genetica per scienze naturali a.a. 04-05 prof s. presciuttini 1. hemoglobinopathies...
TRANSCRIPT

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
1. HemoglobinopathiesHemoglobinopathies occupy a special place in Hemoglobinopathies occupy a special place in humanhuman genetics for many genetics for many reasonsreasons::
They are by far the most common serious Mendelian diseases on a worldwide They are by far the most common serious Mendelian diseases on a worldwide scalescale
Globins illuminate important aspects of evolution of the genome and of diseases Globins illuminate important aspects of evolution of the genome and of diseases in populationsin populations
Developmental controls are probably better understood for globins than for any Developmental controls are probably better understood for globins than for any other human genesother human genes
More mutations and more diseases are described for hemoglobins than for any More mutations and more diseases are described for hemoglobins than for any other gene familyother gene family
Clinical symptoms follow very directly from malfunction of the protein, which Clinical symptoms follow very directly from malfunction of the protein, which at 15 g per 100 ml of blood is easy to study, so that the relationship between at 15 g per 100 ml of blood is easy to study, so that the relationship between molecular and clinical events is clearer for the hemoglobinopathies than for molecular and clinical events is clearer for the hemoglobinopathies than for most other diseasesmost other diseases
Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
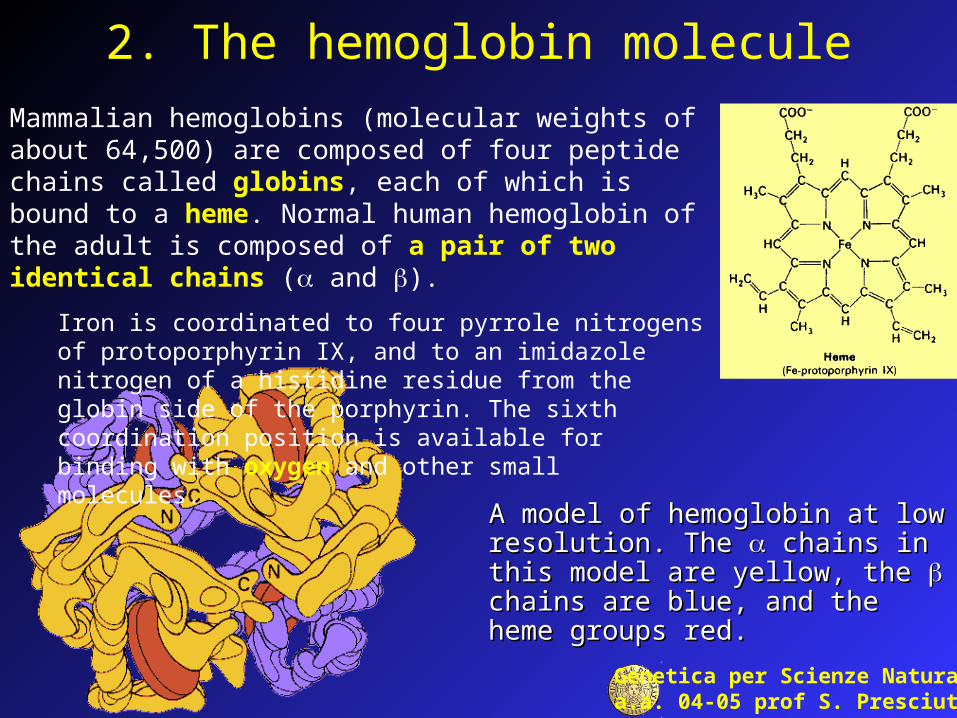
2. The hemoglobin molecule
A model of hemoglobin at low A model of hemoglobin at low resolution. The resolution. The chains in this chains in this model are yellow, the model are yellow, the chains are chains are blue, and the heme groups red. blue, and the heme groups red.
Mammalian hemoglobins (molecular weights of about 64,500) are composed of four peptide chains called globins, each of which is bound to a heme. Normal human hemoglobin of the adult is composed of a pair of two identical chains ( and ).
Iron is coordinated to four pyrrole nitrogens of protoporphyrin IX, and to an imidazole nitrogen of a histidine residue from the globin side of the porphyrin. The sixth coordination position is available for binding with oxygen and other small molecules.

