
COMPREHENSIVE INVITED REVIEW
Understanding the pKa of Redox Cysteines:The Key Role of Hydrogen Bonding
Goedele Roos,1–4 Nicolas Foloppe,5 and Joris Messens2–4
Abstract
Many cellular functions involve cysteine chemistry via thiol–disulfide exchange pathways. The nucleophilic cys-teines of the enzymes involved are activated as thiolate. A thiolate is much more reactive than a neutral thiol.Therefore, determining and understanding the pKas of functional cysteines are important aspects of biochemistryand molecular biology with direct implications for redox signaling. Here, we describe the experimental andtheoretical methods to determine cysteine pKa values, and we examine the factors that control these pKas. Drawinglargely on experience gained with the thioredoxin superfamily, we examine the roles of solvation, charge–charge,helix macrodipole, and hydrogen bonding interactions as pKa-modulating factors. The contributions of these factorsin influencing cysteine pKas and the associated chemistry, including the relevance for the reaction kinetics andthermodynamics, are discussed. This analysis highlights the critical role of direct hydrogen bonding to the cysteinesulfur as a key factor modulating the equilibrium between thiol S–H and thiolate S - . This role is easily understoodintuitively and provides a framework for biochemical functional insights. Antioxid. Redox Signal. 18, 94–127.
I. Introduction 94II. pKa Determination Methods 95
A. Experimental approaches 105B. Computational methods 107C. Future perspective for pKa calculations applied to cysteines 110
III. Factors That Control the pKa Values of Cysteine Thiols in Proteins 111A. Limited role of charged side chains and long-range electrostatics 111B. The strong influence of direct hydrogen bonds on the pKa of cysteines 113C. Reinterpretation of the helical effect on the pKas of cysteines 115D. How general are the mechanisms modulating the pKa of cysteines? 118
IV. Functional Properties Influenced by the Cysteine pKas 119V. Conclusions 121
I. Introduction
Cysteine residues are one of the least-abundant aminoacids, but are actively involved in many ways in protein
functions (109, 110). Consistent with their functional role andability to react chemically, cysteines are frequently foundconserved. They are critical for the activity in oxidases, re-
ductases, disulfide isomerases, and peroxidases (32) (Fig. 1).These enzymes play an important role in the redox homeo-stasis of cells. They are involved in the thiol–disulfide ex-change reactions during oxidative protein folding, and inantioxidant defense mechanisms of the cell. Cysteine thiolsare also essential in cell-cycle-regulating enzymes, like phos-phatases and cysteine proteases. Thus, numerous enzymes
Reviewing Editors: Claudia Blindauer, Sharom L. Campbell, Jeffrey Dickhout, James Fishbein, Cristina Furdui, Vadim Gladyshev, KristineJensen, John Mieyal, Corinne Sebban-Kreuzer, and Mark Wilson.
1General Chemistry, Vrije University Brussel, Brussels, Belgium.2Department of Structural Biology, Vlaams Instituut voor Biotechnologie (VIB), Brussels, Belgium.3Structural Biology Brussels, Vrije University Brussel, Brussels, Belgium.4Brussels Center for Redox Biology, Brussels, Belgium.551 Natal Road, Cambridge, United Kingdom.
ANTIOXIDANTS & REDOX SIGNALINGVolume 18, Number 1, 2013ª Mary Ann Liebert, Inc.DOI: 10.1089/ars.2012.4521
94

depend on redox-active cysteines, the pKa of which is a de-termining factor for their reactivity and nucleophilicity (183).The pKa of a cysteine represents the balance of the equilibriumthiol S–H % thiolate S - . Since thiolates are much more re-active than neutral thiols, they are critical to the function ofmany cysteines. It is therefore important to characterize thepKas of cysteines, not only experimentally but also compu-tationally. These tasks remain challenging; however, muchprogress has been achieved in these areas and in trying tounderstand the factors that modulate the cysteine pKas.
The intrinsic pKa value for the free cysteine thiol–thiolateequilibrium in an aqueous solution is close to 8.6 (26, 39, 82,89, 169). In folded proteins, this pKa can be shifted by the in-fluence of the three-dimensional protein structure (13). Polargroups (charged or neutral) in the vicinity of a cysteine, and/ora different solvation environment compared to an aqueoussolution, can influence the pKa of a cysteine thiol. Underphysiological conditions (pH *7), a thiol with a pKa valuebelow 7 will exist mostly as a more reactive thiolate, critical forcatalysis (27). Note that physiological conditions are not alwaysat pH *7, since different cellular organelles have different pHvalues. Lowered pKa values of catalytic cysteines influence thereaction kinetics and thermodynamics, and strongly influencethe catalytic efficiency of an enzyme during thiol–disulfideexchange reactions. The effect of pKa lowering on reaction rateenhancement will in general be most significant when the pKa
values are close to the solution pH (78). Perturbed pKa valuesalso influence protein stability (189).
With accurate cysteine thiol pKa values, one gains insightsinto catalytic mechanisms and into the factors influencing thepKa values. It has long been known that the pKa values of cat-alytic cysteines in thiol–disulfide oxidoreductases of the thior-edoxin (Trx) superfamily can adopt a wide range of values inproteins with a similar structural fold (19). Therefore, this su-perfamily of enzymes provides a paradigm to study the factorsinfluencing and modulating the pKa of thiol groups (35, 43, 45,49, 56, 61, 77, 117, 129, 132, 142, 179). A thorough compilation ofexperimentally measured pKa values for these enzymes (andrelated model systems) should be a helpful resource (Table 1).
II. pKa Determination Methods
The pKa of a cysteine thiol group can be obtained from theequilibrium constant Ka for the deprotonation reaction:
CysSH%CysS� þHþ (Eq: 1)
with
Ka¼[Hþ ][CysS� ]
[CysSH]and pKa¼ � logKa (Eq: 2)
in which [H + ], [CysS - ], and [CysSH] are the equilibriumconcentrations given in mol/l.
FIG. 1. Cysteines in thiol–disulfide exchange reactions catalyzed by Trx-fold enzymes. Cysteines present in a thiolateform at physiological pH are more sensitive to reactive oxygen species (ROS). Exposure to hydrogen peroxide (H2O2) leads to theoxidation of the thiol group into the reversible sulfenic acid, whereas further exposure leads to irreversible cysteine oxidationstates: sulfinic acid (-SO2H) and sulfonic acid (-SO3H). These higher oxidation states are considered as irreversible, since no generalsulfinic or sulfonic acid reductase enzymes have been identified yet (144). Human sulfiredoxin is the only known exception (186).Sulfenic acids are protected from irreversible oxidation by different mechanisms: disulfide formation with another cysteine, andmixed disulfide formation with low-molecular-weight thiols (LMW thiols; e.g., S-glutathionylation). Disulfide bond formationdoes not always proceed via a sulfenic acid intermediate, but can also result from the oxidation of two cysteine residues byoxidative protein-folding catalysts (e.g., disulfide-binding protein A [DsbA]). Protection with the cysteine side chain by reactionwith a backbone amide nitrogen to form a sulfonamide is not shown. The cysteines of the enzymes with a thioredoxin (Trx)-foldare essential to catalyze the disulfide bond formation and reduction. (To see this illustration in color, the reader is referred to theweb version of this article at www.liebertpub.com/ars.)
PKA OF REDOX CYSTEINES 95

Ta
bl
e1.
Ex
pe
rim
en
ta
ll
yM
ea
su
re
dpK
as
fo
rT
hio
lG
ro
up
sin
Pr
ot
ein
so
ft
he
Th
io
re
do
xin
Su
pe
rfa
mil
ly
Sy
stem
Res
idu
eR
epor
ted
pK
av
alu
eaE
xp
erim
enta
lm
eth
odR
efer
ence
sbC
omm
ents
Trx
wil
dty
pe
Esc
her
ich
iaco
liT
rxw
ild
-ty
pe
(-C
32-G
33-P
34-C
35-)
Cy
s32
6.7
pH
dep
end
ence
of
enzy
me
reac
tio
nw
ith
iod
oac
etic
acid
and
iod
oac
etam
ide
(82)
Th
ep
Ka
of
Cy
s35
app
eare
dto
be
abo
ve
(bu
tcl
ose
to)
9
E.
coli
Trx
wil
d-t
yp
e(-
C32
-G33
-P34
-C35
-)C
ys3
27.
1R
aman
spec
tro
sco
py
(101
)R
aman
spec
tro
sco
py
use
dto
pro
be
cyst
ein
eti
trat
ion
s,re
do
xst
ate,
and
con
form
atio
ns
E.
coli
Trx
wil
d-t
yp
e(-
C32
-G33
-P34
-C35
-)C
ys3
57.
9R
aman
spec
tro
sco
py
(101
)R
aman
spec
tro
sco
py
use
dto
pro
be
cyst
ein
eti
trat
ion
s,re
do
xst
ate,
and
con
form
atio
ns
E.
coli
Trx
wil
d-t
yp
e(-
C32
-G33
-P34
-C35
-)C
ys3
2an
dC
ys3
59–
10p
Hd
epen
den
ceo
feq
uil
ibri
um
reac
tio
nw
ith
glu
tath
ion
ean
dd
irec
tU
Vab
sorb
ance
(166
)T
he
pK
as
infe
rred
inth
isst
ud
yh
ave
bee
nco
ntr
ov
ersi
al,
and
are
inte
rpre
tati
on
of
thes
ere
sult
sis
no
wg
ener
ally
acce
pte
d(3
8).
E.
coli
Trx
wil
d-t
yp
e(-
C32
-G33
-P34
-C35
-)C
ys3
27.
4p
Hd
epen
den
ceo
fN
MR
chem
ical
shif
ts(7
7)C
on
sist
ent
wit
hp
Ka
der
ived
fro
mU
Vm
easu
rem
ents
E.
coli
Trx
wil
d-t
yp
e(-
C32
-G33
-P34
-C35
-)C
ys3
59.
5p
Hd
epen
den
ceo
fN
MR
chem
ical
shif
ts(7
7)C
on
sist
ent
wit
hp
Ka
der
ived
fro
mU
Vm
easu
rem
ents
E.
coli
Trx
wil
d-t
yp
e(-
C32
-G33
-P34
-C35
-)C
ys3
27.
1U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(38)
Co
nsi
sten
tw
ith
pK
ad
eriv
edfr
om
NM
Rm
easu
rem
ents
E.
coli
Trx
wil
d-t
yp
e(-
C32
-G33
-P34
-C35
-)C
ys3
59.
9U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(38)
Co
nsi
sten
tw
ith
pK
ad
eriv
edfr
om
NM
Rm
easu
rem
ents
E.
coli
Trx
wil
d-t
yp
e(-
C32
-G33
-P34
-C35
-)C
ys3
27.
13p
Hd
epen
den
ceo
fre
acti
on
wit
hio
do
acet
amid
e(1
17)
Val
idat
edfu
rth
erth
em
eth
od
rely
ing
on
the
pH
-dep
end
ent
reac
tio
nw
ith
iod
oac
etam
ide,
toes
tim
ate
thio
lp
Kas
E.
coli
Trx
wil
d-t
yp
e(-
C32
-P33
-Y34
-C35
-)C
ys3
27.
0U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(139
)T
oco
mp
are
toth
eI7
5Tm
uta
nt
inth
esa
me
stu
dy
E.
coli
Trx
2w
ild
-ty
pe
(-C
64-G
65-P
66-C
67-)
Cy
s64
5.1
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(4
0)T
he
pK
ao
fth
eac
tiv
e-si
ten
ucl
eop
hil
iccy
stei
ne
of
Trx
2is
low
erth
anin
Trx
1,co
nsi
sten
tw
ith
Trx
2b
ein
gm
ore
ox
idiz
ing
than
Trx
1.
Trx
mu
tan
ts
E.
coli
Trx
mu
tan
tD
26A
Cy
s32
8.0
pH
dep
end
ence
of
NM
Rch
emic
alsh
ifts
(38)
D26
isco
nse
rved
,b
uri
edn
ear
the
Trx
acti
ve
site
,an
dth
ou
gh
tto
be
imp
ort
ant
for
Trx
fun
ctio
n
E.
coli
Trx
mu
tan
tD
26A
Cy
s32
7.8
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(3
8)D
26is
con
serv
ed,
bu
ried
nea
rth
eT
rxac
tiv
esi
te,
and
tho
ug
ht
tob
eim
po
rtan
tfo
rT
rxfu
nct
ion
E.
coli
Trx
mu
tan
tK
57M
Cy
s32
8.0
pH
dep
end
ence
of
NM
Rch
emic
alsh
ifts
(38)
K57
form
sa
bu
ried
salt
bri
dg
ew
ith
D26
inth
ev
icin
ity
of
the
acti
ve
site
E.
coli
Trx
mu
tan
tK
57M
Cy
s32
8.1
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(3
8)K
57fo
rms
ab
uri
edsa
ltb
rid
ge
wit
hD
26in
the
vic
init
yo
fth
eac
tiv
esi
te
(con
tin
ued
)
96
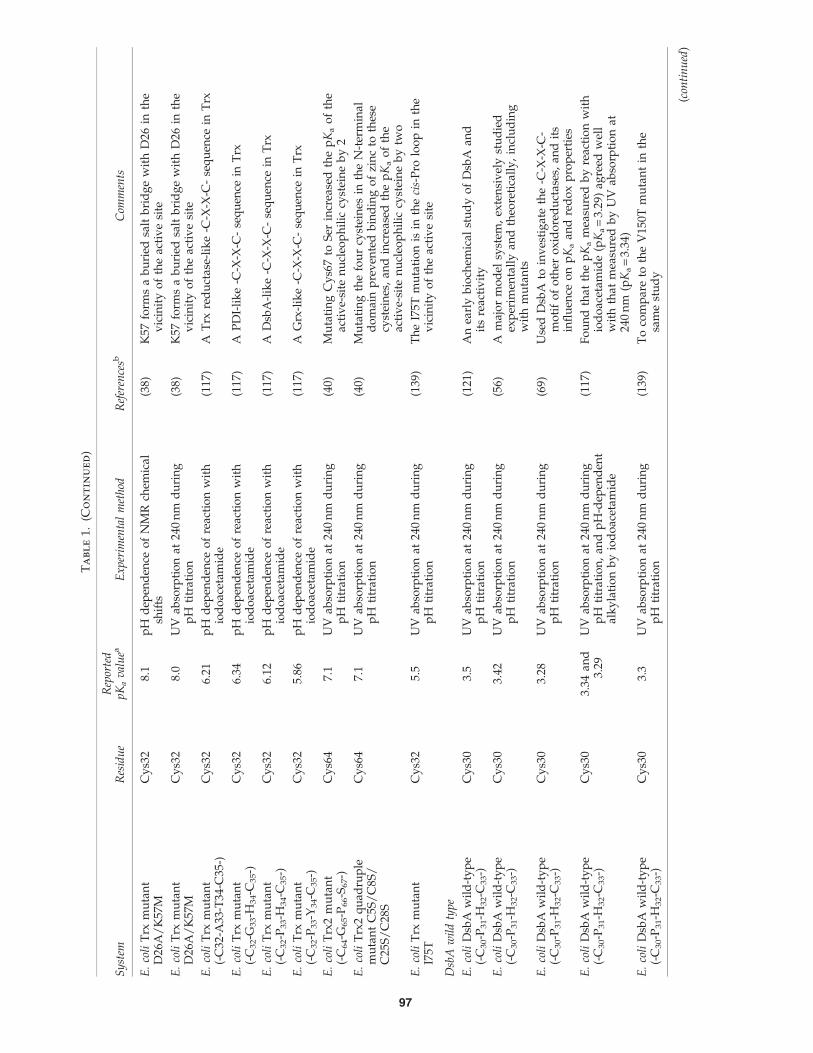
Ta
bl
e1.
(Co
nt
in
ue
d)
Sy
stem
Res
idu
eR
epor
ted
pK
av
alu
eaE
xp
erim
enta
lm
eth
odR
efer
ence
sbC
omm
ents
E.
coli
Trx
mu
tan
tD
26A
/K
57M
Cy
s32
8.1
pH
dep
end
ence
of
NM
Rch
emic
alsh
ifts
(38)
K57
form
sa
bu
ried
salt
bri
dg
ew
ith
D26
inth
ev
icin
ity
of
the
acti
ve
site
E.
coli
Trx
mu
tan
tD
26A
/K
57M
Cy
s32
8.0
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(3
8)K
57fo
rms
ab
uri
edsa
ltb
rid
ge
wit
hD
26in
the
vic
init
yo
fth
eac
tiv
esi
te
E.
coli
Trx
mu
tan
t(-
C32
-A33
-T34
-C35
-)C
ys3
26.
21p
Hd
epen
den
ceo
fre
acti
on
wit
hio
do
acet
amid
e(1
17)
AT
rxre
du
ctas
e-li
ke
-C-X
-X-C
-se
qu
ence
inT
rx
E.
coli
Trx
mu
tan
t(-
C3
2-G
33-H
34-C
35-)
Cy
s32
6.34
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(117
)A
PD
I-li
ke
-C-X
-X-C
-se
qu
ence
inT
rx
E.
coli
Trx
mu
tan
t(-
C3
2-P
33-H
34-C
35-)
Cy
s32
6.12
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(117
)A
Dsb
A-l
ike
-C-X
-X-C
-se
qu
ence
inT
rx
E.
coli
Trx
mu
tan
t(-
C3
2-P
33-Y
34-C
35-)
Cy
s32
5.86
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(117
)A
Grx
-lik
e-C
-X-X
-C-
seq
uen
cein
Trx
E.
coli
Trx
2m
uta
nt
(-C
64-G
65-P
66-S
67-)
Cy
s64
7.1
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(4
0)M
uta
tin
gC
ys6
7to
Ser
incr
ease
dth
ep
Ka
of
the
acti
ve-
site
nu
cleo
ph
ilic
cyst
ein
eb
y2
E.
coli
Trx
2q
uad
rup
lem
uta
nt
C5S
/C
8S/
C25
S/
C28
S
Cy
s64
7.1
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(4
0)M
uta
tin
gth
efo
ur
cyst
ein
esin
the
N-t
erm
inal
do
mai
np
rev
ente
db
ind
ing
of
zin
cto
thes
ecy
stei
nes
,an
din
crea
sed
the
pK
ao
fth
eac
tiv
e-si
ten
ucl
eop
hil
iccy
stei
ne
by
two
E.
coli
Trx
mu
tan
tI7
5TC
ys3
25.
5U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(139
)T
he
I75T
mu
tati
on
isin
the
cis-
Pro
loo
pin
the
vic
init
yo
fth
eac
tiv
esi
te
Dsb
Aw
ild
typ
e
E.
coli
Dsb
Aw
ild
-ty
pe
(-C
30-P
31-H
32-C
33-)
Cy
s30
3.5
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(1
21)
An
earl
yb
ioch
emic
alst
ud
yo
fD
sbA
and
its
reac
tiv
ity
E.
coli
Dsb
Aw
ild
-ty
pe
(-C
30-P
31-H
32-C
33-)
Cy
s30
3.42
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(5
6)A
maj
or
mo
del
syst
em,
exte
nsi
vel
yst
ud
ied
exp
erim
enta
lly
and
theo
reti
call
y,
incl
ud
ing
wit
hm
uta
nts
E.
coli
Dsb
Aw
ild
-ty
pe
(-C
30-P
31-H
32-C
33-)
Cy
s30
3.28
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(6
9)U
sed
Dsb
Ato
inv
esti
gat
eth
e-C
-X-X
-C-
mo
tif
of
oth
ero
xid
ore
du
ctas
es,
and
its
infl
uen
ceo
np
Ka
and
red
ox
pro
per
ties
E.
coli
Dsb
Aw
ild
-ty
pe
(-C
30-P
31-H
32-C
33-)
Cy
s30
3.34
and
3.29
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n,
and
pH
-dep
end
ent
alk
yla
tio
nb
yio
do
acet
amid
e
(117
)F
ou
nd
that
the
pK
am
easu
red
by
reac
tio
nw
ith
iod
oac
etam
ide
(pK
a=
3.29
)ag
reed
wel
lw
ith
that
mea
sure
db
yU
Vab
sorp
tio
nat
240
nm
(pK
a=
3.34
)
E.
coli
Dsb
Aw
ild
-ty
pe
(-C
30-P
31-H
32-C
33-)
Cy
s30
3.3
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(1
39)
To
com
par
eto
the
V15
0Tm
uta
nt
inth
esa
me
stu
dy
(con
tin
ued
)
97

Ta
bl
e1.
(Co
nt
in
ue
d)
Sy
stem
Res
idu
eR
epor
ted
pK
av
alu
eaE
xp
erim
enta
lm
eth
odR
efer
ence
sbC
omm
ents
Vib
rio
chol
erae
Dsb
Aw
ild
-ty
pe
(-C
49-P
50-H
51-C
52-)
Cy
s49
5.1
Kin
etic
so
fo
xid
atio
no
fa
sub
stra
tep
epti
de
mo
nit
ore
db
ytr
yp
top
han
flu
ore
scen
ce
(146
)T
he
pK
aw
asat
trib
ute
dto
the
acti
ve-
site
reac
tiv
ecy
stei
ne,
and
infe
rred
ind
irec
tly
fro
mk
inet
icm
easu
rem
ents
,w
hic
hm
ayac
cou
nt
for
the
surp
risi
ng
lyh
igh
pK
a
rep
ort
edfo
rC
ys4
9
Dsb
Am
uta
nts
E.
coli
Dsb
Am
uta
nt
(-C
30-S
31-V
32-C
33-)
Cy
s30
4.23
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(5
6)R
and
om
mu
tag
enes
iso
fth
e-C
-X-X
-C-
acti
ve-
site
seq
uen
ceo
fD
sbA
tote
stef
fect
on
red
ox
po
ten
tial
and
pK
ao
fC
ys3
0
E.
coli
Dsb
Am
uta
nt
(-C
30-S
31-F
32-C
33-)
Cy
s30
4.34
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(5
6)R
and
om
mu
tag
enes
iso
fth
e-C
-X-X
-C-
acti
ve-
site
seq
uen
ceo
fD
sbA
tote
stef
fect
on
red
ox
po
ten
tial
and
pK
ao
fC
ys3
0
E.
coli
Dsb
Am
uta
nt
(-C
30-P
31-L
32-C
33- )
Cy
s30
4.42
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(5
6)R
and
om
mu
tag
enes
iso
fth
e-C
-X-X
-C-
acti
ve-
site
seq
uen
ceo
fD
sbA
tote
stef
fect
on
red
ox
po
ten
tial
and
pK
ao
fC
ys3
0
E.
coli
Dsb
Am
uta
nt
(-C
30-S
31-T
32-C
33-)
Cy
s30
4.45
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(5
6)R
and
om
mu
tag
enes
iso
fth
e-C
-X-X
-C-
acti
ve-
site
seq
uen
ceo
fD
sbA
tote
stef
fect
on
red
ox
po
ten
tial
and
pK
ao
fC
ys3
0
E.
coli
Dsb
Am
uta
nt
(-C
30-Q
31-L
32-C
33-)
Cy
s30
4.59
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(5
6)R
and
om
mu
tag
enes
iso
fth
e-C
-X-X
-C-
acti
ve-
site
seq
uen
ceo
fD
sbA
tote
stef
fect
on
red
ox
po
ten
tial
and
pK
ao
fC
ys3
0
E.
coli
Dsb
Am
uta
nt
(-C
30-T
31-R
32-C
33-)
Cy
s30
4.76
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(5
6)R
and
om
mu
tag
enes
iso
fth
e-C
-X-X
-C-
acti
ve-
site
seq
uen
ceo
fD
sbA
tote
stef
fect
on
red
ox
po
ten
tial
and
pK
ao
fC
ys3
0
E.
coli
Dsb
Am
uta
nt
(-C
30-L
31-T
32-C
33-)
Cy
s30
4.86
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(5
6)R
and
om
mu
tag
enes
iso
fth
e-C
-X-X
-C-
acti
ve-
site
seq
uen
ceo
fD
sbA
tote
stef
fect
on
red
ox
po
ten
tial
and
pK
ao
fC
ys3
0
E.
coli
Dsb
Am
uta
nt
(-C
30-P
31-P
32-C
33-)
Cy
s30
6.73
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(5
6)R
and
om
mu
tag
enes
iso
fth
e-C
-X-X
-C-
acti
ve-
site
seq
uen
ceo
fD
sbA
tote
stef
fect
on
red
ox
po
ten
tial
and
pK
ao
fC
ys3
0
E.
coli
Dsb
Am
uta
nt
(-C
30-P
31-G
32-C
33-)
Cy
s30
4.85
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(6
9)H
is32
Gly
mu
tati
on
intr
od
uce
dto
test
the
infl
uen
ceo
fel
ectr
ost
atic
sas
soci
ated
wit
hH
is32
E.
coli
Dsb
Am
uta
nt
(-C
30-G
31-H
32-C
33-)
Cy
s30
3.71
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(6
9)A
PD
I-li
ke
-C-X
-X-C
-se
qu
ence
inD
sbA
E.
coli
Dsb
Am
uta
nt
(-C
30-A
31-T
32-C
33-)
Cy
s30
4.34
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(6
9)A
Trx
red
uct
ase-
lik
e-C
-X-X
-C-
seq
uen
cein
Dsb
A
(con
tin
ued
)
98

