3A).
However, the hybridizing bands for the DsRed transcript haddifferent intensities. The strongest signal was obtained fromred colonies, while in pink and colorless colonies there was aclear reduction in the DsRed transcript level. The most signif-icant reduction was seen in transformants T180 and T131, bothof which appeared to be white on solid media. The gpd probewas used as a reference and gave almost identical signals withRNA from all transformants tested. Comparable results wereobtained when A3/2:DsRed was used as a recipient (data notshown).
More detailed quantification was achieved by real-time PCRusing oligonucleotides that hybridized only to the 240-bp 3�part of the DsRed gene, as described above. For normalization,small-subunit rRNA levels were determined as previously de-scribed (29). As a typical example of this analysis, the log2
ratios for individual transformants compared to the ATCC:DsRed recipient strain are shown in Fig. 3B. These data are ingood accordance with the Northern blot hybridization datashown in Fig. 3A. The colorless transformant T180 showed the
FIG. 1. Schematic map of the pREDi vector and the derived recombinant plasmid pPCBCi. pREDi is a derivative of plasmid pSilent-1, carryinginversely oriented DsRed gene fragments. In the case of plasmid pPCBCi, the pcbC gene from A. chrysogenum was introduced into singularrestriction sites of the pREDi vector (indicated by asterisks). Genes are not drawn to scale. For details concerning construction, see the text.Abbreviations: DsRed, DsRed gene from Discosoma sp.; IT, intron 2 of the cutinase gene from M. oryzae; nat1, nourseothricin acetyltransferasegene from Streptomyces noursei; pcbC, isopenicillin N synthase gene from A. chrysogenum; Pgpd, A. nidulans gpd promoter; PtrpC, A. nidulans trpCpromoter; TtrpC, A. nidulans trpC terminator.
FIG. 2. Phenotypes of three representative colonies of A. chrysoge-num transformants (ATCC:DsRedi) on solid medium, showing differ-ent colors due to silencing of the DsRed gene.
VOL. 73, 2007 FUNGAL RNA-SILENCING SYSTEM USING DsRed REPORTER GENE 965

most drastic change, with a 16-fold reduction in the DsRedtranscript level.
Finally, DsRed gene expression was measured on the proteinlevel by using a simple and reproducible spectrofluorometricmeasurement for quantification. The results of spectrofluoro-metric DsRed protein quantification for the crude mycelialextracts are shown in Fig. 3C and demonstrate that the redtransformants ATCC:DsRedi T25 and T43 had an increasedamount of DsRed compared to the amount in the recipientstrain. All pink transformants gave intermediate values forDsRed, while the white transformants clearly had the lowestlevels of DsRed. Once again, the most remarkable examplesare ATCC:DsRedi T54 and T180, which had a DsRed synthe-sis rate close to zero. The wild-type strain (ATCC 14553)served as a negative control which demonstrated that no back-ground fluorescence was observed in this analysis.
Our results for the spectrofluorometric measurements areconsistent with the results of the RNA analysis. To the best ofour knowledge, this is the first study using spectrofluorometricmeasurement of the DsRed protein in microbial transfor-mants, an approach that also has the advantage of being a fastand efficient procedure for protein quantification. Taken to-gether, our data clearly show that the RNA-silencing approachdescribed here for A. chrysogenum is effective and can be usedfor targeted inhibition of genes.
Cosilencing of DsRed and the endogenous pcbC gene. Toexamine the effect of RNAi on the expression of an endoge-nous fungal gene product, an interference construct was gen-erated to inhibit specifically secondary metabolism in A.chrysogenum. The pcbC gene encodes the isopenicillin N syn-thase and is strongly expressed on the transcriptional levelcompared with the expression of other cephalosporin C bio-synthesis genes. Previously, inactivation of pcbC has resulted instrains having a reduced antibiotic titer (45) and thus can beused as a representative marker to demonstrate downregula-tion of the cephalosporin C biosynthesis pathway.
To achieve this, pcbC was silenced simultaneously withDsRed, which was used as a marker for selecting knockdowntransformants. Starting with plasmid pREDi, inversely ori-ented pcbC fragments, which were about 520 bp long, werecloned using SnaBI/HindIII and BglII/StuI restriction sites forthe pcbCanti and pcbCsense fragments, respectively. In relationto the DsRed gene, each fragment was inserted in the oppositeorientation. A linear map of the resulting plasmid, pPCBCi, isshown in Fig. 1.
In the next step, plasmid pPCBCi was transformed intorecipient strain A3/2:DsRed. As shown in Table 4, about 70%of the transformants had an altered phenotype compared tothe phenotype of the recipient strain. From 68 transformants,
we selected 4 that had different phenotypes for further analy-sis. A3/2:DsRed and A3/2:DsRedi T27 were used as appropri-ate controls. A3/2:DsRedi T27 was generated as describedabove and carried plasmid pREDi instead of pPCBCi. Asshown in Fig. 4A, RNA from the six strains described abovewas used for Northern blot analysis. Similar to the previoushybridization analysis, we generated a probe that detected onlythe full-size DsRed transcript. Similarly, the mature pcbC tran-script hybridized with a probe that was complementary to theresidual 500 bp of the 3� region of pcbC and did not exhibit anyhomology to plasmid pPCBCi. As shown in Fig. 4A, we foundthat there was a good correlation between the color of thecolonies and the RNA transcript levels. In T10, which is a redstrain, neither the level of pcbC nor the level of DsRed mRNAwas reduced. The opposite was observed for T4, T8, and T9. Inthese cases, the DsRed expression levels, as well as the pcbCexpression levels, were reduced. Remarkably, T8 showed thesmallest reduction in the pcbC mRNA level, a finding that isconsistent with data obtained later in protein quantificationand bioassay analyses. On the other hand, the results for T27,which was used as a control, confirmed that downregulation ofDsRed itself was not sufficient to reduce the amount of thepcbC mRNA transcript.
Protein analysis of silenced transformants. To further cor-relate the levels of mRNA and protein synthesis in silencedtransformants, we performed spectrofluorometric measure-ment and Western blot analyses. For the spectrofluorometricanalysis, we used crude protein extracts from all of the trans-formants described above. As shown in Fig. 4B, the amount ofthe DsRed protein correlated perfectly with the intensity of thesignals obtained in the Northern blot analysis. As expected, theA3/2 producer strain exhibited no background fluorescence(data not shown), while for all silenced strains values less than0.05 ng DsRed/�g protein were obtained.
In parallel, we detected isopenicillin N synthase using apolyclonal antibody that was described previously (13). Asshown in Fig. 4C, all control strains, as well as the red knock-down strain T10, gave a strong signal in the Western blotanalysis. Unexpectedly, the colorless strain T8 also showed astrong signal in the Western analysis. However, considering theresults of the Northern hybridization experiment, this can beexplained by the amount of the pcbC mRNA transcript, whichwas detectable in this transgenic strain. The most dramaticreduction in the level of the polypeptide encoded by pcbC wasobserved in T4, a result that correlates perfectly with theamount of pcbC mRNA transcript detected.
Bioassay to measure cephalosporin C titers. The antibioticcephalosporin C inhibits the growth of gram-positive as well asgram-negative bacteria (for a review, see reference 37). During
TABLE 4. Summary of RNAi transformants having different colors of colonies
Recipient strainNo. (%) of transformants
Red Pink Colorless Heterokaryons Total
ATCC:DsRed(pREDi)a 89 (18.6) 194 (40.6) 159 (33.3) 36 (7.5) 478 (100)A3/2:DsRed(pREDi)a 22 (14.3) 64 (41.6) 54 (35) 14 (9.1) 154 (100)A3/2:DsRed(pPCBCi)b 17 (25) 29 (42.6) 18 (26.5) 4 (5.9) 68 (100)
a Recipient strains ATCC:DsRed and A3/2:DsRed were transformed with vector pREDi to silence the DsRed gene.b Transformants were obtained when recipient strain A3/2:DsRed was transformed with pPCBCi in order to cosilence the DsRed and pcbC genes.
966 JANUS ET AL. APPL. ENVIRON. MICROBIOL.
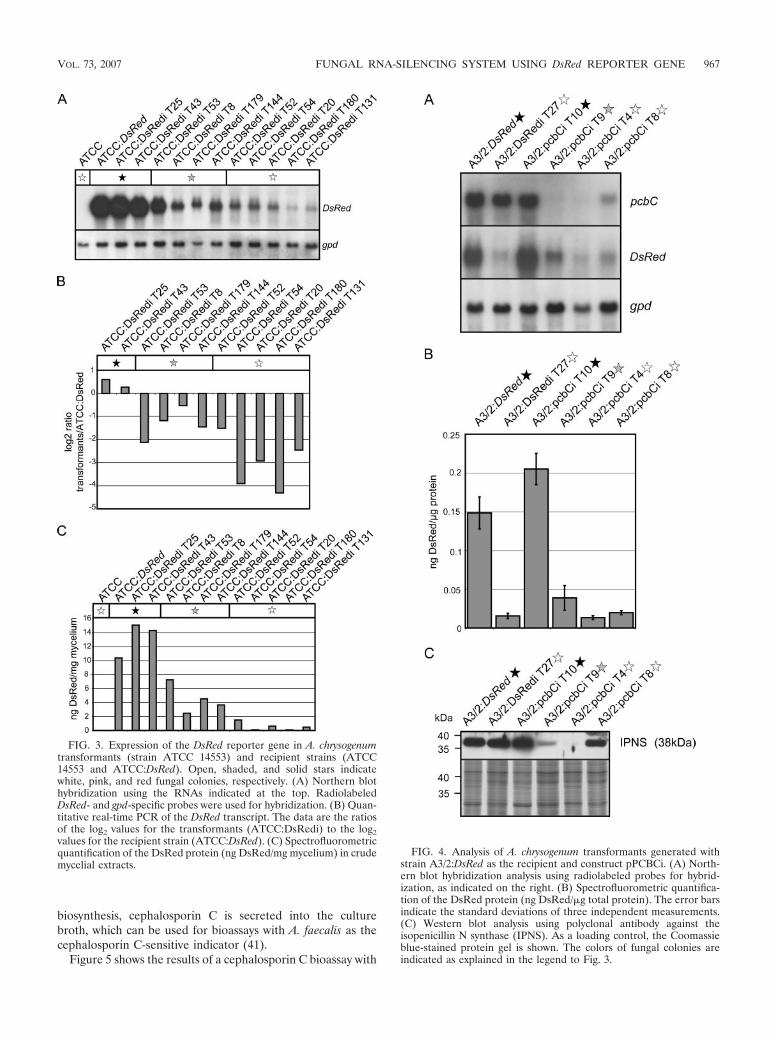
biosynthesis, cephalosporin C is secreted into the culturebroth, which can be used for bioassays with A. faecalis as thecephalosporin C-sensitive indicator (41).
Figure 5 shows the results of a cephalosporin C bioassay with
FIG. 3. Expression of the DsRed reporter gene in A. chrysogenumtransformants (strain ATCC 14553) and recipient strains (ATCC14553 and ATCC:DsRed). Open, shaded, and solid stars indicatewhite, pink, and red fungal colonies, respectively. (A) Northern blothybridization using the RNAs indicated at the top. RadiolabeledDsRed- and gpd-specific probes were used for hybridization. (B) Quan-titative real-time PCR of the DsRed transcript. The data are the ratiosof the log2 values for the transformants (ATCC:DsRedi) to the log2values for the recipient strain (ATCC:DsRed). (C) Spectrofluorometricquantification of the DsRed protein (ng DsRed/mg mycelium) in crudemycelial extracts.
FIG. 4. Analysis of A. chrysogenum transformants generated withstrain A3/2:DsRed as the recipient and construct pPCBCi. (A) North-ern blot hybridization analysis using radiolabeled probes for hybrid-ization, as indicated on the right. (B) Spectrofluorometric quantifica-tion of the DsRed protein (ng DsRed/�g total protein). The error barsindicate the standard deviations of three independent measurements.(C) Western blot analysis using polyclonal antibody against theisopenicillin N synthase (IPNS). As a loading control, the Coomassieblue-stained protein gel is shown. The colors of fungal colonies areindicated as explained in the legend to Fig. 3.
VOL. 73, 2007 FUNGAL RNA-SILENCING SYSTEM USING DsRed REPORTER GENE 967
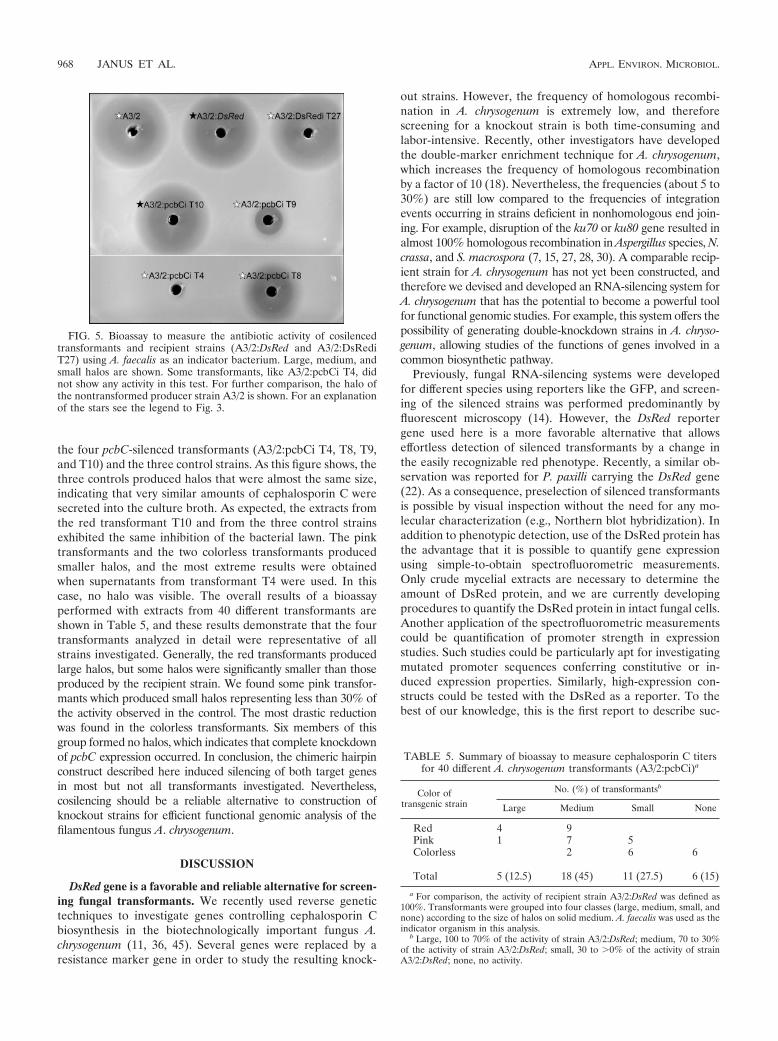
the four pcbC-silenced transformants (A3/2:pcbCi T4, T8, T9,and T10) and the three control strains. As this figure shows, thethree controls produced halos that were almost the same size,indicating that very similar amounts of cephalosporin C weresecreted into the culture broth. As expected, the extracts fromthe red transformant T10 and from the three control strainsexhibited the same inhibition of the bacterial lawn. The pinktransformants and the two colorless transformants producedsmaller halos, and the most extreme results were obtainedwhen supernatants from transformant T4 were used. In thiscase, no halo was visible. The overall results of a bioassayperformed with extracts from 40 different transformants areshown in Table 5, and these results demonstrate that the fourtransformants analyzed in detail were representative of allstrains investigated. Generally, the red transformants producedlarge halos, but some halos were significantly smaller than thoseproduced by the recipient strain. We found some pink transfor-mants which produced small halos representing less than 30% ofthe activity observed in the control. The most drastic reductionwas found in the colorless transformants. Six members of thisgroup formed no halos, which indicates that complete knockdownof pcbC expression occurred. In conclusion, the chimeric hairpinconstruct described here induced silencing of both target genesin most but not all transformants investigated. Nevertheless,cosilencing should be a reliable alternative to construction ofknockout strains for efficient functional genomic analysis of thefilamentous fungus A. chrysogenum.
DISCUSSION
DsRed gene is a favorable and reliable alternative for screen-ing fungal transformants. We recently used reverse genetictechniques to investigate genes controlling cephalosporin Cbiosynthesis in the biotechnologically important fungus A.chrysogenum (11, 36, 45). Several genes were replaced by aresistance marker gene in order to study the resulting knock-
out strains. However, the frequency of homologous recombi-nation in A. chrysogenum is extremely low, and thereforescreening for a knockout strain is both time-consuming andlabor-intensive. Recently, other investigators have developedthe double-marker enrichment technique for A. chrysogenum,which increases the frequency of homologous recombinationby a factor of 10 (18). Nevertheless, the frequencies (about 5 to30%) are still low compared to the frequencies of integrationevents occurring in strains deficient in nonhomologous end join-ing. For example, disruption of the ku70 or ku80 gene resulted inalmost 100% homologous recombination in Aspergillus species, N.crassa, and S. macrospora (7, 15, 27, 28, 30). A comparable recip-ient strain for A. chrysogenum has not yet been constructed, andtherefore we devised and developed an RNA-silencing system forA. chrysogenum that has the potential to become a powerful toolfor functional genomic studies. For example, this system offers thepossibility of generating double-knockdown strains in A. chryso-genum, allowing studies of the functions of genes involved in acommon biosynthetic pathway.
Previously, fungal RNA-silencing systems were developedfor different species using reporters like the GFP, and screen-ing of the silenced strains was performed predominantly byfluorescent microscopy (14). However, the DsRed reportergene used here is a more favorable alternative that allowseffortless detection of silenced transformants by a change inthe easily recognizable red phenotype. Recently, a similar ob-servation was reported for P. paxilli carrying the DsRed gene(22). As a consequence, preselection of silenced transformantsis possible by visual inspection without the need for any mo-lecular characterization (e.g., Northern blot hybridization). Inaddition to phenotypic detection, use of the DsRed protein hasthe advantage that it is possible to quantify gene expressionusing simple-to-obtain spectrofluorometric measurements.Only crude mycelial extracts are necessary to determine theamount of DsRed protein, and we are currently developingprocedures to quantify the DsRed protein in intact fungal cells.Another application of the spectrofluorometric measurementscould be quantification of promoter strength in expressionstudies. Such studies could be particularly apt for investigatingmutated promoter sequences conferring constitutive or in-duced expression properties. Similarly, high-expression con-structs could be tested with the DsRed as a reporter. To thebest of our knowledge, this is the first report to describe suc-
FIG. 5. Bioassay to measure the antibiotic activity of cosilencedtransformants and recipient strains (A3/2:DsRed and A3/2:DsRediT27) using A. faecalis as an indicator bacterium. Large, medium, andsmall halos are shown. Some transformants, like A3/2:pcbCi T4, didnot show any activity in this test. For further comparison, the halo ofthe nontransformed producer strain A3/2 is shown. For an explanationof the stars see the legend to Fig. 3.
TABLE 5. Summary of bioassay to measure cephalosporin C titersfor 40 different A. chrysogenum transformants (A3/2:pcbCi)a
Color oftransgenic strain
No. (%) of transformantsb
Large Medium Small None
Red 4 9Pink 1 7 5Colorless 2 6 6
Total 5 (12.5) 18 (45) 11 (27.5) 6 (15)
a For comparison, the activity of recipient strain A3/2:DsRed was defined as100%. Transformants were grouped into four classes (large, medium, small, andnone) according to the size of halos on solid medium. A. faecalis was used as theindicator organism in this analysis.
b Large, 100 to 70% of the activity of strain A3/2:DsRed; medium, 70 to 30%of the activity of strain A3/2:DsRed; small, 30 to �0% of the activity of strainA3/2:DsRed; none, no activity.
968 JANUS ET AL. APPL. ENVIRON. MICROBIOL.

cessful quantification of the DsRed protein in crude proteinextracts from microbes using a spectrofluorometry approach.Comparable approaches to quantify an autofluorescent proteinspectrofluorometrically were described recently for the GFP intransgenic yeasts, filamentous fungi, and plants (3, 26, 33).Beyond this, our RNAi system offers the possibility of devel-oping a high-throughput screening procedure for functionalgenomics. With the DsRed fluorescent marker, clearly visiblephenotypes were generated that should drastically reduce thescreening workload, as well as increase our current under-standing of fungal functional genomics. As an alternative touse of the DsRed protein, workers may consider using themonomeric red fluorescent protein having accelerated foldingproperties. This fluorescent protein has already been used suc-cessfully in filamentous fungi, but compared with the bright-ness of DsRed, the brightness of the fully mature protein isreduced (38). Further improvements might be obtained withsecond-generation fluorescent proteins, such as tdTomato (38,39); however, so far there have been no successful applicationsof these proteins in filamentous fungi.
Simultaneous silencing of multiple targets occurs with vari-able efficiencies. Recently, it has been demonstrated that vec-tors generating hairpin RNAs are highly effective in induc-ing silencing (10, 14, 21, 26, 40). For this purpose, plasmidpSilent-1, which was shown previously to provide stable silenc-ing in different filamentous fungi (26), was used for simulta-neous inhibition of two target genes in A. chrysogenum.
Our quantitative and qualitative data demonstrate that theDsRed and pcbC genes were successfully downregulated. Si-multaneous silencing of multiple target genes has several ad-vantages. First, only a single silencing construct is needed toachieve simultaneous silencing of multiple target genes, andthis minimizes the number of selectable marker genes re-quired. In contrast, multiple knockouts in a single strain re-quire use of different selectable markers in order to monitorthe integration process. Consequently, the number of genesthat can be deleted is limited by the fact that only a fewdominant resistance marker genes are available (46). The ap-plicability of the RNA-silencing system can be extended fur-ther by using vectors with inducible promoters. In this case,gene expression can be controlled in specific stages duringdevelopment, or alternatively, functional analysis can be per-formed for genes indispensable for fungal growth (24, 26).
Another important result of our study is the discovery thatthe two target genes (pcbC and DsRed) are not cosilenced tothe same extent. Table 4 shows that the red phenotype wassilenced with the same efficiency irrespective of whether it wasused in cosilencing experiments. Using hairpin-expressing plas-mid pPCBCi, we obtained different levels of repression of thetwo target genes. Some transformants with an almost com-pletely downregulated DsRed gene, for example, showed amoderate reduction in cephalosporin C synthesis. Neverthe-less, all colorless transformants in this experiment had a moreor less reduced rate of pcbC expression. This effect has beenreported previously for other RNAi systems suppressing twotarget genes simultaneously. For example, in Venturia inaequa-lis, all transformants showed a cosilencing effect on the twotarget genes encoding GFP and trihydroxynaphthalene reduc-tase (thn); however, quantification by real-time PCR indicatedthat RNAi did not occur to the same extent (9). Comparable
results were obtained when Cryptococcus neoformans was usedto cosilence ADE2, which encodes the phosphoribosylamino-imidazole carboxylase, and CAP59, which is required for syn-thesis of the polysaccharide capsule. Although more than 80%of all transformants exhibited reduction of expression of bothgenes, some exhibited only a single silenced phenotype (19).An explanation for this irregular RNAi efficiency could be thatincomplete formation of the hairpin RNA stem derived fromone target gene might lead to a reduction in the length of thedsRNA. This would result in a decrease in the efficiency ofknockdown effects. Recently, an increase in the stem length ofthe hairpin RNA was shown to enhance the number of trans-formants having a mutant phenotype (10).
RNAi system could unravel components regulating second-ary metabolism. The RNA-silencing system developed in thisstudy may make it possible to conduct high-throughput loss-of-function screening to identify novel regulators of secondarymetabolism. It was recently suggested that in order to generatea fungal RNAi hairpin vector library, the Gateway (Invitrogen)in vitro recombination technology could be used for high-throughput cloning (46). In a comparable approach mamma-lian cells were screened successfully with a lentiviral RNAilibrary for genome-scale loss-of-function screening (34). Theuse of RNA silencing with microbial systems provides a poten-tial alternative to conventional strain improvement programsin which UV rays or chemical mutagens are used for construc-tion of strains with novel properties. Preliminary success in thisdirection using the RNAi system has been reported previouslyfor a few filamentous fungi. For example, in Aspergillus andFusarium species it was possible to reduce mycotoxin yields inorder to eliminate secondary product contamination of food(21). In another attempt, fatty acid production was recentlyimproved in Mortierella alpina, the commercially used producerof arachidonic acid, using the RNAi system (42). The authorssuggested that RNA silencing could enable biosynthesis ofpreviously rare and economically valuable compounds.
We have shown previously that disruption of the pcbC generesults in transformants exhibiting no antibiotic activity (45).The same phenotype was found in this investigation in about15% of cosilenced transformants. Thus, secondary metabolismcan be studied by downregulation of genes that directly orindirectly control cephalosporin C biosynthesis. The RNAi ap-proach also provides the possibility of unraveling signal cas-cades in secondary metabolism. Silencing of repressor pro-teins, for example, may result in an increase in cephalosporinC production and thus in construction of strains with an en-hanced production rate.
In conclusion, here we describe a quantitative and qualitativeinvestigation of cosilenced fungal transformants using hairpinvector technology. The successful coinhibition of the heterolo-gous DsRed gene and the endogenous pcbC gene indicates thatRNAi provides an effective tool for studying secondary metabo-lism in A. chrysogenum. Thus, dsRNA-based silencing is a reliablealternative to gene inactivation experiments, which currently havelow rates of efficiency with A. chrysogenum. Our data indicate thatthis RNA-silencing system along with other technologies shouldaccelerate the genome-wide analysis of A. chrysogenum and thuspromote rational strain improvement in this industrial cephalo-sporin C producer.
VOL. 73, 2007 FUNGAL RNA-SILENCING SYSTEM USING DsRed REPORTER GENE 969

