foreign data and the oncology drugs advisory committee … · · 2014-07-24foreign data and the...
TRANSCRIPT

Foreign Data and the Oncology Drugs Advisory Committee
Howard Fingert, MD, FACP Senior Medical Director, Takeda Pharmaceuticals, Inc.
and Industry Representative to the FDA Oncology Drugs Advisory Committee
Clinical Operations in Oncology Trials July, 2014

Disclosures
• I have no financial relationships with products or companies described in this presentation.
• The views and opinions in this talk are my own and do not represent those of Takeda Pharmaceuticals nor of the FDA Oncology Drugs Advisory Committee
• I am an employee of Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceuticals International Co, and I may discuss investigational uses of Takeda products in development for cancer indications. Claims of efficacy and safety can only be made after regulatory review of the data and approval of labeled claims.

Presentation Topics • Background on Foreign Data
• Learnings from ODAC / FDA
• Partnerships and trial networks

Code of Federal Regulations 314.106 Foreign Data and Multi-Regional Clinical Trials (MRCT)
• An application based solely on foreign clinical data meeting U.S. criteria for marketing approval may be approved if: (1) The foreign data are applicable to the U.S. population and U.S. medical practice; (2) the studies have been performed by clinical investigators of recognized competence; and (3) …FDA is able to validate the data through an on-site inspection or other appropriate means.
• (4) Consultation between FDA and applicants. Applicants are encouraged to meet with agency officials in a “presubmission” meeting when approval based solely on foreign data will be sought.
• Considerations about foreign data, published by FDA staff: Khin et al, Regulatory and Scientific Issues Regarding Use of Foreign Data in Support of New Drug Applications in the United States: An FDA Perspective. Clinical Pharmacology & Therapeutics 94:230-242 August 2013

Why Oncology trials enroll outside the US Major reasons described by Oncology sponsors
different from common perception: “its all about cost”
• Enrollment restricted to US would delay time to complete trials – Pre-approval registration trials & NDA submission – Post approval commitment studies with target date for data submission – Confirmatory trials for accelerated/breakthrough product approvals – Uncommon tumors and pediatric indications
• Competition by US NCI Coop Groups & Independent US investigators
– Motivation for investigator-led research and publications – NCI mandate to improve enrollment metrics - Op Efficiency Work Group
• Many ex-US submissions require minimal enrollment from that country
(Japan, China, EU, etc)
• For recent Oncology trials, cost reduction not uniform determinant

US Product Approvals based totally on foreign data in pivotal trial
• Bedaquiline as part of combination therapy to treat adults with multidrug-resistant pulmonary tuberculosis (Asia, South Africa, South America, and Eastern Europe; US = 0)
• Velaglucerase alfa for type 1 Gaucher disease (Argentina, Paraguay, Israel, Russia, and Tunisia, US = 0)
• Carglumic acid for the treatment of hyperammonemia in patients with N-acetyl-glutamate synthase deficiency of the urea cycle (France, Germany, United Kingdom, US = 0 )

Recent Oncology Product Approvals based largely on foreign data
Oncology Products
• Pertuzumab (Perjeta) neoadjuvant therapy for patients with locally advanced HER2+ breast cancer, combined with Trastuzumab and Docetaxel- Recommended by ODAC 12Sept2013; Approved Oct2013 (0 enrollment from U.S.)
• Afatinib in EGFR-mutation adenocarcinoma of lung “As expected for EGFR mutation positive population, the trial included substantial proportions of females (64.9%), East Asians (71.9%), and never smokers (68.4%).” (FDA Review)
CFR 314.106: (1) The foreign data are applicable to the U.S. population and U.S. medical practice
Recent experience with Oncology product approvals: molecular marker(s) supersedes region to guide what’s “applicable to the US”

Recent ODAC Review of TKI for Renal Cell Cancer pivotal trial based largely on foreign data
Phase 3 Tivozanib vs Sorafenib - ODAC May 2, 2013