Ta
bl
e1.
(Co
nt
in
ue
d)
Sy
stem
Res
idu
eR
epor
ted
pK
av
alu
eaE
xp
erim
enta
lm
eth
odR
efer
ence
sbC
omm
ents
E.
coli
Dsb
Am
uta
nt
(-C
30-P
31-Y
32-C
33-)
Cy
s30
3.75
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(6
9)A
Grx
-lik
e-C
-X-X
-C-
seq
uen
cein
Dsb
A
E.
coli
Dsb
Am
uta
nt
(-C
30-G
31-P
32-C
33-)
Cy
s30
6.21
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(6
9)A
Trx
-lik
e-C
-X-X
-C-
seq
uen
cein
Dsb
A
E.
coli
Dsb
Am
uta
nt
E37
QC
ys3
03.
69U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(66)
Th
em
uta
tio
no
fE
37in
the
vic
init
yo
fth
eac
tiv
esi
ted
idn
ot
resu
ltin
asi
gn
ifica
nt
chan
ge
of
the
pK
ao
fca
taly
tic
Cy
s30
E.
coli
Dsb
Am
uta
nt
E38
QC
ys3
03.
52U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(66)
Th
em
uta
tio
no
fE
38in
the
vic
init
yo
fth
eac
tiv
esi
ted
idn
ot
resu
ltin
asi
gn
ifica
nt
chan
ge
of
the
pK
ao
fca
taly
tic
Cy
s30
E.
coli
Dsb
Am
uta
nt
E37
Q/
E38
QC
ys3
03.
84U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(66)
Mu
tati
on
of
bo
thE
37an
dE
38in
the
vic
init
yo
fth
eD
sbA
acti
ve
site
E.
coli
Dsb
Am
uta
nt
DE
38V
39L
40C
ys3
03.
92U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(66)
Del
etio
no
ftr
ipep
tid
eE
38V
39L
40,
tom
imic
the
corr
esp
on
din
gh
elix
of
Trx
and
Grx
E.
coli
Dsb
Am
uta
nt
DE
38V
39L
40/
H41
PC
ys3
03.
95U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(66)
Del
etio
no
ftr
ipep
tid
eE
38V
39L
40an
dH
41P
mu
tati
on
,to
mim
icev
enm
ore
clo
sely
the
corr
esp
on
din
gh
elix
of
Trx
E.
coli
Dsb
Am
uta
nt
E24
QC
ys3
03.
52U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(74)
Th
ism
uta
tio
nin
the
vic
init
yo
fth
eac
tiv
esi
telo
wer
edth
ep
Ka
of
cata
lyti
cC
ys3
0b
y0.
03re
lati
ve
toth
ew
ild
typ
e
E.
coli
Dsb
Am
uta
nt
K58
MC
ys3
03.
19U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(74)
Th
ism
uta
tio
nin
the
vic
init
yo
fth
eac
tiv
esi
telo
wer
edth
ep
Ka
of
cata
lyti
cC
ys3
0b
y0.
36re
lati
ve
toth
ew
ild
typ
e
E.
coli
Dsb
Am
uta
nt
E37
QC
ys3
03.
69U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(74)
Th
ism
uta
tio
nin
the
vic
init
yo
fth
eac
tiv
esi
tein
crea
sed
the
pK
ao
fca
taly
tic
Cy
s30
by
0.03
rela
tiv
eto
the
wil
dty
pe
E.
coli
Dsb
Am
uta
nt
E24
Q/
K58
MC
ys3
04.
46U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(74)
Th
ism
uta
nt
inth
ev
icin
ity
of
the
acti
ve
site
incr
ease
dth
ep
Ka
of
cata
lyti
cC
ys3
0b
y0.
91re
lati
ve
toth
ew
ild
typ
e
E.
coli
Dsb
Am
uta
nt
E24
Q/
E37
QC
ys3
03.
81U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(74)
Th
ism
uta
nt
inth
ev
icin
ity
of
the
acti
ve
site
incr
ease
dth
ep
Ka
of
cata
lyti
cC
ys3
0b
y0.
26re
lati
ve
toth
ew
ild
typ
e
E.
coli
Dsb
Am
uta
nt
E37
Q/
K58
MC
ys3
03.
50U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(74)
Th
ism
uta
nt
inth
ev
icin
ity
of
the
acti
ve
site
dec
reas
edth
ep
Ka
of
cata
lyti
cC
ys3
0b
y0.
05re
lati
ve
toth
ew
ild
typ
e
E.
coli
Dsb
Am
uta
nt
E24
Q/
E37
Q/
K58
MC
ys3
03.
94U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(74)
Th
ism
uta
nt
inth
ev
icin
ity
of
the
acti
ve
site
incr
ease
dth
ep
Ka
of
cata
lyti
cC
ys3
0b
y0.
39re
lati
ve
toth
ew
ild
typ
e
(con
tin
ued
)
99

Ta
bl
e1.
(Co
nt
in
ue
d)
Sy
stem
Res
idu
eR
epor
ted
pK
av
alu
eaE
xp
erim
enta
lm
eth
odR
efer
ence
sbC
omm
ents
E.
coli
Dsb
Am
uta
nt
E24
Q/
E37
Q/
E38
Q/
K58
MC
ys3
03.
89U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(74)
Th
ism
uta
nt
inth
ev
icin
ity
of
the
acti
ve
site
incr
ease
dth
ep
Ka
of
cata
lyti
cC
ys3
0b
y0.
34re
lati
ve
toth
ew
ild
typ
e
E.
coli
Dsb
Am
uta
nt
V15
0TC
ys3
03.
5U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(139
)T
he
V15
0Tm
uta
nti
on
isin
the
cis-
Pro
loo
pin
the
vic
init
yo
fth
eac
tiv
esi
te
Glu
tare
dox
ins
wil
dty
pe
Hu
man
Grx
1w
ild
typ
eN
D3.
5p
Hd
epen
den
ceo
fio
do
acet
amid
een
zym
ein
acti
vat
ion
(116
)T
he
pK
ao
f3.
5is
mo
stli
kel
yth
ato
fth
en
ucl
eop
hil
icC
ys2
2in
the
acti
ve
site
.T
he
glu
tare
do
xin
isca
lled
thio
ltra
nsf
eras
e.
Hu
man
Grx
1C
7S/
C78
S/
C82
SC
ys2
23.
6p
Hd
epen
den
ceo
fio
do
acet
amid
een
zym
ein
acti
vat
ion
(75)
Th
isco
nst
ruct
may
be
con
sid
ered
rep
rese
nta
tiv
eo
fth
ew
ild
typ
e,si
nce
the
3m
uta
ted
cyst
ein
esar
efa
rfr
om
the
acti
ve
site
.
Yea
stG
rxw
ild
typ
eC
ys2
6<
4p
Hd
epen
den
ceo
fio
do
acet
amid
een
zym
ein
acti
vat
ion
(48)
Th
eg
luta
red
ox
inis
call
edth
iolt
ran
sfer
ase.
E.
coli
Grx
1w
ild
typ
eC
ys1
1<
5E
xp
erim
enta
lp
roto
col
no
tre
po
rted
(9)
Men
tio
ned
asu
np
ub
lish
edw
ork
E.
coli
Grx
3(C
65Y
mu
tan
t)C
ys1
14.
1U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(45)
Th
isco
nst
ruct
isa
rep
rese
nta
tiv
eo
fth
ew
ild
typ
e,si
nce
the
C65
Ym
uta
tio
nis
far
fro
mth
e-C
-X-X
-C-
mo
tif.
Cy
s11
titr
atio
no
ccu
rsco
ncu
rren
tly
wit
hp
rote
inu
nfo
ldin
gat
low
pH
.
Pig
Grx
wil
dty
pe
Cy
s22
2.5
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etic
acid
(47)
Th
ees
tim
ated
pK
av
alu
eo
f2.
5w
assu
bse
qu
entl
yre
vis
edto
be
3.8
in(1
90).
Pig
Grx
Cy
s22
3.8
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(190
)A
theo
reti
cal
stu
dy
has
rati
on
aliz
edm
uch
of
this
exp
erim
enta
lb
ioch
emic
alw
ork
.
Grx
sm
uta
nts
Hu
man
Grx
1C
7S/
C25
S/
C78
S/
C82
SC
ys2
24.
2p
Hd
epen
den
ceo
fio
do
acet
amid
een
zym
ein
acti
vat
ion
(75)
Ref
erre
dto
assi
ng
le-c
yst
ein
eco
nst
ruct
inth
est
ud
y:
SC
-Grx
.T
he
C25
Sm
uta
tio
nis
inth
eac
tiv
esi
te.
Hu
man
Grx
1C
7S/
C25
S/
C78
S/
C82
SK
19L
Cy
s22
4.6
pH
dep
end
ence
of
iod
oac
etam
ide
enzy
me
inac
tiv
atio
n(7
5)R
efer
red
toas
sin
gle
-cy
stei
ne
con
stru
ctin
the
stu
dy
:S
C-G
rx.
Tes
tsth
eef
fect
of
K19
on
the
pK
ao
fca
taly
tic
Cy
s22
Hu
man
Grx
1C
7S/
C25
S/
C78
S/
C82
SK
19Q
Cy
s22
5.0
pH
dep
end
ence
of
iod
oac
etam
ide
enzy
me
inac
tiv
atio
n(7
5)R
efer
red
toas
sin
gle
-cy
stei
ne
con
stru
ctin
the
stu
dy
:S
C-G
rx.
Tes
tsth
eef
fect
of
K19
on
the
pK
ao
fca
taly
tic
Cy
s22
Hu
man
Grx
1C
7S/
C78
S/
C82
SK
19L
Cy
s22
3.7
pH
dep
end
ence
of
iod
oac
etam
ide
enzy
me
inac
tiv
atio
n(7
5)R
efer
red
toas
trip
le-m
uta
nt
con
stru
ctin
the
stu
dy
:T
M-G
rx.
Tes
tsth
eef
fect
of
K19
on
the
pK
ao
fca
taly
tic
Cy
s22
(con
tin
ued
)
100

Ta
bl
e1.
(Co
nt
in
ue
d)
Sy
stem
Res
idu
eR
epor
ted
pK
av
alu
eaE
xp
erim
enta
lm
eth
odR
efer
ence
sbC
omm
ents
Hu
man
Grx
1C
7S/
C78
S/
C82
SK
19Q
Cy
s22
3.7
pH
dep
end
ence
of
iod
oac
etam
ide
enzy
me
inac
tiv
atio
n(7
5)R
efer
red
toas
trip
le-m
uta
nt
con
stru
ctin
the
stu
dy
:T
M-G
rx.
Tes
tsth
eef
fect
of
K19
on
the
pK
ao
fca
taly
tic
Cy
s22
Pig
Grx
mu
tan
tK
27Q
Cy
s22
4.3
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(190
)A
theo
reti
cal
stu
dy
has
rati
on
aliz
edm
uch
of
the
exp
erim
enta
lb
ioch
emic
alw
ork
Pig
Grx
mu
tan
tC
78S
/C
82S
Cy
s22
4.4
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(190
)A
theo
reti
cal
stu
dy
has
rati
on
aliz
edm
uch
of
the
exp
erim
enta
lb
ioch
emic
alw
ork
Pig
Grx
mu
tan
tC
25S
Cy
s22
4.9
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(190
)A
theo
reti
cal
stu
dy
has
rati
on
aliz
edm
uch
of
the
exp
erim
enta
lb
ioch
emic
alw
ork
Pig
Grx
mu
tan
tC
25A
Cy
s22
5.9
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(190
)A
theo
reti
cal
stu
dy
has
rati
on
aliz
edm
uch
of
the
exp
erim
enta
lb
ioch
emic
alw
ork
Pig
Grx
mu
tan
tR
26V
Cy
s22
ND
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(190
)T
he
pK
ao
fC
ys2
2co
uld
no
tb
em
easu
red
wit
hth
eR
26V
mu
tan
t,d
ue
tola
cko
fen
zym
atic
acti
vit
y.
Pig
Grx
mu
tan
tR
26V
/K
27Q
Cy
s22
ND
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
(190
)T
he
pK
ao
fC
ys2
2co
uld
no
tb
em
easu
red
wit
hth
eR
26V
/K
27Q
mu
tan
t,d
ue
tola
cko
fen
zym
atic
acti
vit
y.
E.
coli
Grx
3(C
14A
/C
65Y
mu
tan
t)C
ys1
15
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(4
5)T
ote
stef
fect
of
acti
ve-
site
C14
Am
uta
tio
no
np
Ka
of
nu
cleo
ph
ilic
Cy
s11
E.
coli
Grx
3(K
8A/
C65
Ym
uta
nt)
Cy
s11
4.2
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(4
5)T
ote
stef
fect
of
acti
ve-
site
K8A
mu
tati
on
on
pK
a
of
nu
cleo
ph
ilic
Cy
s11
PD
Iw
ild
typ
e
Bo
vin
eP
DI
Cy
s35
and
Cy
s379
6.7
pH
dep
end
ence
of
enzy
me
inac
tiv
atio
nw
ith
iod
oac
etam
ide
(64)
Th
ep
Ka
of
6.7
was
ten
tati
vel
yas
sig
ned
toC
ys1
inth
etw
o-C
ys 1
-Gly
-His
-Cy
s 2-
mo
tifs
of
PD
I.T
he
tru
ev
alu
eso
fth
ose
pK
as
are
no
wth
ou
gh
tto
be
low
er(8
3).
Bo
vin
eP
DI
ND
5.6
Kin
etic
so
fo
xid
atio
no
fa
sub
stra
tep
epti
de
mo
nit
ore
db
ytr
yp
top
han
flu
ore
scen
ce
(146
)T
he
pK
aw
asat
trib
ute
dto
anac
tiv
e-si
te-
reac
tiv
ecy
stei
ne,
and
infe
rred
ind
irec
tly
fro
mk
inet
icm
easu
rem
ents
,w
hic
hm
ayac
cou
nt
for
this
com
par
ativ
ely
surp
risi
ng
lyh
igh
pK
a.
PD
Im
uta
nts
Hu
man
PD
IC
56S
Cy
s53
4.81
pH
dep
end
ence
of
the
rate
of
reac
tio
nw
ith
Ell
man
’sre
agen
t(8
3)S
tud
yo
fth
e-C
ys 5
3-G
ly5
4-H
is5
5-C
ys 5
6-
mo
tif
ina
cata
lyti
cd
om
ain
acti
ve
site
,to
inv
esti
gat
eP
DI
reac
tio
nm
ech
anis
ms
Hu
man
PD
IC
56S
/R
120Q
Cy
s53
4.84
pH
dep
end
ence
of
the
rate
of
reac
tio
nw
ith
Ell
man
’sre
agen
t(8
3)A
pp
aren
tly
lim
ited
role
of
Arg
120
on
the
pK
a
of
Cy
s53
in-C
ys 5
3-G
ly5
4-H
is5
5-C
ys 5
6-
(con
tin
ued
)
101

Ta
bl
e1.
(Co
nt
in
ue
d)
Sy
stem
Res
idu
eR
epor
ted
pK
av
alu
eaE
xp
erim
enta
lm
eth
odR
efer
ence
sbC
omm
ents
Hu
man
PD
IC
53M
Cy
s56
8.60
pH
dep
end
ence
of
the
rate
of
reac
tio
nw
ith
Ell
man
’sre
agen
t(8
3)T
oin
ves
tig
ate
the
pK
ao
fC
ys5
6in
-Cy
s 53-G
ly5
4-
His
55-C
ys 5
6-,
wit
hC
53M
mim
ick
ing
atr
ansi
ent
mix
edd
isu
lfid
e
Hu
man
PD
IC
53M
/R
120Q
Cy
s56
9.14
pH
dep
end
ence
of
the
rate
of
reac
tio
nw
ith
Ell
man
’sre
agen
t(8
3)In
terp
rete
das
evid
ence
that
R12
0lo
wer
sth
ep
Ka
of
Cy
s56
in-C
ys 5
3-G
ly5
4-H
is5
5-C
ys 5
6-,
wit
hp
oss
ible
mec
han
isti
cim
pli
cati
on
s
Hu
man
PD
IC
53M
/R
120D
Cy
s56
9.22
pH
dep
end
ence
of
the
rate
of
reac
tio
nw
ith
Ell
man
’sre
agen
t(8
3)In
terp
rete
das
evid
ence
that
R12
0lo
wer
sth
ep
Ka
of
Cy
s56
in-C
ys 5
3-G
ly5
4-H
is5
5-C
ys 5
6-,
wit
hp
oss
ible
mec
han
isti
cim
pli
cati
on
s
Res
Aw
ild
typ
e
Bac
illu
ssu
btil
isR
esA
wil
d-
typ
e(-
C7
4-E
75-P
76-C
77-)
Cy
s74
8.8
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(100
)A
pp
aren
tly
un
usu
ally
hig
hp
Ka
for
anN
-ter
min
alcy
stei
ne
ina
-C-X
-X-C
-m
oti
f.X
-ray
stru
ctu
reo
fre
du
ced
Res
Ais
PD
Ben
try
1SU
9
B.
subt
ilis
Res
Aw
ild
-ty
pe
(-C
74-E
75-P
76-C
77-)
Cy
s77
8.2
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(100
)T
he
pK
ao
fth
isC
-ter
min
alcy
stei
ne
app
eare
dh
igh
erth
anth
ep
Ka
for
the
N-t
erm
inal
cyst
ein
e,w
hic
his
aty
pic
al
Res
Am
uta
nts
B.
subt
ilis
Res
A(-
C7
4-E
75-P
76-A
77-)
Cy
s74
8.48
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(100
)A
pp
aren
tly
un
usu
ally
hig
hp
Ka
for
anN
-ter
min
alcy
stei
ne
ina
-C-X
-X-C
-m
oti
f.X
-ray
stru
ctu
reis
PD
Ben
try
2H19
B.
subt
ilis
Res
A(-
A7
4-E
75-P
76-C
77-)
Cy
s77
8.36
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(100
)X
-ray
stru
ctu
reis
PD
Ben
try
2H1A
B.
subt
ilis
Res
A(-
A7
4-E
75-P
76-C
77-)
Cy
s77
8.3
pH
dep
end
ence
of
reac
tio
nra
tew
ith
iod
oac
etat
e(1
00)
X-r
ayst
ruct
ure
isP
DB
entr
y2H
1A
B.
subt
ilis
Res
A(-
C7
4-E
75-P
76-C
77-)
E80
QC
ys7
77.
4p
Hd
epen
den
ceo
fre
acti
on
rate
wit
hal
ky
lati
ng
agen
tb
adan
,m
on
ito
red
by
flu
ore
scen
ce
(100
)X
-ray
stru
ctu
reis
PD
Ben
try
2H1B
B.
subt
ilis
Res
A(-
C7
4-P
75-P
76-C
77-)
Cy
s74
7.0
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(99)
Tes
tsth
ein
flu
ence
of
E75
on
the
pK
as
of
Cy
s74
and
Cy
s77
B.
subt
ilis
Res
A(-
C7
4-P
75-P
76-C
77-)
Cy
s77
6.6
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(99)
Tes
tsth
ein
flu
ence
of
E75
on
the
pK
as
of
Cy
s74
and
Cy
s77
B.
subt
ilis
Res
A(-
C7
4-E
75-H
76-C
77-)
Cy
s74
7.4
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(99)
H76
inth
isR
esA
mu
tan
tm
imic
ks
H32
inD
sbA
.X
-ray
stru
ctu
reis
PD
Ben
try
3C73
(con
tin
ued
)
102