ACKNOWLEDGMENTS
We thank Kerstin Kalkreuter, Stefanie Mertens, and Ingeborg Go-dehardt for their excellent technical assistance, Minou Nowrousian foradvice concerning the quantitative real-time PCR, Gabriele Frenßen-Schenkel for the artwork, and E. Friedlin and H. Kurnsteiner (Kundl,Austria) for their interest and support. We acknowledge receipt of thevector pSilent-1 from H. Nakayashiki (Kobe, Japan). Protein quanti-fication by spectrofluorometric measurement was done in the collab-orative research center (SFB 480, projects A1 and C6).
This work was supported by Sandoz GmbH (Austria).
REFERENCES
1. Allen, R. S., A. G. Millgate, J. A. Chitty, J. Thisleton, J. A. Miller, A. J. Fist,W. L. Gerlach, and P. J. Larkin. 2004. RNAi-mediated replacement ofmorphine with the nonnarcotic alkaloid reticuline in opium poppy. Nat.Biotechnol. 22:1559–1566.
2. Bullock, W. O., J. M. Fernandez, and J. M. Short. 1987. XL1-Blue: a highefficiency plasmid transforming recA Escherichia coli strain with beta-galac-tosidase selection. BioTechniques 5:376–378.
3. Cha, H. J., N. G. Dalal, and W. E. Bentley. 2004. In vivo monitoring ofintracellular expression of human interleukin-2 using green fluorescent pro-tein fusion partner in Pichia pastoris. Biotechnol. Lett. 26:1157–1162.
4. Chaveroche, M. K., J. M. Ghigo, and C. d’Enfert. 2000. A rapid method forefficient gene replacement in the filamentous fungus Aspergillus nidulans.Nucleic Acids Res. 28:E97.
5. Cogoni, C., J. T. Irelan, M. Schumacher, T. J. Schmidhauser, E. U. Selker,and G. Macino. 1996. Transgene silencing of the al-1 gene in vegetative cellsof Neurospora is mediated by a cytoplasmic effector and does not depend onDNA-DNA interactions or DNA methylation. EMBO J. 15:3153–3163.
6. Colot, H. V., G. Park, G. E. Turner, C. Ringelberg, C. M. Crew, L.Litvinkova, R. L. Weiss, K. A. Borkovich, and J. C. Dunlap. 2006. A high-throughput gene knockout procedure for Neurospora reveals functions formultiple transcription factors. Proc. Natl. Acad. Sci. USA 103:10352–10357.
7. da Silva Ferreira, M. E., M. R. Kress, M. Savoldi, M. H. Goldman, A. Hartl,T. Heinekamp, A. A. Brakhage, and G. H. Goldman. 2006. The akuB(KU80)mutant deficient for nonhomologous end joining is a powerful tool foranalyzing pathogenicity in Aspergillus fumigatus. Eukaryot. Cell 5:207–211.
8. de Jong, J. F., H. J. Deelstra, H. A. Wosten, and L. G. Lugones. 2006.RNA-mediated gene silencing in monokaryons and dikaryons of Schizophyl-lum commune. Appl. Environ. Microbiol. 72:1267–1269.
9. Fitzgerald, A., J. A. van Kan, and K. M. Plummer. 2004. Simultaneoussilencing of multiple genes in the apple scab fungus, Venturia inaequalis, byexpression of RNA with chimeric inverted repeats. Fungal Genet. Biol.41:963–971.
10. Goldoni, M., G. Azzalin, G. Macino, and C. Cogoni. 2004. Efficient genesilencing by expression of double stranded RNA in Neurospora crassa. Fun-gal Genet. Biol. 41:1016–1024.
11. Hoff, B., E. K. Schmitt, and U. Kuck. 2005. CPCR1, but not its interactingtranscription factor AcFKH1, controls fungal arthrospore formation in Acre-monium chrysogenum. Mol. Microbiol. 56:1220–1233.
12. Jekosch, K., and U. Kuck. 2000. Glucose dependent transcriptional expres-sion of the cre1 gene in Acremonium chrysogenum strains showing differentlevels of cephalosporin C production. Curr. Genet. 37:388–395.
13. Jekosch, K., and U. Kuck. 2000. Loss of glucose repression in an Acremo-nium chrysogenum beta-lactam producer strain and its restoration by multi-ple copies of the cre1 gene. Appl. Microbiol. Biotechnol. 54:556–563.
14. Kadotani, N., H. Nakayashiki, Y. Tosa, and S. Mayama. 2003. RNA silencingin the phytopathogenic fungus Magnaporthe oryzae. Mol. Plant-Microbe In-teract. 16:769–776.
15. Krappmann, S., C. Sasse, and G. H. Braus. 2006. Gene targeting in Aspergil-lus fumigatus by homologous recombination is facilitated in a nonhomolo-gous end-joining-deficient genetic background. Eukaryot. Cell 5:212–215.
16. Krugel, H., G. Fiedler, C. Smith, and S. Baumberg. 1993. Sequence andtranscriptional analysis of the nourseothricin acetyltransferase-encodinggene nat1 from Streptomyces noursei. Gene 127:127–131.
17. Kuck, U., and B. Hoff. 2006. Application of the nourseothricin acetyltrans-ferase gene (nat1) as a dominant marker for the transformation of filamen-tous fungi. Fungal Genet. Newsl. 53:9–11.
18. Liu, G., J. Casqueiro, O. Banuelos, R. E. Cardoza, S. Gutierrez, and J. F.Martın. 2001. Targeted inactivation of the mecB gene, encoding cystathione-gamma-lyase, shows that the reverse transsulfuration pathway is required forhigh-level cephalosporin biosynthesis in Acremonium chrysogenum C10 but notfor methionine induction of the cephalosporin genes. J. Bacteriol. 183:1765–1772.
19. Liu, H., T. R. Cottrell, L. M. Pierini, W. E. Goldman, and T. L. Doering.2002. RNA interference in the pathogenic fungus Cryptococcus neoformans.Genetics 160:463–470.
20. Matz, M. V., A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L.
Markelov, and S. A. Lukyanov. 1999. Fluorescent proteins from nonbio-luminescent Anthozoa species. Nat. Biotechnol. 17:969–973.
21. McDonald, T., D. Brown, N. P. Keller, and T. M. Hammond. 2005. RNAsilencing of mycotoxin production in Aspergillus and Fusarium species. Mol.Plant-Microbe Interact. 18:539–545.
22. Mikkelsen, L., S. Sarrocco, M. Lubeck, and D. F. Jensen. 2003. Expressionof the red fluorescent protein DsRed-Express in filamentous ascomycetefungi. FEMS Microbiol. Lett. 223:135–139.
23. Minuth, W., P. Tudzynski, and K. Esser. 1982. Extrachromosomal geneticsof Cephalosporium acremonium. Curr. Genet. 5:227–231.
24. Mouyna, I., C. Henry, T. L. Doering, and J. P. Latge. 2004. Gene silencingwith RNA interference in the human pathogenic fungus Aspergillus fumiga-tus. FEMS Microbiol. Lett. 237:317–324.
25. Nakayashiki, H. 2005. RNA silencing in fungi: mechanisms and applications.FEBS Lett. 579:5950–5957.
26. Nakayashiki, H., S. Hanada, B. Q. Nguyen, N. Kadotani, Y. Tosa, and S.Mayama. 2005. RNA silencing as a tool for exploring gene function inascomycete fungi. Fungal Genet. Biol. 42:275–283.
27. Nayak, T., E. Szewczyk, C. E. Oakley, A. Osmani, L. Ukil, S. L. Murray, M. J.Hynes, S. A. Osmani, and B. R. Oakley. 2006. A versatile and efficientgene-targeting system for Aspergillus nidulans. Genetics 172:1557–1566.
28. Ninomiya, Y., K. Suzuki, C. Ishii, and H. Inoue. 2004. Highly efficient genereplacements in Neurospora strains deficient for nonhomologous end-join-ing. Proc. Natl. Acad. Sci. USA 101:12248–12253.
29. Nowrousian, M., C. Ringelberg, J. C. Dunlap, J. J. Loros, and U. Kuck. 2005.Cross-species microarray hybridization to identify developmentally regulatedgenes in the filamentous fungus Sordaria macrospora. Mol. Genet. Genomics273:137–149.
30. Poggeler, S., and U. Kuck. 2006. Highly efficient generation of signal trans-duction knockout mutants using a fungal strain deficient in the mammalianku70 ortholog. Gene 378:1–10.
31. Poggeler, S., S. Masloff, B. Hoff, S. Mayrhofer, and U. Kuck. 2003. VersatileEGFP reporter plasmids for cellular localization of recombinant gene prod-ucts in filamentous fungi. Curr. Genet. 43:54–61.
32. Radzio, R., and U. Kuck. 1997. Efficient synthesis of the blood-coagulationinhibitor hirudin in the filamentous fungus Acremonium chrysogenum. Appl.Microbiol. Biotechnol. 48:58–65.
33. Richards, H. A., M. D. Halfhill, R. J. Millwood, and C. N. Stewart, Jr. 2003.Quantitative GFP fluorescence as an indicator of recombinant protein syn-thesis in transgenic plants. Plant Cell Rep. 22:117–121.
34. Root, D. E., N. Hacohen, W. C. Hahn, E. S. Lander, and D. M. Sabatini.2006. Genome-scale loss-of-function screening with a lentiviral RNAi li-brary. Nat. Methods 3:715–719.
35. Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual,3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Habor, NY.
36. Schmitt, E. K., A. Bunse, D. Janus, B. Hoff, E. Friedlin, H. Kurnsteiner, andU. Kuck. 2004. Winged helix transcription factor CPCR1 is involved inregulation of beta-lactam biosynthesis in the fungus Acremonium chrysoge-num. Eukaryot. Cell 3:121–134.
37. Schmitt, E. K., B. Hoff, and U. Kuck. 2004. Regulation of cephalosporinbiosynthesis. Adv. Biochem. Eng. Biotechnol. 88:1–43.
38. Shaner, N. C., R. E. Campbell, P. A. Steinbach, B. N. Giepmans, A. E.Palmer, and R. Y. Tsien. 2004. Improved monomeric red, orange and yellowfluorescent proteins derived from Discosoma sp. red fluorescent protein.Nat. Biotechnol. 22:1567–1572.
39. Shu, X., N. C. Shaner, C. A. Yarbrough, R. Y. Tsien, and S. J. Remington.2006. Novel chromophores and buried charges control color in mFruits.Biochemistry 45:9639–9647.
40. Smith, N. A., S. P. Singh, M. B. Wang, P. A. Stoutjesdijk, A. G. Green, andP. M. Waterhouse. 2000. Total silencing by intron-spliced hairpin RNAs.Nature 407:319–320.
41. Stauffer, J. F., L. J. Schwartz, and C. W. Brady. 1966. Problems and pro-gression in a strain selection program with cephalosporin-producing fungi.Dev. Ind. Microbiol. 7:104–113.
42. Takeno, S., E. Sakuradani, A. Tomi, M. Inohara-Ochiai, H. Kawashima, T.Ashikari, and S. Shimizu. 2005. Improvement of the fatty acid compositionof an oil-producing filamentous fungus, Mortierella alpina 1S-4, throughRNA interference with �12-desaturase gene expression. Appl. Environ. Mi-crobiol. 71:5124–5128.
43. Toews, M. W., J. Warmbold, S. Konzack, P. Rischitor, D. Veith, K. Vienken,C. Vinuesa, H. Wei, and R. Fischer. 2004. Establishment of mRFP1 as afluorescent marker in Aspergillus nidulans and construction of expressionvectors for high-throughput protein tagging using recombination in vitro(GATEWAY). Curr. Genet. 45:383–389.
44. Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer ofproteins from polyacrylamide gels to nitrocellulose sheets: procedure andsome applications. Proc. Natl. Acad. Sci. USA 76:4350–4354.
45. Walz, M., and U. Kuck. 1993. Targeted integration into the Acremoniumchrysogenum genome: disruption of the pcbC gene. Curr. Genet. 24:421–427.
46. Weld, R. J., K. M. Plummer, M. A. Carpenter, and H. J. Ridgway. 2006.Approaches to functional genomics in filamentous fungi. Cell Res. 16:31–44.
970 JANUS ET AL. APPL. ENVIRON. MICROBIOL.

IV. JANUS ET AL. 2007B
24
Identification of a minimal cre1 promoter sequence promoting glucose-
dependent gene expression in the β-lactam producer Acremonium chrysogenum
Janus D, Hortschansky P, Kück U (2007)
(prepared for submission)

Janus et al. 2007B
1
TITLE:
Identification of a minimal cre1 promoter sequence promoting glucose-dependent
gene expression in the ß-lactam producer Acremonium chrysogenum
Danielle Janus1, Peter Hortschansky2, and Ulrich Kück1*
Lehrstuhl für Allgemeine und Molekulare Botanik, Ruhr-Universität, Universitätsstr. 150,
D-44780 Bochum, Germany1; and Leibniz-Institute for Natural Product Research and
Infection Biology (HKI) and Friedrich-Schiller-University, Department of Molecular and
Applied Microbiology, D-07745 Jena, Germany2
* Corresponding author: Prof. Dr. Ulrich Kück, Lehrstuhl für Allgemeine und Molekulare
Botanik, Ruhr-Universität, Universitätsstr. 150, D-44780 Bochum, Germany,
Tel.: 0049-234-32 26212, Fax.: 0049-234-32 14184,
E-mail: [email protected]
Keywords
cre1 promoter, glucose-dependent gene expression, β-lactam biosynthesis, Acremonium
chrysogenum

Janus et al. 2007B
2
ABSTRACT:
Fungal CRE repressors regulate glucose-dependent gene expression via binding consensus
sequences in target gene promoters, including their own cre promoter sequence. Here, we
report the detailed molecular characterization of the cre1 promoter sequence with 15
putative CRE1 binding sites (BS1 to BS15) from the β-lactam producer Acremonium
chrysogenum. We fused cre1 promoter deletion derivatives with the DsRed reporter gene
for comparative gene expression analysis. Plate assays identified the minimal D4 promoter
sequence that promoted glucose-dependent expression. This was further verified by
Northern blot analysis with RNA from low and multi copy transformants grown on media
with or without glucose. Spectrofluorometric quantification of the DsRed amount finally
confirmed the data from the transcript analysis. Truncated recombinant CRE1 synthesized
in E. coli interacted with D4 in electromobility shift analysis (EMSA) and these binding
studies were further extended with two oligonucleotides, carrying putative CRE binding
sites B14 and B15. Surface plasmon resonance analysis (SPR) was performed using BS14
and BS15, along with four derivatives containing two or four base pair substitutions within
BS14 and BS15, respectively. Substitutions within BS14 abolished the high affinity
interaction with CRE1, while mutations in BS15 only marginally diminished the affinity
with CRE1. In vivo analysis of a modified D4 sequence with substitutions in the two
binding sites confirmed the in vitro binding results and still promoted glucose-dependent
gene expression. Our results will contribute to the construction of versatile expression
vectors carrying a minimal cre1 promoter sequence that still confers glucose-dependent
induction of gene expression.

Janus et al. 2007B
3
INTRODUCTION:
Glucose is a readily metabolizable carbon source, which leads to enhanced growth rates
during fermentation processes. Glucose also causes repression of several enzymes and
secondary metabolites by a phenomenon called carbon catabolite repression (CCR) that
has been observed in diverse prokaryotic and eukaryotic microorganisms (Martín et al.
1999, Warner and Lolkema 2003). In the yeast S. cerevisiae, CCR is mediated by the zinc-
finger protein MIG1 that, via binding a consensus sequence (5’-SYGGRG-3’) in several
glucose-repressible promoters, interacts with the TUP1/SSN6 general repressor complex to
form an active complex and represses gene expression (reviewed by Gancedo 1998). In
filamentous fungi, the homologue of the MIG1 repressor (CREA or CRE1) was first
isolated from A. nidulans (Dowzer and Kelly 1989) and later shown to function in diverse
pathways, e.g. regulation of proline, ethanol, xylan, and arabinan utilization (reviewed by
Ruijter and Visser 1997). To avoid any confusion, we will refer always to cre1 gene,
irrespectively whether it has been named as cre1 or creA in the original literature. In
Trichoderma reesei, the industrially important producer of cellulases and hemicellulases,
the CRE1 homologue is partially responsible for the inhibition of cellulase expression and
also represses the expression of the xylanase I (xyn1) gene (Ilmén et al. 1996, Mach et al.
1996).
The CRE proteins function either by directly competing for binding sites of activator
proteins or by an indirect mechanism that prevents the action of an activator (Ronne 1995).
Characterization of the promoters of many glucose-repressible target genes led to the
identification of CRE binding consensus sequences (5’-SYGGRG-3’) (e.g. Cubero and
Scazzocchio 1994, Mathieu et al. 2000, Rauscher et al. 2006). Furthermore, CRE1 does not
always directly regulate transcription by interacting with binding sites in promoter
sequences, but also indirectly regulates transcription by affecting nucleosome positioning,
such as in the 5’-regulatory sequence of the cbh2 gene (Zeilinger et al. 2003).
CRE1 binding sites are not only found in the promoters of target genes but also in the
promoters of cre1 genes from diverse filamentous fungi. The best molecularly
characterized cre1 promoter is the A. nidulans promoter; which contains four CRE1
binding sequences and large blocks of CT-rich sequences, including long stretches of
polydT sequence (Dowzer and Kelly 1989, Kulmburg et al. 1993, Cubero and Scazzocchio
1994). Two of these CRE1 consensus binding sequences were previously characterized in
A. nidulans and shown to function as an autoregulation motif in vivo. The authors showed
that the glucose pulse-mediated downregulation of cre1 transcript levels is abolished when

Janus et al. 2007B
4
the cre1 gene was fused with a promoter carrying mutated CRE1 recognition sequences
(Strauss et al. 1999).
In this study, we provide a functional characterization of the cre1 promoter from the β-
lactam antibiotic producing fungus Acremonium chrysogenum, in which antibiotic
biosynthesis is repressed by glucose (reviewed by Schmitt et al. 2004a). Detailed transcript
analysis revealed that the transcript levels of pcbC and cefEF, encoding isopenicillin N
synthase and deacetoxycephalosporin C/deacetylcephalosporin C synthetase, respectively,
were reduced in the wild type but not in a producer strain (Jekosch and Kück 2000b). The
cre1 gene from A. chrysogenum was previously isolated in order to analyze glucose-
dependent gene expression, and the CRE1 protein sequence shows high similarity to other
glucose repressor proteins from filamentous fungi, suggesting a similar regulation and
related role in carbon catabolite repression. Analysis of the cre1 transcript levels in two A.
chrysogenum strains showing different production rates of the antibiotic cephalosporin C
displayed a different glucose regulation of the cre1 transcript in both strains. The cre1
transcript level in the wild type strain was increased six-fold during growth in glucose-
containing, pH-buffered medium, whereas the transcript of the producer strain was
unaffected, indicating a positive autoregulation of cre1 in the wild type strain and an
absence of glucose regulation in the producer strain.
In order to measure the cre1 promoter-driven gene expression in A. chrysogenum, we used
the DsRed reporter gene encoding the red fluorescent protein from Discosoma spec. We
recently have shown that the DsRed gene is a valuable reporter not only for quantifying
promoter strengths but also for the phenotypic detection of robustly expressed genes on
Petri dishes (Janus et al. 2007). Previous sequence analysis of the 1030 bp cre1 promoter
sequence identified 15 putative CRE1 binding sites (BS1 to BS15) (Jekosch and Kück
2000a, Jekosch and Kück 2000b) that are further characterized in this investigation. This
includes in vivo expression studies with different cre1 promoter derivatives as well as in
vitro binding studies of CRE1 with putative CRE1 binding sites using electromobility shift
analysis and surface plasmon resonance analysis. Our experiments are complemented by
investigating transformants that carry modified cre1 promoters with mutated CRE1
binding sites. This study identifies a 114 bp minimal promoter with two CRE1 binding
sites, the first minimal regulatory promoter sequence for the β-lactam antibiotic producing
fungus.

Janus et al. 2007B
5
MATERIALS AND METHODS
Strains and culture conditions. Escherichia coli strain K-12 XL1-blue (Stratagene) was
used for plasmid construction and maintenance (Bullock et al. 1987). As already described,
liquid cultures of A. chrysogenum strains were grown at 27 °C and 180 rpm for 3-5 days in
CCM medium, CCM complemented with 100 mM HEPES (CCMH), or CCMH with the
addition of 6.3 % (w/v) glucose (CCMHS) (Walz and Kück 1991, Jekosch and Kück
2000a).
Transformation of A. chrysogenum strain ATCC 14553 was done according to
conventional transformation procedures (Walz and Kück 1993, Radzio and Kück 1997).
All strains used in this study are listed in Table 3 and their genotypes were verified by
Southern blot analysis (data not shown) using gene-specific probes for hybridization of
genomic DNA. Resulting transformants were selected on media containing hygromycin B
at concentrations as previously reported (Walz and Kück 1993, Radzio and Kück 1997).
Construction of the cre1 promoter derivatives. For amplifying the cre1 promoter from
plasmid pCRE13/2 (Jekosch and Kück 2000a) primers 3148 and 3149 were used. The PCR
fragment was ligated into plasmid pDrive (Qiagen) and subsequently sequenced. The
resulting plasmid was hydrolyzed with AgeI and partially digested with NcoI. The
corresponding DNA fragment was used for ligation with the AgeI-NcoI-restricted fragment
of plasmid pRHN1. The reporter gene plasmid pRHN1 carries the full-size DsRed gene
(DsRed-Express; BD Biosciences Clontech, Heidelberg, Germany) under the control of the
gpd promoter and trpC terminator of A. nidulans and an hph-resistance cassette (Godehardt
and Kück, unpublished data). The resulting recombinant plasmid was designated pRPcre1
carrying the DsRed gene under control of the cre1 promoter (Pcre1).
The construction of the cre1 promoter deletion derivatives was performed by conventional
PCR or fusion PCR based on modified protocols described previously (Shevchuk et al.
2004). The cre1 promoter fragments for generating deletion derivatives D1, D3, and D4
were amplified by using primers Pcre1-D1/ Pcre1-D3/ Pcre1-D4 and 3149, respectively,
and plasmid pCRE13/2 as template. Using primers 3148 and 3149 and plasmid
pCRECEFD2, promoter deletion fragment D2 was amplified to generate plasmid pRPcre1-
D2. pCRECEFD2 carries the full-size A. chrysogenum cefEF gene controlled by a
truncated version of the cre1 promoter (Δ bp 488-687). cre1 promoter derivatives D5, D6
and D7 harbor internal deletions and were therefore made by a three step fusion PCR. In a
first step, both fragments which should be fused, were amplified using A. chrysogenum

Janus et al. 2007B
6
genomic DNA as template and primers cre1ko5 and D5-anti/D6-anti/D7-anti, and cre1-as
and D5-sense/D6-sense/D7-sense, respectively. For deletion derivative D4*, primers cre1-
as and Pcre1-D4*-s, and Pcre1-D3 and Pcre1-D4*-as were used to amplify the first two
PCR fragments of the promoter. In a next step the two fragments that correspond to one
deletion fragment were fused by a further PCR step. In the final step, the cre1 promoter
deletion fragments were amplified using nested primers 3148 and 3149 (deletion
derivatives D5, D6, and D7), and primers 3149 and Pcre1-D4 (deletion derivative D4*) to
attach restriction sites AgeI and NcoI. The corresponding cre1 promoter fragments were
ligated into pDrive (Qiagen) and sequenced. In the next step, all obtained fragments were
digested and inserted into AgeI-NcoI restriction sites in plasmid pRHN1 to generate the
different promoter deletion constructs (Table 2). The sequences of all oligonucleotides and
the recombinant plasmids are given in Table 1 and Table 2, respectively.
Preparation and analysis of RNA. RNA was isolated as described by Jekosch and Kück
(Jekosch and Kück 2000a), and the integrity of all RNAs was verified by agarose gel
electrophoresis and Northern blot analyses (Sambrook and Russell 2001). The blots were
hybridized with 32P-radiolabeled DNA probes as specified in the Results section.
Protein purification. The purification of fungal proteins was carried out as described
previously (Schmitt et al. 2004b), with the modification that the mycelium was
resuspended in 2 ml of buffer A (0.1 M MOPS (morpholinepropanesulfonic acid, pH 7.5),
0.2 M KCl, 10 mM MgCl2, 1 mM EDTA, 10 mM dithiothreitol, 4.2 mM
phenylmethylsulfonyl fluoride, 40 % [w/v] glycerol).
Spectrofluorometric measurements. The fluorescence of the DsRed protein was
measured at 576 nm with the excitation set at 554 nm using a JASCO FP-6500
spectrofluorometer (JASCO, Tokyo, Japan) as previously described (Janus et al. 2007). A
calibration curve using purified DsRed protein (BD Biosciences) was produced and served
as control to quantify DsRed levels in A. chrysogenum protein extracts.
Synthesis of recombinant CRE1 polypeptide in E. coli. GST and GST fusion proteins
were purified from E. coli BL21 (DE3) cells (Stratagene). The gene sequence for the first
183 amino acids from CRE1 were ligated in frame to the coding sequence of GST by using
restriction sites EcoRI and NotI of pGEX-4T1 (Amersham Bioscience). The resulting

Janus et al. 2007B
7
plasmid named pGEXcre1_183 was transformed into E. coli expression host BL21 (DE3).
Gene expression was induced at midlog phase by adding 1 mM isopropyl β-D-
thiogalactoside and the cells were incubated for one hour at 30 °C. GST and GST fusion
proteins were purified by affinity chromatography using a 50 % slurry of Glutathione
Sepharose beads according to the supplier’s manual (Amersham Biosciences). Purified
proteins were stored at –80 °C until used for electrophoretic mobility shift assays.
Electromobility shift assays (EMSA). Gel shift experiments were performed using either
radiolabeled PCR fragments or annealed oligonucleotides from the cre1 promoter. The
promoter fragments were amplified with oligonucleotides listed in Table 1 and
radiolabeled during PCR as described previously (Schmitt et al. 2004b). The PCR mixture
contained 90 ng of pCRE13/2 as template, 40 ng of each primer, 4 µM dATP, 4µM dTTP,
4µM dGTP, 0.2 µM dCTP, 8.33 x 10-12 mol of [α-32P]dCTP, and 1U Taq polymerase
(Eppendorf) in a volume of 50 µl. To remove free nucleotides from the samples, an
electrophoresis was performed in a 5 % polyacrylamid gel with 0.5 x Tris-borate-EDTA
buffer. Single bands were extracted, incubated with elution buffer (0.5 M ammonium
acetate, 1mM EDTA, 0.1 % SDS) and precipitated with ethanol. The precipitate was
finally dissolved in 100 µl H2O. Labeling of annealed oligonucleotides was achieved by
5’-end labeling using polynucleotide kinase (Roche) and [γ-32P]-dATP. To investigate the
binding of the CRE1 polypeptide, 10-30 fmol of various radiolabeled promoter fragments
or oligonucleotides were incubated with varying protein concentrations (indicated in the
corresponding figures) in the presence of 2 µl binding buffer (250 mM Tris/HCl pH 8.0,
1M KCl, 50 % glycerin) and 1 µg of poly(dI-dC) • poly(dI-dC) in a total volume of 20 µl.
After 20-min incubation at room-temperature, the samples were electrophoresed in a 5 %
polyacrylamide gel at 4 °C with Tris/glycine buffer.
Protein purification of CRE1 for Biacore measurements.
GST-tagged CRE1(1-183) was produced by autoinduction in E. coli Rosetta 2 (DE3) cells
grown at 30 °C in 1 l Overnight Express Instant TB Medium (Novagen) (Studier 2005). 15
g cells were collected by centrifugation, resuspended in 200 ml lysis buffer (20 mM
NaH2PO4, 150 mM NaCl, 1 mM PMSF, pH 7.4) and homogenized using an Emulsiflex C5
high pressure homogenizer (Avestin). Clarified cellular extract was loaded onto a 20 ml
Glutathione Sepharose 4 FF column (GE Healthcare) followed by an overnight on-column
cleavage at room temperature using 320 units human thrombin (Sigma). CRE1 was eluted

Janus et al. 2007B
8
with 50 mM Tris/HCl, 10 mM reduced glutathione, pH 8.0, transferred to 50 mM
NaH2PO4, 150 mM NaCl, pH 7.4 and applied to a 5 ml Heparin Sepharose HP column (GE
Healthcare), followed by elution with a salt gradient up to 2 M NaCl. CRE1 containing
fractions were concentrated using a Vivaspin 10 kD concentrator (Vivascience) and
purified to homogeneity by gel filtration chromatography on a Superdex PG 200 column
(GE Healthcare) with 50 mM NaH2PO4, 150 mM NaCl, 1 mM DTT, pH 7.4 as running
buffer.
Binding analysis by surface plasmon resonance (SPR). Real-time analysis of protein-
DNA-interaction was performed on a Biacore 2000 system at 25 °C. A commercially
available SA sensor chip (Biacore) was used having a layer of a thin gold film coated with
carboxymethyldextran hydrogel matrix to which streptavidin was cross-linked. Data were
processed with the BIAevaluation software version 4.1 (Biacore). The running buffer used
for DNA immobilization and SPR assay was 10 mM HEPES pH 7.4, 0.15 M NaCl, 1 mM
DTT, 0.005 % (v/v) surfactant P20. The buffer was freshly prepared, filtered through a
0.22 µm membrane and degassed prior to use. Refractive index errors due to bulk solvent
effects were corrected by substracting responses from the non-coated flow cell 1. 5’-
biotinylated DNA duplexes (5 nM) containing CRE1 wild type and mutant binding sites
were immobilized on a SA sensor chip on flow cells 2, 3 and 4 by injection at a flow rate
of 10 µl/min until 80 response units (RU) were bound. The purified CRE1 protein was
diluted to nanomolar concentrations in running buffer and a concentration series from 12.5
nM to 400 nM was examined for each DNA duplex. Sample injection and dissociation
time was set to 2.5 minutes at a flow rate of 20 µl/min. The chip surface was regenerated
with an injection of running buffer containing 0.5 M NaCl for one minute. Dissociation
constants were calculated from the concentration-dependent steady-state binding using the
1:1 steady-state affinity model.