Tivozanib Phase 3 Protocol Design
Dominant enrollment from foreign centers with inconsistent access to TKIs or standard care, which can influence OS outcomes
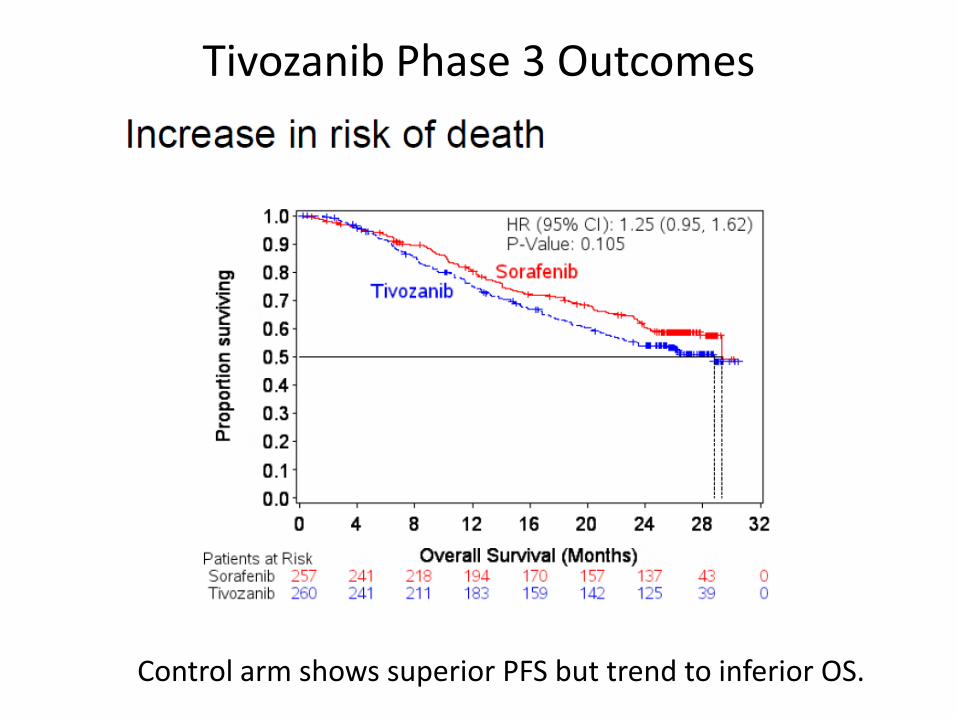
Tivozanib Phase 3 Outcomes
Control arm shows superior PFS but trend to inferior OS.

Major FDA Concerns at Tivozanib ODAC Excerpt from FDA Slides, ODAC May 2, 2013
“Inconsistent PFS and OS results and imbalance in post study treatments… results inconclusive when making a risk-benefit assessment …for approval Has the Applicant demonstrated favorable benefit to risk evaluation for treatment in an adequate and well-controlled trial? “

Perjeta Pivotal Study “Neosphere” BLA approved and based largely on ex-US Data

FDA Describes Enrollment by Region & Race North America included Canada - but not US
13

Common Question and Slide from FDA Excerpt from FDA Questions for ODAC 12Sept2013
A related question: if approved how would treatment be used in US clinical practice?

Perspectives from Prescribing Physician Common topic for ODAC: if approved, how would drug be used in practice?
1) Need exists for alternative treatment options, supported by current data, an informative product label, even if other options are available on or off label.
2) Oncology patient care (including use of drugs) is a dynamic, changing process for many situations. The initial Prescribing information is a good start, but also recognized value of emerging data coupled with initiatives to improve product label & patient guidance
3) Given broad, established professional networking in the Oncology community, effective learning happens about proper use & limits through publications by KOLs and FDA staff, responsible Sponsor education, numerous guidances (NCCN, ASCO, ASH) which are also updated and re-reviewed on a regular basis by prescribers.
4) Appropriate product use, monitoring, patient care can succeed (without restrictions to medical practice, imposed by labeling or REMS certification programs, etc.)
5) Clinical trials conducted outside the US are common in Oncology, and US oncologists can often understand context & limitations in making decisions about local patient care.
6) Patient preferences can also influence product use, but must not be overly affected by marketing, incentives, biases, false assumptions about risks and benefits.
7) Medical/scientific considerations a) Treatment failure or intolerance to standard of care (SOC), used for same indication b) Patients who are not be optimal candidates for available SOC
8) Also value to consider when NOT to use: 1) If appropriate, patients with (asymptomatic/indolent?) disease or others who are better
candidates for available alternatives 2) Avoid diverting patients from more appropriate therapeutic options, or no active therapy
but ‘watch and wait’ when appropriate

