genética das leucoencefalopatias -...
TRANSCRIPT

Jaime Lin Médico neuropediatra
Professor de Neuropediatria – UNISUL
Presidente do Departamento de Neuropediatria - SCP
Genética das leucoencefalopatias

Introdução
Genética das leucoencefalopatias
Leucodistrofias: grupo heterogêneo de doenças
com quadro clínico altamente variável e
mecanismos fisiopatológicos diversos –
tecnicamente refere-se a “perda” (distrofia) da
substância branca (leuko).
Vanderver, A: Mol Genet Metab. (2015)
Leucoencefalopatias: englobam insultos tóxicos,
adquiridos, de origem vascular ou infecciosos.
Hipomielinização, desmielinização,
dismielinização
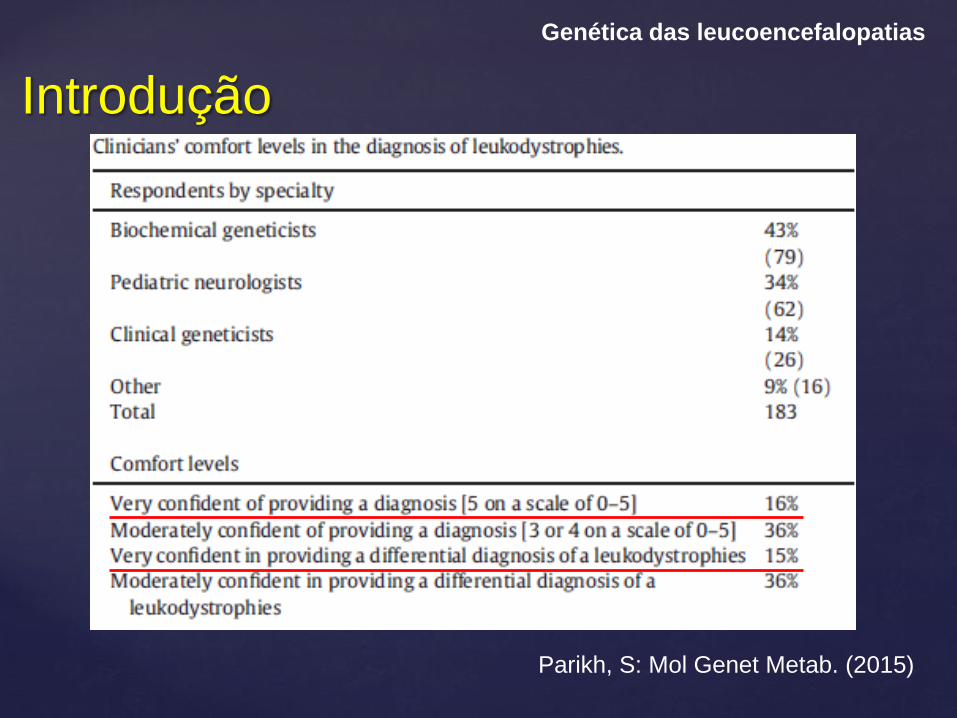
Introdução
Genética das leucoencefalopatias
Parikh, S: Mol Genet Metab. (2015)

Definição
Genética das leucoencefalopatias
Leucodistrofias: doenças hereditárias que afetam a
substância branca do SNC com ou sem
envolvimento periférico.
Vanderver, A: Mol Genet Metab. (2015)
Anormalidades em células gliais (envolvimento
de astrócitos e oligodendrócitos) e na bainha de
mielina.
RM de crânio: hiperintensidade em T2, sinal em
T1 variável (iso – hiperintenso = hipomielinizante;
hipointenso = desmielinizante).

Definição
Genética das leucoencefalopatias
Leucoencefalopatias genéticas: doenças
hereditárias e que resultam em anormalidade da
substância branca, mas não preenchem todos os
critérios para leucodistrofia. Pode haver um
envolvimento neuronal, vascular ou sistêmico
primário em que o comprometimento da
substância branca é secundário.
Vanderver, A: Mol Genet Metab. (2015)
Leucodistrofias são leucoencefalopatias
genéticas, mas nem todas as leucoencefalopatias
genéticas são leucodistrofias.

Epidemiologia
Genética das leucoencefalopatias
Estimativas de incidência variam enormemente:
• 1: 50.000 a 1:7.663
Parikh, S: Mol Genet Metab. (2015)
Na grande maioria dos casos, o diagnóstico
definitivo depende de uma miríade de ferramentas:
clínicas, bioquímicas, patológicas, radiológicas e
moleculares.

Algoritmo diagnóstico
Genética das leucoencefalopatias
Sinal da subst. branca anormal na RM
Parikh, S: Mol Genet Metab. (2015)
Provavelmente adquirida
(infecção/trauma/imunológica)
Tratar de forma apropriada
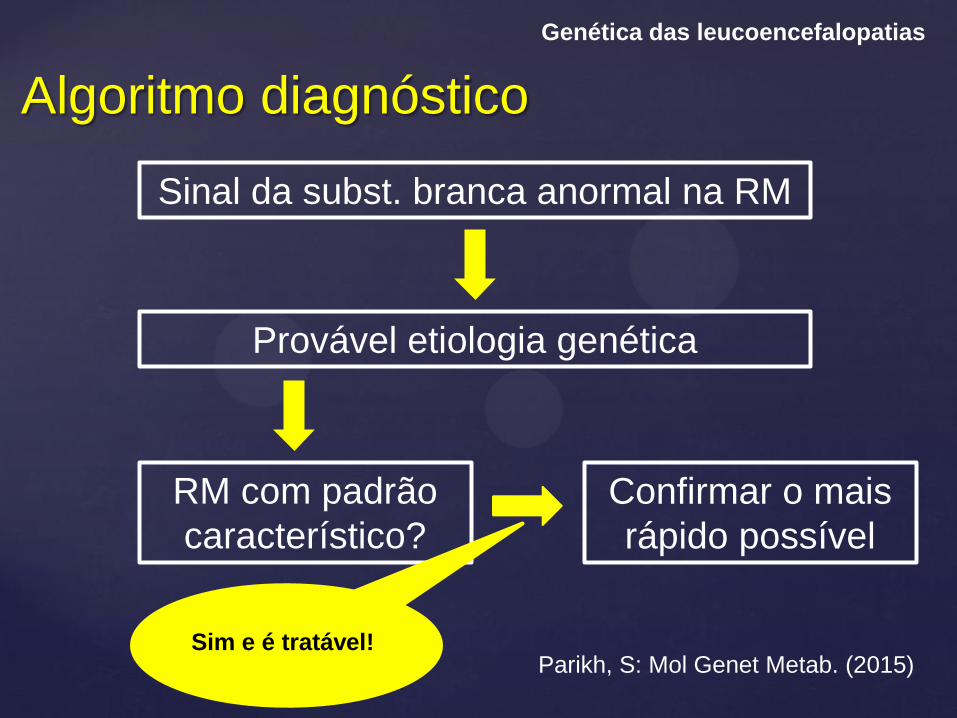
Algoritmo diagnóstico
Genética das leucoencefalopatias
Sinal da subst. branca anormal na RM
Parikh, S: Mol Genet Metab. (2015)
Provável etiologia genética
RM com padrão
característico?
Confirmar o mais
rápido possível
Sim e é tratável!