Janus et al. 2007B
9
RESULTS AND DISCUSSION
cre1 promoter directed DsRed expression. Transcriptional expression of the glucose-
repressor gene cre1 is detectably up regulated in the presence of glucose in the wild type
Acremonium chrysogenum strain (Jekosch and Kück 2000a). We examined the 5’-
upstream region of the cre1 gene and detected several putative CRE1 consensus binding
sites (5’-SYGGRG-3’), suggesting a positive autoregulation of cre1 expression by its own
gene product. We discovered 15 CRE1 consensus sequences (BS1 to BS15) within the
1030 bp upstream of the cre1 transcription start site, which is preceded by an 18 bp T-
stretch as well as a CT-rich region. In order to study cre1 promoter-driven expression in
vivo, we used a recently developed DsRed gene based reporter system. Expression of the
gene encoding the red fluorescent protein (DsRed) results in fungal transformants
displaying a red coloured phenotype on solid medium. Moreover, variations in DsRed
synthesis can be quantified with spectrofluorometric measurements (Janus et al. 2007).
For our expression studies, we constructed plasmid pRPcre1, which contains the DsRed
gene under control of the full-length cre1 promoter (Pcre1). Transformation of pRPcre1
into A. chrysogenum resulted in several differently coloured transformants obtained on
agar plates. Of the total 19 transformants, seven showed a red or pink coloured phenotype
and were further analyzed for plasmid copy number and glucose-induced gene expression.
We identified low (T2) and multi copy number transformants (T13, T17), and glucose-
induction was further analyzed when the strains were grown in the presence (+G) or
absence (-G) of glucose. From these various strains in different growth conditions, RNA
was isolated and subjected to Northern blot hybridizations. As presented in Figure 1A, the
DsRed transcript markedly increases in all transformants when grown in glucose-
containing medium. Transcript levels are barely detectable when the strains were grown in
the absence of glucose. Only T17, which is a multi copy strain, showed detectable levels of
DsRed in the absence of glucose.
We next examined the time-course of DsRed expression. Strains were cultured for seven
days and protein extracts were obtained at specific time points. To guarantee sufficient
cellular material for the quantification of DsRed, the measurement began two days after
inoculation, and, as recently described, spectrofluorometric measurements were conducted
to determine the amount of DsRed in each sample (Janus et al. 2007). As shown in Figure
1B, all transformants showed the highest amount of DsRed in a copy-dependent manner
when grown in the presence of glucose. Maximal expression was observed after five to six
days of growth in all cases, confirming the usefulness of the DsRed reporter gene system to

Janus et al. 2007B
10
study regulated gene expression in A. chrysogenum. Furthermore, the expression data
presented here supports the observation in our previous study that the DsRed protein does
not interfere with any endogenous gene products or cellular growth, and thus is a reliable
marker to measure gene expression in quality and quantity. Other reporter gene products,
such as the E. coli β-galactosidase, may interfere with endogenous enzyme activity,
previously shown in A. chrysogenum, when endogenous β-galactosidase activity was
repressed by the addition of glucose (Menne et al. 1994). Usually, a locus-dependent
integration accompanied by homologous recombination of a marker gene is the desired
system to study reproducible expression rates in filamentous fungi. This has, for example,
been established for the biotechnically relevant fungus A. niger, using the orotidine-5’-
phosphate decarboxylase (pyrG) gene as a marker (van Hartingsveldt et al. 1987). In A.
chrysogenum, however, a comparable system has not yet been established and a larger
number of transformants carrying ectopically integrated plasmids with variable gene
expression patterns must be investigated in separate experiments. For this purpose, the
DsRed gene is an ideal reporter system, allowing simple plate tests to distinguish
transformants showing high, medium, and low expression rates.
Expression studies with cre1 promoter deletion derivatives. To detect critical sequences
within the cre1 promoter region, we performed functional analysis of seven promoter
deletion mutants that lack at least one of the putative CRE1 binding sites (Figure 2). All
promoter constructs were fused upstream of DsRed and analyzed phenotypically.
Transformation of A. chrysogenum with plasmid pRPcre1 in which DsRed expression is
driven by the wild type cre1 promoter resulted in a total of 19 analyzed transformants,
seven of which displayed clear red or pink colour. In comparison, the panel of promoter
mutants displayed a diverse range of DsRed regulation. As discussed above, ectopic
integration of transformed DNAs into the genome, such as during DNA-mediated fungal
transformation, leads to modification of the plasmid-derived genes expression (Janus et al.
2007). Thus, the phenotypic characterization of fungal transformants on Petri dishes is a
rough estimate of promoter-driven reporter gene expression. From the data presented in
Figure 2, the T-stretch and the CT-rich region, both comprising a region of about 149 bp,
are essential for gene expression, exemplified by the high frequency of reddish
transformants, carrying either D1, D3, D4, or D6. In contrast, D5 and D7 showed no
reddish transformants and lack the T-stretch as well as the CT-rich region. One exception
seemed to be the D2 mutant that carries a deletion just upstream of the T-stretch,

Janus et al. 2007B
11
suggesting that the upstream region contains binding sites for further activator or repressor
proteins that modulate gene expression. In separate experiments, we constructed promoter
derivatives lacking the 5’-transcribed but not translated region. None of these mutants
showed a detectable expression of the reporter gene (data not shown).
To quantify the observed variations in transcriptional expression, 10 to 14 randomly
selected transformants obtained with each promoter derivative were selected and analyzed
spectrofluorometrically. After five days of growth, the DsRed protein content was
determined compared to total protein amount (Supplementary Data Table 1). The results of
the spectrofluorometric measurements are consistent with the data shown in Figure 2.
Transformation with promoter derivatives that failed to yield any coloured transformants
correspondingly had barely detectable DsRed protein, while among the derivatives that
generated a range of phenotypes, we observed a correlation between DsRed protein content
and copy number. Notably, the D4 construct generated the most distinct protein yields
(Supplementary Data Table 1).
We further analyzed DsRed expression in both, low and multi copy transformants
harbouring the promoter derivatives D2, D4, D5, D6, and D7, in the absence and presence
of glucose. Northern blot analysis showed that cells harbouring DsRed driven by the full
length cre1 promoter markedly induced DsRed expression in the presence of glucose, and
levels correlated well with copy number (Figure 3A). A glucose-dependent transcriptional
induction was generally observed among all transformants investigated, except D5 and D7,
which acted similar to a negative control. The derivatives D4 and D6 induced strong
DsRed levels similar to that obtained with the full-length promoter Pcre1. Interestingly, in
cells with the full-length promoter, a basal expression was detectable in the absence of
glucose, while D4 showed no detectable transcript at all. These transcriptional expression
data were further verified by spectrofluorometric measurement of the DsRed protein level
(Figure 3B). Glucose-dependent protein levels were observed, with the highest yield
generated by the multi copy transformant carrying the D4 derivative (D4 T9 +G). The wild
type strains served as negative control to measure background fluorescence in all cases.
Our expression analysis data strongly suggests that the 114 bp CT-rich sequence in the
non-transcribed upstream region is required to trigger glucose-dependent gene expression.
CT-rich regions are frequently found in promoters of strongly transcribed genes in
filamentous fungi, mostly in genes lacking conventional TATA and CAAT boxes (Kapoor
et al. 1995). This indicates that CT-rich regions are putative activator sequences in close
proximity to transcription starts sites (Gurr et al. 1987, Yamazaki et al. 2000, Sirand-

Janus et al. 2007B
12
Pugnet et al. 2003, Fei et al. 2006, Neveu et al. 2007). For example, deletion of the CT-rich
region upstream of the transcription start point in different Aspergillus gpdA and oliC
genes, encoding glyceraldehyde-3-phosphate dehydrogenase and oligomycin-resistant ATP
synthase subunit 9, respectively, resulted in an altered transcription initiation. This
supported the involvement of this region in correct transcription initiation and determining
the frequency of initiation (Ballance 1986, Ward and Turner 1986, Ward et al. 1988, Punt
et al. 1990). In plants, the CT-rich regions flanking the 5’-end of plant genes function
similarly, since mutagenesis of this sequence resulted in a significant decrease of transgene
expression (Bolle et al. 1994).
Gel shift analysis of cre1 promoter sequences with CRE1. To determine whether the
CRE1 protein could bind fragments of the cre1 promoter carrying putative CRE1 binding
sites, we performed in vitro binding studies using EMSA. As detailed in Materials and
Methods, we generated a recombinant GST::CRE1 fusion protein consisting of the GST-
tag and the first 183 amino acids of the CRE1 polypeptide and synthesized the fusion
protein in E. coli. Different DNA fragments that were generated with individual primer
pairs (Figure 4A) were incubated with GST::CRE1, and in all these cases, almost every
fragment was bound by the CRE1 protein (data not shown). As an example, incubation of
two labelled DNA fragments with recombinant CRE1 resulted in a high molecular weight
DNA-protein complex only with fragment 21-24, which contains two putative CRE1
binding sites, while fragment 1-2, which lacks binding sites, and the GST alone were both
unable to form any complex (Figure 4B). Increasing the amount of CRE1 (0.5 – 10 µg)
intensified the binding, suggesting strong interaction between CRE1 and fragment 21-24.
Interestingly, multiple bands were observed when the amount of the CRE1 protein was
increased, indicating consecutive binding of different sequences with the CRE1
polypeptide (Figure 4C).
To more closely analyze CRE1 binding, the two putative binding sites (BS14, BS15), part
of the 114 bp DNA sequence of promoter derivative D4 triggering glucose dependent gene
expression, were used for gel shift analysis (Figure 5A). BS14 contains the putative
binding site 14 flanked by ten base pairs on both sites, while BS15 carries binding site 15
and is flanked by seven and 13 base pairs. Gel shift analysis showed that both sequences
interact with CRE1, and that the protein binds BS15 more intensely (Figure 5B).
Increasing amounts of CRE1 in the binding analysis produced only a single complex with
BS14 and BS15 (Figure 5C). These results are consistent with those obtained in the

Janus et al. 2007B
13
investigation of autoregulation of CRE1 in A. nidulans, which showed that two closely
spaced putative CRE1 binding sites in the cre1 promoter are necessary to mediate
autoregulation. Furthermore, gel shift analysis with a recombinant GST::CRE1 protein and
cell-free extracts showed direct interaction of the CRE1 protein to binding sequences in the
cre1 promoter (Strauss et al. 1999).
Interactions between CRE1 and binding sites BS14 and BS15 determined by SPR
analysis. To further characterize the binding specificity of CRE1, surface plasmon
resonance (SPR) analyses of protein-DNA interactions were carried out on a streptavidin-
coated Biacore SA sensor chip using oligonucleotides BS14 and BS15, as well as mutant
oligonucleotides with two to four substitutions (BS14m1, BS14m2, BS15m1, BS15m2)
(see Table 1) within the consensus binding sites as shown in Figure 5A. The non-coated
flow cell 1 served as a reference in order to reduce bulk solvent effects. The RU- values of
different DNA loaded on each flow cell were about 80 RU. As detailed in the Materials
and Methods section, a highly purified CRE1 protein detached from the GTS-tag was
synthesized for SPR analysis. The purified truncated CRE1 protein (CRE1 1-183) used in
these studies showed a single band on SDS-polyacrylamide gels (Figure 6A).
SPR sensorgrams for equilibrium binding of CRE1 to immobilized DNA-duplexes were
recorded (data not shown) and their corresponding steady-state affinity analyses are shown
in Figures 6B and 6C. Binding of the CRE1 polypeptide to BS14 compared to the two
mutant DNA-duplexes BS14m1 and BS14m2 is shown in Figure 6B. Interaction of 400
nM CRE1 with BS14 attained 73.8 RU at equilibrium whereas mutant oligonucleotides
showed no or very marginal interactions at all. SPR analyses using oligonucleotides BS15
and mutant forms (BS15m1, BS15m2) are shown in Figure 6C and demonstrate that the
protein-DNA interaction between CRE1 and BS15 had a high affinity. On the other hand,
mutant oligonucleotides retained lower binding specificities but still showed a weak
affinity, which is reduced by a factor of approximately 3. Consequently, substitution of two
to four base pairs within the consensus binding sequence of BS15 weakened the protein-
DNA complex formation but did not completely inhibit CRE1 binding.
Comparing the calculated KD-values (Table 4) supports the observation that both BS14 and
BS15 are specifically bound by CRE1 but BS14 (with a KD value of 154 nM) interacts
with a lower affinity than BS15 (with a KD value of 85.9 nM). In contrast, mutant
oligonucleotides, BS14m1, BS14m2, BS15m1 and BS15m2, show higher KD-values in
comparison to BS14 and BS15, which refers to a lower binding affinity to the CRE1

Janus et al. 2007B
14
polypeptide. These results are in good agreement with the data obtained with gel
retardation analysis of CRE1 interaction to oligonucleotides BS14 and BS15 (Figures 5B
and 5C).
SPR measurements have been used to determine the interaction of two proteins or protein
complexes with specific DNA sequences (e.g. Hortschansky et al. 2007, Shi et al. 2007).
Recently, the interaction of the Saccharomyces cerevisiae alcohol dehydrogenase 1 protein
with sequences of the alcohol dehydrogenase II gene were analysed by surface plasmon
resonance. The determination of dissociation constants for the interaction of this zinc-
finger protein with the corresponding DNA has been determined with two-digit nanomolar
affinities (Schaufler and Klevit 2003), similar to the approach in our investigation.
Furthermore, our data reveal that SPR analysis is a convenient method to determine the
interaction of a protein to different DNA sequences in real-time without labelling
requirements (Myszka et al. 1998). In addition, the comparison of binding specificities of a
protein to wild type and mutant binding sites can be easily performed and quantified (Hart
et al. 1999). To the best of our knowledge, this is the first example where the CRE1
affinity to consensus binding sites was measured using SPR analysis.
In vivo analysis of substitutions in binding sites BS14 and BS15. The SPR analysis
indicates that BS14m2 lacks almost any binding specificity with CRE1 while BS15m2,
despite four nucleotide substitutions, still retains decent interaction with the CRE1 protein.
This suggests that in vivo a D4 fragment containing the BS14m2, as well as the BS15m2,
substitution could still have a glucose-dependent transcriptional expression. We thus
constructed plasmid pRPcre1_D4* harbouring four base pair substitutions in each of the
BS14 and BS15 CRE1 consensus binding sites, and transformed it into the wild type strain
of A. chrysogenum, resulting in reddish colonies on agar plates. When we tested a total of
35 transformants, we found that 17 had a pink or red colour while the remaining did not
show any expression of the DsRed gene on solid media. This is similar to the distribution
we observed when we used the D4 construct carrying the wild type sequence for fungal
transformation (see also supplementary material Table 1). This rough approach indicates
that, despite the base pair substitutions, the modified D4* promoter sequence is still able to
confer glucose-dependent gene expression. These results were further confirmed when two
randomly selected D4* transformants, a low copy and a multi copy strain, were subjected
to Northern blot analysis and compared to transformants carrying either the full-length
cre1 promoter or the D4 promoter fragment. As shown in Figure 7, the level of the

Janus et al. 2007B
15
transcript clearly increased in all strains investigated under glucose-inducing growth
conditions, and this was further confirmed by spectrofluorometric measurements of DsRed
protein (data not shown). Thus, these data are in good agreement with the results obtained
with the SPR measurements showing a sufficient affinity of CRE1 to the BS15m2
sequence harbouring four T-substitutions, being capable of driving glucose-induced gene
expression. The binding affinity to BS15 seemed to be stronger than interaction with BS14,
as demonstrated by SPR analysis, and this is in line with the in vivo analysis with deletion
derivative D4* still showing strong gene expression and induction by glucose.
Detailed sequence analysis of binding site BS15 (5’-AGACTGGGGGGGGGGG-3’) and
mutant binding sites, as well as adjacent sequences, displayed that the consensus binding
site (5’-SYGGRG-3’) in the mutant oligonucleotides shifted to the 3’ end of BS15m2 (5’-
AGACTTTTTGGGGGGG-3’) with only a single nucleotide substitution. Therefore, it is
possible that this incompletely truncated binding site is responsible for the glucose
induction in transformants carrying deletion derivative D4*. Deletion of flanking
sequences from BS15 should be performed to prevent further binding of CRE1 to the
displaced binding site and to further determine whether CRE1 is the only regulator
responsible for glucose induction directed by promoter derivative D4. Another explanation
could be the interaction of other proteins and possible transcriptional modulators directed
by the cre1 promoter. Similarly, in Phytophthora infestans, two still uncharacterized 32
and 43 kDa proteins interact with CT-elements in the promoter of the piypt1 gene,
encoding a monomeric G-protein (Chen and Roxby 1997).
CONCLUSION:
The cre1 promoter from A. chrysogenum displays a positive autoregulation upon glucose
induction, and our identification of a 114 bp minimal promoter with two CRE1 binding
sites is the first example of a minimal regulatory promoter sequence for this β-lactam
antibiotic producing fungus. Two CRE1 consensus binding sites are located within the
minimal promoter sequence that are tightly bound by the CRE1 protein, and this
interaction was quantified using the Biacore system and represents the first
characterization of a CRE1-DNA-interaction by SPR analysis. Importantly, the minimal
cre1 promoter identified here will be a functionally useful tool for the construction of
compact fungal expression vectors suitable for biotechnically relevant filamentous fungi.
For example, normal glucose-dependent repression of cephalosporin C biosynthesis genes

Janus et al. 2007B
16
could be converted into expressional activation by fusing biosynthesis genes to the
minimal cre1 promoter.
ACKNOWLEDGMENTS
We thank Kerstin Kalkreuter (Bochum), Sylke Fricke (Jena), and Ingeborg Godehardt
(Bochum) for excellent technical assistance, Prof. Eckhard Hofmann (Bochum) for the use
of the JASCO spectrofluorometer, Drs. E. Friedlin and H. Kürnsteiner (Kundl) for fruitful
discussions, and Prof. A. Brakhage (Hans-Knöll Institute, Jena) for institutional support.
This work was funded by Sandoz GmbH (Austria, Kundl).

Janus et al. 2007B
17
REFERENCES
Ballance DJ (1986) Sequences important for gene expression in filamentous fungi. Yeast 2: 229-236
Bolle C, Sopory S, Lubberstedt T, Herrmann RG, Oelmuller R (1994) Segments encoding
5'-untranslated leaders of genes for thylakoid proteins contain cis-elements essential for transcription. Plant J 6: 513-523
Bullock WO, Fernandez JM, Short JM (1987) XL1-Blue: a high efficiency plasmid
transforming recA Escherichia coli strain with beta-galactosidase selection. BioTechniques 5: 376-378
Chen Y, Roxby R (1997) Identification of a functional CT-element in the Phytophthora
infestans piypt1 gene promoter. Gene 198: 159-164 Cubero B, Scazzocchio C (1994) Two different, adjacent and divergent zinc finger binding
sites are necessary for CREA-mediated carbon catabolite repression in the proline gene cluster of Aspergillus nidulans. EMBO J 13: 407-415
Dowzer CEA, Kelly JM (1989) Cloning ot the creA gene form Aspergillus nidulans: a
gene involved in carbon catabolite repression. Curr Genet 15: 457-459 Fei X, Zhao MW, Li YX (2006) Cloning and sequence analysis of a glyceraldehyde-3-
phosphate dehydrogenase gene from Ganoderma lucidum. J Microbiol 44: 515-522 Gancedo JM (1998) Yeast carbon catabolite repression. Microbiol Mol Biol Rev 62: 334-
361 Gurr SJ, Uncles SE, Kinghorn JR (1987) The structure and organization of nuclear genes
in filamentous fungi. In: Kinghorn JR (eds.) Gene structure in eukaryotic microbes. IRL Press, Oxford pp. 93-139
Hart DJ, Speight RE, Cooper MA, Sutherland JD, Blackburn JM (1999) The salt
dependence of DNA recognition by NF-kappaB p50: a detailed kinetic analysis of the effects on affinityand specificity. Nucleic Acids Res 27: 1063-1069
Hortschansky P, Eisendle M, Al-Abdallah Q, Schmidt AD, Bergmann S, Thon M,
Kniemeyer O, Abt B, Seeber B, Werner ER, Kato M, Brakhage AA, Haas H (2007) Interaction of HapX with the CCAAT-binding complex-a novel mechanism of gene regulation by iron. Embo J
Ilmén M, Thrane C, Penttilä M (1996) The glucose repressor gene cre1 of Trichoderma:
isolation and expression of a full-length and a truncated mutant form. Mol Gen Genet 251: 451-460
Janus D, Hoff B, Hofmann E, Kück U (2007) An efficient fungal RNA-silencing system
using the DsRed reporter gene. Appl Environ Microbiol 73: 962-970 Jekosch K, Kück U (2000a) Glucose dependent transcriptional expression of the cre1 gene
in Acremonium chrysogenum strains showing different levels of cephalosporin C production. Curr Genet 37: 388-395

Janus et al. 2007B
18
Jekosch K, Kück U (2000b) Loss of glucose repression in an Acremonium chrysogenum
beta-lactam producer strain and its restoration by multiple copies of the cre1 gene. Appl Microbiol Biotechnol 54: 556-563
Kapoor M, Curle CA, Runham C (1995) The hsp70 gene family of Neurospora crassa:
cloning, sequence analysis, expression, and genetic mapping of the major stress-inducible member. J Bacteriol 177: 212-221
Kulmburg P, Mathieu M, Dowzer C, Kelly J, Felenbok B (1993) Specific binding sites in
the alcR and alcA promoters of the ethanol regulon for the CREA repressor mediating carbon catabolite repression in Aspergillus nidulans. Mol Microbiol 7: 847-857
Mach RL, Strauss J, Zeilinger S, Schindler M, Kubicek CP (1996) Carbon catabolite
repression of xylanase I (xyn1) gene expression in Trichoderma reesei. Mol Microbiol 21: 1273-1281
Martín JF, Casqueiro J, Kosalková K, Marcos AT, Gutiérrez S (1999) Penicillin and
cephalosporin biosynthesis: mechanism of carbon catabolite regulation of penicillin production. Antonie Van Leeuwenhoek 75: 21-31
Mathieu M, Fillinger S, Felenbok B (2000) In vivo studies of upstream regulatory cis-
acting elements of the alcR gene encoding the transactivator of the ethanol regulon in Aspergillus nidulans. Mol Microbiol 36: 123-131
Menne S, Walz M, Kück U (1994) Expression studies with the bidirectional pcbAB-pcbC
promoter region from Acremonium chrysogenum using reporter gene fusions. Appl Microbiol Biotechnol 42: 57-66
Myszka DG, Jonsen MD, Graves BJ (1998) Equilibrium analysis of high affinity
interactions using BIACORE. Anal Biochem 265: 326-330 Neveu B, Belzile F, Bélanger RR (2007) Cloning of the glyceraldehyde-3-phosphate
dehydrogenase gene from Pseudozyma flocculosa and functionality of its promoter in two Pseudozyma species. Antonie Van Leeuwenhoek
Punt PJ, Dingemanse MA, Kuyvenhoven A, Soede RD, Pouwels PH, van den Hondel CA
(1990) Functional elements in the promoter region of the Aspergillus nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 93: 101-109
Radzio R, Kück U (1997) Efficient synthesis of the blood-coagulation inhibitor hirudin in
the filamentous fungus Acremonium chrysogenum. Appl Microbiol Biotechnol 48: 58-65
Rauscher R, Wurleitner E, Wacenovsky C, Aro N, Stricker AR, Zeilinger S, Kubicek CP,
Penttilä M, Mach RL (2006) Transcriptional regulation of xyn1, encoding xylanase I, in Hypocrea jecorina. Eukaryot Cell 5: 447-456
Ronne H (1995) Glucose repression in fungi. Trends Genet 11: 12-17

Janus et al. 2007B
19
Ruijter GJ, Visser J (1997) Carbon repression in Aspergilli. FEMS Microbiol Lett 151:
103-114 Sambrook J, Russell DW (2001) Molecular cloning. A laboratory manual. In: (eds.) Cold
Spring Harbor Laboratory Press, Cold Spring Habor, New York pp. Schaufler LE, Klevit RE (2003) Mechanism of DNA binding by the ADR1 zinc finger
transcription factor as determined by SPR. J Mol Biol 329: 931-939 Schmitt EK, Hoff B, Kück U (2004a) Regulation of cephalosporin biosynthesis. In:
Brakhage AA (eds.) Adv Biochem Engin/Biotechnol. Springer Verlag, Berlin, Heidelberg pp. 1-43
Schmitt EK, Bunse A, Janus D, Hoff B, Friedlin E, Kürnsteiner H, Kück U (2004b)
Winged helix transcription factor CPCR1 is involved in regulation of beta-lactam biosynthesis in the fungus Acremonium chrysogenum. Eukaryot Cell 3: 121-134
Shevchuk NA, Bryksin AV, Nusinovich YA, Cabello FC, Sutherland M, Ladisch S (2004)
Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res 32: e19
Shi C, Kaminskyj S, Caldwell S, Loewen MC (2007) A role for a complex between
activated G protein-coupled receptors in yeast cellular mating. Proc Natl Acad Sci U S A 104: 5395-5400
Sirand-Pugnet P, Santos C, Labarere J (2003) The Aa-Pri4 gene, specifically expressed
during fruiting initiation in the Agrocybe aegerita complex, contains an unusual CT-rich leader intron within the 5' uncoding region. Curr Genet 44: 124-131
Strauss J, Horvath HK, Abdallah BM, Kindermann J, Mach RL, Kubicek CP (1999) The
function of CreA, the carbon catabolite repressor of Aspergillus nidulans, is regulated at the transcriptional and post-transcriptional level. Mol Microbiol 32: 169-178
Studier FW (2005) Protein production by auto-induction in high density shaking cultures.
Protein Expr Purif 41: 207-234 van Hartingsveldt W, Mattern IE, van Zeijl CM, Pouwels PH, van den Hondel CA (1987)
Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet 206: 71-75
Walz M, Kück U (1991) Polymorphic karyotypes in related Acremonium strains:. Curr
Genet 19: 73-76 Walz M, Kück U (1993) Targeted integration into the Acremonium chrysogenum genome:
disruption of the pcbC gene. Curr Genet 24: 421-427 Ward M, Turner G (1986) The ATP synthase subunit 9 gene of Aspergillus nidulans:
sequence and transcription. Mol Gen Genet 205: 331-338

Janus et al. 2007B
20
Ward M, Wilson LJ, Carmona CL, Turner G (1988) The oliC3 gene of Aspergillus niger: isolation, sequence and use as a selectable marker for transformation. Curr Genet 14: 37-42
Warner JB, Lolkema JS (2003) CcpA-dependent carbon catabolite repression in bacteria.
Microbiol Mol Biol Rev 67: 475-490 Yamazaki T, Hasebe T, Kajiwara S, Shishido K (2000) Structure and function of a
pyrimidine/purine-biased sequence from the 5'-flanking region of the basidiomycete Lentinus edodes gene priA. Mol Gen Genet 263: 262-270
Zeilinger S, Schmoll M, Pail M, Mach RL, Kubicek CP (2003) Nucleosome transactions
on the Hypocrea jecorina (Trichoderma reesei) cellulase promoter cbh2 associated with cellulase induction. Mol Genet Genomics 270: 46-55
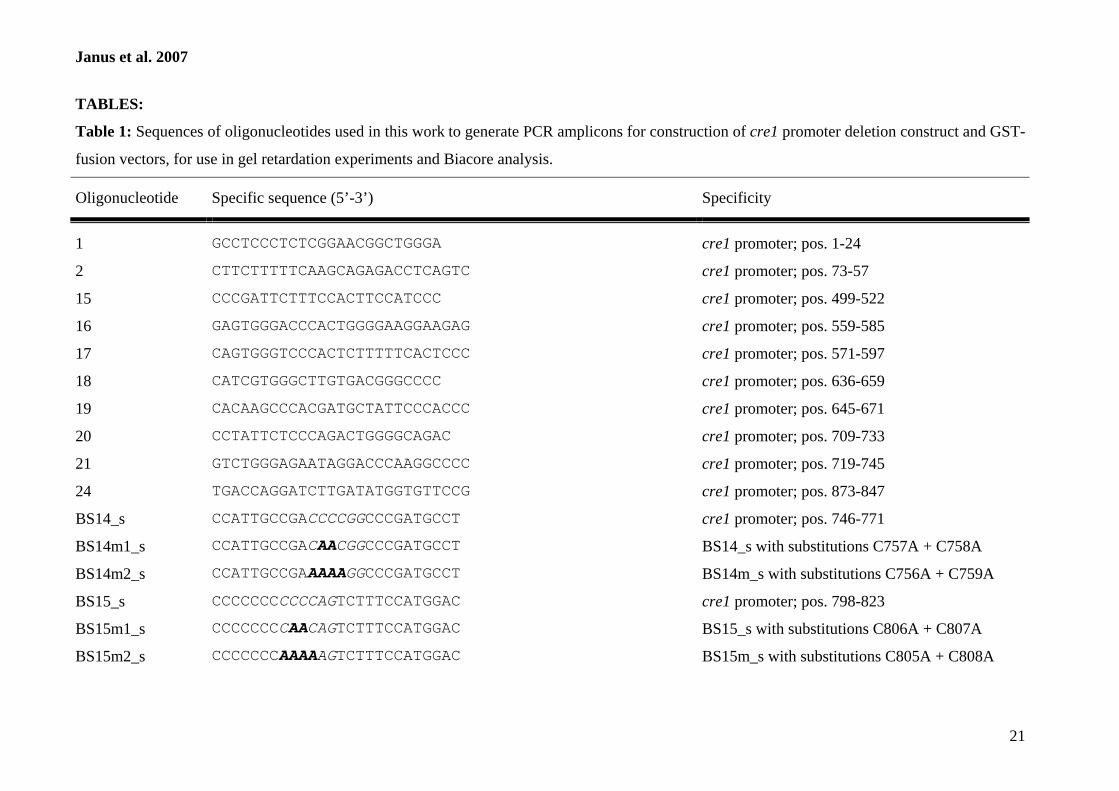
Janus et al. 2007
21
TABLES:
Table 1: Sequences of oligonucleotides used in this work to generate PCR amplicons for construction of cre1 promoter deletion construct and GST-
fusion vectors, for use in gel retardation experiments and Biacore analysis. Oligonucleotide
Specific sequence (5’-3’)
Specificity
1
GCCTCCCTCTCGGAACGGCTGGGA
cre1 promoter; pos. 1-24
2 CTTCTTTTTCAAGCAGAGACCTCAGTC cre1 promoter; pos. 73-57
15 CCCGATTCTTTCCACTTCCATCCC cre1 promoter; pos. 499-522
16 GAGTGGGACCCACTGGGGAAGGAAGAG cre1 promoter; pos. 559-585
17 CAGTGGGTCCCACTCTTTTTCACTCCC cre1 promoter; pos. 571-597
18 CATCGTGGGCTTGTGACGGGCCCC cre1 promoter; pos. 636-659
19 CACAAGCCCACGATGCTATTCCCACCC cre1 promoter; pos. 645-671
20 CCTATTCTCCCAGACTGGGGCAGAC cre1 promoter; pos. 709-733
21 GTCTGGGAGAATAGGACCCAAGGCCCC cre1 promoter; pos. 719-745
24 TGACCAGGATCTTGATATGGTGTTCCG cre1 promoter; pos. 873-847
BS14_s CCATTGCCGACCCCGGCCCGATGCCT cre1 promoter; pos. 746-771
BS14m1_s CCATTGCCGACAACGGCCCGATGCCT BS14_s with substitutions C757A + C758A
BS14m2_s CCATTGCCGAAAAAGGCCCGATGCCT BS14m_s with substitutions C756A + C759A
BS15_s CCCCCCCCCCCAGTCTTTCCATGGAC cre1 promoter; pos. 798-823
BS15m1_s CCCCCCCCAACAGTCTTTCCATGGAC BS15_s with substitutions C806A + C807A
BS15m2_s CCCCCCCAAAAAGTCTTTCCATGGAC BS15m_s with substitutions C805A + C808A

