fabrication of an electrochemical l -cysteine sensor based on graphene nanosheets decorated...
TRANSCRIPT

Fabrication of an Electrochemical l-Cysteine Sensor Based onGraphene Nanosheets Decorated Manganese OxideNanocomposite Modified Glassy Carbon Electrode
Samira Mansouri Majd,a Hazhir Teymourian,a Abdollah Salimi*a, b
a Department of Chemistry, University of Kurdistan, 66177-15175, Sanandaj, Irantel:+98-871-6624001, fax:+98-871-6624008
b Research Center for Nanotechnology, University of Kurdistan, 66177-15175, Sanandaj, Iran*e-mail: [email protected]; [email protected]
Received: May 25, 2013Accepted: July 8, 2013Published online: && &&, 2013
AbstractThe graphene nanosheets/manganese oxide nanoparticles modified glassy carbon electrode (GC/GNSs/MnOx) wassimply prepared by casting a thin film of GNSs on the GC electrode surface, followed by performing electrodeposi-tion of MnOx at applied constant potential. The GC/GNSs/MnOx modified electrode shows high catalytic activitytoward oxidation of l-cysteine. Hydrodynamic amperometry determination of l-cysteine gave linear responses overa concentration range up to 120 mM with a detection limit of 75 nM and sensitivity of 27 nA mM�1. The GC/GNSs/MnOx electrode appears to be a highly efficient platform for the development of sensitive, stable and reproduciblel-cysteine electrochemical sensors.
Keywords: Electrochemical sensors, Manganese oxide nanoparticles, Graphene nanosheets, l-Cysteine
DOI: 10.1002/elan.201300245
Supporting Information for this article is available on the WWW under http://dx.doi.org/10.1002/elan.201300245.
1 Introduction
Cysteine, a sulfur-containing amino acid, plays a crucialrole in biological systems [1], because of its chemical ac-tivity in the formation of complexes with various ionicspecies and biomolecules [2]. It is critical for the propermetabolism of a number of essential biochemicals, suchas co-enzyme A, heparin, biotin, lipoid acid and gluta-thione, and has been used as a prospective radiation pro-tector and cancer indicator in a number of pathologicalconditions, including Alzheimer�s and Parkinson�s diseas-es as well as autoimmune deficiency syndrome [3]. l-Cys-teine is also used in some proprietary antibiotic prepara-tions used for the treatment of skin damages [4], and asantioxidant in food industry [5]. Thus, the sensitive andselective assessment of l-cysteine in many fields such asfood processing, biochemistry, pharmaceuticals and clini-cal analysis is highly demanded [6].
Various analytical methods have been applied to deter-mine l-cysteine, such as high-performance liquid chroma-tography (HPLC) with fluorescence, mass spectrometric(MS) or ultraviolet (UV) detection [5, 7,8], capillary elec-trophoresis (CE) with amperometric or laser-inducedfluorescence detection [9,10], gas chromatography (GC)with fluorescence or MS [11, 12] and ion exchange chro-matography-fluorescence detection [13]. In addition,spectroscopy [14,15] and electrochemical detection [16]
were applied to determine l-cysteine. Among these, elec-trochemical analysis has inherent advantages of simplicity,rapid detection, ease of miniaturization, high sensitivity,as well as cost efficiency [17].
Although sulfhydryl compounds are known to undergoelectrochemical processes at solid electrodes, their oxida-tion occurs at relatively high overpotentials with narrowlinear range [18,19]. To overcome these problems, modi-fication of electrodes with suitable electron transfer medi-ators and/or other materials have been developed for theoxidation and detection of l-cysteine at lower overpoten-tials with higher sensitivities [20]. For example, metalliccompound materials [21,22], carbon paste electrodemodified with cobalt(II) and salophen [22], carbon ce-ramic electrodes modified with Ru-complex [24, 25], andconductive polymers modified electrodes [26] have beenapplied for electrochemical determination of cysteine.However, these electrodes contain certain disadvantages,such as leaching of modified materials, irreversible ad-sorption of substrates onto the electrode surface ornarrow detection range [27].
In recent years, with the advent of nanoscience andnanotechnology, the introduction of nanomaterials in theelectrochemical field as a sensing interface has revolu-tionized the concepts and application of original electro-
Electroanalysis 2013, 25, No. & , 1 – 10 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim &1&
These are not the final page numbers! ��
Full Paper

chemical sensors [28,29]. As the most recent member ofmultidimensional nanostructures in the carbon family,graphene has unique electronic, mechanical and thermalproperties, which are different from those of other kindsof carbon structures such as diamond, fullerene, andcarbon nanotubes [30]. Graphene has been the majorfocus of recent research to exploit an sp2 hybrid carbonnetwork in applications such as capacitors, sensors, drugdelivery, and solar cells [31–33]. In addition, graphene isan ideal material for electrochemistry because of its verylarge 2-D electrical conductivity (550 S cm�1), large sur-face area (2630 m2 g�1) and the large number of electro-chemically favorable edge carbons per mass of graphenefacilitate electron transfer between molecules to an elec-trode substrate with a low overpotential [34,35]. It hasmost of the advantageous properties of carbon nanotubes(such as homogeneous distribution of electrochemicallyactive sites on a nanometer scale) without carrying themost challenging element of carbon nanotube materials,that is, residual metallic impurities [36]. Moreover, gra-phene bears advantages in terms of its easy preparationfrom inexpensive graphite [37]. Besides the numerous ap-plications of graphene, nanocomposites containing gra-phene nanosheets and inorganic nanoparticles have beenintensively developed, and found to exhibit a range ofunique and useful properties, which are attracting moreand more attention from researchers [38]. There aremany reports in which graphene has been exploited asa two-dimensional carbon network to anchor metal andsemiconductor nanoparticles such as Pd-Pt alloy nanopar-ticles [39], Co3O4 [40], Fe3O4 [41], and Mn3O4 [42].