Adrenoleucodistrofia ligada ao X
Genética das leucoencefalopatias
Doença peroxissomal mais comum
1:17.000 nascidos vivos
Engelen, M: Orphanet J. Rare. Dis. (2012)
Forma cerebral infantil:
• Antes dos 10 anos (média 7 anos);
• Deterioração cognitiva e comportamental, alterações visuais,
tetraparesia espástica, insuficiência adrenal (maioria), epilepsia (20%).
Doença de Addison isolada:
• Entre 2 anos e idade adulta (geralmente antes dos 7 anos);
• Responsável por 20% dos casos de Dça de Addison idiopática
(vômitos, fraqueza, coma, hiperpigmentação cutânea por
hipersecreção de ACTH)
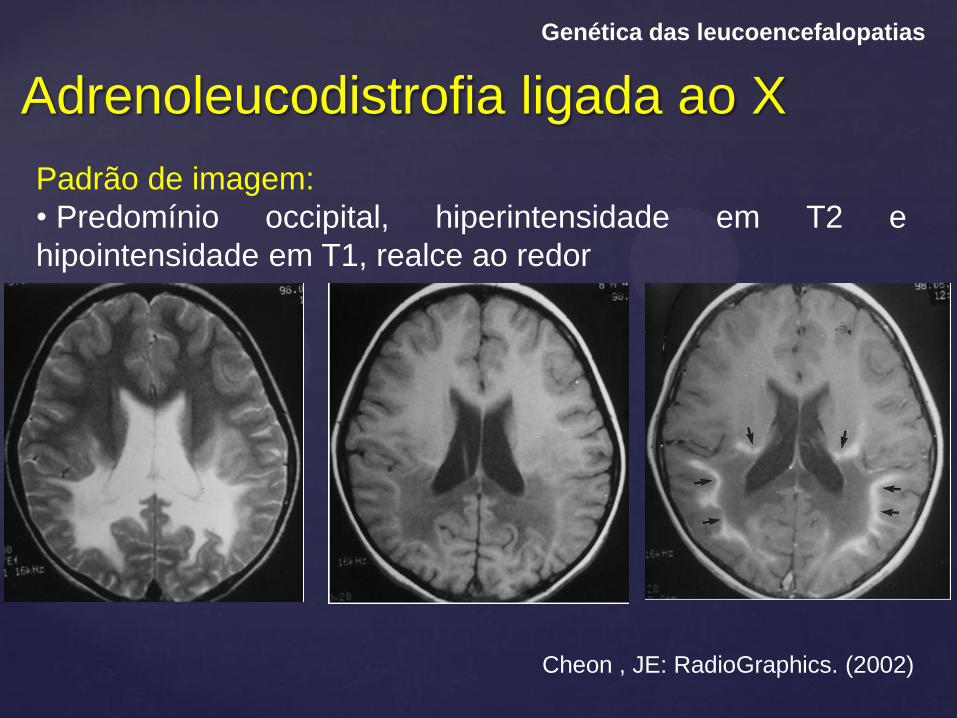
Adrenoleucodistrofia ligada ao X
Genética das leucoencefalopatias
Padrão de imagem:
• Predomínio occipital, hiperintensidade em T2 e
hipointensidade em T1, realce ao redor
Cheon , JE: RadioGraphics. (2002)

Adrenoleucodistrofia ligada ao X
Genética das leucoencefalopatias
Diagnóstico:
Dosagem de ácidos graxos de cadeia muito longa
no plasma.
Engelen, M: Orphanet J. Rare. Dis. (2012)
Diagnóstico pré-sintomático:
Screening neonatal tecnicamente factível. Dosagem
de C26:0 lisofosfatidilcolina em papel filtro.

Adrenoleucodistrofia ligada ao X
Genética das leucoencefalopatias
Genética:
Mutação no gene ABCD1.
Engelen, M: Orphanet J. Rare. Dis. (2012)
Gene ABCD1:
• Localizado no cromossomo Xq28;
• Codifica a proteína ALDP, proteína peroxissomal
transmembrana envolvida no transporte de ésteres de
VLCFA-CoA do citosol para o interior do peroxissomo;
• Impede beta-oxidação de VLCFA. Acúmulo de VLCFA é
tóxico e seu excesso diminui a liberação de cortisol pelas
células adrenais e causa morte celular de astrócitos e
oligodendrócitos.