Janus et al. 2007
22
neg_Pcre1_s GAGAGCTGACTGGACTGCTGACTGAG cre1 promoter (pos. 38-63)
3148 ACCGGTGCCTCCCTCTCGGAACGGCTGGG cre1 promoter (pos. 1- 23) + AgeI
3149 CCATGGGTGGTCCACCAGCTCTATTTGCG cre1 promoter (pos. 1008-1030) + NcoI
Pcre1-D1 ACCGGTTCACAAGCCCACGATGCTA cre1 promoter (pos. 644-662) + AgeI
Pcre1-D3 ACCGGTGTACTGCCTCCCGATTCTTTC cre1 promoter (pos. 490-510) + AgeI
Pcre1-D4 ACCGGTTGGGAGAATAGGACCCAAG cre1 promoter (pos. 722-740) + AgeI
cre1-as GACTGCGATCGTTGCGGCAT cre1 gene (pos. 1-20)
cre1ko-5 CTCATTACCACACATCCAGTTTCG A.c. genomic DNA in front of cre1 promoter
Pcre1-D5-s CTATTCCCACCCCCCGCGCACGTTCTGGCCGGAACACCATATCAAG cre1 promoter (pos. 660-677 + 836-863)
Pcre1-D5-as GTGTTCCGGCCAGAACGTGCGCGGGGGGTGGGAATAGCATCGTGGGC cre1 promoter (pos. 650-677 + 836-854)
Pcre1-D6-s CCCCGCGCTTTTGCATTCAAGTCTGCCCCAGTCTGGGAGAATAGGACC cre1 promoter (pos. 671-688 + 707-736)
Pcre1-D6-as CCCAGACTGGGGCAGACTTGAATGCAAAAGCGCGGGGGGTGGGAATAGC cre1 promoter (pos. 659-688 + 707-725)
Pcre1-D7-s TTTTTTTTTTTAAGTCTGCCACGTTCTGGCCGGAACACCATATCAAG cre1 promoter (pos. 696-714 + 836-863)
Pcre1-D7-as TGTTCCGGCCAGAACGTGGCAGACTTAAAAAAAAAAAAAAAAAAGAATGC cre1 promoter (pos. 683-714 + 836-853)
Pcre1-D4*-s CGCTCTTTTTAAACCACCACGTCCCCCCCAAAAAGTCTTTCCATGGACC cre1 promoter (pos.776-824)
Pcre1-D4*-as GGGACGTGGTGGTTTAAAAAGAGCGTGGGAGGCATCGGGCCTTTTTCGGC cre1 promoter (pos.800-751)
3074 GAATTCATGCCGCAACGATCGCAGTG cre1 gene (pos. 1-20) + EcoRI
cre1_NotI GCGGCCGCTTAGACAAATGACGAGTACGTG cre1 gene (pos. 1561-1579) + stop codon + NotI
3011 TGTCGATCGACCACGACGACCACC cre1 promoter (pos. 933-956)
CRE1koanti GACGCCCTGGTGCTGAGGAGCGATGCCC cre1 gene (pos. 663-636) aNucleotide positions are from accession no. AJ245727. b Only the sense strand is given for each double-stranded DNA probe, used in gel retardation analysis and Biacore assays. Restriction sites are underlined. Nucleotide substitutions are shown in bold face. CRE1 consensus binding sites (SYGGRG) are shown in italics.
Janus et al. 2007
23
Table 2: Plasmids used for in vivo reporter gene assays and protein synthesis. Plasmid
Characteristics Reference
pRHN1
gpd promoter, DsRed gene (aa 1-226) separated by a NcoI site and trpC terminator of A. nidulans
Godehardt and Kück (unpublished data)
pCRE13/2
cre1 promoter and cre1 gene (aa 1-406) of Acremonium chrysogenum
Jekosch and Kück (2000)
pCRECEFD2
cre1 promoter with deletion of bp 488-687 and cefEF gene of A. chrysogenum
Ogrodowczyk and Kück (unpublished data)
pRPcre1
cre1 promoter, DsRed gene (aa 1-226) separated by a NcoI site and trpC terminator of A. nidulans
This work
pRPcre1_D1
pRPcre1 with deletion of bp 1-643 of the cre1 promoter
This work
pRPcre1_D2
pRPcre1 with deletion of bp 488-687 of the cre1 promoter
This work
pRPcre1_D3
pRPcre1 with deletion of bp 1-489 of the cre1 promoter
This work
pRPcre1_D4
pRPcre1 with deletion of bp 1-721 of the cre1 promoter
This work
pRPcre1_D5
pRPcre1 with deletion of bp 678-835 of the cre1 promoter
This work
pRPcre1_D6
pRPcre1 with deletion of bp 689-706 of the cre1 promoter
This work
pRPcre1_D7
pRPcre1 with deletion of bp 715-835 of the cre1 promoter
This work
pRPcre1_D4*
pRPcre1_D4 with substitutions in BS14 (bp 756-759) and BS15 (bp 805-808)
This work
pGEXcre1_183
cre1 fragment (aa 1-182) in pGEX-4-T1 (GE Healthcare / Amersham Biosciences)
This work

Janus et al. 2007
24
Table 3: Recipient and transgenic strains used in this study.
Strain
Characteristics
References
ATCC 14553
Wild-type strain
-
ATCC:pRPcre1 cre1(p)::DsRed::trpC(t); trpC(p)::hph This work
ATCC:D1 cre1(p)Δ1-643::DsRed::trpC(t); trpC(p)::hph This work
ATCC:D2 cre1(p)Δ488-687::DsRed::trpC(t); trpC(p)::hph This work
ATCC:D3 cre1(p)Δ1-489::DsRed::trpC(t); trpC(p)::hph This work
ATCC:D4 cre1(p)Δ1-721::DsRed::trpC(t); trpC(p)::hph This work
ATCC:D4* cre1(p)Δ1-721_C756-759A_C805-808A::DsRed::trpC(t); trpC(p)::hph
This work
ATCC:D5 cre1(p)Δ678-835::DsRed::trpC(t); trpC(p)::hph This work
ATCC:D6 cre1(p)Δ689-706::DsRed::trpC(t); trpC(p)::hph This work
ATCC:D7 cre1(p)Δ715-835::DsRed::trpC(t); trpC(p)::hph This work
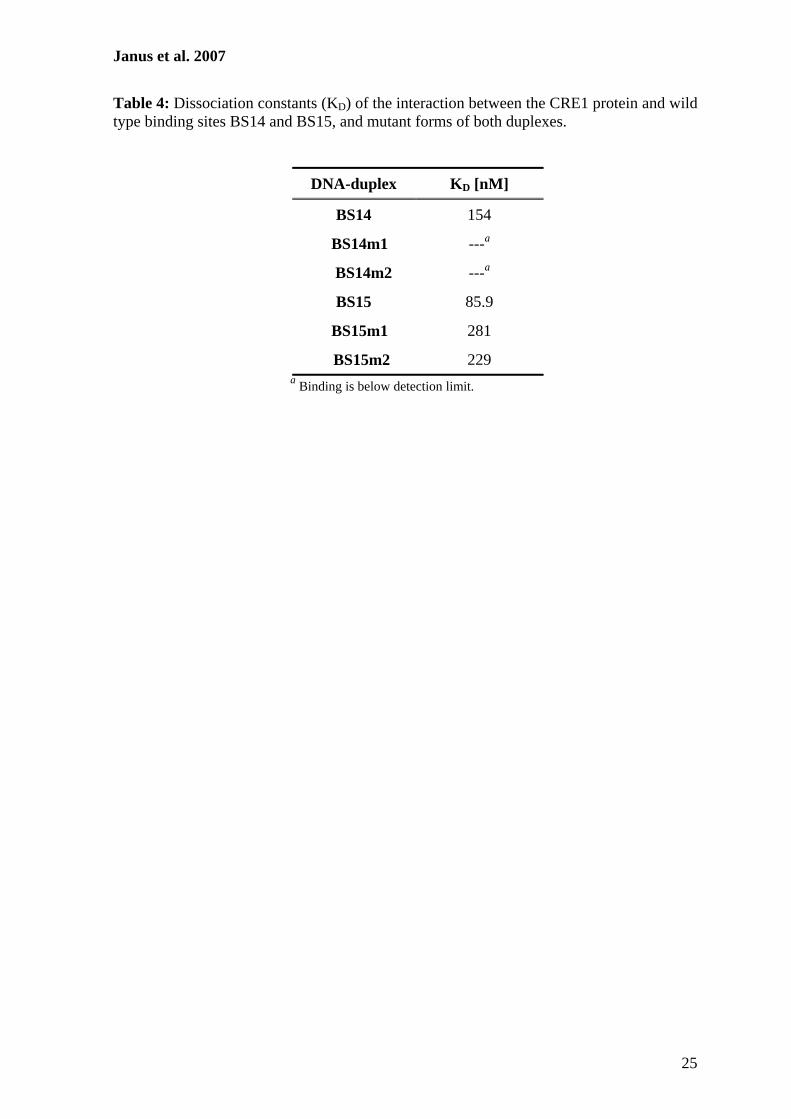
Janus et al. 2007
25
Table 4: Dissociation constants (KD) of the interaction between the CRE1 protein and wild type binding sites BS14 and BS15, and mutant forms of both duplexes.
DNA-duplex
KD [nM]
BS14
154
BS14m1
---a
BS14m2
---a
BS15
85.9
BS15m1
281
BS15m2
229
a Binding is below detection limit.
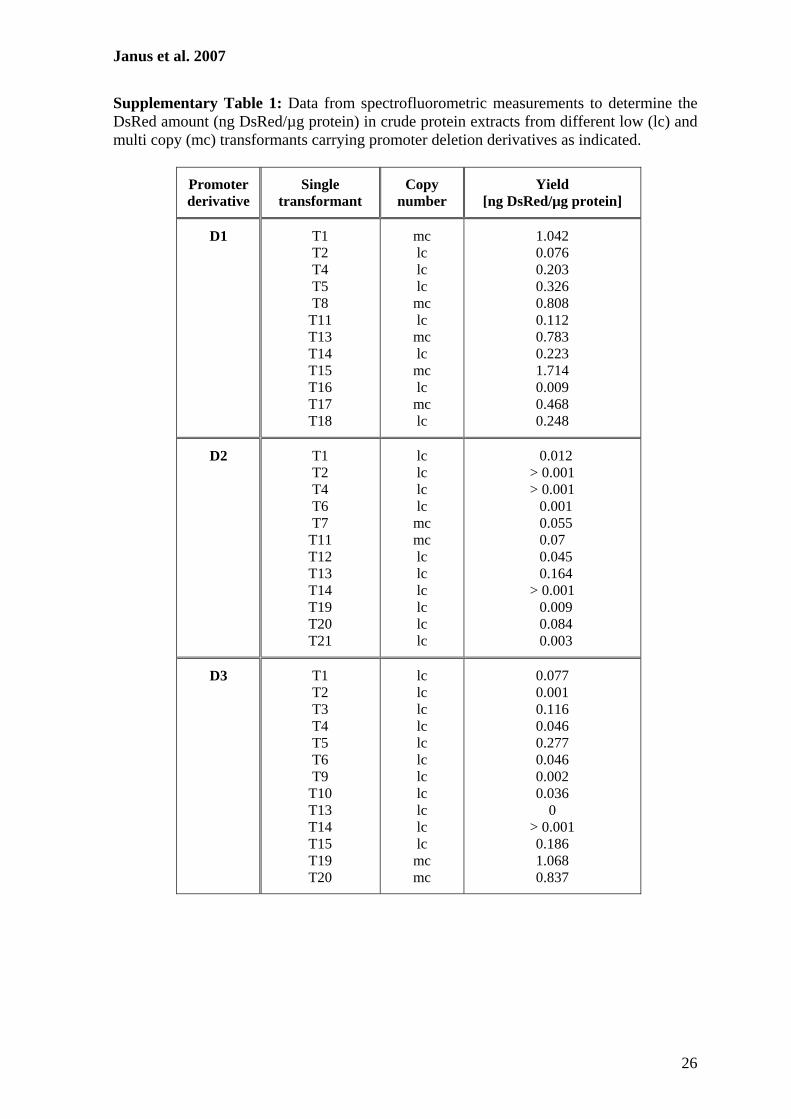
Janus et al. 2007
26
Supplementary Table 1: Data from spectrofluorometric measurements to determine the DsRed amount (ng DsRed/µg protein) in crude protein extracts from different low (lc) and multi copy (mc) transformants carrying promoter deletion derivatives as indicated.
Promoter derivative
Single
transformant
Copy
number
Yield
[ng DsRed/µg protein]
D1
T1 T2 T4 T5 T8
T11 T13 T14 T15 T16 T17 T18
mc lc lc lc mc lc mc lc mc lc mc lc
1.042 0.076 0.203 0.326 0.808 0.112 0.783 0.223 1.714 0.009 0.468 0.248
D2
T1 T2 T4 T6 T7
T11 T12 T13 T14 T19 T20 T21
lc lc lc lc mc mc lc lc lc lc lc lc
0.012 > 0.001 > 0.001 0.001 0.055
0.07 0.045 0.164 > 0.001 0.009 0.084 0.003
D3
T1 T2 T3 T4 T5 T6 T9
T10 T13 T14 T15 T19 T20
lc lc lc lc lc lc lc lc lc lc lc mc mc
0.077 0.001 0.116 0.046 0.277 0.046 0.002 0.036
0 > 0.001 0.186 1.068 0.837
Janus et al. 2007
27
D4
T1 T2 T3 T4 T5 T6 T7 T9
T10 T11 T13 T14
lc lc mc mc lc lc lc mc mc mc lc lc
0.098 0.574
2.12 0.477 0.003 0.001 0.345
1.98 1.86
0.522 0.087
0.75
D5
T1 T2 T5 T7 T8
T10 T11 T12 T13 T15 T16 T23
mc mc lc lc lc mc lc lc lc lc mc lc
0
0.022 0.001
0 0
0.012 0 0
0.004 0.019
0 0.001
D6
T1 T3 T4 T5 T6 T8
T10 T11 T12 T13 T16 T18 T19 T20
lc lc lc lc mc lc lc lc lc mc mc lc mc lc
0.16
0.171 0.176 0.127 0.633 0.113 0.001 0.011 0.057
0.86 0.663 0.014 1.267 0.518
D7
T2 T3 T4 T5 T6 T7 T8 T9
T10 T11
lc lc lc mc mc mc lc lc lc lc
> 0.001 0.003
0 0.027 0.051 0.034 0.013 0.013 0.005 0.006

Janus et al. 2007
28
FIGURES:
Figure 1: Expression of the DsRed reporter gene under control of the cre1 promoter in
multi (T13, T17) and low (T2) copy transformants. (A) Northern blot hybridization was
performed using the DsRed gene as a probe. (B) Spectrofluorometric measurements of the
DsRed protein in A. chrysogenum total protein extracts. +G and -G indicate that strains
were grown in the presence or absence of glucose. The wild type strain served as a
negative control.
Figure 2: Schematic drawing of the wild type cre1 promoter (Pcre1) and different
promoter deletion derivatives. All promoter fragments were fused to the DsRed reporter
gene. Numbers on the right represent red coloured transformants (1st number) within the
total number of analyzed transformants (2nd number). The putative CRE1 binding sites
(bars), a T-stretch (T), a CT-rich region (CT), as well as the transcriptional start point
(arrow) are indicated.
Figure 3: Impact of glucose on DsRed expression. (A) Northern blot hybridization
analysis with RNAs from low (lc) and multi copy (mc) transformants grown with (+G) and
without (-G) glucose. Radiolabeled DsRed served as hybridization probe and the rRNA
served for standardization for the RNA loaded. (B) Spectrofluorometric measurements of
the DsRed amount in the transformants as indicated. Strains were grown in the presence
(black bars) or absence (grey bars) of glucose.
Figure 4: Gel shift analysis to determine CRE1 binding sites. (A) Schematic map of the
cre1 promoter region as already shown in Figure 2. PCR fragments used in electromobility
shift analysis (EMSA) with the CRE1 protein are indicated with numbers that represent
primers for PCR amplification. (B) Gel retardation analysis of the CRE1 protein together
with PCR fragments 1-2 and 21-24. The recombinant GST::CRE1 (CRE1) protein purified
from E. coli and an E. coli control protein (GST) were incubated with radiolabeled PCR
fragments. (C) EMSA with PCR fragment 21-24 and increasing amounts of the
GST::CRE1 fusion protein.
Figure 5: Electromobility shift analysis with primers BS14 and BS15. (A) 5’-sequence of
deletion derivative D4. Putative CRE1 binding sites BS14 and BS15 are underlined, the
transcriptional start point is marked by an arrow and the start codon of the open reading

Janus et al. 2007
29
frame is shown in italics. Shaded sequences were used for gel retardation analysis and SPR
assays, and substitutions of the putative binding sequence are given below the sequence.
(B) Analysis of protein-DNA interactions with radiolabeled oligonucleotides BS14 and
BS15 and the recombinant GST::CRE1 protein. (C) Gel retardation analysis with
oligonucleotides BS14 and BS15 with increasing amounts of recombinant protein as
indicated.
Figure 6: Surface plasmon resonance analysis. (A) Coomassie blue-stained protein gel
with highly purified CRE1 protein (lane 1). Molecular weight markers are indicated on the
site. (B) Analysis of steady state affinity of CRE1 binding to BS14, BS14m1 and BS14m2
determined using SPR measurements. (C) Steady state affinity of CRE1 binding to BS15,
BS15m1 and BS15m2 determined using SPR measurements. The graphs show the
response units (RU) in comparison to the protein concentration.
Figure 7: Impact of nucleotide substitutions within CRE1 binding sites on DsRed
expression. Northern blot hybridization with RNA from the wild type strain and from
selected transformants, harbouring plasmids pRPcre1 (Pcre), pRPcre1_D4 (D4) or
pRPcre1_D4* (D4*). RNA was isolated from strains that were grown in the presence (+G)
or absence of glucose (-G). Hybridization was done with a radiolabelled DsRed probe.
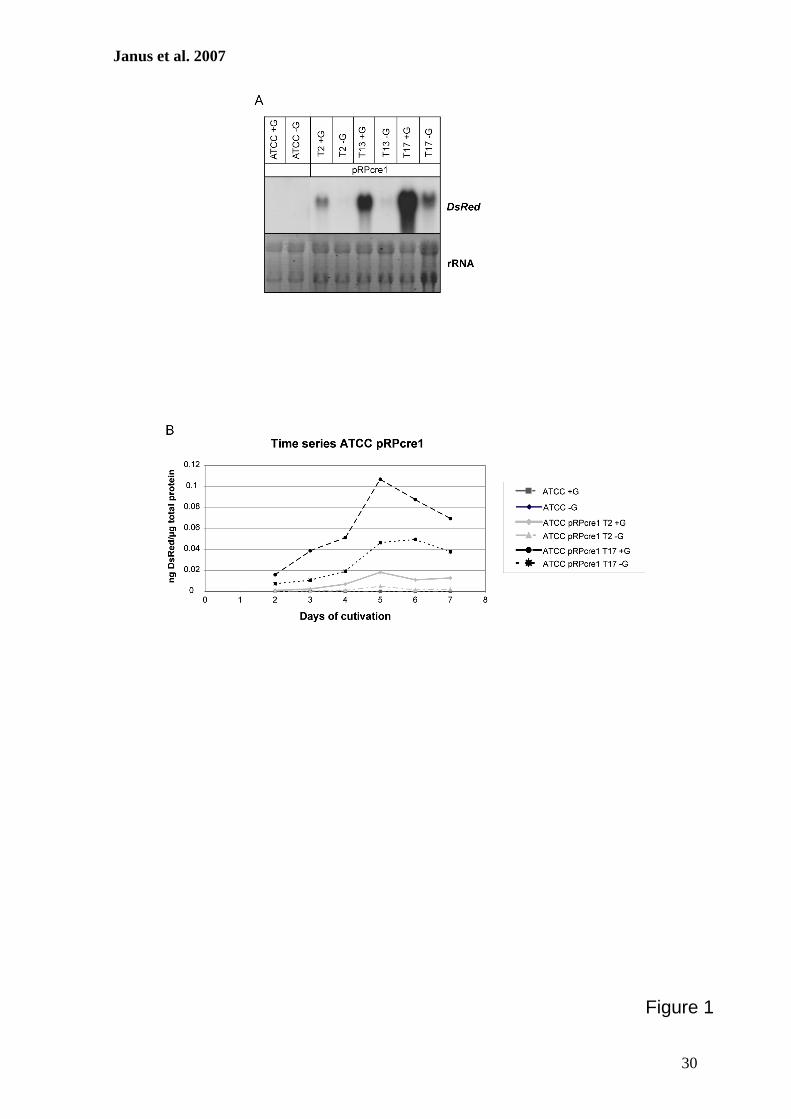
Janus et al. 2007
30
Figure 1