Perjeta for Neoadjuvant Treatment of Locally Advanced Breast Cancer
Excerpts from Genentech Briefing Book
• No U.S. patients; N American (Canada) 6.7%
• Enrollment from Europe 58.8%, Asia 22.8%, S America 11.5%
• “The population of patients enrolled in NEOSPHERE was typical of patients with HER2-positive LABC, IBC and EBC enrolled in clinical studies of neoadjuvant therapy”
• “…population consistent with NCCN recommendations regarding patients suitable for neoadjuvant therapy”
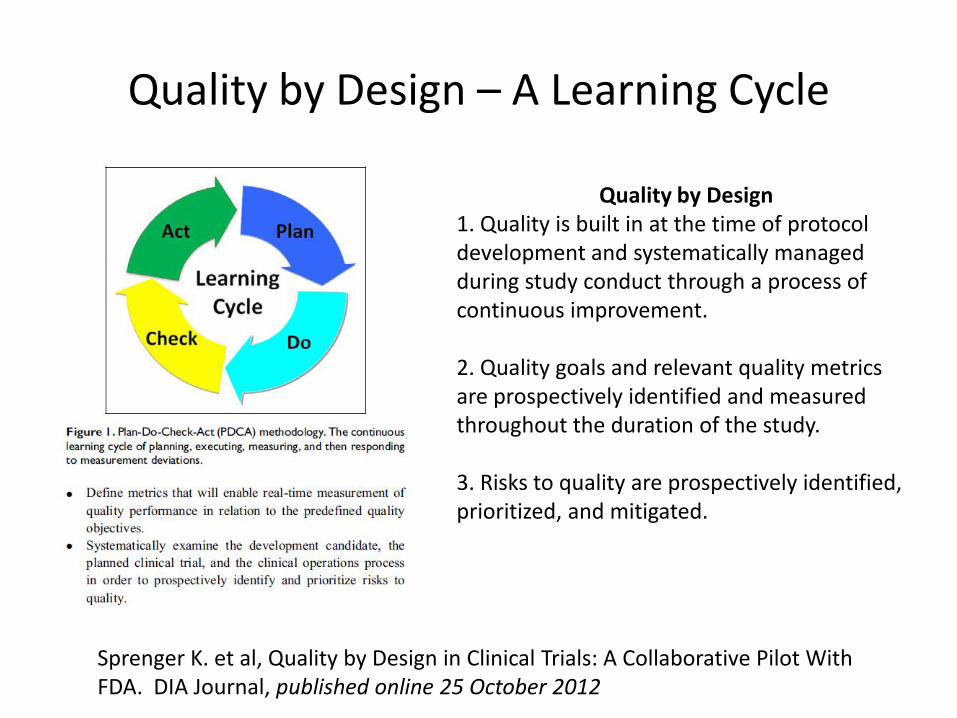
Quality by Design – A Learning Cycle
Quality by Design 1. Quality is built in at the time of protocol development and systematically managed during study conduct through a process of continuous improvement. 2. Quality goals and relevant quality metrics are prospectively identified and measured throughout the duration of the study. 3. Risks to quality are prospectively identified, prioritized, and mitigated.
Sprenger K. et al, Quality by Design in Clinical Trials: A Collaborative Pilot With FDA. DIA Journal, published online 25 October 2012
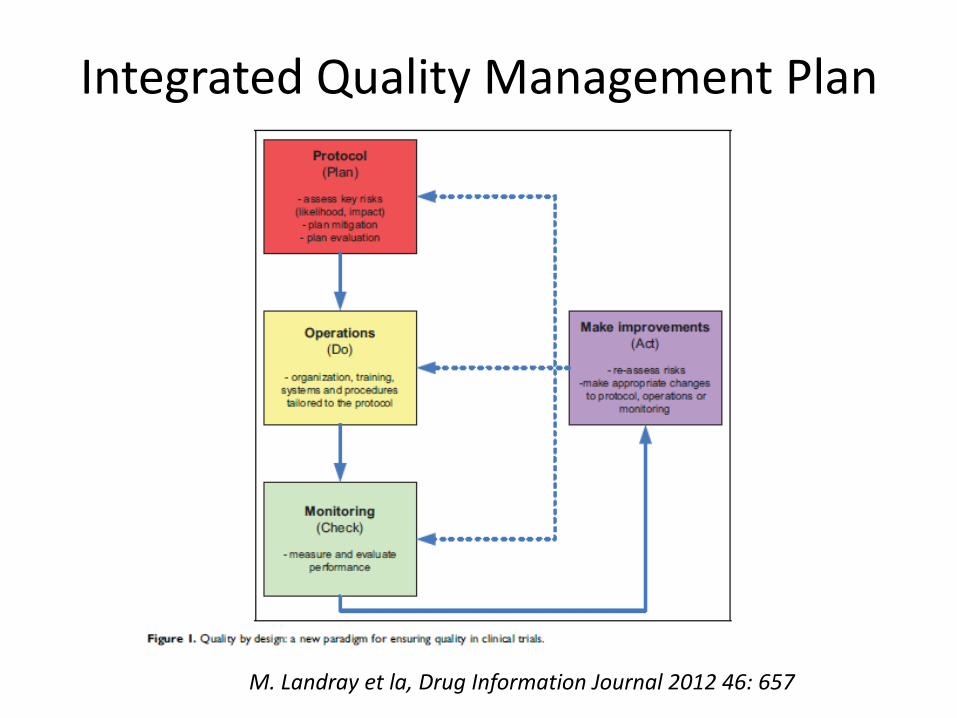
Integrated Quality Management Plan
M. Landray et la, Drug Information Journal 2012 46: 657

