
DOI: 10.1002/cphc.200700635
Photofragmentation of 2-Deoxy-d-Ribose Moleculesin the Gas PhaseGemma Vall-llosera,*[a] Michael A. Huels,[b] Marcello Coreno,[c] Antti Kivim�ki,[d]
Katarzyna Jakubowska,[a] Marek Stankiewicz,[e] and Elisabeth Rachlew[a]
Introduction
Understanding the effects of ionizing radiation on DNA and itscomponents is important from a radiobiological point of view.Ionizing radiation is used for therapeutic purposes; however, itcan also be the cause of cancer diseases. Impinging high-energy photons can produce alterations in the DNA structure,thus leading to base damage, single- or double-strand breaks,and the release or modification of nucleobases.[1] Any of theselesions, if unrepaired, may contribute to mutagenesis, carcino-genesis, ageing, and cell death.
The interaction of secondary particles with the biologicalmedium as a cause of DNA damage has been studied exten-sively by means of chemical and biochemical methods.[1] How-ever, lesions induced directly by ionizing/exciting DNA (directeffect) are thought to be significant (ca. 40–50%),[2] and thus, acomplete understanding of DNA damage induced by ioniza-tion radiation must also include a thorough investigation ofthe primary events. When monochromatic vacuum ultraviolet(VUV) photons interact with biological molecules, processessuch as excitation, ionization, autoionization, dissociative pho-toionization, fluorescence or ion-pair formation can be ob-served. Some of the processes lead to simple or complex frag-mentation of the molecules.
The monosaccharide 2-deoxy-d-ribose (dR) contains fiveC atoms and is classified as a pentose. Chemically, it can befound in three different conformations, namely, a linear form, afive-membered ring, and a six-memebered ring. In DNA, thesugar molecule is in its furanose form (five-membered ring)and it occupies a central position by linking the phosphate
sugar backbone to the DNA bases.[3] Thus, dR has no free OHgroups in the cellular DNA network. In the gas phase, Guleret al.[4] studied the reactivity of monosaccharides with twophosphenium ions. Their results indicated that free monosac-charides are cyclic molecules and that they maintain their crys-talline structure, the pyranose form (six-membered ring, seeFigure 1), throughout the evaporation procedure.
The type and abundance of ions in a mass spectrum dependgreatly on the type of excitation source. In this manner, ion,electron or photon impact will show different yields for differ-ent created fragments. Electromagnetic radiation below 10 keVcreates more complex and clustered molecular damage in cel-
We have measured the synchrotron-induced photofragmentationof isolated 2-deoxy-d-ribose molecules (C5H10O4) at four photonenergies, namely, 23.0, 15.7, 14.6, and 13.8 eV. At all photon ener-gies above the molecule’s ionization threshold we observe theformation of a large variety of molecular cation fragments, in-cluding CH3
+ , OH+ , H3O+ , C2H3
+ , C2H4+ , CHxO
+ACHTUNGTRENNUNG(x=1,2,3),
C2HxO+ACHTUNGTRENNUNG(x=1–5), C3HxO
+ACHTUNGTRENNUNG(x=3–5), C2H4O2
+ , C3HxO2+ (x=1,2,4–6),
C4H5O2+ , C4HxO3
+ACHTUNGTRENNUNG(x=6,7), C5H7O3
+ , and C5H8O3+ . The formation
of these fragments shows a strong propensity of the DNA sugarto dissociate upon absorption of vacuum ultraviolet photons. Theyields of particular fragments at various excitation photon ener-gies in the range between 10 and 28 eV are also measured andtheir appearance thresholds determined. At all photon energies,the most intense relative yield is recorded for the m/q=57 frag-
ment (C3H5O+), whereas a general intensity decrease is observed
for all other fragments— relative to the m/q=57 fragment—with decreasing excitation energy. Thus, bond cleavage dependson the photon energy deposited in the molecule. All fragmentsup to m/q=75 are observed at all photon energies above theirrespective threshold values. Most notably, several fragmentationproducts, for example, CH3
+ , H3O+ , C2H4
+ , CH3O+ , and C2H5O
+ ,involve significant bond rearrangements and nuclear motionduring the dissociation time. Multibond fragmentation of thesugar moiety in the sugar–phosphate backbone of DNA results incomplex strand lesions and, most likely, in subsequent reactionsof the neutral or charged fragments with the surrounding DNAmolecules.
[a] G. Vall-llosera, K. Jakubowska, Prof. Dr. E. RachlewDepartment of Physics, School of Engineering SciencesRoyal Institute of Technology, SE-10691 Stockholm (Sweden)Fax: (+46)855378216E-mail : [email protected]
[b] Prof. Dr. M. A. HuelsIon Reaction LaboratoryDepartment of Nuclear Medicine and RadiobiologyFaculty of Medicine and Health SciencesUniversity of Sherbrooke, Sherbrooke, Quebec, J1H5N4 (Canada)
[c] Dr. M. CorenoCNR-IMIP, Montelibretti, 00016 Rome (Italy)
[d] Dr. A. KivimDkiCNR-INFM, Laboratorio Nazionale TASC, 34012 Trieste (Italy)
[e] Prof. Dr. M. StankiewiczInstytut Fizyki im. Mariana SmoluchowskiegoUniwersytet Jagiellonski, ul. Reymonta 4, 30-059 KrakFw (Poland)
1020 www.chemphyschem.org ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemPhysChem 2008, 9, 1020 – 1029

lular DNA because it producesdenser low-energy electron tracksper absorbed dose relative to hardX-rays or g rays. These electronsbecome thermalized by loosing theirenergy during inelastic collisions inthe medium, and it has been shownthat they induce strand breaks inplasmids[5] as well as ionic and neu-tral fragment formation in oligonu-
cleotides by dissociative electron attachment and dipolar dis-sociation.[6] Studies of DNA damage upon core ionization atthe carbon, nitrogen, oxygen,[7] and phosphorous[8] 1s edgeshave focused on both direct decomposition of DNA due to thecore hole created and the effect of the fast Auger electronsejected in a secondary process. Raju et al.[9] studied subsequentcellular responses, such as cell killing caused by soft X-rays.Fujii et al.[10,11] used monochromatic soft X-rays (O K-edge) todesorb positive ions from thin films of DNA components, in-cluding 2-deoxy-d-ribose. Their unresolved mass spectra ofthymidine and thymidine 5’-monophosphate were qualitativelysimilar to that of 2-deoxy-d-ribose, thus indicating that a greatnumber of ions was generated at the sugar site and that thesugar moiety is more fragile than the thymine base. Also, athigher energies (2–20 keV), Alvarado et al.[12] used high-energyion beams to ionize gas-phase 2-deoxy-d-ribose molecules.From their measurements of fragmentation patterns for threedifferent ion beams, they concluded that the change in masspeak intensity with projectile implies that certain fragmentsoriginate from scission of rather strong bonds whilst fragmentsformed in more gentle collisions are not stable after highenergy depositions in the molecule. At lower energies, Hieda[13]
found that for photon energies ranging from 8–2000 eV,single-strand break damage in plasmid DNA is more probablethan double-strand break. In particular, for the lowest energies,ionizing-radiation damage below 15 eV has an upward tenden-cy (i.e. the higher the photon energy the higher the damageyield) whereas photons above 15 eV give an almost constantvalue of single- strand breaks. Ptasinska et al.[14] showed that inthe gas phase, a large number of ionic fragments from2-deoxy-d-ribose are formed when the incoming beam of elec-trons has a kinetic energy larger than the ionization energy ofthe molecular target. The same study demonstrated that thesugar is extremely fragile compared to the DNA bases, andthus, the sugar is believed to be a fragile moiety in DNA; forexample, abstraction of a hydrogen atom from deoxyriboseproduces a carbon-based sugar radical that can rearrange,thereby culminating in scission of the nucleic-acid strand.[15]
Deng et al. and Bald et al. studied DNA components—2-deoxy-d-ribose among them—in the condensed phase andupon low-energy He+ [16] and Ar+ [17] ion impact. They observedthe complete destruction of the molecules via fragmentationof the parent. The damage was mainly attributed to the kineticenergy and potential energies of the incident projectiles. Inthat study they also found that the formation of the H3O
+ ionis related to the presence of hydroxyl groups in the sugar mol-ecule and is associated with additional hydrogen loss from the
parent or adjacent molecules via hydrogen abstraction orproton transfer.