Janus et al. 2007
31
Figure 2
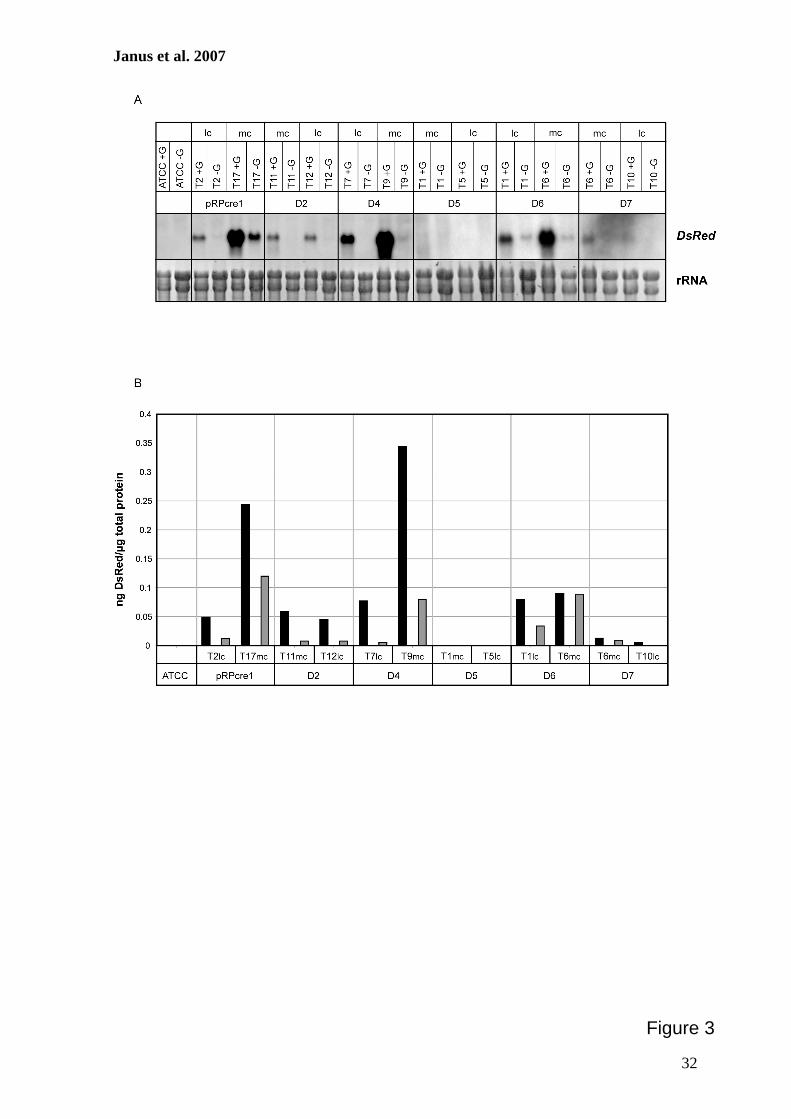
Janus et al. 2007
32
Figure 3
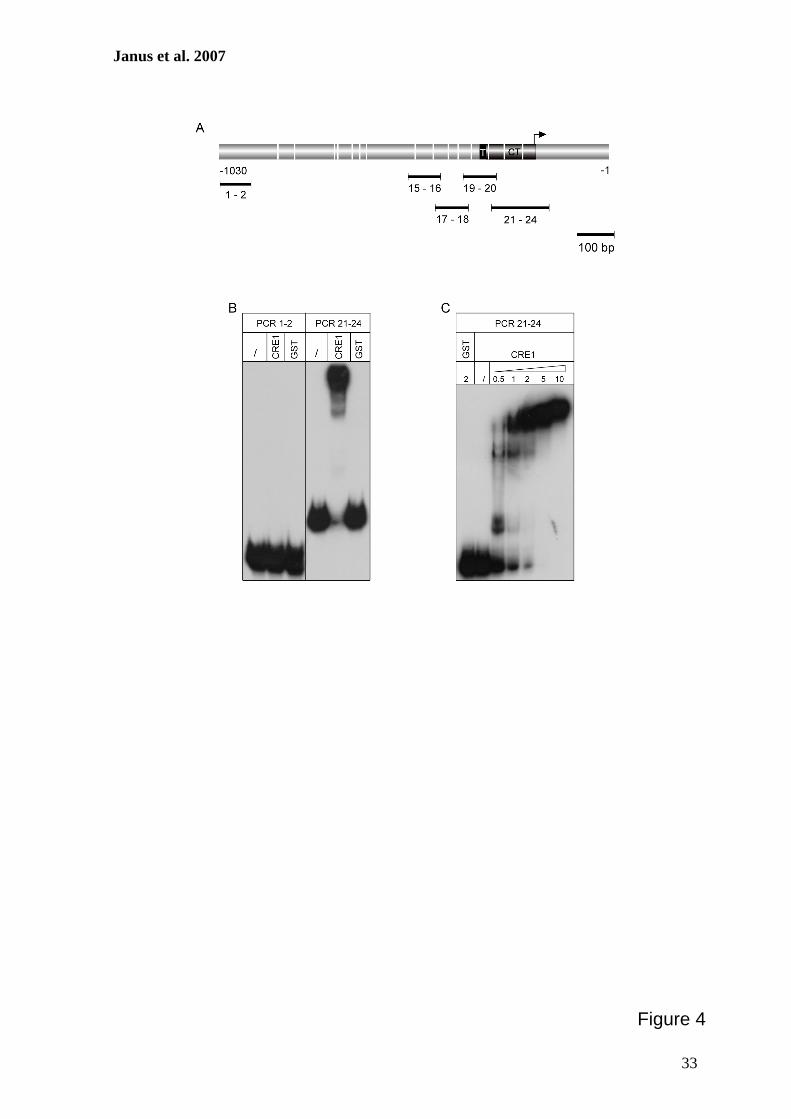
Janus et al. 2007
33
Figure 4
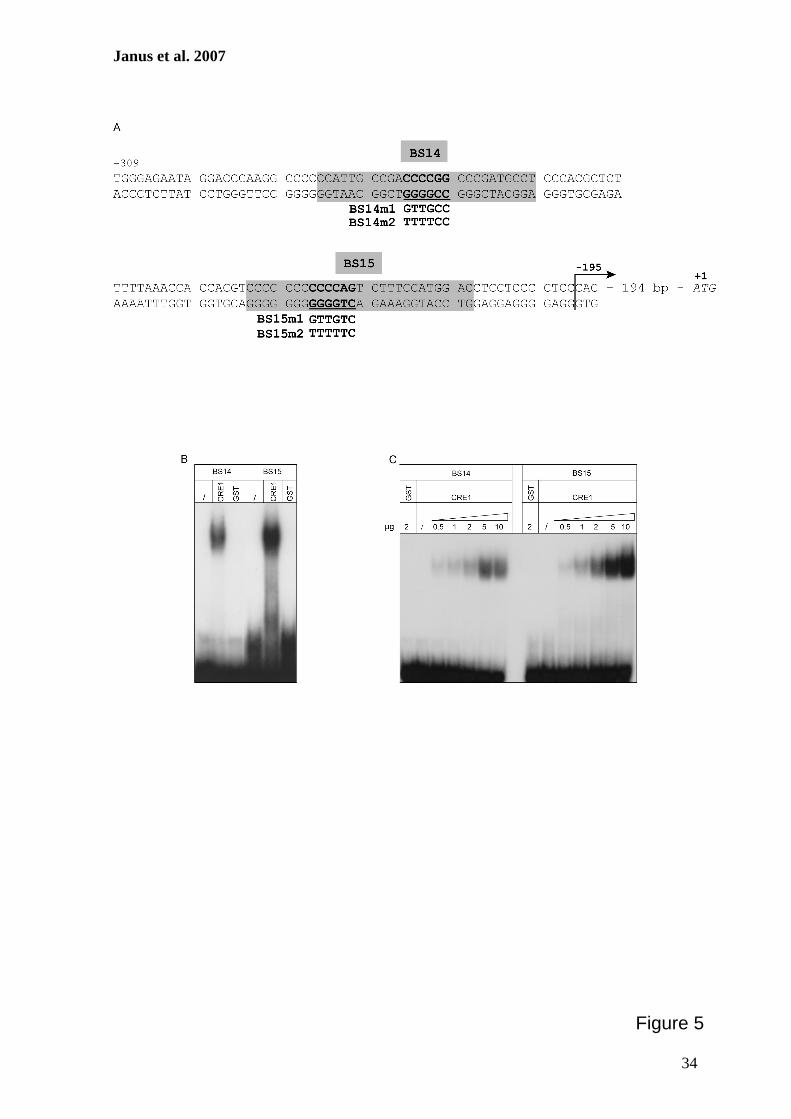
Janus et al. 2007
34
Figure 5
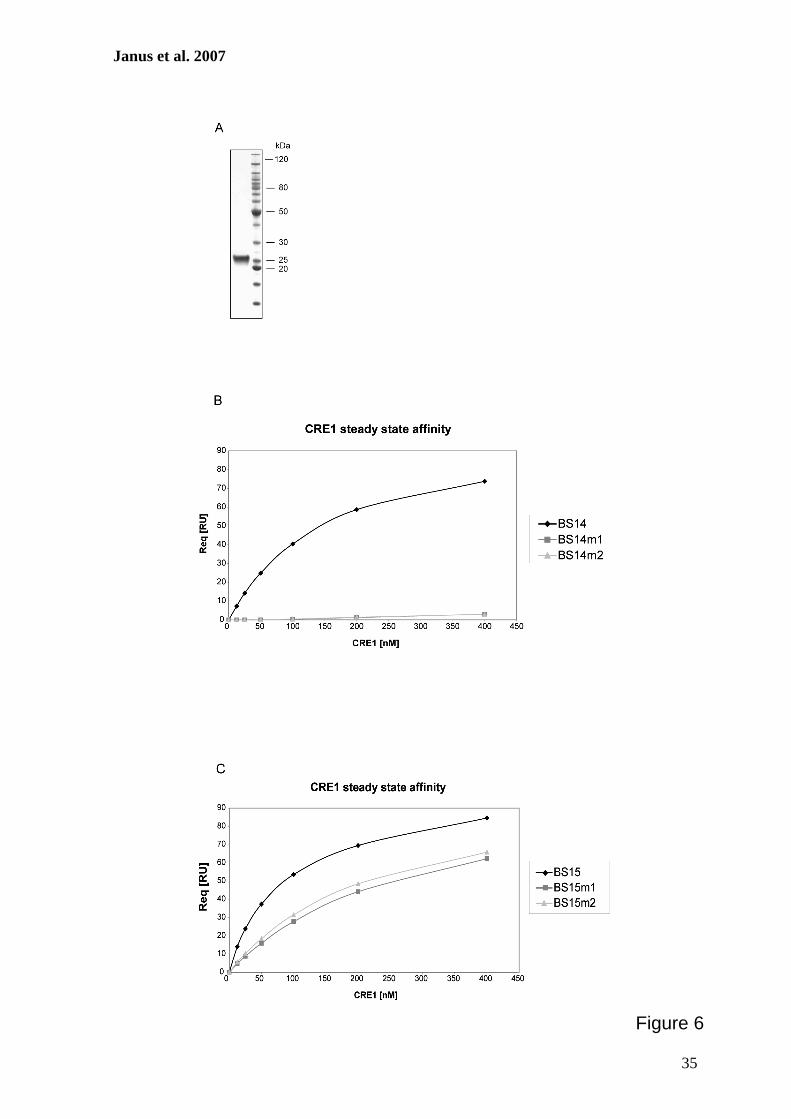
Janus et al. 2007
35
Figure 6

Janus et al. 2007
36
Figure 7

V. GESAMTDISKUSSION
25
V. GESAMTDISKUSSION Der imperfekte Hyphenpilz Acremonium chrysogenum hat aufgrund seiner Fähigkeit zur
Synthese des β-Laktam-Antibiotikums Cephalosporin C eine große wirtschaftliche
Bedeutung in der Biotechnologie und Pharmazie. Daher besteht eine gesteigerte
Motivation zur gezielten Stammoptimierung, im Speziellen zur Erhöhung der Synthese-
leistung und Verbesserung der Anzuchtbedingungen. Zahlreiche Komponenten bilden
hierbei ein komplexes Regulationssystem, somit steht die Analyse regulatorischer Faktoren
der Biosynthese im Vordergrund. Mit der Etablierung des RNAi-Systems sowie der
detaillierten Analyse des Promotors des Transkriptionsfaktor-kodierenden cre1-Gens unter
Verwendung des DsRed-Gens als Reporter stellt diese Arbeit einen Beitrag zur
Erweiterung des angewandten Methodenspektrums dar. Im Rahmen der Gesamtdiskussion
sollen die in dieser Arbeit vorgestellten Inhalte in den Kontext der aktuellen Forschung
gestellt werden. Zudem werden Perspektiven dargestellt, die sich aus den erzielten Daten
ergeben.
1. Etablierung eines RNAi-Systems in A. chrysogenum Die Analyse von Genfunktionen in Hyphenpilzen ist in den letzten Jahren stark
fortgeschritten. Viele verschiedene Ansätze zur Zerstörung eines Zielgens durch homologe
Rekombination wurden hierzu etabliert (Weld et al. 2006). In den meisten Hyphenpilzen
erfolgt die Substitution eines Zielgens durch homologe Rekombination aufgrund einer
bevorzugten ektopischen Integration der zu transformierenden DNA eher selten. Für
Wildtyp-Stämme von A. nidulans, N. crassa und S. macrospora wurden homologe
Rekombinationsraten von 0,1 bis 5 % beschrieben (Chaveroche et al. 2000, Colot et al.
2006, Pöggeler und Kück 2006). In den letzten Jahren konnten einige knockout-Stämme in
A. chrysogenum generiert werden (Walz und Kück 1993, Schmitt et al. 2004c, Hoff et al.
2005, Dreyer et al. 2007). Allerdings war dies aufgrund der sehr geringen Frequenz
(< 0,1 – 1 %) homologer Rekombination immer eine sehr zeitaufwändige und
arbeitsintensive Aufgabe. Die Herstellung von sogenannten multiplen knockout-Stämmen
benötigt weiterhin mehrere Resistenzmarker zur Selektion individueller Transformanten,
die homologe Rekombinationen aufweisen (Fitzgerald et al. 2004). Die Entdeckung und
fortwährende Entwicklung von RNAi-Systemen bietet aus diesem Grund eine sichere
Alternative zur Analyse von Genfunktionen, auch in größerem Maßstab. In C. elegans

V. GESAMTDISKUSSION
26
konnte beispielsweise durch die Verwendung von 20.326 dsRNAs eine Genom-weite
RNAi-Analyse zur Identifizierung von Genen, die an der Embryogenese beteiligt sind,
durchgeführt werden (Sönnichsen et al. 2005). Die RNAi-Methode bietet viele Vorzüge im Vergleich zur Herstellung eines knockout-
Stammes. So können mit Hilfe von RNAi-Stämmen, Gene analysiert werden, deren
vollständiger Verlust letal wirken kann. Die Reduktion der Expression mit Hilfe von RNAi
ermöglicht somit ein Überleben des Pilzes und gleichzeitig die Analyse dieses essentiellen
Gens. Um diese Vorzüge auch für A. chrysogenum zu nutzen, wurde im Rahmen dieser
Arbeit ein einfaches und zuverlässiges RNAi-System erfolgreich für A. chrysogenum
etabliert (siehe Kapitel III). Die Analyse der Genfunktion mittels RNAi benötigt eine
Vorselektion von Transformanten, da diese häufig variable RNAi-Effizienzen aufweisen
(Liu et al. 2002). Zur Vereinfachung der Selektion werden daher häufig Vektorsysteme
konstruiert, die das simultane Silencing eines Reportergens zusammen mit dem zu
analysierenden Gen ermöglichen. Solche RNAi-Systeme nutzen entweder stamm-
spezifische Marker, die phänotypisch identifiziert werden können, wie beispielsweise das
pks-Gen, dessen Genprodukt an der Melaninbiosynthese beteiligt ist (Liu et al. 2002,
Goldoni et al. 2004, Moriwaki et al. 2007), oder alternativ das gfp-Reportergen, welches
häufig mittels Fluoreszenzmikroskopie nachgewiesen wurde (Fitzgerald et al. 2004, de
Jong et al. 2006). Im Fall des Versuchsorganismus A. chrysogenum wurde ein RNAi-
System basierend auf dem DsRed-Reportergen entwickelt, da die Expression des DsRed-
Gens anhand der Färbung des Myzels beobachtet werden kann. Insofern stellt dieses
Reportergen einen geeigneten Marker zur Identifizierung von Transformanten, die einen
RNAi-Effekt aufweisen, dar.
Zur Herstellung eines RNAi-Vektors, der eine spezifische Herunterregulation der
Expression des DsRed-Reportergens in einem DsRed-exprimierenden Rezipientenstamm
von A. chrysogenum bewirkt, wurden ca. 430 bp des Gens in „sense“ und „antisense“
Orientierung in den Vektor pSilent-1 kloniert. Dieser RNAi-induzierende Vektor wurde
bereits erfolgreich in den Hyphenpilzen M. oryzae und C. lagenarium eingesetzt. Multiple
Klonierungsstellen, die zur Integration von Ziel-DNA-Fragmenten genutzt werden können,
flankieren hier das Cutinase-Intron 2 aus M. oryzae. Während der Erstellung des Vektors
pSilent-1 wurden verschiedene Intron-Bereiche als Abstandshalter zwischen invers
orientierten Fragmenten von Zielgenen getestet, aber Vektoren mit dem Cutinase-Intron 2

V. GESAMTDISKUSSION
27
zeigten die effizientesten RNAi-Effekte (Nakayashiki et al. 2005). Generell steigert die
Anwesenheit von Intron-Sequenzen in verschiedenen RNAi-induzierenden Vektoren die
Effektivität des Silencing und fördert die Stabilität des Vektors während der Klonierung in
E. coli (Smith et al. 2000, Goldoni et al. 2004, Nakayashiki et al. 2005). Es wird vermutet,
dass das Entfernen des Introns vom Konstrukt durch das Spliceosome dazu beiträgt, das
Zusammenfügen der beiden komplementären RNA-Stränge innerhalb der Haarnadel-
struktur zu fördern (Smith et al. 2000). Ein genereller Vorteil von RNAi-Systemen ist, dass kurze Sequenzhomologien
ausreichend sind, um ein Silencing von Zielgenen zu erzeugen. So reichen in C. elegans
Oligonukleotide von 25 bp Länge aus, um RNAi zu induzieren (Sönnichsen et al. 2005). In
Pilzen werden 132 – 500 bp verwendet, um einen effizienten RNAi-Effekt zu erhalten
(Cogoni et al. 1996, Liu et al. 2002, Weld et al. 2006). Das bedeutet, dass selbst
Sequenzdaten aus EST-Datenbanken verwendet werden können, um RNAi-Vektoren
herzustellen, da die Identifizierung flankierender Bereiche, wie Promotor und Terminator,
nicht benötigt wird (McDonald et al. 2005). Im Vergleich dazu werden zu Herstellung von
knockout-Stämmen meist wesentlich mehr Sequenzdaten (> 1 kb), vor allem auch aus der
flankierenden Region, zur homologen Rekombination benötigt (Weld et al. 2006). In dieser
Arbeit konnte gezeigt werden, dass 430 bp des DsRed-Gens zu einem starken Silencing-
Effekt in dem A. chrysogenum Rezipientenstamm führen. Von den insgesamt 478
erhaltenen Transformanten zeigten mehr als 70 % eine Reduktion des roten Phänotyps zu
rosafarbenen oder „farblosen“ Stämmen. Vergleichende Analysen von DsRed-mRNA und
DsRed-Protein ließen die Schlussfolgerung zu, dass der Farb-Phänotyp gut mit der
Genexpression auf Transkript- und Proteinebene korreliert. Transformanten mit rotem
Phänotyp wiesen die stärksten DsRed-Transkriptlevel auf, wohingegen „farblose“
Transformanten die geringste DsRed-Synthese zeigten (siehe Kapitel III).
Auf Grundlage dieser Ergebnisse wurde anschließend analysiert, ob auch die Expression
endogener Gene mittels RNAi beeinflusst werden kann. Hierzu wurde ein RNAi-Vektor
konstruiert, der sowohl Fragmente des DsRed-Reportergens, als auch des pcbC-
Biosynthesegens trägt, das für die Isopenicillin N-Synthase aus A. chrysogenum kodiert.
Vorausgegangene Analysen haben verdeutlicht, dass die Inaktivierung des pcbC-Gens zur
signifikanten Reduktion des Cephalosporin C-Titers führt (Walz und Kück 1993).

V. GESAMTDISKUSSION
28
Das Antibiotikum Cephalosporin C inhibiert das Wachstum von sowohl gram-positiven,
als auch gram-negativen Bakterien (Schmitt et al. 2004a). Während der Biosynthese wird
Cephalosporin C in den Kulturüberstand sezerniert, so dass dieser für einen Hemmhoftest
mit dem Indikatorbakterium Alcaligenes faecalis verwendet werden kann. Somit stellt das
pcbC-Gen einen repräsentativen Indikator dar, um die Frage zu klären, ob mit Hilfe des
RNAi-Systems endogene Gene aus A. chrysogenum analysiert werden können. Der für
diese Analysen hergestellte RNAi-induzierende Vektor führte ebenfalls zur Identifizierung
von Transformanten, die sowohl „farblos“, als auch rosa und rote Färbung zeigten.
Kulturüberstände verschiedenfarbiger RNAi-Transformanten wurden auf ihren
Cephalosporin C-Gehalt mit Hilfe des Hemmhoftests überprüft. Im Allgemeinen zeigte
sich, dass rote Transformanten die größten Hemmhöfe ausbilden, allerdings gab es auch
einige, die im Vergleich zum Hemmhof des Rezipientenstammes reduziert waren.
Rosafarbene Transformanten bildeten Hemmhöfe in allen Größen aus. Die drastischsten
Effekte konnten bei „farblosen“ Transformanten beobachtet werden. Hier wurden sechs
Transformanten ermittelt, die nicht mehr in der Lage waren, das Wachstum des Bakteriums
zu inhibieren. Das bedeutet, dass hier ein vollständiger Verlust der pcbC-Expression
stattgefunden hat. Während der Untersuchung der RNAi-Transformanten wurde
beobachtet, dass die Verringerung der Expression beider Gene (DsRed, pcbC) nicht immer
im gleichen Maße erfolgt, es konnte jedoch eine gewisse Korrelation zwischen Phänotyp
und RNAi-Effizienz ermittelt werden (siehe Abschnitt III). Dabei ist festzuhalten, dass alle
„farblosen“ Transformanten eine mehr oder weniger reduzierte pcbC-Expressionsrate und
IPNS-Synthese besitzen. Ähnliche Effekte zeigten sich auch in anderen Pilzen. In Venturia
inaequalis erfolgte in allen Transformanten eine Suppression der Zielgene, die für das GFP
und die Trihydroxynaphthalenreduktase der Melanin-Biosynthese kodieren. Real-time
PCR Analysen verdeutlichten jedoch, dass die Expressionsraten der beiden Gene auch hier
nicht im selben Umfang reduziert wurden (Fitzgerald et al. 2004). Obwohl in Cryptococcus
neoformans 80 % aller Transformanten eine reduzierte Expression beider Zielgene (ADE2,
CAP59) zeigten, gab es einige Transformanten, die nur einen einzigen regulierten
Phänotyp besaßen (Liu et al. 2002). Eine mögliche Erklärung dieser ungleichmäßigen
RNAi-Effizienzen ist eine mangelhafte Ausbildung des Stammes der Haarnadelstruktur
innerhalb der RNA. Dies führt zu einer Reduktion der Länge der dsRNA und folglich zu
Minderung der Effizienz des knockdown-Effektes. Kürzlich konnte am Beispiel von

V. GESAMTDISKUSSION
29
N. crassa gezeigt werden, dass Verlängerungen des Stammes der Haarnadelstruktur der
albino-1 RNA zu einer Steigerung der Anzahl von Transformanten mit veränderten
Phänotypen führen (Goldoni et al. 2004). Die erzielten Ergebnisse verdeutlichen, dass das DsRed-Reportergen ein guter Indikator
der RNA-Interferenz in A. chrysogenum darstellt. Zudem konnte verdeutlicht werden, dass
die entwickelte Methode das simultane Silencing mehrerer Gene zulässt und eine effiziente
Vorselektion von geeigneten Transformanten ermöglicht. Simultanes Silencing
verschiedener Gene hat den Vorteil, dass beispielsweise nur ein einziges Konstrukt zur
Transformation benötigt und somit die Anzahl der benötigten Selektionsmarker gering
gehalten wird. Ein weiterer Vorteil des RNAi-Systems besteht darin, dass beispielsweise
mehrere unterschiedliche Gene einer Genfamilie zeitgleich reguliert werden können.
Hierbei werden zur Erstellung von RNAi-induzierenden Vektoren Sequenzen verwendet,
die einzigartig für diese Genfamilie sind und alle Mitglieder teilen. So konnte im Mohn,
Papaver somniferum, beispielsweise das vorletzte Enzym der Morphin-Biosynthese,
welches von einer Multigenfamilie kodiert wird, effektiv durch die Anwendung eines
RNAi-induzierenden Vektors beeinflusst werden (Allen et al. 2004).
2. Das RNAi-System kann zur Aufklärung regulatorischer Komponenten des
Sekundärmetabolismus beitragen Das während dieser Arbeit etablierte RNAi-System gewährleistet die schnelle Analyse
neuartiger Regulatoren des Sekundärmetabolismus und bietet das Potential, viele Faktoren
sukzessiv funktionell zu untersuchen. Das Anlegen sogenannter RNAi Vektor-
Bibliotheken kann zur Genom-weiten Untersuchung von großem Nutzen sein. Zur
schnellen Herstellung dieser Bibliotheken können kommerziell erhältliche in vitro-
Rekombinationstechnologien zur Hochdurchsatz-Klonierung verwendet werden (Weld et
al. 2006). In vergleichbaren Ansätzen in Säugerzellen wurden lentivirale RNAi-
Bibliotheken zur „loss-of-function“ Rasterung im Genom-weiten Maßstab genutzt. Die
Effektivität dieser Bibliothek konnte verdeutlicht werden, in dem mit Hilfe von 1.000
Genen aus der Bibliothek nach Regulatoren der mitotischen Progression in humanen
Dickdarm-Krebszellen gesucht wurde (Root et al. 2006).
Der Einsatz von RNA-Silencing in Mikroorganismen bietet somit eine potentielle
Ergänzung zu konventionellen Stammoptimierungsprogrammen, bei denen UV-Strahlung

V. GESAMTDISKUSSION
30
oder chemische Reagenzien zur Herstellung von Stämmen mit neuen Eigenschaften
genutzt werden. Erste Erfolge in dieser Richtung konnten mit Hilfe des RNAi-Systems
bereits in einigen Hyphenpilzen erzielt werden. In Mortierella alpina, dem kommerziellen
Produzenten der Arachidonsäure, konnte beispielsweise durch Anwendung von RNAi die
Menge der Zwischenprodukte der Fettsäureproduktion gesteigert werden. Das Ziel dieser
Untersuchung war das Δ-12-Desaturase-Gen, welches für ein Schlüsselenzym des
Biosyntheseweges der Arachidonsäure kodiert. Das Herunterregulieren dieses Gens führte
zur Akkumulation von n-9 Fettsäuren, die für gewöhnlich nicht in Wildtyp-Stämmen
gemessen werden können. Dadurch wird postuliert, dass RNAi das Potential besitzt die
Biosynthese von seltenen und wirtschaftlich nützlichen Komponenten vorantreiben zu
können. Hierzu könnten mit Hilfe von RNAi verschiedene Gene für Enzyme des Fettsäure-
Biosynthesewegs herunterreguliert werden, was möglicherweise zur Synthese seltener
Zwischenprodukten führt (Takeno et al. 2005).
In verschiedenen Aspergillus- und Fusarium-Spezies war es möglich die Mykotoxinmenge
zu reduzieren, um Kontaminationen von Lebensmitteln zu vermeiden. Die Transformation
von RNAi-induzierenden Vektoren, die Sequenzen von Genen von Mykotoxin-
spezifischen Regulatoren führte zur Suppression der Mykotoxinproduktion (McDonald et
al. 2005).
Pflanzenpathogene Hyphenpilze produzieren während der Infektion von Getreide und Hülsenfrüchten oft toxische Sekundärmetabolite, so genannte Mykotoxine, die zu direkten (z.B. Ertragsverlust) und indirekten (z.B. Gesundheitsverlust) Kosten in der Land-wirtschaft, Tierzucht und menschlichen Gesundheit beitragen. Diese Mykotoxine werden durch den Verzehr kontaminierter Produkte, wie z.B. Getreideprodukte aufgenommen (McDonald et al. 2005).
Die in dieser Arbeit erzielten Ergebnisse verdeutlichen, dass der Sekundärmetabolismus
des Hyphenpilzes A. chrysogenum durch Verminderung der Expression von Genen, welche
die Cephalosporin C-Biosynthese kontrollieren, analysiert werden kann (siehe Kapitel III).
In vorausgegangenen Arbeiten konnte durch die Deletion des pcbC-Gens eine Mutante
erhalten werden, die keinerlei antibiotische Aktivität aufwies (Walz und Kück 1993). Der
gleiche Phänotyp wurde während dieser Arbeit bei 15 % der Transformanten, die durch
„Cosilencing“ entstanden sind, erzielt. So ist vorstellbar, dass das RNAi-System
signifikant dazu beitragen kann, Signalkaskaden des Sekundärmetabolismus in
A. chrysogenum zu entschlüsseln. Die Hemmung der Expression von Repressoren könnte
in einer gesteigerten Synthese des Antibiotikums und folglich zu Stämmen mit erhöhter

V. GESAMTDISKUSSION
31
Produktivität führen. Die kontrollierte Expression von RNAi-Kassetten, z.B. durch
induzierbare Promotoren, kann zur Verringerung der Expression von Zielgenen während
spezifischer Stadien der Entwicklung oder der Biosynthese genutzt werden. So können
entsprechende RNAi-Stämme zunächst „normal“ wachsen, ab einem bestimmten Zeitpunkt
der Biosynthese oder bestimmter Stadien der Entwicklung erfolgt dann die Induktion des
Promotors der RNAi-Kassette, so dass erst ab diesem definierten Zeitpunkt RNAi
hervorgerufen wird (Goldoni et al. 2004). Ein effektives, induzierbares System bestehend
aus einem RNAi-Vektor, dessen Expression vom Promotor des Cellobiohydrolase B-Gens
aus Aspergillus fumigatus vermittelt wird, konnte hierzu kürzlich beschrieben werden.
Dieser Promotor zeigt in Gegenwart von Carboxymethylcellulose eine Aktivierung der
Genexpression, wodurch es zur RNAi-Induktion kommt. Entsprechende Transformanten
zeigten verringerte Transkriptmengen des Zielgens bis hin zu „loss-of-function“-
Phänotypen. In Anwesenheit von Glukose erfolgt eine Repression der Expression des
RNAi-Konstruktes, so dass die Transformanten Wildtyp-ähnliche Transkriptmengen und
Phänotypen aufweisen (Bromley et al. 2006).
Neben den hier dargestellten Vorteilen von RNAi-Systemen konnte ferner beobachtet
werden, dass selbst Gene mit geringen Expressionsraten mit Hilfe von RNAi effizient
herunterreguliert werden können (Goldoni et al. 2004). Der größte Vorteil des RNAi-
Systems wird jedoch durch die Analyse von Genen, die unerlässlich für das pilzliche
Wachstum sind, geboten. In solchen Fällen kann ein fast vollständiges Silencing der
Genexpression von Zielgenen zum Überleben der Zellen und zur funktionellen Analyse der
Gene beitragen, wohingegen der vollständige Verlust des Gens letal wirken würde. Dies
kann z.B. zum Verständnis der Funktion globaler und vor allem essentieller Faktoren
beitragen, die beispielsweise an der Zellwandbiosynthese oder der Morphogenese beteiligt
sind und gegebenenfalls einen Einfluss auf die Cephalosporin C-Biosynthese haben. Zusammenfassend konnte während dieser Arbeit eine quantitative und qualitative
Untersuchung pilzlicher Transformanten durchgeführt werden, bei denen die Expression
mehrerer Gene zeitgleich herunterreguliert wurde. Die erfolgreiche, simultane Inhibierung
des heterologen DsRed- und des endogenen pcbC-Gens zeigt, dass das RNAi-System ein
wirkungsvolles „Werkzeug“ zur Analyse des Sekundärmetabolismus in A. chrysogenum
darstellt. Ferner ist es eine zuverlässige Alternative zu Gen-knockout-Experimenten, die
gegenwärtig nur mit geringer Effizienz in A. chrysogenum durchgeführt werden können.