Critical to Quality = CTQ Examples about SAE Reporting Compliance, CRA Training metrics
Sprenger K et al, 2012
CTQ Factor
CTQ Measure
SAEs and other reportable information are
reported from site to sponsor in a timely manner
Percentage of SAEs and other reportable
information reported from site to sponsor within
24 hours of investigator awareness
Trials are overseen to monitor existing and/or
identify new/emerging safety signals
Safety review frequency as per SOP
Investigators (principal and sub-investigators) are
appropriately trained prior to performing any
subject-related activities
Percentage of investigators (principal and
subinvestigators) trained on
study-specific requirements for each study
CRAs/monitors are trained prior to performing any study related activities
Percentage of CRAs/monitors trained on study-specific requirements for each study to which they are assigned
FDA advocates pro-active over reactive: “CDER Perspective: Existing practices are generally reactive and resource intensive, not risk adapted. Moreover, they may be poorly suited to preventing or mitigating all critical risks…Modernizing clinical trial oversight is a key initiative of the FDA. …identifying and analyzing risks to trial quality and subject safety and in focusing their resources on addressing the most significant risks. Importantly…free sponsors from the perceived need to mitigate every risk, particularly those risks…expected to have no or minimal impact on data integrity and subject safety.”

Foreign Data- Points to Consider for Sponsor & CRO Recently Published Guidance by FDA Staff
with Quality at Top of List
Khin et al, Clinical Pharmacology & Therapeutics 94:230-242 August 2013

FDA Conducts Its Own Meta-analysis of Outcome by Region Psychiatric Drugs - Khin et al, 2013

Expanding Considerations about CTQ in context of MRCTs
Advice by FDA authors go beyond SAE and CRA training metrics
1) Focus on aspects critical to quality = if not performed correctly, would (a) decrease the trial’s ability to reach reliable conclusions or (b) would materially impact subject safety.
2) Identifying and evaluate important risks critical to quality 3) Modify protocol and tailor implementation & monitoring activities 4) Select investigators with qualifications, training, and resources necessary 5) Early evaluation of feasibility 6) Identify differences in medical practice
7) Identify regional practices that might alter data collection
8) Take steps to minimize the impact of such differences
Khin et al, Regulatory and Scientific Issues Regarding Use of Foreign Data in Support of New Drug Applications in the United States: An FDA Perspective. Clinical Pharmacology & Therapeutics 94:230-242 August 2013