Adrenoleucodistrofia ligada ao X
Genética das leucoencefalopatias
Genética:
• Todas as filhas de um homem afetado serão carregadoras
enquanto que os filhos nunca serão afetados;
• Quando a mulher carrega do gene, existe 50% de
probabilidade de transmissão do gene;
• 4% dos casos ocorrem por mutação de novo;
• Sequenciamento completo do gene ABCD1 (93%); PCR,
MLPA e Southern Blot para detecção de deleções,
duplicações ou rearranjos;
• Sequenciamento de EXOMA.
Wiesinger, C: The Application of clinical genetics. (2015)
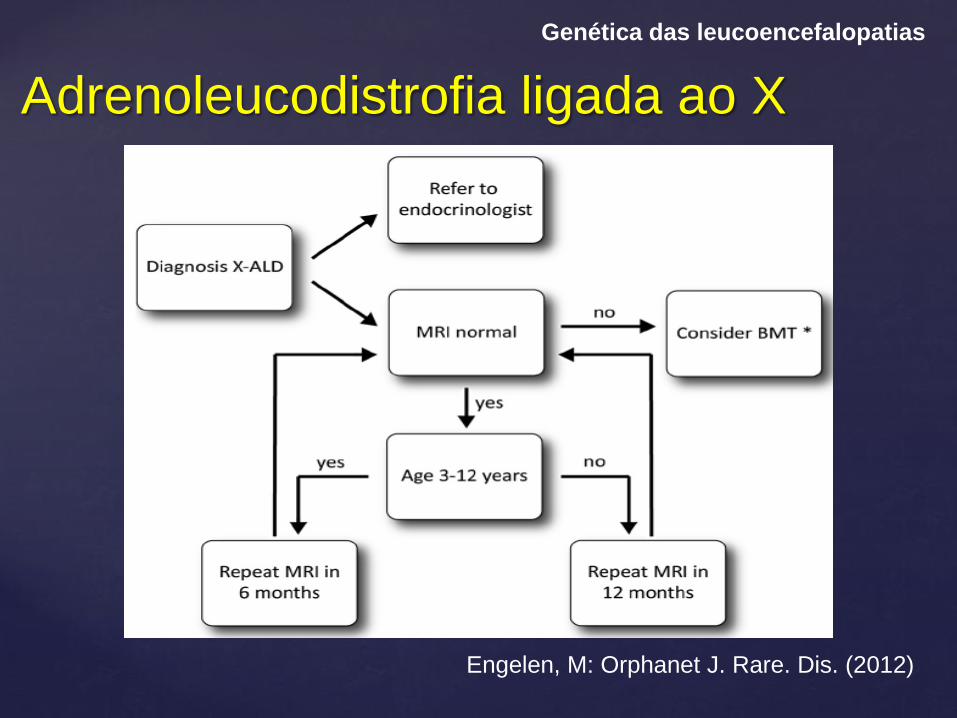
Adrenoleucodistrofia ligada ao X
Genética das leucoencefalopatias
Engelen, M: Orphanet J. Rare. Dis. (2012)

Doença de Krabbe
Genética das leucoencefalopatias
1:100.000 nascidos vivos
Sano, TS: Einstein. (2012)
Forma clássica infantil:
• Fase I: Início com 6 meses (desenvolvimento inicial normal);
irritabilidade, hipertonia, envolvimento de nervos periféricos;
• Fase II: Deterioração rápida, descerebração, opistótono;
• Fase III: Deterioração lenta, atrofia óptica, epilepsia, estado
vegetativo.
Outras formas:
• Infantil tardia: após 6 meses, perda visual, regressão, espasticidade;
• Juvenil e adulta: após 4 anos, clínica variável, alteração de marcha,
perda visual, epilepsia, alteração comportamental e cognitiva.
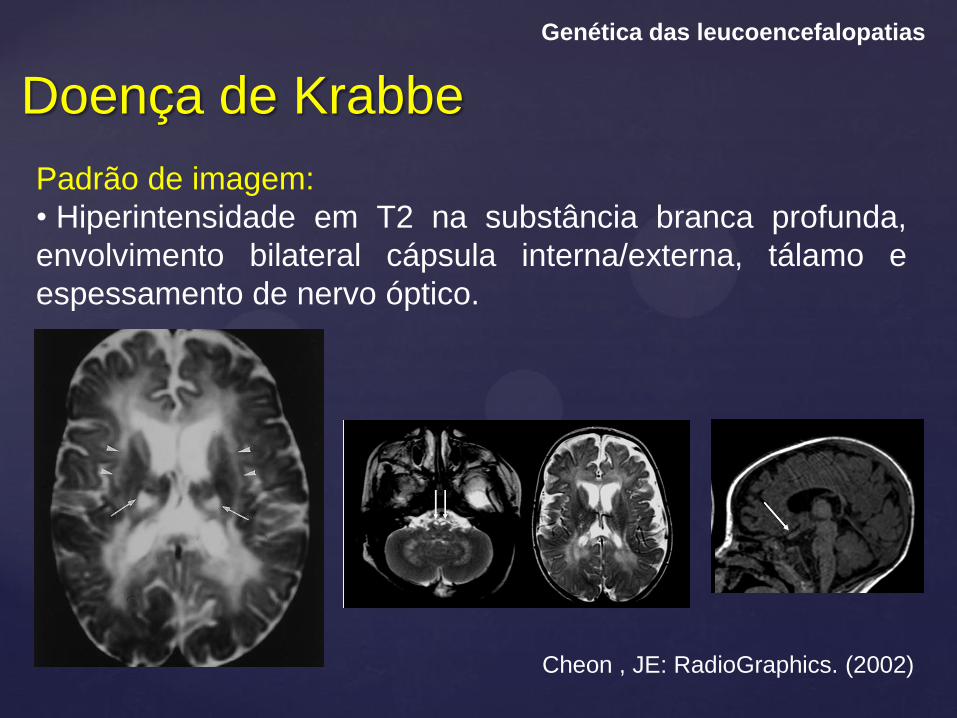
Doença de Krabbe
Genética das leucoencefalopatias
Padrão de imagem:
• Hiperintensidade em T2 na substância branca profunda,
envolvimento bilateral cápsula interna/externa, tálamo e
espessamento de nervo óptico.
Cheon , JE: RadioGraphics. (2002)

Doença de Krabbe
Genética das leucoencefalopatias
Diagnóstico: Dosagem de atividade da enzima galactosilceramidase
(GALC) em leucócitos e fibroblastos.
• em indivíduos sintomáticos 0 a 5% de atividade
• em indivíduos com 8 a 20% de atividade sem apresentação
clínica clássica requerem confirmação molecular.
Puckett, RL: Mol Genet Metabol.. (2012)

Doença de Krabbe
Genética das leucoencefalopatias
Genética:
Mutação no gene GALC.
Won, J-S: Journal of Neuroscience Research. (2016)
Gene GALC:
• Localizado no cromossomo 14;
• A deficiência da galactocerebrosidase leva ao acúmulo de
galactosilsfingosina (lipídeo citotóxico e produto do
metabolismo deficiente de esfingolípides) nos macrófagos
levando a perda progressiva de mielina.