Herein, we present a mass-resolved fragmentation study of2-deoxy-d-ribose (m/q=134) in the gas phase after excitationwith VUV synchrotron photons. We show that absorption ofVUV photons may lead to destruction of the molecule. Wehave assigned each fragment, thereby discussing its probableformation. At all photon energies, the most intense relativeyield was recorded for the m/q=57 fragment (C3H5O
+), whilea general intensity decrease, relative to the m/q=57 fragment,was observed for all other fragments with decreasing excita-tion energy. Thus, bond cleavage was found to be dependenton the photon energy deposited in the molecule. All frag-ments up to m/q=75 were clearly observed at all photon en-ergies; however, the parent cation was below the detectionlimit of the quadrupole used here for the chosen energies. Theappearance energy thresholds were also measured for some ofthe fragments and compared to those obtained in previousworks.
Experimental Section
The experiments were performed at the beamline 52 of the MAX Istorage ring at the MAX-laboratory in Lund, Sweden. The source ofVUV radiation was a bending magnet which—together with a 1 mnormal incidence monochromator—enabled us to work in thephoton energy range of 5 to 30 eV.[18] The entrance and exit slits ofthe monochromator were set to 400/400 mm for the whole experi-ment, thereby giving a resolution of 0.13 eV at hn=22.5 eV.
The experimental setup was built around the vacuum chambercomposed of three crosses (see Figures 2 and 3). Two five-waycross chambers were arranged such that the first one was attachedto the end of the beamline and connected to the second one via aglass capillary (with an inner diameter of 2 mm) in order to guidethe beam to the interaction region, and also to act as a differentialpumping stage. The interaction region was located in the centre ofthe third chamber, a six-way cross. A quadrupole mass spectrome-ter (QMS, VG-300SPX) was placed on the top flange, and a repellerplate was kept under a constant positive voltage (of 40 V) at thebottom port, facing the QMS, to drive the ions from the interactionregion towards the QMS entrance. The QMS was used in two dif-ferent modes, namely, the scanning mode, where the yield of ionicfragments was recorded as a function of the ion mass/charge ratioat a fixed exciting photon energy, and the fixed-mass mode, wherethe QMS was held at a constant mass, the ion intensity being re-corded while varying the photon energy (yield of the fragment). Inthis cross-beam geometry, and perpendicular to the plane formed
Figure 1. Molecular struc-ture of 2-deoxy-d-ribose(in the pyranose form) inthe gas phase.
Figure 2. Schematic top view of the experimental setup: A) Indicates a turbomolecular pump. A mini-oven and a thermocouple feedthrough are placedat (B). C) Accommodates a pirani gauge. D) represents a cooling plate withan LN2 feedthrough. A Si diode measured the photocurrent after the interac-tion region.
ChemPhysChem 2008, 9, 1020 – 1029 ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemphyschem.org 1021
Photofragmentation of the DNA Sugar
by the synchrotron photon beam and the central axis of the QMS,there was a home-made mini-oven to evaporate the moleculesand an LN2-cooled plate to recondense the molecular vapor out-side the interaction region and maintain a low pressure in thevacuum system.
The sugar 2-deoxy-d-ribose (with a stated purity of 99%) was pur-chased from Sigma–Aldrich and heated (at ca. 30 8C) for about onehour to degas the compound. Due to the relatively high volatilityof the sample we had to reload the oven every 24 h. The workingtemperature—measured by a thermocouple placed very near theaperture of the oven—was carefully monitored to be 100oC (orbelow), which is well below the point where decompositionoccurs. In the study of Ptasinksa et al. ,[14] the presence of one spe-cific cation fragment, namely, C5H6O2
+ (equivalent to the parentcation minus two neutral water molecules), suggested the possiblethermal decomposition of dR at the highest temperatures (e.g.110 8C), mainly because this fragment appeared at an electronenergy below the ionization potential (IP) of the parent molecule.However, the authors attributed their results to a linear conformerof the parent molecule at higher temperatures, in part because theelectron energy of this fragment did not depend on the evapora-tion temperature. In any case, here a) we observe no cation frag-ments at photon energies below the parent molecule’s IP, andb) the fragment C5H6O2
+ (equivalent to the parent cation minustwo neutral water molecules) is not seen in our mass spectra,which therefore suggests no decomposition of dR, or presence ofa linear conformer. Moreover, the presence of both H2O
+ , and thecomplementary cation fragment consisting of (parent-H2O)+ rein-forces the notion that ionic water and its complementary cationfragment are formed by two alternate dissociation pathways of theionized parent molecule after photon absorption.
A thermocouple was used to read the temperature at the apertureof the oven, which was set to 100 8C for all experiments. Measure-ments were performed after stabilizing the ambient pressure inthe chamber to a value near 8O10�6 mbar upon heating of theoven. Since the molecular beam from the oven was recondensedon a cold plate right outside the interaction region, the pressure ofthe vapor at the interaction region is expected to be about tentimes higher than the background vapor pressure. The results pre-sented herein were corrected for variations in the incident photonflux measured by an Au mesh located prior to our experimentalchamber. Residual gas spectra were acquired and subtracted fromour measured mass spectra. Despite subtraction of the back-ground-gas spectrum from all measurements, the measured signalof OH and H2O may still include some small contributions from the
background gas. However, H3O+ has never been reported to be
formed by e� or photon impact in the gas phase from water, andhere, we assume that all H3O
+ yield is due to photon interactionwith dR. The absolute transmission through the QMS is notknown; thus, unless stated otherwise, any comparison betweenpeaks in the same spectrum is always relative to the most intensepeak in each spectrum.