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
3. A problem of development TThe he mammalian mammalian fetus obtain oxygen from maternal bloodfetus obtain oxygen from maternal blood (in the placenta), not (in the placenta), not
from air.from air. How can fetus’s blood accomplish this?How can fetus’s blood accomplish this? The solution involves the development of a The solution involves the development of a fetal hemoglobinfetal hemoglobin. Two of the four . Two of the four
peptides of the fetal and adult hemoglobin chains are identicalpeptides of the fetal and adult hemoglobin chains are identical, , the alpha (the alpha () chains) chains, , but adult hemoglobin has two beta (but adult hemoglobin has two beta () chains, while the fetus has two gamma () chains, while the fetus has two gamma () ) chains.chains. As a consequence, fetal hemoglobin can bind oxygen more efficiently than As a consequence, fetal hemoglobin can bind oxygen more efficiently than can adult hemoglobin. can adult hemoglobin. This small difference in oxygen affinity mediates the transfer This small difference in oxygen affinity mediates the transfer of oxygen from the mother to the fetus.of oxygen from the mother to the fetus. Within the fetus, the myoglobin of the fetal Within the fetus, the myoglobin of the fetal muscles has an even higher affinity for oxygen, so oxygen molecules pass from muscles has an even higher affinity for oxygen, so oxygen molecules pass from fetal hemoglobin for storage and use in the fetal muscles.fetal hemoglobin for storage and use in the fetal muscles.
In the placenta, there is a net flow (arrow) of oxygen In the placenta, there is a net flow (arrow) of oxygen from the mother's blood (which gives up oxygen to the from the mother's blood (which gives up oxygen to the tissues at the lower oxygen pressure) to the fetal blood, tissues at the lower oxygen pressure) to the fetal blood, which is still picking it upwhich is still picking it up

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
4. Fetal hemoglobinsIIn n human human fetuses, until birth, about 80 percent of fetuses, until birth, about 80 percent of chains are substituted by a related chains are substituted by a related chain. These two polypeptide chains are 75 percent identical, and the gene for the chain. These two polypeptide chains are 75 percent identical, and the gene for the chain is close to the b-chain gene on chromosome 11 and has an identical intron-exon chain is close to the b-chain gene on chromosome 11 and has an identical intron-exon structure. This developmental change in globin synthesis is part of a larger set of structure. This developmental change in globin synthesis is part of a larger set of developmental changes that are shown in Figure developmental changes that are shown in Figure belowbelow. The early embryo begins with . The early embryo begins with and and chains and, after about 10 weeks, the chains and, after about 10 weeks, the and and are replaced by are replaced by and and . Near birth, . Near birth, replaces replaces and a small amount of yet a sixth globin, and a small amount of yet a sixth globin, , is produced, is produced. . The The normal adult hemoglobin profile is 97% normal adult hemoglobin profile is 97% 2222, 2, 2--3% 3% 2222, and 1% , and 1% 2222..
Developmental changes in the synthesis of the -like and -like globins that make up human hemoglobin.

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
5. Chromosomal locations of globin genesChromosomal distribution of the genes for the family of globins on chromosome 16 and the family of globins on chromosome 11 in humans.
Gene structure is shown by black bars (exons) and colored bars (introns).