Proactive Options for CTQ - The Sponsor’s Role More than efforts to ‘minimize the impact’ (after the fact)
Case study from Oncology MRCTs Registration protocol designed for product in ER+ Breast cancer – US Standard of Care includes ER status known in >90% patients
1) CRO passes all standard ‘checklist’ based on prior trial experiences, and promotes its working practices for rapid enrollment & data capture
2) Full enrollment enrolled on time, largely ex-US
3) Final pivotal trial shows 30% missing ER status
4) FDA determines that NDA not acceptable
“Molecular marker(s) supersedes region” = a guide to: 1) Applicable to the US 2) A feature Critical to Quality so proactive planning to assure compliance & regulatory/technical validation

Proactive Options for CTQ - The Sponsor’s Role More than efforts to ‘minimize the impact’ (after the fact)
Alternative proactive options 1) Sponsor determines ER Status to be a priority CTQ –
(not urine color nor respiratory rate)
2) Early results show emerging issue with CTQ issues;
3) Investigator Meeting ; ARS test results; prior trials with same sites show CTQ issue with some sites, investigators, coordinators
4) Proactive action plan implemented - long prior to full phase 3 enrollment
5) Final NDA shows < 10% missing ER status; NDA accepted for review
“Molecular marker(s) supersedes region” = a guide to: 1) Applicable to the US 2) A feature Critical to Quality so proactive planning to assure compliance & regulatory/technical validation

Expanding Considerations for CTQ
Case Study from Oncology MRCTs: Benefit-risk assessment based on accurate safety data in eligible pts
1) Pivotal study eligibility requires ECOG 0-1-2 Performance Status
2) Low frequency, high severity (G3-4) organ dysfunction (kidney) reported
without detailed evaluation of baseline characteristics & eligibility
3) Higher event rate in experimental arm v control, perceived by FDA and ODAC to represent direct organ (kidney) toxicity -unrelated to manageable, expected events such as infection, dehydration
4) Totality of data perceived to be insufficient for product approval

Expanding Considerations for CTQ
Alternative proactive options:
1) Earlier trials in same sites show “Renal failure” reports are lab findings (creatinine elevation), diagnostic of dehydration, infection
2) Some pts with prior history of multiple bed sores – ---- reflecting ECOG 3 (ineligible)
3) Site training includes concrete examples of ECOG PS, ARS confirmation 4) Enrolment form requires details to demonstrate proper ECOG Perf Status 5) Site selection avoids investigators enrolling ineligible or unevaluable patients
6) Investigators and coordinators enroll eligible pts with far fewer G3-4 events
7) Reported events more accurately reported and understood by reviewers to be
manageable and expected (Infection, Dehydration) and not organ dysfunction

Performance Status (and its accuracy) Can Influence Treatment Outcomes
Randomized Phase 2 trial Axitinib + Gemcitabine vs Gemcitabine Spano et al, Lancet 371:2101-8, 2008

Initiatives to Improve Quality and Experience in Underdeveloped Regions
In addition to Industry and Regulators, improving quality and conduct of multi-regional clinical trials is a global health, academic, and public concern

MRCT Current Focus Areas See http://globalhealth.harvard.edu/multi-regional-clinical-trials-mrct-center

Initiatives to Broaden US Enrollment Institute of Medicine: Developing Clinical Trials Infrastructure in the US
April 13, 2012
• Ongoing engagement by J Sigal and Friends of Cancer Research • Co-authored by Janet Woodcock, FDA • Topics include standards for contracts, reimbursed medical expenses, privacy, central IRB, regulatory issues, patient education and recruitment
Reference: http://www.ncbi.nlm.nih.gov/books/NBK114674/