V. GESAMTDISKUSSION
32
Das hier vorgestellte RNAi-System in Kombination mit anderen Methoden kann zur
Beschleunigung der Genom-weiten Analysen von A. chrysogenum beitragen und
Stammoptimierungen dieses industriell bedeutsamen Hyphenpilzes fördern.
3. Die CT-reiche Region des cre1-Promotors ist verantwortlich für eine
erhöhte Genexpression und interagiert mit dem Glukoserepressor CRE1 In einem zweiten Ansatz sollte im Rahmen dieser Arbeit die Promotorregion des cre1-
Gens mit Hilfe des DsRed-Reportergens funktionell analysiert werden. Das cre1-Gen aus
A. chrysogenum kodiert für einen Glukoserepressor, der unter anderem für die Repression
der Transkription verschiedener Gene des Cephalosporin C-Biosyntheseweges
verantwortlich ist. Vorausgegangene Analysen in A. chrysogenum führten des weiteren zu
der Vermutung, dass die cre1-Expression selbst autoreguliert ist. Eine erhöhte cre1-
Transkription erfolgt, wenn Glukose im Kulturmedium vorhanden ist, somit scheint es sich
hierbei um eine positive Autoregulation zu handeln (Jekosch und Kück 2000a, Jekosch und
Kück 2000b). Letzteres widerspricht Ergebnissen, die in Trichoderma reesei und
Aspergillus nidulans beobachtet werden konnten. Der Transkriptlevel der cre1/creA-
Expression ist in diesen Pilzen in Gegenwart von Glukose reduziert. Anhand von Protein-
DNA-Interaktionsstudien konnte experimentell gezeigt werden, dass diese negative
Autoregulation durch die Bindung der CRE-Proteine an mehrere Konsensusbindestellen in
ihren eigenen Promotoren vermittelt wird (Ilmén et al. 1996a, Strauss et al. 1999).
Die Promotoren von cre1-Orthologen aus verschiedenen Pilz-Spezies weisen einige
Übereinstimmungen in den entsprechenden Sequenzen auf. So beinhalten die creA-
Promotoren aus A. niger und A. nidulans große Bereiche CT-reicher Sequenzen
einschließlich langer Abschnitte von polyT-Abfolgen. Zusätzlich finden sich mehrere
CREA-Bindestellen mit der für CRE-Proteine identifizierten Konsensussequenz
5’-SYGGRG-3’. Diese CRE-Konsensussequenz wurde während der Analyse von Genen,
die an dem Ethanol-Metabolismus von A. nidulans beteiligt sind, mit Hilfe von DNase I
footprinting-Untersuchungen identifiziert. Sie können ebenfalls in weiteren Promotoren
von Genen, die eine Glukose-abhängige Expression aufweisen, vorgefunden werden
(Dowzer und Kelly 1989, Kulmburg et al. 1993, Cubero und Scazzocchio 1994).
CT-reiche Regionen wurden auch in den cre1-Promotoren verschiedener Trichoderma-
Spezies entdeckt. Sie befinden sich direkt stromabwärts von putativen TATA-Boxen, die

V. GESAMTDISKUSSION
33
Anwesenheit von polyT-Abfolgen konnte hier allerdings nicht beobachtet werden. Die
Konsensussequenzen für die Bindung des CRE1-Proteins erstrecken sich über einen
Bereich von 700 bis 950 bp stromaufwärts der Protein-kodierenden Region (Ilmén et al.
1996a).
Ähnliche Sequenzcharakteristika konnten auch in der Sequenz des cre1-Promotors von
A. chrysogenum identifiziert werden. Dazu gehören eine CT-reiche Region von ca. 150 bp,
sowie ein charakteristischer 18 bp langer T-Bereich. Zusätzlich konnten 15 putative CRE1-
Bindestellen mittels in silico-Analysen ermittelt werden, die über einen 1030 bp langen
Promotorbereich verteilt vorliegen (Jekosch und Kück 2000a). Zur Identifizierung, von für
die transkriptionelle Aktivität essentiellen Regionen und zur näheren Charakterisierung der
cre1-Autoregulation in A. chrysogenum wurden im Rahmen dieser Arbeit molekular-
biologische Analysen der 5’-flankierenden Region des cre1-Gens durchgeführt. Hierzu
wurden sieben Deletionsderivate angefertigt, bei denen eine unterschiedliche Anzahl
verschiedener CRE1-Konsensussequenzen und charakteristischer Bereiche, wie die CT-
reiche Region und die T-Abfolge, entfernt wurden (siehe Abb. 2). Alle Promotorderivate
wurden mit dem DsRed-Gen als Reporter fusioniert und die Konstrukte in A. chrysogenum
transformiert.
Für gewöhnlich erfolgt während der DNA-vermittelten Transformation von Hyphenpilzen
die ektopische Integration der zu transformierenden DNA an verschiedene Orte und in
multiplen Kopien ins Genom. Während der Analyse von Genexpressionen darf daher nicht
außer Acht gelassen werden, dass diese sogenannten Positionseffekte die Ergebnisse der
Analysen verfälschen können. Um diesen Effekt zu vermeiden können
Transformationssysteme verwendet werden, die eine Lokus-abhängige Insertion der zu
transformierenden DNA ins Genom gewährleisten. So ein homologes Transformations-
system konnte beispielsweise mit Hilfe des Gens der Orotidin-5’-Phosphat Carboxylase
(pyrG) in A. niger etabliert werden (van Hartingsveldt et al. 1987).
Dieses System basiert auf einen Stamm, der eine Mutation im pyrG-Gen aufweist und einen Vektor, der das funktionelle pyrG-Gen als Selektionsmarker trägt. Eine Integration der DNA an den pyrG-Lokus erfolgte in 90 % der Fälle, wobei entweder das mutierte durch das Wildtyp-Allel ersetzt (60 %) oder ein bis mehrere Kopien an den Ort integriert wurden (40 %) (van Hartingsveldt et al. 1987).
Eine weitere Möglichkeit bietet die Lokus-unabhängigen Integration von Plasmid-DNA
durch die Verwendung von selbst-replizierenden Vektoren, die sogenannte AMA1

V. GESAMTDISKUSSION
34
(autonomous maintenace in Aspergillus) replizierende Elemente aus A. nidulans tragen
(Gems et al. 1991). Diese Sequenzen gewährleisten die extrachromosomale Replikation
von Plasmid-DNA, so dass Positionseffekte ausgeschlossen werden können.
Die Stabilität dieser Vektoren liegt bei 40 bis 60 % auf Selektionsmedium. Vektoren mit A. nidulans AMA1-Sequenzen konnten schon für eine Vielzahl anderer Hyphenpilze, wie verschiedener Aspergillus- und Penicillium-Spezies, Giberella fujikuroi und T. reesei, etabliert werden (Brückner et al. 1992, Aleksenko et al. 1995, Aleksenko und Clutterbuck 1997, Kubodera et al. 2002, Fierro et al. 2004). Erst kürzlich konnte ein episomaler RNAi-Vektor für A. fumigatus konstruiert werden, der AMA1-Sequenzen trägt. Es zeigte sich, dass dieses frei-replizierende Konstrukt in der Lage ist RNAi in A. fumigatus erfolgreich zu induzieren (Khalaj et al. 2007).
Da eine Lokus-abhängige Transformation bzw. freireplizierende Plasmide für
A. chrysogenum bis zum jetzigen Zeitpunkt aber nicht beschrieben sind, musste im
Rahmen dieser Arbeit für jedes Reportergenkonstrukt eine große Anzahl jeweils
unterschiedlicher Transformanten mit ektopisch integrierten Vektoren parallel analysiert
werden (siehe Kapitel IV). Hier stellt das DsRed-Gen einen idealen Reporter dar, der mit
Hilfe einfacher Plattentests die Einteilung der Transformanten mit starker, mittlerer und
geringer Expressionsrate zulässt. Die Analyse der Promotorderivate zeigte, dass Transformanten, bei denen die CT-reiche
Region deletiert vorliegt, keine Glukose-induzierbare Expression des Reporters DsRed
aufweisen (siehe Abb. 2), wohingegen das Deletionsderivat D4, das nur die CT-reiche
Region umfasst, eine starke Glukose-Induzierbarkeit zeigt. Diese Analysen verdeutlichen,
dass die entsprechende Region essentiell für eine Glukose-induzierbare Genexpression ist
(siehe Kapitel IV). Ähnliche Ergebnisse konnten auch für 5’-flankierende Regionen
pflanzlicher Gene beobachtet werden. Eine Mutagenese der CT-reichen Region führte in
vivo zur Reduzierung der Transkription von Transgenen (Bolle et al. 1994). Weiterhin
finden sich CT-reiche Regionen in Promotoren von Hyphenpilzen von stark exprimierten
Genen wie z.B. das gpdA-Gen (Glyceraldehyd-3-Phosphat Dehydrogenase) verschiedener
Aspergillus-Spezies und in 5’-flankierenden Regionen von Genen, denen die klassischen
TATA- und CAAT-Boxen fehlen. Demnach wird den CT-reichen Regionen eine mögliche
Funktion als Transkriptionsaktivatorsequenzen zugesprochen (Punt et al. 1990, Chen und
Roxby 1997, Neveu et al. 2007). Möglicherweise trifft dies auch auf den cre1-Promotor
aus A. chrysogenum zu. Zwar wurden mittels in silico-Analysen von Jekosch und Kück
(2000a) putative TATA- und CAAT-Boxen identifiziert, ihre Position nahe des

V. GESAMTDISKUSSION
35
Translationsstartpunktes ließ jedoch die Vermutung zu, dass sie nicht funktionell sind
(Jekosch und Kück 2000a). Mit der eindeutigen Identifizierung des Transkriptionsstarts
des cre1-Gens durch Primer-Extension-Analysen, welcher 195 bp vor dem
Translationsstart liegt, scheint sich diese Vermutung zu bestätigen (siehe Abb. 2,
Ogrodowczyk 2002). CT-reiche Regionen befinden sich obendrein häufig in der Nähe des
Transkriptionsstartpunktes (Gurr et al. 1987, Yamazaki et al. 2000, Sirand-Pugnet et al.
2003, Fei et al. 2006, Neveu et al. 2007), was auch beim A. chrysogenum cre1-Promotor
ermittelt werden konnte (siehe Abb. 2, Ogrodowczyk 2002).
Abb. 2: Schematische Darstellung des Wildtyp cre1-Promotors (Pcre1) und verschiedener Promotordeletionsderivate (D1 – D7). Alle Promotorfragmente wurden mit dem DsRed-Reportergen fusioniert. Auf der rechten Seite ist die Induzierbarkeit (+/-) durch Glukose angegeben. Weiße Striche verdeutlichen putative CRE1-Bindestellen und schwarze Striche geben putative TATA- und CAAT-Boxen an. Die CT-reiche Region (CT) und der 18 bp T-Bereich (T) sind hervorgehoben (Jekosch und Kück 2000a). Der Transkripstart (-195) ist durch einen Pfeil gekennzeichnet (Ogrodowczyk 2002). Abkürzung: MP: Minimalpromotor

V. GESAMTDISKUSSION
36
In verschiedenen Aspergillus-Spezies führte die Deletion der CT-reichen Region
stromaufwärts des Transkriptionsstarts der Gene gpdA und oliC (Oligomycin-resistente
ATP-Synthase Untereinheit 9) zu einer veränderten Transkriptionsinitiation. Es wird
angenommen, dass diese Region an der ordnungsgemäßen Initiation der Transkription und
an der Bestimmung der Häufigkeit der Transkriptionsinitiation beteiligt ist (Ballance 1986,
Ward und Turner 1986, Ward et al. 1988, Punt et al. 1990). Zusammenfassend konnte
verdeutlicht werden, dass die hier dargestellten Analysen zur Identifizierung eines 114 bp
großen Core-Promotors geführt haben, der sowohl eine starke Expression als auch eine
Glukose-Induzierbarkeit aufweist. Dieser Core-Promotor umfasst einen großen Teil der
CT-reichen Region inklusive zwei putativer CRE1-Konsensussequenzen (siehe Abb. 3).
Die Glukose-Induzierbarkeit impliziert die Autoregulation durch das CRE1-Protein, daher
wurden weitere Analysen des 114 bp großen Core-Promotors zur Identifikation von
möglichen funktionellen CRE1-Bindestellen durchgeführt (siehe Kapitel IV). Die im Rahmen dieser Arbeit durchgeführten Surface Plasmon Resonance (SPR)- und
Gelretentionsanalysen haben verdeutlicht, dass das A. chrysogenum CRE1-Protein direkt
mit der CT-reichen Region im eigenen Promotor interagiert. Innerhalb dieses cre1-
Minimalpromotors konnten zwei spezifische, funktionelle CRE1-Konsensusbindestellen
(BS14 & BS15) ermittelt werden, die 43 bp voneinander entfernt liegen und anscheinend
entscheidend für die Glukose-induzierte Autoregulation sind. Zur Bestimmung der
Spezifität dieser Interaktionen wurden SPR-Analysen mit Oligonukleotiden dieser
Bindestellen durchgeführt, die Substitution von zwei bis vier Basenpaaren von Guanin zu
Thymin innerhalb der Konsensussequenzen aufwiesen. Im Fall von Bindestelle BS14
führte dies zu einem totalen Verlust der Interaktion des CRE1-Proteins und der DNA,
wohingegen die Interaktion von CRE1 mit der Bindestelle BS15 nicht komplett inhibiert
wurde, es konnte allerdings eine Reduktion der Affinität um den Faktor drei ermittelt
werden (siehe Kapitel IV). Detaillierte Sequenzanalysen der Bindestelle BS15 und der
mutierten Bindestelle BS15*, sowie benachbarter Sequenzen verdeutlichten, dass durch die
Substitutionen eine Verlagerung der Konsensusbindestelle im mutierten Oligonukleotid
zum 3’-Ende erfolgte, wobei nur noch ein Nukleotidaustausch erhalten bleibt (siehe
Abb. 3). Die Integration aller Basenpaarsubstitutionen in den cre1-Minimalpromotor D4
und die anschließende Transformation des entstandenen Vektors D4* in A. chrysogenum
führte zu Transformanten, deren DsRed-Expression immer noch durch Glukose induzierbar

V. GESAMTDISKUSSION
37
ist. Diese Analysen verdeutlichen, dass selbst die verschobene „semi-konservierte“
Bindestelle BS15* noch die Glukose-Induktion zulässt (siehe Kapitel IV). Deletionen der
flankierenden Bereiche der Bindestelle BS15 könnten durchgeführt werden, um weitere
Interaktionen des CRE1-Proteins zu verhindern und um einen eindeutigen Beweis zu
liefern, dass das CRE1-Protein der einzige Regulator ist, der die Glukose-Induktion im
Minimalpromotorfragment hervorruft. Die Daten der Protein-DNA-Interaktionsstudien und
der in vivo-Analyse des Minimalpromotorfragmentes D4* stimmen nicht mit Ergebnissen
überein, die während der Analyse der CREA-Interaktion mit dem creA-Promotor in
A. nidulans erzielt wurden. Hier zeigte sich, dass die Glukose-spezifische Abnahme der
creA-Transkription durch eine CREA-abhängige negative Autoregulation aufgrund der
Bindung des Polypeptids an zwei eng benachbarte CREA Bindstellen im creA-Promotor
vermittelt wird (Strauss et al. 1999). Für T. reesei wurde ebenfalls diskutiert, dass nur
Doppel-CRE1-Bindestellen Glukose-Repression vermitteln (Zeilinger et al. 2003). So
konnten für die Promotoren der Xylanase- (xyn1) und Cellobiohydrolase-Gene (cbh1)
jeweils zwei Konsensusbindestellen identifiziert werden, die für die Glukose-Repression
der Genexpression verantwortlich zu sein scheinen (Ilmén et al. 1996b, Mach et al. 1996).
Die im Rahmen dieser Arbeit ermittelten Daten verdeutlichen, dass die CRE1-Interaktion
mit der verschobenen „semi-konservierten“ Bindestelle BS15* (Abb. 3, gelb) zur Glukose-
Induktion der Genexpression ausreicht.
Abb. 3 : Schematische Darstellung der Interaktion des CRE1-Proteins mit den Bindestellen BS14 und BS15 (weiße Striche) des Minimalpromotorfragmentes D4 und dem Minimalpromotor-fragment D4*, das Basenpaarsubstitutionen in den BS14 und BS15 aufweist (rote Striche). Die verschobene „semi-konservierte“ Bindestelle BS15* ist durch einen gelben Balken dargestellt. Abkürzung: CT: CT-reiche Region

V. GESAMTDISKUSSION
38
Es ist nicht auszuschließen, dass weitere Proteine mit der CT-reichen Region des cre1-
Promotors interagieren und somit an der Regulation der Expression des cre1-Gens beteiligt
sind. Für den Promotor des piypt1-Gens, welches für ein monomeres G-Protein aus
Phytophthora infestans kodiert, wurde beschrieben, dass die CT-reiche Region von zwei
verschiedenen, nicht genauer identifizierten Proteinen gebunden wird (Chen und Roxby
1997). Ein Hinweis, dass zusätzliche Regulatoren auch mit dem cre1-Promotor in
A. chrysogenum interagieren und regulierend wirken, wird durch die Tatsache verstärkt,
dass das Deletionsderivat D2, dem nur ca. 200 bp vor der CT-reichen Region fehlen, eine
eingeschränkte Expressionsstärke aufweist, obwohl die CT-reiche Region selbst in diesem
Konstrukt vorhanden ist (siehe Kapitel IV). Diese veränderte Regulation kann
beispielsweise dadurch zustande kommen, dass die durch die Mutation eingefügte
Sequenzveränderung zur Herstellung konservierter neuer Bindesequenzen von Proteinen
führt, die mit dem Wildtyp cre1-Promotor eigentlich nicht interagieren würden und somit
auch normalerweise nicht auf die Expression des cre1-Gens wirken. Durch die Deletion im
Promotorderivat D2 kann ferner eine Veränderung der Chromatin-Struktur hervorgerufen
werden, die möglicherweise nicht auftritt, wenn größere Bereiche (wie z.B. in
Deletionsderivat D4) deletiert werden. Für den T. reesei Cellulase-Promotor (cbh2) konnte
der Einfluss des CRE1-Polypeptids auf die Positionierung der Nucleosomen in der
5’-regulatorischen Region des cbh2-Gens ermittelt werden (Zeilinger et al. 2003), so dass
nicht auszuschließen ist, dass das A. chrysogenum CRE1-Protein an der Nucleosomen-
Positionierung im cre1-Promotor verantwortlich sein kann. Obwohl CRE-Proteine gleiche Bindeeigenschaften aufweisen, werden sie unterschiedlich
reguliert. Der genaue Mechanismus in A. chrysogenum ist hierzu nicht bekannt. Eine
mögliche Erklärung wäre jedoch, dass die gegensätzliche Autoregulation im Vergleich zu
anderen CRE-Proteinen durch unterschiedliche Interaktionspartner hervorgerufen wird.
Weitere Analysen der cre1-Promotordeletionsderivate, vor allem des Minimal-
promotorfragmentes, sowie der CRE1-Bindestellen, in einem cre1-Deletionsstamm
würden weiteren Aufschluss darüber liefern, ob das CRE1-Protein tatsächlich der
Hauptregulator seiner Glukose-induzierten Autoregulation ist. Alle Bemühungen einen
cre1-Disruptionsstamm herzustellen, sind bisher jedoch fehlgeschlagen (Janus und Kück,
unveröffentlicht). Die Herstellung von creA-Deletionsstämmen in A. nidulans führte
zumeist nur zu Transformanten, die Deletionen im creA-Gen aufwiesen, jedoch nicht zum

V. GESAMTDISKUSSION
39
totalen Verlust des Gens. Diese Transformanten zeigen im Vergleich zum Wildtyp-Stamm
eine streng reduzierte Lebensfähigkeit, was sich in einer verminderten Wachstumsrate und
reduzierter Konidienbildung darstellt (Dowzer und Kelly 1991, Shroff et al. 1997). So liegt
die Vermutung nahe, dass eine Deletion des cre1-Gens in A. chrysogenum zur Letalität des
Pilzes führen kann.
Zusammenfassend konnte in diesem Teil der Arbeit durch in vivo-Analysen von sieben
deletierten Promotorfragmenten die Identifizierung eines cre1-Minimalpromotors erfolgen,
dessen Aktivität Glukose-induzierbar ist. Ferner zeigte sich in vitro eine direkte Interaktion
des CRE1-Proteins mit Bereichen dieses Promotorfragments.
4. Der cre1-Promotor besitzt das Potential zur modifizierten Expression von
Cephalosporin C-Biosynthesegenen Kohlenstoffquellen haben einen großen Einfluss auf das pilzliche Wachstum und den
Sekundärmetabolismus von A. chrysogenum. Glukose beispielsweise stellt einen leicht
metabolisierbaren Zucker dar, der zu einem erhöhten Wachstum und einer verringerten
Produktion von Cephalosporin C führt. Die meisten Promotoren der Cephalosporin C-
Biosynthesegene weisen putative Konsensusbindestellen des Glukoserepressors CRE1 auf
(Übersicht bei Schmitt et al. 2004a), des weiteren ist bekannt, dass in Gegenwart von
Glukose die Expression einiger Biosynthesegene reprimiert wird (Jekosch und Kück
2000b). Aufgrund dieser Tatsache werden Fermentationsprozesse häufig in Gegenwart von
Kohlenstoffquellen durchgeführt, die keine Glukose-Repression hervorrufen aber zu einem
langsameren Wachstum des Pilzes führen (Martín et al. 1999).
Die Verwendung des cre1-Promotors stellt neue Ansätze dar, die Regulation der
Cephalosporin C-Biosynthesegene zu verändern. Die im Rahmen dieser Arbeit
durchgeführten Analysen der cre1-Promotordeletionsderivate verdeutlichen, dass die CT-
reiche Region sowohl notwendig, wie auch ausreichend ist, um eine erhöhte Glukose-
induzierbare Genexpressionen zu vermitteln (siehe Kapitel IV). Somit lässt dieses
Minimalpromotorfragment die Möglichkeit zu, Veränderungen der Expression von
Cephalosporin C-Biosynthesegenen durchzuführen, in dem die entsprechende kodierende
Sequenz der Biosynthesegene mit dem cre1-Promotor fusioniert wird. Die cre1-Promotor-
vermittelte Glukose-induzierbare Expression der Cephalosporin C-Biosynthesegene könnte
somit in einer verstärkten Produktivität resultieren, da die Anzucht in Glukose-haltigem

V. GESAMTDISKUSSION
40
Medium zu erhöhten Wachstumsraten führt und sich somit die Biomasse des Pilzes und
daraus resultierend möglicherweise die Ausbeute an sezerniertem Antibiotikum steigert.
Die Repression der endogenen Biosynthesegene könnte eventuell durch die starke,
Glukose-induzierte Expression zusätzlicher Genkopien überdeckt werden. In einer
vorausgegangenen Diplomarbeit, auf der die hier durchgeführten Analysen des cre1-
Promotors aufbauen, konnte bereits verdeutlicht werden, dass die eigentliche Repression
der cefEF-Expression durch den von Glukose aktivierten cre1-Promotor umkehrbar ist
(Ogrodowczyk 2002).
Stammoptimierungen, die auf das Einbringen multipler Kopien verschiedener
Cephalosporin C-Biosynthesegene beruhen, wurden bereits durchgeführt und brachten
unterschiedliche Ergebnisse hervor. Zum einen erfolgte die Transformation und multiple
Integration des pcbC-Gens, hierbei zeigten sich keine signifikanten Effekte auf die
Produktion des Sekundärmetaboliten Cephalosporin C. Folglich handelt es sich hierbei
nicht um einen limitierenden Schritt in der Biosynthese (Skatrud und Queener 1989). Zum
anderen wurden Stämme mit multiplen Kopien der cefEF- und cefG-Gene hergestellt,
wobei eine verstärkte Produktion des Antibiotikums beobachtet wurde. Des weiteren
konnte in diesem Fall eine Korrelation zwischen der cefG-Kopienzahl und dem
Cephalosporin C-Titer detektiert werden. So zeigten vergleichende Analysen mit einem
A. chrysogenum Wildtyp-Stamm und einer Transformante, die fünf Kopien des cefG-Gens
trägt, einen Anstieg der Produktivität um den Faktor drei. Es wurde geschlussfolgert, dass
die Aktivität der vom cefG-Gen kodierten Acetyl-Transferase einen limitierenden Schritt
der Biosynthese darstellt (Mathison et al. 1993). Eine weitere Forschergruppe führte
ähnliche Analysen mit dem cefG-Gen unter Kontrolle des homologen und anderer
heterologer Promotoren in einem Produzentenstamm durch. Auch hier wurde von einer
erhöhten Cephalosporin C-Synthese berichtet (Gutiérrez et al. 1997).
Die Identifizierung des Minimalpromotors mit zwei CRE1-Bindestellen ist das erste
Beispiel einer minimalen induzierbaren regulatorischen Sequenz für den β-Laktam-
Antibiotika-Produzenten A. chrysogenum. Der hier ermittelte cre1-Minimalpromotor stellt
ein ideales Hilfsmittel für die Konstruktion kompakter pilzlicher Expressionsvektoren für
biotechnologisch relevante Pilze dar. Wie bereits erwähnt, kann mit Hilfe dieses
Minimalpromotors die Repression der Cephalosprin C-Biosynthesegene in Gegenwart von
Glukose in eine expressionelle Aktivierung konvertiert werden.

V. GESAMTDISKUSSION
41
5. Das rot-fluoreszierende Protein (DsRed) - eine zuverlässige Alternative zur
Selektion pilzlicher Transformanten Das DsRed-Gen aus Discosoma spec. bietet viele Vorteile, wenn es als Reporter in
Hyphenpilzen verwendet wird. In der vorliegenden Arbeite konnte verdeutlicht werden,
dass dieses Protein einen nützlichen Reporter in RNAi-Systemen und ebenfalls in
Genexpressionsanalysen darstellt (siehe Kapitel III & IV). Die Tatsache, dass die Synthese
des DsRed in A. chrysogenum durch die Färbung des Myzels bereits auf Festmedium
nachvollzogen werden kann, macht es zu einem attraktiven „Werkzeug“ für
weiterführende Analysen. Die Selektion von Transformanten kann anhand der Färbung des
Myzels somit schnell und mühelos, ohne zusätzliche Anwendung molekularbiologischer
Methoden, wie z.B. Northern-Blot Hybridisierungen, durchgeführt werden. Dies ist vor
allem von Vorteil, wenn bei RNAi-Analysen eine visuelle Vorselektion der
Transformanten erfolgen kann, so dass sich die Anzahl zu analysierender Transformanten
reduziert. Die Menge des DsRed-Proteins kann weiterhin einfach mit Hilfe von
Fluoreszenzspektrometrie quantifiziert werden. Hierzu reichen eine schnelle Aufarbeitung
des Gesamtproteinextraktes oder selbst Myzelrohextrakte aus.
Als Alternative könnten auch andere, schneller reifende rot-fluoreszierende Proteine, wie
z.B. das monomere rot fluoreszierende Protein (mRFP), verwendet werden, da die
Expression des mRFP-Gens in A. chrysogenum ebenfalls zur Ausbildung eines roten
Phänotyps führt (Janus und Kück, unveröffentlicht). Allerdings zeigen einige dieser
Varianten im Vergleich zum DsRed eine reduzierte Leuchtkraft oder ihr Einsatz in
Hyphenpilzen ist bisher noch nicht etabliert (z.B. tdTomato) (Shaner et al. 2004, Shu et al.
2006).
Auch bei Promotoranalysen hat sich das DsRed als sinnvoller und zuverlässiger Reporter
erwiesen (siehe Kapitel IV). Untersuchungen von Deletionskonstrukten eines Promotors
können zur Identifizierung von Regionen, die essentiell für die leistungsfähige
Promotoraktivität sind, beitragen, indem zunächst phänotypische Vergleichsanalysen der
Transformanten mit Deletionsderivaten und mit dem Volllängenpromotor durchgeführt
werden. Zusätzlich kann die durch einen schwachen Promotor vermittelte Expression des
DsRed-Gens einfach mit Hilfe sensitiver spektrofluorometrischer Messungen bestimmt
werden. Ein weiterer Vorteil besteht darin, dass kein endogenes Genprodukt mit den
Extinktions- und Emissionsspektren des DsRed interferiert. Bei anderen Reportersystemen,

V. GESAMTDISKUSSION
42
wie z.B. dem lacZ-Gen aus E. coli, wurde oft berichtet, dass endogene β-Galaktosidase-
aktivitäten von Hyphenpilzen störend wirken können. A. chrysogenum weist eine relativ
hohe endogene β-Galaktosidaseaktivität auf, die Enzymsynthese kann jedoch durch
Zugabe von Glukose ins Medium unterdrückt werden (Menne et al. 1994). Folglich wäre
das lacZ-Gen als Reporter für die Glukose-induzierte Genexpression des cre1-Promotors
nicht verwendbar gewesen.
Im Allgemeinen weist die Anwendung von Fluoreszenzproteinen als Reporter mehrere
Vorteile auf. Die Expression des Reporters kann in vivo z.B. mit Hilfe der
Fluoreszenzmikroskopie verdeutlicht werden. Zusätzlich besitzen diese Proteine eine
geringe Größe und benötigen keine Kofaktoren oder Substrate, somit kann die Expression
direkt analysiert werden, wohingegen die Verwendung von Reportergensystemen mit
Enzym-kodierenden Reportergenen in den meisten Fällen eher zeitaufwendig ist (Lorang et
al. 2001). Die Weiterentwicklung verschiedener Varianten von Fluoreszenzproteinen
ermöglicht eine gleichzeitige Verwendung mehrerer Fluoreszenzproteine mit
unterschiedlichen spektralen Eigenschaften, um beispielsweise zeitgleich bidirektionale
Promotoren zu analysieren oder mehrere Proteine innerhalb einer Zelle zu lokalisieren.
In der vorliegenden Arbeit konnte verdeutlicht werden, dass mit Hilfe des DsRed-
Reportergens die phänotypische Charakterisierung pilzlicher Transformanten auf Fest-
medium eine schnelle, erste Bewertung der Reportergenexpression zulässt. Es eignet sich
sowohl als Marker von RNAi-Effekten, als auch zur Analyse von Promotoraktivitäten.