Ta
bl
e1.
(Co
nt
in
ue
d)
Sy
stem
Res
idu
eR
epor
ted
pK
av
alu
eaE
xp
erim
enta
lm
eth
odR
efer
ence
sbC
omm
ents
B.
subt
ilis
Res
A(-
C7
4-E
75-H
76-C
77-)
Cy
s77
7.5
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(99)
H76
inth
isR
esA
mu
tan
tm
imic
ks
H32
inD
sbA
.X
-ray
stru
ctu
reis
PD
Ben
try
3C73
B.
subt
ilis
Res
A(-
C7
4-P
75-H
76-C
77-)
Cy
s74
6.3
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(99)
H76
inth
isR
esA
mu
tan
tm
imic
ks
H32
inD
sbA
.X
-ray
stru
ctu
reis
PD
Ben
try
3C73
B.
subt
ilis
Res
A(-
C7
4-P
75-H
76-C
77-)
Cy
s77
5.7
pH
dep
end
ence
of
reac
tio
nra
tew
ith
alk
yla
tin
gag
ent
bad
an,
mo
nit
ore
db
yfl
uo
resc
ence
(99)
H76
inth
isR
esA
mu
tan
tm
imic
ks
H32
inD
sbA
.X
-ray
stru
ctu
reis
PD
Ben
try
3C73
Dsb
Dw
ild
typ
e
E.
coli
Dsb
DC
-ter
min
ald
om
ain
(-C
46
1-V
46
2-A
46
3-C
46
4-)
Cy
s461
10.5
NM
Rch
emic
alsh
ifts
det
erm
ined
asa
fun
ctio
no
fp
H(1
12)
pK
afo
rC
ys4
61in
the
iso
late
dC
-ter
min
ald
om
ain
of
Dsb
D.
Un
usu
ally
hig
hp
Ka
for
anN
-ter
min
alcy
stei
ne
ina
-C-X
-X-C
-m
oti
f.
E.
coli
Dsb
DC
-ter
min
ald
om
ain
(-C
46
1-V
46
2-A
46
3-C
46
4-)
Cy
s464
>12
.2N
MR
chem
ical
shif
tsd
eter
min
edas
afu
nct
ion
of
pH
(112
)p
Ka
for
Cy
s461
inth
eis
ola
ted
C-t
erm
inal
do
mai
no
fD
sbD
.
E.
coli
Dsb
D-g
amm
ad
om
ain
(-C
46
1-V
46
2-A
46
3-C
46
4-)
Cy
s461
9.3
pH
dep
end
ence
of
reac
tio
nw
ith
iod
oac
etam
ide
and
UV
abso
rpti
on
at24
0n
m
(161
)U
nu
sual
lyh
igh
pK
afo
ran
N-t
erm
inal
cyst
ein
ein
a-C
-X-X
-C-
mo
tif.
X-r
ayst
ruct
ure
isP
DB
entr
y2F
WF
Dsb
Dm
uta
nts
E.
coli
Dsb
DC
-ter
min
ald
om
ain
(-C
46
1-V
46
2-A
46
3-C
46
4-)
E46
8QC
ys4
619.
9N
MR
chem
ical
shif
tsd
eter
min
edas
afu
nct
ion
of
pH
(112
)T
ote
stth
eef
fect
of
E46
8o
nth
ep
Ka
of
Cy
s461
E.
coli
Dsb
DC
-ter
min
ald
om
ain
(-C
46
1-V
46
2-A
46
3-C
46
4-)
D45
5NC
ys4
619.
3N
MR
chem
ical
shif
tsd
eter
min
edas
afu
nct
ion
of
pH
(112
)T
ote
stth
eef
fect
of
D45
5o
nth
ep
Ka
of
Cy
s461
E.
coli
Dsb
DC
-ter
min
ald
om
ain
(-C
46
1-V
46
2-A
46
3-C
46
4-)
D45
5N/
E46
8Q
Cy
s461
8.6
NM
Rch
emic
alsh
ifts
det
erm
ined
asa
fun
ctio
no
fp
H(1
12)
To
test
the
com
bin
edef
fect
of
D45
5Nan
dE
468
on
the
pK
ao
fC
ys4
61
Dsb
C
E.
coli
Dsb
Cw
ild
typ
e(-
C9
8-G
99-Y
10
0-C
10
1-)
Cy
s98
4.1
pH
dep
end
ence
of
UV
abso
rpti
on
at24
0n
m(1
63)
Th
isp
Ka
inw
ild
-ty
pe
dim
eric
Dsb
Cw
asfo
un
dto
be
ver
ysi
mil
arto
its
cou
nte
rpar
tin
aC
-ter
min
alfr
agm
ent
of
Dsb
C(r
esid
ues
66–2
16)
E.
coli
Dsb
CC
-ter
min
alfr
agm
ent
(-C
98-G
99-
Y1
00-C
10
1-)
Cy
s98
4.3
pH
dep
end
ence
of
UV
abso
rpti
on
at24
0n
m(1
63)
Th
isC
-ter
min
alfr
agm
ent
of
Dsb
Cco
nta
ined
on
lyre
sid
ues
66–2
16an
dw
asm
on
om
eric
E.
coli
Dsb
C(-
C9
8-G
99-
Y1
00-C
10
1-)
Mu
tan
tT
182V
Cy
s98
5.8
pH
dep
end
ence
of
UV
abso
rpti
on
at24
0n
m(1
39)
Th
em
uta
tio
nT
182V
isin
the
cis-
Pro
loo
p,
inth
ev
icin
ity
of
the
acti
ve
site
(con
tin
ued
)
103

Ta
bl
e1.
(Co
nt
in
ue
d)
Sy
stem
Res
idu
eR
epor
ted
pK
av
alu
eaE
xp
erim
enta
lm
eth
odR
efer
ence
sbC
omm
ents
E.
coli
Dsb
Cw
ild
-ty
pe
(-C
98-G
99-Y
10
0-C
10
1-)
Cy
s98
4.6
pH
dep
end
ence
of
UV
abso
rpti
on
at24
0n
m(1
39)
To
com
par
eto
the
pK
ain
the
T18
2Vm
uta
nt
ob
tain
edin
the
sam
est
ud
yD
sbG
E.
coli
Dsb
Gw
ild
-ty
pe
(-C
10
9-P
11
0-Y
11
1-C
11
2-)
Cy
s109
3.5
pH
dep
end
ence
of
UV
abso
rpti
on
at24
0n
m(1
39)
Th
isst
ud
yal
sod
eter
min
edth
ere
do
xp
ote
nti
al,
bu
tn
ot
the
pK
a,
for
two
mu
tan
tso
fD
sbG
Try
par
edox
in
Try
pan
osom
abr
uce
iT
ryp
ared
ox
in(-
C4
0-P
41-P
42-C
43-)
Cy
s40
7.2
pH
dep
end
ence
of
UV
abso
rpti
on
at24
0n
m(1
38)
Wil
d-t
yp
etr
yp
ared
ox
in
T.
bru
cei
Try
par
edo
xin
(-C
40-G
41-P
42-C
43-)
Cy
s40
7.2
pH
dep
end
ence
of
UV
abso
rpti
on
at24
0n
m(1
38)
Mu
tan
ttr
yp
ared
ox
inw
ith
the
-C
-X1-X
2-C
-m
oti
fo
fa
typ
ical
Trx
T.
bru
cei
Try
par
edo
xin
(-C
40-P
41-Y
42-C
43-)
Cy
s40
£4
pH
dep
end
ence
of
UV
abso
rpti
on
at24
0n
m(1
38)
Mu
tan
ttr
yp
ared
ox
inw
ith
the
-C-X
1-X
2-
C-
mo
tif
of
typ
ical
Grx
Pep
tid
em
odel
syst
ems
Cy
stei
ne
inra
nd
om
-co
ilp
epti
des
Var
iou
sse
qu
ence
s8.
48–8
.90
UV
abso
rpti
on
at24
0n
md
uri
ng
pH
titr
atio
n(8
9)P
rov
ides
refe
ren
cev
alu
esfo
rth
ep
Ka
of
cyst
ein
esin
dis
ord
ered
(un
fold
ed)
pep
tid
es
Ala
nin
ep
enta
pep
tid
eN
A8.
55P
ote
nti
om
etry
(169
)S
tud
yd
esig
ned
top
rov
ide
refe
ren
ce,
un
per
turb
ed(i
ntr
insi
c),
pK
av
alu
esfo
rti
trat
able
gro
up
sin
pro
tein
s
Cy
stei
ne
in16
mo
del
pep
tid
esV
ario
us
seq
uen
ces
7.35
–9.0
8p
Hd
epen
den
ceo
fre
acti
on
rate
sw
ith
iod
oac
etam
ide
or
of
UV
abso
rpti
on
at24
0nm
(17)
Car
efu
lly
des
ign
edm
od
elsy
stem
s,to
mo
del
cyst
ein
eso
fB
PT
Io
ro
fm
emb
ers
of
the
Trx
sup
erfa
mil
y
Cy
stei
ne
atN
-ter
min
us
of
hel
ical
pep
tid
esV
ario
us
seq
uen
ces
7.20
–7.6
3U
Vab
sorp
tio
nat
240
nm
du
rin
gp
Hti
trat
ion
(89)
Car
efu
lly
des
ign
edm
od
elsy
stem
s,te
stin
gth
ein
flu
ence
of
pep
tid
eh
elic
ity
and
seq
uen
ceo
nth
ep
Ka
ath
elic
alN
-ter
min
i
CA
AC
atN
-ter
min
us
of
a-h
elic
alp
epti
de
Nca
pan
dN
3Cy
s6.
74E
llip
tici
tym
on
ito
red
by
circ
ula
rd
ich
rois
mat
222
nm
(73)
On
lyan
app
aren
tp
Ka
was
det
erm
ined
,si
nce
the
pK
as
of
the
ind
ivid
ual
cyst
ein
esco
uld
no
tb
ese
par
ated
.
aE
xp
erim
enta
lly
mea
sure
dp
Ka
val
ues
rep
ort
edin
the
lite
ratu
re;
thes
ev
alu
esar
eli
sted
her
ew
ith
the
sam
ep
reci
sio
nas
inth
eo
rig
inal
pu
bli
cati
on
.bF
or
clar
ity
,o
nly
refe
ren
ces
toex
per
imen
tal
(no
nco
mp
uta
tio
nal
)w
ork
are
cite
dh
ere;
see
mai
nte
xtfo
rre
fere
nce
sto
theo
reti
cal/
com
pu
tati
on
alw
ork
.D
sbA
,d
isu
lfid
e-b
ind
ing
pro
tein
A;
Grx
,g
luta
red
oxi
n;
NA
,n
ot
app
lica
ble
;N
D,
no
td
eter
min
ed;
NM
R,
nu
clea
rm
agn
etic
reso
nan
ce;
PD
B,
Pro
tein
Dat
aB
ank
;P
DI,
pro
tein
dis
ulfi
de
iso
mer
ase;
Trx
,th
iore
do
xin
.
104

The central equation for this equilibrium is the Henderson–Hasselbalch equation (Eq. 3), which is a convenient re-arrangement of the Ka equilibrium equation:
log Ka¼ log[Hþ ][CysS� ]
[CysSH]¼ log[Hþ ]þ log
[CysS� ]
[CysSH]
� log[Hþ ]¼ � log Kaþ log[CysS� ]
[CysSH]
pH¼ pKaþ log[CysS� ]
[CysSH]
10(pKa� pH)¼ [CysSH]
[CysS� ](Eq: 3)
In this section, we briefly describe the experimental andcalculation methods to obtain pKa values. We focus on thecurrent widely used techniques to obtain thiol pKa values inproteins.
A. Experimental approaches
Traditional methods used to determine pKa values of cys-teines in proteins are based on UV absorption spectroscopy,rate constant determination, microcalorimetry, and in somecases, nuclear magnetic resonance (NMR) spectroscopy. Ra-man spectroscopy (101), quantitative mass spectrometry(122), and potentiometric titrations (169) have also been used.
A well-established spectroscopic method is based on thegreater absorption of ultraviolet light by the thiolate versus thethiol at 240 nm (Ared) (Fig. 2). The difference in the ultravioletextinction coefficient of the thiol and the thiolate anion isabout 4000 M - 1 cm - 1 at 240 nm (14, 131), which has beenused to determine the pKa values to the thiols of disulfide-binding protein A (DsbA) (121) and in several wild-type andactive-site mutants of Trx (40, 142, 166) and related proteins(Table 1). The protein in a buffer solution with high pH istitrated with HCl, and Ared is followed at decreasing pH
during the titration. However, several corrections on Ared areneeded. For correction of the varying protein concentrationdue to the increased volume during to addition of HCl, theabsorption at 240 nm needs to be corrected for absorptions at280 nm, the wavelength at which one can spectroscopicallydetermine the protein concentration (A240red/A280red). Mainly,the side chains of Phe, Tyr, and Trp will also absorb at 240 nm,and their absorbance might change due to changes in theenvironment during the titration. This source of variability on(A240red/A280red), which is thiolate/thiol independent, can becorrected by measuring the absorption of the same protein inwhich the thiol group is absent (A240oxid/A280oxid). Therefore,the same protein in which the cysteines are alkylated withiodoacetamide (IAM) can be used, which will not contributeto the thiolate absorbance at 240 nm. Alternatively, an en-gineered variant in which the cysteine is mutated to a serinecan be used. In case of a protein with a - C-X-X-C- active-sitemotif, the oxidized form with a disulfide between the twocysteines can be used. To make each experimentally obtainedabsorption ratio (A240red/A280red) pH independent, one nor-malizes by dividing the (A240red/A280red) value of the thiolateform by the absorption (A240oxid/A280oxid), where no thiol orthiolate is present. The (A240red/A280red)/(A240oxid/A280oxid) ratiois determined at varying pH by titration with HCl and plottedagainst pH (Fig. 2). The pKa is obtained from a nonlinear least-square fitting with the rearranged Henderson–Hasselbalchequation:
Aexp¼ASH þ(As� �ASH)
1þ 10(pKa� pH)(Eq: 4)
in which Aexp is (A240red/A280red)/(A240oxid/A280oxid) for the ex-perimentally determined value; ASH is the A240/A280 value forthe protonated thiol form; and As- is the A240/A280 for thedeprotonated thiolate form.
This rearranged equation has been deduced from the rela-tion between the fraction Aexp and the absorption of the pro-tonated and deprotonated form of the cysteine (Fig. 2),according to the following expression:
Aexp¼As� RþASH(1�R)
R¼Aexp�ASH
As� �ASH
Aexp�ASH ¼R(As� �ASH) (Eq: 5)
in which R is a fraction of the total absorption difference be-tween totally deprotonated and protonated cysteine. By usingequation (3), this fraction R can be written as:
R¼ [CysS� ]
[CysS� ]þ [CysSH]¼ 1
1þ [CysSH]=[CysS� ]
¼ 1
1þ 10(pKa� pH)(Eq: 6)
which then finally results in the rearranged Henderson–Hasselbalch equation:
Aexp¼ASH þ(As� �ASH)
1þ 10(pKa� pH)using (Eq: 5) and (Eq: 6)
In some cases, the protein structural environment aroundthe cysteine or its local dynamics is affected by the pH, which
FIG. 2. Experimental pKa titration curve for a cysteinethiol. The curve has been fitted to the rearranged Hender-son–Hasselbalch equation (Eq. 4). Similar curves may beobtained for UV spectroscopy results, isothermal titration,nuclear magnetic resonance, and second-order rate con-stants.
PKA OF REDOX CYSTEINES 105

in turn influences the cysteine absorbance. In these situations,it is difficult to evaluate the transition between protonatedand deprotonated cysteine at 240 nm.
Alternatively, thiol pKas in proteins and peptides can bedetermined based on the rates of alkylation as a function ofpH (17, 83, 93, 106, 108), with IAM, with fluorescent IAM de-rivatives, with 5,5¢-dithiobis(2-nitrobenzoate) (DTNB), with2,2¢-dipyridyl disulfide (2DPS), or with monobromobimane.The alkylation rates are evaluated over a certain pH range byevaluating the residual enzyme activity as a function of time.The pH-dependent degree of alkylation can also be used whenthere is no enzymatic assay to monitor direct changes in thereaction rate, typically when the cysteine of interest is not in anenzyme active site. Then, the amount of alkylated cysteine maystill be determined as a function of time, for example, bychromatographic retention times or fluorescence of the alky-lating agent linked to the cysteine. Nowadays, new superiorfluorescent probes are being developed, which allows highlyspecific thiol labeling at low pH (127).
At each pH, the pseudo-first-order rate constants kobs aredetermined from the slope of the plots of ln (E0/Et) versus time,with E0 the initial enzyme activity (or nonalkylated amount ofprotein) and Et the enzyme activity (or amount of alkylatedprotein) at time t.
The second-order kinetic constant (k2) is then calculated bydividing kobs by the concentration of alkylating agent at eachpH, and then fitting to the following equations to determinethe best-fit pKapp.
For a one-pKa model:
k2¼ k¢�
1
1þ 10(pKapp � pH)
�
or for a two-pKa model (if another protein titratable side chaininfluences the reactivity of the nucleophilic cysteine):
k2¼ k¢�
1
1þ 10(pKapp � pH)
��1
1þ 10(pKapp2 � pH)
�
þ k¢¢�
1
1þ 10(pKapp2 � pH)
�
with k¢ and k¢¢, the two different second-order constants for thethiolate form.
This method has been applied for Trx (82), thiol/disulfideoxidoreductases (117), glyceraldehyde 3-phosphate dehy-drogenase (108), yeast glutaredoxins (Grxs) (36), and perox-iredoxin 6 from Arenicola marina (106). For Mt_AhpE, an alkylhydroperoxide reductase of Mycobacterium tuberculosis, thepKa of oxidation and overoxidation (Fig. 1) of the peroxidaticthiol group were measured by determining the rate constantsof peroxynitrite and H2O2-mediated oxidation and over oxi-dation using the difference in intrinsic fluorescence (70). Forhighly reactive cysteines, kinetic measurements need to becarried out in a stopped-flow apparatus. Overall, the chemicalmodification method has been very successful with enzymesof the Trx superfamily, since in these enzymes, the maincysteine of interest is catalytically active and solvent exposed.This contributes to explain the wealth of pKa data on the Trxsuperfamily.
This method of chemical modification is however nottrivial and certainly not applicable to all proteins, as it as-
sumes that the cysteine side chain of interest is accessible foralkylation. Further, the pKa value of the cysteine is rightlyassumed to be the principal determinant of its alkylation rate.However, this is only the case in an idealized situation, be-cause some cysteine side chains are partially or fully protectedby the protein structure; their alkylation rates can also beaffected by steric accessibility, conformational stability, anddynamics. Protected cysteines can only react with alkylatingreagents when they are transiently exposed to bulk solvent byglobal or local unfolding events. Because protected cysteinesprimarily (or only) react in transient unfolded (open) states,they will exhibit seemingly unperturbed pKa values (*8.6).However, the actual pKa value of the cysteine in its protected(folded) state could be very different.
Another method that should be applicable to low-solubilityproteins is isothermal titration microcalorimetry (ITC) (165).ITC has been used to infer pKa values of reactive residues ofenzyme–substrate complexes by measuring the substrate-binding enthalpy DHbinding as a function of pH and buffercomposition (162). Tajc and coworkers (165) used ITC tomonitor directly the covalent reaction of IAM with a thiolateto produce a thioether, as a function of pH. The ITC instru-ment is set up in a single-injection mode, and sufficient IAM(up to 300 mM) is introduced to alkylate all the thiolategroups. The produced heat change upon injection qQ=qt isproportional to the reaction rate. The maximum absolute va-lue of qQ=qt is proportional to the initial rate, which accordingto the law of mass action, is proportional to the initial con-centration of the thiolate. So, the maximum absolute value(qQ=qt)max, obs is determined at varying pHs. The pKa can becalculated from the (qQ=qt)max-versus-pH plot using theHenderson–Hasselbalch equation:
(qQ=qt)max, obs¼ (qQ=qt)max, low
þ(qQ=qt)max, high� (qQ=qt)max, low
1þ 10(pKa � pH)
with (qQ=qt)max, low, the value at low pH (protonated cysteine),and (qQ=qt)max, high at high pH (deprotonated cysteine)(Fig. 2).
NMR spectroscopy can also be used to determine the pKa ofa cysteine (38, 44, 77, 89, 112, 129). This method is expensive,requires a sophisticated instrumentation, and is labor inten-sive (resonance assignment). The presence of more than onecysteine in a protein does not limit in principle the determi-nation of their pKas by NMR; ideally, the pKa values of allcysteine residues may be determined in the same NMR ex-periment. For the cysteine thiol ionization measured by 1D 1HNMR spectroscopy, the cysteine Ca and Cb protons can beused as probes for the ionization state of the cysteine thiolgroup (89). A problem that might occur is that the resonancesof the Cys Ca proton might not be followed over the entire pHrange because of signal overlap or low intensity of the signal.Also, one potential complicating factor is that pH-dependentchanges in a resonance chemical shift may occur due to pH-dependent changes in other surrounding residues. Becausethe chemical shift is particularly sensitive to the local envi-ronment, disentangling the effects of ionization at the moni-tored residue versus other changes in proximal residues can benontrivial. With more expensive 13C-labeled material, thechemical shifts of the 13Cb resonance can also be followed as afunction of the pH (38). The obtained chemical shifts can then
106 ROOS ET AL.

be plotted against pH and fitted, again with the Henderson–Hasselbalch equation:
d¼ dSH þ(ds� � dSH)
1þ 10(pKa � pH)
with d, the chemical shift of a resonance as function of pH, andds- and dSH, the chemical shifts at high and low extremes of thepH, respectively. The pKa values obtained by NMR seem toagree well with those obtained by other spectroscopic meth-ods (38, 77, 89).
Since proteins tend to have a solubility minimum at theirisoelectric point (pI), this might undermine the measurementof the pKa during the pH titration. In addition, proteins mayunfold when a titration changes a protonation state importantfor the stability of the folded protein. When proteins start toaggregate, unfold, or precipitate, the experimental measure-ments become difficult and unreliable. These limitations andother considerations have emphasized the growing interest inthe development of computational methods to calculate thepKas.
B. Computational methods
In proteins, there are many protonation sites, and in manycases, even several cysteine residues, complicating accurateexperimental pKa measurements and interpretation. There-fore, there is an increasing interest in supplementing, guiding,and interpreting experimental approaches with computa-tional methods for pKa rationalization/prediction (2, 98).Calculations can also help assign accurately measured pKas toparticular residues.
The basis for pKa calculation techniques that can be used onlarge systems like proteins relies on estimates of energy terms,which influence the relative populations of the protonatedand deprotonated states of the titratable groups. For the rel-evant chemical functionalities, those terms typically includethe desolvation energy, the background interaction energy,and the site–site interaction energies. The desolvation energyis the energy change when transferring the titratable groupfrom an aqueous solvent into the protein environment. Thebackground interaction energy is the interaction energy be-tween the titratable group and the protein, when all othertitratable groups are considered as neutral entities. The maincontribution to this term comes from hydrogen-bonding in-teractions between the titratable group and neutral polargroups. The site–site interaction energy describes the elec-trostatic interactions between pairs of charged titratablegroups. In principle, one has to consider the interactionsamong all the charged groups simultaneously, a challengewhen the titrations of several groups are coupled. These en-ergy contributions can be addressed in various theoreticalframeworks (2, 98), usually categorized as classical electro-statics with continuum solvation models, physics-based fullymicroscopic models, or more empirical approaches. Allcomputational methods require a good-quality structuralmodel of the protein, which determines for a large part theoutcome of the calculations.
The Poisson-Boltzmann (PB) approach (4, 13, 29, 31, 35, 45,68, 126, 161, 179, 188) is maybe the most representative andbest-developed approach to address the pKa shifts in proteinsin the framework of classical electrostatics (68, 154). The cal-
culation of pKa shifts in proteins in the PB framework has beendescribed in detail (13, 42). In summary, the pKa in a proteinenvironment is calculated as a shift relative to the referenceintrinsic pKa of the same residue free in aqueous solution. Thereference pKa value is known from measurements on modelsystems, typically peptides in solution (bottom of Table 1). Forthe cysteine thiol, a reference unperturbed pKa of 8.3–8.6 iscommonly accepted, with small variations in the corre-sponding measurements (17, 26, 39, 82, 89, 169). The range ofcysteine pKas measured in peptides is much narrower than infolded proteins (Table 1). Yet, some pKa differences are ob-served in peptides, although the reasons for such spread areobscure, since there are no structures for these flexible pep-tides. One cannot say for sure if the differences between pKa
values of cysteines in peptides can be rationalized by hydro-gen-bonding differences. The differences in measured pKasalso probably reflect different experimental protocols (169).Reference pKa values obtained recently with pentapeptideswith neutral (blocked) termini (169) are of special interest,since they minimize the influence of secondary structure andhydrogen bonding, which can occur in larger peptides, andsuggest a reference pKa value of 8.6 for cysteines. Note that theeffect of the bulk aqueous solvent on the pKa is encapsulatedin the reference pKa. Estimating the influence of the proteinrelative to such known reference pKa is the approach currentlyadopted with all methods.
The thiol of cysteines is treated like any other titratablegroup. Therefore, the pKa shift of a particular thiol is calcu-lated by estimating the electrostatic interactions between thecysteine thiolate (and thiol) and the rest of the protein whiletaking into account the desolvation energies. In practical ap-plications, neglecting electrostatic interactions betweencharged titratable groups which are far apart tends to be avalid working hypothesis (53). The desolvation correspond-ing to the transfer of a charge (e.g., a thiolate) from bulk waterinto the protein interior is calculated by representing the bulksolvent as a continuous high-dielectric-constant medium andthe protein interior as a region of lower dielectric constant (13,31, 55, 68). Such desolvation is largely electrostatic in nature. Itdestabilizes a charge, contributing to pKa shifts, for instance,by destabilizing the charged form of a titratable group. Thus,one expects reactive cysteines to be mostly solvent accessiblebecause of steric requirements allowing encounters with thesubstrate, but also because proximity to the water contributesto stabilize their thiolate. On the other hand, cysteine residuesare very vulnerable to oxidation, which might explain thelimited occurrence of nonactive cysteine residues on proteinsurfaces (109).
Representing the protein interior by a single dielectricconstant can only be an approximate, and sometimes crude,treatment. For extended discussions on this subject, see thefollowing references (4, 31, 45, 55, 154, 158, 159). With a sys-tem comprised of different dielectric regions (solvent andsolute interior), the straightforward use of Coulomb’s law inits well-known, simplest form is not applicable, and trying totreat the system in this framework leads to a mathematicaldescription too complex to solve. Therefore, one uses the PBequation instead to obtain the electrostatic properties (54,154), usually in its linearized form (68). The physical param-eters that need to be supplied by the user include the dielectricconstant of the protein interior and of the aqueous solvent, thetemperature, partial charges, and radii for the protein atoms,
PKA OF REDOX CYSTEINES 107