Results
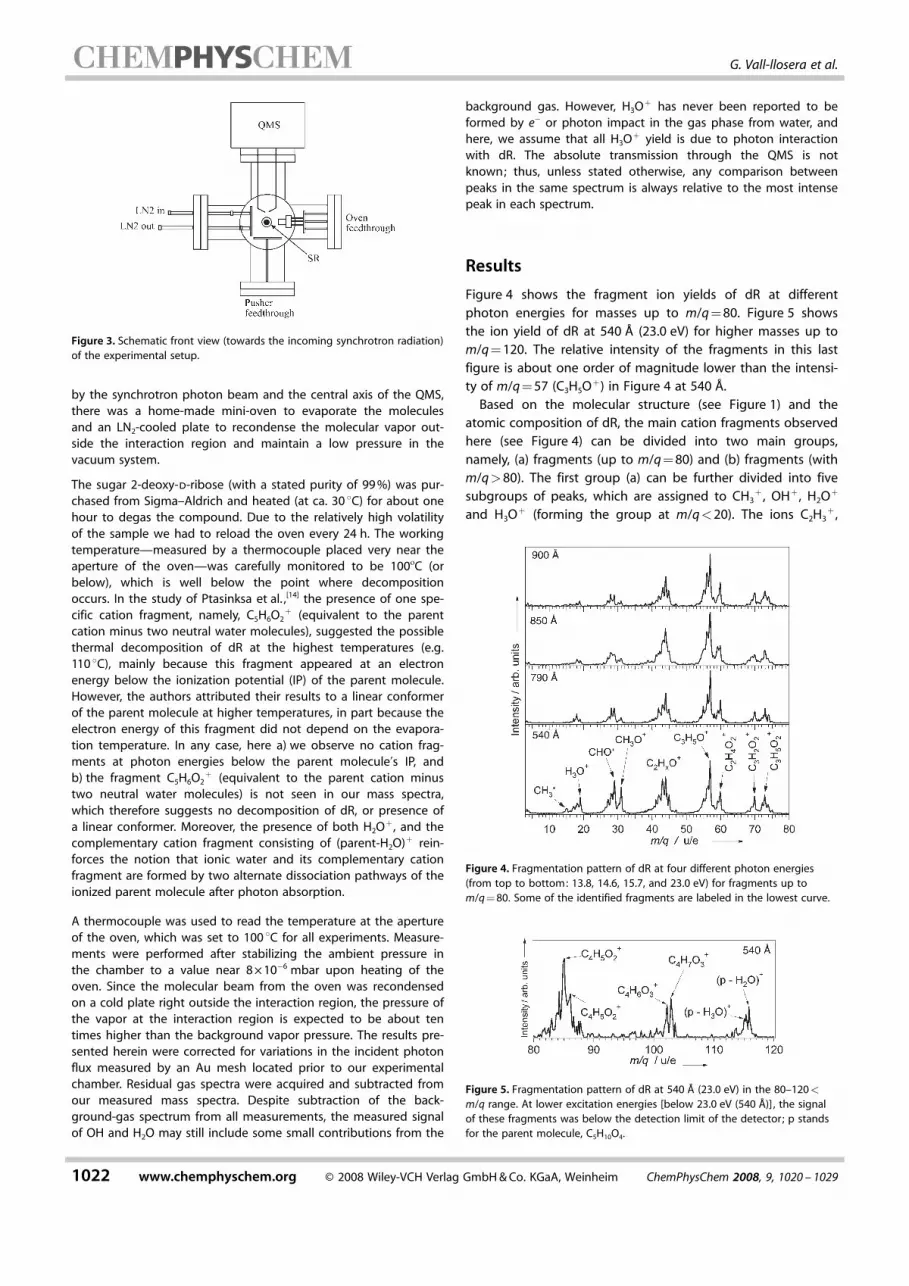
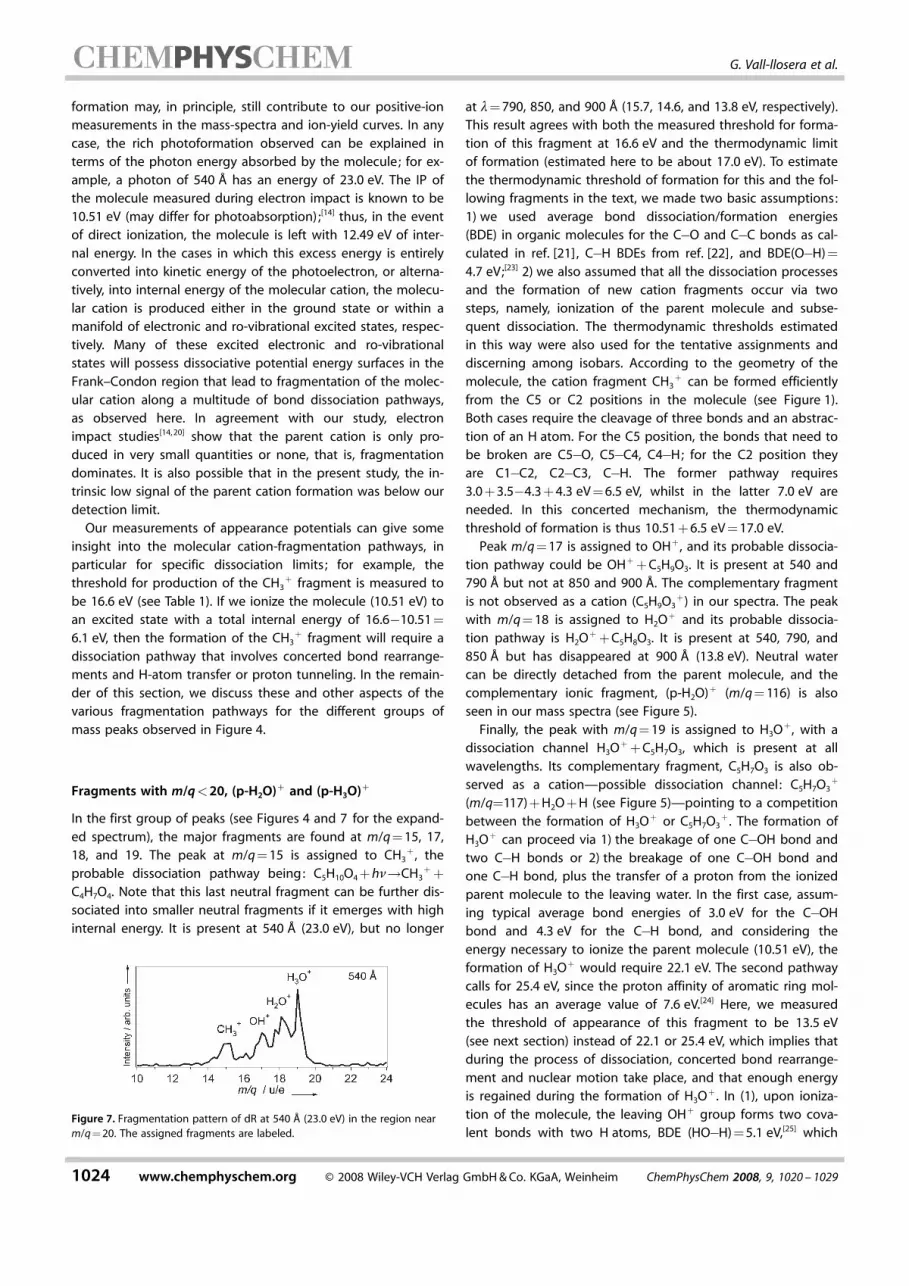
Figure 4 shows the fragment ion yields of dR at differentphoton energies for masses up to m/q=80. Figure 5 showsthe ion yield of dR at 540 P (23.0 eV) for higher masses up tom/q=120. The relative intensity of the fragments in this lastfigure is about one order of magnitude lower than the intensi-ty of m/q=57 (C3H5O
+) in Figure 4 at 540 P.Based on the molecular structure (see Figure 1) and the
atomic composition of dR, the main cation fragments observedhere (see Figure 4) can be divided into two main groups,namely, (a) fragments (up to m/q=80) and (b) fragments (withm/q>80). The first group (a) can be further divided into fivesubgroups of peaks, which are assigned to CH3
+ , OH+ , H2O+
and H3O+ (forming the group at m/q<20). The ions C2H3
+ ,
Figure 3. Schematic front view (towards the incoming synchrotron radiation)of the experimental setup.