VI. ZUSAMMENFASSUNG
43
VI. ZUSAMMENFASSUNG Die Analyse regulatorischer Elemente des Sekundärmetabolismus biotechnologisch
relevanter Hyphenpilze ist Gegenstand der aktuellen Forschung. Hierbei steht vor allem die
Identifizierung und Modifikation von Faktoren im Vordergrund, die zur Steigerung der
Syntheseleistung beitragen. Der Hyphenpilz Acremonium chrysogenum ist der Haupt-
produzent des β-Laktam-Antibiotikums Cephalosporin C. Aufgrund der geringen
homologen Rekombinationsrate können Genom-verändernde Maßnahmen nur unter
großem Aufwand durchgeführt werden. Die Etablierung neuer Methoden, die zur
vereinfachten Analyse und Manipulation beitragen, verhelfen zur schnelleren
Identifizierung wichtiger, regulatorischer Parameter und können folglich zu effizienteren
Stammoptimierungsprozessen führen.
Zur Etablierung eines RNAi-Systems in A. chrysogenum erfolgte zunächst die Generierung
eines Stammes, der eine starke Expression des DsRed-Gens aufweist, was in der
Rotfärbung des Myzels resultiert. Dieser Stamm wurde anschließend als Rezipient für
RNAi-induzierende Vektoren verwendet. Die Transformation dieses Rezipienten mit
einem RNAi-induzierenden Vektor, der invers orientierte DsRed-Fragmente trägt, führte
zur Identifizierung von Transformanten, die einen Silencing-Effekt in der DsRed-
Expression aufwiesen. Dies zeigte sich in einer Reduktion des roten Phänotyps zu
rosafarbenen und „farblosen“ Transformanten. Mit Hilfe von vergleichenden Northern
Blot-Hybridisierungen, Real-time PCR und spektrofluorometrischen Messungen zur
Bestimmung der DsRed-Proteinmenge konnte bestätigt werden, dass RNAi-Reaktionen in
unterschiedlichen Ausprägungen vorkommen, und dass die RNA-, die Proteinmengen und
der Phänotyp miteinander korrelieren. In weiterführenden Experimenten wurde die
simultane Reduktion der Transkriptlevel des DsRed- und des pcbC-Gens durchgeführt. Das
pcbC-Gen kodiert für die Isopenicillin N Synthase, einem Enzym des Cephalosporin C
Biosyntheseweges. Die Abnahme des pcbC-Transkriptniveaus und daraus resultierend die
Veränderung des Cephalosporin C-Titers konnte einfach mit sogenannten Hemmhoftests
beobachtet werden. Zusätzlich wurden Northern Blot-Hybridisierungen, Real-time PCR
und Wester Blot-Analysen zum Nachweis der Isopenicillin N-Synthase durchgeführt. Die
erzielten Daten zeigten eine ungefähre Korrelation zwischen der phänotypischen
Einteilung der Transformanten (rot, rosafarben, „farblos“) und den Expressionsdaten.
Transformanten, die einen „farblosen“ Phänotyp aufwiesen, zeigten ebenfalls eine

VI. ZUSAMMENFASSUNG
44
Reduktion des pcbC-Transkriptlevels, allerdings erfolgt die Reduktion nicht immer im
gleichen Ausmaß. Das DsRed-Reportergen konnte ferner zur Analyse des cre1-Promotors verwendet werden.
Das cre1-Gen kodiert für einen Glukoserepressor, der in Gegenwart von Glukose
regulierend auf die Expression verschiedener Cephalosporin C-Biosynthesegene wirkt. Die
Transkription des cre1-Gens selbst wird in Gegenwart von Glukose hochreguliert, so dass
Vermutungen über eine positive Autoregulation entstanden. Zur genaueren Bestätigung
dieser Vermutung und zur Identifizierung weiterer regulatorischer Bereiche innerhalb der
cre1-Promotorregion wurden sieben Deletionsderivate hergestellt, in denen
unterschiedliche CRE1-Bindestellen, sowie prägnante Sequenzen (z.B. CT-reiche Region
und T-Abschnitt) entfernt wurden. Die entsprechenden Promotorfragmente kontrollierten
die Expression des DsRed-Reportergens in A. chrysogenum. In vivo-Analysen dieser sieben
Derivate zeigten, dass die CT-reiche Region des cre1-Promotors einen Minimalpromotor
darstellt, der sowohl essentiell als auch ausreichend für die Induzierbarkeit durch Glukose
ist. Um die Fragen zu klären, ob CRE1 an der Glukose-Induzierbarkeit beteiligt ist und
dafür mit Regionen des Minimalpromotors interagiert, wurde ein rekombinantes CRE1-
Protein in E. coli synthetisiert und in Surface Plasmon Resonance (SPR)- und
Gelretentionsanalysen mit PCR-Fragmenten oder Oligonukleotiden eingesetzt. Die
Ergebnisse dieser Untersuchungen verdeutlichten eine direkte Interaktion des CRE1-
Polypeptids mit zwei Bindestellen (BS14 & BS15) innerhalb des cre1-Minimalpromotors. In der vorliegenden Arbeit wurden mit Hilfe des DsRed-Reporters verschiedene Ansätze
zur Analyse und Manipulation des biotechnologisch relevanten Hyphenpilzes Acremonium
chrysogenum etabliert. Ferner konnte ein Minimalpromotorfragment identifiziert werden,
das durch Glukose aktivierbar ist. Das hier ebenfalls vorgestellte RNAi-System wird zur
weiteren Charakterisierung und Modifizierung der Cephalosporin C-Biosynthese und ihrer
Regulatoren großen Beitrag leisten können.

VII. SUMMARY
45
VII. SUMMARY The analysis of regulatory elements of secondary metabolism in biotechnically relevant
filamentous fungi is a major aspect of current research. One focus lies on the identification
and modification of factors that lead to an increase in productivity of the fungi. The
filamentous fungus Acremonium chrysogenum is the main producer of the β-lactam
antibiotic cephalosporin C. Because of a low homologous recombination rate in
A. chrysogenum genome-altering methods are difficult to apply. The establishment of new
techniques leading to an easier analysis and manipulation will contribute to the expeditious
identification of regulatory parameters und will consequently lead to efficient strategies for
strain improvement.
To establish an RNAi system in A. chrysogenum, a strain showing a high level of DsRed
expression was constructed, resulting in red fungal colonies. This strain served as recipient
for RNAi-inducing vectors. The transformation of a silencing vector carrying fragments of
the DsRed gene into this recipient strain resulted in the identification of transformants
showing efficient silencing of DsRed expression. These transformants exhibited at least
three different types of colouring (red, pink, colourless). Using Northern blot
hybridization, real-time PCR and spectrofluorometric measurements to quantify the
amount of DsRed RNA and DsRed protein, respectively, it was demonstrated that
downregulation of gene expression can be observed at different expression levels. The
RNA levels correlated well with the protein levels demonstrating the usefulness of the
DsRed reporter gene. Further experiments showed successful co-silencing of two target
genes, the heterologous DsRed and the endogenous pcbC gene, encoding the isopenicillin
N synthase, an enzyme of the cephalosporin C pathway. The reduction of the pcbC
transcript level and hence alterations in the cephalosporin C titre were analysed with
simple-to-obtain bioassays. In addition, the presence of the isopenicillin N synthase was
investigated by Western blot hybridization. These data clearly demonstrate a rough
correlation between the phenotypic classification (red, pink, colourless) and data from
expression analysis. All transformants having a colourless phenotype showed simultaneous
downregulation of the pcbC gene, albeit at different levels. The DsRed reporter gene was also used to analyse the cre1 promoter. The cre1 gene
encodes a glucose repressor (CRE1) that regulates the expression of several genes from the
cephalosporin C biosynthetic pathway in the presence of glucose. The transcription of the

VII. SUMMARY
46
cre1 gene itself is upregulated in the presence of glucose, leading to the assumption of a
positive autoregulation. Seven promoter deletion derivatives with removals of putative
CRE1 binding sites and other putative regulatory sequences have been generated to test
this model and to further identify regulatory regions within the cre1 promoter. The
corresponding promoter fragments direct the expression of the DsRed reporter gene in
A. chrysogenum. In vivo analysis of these seven promoter derivatives revealed that a
minimal promoter containing the CT-rich region is both necessary and sufficient for
glucose induction. To elucidate whether the CRE1 protein is involved in glucose induction
and therefore interacts with sequences of the minimal promoter, a recombinant CRE1
protein was synthesized in E. coli and used for surface plasmon resonance (SPR) and
gel retardation analysis with PCR fragments or oligonucleotides. The results of these
investigations demonstrated a direct interaction of the CRE1 polypeptide with two binding
sites within the minimal promoter (BS14 & BS15). In the present thesis, the DsRed reporter gene has been used to establish novel techniques
for the analysis and manipulation of the biotechnologically important filamentous fungus
Acremonium chrysogenum. Furthermore, a minimal promoter fragment was identified, that
can be activated by the addition of glucose. The RNAi system presented here will
contribute to further characterization and modification of the cephalosporin C biosynthetic
pathway, as well as its regulators.

VIII. LITERATUR
47
VIII. LITERATUR
Das Literaturverzeichnis beinhaltet nur Zitate der Kapitel I, II und V. Agrawal N, Dasaradhi PV, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK (2003) RNA
interference: biology, mechanism, and applications. Microbiol Mol Biol Rev 67: 657-685 Alcocer MJ, Furniss CS, Kroon PA, Campbell M, Archer DB (2003) Comparison of modular and non-
modular xylanases as carrier proteins for the efficient secretion of heterologous proteins from Penicillium funiculosum. Appl Microbiol Biotechnol 60: 726-732
Aleksenko A, Clutterbuck AJ (1997) Autonomous plasmid replication in Aspergillus nidulans: AMA1 and
MATE elements. Fungal Genet Biol 21: 373-387 Aleksenko AY, Makarova NA, Nikolaev IV, Clutterbuck AJ (1995) Integrative and replicative
transformation of Penicillium canescens with a heterologous nitrate-reductase gene. Curr Genet 28: 474-477
Allen RS, Millgate AG, Chitty JA, Thisleton J, Miller JA, Fist AJ, Gerlach WL, Larkin PJ (2004)
RNAi-mediated replacement of morphine with the nonnarcotic alkaloid reticuline in opium poppy. Nat Biotechnol 22: 1559-1566
Baird GS, Zacharias DA, Tsien RY (2000) Biochemistry, mutagenesis, and oligomerization of DsRed, a
red fluorescent protein from coral. Proc Natl Acad Sci U S A 97: 11984-11989 Baldrian P (2006) Fungal laccases - occurrence and properties. FEMS Microbiol Rev 30: 215-242 Ballance DJ (1986) Sequences important for gene expression in filamentous fungi. Yeast 2: 229-236 Bassford P, Beckwith J, Berman M, Brickman E, Casadaban M, Guarente L, Saint-Girons I, Sarthy A,
Schwartz M, Shuman H, Silhavy T (1978) Genetic fusions of the lac operon: a new approach to the study of biological processes. In: Miller JH, Reznikoff WS (eds.) The Operon. Cold Spring Harbor Laboratory Press, New York pp. 245-261
Beckwith JR (1970) Lac: The genetic system. In: Beckwith JR, Zipser D (eds.) The lactose operon. Cold
Spring Harbor Laboratory Press, New York pp. 5-26 Bevis BJ, Glick BS (2002) Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed). Nat
Biotechnol 20: 83-87 Bhatti HN, Madeeha M, Asgher M, Batool N (2006) Purification and thermodynamic characterization of
glucose oxidase from a newly isolated strain of Aspergillus niger. Can J Microbiol 52: 519-524 Bollag JM, Leonowicz A (1984) Comparative Studies of Extracellular Fungal Laccases. Appl Environ
Microbiol 48: 849-854 Bolle C, Sopory S, Lubberstedt T, Herrmann RG, Oelmuller R (1994) Segments encoding 5'-untranslated
leaders of genes for thylakoid proteins contain cis-elements essential for transcription. Plant J 6: 513-523
Brakhage AA, Caruso ML (2004) Biotechnical Genetics of Antibiotic Biosynthesis. In: Kück U (eds.) The
Mycota II. Springer Press, Berlin, Heidelberg, New York pp. Brakhage AA, Spröte P, Al-Abdallah Q, Gehrke A, Plattner H, Tüncher A (2004) Regulation of
penicillin biosynthesis in filamentous fungi. Springer Verlag, Berlin, Heidelberg

VIII. LITERATUR
48
Bromley M, Gordon C, Rovira-Graells N, Oliver J (2006) The Aspergillus fumigatus cellobiohydrolase B (cbhB) promoter is tightly regulated and can be exploited for controlled protein expression and RNAi. FEMS Microbiol Lett 264: 246-254
Bronstein I, Fortin J, Stanley PE, Stewart GS, Kricka LJ (1994) Chemiluminescent and bioluminescent
reporter gene assays. Anal Biochem 219: 169-181 Brotzu G (1948) Richerche su di un nuovo antibiotico. Lav Ist d`Igiene Cagliari 1948 1-11 Brückner B, Unkles SE, Weltring K, Kinghorn JR (1992) Transformation of Gibberella fujikuroi: effect
of the Aspergillus nidulans AMA1 sequence on frequency and integration. Curr Genet 22: 313-316 Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY (2002) A
monomeric red fluorescent protein. Proc Natl Acad Sci U S A 99: 7877-7882 Casselton LA (2002) Mate recognition in fungi. Heredity 88: 142-147 Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC (1994) Green fluorescent protein as a marker for
gene expression. Science 263: 802-805 Chan TW, Bruice TC (1977) One and two electron transfer reactions of glucose oxidase. J Am Chem Soc
99: 2387-2389 Chaveroche MK, Ghigo JM, d'Enfert C (2000) A rapid method for efficient gene replacement in the
filamentous fungus Aspergillus nidulans. Nucleic Acids Res 28: E97 Chen Y, Roxby R (1997) Identification of a functional CT-element in the Phytophthora infestans piypt1
gene promoter. Gene 198: 159-164 Chiu W, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J (1996) Engineered GFP as a vital reporter in
plants. Curr Biol 6: 325-330 Cody CW, Prasher DC, Westler WM, Prendergast FG, Ward WW (1993) Chemical structure of the
hexapeptide chromophore of the Aequorea green-fluorescent protein. Biochemistry 32: 1212-1218 Cogoni C, Irelan JT, Schumacher M, Schmidhauser TJ, Selker EU, Macino G (1996) Transgene
silencing of the al-1 gene in vegetative cells of Neurospora is mediated by a cytoplasmic effector and does not depend on DNA-DNA interactions or DNA methylation. Embo J 15: 3153-3163
Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, Weiss RL, Borkovich KA,
Dunlap JC (2006) A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A 103: 10352-10357
Cormack B, Bertram G, Egerton M, Gow N, Falkow S, Brown A (1997) Yeast-enhanced green
fluorescent protein (yEGFP) a reporter of gene expression in Candida albicans. Microbiology 143: 303-311
Cormack BP, Valdivia RH, Falkow S (1996) FACS optimized mutants of the green fluorescent protein
(GFP). Gene 173: 33-38 Cubero B, Scazzocchio C (1994) Two different, adjacent and divergent zinc finger binding sites are
necessary for CREA-mediated carbon catabolite repression in the proline gene cluster of Aspergillus nidulans. EMBO J 13: 407-415
Czymmek KJ, Bourett TM, Howard RJ (2004) Fluorescent Protein Probes in Fungi. Methods in Microbiology. In: Charalabos TSaP (eds.) Microbial Imaging. Academic Press, Amsterdam pp.
27-62

VIII. LITERATUR
49
Davenport D, Nicol JAC (1955) Luminescence in Hydromedusae. Proc Roy Soc Ser B 144: 399-411 Davis AS, Davey MR, Clothier RC, Cocking EC (1992) Quantification and comparison of
chloramphenicol acetyltransferase activity in transformed plant protoplasts using high-performance liquid chromatography- and radioisotope-based assays. Anal Biochem 201: 87-93
de Jong JF, Deelstra HJ, Wösten HA, Lugones LG (2006) RNA-mediated gene silencing in monokaryons
and dikaryons of Schizophyllum commune. Appl Environ Microbiol 72: 1267-1269 de Wet JR, Wood KV, DeLuca M, Helinski DR, Subramani S (1987) Firefly luciferase gene: structure
and expression in mammalian cells. Mol Cell Biol 7: 725-737 Dementhon K, Iyer G, Glass NL (2006) VIB-1 is required for expression of genes necessary for
programmed cell death in Neurospora crassa. Eukaryot Cell 5: 2161-2173 Dowzer CE, Kelly JM (1991) Analysis of the creA gene, a regulator of carbon catabolite repression in
Aspergillus nidulans. Mol Cell Biol 11: 5701-5709 Dowzer CEA, Kelly JM (1989) Cloning ot the creA gene form Aspergillus nidulans: a gene involved in
carbon catabolite repression. Curr Genet 15: 457-459 Dreyer J, Eichhorn H, Friedlin E, Kürnsteiner H, Kück U (2007) A homologue of the Aspergillus velvet
gene regulates both cephalosporin C biosynthesis and hyphal fragmentation in Acremonium chrysogenum. Appl Environ Microbiol 73: 3412-3422
Elander RP (1999) Two decades of strain development in antibiotic-producing microorganisms. J Ind
Microbiol Biotechnol 22: 241-253 Elander RP (2003) Industrial production of beta-lactam antibiotics. Appl Microbiol Biotechnol 61: 385-392 Enke C, Zekert N, Veith D, Schaaf C, Konzack S, Fischer R (2007) Aspergillus nidulans Dis1/XMAP215
protein AlpA localizes to spindle pole bodies and microtubule plus ends and contributes to growth directionality. EukaryotCell 6: 555-562
Fantes PA, Roberts CF (1973) Beta-galactosidase activity and lactose utilization in Aspergillus nidulans. J
Gen Microbiol 77: 471-486 Fei X, Zhao MW, Li YX (2006) Cloning and sequence analysis of a glyceraldehyde-3-phosphate
dehydrogenase gene from Ganoderma lucidum. J Microbiol 44: 515-522 Fekete E, Karaffa L, Sandor E, Seiboth B, Biro S, Szentirmai A, Kubicek CP (2002) Regulation of
formation of the intracellular beta-galactosidase activity of Aspergillus nidulans. Arch Microbiol 179: 7-14
Fernández-Ábalos JM, Fox H, Pitt C, Wells B, Doonan JH (1998) Plant-adapted green fluorescent protein
is a versatile vital reporter for gene expression, protein localization and mitosis in the filamentous fungus, Aspergillus nidulans. Mol Microbiol 27: 121-130
Fierro F, Laich F, Garcia-Rico RO, Martín JF (2004) High efficiency transformation of Penicillium
nalgiovense with integrative and autonomously replicating plasmids. Int J Food Microbiol 90: 237-248
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic
interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806-811 Fitzgerald A, van Kan JA, Plummer KM (2004) Simultaneous silencing of multiple genes in the apple
scab fungus, Venturia inaequalis, by expression of RNA with chimeric inverted repeats. Fungal Genet Biol 41: 963-971

VIII. LITERATUR
50
Fowler AV, Zabin I (1977) The amino acid sequence of beta-galactosidase of Escherichia coli. Proc Natl Acad Sci U S A 74: 1507-1510
Fuhrmann M, Oertel W, Hegemann P (1999) A synthetic gene coding for the green fluorescent protein
(GFP) is a versatile reporter in Chlamydomonas reinhardtii. Plant J 19: 353-361 Geisen R (1995) Expression of the Aspergillus niger glucose oxidase gene in Penicillium nalgiovense. World
J Micribiol Biotechnol 11: 322-325 Gems D, Johnstone IL, Clutterbuck AJ (1991) An autonomously replicating plasmid transforms
Aspergillus nidulans at high frequency. Gene 98: 61-67 Givaudan A, Effosse A, Faure D, Potier P, Bouillant M, Bally R (1993) Polyphenol oxidase in
Azospirillum lipoferum isolated from rice rhizophere: Evidence for laccase activity in non-motile strains of Azospirillum lipoferum. FEMS Microbiol Lett 108: 205-210
Goldoni M, Azzalin G, Macino G, Cogoni C (2004) Efficient gene silencing by expression of double
stranded RNA in Neurospora crassa. Fungal Genet Biol 41: 1016-1024 Gordon CL, Khalaj V, Ram AFJ, Archer DB, Brookman JL, Trinci APJ, Jeenes DJ, Doonan JH,
Wells B, Punt PJ, van den Hondel CAMJJ, Robson GD (2000) Glucoamylase::green fluorescent protein fusions to monitor protein secretion in Aspergillus niger. Microbiology 146: 415-426
Gouka RJ, Punt PJ, van den Hondel CA (1997) Efficient production of secreted proteins by Aspergillus:
progress, limitations and prospects. Appl Microbiol Biotechnol 47: 1-11 Greer LF, 3rd, Szalay AA (2002) Imaging of light emission from the expression of luciferases in living
cells and organisms: a review. Luminescence 17: 43-74 Gurr SJ, Uncles SE, Kinghorn JR (1987) The structure and organization of nuclear genes in filamentous
fungi. In: Kinghorn JR (eds.) Gene structure in eukaryotic microbes. IRL Press, Oxford pp. 93-139 Gutiérrez S, Velasco J, Marcos AT, Fernández FJ, Fierro F, Barredo JL, Diéz B, Martín JF (1997)
Expression of the cefG gene is limiting for cephalosporin biosynthesis in Acremonium chrysogenum. Appl Microbiol Biotechnol 48: 606-614
Haas J, Park EC, Seed B (1996) Codon usage limitation in the expression of HIV-1 envelope glycoprotein.
Curr Biol 6: 315-324 Heim R, Tsien RY (1996) Engineering green fluorescent protein for improved brightness, longer
wavelengths and fluorescence resonance energy transfer. Curr Biol 6: 178-182 Heim R, Prasher DC, Tsien RY (1994) Wavelength mutations and posttranslational autoxidation of green
fluorescent protein. Proc Natl Acad Sci U S A 91: 12501-12504 Heim R, Cubitt AB, Tsien RY (1995) Improved green fluorescence. Nature 373: 663-664 Hodgkins M, Mead D, Ballance DJ, Goodey A, Sudbery P (1993) Expression of the glucose oxidase gene
from Aspergillus niger in Hansenula polymorpha and its use as a reporter gene to isolate regulatory mutations. Yeast 9: 625-635
Hoff B, Kück U (2005) Use of bimolecular fluorescence complementation to demonstrate transcription
factor interaction in nuclei of living cells from the filamentous fungus Acremonium chrysogenum. Curr Genet 47: 132-138
Hoff B, Schmitt EK, Kück U (2005) CPCR1, but not its interacting transcription factor AcFKH1, controls
fungal arthrospore formation in Acremonium chrysogenum. Mol Microbiol 56: 1220-1233

VIII. LITERATUR
51
Hoffmeister D, Keller NP (2007) Natural products of filamentous fungi: enzymes, genes, and their regulation. Nat Prod Rep 24: 393-416
Holm KA (1986) Automatic spectrometric determination of aminoglucosidase activity using p-nitrophenol-
D-glucopyranoside and a flow injection analyzer. Analyst 111: 927-929 Hu CD, Chinenov Y, Kerppola TK (2002) Visualization of interactions among bZIP and Rel family
proteins in living cells using bimolecular fluorescence complementation. Mol Cell 9: 789-798 Ilmén M, Thrane C, Penttilä M (1996a) The glucose repressor gene cre1 of Trichoderma: isolation and
expression of a full-length and a truncated mutant form. Mol Gen Genet 251: 451-460 Ilmén M, Onnela ML, Klemsdal S, Keränen S, Penttilä M (1996b) Functional analysis of the
cellobiohydrolase I promoter of the filamentous fungus Trichoderma reesei. Mol Gen Genet 253: 303-314
Inouye S, Tsuji FI (1994) Aequorea green fluorescent protein. Expression of the gene and fluorescence
characteristics of the recombinant protein. FEBS Lett 341: 277-280 Jakobs S, Subramaniam V, Schonle A, Jovin TM, Hell SW (2000) EFGP and DsRed expressing cultures
of Escherichia coli imaged by confocal, two-photon and fluorescence lifetime microscopy. FEBS Lett 479: 131-135
Janus D, Hoff B, Hofmann E, Kück U (2007) An efficient fungal RNA-silencing system using the DsRed
reporter gene. Appl Environ Microbiol 73: 962-970 Jefferson RA, Burgess SM, Hirsh D (1986) beta-Glucuronidase from Escherichia coli as a gene-fusion
marker. Proc Natl Acad Sci U S A 83: 8447-8451 Jefferson RA, Kavanagh TA, Bevan MW (1987a) GUS fusions: beta-glucuronidase as a sensitive and
versatile gene fusion marker in higher plants. EMBO J 6: 3901-3907 Jefferson RA, Klass M, Wolf N, Hirsh D (1987b) Expression of chimeric genes in Caenorhabditis elegans.
J Mol Biol 193: 41-46 Jekosch K, Kück U (2000a) Glucose dependent transcriptional expression of the cre1 gene in Acremonium
chrysogenum strains showing different levels of cephalosporin C production. Curr Genet 37: 388-395
Jekosch K, Kück U (2000b) Loss of glucose repression in an Acremonium chrysogenum beta-lactam
producer strain and its restoration by multiple copies of the cre1 gene. Appl Microbiol Biotechnol 54: 556-563
Karmali K, Karmali A, Teixeira A, Curto MJ (2004) Assay for glucose oxidase from Aspergillus niger
and Penicillium amagasakiense by Fourier transform infrared spectroscopy. Anal Biochem 333: 320-327
Khalaj V, Eslami H, Azizi M, Rovira-Graells N, Bromley M (2007) Efficient downregulation of alb1 gene
using an AMA1-based episomal expression of RNAi construct in Aspergillus fumigatus. FEMS Microbiol Lett 270: 250-254
Kiiskinen LL, Viikari L, Kruus K (2002) Purification and characterisation of a novel laccase from the
ascomycete Melanocarpus albomyces. Appl Microbiol Biotechnol 59: 198-204 Kleanthous C, Shaw WV (1984) Analysis of the mechanism of chloramphenicol acetyltransferase by
steady-state kinetics. Evidence for a ternary-complex mechanism. Biochem J 223: 211-220