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
6. Organization of globin gene family in human The The , and , and chains all belong to a " chains all belong to a "-like" group; they have very similar -like" group; they have very similar
amino acid sequences and are encoded by genes of identical intron-exon structure amino acid sequences and are encoded by genes of identical intron-exon structure that are all contained in a 60-kb stretch of DNA on chromosome 11.that are all contained in a 60-kb stretch of DNA on chromosome 11.
The The and and chains belong to an " chains belong to an "-like" group and are encoded by genes contained -like" group and are encoded by genes contained in a 40-kb region on chromosome 16. Two slightly different forms of the a chain are in a 40-kb region on chromosome 16. Two slightly different forms of the a chain are encoded by neighboring genes with identical intron-exon structure, as are two forms encoded by neighboring genes with identical intron-exon structure, as are two forms of the of the chain. chain.
In addition, both chromosome 11 and chromosome 16 In addition, both chromosome 11 and chromosome 16 carrycarry pseudogenes, labeled pseudogenes, labeled and and . These pseudogenes are duplicate copies of the genes that did not . These pseudogenes are duplicate copies of the genes that did not acquire new functions but accumulated random mutations that render them acquire new functions but accumulated random mutations that render them nonfunctional.nonfunctional.
At every moment in development, hemoglobin molecules consist of two chains At every moment in development, hemoglobin molecules consist of two chains from the "from the "-like" group and two from the "-like" group and two from the "-like" group, but the specific members -like" group, but the specific members of the groups change in embryonic, fetal, and newborn life. What is even more of the groups change in embryonic, fetal, and newborn life. What is even more remarkable is that the order of genes on each chromosome is the same as the remarkable is that the order of genes on each chromosome is the same as the temporal order of appearance of the globin chains in the course of development.temporal order of appearance of the globin chains in the course of development.

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
7. Globin genes and hemoglobin molecules
The various forms of The various forms of hemoglobin molecules hemoglobin molecules and the genes from and the genes from which they are codedwhich they are coded

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
8. Two groups of hemoglobinopathiesHemoglobinopathies are classified into two main groups:Hemoglobinopathies are classified into two main groups: The The thalassemiasthalassemias are are generally generally caused by caused by inadequate quantitiesinadequate quantities of of
thethe polypeptide chains that form hemoglobin polypeptide chains that form hemoglobin.. The most frequent forms of thalassemia are therefore the The most frequent forms of thalassemia are therefore the -- and and -talassemias-talassemias Alleles are classified into those producing no product (Alleles are classified into those producing no product (00, , 00) and those ) and those
producing reduced amounts of product (producing reduced amounts of product (+, +, +). +).
Abnormal hemoglobins with Abnormal hemoglobins with amino acid changesamino acid changes cause a variety of cause a variety of problems, of which problems, of which sickle cell disease sickle cell disease is the best known.is the best known. In sickle cell disease, a missense mutation In sickle cell disease, a missense mutation (glutammic acid to valine at codon 6) (glutammic acid to valine at codon 6)
replaces a polar by a neutral amino acid on the outer surface of the replaces a polar by a neutral amino acid on the outer surface of the -globin -globin molecule. molecule.
Other amino acid changes can cause anemia, cyanosis, polycythemia (excessive Other amino acid changes can cause anemia, cyanosis, polycythemia (excessive numbers of red cells), methemoglobinemia (conversion of the iron from the numbers of red cells), methemoglobinemia (conversion of the iron from the ferrous to the ferric state), etc.ferrous to the ferric state), etc.