Figure 4. Fragmentation pattern of dR at four different photon energies(from top to bottom: 13.8, 14.6, 15.7, and 23.0 eV) for fragments up tom/q=80. Some of the identified fragments are labeled in the lowest curve.
Figure 5. Fragmentation pattern of dR at 540 P (23.0 eV) in the 80–120<m/q range. At lower excitation energies [below 23.0 eV (540 P)] , the signalof these fragments was below the detection limit of the detector; p standsfor the parent molecule, C5H10O4.
1022 www.chemphyschem.org ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemPhysChem 2008, 9, 1020 – 1029
G. Vall-llosera et al.

C2H4+ , CHO+ , and CH3O
+ formthe group at 25<m/q<32. Be-tween 40 and 45 we findC2HxO
+ (x=1–5), and from 50 to60 we assign C3HxO
+ (x=3–5),C2H4O2
+ . The five fragments atm/q=69, 70, 72, 73, and 74 areassigned to C3HxO2
+ (x=1, 2,and 4–6). In the high-massregion (b), the major fragmentsobserved are assigned toC4H5O2
+ , C4HxO3+ (x=6, 7) and
C5HxO3+ (x=7, 8, see Figure 5).
The presence of all these ionsdemonstrates that the moleculeexposed to VUV photons under-goes very significant fragmenta-tion. The results reveal severalcharacteristics, namely, a maxi-mum yield for m/q=57common to all energies, and anoverall peak intensity decreasewith increasing m/q for m/q>70. All the assigned fragments(up to m/q=75) are present inthe mass spectra at the four ex-citation energies used here;however, the larger cation frag-ments (m/q>80) are not ob-served for wavelengths >540 P(23.0 eV).
We also measured the ionyields for the m/q=15, 19, 31,57, and 73 fragments as a func-tion of the incident photonenergy. From the recordedcurves we determined the ap-pearance energies (AE) by fittinga Wannier-type threshold law [f(x)=b+ax1.12689][19] near thethreshold of formation, as exemplified by the results for m/q=
31 (shown in Figure 6 and summarized in Table 1).
Discussion
Mass Spectra
After the absorption of a photon, the molecule can either di-rectly ionize, or it can be left in an excited state that can sub-sequently autoionize, dissociate into neutral species, or relaxvia ion-pair formation as shown in the following reactions (1):
Mþ hn! Mþ þ e�
! Mþ þ e� ! Aþ þ B
! M* ! Aþ þ B�
! M* ! Aþ þ e� þ B
! M* ! Aþ B
! M*þ þ e� ! Aþ þ Bþ e�
ð1Þ
In our experiments neither negative ions nor neutral frag-ments were detected, so that the neutral fragmentation chan-nels shown above are not discussed here. However, ion-pair
Figure 6. Excitation function for the CH3O+ fragment (m/q=31). The data
were fitted to a Wannier-type threshold law to extract the energy thresholds(shown in the curve). The same procedure was followed for the rest of thefragments for which formation thresholds were measured.
Table 1. Identified masses and tentative assignment of the fragments. The fragments observed in this workare compared to those from previous measurements (reported in the literature).[a] The appearance thresholdsfor some of the fragments are also shown and, when available, compared to those for electron impact.[b]
m/qACHTUNGTRENNUNG[u/e]
Comparison toother works[a]
Tentativeassignment(this work)
Dissociationchannel
Appearance threshold [eV]hn (this work) e� [14]
116 ce C5H8O3+ p-H2O
115 e C5H7O3+ p-H3O
103 ce C4H7O3+ p-CH3O
102 C4H6O3+ p-CH4O
C4H7O3�H86 ce C4H6O2
+ C4H7O3�OH85 ce C4H5O2
+ C4H7O3�H2O74 ce C3H6O2
+ p-C2H4O2
73 abce C3H5O2+ p-C2H4O2�H 12.1�0.4 10.74�0.03
72 C3H4O2+ C3H6O2�2H
70 ce C3H2O2+ C3H6O2�4H
69 abce C3HO2+ C3H6O2�5H
60 abce C2H4O2+ p-C3H6O2
p-C3H5O�OH57 abcde C3H5O
+ p-OH�C2H4O2 10.9�0.2 11.63�0.0656 cde C3H4O
+ C3H5O�H55 abcd C3H3O
+ C3H5O�2H45 abcde C2H5O
+ C2H4O+H44 abcde C2H4O
+ p-C3H6O3
43 abcde C2H3O+ C2H4O�H
42 abcde C2H2O+ C2H4O�2H
41 abcde C2HO+ C2H4O�3H31 abce CH3O
+ACHTUNGTRENNUNG(p-C4H8O3)+H 13.9�0.2 12.36�0.14
29 abcde CHO+ p-C2H8O3�H28 abcde C2H4
+ p-C3H5O3�OHp-C3H6O3�O
27 abcde C2H3+ p-C3H6O3�OH
19 abce H3O+ p-C5H7O3 13.5�0.3
18 ce H2O+ p-C5H8O3
17 ce OH+ p-C5H9O9
p-C3H5O�C2H4O2
15 abcde CH3+ p-C4H7O4 16.6�0.5 13.31�0.50
[a] a) 10–100 eV Ar+ ion impact in the condensed phase;[17] b) 10–100 eV He+ ion impact in the condensedphase;[16] c) high-energy (keV) He2+ ion impact in the gas phase;[12] d) soft-X rays on thin films;[11] e) 0–20 eVelectron impact in the gas phase.[14] [b] The uncertainties in the measurements were estimated from propaga-tion of error in the fitting procedure and the photon bandwidth (see ’Appearance Energies’ Section).
ChemPhysChem 2008, 9, 1020 – 1029 ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemphyschem.org 1023
Photofragmentation of the DNA Sugar
formation may, in principle, still contribute to our positive-ionmeasurements in the mass-spectra and ion-yield curves. In anycase, the rich photoformation observed can be explained interms of the photon energy absorbed by the molecule; for ex-ample, a photon of 540 P has an energy of 23.0 eV. The IP ofthe molecule measured during electron impact is known to be10.51 eV (may differ for photoabsorption) ;[14] thus, in the eventof direct ionization, the molecule is left with 12.49 eV of inter-nal energy. In the cases in which this excess energy is entirelyconverted into kinetic energy of the photoelectron, or alterna-tively, into internal energy of the molecular cation, the molecu-lar cation is produced either in the ground state or within amanifold of electronic and ro-vibrational excited states, respec-tively. Many of these excited electronic and ro-vibrationalstates will possess dissociative potential energy surfaces in theFrank–Condon region that lead to fragmentation of the molec-ular cation along a multitude of bond dissociation pathways,as observed here. In agreement with our study, electronimpact studies[14,20] show that the parent cation is only pro-duced in very small quantities or none, that is, fragmentationdominates. It is also possible that in the present study, the in-trinsic low signal of the parent cation formation was below ourdetection limit.
Our measurements of appearance potentials can give someinsight into the molecular cation-fragmentation pathways, inparticular for specific dissociation limits ; for example, thethreshold for production of the CH3
+ fragment is measured tobe 16.6 eV (see Table 1). If we ionize the molecule (10.51 eV) toan excited state with a total internal energy of 16.6�10.51=
6.1 eV, then the formation of the CH3+ fragment will require a
dissociation pathway that involves concerted bond rearrange-ments and H-atom transfer or proton tunneling. In the remain-der of this section, we discuss these and other aspects of thevarious fragmentation pathways for the different groups ofmass peaks observed in Figure 4.