Doença de Krabbe
Genética das leucoencefalopatias
Engelen, M: Orphanet J. Rare. Dis. (2012)
Tratamento:
• Transplante de medula óssea em pacientes pré-
sintomáticos.

Leucodistrofia metacromática
Genética das leucoencefalopatias
1,4 a 1,8:100.000 nascidos vivos
Van Rapard D: Best Practice & Research Clinical Endocrinology and Metabolism. (2014)
Forma infantil tardia:
• 1 a 2 anos;
• Distúrbios de marcha, ataxia, espasticidade, distonia e polineuropatia
periférica, declínio cognitivo.
Forma juvenil:
• 5 a 12 anos;
• Pode-se iniciar com sintomas motores ou cognitivos.
Forma adulta:
• Adolescentes e adultos;
• Sinais cognitivos precoces (incluindo psicose), declínio motor.

Leucodistrofia metacromática
Genética das leucoencefalopatias
Padrão de imagem:
• Em imagens ponderadas em T2: áreas de
hiperintensidade cofluentes e simétricas na sustância
branca periventricular poupando fibras U.
Cheon , JE: RadioGraphics. (2002)

Leucodistrofia metacromática
Genética das leucoencefalopatias
Diagnóstico:
Dosagem de atividade da enzima arilsulfatase A
(ASA) em leucócitos e fibroblastos.
• Níveis de atividade ASA baixos: pseudodeficiência
• Níveis de atividade ASA normais: deficiência de saposina B
Van Rapard D: Best Practice & Research Clinical Endocrinology and Metabolism. (2014)

Leucodistrofia metacromática
Genética das leucoencefalopatias
Genética:
Mutação no gene ARSA
Engelen, M: Orphanet J. Rare. Dis. (2012)
Gene ARSA:
• Localizado no cromossomo 22q13.33
Tratamento:
• Transplante de medula óssea em pacientes pré-
sintomáticos ou oligossintomáticos.

Xantomatose cerebrotendínea
Genética das leucoencefalopatias
Doença peroxissomal.
5:100.000 nascidos vivos
Nie, S: Orphanet J. Rare. Dis. (2014)
Quadro clínico:
• Adolescência e idade adulta (média 19 anos);
• Epilepsia, parkinsonismo, sintomas cerebelares, mielopatia crônica,
deficiência intelectual, demência, sintomas psiquiátricos, sintomas
piramidais, ataxia progressiva, distonia e mioclonia palatal;
• Envolvimento de múltiplos sistemas: catarata, arterioesclerose
precoce, xantomas tendíneos.

Xantomatose cerebrotendínea
Genética das leucoencefalopatias
Padrão de imagem:
• Em imagens ponderadas em T2: hiperintensidade bilateral
e simétrica na substância branca periventricular e
cerebelar. Atrofia cerebelar e cortical.
Vanrietvelde , F: Eur Radiol. (2000)

Xantomatose cerebrotendínea
Genética das leucoencefalopatias
Diagnóstico:
Elevação dos níveis plasmáticos de colestanol e da relação
colestanol/colesterol.
Nie, S: Orphanet J. Rare. Dis. (2014)

Xantomatose cerebrotendínea
Genética das leucoencefalopatias
Genética:
Mutação no gene CYP27A1.
Gene CYP27A1:
• Localizado no cromossomo 2q33-qter;
• Várias mutações detectadas: 50% (exons 6-8); 16% (2) e
14% (4);
• 45% (missense); 20% (nonsense); 14% (deleção) e 2%
(inserção).
Nie, S: Orphanet J. Rare. Dis. (2014)

Xantomatose cerebrotendínea
Genética das leucoencefalopatias
Nie, S: Orphanet J. Rare. Dis. (2014)

Xantomatose cerebrotendínea
Genética das leucoencefalopatias
Tratamento:
• Ácido quenodesoxicólico 750mg/d
• Ácido quenodesoxicólico 300mg/d + pravastatina 10mg/d
Nie, S: Orphanet J. Rare. Dis. (2014)
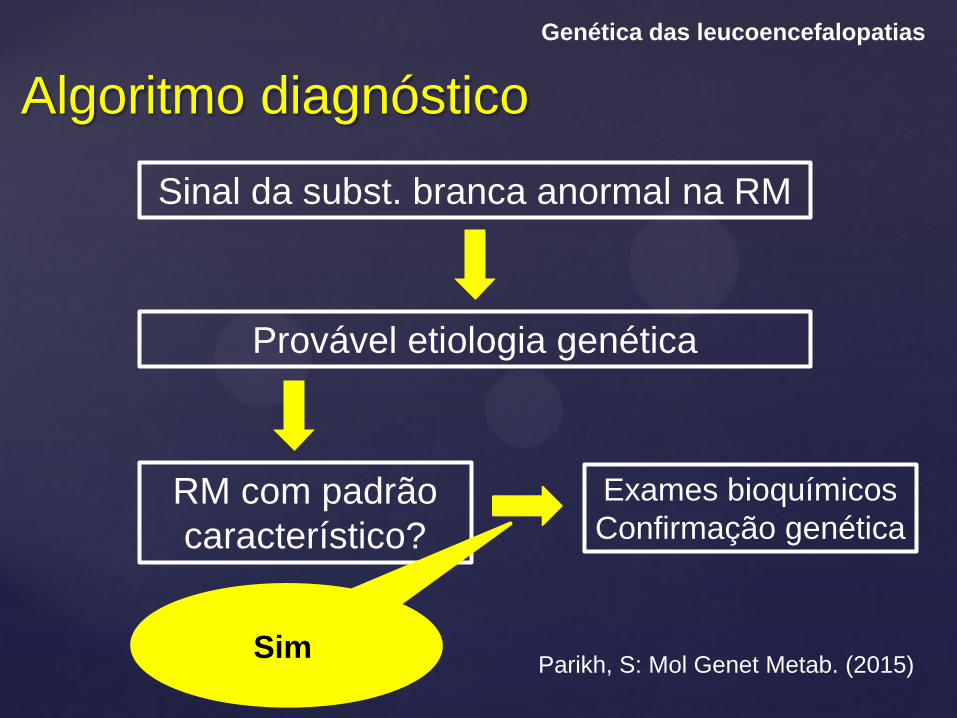
Algoritmo diagnóstico
Genética das leucoencefalopatias
Parikh, S: Mol Genet Metab. (2015)
Sinal da subst. branca anormal na RM
Provável etiologia genética
RM com padrão
característico?
Exames bioquímicos
Confirmação genética
Sim