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
9. Major and minor thalassemia In 1925, Thomas Cooley, a In 1925, Thomas Cooley, a USUS pediatrician, described a severe type of anemia in pediatrician, described a severe type of anemia in
children of Italian origin.children of Italian origin. He noted abundant He noted abundant nucleatednucleated red blood cells in the peripheral blood and initially thought red blood cells in the peripheral blood and initially thought
that he was dealing with erythroblastic anemia, described earlier. Before long, Cooley that he was dealing with erythroblastic anemia, described earlier. Before long, Cooley realized that erythroblastemia is neither specific nor essential in this disorderrealized that erythroblastemia is neither specific nor essential in this disorder . He noted a . He noted a number of infants who became seriously anemic and developed splenomegaly number of infants who became seriously anemic and developed splenomegaly (enlargement of the spleen) during their first years of life. The disease was deadly, (enlargement of the spleen) during their first years of life. The disease was deadly, usually before age 10.Very soon, the disease was named after him, Cooley's anemia.usually before age 10.Very soon, the disease was named after him, Cooley's anemia.
In the same years, in Europe, Riette described Italian children with unexplained In the same years, in Europe, Riette described Italian children with unexplained mild hypochromic and microcytic anemiamild hypochromic and microcytic anemia, and other authors in the United States , and other authors in the United States reported a mild anemia in reported a mild anemia in both parentsboth parents of a child with Cooley anemia; this anemia of a child with Cooley anemia; this anemia was similar to that described by Riette in Italy.was similar to that described by Riette in Italy.
In 1936, it was realized that all disorders designated diversely as von Jaksch's In 1936, it was realized that all disorders designated diversely as von Jaksch's anemia, splenic anemia, Cooley's anemia, erythroblastosis, and Mediterranean anemia, splenic anemia, Cooley's anemia, erythroblastosis, and Mediterranean anemia, were in fact a single entity, mostly seen in patients who came from the anemia, were in fact a single entity, mostly seen in patients who came from the Mediterranean area, hence to name the disease they proposed 'thalassemia' derived Mediterranean area, hence to name the disease they proposed 'thalassemia' derived from the Greek word from the Greek word , meaning 'the sea'. It was also recognized that , meaning 'the sea'. It was also recognized that Cooley severe anemia was the Cooley severe anemia was the homozygous formhomozygous form of the of the mild anemiamild anemia described by described by Riette and Wintrobe. The severe form then was labeled as Riette and Wintrobe. The severe form then was labeled as thalassemia majorthalassemia major and and the mild form as the mild form as thalassemia minorthalassemia minor. .

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
10. Complexity of thalassemias The fundamental abnormality in thalassemia is impaired production of The fundamental abnormality in thalassemia is impaired production of
either the either the or or hemoglobin chain. Thalassemia is a difficult subject hemoglobin chain. Thalassemia is a difficult subject to explain, since the condition is not a single disorder, but a group of to explain, since the condition is not a single disorder, but a group of defects with similar clinical effects. More confusion comes from the defects with similar clinical effects. More confusion comes from the fact that the clinical descriptions of thalassemia were coined before fact that the clinical descriptions of thalassemia were coined before the molecular basis of the thalassemias were uncovered.the molecular basis of the thalassemias were uncovered.
The initial patients The initial patients with Cooley’s disease with Cooley’s disease are now recognized to have are now recognized to have been afflicted with been afflicted with thalassemia. In the following few years, thalassemia. In the following few years, different types of thalassemia involving polypeptide chains other than different types of thalassemia involving polypeptide chains other than beta chains were recognized and described in detail. beta chains were recognized and described in detail.
In recent years, the molecular biology and genetics of the thalassemia In recent years, the molecular biology and genetics of the thalassemia syndromes have been described in detail, revealing the wide range of syndromes have been described in detail, revealing the wide range of mutations encountered in each type of thalassemia. Beta thalassemia mutations encountered in each type of thalassemia. Beta thalassemia alone can arise from any of more than 150 mutations.alone can arise from any of more than 150 mutations.

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
11. Gene dosage The two chromosomes #11 have one beta globin gene each (for a total of two The two chromosomes #11 have one beta globin gene each (for a total of two
genes). The two chromsomes #16 have two alpha globin genes each (for a total of genes). The two chromsomes #16 have two alpha globin genes each (for a total of four genes). Hemoglobin protein has two alpha subunits and two beta subunits. four genes). Hemoglobin protein has two alpha subunits and two beta subunits. Each alpha globin gene produces only about half the quantity of protein of a single Each alpha globin gene produces only about half the quantity of protein of a single beta globin gene. This keeps the production of protein subunits equal. Thalassemia beta globin gene. This keeps the production of protein subunits equal. Thalassemia occurs when a globin gene fails, and the production of globin protein subunits is occurs when a globin gene fails, and the production of globin protein subunits is thrown out of balance.thrown out of balance.
If only one If only one beta globin genebeta globin gene is is defective, the other gene supply defective, the other gene supply almost enough protein, though almost enough protein, though people may show mild anemia people may show mild anemia symptoms (thalassemia minor); symptoms (thalassemia minor); the severe b-thalassemia disease the severe b-thalassemia disease (thalassemia major) arise when (thalassemia major) arise when both homologous genes are both homologous genes are defectivedefective