On the other hand, manganese oxide (MnOx) nanoma-terials are one of the most attractive candidates for elec-trochemical electrode materials. Because of their lowcost, environmental friendliness, nontoxic, natural abun-dance, relatively high energy density and high activity inalkaline media [43,44], MnOx nanomaterials have beenextensively utilized to construct chemical sensors or bio-sensors [45]. For example, manganese oxide nanorodsmodified electrodes for the enhanced water electrolysis[46], MnOOH nanorods modified Pt electrodes for elec-trocatalytic oxidation of methanol [47], gold nanoparti-cles/chitosan and MnO2 nanoparticles composite mem-brane/Prussian blue modified gold electrode as immuno-sensor [48], cholesterol biosensor based on MWCNTs-MnO2 nanoparticles using cyclic voltammetry, and MnO2-based nanostructures as catalysts for electrochemicaloxygen reduction in alkaline media [49]. In addition,some enzyme sensors using nano-MnO2 have been de-scribed in which nano-MnO2 with good biocompatibilityprovide a rigid support and an electron transferring pathto the electrode surface providing improved charge ex-change efficiency and stability during redox cycling [50].
Herein, we cast graphene nanosheets (GNSs) on thesurface of the glassy carbon (GC) electrode, and subse-quently electrodeposited MnOx on the GNSs modifiedGC electrode to fabricate the GC/GNSs/MnOx electrode.In the as-prepared nanocomposite, GNSs serve mainly as
highly conductive support, which can also provide a largesurface for the deposition of nanoscale MnOx particles.The excellent interfacial contact and increased contactarea between MnOx and graphene can significantly pro-mote the electrical conductivity of the electrode due tothe high electrical conductivity of graphene. The electro-chemical properties, stability, pH effect and kinetic pa-rameters of the redox active film were evaluated by elec-trochemical techniques. The fabricated electrode exhibit-ed excellent electrochemical response to l-cysteine at re-duced overpotential in acetate buffer solution (pH 6).Combining the benefits of GNSs and MnOx nanoparticlestogether with the easily fabrication procedure can bringmany advantages to the sensor, such as simplicity, lowcost, rapid response, high sensitivity and stability.
2 Experimental
2.1 Chemicals and Reagents
Natural graphite powders were supplied from BayCarbon (SP-1. MI. USA). Monohydrate manganese sul-fate (MnSO4·H2O), and l-cysteine were obtained fromMerck. All other chemicals were purchased from Merckand used as supplied (analytical or HPLC grade) withoutfurther purification. Acetate (0.1 M) and phosphate(0.1 M) buffer solutions with different pHs were preparedwith acetic acid and sodium acetate, sodium dihydrogenphosphate and disodium hydrogen phosphate, respective-ly, and used as supporting electrolytes in electrochemicalstudies. All aqueous solutions were prepared with ultra-pure water (>18 MW cm) obtained from a Milli-Q Plussystem (Millipore).
2.2 Apparatus
The surface morphology was examined using a Vega-Tescan electron microscope. The Raman spectra were col-lected on a Bruker Senterra Dispersive Raman Micro-scope spectrometer, the laser excitation wavelength wasfixed at 785 nm and the spectral resolution was <3 cm�1.Cyclic voltammetry (CV) was performed on an AUTO-LAB modular electrochemical system (ECO Chemie,Utrecht, The Netherlands) equipped with a PGSTAT 101module and driven by GPES software (ECO Chemie) inconjunction with a conventional three-electrode systemand a personal computer for data storage and processing.An unmodified or modified GC electrode (2.0 mm) wasemployed as the working electrode and a platinum wireas the counter electrode. All potentials were referred toan Ag/AgCl/KCl (3 M) electrode. Amperograms werecarried out with a Metrohm multipurpose instrumentmodel 693 VA Processor, equipped with a 694 VA Stand.A Metrohm drive shaft was used to rotate working elec-trodes during amperometric detection. All electrochemi-cal measurements are performed at room temperature.
&2& www.electroanalysis.wiley-vch.de � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Electroanalysis 2013, 25, No. & , 1 – 10
�� These are not the final page numbers!
Full Paper S. Mansouri Majd et al.

2.3 Synthesis of GNSs
Natural graphite powder is oxidized to graphite oxideusing a modified Hummers method [51]. Briefly, 1 ggraphite powder is put into a mixture of 3 mL concentrat-ed HNO3 and 70 mL H2SO4 in an ice bath. Then, 3 g ofKMnO4 was gradually added. The mixture is stirred for2 h and then diluted with water. Then, 5 % H2O2 is addedinto the solution until the color of the mixture is changedto brilliant yellow indicating fully oxidized graphite. Theas-obtained graphite oxide slurry was redispersed inwater and then exfoliated to generate graphene oxidenanosheets by ultrasonication. Then, the mixture was fil-tered and washed with diluted HCl solution to removemetal ions. Finally, the product was washed with water toremove the acid. Afterwards, the graphene oxide nano-sheets (GONs) have been reduced by hydrazine as re-ported previously [52]. In a typical process, 50 mg of solidproduct obtained is redispersed in 20 mL of H2O viaultrasonication. Then, 100 mg of poly (sodium 4-styrene-sulfonate) (PSS, C8H7NaOS)n, Mw =70,000) and 2.5 mLof hydrazine (NH2NH2) were added into the GONs dis-persion. After stirring for 30 min, the mixture was trans-ferred to a teflon lined autoclave and held in an oven at110 8C for 1 h. During the hydrothermal reaction, gra-phene oxide reacted with PSS and was simultaneously re-duced by hydrazine to GNSs-PSS. The reaction productwas centrifuged and then washed with water and ethanolto remove the unreacted PSS Surfactant. The final prod-uct was dried in a vacuum oven at 60 8C.
2.4 Preparation of GC/GNSs/MnOx Electrode
Prior to coating, GC electrode was carefully polishedwith 3.0 mm alumina powder on polishing cloth and soni-cated successively in ethanol and doubly distilled water inorder to remove adsorbed particles. Then, 10 mL of GNSssuspension (1 mg mL�1) in ethanol was cast on the result-ing GC electrode surface andallowed to dry at room tem-perature. After rinsing the electrode with distilled water,-the potential of GC/GNSs modified electrode is cycled(20 cycles) between 0.0 and +1.5 V at scan rate of100 mV s�1 in phosphate buffer solution(pH7). During po-tential cycling, the aromatic species or functional groupscontaining p bond (�COOH, �COH, �CO�) produced atGC electrode. The p–p staking and hydrophobic interac-tions between the aromatic or p-containing functionalgroups and sp2 networks of graphene increased the stabil-ity of immobilized graphene onto GC surface [49]. After-wards, the as-obtained GC/GNSs electrode was modifiedby MnOx nanoparticles by using electrodeposition at con-stant potential of +1.4 V for 200 s. The aqueous electro-lytic solution used during the electrodeposition step con-tained MnSO4·H2O (1.0 mM) and its pH was adjusted to1.8 using concentrated H2SO4. The as-obtained electrode(denoted as GC/GNSs/MnOx) was washed with waterand stored at ambient temperature (25 8C) before beingused in experiments. The effective area of the electrode
modified with GNSs/MnOx nanocomposite was deter-mined to be 0.057 cm2 from CV of 5 mM [Fe(CN)6]
3�/[Fe(CN)6]
4� in phosphate buffer solution (pH 7).