and the ionic strength. The atomic radii are critical, since theyunderpin the boundary between the dielectric regions. Theionic strength represents the screening of electrostatic inter-actions by counter ions, but the calculated pKa values tend notto be highly sensitive to this parameter. The general experi-ence is that the calculated pKa values tend to be much moresensitive to the value of the protein dielectric constant. Cal-culations with the PB method are more time consuming thanempirical methods (e.g., PROPKA); however, they are quitetractable on commodity computers; a PB pKa calculation on amedium-size protein takes less than a minute.
The best practices regarding PB-based pKa calculations arestill an evolving field (2). To gain productive insights from suchcalculations, one requires a good grasp of the underlyingmethod and approximations. Accordingly, no standard genericsafe protocol can be recommended. Yet, the physical principles(desolvation, Coulombic interactions, and hydrogen bonds)underlying the PB calculations are intuitively easy to under-stand, and the PB framework should make the notion of pKa
calculations accessible to the nonexpert, at least in terms ofgeneral ideas and as a basis for discussions. Of several softwarepackages available, none has emerged as systematically moreaccurate or popular (2). Nevertheless, the software to performPB pKa calculations is freely downloadable; see Lee and Crip-pen (98) and Fitch and Garcia-Moreno (42).
As far as we know, no PB-based study has focused on thecalculations of cysteine pKas across proteins systematically.Instead, computational analyses of cysteine pKas werereported in case studies of individual enzymes. The PBmethodology was used to calculate the cysteine pKa values ofwild-type and mutant human Trx and Escherichia coli DsbA(49, 119). For the nucleophilic cysteine of wild-type Trx andDsbA, pKa values of, respectively, 7.1 and 2.6 were calculated,in good agreement with the experimental values of 6.9 and3.4. Also for the -X1X2- mutants in the -Cys1-X1-X2-Cys2- ac-
tive-site motif of Trx and DsbA, the calculated pKa valueswere close to their experimental counterpart (119). Only aslightly better agreement with the experimental pKa valueswas obtained after conformational relaxation of the flexibleionizable groups. For E. coli Grxs 1 and 3, as well as pig Grx(and its mutants), the PB methodology calculated the relevantcysteine pKa values in reasonable agreement with experiment(43, 44). In particular, the PB calculations account for the largedownshift of the pKa of Cys1 in the -Cys1-Pro-Tyr/Phe-Cys2-motif of Grxs (pKa £ 5) and correctly assign a significantlyhigher pKa to Cys2. Crucially, these studies provided a de-tailed theoretical analysis of the factors that lower the pKa ofthe catalytic cysteine. This analysis was strengthened by theformulation of true predictions that were subsequently tested,and experimentally supported (45). Small structural differ-ences in the active site of enzymes of the Trx superfamily canlargely rationalize the pKa variations for the catalytic cysteineacross this superfamily (Fig. 3). In addition, these local inter-actions were found to be sensitive to the protein dynamics.This was already apparent in a PB-based study of the pKa ofCys32 in an NMR structure of E. coli Trx (35).
Importantly, the output of PB pKa calculations provides adecomposition of the energetic components contributing to apKa shift, which allows for interpretation of the calculations.Such decomposition has provided much of the increasingevidence that the pKas of many titratable groups are primarilyinfluenced by short-range polar interactions such as hydrogenbonds (6, 44, 45, 49, 103, 134, 141, 143). It strongly supports theview that the pKas are in general very sensitive to the details ofthe protein structure and dynamics (35, 43–45, 134). The in-fluence of local structural differences on the pKa calculationswas also apparent when comparing pKas calculated with bothX-ray and NMR structures of the same proteins (5, 86). In-cidentally, there was no clear systematic improvement betweencalculated and measured pKas depending on the technique (X-
FIG. 3. Thiolate stabilization with hydrogen bonds: a unifying theme across the Trx superfamily. Comparison of thehydrogen bond networks (green dotted lines) stabilizing the thiolate in reduced active sites of representative enzymes in theTrx superfamily (left: human Trx, Protein Data Bank [PDB] entry 1ERT; middle: Escherichia coli glutaredoxin [Grx] 3, PDB entry1ILB; right: E. coli DsbA, PDB entry 1A2L). For each enzyme, only the -Cys1-X1-X2-Cys2- motif is shown, that is, -Cys1-Gly-Pro-Cys2- in Trx, -Cys1-Pro-Tyr-Cys2- in Grx, and -Cys1-Pro-His-Cys2- in DsbA. The sequence numbering of Cys1 and Cys2 isshown, with the thiol of Cys2 donating a hydrogen bond to the nucleophilic thiolate of Cys1. The number of hydrogen bondsdonated to the thiolate depends on the -Cys1-X1-X2-Cys2- sequence. In particular, note that the proline in Trx removes abackbone N-H hydrogen bond donor. The more hydrogen bonds can stabilize the thiolate, the lower its experimentallymeasured pKa, as illustrated by comparing Trx (pKa *7.1, two hydrogen bonds), Grx (pKa *4.0, three hydrogen bonds), andDsbA (pKa *3.5, four hydrogen bonds). Modulation of the cysteine pKa by local hydrogen bonds also explains why the pKa
of Cys1 is lowered, but not that of Cys2. (To see this illustration in color, the reader is referred to the web version of this articleat www.liebertpub.com/ars.)
108 ROOS ET AL.

ray or NMR) used to build the structural model. In addition, theraw X-ray or NMR coordinates may have to be refined beforepKa calculations, to orient protein side-chain amide and imid-azole groups, as well as conserved water molecules, accordingto the most likely hydrogen-bonding network (123, 126). Thus,a careful preparation of the protein structure is critical for pKa
calculations, regardless of the pKa calculation method.Preparing a relevant system for pKa investigations might be
even more sophisticated, if the pKa depends on the bindingbetween two or more molecular species. Thus, one may haveto consider how the calculated pKa might be influenced by thebinding of cofactors (ions and organic ligands) or by the for-mation of a protein–protein complex. Since low cysteine pKasare frequently involved in enzymes and their reactions withsubstrates, one can imagine situations where building a rele-vant enzyme–substrate complex may be a prerequisite topertinent pKa calculations. Some of these aspects have beenillustrated in a study of a covalent complex between Trx andarsenate reductase (ArsC) (141). Another interesting system tostudy a cysteine pKa influenced by complexation is DsbD. Inthe isolated C-terminal domain (gamma-domain), the pKa ofthe reactive Cys461 is unusually high, with a pKa of 9.3–10.5(112, 161). Yet, there is indirect evidence that the pKa ofCys461 is lowered upon complexation with the substrate N-terminal domain of DsbD (113). A model in which substratebinding enhances the reactivity of the active-site cysteines hasalso been proposed for ResA (28). It has been proposed thatchanges of the relevant pKas during complexation allowcontrolled activation of reactive cysteines upon binding ofcognate substrates, to restrict the reactivity toward thosesubstrates (28, 113). Macromolecular crowding in the cell mayalso lead to nonspecific, but significant, interactions, whichmight alter protein structures, and therefore the pKa of reac-tive amino acids. However, these nonspecific effects are ex-pected to be even more challenging to characterize than thosearising in the formation of specific complexes.
Empirical pKa calculation methods use rules derived fromexperimental observations to predict pKas and have the ad-vantage to be very fast (seconds per protein conformation).
They also lend themselves to updates of the underlying modeland associated terms, driven to maximize pragmatically theagreement between experiment and calculations withouthaving to abide by a constraining or interpretable theoreticalframework. An overview of the available empirical pKa pre-diction methods is given in reference (98). A currently popularempirical method is PROPKA (12, 30, 88, 103, 111, 130, 133,134, 149, 160), which is available (http://propka.ki.ku.dk/).PROPKA considers an environmental pKa perturbation DrMc
to the unperturbed, solution pKa, of the titratable group.The DrMc term includes desolvation effects, hydrogen
bonding, and charge–charge interactions via empirical rela-tions. The first version, PROPKA1, was developed using a testset of 314 experimental pKas, which contained only 12 cyste-ine pKas. For these cysteine residues, the root mean squaredeviation between experimental and calculated pKa valueswas 1.39. For oxidized (bonded) cysteine thiol groups, no pKa
calculation is performed (flagged by returning a value of 99.99instead of an estimated pKa value), with the exception ofproteins from the Trx superfamily. Even when the cysteines ofthe conserved -Cys1-X1-X2-Cys2- motifs are in the oxidizedform (i.e., when a disulfide bond is formed between Cys1 andCys2), the pKa values of the reduced cysteines are evaluated.
PROPKA has been updated twice. In PROPKA1, no pKa
shifts due to ligands, ions, and structural water moleculeswere considered. These effects were incorporated in PROP-KA2 (12). In PROPKA3 (130), residues are no longer classifiedas either buried or surface residues, but an interpolation be-tween these two extremes is used. This results in a burial ratioby which Coulomb interactions are no longer strictly eitherturned off (surface residues) or turned on (buried residues). Alinear interpolation between the two extremes is made via aposition-dependent weight function that depends on thenumber of heavy atoms within a sphere of 15 A around thecharge center.
A benchmark study (110) of PROPKA on cysteine residuesfor several Trx and ArsC proteins revealed a fair correlation(R2 = 0.74 with an average deviation from experimental valueof 0.88 pKa units) with experimental pKas (Table 2). Before pKa
Table 2. Comparison of Some Calculated and Experimentally Measured Cysteine pKas
Species PDB
Cysteineresiduesurface
(S)/buried (B) PROPKA3.0 PROPKA2
PROPKA withCHARMMminimizedstructures
NPA-pKa
correlation(141)
Experimentallyobtained pKa
E. coli Trx1 1XOB (76) Cys32 (S) 9.11 6.64 6.6a 6.5 7.1 (38)Staphylococcus aureus
Trx1 (P31T C32S)2O89 (142) Cys29 (S) 7.61 3.86 4a 6.5 6.4 (142)
Rhodobacter capsulatus Trx2 2PPT (191) Cys73 (S) 8.30 5.84 5.7a 4.8 5.2b (40)B. subtilis resA 1SU9 (28, 99) Cys76 (B) 13.19 15.83 10a 8.1 8.2 (28, 99)S. aureus ArsC 1LJL (194) Cys89 (S) 10.32 9.21 9.2a 10.0 9.5c (141)S. aureus ArsC 1LJL (194) Cys10 (B) 8.85 - 0.41 6.8a 6.9 6.8d (141)B. subtilis Trx 2GZY (104) Cys29 (S) 8.37 5.91 5.7a 5.5 ND
Cys32 (S) 11.30 8.99 ND 8.2 NDE. coli Grx3 1ILB Cys11 (S) 7.59 4.29 ND 5.0 < 5.5
Cys14 (S) 10.92 7.71 ND 14.1 > 10.5
aSee (110).bpKa value obtained from E. coli Trx2.cpKa value obtained from C15A/C10S/C82A Sa_ArsC.dpKa value obtained from oxidized C15A Sa_ArsC.ArsC, arsenate reductase; NPA, natural population analysis.
PKA OF REDOX CYSTEINES 109

calculation, the raw X-ray structures were energy minimizedwith CHARMM. For comparison, Table 2 reports the PROP-KA2 values for nonminimized structures, giving in somecases a notably degraded performance of PROPKA. Thisstrengthens the notion that a careful preparation of the proteinstructure is critical for pKa calculations. The performance ofPROPKA3 with cysteines appears to be less accurate than theperformance of PROPKA2 (Table 2). The advantage ofPROPKA is its balance between speed and performance, andespecially its Web-based ease of use. Therefore, PROPKA is avaluable tool to give initial insights in the protonation state ofa cysteine. PROPKA illustrates how the development andsuccessive adjustments of empirical models rely on aug-mented training sets of experimental data. Indeed, the con-tinued experimental determination of pKa values for cysteinesis very valuable for an improved calibration of empirical pKa
calculators.The observation that pKas in proteins may be primarily
influenced by short-range interactions (35, 43–45, 49, 141)suggests that one may not need to include the full protein inthe calculations. This has led to the development of a pKa
calculation protocol in which calculations of site–site electro-static interaction energies were omitted for pairs of titratablegroups beyond a distance cut-off value (125). The immediateenvironment of the titratable group is treated in detail, whilethe rest of the protein is described less accurately, whichlowers the calculation time significantly without apparentlydegrading the quality of the calculated pKa values. Thesefindings suggest that protein active sites might be treated asindependent units with respect to pKa calculations. It opensthe possibility to apply computationally demanding quantummechanical methods to the calculation of pKas in their proteinmicroenvironment. A possibility is to use a model in whichonly the titratable group plus its directly interacting proteinenvironment is represented. The rest of the protein is re-presented by a bulk dielectric constant or treated by molecularmechanics in a combined quantum mechanical (QM)–molec-ular mechanics approach. Alternatively, the bulk proteinaround the region of interest can be neglected altogether,without being included in the calculations; examples can befound in Li et al. (102), Zheng et al. (195), and Ullmann et al.(170). The combination of a model system for the protein siteof interest and a surrounding dielectric constant was used byRoos et al. (141). The pKa values were predicted via the linearrelationship between the natural population analysis (NPA)charge calculated quantum mechanically on the sulfur atomof the deprotonated thiolate, and compared with the experi-mental pKa values. The more negative the NPA charge on thesulfur atom, the higher the tendency to bind a proton and themore basic (higher pKa) the thiol is. For Trx and ArsC systems,this linear relation works very well (Fig. 4 and Table 2) (141).
C. Future perspective for pKa calculations appliedto cysteines
The continued development of computational methods hasproduced powerful tools to calculate pKa values for each ti-tratable group in a protein. When used wisely, such tools helpto formulate specific working hypotheses about the pKa val-ues of interest and the factors influencing these pKas. It pro-vides a basis for a productive interplay with experimentalapproaches, including mutagenesis. However, theoretical pKa
prediction methods still suffer from a number of limitations(2, 30, 98). Large errors can be encountered for residues withunusual pKa values (2, 31, 98, 126, 157), which tend to be ofparticular interest, because they are often involved in catalyticactivities. Ionizable side chains partially or fully sequesteredfrom bulk solvent by burial in the protein core are particularlyprone to marked discrepancies between calculated and mea-sured pKas. In a recent blind calculation of 77 pKas with un-disclosed experimental values, by 12 groups using a range ofmethods (www.pkacoop.org), it was found that no methodperformed significantly better than the others. Each methodhad successful and unsuccessful predictions, indicating thateach method suffers limitations (2). Hence, there is a clearneed to improve the calculations methods, which could takeseveral directions.
One avenue would be to develop more physically completemodels with all atomic details at all stages of the calculations.For instance, one could aim to treat the aqueous solvent withdiscrete and dynamic water molecules rather than with acontinuum dielectric. One can also envisage an increasing rolefor reliable quantum chemistry to quantify interactions. At theother extreme, one could may be improve empirical modelsby increasing the training sets on which they are based. Thereis also room to improve the current theoretical models, forinstance, with respect to the protein structural relaxation anddynamics, or the underlying force fields. Polarizable forcefields may improve the electrostatic energies when switchingbetween neutral and charged state for titratable groups (23).
A common issue with pKa calculations is the assumptionthat a protein structure is rigid and identical to a representa-tive crystal structure. For example, the same structure is fre-quently used to evaluate the energetics of both the chargedand neutral form of a titratable group, although one wouldexpect the microenvironment of this group to relax accordingto its charge state. This suggests that different conformersand tautomers should be included in the pKa calculations (51,91, 192). Sampling of the hydrogen locations in response
FIG. 4. Correlation between experimental pKa values ofselected cysteine thiols and the calculated natural popula-tion analysis (NPA) charge. The selected cysteines wereCys89 Staphylococcus aureus arsenate reductase (ArsC) (141)(a), Cys76 Bacillus subtilis resA (28, 99) (b), Cys10 S. aureusArsC (141) (c), Cys32 E. coli Trx1 (38) (d), Cys29 S. aureusP31T C32S Trx1 (142) (e), and Cys73 Rhodobacter capsulatusTrx2 (40) (f). Reprinted with permission of Roos et al. (141).
110 ROOS ET AL.

to protonation states already yields improvements (91). Withthe multiconfigurational continuum electrostatic method(MCCE) (51), the sampling was extended to the side-chainorientations. The MCCE approach gives pKa values in betteragreement with experiment than the single-configurationalcontinuum electrostatic method. Another option is to precedethe pKa calculation with a substantially long molecular dy-namics (MD) run (43–45, 90, 161, 173). There is no recipe todetermine the appropriate length of a simulation, but at leasttens of nanoseconds (and ideally more) appear required, forexample, to sample the conformations of long, flexible,charged side chains. In addition, MD simulations haveproved of special interest to refine NMR structural modelsbefore the pKa calculations (43). Using the MD snapshots asinput, the pKas can be calculated for regularly spaced snap-shots, and then averaged (since the Boltzmann weighting ofconformers is in principle implicitly contained in the simu-lated populations). This does yield not only the averagedcalculated pKa values but also the instantaneous pKa fluctu-ations along the simulation. Examination of the instantaneouspKa values and their variation can help interpret the structuralfactors, which affect the calculated pKa, with full microscopicdetails. It may reveal that the averaged pKa (equivalent to itsmeasured counterpart) is a composite of distinct pKa perstructural microstates. Such microstates may be associated todifferent reactivities and may lead to mechanistic insights.When the MD runs are too short, it may produce a bias thatcan sometimes strongly influence the relative occurrence ofthe different populations and consequently the outcome of thepKa calculations (90), for instance, when the pKa of asemiburied residue depends on the motion of flexible chargedside chains at the protein surface (43). Indeed, MD simulationsdo not always improve subsequent pKa calculations (125,185). For instance, MD simulations are still typically per-formed while keeping the initially assigned protonation statesconstant, in which case those states might be overstabilized inthe MD conformational ensemble, compared to alternativeprotonation states. This is being addressed with the devel-opment of MD simulations at constant pH (85). Alternatively,one can perform simulations at various predefined relevantprotonation states (128).
Another way to improve pKa calculations is the collection ofadditional reliable experimental reference data. That may beparticularly the case for cysteine residues for which there arestill only limited experimentally measured pKas, and not en-ough structural information for redox active cysteines at thevarious stages of their redox cycles. Crystallizing the reducedform of some enzymes has proved particularly problematic(72), as the nucleophilic cysteines have the tendency to oxidizeto sulfenic, sulfinic, and sulfonic acids (144). This heteroge-neous population of partially oxidized proteins may hampercrystallization (10). Therefore, additional good-quality experi-mental data on thiol pKas in proteins combined with deeperinsights in the factors controlling pKa values (e.g., regardingprotein dynamics) should pave the way toward more accuratepKa predictors. Meeting these challenges will require genuinecollaborative efforts. In this light, the pKa cooperative wasfounded to improve pKa calculations with proteins(www.pkacoop.org). To support this initiative, we depositedthe present Table 1 on the Website of the cooperative. In themeantime, the state of the art of pKa calculations is alreadymature enough to gain important insights, which can hardly be
accessed otherwise. Although the data set of measured cysteinepKas is relatively small, enough evidence has been accumu-lated to comment on the factors that modulate these pKas.
III. Factors That Control the pKa Values of CysteineThiols in Proteins
The propensity of a chemical functionality to ionize de-pends on its environment, in particular on factors that affectthe stability of the charged form of the functionality. There-fore, the pKa of a cysteine thiol can be strongly influenced byits microenvironment in a protein. Charge–charge interac-tions, hydrogen bonds, aqueous desolvation, and helix–dipole effects are generally invoked to rationalize perturbedpKas of cysteine residues (62, 124). However, it has becomeincreasingly clear that residues in the immediate environmentof a titratable group play a prominent role (45, 153). In thissection, we discuss the limited role of charged side chains andlong-range electrostatic interactions on the cysteine pKas inoxidoreductases and the direct link between thiol pKa andhydrogen bonds. These ideas were initially inspired bystudies of the catalytic cysteine in enzymes of the Trx super-family (45). However, since these notions emerged from an-alyses rooted in physical chemistry, one expects that they canbe generalized to a good extent (103, 134), as confirmed byinspection of a variety of systems (section III.D).
A. Limited role of charged side chainsand long-range electrostatics
It has long been known that many enzymes have a thiolatecysteine in their active site with a pKa clearly lower than thepKa of free cysteine in an aqueous solution (Table 1). For ex-ample, proteins of the Trx superfamily have a recurrent -Cys1-X1-X2-Cys2- active-site motif within a conserved structuralframework, where the pKa of Cys1 is dramatically lowered.Considerable efforts have been dedicated to characterize en-zymes of this superfamily, which includes DsbA, Grx, andTrx, and many variants (Table 1). In these enzymes, the pKa ofCys1 covers a broad range of values, from *3.5 in DsbA to*7.1 in Trx, to > 8.0 in atypical Trxs such as ResA (35, 38, 43,45, 46, 49, 56, 61, 77, 82, 117, 129, 142, 179). Mutagenesisstudies have shown that the pKa of Cys1 is strongly influencedby the X1-X2 residues (56, 117).
The discovery of the very low pKas of Cys1 initially fuelledthe speculation that this pKa would probably be stabilized bypositively charged side chains. This hypothesis has now beentested with a number of systems for which it has been mostlydispelled. However, it formed the basis for much mutagenesiswork, for example, with pig Grx (190), E. coli Grx3 (45, 129),E. coli DsbA (56, 66, 117), and human Grx1 (75). Early worksuggested that Arg26 in pig Grx may be the key for a directstabilization of the thiolate of Cys22, but structural work hassince shown that formation of a salt bridge between these twoside chains is sterically precluded (44). In reduced E. coli Grx3,His15 was mutated to Val (129), assuming that His15 wouldbe positively charged and thereby stabilizing the thiolate ofCys11. Yet, His15 was subsequently shown to be neutral atphysiological pH and cannot stabilize the thiolate via long-range electrostatics (45). The other candidate basic side chainfor stabilization of the thiolate in E. coli Grx3 is Lys8. In MDsimulations of E. coli Grx3 (43, 45), the side chain of Lys8 wasfound to be very flexible with its amino group spending most
PKA OF REDOX CYSTEINES 111