Hipomielinização
Genética das leucoencefalopatias
• Moderada hiperintensidade em T2 / hiperintensidade ou
sinal normal em T1;
• Padrão inalterado de mielinização deficiente em 2 RM com
6 meses de intervalo após 1 ano de idade.
• As doenças hipomielinizantes representam,
individualmente, a maior categoria dentro das
leucoencefalopatias não caracterizadas.
Engelen, M: Orphanet J. Rare. Dis. (2012)

Sem atrofia / Gg base normais
Genética das leucoencefalopatias
Doença de Pelizaeus Merzbacher
Regis, S: Clin Genet. (2008)
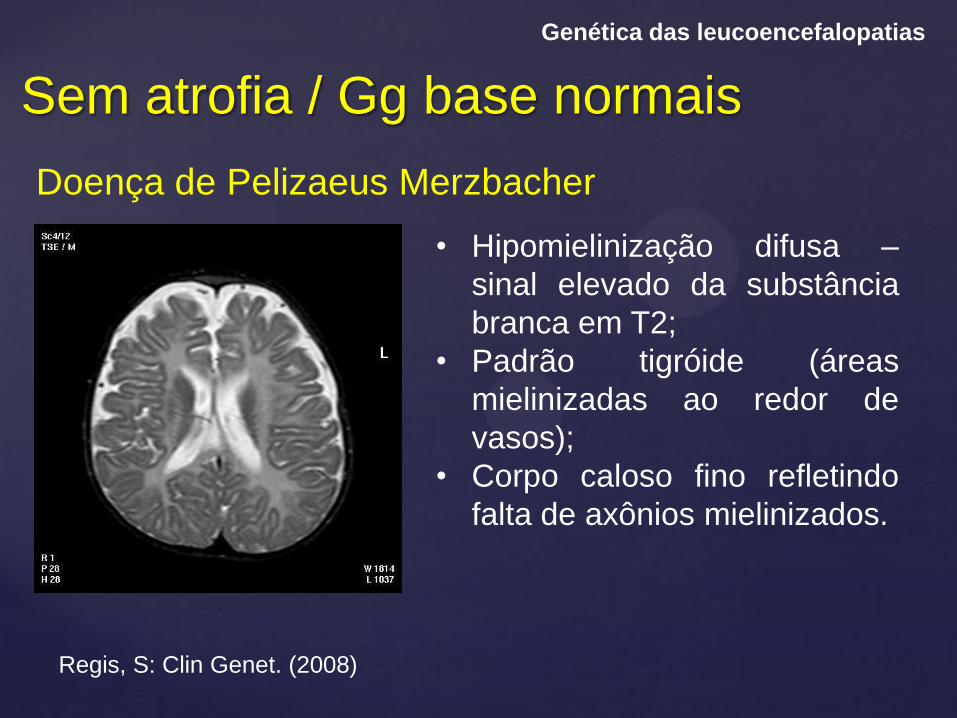
Sem atrofia / Gg base normais
Genética das leucoencefalopatias
Doença de Pelizaeus Merzbacher
Regis, S: Clin Genet. (2008)
• Hipomielinização difusa –
sinal elevado da substância
branca em T2;
• Padrão tigróide (áreas
mielinizadas ao redor de
vasos);
• Corpo caloso fino refletindo
falta de axônios mielinizados.

Sem atrofia / Gg base normais
Genética das leucoencefalopatias
Doença de Pelizaeus Merzbacher
Regis, S: Clin Genet. (2008)
• Gene: PLP1 • Codifica o principal componente proteico da mielina
PLP1/DM20;
• Mutações de ponto são responsáveis por 20% dos casos,
apresentando quadro clínico extremamente variável, desde
a forma clássica até a paraplegia espástica tipo 2;
• Deleções completas do gene PLP1 causam formas
paraplégicas mais brandas da doença;
• Duplicações são responsáveis por 70% dos casos de PMD,
usualmente em sua forma clássica (início com 5 anos de
vida, nistagmo, ataxia e deficiência intelectual).

Sem atrofia / Gg base normais
Genética das leucoencefalopatias
Doença de Pelizaeus Merzbacher - Like
Hobson, G: Sem Neurol. (2012)
• Fenotipicamente similar a doença de Pelizaeus-
Merzbacher;
• Autossômica recessiva;
• Nistagmo de início precoce, ataxia e espasticidade;
• Gene: GJC2 • Causada por mutações no gene da gap junction protein –
gamma2 (GJC2), que codifica proteínas da família das
conexinas. Expressam-se nos oligodendrócitos e são
importantes para a manutenção da integridade da bainha
de mielina.

Sem atrofia / Gg base normais
Genética das leucoencefalopatias
Diagnósticos diferenciais
Doença de Salla
HEMS
HCC
SLC17A5
PLP1 (exon 3B)
DRCTNNB1A / FAM126A
Varho TT, G: Pediatr Neurol. (2002)
Steenweg, ME. Arch Neurol (2012)
Kevelam SH. Annals of Clinical and Translational Neurology (2016)
Rossi, A. Am j. Neuroradiol (2008)
Gazzerro, E. PLOS One, 2012

Com anormalidades em Gg da base
Genética das leucoencefalopatias
Hipomielinização com atrofia de Gg base e cerebelo
Hamilton , EM: Brain. (2014)
• Atraso no desenvolvimento
neurológico, hipotonia, nistagmo e
deterioração motora. Sintomas
extrapiramidais são comuns:
distonia, rigidez e, mais raramente,
coreoatetose.
• Hiperintensidade em T2; atrofia
cerebelar; atrofia de gânglios da
base.