3 Results and Discussion
3.1 Electrodeposition of MnOx Nanoparticles
Among the various electrochemical techniques allowingthe electrodeposition of very adherent MnOx thin films,electrolysis at a constant potential was chosen in thiswork preferentially for its easy and quick use. Variationof current as a function of time observed for a +1.4 Velectrolysis potential (which is the value measured for theanodic peak potential (figure not shown))was measuredexperimentally (Figure S1, Supporting Information (SI)).After a sharp peak followed by a fast drop correspondingto double layer charging and simultaneous nucleation/growth process of the manganese dioxide thin film, thecurrent reaches a nonzero steady value with an extremelyslow kinetics. One should also point out that this poten-tial (1.4 V) was shown to be the foot potential of the oxi-dation wave related to water oxidation and/or to surfaceactivation of the GC surface in the absence of Mn2+cat-ions in the electrolytic solution. In the first case, oxygenproduction may produce direct oxidation of Mn2+ at theGC electrode according to [54]:
Mn2þ þ 1=4O2 þ 3=2H2O!MnOOHþ 2Hþ ð1Þ
Due to the very low solubility product, one mayassume that as soon as Mn3+ is formed with a detectableconcentration at the interface, MnOOH precipitates andforms an insulating thin film, through which Mn2+ diffu-sion is strongly slowed down and therefore the currentkeeps a negligible value. Once the potential reachesa value at which forward below reaction:
MnOOH!MnO2 þHþ þ e� ð2Þ
becomes quantitative, MnOOH is rapidly transformedinto MnO2 which has sufficient electronic conductivity toensure current at its surface. MnOOH appears then asa minor intermediate, which does not limit the current.When pH domain becomes more acidic, MnOOH be-comes unstable and disproportionation may occur [55]:
2MnOOHþ 2Hþ !MnO2 þMn2þ þ 2H2O ð3Þ
In order to confirm that the GO sheets has been re-duced to GNSs, the Raman spectrum of GNSs was shownin Figure S2 (Supporting Information). As shown, theRaman spectrum of GNSs exhibited a D-band peak at1341 cm�1 due to the breathing mode of k-point phononsof A1g symmetry [56] and a G-band peak at 1588 cm�1
that corresponds to the first-order scattering of the E2g
phonons [57]. Our results are in close agreement with the
Electroanalysis 2013, 25, No. & , 1 – 10 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.electroanalysis.wiley-vch.de &3&
These are not the final page numbers! ��
Fabrication of an Electrochemical l-Cysteine Sensor

previously reported results [58]. The surface morpholo-gies of the GC/GNSs and GC/GNSs/MnOx electrodeswere investigated by SEM and the results were presentedin Figure 1. The distinctive layered structure of GNSs canbe obviously distinguished (Figure 1A). The GNSs havelateral dimension ranged from a few hundred nanometersto several micrometers. It is evident from Figure 1B thatafter the formation of MnOx nanoparticles on the elec-trode surface, two-dimensional GNSs are well decoratedby a large quantity of spherical MnOx nanostructures(with average size of ~50 nm and rather good size distri-bution) and both the outline of GNSs and MnOx nano-particles can be clearly observed.
The formation of a MnOx layer on the electrode sur-face was checked by recording CVs of the unmodifiedand modified electrodes in acetate buffer solution (pH 6)without manganese ions (Figure 2). As shown for thebare GC and GC/GNSs electrodes, no redox peaks wereobserved (voltammograms “a and b”) at a potential rangeof 0.0 to 1.2 V, which provides a potential window for in-
vestigating the voltammetric behavior of MnOx. For vol-tammogram c, oxidation peaks were observed at 0.60 V(I) and 0.71 V (II) during the positive potential scan thatcan be attributed to the oxidation of MnOOH to MnO2
(Equation 4) and Mn2+ to MnOOH (Equation 5), respec-tively. In the reverse cathodic sweep, two cathodic peaksare observed at 0.54 V (III) and 0.4 V (IV), correspond-ing to the reduction of the MnOOH to Mn2+ (Equa-tion 6) and MnO2 to MnOOH (Equation 7) [59].
e� þMnOOH!MnO2 þHþ ð4Þ
MnðOHÞ2 !MnOOHþHþ þ e� ð5Þ
MnOOHþ 3Hþ þ e� !Mn2þ þ 2H2O ð6Þ
MnO2 þHþ þ e� !MnOOH ð7Þ
3.2 Electrochemical Behavior of the GC/GNSs/MnOxElectrode
In order to study the kinetic parameters, CVs of the GC/GNSs/MnOx electrode at different scan rates were re-corded (Figure 3). The peak currents of the redox systemare directly proportional to the scan rate for sweep ratesbelow 0.1 Vs�1 (Inset A in Figure 3) which confirms theadsorption nature of the electrochemical process. Athigher sweep rates, the plot of peak currents vs. scan ratedeviates from linearity and the peak current becomesproportional to the square root of the scan rate, indicat-ing a diffusion-controlled process. The slow diffusion ofcounter ions (H+) into the electrode surfaces controlledthe electrochemical reaction. Moreover, at higher sweeprates (>1 V s�1), peak separations begin to increase (InsetB in Figure 3). This indicates the limitation arising fromcharge transfer kinetics. Based on Laviron theory [60],the electron transfer rate constant (ks) and charge trans-fer coefficient (a) can be determined by measuring thevariation of peak potential with scan rate. The values ofpeak potentials were proportional to the logarithm of thescan rate for scan rates higher than 1 V s�1. The calculatedvalues for ks and a were about 0.578 s�1 and 0.3, respec-tively. The large value of ks indicates the high ability ofMnOx nanoparticles for promoting electron transfer atthe electrode surface. The surface concentration of elec-troactive species, G, can be calculated from the slope ofthe plot of Ipa vs. scan rate (v <100 mV s�1) by the follow-ing equation [61]:
I ¼ n2F2v AG=4RT ð8Þ
where v is the sweep rate, A is the surface area and theother symbols have their usual meaning. The value of G
calculated to be approximately 5.36� 10�9 molcm�2 forGC/GNSs/MnOx.