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
12. Summary of genetic defect in -thalassemia ++ : : reduced beta-globin chain synthesis reduced beta-globin chain synthesis 00 : : no beta-globin chain synthesis no beta-globin chain synthesis More than 100More than 100 point mutations and several deletional mutations have point mutations and several deletional mutations have
been identified within and around the beta-globin chain gene all been identified within and around the beta-globin chain gene all affecting the expression of the beta-globin chain gene resulting in affecting the expression of the beta-globin chain gene resulting in defects in activation, initiation, transcription, processing, splicing, defects in activation, initiation, transcription, processing, splicing, cleavage, translation, and/or termination cleavage, translation, and/or termination
genetic defectgenetic defect:: abnormal or no synthesis of the beta-globin chain -> bone marrow fails to abnormal or no synthesis of the beta-globin chain -> bone marrow fails to
produce adequate produce adequate erythrocyteserythrocytes and increased hemolysis of circulating and increased hemolysis of circulating erythrocyteserythrocytes -> anemia -> medullary hematopoiesis and extramedullary -> anemia -> medullary hematopoiesis and extramedullary hematopoiesis (hepatosplenomegaly, lymphadenopathy)hematopoiesis (hepatosplenomegaly, lymphadenopathy)

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
-thalassemia In α-thalassemia, there is deficient synthesis of α-chains. The In α-thalassemia, there is deficient synthesis of α-chains. The
resulting excess of β-chains bind oxygen poorly, leading to a low resulting excess of β-chains bind oxygen poorly, leading to a low concentration of oxygen in tissues (hypoxemia).concentration of oxygen in tissues (hypoxemia).
Deletions of HBA1 and/or HBA2 tend to underlie most cases of α-Deletions of HBA1 and/or HBA2 tend to underlie most cases of α-thalassemia. The severity of symptoms depends on how many of these thalassemia. The severity of symptoms depends on how many of these genes are lost. genes are lost.
Reduced copy numbers of α-globin genes produce successively more Reduced copy numbers of α-globin genes produce successively more severe effects. Most people have four copies of the α-globin gene severe effects. Most people have four copies of the α-globin gene (αα/αα). People with three copies (αα/α-) are healthy; those with two (αα/αα). People with three copies (αα/α-) are healthy; those with two (whether the phase is α-/α- or αα/--) suffer mild α-thalassemia; those (whether the phase is α-/α- or αα/--) suffer mild α-thalassemia; those with only one gene (α-/--) have severe disease, while lack of all four α with only one gene (α-/--) have severe disease, while lack of all four α genes (--/--) causes lethal hydrops fetalis.genes (--/--) causes lethal hydrops fetalis.