Initiatives to Improve Efficiency of US Enrollment through NCI-Sponsored Cooperative Groups
OPERATIONAL EFFICIENCY WORKING GROUP = OEWG
(1) Improved opportunities for (collaborative) trials and enrollment in US
(2) Greater efficiency, improved study conduct & completion
• Common Data Management System & Harmonized/Shared Policies and Procedures
– Ease administrative challenges by 1 general approach; aid transformation to a Network
– Increase ability to address issues related to FDA, other regulatory entities, AE reporting,
common CRFs in a unified manner
• Increased Operational Efficiency and Shared Goals for Accruing to National Trials
– Provide greater confidence regarding commitment to trials by the entire US network reducing
“risk” for industry & other collaborators
– Provide help in key areas of delay in development
(3) Enrollment to uncommon indications (reduced need for foreign data)
• Increased Ability to Conduct Trials in Rare Tumors and Patient Populations within a Transformed
System. Example: Trials in adolescent population between COG & adult Groups
• Opportunities for enrollment of Special Populations, including minority groups

Overview of the Current Program
3,100
Institutions
14,000
Investigators
About
25,000 pts
enrolled on
tx trials
annually
Trials FY2006 FY2007 FY2008 FY2009 FY2010 FY2011
All Phases:
Treatment
Trials
27,263
24,289
25,540
29,063
23,299
19,462
Accrual
Distribution
FY06 – FY10: Phase 3: 82.0%
Phase 2: 15.3%
Phase 1/Pilot: 2.6%
BROAD NATIONAL REPRESENTATION by NCI COOP GROUPS

Improving Minority Enrollment a challenge for dominant enrollment from foreign sites
and developing countries
• Industry options: – CROs mandated to improve recruitment and retention
– Minority focused CROs- Anaclim, Indianapolis, IN
– Direct partnering with minority organizations
• Partnering with the National Medical Association (NMA) and Intermediacy College of Physicians and Surgeons (ICPS)
– Support for NCI initiatives in minority populations e.g. MCCOP
– Provide additional funding with each trial
– Internal initiatives - dedicated to diversity and enrollment
– Track minority enrollment and encourage during site & CRO selection process
– Built-in strategies for retention with each trial
• Prospective data gathering for opinions regarding barriers

Minority Enrollments: 2000-2007 at Ohio State A desired outcome - best achieved when measured.
0
20
40
60
80
100
2000 2001 2002 2003 2004 2005 2006 2007*
Pati
en
ts o
n T
hera
peu
tic T
rials
* projected

Operational Questions
• Will study enroll sufficient proportion from US? – Especially concerns if << 30% treated population
– Option for consultation with FDA review division
• Will sites enroll demographics (incl. race) relevant to US? – Development plans to enrich for special populations and minorities
in same or supplemental trial(s)
• Is indication defined by molecular marker? and does ‘marker supersede region’ in context of therapy
– If yes, proactive plans for ‘quality by design’ to include marker compliance and testing
• Will regional differences in care influence key outcomes? – Proactive approach to CTQ, bias, post-treatment care

Conclusions • Clinical Operations and Foreign Data
– Industry-sponsored trials will continues to enroll from multiple regions outside the US
– More than CRO, the Sponsor needs to be proactively engaged to identify and manage key issues – especially with Foreign Data
• ODAC sessions provide learning opportunities – Gain understanding about what is Critical to Quality for Oncology – Enrollment of special populations, minorities
• Growing partnerships and trial networks – Expand enrollment in the US; and – Strengthen quality of study conduct outside the US

Opportunities for additional learning
39
Pharmaceutical Education & Research Institute (PERI) at PERI Headquarters, Arlington, VA Course title: Cancer: Pathophysiology, Current Therapies, Clinical Trials and Drug Development Planned Topics for October 15-17, 2014 • Conduct of Oncology protocols outside US • Major elements of protocol design and conduct • Summary of recent ODAC sessions, consensus, considerations for benefit-risk and US approval & label • Faculty from FDA, CROs, Industry, NCI • Experience with new targeted therapies, immunotherapies (PD1, CTLA4, vaccines, antibody-drug conjugates)
• See www.peri.org for full agenda