The effect of pH on the electrochemical behavior ofthe GC/GNSs/MnOx electrode was investigated in the
Fig. 1. SEM images of (a) GNSs and (b) GNSs/MnOx nano-composite.
&4& www.electroanalysis.wiley-vch.de � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Electroanalysis 2013, 25, No. & , 1 – 10
�� These are not the final page numbers!
Full Paper S. Mansouri Majd et al.
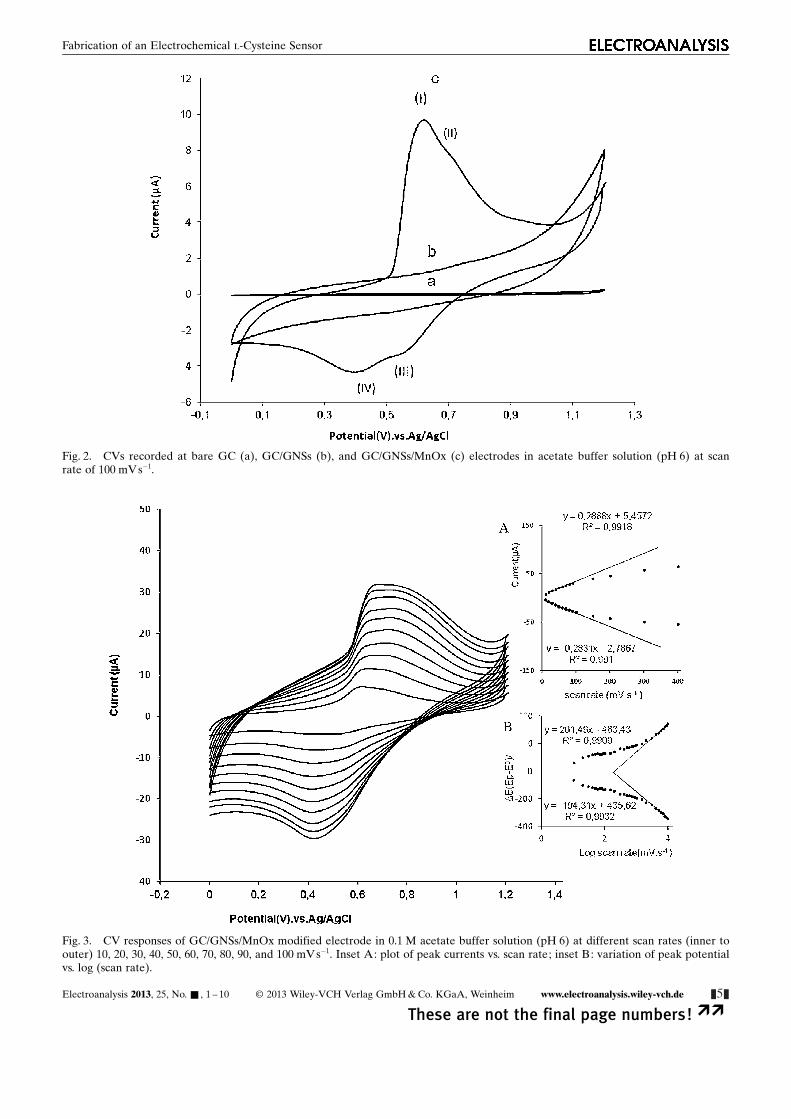
Fig. 2. CVs recorded at bare GC (a), GC/GNSs (b), and GC/GNSs/MnOx (c) electrodes in acetate buffer solution (pH 6) at scanrate of 100 mV s�1.
Fig. 3. CV responses of GC/GNSs/MnOx modified electrode in 0.1 M acetate buffer solution (pH 6) at different scan rates (inner toouter) 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100 mV s�1. Inset A: plot of peak currents vs. scan rate; inset B: variation of peak potentialvs. log (scan rate).
Electroanalysis 2013, 25, No. & , 1 – 10 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.electroanalysis.wiley-vch.de &5&
These are not the final page numbers! ��
Fabrication of an Electrochemical l-Cysteine Sensor

pH range of 2–8 (Figure 4). As is obvious, CVs of themodified electrode show a strong dependence on pH. Apair of redox peaks were observed for adsorbed MnOxnanoparticles in different pH values. The peak currentsincreased with increasing pH values from pH 2 to pH 6and the maximum current was obtained at pH 6.
Long-term stability is one of the most important prop-erties for sensors, biosensors and bioreactors. The stabilityof the GC/GNSs/MnOx electrode was investigated byusing CV. The stability of the modified electrode wasverified by monitoring the remaining amount of activesubstance after 200 successive sweeps with scan rate of100 mV s�1 in potential range of 0.0 to 1.2 V (SupportingInformation Figure S3). The peak height and peak poten-tial of the immobilized MnOx remained nearly un-changed. In addition, no significant decrease can be seenafter replacing the electrolyte that has been used for 200repetitive cycles with fresh buffer solution. The observedhigh stability of modified electrode can be related to thechemical and mechanical stability and good adherence ofthe nanocomposite. Therefore, the GNSs/MnOx modifiedGC electrode can be used as a sensor due to its long-termstability and excellent electron transfer rate constant.
3.3 Electrocatalytic Oxidation of l-Cysteine at GC/GNSs/MnOx Electrode
In order to study the electrocatalytic activity of the modi-fied electrode for l-cysteine oxidation, the electrochemi-cal experiments in the absence and presence of l-cysteinewere carried out. Figure 5 compares the recorded CVs of
bare GC (a, b), GC/GNSs (c, d) and GC/GNSs/MnOx (e,f) electrodes in acetate buffer solution (pH 5) in the ab-sence (a, c, e) and presence (b, d, f) of 0.2 mM cysteine.As shown, while for bare GC and GC/GNSs electrodes,no redox response can be seen in the potential rangefrom 0.0 to 1.2 V upon l-cysteine addition (voltammo-grams b, d), at the GC/GNSs/MnOx electrode, the oxida-tion current was greatly increased due to the catalytic oxi-dation of cysteine. In fact, the diminution of overvoltageand increased peak current of cysteine oxidation confirmthat GNSs/MnOx nanocomposite has high catalytic abilityfor cysteine oxidation and thus, it can act as a suitablemediator to shuttle electrons between cysteine and work-ing electrode and facilitate electrochemical regenerationfollowing electron exchange with cysteine.