of the time solvated by water, away from the thiolate of Cys11(Fig. 5). In this situation, water screens the electrostatic in-teractions between the thiolate and the lysine side chain, andthe long-range Coulomb interaction between these groupswas found to be negligible for the thiolate stabilization (43,45). The amino group of Lys8 comes occasionally into contactwith the thiolate in the computer simulations, reflected in atransient further drop in the calculated pKa of Cys11 of E. coliGrx3. However, the formation of this salt bridge was short-lived and predicted to contribute little to the overall stabili-zation of the thiolate (45), as subsequently confirmed experi-mentally (45). Similar arguments probably also explain whyLys19 has only a marginal influence on the pKa of Cys22 inhuman Grx1 (75).
The extensive mutagenesis work with E. coli DsbA alsosupports the notion of a limited influence of the surroundingcharged side chains on the pKa of its catalytic Cys30. There aremany mutants of residues X1 and X2 in the -Cys30-X1-X2-Cys33- active-site motif of DsbA (56, 66, 117), for which thepKa of Cys30 has been measured (Table 1). In the majority ofthe DsbA mutants, the pKa of Cys30 remains below 5.0 (Table1 and (56, 117)), suggesting that the side chains of residues X1
and X2 are not the main factors decreasing the pKa of Cys30.Indeed, including a basic arginine in mutant -C30-T31-R32-C33- gave a pKa of 4.76 for Cys30 (56), higher than the pKa of*3.5 for the wild-type sequence -C30-P31-H32-C33-. This sup-ports the view that positioning basic side chains in the vicinityof a cysteine is not sufficient to lower its pKa. It is consistent
FIG. 5. Importance of exploiting dynamical structural protein models for pKa calculations. Conformational dynamics ofcationic flexible side chains in the vicinity of the -Cys11-Pro12-Tyr13-Cys14- motif in E. coli Grx3 (A) and E. coli Grx1 (B),obtained during molecular dynamics (MD) simulations (43) in explicit solvent for 50 ns (Grx3) and 75 ns (Grx1). The catalyticnucleophilic cysteine Cys11 (in both Grx3 and Grx1) was treated as a thiolate with both proteins. (A) Conformational spreadadopted by Lys8 relative to Cys11 during the simulation of Grx3. Lys8 only rarely comes close to Cys11 and rarely hydrogenbonds its thiolate (C). Instead, Lys8 spends most of the time pointing in solvent (for clarity, the surrounding water moleculesare not shown), separated from Cys11. (B) Lys8 is replaced by Arg8 in Grx1. The guanidinium group of Arg8 spends moretime hydrogen bonding the thiolate of Cys11 in Grx1 than Lys8 in Grx3. (C) Distance between the thiolate sulfur of Cys11 and(i) the Lys8 side chain amino nitrogen (blue, Grx3), and (ii) the Arg8 side chain nitrogen Ng2 (red, Grx1). Distances around 3Acorrespond to hydrogen bonds between the thiolate and the cationic side chain. The pKa of Cys11 depends on how frequentlysuch hydrogen bonds are formed. Importantly, both Lys8 and Arg8 are very flexible, and it is therefore difficult to infer thestructural and electrostatic roles of such side chains without the insights provided by simulations in explicit solvent. In turn,such computational models can inform experimental testing (155). (To see this illustration in color, the reader is referred to theweb version of this article at www.liebertpub.com/ars.)
112 ROOS ET AL.

with the observation that the pKa of Cys30 is virtually in-sensitive to the E37Q and E38Q mutations in the surroundingof the DsbA active site (66). In fact, elimination of all chargedresidues in the neighborhood of the active site of DsbA had anegligible impact on the pKa of its nucleophilic cysteine (74).Instead, in DsbA as well as in other members of the Trx su-perfamily, there is evidence that interactions between thethiolate and the backbone of residues X1 and X2 are critical formodulation of the thiolate pKa (45).
In general, computational and experimental results indi-cate that the surrounding charged side chains do not pri-marily control the thiolate pKa in the Trx superfamily. Aninteresting possible exception, however, has been uncoveredwith Arg8 in reduced E. coli Grx1 (43). A comparative study ofhomologous E. coli Grx1 and Grx3 by MD and pKa calcula-tions convincingly suggested that Arg8 forms a salt bridge(including a hydrogen bond) with Cys11 relatively frequentlyin Grx1 (43), in contrast to Lys8 and Cys11 in Grx3 (Fig. 5).This could be explained in terms of subtle differences betweenthe conformational dynamics of Arg8 versus Lys8. Therefore,it was proposed that Arg8 contributes to lower the pKa ofCys11 in Grx1 (43), but not Lys8 in Grx3. These computationalinsights have recently received indirect independent experi-mental support (155). Thus, peripheral side chains cansometimes influence the stability of the catalytic thiolate, andsuch mechanism may play a role in other systems (83, 96, 100).The extracytoplasmic atypical Trx ResA provides anotherexample, for which it has been reported that Glu80, locatedclose to the active site, but not forming hydrogen bonds withthe active thiolates, plays a role in controlling the acid–baseproperties of both active-site cysteines (100). Another inter-esting case is that of protein disulfide isomerase (PDI), wherethe pKa values of cysteine residues play a crucial role duringoxidative protein folding (83). In PDI, Arg120 was reported tolower the pKa of the C-terminal cysteine from 9.2 to 8.6 (96). Amovement of Arg120 in the active site was reported, but notdescribed in details (83), so it is unclear if its effect on the C-terminal cysteine is mediated by a direct hydrogen bond orlonger-range interactions. Overall, however, the role of pe-ripheral side chains for thiolate stabilization in redox enzymesof the Trx superfamily appears limited.
If the charged side chains form hydrogen bonds with thethiolate, it stabilizes the thiolate. Apart from Grx1, this wasalso demonstrated in human peroxiredoxin, where a con-served arginine residue (Arg127) is hydrogen bonded to theredox-active cysteine Cys51 and strongly diminishes theproton affinity of the thiolate form of Cys51 (16). The protonaffinity is related to the Cys51 pKa. Another conserved argi-nine molecule (Arg150), which is also a part of the active sitewith Cys51, but is not hydrogen bonded to Cys51, has a muchsmaller influence on the Cys51 proton affinity. So, hydrogenbond contacts seem important to mediate the influence ofcharged side chains on cysteine pKas.
Considering the enzymatic functional requirements, it ismaybe not surprising that nature has not selected flexiblecharged side chains as the main mechanism for thiolate sta-bilization in the Trx superfamily and its solvent-exposed ac-tive site. First, peripheral side chains have to be long (Arg andLys), and therefore flexible, to reach to the thiolate. Thus, astable ionic contact with the thiolate would incur an entropiccost. Second, a side chain approaching the thiolate on itssolvent-exposed side may occlude the active site and sterically
prevent the approach between the thiolate and its substrate.Third, when charges are exposed to a high-dielectric mediumsuch as water, the electrostatic interaction between charges isscreened and strongly diminished. Therefore, water mole-cules in a solvent-exposed active site could easily disrupt thesalt bridge. Fourth, the stabilization of the thiolate by chargedside chains would expose the enzymatic activity to the influ-ence of ionic strength. In contrast, this ionic strength effect isminor when the thiolate is stabilized by hydrogen bonds withneutral groups, for example, with backbone amides (44, 116).In addition, controlling a cysteine pKa by local hydrogenbonds means that the peripheral ionized side chains canevolve independently of the maintenance of this pKa (44). Thatleaves the peripheral side chains free to evolve under differentselection pressures, guided may be instead, by substrate rec-ognition. Also, hydrogen bonds allow a much more precisemolecular control of the enzyme chemistry, for instance,by discriminating between the active-site N-terminal and C-terminal cysteines in the -C-X-X-C- active-site motif of en-zymes (Fig. 3). Indeed, it is difficult to imagine how long-range electrostatic interactions with flexible peripheralcharged side chains would discriminate between the active-site N-terminal and C-terminal cysteines, since these twocysteines are very close in space. Instead, very directionallocalized hydrogen bonds can dramatically decrease the pKa
of the N-terminal cysteine without affecting the neighboringC-terminal cysteine, as discussed in the next section.
B. The strong influence of direct hydrogen bondson the pKa of cysteines
Before discussing how hydrogen bonds perturb the pKa ofcysteine side chains, it is helpful to review the informationpertaining to hydrogen bonds involving sulfur. Hydrogenbonds are energetically favorable interactions formed be-tween a donor group (D - H) and an acceptor atom (A). D is anelectronegative atom that polarizes the Dd - - Hd + bond, re-sulting in a partial positive charge on the hydrogen atom. Thehydrogen positive charge interacts with the (partial) negativecharge of the acceptor atom, resulting in a favorable electro-static interaction. Thus, hydrogen bonds are largely electro-static interactions in nature, resulting in the well-knownpattern: Dd - - Hd + –A(d) - .
Since sulfur is less electronegative than oxygen or nitrogen,the role of sulfur in hydrogen bonding has been a matter ofdebate. Compared to oxygen, sulfur has a lower electroneg-ativity and a larger radius, which reduce the ability of sulfurto participate in hydrogen-bonding interactions, as a donor oras an acceptor. However, there is now evidence that the thiolgroup can act as a moderately strong hydrogen bond donor oracceptor, and the thiolate is a hydrogen bond acceptor (1, 37,39, 45, 57, 84, 129, 137, 183, 196). Hydrogen bonds with sulfurare longer than those with nitrogen or oxygen because of thesize of the sulfur atom and its more diffuse electron cloud (57,196). A statistical analysis on more than 500 high-resolutionprotein crystal structures indicated a 5:1 donor:acceptor ratiofor sulfur in protein thiol groups (196), suggesting that sulfurin (neutral) thiols has a greater propensity to donate a hy-drogen bond than to accept one. Yet, some QM calculationshave suggested that sulfur may, at least sometimes, be almostas strong a hydrogen-bond acceptor as oxygen (137, 182). Inthe reduced -Cys1-X1X2-Cys2- motif of the Trx superfamily,
PKA OF REDOX CYSTEINES 113

the cysteine thiols act as hydrogen bond donors and acceptors(39, 45, 129).
The perception of hydrogen bonds by sulfur is probablydominated by crystallographic observations of distances andangles between sulfur atoms and potential donor or acceptorgroups (1, 37, 57, 84, 196). A crystallography-based view of thegeometric parameters of various types of hydrogen bondsinvolving sulfur in proteins was recently presented (196). Toconsider hydrogen bonds that stabilize thiolate anions, weconcentrate on the geometric parameters of sulfur as a hy-drogen bond acceptor. In protein crystal structures of NH–Ssystems, the distance between the donor nitrogen and theacceptor sulfur ranged from 3.25 to 3.55 A, with deviationsfrom linearity of the NH–S angle up to 25� (1, 37). An analysisof 151 high-resolution protein X-ray structures identified amean distance of 3.54 A and a mean angle of 138� for hy-drogen bonds with cysteine sulfur atoms as hydrogen bondacceptors (196). Table 3 summarizes the mean distances andangles for cysteine sulfur accepting hydrogen bonds fromdifferent conventional donor types (196). In principle, oneshould also consider nonconventional, weaker, C - H hydro-gen bonds to the sulfur. Indeed, there is now ample evidencethat the C - H bond can be polarized enough to form inter-actions with a marked hydrogen bond character (33, 59, 167,174). In proteins, examples of C - H hydrogen bond donorsinclude the backbone Ca - H group, and aromatic C - H vec-tors provided by aromatic side chains (18, 71, 168, 181). Al-though the C - H groups are weaker hydrogen bond donorsthan conventional donors, they could still contribute to tunethe pKa of some thiols. One such example has been proposedwith pig Grx (Fig. 6), for which a detailed analysis (44) sug-gested that an aromatic C - H from a phenylalanine contrib-utes to stabilize the thiolate. However, favorable interactionsbetween sulfur acceptor and nonconventional C - H hydro-gen bond donors have barely been explored. Thus, our anal-ysis concentrates on conventional hydrogen bonds to thesulfur.
For redox cysteines, the geometric hydrogen-bonding pa-rameters are likely to depend whether the accepting sulfur isneutral or in thiolate form. A thiolate acceptor may allow abroader range of angles for interaction with a hydrogen bonddonor. In addition, one has to consider the situation wheresulfur receives a hydrogen bond from another thiol, whichlengthens the hydrogen bond distance. Thus, pragmatically,the following geometric criteria are frequently suitable to
capture hydrogen bonds between sulfur and a donor atom: S–D distance < 4 A and S–H - D angle > 90� (141, 196). In thefollowing paragraphs, we discuss some examples that dem-onstrate the influence of hydrogen bond interactions on thiolpKa in redox enzymes of the Trx superfamily.
For the conserved -Cys1-X1-X2-Cys2- motif of proteins in theTrx superfamily, observations link the pKa of Cys1 to thenumber of hydrogen bonds received by the acceptor sulfur ofCys1. Structural analysis revealed that in reduced Trx, twohydrogen bonds are formed with Cys1; three in reduced Grx;and four in reduced DsbA (45) (Fig. 5). This is consistent withthe pKa of 3.5 for Cys1 in DsbA, of 4.0 to 5.0 in Grx, andranging from 6.3 to 7.1 in Trx (38, 46, 77, 82). Note the im-portance of the hydrogen bonds with the backbone N–Hgroups, which may also rationalize the largest pKa variationsacross the DsbA X1-X2 mutants. The predominant role of di-rect hydrogen bonding in stabilizing the thiolate in the Trxsuperfamily is consistent with the lack of effect of increasedionic strength (0.05–2 M) on the pKa of Cys22 of human Grx1(44, 116). Using PB-based calculations, the pKa of Cys1 in Grxwas estimated with the hydrogen bonds to the thiolate formedor not, depending on the conformation adopted by the sidechain of Cys1 (45). Simply changing the rotamer of Cys1 canswitch the hydrogen bonds to the sulfur on or off, whichshowed that disrupting the hydrogen bonds clearly increasesthe calculated thiol pKa. In general, the more hydrogen bondsto the sulfur that were present, the lower the pKa of the thiol inPB calculations (35, 43, 45). The influence of hydrogen bondson the pKa of Cys1 is also seen in calculations with PROPKA1(103). PROPKA1 calculates a Cys1 pKa of 3.4, 4.4, and 5.5 in,respectively, DsbA (Protein Data Bank [PDB] entry 1DSB),human protein disulfide isomerase (hDPI, PDB entry 1MEK),and E. coli Grx 3 (PDB entry 3GRX). PROPKA1 attributesthese low pKas to hydrogen bond interactions between thethiolate form of Cys1 and surrounding residues (103).
There is also evidence that controlling the thiol pKas withhydrogen bonds plays a role during thiol–disulfide exchangereaction mechanisms, as suggested for Trx and Staphylococcusaureus pI258 arsenate reductase (Sa_ArsC) (Fig. 7). Sa_ArsC isone of the endogenous substrates of the powerful reductaseTrx (8, 21, 115). Trx reduces oxidized ArsC via the nucleophilicattack of Cys29 of Trx (Cys1 in Cys1-X1-X2-Cys2) on the ArsCdisulfide (Cys82–Cys89), leading to the release of Cys82 andformation of the Trx-ArsC Cys29–Cys89 mixed disulfide(115). In a subsequent step, this mixed disulfide needs to be
Table 3. Geometric Characterization of Hydrogen Bonds Accepted by the Sulfur of Cysteines
in Proteins with Different Donor Types
Donor type Number of structures d (S–H) A d (S–X) A y (S–H–X)�
Backbone N 95 2.79 (0.23)a 3.58 (0.19) 143.5 (24.6)Amide N 12 2.87 (0.25) 3.61 (0.23) 136.8 (25.0)Charged N 18 2.78 (0.33) 3.44 (0.23) 128.2 (22.2)Aromatic N 8 3.05 (0.12) 3.50 (0.18) 109.4 (15.4)Hydroxyl O 18 2.76 (0.35) 3.45 (0.24) 133.1 (27.8)All 151 2.80 (0.26) 3.54 (0.21) 138.1 (25.6)
aGeometric mean with standard deviation in brackets.d (S–H): distance between the acceptor S and donor H atom; d (S–X): distance between the acceptor S and the donor X atom; h (S–H–X):
angle between sulfur, hydrogen, and donor.Part of the data used in this table are reproduced from reference (196).
114 ROOS ET AL.

reduced to release reduced ArsC (141). In this process, Cys32of Trx (Cys2 in Cys1-X1-X2-Cys2) attacks Cys29 of Trx in themixed disulfide, and Trx becomes oxidized (Cys29–Cys32disulfide formed). Thus, the dissociation of the Trx-ArsCmixed disulfide proceeds via the nucleophilic attack of Cys32of Trx on Cys29 of the Cys29Trx–Cys89ArsC disulfide. In iso-
lated, reduced Trx, Cys32 has a high pKa (pKa > 9), and ispresent in its thiol form. In the mixed disulfide, the resolvingcysteine Cys32 needs to be activated to its nucleophilic thio-late form. A detailed study of the Trx-ArsC mixed disulfidecomplex (141) suggested that two hydrogen bonds betweenthe Cys32 sulfur and backbone amides of Cys29 and Trp28 ofTrx stabilize the thiolate form of Cys32. In the presence ofthese hydrogen bonds, the pKa of Cys32 drops to *7.7 (141),which activates Cys32 for its nucleophilic attack on Cys29.Formation of these hydrogen bonds to Cys32 was uncoveredby MD simulations after localized conformational re-arrangements around Cys32 (141). This illustrates how hy-drogen bonds control the reactivity of a thiol via small, butprecise, structural rearrangements.
The above examples, and others (see section III.D), supportthe notion that hydrogen bonds may be the primary factorsmodulating thiol pKas. It follows that a first indication of adecreased thiol pKa could be gained from structural infor-mation, simply by counting the hydrogen bonds to sulfuratoms in the structural model. Since the influence of hydrogenbond interactions is at short range, they are very sensitive tothe details of the protein structure and dynamics (43–45).Consequently, the details of the structural model used forvisual interpretation and pKa calculations are important, andapparently, minor structural changes may strongly affect thecalculated pKa values of interest (3, 35, 43–45, 192). Hence, adegree of manipulation of the raw X-ray coordinates may berequired to orientate protein side-chain amide and imidazolegroups, as well as conserved water molecules, according tothe most likely hydrogen-bonding network (123, 126). Also,NMR structures present special challenges, since their detailscan be prone to uncertainties, which affect the outcome of pKa
calculations (35, 43, 134).Apart from hydrogen bonds, another putative influence on
the pKa needs further consideration. In the Trx superfamilyactive sites, Cys1 is located at the N-terminus of an a-helix.Helices have long been perceived to decrease the pKa of res-idues at their N-terminus. For example, a pKa decrease of theN-terminal cysteine of, respectively, 1.8 and 2.0 units wasmeasured in rhodanese (151) and human Trx (46). The mea-sured pKa of a N-terminal aspartate in a helical dodecapeptidewas suppressed by 0.6 units (81). Earlier quantum chemicalstudies on papain have shown that a helix near the active sitefacilitates the proton transfer from the N-terminal cysteine tothe histidine residue of the catalytic dyad (171). The origin ofthis effect is explored in the following section.
C. Reinterpretation of the helical effect on the pKasof cysteines
The decreased pKas of residues at the N-terminus of heliceshave been attributed for a long time to a helix–macrodipoleeffect, which would originate from the vector sum of the mi-crodipole moments of the individual peptide units, andwould be oriented along the helix-axis (67, 135, 150, 156, 176,180). However, recent PB calculations on several model heli-ces stressed that the helix dipole depends on the geometry andon the solvent exposure of the helix termini (152). Therefore,the helix macrodipole is not a simple vector sum of the indi-vidual dipoles. In vacuum, the helix dipole increases with thehelical length, but in transmembrane helices in which bothhelical termini are solvent exposed, the helical dipole was
FIG. 6. Nonconventional hydrogen bonds may contributeto stabilize a thiolate. The figure shows snapshots of re-duced pig glutaredoxin (pGrx) taken every nanosecond froman MD simulation of 120 ns (44), overlaid on the -Cys22-Pro23-Phe24-Cys25- motif. Three hydrogen bonds between thethiolate of Cys22 and conventional hydrogen bond donors(backbone N - H groups of Phe24 and Cys25, and thiol ofCys25) are shown with green dotted lines. A nonconventionalhydrogen bond between the thiolate and an aromatic C - Hgroup of the phenyl of Phe24 is also suggested (magentadotted line). The significance of this interaction was discussedin details (44), providing evidence that this interaction alsocontributes to stabilize the thiolate, in addition to conven-tional hydrogen bonds. Indeed, some C - H dipoles can beregarded as weak, but significant, hydrogen bond donors(33, 59, 167, 174). The C - H bonds of aromatic rings havemarked dipoles, with an excess of positive charge on theproton (18, 71, 168, 181). In pGrx, one of the C - H dipoles ofPhe24 was frequently positioned to point to the thiolatesulfur, with geometries consistent with some hydrogen-bonding character. The Phe24 side chain is at the proteinsurface, where it could adopt alternative conformations.However, the conformation with a stabilizing electrostaticcontact between the phenyl group and the thiolate was fa-vored. This is consistent with the conservation of an aromaticside chain at the X2 position in the -Cys1-X1-X2-Cys2- motif ofGrxs, where X2 is either Tyr or Phe. An aromatic side chain ispresent at this position even in nonclassical Grx sequences,for example, -Cys14-Val15-Tyr16-Cys15- in phage T4 Grx or -Cys30-Gly31-Phe32-Ser33- in E. coli Grx4. (To see this illustra-tion in color, the reader is referred to the web version of thisarticle at www.liebertpub.com/ars.)
PKA OF REDOX CYSTEINES 115

reported to decrease with the helical length (152). For solvent-exposed helices, the effective dipole is strongly dependent on theorientation of the helix relative to the aqueous medium (152).When aqueous solvent is present, the helix macrodipole iscounteracted by the solvent reaction field, which drastically re-duces the long-range effects of the helix macrodipole. So, inmany situations, there is effectively no helix macrodipole atwork. Thus, although the older helix macrodipole hypothesishas been widely influential (19), there is strong evidence that theorigin of the so-called helical effect can be explained and re-interpreted without needing to invoke a helical macrodipole.These new insights came from several studies, both computa-tional and experimental (6, 45, 49, 66, 89, 103, 143, 147).
Early new insights into the helical effect on pKas came fromcomputational studies on papain, for which more than half ofthe helical effect was attributed to hydrogen bonds with thebackbone rather than to the macrodipole (147). This empha-sized localized short-range interactions in addition to long-range electrostatic interactions. With sulfate-binding protein,Aqvist et al. (6) calculated the electrostatic contribution to thefree energy of interaction between the helix and the substrateSO4
2 - bound at its N-terminus, and concluded that thecharge-stabilizing effect of the a-helix can be best explained byshort-range interactions with individual peptide bond N–Hdipoles at the N-terminus of the helix. They found that the firsttwo helical turns account for 95% of the overall helical effect.The minimal influence of the helix macrodipole was sup-ported by investigating the effect of introducing a positive
charge by mutation at the helical C-terminus. Such chargewould have neutralized the dipole charge, but its introductionhad a negligible effect according to the calculations (6).
It has also been possible to address the helical effect ex-perimentally, although it is more difficult to control thepresence and geometry of a helix experimentally. DsbA of-fered an opportunity to study experimentally the effect of thehelix dipole on the pKa of its catalytic Cys30 by manipulatinga kink in the relevant helix (66). Mutants designed to alter thehelical kink were expected to affect the overall helix dipole;however, only a minor effect on the pKa of Cys30 at the N-terminus of this helix was observed.
Kortemme and Creighton explored experimentally andsystematically the nature of the helical effect (89), by moni-toring the pKa of a cysteine at the N- or C-terminus of model a-helical peptides. They observed that a thiol pKa at the N-terminus of a peptide with high helical content was decreasedby up to 1.6 pKa units (pKa values from 7.20 to 7.63) relative toa normal thiol pKa measured in an unfolded peptide. Theinterpretation was that a combination of electrostatic charge–helix dipole and hydrogen bonding interactions contribute tothe pKa-lowering effect (89). Yet, several observations wereconsistent with the particular importance of hydrogen bondsand local effects. Thus, variation of the (neutral) amino acidsat the peptide N-terminus had an impact on the thiol pKa atthis N-terminus, pointing to the importance of local confor-mational effects and geometries, compatible with the pKa
being decreased by hydrogen bonds. The same study varied
FIG. 7. Reaction mechanism of disulfide reduction by Trx. A schematic representation of the reaction mechanism is shownon top of the structures of Trx at each step of the reaction. The reaction takes off with a nucleophilic attack of the N-terminalcysteine of the conserved -C-G-P-C- motif targeting the disulfide (1). The thiolate of the nucleophilic cysteine is stabilized bytwo hydrogen bonds with the - NH of the glycine and the - SH of the C-terminal cysteine [PDB code: 1TRV(136)] (A). As aresult, an intermediate mixed disulfide complex is formed between Trx and the substrate protein, which in turn is reduced bya nucleophilic attack of the C-terminal cysteine of the -C-G-P-C- motif (2). The C-terminal cysteine is primed for nucleophilicattack in the Trx–protein mixed disulfide complex. Selected snapshots from an MD simulation of the B. subtilis Trx and ArsCcomplex show that the thiolate on the C-terminal cysteine is stabilized with two backbone amide hydrogen bonds, whichlowers its pKa to 7.4 (B) (141). Further, the N-terminal cysteine of Trx has been found to be more susceptible for thenucleophilic attack of the C-terminal cysteine and is also sterically closer to the C-terminal cysteine (141). One single catalyticreduction cycle stops with the release of a reduced substrate protein and oxidized Trx [PDB code: 1TRU (136)] (C). The figureswere generated using MacPyMol (Delano Scientific LLC 2006). Reproduced with the permission of (24). (To see this illus-tration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
116 ROOS ET AL.