Fragments with m/q<20, (p-H2O)+ and (p-H3O)
+
In the first group of peaks (see Figures 4 and 7 for the expand-ed spectrum), the major fragments are found at m/q=15, 17,18, and 19. The peak at m/q=15 is assigned to CH3
+ , theprobable dissociation pathway being: C5H10O4+hn!CH3
+ +
C4H7O4. Note that this last neutral fragment can be further dis-sociated into smaller neutral fragments if it emerges with highinternal energy. It is present at 540 P (23.0 eV), but no longer
at l=790, 850, and 900 P (15.7, 14.6, and 13.8 eV, respectively).This result agrees with both the measured threshold for forma-tion of this fragment at 16.6 eV and the thermodynamic limitof formation (estimated here to be about 17.0 eV). To estimatethe thermodynamic threshold of formation for this and the fol-lowing fragments in the text, we made two basic assumptions:1) we used average bond dissociation/formation energies(BDE) in organic molecules for the C�O and C�C bonds as cal-culated in ref. [21], C�H BDEs from ref. [22], and BDE ACHTUNGTRENNUNG(O�H)=4.7 eV;[23] 2) we also assumed that all the dissociation processesand the formation of new cation fragments occur via twosteps, namely, ionization of the parent molecule and subse-quent dissociation. The thermodynamic thresholds estimatedin this way were also used for the tentative assignments anddiscerning among isobars. According to the geometry of themolecule, the cation fragment CH3
+ can be formed efficientlyfrom the C5 or C2 positions in the molecule (see Figure 1).Both cases require the cleavage of three bonds and an abstrac-tion of an H atom. For the C5 position, the bonds that need tobe broken are C5�O, C5�C4, C4�H; for the C2 position theyare C1�C2, C2�C3, C�H. The former pathway requires3.0+3.5�4.3+4.3 eV=6.5 eV, whilst in the latter 7.0 eV areneeded. In this concerted mechanism, the thermodynamicthreshold of formation is thus 10.51+6.5 eV=17.0 eV.
Peak m/q=17 is assigned to OH+ , and its probable dissocia-tion pathway could be OH+ +C5H9O3. It is present at 540 and790 P but not at 850 and 900 P. The complementary fragmentis not observed as a cation (C5H9O3
+) in our spectra. The peakwith m/q=18 is assigned to H2O
+ and its probable dissocia-tion pathway is H2O
+ +C5H8O3. It is present at 540, 790, and850 P but has disappeared at 900 P (13.8 eV). Neutral watercan be directly detached from the parent molecule, and thecomplementary ionic fragment, (p-H2O)+ (m/q=116) is alsoseen in our mass spectra (see Figure 5).
Finally, the peak with m/q=19 is assigned to H3O+ , with a
dissociation channel H3O+ +C5H7O3, which is present at all
wavelengths. Its complementary fragment, C5H7O3 is also ob-served as a cation—possible dissociation channel: C5H7O3
+
ACHTUNGTRENNUNG(m/q=117)+H2O+H (see Figure 5)—pointing to a competitionbetween the formation of H3O
+ or C5H7O3+ . The formation of
H3O+ can proceed via 1) the breakage of one C�OH bond and
two C�H bonds or 2) the breakage of one C�OH bond andone C�H bond, plus the transfer of a proton from the ionizedparent molecule to the leaving water. In the first case, assum-ing typical average bond energies of 3.0 eV for the C�OHbond and 4.3 eV for the C�H bond, and considering theenergy necessary to ionize the parent molecule (10.51 eV), theformation of H3O
+ would require 22.1 eV. The second pathwaycalls for 25.4 eV, since the proton affinity of aromatic ring mol-ecules has an average value of 7.6 eV.[24] Here, we measuredthe threshold of appearance of this fragment to be 13.5 eV(see next section) instead of 22.1 or 25.4 eV, which implies thatduring the process of dissociation, concerted bond rearrange-ment and nuclear motion take place, and that enough energyis regained during the formation of H3O
+ . In (1), upon ioniza-tion of the molecule, the leaving OH+ group forms two cova-lent bonds with two H atoms, BDE (HO�H)=5.1 eV,[25] which
Figure 7. Fragmentation pattern of dR at 540 P (23.0 eV) in the region nearm/q=20. The assigned fragments are labeled.
1024 www.chemphyschem.org ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemPhysChem 2008, 9, 1020 – 1029
G. Vall-llosera et al.

will contribute 10.2 eV to the total reaction energy; this sug-gests a thermodynamic threshold of 11.9 eV (22.1�10.2=
11.9 eV) for the formation of H3O+ . In (2), the neutral OH group
forms one covalent bond with an H atom, thus gaining 5.1 eVand, simultaneously, the proton is transferred from the dissoci-ating parent molecule to the neutral water molecule (atomscrambling[26]), which will contribute 7.3 eV to the total reac-tion energy based on the proton affinity of water.[27] In thelatter case, a total energy of 12.4 eV is regained, thus suggest-ing a thermodynamic threshold of 25.4�12.4=13.0 eV. Bothcases, (1) and (2), are below the measured threshold, but it islikely that a certain activation energy is required for this con-certed reaction process, which involves nuclear motion, sincethe excitation of vibrational states involving H atoms isneeded.
The formation of H3O+ is also seen in the condensed-phase
results of Deng et al.[28] and in the gas-phase results of Alvara-do et al.[12] According to Deng, the production of this fragmentis the signature of an indirect effect, that is, the abstraction ofthe hydrogen atoms from adjacent molecules by a highly reac-tive OH+ ion. However, Alvarado’s and Ptasinska’s studies inthe gas phase[12, 14] show that this fragment can be directlyformed from isolated dR via hydrogen abstraction from themolecule. This observation is also supported in our study.
Fragments with 25<m/q<32, m/q =85, 86, 102, and 103
In the second group of peaks, near m/q=29 (see Figures 4and 8 for the expanded spectrum), the same number of frag-ments is present at the four excitation energies, but with alower relative intensity for the wavelengths >540 P (23.0 eV)relative to the strongest peak (m/q=57).
The peak with m/q=27 is assigned to C2H3+ , its probable
formation pathway being: C5H10O4+hn!C2H3+ +C3H6O3+OH.
If we look at the energy balance again, we find that two C�Obonds (average energy per bond: 3.0 eV) and one C�C bond(average energy: 3.5 eV) need to be broken. Thermodynamical-ly, the threshold would be at 20.0 eV (10.51+9.5 eV); however,this peak is clearly seen in the mass spectra at 790, 850, and900 P (15.7, 14.6, and 13.8 eV, respectively). In analogy to thecase of H3O
+ , this suggests concerted bond rearrangementsand nuclear motion during the formation of C2H3
+ . Accordingto the geometry of the molecule [see Figure 9 (1)] , while theparent cation dissociates along, for example, the C2�C3, C1�O,and C1�OH bonds, the C1 and C2 atoms of the emerging
C2H3+ fragment may form a double bond which contributes
7.6/2 eV=3.8 eV (7.6 eV for ethene[21]). Therefore, the thermo-dynamic threshold is 20.0�3.8 eV=16.2 eV. Yet, this value ishigher than three of the photon energies used here to acquirethe mass spectra in Figure 4. To justify the presence of thepeak in the spectra at l>540 P, we have to take into accountthe energy of formation of the neutral complementary frag-ment, C3H6O3. According to ref. [20], this neutral fragment canadopt the glyceraldehyde form [see Figure 9 (1)] , and thus, adouble bond C=O could be formed (BDE=5.9 eV[21]), thus low-ering the thermodynamic threshold to 16.2�2.9 eV=13.3 eV,which is lower than the lowest photon energy used here, thatis, 13.8 eV (900 P).