Com anormalidades em Gg da base
Genética das leucoencefalopatias
Hipomielinização com atrofia de Gg base e cerebelo
Hamilton , EM: Brain. (2014)
Gene: TUBB4A
• Codifica um membro da família das Beta-tubulinas;
• As Beta-tubulinas formam heterodímeros com as alfa-tubulinas –
essas proteínas alfa e beta se alternam formando co-polímeros que
se unem em microtúbulos. Microtúbulos são componentes
essenciais do citoesqueleto e determinam uma matriz para o
formato celular;
• Mutações no gene TUBB4A causam alterações na dinâmica dos
microtúbulos e na sua estabilidade. Alterações nos microtúbulos
comprometem o transporte axonal, impedindo a mielinização
adequada;
• Quanto ao cerebelo e gg da base – o TUBB4A é um gene que se
expressa exclusivamente no cérebro, sendo a sua expressão maior
no putâmen e no cerebelo.

Desmielinização
Genética das leucoencefalopatias
• Hiperintensidade proeminente em T2;
• Hipointensidade em T1.
Purkih, S: Molecular Genetics and Metabolism. (2015)

Predominantemente periventricular
Genética das leucoencefalopatias
Leucoencefalopatia megalencefálica com cistos
Koç, K: Neuroradiology Jornal. (2015)
• Doença autossômica recessiva;
• Macrocefalia pós-natal é o sinal predominante (nenhum
caso de MLC sem macrocrania foi molecularmente
identificado até o momento);
• O perímetro cefálico se estabiliza usualmente após o
primeiro ano de idade (4 a 6 desvios-padrão acima da
média);
• Atraso no desenvolvimento neuromotor e cognitivo;
• Posteriormente os pacientes evoluem com espasticidade,
ataxia, epilepsia.
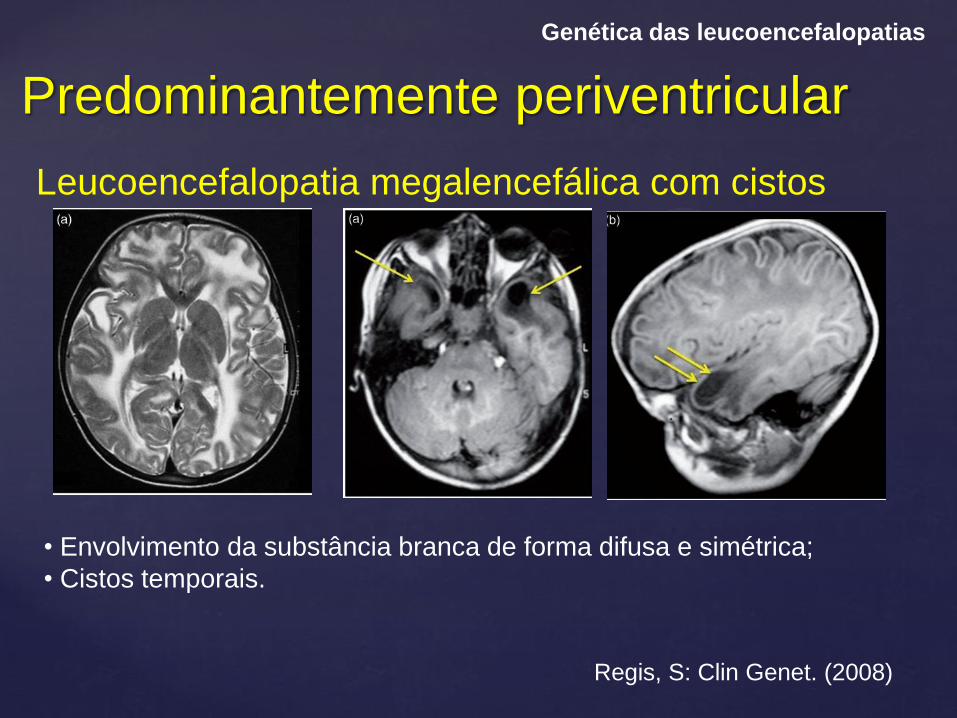
Predominantemente periventricular
Genética das leucoencefalopatias
Leucoencefalopatia megalencefálica com cistos
Regis, S: Clin Genet. (2008)
• Envolvimento da substância branca de forma difusa e simétrica;
• Cistos temporais.

Predominantemente periventricular
Genética das leucoencefalopatias
Leucoencefalopatia megalencefálica com cistos
Brignone, MS: Neurobaiology Disease. (2014)
Gene: MLC1
• Gene MLC1 se expressa em organelas intracelulares nos astrócitos
perivasculares;
• A proteína MLC1 está envolvida na resposta astrocítica, nas
alterações osmóticas e na regulação do volume celular;
• Além disso, regula o pH intracelular e a reciclagem de proteínas em
situações de stress celular reciclagem anormal de proteínas de
adesão poderiam comprometer a integridade celular levando a
formação de vacúolos;
• Endocitose e reciclagem proteica são processos essenciais na
expressão de moléculas de superfície que medeiam a diferenciação
celular durante o desenvolvimento neurológico modificações
resultam em edema e distúrbios na formação da mielina.

Predominantemente subcortical
Genética das leucoencefalopatias
Doença de Canavan
Sreenivasan, P: Indian J. Pediatr. (2013)
• Desenvolvimento normal até os 3-6 meses de idade;
• Hipotonia, perda do controle cefálico, irritabilidade, macrocrania,
espasticidade; atrofia óptica, movimentos extra-piramirais;
epilepsia;
• Neuroimagem: substância branca subcortical; degeneração
espongiforme.