the ionic strength to distinguish between stabilization of thethiolate by hydrogen bonds or by a more-diffuse electrostaticinteraction between the thiolate and a helix macrodipole.Hydrogen-bonding interactions should be much less sensitiveto screening by salt than charge–helix dipole interactions.Indeed, the thiol pKa was only weakly affected by changes inionic strength, suggesting a dominant role for hydrogenbonding. Further evidence for a very weak interaction be-tween the thiolate charge and a helix macrodipole came frompKas measured for thiols at the C-terminus of peptides withhigh helical content (89). Such pKa increased by only 0.2 pKa
units relative to a normal cysteine pKa. The asymmetry of themagnitude of the pKa shifts observed at the N- versus C-terminus is difficult to reconcile with a strong overall helixmacrodipole. However, this asymmetry can be explained interms of local hydrogen bond interactions, since the helical N-terminus, but not the C-terminus, presents amide N–H donorgroups for interactions with a negative charge.
Recent work showed that the main influence on the pKa ofCys1 of the -Cys1-X1-X2-Cys2- motif located at the N-terminusof an a-helix in proteins of the Trx superfamily is from hy-drogen bond interactions. In the PB study of E. coli Grx3 (45),the effect of the entire helix on Cys1 (Cys11) was investigatedby turning off incrementally the charges on the peptide bonddipoles one after another, starting with the amide groupsclosest to the cysteine of interest (Fig. 8). This revealed that thefirst two helical amide groups decreased the Cys11 pKa by*1.5 units each; the third and fourth amide groups decreasedthe pKa by *0.5 and *0.2 units, respectively; with the sixthamide group, a decrease of only 0.09 units was found. So, thepKa value of Cys1 in the -Cys1-X1-X2-Cys2- motif could bepredicted in satisfactory agreement with an experimentwithout invoking the helix–macrodipole effect (45). This re-sult was confirmed during the development of PROPKA(103), when it was found that pKa values can be reproducedbased on the numbers of hydrogen bonds formed with thecysteine sulfur without intervention of a helix macrodipole(there is no such parameter in the PROPKA model).
Further evidence came from a computational quantummechanical study on the effect of the helix length on the pKa ofa cysteine thiol located at the N-terminus of model helices(143). Both 310- and a-helices of increasing lengths were tested,made of (neutral) alanine residues (143). The pKas were cal-culated with the NPA method (see section II.B). An initialdecrease of the cysteine pKa with the first helical turns wasfound, but this effect weakened quickly after a few addedresidues. This pKa decrease was accompanied by the increaseof the backbone amide–ScCys hydrogen bond strength. Thefirst Ala residue decreases the cysteine pKa by 1.5 or 0.6 unitsin an a- or 310-helix, respectively. For the second residue, anextra decrease of, respectively, 0.2 or 0.1 pKa units was foundin a- or 310-helices. The third residue was responsible for anextra decrease of 0.1 units (Table 4). As such, the pKa decreasediminished for every additional helical residue. Thus, oncethe hydrogen bonds to the sulfur are formed (typically asso-ciated with the first helical residues i.e., cysteine plus one al-anine), the next residues strengthen these hydrogen bonds,but this has only a secondary effect on pKa perturbation.These QM calculations (143) pointed out that the lowering ofthe pKa of the N-terminal cysteine of an a- or 310-helix is lar-gely due to the hydrogen bonds formed between the cysteinesulfur and the helical N-terminus.
FIG. 8. Effect of the helix electrostatics on the pKa of acysteine thiol at the helix N-terminus. (A) Shows the re-duced Cys11 (space-filling, sulfur in yellow) of E. coli Grx3, atthe N-terminus of a helix comprising residues 12–24 (PDBentry 1ILB). For clarity, only the helix backbone and selectedhydrogens are shown. The amide N - H groups are depictedin ball and stick. The two direct hydrogen bonds between thehelix backbone amide N - H groups and the sulfur of Cys11are shown as green dotted lines. (B) Shows how the pKa ofCys11 is influenced by the electrostatics of the helix back-bone, represented by the partial charges of its backboneamide groups. The pKa was calculated with the Poisson-Boltzmann method in the context of the full protein, as de-scribed (45). This approach allows calculating the pKa whilethe charges (dipoles) for specific amide group of the helix areremoved (i.e., turned off) from the calculation. The plot in (B)shows how the pKa of Cys11 increases by removing cumu-latively the partial charges on the helix backbone amidegroups, from the helix N-terminus to its C-terminus. Thecalculated pKa with charges on all residues included (value 0on the X-axis) of Cys11 was *5.2. By removing the chargeson the first amide group at the helix, the N-terminus in-creased the pKa to *6.7. This corresponds to removing ahydrogen bond between a backbone N - H group and thesulfur. Removing also the charges on the next backboneamide group removes the second hydrogen bond to thesulfur and increased the pKa to *8.2. Importantly, removingthe electrostatic influence of further backbone amide groupsonly has a limited impact on the pKa. Thus, most of the pKa
downshift due to the helix can be attributed to its two N - Hgroups directly hydrogen bonding the sulfur. The backbonedipoles further away from the helix N-terminus have an in-creasingly minor contribution to the pKa shift, arguingagainst a significant role for a helix macrodipole. (To see thisillustration in color, the reader is referred to the web versionof this article at www.liebertpub.com/ars.)
PKA OF REDOX CYSTEINES 117

Overall, the above-mentioned studies consistently showthat direct hydrogen bonds between the cysteine sulfur andthe helical N-terminus are essentially sufficient to account forthe thiol pKa downshifts. To explain such pKa shifts, the no-tion of helix macrodipole is not required. This further sup-ports the idea developed in the previous section thathydrogen bonds to the cysteine sulfur are the main factorsinfluencing the pKa values of the corresponding thiol groups.
D. How general are the mechanisms modulating thepKa of cysteines?
Apart from their function in the redox biochemistry, cys-teines play important roles in the catalytic processes of a wide
variety of enzymes (80, 109, 110) like proteases, transferases,kinases, phosphatases, and isomerases. Cysteine residuescoordinate metallic redox centers as in iron–sulfur clusters.Coordination of metals by cysteines can also play structuralroles, such as zinc coordination in Zn-finger domains (94). Inaddition, the structural organization and oxidative folding ofproteins rely on disulfide bonds. One can surmise that thestructural and physical principles modulating the cysteinethiol pKa in redox enzymes, as discussed in the previoussections, are likely to be largely transferable to cysteines inother protein families.
For example, the reactive cysteine (Cys106) in human DJ-1 [aprotein linked to Parkinson’s disease and member of the class Iglutamine amidotransferase-like superfamily (107)] has a de-creased pKa of 5.4. A marked contribution to this pKa shift hasbeen attributed to a hydrogen bond interaction with a con-served protonated glutamic acid Glu18, accounting for a pKa
decrease of 1.0 unit (184). On the other hand, the sulfate anionat 5.9 A of the reactive cysteine only increases the thiol pKa by0.4 units (184). This further illustrates the limited role of long-range electrostatics and identifies hydrogen bonding as a keyfactor determining the pKa of Cys106. Another example ofhydrogen bonds determining a cysteine pKa is found in humanmuscle creatine kinase. In this enzyme, the pKa of cysteine 282was measured to be 5.8 (177). QM calculations pointed out thatthe main determinants of this low pKa are the hydrogen bondsbetween Cys282 and the - OH group of a serine side chain anda backbone amide (120, 177). Each hydrogen bond lowers thepKa by, respectively, 0.8 and 1.5 units (120).
In human a-antitrypsin, the pKa of the active Cys323 wasmeasured to be 6.9 (58). QM studies indicated that a hydrogenbond with the amide group of a neighboring Leu residuedecreases the pKa of Cys323 by 1 (120). In ArsCs with a low-molecular-weight tyrosine phosphatase fold from S. aureus(145) and Corynebacterium glutamicum (175), a hydrogen bondnetwork decreases the pKa of the nucleophilic cysteine (Fig. 9).
Table 4. Electrostatic Properties of Model
310-Helices and a-Helices, and Influence
on the pKa
of Cysteines at the N-Terminus,
Calculated Quantum Mechanically
Dipole(D)
NPA Sg(a.u.) pKa
Additional pKa
decrease peramino acid
310-helicesCysteine 6.45 - 0.824 8.3 —S_310_2 14.33 - 0.791 8.06 - 0.64S_310_3 14.38 - 0.786 7.92 - 0.14S_310_4 17.97 - 0.782 7.80 - 0.12
a-helicesS_a_2 11.64 - 0.763 7.22 - 1.48S_a_3 12.77 - 0.758 7.04 - 0.18S_a_4 15.32 - 0.753 6.90 - 0.14S_a_6 26.65 - 0.754 6.93 + 0.03
Helix–macrodipole, NPA charge and pKa of N-terminal ScCysobtained in aqueous solution in 310-helices and in a-helices.
Table adapted from reference (143).
FIG. 9. A hydrogen-bonding network in ArsCs with a low-molecular-weight protein tyrosine phosphatase (LMWPTPase) fold is lowering the pKa of the nucleophilic cysteine. (A) View on the active-site P-loop of pI258 ArsC from S.aureus (PDB entry code 1LJL) (114). This potassium-binding site is an interesting feature observed in pI258 Sa_ArsC (95), asbinding of K + stabilizes the structure of Sa_ArsC and increases the specific activity with a factor of 5 (95, 140). Potassium ishere a part of a hydrogen-bonding network (black) that decreases the pKa of the nucleophilic cysteine thiol in pI258 Sa_ArsC(145). Other sulfur amide hydrogen bonds are in blue. (B) A view on the acitve-site P-loop of ArsC1¢ from C. glutamicum (PDBentry code 3T38) (175). The hydrogen-bonding network from the lysine 144 (K144) via the asparagine 91 (N91) and serine 95(S95) to the sulfur of C88 is indicated (black) next to the hydrogen bonds with the backbone amides (blue). The asparagine ofthe active-site P-loop is conserved in the b-conformation of the Ramachandran plot among ArsCs with a LMW-PTPase fold.In ArsC1¢ and ArsC2 (not shown) from C. glutamicum, there is no potassium-binding site. Here, the charged NfH + of aconserved lysine takes over the role of the potassium. The figure was generated using MacPyMol (Delano Scientific LLC2006). (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
118 ROOS ET AL.

Another example is found in Tn501 mercuric ion reductase,the key enzyme involved in reducing Hg2 + to Hg0 in bacteria(11). Its N-terminal heavy-metal-associated domain containstwo cysteines, Cys11 and Cys14, with decreased pKas of 7.7and 7.2, respectively (97). The authors invoke the position ofboth cysteines at the N-terminus of an a-helix as main factorlowering the pKas. This helical effect is not further character-ized, that is, the authors do not give evidence for a helicaldipole effect nor mention hydrogen bonds. Based on sectionIII.C, we propose that the so-called helical effect may be at-tributed to hydrogen bond interactions between Cys11 orCys14 and N-terminal helical residues. This is supported byan examination of the structure with the PDB code 2KT2 (97).Based on this NMR structure, the following hydrogen bondsare found between the thiol side chain of Cys14 and backboneamides: Cys14Sc–NHTyr10: 3.4 A and Cys14Sc–NHCys11:3.7 A, while no hydrogen bonds formed with Cys11 could beobserved. This is consistent with the authors’ statement thatthe lower pKa found for Cys14 compared to Cys11 may beattributed to the more precise positioning of Cys14 in the firstturn of the helix compared to Cys11 located in the moreflexible loop preceding the helix (97). Further, the - OH groupof Tyr62 accounts for an extra pKa decrease of 0.8–1 pKa unitsfor both cysteines, while the electrostatic interaction with thepositively charged His8 side chain influences the pKa of bothcysteines by only 0.2 to 0.4 pKa units (97). Again, it suggeststhat the decreased pKas of Cys11 and Cys14 can be attributedto hydrogen bonding rather than to electrostatic interactionsbetween charged residues.
The lowering of the pKa of the cysteine in glutathione(GSH) is seen in GSH S-transferases. GSH S-transferases areinvolved in cellular detoxification in a wide variety of or-ganisms and catalyze the conjugation of GSH to electrophilicsubstrates by lowering the pKa of the cysteine of GSH. Inrelation to this mechanism, the alpha, mu, pI, sigma, and thetaGSH S-transferase classes are the best documented (7). GSH S-transferases are homodimers with a hydrophilic subunit in-terface and each polypeptide chain consists of two domains,an N-terminal domain with a Trx-fold and a C-terminal a-helical bundle. The N-terminal domain contains the active-sitefunctional group, the hydroxyl group of a tyrosine or serineresidue (79, 87, 105, 178), believed to activate the cysteine ofGSH. The reactive species of GSH in the binary complexes ismost probably the thiolate anion, which accepts a hydrogenbond from the seryl or tyrosyl hydroxyl group (E-OH–SG)and gathers additional stabilization from a positive charge ofan arginine in the class a enzymes (Fig. 10). In some mu-classGSH S-transferases extra stabilization of the GSH thiol comesfrom a second sphere of electrostatic effects in which the p-electron cloud of the tyrosine is involved (PDB code 6GST)(187). Hydrogen bonding and other electrostatic effects lowerthe pKa of the GSH thiol from *9 to *6 in the enzyme–GSHcomplex (20, 105), so that it is predominantly present asthiolate at physiological pH, and more nucleophilic.
It is not surprising that the factors modulating cysteine pKa
values also modulate pKa values of other titratable groups inproteins (103). Local interactions were also proposed to be themain pKa determinants for aspartate and glutamate residuesin turkey ovomucoid third domain (OMTKY3) (102), sincecalculated and experimental pKa values were in agreementwhen considering only interactions in the immediate vicinity(4–5 A) of Asp or Glu. The developers of PROPKA general-
ized these conclusions regarding the pKas of Asp and Gluresidues by identifying hydrogen bond interactions as themain source for their pKa perturbations (103). It is known thatAsp and Glu residues at the N-terminal of an a-helix usuallyhave lower pKa values (133). As with cysteines at helical N-termini, this effect has long been attributed to the helicalmacrodipole (67, 176). However, recent analyses concludedthat hydrogen bonds between helical backbone amides andAsp and Glu residues are the main contributors to their de-creased pKa values (133), instead of a helical macrodipole.These conclusions echo those obtained for cysteines (45, 143).
IV. Functional Properties Influenced by theCysteine pKas
The previous sections summarized techniques for deter-mining the pKas of cysteines and gaining insight into thefactors that modulate those pKas. This was largely illustratedbased on the active-site cysteines of the redox proteins of theTrx superfamily. During a catalytic cycle, those cysteinesundergo oxidation and reduction. The enzymatic activity ofthese proteins is determined by their reaction kinetics andredox potentials. In this section, we discuss how these prop-erties are influenced by the relevant cysteine pKas. This
FIG. 10. Ribbon diagram of the three-dimensional struc-tures of a glutathione (GSH) S-transferase from the alphaclass in complex with GSH. [Figure made with MacPyMol(Delano Scientific LLC 2006) using the structural coordinatesof PDB code 1F3A (60).] A view on the GSH-binding site isshown. GSH S-transferases are homodimers, each consistingof an N-terminal Trx-fold and a C-terminal helical bundle.The polypeptide chains are in red and gray. The thiol of GSHmakes hydrogen bonds with the tyrosine (Y8) in strand b1and the arginine (R14) located at the N-terminus of helix a1in the position of the nucleophilic cysteine in Trxs. The hy-drogen bonds stabilize the thiolate of GSH for nucleophilicattack and transfer of GSH. (To see this illustration in color,the reader is referred to the web version of this article atwww.liebertpub.com/ars.)
PKA OF REDOX CYSTEINES 119

underlines the functional relevance of the pKa of cysteinesinvolved in enzymatic catalysis.
Lowered pKa values of catalytic cysteines influence thereaction kinetics and thermodynamics, and strongly influencethe catalytic efficiency of an enzyme. This is especially true forthiol–disulfide exchange reactions, which are characterizedby a Brøndsted coefficient 0 < bnuc < 1 for the nucleophiliccysteine (17, 78). The b-coefficients (bnuc, blg, and bc) deter-mine the slope of the plot of the logarithm of the second-orderrate constant versus the pKa: [log(ks - ) = bnuc · pKa(nuc) + blg ·pKa(lg) + bc · pKa(c) + C, with C a constant applicable for a spe-cific thiol–disulfide exchange reaction, and blg and bc, theBrøndsted coefficients for respectively the leaving group thioland the central thiol when a thiolate attacks an unsymmetricaldisulfide] (Fig. 11). Brøndsted coefficients characterize thesensitivity of the reaction to the pKa. The coefficient estab-lishes the change in atomic charge as the reaction proceedsfrom the ground state to the transition state (Fig. 11). Com-plete proton transfer to the nucleophile gives a value for bnuc
of 1, and no transfer a value of 0. At those extreme values,changing the pKa has no influence on the reaction rate. Fornucleophilic thiols with a pKa below the solution pH, an in-creased concentration of the thiolate is less significant than thedecreased nucleophilicity resulting from electron withdrawal.The most significant effect in thiol–disulfide exchange reac-tions comes from the decrease of the pKa of the leaving group.As the pKa of the leaving thiolate is decreased by electron-withdrawing substituents or electrostatic effect, the rate con-stants for the reaction increases with a factor of 3.2 to 5 foreach unit decrease in the pKa of the leaving thiolate (blg = - 0.5to - 0.7) (164). Decreasing the pKa of the leaving thiolate from8.5 to 4.5 should increase the rate for thiol–disulfide exchangeby *100-fold to 630-fold (52). Also, the electron withdrawalon the central thiol (bc) will accelerate the thiol–disulfide ex-change with a factor of *2 for each unit decrease in the pKa
(bc = - 0.3) (164). All together, the decrease of the thiolate pKa
in proteins should be more important in the function of thethiolate as a leaving group than in the function of the thiolateas a nucleophile.
Another example on how the pKa determines reaction ratesis the correlation between a cysteine pKa and the rate constantof the H2O2-induced cysteine oxidation to sulfenic acid. Thio-lates react more rapidly with H2O2 than thiols (183); here again,a low pKa means a high reaction rate. Ferrer-Sueta et al. refinedthis overall view (41). Features that act to decrease the pKa mayalso decrease the nucleophilic character of the thiol, and hencemake it less reactive. The effect of lowering pKa on rate en-hancement will in general be most significant when pKa valuesare close to solution pH. A small increase in the concentration ofthe thiolate resulting from a further reduction of pKa will beundermined by the corresponding decreased nucleophilicityresulting from electron withdrawal (Fig. 11).
In the Trx superfamily, the pKa of the nucleophilic cys-teine is related not only to reaction rates but also to thedisulfide reduction potential. When this pKa decreases, theassociated disulfide/thiol reduction potential increases, thatis, the enzyme becomes a stronger oxidant. This wasshown for Trx and DsbA active-site mutants for which alinear correlation between pKa and reduction potential wasfound (69, 142). Empirical relations between the cysteinepKa and the disulfide/thiol reduction potential have beenproposed for Trx-type oxidoreductases (118). Here, we
show a correlation for Trx-fold enzymes of E. coli (Fig. 12).The redox potential increases with *54 mV for each de-creasing pKa unit. The only exception is seen for thegamma-domain of DsbD, which has an unusual high pKa
of more than 9 [9.3 (161) and 10.5 (112)] and a redox po-tential of - 241 mV (25). This makes the nucleophilic cys-teine (Cys461) poorly reactive toward the disulfide ofDsbDa and prevents its nonspecific oxidation (32). How-ever, formation of the complex between DsbDc (C-terminaldomain) and DsbDa (N-terminal domain) seems to lowerthe pKa of Cys461, allowing the transfer of electrons be-tween these two DsbD domains (112, 113).
The importance of pKas with respect to disease and healthcan be gathered from many examples. For example, Trxproteins, the function of which is largely depended on thepKa of the cysteines in the -Cys1-X-X-Cys2- motif, are in-volved in the regulation of diverse proteins like
FIG. 11. The decrease of the thiolate pKa in proteinsshould be more important in its function as leaving groupthan as nucleophile. (A) Thiol–disulfide exchange reactionshowing the ground state and the transition state structures.The nucleophilic attack of a negatively charged thiolate on anunsymmetrical disulfide results in a transition state with thenegative charge distributed on the nucleophilic thiolate(Snuc), the leaving thiolate (Slg), and the central sulfur (Sc). (B)The effect of the electron withdrawal on the rate constants forthiol–disulfide exchange at pH 7.0. The figure was re-produced from the paper of Hiram Gilbert (52) showing aneducative view based on approximate rate constants of LMWthiols calculated using the Brønsted relationship of Szajewskiand Whitesides (164). With a pKa of the nucleophile > 7,lowering the pKa (increasing electron withdrawal) will resultin an increased reactivity at pH 7 due to an increase of thethiolate concentration [Snuc
- ]. With a pKa of the nucleophile< 7, lowering the pKa results in a decreased nucleophilicityand reaction rate due to electron withdrawal. These twoopposing effects result in an optimum pKa around the so-lution pH. As the pKa of the leaving thiolate (Slg
- ) is de-creased by electron withdrawal by electrostatic effects, therate of the reaction increases linearly with decreasing pKa.Both effects are clearly visualized in the log(kobs)-versus-pKa
plot for which the slopes indicate the magnitude of theBrønsted coefficient in the Brønsted equation: log(ks - ) =bnuc · pKa(nuc) + blg · pKa(lg) + bc · pKa(c) + C.
120 ROOS ET AL.