When l-cysteine was added in the test solution, theelectrode modified with MnOx nanoparticles had a bidir-ection electrocatalytic ability toward the reduction/oxida-tion of l-cysteine. It is a phenomenon that not only theoxidation but also the reduction current increase with theconcentration of l-cysteine [62]. In this system, with theaddition of l-cysteine, the oxidation current increasedmost significantly and the reduction current increases lesssignificantly.
The following catalytic scheme describes the reactionsequence in the oxidation of l-cysteine with manganeseoxide [62,63]:
MnO2 þHþ þ e� !MnOOH ð9Þ
2MnOOHþ 2CySH! CyS-SCyþ 2MnðOHÞ2 ð10Þ
Fig. 4. CVs of GC/GNSs/MnOx electrode in solutions with different pH values (from right to left) 2 to 8, scan rate 100 mVs�1.
&6& www.electroanalysis.wiley-vch.de � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Electroanalysis 2013, 25, No. & , 1 – 10
�� These are not the final page numbers!
Full Paper S. Mansouri Majd et al.
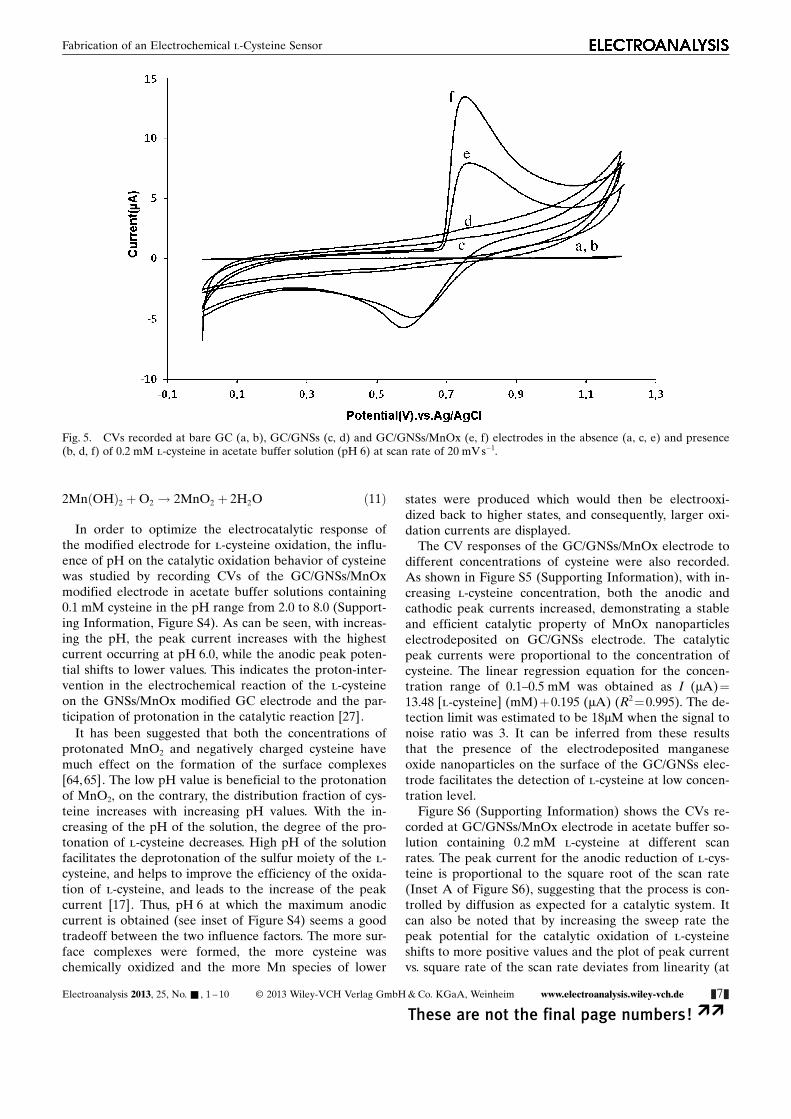
2MnðOHÞ2 þO2 ! 2MnO2 þ 2H2O ð11Þ
In order to optimize the electrocatalytic response ofthe modified electrode for l-cysteine oxidation, the influ-ence of pH on the catalytic oxidation behavior of cysteinewas studied by recording CVs of the GC/GNSs/MnOxmodified electrode in acetate buffer solutions containing0.1 mM cysteine in the pH range from 2.0 to 8.0 (Support-ing Information, Figure S4). As can be seen, with increas-ing the pH, the peak current increases with the highestcurrent occurring at pH 6.0, while the anodic peak poten-tial shifts to lower values. This indicates the proton-inter-vention in the electrochemical reaction of the l-cysteineon the GNSs/MnOx modified GC electrode and the par-ticipation of protonation in the catalytic reaction [27].
It has been suggested that both the concentrations ofprotonated MnO2 and negatively charged cysteine havemuch effect on the formation of the surface complexes[64,65]. The low pH value is beneficial to the protonationof MnO2, on the contrary, the distribution fraction of cys-teine increases with increasing pH values. With the in-creasing of the pH of the solution, the degree of the pro-tonation of l-cysteine decreases. High pH of the solutionfacilitates the deprotonation of the sulfur moiety of the l-cysteine, and helps to improve the efficiency of the oxida-tion of l-cysteine, and leads to the increase of the peakcurrent [17]. Thus, pH 6 at which the maximum anodiccurrent is obtained (see inset of Figure S4) seems a goodtradeoff between the two influence factors. The more sur-face complexes were formed, the more cysteine waschemically oxidized and the more Mn species of lower
states were produced which would then be electrooxi-dized back to higher states, and consequently, larger oxi-dation currents are displayed.