The peak at m/q=28 has two isobars, namely, CO and C2H4+ .
According to the thermodynamic thresholds estimated here,26.2 eV (i.e. 10.5 eV for the ionization of the parent moleculeplus 15.7 eV due to bond breaking) are required for the forma-tion of CO+ , whilst 21.7 eV are needed to form C2H4
+ . Thus,the tentative assignment of m/q=28 here is C2H4
+ . A probablefragmentation pathway for the assigned ionic fragment wouldbe C2H4
+ +C3H5O3+OH. In this case it would relate to the for-mation of C2H3
+ , if in the previous concerted reaction, theFigure 8. Fragmentation pattern of dR at 540 P (23.0 eV) in the region nearm/q=30. The assigned fragments are labeled.
Figure 9. Sketches of some fragment formations, discussed in the text.
ChemPhysChem 2008, 9, 1020 – 1029 ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemphyschem.org 1025
Photofragmentation of the DNA Sugar

C2H3+ picks up a second H atom from an adjacent C atom or
from the OH group in the molecule. The thermodynamicenergy balance for this channel would not be significantly dif-ferent from that for C2H3
+ . On the other hand, C2H4+ formation
could also be the result of the dissociation of the molecularcation into: C2H4
+ +C3H6O3+O. Although the C2H4+ peak is
decreased at energies lower than 540 P (23.0 eV), it is presentin all spectra. As in the previous discussion, the formation ofC2H4
+ at energies below 22.9 eV requires a C=C double bondbeing formed.
While the peak at m/q=29 may also involve the formationof C2H5
+ , here it is more likely the result of fragmentation ofthe molecular cation into CHO+ +C4H8O3+H. It is the highestpeak in the group at 540, 850, and 900 P, and its intensity alsodecreases with decreasing photon energy. For this specific dis-sociation channel we observe a possible competition betweenthe formation of CHO+ , C4H8O3
+ or H+ . While the C4H8O3+
cation is not observed here we do observe the speciesC4H7O3
+ (see Figure 5). Thus, the formation of C4H7O3+ would
come from the loss of one H atom from C4H8O3+ in the pro-
cess of dissociation. Peak m/q=31 is assigned to CH3O+ and
may involve the formation of C4H7O3(=C4H8O3�H), which isalso observed as a positive ion in our spectra (see Figure 5). Itis interesting to notice that CH2O
+ (m/q=30) is only weaklyformed (see Figure 8), which is contrary to what the geometryof simple bond cleavage would predict, for example, the for-mation of HCOH+ at C1 [see Figure 9 (2), fragment B]. Thus,after bond cleavage along the C1�O and C1�C2 bonds, twofragments are formed, namely, C4H8O3 and CH2O, which are la-beled as A and B, respectively, in Figure 9 (2). Fragment B isweakly observed as a cation whereas fragment A is not. Fur-thermore, fragments labeled as AII+ , BI+ , and BII+ inFigure 9 (2) are clearly seen in the mass spectra. This points toa competition between the following dissociation channels :a) CHO+(BI)+ (C4H8O3+H)(AI), b) CH3O
+ACHTUNGTRENNUNG(BII)+C4H7O3 ACHTUNGTRENNUNG(AII), and
c) CH3O ACHTUNGTRENNUNG(BII)+C4H7O3+(AII, m/q=103). The first two channels
lead to the formation of CHO+ and CH3O+ , respectively, as is
clearly seen in Figure 8, whereas the third channel yields theobserved fragment at m/q=103 (see Figure 5).
We note that in the above process of formation of C4H7O3+ ,
either subsequent bond rearrangements or the internal excita-tion energy may cause this molecular cation to loose i) eitheran OH group or an H2O molecule, thus leading to the forma-tion of C4H6O2
+ACHTUNGTRENNUNG(m/q=86) or C4H5O2
+ACHTUNGTRENNUNG(m/q=85), respectively, or
ii) an H atom, thus leading to C4H6O3+ACHTUNGTRENNUNG(m/q=102), all of which
are observed here, see Figure 5. On the other hand, here thecharge may also localize on the leaving OH or H2O moieties,both of which are observed as cations here. Note that BI andBII could also be formed at the C4 and C3 positions; however,the C1 and C5 positions are known to be the weakest in thesugar.[29,30]
Fragments with 40<m/q<46
In the third group of products (40<m/q<46, see Figures 4and 10 for the magnified spectrum), all five fragments are ob-served at all four photon energies. Comparing the relative in-
tensities of the whole group of cation fragments with that ofthe peak at m/q=57, we observe that these ions have similarrelative intensities when formed with photon excitations of540 and 850 P (i.e. 23.0 and 14.6 eV, respectively), but higherrelative intensities than the ions of the same mass formedafter photon absorption of 790 and 900 P (i.e. 15.7 and13.8 eV, respectively). The five observed ions correspond tom/q=41 (C2HO+), m/q=42 (C2H2O
+), m/q=43 (C2H3O+),
m/q=44 (C2H4O+), and m/q=45 (C2H5O
+). The fragment withthe highest relative intensity corresponds to m/q=44 orC2H4O
+ . Similarly to the preceding discussion on concertedbond rearrangements, when C2H4O
+ is formed it can loose ahydrogen atom in favor of C2H3O
+ [see Figure 9 (3)] with equalprobability, judging by the similar intensity of these two peaks(m/q=44 and 43). Alternatively, the C2H4O
+ ion can either ab-stract a hydrogen atom from its neutral complementary frag-ment during the concerted dissociation process, thereby form-ing C2H5O
+ , or loose two H atoms to form C2H2O+ (both pro-
cesses appear to occur with similar probability). The loss ofthree H atoms is also observed (C2HO is formed); however, itappears to be a less favorable process than the previous path-ways, as seen in Figure 10. The complementary neutral frag-ment to C2H4O
+ is C3H6O3, labeled A in Figure 9 (3). This frag-ment is not observed as a cation in our spectra; thus, the pro-posed competition for the formation of the C2H4O
+ , C2H3O+ ,
C2H5O+ , C2H2O
+ , and C2HO+ fragments is drawn in Figure 9 (3).The formation of C2H4O
+ requires the breaking of two bonds(a C�C bond and a C�O bond); it is an endothermic process(by ca. 6.5 eV). Given the IP of the parent molecule, the totalenergy required is about 17.0 eV. While a photon of 23.0 eVcan directly induce the formation of the fragment C2H4O
+ ,photons below 17.0 eV cannot. Thus, similarly to the discussionabove, the presence of this fragment at energies lower than17.0 eV requires a concerted bond rearrangement, for example,the transformation of a C�C bond into a C=C bond, therebyreleasing about 5.9�2.9=3 eV [BDE ACHTUNGTRENNUNG(C=C)�BDEACHTUNGTRENNUNG(C�C!C=C)] .
Fragments with m/q =50–75
In the next group of products m/q=55–60 (see Figures 4 and11 for an enlarged spectrum) we resolve four ionic fragments,one of them (m/q=57) being the highest in relative intensityin all spectra. The peaks with m/q=55, 56, and 57 are assignedto C3HxO
+ (x=3–5), whereas the peak with m/q=60 is as-signed to C2H4O2
+ , based on the structure of the molecule.