VIII. LITERATUR
52
Kubodera T, Yamashita N, Nishimura A (2002) Transformation of Aspergillus sp. and Trichoderma reesei using the pyrithiamine resistance gene (ptrA) of Aspergillus oryzae. Biosci Biotechnol Biochem 66: 404-406
Kulmburg P, Mathieu M, Dowzer C, Kelly J, Felenbok B (1993) Specific binding sites in the alcR and
alcA promoters of the ethanol regulon for the CREA repressor mediating carbon catabolite repression in Aspergillus nidulans. Mol Microbiol 7: 847-857
Lalitha Kumari H, Sirsi M (1972) Purification and properties of laccase from Ganoderma lucidum. Arch
Microbiol 84: 350-357 Lauf U, Lopez P, Falk MM (2001) Expression of fluorescently tagged connexins: a novel approach to
rescue function of oligomeric DsRed-tagged proteins. FEBS Lett 498: 11-15 Lederberg J (1950) The beta-d-galactosidase of Escherichia coli, strain K-12. J Bacteriol 60: 381-392 Leiter E, Marx F, Pusztahelyi T, Haas H, Pocsi I (2004) Penicillium chrysogenum glucose oxidase -- a
study on its antifungal effects. J Appl Microbiol 97: 1201-1209 Leskovac V, Trivic S, Wohlfahrt G, Kandrac J, Pericin D (2005) Glucose oxidase from Aspergillus niger:
the mechanism of action with molecular oxygen, quinones, and one-electron acceptors. Int J Biochem Cell Biol 37: 731-750
Lim CR, Kimata Y, Oka M, Nomaguchi K, Kohno K (1995) Thermosensitivity of green fluorescent
protein fluorescence utilized to reveal novel nuclear-like compartments in a mutant nucleoporin NSP1. J Biochem (Tokyo) 118: 13-17
Lippincott-Schwartz J, Patterson GH (2003) Development and use of fluorescent protein markers in living
cells. Science 300: 87-91 Liu G, Zhang Y, Li Y, Yu SW, Xing M (2007) Analysis of cellulase synthesis mechanism in Trichoderma
reesei using red fluorescent protein. Wei Sheng Wu Xue Bao 47: 69-74 Liu H, Cottrell TR, Pierini LM, Goldman WE, Doering TL (2002) RNA interference in the pathogenic
fungus Cryptococcus neoformans. Genetics 160: 463-470 Lorang JM, Tuori RP, Martinez JP, Sawyer TL, Redman RS, Rollins JA, Wolpert TJ, Johnson KB,
Rodriguez RJ, Dickman MB, Ciuffetti LM (2001) Green fluorescent protein is lighting up fungal biology. Appl Environ Microbiol 67: 1987-1994
Mach RL, Strauss J, Zeilinger S, Schindler M, Kubicek CP (1996) Carbon catabolite repression of
xylanase I (xyn1) gene expression in Trichoderma reesei. Mol Microbiol 21: 1273-1281 Mander GJ, Wang H, Bodie E, Wagner J, Vienken K, Vinuesa C, Foster C, Leeder AC, Allen G,
Hamill V, Janssen GG, Dunn-Coleman N, Karos M, Lemaire HG, Subkowski T, Bollschweiler C, Turner G, Nusslein B, Fischer R (2006) Use of laccase as a novel, versatile reporter system in filamentous fungi. Appl Environ Microbiol 72: 5020-5026
Markwell J, Frakes LG, Brott EC, Osterman J, Wagner FW (1989) Aspergillus niger mutants with
increased glucose oxidase production. Appl Microbiol Biotechnol 30: 166-169 Martín JF, Casqueiro J, Liras P (2005) Secretion systems for secondary metabolites: how producer cells
send out messages of intercellular communication. Curr Opin Microbiol 8: 282-293 Martín JF, Casqueiro J, Kosalková K, Marcos AT, Gutiérrez S (1999) Penicillin and cephalosporin
biosynthesis: mechanism of carbon catabolite regulation of penicillin production. Antonie Van Leeuwenhoek 75: 21-31

VIII. LITERATUR
53
Mathison L, Soliday C, Stepan T, Aldrich T, Rambosek J (1993) Cloning, characterization, and use in strain improvement of the Cephalosporium acremonium gene cefG encoding acetyl transferase. Curr Genet 23: 33-41
Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA (1999)
Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol 17: 969-973 Mayer AM, Staples RC (2002) Laccase: new functions for an old enzyme. Phytochemistry 60: 551-565 Mayrhofer S, Pöggeler S (2005) Functional characterization of an alpha-factor-like Sordaria macrospora
peptide pheromone and analysis of its interaction with its cognate receptor in Saccharomyces cerevisiae. Eukaryot Cell 4: 661-672
McDonald T, Brown D, Keller NP, Hammond TM (2005) RNA silencing of mycotoxin production in
Aspergillus and Fusarium species. Mol Plant Microbe Interact 18: 539-545 Menne S, Walz M, Kück U (1994) Expression studies with the bidirectional pcbAB-pcbC promoter region
from Acremonium chrysogenum using reporter gene fusions. Appl Microbiol Biotechnol 42: 57-66 Mikkelsen L, Sarrocco S, Lübeck M, Jensen DF (2003) Expression of the red fluorescent protein DsRed-
Express in filamentous ascomycete fungi. FEMS Microbiol Lett 223: 135-139 Miller JH (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, New York Minussi RC, Pastore GM, Durán N (2002) Potential applications of laccase in the food industry. Trends
Food Sci Technol 13: 205-216 Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY (1997) Fluorescent
indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388: 882-887 Moriwaki A, Ueno M, Arase S, Kihara J (2007) RNA-mediated gene silencing in the phytopathogenic
fungus Bipolaris oryzae. FEMS Microbiol Lett 269: 85-89 Myszka DG (2000) Kinetic, equilibrium, and thermodynamic analysis of macromolecular interactions with
BIACORE. Methods Enzymol 323: 325-340 Nakayashiki H (2005) RNA silencing in fungi: mechanisms and applications. FEBS Lett 579: 5950-5957 Nakayashiki H, Hanada S, Nguyen BQ, Kadotani N, Tosa Y, Mayama S (2005) RNA silencing as a tool
for exploring gene function in ascomycete fungi. Fungal Genet Biol 42: 275-83 Naylor LH (1999) Reporter gene technology: the future looks bright. Biochem Pharmacol 58: 749-757 Neveu B, Belzile F, Bélanger RR (2007) Cloning of the glyceraldehyde-3-phosphate dehydrogenase gene
from Pseudozyma flocculosa and functionality of its promoter in two Pseudozyma species. Antonie Van Leeuwenhoek
Norouzian D, Akbarzadeh A, Scharer JM, Moo Young M (2006) Fungal glucoamylases. Biotechnol Adv
24: 80-85 Nowrousian M, Frank S, Koers S, Strauch P, Weitner T, Ringelberg C, Dunlap JC, Loros JJ, Kück U
(2007) The novel ER membrane protein PRO41 is essential for sexual development in the filamentous fungus Sordaria macrospora. Mol Microbiol 64: 923-937
Nunberg JH, Meade JH, Cole G, Lawyer FC, McCabe P, Schweickart V, Tal R, Wittman VP,
Flatgaard JE, Innis MA (1984) Molecular cloning and characterization of the glucoamylase gene of Aspergillus awamori. Mol Cell Biol 4: 2306-2315

VIII. LITERATUR
54
Ogrodowczyk C (2002) Genexpressionsanalysen bei dem β-Laktamproduzenten Acremonium chrysogenum. Diplomarbeit, Ruhr-Universität Bochum
Ormö M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ (1996) Crystal structure of the
Aequorea victoria green fluorescent protein. Science 273: 1392-1395 Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW (1997) Use of the green fluorescent protein
and its mutants in quantitative fluorescence microscopy. Biophys J 73: 2782-2790 Pöggeler S, Kück U (2006) Highly efficient generation of signal transduction knockout mutants using a
fungal strain deficient in the mammalian ku70 ortholog. Gene 378: 1-10 Pöggeler S, Masloff S, Hoff B, Mayrhofer S, Kück U (2003) Versatile EGFP reporter plasmids for cellular
localization of recombinant gene products in filamentous fungi. Curr Genet 43: 54-61 Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ (1992) Primary structure of the
Aequorea victoria green-fluorescent protein. Gene 111: 229-233 Punt PJ, Dingemanse MA, Kuyvenhoven A, Soede RD, Pouwels PH, van den Hondel CA (1990)
Functional elements in the promoter region of the Aspergillus nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 93: 101-109
Punt PJ, Greaves PA, Kuyvenhoven A, van Deutekom JC, Kinghorn JR, Pouwels PH, van den Hondel
CA (1991) A twin-reporter vector for simultaneous analysis of expression signals of divergently transcribed, contiguous genes in filamentous fungi. Gene 104: 119-122
Rhemrev-Boom RM, Tiessen RG, Jonker AA, Venema K, Vadgama P, Korf J (2002) A lightweight
measuring device for the continuous in vivo monitoring of glucose by means of ultraslow microdialysis in combination with a miniaturised flow-through biosensor. Clin Chim Acta 316: 1-10
Roberts IN, Oliver RP, Punt PJ, van den Hondel CA (1989) Expression of the Escherichia coli beta-
glucuronidase gene in industrial and phytopathogenic filamentous fungi. Curr Genet 15: 177-180 Rodriguez Couto S, Toca Herrera JL (2006) Industrial and biotechnological applications of laccases: a
review. Biotechnol Adv 24: 500-513 Root DE, Hacohen N, Hahn WC, Lander ES, Sabatini DM (2006) Genome-scale loss-of-function
screening with a lentiviral RNAi library. Nat Methods 3: 715-719 Roth M (1969) Fluorimetric assay of enzymes. Methods Biochem Anal 17: 189-285 Sauer J, Sigurskjold BW, Christensen U, Frandsen TP, Mirgorodskaya E, Harrison M, Roepstorff P,
Svensson B (2000) Glucoamylase: structure/function relationships, and protein engineering. Biochim Biophys Acta 1543: 275-293
Saunders G, Picknett TM, Tuite MF, Ward M (1989) Heterologous gene expression in filamentous fungi.
Trends Biotechnol 7: 283-287 Schmitt EK, Kück U (2000) The fungal CPCR1 protein, which binds specifically to beta-lactam
biosynthesis genes, is related to human regulatory factor X transcription factors. J Biol Chem 275: 9348-9357
Schmitt EK, Kempken R, Kück U (2001) Functional analysis of promoter sequences of cephalosporin C
biosynthesis genes from Acremonium chrysogenum: specific DNA-protein interactions and characterization of the transcription factor PACC. Mol Genet Genomics 265: 508-518
Schmitt EK, Hoff B, Kück U (2004a) Regulation of cephalosporin biosynthesis. In: Brakhage AA (eds.)
Adv Biochem Engin/Biotechnol. Springer Verlag, Berlin, Heidelberg pp. 1-43

VIII. LITERATUR
55
Schmitt EK, Hoff B, Kück U (2004b) AcFKH1, a novel member of the forkhead family, associates with the RFX transcription factor CPCR1 in the cephalosporin C-producing fungus Acremonium chrysogenum. Gene 342: 269-281
Schmitt EK, Bunse A, Janus D, Hoff B, Friedlin E, Kürnsteiner H, Kück U (2004c) Winged helix
transcription factor CPCR1 is involved in regulation of beta-lactam biosynthesis in the fungus Acremonium chrysogenum. Eukaryot Cell 3: 121-134
Schuster E, Dunn-Coleman N, Frisvad JC, Van Dijck PW (2002) On the safety of Aspergillus niger--a
review. Appl Microbiol Biotechnol 59: 426-435 Scorpione RC, De Camargo SS, Schenberg AC, Astolfi-Filho S (1993) A new promoter-probe vector for
Saccharomyces cerevisiae using fungal glucoamylase cDNA as the reporter gene. Yeast 9: 599-605 Seed B, Sheen JY (1988) A simple phase-extraction assay for chloramphenicol acyltransferase activity.
Gene 67: 271-277 Shaner NC, Steinbach PA, Tsien RY (2005) A guide to choosing fluorescent proteins. Nat Methods 2: 905-
909 Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY (2004) Improved
monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22: 1567-1572
Shaw WV (1967) The enzymatic acetylation of chloramphenicol by extracts of R factor-resistant
Escherichia coli. J Biol Chem 242: 687-693 Shaw WV, Leslie AG (1991) Chloramphenicol acetyltransferase. Annu Rev Biophys Biophys Chem 20:
363-386 Shimomura O, Johnson FH (1975) Chemical nature of bioluminescence systems in coelenterates. Proc Natl
Acad Sci U S A 72: 1546-1549 Shroff RA, O'Connor SM, Hynes MJ, Lockington RA, Kelly JM (1997) Null alleles of creA, the
regulator of carbon catabolite repression in Aspergillus nidulans. Fungal Genet Biol 22: 28-38 Shu X, Shaner NC, Yarbrough CA, Tsien RY, Remington SJ (2006) Novel chromophores and buried
charges control color in mFruits. Biochemistry 45: 9639-9647 Simpson C, Jordaan J, Gardiner NS, Whiteley C (2007) Isolation, purification and characterization of a
novel glucose oxidase from Penicillium sp. CBS 120262 optimally active at neutral pH. Protein Expr Purif 51: 260-266
Sirand-Pugnet P, Santos C, Labarere J (2003) The Aa-Pri4 gene, specifically expressed during fruiting
initiation in the Agrocybe aegerita complex, contains an unusual CT-rich leader intron within the 5' uncoding region. Curr Genet 44: 124-131
Skatrud PL, Queener SW (1989) An electrophoretic molecular karyotype for an industrial strain of
Cephalosporium acremonium. Gene 79: 331-338 Sleigh MJ (1986) A nonchromatographic assay for expression of the chloramphenicol acetyltransferase gene
in eucaryotic cells. Anal Biochem 156: 251-256 Smith NA, Singh SP, Wang MB, Stoutjesdijk PA, Green AG, Waterhouse PM (2000) Total silencing by
intron-spliced hairpin RNAs. Nature 407: 319-320 Snaith HA, Samejima I, Sawin KE (2005) Multistep and multimode cortical anchoring of tea1p at cell tips
in fission yeast. Embo J 24: 3690-3699

VIII. LITERATUR
56
Sönnichsen B, Koski LB, Walsh A, Marschall P, Neumann B, Brehm M, Alleaume AM, Artelt J, Bettencourt P, Cassin E, Hewitson M, Holz C, Khan M, Lazik S, Martin C, Nitzsche B, Ruer M, Stamford J, Winzi M, Heinkel R, Röder M, Finell J, Häntsch H, Jones SJ, Jones M, Piano F, Gunsalus KC, Oegema K, Gönczy P, Coulson A, Hyman AA, Echeverri CJ (2005) Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature 434: 462-469
Spellig T, Bottin A, Kahmann R (1996) Green fluorescent protein (GFP) as a new vital marker in the
phytopathogenic fungus Ustilago maydis. Mol Gen Genet 252: 503-509 Spröte P, Brakhage AA (2007) The light-dependent regulator velvet A of Aspergillus nidulans acts as a
repressor of the penicillin biosynthesis. Arch Microbiol 188: 69-79 Stauffer JF, Schwartz LJ, Brady CW (1966) Problems and progression in a strain selection program with
cephalosporin-producing fungi. Dev Ind Microbiol 7: 104-113 Strauss J, Horvath HK, Abdallah BM, Kindermann J, Mach RL, Kubicek CP (1999) The function of
CreA, the carbon catabolite repressor of Aspergillus nidulans, is regulated at the transcriptional and post-transcriptional level. Mol Microbiol 32: 169-178
Suelmann R, Fischer R (2000) Mitochondrial movement and morphology depend on an intact actin
cytoskeleton in Aspergillus nidulans. Cell Motil Cytoskeleton 45: 42-50 Suelmann R, Sievers N, Fischer R (1997) Nuclear traffic in fungal hyphae: in vivo study of nuclear
migration and positioning in Aspergillus nidulans. Mol Microbiol 25: 757-769 Takeno S, Sakuradani E, Tomi A, Inohara-Ochiai M, Kawashima H, Ashikari T, Shimizu S (2005)
Improvement of the fatty acid composition of an oil-producing filamentous fungus, Mortierella alpina 1S-4, through RNA interference with delta12-desaturase gene expression. Appl Environ Microbiol 71: 5124-5128
Talabardon M, Yang ST (2005) Production of GFP and glucoamylase by recombinant Aspergillus niger:
effects of fermentation conditions on fungal morphology and protein secretion. Biotechnol Prog 21: 1389-1400
Temporini ED, Alvarez ME, Mautino MR, Folco HD, Rosa AL (2004) The Neurospora crassa cfp
promoter drives a carbon source-dependent expression of transgenes in filamentous fungi. J Appl Microbiol 96: 1256-1264
Terskikh AV, Fradkov AF, Zaraisky AG, Kajava AV, Angres B (2002) Analysis of DsRed Mutants.
Space around the fluorophore accelerates fluorescence development. J Biol Chem 277: 7633-7636 Thibault G, Guitard M, Daigneault R (1980) A study of the enzymatic inactivation of chloramphenicol by
highly purified chloramphenicol acetyltransferase. Biochim Biophys Acta 614: 339-342 Toews MW, Warmbold J, Konzack S, Rischitor P, Veith D, Vienken K, Vinuesa C, Wei H, Fischer R
(2004) Establishment of mRFP1 as a fluorescent marker in Aspergillus nidulans and construction of expression vectors for high-throughput protein tagging using recombination in vitro (GATEWAY). Curr Genet 45: 383-389
Tsien RY (1998) The green fluorescent protein. Annu Rev Biochem 67: 509-544 Turner SL, Ford GC, Mountain A, Moir A (1992) Selection of a thermostable variant of chloramphenicol
acetyltransferase (Cat-86). Protein Eng 5: 535-541 van Gorcom RF, Punt PJ, Pouwels PH, van den Hondel CA (1986) A system for the analysis of
expression signals in Aspergillus. Gene 48: 211-217

VIII. LITERATUR
57
van Hartingsveldt W, Mattern IE, van Zeijl CM, Pouwels PH, van den Hondel CA (1987) Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet 206: 71-75
Verkhusha VV, Lukyanov KA (2004) The molecular properties and applications of Anthozoa fluorescent
proteins and chromoproteins. Nat Biotechnol 22: 289-296 Walz M, Kück U (1993) Targeted integration into the Acremonium chrysogenum genome: disruption of the
pcbC gene. Curr Genet 24: 421-427 Wang HX, Ng TB (2006) A laccase from the medicinal mushroom Ganoderma lucidum. Appl Microbiol
Biotechnol 72: 508-513 Ward M, Turner G (1986) The ATP synthase subunit 9 gene of Aspergillus nidulans: sequence and
transcription. Mol Gen Genet 205: 331-338 Ward M, Wilson LJ, Carmona CL, Turner G (1988) The oliC3 gene of Aspergillus niger: isolation,
sequence and use as a selectable marker for transformation. Curr Genet 14: 37-42 Weld RJ, Plummer KM, Carpenter MA, Ridgway HJ (2006) Approaches to functional genomics in
filamentous fungi. Cell Res 16: 31-44 Winnard PT, Jr., Kluth JB, Raman V (2006) Noninvasive optical tracking of red fluorescent protein-
expressing cancer cells in a model of metastatic breast cancer. Neoplasia 8: 796-806 Yamazaki T, Hasebe T, Kajiwara S, Shishido K (2000) Structure and function of a pyrimidine/purine-
biased sequence from the 5'-flanking region of the basidiomycete Lentinus edodes gene priA. Mol Gen Genet 263: 262-270
Yang F, Moss LG, Phillips GN, Jr. (1996a) The molecular structure of green fluorescent protein. Nat
Biotechnol 14: 1246-1251 Yang TT, Cheng L, Kain SR (1996b) Optimized codon usage and chromophore mutations provide
enhanced sensitivity with the green fluorescent protein. Nucleic Acids Res 24: 4592-4593 Yang TT, Sinai P, Green G, Kitts PA, Chen YT, Lybarger L, Chervenak R, Patterson GH, Piston DW,
Kain SR (1998) Improved fluorescence and dual color detection with enhanced blue and green variants of the green fluorescent protein. J Biol Chem 273: 8212-8216
Yanushevich YG, Staroverov DB, Savitsky AP, Fradkov AF, Gurskaya NG, Bulina ME, Lukyanov
KA, Lukyanov SA (2002) A strategy for the generation of non-aggregating mutants of Anthozoa fluorescent proteins. FEBS Lett 511: 11-14
Zadra I, Abt B, Parson W, Haas H (2000) xylP promoter-based expression system and its use for antisense
downregulation of the Penicillium chrysogenum nitrogen regulator NRE. Appl Environ Microbiol 66: 4810-4816
Zeilinger S, Schmoll M, Pail M, Mach RL, Kubicek CP (2003) Nucleosome transactions on the Hypocrea
jecorina (Trichoderma reesei) cellulase promoter cbh2 associated with cellulase induction. Mol Genet Genomics 270: 46-55
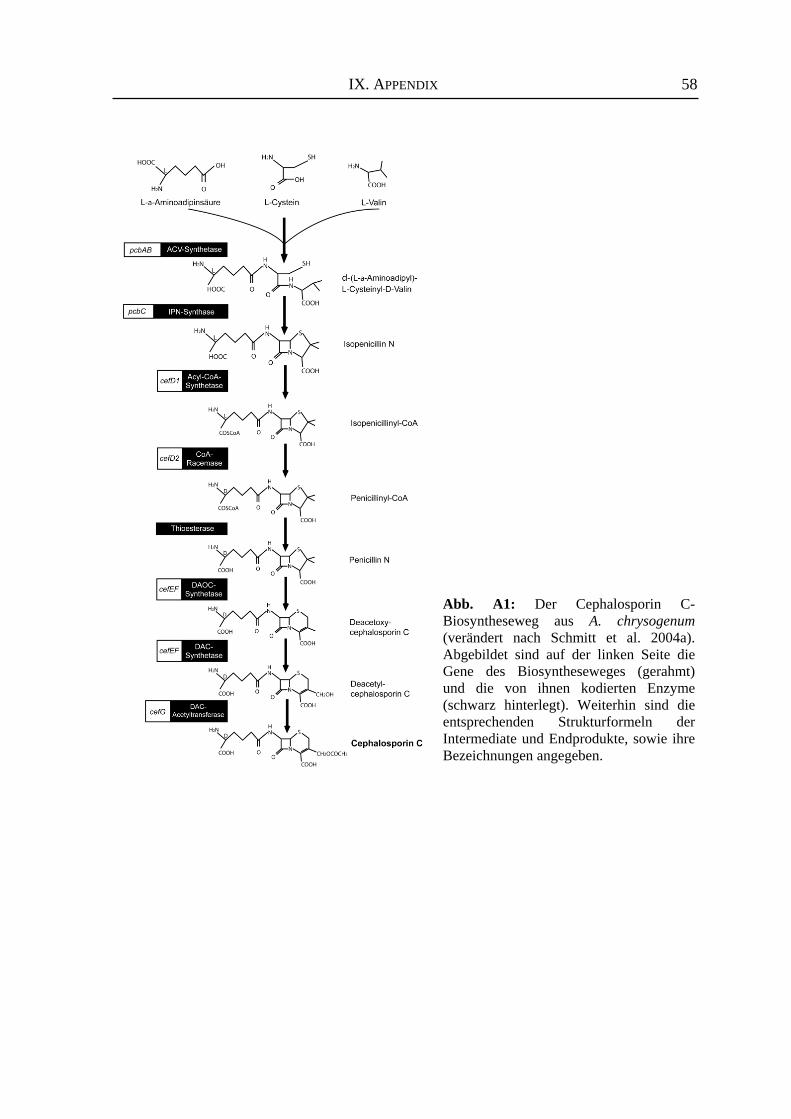
IX. APPENDIX
58
Abb. A1: Der Cephalosporin C-Biosyntheseweg aus A. chrysogenum(verändert nach Schmitt et al. 2004a).Abgebildet sind auf der linken Seite dieGene des Biosyntheseweges (gerahmt)und die von ihnen kodierten Enzyme(schwarz hinterlegt). Weiterhin sind dieentsprechenden Strukturformeln derIntermediate und Endprodukte, sowie ihreBezeichnungen angegeben.

PUBLIKATIONEN
59
PUBLIKATIONEN
ARTIKEL Schmitt EK, Bunse A, Janus D, Hoff B, Friedlin E, Kürnsteiner H, Kück U (2004) The winged helix transcription factor CPCR1 is involved in the regulation of β-lactam biosynthesis in the fungus Acremonium chrysogenum. Eucaryot Cell 3: 121-134 (nicht Teil der Arbeit) Janus D, Hoff B, Hofmann E, Kück U (2007) An efficient fungal RNA-silencing system using the DsRed reporter gene. Appl Environ Microbiol 73 (3): 962-970 Janus D, Hortschansky, Kück U (2007) Identification of a minimal cre1 promoter sequence promoting glucose-dependent gene expression in the β-lactam producer Acremonium chrysogenum (prepared for submission) KONGRESS-BEITRÄGE Hoff B, Schmitt EK, Janus D, Kück U (2003) The winged helix/RFX transcription factor CPCR1 affects both cephalosporin C gene expression and hyphae morphogenesis in Acremonium chrysogenum. Fungal Genetics Newslet. 50 (Supl.) Abst. 217 Hoff B, Janus D, Kück U (2003) Morphogenetic studies of Acremonium chrysogenum, the main producer of the β-lactam antibiotic cephalosporin C. VAAM-Conference Molecular Biology of Fungi, Göttingen, Abstracts, p 45 and 63 Janus D, Hoff B, Schmitt EK, Kück U (2003) Molecular biology of the β-lactam producer Acremonium chrysogenum: Transcription factor CPCR1 controls both cephalosporin biosynthesis and hyphal morphology. VAAM-Conference Molecular Biology of Fungi, Göttingen, Abstracts, p 56 Hoff B, Schmitt EK, Janus D, Kück U (2004) Transcriptionfactors and the regulation of cephalosporin C biosynthesis genes. European Federation of Biotechnology. Physiology of Yeast and Filamentous Fungi, Anglet, France, Abstracts, p 227 Janus D, Schmitt EK, Hoff B, Kück U (2004) Determination of novel CPCR1 binding sites in promoters of cephalosporin C biosynthesis genes. European Federation of Biotechnology. Physiology of Yeast and Filamentous Fungi, Anglet, France, Abstracts, p 255 Hoff B, Schmitt EK, Janus D, Kück U (2004) Transcription factor CPCR1 regulates cephalosporin C biosynthesis in Acremonium chrysogenum. 7th European Conference on Fungal Genetics, Copenhagen, Denmark, Abstracts, p 165

PUBLIKATIONEN
60
Hoff B, Janus D, Schmitt EK, Kück U (2005) The two interacting transcription factors CPCR1 and AcFKH1 control cephalosporin C biosynthesis and morphogenesis in A. chrysogenum. Fungal Genetics Newslet. 52 (Supl.) Abst. 527 Janus D, Kück U (2005) Regulation of the carbon catabolite repressor CRE1 in the β-lactam-antibiotic producer Acremonium chrysogenum. Molecular Biology of Fungi, 7th VAAM-Symposium, Abstracts, p 117 Janus D, Kück U (2006) DsRed as a reporter to study regulation of cre1 gene expression in the β-lactam-antibiotic producer Acremonium chrysogenum. 8th European Conference on Fungal Genetics, Vienna, Austria, Abstracts, p 60 Janus D, Hoff B, Kück U (2007) Use of the DsRed reporter gene to establish an RNAi system for the β-lactam-antibiotic producer Acremonium chrysogenum. Fungal Genetics Newslet. 54 (Supl.) Abst. 268 DIPLOMARBEIT Janus D (2003) Transkriptionsfaktoren bei dem β-Laktam-Produzenten Acremonium chrysogenum: Molekularbiologische Analysen des CPCR1-Proteins. Diplomarbeit, Fakultät für Biologie, Ruhr-Universität Bochum

LEBENSLAUF
61
LEBENSLAUF
PERSÖNLICHE DATEN Danielle Irina Margarete Janus geboren am 31.05.1978 in Herne ledig, keine Kinder
SCHULAUSBILDUNG August 1984 – Juni 1988 Grundschule an der Forellstrasse, Herne August 1988 – Juni 1997 Gymnasium Wanne Abschluss: allgemeine Hochschulreife
HOCHSCHULAUSBILDUNG Oktober 1997 Beginn des Studiums der Biologie Ruhr-Universität Bochum Juni 2002 – April 2003 Anfertigung der Diplomarbeit: „Transkriptionsfaktoren bei
dem β-Laktam-Produzenten Acremonium chrysogenum: Molekularbiologische Analysen des CPCR1-Proteins.“
Lehrstuhl für Allgemeine und Molekulare Botanik Ruhr-Universität Bochum April 2003 Abschluss: Diplom-Biologin, Ruhr-Universität Bochum
BERUFLICHER WERDEGANG Seit April 2003 Ruhr-Universität Bochum, Lehrstuhl für Allgemeine und
Molekulare Botanik; Tätigkeit als Doktorandin, wissenschaftliche Mitarbeiterin Schwerpunkte: Promotion zum Thema „Genexpressionsanalysen bei dem
biotechnologisch relevanten Hyphenpilz Acremonium chrysogenum: Verwendung des autofluoreszierenden Proteins DsRed“
AUSZEICHNUNGEN April 2007 Erhalt eines Posterpreises während der 24. Fungal Genetics
Conference in Asilomar (USA), verliehen von der Genetics Society of America (GSA)

ERKLÄRUNG
Hiermit erkläre ich, dass ich die Arbeit selbständig verfasst und bei keiner anderen Fakultät
eingereicht habe und dass es sich bei der heute von mir eingereichten Dissertation um fünf in
Wort und Bild völlig übereinstimmende Exemplare handelt.
Weiterhin erkläre ich, dass digitale Abbildungen nur die originalen Daten enthalten und in
keinem Fall inhaltsverändernde Bildbearbeitung vorgenommen wurde.
Bochum, den 02. Juli 2007
_________________________________
(Dipl.-Biol. Danielle Janus)