peroxiredoxin, transcription factors, and ribonucleotide re-ductase. As such, Trxs are involved in maintaining the redoxhomeostasis of the cell (22, 24, 63, 78), antioxidant defense,apoptosis and DNA synthesis, and repair. All these factorsare involved in many diseases like diabetes, cardiovasculardiseases, cancer, Alzheimer’s, and Parkinson’s disease. Forexample, proteins involved in protein folding, like PDI, havea direct impact on heart and kidney failure (34). In addition,GSH S-transferases, which catalyze the conjugation of GSHto a broad range of xenobiotic substrates by lowering thepKa of GSH, are well known to influence the metabolism of anumber of drugs (65, 148). Thus, there is ample evidence thatcysteine pKas are critical to many biomolecular mechanismsessential for cellular functions, and are also involved inpharmacological processes. Therefore, understanding themolecular factors that control these pKas will help futureresearch to understand the molecular basis of some diseaseconditions.
V. Conclusions
Empirical and case-by-case determination of the pKa val-ues of catalytic cysteines remains very valuable, since itprovides insights into the chemistry of individual enzymaticreactions. Also, determining additional reliable experimentalpKa data is very important to strengthen the foundations forthe development of theoretical predictive methods. How-ever, there are limits to what one can expect from time-consuming experimental measurements. It is probably un-realistic to hope to be able to measure all interesting pKa
values. Indeed, many relevant pKa values will adjust tran-siently during molecular encounters and complex reactionmechanisms (141). In these situations, it is already clear thatcomputational approaches will become the methods ofchoice. However, for those theoretical methods to be usedroutinely and confidently, their performances need to be
further tested and improved. The results of accurate calcu-lations will provide not only the pKa values themselves butalso precise insights into the factors modulating the pKasand the associated functional properties. Such improvementswill require method developments, which would immenselybenefit from close collaborations between computational andexperimental scientists.
Acknowledgments
JM is group leader Redox Biology at the Vlaams Instituutvoor Biotecnologie (VIB), and thanks NF and GR for thisfruitful collaboration. GR thanks the Fund for Scientific Re-search Flanders (FWO) for a postdoctoral fellowship.We would like to thank all the 10 reviewers for their valuablecomments, which have enormously improved this review.
References
1. Adman E, Watenpaugh KD, and Jensen LH. NH–S hy-drogen bonds in Peptococcus aerogenes ferredoxin, Clos-tridium pasteurianum rubredoxin, and Chromatium highpotential iron protein. Proc Natl Acad Sci U S A 72: 4854–4858, 1975.
2. Alexov E, Mehler EL, Baker N, A MB, Huang Y, Milletti F,Erik Nielsen J, Farrell D, Carstensen T, Olsson MH, ShenJK, Warwicker J, Williams S, and Word JM. Progress in theprediction of pK(a) values in proteins. Proteins 79: 3260–3275, 2011.
3. Alexov EG and Gunner MR. Incorporating protein con-formational flexibility into the calculation of pH-dependentprotein properties. Biophys J 74: 2075–2093, 1997.
4. Antosiewicz J, McCammon JA, and Gilson MK. Predictionof pH-dependent properties of proteins. J Mol Biol 238: 415–436, 1994.
5. Antosiewicz J, McCammon JA, and Gilson MK. The deter-minants of pKas in proteins. Biochemistry 35: 7819–7833, 1996.
6. Aqvist J, Luecke H, Quiocho FA, and Warshel A. Dipoleslocalized at helix termini of proteins stabilize charges. ProcNatl Acad Sci U S A 88: 2026, 1991.
7. Armstrong RN. Structure, catalytic mechanism, and evo-lution of the glutathione transferases. Chem Res Toxicol 10:2–18, 1997.
8. Aslund F, Berndt KD, and Holmgren A. Redox potentialsof glutaredoxins and other thiol-disulfide oxidoreduc-tases of the thioredoxin superfamily determined by directprotein-protein redox equilibria. J Biol Chem 272: 30780–30786, 1997.
9. Aslund F, Ehn B, Miranda-Vizuete A, Pueyo C, andHolmgren A. Two additional glutaredoxins exist in Escher-ichia coli: glutaredoxin 3 is a hydrogen donor for ribonucle-otide reductase in a thioredoxin/glutaredoxin 1 doublemutant. Proc Natl Acad Sci U S A 91: 9813–9817, 1994.
10. Bacik JP and Hazes B. Crystal structures of a poxviralglutaredoxin in the oxidized and reduced states show re-dox-correlated structural changes. J Mol Biol 365: 1545–1558, 2007.
11. Barkay T, Miller SM, and Summers AO. Bacterial mercuryresistance from atoms to ecosystems. FEMS Microbiol Rev27: 355–384, 2003.
12. Bas DC, Rogers DM, and Jensen JH. Very fast predictionand rationalization of pKa values for protein-ligand com-plexes. Proteins 73: 765–783, 2008.
FIG. 12. The redox potential of Trx-fold enzymes corre-lates with the pKa of the active-site cysteines. The redoxpotentials of Trx-fold oxidoreductases from E. coli wereplotted versus their respective experimental determined pKavalue. The redox potential increases with *54 mV for eachdecreasing pKa unit (r2 = 0.9). The pKa values are from Table1. For the redox potentials of the different oxidoreductasesfrom E. coli, we refer for DsbA (56), DsbC (193), DsbD (25),DsbG (15, 172), Trx2 (40), Trx (21, 92), and Grx1 and Grx3 (8)to the respective articles.
PKA OF REDOX CYSTEINES 121

13. Bashford D and Karplus M. pKa’s of Ionizable groups inproteins: Atomic detail from a continuum electrostaticmodel. Biochemistry 29: 10219–10225, 1990.
14. Benesch RE and Benesch R. The acid strength of the -SHGroup in cysteine and related compounds. J Am Chem Soc77: 5877–5881, 1955.
15. Bessette PH, Cotto JJ, Gilbert HF, and Georgiou G. In vivoand in vitro function of the Escherichia coli periplasmic cys-teine oxidoreductase DsbG. J Biol Chem 274: 7784–7792, 1999.
16. Billiet L, Geerlings P, Messens J, and Roos G. The ther-modynamics of thiol sulfenylation. Free Radic Biol Med 52:1473–1485, 2012.
17. Bulaj G, Kortemme T, and Goldenberg DP. Ionization-reactivity relationships for cysteine thiols in polypeptides.Biochemistry 37: 8965–8972, 1998.
18. Burley SK and Petsko GA. Aromatic-aromatic interaction: amechanism of protein structure stabilization. Science 229:23–28, 1985.
19. Carvalho AP, Fernandes PA, and Ramos MJ. Similaritiesand differences in the thioredoxin superfamily. Prog Bio-phys Mol Biol 91: 229–248, 2006.
20. Chen WJ, Graminski GF, and Armstrong RN. Dissection ofthe catalytic mechanism of isozyme 4–4 of glutathione S-transferase with alternative substrates. Biochemistry 27: 647–654, 1988.
21. Cheng Z, Arscott LD, Ballou DP, and Williams CH, Jr. Therelationship of the redox potentials of thioredoxin andthioredoxin reductase from Drosophila melanogaster to theenzymatic mechanism: reduced thioredoxin is the reduc-tant of glutathione in Drosophila. Biochemistry 46: 7875–7885, 2007.
22. Cheng Z, Zhang J, Ballou DP, and Williams CH, Jr. Re-activity of thioredoxin as a protein thiol-disulfide oxidore-ductase. Chem Rev 111: 5768–5783, 2011.
23. Click TH and Kaminski GA. Reproducing basic pKa valuesfor turkey ovomucoid third domain using a polarizableforce field. J Phys Chem B 113: 7844–7850, 2009.
24. Collet JF and Messens J. Structure, function, and mecha-nism of thioredoxin proteins. Antioxid Redox Signal 13:1205–1216, 2010.
25. Collet JF, Riemer J, Bader MW, and Bardwell JC. Recon-stitution of a disulfide isomerization system. J Biol Chem277: 26886–26892, 2002.
26. CRC. Handbook of Chemistry and Physics. Florida: CRCPress, 1994.
27. Creighton TE. Proteins: Structures and Molecular Properties.New York: W.H. Freeman, 1993.
28. Crow A, Acheson RM, Le Brun NE, and Oubrie A. Struc-tural basis of Redox-coupled protein substrate selection bythe cytochrome c biosynthesis protein ResA. J Biol Chem279: 23654–23660, 2004.
29. D’Ambrosio K, Pedone E, Langella E, De Simone G, RossiM, Pedone C, and Bartolucci S. A novel member of theprotein disulfide oxidoreductase family from Aeropyrumpernix K1: structure, function and electrostatics. J Mol Biol362: 743–752, 2006.
30. Davies MN, Toseland CP, Moss DS, and Flower DR.Benchmarking pK(a) prediction. BMC biochemistry 7: 18,2006.
31. Demchuk E and Wade RC. Improving the continuum di-electric approach to calculating pKas of ionizable groups inproteins. J Phys Chem 100: 17373–17387, 1996.
32. Depuydt M, Messens J, and Collet JF. How proteins formdisulfide bonds. Antioxid Redox Signal 15: 49–66, 2011.
33. Desiraju GR and Steiner T. IUCr Monographs on Cry-stallography, Vol. 9. Oxford: Oxford University Press/International Union of Crystallography, 1999.
34. Dickhout JG, Carlisle RE, and Austin RC. Interrelationshipbetween cardiac hypertrophy, heart failure, and chronickidney disease: endoplasmic reticulum stress as a mediatorof pathogenesis. Circ Res 108: 629–642, 2011.
35. Dillet V, Dyson HJ, and Bashford D. Calculations of elec-trostatic interactions and pKas in the active site of Escher-ichia coli thioredoxin. Biochemistry 37: 10298–10306, 1998.
36. Discola KF, de Oliveira MA, Rosa Cussiol JR, Monteiro G,Barcena JA, Porras P, Padilla CA, Guimaraes BG, and NettoLE. Structural aspects of the distinct biochemical propertiesof glutaredoxin 1 and glutaredoxin 2 from Saccharomycescerevisiae. J Mol Biol 385: 889–901, 2009.
37. Donohue J. On N-H–S hydrogen bonds. J Mol Biol 45: 231–235, 1969.
38. Dyson HJ, Jeng MF, Tennant LL, Slaby I, Lindell M, Cui DS,Kuprin S, and Holmgren A. Effects of buried chargedgroups on cysteine thiol ionization and reactivity in Es-cherichia coli thioredoxin: structural and functional charac-terization of mutants of Asp 26 and Lys 57. Biochemistry 36:2622–2636, 1997.
39. Dyson HJ, Tennant LL, and Holmgren A. Proton-transfereffects in the active-site region of Escherichia coli thioredoxinusing two-dimensional 1H NMR. Biochemistry 30: 4262–4268, 1991.
40. El Hajjaji H, Dumoulin M, Matagne A, Colau D, Roos G,Messens J, and Collet JF. The zinc center influences theredox and thermodynamic properties of Escherichia colithioredoxin 2. J Mol Biol 386: 60–71, 2009.
41. Ferrer-Sueta G, Manta B, Botti H, Radi R, Trujillo M, andDenicola A. Factors affecting protein thiol reactivity andspecificity in peroxide reduction. Chem Res Toxicol 24: 434–450, 2011.
42. Fitch CA and Garcia-Moreno EB. Structure-based pKacalculations using continuum electrostatics methods. CurrProt Bioinform 8.11.18.11.22, 2006.
43. Foloppe N and Nilsson L. The glutaredoxin -C-P-Y-C- motif:influence of peripheral residues. Structure 12: 289–300, 2004.
44. Foloppe N and Nilsson L. Stabilization of the catalyticthiolate in a mammalian glutaredoxin: structure, dynamicsand electrostatics of reduced pig glutaredoxin and its mu-tants. J Mol Biol 372: 798–816, 2007.
45. Foloppe N, Sagemark J, Nordstrand K, Berndt KD, andNilsson L. Structure, dynamics and electrostatics of the activesite of glutaredoxin 3 from Escherichia coli: Comparison withfunctionally related proteins. J Mol Biol 310: 449–470, 2001.
46. Forman-Kay JD, Clore GM, and Gronenborn AM. Re-lationship between electrostatics and redox function inhuman thioredoxin: characterization of pH titration shiftsusing two-dimensional homo- and heteronuclear NMR.Biochemistry 31: 3442–3452, 1992.
47. Gan Z and Wells WW. Identification and reactivity of thecatalytic site of pig liver thioltransferase. J Biol Chem 262:6704–6797, 1987.
48. Gan Z-R, Sardana MK, Jacobs JW, and Polokoff MA. Yeastthioltransferase-the active site cysteines display differentialreactivity. Arch Biochem Biophys 282: 110–115, 1990.
49. Gane PJ, Freedman RB, and Warwicker J. A molecularmodel for the redox potential difference between thior-edoxin and DsbA, based on electrostatics calculations. J MolBiol 249: 376–387, 1995.
50. This reference has been deleted.
122 ROOS ET AL.

51. Georgescu RE, Alexov EG, and Gunner MR. Combiningconformational flexibility and continuum electrostatics forcalculating pK(a)s in proteins. Biophys J 83: 1731–1748, 2002.
52. Gilbert HF. Molecular and cellular aspects of thiol-disulfideexchange. Adv Enzymol Relat Areas Mol Biol 63: 69–172, 1990.
53. Gilson MK. Multiple-site titration and molecular model-ing: two rapid methods for computing energies andforces for ionizable groups in proteins. Proteins 15: 266–282,1993.
54. Gilson MK and Honig B. Calculation of the total elec-trostatic energy of a macromolecular system: solvationenergies, binding energies, and conformational analysis.Proteins 4: 7–18, 1988.
55. Gilson MK and Honig BH. The dielectric constant of afolded protein. Biopolymers 25: 2097–2119, 1986.
56. Grauschopf U, Winther JR, Korber P, Zander T, Dallinger P,and Bardwell JC. Why is DsbA such an oxidizing disulfidecatalyst? Cell 83: 947–955, 1995.
57. Gregoret LM, Rader SD, Fletterick RJ, and Cohen FE. Hy-drogen bonds involving sulfur atoms in proteins. Proteins 9:99–107, 1991.
58. Griffiths SW, King J, and Cooney CL. The reactivity andoxidation pathway of cysteine 232 in recombinant humanalpha 1-antitrypsin. J Biol Chem 277: 25486–25492, 2002.
59. Gu Y, Kar T, and Scheiner S. Fundamental Properties of theC-H$$$O Interaction: Is it a True Hydrogen Bond? J AmChem Soc 121: 9411–9422, 1999.
60. Gu Y, Singh SV, and Ji X. Residue R216 and catalytic effi-ciency of a murine class alpha glutathione S-transferasetoward benzo[a]pyrene 7(R),8(S)-diol 9(S), 10(R)-epoxide.Biochemistry 39: 12552–12557, 2000.
61. Guddat LW, Bardwell JC, Glockshuber R, Huber-Wunderlich M, Zander T, and Martin JL. Structuralanalysis of three His32 mutants of DsbA: support for anelectrostatic role of His32 in DsbA stability. Protein Sci 6:1893–1900, 1997.
62. Harris TK and Turner GJ. Structural basis of perturbed pKavalues of catalytic groups in enzyme active sites. IUBMBLife 53: 85–98, 2002.
63. Hatahet F and Ruddock LW. Protein disulfide isomerase: acritical evaluation of its function in disulfide bond forma-tion. Antioxid Redox Signal 11: 2807–2850, 2009.
64. Hawkins HC and Freedman RB. The reactivities and ioni-zation properties of the active-site dithiol groups of mam-malian protein disulphide-isomerase. Biochem J 275 (Pt 2):335–339, 1991.
65. Hayes JD, Flanagan JU, and Jowsey IR. Glutathione trans-ferases. Annu Rev Pharmacol Toxicol 45: 51–88, 2005.
66. Hennecke J, Spleiss C, and Glockshuber R. Influence ofacidic residues and the kink in the active-site helix on theproperties of the disulfide oxidoreductase DsbA. J BiolChem 272: 189–195, 1997.
67. Hol WG, van Duijnen PT, and Berendsen HJ. The alpha-helix dipole and the properties of proteins. Nature 273: 443–446, 1978.
68. Honig B and Nicholls A. Classical electrostatics in biologyand chemistry. Science 268: 1144–1149, 1995.
69. Huber-Wunderlich M and Glockshuber R. A single dipep-tide sequence modulates the redox properties of a wholeenzyme family. Fold Des 3: 161–171, 1998.
70. Hugo M, Turell L, Manta B, Botti H, Monteiro G, Netto LE,Alvarez B, Radi R, and Trujillo M. Thiol and sulfenicacid oxidation of AhpE, the one-cysteine peroxiredoxin
from Mycobacterium tuberculosis: kinetics, acidity con-stants, and conformational dynamics. Biochemistry 48:9416–9426, 2009.
71. Hunter CA and Sanders JKM. The Nature of p - p Interac-tions. J Am Chem Soc 112: 5525–5534, 1990.
72. Ingelman M, Nordlund P, and Eklund H. The structure of areduced mutant T4 glutaredoxin. FEBS Lett 370: 209–211,1995.
73. Iqbalsyah TM, Moutevelis E, Warwicker J, Errington N,and Doig AJ. The CXXC motif at the N terminus of an a-helical peptide. Protein Sci 15: 1945–1950, 2006.
74. Jacobi A, Huber-Wunderlich M, Hennecke J, andGlockshuber R. Elimination of all charged residues in thevicinity of the active-site helix of the disulfide oxidoreduc-tase DsbA. Influence of electrostatic interactions on stabilityand redox properties. J Biol Chem 272: 21692–21699, 1997.
75. Jao SC, English Ospina SM, Berdis AJ, Starke DW, Post CB,and Mieyal JJ. Computational and mutational analysis ofhuman glutaredoxin (thioltransferase): probing the molec-ular basis of the low pKa of cysteine 22 and its role incatalysis. Biochemistry 45: 4785–4796, 2006.
76. Jeng MF, Campbell AP, Begley T, Holmgren A, Case DA,Wright PE, and Dyson HJ. High-resolution solution struc-tures of oxidized and reduced Escherichia coli thioredoxin.Structure 2: 853–868, 1994.
77. Jeng MF, Holmgren A, and Dyson HJ. Proton sharing be-tween cysteine thiols in Escherichia coli thioredoxin: impli-cations for the mechanism of protein disulfide reduction.Biochemistry 34: 10101–10105, 1995.
78. Jensen KS, Hansen RE, and Winther JR. Kinetic and ther-modynamic aspects of cellular thiol-disulfide redox regu-lation. Antioxid Redox Signal 11: 1047–1058, 2009.
79. Ji X, von Rosenvinge EC, Johnson WW, Tomarev SI, Pia-tigorsky J, Armstrong RN, and Gilliland GL. Three-dimensional structure, catalytic properties, and evolutionof a sigma class glutathione transferase from squid, a pro-genitor of the lens S-crystallins of cephalopods. Biochemistry34: 5317–5328, 1995.
80. Jones DP and Go YM. Mapping the cysteine proteome:analysis of redox-sensing thiols. Curr Opin Chem Biol 15:103–112, 2011.
81. Joshi HV and Meier MS. The effect of a peptide helixmacrodipole on the pKa of an Asp side chain carboxylate. JAm Chem Soc 118: 12038–12044, 1996.
82. Kallis GB and Holmgren A. Differential reactivity of thefunctional sulfhydryl groups of cysteine-32 and cysteine-35present in the reduced form of thioredoxin from Escherichiacoli. J Biol Chem 255: 10261–10265, 1980.
83. Karala AR, Lappi AK, and Ruddock LW. Modulation of anactive-site cysteine pKa allows PDI to act as a catalyst ofboth disulfide bond formation and isomerization. J Mol Biol396: 883–892, 2010.
84. Kerr KA, Ashmore JP, and Koetzle TF. A neutron diffrac-tion study of L-cysteine. Acta Cryst B31: 2022–2026, 1975.
85. Khandogin J and Brooks CL, 3rd. Constant pH molecular dy-namics with proton tautomerism. Biophys J 89: 141–157, 2005.
86. Khare D, Alexander P, Antosiewicz J, Bryan P, Gilson M,and Orban J. pKa measurements from nuclear magneticresonance for the B1 and B2 immunoglobulin G-bindingdomains of protein G: comparison with calculated valuesfor nuclear magnetic resonance and X-ray structures. Bio-chemistry 36: 3580–3589, 1997.
87. Kolm RH, Sroga GE, and Mannervik B. Participation of thephenolic hydroxyl group of Tyr-8 in the catalytic mecha-
PKA OF REDOX CYSTEINES 123