Figure 10. Fragmentation pattern of dR at 540 P (23.0 eV) in the region nearm/q=43. The assigned fragments are labeled.
1026 www.chemphyschem.org ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemPhysChem 2008, 9, 1020 – 1029
G. Vall-llosera et al.

The dissociation channel leading to the formation of the latteris shown in Figure 9 (4). When the molecular cation rupturesalong the C5�O and C3�C2 bonds, two fragments are formed,namely, A (C3H6O2) and B (C2H4O2). If the charge localizes on B,C2H4O2
+ is formed, and if the charge remains on A (B’s comple-mentary fragment) C3H6O2
+ACHTUNGTRENNUNG(m/q=74) is formed. This last frag-
ment is also observed as a cation in our spectra, and can sub-sequently decompose to other cations, namely, m/q=73, 72,70, and 69 (C3H5O2
+ , C3H4O2+ , C3H2O2
+ and C3HO2+ , respec-
tively) by successively loosing H atoms (see Figure 12). The
suggested dissociation channel for the formation of theC3H5O
+ fragment is as follows: C3H5O+C2H4O2+OH. If thecharge is localized in the OH group, then ions with m/q=17are formed, as observed earlier. If the charge remains onC2H4O2, then this channel competes with the formation ofC3H5O
+ . If the latter is formed with sufficient internal energy, itmay successively loose H atoms, thus leading to the formationof C3H4O
+ACHTUNGTRENNUNG(m/q=56) and C3H3O
+ACHTUNGTRENNUNG(m/q=55).
Appearance Energies
As mentioned earlier in the text, we found that CH3+ (m/q=
15) had a formation threshold close to 16.6 eV. The same frag-ment was measured by Ptasinska et al. ,[14] who obtained a for-mation threshold of 13.31 eV upon electron impact. Here,using average bond dissociation energies in organic molecules,and invoking concerted bond rearrangements, we estimatedthe minimum thermodynamic threshold for formation of thisfragment to be near 17 eV. This value is in reasonable agree-ment with the present results, taking account of thermal
broadening (see below), but disagrees with the electronimpact measurements. The low value recorded in ref. [14]might be due to the regained energy from bond rearrange-ments occurring in the neutral complementary fragment. Weobserve here that absorption of VUV photons leads to a con-certed fragmentation pathway that is likely to be different tothat of electron impact and electron attachment. This is mainlybecause the physical processes that lead to the fragmentationof dR after electron impact/attachment and photon absorptionare different. In the case of photoabsorption, the photon deliv-ers all its energy quanta to the molecule, whilst in the case ofelectron impact, the electron might only loose a fraction of itskinetic energy in the inelastic collision. In either case, theenergy absorbed by the molecule may go into the dissociation,ionization, and/or kinetic energy of the electron or fragments,as well as into the internal energy of the products.
Herein, the species with m/q=19 (hydronium) has a mea-sured threshold at 13.5 eV, whereas that with m/q=31 (CH3O
+)has a threshold of formation at about 13.9 eV, a value which iscomparable to that of 12.36 eV obtained from electron-impactstudies.[14] We also found that the threshold of the m/q=57(C3H5O
+) species was about 10.9 eV and that of the m/q=73species about 12.1 eV. Finally, we also estimated the AE form/q=44 and 60 to be below, namely, 10.2 and 11.5 eV, respec-tively. These threshold values represent upper limits, since theintensity yield for these two fragments was very low comparedto the background signal. Three principal factors contribute tothe experimental uncertainties in our appearance-energymeasurements: 1) the bandwidth of the photon beam, 2) theerror introduced by the Wannier threshold law fitting, and3) thermal broadening of dR (at 100 8C). Uncertainties (1) and(2) were estimated with the error-propagation approach, andthe results are shown in Table 1. The thermal broadening (3)was calculated according to the mean internal energy equationfor molecules (equipartition theorem) as a function of temper-ature, E= (n/2)kT, where n are the internal degrees of freedomof the target molecule at a specific temperature (vibrationaland rotational degrees of freedom), k is the Boltzmann con-stant, and T the temperature of the molecule in Kelvin. For amolecule with 30 degrees of freedom, for example, 30 normalmodes of vibration alone in benzene (not counting rotationaldegrees of freedom), the thermal broadening can be as highas 0.49 eV. The effect of thermal broadening is observed inFigure 6 as a small signal below the threshold determined bythe Wannier fitting.
Comparison to Other Studies
We compared our results with those of previous studies, asummary of which is shown in Table 1. We conclude that thefragmentation patterns most similar to ours are those recordedduring low-energy electron impact.[14] The fragmentation pat-tern after electron impact shows the same trend of groupingsthat we see here. The same fragments are identified, exceptfor m/q=72 and 102, which are exclusively observed in thepresent work. In Alvarado’s work[12] we also note certain group-ings of fragmentation patterns induced by high-energy ion
Figure 11. Fragmentation pattern of dR at 540 P (23.0 eV) in the region nearm/q=50. The assigned fragments are labeled.
Figure 12. Fragmentation pattern of dR at 540 P (23.0 eV) in the region nearm/q=70. The assigned fragments are labeled.
ChemPhysChem 2008, 9, 1020 – 1029 ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemphyschem.org 1027
Photofragmentation of the DNA Sugar

impact in the gas phase: while all the fragments seen in ourwork are also resolved in their mass spectra, the exceptionsare fragments with m/q=72, 102, and 115. Again, two of thesefragments are only seen in our study, that is, m/q=72 and 102,while the m/q=115 fragment observed here is only observedafter e� impact. A fragmentation study made with softX-rays[11] also shows certain global fragmentation patterns, thegrouping pattern in their unresolved mass spectra can betraced in our higher resolution mass spectra up to m/q=80. Atlow energies, ion-impact studies in condensed films of DNAsugars with both He+ [16] and Ar+ [17] ions also show a compara-ble cracking pattern. After hyperthermal He+ impact we findthat 16 fragments are in common with those observed in ourstudy, namely, m/q=15, 19, 27–29, 31, 41–45, 55, 57, 60, 69,and 73. These fragments are the same as those recorded withAr+ ion impact.[17] Thus, we conclude that after VUV photoab-sorption, certain specific new fragments are formed, that is,specific new dissociation channels are opened which are notobserved in ion, electron, or high-energy photon impact.