The CV responses of the GC/GNSs/MnOx electrode todifferent concentrations of cysteine were also recorded.As shown in Figure S5 (Supporting Information), with in-creasing l-cysteine concentration, both the anodic andcathodic peak currents increased, demonstrating a stableand efficient catalytic property of MnOx nanoparticleselectrodeposited on GC/GNSs electrode. The catalyticpeak currents were proportional to the concentration ofcysteine. The linear regression equation for the concen-tration range of 0.1–0.5 mM was obtained as I (mA)=13.48 [l-cysteine] (mM)+0.195 (mA) (R2 =0.995). The de-tection limit was estimated to be 18mM when the signal tonoise ratio was 3. It can be inferred from these resultsthat the presence of the electrodeposited manganeseoxide nanoparticles on the surface of the GC/GNSs elec-trode facilitates the detection of l-cysteine at low concen-tration level.
Figure S6 (Supporting Information) shows the CVs re-corded at GC/GNSs/MnOx electrode in acetate buffer so-lution containing 0.2 mM l-cysteine at different scanrates. The peak current for the anodic reduction of l-cys-teine is proportional to the square root of the scan rate(Inset A of Figure S6), suggesting that the process is con-trolled by diffusion as expected for a catalytic system. Itcan also be noted that by increasing the sweep rate thepeak potential for the catalytic oxidation of l-cysteineshifts to more positive values and the plot of peak currentvs. square rate of the scan rate deviates from linearity (at
Fig. 5. CVs recorded at bare GC (a, b), GC/GNSs (c, d) and GC/GNSs/MnOx (e, f) electrodes in the absence (a, c, e) and presence(b, d, f) of 0.2 mM l-cysteine in acetate buffer solution (pH 6) at scan rate of 20 mV s�1.
Electroanalysis 2013, 25, No. & , 1 – 10 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.electroanalysis.wiley-vch.de &7&
These are not the final page numbers! ��
Fabrication of an Electrochemical l-Cysteine Sensor
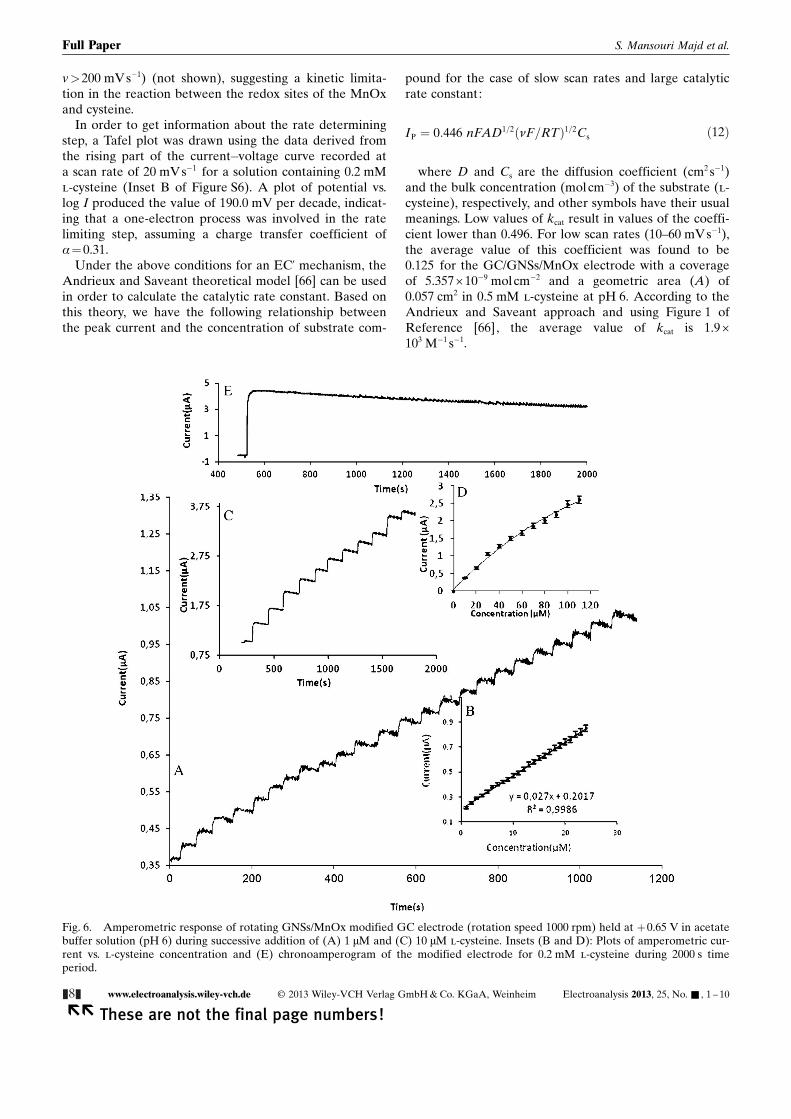
v>200 mVs�1) (not shown), suggesting a kinetic limita-tion in the reaction between the redox sites of the MnOxand cysteine.
In order to get information about the rate determiningstep, a Tafel plot was drawn using the data derived fromthe rising part of the current�voltage curve recorded ata scan rate of 20 mVs�1 for a solution containing 0.2 mMl-cysteine (Inset B of Figure S6). A plot of potential vs.log I produced the value of 190.0 mV per decade, indicat-ing that a one-electron process was involved in the ratelimiting step, assuming a charge transfer coefficient ofa=0.31.
Under the above conditions for an EC’ mechanism, theAndrieux and Saveant theoretical model [66] can be usedin order to calculate the catalytic rate constant. Based onthis theory, we have the following relationship betweenthe peak current and the concentration of substrate com-
pound for the case of slow scan rates and large catalyticrate constant:
IP ¼ 0:446 nFAD1=2ðnF=RTÞ1=2Cs ð12Þ
where D and Cs are the diffusion coefficient (cm2 s�1)and the bulk concentration (molcm�3) of the substrate (l-cysteine), respectively, and other symbols have their usualmeanings. Low values of kcat result in values of the coeffi-cient lower than 0.496. For low scan rates (10–60 mVs�1),the average value of this coefficient was found to be0.125 for the GC/GNSs/MnOx electrode with a coverageof 5.357 � 10�9 molcm�2 and a geometric area (A) of0.057 cm2 in 0.5 mM l-cysteine at pH 6. According to theAndrieux and Saveant approach and using Figure 1 ofReference [66], the average value of kcat is 1.9 �103 M�1 s�1.