Predominantemente subcortical
Genética das leucoencefalopatias
Doença de Canavan
Baslow, MH: Biochimie. (2013)
Gene: ASPA – braço curto do cromossomo 17 (17p13-ter)
• Mutações neste gene comprometem a atividade da enzima ASPA,
causando acúmulo do NAA;
• Hipótese osmótica: A inabilidade em se hidrolizar o NAA levaria a
um desequilíbrio osmótico que seria responsável por: hidrocefalia,
megalencefalia, edema dos astrócitos com formação de vacúolos
esféricos preenchidos por fluidos, levando ao aspecto
“espongiforme” característico da doença.
• Hipótese Acetato-lipídeo-mielina: O NAA deve ser hidrolizado pela
enzima ASPA em acetato, por sua vez, este é requisitado pelos
oligodendrócitos em mielinização, a fim de sintetizar
adequadamente os lipídeos necessários para o envolvimento dos
axônios pela bainha de mielina.
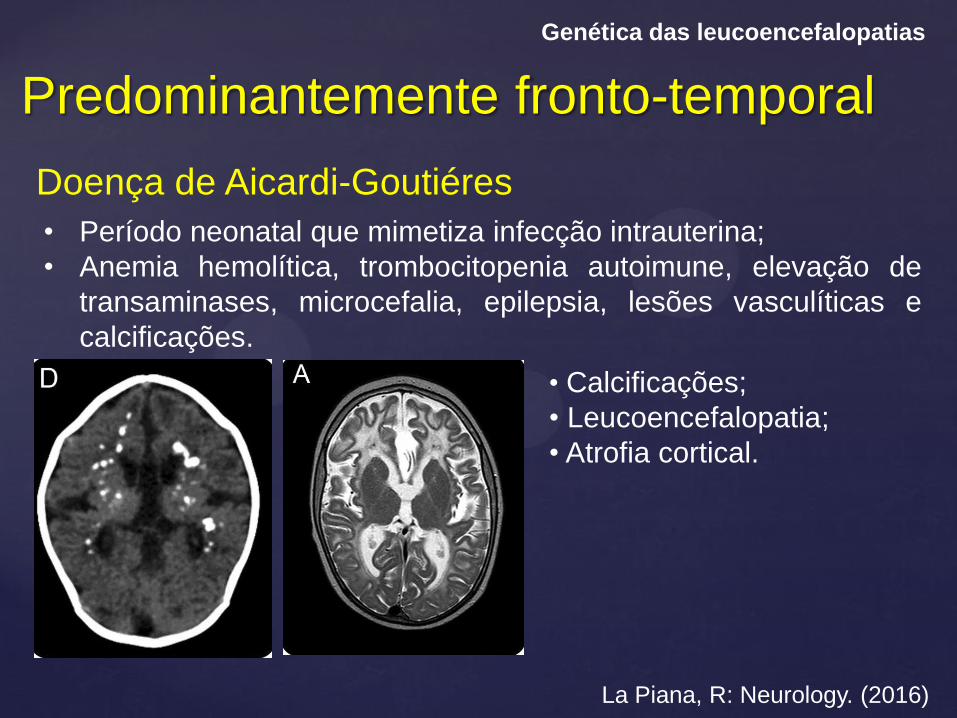
Predominantemente fronto-temporal
Genética das leucoencefalopatias
Doença de Aicardi-Goutiéres
La Piana, R: Neurology. (2016)
• Período neonatal que mimetiza infecção intrauterina;
• Anemia hemolítica, trombocitopenia autoimune, elevação de
transaminases, microcefalia, epilepsia, lesões vasculíticas e
calcificações.
• Calcificações;
• Leucoencefalopatia;
• Atrofia cortical.

Predominantemente fronto-temporal
Genética das leucoencefalopatias
Doença de Aicardi-Goutiéres
La Piana, R: Neurology. (2016)
Genes: RNASEH2A, RNASEH2B, RNASEH2C, TREX1, SAMHD1,
ADAR1, IFIH1
• TREX1: Associado a quadros de predomínio frontotemporal,
presença de calcificações graves e presença de cistos fora da
região frontotemporal. Quadro clínico de início mais precoce, antes
dos 3 meses de idade;
• RNASEH2B: Quadro clínico de início precoce, com atraso na
mielinização sem calcificações;
• RNASEH2A e SAMHD1: leucoencefalopatia predominantemente
periventricular.

Predominantemente fronto-tronco-cerebelo e
pedúnculos cerebelares
Genética das leucoencefalopatias
Doença de Alexander
Sawaishi, Y: Brain & Development. (2016)
Forma infantil de início antes dos 2 anos de idade.
• Quadros de epilepsia, sintomas piramidais e extra-
piramidais, perda do desenvolvimento motor na primeira
década de vida;
• Sintomas bulbares e hidrocefalia podem estar presentes.
Forma juvenil (2 a 12 anos de idade)
• Predomínio de disfunções motoras com comprometimento
progressivo da marcha e espasticidade;
• Sintomas bulbares como mioclonia palatal podem ser
altamente sugestivos.
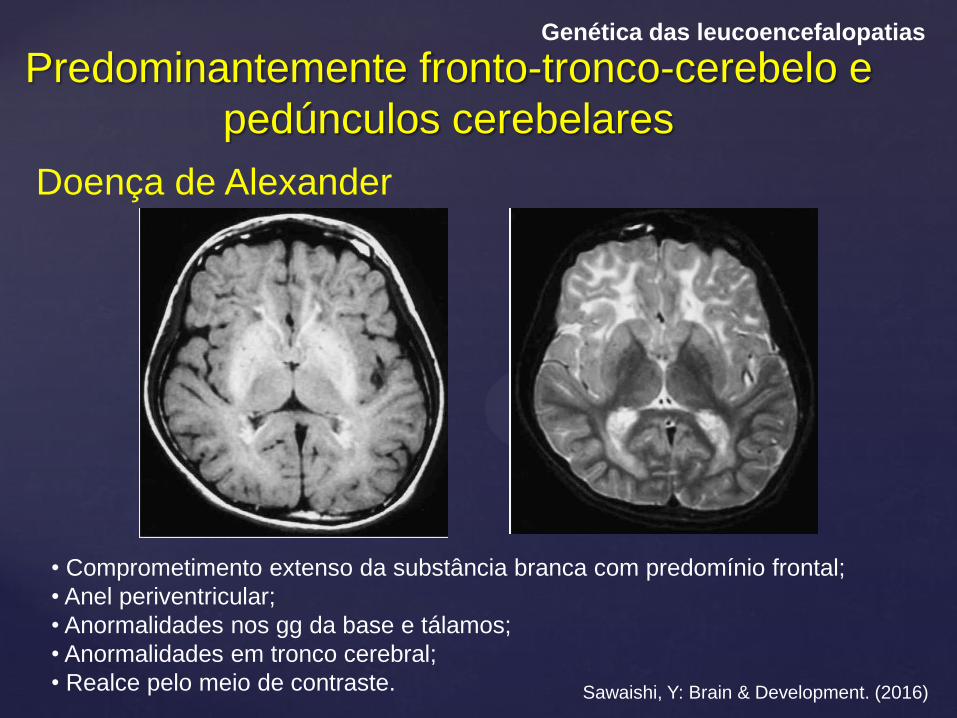
Predominantemente fronto-tronco-cerebelo e
pedúnculos cerebelares
Genética das leucoencefalopatias
Doença de Alexander
Sawaishi, Y: Brain & Development. (2016)
• Comprometimento extenso da substância branca com predomínio frontal;
• Anel periventricular;
• Anormalidades nos gg da base e tálamos;
• Anormalidades em tronco cerebral;
• Realce pelo meio de contraste.