Conclusions
We presented a fragmentation study of 2-deoxy-d-ribose inthe gas phase as a result of VUV photoabsorption. The ob-served fragments were discussed on the basis of the energydeposited into the molecules and compared with fragmentscreated via ion and electron impact. At high masses (m/q>80)the relative intensity of the fragments is reduced by about oneorder of magnitude, and no m/q fragments greater than 80 areobserved for excitation energies below 540 P. At low massesthe spectra can be divided into five groups of endocyclic frag-ments, which correspond to m/q<20, 25<m/q<32,40<m/q<46, m/q=50–65, and m/q=65–75. The most in-tense fragment at all studied energies was m/q=57, whichcorresponds to C3H5O
+ . The least intense cations in our massspectra are those corresponding to m/q>80, which suggeststhat upon photoionization, the DNA sugar efficiently frag-ments. In double-stranded DNA, the fragmentation of thesugar, as observed here, would imply at least a complexsingle-strand break. Of all the studies at comparable particleenergies, the one that shows closest resemblance to the frag-mentation patterns observed here are those involving e�
impact: the same trend of fragment grouping is observed, andonly two fragments are not in common showing that new dis-sociation channels are opened. From the grouping pattern ob-served in this study we conclude that not only abundant ionicfragments are produced by the absorption of VUV photons,but also that concerted intramolecular reaction pathwaysoccur during disintegration of the DNA sugar, thereby involv-ing complex bond rearrangements and nuclear motion. More-over, we find that unlike in DNA bases,[31] upon photon absorp-tion by the DNA sugar, fragmentation clearly dominates oversimple ionization (parent cation formation). This result demon-strates that the DNA sugar in the gas phase is a very fragilemoiety which, after ionization, will rapidly disintegrate into nu-merous radical-cation and neutral-radical fragments. In DNA,these fragments will likely react with adjacent molecules, thus
leading to additional damage to the sugar backbone. In thatsense, our work may explain—at least in part—the surprisingresults presented by Hieda,[13] who observed that in photon-ir-radiated plasmid DNA, the single-strand-break (SSB) yield risesfrom threshold up to about 15 eV, after which it remains con-stant for photon energies up to 2 keV. If, as shown here, ab-sorption of a 15 eV photon already leads to disintegration ofthe sugar (i.e. a complex SSB in DNA), increasing the absorbed-photon’s energy will produce no additional damage to thesugar, and thus, no increase in the SSBs. However, the double-strand-break (DSB) yield will still rise somewhat with increasingphoton energy, since it is believed to be mediated largely bybase damage in the soft X-ray region.[13] Finally, the damageobserved here may even be different from that induced by dis-sociative electron attachment (DA) of ballistic secondary elec-trons (e.g. those produced by VUV photoionization at 20 eV).While 10–12 eV electrons can lead to both SSB and DSB inDNA (via simple bond cleavages initiated by DA to compo-nents of DNA),[5] the absorption of a similarly energetic photonwill lead to the complete decomposition of the sugar, asshown here.
Acknowledgements
Financial support from the Swedish Research Council (VR) andthe EU program—Access to Research Infrastructure action—(through the MAX laboratory) is gratefully acknowledged. M.A.H.acknowledges funding from the Natural Science and EngineeringResearch Council of Canada and the Canadian Space Agency. Weacknowledge Stefano Varas and Fabio Suran from the technicalelectronic services of CNR-INFM, Laboratorio Nazionale TASC,(Trieste, Italy), for their contribution in developing the QMS dataacquisition system.
Keywords: cleavage reactions · DNA damage · gas-phasereactions · mass spectrometry · photolysis
[1] C. von Sonntag, The Chemical Basis of Radiation biology, Taylor & Fran-cis, London, 1987, pp. 94–215.
[2] D. Becker, M. D. Sevilla, Adv. Radiat. Biol. 1993, 17, 121–180.[3] M. Levitt, A. Warshel, J. Am. Chem. Soc. 1978, 100, 2607–2613.[4] L. P. Guler, Y.-Q. Yu, H. I. Kentt�maa, J. Phys. Chem. A 2002, 106, 6754–
6764.[5] B. BoudaRffa, P. Cloutier, D. Hunting, M. A. Huels, L. Sanche, Science
2002, 287, 1658–1660.[6] H. Abdoul-Carime, L. Sanche, Int. J. Radiat. Biol. 2002, 78, 89–99.[7] A. Yokoya, R. Watanabe, T. Hara, J. Radiat. Res. 1999, 40, 145–158.[8] K. Hieda, T. Hirono, A. Azami, M. Suzuki, Y. Furusawa, H. Maesawa, N.
Usami, A. Yokoya, K. Kobayashi, Int. J. Radiat. Biol. 1996, 70, 437–445.[9] M. R. Raju, S. G. Carpenter, J. J. Chmielewski, M. E. Schillaci, M. E. Wilder,
J. P. Freyer, N. F. Johnson, P. L. Schor, R. J. Sebring, D. T. Goodhead,Radiat. Res. 1987, 110, 396–412.
[10] K. Fujii, K. Akamatsu, A. Yokoya, Surf. Sci. 2003, 528, 249–254.[11] K. Fujii, K. Akamatsu, A. Yokoya, Radiat. Res. 2004, 161, 435–441.[12] F. Alvarado, S. Bari, R. Hoekstra, T. Schlathçlter, Phys. Chem. Chem. Phys.
2006, 8, 1922–1928.[13] K. Hieda, Int. J. Radiat. Biol. 1994, 66, 561–567.[14] S. Ptasinska, S. Denifl, P. Scheier, T. D. M�rk, J. Chem. Phys. 2004, 120,
8505–8511.[15] W. K. Pogozelski, T. D. Tullius, Chem. Rev. 1998, 98, 1089–1106.
1028 www.chemphyschem.org ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemPhysChem 2008, 9, 1020 – 1029
G. Vall-llosera et al.

[16] Z. Deng, M. Imhoff, I. Bald, E. Illenberger, M. A. Huels, Phys. Rev. A 2006,74, 012716.
[17] I. Bald, Z. Deng, E. Illenberger, M. A. Huels, Phys. Chem. Chem. Phys.2006, 8, 1215–1222.
[18] S. L. Sorensen, B. J. Olsson, O. Widlund, S. Huldt, S.-E. Johansson, E. Ra-chlew, A. E. Nilsson, R. Hutton, U. LitzUn, A. Svensson, Nucl. Instrum.Methods Phys. Res. Sect. A 1990, 297, 296–300.
[19] G. H. Wannier, Phys. Rev. 1953, 90, 817–825.[20] NIST, Chemistry Web Book, http://webbook.nist.gov, 2007.[21] Y. R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds,
CRC, Boca Raton, 2003.[22] M.-J. Li, L. Liu, Y. Fu, Q.-X Guo, J. Phys. Chem. B 2005, 109, 13818–13826.[23] T. L. Cottrell, The Strengths of Chemical Bonds Second Edition, Butter-
worths, London, 1958.[24] T. Nakanaga, K. Buchold, F. Ito, Chem. Phys. 2003, 288, 69–76.[25] N. W. Alcock, Bonding and Structure : Structural Principles in Inorganic
and Organic Chemistry, Ellis Horwood, New York, 1990.
[26] M. Stepanovic, Y. Praiat, M. Allan, J. Chem. Phys. 1999, 110, 11376–11382.
[27] S.-L. Chong, R. A. Myers, Jr. , J. L. Franklin, J. Chem. Phys. 1972, 56, 2427–2430.
[28] Z. Deng, I. Bald, E. Illenberger, M. A. Huels, Phys. Rev. Lett. 2005, 95,153201.
[29] A. Diaz, R. D. Doepker, J. Phys. Chem. 1978, 82, 10–15.[30] A. A. Scala, E. W. G. Diau, Z. H. Kim, A. H. Zewail, J. Chem. Phys. 1998,
108, 7933–7936.[31] H. W. Jochims, M. Schwell, H. Baumg�rtel, S. Leach, Chem. Phys. 2005,
314, 263–282.
Received: September 20, 2007
Revised: December 12, 2007
Published online on March 20, 2008
ChemPhysChem 2008, 9, 1020 – 1029 ? 2008 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemphyschem.org 1029
Photofragmentation of the DNA Sugar






![2-deoxy-2-[18F]fluoro-D-mannose positron emission tomography imaging in atherosclerosis](https://cdn.vdocuments.mx/doc/165x107/63364edd02a8c1a4ec022316/2-deoxy-2-18ffluoro-d-mannose-positron-emission-tomography-imaging-in-atherosclerosis.jpg)