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
14. Mechanism of -globin gene deletion
Deletions of α-globin genes in α-thalassemia. Normal copies of chromosome 16 Deletions of α-globin genes in α-thalassemia. Normal copies of chromosome 16 carry two active α-globin genes and an inactive pseudogene arranged in tandem. carry two active α-globin genes and an inactive pseudogene arranged in tandem. Repeat blocks (labeled X and Z) may misalign, allowing unequal crossover. The Repeat blocks (labeled X and Z) may misalign, allowing unequal crossover. The diagram shows unequal crossover between mis-aligned Z repeats producing a diagram shows unequal crossover between mis-aligned Z repeats producing a chromosome carrying only one active α gene. Unequal crossovers between X chromosome carrying only one active α gene. Unequal crossovers between X repeats have a similar effect. Unequal crossovers between other repeats (not shown) repeats have a similar effect. Unequal crossovers between other repeats (not shown) can produce chromosomes carrying no functional α gene. Individuals may thus have can produce chromosomes carrying no functional α gene. Individuals may thus have any number from 0 to 4 or more α-globin genes. The consequences become more any number from 0 to 4 or more α-globin genes. The consequences become more severe as the number of α genes diminishes.severe as the number of α genes diminishes.
Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
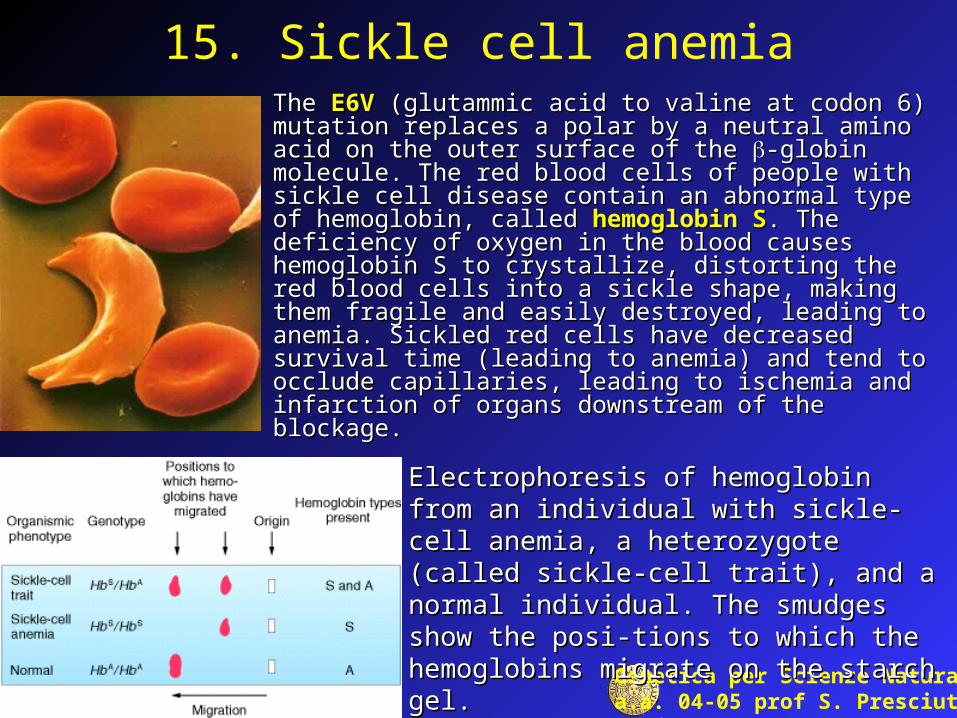
15. Sickle cell anemiaThe The E6VE6V (glutammic acid to valine at codon 6) (glutammic acid to valine at codon 6) mutation mutation replaces a polar by a neutral amino acid on the outer surface of replaces a polar by a neutral amino acid on the outer surface of the the -globin molecule. The red blood cells of people with -globin molecule. The red blood cells of people with sickle cell disease contain an abnormal type of hemoglobin, sickle cell disease contain an abnormal type of hemoglobin, called called hemoglobin Shemoglobin S. The deficiency of oxygen in the blood . The deficiency of oxygen in the blood causes hemoglobin S to crystallize, distorting the red blood causes hemoglobin S to crystallize, distorting the red blood cells into a sickle shape, making them fragile and easily cells into a sickle shape, making them fragile and easily destroyed, leading to anemia. Sickled red cells have decreased destroyed, leading to anemia. Sickled red cells have decreased survival time (leading to anemia) and tend to occlude survival time (leading to anemia) and tend to occlude capillaries, leading to ischemia and infarction of organs capillaries, leading to ischemia and infarction of organs downstream of the blockage. downstream of the blockage.
Electrophoresis of hemoglobin from an individual Electrophoresis of hemoglobin from an individual with sickle-cell anemia, a heterozygote (called with sickle-cell anemia, a heterozygote (called sickle-cell trait), and a normal individual. The sickle-cell trait), and a normal individual. The smudges show the posi-tions to which the smudges show the posi-tions to which the hemoglobins migrate on the starch gel. hemoglobins migrate on the starch gel.