Fig. 6. Amperometric response of rotating GNSs/MnOx modified GC electrode (rotation speed 1000 rpm) held at +0.65 V in acetatebuffer solution (pH 6) during successive addition of (A) 1 mM and (C) 10 mM l-cysteine. Insets (B and D): Plots of amperometric cur-rent vs. l-cysteine concentration and (E) chronoamperogram of the modified electrode for 0.2 mM l-cysteine during 2000 s timeperiod.
&8& www.electroanalysis.wiley-vch.de � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Electroanalysis 2013, 25, No. & , 1 – 10
�� These are not the final page numbers!
Full Paper S. Mansouri Majd et al.

The stability of the electrocatalytic activity of the modi-fied electrode toward l-cysteine oxidation was examinedby repetitive scanning at a scan rate of 20 mV s�1 (notshown). In the first three scans, the electrocatalytic cur-rents decreased with scan number and then the currentremained at 90% of its initial value after 50 cycles, indi-cating the high stable response of the sensor for l-cys-teine detection.
3.4 Amperometric Detection of l-Cysteine at the GC/GNSs/MnOx Electrode
Since amperometry under stirred condition is much morecurrent sensitive than cyclic voltammetry, this methodwas employed in order to estimate the lower detectionlimit of the sensor. The amperometric response of thesensor to l-cysteine was examined by successively in-creasing the concentration of l-cysteine under the opti-mized conditions. As shown in Figure 6, a well-defined re-sponse was observed during successive addition of 1 mMand 10 mM of l-cysteine (Figures 6A and C). There isa linear relationship between response current and l-cys-teine concentration in the range of 1 mM–120 mM (Fig-ure 6B), while for higher concentrations, the plot of thecurrent vs. analyte concentration deviates from linearity(Figure 6D). A linear relationship between anodic currentand the concentration of l-cysteine was observed, I(mA)=0.027 [l-cysteine]+0.201 (mA), with a correlationcoefficient of 0.998 when the concentration of l-cysteinewas between 1 and 24 mM, indicating that the regressionline is well fitted with the experimental data. The detec-tion limit (S/N=3) and sensitivity were 75 nM and27 nAmM�1, respectively. The detection limit, linearcali-bration range and sensitivity for l-cysteine determinationat this modified electrode are comparable with those ob-tained by using other modified electrodes.
Another attractive feature of our proposed electrodewas its highly stable amperometric response to l-cysteine.Figure 6E presents the amperometric response of GC/GNSs/MnOx electrode to 0.2 mM l-cysteine recordedover a continuous period of 2000 s. As can be seen, theresponse of the modified electrode remained nearlystable with only 20% current diminutions after 2000 s.Thus, this electrode can be used as an excellent electroca-talytic material for sensitive and stable amperometric de-tection of l-cysteine.
4 Conclusions
In summary, nanostructured MnOx thin films were elec-trodeposited on a GNSs modified GC electrode ina highly reproducible and fast manner by employing elec-trolysis at a constant potential. Due to the superior elec-trical conductivity and high specific surface area of GNSs,it can act as an excellent support material to anchorMnOx nanoparticles. After modifying the electrode withthis nanocomposite, two pairs of well-defined peaks with
low background and high peak current appeared. TheGC/GNSs/MnOx electrode showed excellent electrocata-lytic effect toward l-cysteine oxidation. The sensitivityand detection limit of the sensor are 27 nA mM�1 and75 nM at a concentration range up to 120 mM. Due to thesimple immobilization procedure, stability and reversibili-ty of adsorbed redox system at different pH solutions,this method was advantageous compared with other im-mobilization procedures. The proposed sensor displayedexcellent characteristics, such as simplicity, low cost, highsensitivity, rapid analysis procedures, low detection limitand long-term stability. Thus, this study suggests that theproposed GC/GNSs/MnOx modified electrode can bea promising candidate for sensors design.
Acknowledgements
This research was supported by the Iranian Nanotechnol-ogy Initiative and the Research Office of the University ofKurdistan.
References
[1] Y. H. Bai, J. J. Xu, H. Y. Chen, Biosens. Bioelectron. 2009,24, 2985.
[2] B. Yosypchuk, I. Novotny, Talanta 2002, 56, 971.[3] B. Filanovsky, Anal. Chim. Acta 1999, 394, 91.[4] S. A. Wring, J. P. Hart, B. J. Birch, Analyst 1989, 114, 1571.[5] Y. Dale, V. Mackey, R. Mushi, A. Nyanda, M. Maleque, J.
Ike, J. Chromatogr. B 2003, 788, 1.[6] X. Tang, Y. Liu, H. Hou, T. You, Talanta 2010, 80, 2182.[7] D. Kutlan, I. Molnar-Perl, J. Chromatogr. A 2003, 987, 311.[8] J. You, Y. Shan, L. Zhen, L. Zhang, Y. Zhang, Anal. Bio-
chem. 2003, 313, 17.[9] J. Wang, S. Mannino, C. Camera, M. P. Chatrathi, M. Scam-
picchio, J. Zima, J. Chromatogr. A 2005, 1091, 177.[10] S. Zhao, Y. Song, Y. Liu, Talanta 2005, 67, 212.[11] M. J. Nozal, J. L. Bernal, M. L. Toribio, J. C. Diego, A. Ruiz,
J. Chromatogr. A 2004, 1047, 137[12] S. G. Villas-Boas, D. G. Delicado, M. Qkesson, J. Nielsen,
Anal. Biochem. 2003, 322, 134.[13] G. Ravindran, W. L. Bryden, Food Chem. 2005, 89, 309.[14] A. L. Jenkins, R. A. Larsen, T. B. Williams, Spectrochim.