nism of human glutathione transferase P1-1. Biochem J 285(Pt 2): 537–540, 1992.
88. Kongsted J, Ryde U, Wydra J, and Jensen JH. Predictionand rationalization of the pH dependence of the activityand stability of family 11 xylanases. Biochemistry 46: 13581–13592, 2007.
89. Kortemme T and Creighton TE. Ionisation of cysteine res-idues at the termini of model alpha-helical peptides. Re-levance to unusual thiol pKa values in proteins of thethioredoxin family. J Mol Biol 253: 799–812, 1995.
90. Koumanov A, Karshikoff A, Friis EP, and Borchert TV.Conformational averaging in pK calculations: improve-ment and limitations in prediction of ionization propertiesof proteins. J Phys Chem B 105: 9339–9344, 2001.
91. Koumanov A, Ruterjans H, and Karshikoff A. Continuumelectrostatic analysis of irregular ionization and proton al-location in proteins. Proteins 46: 85–96, 2002.
92. Krause G and Holmgren A. Substitution of the conservedtryptophan 31 in Escherichia coli thioredoxin by site-directedmutagenesis and structure-function analysis. J Biol Chem266: 4056–4066, 1991.
93. Krautwurst H, Berti M, Encinas MV, and Frey PA. Reactionof wild-type C365S, and C458S saccharomyces cerevisiaephosphoenolpyruvate carboxykinases with fluorescent io-doacetamide derivatives. Arch Biochem Biophys 327: 123–130, 1996.
94. Kroncke KD and Klotz LO. Zinc fingers as biologic redoxswitches? Antioxid Redox Signal 11: 1015–1027, 2009.
95. Lah N, Lah J, Zegers I, Wyns L, and Messens J. Specificpotassium binding stabilizes pI258 arsenate reductase fromStaphylococcus aureus. J Biol Chem 278: 24673–24679, 2003.
96. Lappi AK, Lensink MF, Alanen HI, Salo KE, Lobell M,Juffer AH, and Ruddock LW. A conserved arginine plays arole in the catalytic cycle of the protein disulphide isom-erases. J Mol Biol 335: 283–295, 2004.
97. Ledwidge R, Hong B, Dotsch V, and Miller SM. NmerA ofTn501 mercuric ion reductase: structural modulation of thepKa values of the metal binding cysteine thiols. Biochem-istry 49: 8988–8998, 2010.
98. Lee AC and Crippen GM. Predicting pKa. J Chem Inf Model49: 2013–2033, 2009.
99. Lewin A, Crow A, Hodson CT, Hederstedt L, and Le BrunNE. Effects of substitutions in the CXXC active-site motif ofthe extracytoplasmic thioredoxin ResA. Biochem J 414: 81–91, 2008.
100. Lewin A, Crow A, Oubrie A, and Le Brun NE. Molecularbasis for specificity of the extracytoplasmic thioredoxinResA. J Biol Chem 281: 35467–35477, 2006.
101. Li H, Hanson C, Fuchs JA, Woodward C, and Thomas GJ.Determination of the pKa values of active-center cysteines,cysteines-32 and - 35, in Escherichia coli thioredoxin byRaman spectroscopy. Biochemistry 32: 5800–5808, 1993.
102. Li H, Robertson AD, and Jensen JH. The determinants ofcarboxyl pKa values in turkey ovomucoid third domain.Proteins 55: 689–704, 2004.
103. Li H, Robertson AD, and Jensen JH. Very fast empiricalprediction and rationalization of protein pKa values. Pro-teins 61: 704–721, 2005.
104. Li Y, Hu Y, Zhang X, Xu H, Lescop E, Xia B, and Jin C.Conformational fluctuations coupled to the thiol-disulfidetransfer between thioredoxin and arsenate reductase inBacillus subtilis. J Biol Chem 282: 11078–11083, 2007.
105. Liu S, Zhang P, Ji X, Johnson WW, Gilliland GL, andArmstrong RN. Contribution of tyrosine 6 to the catalytic
mechanism of isoenzyme 3–3 of glutathione S-transferase. JBiol Chem 267: 4296–4299, 1992.
106. Loumaye E, Ferrer-Sueta G, Alvarez B, Rees JF, Clippe A,Knoops B, Radi R, and Trujillo M. Kinetic studies of per-oxiredoxin 6 from Arenicola marina: rapid oxidation byhydrogen peroxide and peroxynitrite but lack of reductionby hydrogen sulfide. Arch Biochem Biophys 514: 1–7, 2011.
107. Macedo MG, Anar B, Bronner IF, Cannella M, Squitieri F,Bonifati V, Hoogeveen A, Heutink P, and Rizzu P. The DJ-1L166P mutant protein associated with early onset Par-kinson’s disease is unstable and forms higher-order proteincomplexes. Hum Mol Genet 12: 2807–2816, 2003.
108. Marchal S and Branlant G. Evidence for the chemicalactivation of essential cys-302 upon cofactor binding tononphosphorylating glyceraldehyde 3-phosphate dehy-drogenase from Streptococcus mutans. Biochemistry 38:12950–12958, 1999.
109. Marino SM and Gladyshev VN. Cysteine function governsits conservation and degeneration and restricts its utiliza-tion on protein surfaces. J Mol Biol 404: 902–916, 2010.
110. Marino SM and Gladyshev VN. Analysis and functionalprediction of reactive cysteine residues. J Biol Chem 287:4419–4425, 2012.
111. Mason AC and Jensen JH. Protein-protein binding is oftenassociated with changes in protonation state. Proteins 71:81–91, 2008.
112. Mavridou DA, Stevens JM, Ferguson SJ, and Redfield C.Active-site properties of the oxidized and reduced C-terminaldomain of DsbD obtained by NMR spectroscopy. J Mol Biol370: 643–658, 2007.
113. Mavridou DA, Stevens JM, Goddard AD, Willis AC, Fer-guson SJ, and Redfield C. Control of periplasmic inter-domain thiol:disulfide exchange in the transmembraneoxidoreductase DsbD. J Biol Chem 284: 3219–3226, 2009.
114. Messens J, Martins JC, Van Belle K, Brosens E, Desmyter A,De Gieter M, Wieruszeski JM, Willem R, Wyns L, and Ze-gers I. All intermediates of the arsenate reductase mecha-nism, including an intramolecular dynamic disulfidecascade. Proc Natl Acad Sci U S A 99: 8506–8511, 2002.
115. Messens J, Van Molle I, Vanhaesebrouck P, Limbourg M,Van Belle K, Wahni K, Martins JC, Loris R, and Wyns L.How thioredoxin can reduce a buried disulphide bond. JMol Biol 339: 527–537, 2004.
116. Mieyal JJ, Starke DW, Gravina SA, and Hocevar BA.Thioltransferase in human red blood cells: kinetics andequilibrium. Biochemistry 30: 8883–8891, 1991.
117. Mossner E, Huber-Wunderlich M, and Glockshuber R.Characterization of Escherichia coli thioredoxin variantsmimicking the active-sites of other thiol/disulfide oxido-reductases. Protein Sci 7: 1233–1244, 1998.
118. Mossner E, Iwai H, and Glockshuber R. Influence of thepK(a) value of the buried, active-site cysteine on the redoxproperties of thioredoxin-like oxidoreductases. FEBS Lett477: 21–26, 2000.
119. Moutevelis E and Warwicker J. Prediction of pKa and re-dox properties in the thioredoxin superfamily. Protein Sci13: 2744–2752, 2004.
120. Naor MM and Jensen JH. Determinants of cysteine pKavalues in creatine kinase and alpha1-antitrypsin. Proteins57: 799–803, 2004.
121. Nelson JW and Creighton TE. Reactivity and ionization ofthe active site cysteine residues of DsbA, a protein requiredfor disulfide bond formation in vivo. Biochemistry 33: 5974–5983, 1994.
124 ROOS ET AL.

122. Nelson KJ, Day AE, Zeng BB, King SB, and Poole LB.Isotope-coded, iodoacetamide-based reagent to determineindividual cysteine pK(a) values by matrix-assisted laserdesorption/ionization time-of-flight mass spectrometry.Anal Biochem 375: 187–195, 2008.
123. Nielsen JE, Andersen KV, Honig B, Hooft RW, Klebe G,Vriend G, and Wade RC. Improving macromolecularelectrostatics calculations. Protein Eng 12: 657–662, 1999.
124. Nielsen JE, Borchert TV, and Vriend G. The determinantsof alpha-amylase pH-activity profiles. Protein Eng 14: 505–512, 2001.
125. Nielsen JE and McCammon JA. Calculating pKa values inenzyme active sites. Protein Sci 12: 1894–1901, 2003.
126. Nielsen JE and Vriend G. Optimizing the hydrogen-bondnetwork in Poisson-Boltzmann equation-based pKa calcu-lations. Proteins 43: 403–412, 2001.
127. Nielsen JW, Jensen KS, Hansen RE, Gotfredsen CH, andWinther JR. A fluorescent probe which allows highly specificthiol labeling at low pH. Anal Biochem 421: 115–120, 2012.
128. Nilsson L and Karshikoff A. Multiple pH regime moleculardynamics simulation for pK calculations. PLoS One 6:e20116, 2011.
129. Nordstrand K, Aslund F, Meunier S, Holmgren A, OttingG, and Berndt KD. Direct NMR observation of the Cys-14thiol proton of reduced Escherichia coli Glutaredoxin-3supports the presence of an active site thiol-thiolate hy-drogen bond. FEBS letters 449: 196–200, 1999.
130. Olsson HMM, Søndergaard CR, Rostkowski M, and JensenJH. PROPKA3: Consistent treatment of internal and surfaceresidues in empirical pKa predictions. J Chem Theory Com-put 7: 525–537, 2011.
131. Polgar L. Mercaptide-imidazolium ion-pair: the reactivenucleophile in papain catalysis. FEBS Lett 47: 15–18, 1974.
132. Porat A, Lillig CH, Johansson C, Fernandes AP, Nilsson L,Holmgren A, and Beckwith J. The reducing activity of glu-taredoxin 3 toward cytoplasmic substrate proteins is re-stricted by methionine 43. Biochemistry 46: 3366–3377, 2007.
133. Porter MA, Hall JR, Locke JC, Jensen JH, and Molina PA.Hydrogen bonding is the prime determinant of carboxylpKa values at the N-termini of alpha-helices. Proteins 63:621–635, 2006.
134. Powers N and Jensen JH. Chemically accurate proteinstructures: Validation of protein NMR structures by com-parison of measured and predicted pKa values. J BiomolNMR 35: 39–51, 2006.
135. Presnell SR and Cohen FE. Topological distribution offour-alpha-helix bundles. Proc Natl Acad Sci U S A 86: 6592–6596, 1989.
136. Qin J, Clore GM, and Gronenborn AM. The high-resolutionthree-dimensional solution structures of the oxidized and re-duced states of human thioredoxin. Structure 2: 503–522, 1994.
137. Rablen PR, Lockman JW, and Jorgensen WL. Ab initiostudy of hydrogen-bonded complexes of small organicmolecules with water. J Phys Chem A 102: 3782–3797, 1998.
138. Reckenfelderbaumer N and Krauth-Siegel RL. Catalyticproperties, thiol pK value, and redox potential of Trypano-soma brucei tryparedoxin. J Biol Chem 277: 17548–17555, 2002.
139. Ren G, Stephan D, Xu Z, Zheng Y, Tang D, Harrison RS,Kurz M, Jarrott R, Shouldice SR, Hiniker A, Martin JL,Heras B, and Bardwell JC. Properties of the thioredoxinfold superfamily are modulated by a single amino acidresidue. J Biol Chem 284: 10150–10159, 2009.
140. Roos G, Buts L, Van Belle K, Brosens E, Geerlings P, LorisR, Wyns L, and Messens J. Interplay between ion binding
and catalysis in the thioredoxin-coupled arsenate reductasefamily. J Mol Biol 360: 826–838, 2006.
141. Roos G, Foloppe N, Van Laer K, Wyns L, Nilsson L,Geerlings P, and Messens J. How thioredoxin dissociates Itsmixed disulfide. PloS Comp Biol 5: e1000461, 2009.
142. Roos G, Garcia-Pino A, Van Belle K, Brosens E, Wahni K,Vandenbussche G, Wyns L, Loris R, and Messens J. The con-served active site proline determines the reducing power ofStaphylococcus aureus thioredoxin. J Mol Biol 368: 800–811, 2007.
143. Roos G, Loverix S, and Geerlings P. Origin of the pK(a)perturbation of N-terminal cysteine in alpha- and 3(10)-helices: a computational DFT study. J Phys Chem B 110:557–562, 2006.
144. Roos G and Messens J. Protein sulfenic acid formation:from cellular damage to redox regulation. Free Radic BiolMed 51: 314–326, 2011.
145. Roos G, Messens J, Loverix S, Wyns L, and Geerlings P. Acomputational and conceptual DFT study on the Michaeliscomplex of pI258 arsenate reductase. Structural aspects andactivation of the electrophile and nucleophile. J Phys ChemB 108: 17216–17225, 2004.
146. Ruddock LW, Hirst TR, and Freedman RB. pH-dependenceof the dithiol-oxidizing activity of DsbA (a periplasmicprotein thiol:disulphide oxidoreductase) and protein dis-ulphide-isomerase: studies with a novel simple peptidesubstrate. Biochem J 315 (Pt 3): 1001–1005, 1996.
147. Rullmann JA, Bellido MN, and van Duijnen PT. The activesite of papain. All-atom study of interactions with proteinmatrix and solvent. J Mol Biol 206: 101–118, 1989.
148. Rushmore TH and Pickett CB. Glutathione S-transferases,structure, regulation, and therapeutic implications. J BiolChem 268: 11475–11478, 1993.
149. Sanchez R, Riddle M, Woo J, and Momand J. Prediction ofreversibly oxidized protein cysteine thiols using proteinstructure properties. Protein Sci 17: 473–481, 2008.
150. Sancho J, Serrano L, and Fersht AR. Histidine residues atthe N- and C-termini of alpha-helices: perturbed pKas andprotein stability. Biochemistry 31: 2253–2258, 1992.
151. Schlesinger P and Westley J. An expanded mechanism forrhodanese catalysis. J Biol Chem 249: 780–788, 1974.
152. Sengupta D, Behera RN, Smith JC, and Ullmann GM. Thealpha helix dipole: screened out? Structure 13: 849–855, 2005.
153. Sham YY, Chu ZT, and Warshel A. Consistent calculationsof pKa’s of ionizable residues in proteins: Semi-microscopicand microscopic approaches. J Phys Chem B 101: 4458–4472, 1997.
154. Sharp K. Electrostatic interactions in macromolecules. CurrOpin Struct Biol 4: 234–239, 1994.
155. Shekhter T, Metanis N, Dawson PE, and Keinan E. Aresidue outside the active site CXXC motif regulates thecatalytic efficiency of Glutaredoxin 3. Mol BioSyst 6: 241–248, 2010.
156. Sheridan RP, Levy RM, and Salemme FR. alpha-Helix di-pole model and electrostatic stabilization of 4-alpha-helicalproteins. Proc Natl Acad Sci U S A 79: 4545–4549, 1982.
157. Simonson T, Carlsson J, and Case DA. Proton binding toproteins: pK(a) calculations with explicit and implicit sol-vent models. J Am Chem Soc 126: 4167–4180, 2004.
158. Simonson T and Perahia D. Dielectric properties of proteinsfrom simulations: tools and techniques. Comput Phys Com-mun 91: 291–303, 1995.
159. Simonson T and Perahia D. Internal and interfacial dielec-tric properties of cytochrome c from molecular dynamics inaqueous solution. Proc Natl Sci U S A 92: 1082–1086, 1995.
PKA OF REDOX CYSTEINES 125

160. Søndergaard CR, Olsson HMM, Rostkowski M, and JensenJH. Improved treatment of ligands and coupling effects inempirical calculation and rationalization of pKa values. JChem Theory Comput 7: 2284–2295, 2011.
161. Stirnimann CU, Rozhkova A, Grauschopf U, BockmannRA, Glockshuber R, Capitani G, and Grutter MG. High-resolution structures of Escherichia coli cDsbD in differentredox states: a combined crystallographic, biochemical andcomputational study. J Mol Biol 358: 829–845, 2006.
162. Subramanian S and Ross PD. The enthalpy of protolysis ofliver alcohol dehydrogenase upon binding nicotinamideadenine dinucleotide. J Biol Chem 254: 7826–7830, 1979.
163. Sun XX and Wang CC. The N-terminal sequence (residues1–65) is essential for dimerization, activities, and peptidebinding of Escherichia coli DsbC. J Biol Chem 275: 22743–22749, 2000.
164. Szajewski RP and Whitesides GM. Rate constants andequilibrium constants for thiol-disulfide interchange reac-tions involving oxidized glutathione. J Am Chem Soc 102:2011–2025, 1980.
165. Tajc SG, Tolbert BS, Basavappa R, and Miller BL. Directdetermination of thiol pKa by isothermal titration micro-calorimetry. J Am Chem Soc 126: 10508–10509, 2004.
166. Takahashi N and Creighton TE. On the reactivity andionization of the active site cysteine residues of Escherichiacoli thioredoxin. Biochemistry 35: 8342–8353, 1996.
167. Taylor R and Kennard O. Crystallographic evidence for theexistence of C-H.O, C-H.N, and C-H.Cl hydrogenbonds. J Am Chem Soc 104: 5063–5070, 1982.
168. Thomas KA, Smith GM, Thomas TB, and Feldmann RJ.Electronic distributions within protein phenylalanine aromaticrings are reflected by the three-dimensional oxygen atom en-vironments. Proc Natl Acad Sci U S A 79: 4843–4847, 1982.
169. Thurlkill RL, Grimsley GR, Scholtz JM, and Pace CN. pKvalues of the ionizable groups of proteins. Protein Sci 15:1214–1218, 2006.
170. Ullmann GM, Noodleman L, and Case DA. Density func-tional calculation of p K(a) values and redox potentials inthe bovine Rieske iron-sulfur protein. J Biol Inorg Chem 7:632–639, 2002.
171. van Duijnen PT, Thole BT, and Hol WG. On the role of theactive site helix in papain, an ab initio molecular orbitalstudy. Biophys Chem 9: 273–280, 1979.
172. van Straaten M, Missiakas D, Raina S, and Darby NJ. Thefunctional properties of DsbG, a thiol-disulfide oxidore-ductase from the periplasm of Escherichia coli. FEBS Lett428: 255–258, 1998.
173. van Vlijmen HW, Schaefer M, and Karplus M. Improvingthe accuracy of protein pKa calculations: conformationalaveraging versus the average structure. Proteins 33: 145–158, 1998.
174. Vargas R, Garza D, Dixon A, and Hay BP. How strong isthe Ca-H.O = C hydrogen bond? J Am Chem Soc 122: 4750–4755, 2000.
175. Villadangos AF, Van Belle K, Wahni K, Tamu Dufe V,Freitas S, Nur H, De Galan S, Gil JA, Collet JF, Mateos LM,and Messens J. Corynebacterium glutamicum survives arsenicstress with arsenate reductases coupled to two distinct re-dox mechanisms. Mol Microbiol 82: 998–1014, 2011.
176. Wada A. The alpha-helix as an electric macro-dipole. AdvBiophys 1–63, 1976.
177. Wang PF, McLeish MJ, Kneen MM, Lee G, and Kenyon GL.An unusually low pK(a) for Cys282 in the active site of humanmuscle creatine kinase. Biochemistry 40: 11698–11705, 2001.
178. Wang RW, Newton DJ, Huskey SE, McKeever BM, PickettCB, and Lu AY. Site-directed mutagenesis of glutathione S-transferase YaYa. Important roles of tyrosine 9 and asparticacid 101 in catalysis. J Biol Chem 267: 19866–19871, 1992.
179. Warwicker J and Gane PJ. Calculation of Cys 30 DpKa’sand oxidising power for DsbA mutants. FEBS Lett 385: 105–108, 1996.
180. Warwicker J and Watson HC. Calculation of the electricpotential in the active site cleft due to alpha-helix dipoles. JMol Biol 157: 671–679, 1982.
181. Waters ML. Aromatic interactions in model systems. CurrOpin Chem Biol 6: 736–741, 2002.
182. Wennmohs F, Staemmler V, and Schindler M. Theoreticalinvestigation of weak hydrogen bonds to sulfur. J ChemPhys 119: 3208–3218, 2003.
183. Winterbourn CC and Metodiewa D. Reactivity of bio-logically important thiol compounds with superoxideand hydrogen peroxide. Free Radic Biol Med 27: 322–328,1999.
184. Witt AC, Lakshminarasimhan M, Remington BC, Hasim S,Pozharski E, and Wilson MA. Cysteine pKa depression bya protonated glutamic acid in human DJ-1. Biochemistry 47:7430–7440, 2008.
185. Wlodek ST, Antosiewicz J, and McCammon JA. Predictionof titration properties of structures of a protein derivedfrom molecular dynamics trajectories. Protein Sci 6: 373–382, 1997.
186. Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS,and Rhee SG. Reduction of cysteine sulfinic acid by sulfir-edoxin is specific to 2-cys peroxiredoxins. J Biol Chem 280:3125–3128, 2005.
187. Xiao G, Liu S, Ji X, Johnson WW, Chen J, Parsons JF, Ste-vens WJ, Gilliland GL, and Armstrong RN. First-sphereand second-sphere electrostatic effects in the active site of aclass mu gluthathione transferase. Biochemistry 35: 4753–4765, 1996.
188. Yang A-S, Gunner MR, Sampogna R, Sharp K, and HonigB. On the Calculation of pKas in Proteins. Proteins 15: 252–265, 1993.
189. Yang AS and Honig B. On the pH dependence of proteinstability. J Mol Biol 231: 459–474, 1993.
190. Yang YF and Wells WW. Identification and characteriza-tion of the functional amino acids at the active center of pigliver thioltransferase by site-directed mutagenesis. J BiolChem 266: 12759–12765, 1991.
191. Ye J, Cho SH, Fuselier J, Li W, Beckwith J, and RapoportTA. Crystal structure of an unusual thioredoxin proteinwith a zinc finger domain. J Biol Chem 282: 34945–34951,2007.
192. You TJ and Bashford D. Conformation and hydrogen iontitration of proteins: a continuum electrostatic model withconformational flexibility. Biophys J 69: 1721–1733, 1995.
193. Zapun A, Missiakas D, Raina S, and Creighton TE. Struc-tural and functional characterization of DsbC, a proteininvolved in disulfide bond formation in Escherichia coli.Biochemistry 34: 5075–5089, 1995.
194. Zegers I, Martins JC, Willem R, Wyns L, and Messens J.Arsenate reductase from S. aureus plasmid pI258 is aphosphatase drafted for redox duty. Nat Struct Biol 8: 843–847, 2001.
195. Zheng F, Zhan C-G, and Ornstein RL. Theoretical deter-mination of two structural forms of the active site in cad-mium-containing phosphotriesterases. J Phys Chem B 106:717–722, 2002.
126 ROOS ET AL.

196. Zhou P, Tian F, Lv F, and Shang Z. Geometric character-istics of hydrogen bonds involving sulfur atoms in proteins.Proteins 76: 151–163, 2009.
Address correspondence to:Dr. Nicolas Foloppe
51 Natal RoadCambridge CB1 3NY
United Kingdom
E-mail: [email protected]
Dr. Joris MessensStructural Biology Brussels
Vrije Universiteit BrusselBuilding ERoom 4.16Pleinlaan 2
1050 BrusselBelgium
E-mail: [email protected]
Date of first submission to ARS Central, January 16, 2012; dateof final revised submission, June 3, 2012; date of acceptance,July 1, 2012.
Abbreviations Used
d¼ chemical shift2DPS¼ 2,2¢-dipyridyl disulfide
A¼ absorptionArsC¼ arsenate reductaseDsbA¼disulfide-binding protein A
DTNB¼ 5,5¢-dithiobis(2-nitrobenzoate)Grx¼ glutaredoxin
GSH¼ glutathioneIAM¼ iodoacetamideITC¼ isothermal titration microcalorimetry
LMW PTPase¼ low-molecular-weight protein tyrosinephosphatase
LMW thiols¼ low-molecular-weight thiolsMCCE¼multiconfigurational continuum
electrostatic methodMD¼molecular dynamics
NMR¼nuclear magnetic resonanceNPA¼natural population analysis
PB¼Poisson-BoltzmannPDB¼Protein Data BankPDI¼protein disulfide isomerase
pI¼ isoelectric pointQM¼ quantum mechanicalTrx¼ thioredoxin
PKA OF REDOX CYSTEINES 127




![Chapter-3, [Chemical bonding (CHM-117)]](https://cdn.vdocuments.mx/doc/165x107/632508f37fd2bfd0cb03402b/chapter-3-chemical-bonding-chm-117.jpg)