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
16. Summary of hemoglobin types There are hundreds of hemoglobin variants that involve involve genes There are hundreds of hemoglobin variants that involve involve genes
both from the alpha and beta gene clusters. The list that follows both from the alpha and beta gene clusters. The list that follows touches on some of the more common normal and abnormal touches on some of the more common normal and abnormal hemoglobin variants. hemoglobin variants.
Normal HemoglobinsNormal Hemoglobins Hemoglobin AHemoglobin A. This is the designation for the normal hemoglobin that . This is the designation for the normal hemoglobin that
exists after birth. Hemoglobin A is a tetramer with two alpha chains and two exists after birth. Hemoglobin A is a tetramer with two alpha chains and two beta chains (beta chains (2222). ).
Hemoglobin A2Hemoglobin A2. This is a minor component of the hemoglobin found in red . This is a minor component of the hemoglobin found in red cells after birth and consists of two alpha chains and two delta chains (cells after birth and consists of two alpha chains and two delta chains (2222). ).
Hemoglobin A2 generally comprises less than 3% of the total red cell Hemoglobin A2 generally comprises less than 3% of the total red cell hemoglobin. hemoglobin.
Hemoglobin FHemoglobin F. Hemoglobin F is the predominant hemoglobin during fetal . Hemoglobin F is the predominant hemoglobin during fetal development. The molecule is a tetramer of two alpha chains and two development. The molecule is a tetramer of two alpha chains and two gamma chains (gamma chains (2222).).

Genetica per Scienze Naturalia.a. 04-05 prof S. Presciuttini
17. Some clinically significant variant hemoglobins Hemoglobin SHemoglobin S ( (22SS
22, severe). This the predominant hemoglobin in people with , severe). This the predominant hemoglobin in people with sickle cell disease. The molecule structure is. sickle cell disease. The molecule structure is.
Hemoglobin CHemoglobin C ( (22CC22, relatively benign). This results from a mutation in the beta , relatively benign). This results from a mutation in the beta
globin gene and is the predominant hemoglobin found in people with hemoglobin C globin gene and is the predominant hemoglobin found in people with hemoglobin C disease.disease.
Hemoglobin EHemoglobin E ( (22EE22 , benign). This variant results from a mutation in the , benign). This variant results from a mutation in the
hemoglobin beta chain. People with hemoglobin E disease have a mild hemolytic hemoglobin beta chain. People with hemoglobin E disease have a mild hemolytic anemia and mild splenomegaly. Hemoglobin E is common in S.E. Asia.anemia and mild splenomegaly. Hemoglobin E is common in S.E. Asia.
Hemoglobin Constant SpringHemoglobin Constant Spring (named after isolation in a Chinese family from the (named after isolation in a Chinese family from the Constant Spring district of Jamaica). (severe). In this variant, a mutation in the Constant Spring district of Jamaica). (severe). In this variant, a mutation in the alpha globin gene produces an alpha globin chain that is abnormally long. Both the alpha globin gene produces an alpha globin chain that is abnormally long. Both the mRNA and the alpha chain protein are unstable.mRNA and the alpha chain protein are unstable.
Hemoglobin HHemoglobin H. (. (44, mild). This is a tetramer composed of four beta globin chains: , mild). This is a tetramer composed of four beta globin chains: it occurs only with extreme limitation of alpha chain availability. Hemoglobin H it occurs only with extreme limitation of alpha chain availability. Hemoglobin H forms in people with three-gene alpha thalassemia as well as in people with the forms in people with three-gene alpha thalassemia as well as in people with the combination of two-gene deletion alpha thalassemia and hemoglobin Constant combination of two-gene deletion alpha thalassemia and hemoglobin Constant Spring.Spring.
Hemoglobin BartsHemoglobin Barts ( (44, lethal). With four-gene deletion alpha thalassemia no alpha , lethal). With four-gene deletion alpha thalassemia no alpha chain is produced. The gamma chains produced during fetal development combine chain is produced. The gamma chains produced during fetal development combine to form gamma chain tetramers. Individuals with four-gene deletion thalassemia and to form gamma chain tetramers. Individuals with four-gene deletion thalassemia and consequent hemoglobin Barts die in utero (hydrops fetalis).consequent hemoglobin Barts die in utero (hydrops fetalis).