Acta A 2005, 61, 1585.[15] J. W. Costin, P. S. Francis, S. W. Lewis, Anal. Chim. Acta
2003, 480, 67.[16] N. S. Lawrence, E. L. Beckett, J. Davis, R. G. Compton,
Anal. Biochem. 2002, 303, 1.[17] O. Nekrassova, N. S. Lawrence, R. G. Compton, Talanta
2003, 60, 1085.[18] J. P. Hart, I. C. Hartley, Analyst 1994, 119, 259.[19] H. Razmi, H. Heidari, Anal. Biochem. 2009, 388, 15.[20] J. Kulys, A. Drungiliene, Anal. Chim. Acta 1991, 243, 287.[21] K. S. Prasad, G. Muthuraman, J. M. Zen, Electroanalysis
2008, 20, 1167.[22] A. Salimi, M. Roushani, Electroanalysis 2006, 18, 2129.[23] M. K. Amini, J. H. Khorasani, S. S. Khaloo, S. Tangestanine-
jad, Anal. Biochem. 2003, 320, 32.[24] A. Salimi, S. Pourbayram, Talanta 2003, 60, 205.[25] A. Salimi, R. Hallaj, M. K. Amini, Anal. Chim. Acta 2005,
534, 335.
Electroanalysis 2013, 25, No. & , 1 – 10 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.electroanalysis.wiley-vch.de &9&
These are not the final page numbers! ��
Fabrication of an Electrochemical l-Cysteine Sensor

[26] W. Y. Tao, Y. J. Liu, D. W. Pan, L. H. Nie, S. Z. Yao, Bioelec-trochemistry 2004, 65, 51.
[27] O. Nekrassova, N. S. Lawrence, R. G. Compton, Electroanal-ysis 2003, 15, 1655.
[28] L. Ding, H. Ju, J. Mater. Chem .2011, 21, 18154.[29] C. Hu, D. P. Yang, K. Xu, H. Cao, B. Wu, D. Cui, N. Jia,
Anal. Chem. 2012, 84, 10324.[30] D. Li, M. B. Muller, S. Gilje, R. B. Kaner, G. G. Wallace,
Nat. Nanotechnol. 2008, 3, 101.[31] M. I. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An, R. S. Ruoff,
Nano Lett. 2008, 8, 3498.[32] X. Wang, L. J. Zhi, K. Mullen, Nano Lett. 2008,8, 323.[33] A. Navaee, A. Salimi, H. Teymourian, Biosens. Bioelectron.
2012, 31, 205.[34] T. J. Davies, M. E. Hyde, R. G. Compton, Angew. Chem.
2005, 117, 5251.[35] A. Ambrosi, A. Bonanni, M. Pumera, Nanoscale 2011, 3,
2256.[36] M. Pumera, Chem. Rec.2009, 9, 211.[37] S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohl-
haas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen,R. S. Ruoff, Nature 2006, 442, 282.
[38] S. Bai, X. Shen, RSC Advances 2012, 2, 64.[39] W. He, H. Jiang, Y. Zhou, S. Yang, X. Xue, X. Zhang, D. L.
Akins, H. Yang, Z. Zou, Carbon 2012, 50, 265.[40] Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P.
Chen, G. M. Zhou, F. Li, H. M. Cheng, ACS Nano 2010, 4,3187.
[41] A. Salimi, H. Teymourian, S. Khezrian, Biosens. Bioelectron.2013, 49, 1.
[42] H. L. Wang, L. F. Cui, Y. A. Yang, H. S. Casalongue, J. T.Robinson, Y. Y. Liang, Y. Cui, H. J. Dai, J. Am. Chem. Soc.2010, 132, 13978.
[43] J. Zhang, W. Chu, J. Jiang, X. S. Zhao, Nanotechnology2011, 22, 125703.
[44] W. Xiao, D. Wang, X. W. Lou, J. Phys. Chem. C 2010, 114,1694.
[45] X. L. Luo, J. J. Xu, W. Zhao, H. Y. Chen, Biosens. Bioelec-tron. 2004, 19, 1295.
[46] S. Thiagarajan, T. Hsuan Tsai, S. M. Chen, Int. J. Electro-chem. Sci. 2011, 6, 2235.
[47] M. S. El-Deab, Int. J. Electrochem. Sci. 2009, 4, 1329.[48] S. Ling, R. Yuan, Y. Chai, T. Zhang, Bioproc. Biosyst. Eng.
2009, 32, 407.[49] F. Cheng, Y. Su, J. Liang, Z. Tao, J. Chen, Chem. Mater.
2010, 22, 898.[50] K. Wang, J. J. Xu, H. Y. Chen, Sens. Actuators B 2006, 114,
1052.[51] W. S. Hummers Jr., R. E. Offeman, J. Am. Chem. Soc. 1958,
80, 1339.[52] G. Wang, X. Shen, B. Wang, J. Yao, J. Park, Carbon 2009,
47, 1359.[53] D. R. Dreyer, S. Park, C. W. Bielawski, R. S. Ruoff, Chem.
Soc. Rev. 2010, 39, 228.[54] N. Cherchoura, C. Deslouis, B. Messaoudi, A. Pailleret,
Electrochim. Acta 2011, 56, 9746.[55] Z. Rogulski, H. Siwek, I. Paleska, A. Czerwinski, J. Elec-
troanal. Chem. 2003, 543, 175.[56] F. Tuinstra, J. L. Koenig, J. Chem. Phys. 1970, 53, 1126.[57] N. A. Kotov, I. Dekany, J. H. Fendler, Adv. Mater. 1996, 8,
637.[58] S. Alwarappan, A. Erdem, C. Liu, C.-Z Li, J. Phys. Chem. C
2009, 113, 8853.[59] Z. Rogulski, H. Siwek, I. Paleska, A. Czerwinski, J. Elec-
troanal. Chem. 2003, 543, 175.[60] E. Laviron, J. Electroanal. Chem. 1974, 52, 355.[61] J. Wang, Analytical Electrochemistry, VCH, New York 1994.[62] Y-H. Bai, H. Zhang, J-J. Xu, H-Y. Chen, J. Phys. Chem. C
2008, 112, 18984.[63] S. S. Khaloo, M. K. Amini, S. Tangestaninejad, S. Shahro-
khian, R. Kia, J. Iran. Chem. Soc. 2004, 1, 128.[64] M. Zhou, J. Ding, L. P. Guo, Q. K. Shang, Anal. Chem.
2007, 79, 5328.[65] F. A. Cotton, G. Wilkinson, Advanced Inorganic Chemistry,
2nd ed. 1967, p. 530.[66] C. P. Andriex, J. M. Saveant, J. Electroanal. Chem. 1978, 93,
163.
&10& www.electroanalysis.wiley-vch.de � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Electroanalysis 2013, 25, No. & , 1 – 10
�� These are not the final page numbers!
Full Paper S. Mansouri Majd et al.