Predominantemente fronto-tronco-cerebelo e
pedúnculos cerebelares
Genética das leucoencefalopatias
Doença de Alexander
Sawaishi, Y: Brain & Development. (2016)
Gene: GFAP
• Localizado no cromossomo 17q21 e codifica a proteína acídica
fribliar glial (GFAP);
• Mutações neste gene promovem uma diminuição na solubilidade da
proteína acídica fibrilar glial, resultando no acúmulo dessas
proteínas e formando as fibras de Rosenthal;
• A localização das anormalidades na RM geralmente se correlaciona
com áreas de maciço acúmulo de fibras de Rosenthal.
• Especula-se que a leucodistrofia na doença de Alexander é um
efeito secundário ao comprometimento da função dos astrócitos,
levando a desregulação dos sinais de mielinização derivados dos
astrócitos.
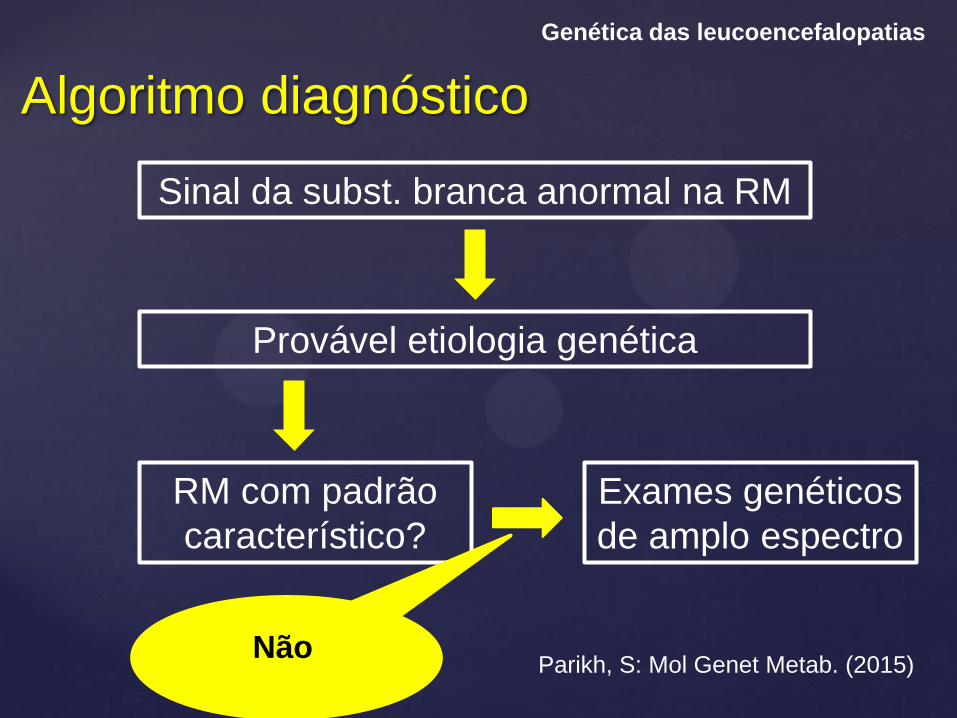
Algoritmo diagnóstico
Genética das leucoencefalopatias
Parikh, S: Mol Genet Metab. (2015)
Sinal da subst. branca anormal na RM
Provável etiologia genética
RM com padrão
característico?
Exames genéticos
de amplo espectro
Não

Genética das leucoencefalopatias
Para os pacientes que apresentam anormalidades
na substância branca sem um padrão sugestivo de
sua etiologia e que apresentem um quadro
claramente genético, recomenda-se investigação
genética de amplo espectro – sequenciamento de
exoma.
Parikh, S: Mol Genet Metab. (2015)

Genética das leucoencefalopatias
Parikh, S: Mol Genet Metab. (2015)
123 genes

Conclusão
Genética das leucoencefalopatias
A heterogeneidade clínica das leucoencefalopatias impede a
utilização de um único método diagnóstico;
Vanderver, A: Mol Genet Metab. (2015)
Achados de neuroimagem muitas vezes são inespecíficos e
patologias previamente agrupadas pelo padrão de imagem
similar se provaram doenças completamente heterogêneas;
A identificação dos defeitos genéticos ampliou o
entendimento fisiopatológico das leucoencefalopatias.

Jaime Lin Médico neuropediatra
Professor de Neuropediatria – UNISUL/SC
Presidente do Departamento de Neuropediatria – SCP
Genética das leucoencefalopatias
MUITO OBRIGADO!